
BACKGROUND
The complicated metabolic pathways of the human body present multiple opportunities for poisoning. Toxins can inhibit essential pathways in many ways, such as by blocking enzymes or overwhelming a normal pathway with toxic metabolites. Although there are many known toxins that affect various metabolic pathways, this chapter focuses on those that affect the mitochondria and cause metabolic acidosis, namely, aspirin, cyanide, methanol, and metformin.
ASPIRIN
Aspirin is a common over-the-counter medication used as a cardioprotective agent as well as a pain and fever reducer. Although it is used therapeutically to inhibit platelets and prostaglandins, in overdose, it has neurotoxic effects.
Aspirin is a weak acid with a pKa of 3.5, which means that in a solution with a pH of 3.5, 50% of the aspirin is in an ionized form. When the pH is lower than the pKa, more of the aspirin is in the nonionized form, which can move more freely through the lipid bilayer of cellular and subcellular membranes. The implication is that at the physiologic pH of tissues and blood, most aspirin is ionized and does not move easily between compartments. However, even at a pH of 7.4, a small amount of aspirin is nonionized (0.004%) and can travel into the brain, and this percentage increases with acidemia.1 In experimental models, lowering the blood pH produces a shift of salicylate into the tissues, especially the brain2; increasing the blood pH with sodium bicarbonate produces a shift in salicylate out of the tissues and into the blood.2–4 This is a key concept used to manage patients with aspirin toxicity.
In the mitochondria, ionized aspirin binds the hydrogen ions trapped in the intermembrane space and then exits the mitochondria in the nonionized form, preventing the proton motive force from fueling ATP formation (by uncoupling oxidative phosphorylation). Heat, but not energy, is therefore generated, and a low-grade temperature can be seen in patients with significant aspirin toxicity. The brain depends on ATP to pump water out of neurons, and in the absence of ATP, loss of oxidative phosphorylation leads to cerebral edema.
Therapeutic salicylate serum concentrations typically range from 15 to 30 mg/dL. At a serum concentration of approximately 35 mg/dL, aspirin stimulates the brainstem's respiratory center, causing hyperventilation that produces respiratory alkalosis. Some of the earliest signs of aspirin intoxication are tachypnea and hyperpnea; the physical exam of a suspected aspirin toxic patient should include careful attention to the rate and depth of breathing. As a weak acid, aspirin itself causes an anion gap metabolic acidosis at supratherapeutic concentrations. In addition, because of aspirin's ability to impair aerobic metabolism and increase fatty acid metabolism, other organic acids and ketoacids accumulate. The classic blood gas in patients with aspirin toxicity, or salicylism shows a mixed process of respiratory alkalosis with a metabolic acidosis (as opposed to a respiratory compensation for a metabolic acidosis). This acid–base pattern may not be as evident in pediatric exposures, because of their limited ventilatory reserve. Adults who show a “normalization” of their serum pH due to a decreasing respiratory alkalosis are extremely concerning, as this is a sign that respiratory fatigue or lung injury is preventing proper ventilation. Patients with acidemia late in the course of salicylate toxicity are at high risk of permanent neurologic damage and death, as the lowered pH will allow more aspirin to enter the brain.
Tinnitus or the sensation of hearing loss can occur early in toxicity. In the era before immune-modulating drugs, patients with rheumatologic disease were often instructed to titrate their aspirin doses to just below the amount that induced tinnitus.
The most concerning and consequential sign of toxicity is alteration in mental status. This is a sign of cerebral edema and an indication for more aggressive management such as dialysis. Seizures are often a preterminal event. Neuroglycopenia may occur and cause mental status abnormalities even when the serum glucose is within normal limits. The glucose concentration in the cerebral spinal fluid is depleted as salicylate-poisoned neurons utilize more glucose to compensate for the loss of ATP due to uncoupling of oxidative phosphorylation.5
The signs and symptoms of chronic salicylism may be more difficult to appreciate, which can delay detection. Furthermore, chronic salicylism is often found in elderly patients, in whom altered mental status, tachypnea, tachycardia, and mild anion gap acidosis may be misinterpreted as resulting from infection, malnourishment, cardiopulmonary disease, or dementia. In one study of 73 consecutive adults hospitalized with salicylate poisoning, 27% were not correctly diagnosed for as long as 72 hours after admission, and the mortality rate associated with this delayed diagnosis was 25%. Many of these patients had neurology consults for altered mental status prior to correct diagnosis.6
Aspirin toxicity can increase pulmonary capillary permeability, leading to acute respiratory distress syndrome (ARDS). This pulmonary toxicity can impair ventilation, reducing the respiratory alkalosis and worsening the clinical effects of salicylate toxicity. It can also limit the ability to use sodium bicarbonate infusion as a therapy, since patients with ARDS may not be able to tolerate the fluid load.
Salicylate-poisoned patients can decompensate rapidly if not carefully managed. The hyperpnea, tachypnea, and diaphoresis associated with aspirin toxicity cause a large amount of insensible losses, so fluid status should be monitored and repletion with normal saline should be initiated on arrival. Every patient with suspected salicylate poisoning requires testing of serum salicylate concentration, and repeat levels are needed if the initial concentration is elevated. The frequency of retesting should be based on clinical findings and trends in serum concentration. Additional laboratory testing should include a blood gas (arterial or venous) to monitor pH and pCO2, serum potassium, serum acetaminophen concentration (in patients with toxicity thought to be due to self-harm), and urine pH every 1 to 2 hours (see Alkalinization below). Given the acid/base physiology of aspirin, a “normal” pH and pCO2 are not reassuring.
Activated charcoal should be administered unless there is a concern for aspiration in a vomiting or altered patient. Whole bowel irrigation should be avoided as it may solubilize an aspirin bezoar and facilitate absorption. If there is concern for a bezoar or the serum salicylate level has plateaued despite alkalinization, multiple doses of activated charcoal may be indicated.
There is no specific antidote for salicylism, but alkalinization is a mainstay of treatment. Alkalinization of the serum promotes trapping of the ionized salicylate outside the brain. Furthermore, alkaline urine promotes elimination of salicylate; and this effect increases logarithmically as the pH of urine increases from five to eight.7 There is no specific uptake mechanism in the kidney for salicylate, and passive reabsorption of charged molecules is very limited. Under therapeutic conditions, approximately 10% of salicylates are excreted in the urine as salicylic acid, while the majority of the salicylate is metabolized into a conjugated form in the liver prior to renal elimination. In overdose, the enzymes involved in hepatic metabolism become saturated, and the amount of urinary unconjugated salicylic acid increases. Urine alkalinization does not affect elimination of conjugated salicylate, so serum clearance is less affected. To alkalinize the serum and the urine, sodium bicarbonate is typically dosed as a 1 to 2 mEq/kg bolus, followed by 150 mEq mixed in D5 water at twice maintenance. Titrate with goal of serum pH around 7.5 to 7.55 and urine pH around 8.
Alkalinization will cause potassium to shift intracellularly in order to release hydrogen ions into the serum. The kidneys sense this relative hypokalemia and will begin to reabsorb potassium in exchange for hydrogen ions, preventing proper urine alkalinization despite the sodium bicarbonate infusion. Serum potassium should be repleted to normal levels in order to suppress this physiologic response and maintain urinary alkalinization.
Although many patients with altered mental status are intubated for airway protection, in the case of salicylism, inadequate matching of hyperventilation and subsequent CO2 retention can be catastrophic because the resulting acidemia will shift more salicylate into the brain.8 Sedation should be avoided unless the patient is carefully monitored or receiving ventilatory support. In patients receiving mechanical ventilation, every effort should be made to prevent a falling pH and rising PCO2, which may be done by matching the ventilator settings with the patient's pre-intubation minute ventilation. An experienced operator should perform the intubation, and sodium bicarbonate, 1 to 2 mEq/kg bolus, should be given just prior to intubation to ensure alkalemia during rapid sequence intubation. Patients will require large tidal volumes and a high respiratory rate with the goal of a minute ventilation of 20 to 30 L/min. Despite these steps, patients still may be unable to maintain an appropriate serum pH or may suffer ventilator-associated barotrauma, in which case hemodialysis is indicated.
Although some resources and textbooks cite a serum aspirin concentration >100 mg/dL as an absolute indication for dialysis, this does not mean that a patient may not need extracorporeal elimination at lower concentrations. Hemodialysis is indicated when there are signs of end-organ injury or when pulmonary edema and lung injury prevents further use of sodium bicarbonate. This is particularly true in the case of the patient with altered mental status, as serum concentrations may underestimate central nervous system (CNS) concentrations.
CYANIDE
Cyanide is a chemical asphyxiant. It is most commonly encountered clinically in patients who were victims of fires, especially involving the combustion of fabrics and plastics. However, cyanide should be on the differential of a sudden death in an otherwise healthy person because it is such a fast-acting, lethal, and potentially treatable toxin. Cyanide salts such as sodium cyanide and potassium cyanide are used in jewelry making, plastic manufacturing, photography, and other industries. They react with water to form hydrogen cyanide, a gas. Organic compounds containing cyanide also exist. Acetonitrile is methyl cyanide and is commonly found in acrylic nail glue remover and other similar cosmetics. When ingested, it is metabolized by the P450 system to hydrogen cyanide and formaldehyde, causing delayed toxicity.
Iatrogenic cyanide poisoning can occur when nitroprusside is used for the treatment of hypertension. Each nitroprusside molecule contains five cyanide molecules, which may be liberated. After rapid or prolonged infusion, or in malnourished patients, cyanide or its metabolite (thiocyanate) toxicity may occur.
Acute cyanide toxicity can occur via inhalational, oral, dermal, and parenteral routes. The dose of cyanide required to produce toxicity is dependent on the form of cyanide and the duration and route of exposure. Hydrogen cyanide gas at concentrations above 270 ppm can be immediately fatal, and ingestion of 200 mg of KCN salt can be fatal within minutes.9 Cyanide is a very potent toxin and is on a short list of rapidly acting, fatal exposures.
Cyanide is eliminated from the body by multiple pathways. The major route is the enzymatic conversion to thiocyanate by rhodanese (thiosulfate–cyanide sulfurtransferase). This enzyme catalyzes the transfer of a sulfur group from a sulfur donor, such as thiosulfate, to cyanide to form thiocyanate. In acute poisoning, the ability of rhodanese to detoxify cyanide is limited by the endogenous amount of sulfur donor, which is rapidly depleted. Thiocyanate has relatively little inherent toxicity and is eliminated in the urine.
Cyanide inhibits many enzymes, but its most consequential effect is the inhibition of cytochrome oxidase in the mitochondria. Cytochrome oxidase is a key enzyme of the electron transport chain, and oxidative phosphorylation cannot occur without it. Cyanide acts at the cytochrome a3 portion of complex IV of the electron transport chain. As a result, hydrogen ions cannot combine with oxygen to form water, ATP cannot be generated, and oxygen utilization by the tissues is decreased. Cellular asphyxiation occurs despite normal blood oxygen tension, and the excess hydrogen ions cause acidemia. Lactate accumulates because the cessation of the electron transport chain prevents the conversion of nicotinamide adenine dinucleotide (NADH) back into NAD+ and H+, and this favors the conversion of pyruvate to lactate.
Cyanide toxicity can cause rapid and severe neurologic dysfunction and hemodynamic instability. When organic cyanogenic compounds such as acetonitrile are ingested, however, symptoms may be delayed for hours because the parent compound must be metabolized to release cyanide. Cyanide toxicity from a nitroprusside infusion may take hours to days to become clinically apparent.
CNS signs and symptoms of cyanide toxicity are typical of those associated with progressive hypoxia and include headache, anxiety, agitation, confusion, lethargy, seizures, and coma. Centrally mediated tachypnea occurs initially and is followed by bradypnea. Cardiovascular signs can vary early in the clinical course, but bradycardia and hypotension are usually the preterminal findings.
Cyanide victims have classically been described as having cherry red skin coloration, due to increased oxygenation of the venous blood. Cyanide does not typically cause cyanosis despite the similar-sounding names. The word cyanide is derived from the Greek word for blue kyanos, due to its liberation from Prussian blue (ferric hexacyanoferrate) upon heating.
Clinicians should have high suspicion for cyanide poisoning in hemodynamically unstable or comatose fire victims, industrial or laboratory workers with sudden collapse, and suicidal patients with rapid collapse and metabolic acidosis following ingestion. Cyanide toxicity should also be considered a potential diagnosis in patients on nitroprusside infusions that develop altered mental status, metabolic acidosis, and abnormal vital signs.
Nitroprusside is a nitric oxide–releasing drug and is used as a vasodilator. The standard infusion rate is 3 mcg/kg/min (0.25 mcg/kg/min to 10 mcg/kg/min). The nitroprusside molecule contains five cyanide radicals that are slowly liberated and rapidly metabolized to thiocyanate. In healthy individuals, cyanide detoxification occurs at a rate of about 1 g/kg/min, which corresponds to a sodium nitroprusside infusion rate of 2 g/kg/min.10 However, critical illness and malnutrition can deplete sulfur stores, so ICU patients are at increased risk for cyanide toxicity. An infusion of nitroprusside at a rate of more than 15 mcg/kg/min administered over a few hours or more than 4 mcg/kg/min for more than 12 hours may overwhelm the capacity of rhodanese for detoxifying cyanide.11
Sodium thiosulfate is sometimes coadministered with nitroprusside in order to prevent cyanide toxicity. Dosing of 1 g sodium thiosulfate for every 100 mg of nitroprusside is typically sufficient to prevent cyanide accumulation.11 However, it is important to note that thiocyanate is renally eliminated and may accumulate in patients with impaired renal function, causing toxicity. The symptoms of thiocyanate toxicity are nonspecific and may include nausea, vomiting, fatigue, dizziness, confusion, delirium, and seizures. Extremely elevated thiocyanate concentrations (>200 g/mL) may produce life-threatening effects, such as hypertension and intracranial pressure elevation. Anion gap metabolic acidosis does not occur with thiocyanate toxicity. Hemodialysis clears thiocyanate from the serum and should be strongly considered in patients with severe clinical manifestations of thiocyanate toxicity.
Laboratory testing for cyanide is not readily available in most clinical settings. In general, the patient's history and physical exam and other ancillary testing will guide management. Expected laboratory findings include an anion gap metabolic acidosis, an elevated lactate concentration, and an elevated venous oxygen saturation.12 However, none of these findings are specific for cyanide. Other metabolic inhibitors such as carbon monoxide, hydrogen sulfide, and sodium azide, as well as medical conditions such as sepsis, high-output cardiac syndromes, and left-to-right intracardiac shunts, can reduce oxygen extraction. Simultaneous arterial and venous blood gases may show a reduced difference in arterial and venous oxygenation saturation (<10 mm Hg).13
A significant association exists between blood cyanide and serum lactate concentrations. In a small group of patients with a strongly suggestive history of cyanide ingestion, a serum lactate concentration above 8 mmol/L was associated with sensitivity of 94%, specificity of 70%, positive predictive value of 64%, and negative predictive value of 98% for a blood cyanide concentration above 1.0 g/mL, which is a toxic concentration.14 In a case–control study of fire victims, a lactate over 10 mmol/L was a sensitive indicator of cyanide intoxication.15
Since cyanide can be so rapidly fatal, there is a limited window to initiate resuscitation. In most cases, empiric administration of antidotes will be required, based on history and clinical appearance. Resuscitation with a focus on airway, breathing, and circulation is the mainstay of treatment, but timely administration of the antidotes is paramount. Patients should be given 100% oxygen. Patients with altered mental status or fire victims with signs of oropharyngeal burns may require intubation in order to protect the airway. Vasopressors may be required to treat persistent hypotension despite adequate intravenous volume resuscitation.
Although some in vitro studies suggest that cyanide does not have significant adsorption to activated charcoal, it remains reasonable to administer charcoal to a patient with a protected airway in the setting of potentially toxic ingestion.
Either hydroxycobalamin or a cyanide antidote kit should be administered as soon as cyanide poisoning is suspected. Hydroxycobalamin, a vitamin B12 precursor, directly binds cyanide (1:1) to form cyanocobalamin (vitamin B12). Hydroxycobalamin has few adverse effects, including a reddish discoloration of the skin, mucous membranes, and urine that can last a few days.16 Colorimetric laboratory testing can be affected by the red color, and common lab tests, such as serum lactate, may yield inaccurate results. For this reason, blood specimens should be taken for laboratory analysis prior to administering hydroxycobalamin. The package insert lists the lab tests commonly affected and for how long the interference can last. Adult dosing for hydroxycobalamin is 5 g administered as an IV infusion over 15 minutes. Depending on the severity of the poisoning and the clinical response, an additional 5 g may be administered (total dose of 10 g).
The cyanide antidote kit contains three components: amyl nitrite, sodium nitrite, and sodium thiosulfate. Both thiosulfate and nitrite have antidotal efficacy when given alone in animal models of cyanide poisoning, but they have even greater benefit when they are given in combination.17 Thiosulfate donates the sulfur atoms necessary for rhodanese to convert cyanide to thiocyanate. The nitrites generate methemoglobin, which cyanide binds preferentially over cytochrome a3, leading to improved cytochrome oxidase function. Amyl nitrite is contained within glass pearls that are crushed and intermittently inhaled or introduced into the ventilator. IV sodium nitrite is preferred, and the use of amyl nitrite pearls is reserved for cases in which IV access is delayed or not possible. It is important to note that standard testing for methemoglobin does not detect cyanomethemoglobin. Therefore, it may be difficult to define the optimal methemoglobin concentration to bind cyanide without causing further hypoxia. In addition to excessive methemoglobin formation, other adverse effects of nitrites include hypotension and tachycardia because of its vasodilatory effects. Avoiding rapid infusion, monitoring blood pressure, and adhering to dosing guidelines limit adverse effects.
Sodium thiosulfate is the second component of the cyanide antidote kit, and it works synergistically with both nitrites and hydroxycobalamin in the detoxification of cyanide. Because sodium thiosulfate does not cause methemoglobinemia, it can be used without nitrites in circumstances when the creation of methemoglobinemia would be concerning, such as in patients with high carboxyhemoglobin concentrations. (Adult dosing for sodium thiosulfate: 12.5 g IV over 10 to 30 minutes, adult dosing for amyl nitrite [only if no IV access]: break one ampule in front of mouth and hold for 15 seconds, remove for 15 seconds and repeat as needed until sodium nitrate infusion is begun [if needed]. Adult dosing for sodium nitrite [NaNO2] 3% [30 mg/mL]: 10 mL [300 mg] IV over 2 to 4 minutes.)
METHANOL
Methanol is a common industrial and household product. It can be found in windshield washer fluid, cooking fuel gels for camping and buffet platters (Sterno), gas line antifreeze, photocopier ink, and perfumes. Large outbreaks occur when improper fermentation occurs in illegal ethanol production. Management is often complicated by the inability to obtain serum concentrations in a timely manner.
Methanol is rapidly absorbed when ingested. It can also be inhaled and dermally absorbed, but these latter routes are uncommon. Although methanol is not eliminated renally, it can be exhaled—a slow exit route that explains the elimination half-life of almost 30 hours.
Methanol is slowly metabolized to formate through successive oxidation by alcohol dehydrogenase (ADH) and aldehyde dehydrogenase, each of which is coupled to the reduction of NAD+ to NADH and H+.18 Formate is a mitochondrial toxin that inhibits cytochrome oxidase, interfering with oxidative phosphorylation. The retinal epithelium and optic nerve are especially sensitive to methanol, and affected patients may experience visual impairment ranging from blurry or hazy vision to “snowfield vision” or total blindness.
Toxic alcohols, such as ethylene glycol and methanol, are on the differential diagnosis for anion gap metabolic acidosis. Like ethanol, methanol can cause inebriation, but animal studies suggest that its lower molecular weight makes it less inebriating than other alcohols.19 Methanol levels of 25 to 50 mg/dL can be toxic, but may not be high enough to cause inebriation, especially in an ethanol-tolerant individual. Lack of inebriation, however, should not be used to rule out toxicity.
The basal ganglia are also uniquely sensitive to formate. Methanol is on a short list of toxins that can cause isolated basal ganglia lesions on CT and MRI.20 In a comatose patient with an acidemia, isolated basal ganglia infarcts may point to a methanol exposure. In one series, typical radiologic lesions were present in six of nine cases. Other CNS lesions reported include necrosis of the corpus callosum and intracranial hemorrhage.21
Local laboratory capabilities greatly affect management of methanol-intoxicated patients. If methanol concentrations can be determined within a clinically reasonable time frame (e.g., within the same day), management and disposition is straightforward. In institutions in which report of serum concentrations takes days to return, physicians often use surrogate tests, which may have significant limitations, to stratify patients.
Methanol and other toxic alcohols are often first considered in the differential of a patient with an unexplained anion gap metabolic acidosis. To help exclude other diagnoses, a serum lactate concentration, serum or urine ketones, salicylate concentration, ethanol concentration, and renal function tests should be assessed. Traditionally, toxic alcohols cause an anion gap acidosis with normal lactate and negative ketones. Once a toxic alcohol is considered on the differential diagnosis, serum concentrations should be sent. Even if the result will not return for days, it can still help guide management.
Often, the diagnostic dilemma in an alcohol user is distinguishing toxic alcohol poisoning from alcoholic ketoacidosis (AKA), since in both cases, an anion gap metabolic acidosis is present. One means of identifying AKA is to note an improvement in the anion gap after administration of intravenous fluids, thiamine, and glucose.
Checking an osmol gap historically has been considered an essential part of the evaluation of a potentially toxic alcohol-poisoned patient. However, there are several critical limitations to the test. The osmol gap is defined as the difference between the values for the measured osmolality and the calculated osmolarity. The formula to calculate osmolarity is as follows:
It is helpful to account for any ethanol in the osmolarity because it may explain the presence of unaccounted osmols:
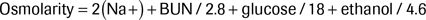
In methanol (or any alcohol) poisoning, the toxic molecule has osmotic activity that is measured but not calculated, which creates the osmol gap. The anion gap does not increase until methanol, for example, is metabolized to formate. Although the formate metabolite also has osmotic activity, its activity is accounted for by the sodium ion in the osmolarity calculation because it exists as dissociated sodium formate in solution. As a result, there will be an elevated osmol gap and a normal anion gap initially after the exposure; but as time progresses, the anion gap will increase and the osmol gap will decrease.
A normal osmol gap is approximately −2 ± 6; the range to account for 95% of patient populations is −10 to +14.22 Note that a normal gap measurement can be misleading, since the patient's baseline osmol gap is unknown. For example, a patient with a normal osmol gap of −5, who presents with an osmol gap of 9, has in reality an osmol gap of 14. Since a patient's baseline osmol gap is unknown, it is impossible to know whether the calculated gap during their initial presentation is elevated or not.
Finally, although large osmol gaps may be suggestive of toxic alcohol ingestions, common conditions such as alcoholic ketoacidosis, lactic acidosis, renal failure, and shock are all associated with elevated osmol gaps. ICU patients, regardless of the underlying diagnosis, often have osmol gaps near 20. As a result, a normal osmol gap cannot safely exclude a toxic alcohol exposure, and a mildly elevated one is not specific enough for confirmation. However, a very high osmol gap (>30 mOsm/L) is very suggestive of toxic alcohol poisoning.
Blocking ADH is the mainstay of treatment in methanol poisoning, since this prevents the production of formate, the toxic metabolite.18 Blocking ADH can be achieved by the administration of either ethanol or fomepizole. ADH has greater affinity for ethanol than for methanol, and complete blockade occurs with serum ethanol concentrations of approximately 100 mg/dL. Fomepizole is a competitive inhibitor of ADH. Intravenous ethanol is no longer readily available in the United States, but in extreme cases when no other antidote is available and dialysis is delayed, oral ethanol may be used.
Traditionally, intravenous ethanol was the antidote of choice, but its use necessitated an ICU bed, and its administration was complicated by mental status changes, potential loss of airway, and electrolyte changes. The goal of either oral or intravenous administration of ethanol is a serum concentration of 100 mg/dL, which can be inebriating to those without any tolerance to ethanol. Fomepizole is not associated with the mental status or electrolyte changes commonly seen with ethanol administration, and its use may not require an ICU admission.23 Fomepizole is given as a loading dose of 15 mg/kg, followed by doses of 10 mg/kg every 12 hours for four doses. Importantly, this dosing regimen of fomepizole is based on the pharmacokinetics of ethylene glycol, not methanol. The main limitation of fomepizole is that the half-life of methanol, once ADH is blocked, reaches 50 hours. Methanol is not renally eliminated, but rather is eliminated via exhalation. As a result, a patient may require a week-long course of fomepizole, which can be expensive and require complicated dosing regimens. Fomepizole induces its own metabolism after 48 hours of use by activating the CYP 450 enzyme 2E1. As a result, higher doses (15 mg/kg) may be required when using beyond 48 hours.24 In this instance, hemodialysis may be a preferred method of treatment.
Hemodialysis is indicated in patients with severe acidemia, signs of end-organ injury such as coma or renal failure, and those with methanol concentrations >50 mg/dL. Hemodialysis can clear toxic alcohols and their metabolites and correct any acid–base disturbances. A nephrology consult should be obtained early in the clinical course of any toxic alcohol patient so that that the proper resources can be obtained in a timely manner if needed. In cases of large ingestions resulting in high concentrations of methanol or ethylene glycol, multiple rounds of hemodialysis as well as administration of fomepizole in between sessions may be indicated. Patients should be monitored for recurrent acidosis, abnormal vision changes, and renal failure (in cases of ethylene glycol poisoning) post-dialysis.
Folate should be administered to any patient with suspected methanol toxicity. Folate is an inexpensive, water-soluble vitamin with minimal associated adverse reactions. Folinic acid (leucovorin) has also been shown to be effective. Animal models show that folic acid and folinic acid enhance formate elimination.25 Scant human case reports also suggest a benefit. Formate is bound by tetrahydrofolate and then undergoes metabolism by 10-formyltetrahydrofolate dehydrogenase to carbon dioxide and water.
METFORMIN
Metformin is an oral antihyperglycemic agent commonly used to treat diabetes mellitus. Its mechanism of action is inhibition of gluconeogenesis and decreased hepatic glucose production. However, it also enhances peripheral glucose uptake by the GLUT transporters in muscle and adipose cells. Metformin overdose should not cause hypoglycemia unless the patient has increased metabolic demands from being critically ill. The most concerning toxicity involves hyperlactemia and metabolic acidosis, commonly referred to as MALA—metformin-associated lactic acidosis.
It is possible for MALA to develop after a single acute overdose of metformin.26 More commonly, MALA occurs in patients who are therapeutically on metformin and develop renal impairment. Patients who have unintentional metformin intoxication do poorly compared to those with intentional metformin overdose.27 This may be due to a delay to diagnosis, inciting medical illness-causing tissue hypoxia or renal failure, or other comorbidities. Patients who are managed on metformin are advised to hold their medication for 72 hours following the administration of iodinated contrast to prevent MALA; however, some authors argue that only diabetics with impaired renal function prior to receiving IV contrast are at risk.28
Recent animal and in vitro studies show that metformin is a mitochondrial toxin. Metformin decreases lactate uptake and consumption in the hepatocyte. However, metformin also decreases global oxygen consumption and causes mitochondrial dysfunction in nonhepatic tissues as well.29,30
The diagnosis of MALA is controversial. The Cochrane Review disputes its existence, but that is likely because any data regarding MALA are derived from case reports and case series rather than randomized controlled trials.31 Randomized controlled trials of metformin exclude patients with kidney disease and are not assessing for the effects of overdose, so the incidence of MALA in those trials is essentially nonexistent. Based on case reports, case series, and animal models, evidence is overwhelmingly supportive of the existence of MALA, and it should be considered in patients with anion gap metabolic acidosis and elevated lactate concentrations.
MALA can be a fatal, but easily missed, diagnosis. Initial symptoms—which include nausea, lethargy, vomiting, and abdominal pain—can be nonspecific. Careful history should assess for etiology of renal impairment such as dehydration, recent infection, new medication, or a recent IV contrast study. Patients can develop a severe metabolic acidosis and multiorgan dysfunction.
Although a sodium bicarbonate infusion may be indicated in patients who have a serum bicarbonate concentration <5 mEq/L, it will likely be insufficient to correct the acid–base abnormalities associated with MALA. Hemodialysis is the mainstay of treatment in those with severe acidemia. Hemodialysis does not effectively remove metformin, but it corrects the acid–base disorder and possibly the renal complications.
CONCLUSION
Mitochondrial toxins can cause severe disruptions in oxidative phosphorylation and ultimately lead to multiorgan failure and death. Initial symptoms are often nonspecific and can be easily overlooked for nontoxicologic etiologies. However, any patient with an anion gap metabolic acidosis, elevated lactate, or suspicious history should be rapidly evaluated for these toxins with judicious use of ancillary testing and antidotes.
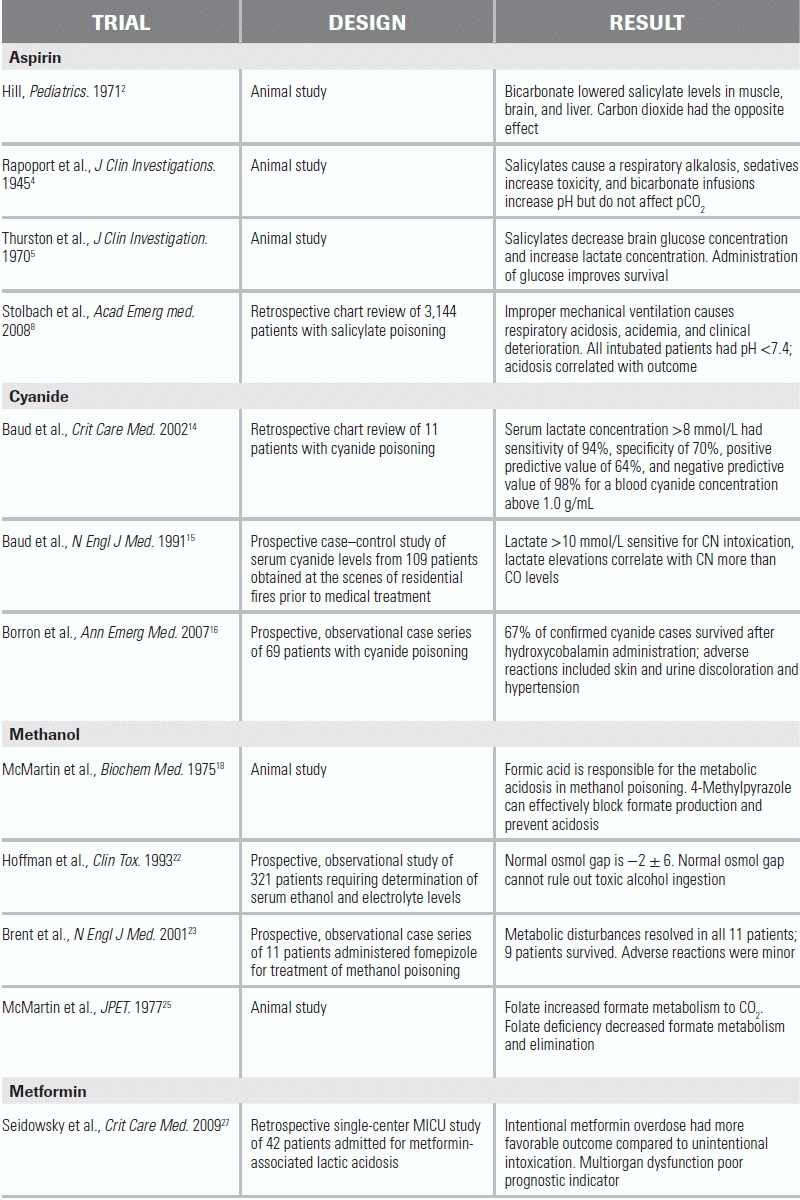
LITERATURE TABLE

1.Flomenbaum NE. Salicylates. In: Nelson LS, Howland MA, Hoffman RS, et al., eds. Goldfrank's Toxicologic Emergencies. 9th ed. New York: McGraw-Hill; 2011.
2.Hill JB. Experimental salicylate poisoning: observations on the effects of altering blood pH on tissue and plasma salicylate concentrations. Pediatrics. 1971;47(4):658–665.
3.Hill JB. Salicylate intoxication. N Engl J Med. 1973;288(21):1110–1113.
4.Rapoport S, Guest GM. The effect of salicylates on the electrolyte structure of the blood plasma. I. Respiratory alkalosis in monkeys and dogs after sodium and methyl salicylate; the influence of hypnotic drugs and of sodium bicarbonate on salicylate poisoning. J Clin Invest. 1945;24(5):759–769.
5.Thurston JH, Pollock PG, Warren SK, et al. Reduced brain glucose with normal plasma glucose in salicylate poisoning. J Clin Invest. 1970;49(11):2139–2145.
6.Anderson RJ, Potts DE, Gabow PA, et al. Unrecognized adult salicylate intoxication. Ann Intern Med. 1976;85(6):745–748.
7.Kallen RJ, Zaltzman S, Coe FL, et al. Hemodialysis in children: technique, kinetic aspects related to varying body size, and application to salicylate intoxication, acute renal failure and some other disorders. Medicine. 1966;45(1):1–50.
8.Stolbach AI, Hoffman RS, Nelson LS. Mechanical ventilation was associated with acidemia in a case series of salicylate-poisoned patients. Acad Emerg Med. 2008;15(9):866–869.
9.Kirk MA HC, Isom GE. Cyanide. In: Nelson LS, Howland MA, Hoffman RS, et al., eds. Goldfrank's Toxicologic Emergencies. 9th ed. New York: McGraw-Hill; 2011.
10.Schulz V. Clinical pharmacokinetics of nitroprusside, cyanide, thiosulphate and thiocyanate. Clin Pharmacokinet. 1984;9:239–251.
11.Rindone JP, Sloane EP. Cyanide toxicity from sodium nitroprusside: risks and management. Ann Pharmacother. 1992;26:515–519.
12.Johnson RP, Mellors JW. Arteriolization of venous blood gases: a clue to the diagnosis of cyanide poisoning. J Emerg Med. 1988;6(5):401–404.
13.Nelson L. Acute cyanide toxicity: mechanisms and manifestations. J Emerg Nurs. 2006;32(4 Suppl):S8–S11.
14.Baud FJ, Borron SW, Megarbane B, et al. Value of lactic acidosis in the assessment of the severity of acute cyanide poisoning. Crit Care Med. 2002;30(9):2044–2050.
15.Baud FJ, Barriot P, Toffis V, et al. Elevated blood cyanide concentrations in victims of smoke inhalation. N Engl J Med. 1991;325(25):1761–1766.
16.Borron SW, Baud FJ, Barriot P, et al. Prospective study of hydroxocobalamin for acute cyanide poisoning in smoke inhalation. Ann Emerg Med. 2007;49(6):794–801, 801 e791–e792.
17.Chen KK, Rose CL. Nitrite and thiosulfate therapy in cyanide poisoning. J Am Med Assoc. 1952;149(2):113–119.
18.McMartin KE, Makar AB, Martin G, et al. Methanol poisoning. I. The role of formic acid in the development of metabolic acidosis in the monkey and the reversal by 4-methylpyrazole. Biochem Med. 1975;13(4):319–333.
19.Wallgren H. Relative intoxicating effects on rats of ethyl, propyl and butyl alcohols. Acta pharmacol Toxicol. 1960;16:217–222.
20.Hantson P, Duprez T, Mahieu P. Neurotoxicity to the basal ganglia shown by magnetic resonance imaging (MRI) following poisoning by methanol and other substances. J Toxicol Clin Toxicol. 1997;35(2):151–161.
21.Sefidbakht S, Rasekhi AR, Kamali K, et al. Methanol poisoning: acute MR and CT findings in nine patients. Neuroradiology. 2007;49(5):427–435.
22.Hoffman RS, Smilkstein MJ, Howland MA, et al. Osmol gaps revisited: normal values and limitations. J Toxicol Clin Toxicol. 1993;31(1):81–93.
23.Brent J, McMartin K, Phillips S, et al. Fomepizole for the treatment of methanol poisoning. N Engl J Med. 2001;344(6):424–429.
24.Wiener SW. Toxic alcohols. In: Nelson LS, Lewin N, Howland MA, Hoffman RS, et al., eds. Goldfrank's Toxicologic Emergencies. 9th ed. New York: McGraw-Hill; 2011.
25.McMartin KE, Martin-Amat G, Makar AB, et al. Methanol poisoning. V. Role of formate metabolism in the monkey. J Pharmacol Exp Therap. 1977;201(3):564–572.
26.Teale KF, Devine A, Stewart H, et al. The management of metformin overdose. Anaesthesia. 1998;53(7):698–701.
27.Seidowsky A, Nseir S, Houdret N, et al. Metformin-associated lactic acidosis: a prognostic and therapeutic study. Crit Care Med. 2009;37(7):2191–2196.
28.Nawaz S, Cleveland T, Gaines PA, et al. Clinical risk associated with contrast angiography in metformin treated patients: a clinical review. Clin Radiol. 1998;53(5):342–344.
29.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–614.
30.Protti A, Fortunato F, Monti M, et al. Metformin overdose, but not lactic acidosis per se, inhibits oxygen consumption in pigs. Crit Care (London, England). 2012;16(3):R75.
31.Salpeter SR, Greyber E, Pasternak GA, et al. Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitus. Cochrane Database Syst Rev (Online). 2010(4):CD002967.