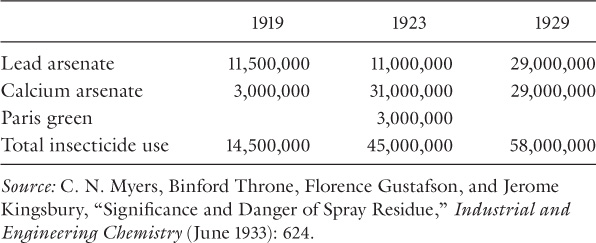![]()
In 1893 the city of Chicago announced to the world its arrival as a major metropolis through the Columbian Exposition. That same year, the British medical journal Lancet deployed a public health doctor to report on the state of the Chicago stockyards, which in terms of size and productivity were among the largest in the world. In the Chicago stockyards, slaughterhouses became industrial meat factories that rendered thousands of cattle into meat products to be transported via refrigerated railcar across the United States and shipped around the world. With the industrialization of meat production came spectacular demand, and the Chicago packinghouses refined meat production to the essence of efficiency. Nothing mattered save enhanced productivity and profit. Safeguards fell by the wayside, endangering workers and consumers alike.1 Technical reports failed to excite concern, but when Upton Sinclair published The Jungle in February of 1906, the conditions in the Chicago stockyards caught the attention of legislators, and even the president responded. Having languished in the form of several related bills for nearly two decades, the Pure Food and Drug Act (PFDA) was finally passed in June of 1906. Primarily directed at product labeling, the PFDA prohibited interstate transport of unlawful food and drugs.
The industrial principles of efficiency that infused the Chicago packing-houses extended to other dimensions of agriculture as well. Novel technologies facilitated the expansion of monoculture as the preferred and most profitable method, but for the threat of insect invasion. Paris green, and later lead arsenate, provided a technological fix to this problem. Adelynne Whitaker, historian of pesticide legislation, argued that agricultural chemists and entomologists during the early twentieth century were primarily concerned with “the economic aspects of adulterated and ineffective insecticides,” but “the scientists were not unaware of the public health implications of their work.”2 Just as the Chicago packers found an insatiable demand for their low-cost meat products, farmers found virtually unlimited demand for their produce. In both cases, industrializing modes of production resulted in previously un-imagined levels of productivity. As production became concentrated in Chicago and points farther west, Americans increasingly found themselves separated from the sources of their food. The PFDA and the Insecticide Act of 1910 reassured Americans that despite such detachment they could still expect a safe and healthy food supply. During the next three decades the foundations of such presumptions were shaken as Americans found their health and welfare threatened by pesticides and adulterated drugs. Yet it took a national tragedy to shake American consumers and regulators out of their complacency.
There is no question that the PFDA represented a watershed moment in the history of regulation in the United States. Yet legislators recognized that the act incorporated compromises that left American public health vulnerable to corporate deceit and malfeasance. By the 1930s, the president had called on Congress to revisit food and drug legislation and forge a law with greater power to protect Americans. As the House and the Senate debated health legislation, several crises accentuated the limitations of the 1906 law, notably its inability to prosecute abuses that led to injury and even death. In addition to legislators, a new generation of consumer advocates took up the cause, writing books and articles to alert Americans to the failings of existing legislation and to the flagrant violations that exposed them to significant risks. Among the many examples that failed to elicit regulation, the case of Jamaica ginger (“ginger jake”) paralysis, in which many thousands were poisoned after unwittingly consuming a highly toxic alternative to alcohol during Prohibition, was particularly egregious. The pesticide residues of arsenic and lead on fruit also failed to motivate consumers or legislators. Both ginger jake and pesticide residues received considerable coverage in the media (and in the books and articles of consumer advocates), but not until the Elixir Sulfanilamide tragedy, in which ninety-three individuals died after ingesting a contaminated drug, did legislators pass the Federal Food, Drug, and Cosmetic Act of 1938 (FFDCA), thereby revising the 1906 law.
The Elixir Sulfanilamide tragedy, similar incidents, and the legislation that followed gave toxicology new standards and food and drug laws increased leverage.3 Moreover, several scientists and regulators launched their lengthy careers with the study of Elixir Sulfanilamide. Through their work on Elixir Sulfanilamide they learned vital lessons and developed new approaches to toxicology that they would continue to apply to pharmaceuticals and other new chemicals, such as insecticides, throughout their careers in government, industry, and research universities. Such cases also affected popular perceptions of risk in America. In addition to an expectation that government would provide safety standards for pharmaceuticals, Americans were becoming accustomed to the use of powerful chemicals in medicine as therapies. Similarly, new technologies, including chemical insecticides, accelerated the industrialization of agriculture in America during the first decades of the twentieth century.
The scale of meatpacking drove intensification of production, but meat was not the only agricultural commodity that was industrialized during the last half of the nineteenth century. The agricultural revolution saw the introduction of new technology in form of steel plows, seed drills, cultivators, and reapers, which greatly reduced the need for a large labor force. Moreover, methods of crop rotation and the application of fertilizers significantly enlarged yields of many crops. Mechanization and fertilization meant that established farmers could plant extensive crops consisting of monocultures. Successful harvests could be spectacularly profitable. Nevertheless, monoculture left crops profoundly vulnerable to insect invasions, which could quickly bankrupt ambitious farmers. Historian James Whorton placed these agricultural developments in context: “The favorable insect environment created by monoculture was further enhanced in America by westward expansion. The fulfillment of Manifest Destiny not only involved an enormous increase in the area of land under cultivation but, also, by the prerequisite clearing of forests in many areas, frequently destroyed predators of insects while forcing the insects themselves to turn to a domestic food supply.”4 Even the novel technologies, such as railroads and trans-Atlantic ships, that facilitated a related revolution in transportation contributed to the problem of insect invasions by transferring the culprits around the country.
Farmers became desperate for effective means to control insect infestations. Economic entomologists, hoping to escape unfavorable stereo-types as ineffectual, disengaged scientists preoccupied with some of the smallest and most inconsequential members of the animal kingdom, answered farmers’ hopes. Particularly promising was an insecticide extracted from the pyrethrum flower, a chrysanthemum. Drying and crushing the stamens of the flowers produced a powerful insecticide. Pyrethrum, as the insecticide became known, was prohibitively expensive because farmers in the Caucasus guarded their monopoly on the plant.5
Economic entomologists sought a synthetic insecticide as effective as pyrethrum. In 1867, farmers received the answer to their prayers in the form of Paris green, a copper acetoarsenite. Some journalists warned against adding arsenic to agriculture, but farmers soon adopted Paris green to fight a range of insect pests. In the five years after its introduction, Paris green became “the ally of first resort whenever death must be dealt to any pest.”6 Paris green was popular with farmers because it was inexpensive and effective against a variety of insects. The wonder insecticide met its match after a Harvard astronomer with an interest in silk production imported the gypsy moth, which took flight in 1869. Their caterpillars soon stripped trees around Medford, Massachusetts, of their leaves, and the moths expanded their range around New England. Heroic efforts at control included setting caterpillars aflame with kerosene. By 1890, gypsy moth caterpillars threatened orchards and forests throughout New England.7 Surprisingly, Paris green failed to control the resistant caterpillars. In 1892, F. C. Moulton, a Gypsy Moth Commission chemist, had introduced the solution in the form of lead arsenate. Its effectiveness outweighed its expense, and as an added benefit it was gentler on the foliage on which it was sprayed. At the turn of the century, lead arsenate had become the preferred insecticide, a position it would continue to occupy until the introduction of DDT after World War II. Despite the proliferation of Paris green and lead arsenate, regulation of pesticides languished until the passage of PFDA in 1906 and, more important, the Insecticide Act of 1910.
Once the PFDA established labeling standards for drugs and foods in the U.S. in 1906, prospects for comparable standards for insecticides (and fungicides) began to look very promising. E. Dwight Sanderson reported that one of the nation’s largest insecticide manufacturers agreed to include an analysis of its goods on all labels. As the director and entomologist of the New Hampshire Experiment Station and the head of the Standing Committee on Proprietary Insecticides of the Association of Economic Entomologists, Sanderson interpreted the agreement as a promising sign that the insecticides industry would support his committee’s resolution for national labeling legislation for insecticides and fungicides.8 Harvey Wiley, director of the Bureau of Chemistry and a staunch advocate for the regulation of foods and drugs, encouraged Sanderson to lobby Congress for such legislation for three reasons: the PFDA did not extend to insecticides, an amendment to the PFDA was not feasible, and labeling of insecticides was critically important. When compared to the tortuous path of the food and drug act, passage of the insecticide act was both speedier and more direct. The passage of the Insecticide Act in April 1910 confirmed Sanderson’s impression that the insecticide industry was ready for regulation. Historian Adelynne Whitaker argued that the quick passage of Insecticide Act hinged on industry acceptance, but insecticides producers witnessed how the food and drug act benefited responsible and reliable producers of food and drugs, whereas producers of adulterated goods were forced out of the market.9
As the PFDA set labeling standards for foods and drugs, the Insecticide Act of 1910 established similar standards for insecticides. The manufacture and sale of adulterated and misbranded insecticides and fungicides in interstate commerce became illegal under the insecticide law. It codified legislative standards for insecticides in general and specifically the two insecticides that were most commonly used in the United States: lead arsenate and Paris green. Regulators expected specific standards to reduce problems with enforcement, which had posed difficulties for the USDA.10 Enforcement of the federal Insecticide Act fell to the Insecticide and Fungicide Board within the Bureau of Chemistry. Early in the act’s history, few if any noted the potential conflict of interest, but by the mid-1920s, the USDA’s dual role as protector of both farmers and consumers garnered criticism, particularly as the problem of pesticide residues on produce became a matter for regulatory concern.11 Understanding of the risks posed by heavy metals contaminants, such as lead and arsenic, owed much to the pioneering work of a few researchers who were establishing the new study of industrial hygiene.
The history of industrial hygiene (later, occupational medicine) owes much to the sweeping research of the social historian of medicine Christopher Sellers.12 Other historians have made important contributions with respect to specific toxins, diseases, and companies.13 In general, the U.S. lagged behind Britain and Germany in the evolution of industrial hygiene. Sellers attributed the lag to several factors, including the ambiguity of indeterminate symptomology of industrial diseases, delayed onset of diseases, a worker culture that dictated stoicism and personal fortitude in the face of workplace hazards, rapid turnover in employment, fear and mistrust of company physicians and orthodox medicine in general, financial barriers to medical care, and a general lack of knowledge of industrial diseases (exacerbated by the increasing occupational specialization).14 In Great Britain and Germany, pioneer researchers in academic or government posts began to study occupational disease in the late nineteenth century. Particularly noteworthy were the efforts of Karl Lehmann as the director of the Hygienic Institute in Wurzburg. Lehmann began studies the effects of gases and vapors on cats.15
In the U.S., systematic study of occupational disease took form in Alice Hamilton’s studies of working conditions in Illinois factories beginning in 1910. Hamilton (1869–1970) meticulously documented the hazards of various occupations, most notably the lead industry. Remarkably, without specific authority, she depended on the good faith of lead companies to allow her to survey workers and determine their illnesses and the causes thereof. After surveying several companies, Hamilton was able to compare lead poisoning rates among their workers. Such comparison led to informal competition between companies as they tried to lower their disease rates below those of their rivals. Hamilton later expressed mixed feelings regarding the effect of such competition and hoped that the companies sought the moral high ground rather than favorable cost-benefit ratios.16
Hamilton’s research documented effects of lead poisoning, but it remained for other researchers to determine pathways of toxic insult. In 1921, several researchers at Harvard Medical School, where Hamilton had joined the faculty as professor, launched what became known as the “lead study.” Cecil Drinker and Joseph Aub, the Harvard faculty members who led the lead study, envisioned a study of occupation disease grounded in science. Both strove to distinguish their work from Hamilton’s socially conscious efforts. Although David Edsall, Drinker, and Hamilton negotiated with the lead companies for “full scientific liberty,” Aub never forgot his obligations to the companies he studied, even as he developed a clinical and scientific research agenda. In a critical early move, Aub called upon a chemist—Lawrence Fairhall—to develop a more reliable method of analyzing minute amounts of lead in biological material. In addition to the chemist, Aub would soon incorporate a pathologist and physiologists into the research of the lead study, which anticipated the spirit of interdisciplinary collaboration that exemplified the study of toxicology. Moreover, Sellers argued, the lead study placed the toxicological approach at the core of studies of industrial disease.17 The Harvard study shaped the toxicological approach to the study of industrial disease in other important ways. Fairhall’s method of quantitative analysis provided a uniform basis for comparing lead levels in workers to the levels in their surroundings. Researchers could compare lead levels within factories and between industries. With the development of accurate techniques of analyzing other chemical hazards, dangers could be placed along single quantitative scales, typically in the same units (milligrams per ten cubic meters). Aub and the Harvard researchers also incorporated laboratory experiments into the lead study. These experiments, using both humans (volunteers) and animals, enabled the researchers to isolated specific workplace causes and effects. Such experiments transferred the research out of the workplace, to laboratories where researchers could study the more dangerous effects of lead and other toxins on cats or rabbits including pathological examination, which was unthinkable for humans, even volunteers. Finally, even as Aub and the other researchers in the Harvard lead study shifted the locus of the study from the workplace to the laboratory, they maintained strong ties to managers and doctors at some of the largest corporations in the U.S. Sellers argued somewhat ironically that the independent course of their research depended on continued corporate support.18
One of the first corporations to develop its own toxicological laboratory happened to be the largest chemical company in the U.S.: DuPont. Wilhelm C. Hueper (1894–1978) lobbied for what became the Haskell Laboratory at DuPont. Hueper made many important contributions to the study of occupational and environmental carcinogenicity over the course of a long career.19 After completing his medical education in Germany, Hueper immigrated to the United States, where he held various posts in academia, industry, and government. As chief pathologist at the University of Pennsylvania’s Cancer Research Laboratory and director of pathology at the American Oncologic Hospital, he asked to visit the DuPont Dye Works at Deepwater, New Jersey. At the factory he discovered in use aromatic amines that he knew, based on his experience in German factories, to cause bladder cancer. Through channels, Hueper notified Irénée du Pont, vice chairman of the board, of this potential hazard and recommended that DuPont establish an in-house biological laboratory to conduct toxicity studies. Internally, George Gehrmann advocated for a new lab, and DuPont’s Haskell Laboratory open on January 22, 1935, under the directorship of Wolfgang F. von Oettingen. Hueper joined Haskell after the University of Pennsylvania declined to renew his contract in the spring of 1934. Dow and Union Carbide established similar laboratories in the same year.20
Oettingen set as the first priority to determine the mechanism of the formation of bladder tumors. By late 1935, seventy DuPont workers had developed bladder tumors, and Hueper and his colleagues Frank Wiley and Humphrey D. Wolfe launched a long-term experimental study of dogs that were fed chemicals, including beta-naphthylamine (“beta”). After three years, the beta-fed dogs developed tumors, whereas those fed the other chemicals remained tumor free. Hueper and his colleagues published their findings, much to the chagrin of the DuPont board, which soon barred Hueper and other Haskell scientists from publishing their results. In a productive three years, Hueper conducted animal studies on a range of DuPont products, including seed grain vermicides, carbon disulfide, ethylene glycol and related solvents, refrigerant gases such as Freon, and Teflon coatings for kitchen utensils, but DuPont management fired him in November 1937 and stipulated that he never publish his research from Haskell. Hueper refused to comply with Du-Pont’s stipulation. Historian Robert Proctor has argued that, as a result of his refusal, DuPont hounded Hueper for the rest of his career, even threatening him with a lawsuit when he was invited to speak before the International Union against Cancer.21
Nevertheless, Hueper set to work on his magnum opus in 1938 and published his authoritative Occupational Tumors and Allied Diseases in 1942. Although Hueper recognized that acute poisonings represented a significant problem in the workplace, he argued that long-term chronic effects, like cancer, were far more important. But since chronic effects often did not manifest until months or years after exposure, physicians might not consider the particular cause of a particular disorder. In addition, Hueper suggested that most occupational cancers were preventable with proper procedures to protect workers, but he doubted that society would accept the necessary precautions to prevent cancer in the workplace.22 Still, support and participation of U.S. corporations proved to be critical to the development of toxicology and, as Sellers has shown, the toxicological approach to occupational medicine. Notwithstanding these developments in corporations, regulators and consumer advocates found recent legislation lacking.
Despite the general sense of satisfaction with the passage of the 1906 food and drug law and the 1910 insecticide law, as well as the advances in toxicology emerging from research in industrial hygiene, regulators soon confronted the limitations of the legislation. By 1933, several distinct groups questioned the efficacy of the laws. As chief of the Food and Drug Administration, Walter Campbell struggled with the law, particularly because it often fell to him to explain its deficiencies. In one case, the assistant secretary of agriculture, Rexford Tugwell, returned a routine spray-residue letter with a question along the lines of, if lead arsenate was a poison, why didn’t the FDA prohibit its use? Reflecting on the moment years later, Campbell’s assistant, Paul Dunbar, wrote: “The effect on all of us after these long years of fighting a lone battle against spray residues was like a kick in the teeth.”23 No one was more certain of the FDA’s inability to ban lead arsenate based on the 1906 law than its chief. Regulators could not address adequately cosmetics, patent medicines, adulteration of food, and even false advertising under the provisions of the current statute.24
In the 1930s a new generation of consumer advocates emerged, voicing a sharp critique of the inadequacy of federal food and drug law. In 1933, Arthur Kallet and F. J. Schlink, both with Consumers’ Research, Inc., wrote 100,000,000 Guinea Pigs: Dangers in Everyday Foods, Drugs, and Cosmetics, which constituted a broad indictment of the gaps in existing policy. The title suggested that food, drug, and cosmetic producers treated the 100,000,000 Americans like guinea pigs by exposing them to unknown and unrecognized risks. Kallet and Schlink cited numerous cases that the FDA could not (or would not) prosecute, ranging from food residues to prescription drugs to cosmetics, not to mention cases of false advertising. They split their critique between the 1906 law and the companies that flouted the spirit (if not the letter) of the law. They wrote: “Using the feeble and ineffective pure food and drug laws as a smokescreen, the food and drug industries have been systematically bombarding us with falsehoods about purity, healthfulness, and safety of their products, while they have been making profits by experimenting on us with poisons, irritants, harmful chemical preservatives, and dangerous drugs.”25
The book 100,000,000 Guinea Pigs became a model for other consumer advocates, among them Ruth deForest Lamb, who published American Chamber of Horrors: The Truth about Food and Drugs in 1936. Lamb ratcheted up the level of concern, focusing particularly on American housewives and their families. In her first chapter, “Why Doesn’t the Government Do Something about It?,” Lamb commanded her readers’ attention right from the opening paragraph: “You’ve been told you take your life in your mouth every time you bite into an apple or brush your teeth. All of your food is injurious, and your drugs and cosmetics are dripping with poisons. Anesthetic ether is always adulterated, and the ergot on which physicians depend to stop the hemorrhages of childbirth is impotent—unless, of course, it comes from Spain.”26 Thus she asserted that American consumers faced grave dangers through callous abuses on the part of the companies that produced foods, drugs, and cosmetics. Residues of pesticides on fruit were among the most worrisome to scientists and advocates.
As we have seen, chemical insecticides such as lead arsenate and Paris green proliferated during the nineteenth century. Use of these heavy metal insecticides rose dramatically during the first three decades of the twentieth century (table 1). Agricultural applications of arsenates quadrupled in the decade between 1919 and 1929. The arsenates appealed to farmers as broad-spectrum insecticides, which is to say, they were very effective against a wide range of insects. In Before Silent Spring, Whorton analyzed the toxicological studies of insecticide residues conducted between 1900 and 1920. Entomologists found themselves caught in a tug of war between the need to protect people from poisons while simultaneously protecting them (and their foods) from insects. The insect threat could be measured in precise fiscal terms, but the language of toxicology lacked the sense of immediacy and precision. Thus, Whorton concluded, “The imbalance between these opposed considerations easily tipped entomologists toward optimistic conclusions, and spray residues were generally dismissed as being considerably less dangerous than they actually were.”27 English regulators had been far more aggressive about limiting exposures to arsenic residues, limiting arsenic to 1/100 of a grain per pound (equivalent to 1.43 mg/kg)28 on fruit in 1903 in direct response to the findings of a Royal Commission on Arsenical Poisoning, which was led by Lord Kelvin.29 U.S. regulators ignored this standard until 1925, when the English threatened to ban American fruit imports. In 1927, the FDA restricted apples intended for export to 1/100 of a grain per pound of arsenic trioxide (the 1903 British standard), but apples intended for domestic consumption could have as twice as much arsenic per pound.30
In 1933, C. N. Myers, a physiologist, and Binford Throne, a clinician, jointly presented the results of several years of research into the risks of spray residues at the eighty-fifth meeting of the American Chemical Society. The researchers acknowledged the considerable costs of international legislation regulating pesticide residues, but they wondered about the costs in terms of human life and health. Previous studies had quantified the arsenic residues on fruit from 0.08 to 0.77 mg per apple and concluded, “No fruit carried sufficient lead arsenate to cause fatal poisoning through the consumption of one piece.” But such a statement regarding acute toxicity (resulting in death) neatly sidestepped the problem of chronic toxicity as did subsequent comments, for example, “The case is not clear as to the possible injurious effects from long continued daily consumption of fruits carrying relatively small residue.”31 Although there were cases of acute poisoning in which victims died within days of consuming contaminated fruits or vegetables, Myers and his collaborators worked to sharpen the picture of risks associated with increased arsenic use in a variety of products, among them glucose, drugs, lotions, tobacco, foods, fruits, vegetables, larvicides, and especially insecticides. The risks included eczema, keratosis, peripheral neuritis, disturbance of vision, and neurological symptoms. In addition to the risks borne by humans, Myers noted the destruction of large numbers of bees and birds as a direct result of lead arsenate spraying.32
In 100,000,000 Guinea Pigs, Kallet and Schlink decried spray residues and condemned the FDA for apparent indifference to the problem. The Guinea Pigs authors cited arsenic poisonings in the U.S. and England and wondered why the U.S. had been so slow to adopt stricter standards for arsenic residues on fruits and vegetables despite ongoing scientific research and congressional hearings. Of greater concern, however, were lead residues (concern about lead exposures drove the development of occupational medicine, see below). The FDA recognized the considerable hazard posed by lead and banned from the market fruits and vegetables containing residues of lead. However, there was little evidence of enforcement of this strict standard. One researcher tested apples for residues of lead and arsenic and found that none of the forty-five samples were free of either chemical. More troubling, some of the apples carried sixty times as much lead as arsenic trioxide.33
It is this sorry state of affairs that returns us to the frustration of Walter Campbell, administrator of the FDA. When the new assistant secretary of agriculture, Rexford Tugwell, queried him on the FDA’s failure to ban spray residues, Campbell responded that the department’s commitment to the agricultural industry forced the FDA to adopt a lenient policy with respect to growers. The surprisingly receptive Tugwell agreed with Campbell that the 1906 food and drug law needed revision and within hours secured approval from the president for a revision of the act.34 Tugwell promptly introduced a complete revision of the 1906 law into Congress, but industry representatives universally condemned the bill, particularly a provision that would have required drugs to be licensed. A conservative homeopathic physician named Royal S. Copeland, however, strove to forge a compromise, which would produce a bill with the potential to pass through Congress. The compromise bill passed the Senate but remained stuck in a House committee.
Meanwhile, by 1930 insecticide use in the United States had exploded to unprecedented levels. Farmers sprayed nearly sixty million pounds (27,215,542.2 kg) of the two most popular insecticides (calcium arsenate and lead arsenate) on crops in 1929. Only rarely did spray residues result in cases of acute poisoning and death, but scientists and physicians began to link common ailments like eczema and stomach upset with chronic arsenic and lead toxicity. International regulation of spray residues on fruit imported from the United States prompted review and revision of standards, but levels remained much higher for fruit sold in the U.S.35 Even Kallet and Schlink’s exposé provoked a relatively minor protest. Most Americans remained complacent, assuming that the 1906 law protected them from contaminated food and drugs, and farmers relied on the Insecticide Act of 1910 to keep insecticides free from adulterants. Nevertheless, by 1933 regulators had received executive approval to revise the pure food law. Tragically, an epidemic of poisonings failed to accelerate the legislative process.
During Prohibition following passage of the Eighteenth Amendment, many people sought alternatives to alcohol. Alcoholic extract of ginger had been available since the nineteenth century as a patent medicine and as such it provided a source of ethanol that could be marketed legally. “Jamaica ginger” or “jake” referred to a fluid extract of ginger that was sold widely during Prohibition. The United States Pharmacopoeia (USP), the official body established to monitor patent medicines, attempted to curb abuse of Jamaica ginger by requiring that the content of the extract contain five grams of ginger per one milliliter of solvent, which was usually ethanol. But this formulation tasted so bitter that consumers rejected it as non-potable. Pharmacies and roadside stands sold Jamaica ginger, typically in two-ounce bottles, as a remedy for a host of ills. The high alcohol content (up to 80 percent) meant that consumers could buy a two-ounce (56.7 g) bottle of jake for thirty-five cents and mix it with a soft drink, thus creating an inexpensive intoxicating beverage. USDA agents monitored producers by boiling samples of jake and weighing the remaining solids to ascertain that they conformed to the proportion dictated by the USP. Jake manufacturers cut costs by substituting various adulterants, such as castor oil, glycerin, and molasses, for the more expensive ginger solids.36
Triorthocresyl phosphate or TOCP appealed to some jake producers as an additive because, unlike other compounds, it would not evaporate away upon analysis by USDA agents (thereby upsetting the ratio of five grams of ginger per one milliliter of solvent). One researcher referred to TOCP as one of the most stable esters used in commercial organic chemistry. It was used in large quantities under various trade names as a liquid plasticizer and in lacquers, leather dopes, and even airplane finishes. Most manufacturers used castor oil as the adulterant to produce a palatable ginger extract that maintained the appropriate ratio of key ingredients. But Harry Gross, president and general manager of Hub Products Corporation, sought alternatives when the price for castor oil climbed at the end of the 1920s. Gross consulted a Boston chemical wholesaler for suggestions of other stable solvents. The wholesaler initially recommended ethylene glycol, which Gross rejected as too volatile, having subjected the resulting Jamaica ginger compound to mock testing. Next, Gross tried diethylene glycol, which produced similar results. Note: Gross rejected ethylene glycol and diethylene glycol on the grounds that the two compounds were too volatile rather than for their toxicity (see below for detailed discussion of diethylene glycol and the Elixir Sulfanilamide tragedy) Finally, Gross settled on Lyndol, which was a mixture of TOCP. When it passed the volatility test, Gross asked the chemical wholesaler about its toxicity. The wholesaler relayed this question to the Celluloid Corporation, which produced Lyndol, and the chemical company noted that it was presumably nontoxic. Gross purchased 135 gallons (511 liters) of Lyndol and began mixing and shipping the new jake.37
Near the beginning of 1930, physicians and the media began to report a strange illness that was attacking many people across the southern states. By spring the disease had become an epidemic. In Cincinnati, for example, more than four hundred people checked in to the Cincinnati General Hospital with muscular pain, weakness in both the upper and lower extremities, and rather minimal sensory findings. Since most, if not all, of the victims associated the disease with recent consumption of Jamaica ginger, they referred to it as “ginger jake paralysis” or “jake leg.” In February 1930, Ephraim Goldfain described a man who had progressive bilateral foot drop, thus becoming the first physician to recognize the disease. That same day, he saw another case and soon developed a list of sixty-five individuals.38 Over the course of the next few months, thousands of people suffered ginger jake paralysis. So prevalent was the disease across the southern states (especially Tennessee, Oklahoma, Kentucky, and Mississippi) that “jake leg” developed into a significant theme in blues music of the 1930s.39 Later there were smaller epidemics in Massachusetts and California.
Two researchers with the U.S. Public Health Service, Maurice Smith and Elias Elvolve, isolated the toxic compound as a phenolic compound (triorthocresyl phosphate—TOCP). Moreover, they were able to determine that the poisoned extract of ginger must have originated at a single source based on the fact that it was sold under at least eight different brands in Cincinnati, Ohio, and at least four brands in Johnson City, Tennessee. To reach this conclusion, they needed to develop a toxicological profile for the contaminated samples of ginger jake. Smith and Elvolve first ruled out poisoning by heavy metals (arsenic and lead), which, as we have seen, occurred frequently during the 1920s. The two researchers eventually obtained thirteen samples of the ginger extract. Five of those were almost definitely paralytic. A test for phenols indicated that every sample that contained phenols caused paralysis. To supplement the chemical analysis and correlation with paralytic samples, the researchers administered those samples that tested positive for phenols to rabbits, which exhibited a symptom complex that included muscular tremors, hyperexcitability, and spastic rigidity. General muscular weakness and flaccid paralysis of all the extremities followed. Treated rabbits died of respiratory failure. Smith and Elvolve conducted similar tests with monkeys and dogs only to find that they did not react like the rabbits. Further experiments with a solution that included the same elements yielded essentially the same results, but Smith and Elvolve could not explain the specific relation of the phenolic compound to the various neurological effects in ginger jake victims.
In the second report on ginger paralysis, Smith considered the pharmacological action of phenol esters. The comparison of TOCP with several other phenolic esters revealed the Jamaica ginger adulterant to be far more toxic than other similar compounds. The minimum lethal dose for TOCP in rabbits was 100 mg/kg, but as little as 50 mg/kg produced definite symptoms, which occasionally led to death. It was possible to replicate these results in two monkeys by administering the TOCP subcutaneously rather than orally. Pharmacological testing procedures had not been standardized by 1930, so Smith also tested TOCP on calves and chickens, with similar results to those produced in rabbits and monkeys.
Oddly, unlike other phenols or cresyls, the systemic action of TOCP developed slowly. The initial effects of a lethal dose appeared to be limited to the effects of the alcohol in which it was administered. The characteristic group of symptoms developed after an interval of one to several days. The symptoms of TOCP poisoning combined the manifestations of mild strychnine poisoning with some aspects of phenol poisoning. After reproducing symptoms in four experimental species, Smith concluded that TOCP was “capable of producing specific paralysis of the motor nerves of the extremities in certain species of animals and under certain conditions more or less exactly the same as occurred in thousands of human victims traceable to an adulterated fluid extract of ginger.”40 The fact that the pharmacological action of TOCP did not follow any known rule or law inspired Smith to call for further investigations into the pharmacological actions of chemicals.
By summer 1930 estimates of the number of ginger jake paralysis victims had reached as high as twenty thousand. Subsequent estimates significantly increased the number to more than fifty thousand victims.41 However, since many of the victims were impoverished minorities, researchers suspected underreporting. Despite the extent of illness, the response of the FDA and other federal offices verged on nonexistent. During congressional hearings, a senator criticized the FDA’s administrator for the lack of response to ginger paralysis:
SENATOR WHEELER: There was only one thing you could do under the law and should do, and that was to seize that product.
MR. CAMPBELL: And we did, promptly.
SENATOR WHEELER: You did, promptly—two or three months afterwards.42
Kallet and Schlink, in 100,000,000 Guinea Pigs, cited the case as a prime example of the failing of the 1906 law: by 1932, only three companies had been fined, but only $50 in two cases and $150 in the third. Later, Gross delayed federal prosecution by promising to cooperate and implicate the “real poisoners.” Before a trial could proceed, Gross (and his associates) pled guilty in front of a Boston judge, who released him on two years’ probation and with a fine of $1,000.
The ginger paralysis epidemic raised a number of questions regarding food and drug legislation, the responsibility of producers, and the role of toxicology. First, the practice of adulterating products with substances to deliberately change the taste or appearance appears to have received at least tacit sanction. If a manufacturer could replace a harmless substance with one of unknown toxicity, consumers were at considerable risk of exposure. In his search for a solvent to replace castor oil, Gross considered ethylene glycol, diethylene glycol, and TOCP. None of these compounds had clear toxicological profiles. In developing the pharmacology of TOCP, the Public Health Service (PHS) scientists first ruled out arsenic and lead, presumably because these were two of the most prevalent poisons that consumers encountered. Tests of samples of ginger jake on rabbits revealed effects similar to those human victims suffered, but monkeys seemed unaffected. Further tests of phenol and several cresols focused on the minimum lethal dose. Compared to similar compounds, TOCP exhibited a much greater toxicity (100 mg/kg and even 50 mg/kg resulted in symptoms and sometimes death). By administering doses to monkeys subcutaneously, researchers produced symptoms comparable to those experienced by humans and rabbits, but the fact that simian system cleared oral doses without toxicity underscored the importance of multiple test species. Chickens and calves also exhibited symptoms. Nevertheless, a clear toxicological profile for TOCP did not facilitate prosecution of litigation against the manufacturers or distributors, who claimed ignorance. Nor did it follow from the ginger paralysis epidemic that manufacturers should be required by federal statute to conduct toxicity tests on compounds before releasing them for public consumption. The fines levied against the companies that poisoned fifty thousand victims struck contemporaries as trivial. Rather than look to the federal government for protection or consideration, jake leg victims turned to music, finding a measure of comfort in the blues.
The best-known instance of public exposure to a lethal compound was the Elixir Sulfanilamide tragedy. The therapeutic properties of sulfanilamide first came to light in 1932, when two German laboratory scientists developed a red dye by combining sulfanilamide to a naphthalene-containing chemical that they called Prontosil. For the next three years, physicians experimented with the new drug for possible applications in their clinics. The real breakthrough came in 1935 when researchers treated streptococci-infected mice with Prontosil with allegedly remarkable therapeutic results. Gerhardt Domagk, who was a researcher for I. G. Farbenindustrie, announced the discovery of Prontosil, but it was largely unheralded due to the limited details of dubiously perfect results. Although cultures of streptococci did not respond to Prontosil in vitro, scientists at the Pasteur Institute in Paris discovered that the mouse systems broke the bond between the naphthalene and sulfanilamide, leaving sulfanilamide to destroy the streptococcus germs. In making this discovery, the Pasteur Institute clarified that it was sulfanilamide that produced therapeutic effects. Moreover, the French results undermined any hopes that I. G. Farbenindustrie may have held that Prontosil could be patented (sulfanilamide’s patent of 1909 had expired).43
Americans had also participated in early tests on sulfanilamide. The weak bond between naphthalene and sulfanilamide that characterized Prontosil had not met the standards of earlier researchers. During the 1920s, Rockefeller Institute scientists successfully produced a strong bond between sulfanilamide and quinine, but the bond was so strong that sulfanilamide was never released into the system and thus had no effect on disease. This apparent success was ultimately a significant failure, in that it delayed for nearly a decade research on the chemotherapeutic effects of sulfanilamide, until the German researchers utilized the naphthalene combination.44
The quest for new chemical therapies following the success of Paul Ehrlich’s Salvarsan inspired a campaign to discover new applications and solutions for sulfanilamide.45 By 1937 more than one hundred firms manufactured proprietary forms of the drug. When the public became aware that sulfanilamide might be a cure for gonorrhea, people began using the medication independently of a doctor’s orders, thereby raising the problem of self-dosing. Compounding this problem was the low cost of sulfanilamide on a daily basis. Whereas the most popular patented medicine for treatment of infections like streptococcus cost up to six dollars each day, the cost of sulfanilamide was only thirty cents daily.46
The search for chemicals to combine with sulfanilamide in order to create flexible drug-delivery systems led directly to the Elixir Sulfanilamide tragedy. One of the problems with sulfanilamide was the size of the tablets necessary to contain the correct dosage. Because children were most frequently infected by streptococcus, pharmaceutical companies sought a liquid form of the drug. After a few days of research in July 1937, Harold Cole Watkins, the chief chemist and pharmacist at the S. E. Massengill Company, appeared to have solved the problem by mixing sulfanilamide with diethylene glycol, which the Massengill Company had employed successfully in other drugs in extremely small amounts. Ultimately, Watkins determined that forty grains of sulfanilamide per ounce (91.4 g/kg) of diethylene glycol was the optimum proportion, and he prepared a mixture of eighty gallons (302.8 l) of water, sixty gallons (227.1 l) of diethylene glycol, and fifty-eight pounds (26.3 kg) of powdered sulfanilamide, as well as small amounts of the following flavor-enhancing substances: elixir flavor, saccharin, caramel, amaranth solution, and raspberry extract.
After calling the new drug Elixir Sulfanilamide, Watkins sent it to the Massengill laboratory, where it underwent examination for appearance, flavor, and fragrance. Notably, no one tested the new solution for toxicity.47 In Watkins’s view, Elixir Sulfanilamide was exempt from such testing since, he believed, “The glycols, related to glycerine, had been widely used by drug companies, and, Watkins averred, were well known not to be toxic.”48 With quality control completed, Massengill began commercial distribution of Elixir Sulfanilamide on September 4, 1937, a scant two months after Watkins had initiated experiments with diethylene glycol.
As the Massengill Company was distributing Elixir Sulfanilamide around the United States and Canada, the Journal of the American Medical Association published an editorial by Morris Fishbein, the journal’s editor. The editorial opened with a generous appraisal of sulfanilamide in general: “Seldom has any new drug introduced in medical practice aroused the enthusiasm that has developed for sulfanilamide. Much of this enthusiasm is warranted. The drug is truly remarkable, as indicated by startling results reported in the treatment of various infections.”49 Yet Fishbein expressed concern that chemical companies were studying similar and associated preparations in the search for therapeutic agents that they could market as new products superior to sulfanilamide. Fishbein warned the medical profession to proceed with caution regarding new preparations of sulfanilamide. His particular concern was that the therapeutic and toxic properties of new drugs could not be predicted from their chemical formulas: “Many months of investigations of the pharmacology, toxicology, and clinical application of new preparations under carefully controlled conditions are needed to provide evidence of clinical value.”50 There had already been reports of toxic reactions to the self-medication brought about by rumors that sulfanilamide could cure gonorrhea in forty-eight hours. (Fishbein placed considerable blame for such incidents on unscrupulous pharmacists who willingly sold drugs to anyone over the counter.) In a statement that proved to be tragically prophetic, Fishbein concluded, “Sulfanilamide should not be administered in association with other drugs until definite information is available as to toxic effects.”51
Given the early concern of the American Medical Association (AMA) with the risks of sulfanilamide, it was appropriate that the AMA was one of the first official bodies to learn of the Elixir Sulfanilamide crisis. Two doctors from the Springer Clinic in Tulsa, Oklahoma, sent a telegram detailing clinical and pathologic effects of poisonings in Tulsa to the AMA on October 15. Even allowing for the constraints of communication by telegram and the restrictions of medical reports, the following excerpt seemed particularly stark:
Total of ten cases. Eight dead. One recovered. One critical. Ages from eleven months to twenty-six years. All received Elixir Sulfanilamide in amounts varying from one-half to seven ounces. Characteristic onset with nausea, vomiting, occasional diarrhea, malaise, later pain over kidney region and abdomen. All developed anuria within two to five days after beginning medication. Indications for the use of sulfanilamide were varied. Nine cases hospitalized.52
It was in fact this report and the cluster of cases it represents that prompted action by the AMA and the FDA.
On October 16, 1937, a representative of the Kansas City Station of the FDA who had been dispatched to Tulsa, Oklahoma, confirmed the doctors’ report by telegram to the FDA headquarters, reporting the deaths of nine individuals attributable to a preparation of sulfanilamide. Of these nine, eight of the victims had been children with streptococcic sore throat and one an adult with gonorrhea. All had taken Elixir Sulfanilamide. The FDA immediately sent inspectors to the Massengill Company headquarters in Bristol, Tennessee, and to distribution centers in Kansas City, New York, and San Francisco. Upon receiving news of the deaths, the Massengill Company issued more than a thousand telegrams recalling all outstanding shipments. These messages warned consumers, salesmen, druggists, and doctors not to use Elixir Sulfanilamide and to return all stocks for credit at the manufacturer’s expense.53 While the telegrams conveyed Massengill’s desire to collect all outstanding quantities of Elixir Sulfanilamide, they gave no indication of the dangerous character of the product or the emergency that necessitated the recall of the drug. On October 19 the FDA inspector on location in Bristol issued another telegram to all persons known to have received shipments of the drug from Bristol: “Imperative you take up immediately all elixir sulfanilamide you dispensed. Product may be dangerous to life. Return our expense.”54 On the insistence of FDA field agents, similar telegrams were sent from the Massengill branches in Kansas City, San Francisco, and New York, also on October 19 or shortly thereafter.
Despite the return of numerous shipments to the four distribution centers, the FDA faced the considerable task of confiscating all outstanding supplies of Elixir Sulfanilamide. To accomplish this, the FDA diverted most of its field force of inspectors and chemists to review the thousands of order slips in each of the four distribution centers as well as in wholesale and retail drug stores. Complicating matters was the fact that many of the sales of Elixir Sufanilamide were over-the-counter (not by prescription) to unknown individuals. In the most problematic cases, doctors had either no names or fictitious names for individuals who received the drug. Massengill salesmen also proved hard to locate, and at least one went to jail rather than cooperate with the FDA officials. Most doctors and pharmacists contributed willingly to the confiscation campaign, but a few refused to cooperate and even denied the adverse effects of the drug. One South Carolina doctor admitted to inspectors that he had dispensed just under two pints of the elixir to three white patients and two black patients, none of whom died. Further investigation revealed that in fact the doctor administered the elixir to seven patients, of whom four had died: one white man, one white girl, and two black men. The inspector tracked the cause of death to the gravestone of one of the black victims, where a grieving relative had left a small bottle of Elixir Sulfanilamide with the prescription label of the doctor’s office still intact.55
Notwithstanding the considerable effort of the FDA, as well as state and local food, drug, and health authorities, Elixir Sulfanilamide caused numerous deaths. The tragedy progressed rapidly throughout the Southeast with deaths reported in Mississippi, Alabama, Georgia, South Carolina, and Texas. On November 1, the AMA Chemical Laboratory calculated the total number to be sixty-one. Later that month, the New York Times reported that ninety-three individuals had died from Elixir Sulfanilamide, but the official toll stood at seventy-three as a direct result of the elixir and twenty more deaths associated with the drug, according to the report of the secretary of agriculture regarding the sulfanilamide tragedy. Deaths occurred in fifteen states from Ohio to Texas. Most of the deaths were in southeastern states, perhaps due to the proximity of the Massengill headquarters in Bristol, Tennessee. The AMA Chemical Laboratory mapped the epidemiology of the tragedy.56 Nevertheless, by seizing 228 gallons (863.0 l) of the 240 gallons (908.5 l) manufactured and more than half of the 11 gallons dispensed in prescriptions or over-the-counter sales the FDA surely avoided a much greater disaster.57
While the FDA was containing and controlling the immediate crisis, the AMA began a detailed analysis of the chemistry, pharmacology, pathology, and necropsy of Elixir Sulfanilamide. Three scientists at the University of Chicago performed the chemical examination. Their analysis revealed Elixir Sulfanilamide to be a reddish, somewhat viscous liquid with an aromatic odor resembling raspberry and anise and a sweet taste. The drug resembled glycerin in general physical character. Further analysis yielded the proportions of the major ingredients of the drug:
Diethylene glycol |
72 percent |
Sulfanilamide |
10 percent weight/volume |
Water |
15.6 percent |
Furthermore, the chemists conducted spectrographic examination that failed to reveal the presence of known poisonous substances, such as lead, bismuth, mercury, or arsenic. In essence, the chemical analysis of Elixir Sulfanilamide confirmed the statements of the Massengill Company regarding the composition of the drug.58
E. M. K. Geiling, chair and professor in the Department of Pharmacology at the University of Chicago, also conducted toxicity studies of Elixir Sulfanilamide. Geiling determined the toxic agent by carrying out toxicity experiments on rats, rabbits, and dogs using the following substances: pure diethylene glycol, pure sulfanilamide, Elixir Sulfanilamide-Massengill, and “synthetic” elixir of sulfanilamide (produced by the AMA Chemical Laboratory with pure substances in approximately the same proportions as found in the Massengill elixir). Through a series of experiments, Geiling hoped to determine three things:
1. The toxic and lethal doses of each of the substances when given in relatively small doses three times daily. This information seems particularly necessary since we were not able to find any data in the literature on this specific point.
2. Our experiments were further planned with the hope of being able to reproduce in healthy experimental animals, in about the same time, the clinical and pathologic picture as presented by patients who had taken fatal doses of the Elixir of Sulfanilamide-Massengill.
3. Through our experiments we hoped to discern the toxic ingredient in the Massengill elixir.59
Such a strategy laid the foundation for future toxicological investigations. Geiling’s results left no doubt as to the cause of the deaths. In rats fed via a stomach tube three times daily, the mortality rate rose to 100 percent after eight or nine doses of two cc’s of diethylene glycol, Elixir Sulfanilamide, or synthetic elixir. By contrast, none of the rats treated with sulfanilamide or water died during the experiment. Thus Geiling and his collaborators concluded that diethylene glycol was the toxic agent in Elixir of Sulfanilamide-Massengill since animals treated with the chemical exhibited the same symptoms as animals treated with Elixir Sulfanilamide and a synthetic elixir formulated from the same ingredients in the same proportions.60
As further confirmation, they noted that sulfanilamide alone did not prove fatal to rats, rabbits, or dogs, but the drug did cause convulsions in some of the animals, and the researchers raised the possibility that the drug contributed to tissue damage in animals or human beings with impaired renal function. The final element of the conclusion contained significant implications for future policy, specifically premarket testing on animals: “Our experiments emphasize the importance of administering drugs in divided doses to experimental animals when it becomes necessary to know whether or not a drug has cumulative effects. Errors resulting from an oversight of this important pharmacologic principle may be costly to human lives.”61
Given that drug therapy often required repeated daily doses, Geiling believed that testing new therapies should reflect such procedures if cumulative effects were to be understood. Such a view began to broaden pharmacologists’ perspective to include chronic effects. Geiling further clarified his point: “We can confirm the finding of Haag and Ambrose that the ingestion of 15 cc. of diethylene glycol per kilogram in a single dose by stomach tube proves fatal to rats. This figure, however, is no index of the toxic and possible fatal effects of the drug, if administered in small divided doses, especially since neither the fate nor the mechanism of detoxification is known.”62 The earlier research had isolated the lethal dose of diethylene glycol, which is to say, its acute toxicity, but Geiling distinguished this piece of data from the equally important effects of repeated doses, or chronic toxicity. In this sense, the toxicity of diethylene glycol had broader implications for the science and policy of newly introduced drugs.
Finally, Paul R. Cannon, M.D., also from the University of Chicago, conducted a pathological evaluation of rats, dogs, and rabbits that had died following toxic doses of diethylene glycol, Elixir of Sulfanilamide-Massengill, synthetic elixir, and sulfanilamide alone. Cannon found a remarkable similarity between the pathological effects of a toxic dose of diethylene glycol, the synthetic elixir, or Elixir of Sulfanilamide-Massengill in animals and a lethal dose of Elixir Sulfanilamide in humans.63 In addition to the research of Geiling and Cannon, Edwin P. Laug and his colleagues at the FDA published the first toxicological benchmark: the LD50 (see below).
An appreciation of the impact of the Elixir Sulfanilamide case on the evolution of toxicology in the United States requires a review of the history of the Division of Pharmacology at the FDA. The FDA was established in 1927, when the activities of the Bureau of Chemistry within the USDA became distinct from agricultural chemistry and the work of the department as a whole. Regulators and regulatory chemists were moved to a separate agency called the Food, Drug, and Insecticide Administration (later, the FDA).64 However, jurisdiction for the Federal Insecticide Act remained under the USDA. This move, noted Christopher Bosso, separated regulatory activities on behalf of consumers from those on behalf of farmers.65 The first commissioner of the FDA, Walter Campbell was determined to examine the toxicology of lead and arsenic in response to the widespread use of these chemicals as agricultural insecticides. To oversee this project, he appointed Erwin Nelson, a pharmacologist on leave from the University of Michigan, as the acting chief of the new division.66
In 1935 Nelson devoted himself to raising pharmacology, hitherto part of the Division of Medicine at the FDA, to division status. To accomplish his goal, Nelson canvassed various universities for experts in critical aspects of toxicology. Several individuals from the original branch of pharmacology made up the core of the new division: Harold Morris, Herman Morris, Howard Lightbody (a biochemist specializing in the study of enzymes), and W. T. McCloskey. Nelson recruited several additional scientists for the new division: Edwin P. Laug, from the University of Pennsylvania; Lloyd C. Miller, a lipid biochemist trained at the University of Rochester; and Herbert Braun, from the University of Wisconsin. Rather than selecting trained pharmacologists, Nelson sought scientists whose specific expertise could contribute to the toxicological analysis of lead and arsenic. Thus he assembled experts in analytical work, enzymology, animal studies, and pathology.67
Herbert O. Calvery joined the Division of Pharmacology as Nelson’s replacement as acting chief. Other scientists joined the new division. Geoffrey Woodard entered the division as a laboratory apprentice, but he played an important role in acute toxicity studies. Harold Morris moved to the National Cancer Institute, where he became widely respected for his research on cancer. O. Garth Fitzhugh, a specialist in the study of chronic toxicity, replaced him in the area of chronic toxicology in 1938. These individuals transformed toxicology from the study of the effect of a single dose on a single animal to the sophisticated statistical analysis of dose response curves necessary to understand the toxic effects of drugs and other chemicals on various animals and humans. Such precision arose as a direct response to the Elixir Sulfanilamide tragedy.68
The FDA mobilized its field scientists against Elixir Sulfanilamide and now through the new Division of Pharmacology it could also respond through laboratory analysis. In reviewing the existing literature, including Geiling’s research, Edwin Laug and the other FDA toxicologists realized that no one had developed a method for comparing the toxicity of one substance to another. Although Laug and others suspected that diethylene glycol was the cause of the many deaths associated with Elixir Sulfanilamide, they had to develop an approach that would confirm their suspicions statistically. To do this, the FDA toxicologists were assisted by the ground-breaking research of Chester I. Bliss, who was brought to the FDA as a part-time consultant by Herbert O. Calvery. Bliss had studied with R. A. Fisher, the great English bio-statistician who developed and promoted the use of statistics in biology.
Bliss had published a seminal paper, “The Calculation of the Dosage-Mortality Curve,” in which he demonstrated the sigmoid character of the typical dosage-mortality curve as established by numerous toxicological studies of a large number of organisms by many biologists. Acknowledging his debt to R. A. Fisher and the trends in biostatistics Fisher inspired, Bliss selected his procedures on the basis of their statistical accuracy and efficiency. Bliss intended to present his techniques for calculating the transformed dosage-mortality curve in sufficient detail so that biologists with limited knowledge of statistics could use them. The value of the dosage-mortality curve to biologists in general, and toxicologists in particular, arose from its ability to describe the variation in susceptibility between individuals of a population. In line with the theory that any given population would show a range of susceptibilities to any given toxin, Bliss demonstrated that mortality could be plotted against dose in a way that would indicate what percentage of any given population would be killed by a particular dose. This experimental technique could determine the precise minimum lethal dose for each organism, since a dosage above minimum effectively killed susceptible individuals. Bliss recommended exposing a series of sample groups of organisms to graduated doses and recording the percentage killed for each sample group. When Bliss plotted this data, it resulted in a sigmoid curve (exactly as predicted). Thus he concluded: “The sigmoid dosage-mortality curve, secured so commonly in toxicity tests upon multicellular organisms, is interpreted as a cumulative normal frequency distribution of the variation among the individuals of a population in their susceptibility to a toxic agent, which susceptibility is inversely proportional to the logarithm of the dose applied.”69 Bliss’s explanation and technique were enhanced by a note from Fisher. It examined the case in which there were few or no survivors and appended a method to address this problem.
Bliss’s paper appeared in 1935, but the FDA pharmacologists were already familiar with his work, since he worked in their laboratory. The dosage-mortality curve provided an excellent method to evaluate the toxicity of a given chemical. Laug and his colleagues in the Division of Pharmacology were among the first to apply Bliss’s method to an actual case when they evaluated the toxicology of glycols and derivatives in response to the Elixir Sulfanilamide disaster. Prior to this study, toxicologists had estimated the doses that were lethal to 10 percent, 50 percent, and 90 percent of a given population, but the results were unsatisfactory, particularly when toxicologists used such data to interpolate a sigmoid curve through a number of points. Laug and the FDA pharmacologists used Bliss’s method to calculate and plot the dose-mortality curve for rats, mice, and guinea pigs. They determined that the most useful parameter was the lethal dose for 50 percent of the population (LD50), as this value required the fewest animals for calculation (a minimum of 10). In contrast, arriving at the same level of precision for 99 percent deaths required at least 103 animals. Moreover, Laug and his team calculated standard errors for the LD50 and found they could determine the dosage at which 50 percent of the population would be killed in nineteen out of twenty experiments.
Citing the studies of Geiling and others at the University of Chicago, Laug reflected on the importance and probable significance of his team’s findings for humans. Although the FDA researchers had confirmed the findings of Geiling and others, Laug advised restraint in applying the results to humans: “It seems proper at this point to reemphasize the inadvisability of attempts to interpret experimental data on laboratory animals directly in terms of man. It is entirely too dangerous. The present investigation confirms again the wide variations that may occur between species.”70
Although they presented the derivation of LD50 as a valuable new tool in toxicology, the FDA researchers acknowledged that neither acute nor chronic toxicity gave a complete picture and scientists should assess both dimensions of toxicology. They argued for establishing the most complete toxicological profile possible. Ideally, such a profile would include both acute and chronic toxicity studies as well as pathological evaluations. In addition, certain glycols produced acute effects at high doses without producing chronic effects at lower doses. Laug contrasted this toxicological behavior with that of the well-studied heavy metals and narcotics: “Again, although small doses of lead, mercury, selenium, fluorine, narcotics, etc., are acutely toxic, it is the insidious character of their chronic toxic effects that makes them even more dangerous.”71 As of the 1930s, the study of chronic toxicity had not progressed as far as the study of acute toxicity, except in the case of heavy metals and certain drugs.72 Nevertheless, the model of chronic toxicity provided by heavy metals was hardly universal, as Laug noted in the statement above.
Methods for deriving dose-response curves and LD50s were certainly among the most important legacies of the Elixir Sulfanilamide tragedy. Not only did the approach transform regulatory science as it was conducted at the FDA, industry adopted the same procedures for its analyses of new chemicals (at the FDA’s direction). Long after he retired, Laug would recall the significance of the division’s initial exploration of LD50 as one of the first successful applications of statistical approach to toxicology: “I think it was the most significant thing that we did … in those days there was not much precision when determining toxicity. And what we did was by the use of statistics, we made it possible that when you treated animals with something toxic, you could create a curve, a slope, and the significance of that was that you could then compare it to something else and that was the point, LD50.”73 A small group of FDA toxicologists in the Division of Pharmacology transformed the field of toxicology. More than any other single procedure, LD50 became the benchmark in most initial studies of toxicity for pharmaceuticals and environmental chemicals.
Still, the question remains: Why did the Division of Pharmacology, which was established to evaluate the hazards of lead and arsenic in insecticides, develop the LD50 in response to the Elixir Sulfanilamide disaster? The Division of Pharmacology lost its funds to investigate lead and arsenic in 1937 or 1938. According to Geoffrey Woodard, another FDA pharmacologist, “Well, the insecticide problem was the original basis for having set up a division. Now as Ed [Laug] said, it was a political football and I remember we worked through ’37 or ’38 until New Year’s Eve—right up until midnight on New Year’s. Because Congress cut off our funds and said there was to be no more work on lead and arsenic.”74 In Woodard’s view, which was shared by the other FDA pharmacologists, lead and arsenic insecticides had become politically charged.75 In response to increased scrutiny, apple growers had appealed to their congressmen, who voted to transfer the authority for the examination of insecticides to the Public Health Service (PHS) at the National Institutes of Health (NIH) by revising the appropriation act of the FDA so that none of the funds could be used for the study of toxicity of lead and arsenic. The restriction of funds did not stop Calvery and Laug from publishing on the risks of lead compounds, however.76 Deprived of its work on the toxicology of lead and arsenic insecticides, the Division of Pharmacology concentrated its considerable effort on the study of Elixir Sulfanilamide, the glycols, and eventually other chemicals. Christopher Bosso revealed that shifting research on insecticides from the FDA to the PHS meant a move away from long-term laboratory studies based on experiments with laboratory animals to extrapolate chronic effects. In contrast, the PHS emphasized field surveys that questioned farmers about their health, which could reveal acutely toxic effects, but for the FDA, the PHS approach was inadequate for determining longer-term chronic effects.77
While the FDA strove to eliminate the risks posed by Elixir Sulfanilamide, and the AMA with the assistance of Geiling and other University of Chicago faculty conducted the chemical and toxicological analysis of the elixir, Massengill attempted to defend its product. The company had earlier been convicted and paid fines for violations of the Food and Drugs Act, in September 1934 and March 1937. H. C. Watkins, Massengill’s chief chemist, had been cited by the solicitor of the Post Office for distributing a medicine alleged to reduce weight, to bring about “perfect slenderness,” and to cause the body to acquire “a trim, youthful, athletic look.” To avoid charges of fraud, Watkins filed a stipulation agreeing that the sale of the product would be abandoned and not resumed at any future time.78 Unfortunately, under the PFDA, the only basis for action against the interstate distribution of the “elixir” was the allegation that the word implied an alcoholic solution, whereas the product was a glycol solution. It was for this reason that S. E. Massengill, owner of the eponymous company, could claim in a letter to the AMA, “I have violated no law.” Although Massengill’s statement conformed to the letter of the law, regulatory bodies generally believed that most drug manufacturers recognized a greater responsibility to the public. Such ethical obligations did not concern Massengill. He refused to take responsibility for the deaths, instead blaming them on the “bad effects” of sulfanilamide.79 Massengill’s statement betrayed his complete lack of knowledge regarding the toxicity of Elixir Sulfanilamide. Even in the absence of controlled animal experiments, like those conducted at the University of Chicago and the FDA, a simple literature review would have revealed a careful analysis of ethylene glycol (the active ingredient in antifreeze) and some of its derivatives, including diethylene glycol.
Specifically, Massengill and Watkins would have discovered a paper written by Oettingen, one of the fathers of pharmacology in America. Oettingen, who at the time taught pharmacology at Western Reserve University in Cleveland (and later served as the director of the Haskell Laboratory), tested the toxicity of the glycols on various animals, including rats and frogs.80 Regarding the therapeutic potential of diethylene glycol and ethylene glycol, Oettingen wrote: “Ethylene glycol and diethylene glycol may be of interest for therapeutic use, as solvent and as vehicle. With these substances the local irritation is comparatively small. Their application to the skin seems to be without risk. Given orally in larger doses, they may produce severe gastro enteritis and systemic symptoms.”81 Still, even if they had completed a literature review, Massengill and Watkins might have argued that the benefit of the drug sulfanilamide outweighed the unknown risk of gastro enteritis. It is also possible that they had experience with one of the nontoxic glycols, such as polyethylene glycol, which is nontoxic and an effective laxative (widely marketed today as an over-the-counter therapy).
Given their academic and professional experience, Massengill and Watkins should have been qualified to appreciate the inherent risks associated with their enterprise. Yet FDA investigators found Watkins to be particularly unconcerned about the hazardous effects of his products: “What impressed [Theodore G.] Klumpp and [William T.] Ford most about Watkins was what they deemed a certain callousness in his conversation. He spoke of a preparation of colloidal sulfur he had devised. When marketed, this compound resulted in the death of a number of people. ‘Mr. Watkins told about this event,’ Klumpp wrote, ‘as if it were an ordinary incident in the business of making and marketing pharmaceuticals.’ ”82 Indeed, Watkins seemed to doubt the possibility of toxic effects altogether. In his interviews with FDA officials, he claimed that when he first heard reports the Elixir Sulfanilamide had been linked to numerous deaths, he had personally taken a huge oral dose of diethylene glycol without ill effects. FDA officials doubted this story and dismissed its claim as a “futile heroic gesture,” which could not make up for Watkins’s failure to test the drug properly before its distribution.83
Although generally regarded as inadequate, the PFDA was the only relevant legislation available to FDA in bringing charges against the S. E. Massengill Company in the Elixir Sulfanilamide case. As owner of the company, S. E. Massengill was particularly susceptible to the sanctions of this act because the two earlier prosecution charges exposed the company to more severe penalties as a second offender. For his part, Massengill continued to proclaim his innocence, arguing that Elixir Sulfanilamide had helped more people than it had harmed. The company did fire Watkins, and it admitted a level of moral responsibility when it settled numerous suits brought by the families of those who had died as a result of the elixir.84 Nevertheless, the FDA mounted a strong case against the company, confirming that diethylene glycol was the agent of toxicity in the elixir and assembling a team of experts to testify to the toxicity of the drug. These included Perrin H. Long of Johns Hopkins, who later wrote a monograph on the clinical and experimental use of sulfanilamide, as well as Geiling and Cannon of the University of Chicago. In addition, FDA scientists prepared testimony and exhibits for the trial. On the first day of the scheduled trial (October 3, 1938), Massengill appeared and revised his plea to guilty, and he received a fine of $150 on each of 174 counts amounting to a total of $26,100, the largest fine ever levied under the provisions of the 1906 act. Adding to the tragedy, Harold Watkins avoided further condemnation by committing suicide before the trial. Nevertheless, it was the calamity of Elixir Sulfanilamide that finally motivated Congress to pass new legislation regarding food and drugs.
As the supervising agent, Henry A. Wallace, the secretary of agriculture, completed an extensive review and analysis of the deaths attributed to Elixir Sulfanilamide. The report responded to two resolutions in the United States Congress: House Resolution 352 of November 18, 1937, and Senate Resolution 194 of November 18, 1937. These resolutions mandated an investigation into the Elixir Sulfanilamide debacle and its implications for food and drug legislation. After thoroughly reviewing the case, Wallace developed four broad recommendations for legislation. First, he recommended the licensed control of new drugs to insure that they would not be generally distributed until experimental and clinical tests showed them to be safe for use. As a corollary to this recommendation, the secretary defined exactly what constituted a “new drug.” In order to justify drug licensure, he noted the importance of safety: “In the interest of safety, society has required that physicians be licensed to practice the healing art. Pharmacists are licensed to compound and dispense drugs. Electricians, plumbers and steam engineers pursue their respective trades under license. But there is no such control to prevent incompetent drug manufacturers from marketing any kind of lethal potion.”85 Another provision of the recommendations called for the prohibition of drugs that were dangerous to health when administered in accordance with the manufacturer’s directions. As self-evident as this stipulation seems, recall that the only grounds on which to prosecute S. E. Massengill Company was the minor issue that the drug was not technically an elixir as it had not been combined with alcohol. The secretary also stipulated that drug labels bear appropriate directions for use and warnings against probable misuse. Finally, he called for the prohibition of secret remedies by requiring that labels disclose fully the composition of drugs. The new language would broaden the legislative authority of the FDA, enabling the prosecution of manufacturers of dangerous drugs then on the market. Under the Pure Food and Drug Act of 1906, the FDA could not bring even the most trivial of charges against many of the dangerous drugs. In a recent analysis of the role of the tragedy in food and drug legislation, the government scholar Daniel Carpenter indicated the significance of the efforts of Wallace and FDA Chief Campbell, “Yet it is no understatement to say that Campbell and Wallace’s document forms the originary basis of modern pharmaceutical regulation the United States and much of the industrialized world.”86
As a result of the Elixir Sulfanilamide disaster, pharmacologists developed new standards for the approval of drugs for public use. Of the many scientists who analyzed the toxicity of Elixir Sulfanilamide, one of the most outspoken was E. M. K. Geiling. It was Geiling’s conviction that drugs needed to undergo a thorough toxicological examination before release. Based on his work with Elixir Sulfanilamide, Geiling developed a nine-part analysis utilizing animal experiments for new drugs. Geiling’s proposed method required knowledge of the exact composition (qualitative and quantitative) or the detailed method of preparation of the product. Repeatedly, scientists and legislators cited lack of knowledge of the contents of Elixir Sulfanilamide and its potential effects as a serious flaw in the PFDA. The new drug analysis demanded acute and chronic toxicity studies of animals of different species at varying dosage levels given that studies of a single species at constant dosage levels could be misleading. During the course of chronic toxicity studies, Geiling encouraged careful and frequent observations of the animals in order to establish a composite picture of the clinical course of the drug. Geiling noted that the data on many drugs were very deficient in this respect.
Drug analysis also called for careful pathologic examination of the tissues with appropriate stains. Animal experiments would also facilitate the study of the effects of experimental lesions of various important excretory or detoxifying organs, particularly the kidneys and liver, on the action of the drug. Related to this requirement is the determination of the rate of absorption and elimination of the drug, its path and manner of excretion, and the concentration levels in the blood and tissues at varying times after administration. Another strategy that Geiling recommended was the analysis of potential interactions between the drug and foods or other drugs (as an example, Geiling offered the contraindication of sulfanilamide and magnesium sulfide). Finally, Geiling called for careful examination for idiosyncrasies or untoward reactions of new drugs.87
With this method of analysis, Geiling defined the toxicological assessment of drugs, but he acknowledged that his expectations probably exceeded the dedication of pharmaceutical chemists: “It is recognized that some will consider these safeguards to be too rigid and that they may simply be considered an ideal. It can correctly be charged, in fact, that some of the pharmacopeia drugs have not been studied along such lines.”88 Geiling intended, however, to elevate the standards of drug approval to new levels and thereby avoid other disasters: “Admitting this, it is nevertheless regrettably true that many human lives have been sacrificed by the failure to meet the standards of these preliminary tests and that many more lives will be sacrificed if such standards are not put into effect. Any essential compromise with these requirements will inevitably exact a toll of deaths or injuries among the public. The life and safety of the individual should not be subordinated to the competitive system of drug exploitation.”89 Geiling believed that such standards would protect the public from untested drugs with unknown effects. Finally, Geiling hoped that new standards would direct drug manufacturers toward the development of new and genuinely valuable agents in the treatment of disease in place of spending enormous sums on advertisements.90
Industry representatives contradicted Geiling’s notion of the role of advertising in drug manufacture and public health. A former president of the National Association of Insecticide and Disinfectant Manufacturers, Lee H. Bristol (vice president of the Bristol-Meyers Company) claimed that manufacturers of proprietary drug products favored drastic control over untried potentially dangerous drugs. He also noted that the sulfanilamide elixir was not a consumer-proprietary product intended for self-medication, but one prescribed by physicians. The responsibility for the calamity rested with the drug manufacturer who used an inadequately tested and dangerous solvent for a potent drug and the physician who prescribed or used the elixir without being familiar with its therapeutic effects. The New York Times reported an interesting inversion of Geiling’s statements cited above: Bristol argued that “advertising was the greatest safety measure ever devised for the protection of the consumer” and pointed out that Elixir Sulfanilamide was an unadvertised product belonging to a group of so-called ethical specialties.91 Bristol went on to concede that government should have control over such potent drugs as sulfanilamide elixir and said that no reputable manufacturer of packaged medicine stands in the way or opposes legislation to protect the public from such calamities. He added the following qualification to his statements, however, “Experienced and responsible manufacturers of carefully compounded products should not be penalized simply because their products are advertised to the public.”92
Bristol’s comments were particularly significant in light of his association with the National Association of Insecticide and Disinfectant Manufacturers for his statements applied equally well to insecticides and drugs. The difference between Geiling’s perspective and Bristol’s was one of emphasis. Whereas Geiling believed that advertising drugs necessarily detracted from meticulous evaluation of the toxicology and pharmacology of new drugs, Bristol argued that advertising served to inform consumers as to the qualities of new pharmaceutical agents. Neither position was wholly sustainable independent of a deeper understanding of how pharmaceutical companies conducted their research, development, production, and advertising. Geiling and Bristol both clearly recognized that the Elixir Sulfanilamide case revealed an urgent need for change in food and drug law but differed as to the precise form of such legislation.
Other calls for new legislation were more explicit. In an editorial for Hygeia, a family health journal, Fishbein argued that the PFDA of 1906 was passed at a time when modern advertising was in its infancy and that more food and drugs were sold in 1937 by advertising than by claims made in other ways. More troubling was Hygeia’s contention that present food and drugs law provided no potent weapon against false and fraudulent advertising of foods, drugs, devices, or cosmetics. At its core, the 1906 Act did not provide for standards of purity, potency, wholesomeness, or labeling of foods and drugs. Geiling added toxicity standards to this list. Hygeia also decried the many loopholes contained in existing legislation.93 More than any other incident, the Elixir Sulfanilamide disaster crystallized ideas regarding food and drug legislation.
To this point in the story, we have seen how several health tragedies, particularly the Elixir Sulfanilamide disaster, transformed toxicology. Moreover, we have seen how researchers at universities, government agencies, and industry refined the toxicological approach. But how did such developments influence food and drug law? The journalist Philip J. Hilts argued that there were two basic requirements for novel legislation: “A bill must already be present in Congress, and legislators and significant elements of the public must already be educated and paying attention when the crisis hits.”94 He added the corollary that the crisis must also involve children. When the Elixir Sulfanilamide disaster first appeared in newspapers, and 100,000,000 Guinea Pigs reached bookstores and libraries, Congress was reviewing the Pure Food and Drug Act of 1906, which regulators and the public regarded as inadequate protection for consumers. Republican control of Congress during the 1920s was not conducive to legislative reform, but the arrival of Franklin Delano Roosevelt and the New Deal promised change. As we have seen, when the story of the Elixir Sulfanilamide tragedy broke in 1937, a compromise bill combining elements of the Tugwell bill and the Copeland bill had passed the Senate but remained stalled in a House committee.
It was the sulfanilamide tragedy that prompted sharp demands for more effective legislation from Campbell, Wallace, and Fishbein of the AMA. Reinforcing these demands were those of the media and the public. Congress reopened its consideration of revising the PFDA. Although drug licensing authority and continuous inspection of drug manufacturing did not become part of the new law as the FDA had hoped, Congress did include a “new drugs” provision: Section 505. This provision clearly defined a new drug and forbade the sale of new drugs in interstate commerce before their manufacturers had demonstrated to the FDA that they were safe. In their application to the FDA, manufacturers had to include samples of the drug, a description of the methods of manufacture, and full reports of investigations made to determine whether or not the drug was safe for use. In the absence of protest by the FDA within a specified amount of time, the drug could be released for interstate sale. Alternatively, the FDA reserved the right to refuse an application, effectively barring a drug from interstate commerce pending appeal. Thus the Federal Food, Drugs, and Cosmetics Act of 1938 (FFDCA) significantly promoted the regulatory authority of the FDA, albeit still falling short of licensing.95 In an insightful analysis of the bureaucratic history of the FDA’s efforts toward revision of food and drug legislation, the historian of medicine Gwen Kay concluded: “Ultimately, one tragedy did what five years of public relations by the FDA could not do, which was to convince members of the House of Representatives that revision of the 1906 act was important.”96
Although arsenic and lead spray residues and the ginger jake paralysis epidemic affected thousands of lives and caught the attention of regulators and consumer advocates, it was the Elixir Sulfanilamide tragedy that ultimately set the stage for the development of a scientific understanding of environmental risk as posed by pesticides. Discontinuation of congressional appropriations restricted the FDA from the study of lead and arsenic insecticides, but this shift proved to be inspirational. The Division of Pharmacology at the FDA developed the LD50 in order to determine the toxicity of glycols and derivatives. Meanwhile, at the University of Chicago, E. M. K. Geiling developed a broad-based approach to the toxicology of new and unfamiliar chemicals. The Division of Pharmacology at the FDA and the Department of Pharmacology at the University of Chicago served as centers for toxicological research during World War II. The research programs at the two institutions diverged after the Elixir Sulfanilamide tragedy, but they would converge in time. In addition, the study of industrial disease had developed as researchers studied workers in lead factories. This research led to the establishment of in-house toxicology laboratories at DuPont, Dow, and Union Carbide. Meanwhile, partially in response to consumer advocacy and partially in response to the Elixir Sulfanilamide tragedy, federal legislators significantly revised the PFDA to produce the Food, Drugs, and Cosmetics Act of 1938, which would define pesticides regulation for several decades. The FFDCA expanded the FDA’s authority in regulating insecticides but failed to provide a provision for licensing. During and after World War II, the Division of Pharmacology at the FDA applied the new techniques of toxicology to DDT, a new insecticide introduced for the control of insect-borne disease in World War II.