Kidney and urinary tract disease
M Magdi Yaqoob, Neil Ashman
Anatomy and Physiology of the Kidney and Urinary Tract
Functional anatomy
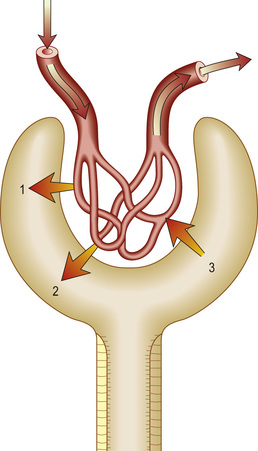
The kidneys are paired organs, 11–14 cm in length in adults, 5–6 cm in width and 3–4 cm in depth. They lie in the retroperitoneum, on either side of the vertebral column at the level of T12–L3 (the right kidney lies lower than the left, pushed down by the liver). Each kidney is enclosed in a fibrous capsule, and has an outer cortex and an inner medulla (Fig. 20.1). There are about 1 million nephrons in each kidney. Each nephron contains a glomerulus, proximal tubule, loop of Henle, distal tubule and collecting duct. All glomeruli lie in the cortex, and tubules dip in and out of the medulla, where the collecting ducts merge to form the ducts of Bellini, emptying at a papilla at the apex of renal pyramid into a calyx. Urine then flows through merging calyces into the renal pelvis, ureters and bladder.
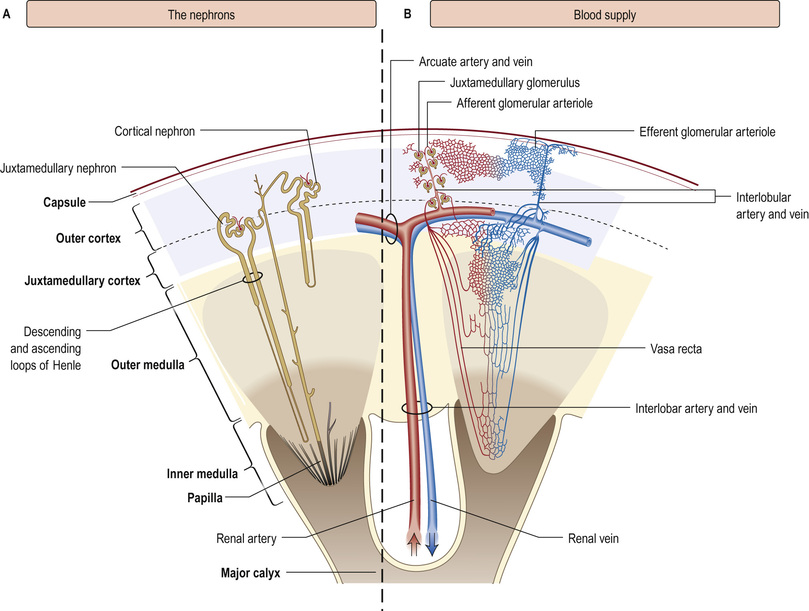
The renal arteries branch off the abdominal aorta, dividing into smaller branches until arterial blood reaches the glomerulus. About 25% of people have dual or multiple renal arteries on one or both sides. Afferent glomerular arterioles arise from interlobular branch arteries to supply the glomerular capillary tuft, which drains into efferent glomerular arterioles. Efferent arterioles from (outer) cortical glomeruli drain into a peritubular capillary network within the renal cortex and then into increasingly large and more proximal branches of the renal vein. By contrast, blood from the (inner) juxtamedullary glomeruli passes via vasa recta in the medulla and returns via the cortex to renal veins that drain into the inferior vena cava.
The renal capsule and ureters are innervated via T10–12 and L1 nerve roots, and renal pain is felt over the corresponding dermatomes.
The nephron
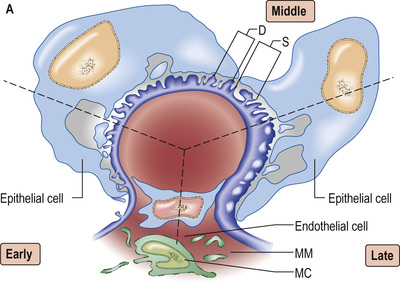
A ball of capillaries makes up the glomerular tuft, enclosed by Bowman's capsule, a chamber lined with specialized parietal epithelial cells that marks the origin of the tubule (Fig. 20.2). The tuft, held together and regulated by mesangial cells, then serves as the filtration barrier, allowing filtrate from plasma to move into the urinary (Bowman's) space. The rate of glomerular filtration is influenced by changes in the contractile tone in either the afferent or the efferent arterioles; for example, efferent vasoconstriction will increase the transglomerular capillary pressure, and increase filtration.
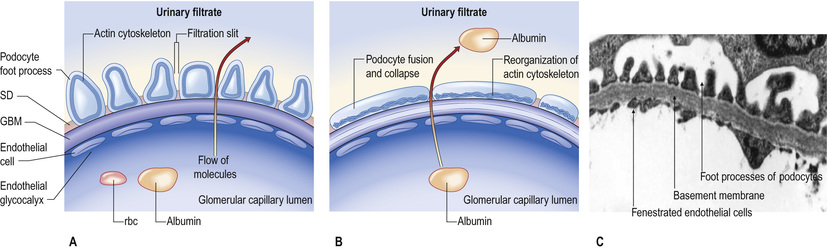
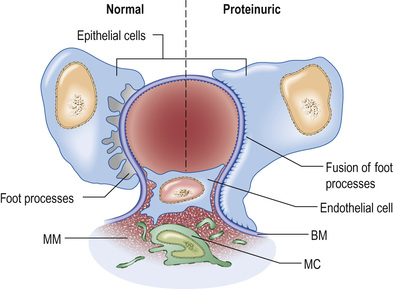
Glomerular capillaries are lined with endothelial cells, fenestrated with 60–80-nm pores, and covered with charged glycocalyx. The glomerular basement membrane (GBM), about 300 nm thick and made of type IV collagen, laminin and heparin sulphate, separates endothelium from podocytes (or visceral epithelial cells). Podocytes anchor on to the GBM by means of an extensive trabecular network of foot processes, and hang into Bowman's space. The interdigitating foot processes of podocytes then form the 40-nm filtration slit, a narrow potential space traversed by a protein ‘zipper’ that may prevent the passage of larger molecules (such as albumin) into the urinary space, and regulates the architecture and function of podocytes.
Mesangial cells (thought to be related to macrophages) sit within the tuft, able to contract and relax to control blood flow and the filtration surface area along the glomerular capillaries in response to a host of mediators. They also secrete the mesangial matrix, which provides the scaffolding for glomerular capillaries.
The renal tubules are lined by epithelial cells, which alter the composition of filtrate to form urine eventually. Proximal tubular cells have a luminal brush border to increase (by 30-fold) the surface area exposed to filtrate, rich in transporters and channels. The loop of Henle is lined with squamous cells, which are more permeable to water than solute, and cuboidal epithelium, when the reverse is true. The distal tubule regulates electrolytes and pH through cuboidal epithelium, and the cortical portion of the collecting ducts contains two cell types with different functions: principal cells (sodium, potassium and water) and intercalated cells (acid–base; see p. 153). Finally, resident interstitial fibroblast-like cells in the renal cortex produce erythropoietin in response to hypoxia (see p. 728).
The juxtaglomerular apparatus
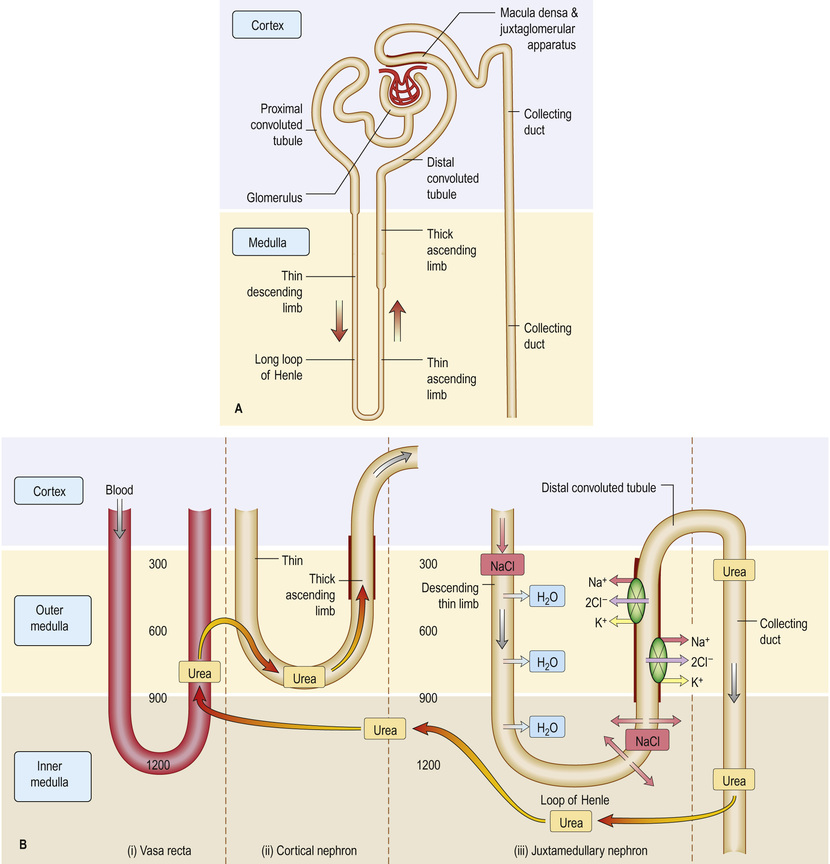
The juxtaglomerular apparatus regulates flow and filtration in each individual nephron. Columnar epithelium in the macula densa (Fig. 20.3) senses the concentration of tubular fluid sodium (higher filtrate flow means more delivered sodium), triggering adenosine-mediated vasoconstriction of the afferent arteriole to drop glomerular filtration (so-called tubule–glomerular feedback). Juxtaglomerular cells secrete renin, able to induce aldosterone release, allowing the apparatus to monitor flow, and respond when necessary to drop glomerular filtration rate (GFR) and retain salt to maintain fluid balance.
Physiology
A hydrostatic pressure gradient of approximately 10 mmHg (a capillary pressure of 45 mmHg minus 10 mmHg of pressure within Bowman's space and 25 mmHg of plasma oncotic pressure) provides the driving force for ultrafiltration of virtually protein-free and fat-free fluid across the glomerular filter into Bowman's space and so into the renal tubule (Fig. 20.4).
The ultrafiltration rate (GFR) varies with age and sex but is approximately 120–130 mL/min per 1.73 m2 surface area in adults. This means that, each day, ultrafiltration of 170–180 L of water and unbound small-molecular-weight constituents of blood occurs. If these large volumes of ultrafiltrate were excreted unchanged as urine, it would be necessary to drink huge amounts of water and salts to stay in balance.
Absorption of solutes
Essential electrolytes and other blood constituents, such as glucose and amino acids, are absorbed from filtrate in transit along the long course of the nephron (see Fig. 20.3).
Sodium filters freely, and 60–80% of filtered sodium (and water) is reabsorbed in the late proximal tubule, where the apical membrane Na+/H+ exchanger (NHE3) trades hydrogen ions into the lumen for absorbed sodium, with anionic chloride (Cl−) accompanying sodium to maintain electric neutrality. Secreted H+ allows bicarbonate (HCO3−), formed from water and CO2 by cellular and luminal carbonic anhydrase, to exit the basolateral membrane with absorbed Na+. Around 25% is then absorbed in the thick ascending limb of the loop of Henle, as NaCl by the Na+/K+/2Cl− co-transporter (NKCC). The remaining 5% is absorbed by the thiazide-sensitive NaCl co-transporter and the epithelial sodium channel (ENaC), in the principal cell of the collecting duct. Of the recommended allowance of <6 g/day of salt (equivalent to around 2 g Na+), around 5–10% will be excreted in urine, stool and sweat.
Potassium is freely filtered at the glomerulus, largely reabsorbed in the proximal tubule, and secreted in the distal tubule and collecting ducts. A clinical consequence of this is that the ability to eliminate unwanted potassium is less dependent on GFR than is the elimination of urea or creatinine. Virtually all bicarbonate, glucose and amino acids (Fig. 20.3B) are absorbed in the proximal tubule, making use of the large surface area of this segment of the nephron.
The proximal tubule has a maximal absorptive capacity for many compounds. For instance, if blood glucose levels are elevated above the normal range, the amount filtered (filtered load = GFR × plasma concentration) may exceed the maximal absorptive capacity of the proximal tubule and glucose will ‘spill over’ into the urine as glycosuria. Inherited or acquired defects in tubular function may also lead to incomplete absorption of a normal filtered load, and substances such as glucose (renal glycosuria), amino acids, phosphate, sodium, potassium and calcium will appear in the urine, either singly or in combination. Examples include cystinuria (a defect of a specific amino acid transporter) or Fanconi syndrome (see pp. 1286–1287).
Other compounds filtered and reabsorbed or secreted to a variable extent include urate, many organic acids and many drugs or their metabolic breakdown products. The more tubular secretion of a compound that occurs, the less dependent elimination is on the GFR.
Absorption of water
Urine is concentrated by the countercurrent multiplier mechanism in the loop of Henle, the medullary interstitium, medullary blood vessels (vasa recta) and, finally, the collecting ducts (see pp. 153–154). Sodium and urea in water flow as filtrate into the descending loop, permeable to water and impermeable to sodium. In the thick ascending limb (impermeable to water), active absorption of sodium by the Na+/K+/2Cl− co-transporter into the interstitium increases the tissue osmolarity. Because of the hairpin nature of the loop, water from the permeable descending limb enters the interstitium by osmosis, where the vasa recta return fluid to the circulation. Constant active absorption of sodium in the ascending loop multiplies this process over time, and the solute concentration at the tip of the loop is fourfold that of extracellular fluid. By this mechanism, salt and water are returned to the circulation. Filtrate entering the collecting duct is increasingly concentrated by the action of antidiuretic hormone (ADH, or vasopressin), leading to the release of intracellular aquaporin water channels, which insert themselves across the apical membrane. Water then enters the cell along an osmotic gradient. When the effect of ADH wears off, water channels return to the cell cytoplasm (see Fig. 9.5). The final urine volume is 1–2 L daily.
Glomerular filtration rate
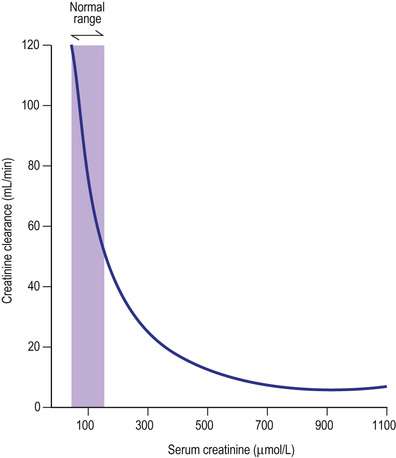
In health, the GFR remains constant owing to intrarenal regulatory mechanisms. In disease (e.g. a reduction in intrarenal blood flow, damage to or loss of glomeruli or obstruction to the free flow of ultrafiltrate along the tubule), the GFR will fall. The ability to eliminate wastes and to regulate the volume and composition of body fluid will decline. This is measured as a rise in the plasma urea or creatinine, or as a reduction in measured GFR.
The concentration of urea or creatinine in plasma represents the dynamic equilibrium between production and elimination. In healthy subjects, there is an enormous reserve of renal excretory function, and serum urea and creatinine do not rise above the normal range until there is a reduction of 50–60% in the GFR (Fig. 20.5). Thereafter, the level of urea depends on both the GFR and its production rate (Box 20.1). The latter is heavily influenced by protein intake and tissue catabolism. The level of creatinine is much less dependent on diet but is more related to age, sex and muscle mass. Once it is elevated, serum creatinine is a better guide to GFR than urea and is widely used to monitor further deterioration in the GFR.
 Box 20.1
Box 20.1
Factors influencing serum urea levels
| Production | Elimination |
| Increased by | |
| Elevated GFR, e.g. pregnancy | |
| Decreased by | |

Measuring or estimating the GFR
If a substance is filtered, and then unmodified by the tubules as it passes along the nephron, the concentration of that substance per mL will be the same in blood and urine. Accurate calculations of the GFR, particularly in cases where the urea and creatinine may be in the normal range or near normal, can be assessed by cystatin C and creatinine clearance. Alternatives for quantifying the true GFR include measuring iohexol or inulin clearance, or radio-isotope (51Cr-EDTA) GFR. Neither is practical for daily clinical practice.
Cystatin C
This is a freely filtered, low-molecular-weight protein that appears to be a more accurate marker of kidney function than creatinine. Blood concentrations are less affected by muscle mass, age and gender, but measurement of cystatin C has not widely entered clinical practice as yet.
Creatinine clearance
Daily production of creatinine (principally from muscle cells) is fairly constant and little affected by protein intake. Serum creatinine and excreted (urinary) creatinine vary little throughout the day. Small quantities of creatinine are secreted into the tubule but this does not usually affect the assay. Urine is collected over 24 hours for measurement of urinary creatinine. A single plasma level of creatinine is measured some time during the 24-hour period, and a creatinine clearance calculated as U × VP, where U = urine concentration of creatinine, V = rate of urine flow in mL/min, and P = plasma concentration of creatinine. Normal ranges are 90–140 mL/min in men and 80–125 mL/min in women.
Estimated or calculated GFR – the eGFR
Measurement of GFR is cumbersome and time-consuming, and may be inaccurate if 24-hour urine collections are incomplete. Several formulae have been developed that predict creatinine clearance or GFR from serum creatinine and patient characteristics, often derived from large trials. Variables might include age, weight, gender and ethnicity. Commonly used equations are displayed in Box 20.2. All have their shortcomings, and are less accurate the closer a GFR is to normal (so patients with seemingly abnormal calculated GFRs may have normal kidney function). Of the equations, the CKD-EPI equation is more accurate than the modification of diet in renal disease (MDRD) study equation overall and is most reliable for predicting eGFR >60 mL/min/1.73 m2.
Box 20.2
Estimation of glomerular filtration rate (GFR)
To convert creatinine values in µmol/L to mg/dL, multiply by 0.0113.
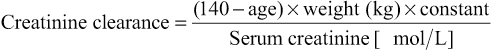
Modification of diet in renal disease (MDRD) equation
Calculation of estimated GFR by four variables:
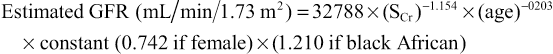
CKD Epidemiology (CKD-EPI) Collaboration equation
GFR (mL/min/1.73 m2) = 141 × min (serum creatinine/κ, 1)α × max (serum creatinine/κ, 1)−1.209 × 0.993Age × 1.018 [if female] × 1.159 [if black]
where κ is 0.7 for females and 0.9 for males, α is −0.329 for females and −0.411 for males, min indicates the minimum or serum creatinine/κ or 1, and max indicates the maximum of serum creatinine/κ or 1.
(Adapted from MacGregor MS, Boag DE, Innes A. Chronic kidney disease. Quarterly Journal of Medicine 2006; 99:365–375.)
All these equations have not, however, been fully validated across all ranges of renal impairment, weights or body mass index (BMI), or in all ethnic groups. However, for monitoring patients with acute or chronic kidney disease, the convenience and ease of the eGFR has led to its widespread adoption.
Drugs, toxins, proteins and the kidney
A substantial fraction of prescription drugs is handled and eliminated by the kidney. Many of these medications (e.g. penicillins, cephalosporins, diuretics, non-steroidal anti-inflammatory drugs (NSAIDs) and antivirals) circulate in the plasma as small organic anions, and are actively eliminated in the proximal tubule by a specific organic anion transporter (OAT) system.
The kidney is also a major site for the catabolism (and so elimination) of many small-molecular-weight proteins and polypeptides, including many hormones such as insulin, parathyroid hormone (PTH) and calcitonin, by endocytosis carried out by the megalin–cubilin complex in the brush border of proximal tubular cells. In chronic kidney disease (CKD), the metabolic clearance of these substances is reduced and their half-life is prolonged. This accounts, for example, for the reduced insulin requirements of patients with diabetes as their renal function declines.
Endocrine function
Renin–angiotensin system
(See Fig. 20.6.) The juxtaglomerular apparatus (JGA) regulates flow and filtration in each individual nephron. Columnar epithelium in the macula densa (see Fig. 20.3) senses the concentration of tubular fluid sodium (higher filtrate flow means more delivered sodium), triggering adenosine-mediated vasoconstriction of the afferent arteriole to drop glomerular filtration (tubuloglomerular feedback). Juxtaglomerular cells secrete renin, able to induce aldosterone release, allowing the apparatus to monitor flow, and respond when necessary to drop GFR and retain salt to maintain fluid balance.
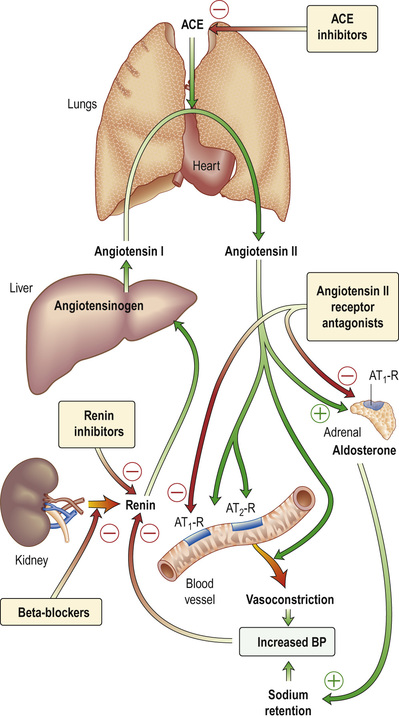
Pro-renin, synthesized by specialized arteriolar smooth muscle cells in the JGA, is cleaved into the active proteolytic enzyme, renin. Active renin is stored in the JGA, and released in response to triggers related to a falling intravascular volume, pressure or increased fluid losses via the kidney. Specifically, these are:
• pressure changes in the afferent arteriole
• chloride and osmotic concentration in the distal tubule via the macula densa (see Fig. 20.3A) – tubuloglomerular feedback
In the blood, renin converts angiotensinogen, an α2-globulin of hepatic origin, to inactive angiotensin I. Angiotensin I is further cleaved by angiotensin-converting enzyme (ACE, present in lung and vascular endothelium) into active angiotensin II (AII), which has two major actions (mediated by two types of receptor, AT1 and AT2). When AII binds the AT1 subtype (found in the heart, blood vessels, kidney, adrenal cortex, lung and brain), this binding mediates vasoconstriction. AII binding AT2 is probably involved in vascular growth.
AII:
• causes rapid, powerful vasoconstriction
• stimulates the adrenal zona glomerulosa to increase aldosterone production (over hours or days), leading to sodium (and water) absorption in the collecting duct
• causes vasoconstriction of efferent (but also, to a lesser extent, afferent) renal arterioles, resulting in increase of glomerular capillary pressure to maintain GFR.
The net result is that AII has opposing effects on the regulation of GFR:
• an increase in glomerular pressure and consequent rise in GFR
• reduction in renal blood flow and mesangial cell contraction, reducing filtration (see Fig. 20.44).
In renal artery stenosis with resultant low perfusion pressure, AII maintains GFR.
The renin–angiotensin system can be blocked at several points with renin inhibitors, ACE inhibitors and angiotensin II receptor antagonists (AII-RAs). These are useful agents in the treatment of hypertension and heart failure (see pp. 1047 and 985) but have differences in action: ACE inhibitors also block kinin production while AII-RAs are specific for AT1 receptors.
Erythropoietin
Erythropoietin (see also p. 519) is the major stimulus for erythropoiesis, the synthesis of red cells. It is a glycoprotein produced principally by fibroblast-like cells in the renal interstitium.
• Under hypoxic conditions, both the α and β subunits of hypoxia inducible factor 2 (HIF-2) are expressed, forming a heterodimer and causing erythropoietin gene transcription. Erythropoietin, once formed, binds to its receptors on erythroid precursor cells, to maintain normal red cell synthesis.
• Under normal oxygen conditions, only the HIF-2-β subunit is constitutively expressed. The α subunit undergoes proline hydroxylation in the presence of iron and oxygen.
• The hydroxylated HIF-2-α subunit binds to von Hippel–Lindau protein, with the activating ubiquitination (see p. 104) and subsequent degradation of HIF-2-α via proteosomes so that no erythropoietin is transcribed.
This and other hydroxylation steps have an absolute requirement for molecular oxygen; this forms the basis of oxygen sensing.
Loss of renal substance, with decreased erythropoietin production, results in a normochromic, normocytic anaemia. Conversely, erythropoietin secretion may be increased, with resultant polycythaemia, in people with polycystic renal disease, benign renal cysts or renal cell carcinoma.
Recombinant human erythropoietin has been biosynthesized and is available for clinical use, particularly in people with CKD (see p. 778).
Vitamin D metabolism
Naturally occurring vitamin D (see also p. 708; cholecalciferol) requires hydroxylation in the liver at position 25 and again by a renal 1α-hydroxylase enzyme to produce the metabolically active 1,25-dihydroxycholecalciferol (1,25-(OH)2D3).
Activity of 1α-hydroxylase is increased by:
Both 1,25-dihydroxycholecalciferol and 25-hydroxycholecalciferol are degraded in part by being hydroxylated at position 24 by 24-hydroxylase. The activity of this enzyme is reduced by PTH and increased by 1,25-(OH)2D3 (which therefore promotes its own inactivation).
Reduced 1α-hydroxylase activity in diseased kidneys leads to relative deficiency of 1,25-(OH)2D3. As a result, gastrointestinal calcium and, to a lesser extent, phosphate absorption is reduced and bone mineralization impaired. Reduced gut calcium absorption leads to hypocalcaemia, which is sensed by a specific calcium-sensing receptor (CaSR) on parathyroid glands, and in turn induces release of PTH. Receptors for 1,25-(OH)2D3 are also found on parathyroid glands, and reduced receptor binding alters the set-point for release of PTH when plasma calcium falls. This combination contributes to the (common) secondary hyperparathyroidism seen in patients with CKD, even of modest degree.
Autocrine function
Endothelins
The endothelins (ETs) are a family of potent vasoactive peptides that also influence cell proliferation, tissue fibrosis and epithelial solute transport. They do not circulate but act locally. ETs are produced by most types of cell in the kidney. When binding ET-type A receptor, ET mediates vasoconstriction. When binding to ET-type B receptor, ETs cause vasodilatation. Through vasoconstriction by ETA and salt and water retention via ETB receptors, ETs cause hypertension.
Prostaglandins
Prostaglandins are unsaturated, oxygenated fatty acids, derived from the enzymatic metabolism of arachidonic acid, mainly by constitutively expressed cyclo-oxygenase-1 (COX-1) or inducible COX-2 (see Fig. 24.30). COX-1 is highly expressed in the collecting duct, while COX-2 expression is restricted to the macula densa. Both COX isoforms convert arachidonic acid to unstable prostaglandin H2 (PGH2). PGH2 is then converted to:
• PGE2 in the collecting duct, responsible for natriuretic and diuretic effects
• PGD2, which is of undetermined significance, produced in the proximal tubule
• prostacyclin (PGI2), synthesized in the interstitium and vessels
• thromboxane A2, a vasoconstrictor, mainly synthesized in the glomerulus.
They all maintain renal blood flow and GFR in the face of vasoconstrictors such as angiotensin II, catecholamines and α-adrenergic stimulation. Inhibition of prostaglandin synthesis by NSAIDs results in a further reduction in GFR, which is sometimes sufficiently severe as to cause acute kidney injury (AKI), particularly in the elderly, with volume depletion, or where ACE inhibitors or AII-RAs are used.
Natriuretic peptides
Atrial natriuretic peptides (ANPs) are secreted from granules in the cardiac atria in response to atrial stretch (as might be caused by increased venous return or by volume overload). They produce marked sodium and water excretion and increase GFR rate. ANP is also a direct vasodilator, lowering blood pressure; it reduces renin release and aldosterone secretion, and consequently inhibits AII synthesis. Their effects are to reverse salt and water retention. Brain natriuretic peptide (BNP) is found in the ventricle as well as the brain and has sequence homology with ANP. Normally, its circulating level is 25% less than for ANP but may exceed it in congestive cardiac failure (see p. 983).
Nitric oxide and the kidney
Nitric oxide (see Fig. 25.20), a molecular gas, is formed by the action of three isoforms of nitric oxide synthase (NOS), all of which are expressed in the kidney: eNOS is found in vessels, nNOS mainly in the macula densa and inner medullary collecting duct, and iNOS in several tubule segments. Nitric oxide binds soluble guanylate cyclase, enhancing the synthesis of cyclic guanosine monophosphate (cGMP) from guanosine triphosphate (GTP), and mediates the following physiological actions in the kidney:
• regulation of renal perfusion and glomerular pressure
• natriuresis, by inhibiting Na+/K+-ATPase and NHE3
• modulation of tubule–glomerular feedback (see p. 724).
Clinical Approach to the Patient with Kidney and Urinary Tract Disease
Investigation of kidney and urinary tract disease
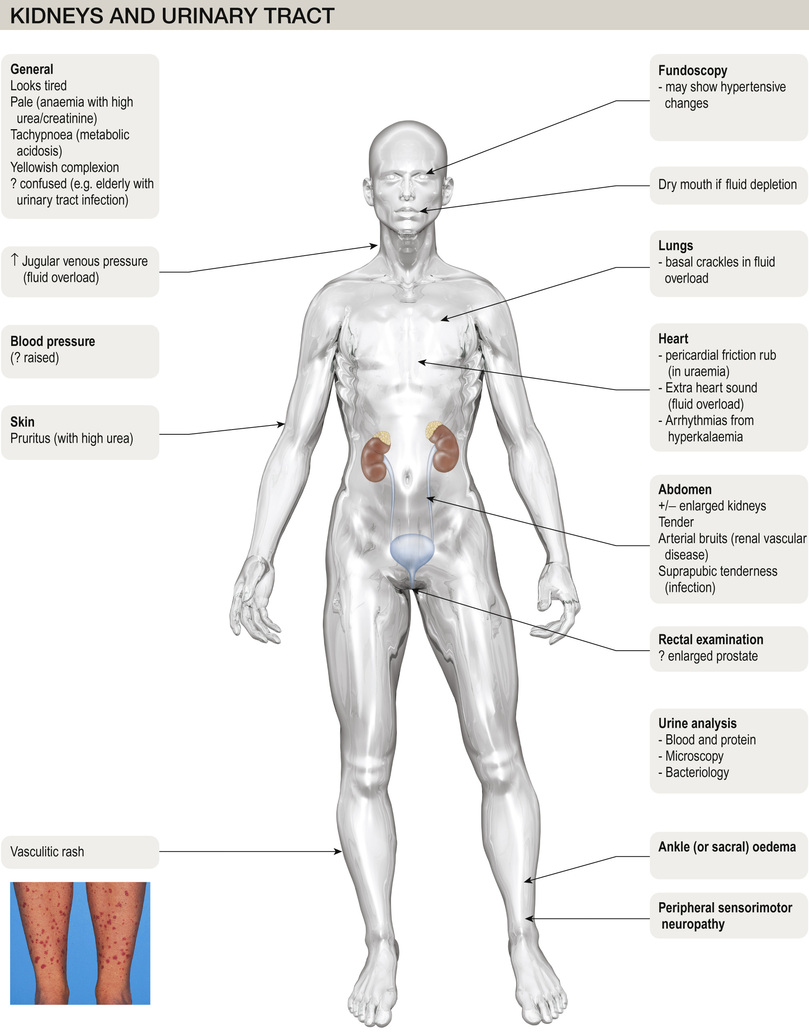
Examination of the urine
Appearance
Unless urine is visibly bloody (or cola-coloured, in cases of myoglobinuria or haemoglobinuria), inspection of the urine is not helpful. Cloudy, offensive-smelling urine may denote infection, but this should be further tested by dipsticks.
Volume
In temperate climates, healthy adults will pass between 800–2500 mL per 24 hours (roughly 1 mL/kg/h). Usually, the minimum urine output capable of maintaining solute excretion for health (without accumulation in the body) is around 650 mL. If the kidney loses concentrating capacity (as occurs in CKD or diabetes insipidus), higher urine volumes are needed for the same daily solute output, and urine outputs may rise well above 2500 mL. Nocturia is a symptom of kidney disease, as patients without the ability to concentrate solute into a small urine volume will waken, needing to pass urine during the night. High daily volumes are also seen with glycosuria or increased protein catabolism following surgery, as the solute load requiring excretion is higher.
Specific gravity and osmolality
Urine specific gravity (SG, where <1.008 is dilute and >1.020 is concentrated) is a measure of the weight of dissolved particles in urine, whereas urine osmolality reflects the number of such particles. Usually, the relationship between the two is close. Measurement of urine SG or osmolality can be helpful in confirming loss of concentrating ability (as might be seen when tubular function fails in acute tubular necrosis or CKD). It can also be helpful in oliguric patients, when a high SG might suggest pre-renal AKI, as opposed to established acute tubular necrosis (see pp. 771–772).
Urinary pH
Measurement of urinary pH (usually 5.5–6.5) is helpful only in the investigation and treatment of renal tubular acidosis (see pp. 177–178).
Dipsticks (chemical testing) and urine microscopy
Dipsticks are cheap and hugely helpful in investigating suspected kidney disease, using reagents fixed on pads that change colour on reacting with specific elements in urine. Microscopy on a mid-stream sample spun at 2000 rpm is essential to understand dipstick findings fully.
Blood
Haematuria may originate from anywhere in the urinary tract. Once found on dipstick, a mid-stream sample should be examined for red cells or casts. Red cells (usually 1–2 per high-power field) are described as dysmorphic if they originate from the early nephron (glomerular bleeding), and may be accompanied by red-cell casts. Casts are cylindrical bodies, moulded in the shape of the distal tubular lumen. Red-cell casts – even if only single – always indicate renal disease. A dipstick that is strongly positive for haematuria with no red cells seen on microscopy might suggest haemoglobinuria or myoglobinuria. Bleeding may come from any site within the urinary tract (Fig. 20.7):
• Overt bleeding from the urethra is suggested when blood is seen at the start of voiding and then the urine becomes clear.
• Blood diffusely present throughout the urine comes from the bladder or above.
• Blood only at the end of micturition suggests bleeding from the prostate or bladder base.
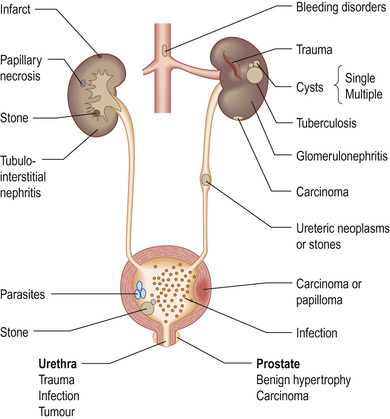
Women will commonly have dipstick-positive haematuria during a period; it is usually worth repeating testing after menstruation.
Protein
Proteinuria is one of the most common signs of renal disease. Dipsticks will detect proteinuria from around 50–150 mg/L, and may be designed to test for either albuminuria or proteinuria. They will not detect light chains or immunoglobulins (Bence Jones proteins). Normal individuals excrete less than 20 µg of albumin per minute (30 mg in 24 h). Dipsticks, however, detect albumin only in a concentration above 200 µg/min (300 mg per 24 h if urine volume is normal). If two separate urine samples find proteinuria on dipstick testing, a random urine protein : creatinine ratio (uPCR) should be measured. This convenient test has largely replaced formal (timed) 24-hour urine collections for proteinuria; an alternative is the albumin : creatinine ratio (ACR). As albumin is usually the dominant protein lost in the urine, patients who have albuminuria always have proteinuria (and the urine PCR will be higher than the urine ACR). Confusingly, the terms are often used interchangeably. For people with diabetes, the urine ACR has particular prognostic significance, and is usually the preferred screening test (see p. 1269) for the microalbuminuria found in early diabetic kidney disease.
Both tests are expressed as mg (protein)/mmol creatinine. Generally, an ACR of 2.5–20 mg/mmol corresponds to albuminuria of 30–300 mg daily (crudely, multiply the result by a factor of 15 – so an ACR of 50 mg/mmol = 750 mg/day albuminuria). Similarly, a uPCR of 3–30 mg/mmol corresponds to 30–300 mg daily (multiplying by a factor of 10 for this test – so a PCR of 50 mg/mmol = 500 mg proteinuria per day). The uPCR assay is relatively cheap and identifies patients whose proteinuria is of tubular and glomerular origin. ACR or PCR levels independently predict all-cause and cardiovascular mortality in the general population in addition to better risk stratifications of patients with CKD for future renal outcomes. Pyrexia, exercise and adoption of the upright posture (postural proteinuria) all increase urinary protein output but are benign. Proteinuria may associate with coarse granular casts seen on urine microscopy.
Glucose
Renal glycosuria is uncommon, so that a positive test for glucose might prompt consideration of diabetes mellitus. Dipsticks for glucose are very sensitive, however.
Bacteria and pus cells
Dipstick tests for bacteriuria are based on the detection of nitrite produced from the reduction of urinary nitrate by bacteria, and also on the detection of leucocyte esterase, an enzyme specific for neutrophils. Although each test on its own has limitations, a positive reaction with both tests has a high predictive value for urinary tract infection (see pp. 763–764). Where positive, a mid-stream sample should be sent for microscopy, culture and sensitivities (MC&S). White blood (pus) cells (WBCs) may be seen on microscopy, as may bacteria. A measurement of ≥10 WBCs/mL in fresh mid-stream urine samples is abnormal and suggests urinary tract infection (UTI). Not all pyuria is UTI, though; pus cells are seen with renal stones, tubule-interstitial nephritis, papillary necrosis, tuberculosis and interstitial cystitis. White cell casts may be seen with (and are more characteristic of) acute pyelonephritis.
Blood and quantitative tests
The use of serum urea, creatinine and GFR as measures of renal function is discussed on pages 726–727. Other quantitative tests of disturbed renal function are described under the relevant disorders, as are diagnostic tests, such as anti-neutrophil cytoplasmic antibody (ANCA), immunofluorescence and complement.
Imaging techniques
Ultrasound of the kidneys and bladder is safe and non-invasive, avoiding ionizing radiation and intravascular contrast medium. In renal diagnosis, it is the imaging method of choice for:
• Assessing renal size and symmetry (normal-sized kidneys with abnormal function suggest an acute cause, as kidneys scar as they fail, and may shrink in length and volume), and allowing directed renal biopsy.
• Ruling out obstruction, either of the bladder and ureters (with unilateral or bilateral hydronephrosis), or of the kidney itself (where pelvicalyceal dilatation may suggest high ureteric or pelvic disease).
• Characterizing renal masses as cystic (either simple cysts or polycystic kidneys) or complex and solid (benign and malignant renal tumours, or infected collections).
• Guiding intervention aimed at relieving obstruction (percutaneous nephrostomy).
• Confirming renal vein patency, and suggesting (but not proving) renal artery disease, in the case of Doppler ultrasonography (duplex).
• Looking for bladder tumours or stones. A scan obtained after voiding (post-micturition) allows bladder emptying to be assessed.
Computed tomography
Unenhanced computed tomography (CT) is the first-line investigation for cases of ureteric colic and suspected renal calculi. It has superseded excretion urography (also known as intravenous urography (IVU) or intravenous pyelography (IVP)).
Multislice detector CT has both improved image resolution and allowed reconstruction of the imaging data in a variety of planes. CT is also used to:
• characterize renal masses that are indeterminate at ultrasonography
• stage renal and bladder tumours
• evaluate the retroperitoneum for tumours, retroperitoneal fibrosis (peri-aortitis) and other causes of ureteric obstruction
Disadvantages include radiation and contrast nephrotoxicity (see p. 774).
Positron emission tomography (PET), using 18-F-fluorodeoxyglucose (FDG), is useful for detection of infection (e.g. in a cyst), inflammation or tumours, and is often used with CT (PET/CT).
Magnetic resonance imaging
Magnetic resonance imaging (MRI) is used as an alternative to CT with no irradiation:
• To stage prostate (but also renal and bladder) cancer.
• To reconstruct the anatomy of the renal arteries using magnetic resonance angiography with gadolinium as contrast medium. In experienced hands, its sensitivity and specificity approach that of renal angiography.
The Food and Drug Administration (FDA) advises not using gadolinium in patients with renal insufficiency because of the development of nephrogenic systemic fibrosis (see pp. 1365 and 781).
Plain X-ray
A plain radiograph of the abdomen may be useful to identify renal calcification or radiodense calculi in the kidney, renal pelvis, and the line of the ureters or bladder (Fig. 20.8).
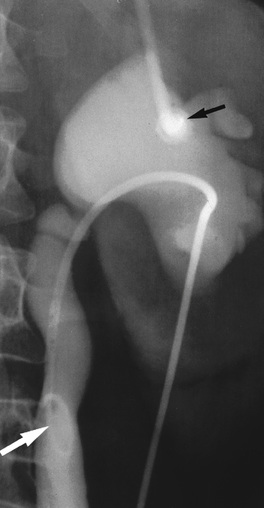
Antegrade pyelography
Antegrade pyelography (Fig. 20.9) involves percutaneous puncture of a pelvicalyceal system with a needle and the injection of contrast medium to outline the pelvicalyceal system and ureter to the level of obstruction. Drains can be sited and stents placed during the procedure. Retrograde pyelography under screening control allows a contrast study of the ureter from the bladder. It is invasive, commonly requires a general anaesthetic, and may result in the introduction of infection.
Micturating cystourethrography
After bladder catheterization, contrast is instilled into the bladder. The catheter is then removed and the patient screened during voiding to check for vesicoureteric reflux and to study the urethra and bladder emptying. Micturating cystourethrography (MCUG) is used in children with recurrent infection (see p. 764). It is rarely appropriate in adults, as with bladder wall hypertrophy in adulthood, vesicoureteric reflux tends to disappear.
Aortography or renal arteriography
Conventional or digital subtraction angiography (DSA) is used both diagnostically, and in cases of suspected renal artery stenosis, to allow therapeutic renal artery balloon angioplasty and stenting. Complications include cholesterol embolization (see pp. 753–754) and contrast-induced kidney damage (contrast nephropathy).
Renal scintigraphy
Isotope studies are helpful for dynamic or static studies of perfusion or excretion. Following venous injection of a bolus of tracer, emissions from the kidney can be recorded by gamma camera. Technetium-labelled diethylenetriaminepenta-acetic acid (99mTc-DTPA) is excreted by glomerular filtration and can be used to confirm renal perfusion. In patients with unilateral renal artery stenosis, there is, typically, a slowed and reduced uptake of tracer with a delay in reaching a peak. Studies carried out before and after administration of an ACE inhibitor may demonstrate a fall in uptake that is suggestive of functional arterial stenosis. Both false-positive and false-negative results are common, particularly in patients with CKD, and renal arteriography remains the ‘gold standard’ in the diagnosis of renal artery stenosis.
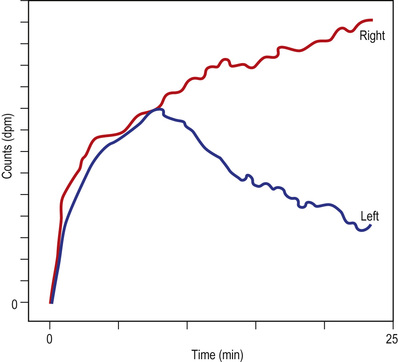
Dimercaptosuccinic acid labelled with technetium (99mTc-DMSA) is filtered by the glomerulus and then binds to proximal tubular cells. Static studies are useful to assess the relative contribution in function of asymmetric kidneys. 99mTc-DMSA is also useful in highlighting ‘photon-deficient’ areas (where isotope is not seen), suggestive of scars or infarction, when compared to healthy tissue uptake. Mercapto-acetyltriglycine (MAG3) labelled with technetium (99mTc) is excreted by renal tubular secretion, so resistance to flow in the pelvis or ureter (with obstruction) prolongs the parenchymal transit of tracer with a delay in emptying the pelvis. On whole-kidney renograms, the time–activity curve fails to fall after an initial peak, or continues to rise (Fig. 20.10), confirming hold-up to flow. Furosemide may be given to exaggerate urine output and emphasize the delay, in order to aid diagnosis.
Transcutaneous renal biopsy
Renal biopsy (Box 20.3) is carried out under ultrasound control in specialized centres and requires interpretation by an experienced pathologist. It is helpful in the investigation of the nephritic and nephrotic syndromes, acute and chronic kidney disease, haematuria after urological investigations and renal graft dysfunction. Native renal biopsy material must be examined by conventional histochemical staining, by electron microscopy, and by immunoperoxidase or immunofluorescence. Techniques like in situ hybridization and polymerase chain reaction (PCR) analysis are also widely used in renal biopsy specimens.
 Box 20.3
Box 20.3
Transcutaneous renal biopsy
The complications of transcutaneous renal biopsy are shown in Box 20.4.
Box 20.4
Complications of transcutaneous renal biopsy
• Macroscopic haematuria – up to 10%
• Pain in the flank, sometimes referred to the shoulder tip
• Arteriovenous aneurysm formation – about 20%, almost always of no clinical significance
• Profuse haematuria demanding blood transfusion – 1–3%
• Profuse haematuria demanding occlusion of the bleeding vessel at angiography or nephrectomy – approximately 1 in 400
Glomerular disease is usually described by kidney biopsy findings. Commonly used terms are shown in Box 20.5.
Box 20.5
Glomerular disease: commonly used terms
• Focal: some, but not all, glomeruli show the lesion
• Diffuse (global): most of the glomeruli (>75%) contain the lesion
• Segmental: only a part of the glomerulus is affected (most focal lesions are also segmental, e.g. focal segmental glomerulosclerosis)
• Global: all of the glomerulus is symmetrically involved
• Proliferative: an increase in cell numbers due to hyperplasia of one or more of the resident glomerular cells with or without inflammation
• Membrane alterations: capillary wall thickening due to deposition of immune deposits or alterations in basement membrane
• Crescent formation: epithelial cell proliferation with mononuclear cell infiltration in Bowman's space
The Glomerulus and Glomerular Disease
A glomerulus consists of a collection of capillaries seated within Bowman's capsule in the urinary space. Blood flows in via the afferent arteriole, and exits via the efferent arteriole. Filtrate leaves Bowman's space into the proximal tubule. The capillary tuft is supported by mesangial cells and mesangial matrix. Filtrate moves from the capillary lumen into the urinary space across the glomerular filter (see p. 724). Three elements are involved in allowing or preventing filtration: endothelium, the glomerular basement membrane (GBM) and podocytes (Fig. 20.11).
Filtration barrier (slit diaphragm)
The glomerular filtration barrier (see Fig. 20.2) consists of the fenestrated endothelium, the GBM and the terminally differentiated visceral epithelial cells known as podocytes. Podocytes attach to the GBM by foot processes via adhesion molecules, such as α3β1 and dystroglycans. Adjacent podocytes are joined laterally via their foot process by slit diaphragms, which bridge across the filtration slits. The various proteins comprising the slit diaphragm include nephrin, CD2-associated protein (CD2AP), canonical transient receptor potential channel 6 (TRPC6), podocin, P-cadherin, α- and β-catenin, and zonula occludens-1 (ZO-1). These co-localize within the subcellular domain to function as a molecular sieve. These proteins, in addition to providing structural support to the cytoskeletal proteins like filamentous actin, also have signalling functions in order to maintain the normal function of podocytes. Abnormalities in any of these proteins result in the breakdown of the filtration barrier with consequent torrential leak of macromolecules.
Podocyte changes
The podocyte structure (see above) is maintained by actin, which supports the cytoskeleton (see Fig. 20.2). A rearrangement of the fluid actin cytoskeleton leads to foot process effacement (flattening). As the architecture of the filtration slit is now disrupted, albumin leaks into the urine, and recovery of the cytoskeleton reverses proteinuria. The cytoskeleton can be altered by:
Glomerular disease
Glomerular disease is the third most common cause (after diabetes and hypertension) of end-stage kidney disease (ESKD) in Europe and the USA, accounting for some 10–15% of such patients. These are diseases in which:
• there may be an immunologically mediated inflammatory injury to glomeruli, or structural or functional glomerular damage without inflammation
• renal interstitial damage is a regular accompaniment
• the kidneys are involved symmetrically
• secondary mechanisms of glomerular injury may come into play following an initial immune insult, such as fibrin deposition, platelet aggregation, neutrophil infiltration and free radical-induced damage
• haemodynamic consequences of a primary injury may further disturb glomerular function
• renal lesions may be part of a generalized disease (e.g. systemic lupus erythematosus, SLE).
Describing glomerular disease
The nomenclature for glomerular disease can be confusing, as descriptive terms (as seen on histology) overlap with clinical syndromes and more recent molecular insights into the pathogenesis of disease. If there is predominant inflammation on histology, glomerular disease may be described as a glomerulonephritis. If inflammation is absent, glomerulopathy is more correct. There remains much overlap between the two, and the terms are often (wrongly) used interchangeably. It may be better to think about glomerular disease in terms of the predominant compartment involved, where the GBM separates podocytes from mesangial and endothelial cells.
• Podocytes (in the urinary compartment) are principally involved in glomerular diseases (usually glomerulopathies) that present as the nephrotic syndrome, where proteinuria is often heavy.
• Endothelial and mesangial cells (in the endocapillary compartment) are principally involved in glomerular disease presenting as nephritis (glomerulonephritis), where haematuria, proteinuria and often hypertension are equally evident.
• Podocytes, endothelial and mesangial cells may be equally involved where a glomerulonephritis presents with heavy proteinuria and the nephrotic syndrome as well.
Clinical classification of glomerular disease is also often used, although there is no complete correlation between histopathological types and clinical features. Four major glomerular syndromes are often described:
• Nephrotic syndrome: massive proteinuria (>3.5 g/day), hypoalbuminaemia, oedema, lipiduria and hyperlipidaemia. Podocyte malfunction or injury is often causative.
• Glomerulonephritis (nephritic syndrome):
– Acute glomerulonephritis: abrupt onset of glomerular haematuria (red blood cell casts or dysmorphic red blood cells), non-nephrotic-range proteinuria, oedema, hypertension and transient renal impairment, or
– Rapidly progressive glomerulonephritis: features of acute nephritis, focal necrosis with or without crescents, and rapidly progressive renal failure over weeks.
• Mixed nephritic/nephrotic presentations: where glomerulonephritis is part of a systemic disease (e.g. lupus nephritis, cryoglobulinaemia and Henoch–Schönlein purpura), a nephritic syndrome is often associated with the nephrotic syndrome.
Investigation of glomerular diseases is shown in Box 20.6.
Box 20.6
Investigation of glomerular diseases
| Investigations | Positive findings |
| Urine microscopy | Red cells, red-cell casts |
| Urinary protein | Nephrotic or sub-nephrotic-range proteinuria |
| Serum urea | May be elevated |
| Serum creatinine | May be elevated |
| Culture (throat swab, discharge from ear, swab from inflamed skin) | Nephritogenic organism (not always) |
| Antistreptolysin-O titre | Elevated in post-streptococcal nephritis |
| C3 and C4 levels | May be reduced |
| Antinuclear antibody (ANA) | Present in significant titre in systemic lupus erythematosus |
| Antinuclear cytoplasmic antibody (ANCA) | Positive in some vasculitides |
| Anti-glomerular basement membrane (anti-GBM) | Positive in Goodpasture syndrome |
| Cryoglobulins | Increased in cryoglobulinaemia |
| Chest X-ray | Cardiomegaly, pulmonary oedema (not always) |
| Renal imaging | Usually normal |
| Renal biopsy | Any glomerulopathy |
Nephrotic syndrome
Nephrotic syndrome is characterized by:
 Pathophysiology
Pathophysiology
Hypoalbuminaemia
Loss of urinary protein (largely albumin) of the order ≥3.5 g daily in an adult may lead to hypoalbuminaemia. Normal dietary protein intake in the UK is around 70 g daily and the normal liver can synthesize albumin at a rate of 10–12 g daily. How, then, does a daily urinary protein loss of 3.5 g result in hypoalbuminaemia? This can be partly explained by increased catabolism of reabsorbed protein, largely albumin, in the proximal tubules, even though the rate of albumin synthesis is increased.
Proteinuria
Proteinuria occurs partly because structural damage to the glomerular barrier (podocytes, basement membrane, fenestrated endothelium and endothelial charge) allows the passage of more and larger molecules. The filtration slit between podocytes and normal podocyte architecture, and interdigitating podocyte foot processes are critical to maintaining a barrier to protein loss into the urinary space, as is a functional GBM and healthy capillary endothelium (and its charge).
Hyperlipidaemia
This is a consequence of increased synthesis of lipoproteins (such as apolipoprotein B, C-III lipoprotein (a)), as a direct consequence of a low plasma albumin. Low-density lipoprotein (LDL) increases, partly due to upregulation of a liver serine protease, pro-protein convertase subtilisin kexin-9 (PCSK9), which causes internalization of LDL receptors. Very-low-density lipoprotein (VLDL) and/or intermediate-density lipoprotein (IDL) fractions increase, with no change (or a decrease) in HDL (the LDL/HDL cholesterol ratio increases). There is also reduced clearance of the principal triglycerides bearing lipoprotein (chylomicrons and VLDL), as high plasma levels of free fatty acid (FFA) trigger release of appropriately sialylated angiopoietin-like 4 (Angptl4) protein from adipose tissue, heart and skeletal muscles, inhibiting lipoprotein lipase and resulting in hypertriglyceridaemia.
Oedema in hypoalbuminaemia
See page 155.
 Management
Management
General measures
• Initial management should be with dietary sodium restriction and a thiazide diuretic (e.g. bendroflumethiazide 5 mg daily). Unresponsive patients require furosemide 40–120 mg daily with the addition of amiloride (5 mg daily; monitor serum potassium concentration regularly). Nephrotic patients may malabsorb diuretics (as well as other drugs) owing to gut mucosal oedema, and intravenous administration may be needed initially. Patients are sometimes hypovolaemic, and moderate oedema may have to be accepted in order to avoid postural hypotension.
• Normal protein intake is advisable. A high-protein diet (80–90 g protein daily) increases proteinuria and can be harmful in the long term.
• Albumin infusion produces only a transient effect. It is only given to patients who are diuretic-resistant and those with oliguria and uraemia in the absence of severe glomerular damage: for example, in minimal-change nephropathy. Albumin infusion is combined with diuretic therapy, and diuresis often continues with diuretic treatment alone.
• Hypercoagulable states predispose to venous thrombosis. The hypercoagulable state is due to loss of clotting factors (e.g. antithrombin) in the urine and an increase in hepatic production of fibrinogen. Prolonged bed rest should be avoided, as thromboembolism is very common (particularly in membranous nephropathy). Long-term prophylactic anticoagulation may be indicated, and if renal vein thrombosis occurs, permanent anticoagulation is required.
• Sepsis is a major cause of death in nephrotic patients. The increased susceptibility to infection is partly due to loss of immunoglobulin in the urine. Pneumococcal infections are particularly common and pneumococcal vaccine should be given. Early detection and aggressive treatment of infections, rather than long-term antibiotic prophylaxis, constitute the best approach.
• Lipid abnormalities are responsible for an increase in the risk of cardiovascular disease in patients with proteinuria. Treatment of hypercholesterolaemia starts with an HMG-CoA reductase inhibitor (a statin).
• Lastly, ACE inhibitors and/or angiotensin II receptor antagonists (AII-RAs) are indicated for their antiproteinuric properties in all types of glomerulonephropathy, but most especially the nephrotic syndrome. These drugs reduce proteinuria by lowering glomerular capillary filtration pressure (a fall in efferent tone drops the transglomerular capillary pressure, and so protein loss into the urinary space); blood pressure and renal function should be monitored regularly.
Specific measures
Treat the underlying cause of any urinary protein leak. Box 20.7 shows the glomerular lesions commonly associated with the nephrotic syndrome.
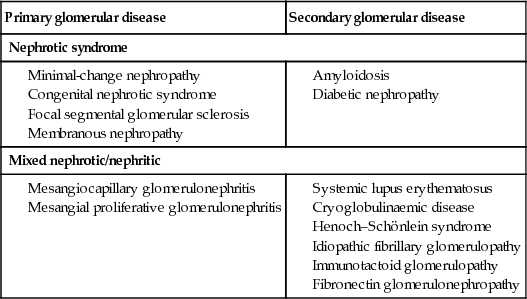
Causes of nephrotic syndrome
 Minimal-change nephropathy (minimal-change disease)
Minimal-change nephropathy (minimal-change disease)
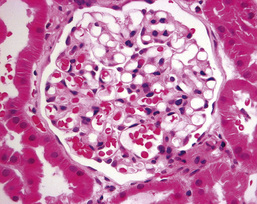
In minimal-change nephropathy (MCN; also called minimal-change disease, MCD), glomeruli appear normal on light microscopy (Fig. 20.12). On electron microscopy, fusion of the foot processes of podocytes is seen, consistent with a disrupted podocyte actin cytoskeleton (see Fig. 20.2B). Neither immune complexes nor anti-GBM antibody can be demonstrated by immunofluorescence on glomerular staining for antibody.
Immature differentiating CD34 stem cells (rather than mature T lymphocytes) appear to be responsible for the pathogenesis of MCN. Podocyte function is also affected by interleukin 13 (IL-13), the production of vascular endothelial growth factor (VEGF), or the upregulation of vascular hyposialylated-angiopoietin-like 4 (ANGPTL4), secreted by the podocytes.
Many drugs have been implicated in MCN, including NSAIDs, lithium, antibiotics (cephalosporins, rifampicin, ampicillin), bisphosphonates and sulfasalazine. Atopy is present in 30% of cases of MCN, and allergic reactions can trigger the nephrotic syndrome. Infections, such as hepatitis C virus (HCV), the human immunodeficiency virus (HIV) and tuberculosis, are rarer causes.
Clinical features
MCN is most common in children, particularly boys, and is responsible for the large majority of cases of nephrotic syndrome in childhood. Proteinuria is usually ‘highly selective’, where albumin, but not higher-molecular-weight proteins such as immunoglobulins, is lost in the urine. Oedema is usual and in children this may present predominantly around the face. The condition accounts for 20–25% of cases of adult nephrotic syndrome. It is often regarded as a condition that does not lead to CKD (but see focal segmental glomerulosclerosis below).
Management
• Manage symptoms with general measures (above).
• High-dose corticosteroid therapy with prednisolone 60 mg/m2 daily (up to a maximum of 80 mg/day) for a maximum of 4–6 weeks, followed by 40 mg/m2 every other day for a further 4–6 weeks, reverses proteinuria in more than 95% of children. Response rates in adults are significantly lower and response may occur only after many months (12 weeks with daily steroid therapy and 12 weeks of maintenance with alternate-day therapy). Spontaneous remission also occurs and steroid therapy should, in general, be withheld if urinary protein loss is insufficient to cause hypoalbuminaemia or oedema. In both children and adults, if remission lasts for 4 years after steroid therapy, further relapse is very rare.
• In children, two-thirds relapse after steroid therapy and further courses of corticosteroids are required. One-third of these children regularly relapse on steroid withdrawal, so that a second-line agent should be added after repeat induction with steroids.
• Cyclophosphamide 1.5–2.0 mg/kg daily is given for 8–12 weeks with prednisolone 7.5–15 mg/day. This increases the likelihood of long-term remission. Steroid-unresponsive patients may also respond to cyclophosphamide. No more than two courses of cyclophosphamide should be prescribed in children because of the risk of side-effects, which include future infertility (azoospermia and premature ovarian failure).
• An alternative to cyclophosphamide is ciclosporin 3–5 mg/kg per day, which is effective but must be continued long-term to prevent relapse on stopping treatment. The antiproteinuric effect of ciclosporin is normally attributed to its immunosuppressive action but may result from the stabilization of the actin cytoskeleton in kidney podocytes. Ciclosporin inhibits the calcineurin-mediated dephosphorylation of synaptopodin (a regulator of actin cytoskeleton) and protects it from cathepsin L-mediated degradation. These results have shed new light on the role of calcineurin signalling in proteinuric kidney diseases. Excretory function and ciclosporin blood levels (recommended trough levels 80–150 ng/mL) must be monitored regularly, as ciclosporin is potentially nephrotoxic.
• Rituximab, a depleting monoclonal antibody directed against CD20, present on B lymphocytes, is showing promise in reducing the number of relapses in frequently relapsing disease, and also in minimizing the immunosuppressant burden in corticosteroid-dependent disease.
• In corticosteroid-dependent children, the anthelminthic agent levamisole 2.5 mg/kg to a maximum of 150 mg on alternate days is also useful in maintenance of remission but its mode of action is unexplained.
Congenital nephrotic syndrome
Congenital nephrotic syndrome (Finnish type) is an autosomal recessively inherited disorder due to mutations in the gene coding for a transmembrane protein, nephrin; it occurs at a frequency of 1 per 8200 live births in Finland. Nephrin is a critical element of the filtration slit, and its loss of function results in massive proteinuria shortly after birth. The disorder can be diagnosed in utero, as increased α-fetoprotein in amniotic fluid is a common feature. Histologically, some glomeruli are small and infantile, whereas others are enlarged and more mature, and have diffuse mesangial hypercellularity. Because of the massive proteinuria, some tubules develop microcysts and are dilated. On electron microscopy, complete effacement of the foot processes of visceral epithelial cells is observed. This condition is characterized by relentless progression to ESKD.
Other inherited nephrotic syndromes involve mutations in other genes that encode other podocyte proteins, such as podocin, α-actinin 4 and Wilms' tumour suppressor gene.
Focal segmental glomerulosclerosis
Focal segmental glomerulosclerosis (FSGS) describes a sclerotic glomerular lesion that affects some (but not all) glomeruli, and some (but not all) segments of each tuft.
Primary focal segmental glomerulosclerosis
Clinical features of primary FSGS
This disease of unknown aetiology usually presents as massive proteinuria (usually non-selective), haematuria, hypertension and renal impairment. The associated nephrotic syndrome is often resistant to steroid therapy. All age groups are affected. It usually recurs in transplanted kidneys, sometimes within days of transplantation, and particularly in patients with aggressive primary renal disease.
Aetiology of primary FSGS
A circulating permeability factor causes the increased protein leak; plasma from patients increases membrane permeability in isolated glomeruli. Kidneys transplanted into murine models of FSGS develop the lesion, but kidneys from FSGS-prone mice transplanted to a normal strain are protected. Removal of this factor by plasmapheresis results in transient amelioration of proteinuria. The identity of the permeability factor remains unknown but recent findings suggest that cardiotrophin-like cytokine 1 is likely a candidate in FSGS. Soluble urokinase-like plasminogen activator receptor (SuPAR) was initially thought to be involved but now appears less likely to be causative, based on recent experimental and clinical evidence. Mutations in the MYO1E gene, which encodes for myosin 1E, found in podocytes, has been described in families with FSGS.
Pathology
Segmental glomerulosclerosis is seen on light microscopy, which later progresses to global sclerosis. The deep glomeruli at the corticomedullary junction are affected first. These may be missed on transcutaneous biopsy, leading to a mistaken diagnosis of MCN (pp. 735–736). A pathogenetic link may exist between MCN and FSGS, as a proportion of cases classified as having the former condition develop progressive CKD, which is unusual. Immunofluorescence shows deposits of C3 and immunoglobulin M (IgM) in affected portions of the glomerulus. The other glomeruli are usually enlarged but may be of normal size. Focal tubular atrophy and interstitial fibrosis are invariably present. Electron microscopy demonstrates primarily foot process effacement, occasionally in a patchy distribution. The degree of podocyte foot process effacement on electron microscopy can help distinguish between ‘primary’ and ‘secondary’ FSGS. If severe foot process effacement is present in normal and sclerosed glomeruli on electron microscopy, primary FSGS is more likely (this is not the case if foot process effacement is largely localized to sclerosed glomeruli alone).
Five histological variants of FSGS exist:
• In classic FSGS (Fig. 20.13A), the involved glomeruli show sclerotic segments in any location of the glomerulus.
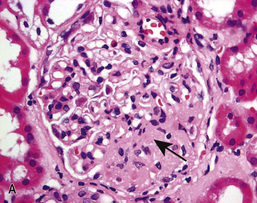
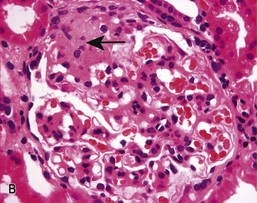
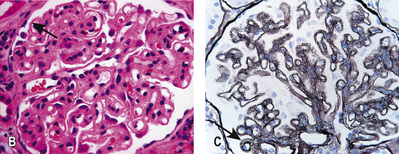
• The glomerular tip lesion is characterized by segmental sclerosis, at the tubular pole of all the affected glomeruli at a very early stage (tip FSGS; Fig. 20.13B). These patients have a more favourable response to steroids and disease runs a more benign course.
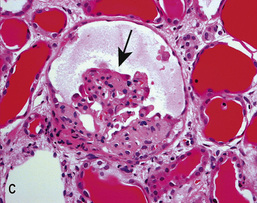
• In collapsing FSGS (Fig. 20.13C), podocytes are usually enlarged and coarsely vacuolated with wrinkled and collapsed capillary walls. Collapsing FSGS is commonly seen in young blacks with HIV infection or disease, and is known as HIV-associated nephropathy (HIVAN; see below).
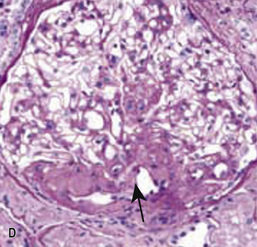
• The perihilar variant (Fig. 20.13D) consists of perihilar sclerosis and hyalinosis in more than 50% of segmentally sclerotic glomeruli. It is frequently observed with secondary FSGS.
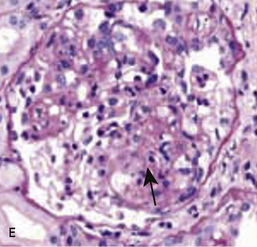
• The cellular variant (Fig. 20.13E) is characterized by at least one glomerulus with segmental hypercellularity (proliferation) that occludes the capillary lumen.
Management
• Prednisolone 0.5–2 mg/kg per day is used in most patients and continued for 6 months before the patient is considered resistant to therapy, which is common.
• Ciclosporin at doses to maintain serum trough levels at 150–300 ng/mL may be effective in reducing or stopping urinary protein excretion (tacrolimus is an alternative). Relapse after reducing or stopping ciclosporin is very common so that long-term use is required.
• Cyclophosphamide, chlorambucil or azathioprine is used for second-line therapy in adults. In patients with FSGS with mesangial hypercellularity and tip lesion, cyclophosphamide 1–1.5 mg/kg per day with 60 mg of prednisolone for 3–6 months, followed by prednisolone and azathioprine, can be used as maintenance therapy.
About 50% of patients progress to ESKD within 10 years of diagnosis, particularly those who are resistant to therapy. The recurrence of this renal lesion following renal transplantation is very high with a poor renal prognosis. Plasmapheresis or immunoabsorption has been the mainstay of treatment in patients with post-transplant recurrence but has had modest results. Anti-CD80 antibodies (abatacept), used in rheumatology, have been tried with success in this condition.
Secondary FSGS
Secondary FSGS with similar glomerular changes is seen as a secondary phenomenon when the number of functioning nephrons is reduced for any reason. Here, FSGS represents the common final glomerular lesion seen in response to subsequent haemodynamic glomerular strain. As nephrons fail, increased flow through the remaining nephrons leads to glomerular hypertrophy and hyperfiltration and hydraulic injury, with the secondary changes of FSGS. It is also described as remnant nephropathy (see p. 776). Associations include:
• reduced nephron number (e.g. nephrectomy, hypertension, gross obesity, ischaemia, sickle nephropathy, reflux nephropathy, chronic allograft nephropathy, IgA nephropathy, and scarring following renal vasculitis)
• mutations in specific podocyte genes
• viruses, e.g. HIV type 1, erythrovirus B19, cytomegalovirus, Epstein–Barr virus and simian virus 40
• drugs such as heroin, all interferons, anabolic steroids, lithium, sirolimus, pamidronate and calcineurin inhibitors, e.g. ciclosporin, which can also cause FSGS
• APOL1 gene mutations in patients of African ancestry, which makes them susceptible to FSGS in response to insults such as hypertension, SLE and HIV.
HIV-associated nephropathy
In HIV-associated nephropathy (HIVAN), glomeruli are characteristically ‘collapsed’ on light microscopy (Fig. 20.13C); podocytes are enlarged, hyperplastic and coarsely vacuolated, containing protein absorption droplets and overlying capillaries with varying degrees of wrinkling and collapse of the walls. Direct podocyte HIV-1 infection is associated with loss of podocyte-specific markers such as Wilms' tumour factor and synaptopodin in HIVAN. HIVAN presents with nephrotic-range proteinuria, oedema and CKD, which can be rapid in progression. Antiretroviral therapy (ART) may reverse the renal lesions seen, and restores renal function if treatment is commenced early.
Membranous glomerulopathy
Idiopathic membranous glomerulopathy is an autoimmune disease that occurs mainly in adults, and predominantly in men. It presents with asymptomatic proteinuria or frank nephrotic syndrome. Microscopic haematuria, hypertension and/or renal impairment may accompany the nephrotic syndrome. As in other glomerular disease, hypertension and a greater degree of renal impairment are poor prognostic signs. In membranous glomerulopathy, almost half of the patients undergo spontaneous or therapy-related remission. Eventually, however, about 40% develop CKD, usually in association with persistent nephrotic-range proteinuria. Younger people, females and those with asymptomatic proteinuria of modest degree at the time of presentation do best.
Pathogenesis
In the primary or idiopathic form (which comprises 75% of the cases), glomerular histology is identical to that seen when membranous glomerulopathy is secondary to another insult. These include:
• drugs (e.g. penicillamine, gold, NSAIDs, probenecid, mercury, captopril)
• autoimmune disease (e.g. SLE, thyroiditis)
• infections (e.g. hepatitis B, hepatitis C, schistosomiasis, Plasmodium malariae)
• cancers (e.g. carcinoma of the lung, colon, stomach, breast and lymphoma)
• other causes (e.g. sarcoidosis, kidney transplantation, sickle cell disease).
A majority of patients (70%) with idiopathic membranous nephropathy have been found to have IgG4-type autoantibodies against phospholipase A2 receptor (PLA2R), a glycoprotein constituent of normal glomeruli. PLA2R is present in normal human podocytes and in immune deposits in patients with idiopathic membranous nephropathy, indicating that it could be a major autoantigen in this disease; it is linked to human leucocyte antigen (HLA)-DQA1. Specific IgG4 autoantibodies to anti-aldose reductase (AR) and anti-manganese superoxide dismutase (SOD2) have also been found in the sera and glomeruli of patients with membranous nephropathy but not in other renal pathologies or normal kidney. Recently, antibodies against a novel antigen, thrombospondin type 1 domain-containing 7A (THSD7A), have been identified in anti-PLA2R-negative patients with membranous nephropathy. This suggests that AR, THSD7A and SOD2 could be additional renal autoantigens of human membranous nephropathy under certain clinical circumstances.
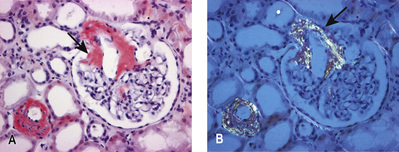
On light microscopy, capillary loops appear thick. Using a periodic acid–Schiff, or silver, stain (which highlights basement membrane), ‘spikes’ of basement membrane are visible. On electron microscopy, small, electron-dense deposits in the sub-epithelial aspects of the capillary walls are seen, encircled by perpendicular basement membrane spikes. Uniform granular capillary wall deposits of PLA2R antigen and IgG subclasses (IgG4 is predominant in idiopathic membranous nephropathy), as well as complement C3, are seen on immunofluorescence. Late in the disease, deposits are completely surrounded by basement membrane and are undergoing resorption, which appears as uniform thickening of the capillary basement membrane on light microscopy (Fig. 20.14).
Management
As many as a third or more of patients will undergo spontaneous remission if watched for at least 6–12 months, particularly if kidney function is normal and proteinuria modest. In general, patients with heavier proteinuria, progressive renal dysfunction and a high titre of anti-PLA2R antibodies are considered for early treatment.
• All patients should receive ACE inhibition at the maximum tolerated dose.
• The alkylating agents, cyclophosphamide (1.5–2.5 mg/kg per day for 6–12 months with 1 mg/kg per day of oral prednisolone on alternate days for the first 2 months) and chlorambucil (0.2 mg/kg per day in months 2, 4 and 6, alternating with oral prednisolone 0.4 mg/kg per day in months 1, 3 and 5), are both effective.
• Ciclosporin or tacrolimus is also of use, though remission is less well sustained and treatment courses are longer.
• Mycophenolate mofetil has demonstrated benefit in smaller studies with short follow-up.
• Anti-CD20 antibodies (rituximab, which ablates B lymphocytes) have been shown to improve renal function, reduce proteinuria and increase the serum albumin; no significant adverse affects have been shown in the short term.
• Oral corticosteroids are of no benefit alone but may be additive. A pilot study has shown promise with subcutaneous administration of adrenocorticotrophic hormone (tetracosactide) twice weekly, demonstrating an improvement in proteinuria. It is believed that it acts directly on podocytes by binding to melanocortin receptors. It is licensed by the FDA for use in nephrotic syndrome of any cause.
Amyloidosis
Amyloidosis (see pp. 1288–1289) is a systemic acquired or inherited disorder of protein folding, in which normally soluble proteins or fragments are deposited extracellularly as abnormal insoluble fibrils, causing progressive organ dysfunction and death.
The abnormal protein may be derived from light chains or immunoglobulin (AL amyloid), or from serum amyloid A protein (AA amyloid). The renal consequences are similar, even if systemic features differ.
Pathology
On light microscopy, widespread eosinophilic deposits are seen in the mesangium, capillary loops and arteriolar walls. Deposits stain pink, with green bi-refringence under polarized light with Congo red (Fig. 20.15). On electron microscopy, the characteristic fibrils of amyloid can be seen. Amyloid consisting of immunoglobulin light chains (AL amyloid) can be identified by immunohistochemistry in only 40% of cases, as compared to almost 100% of patients with protein found in secondary amyloid (AA amyloid).
Diagnosis
The diagnosis can often be made clinically when features of amyloidosis are present elsewhere (see p. 1289). On imaging, the kidneys are often large. Scintigraphy with radiolabelled serum amyloid P (SAP), a technique for quantitatively imaging amyloid deposits in vivo, is used to detect the rate of regression or progression of amyloidosis over a period of time (see p. 1289). Renal biopsy is necessary in all suspected cases of renal involvement.
Management
Treatments that reduce production of the amyloidogenic protein can improve organ function and survival in immunoglobulin-light-chain-related (AL) amyloidosis and hereditary transthyretin-associated (ATTR) amyloidosis (see p. 1288). In AA amyloidosis, production of serum amyloid A can sometimes be decreased by treatment of the underlying inflammatory condition but cannot be completely suppressed.
Renoprotective measures should be started (see (Box 20.9)). The success of dialysis and kidney transplantation depends on the extent of amyloid deposition in extrarenal sites, especially the heart.
Diabetic nephropathy
Diabetic renal disease is the leading cause of ESKD in the Western world, arising largely as a complication of type 2 diabetes mellitus. Diabetic kidney disease occurs in about 20–30% of both type 1 and type 2 diabetics (see pp. 1269–1270); the natural history is similar from the onset of proteinuria, and the histological lesion is the same. Risk factors for nephropathy include poor glycaemic control, hypertension, male gender, ethnicity and social deprivation.
Pathology
Glomerular hyperfiltration (the GFR increases to >150 mL/min/m2) and initial enlargement of kidney volume occur as local vasoactive factors increase flow. The GBM thickens and the mesangium expands. Progressive depletion of podocytes (see p. 733) from the filtration barrier (through apoptosis or detachment) results in podocyturia early in the disease. Proteinuria evolves as filtration pressures rise and the filter is compromised. Later, glomerulosclerosis develops with nodules (Kimmelstiel–Wilson lesion) and hyaline deposits in the glomerular arterioles (Fig. 20.16). Mesangial expansion and hyalinosis are partly due to amylin (β-islet-specific amyloid protein) deposits, with increasingly heavy proteinuria.
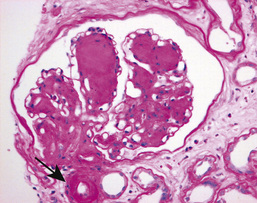
The Renal Pathology Society has developed a consensus classification combining type 1 and type 2 diabetic nephropathies (Box 20.8). This discriminates lesions by various degrees of severity for use in international clinical practice.
Box 20.8
Renal Pathology Society classification of types 1 and 2 diabetic nephropathy
| Class | Name |
| I | Isolated glomerular basement membrane thickening (>395 nm in females, >430 nm in males). No evidence of mesangial expansion, mesangial matrix increase, or global glomerulosclerosis involving >50% of glomeruli |
| IIa | Mild mesangial expansion |
| IIb | Severe mesangial expansion (in a severe lesion, >25% of the total mesangium contains areas of expansion larger than the mean area of a capillary lumen) |
| III | Nodular intercapillary glomerulosclerosis (≥1 Kimmelstiel–Wilson lesion(s)) and <50% global glomerulosclerosis |
| IV | Advanced diabetic glomerulosclerosis and >50% global glomerulosclerosis |
The pathophysiology is discussed on page 1269.
Management
Lifestyle changes (cessation of smoking, attention to salt intake, weight loss and increased exercise) are necessary in preventing progression of any diabetic complication.
• Aim for good (intensive) glycaemic control. If achieved for even a limited period, this reduces the incidence of ESKD and other microvascular complications in the long term (the so-called ‘legacy effect’ in both type 1 and type 2 diabetes mellitus).
• Control blood pressure to <120/80 mmHg with ACE inhibitors or AII-RA; these should be used once microalbuminuria develops, even if blood pressure control is good. Combined use of ACE inhibitors and AII-RA (dual blockade) does not provide additional benefit but is associated with an increased risk of AKI and hyperkalaemia; it is no longer recommended in recent trials.
As in other kidney diseases, however, nearly the entire course of renal injury in diabetes is clinically silent. The aim of medical intervention during this silent phase is renoprotection (Box 20.9), as judged by a slowed loss of glomerular filtration over time. Despite intensified metabolic control and antihypertension treatment in patients with diabetes, a substantial number still go on to develop ESKD.
Box 20.9
Renoprotection
Treatment measures
Patients with chronic kidney disease and proteinuria >1 g/24 h:
• Angiotensin-converting enzyme inhibitor increasing to maximum dose
Angiotensin receptor antagonist if goals are not achieveda
• Addition of diuretic to prevent hyperkalaemia and help to control blood pressure
Addition of calcium-channel blocker (verapamil or diltiazem) if goals are not achieved
Other interventions with a less robust evidence base include:
• Paricalcitol (a selective activator of the vitamin D receptor), added to treatment with ACE inhibitors, reduced albuminuria (a surrogate marker of progressive renal disease) in patients with type 2 diabetes in a randomized controlled trial. Paricalcitol worked best in patients with a high sodium intake in their diet, who are known to respond poorly to ACE inhibitor and angiotensin receptor blocker therapy.
• Bardoxolone methyl is a nuclear 1 factor (erythroid-derived 2)-related factor 2 (NRF-2) activator, an anti-inflammatory known to reduce oxidative stress. A large study failed to demonstrate benefit in patients with type 2 diabetes mellitus and CKD.
• Pentoxyphylline (previously used for peripheral vascular disease) slows the rate of GFR decline and proteinuria (by putatively reducing the production of tumour necrosis factor-alpha, TNF-α). This interesting observation requires external validation.
• Atrasentan (a selective endothelin A receptor (ETAR) antagonist when used with renin–angiotensin system inhibitors) is similarly generally safe and effective in reducing residual albuminuria. This could ultimately translate into improved renal outcomes in patients with type 2 diabetic nephropathy, but needs confimation in long-term follow-up studies.
Isolated proteinuria without haematuria
In asymptomatic patients, this is often an incidental finding. It is usually found at <1 g/day) with normal renal function. Over 50% of these patients have postural proteinuria. The outcome of isolated proteinuria (postural or non-postural) is excellent in the majority of patients, with a gradual decline in proteinuria over time. Occasionally, it may be an early sign of a serious glomerular lesion such as membranous glomerulopathy, IgA nephropathy, FSGS, diabetic nephropathy or amyloidosis (see above). Mild proteinuria may also accompany a febrile illness, congestive heart failure or infectious diseases with no clinical renal significance.
Glomerulonephritis (asymptomatic, acute and rapidly progressive)
Glomerulonephritis (GN) is immunologically mediated, with involvement of cellular immunity (T lymphocytes, macrophages/dendritic cells), humoral immunity (antibodies, immune complexes, complement) and other inflammatory mediators (including cytokines, chemokines and the coagulation cascade). The immune response can be directed against known target antigens, particularly when GN complicates infections, cancers or drugs. The underlying antigenic target is more often unknown. Primary GN may occur in genetically susceptible individuals (usually determined by major histocompatibility complex (MHC) genes like HLA-A1, B8, DR2 and DR3), following environmental insults. Circulating autoantibodies and/or abnormalities in serum complement, and glomerular deposition of antibodies, immune complexes, complement and fibrin characterize the condition. Glomerulonephritis may present as:
The same underlying histology may often present in any of the above ways, and these should be seen as clinical syndromes on a spectrum rather than as distinct diseases.
Asymptomatic urinary abnormalities
Haematuria with or without sub-nephrotic-range proteinuria in an asymptomatic patient may lead to the early discovery of potentially serious glomerular disease such as SLE, Henoch–Schönlein purpura, post-infectious GN or idiopathic hypercalciuria in children. Asymptomatic haematuria is also the primary presenting manifestation of a number of specific glomerular diseases discussed below.
Acute nephritis (nephritic syndrome)
This classically presents as:
• haematuria (macroscopic or microscopic) – with red-cell casts on urine microscopy
Nephritis can present indolently or incidentally, and is usually distinguished from rapidly progressive glomerulonephritis (see below) by the lack of cellular necrosis (and crescent formation) in the glomeruli seen on biopsy, and the rate at which renal decline evolves. These syndromes should be seen as a continuum.
Rapidly progressive glomerulonephritis
Rapidly progressive glomerulonephritis (RPGN) is a syndrome with glomerular haematuria (red blood cell casts or dysmorphic red blood cells), rapidly developing acute kidney failure over weeks to months and focal glomerular necrosis (Fig. 20.17) with or without glomerular crescent development on renal biopsy. The ‘crescent’ is an aggregate of macrophages and epithelial cells in Bowman's space (Fig. 20.17). RPGN can develop with immune deposits (anti-GBM or immune complex type, e.g. SLE) or without immune deposits (pauci-immune, e.g. anti-PR3 and or anti-MPO-ANCA-positive vasculitides). It can also develop as an idiopathic primary glomerular disease, or can be superimposed on secondary glomerular diseases such as IgA nephropathy, membranous GN and post-infective GN. It can be classified based on the pattern of immune complex deposition in glomeruli (seen on immunofluorescence): that is, linear, granular and negative immunofluorescent patterns (Box 20.10).
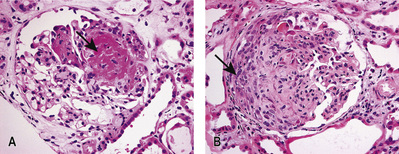
Box 20.10
Types of rapidly progressive glomerulonephritis (RPGN)
Linear immunofluorescent pattern (Fig. 20.20A)
Granular immunofluorescent pattern (immune complex-mediated RPGN; see Fig. 20.20B)