Chapter 45
Excitation—Contraction Coupling in Skeletal Muscle
Chapter Outline
IV. Speed of Skeletal Muscle Activation
V. Membrane Architecture of EC Coupling
VIIA. Distribution, Structure and Isolation of RyRs
I Summary
Excitation–contraction coupling in skeletal muscle is a fast signal transduction process by which depolarization of the sarcolemmal membranes is coupled to the opening of Ca2+ release channels on the sarcoplasmic reticulum (SR). This transduction occurs at specialized triad junctions and is mediated by two key proteins – the t-tubule dihydropyridine receptor (DHPR)/Ca2+ channel and the SR ryanodine receptor (RyR) channel. The DHPR and RyR1 associate with other junctional proteins to form a macromolecular complex which spans the triad junction and interacts to control RyR opening. This interaction produces a fast transient rise in [Ca2+]i which activates the contractile proteins and results in muscle contraction.
II Introduction
Skeletal muscle is activated to contract by a sequence of fast, electrically driven events that are collectively termed excitation–contraction coupling (EC coupling). EC coupling is a highly organized process of signal transduction that utilizes specialized membranes, membrane junctions and ion channels on both the exterior and interior of the cell. This chapter describes the cellular and molecular processes that mediate electrical excitation of the outer plasma membrane and transduces it into a signal for Ca2+ release from the intracellular sarcoplasmic reticulum (SR). The focus is on vertebrate fast-twitch skeletal muscle which is the best characterized.
Skeletal muscle is the fastest contracting of the three major muscle types and is related to cardiac and smooth muscle types evolutionarily and embryonically. The same two key proteins – a voltage-dependent calcium channel on the surface membrane (dihydropyridine receptor Ca2+ channel, DHPRs) and a Ca2+-release channel (ryanodine receptor, RyR) in the sarcoplasmic reticulum – mediate excitation–contraction in cardiac, smooth and skeletal muscle. These two proteins share a high degree of homology but the different muscle types express different isoforms and combinations of these proteins. Importantly, each muscle type has evolved highly differentiated mechanisms of using these proteins to serve its specialized functions. Cardiac and smooth muscle primarily mediate enteric contractile processes that are under autonomic and humoral control, while skeletal muscle primarily mediates rapid, willed bodily movements that are under the control of the central nervous system. Consequently, the defining features of skeletal muscle activation are speed and voluntary control, as needed for fine control of body movement.
III Overview of EC Coupling
The sequence of EC coupling in a vertebrate fast-twitch skeletal muscle fiber is shown in Fig. 45.1. Activation begins when an action potential from a motor neuron arrives at the neuromuscular junction and causes the neurotransmitter acetylcholine (ACh) to be released into the postsynaptic clefts (see chapter on synaptic transmission). The opening of AChRs produces a local depolarization, termed the end-plate potential, which brings the postsynaptic membrane to threshold potential for exciting an action potential (see chapter on synaptic transmission). The action potential rapidly propagates the depolarization along the outer sarcolemma and into the fiber interior via a specialized system of transversely oriented tubular membranes (transverse-tubules or t-tubules). The transverse tubules are continuous with and invaginate transversely from the sarcolemma at periodic intervals to form a planar network. The t-tubules provide the conduit for the action potential to reach the fiber interior and also bring the outer membranes into close proximity with the internal sarcoplasmic reticulum (SR) at specialized intracellular junctions called triads. The triad junctions serve as a platform for assembling sarcolemmal calcium channels (DHPRs), SR Ca2+-release channels (ryanodine receptors) and additional proteins into a macromolecular complex which spans the junctional gap and controls Ca2+ release from the SR. The DHPRs serve as the voltage-sensors in this process. DHPRs contain charged, intramembrane domains which move in response to the action potential depolarization; these molecular rearrangements, termed charge movement, in turn, drive a conformational change on the RyR1 which causes it to open. These concerted molecular interactions occur between cytoplasmic regions of the DHPR and cytoplasmic domains of the RyR1 which face each other at the triad junction. In this way, the opening of an integral SR membrane calcium channel is gated by conformational transitions of a sarcolemmal, voltage-dependent calcium channel. Consequently, Ca2+ release in skeletal muscle remains under tight control of the plasma membrane potential.
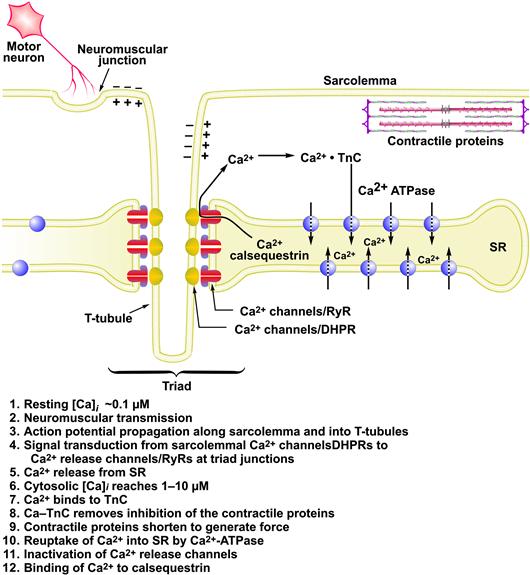
FIGURE 45.1 Sequence of excitation–contraction coupling in skeletal muscle.
The SR is a subcellular membrane compartment that is related evolutionarily to the endoplasmic reticulum. In skeletal muscle, it is a highly specialized for controlling cytosolic Ca2+, [Ca2+]i. The SR actively sequesters Ca2+ in resting muscle via an ATP-dependent Ca2+ pump (SERCA), to maintain the intracellular Ca2+ concentration, [Ca2+]i, at submicromolar levels (typically ≈0.1 μM). Ca2+ is stored in bound form within the SR, complexed to the Ca2+-binding protein calsequestrin. When a muscle is excited to contract, RyRs open and release this Ca2+ into the cytosol at rates approaching 100 μM/ms. This rapid Ca2+ release transiently raises cytosolic [Ca2+]i to micromolar levels (typically 1–10 μM). The released Ca2+ ions diffuse and bind to troponin-C (TnC), the regulatory subunit of troponin, thereby removing troponin’s inhibitory effect on the contractile proteins, actin and myosin, which shorten to generate force (see Chapter 46). Force output in skeletal muscle is directly proportional to cytosolic Ca2+ concentration (Fig. 45.2). Ca2+ release from the SR is brief and terminates quickly. As the t-tubule membrane repolarizes, DHPRs return to their inactive conformation and the RyR is itself inactivated by a Ca2+-dependent mechanism. Multiple mechanisms return cytosolic Ca2+ to resting levels. The Ca2+-ATPase of the SR membrane (SERCA) is stimulated rapidly to pump Ca2+ back into the SR. Cytosolic Ca2+-binding proteins with rapid kinetics buffer Ca2+ transiently while the SERCA pumps return it to the SR. Some Ca2+ is also extruded extracellularly by active Ca2+ transporters and Ca2+ exchangers on the plasma membrane. Force terminates when cytosolic Ca2+ returns to resting levels. Thus, cytosolic [Ca2+]i is the central link between membrane excitation and activation of the contractile proteins (Fig. 45.3). Contraction is inhibited at low resting Ca2+ levels, and proceeds when Ca2+ is elevated transiently in response to membrane excitation.
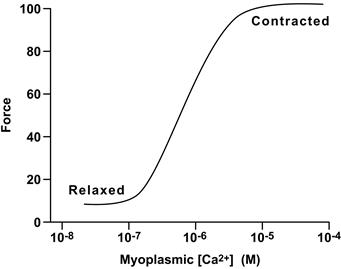
FIGURE 45.2 Relationship between force and myoplasmic Ca2+ concentration.
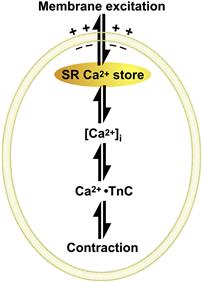
FIGURE 45.3 A rise in cytosolic [Ca2+] links membrane excitation to activation of contractile proteins.
IV Speed of Skeletal Muscle Activation
Activation of skeletal muscle is extremely rapid, occurring in milliseconds. This speed is remarkable given that skeletal muscle fibers are among the largest of mammalian cells. Skeletal muscle fibers are multinucleated cells that form during embryogenesis by fusion of myoblasts. They have a cylindrical shape with diameters from 30 to 150 μM and lengths from millimeters to centimeters. To produce effective mechanical force, the contractile proteins in all regions of a muscle fiber must shorten simultaneously. This synchronous activation is achieved by the propagating action potential which spreads the depolarization along the sarcolemma and into the transverse tubules within milliseconds, and by the anatomical configuration of the triad junctions which directly connect voltage-sensing DHPRs to the RyR Ca2+ release channels. Figure 45.4 and Table 45.1 show the relative timing and durations of key events in excitation contraction coupling for amphibian and mammalian muscle. The entire process, from membrane excitation to RyR1 opening, is complete within milliseconds. Overall, it operates largely as an on/off switch, insuring that Ca2+ release occurs rapidly and synchronously in response to each excitatory input from the motor nerve.
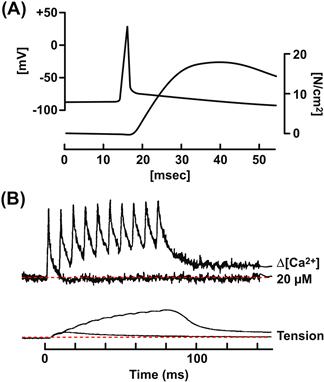
FIGURE 45.4 Speed of EC coupling in amphibian and mammalian skeletal muscles. (A) Time course of the action potential (top trace) and tension development (lower trace) in a fast-twitch frog skeletal muscle fiber at 18°C. (Modified fromHodgkin and Horowicz, 1957.) (B) Time course of EC coupling in a mouse skeletal muscle fiber at 37°C. The top traces record the cytosolic [Ca2+]i change measured with a Ca2+ indicator dye. The lower trace shows tension. The fiber was stimulated at time zero with a single action potential and with repetitive action potentials at high frequency, to simulate a muscle tetanus. (Reproduced from The Journal of General Physiology, 1996, 108, 455–469 by copyright permission of The Rockefeller University Press.)
TABLE 45.1. Duration of Key Steps in the Activation of Fast-Twitch Skeletal Muscle
| EC Coupling Steps | Durationa (ms) |
| Action potential propagation along sarcolemma | 5–10 |
| Action potential propagation to center of fiber along t-tubules | ≈0.7 |
| Signal transduction at triad junction, from t-tubule depolarization to activation of RyR on SR | ≈0.5 |
| Peak rate of Ca2+ release to peak Ca2+ binding to TnC (start of tension) | 2–3 |
| Peak myoplasmic Ca2+ change to peak tension | 15–25 |
a Durations were calculated for a hypothetical frog fiber of 50 μm diameter and 5 cm length, having a central end-plate. Literature values for conduction velocity and duration of intermediate steps (Gonzales-Serratos, 1971; Vergara and Delay, 1986; Jong et al., 1996) were adjusted to 18°C.
V Membrane Architecture of EC Coupling
As noted, the outer and intracellular membranes of skeletal muscle are highly organized to achieve rapid communication between the extracellular sarcolemma and the interior SR membrane (Franzini-Armstrong and Jorgensen, 1994; Franzini-Armstrong and Protasi, 1997). Figure 45.5 illustrates the three-dimensional architecture of the sarcolemma, t-tubules, triad junctions and SR membranes of a fast-twitch frog skeletal muscle fiber and their physical relationship to the contractile proteins. The t-tubules comprise the majority of the sarcolemma membrane, representing 50–80% of the plasma membrane area (Peachey, 1965). They invaginate from the sarcolemma in a transverse plane approximately twice every 1–2 μM (or, at two planes per sarcomere in mammalian muscle). Within this plane they branch extensively to form a planar network that covers the entire cross-section of the muscle fiber (Fig. 45.6). This geometry insures that the Ca2+ binding sites on TnC are less than a few tenths of a micron diffusion distance from the Ca2+ release channels. Up to 80% of the tubular membrane area forms triad junctions with the SR (Peachey, 1965, Dulhunty, 1984). In mammalian skeletal muscles, two planes of transverse tubules form in each sarcomere, at the A-I bands.
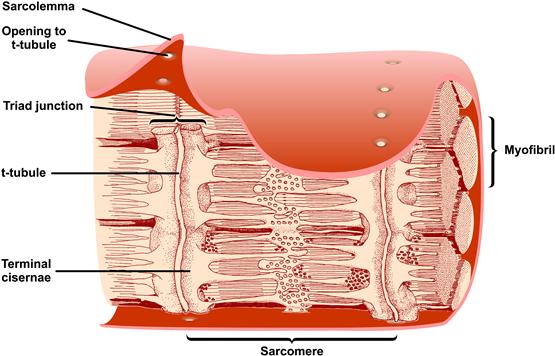
FIGURE 45.5 Three-dimensional reconstruction of a longitudinal section of a skeletal muscle fiber from the frog. The sarcolemma surrounds bundles of contractile proteins called myofibrils. Transverse tubules (t-tubules) invaginate from the sarcolemma in a plane at periodic intervals. The t-tubules form triad junctions with the SR along most of their length. At the triad junctions, the longitudinally oriented SR membranes widen into sacs called terminal cisternae, which closely oppose the t-tubules and contain the Ca2+ release channels/RyRs. The terminal cisternae are positioned near the activating Ca2+ sites on TnC. The functional unit of force generation is the sarcomere, consisting of overlapping actin and myosin filaments anchored at each end. The longitudinally oriented tubular regions of SR contain the Ca2+-ATPase which takes up released Ca2+ all along the sarcomere. (Reproduced from The Journal of Cell Biology, 1965, 25, 209–232 by copyright permission of The Rockefeller University Press.)
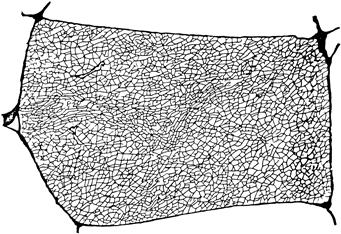
FIGURE 45.6 Reconstruction of the t-tubule network in one cross-sectional plane through a single frog twitch muscle fiber. The reconstruction was made from electron micrographs of transverse serial slices of a muscle fiber perfused with the electron-dense and membrane-impermeant molecule peroxidase, which diffused into the t-tubules. (From Peachey and Eisenberg, 1978.)
The specific positioning of skeletal-type DHPRs in relation to RyR molecules at the triad junction allows the two channels to interact during excitation–contraction coupling (Fig. 45.7). The t-tubule and SR membranes flatten and face each other across a narrow gap of about 15 nm. RyR1s assemble on the junctional surface of the terminal cisternae in an ordered array of two rows with a center-to-center spacing of about 30 nm. Each RyR1 is a tetramer composed of four identical subunits. The RyRs are extremely large molecules (≈5000 amino acids and ≈560 kDa) of approximately 29 × 29 × 12 nm size. An intramembrane region of RyR1 inserts into the SR membrane and a large cytosolic domain extends across the junctional gap and associates with cytosolic domains of the DHPR. Viewed from above, the cytosolic domains of RyR1 roughly resemble a double row of quatrefoil-shaped proteins which interlock with each other in a precisely ordered array. In side view, the RyR1 tetramer assumes a mushroom shape with a large extracellular domain and a smaller transmembrane domain that contains the Ca2+-conducting pore (see Section VII below). Every alternate RyR1 associates with a cluster of four DHPRs, termed a tetrad (shown here as small blue spheres), i.e. each DHPR in a tetrad interacts with one of the four subunits of RyR1 and every other RyR1 is not directly associated with a tetrad of DHPRs. The DHPR tetrads are arranged in a pattern approximately corresponding to the four outer corners of the cytosolic spheres of the RyR. The outline of the DHPR tetrad is larger than the outline of the RyR1 tetramer and this may explain why tetrads associate only with alternate RyR1s. In non-mammalian muscle, a second ryanodine receptor isoform, RyR3 is present on peripheral regions of the SR junctional membrane. RyR3s do not associate with DHPRs.

FIGURE 45.7 (A) Electron micrograph showing triad junctions in a longitudinal section through a fish skeletal muscle. Each t-tubule (outlined in red) is flanked on both sides by terminal cisternae of the SR (outlined in green). Contractile proteins are oriented longitudinally. The RyR1s appear as dense electron opaque material between the gap and were initially named “foot proteins” because of this appearance (FromFranzini-Armstrong and Peachey, 1981.) (B) Three-dimensional reconstruction of a triad showing the spatial organization of the junctional membranes, DHPRs and RyRs. The t-tubule (TT) flattens and apposes a region of SR across a gap of 12–15 nm. RyR1 Ca2+ release channels (red) in the SR align in a double row with a skewed spacing such that each RyR1 makes multiple contact points with adjacent RyR1s. A tetrad of four DHPRs (blue) in the TT couples to every other RyR1. Non-mammals, such as frog and fish, have a second, RyR3 (green), isoform which is localized at the periphery of the SR membrane at the triad junction. RyR3 isoforms are not associated with TT tetrads (red). (Reproduced with permission from Felder and Franzini-Armstrong, 2002.)
Figure 45.7 is based on a fish skeletal muscle fiber. A similar arrangement of tetrads is suggested in triads and peripheral couplings of other skeletal muscles from the frog, rat, chicken, mouse and human (Franzini-Armstrong and Jorgensen, 1994). Variations on this structure can occur in different species. These include dyads, in which a junctional region of terminal cisterna is opposed to a single short segment of t-tubule, and peripheral couplings, in which a junctional region of terminal cisterna is opposed to an approximately circular invagination of the surface membrane. Triads, dyads and peripheral couplings are structurally and functionally equivalent. Each is the intracellular site at which DHPRs and RyR1s interact to control Ca2+ release from the SR.
The triad junction has a strong structural integrity. Early studies demonstrated that purified fractions of t-tubule membrane could form junctions with SR membranes and that triad fractions could be isolated intact, despite stringent fractionation procedures (Caswell et al., 1979). Such strong protein–protein associations at the triad junction are required to preserve the structural integrity of the triad junction as it sustains the strong forces of contraction.
VI The DHPR Protein
The skeletal muscle DHPR is a voltage-gated Ca2+ channel (Cav1.1) belonging to the voltage-dependent superfamily of Ca2+ channels which share common structural and functional motifs (Yu et al., 2005; see also chapter on structure and mechanisms of voltage-gated ion channels). All Cav channels are multisubunit complexes composed of at least three subunits associated in an equimolar ratio: α1, α2-δ and β. The skeletal muscle channel has a fourth γ subunit. Multiple isoforms of the subunits exist and are distributed in a tissue-specific manner. The complete skeletal muscle calcium channel is a heteromultimeric complex consisting of α1s (212 kDa), α2-δ1 (125 kDa), β1a and γ1 subunits (Fig. 45.8A). The individual subunits have been purified, sequenced, cloned and studied in expression systems. The α1s alone can reconstitute a functional Ca2+ ion current. However, all four subunits are required to form a functional skeletal muscle DHPR with properties of the native channel (Suh-Kim et al., 1996).
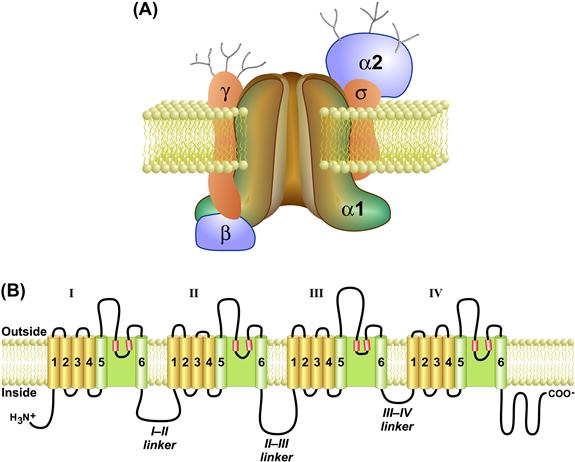
FIGURE 45.8 The skeletal muscle DHPR protein. (A) The skeletal muscle DHPR is composed of four subunits (α1s, α2-δ, β1a, and γ). The α1s subunit contains the key functional domains of the channel, including the voltage-sensing helices (S4) and the pore (S5–S6 extracellular domains). (B) The primary amino acid sequence of α1s contains four homologous transmembrane domains (I–IV) connected together by cytosolic linker sequences (I–II, II–III and III–IV linkers). Each of the four domains is composed of six transmembrane helices (drawn schematically as cylinders) plus two shorter intramembrane helices that fold into the bilayer (S5–S6 pore helices) to form the ion conducting pore. The cytosolic linker between domains II–III is a key interaction domain with the RyR and is required for skeletal-type EC coupling. The β1a subunit interacts with the I–II linker and with RyR1 and is also required for skeletal muscle type EC coupling.
The primary α1s subunit contains the major functional domains of the channel. It is an integral membrane protein (212 kDa) that contains the pore-forming and voltage-sensing regions as well as the binding site for dihydropyridines and other calcium channel blockers. It is composed of four hydrophobic repeat domains – I, II, III and IV (see Fig. 45.8B) – which share a high degree of homology. Domains I–IV assemble to form a transmembrane protein with a central pore. Each domain consists of six transmembrane alpha-helical segments, denoted S1–S6, plus two smaller helices between S5 and S6 (pore helices) which fold into the membrane to line the pore. The S4 transmembrane helix within each domain forms the voltage sensor that moves in response to the action potential depolarization. The S4 alpha helix is highly charged, containing a positively-charged residue on every third amino acid. Upon depolarization, all four S4 helices move in a cooperative fashion to activate the DHPR and, in turn, allosterically activate a coupled RyR1. Movement of the intramembrane S4 helices generates the macroscopic charge movement currents that are detected electrically during EC coupling (Schneider and Chandler, 1973). Domains I–IV are connected at the cytosolic face by segments of more hydrophilic amino acids (denoted I–II, II–III and III–IV linkers). The cytosolic region between domains II and III (II–III linker) plays a critical role in interactions with RyR1. The I–II linker also plays an essential role in EC coupling because it is the site for interaction with the β1a subunit which is required for EC coupling (see Section VIIIC; below). The β1a subunit is a smaller cytoplasmic protein (58 kDa) that associates specifically with the skeletal alpha isoform at an interaction domain on the I–II cytoplasmic linker. The α2-δ1 subunit (125 kDa) may participate in membrane targeting and anchoring, The γ1 subunit is a small glycosylated membrane protein (25 kDa) that associates with the skeletal α1s isoform but not with heart or brain alpha subunits and may play a regulatory role.
The skeletal muscle Ca2+ channel is capable of functioning both as a voltage sensor for EC coupling and as a Ca2+ ion channel. However, as a Ca2+ channel it has a low probability of opening and is characterized by high-voltage activation, brief openings and small single-channel currents. These features result in a very small, slow Ca2+ current through skeletal muscle DHPRs compared to the cardiac isoform (α1c). Therefore, in skeletal muscle, movement of the S4 gating domains may be followed by slower, less steeply voltage-dependent conformational changes that open the pore. It is thought that the early rapid movement of S4 domains, and not the Ca2+ current that follows after a delay of several hundred milliseconds, has been exploited evolutionarily by skeletal muscle to control RyR1 activation. This small, slow Ca2+ influx through skeletal muscle DHPRs is not significant during a single action potential. However, it may accumulate to measurable levels during a tetanus, when a skeletal muscle is activated repetitively at rates of 50–100 Hz to elicit a sustained contraction. In this case, Ca2+ entry may add to and perhaps modulate intracellular Ca2+ release from the SR (Dirksen, 2009).
VII The Ryanodine Receptor
VIIA Distribution, Structure and Isolation of RyRs
The release of Ca2+ ions from the endo/sarcoplasmic reticulum via Ca2+-release channels is a key step in a wide variety of biological functions. The endo/sarcoplasmic reticulum Ca2+-release channels are commonly referred to as ryanodine receptors (RyRs) to distinguish them from other intracellular Ca2+ channels. RyRs have been identified in non-vertebrate and vertebrates including flies, crustaceans, birds and amphibians. In mammals, there are three RyR isoforms. RyR1 is the dominant isoform in skeletal muscle. RyR2 is found in high levels in cardiac muscle. RyR3 was initially identified in brain but is expressed in many tissues including diaphragm, smooth muscle and brain.
The RyR/Ca2+-release channels have been extensively studied with rabbit skeletal muscle as the source for RyRs. Fragmentation of the sarcoplasmic reticulum during homogenization and subsequent fractionation by differential and density-gradient centrifugation yields a heavy SR vesicle fraction which is enriched in Ca2+-release channels and [3H]ryanodine-binding activity and corresponds to the junctional region of the SR (junctional SR) (Meissner, 1984). Another important advance has been the isolation of triads, membrane fractions composed of a t-tubule segment sandwiched between two junctional SR vesicles (Fleischer and Inui, 1989). Microsomal membrane fractions enriched in ryanodine-sensitive Ca2+-release channels have been also isolated from other excitable tissues, including cardiac muscle, smooth muscle and brain. In these cases, however, the membrane fractions are typically of a lower purity than those from skeletal muscle.
The RyRs are 2200 kDa multiprotein complexes that typically span the narrow gap where the SR and t-tubule (and plasmalemma) are within ≈15 nm of each other (Franzini-Armstrong and Protasi, 1997). The mammalian RyRs share ≈70% sequence homology, with the greatest homology in the carboxyl-terminal region. In all isoforms, the C-terminal portion of the protein contains the transmembrane domain. More recent studies suggest that six membrane spanning segments per RyR subunit are sufficient to form a channel. The remaining RyR amino acids form the large catalytic cytoplasmic “foot” structure. Experimental information on the structure of the RyRs has been mainly obtained by cryoelectron microscopy. Three-dimensional reconstruction of electron micrographs of frozen-hydrated RyR specimen shows dimensions of 29 × 29 × 12 nm for the large cytoplasmic assembly with a 7 nm length and 8 nm diameter for the transmembrane domain (Fig. 45.9).
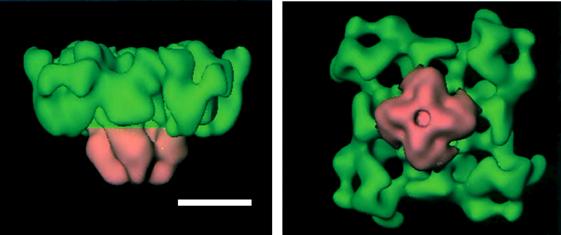
FIGURE 45.9 Dimensional reconstruction of RyR1. The cytoplasmic (green) and transmembrane (pink) domains are shown. Scale bar, 10 nm. (With permission from M. Samso and T. Wagenknecht (1998) J Struct Biol. 121,172–180.)
The isolation of the RyRs has been greatly facilitated by the identification of ryanodine as a channel-specific ligand. Ryanodine is a neutral plant alkaloid that is obtained from the stems of the South American shrub, Ryania speciosa, and is composed of two major compounds: ryanodine and 9,21-didehydroryanodine (Fig. 45.10). Ryanodine is a highly toxic compound. Its pharmacological effects have been most clearly shown in muscle where, depending on muscle type and activity, it can cause either contracture or a decline in contractile force (Sutko and Airey, 1996). Ryanodine activates the channel at low (nanomolar) concentrations, but inhibits the channel at high (micromolar) concentrations (Fig. 45.11). Because the drug binds with high specificity and dissociates slowly from the high-affinity site of the receptor (either membrane-bound or detergent-solubilized), [3H]ryanodine has been found to be an ideal probe in the isolation of the RyRs from a variety of tissues and species.
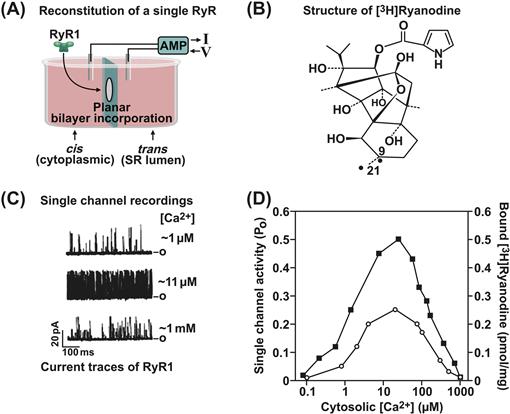
FIGURE 45.10 Measurement of RyR1 activity. (A) Planar lipid bilayer setup. (C) Three current traces of a single RyR1 ion channel recorded in symmetric 250 mM K+ medium. Bars on right represent the closed (c) channel. The effects of three different cis (cytoplasmic) [Ca2+] on channel activity are shown. In the upper trace, the channel is minimally activated by 1 μM Ca2+. Short, often poorly resolved events are seen. Channel activity is increased by increasing cytoplasmic Ca2+ from 1 to 11 μM and decreased when further increasing cytoplasmic Ca2+ to 1 mM. (D) Bimodal Ca2+ dependence of channel activity suggests the presence of high-affinity activating and low-affinity inhibitory Ca2+ binding sites. (B) Structure of [3H]ryanodine. (D) [3H]ryanodine binding measurements yield a Ca2+ dependence in good agreement with single channel measurements.
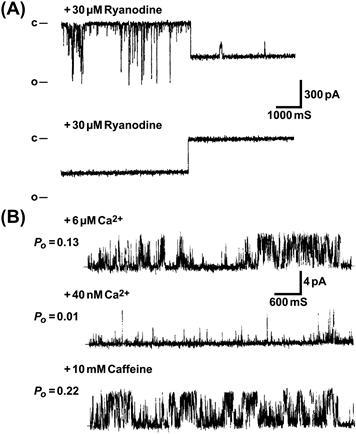
FIGURE 45.11 Effects of ryanodine and caffeine on single skeletal muscle RyRs. 30S purified channel complexes are reconstituted into planar lipid bilayers. (A) Upper trace shows appearance of subconducting channel state with open probability (Po) of ≈1, following several minutes after the addition of 30 μM cis ryanodine. An additional, infrequent substate is also observed. Lower trace illustrates the sudden transition from the subconductance state to a fully closed state 1 min after addition of 2 mM cis (cytoplasmic) ryanodine (from Lai et al., 1989 J Biol Chem. 264, 16776-16785). Note, relatively high ryanodine concentrations were used to shorten the time of binding to RyR1. (B) Single channel activity is with 6 μM free cytoplasmic Ca2+ (upper trace). Upper free Ca2+ is decreased to 40 nM by the addition of a Ca2+ buffer (EGTA) (middle trace). After the addition of 10 mM caffeine to the 40 nM free cytoplasmic Ca2+ medium (bottom trace). In contrast to ryanodine, caffeine activates the channel without changing its conductance. Bars on left represent the closed (c) channel. Po, channel open probability. (From Rousseau et al., Arch Biochem Biophys. 267, 75–86, 1988).
The RyR was first isolated from striated muscle because its role in regulating free cytoplasmic Ca2+ levels was originally recognized in muscle and relatively large amounts of membranes enriched in [3H]ryanodine-binding activity can be obtained from skeletal muscle and cardiac muscle. The membrane-bound Ca2+ release channels were solubilized in the presence of [3H]ryanodine, using the zwitterionic detergent Chaps and high ionic strength (1.0 M NaCl); they can then be purified in essentially one step by density gradient centrifugation through a linear sucrose gradient. Fig. 45.12A shows the [3H]ryanodine pattern on the sucrose gradients and sedimentation profile of the proteins associated with junctional SR vesicles isolated from rabbit skeletal muscle. A single peak of bound radioactivity, co-migrating with a small protein peak possessing an apparent sedimentation coefficient of 30 S, is observed in the lower half of the gradients. Binding to the small protein peak is specific since no radioactivity is present in the lower half of the gradients when the membranes are incubated with an excess of cold ryanodine. A sedimentation coefficient of 30 S revealed that the RyR is a very large protein complex.
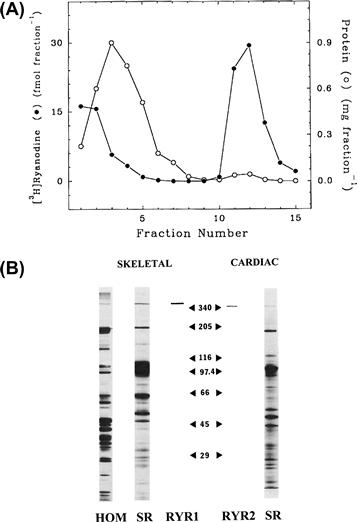
FIGURE 45.12 Sedimentation profile of RyR1 and SDS gel electrophoresis of rabbit skeletal muscle and canine cardiac RyRs. (A) Junctional skeletal muscle SR vesicles were solubilized in Chaps, centrifuged through a linear sucrose gradient and fractionated. Fractions were analyzed for protein and 3H-radioactivity. Unbound [3H]ryanodine and the majority of the solubilized proteins sedimented near the top of the gradient (fractions 1–6), whereas the [3H]ryanodine-labeled RyR comigrated with a small protein peak to the bottom of the gradient (fractions 11–13) (in modified form from Lai et al., Nature. 331, 315–319, 1988). (B) Silver stained SDS-polyacrylamide gel of whole rabbit skeletal muscle homogenate (HOM) and rabbit skeletal and canine cardiac muscle heavy SR membranes (SR) and purified RyRs (RYR1 and RYR2). Sizes of molecular weight standards are shown (×10−3). (From Meissner et al., 1989 Mol. Cell Biochem. 82, 59–65.)
The sucrose gradient centrifugation procedure is relatively simple and straightforward. The procedure results in efficient separation of the large 30 S RyR complex from the other solubilized smaller SR proteins because of its faster sedimentation rate. This method has been used to isolate a functional 30 S RyRs from several species and tissues including skeletal muscle, cardiac muscle, smooth muscle and brain.
SDS polyacrylamide gel electrophoresis shows that the 30 S protein complexes of mammalian skeletal and cardiac muscles are composed of a single major high molecular weight RyR polypeptide and isoform-specific low molecular weight immunophilin (FK506 binding protein) which migrate with apparent Mr > 340 000 (see Fig. 45.12B) and Mr ≈12 000 (not visible on the gels in Fig. 45.12B), respectively. Cloning and sequencing of the complementary DNA of the mammalian skeletal and cardiac muscle RyR isoforms have revealed an open reading frame of about 15 kb and encoding RyR polypeptides of Mr ≈560 000. In contrast, the presence of two immunologically distinct high molecular weight RyR protein bands (corresponding to the mammalian RyR1 and RyR3) has been described for the main skeletal muscles of chicken, frog and fish (Sutko and Airey, 1996). The two RyR isoforms are present as discrete homo-oligomers in amphibian and avian skeletal muscles. The appearance of the RyR1 isoform alone in some very fast-contracting muscle of fish suggests that this isoform is selectively expressed when rapid contraction is required in non-mammalian vertebrate muscles (O’Brien et al., 1993).
VIIB RyR1 Pore Structure
RyRs have large conductances for both monovalent (≈750 pS with 250 mM K+) and divalent (≈150 pS with 50 mM Ca2+) cations, while maintaining divalent vs monovalent selectivity (PCa/PK ≈7). Sequence comparison, cryoelectron microscopy and extensive mutagenesis and single channel studies indicate that the RyRs have a pore structure similar to that of K+ channels whose structure is known (Fig. 45.13). In the RyRs, the residues between the two C-terminal membrane-spanning segments are luminally located and have a pore helix and an amino acid motif (GGGIG) that is similar to the selectivity filter motif (TV/IGYG) of K+ channels whose structure is known. Mutagenesis shows that rings of negative charges in the luminal and cytosolic vestibules maintain the high rates of RyR1 ion fluxes (Xu et al., 2006). Several models of RyR ion permeation based on Eyring rate theory (Tinker et al., 1992), Poisson Nernst Planck-Density Functional Theory (Gillespie et al., 2005) and the solution structure of K+ channels (Fig. 45.13) have been described.
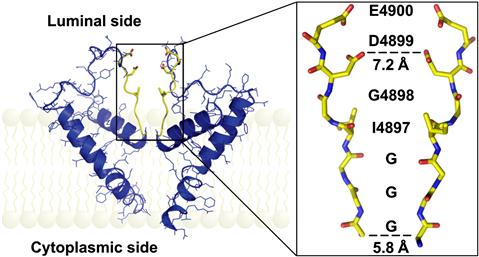
FIGURE 45.13 Pore model of RyR1. (Left panel) Two opposing RyR1 monomers (shown is only one of the six membrane spanning segments per monomer) in cartoon representation. The selectivity filter (4894GGGIGDE) is shown in yellow. (Right panel) Selectivity filter region of two opposing RyR1 monomers in stick representation. The sequence is shown along the path of the ions. (From Ramachandran et al. PLoS Comput Biol. 5, e1000367, 2009.)
VIIC Regulation by Ca2+ and Endogenous Effectors
The in vitro function of RyR ion channels has been studied by measurement of single channels incorporated in planar lipid bilayers and [3H]ryanodine binding. These studies have shown that RyRs are Ca2+-gated channels that are activated by micromolar Ca2+ concentrations and inhibited by millimolar Ca2+ concentrations (see Fig. 45.10) (Lanner et al., 2010). Mg2+ inhibits RyRs by binding to high-affinity and low-affinity Ca2+ binding sites. ATP complexed with Mg2+ modifies regulation of RyRs by Ca2+ indicating that it is an allosteric modulator of Ca2+-gated RyR activity. In addition to cytosolic Ca2+, SR luminal Ca2+ regulates RyRs at several channel sites. Ca2+ binds to luminal channel sites and, by passing through the channel, accesses cytosolic Ca2+-activation and inactivation sites.
VIID RyR-Associated Proteins
A large number of junctional SR membrane proteins and cytosolic and SR luminal proteins have been reported to form a macromolecular complex with the RyRs.
Kinases and phosphatases that are part of the RyR1 and RyR2 multiprotein complexes include protein kinase A (PKA), Ca2+/calmodulin-dependent protein kinase II (CaMKII) and protein phosphatase 1 and protein phosphatase 2A (Zalk et al., 2007). RyR1 has been reported to be phosphorylated at Ser2843. RyR2 is phosphorylated at Ser-2030 by PKA, at Ser-2809 by PKA and CaMKII (corresponding to Ser2843 in RyR1) and at Ser-2815 by CaMKII. Phosphorylation of RyRs by protein kinases has been suggested to result in leaky SR Ca2+ channels (see below).
Calmodulin (CaM) is a small, cytoplasmic Ca2+-binding protein that regulates cellular activities including protein kinases and ion channels. RyRs interact with CaM in the absence and presence of Ca2+ (Balshaw et al., 2002). The Ca2+-free and Ca2+-bound forms of CaM bind to a highly conserved CaM-binding domain of RyRs. CaM inhibits the three mammalian RyR isoforms at micromolar Ca2+ concentrations. At submicromolar Ca2+ concentrations, RyR2 is also inhibited by CaM, but RyR1 and RyR3 are activated to different extents by CaM. Mutagenesis, peptide studies and cryoelectron microscopy suggest that Ca2+-free and Ca2+-bound forms of CaM bind to an overlapping region distal to the effector site (ion pore) (Lanner et al., 2010). This suggests that CaM exerts its effects allosterically over relatively long distances within the RyRs. To determine the physiological importance of CaM regulation of RyR2, a mutant mouse was generated with three amino acid substitutions in the CaM-binding site (Yamaguchi et al., 2007). Mice expressing only the mutant form of RyR2 have cardiac hypertrophy as early as 1 day after birth and die around 2 weeks after birth, which suggests that CaM inhibition of RyR2 is required for normal cardiac function in mice.
Other RyR associated proteins include S100A1, triadin, junctin, JP-45, junctate, juncophilin, homer, sorcin, selenoprotein 1 and calseqestrin.
VIIE Pharmacology of RyRs
A large number of pharmacological reagents modulate the activity of the RyRs. Exogenous effectors found to affect RyR function include ryanodine (see Fig. 45.11A) and other ryanoids, caffeine (see Fig. 45.11B) and other xanthines, toxins, anthraquinones, phenol derivatives, dantrolene, local anesthetics and polycationic and sulfhyryl reacting reagents.
VIIF RyRs and Muscle Disorders
Mutations in RyR1 give rise to muscle diseases, malignant hyperthermia (MH), central core disease (CCD) and multiminicore disease (MmD) (Lanner et al., 2010). MH is an inherited disease that causes a rapid rise in body temperature and muscle contraction when the affected person receives general anesthesia. MH-linked mutations were initially mapped to the NH2 and central domains; however, more recently identified MH mutations appear to be distributed throughout the entire RyR1 coding sequence. CCD is an autosomal dominant congenital myopathy leading to the formation of cores void of mitochondria and oxidative enzymes. In many cases, dominant RyR1 mutations linked to CCD localize to the COOH-terminal domain of the channel. Single channel measurements indicate that CCD mutants lose the ability to conduct Ca2+. MmD is an autosomal recessive congenital myopathy leading to formation of multiple small areas of disorganized sarcomeres. MmD is a genetically heterogeneous disease that is linked to recessive mutations in RyR1 and selenoprotein 1, an RyR1-associated protein that is thought to maintain the receptors normal response to redox active molecules.
RyR2 mutations are associated with catecholaminergic polymorphic ventricular tachycardia and arrhythmogenic right ventricular cardiomyopathy (Lanner et al., 2010). A deficiency in SR lumenal cardiac calsequestrin and missense mutation D307H also result in imbalances of Ca2+ handling and catecholaminergic polymorphic ventricular tachycardia. Overexpression of CSQ2 results in severe cardiac hypertrophy in mice. Abnormal Ca2+ handling is also observed in mice that lack or overexpress RyR-associated proteins including triadin (Eltit et al., 2010) and junctin (Pritchard and Kranias, 2009).
Loss of the small 12 and 12.6 kDa FK506-binding proteins from RyR1 and RyR2 has been suggested to result in leaky Ca2+ channels (Zalk et al., 2007; Kushnir et al., 2010; Lanner et al., 2010). In failing hearts, PKA hyperphosphorylation of RyR2 has been reported to lead to removal of the FKBP12.6 subunit and increased channel activity. In skeletal muscle of animal models with heart failure and patients with heart diseases, exercise is linked to hyperphosphorylation, FKBP12 depletion of the skeletal muscle RyR1, increased RyR1 channel activity and decreased exercise capacity. However, other laboratories have failed to confirm this. In addition to PKA-mediated phosphorylation, mechanisms implicated in generation of “leaky” release channels include increased S-nitrosylation, oxidation and loss of protein phosphatases 1 and 2A and phosphodiesterase 4D from the RyR macromolecular complexes. A 1,4-benzothiazepine derivative, JTV519 (also known as K201), and the more specific derivative S107, have been reported to improve muscle function by stabilizing the RyR–FKBP complexes.
VIII Physiological Interactions Between the DHPR and RyR1
At the triad junction, a unique intracellular signal transduction occurs whereby a voltage-gated Ca2+ channel on an extracellular membrane controls the opening of a separate Ca2+ channel on an intracellular membrane. The unique structure of the RyR1, with a Ca2+ channel domain embedded in the SR and a large cytosolic domain facing cytosolic regions of the DHPR, suggests that opening and closing of RyR1 could be remotely controlled by the DHPR. The essential steps in this coupling are: detection of the action potential depolarization by voltage-sensing DHPRs in the t-tubules, interactions between the DHPR and RyR1 leading to RyR1 channel opening and Ca2+ efflux from the SR.
VIIIA Voltage Sensing
It was recognized early that EC coupling in fast skeletal muscles of higher vertebrates is directly controlled by membrane potential, without a requirement for extracellular Ca2+. This contrasts with EC coupling in the skeletal muscles of some lower invertebrates and cardiac muscle, in which Ca2+ influx through sarcolemmal Ca2+ channels is absolutely required for contraction. In cardiac muscle, the Ca2+ that enters through the cardiac-specific DHPR (α1c) functions as a second messenger to activate the cardiac ryanodine receptor isoform, RyR2. This process in cardiac muscle is termed calcium-induced calcium release. In a classic experiment, it was demonstrated that a frog skeletal muscle stimulated by action potentials is able to twitch continuously in the complete absence of extracellular Ca2+ (Armstrong et al., 1972). Similarly, voltage-clamp experiments demonstrated that a skeletal muscle fiber can develop tension provided only that the membrane is depolarized to a minimum potential and duration, termed the mechanical threshold. That is, when the Na+, K+ and Cl− ion channels which mediate the action potential are blocked and/or their permeant ions are removed, all that is required to activate contraction in skeletal muscle is a suprathreshold change in membrane potential. A precise relationship between tension and membrane potential exists in skeletal muscle (Fig. 45.14); development of tension is steeply voltage-dependent in the range from −60 to −40 mV.
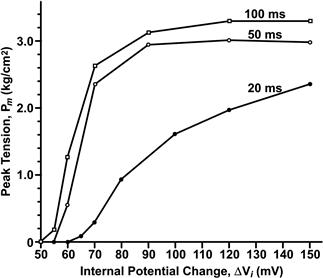
FIGURE 45.14 Voltage-dependence of tension in the absence of an action potential. The fibers were depolarized by holding the membrane potential constant for 20–100 ms with a voltage-clamp. Tension was measured simultaneously with a transducer attached to one tendon. (Reproduced from The Journal of General Physiology, 1984, 84,133–154, by copyright permission of The Rockefeller University Press.)
Subsequently, Schneider and Chandler (1973) discovered the presence in skeletal muscle membranes of mobile intramembrane charges whose movement could be detected electrically as a voltage-dependent dielectric current, termed charge movement (Fig. 45.15). This charge movement was steeply voltage-dependent in the same range of potentials as mechanical activation. Based on the similar kinetics and voltage dependence of charge movement and mechanical activation, they proposed that these gating currents reflected the movement of charged intramembrane domains of a molecule in the t-tubules that functioned as the voltage sensor for EC coupling. These measurements suggested that positively-charged regions of the DHPR undergo a net outward movement upon depolarization, away from RyR1. Therefore, they suggested that the voltage sensor might interact directly with a hypothetical Ca2+-release channel on the SR to cause its opening. This mechanical or allosteric model of EC coupling, formulated before the proteins of the triad junction had been identified, has formed the framework for subsequent investigations into the nature of the coupling mechanism.
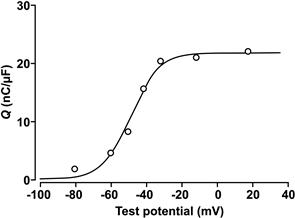
FIGURE 45.15 Voltage-dependence of charge movement (nC/μF vs membrane potential, mV). Charge moved at each potential was obtained by integrating the voltage-dependent capacity current, normalized to the fiber linear capacitance near the resting potential. Symbols represent the mean values of charge moved at the “on” and “off ” of the pulse. The continuous curve is a fit of the data to a two-state Boltzmann function, with parameters: Vmid = −47.7 mV, k = 8 mV and Qmax = 21.5 nC/μF. (From Chandler et al., 1976.)
Several lines of evidence led to the identification of the voltage sensor as the DHPR/t-tubule Ca2+ channel (Rios and Pizarro, 1991, Schneider, 1994, Melzer et al., 1995). A definitive identification came following the cloning of the skeletal muscle Ca2+ channel (Tanabe et al., 1987) and its use in experiments on dysgenic mice (Adams and Beam, 1990). Muscular dysgenesis is a lethal disorder arising from a single point mutation in the gene for the DHPR α1s; it results in loss of DHPR protein, the absence of slow Ca2+ currents and charge movement and failure of EC coupling. Tanabe and colleagues (Tanabe et al., 1988) demonstrated that Ca2+ currents, charge movement and EC coupling could be restored in dysgenic muscle fibers by transfecting the cDNA for the α1s subunit of the DHPR. This demonstration established the DHPR Ca2+ channel as the essential voltage-sensing molecule in EC coupling.
VIIIB The Myoplasmic Ca2+ Release Transient and SR Ca2+ Release Flux
In studies using intact skeletal muscle cells, SR Ca2+ release is detected by monitoring myoplasmic Ca2+ transients using Ca2+ indicator dyes introduced into the myoplasm (as shown in Fig. 45.4B). These measurements record the global Ca2+ change occurring throughout the myoplasm. Much useful information on the kinetics and voltage-dependence of SR Ca2+ release under physiological conditions has come from these studies (Rios and Pizarro, 1991, Melzer et al., 1995). These studies confirmed that a strict relationship exists between the voltage-dependence of Ca2+ release and voltage-dependent charge movement, as expected for a coupling mechanism which is controlled by the t-tubule membrane potential. Importantly, it was shown that SR Ca2+ release can both be turned on by membrane depolarization and turned off by membrane repolarization. Other studies have also demonstrated that considerable cross-talk exists between t-tubule Ca2+ channel functions and SR Ca2+ release (Sheridan et al., 2006; Dirksen, 2009). For example, the RyR1 release channel, through its interaction with calsequestrin, is able to respond to surface depolarization in a manner that depends on the Ca2+ load within the SR. Changes in SR Ca2+ load are reflected in the kinetics of charge movement and the rate of Ca2+ release. In addition, although Ca2+ entry is not required for skeletal muscle type EC coupling, manipulations that alter the RyR1 or the Ca2+ release transient also modify the slow Ca2+ current through DHPRs. Such bidirectional cross-talk between the DHPR and RyR, involving both forward and retrograde interactions, is expected for a mechanism that involves protein–protein interactions within a macromolecular complex.
The global myoplasmic Ca2+ change (see Fig. 45.4) arises from the summation of thousands of individual Ca2+ release events. Discrete, local Ca2+ signals, termed Ca2+ sparks, have been detected in skeletal and cardiac muscle (Fig. 45.16) (Cheng and Lederer, 2008). These discrete Ca2+ changes are limited in time and space and can occur spontaneously or in response to membrane depolarization. The Ca2+ spark does not reflect the unitary opening of a single RyR1 channel, but is proposed to report an elementary unit of SR Ca2+ release arising from a Ca2+-release unit. Calcium-release units consist of a cluster of DHPRs, RyR1s and possibly other proteins acting together as a functional unit (see below). Thus, the global depolarization-triggered Ca2+ transient represents a summation of discrete unitary events. This mechanism may allow a more graded control of Ca2+ release. Some important differences in sparks exist between vertebrate amphibian and mammalian skeletal muscles. Mammalian muscles do not readily show spark activity. Rather, discrete Ca2+ release events, termed embers, with longer duration and constant amplitude are observed (Fig. 45.17) (Zhou et al., 2003, Csernoch et al., 2004; Klein and Schneider, 2006). The voltage dependence of embers is manifested in the duration and latency, but not in the amplitude of the individual events. It is possible that these differences in the elementary Ca2+ release events may reflect the different protein composition of the unitary Ca2+-release units of amphibian and mammalian muscle. Although both species share the same two key proteins – a skeletal DHPR and RYR1 – amphibian muscle has an addition RyR3 isoform that is positioned peripheral to the double rows of RyR1 which oppose the DHPR tetrads. These parajunctional RyR3 channels in amphibian muscle do not associate with DHPR tetrads and may participate in a secondary Ca2+-induced Ca2+ release, to generate sparks with properties similar to those in cardiac muscle. As described above, additional junctional proteins associate with the RyR1 and the molecular composition of the complete, elementary Ca2+-release complex in amphibian and mammalian muscles is not yet defined.
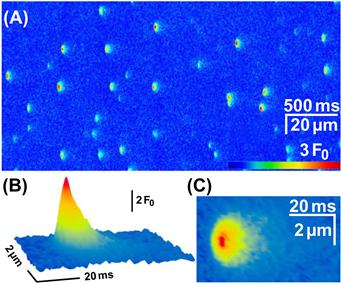
FIGURE 45.16 Spontaneous elementary calcium release events in an amphibian skeletal muscle. (A) A typical line scan image from a permeabilized frog skeletal muscle fiber immersed into a glutamate-based internal solution containing 50 μM fluo-4 as the calcium-sensitive fluorescence indicator. The sparks occur with high frequency and appear relatively homogenous. Time course (B) and spatial distribution (C) of the average spark in amphibian skeletal muscle. (Reproduced with permission from Csernoch et al., 2004.)
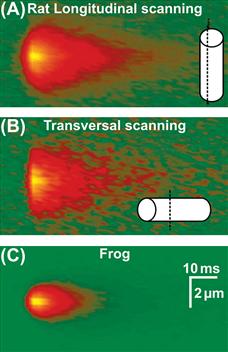
FIGURE 45.17 Elementary Ca2+ release events, termed “embers”, in a mammalian skeletal muscle of the rat (A, B) compared to frog (C). Averages detected in line scans parallel (A) or perpendicular (B) to the fiber axis. Mammalian embers are wider and longer than the frog sparks. (Reproduced with permission from Zhou et al., 2003.)
VIIIC Molecular Interactions Between the DHPR and RyR
The molecular mechanisms by which the pore of RyR1 opens under control of the t-tubule DHPR/Ca2+ channel has been the subject of intense research and is still not entirely resolved. Studies using dysgenic and transgenic mice have provided powerful experimental models for identifying the key proteins that are absolutely required for skeletal type EC coupling (Beam and Bannister, 2010). These are the α1s and β1a subunits of the DHPR and RyR1. Mice null for either the α1s or β1a subunit or the DHPR, or mice lacking the RYR1 protein, show an EC coupling dead phenotype and die perinatally. On the other hand, ablation of the α2-δ1 or γ1 subunits of DHPR has little effect on EC coupling (Obermair et al., 2008).
The muscular dysgenesis mouse (lacking α1s) has been used to identify further regions of DHPR α1s that are critical for interactions with RyR1. The α subunits of the skeletal (α1s) and cardiac (α1c) DHPR share a high degree of homology, but only the α1s can restore a skeletal-type EC coupling. When the skeletal muscle α1s subunit was expressed in dysgenic myotubes, Ca2+ release could be elicited in response to electrical depolarization and did not require Ca2+ entry; whereas, Ca2+ entry was required to elicit Ca2+ release when the cardiac α1c subunit was expressed. The main sequence differences between α1s and α1c reside in the large cytosolic regions at the N- and C-termini and the linker sequences I–II, II–III and III–IV. By constructing chimeric DHPRs with various mixes of cardiac and skeletal sequences, Beam and collaborators (Tanabe et al., 1990) demonstrated that the 138-amino-acid cytoplasmic sequence between domains II and III is a critical determinant of skeletal-type EC coupling. A chimeric DHPR having the cardiac protein backbone but the not critical skeletal II–III linker could elicit skeletal-type EC coupling when expressed in dysgenic myotubes. Further experiments demonstrated that the cytosolic I–II linker, which binds the cytosolic β1a subunit, is also essential for skeletal-type EC coupling. EC coupling is lost in myotubes of transgenic mice when the β subunit gene is knocked out, but is rescued when β1a (but not the cardiac β2b) is expressed. Thus, the interaction of β1a with the I–II linker of the skeletal α1s is also required for skeletal-muscle-type EC coupling.
Other cytosolic regions on the α1s subunit have been implicated in interactions with RyR1. A mutation in the III–IV cytosolic linker of the human DHPR (A1086H) is associated with susceptibility to one form of malignant hyperthermia (MH), a hereditary skeletal muscle disorder that is characterized by an uncontrolled, sustained Ca2+ release (Monnier et al. 1997). Studies using more complex chimeras between α1s and α1c have identified specific sites within the II–III linker which interact with RyR1. Biochemical studies have also demonstrated binding of specific fragments of the II–III sequence to RyR1. However, these studies have not yet yielded a consistent picture of the specific sequences which form interaction sites between DHPR and RyR1.
Similarly, regions of RyR1 critical for skeletal-type EC coupling have been studied using mice having a disrupted RyR1 gene (dyspedic mice). Dyspedic mice lack EC coupling, as expected and, in addition, have a 30-fold lower density of calcium channel current (Nakai et al., 1996). Thus, the absence of RyR also alters DHPR function, as expected if the DHPR and RYR1 associate in a macromolecular complex. Expression of the cDNA for RyR1 in dyspedic myotubes restores EC coupling and enhances the DHPR calcium current. This suggests that RyR1, in addition to receiving the EC coupling signal from the DHPR, also transmits a retrograde signal that enhances the function of skeletal DHPRs as calcium channels. These interactions require the skeletal muscle RyR1 isoform and indicate that the identity of the ryanodine receptor isoform also determines the mode of EC coupling. Expression of the predominant cardiac isoform, RyR2, did not restore skeletal-type EC coupling or enhance DHPR calcium channel activity (Nakai et al., 1997). Other studies combining freeze-fracture and pharmacology demonstrate that the spatial orientation of DHPRs and the conformational state of RyR1 are intimately related, further supporting the idea that the DHPR and RyR1 are capable of bidirectional interactions. The average distance between centers of each adjacent DHPR within a tetrad (≈19.5≈nm) is decreased by ≈2 nm upon application of ryanodine at a concentration (500 μM) which locks RyR1 in a subconductance state (Paolini et al., 2004). Thus, the functional state of RyR1 is able to alter the spatial orientation of DHPRs within a tetrad, a phenomenon that could only be a result of physical coupling between RyR1 and the DHPR.
Biochemical studies of isolated triad membranes and proteins in vitro have also provided compelling evidence for protein–protein interactions between the skeletal DHPR and RyR1 (Meissner and Lu, 1995; Leong and MacLennan, 1998) and confirmed that the skeletal muscle isoforms of both DHPR and RyR1 are essential for skeletal type EC coupling. Peptides composed of either the skeletal or cardiac II–III linker sequence can activate isolated RyR1s (Lu et al., 1995), but do not activate the cardiac RyR2 isoform. Binding assays have also been used to identify regions on RyR1 that interact specifically with the skeletal DHPR. However, these studies have not yet led to a consensus on the specific interaction sites. Taken together, the body of biochemical evidence and results of studies using transgenic mice indicate that a minimal complex containing the skeletal muscle DHPR α1s and β1a subunits plus RyR1 is required to initiate Ca2+ release by the physiologically relevant (independent of Ca2+ entry) mode in fast twitch skeletal muscle. Moreover, the data suggest that the bidirectional interaction between DHPR and RyR1 may involve multiple contact points on both proteins and possibly low affinity interactions (Sheridan et al., 2006; Beam and Bannister, 2010). A more specific identification of the interaction sites will likely become available when detailed structural information becomes available from crystallographic studies.
VIIID Overall Control and Integration of Ca2+-Release Events
There is general agreement that the initial event leading to calcium release in vertebrate skeletal muscle is a depolarization-driven interaction between a DHPR tetrad and at least one RyR1 tetramer. However, the mechanism by which this signal is communicated to adjacent RyRs which do not underlie a DHPR tetrad is less well understood. The alternate disposition of DHPR tetrads opposite every other RyR1 suggests a dual control of RyR1 activation. One RyR1 may be opened by allosteric movements of the associated DHPR tetrad (a coupled RyR1), while a neighboring RyR1 (non-coupled to a tetrad) may be opened by a different mechanism. One early proposal was that the local Ca2+ released through coupled RyR1s binds to and activates adjacent, non-coupled RyR1s (Fig. 45.18), acting as a ligand-gated channel. However, all of the functional evidence indicates that there is little Ca2+-induced-Ca2+ release in the mammal. An alternate proposal is that the conformational changes initiated between a coupled RyR1 and a DHPR tetrad is communicated to non-coupled RyR1s through coordinated interactions between physically linked RyR1s. In this model, both populations of RyR1s are activated by depolarization without requiring Ca2+ as a ligand. This model is suggested by the tight packing of RyR1s in an ordered, interlocking array. In this model, clusters of RyR1s open simultaneously as a functional Ca2+ release unit. Opening of one RyR1 by its coupled DHPR tetrad results in simultaneous opening of all contiguous RyRs in the cluster. The minimal number of RyR1s which constitute an elementary Ca2+-release unit (generating an “ember”) is not yet resolved. Such clusters of RyRs operating as a functional unit is expected to generate fast local Ca2+ release events and a more synchronous global change in myoplasmic Ca2+. An additional mechanism may operate in amphibian muscle where parajunctional RyR3 channels are also present. The RyR3 channels may be opened by a Ca2+-induced-Ca2+-release mechanism, similar to the cardiac RyR2 channel, and generate the more cardiac-like sparks recorded from frog muscle.
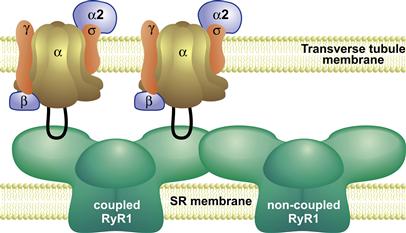
FIGURE 45.18 Integration of Ca2+ release from coupled and non-coupled RyR1s. RyR1s associate with each other on the SR surface of the triad junction in an ordered array. Every alternate RyR1 is coupled to a tetrad of four DHPRs in the transverse tubule membrane. This schematic depicts a cross-sectional plane through two subunits of a RyR1 (left) interacting with two DHPRs. Voltage-driven movements of the DHPR initiate molecular interactions between cytosolic regions of the DHPR (II–III loop) and a coupled RyR1, leading to RyR1 channel opening. This initiating event activates non-coupled RyR1s, possibly via coordinated interactions between adjacent RyR1s, to effect a rapid and synchronous efflux of Ca2+.
VIIIE Modulation of EC Coupling
EC coupling can be modulated in response to different demands of muscle use. Potential regulatory sites on both the DHPR and RyR1 have been identified. The DHPR α1s subunit contains consensus phosphorylation sites including a serine residue, Ser 687, within the critical II–III loop that is rapidly phosphorylated by a cAMP-dependent kinase. Phosphorylation of this residue is required for binding of the II–III loop to RyR1 (but not RyR2), but only the dephosphorylated II–III loop can activate the RyR (Lu et al., 1995). Moreover, Ca2+ release can be both inhibited and activated by Ca2+ acting at specific sites on the RyR1 or α1s (El-Hayek et al., 1995; Meissner, 2004). As noted (see Section VI), RyR1 has both Ca2+-activating and Ca2+-inactivation sites as well as a CaM binding site which can bind either CaM or the soluble protein S100 to modulate Ca2+ release. The complete RyR1 complex includes other associated proteins which contribute to EC coupling and to the structural integrity of the triad junction. Finally, long-term regulation of EC coupling may occur at the level of gene expression. It is known that the expression of α1s can be altered during adaptation to physiological demands, including altered patterns of nerve activity, exercise, changes in the load-bearing state and metabolic or endocrine status.
Acknowledgment
Support by National Institutes of Health grants AR018697 and HL073501 and the Physiology Research Fund of the University of Cincinnati is gratefully acknowledged.
BIBLIOGRAPHY
1. Adams BA, Beam KG. Muscular dysgenesis in mice: a model system for studying excitation-contraction coupling [Review]. FASEB J. 1990;4:2809–2816.
2. Armstrong CM, Bezanilla FM, Horowicz P. Twitches in the presence of ethylene glycol bis(-aminoethyl ether) -N, N’-tetracetic acid. Biochim Biophys Acta. 1972;267:608.
3. Balshaw DM, Yamaguchi N, Meissner G. Modulation of intracellular calcium-release channels by calmodulin. J Membr Biol. 2002;185:1–8.
4. Beam KG, Bannister RA. Looking for answers to EC coupling’s persistent questions. J Gen Physiol. 2010;136:7–12.
5. Caswell AH, Lau YH, Garcia M, Brunschwig JP. Recognition and junction formation by isolated transverse tubules and terminal cisternae of skeletal muscle. J Biol Chem. 1979;254:202–208.
6. Cheng H, Lederer WJ. Calcium sparks. Physiol Rev. 2008;88:1491–1545.
7. Csernoch L, Zhou. J, Stern MD, Brum G, Rios E. The elementary events of Ca2+ release elicited by membrane depolarization in mammalian muscle. J Physiol. 2004;557:43–58.
8. Dirksen RT. Checking your SOCCs and feet: the molecular mechanisms of Ca2+ entry in skeletal muscle. J Physiol. 2009;587:3139–3147.
9. Dulhunty AF. Heterogeneity of t-tubule geometry in vertebrate skeletal muscle fibres. J Muscle Res Cell Motil. 1984;5:333–347.
10. El-Hayek R, Antoniu B, Wang J, Hamilton SL, Ikemoto N. Identification of calcium release-triggering and blocking regions of the II-III loop of the skeletal muscle dihydropyridine receptor. J Biol Chem. 1995;270:22116–22118.
11. Eltit JM, Feng W, Lopez JR, et al. Ablation of skeletal muscle triadin impairs FKBP12/RyR1 channel interactions essential for maintaining resting cytoplasmic Ca2+. J Biol Chem. 2010;285:38453–38462.
12. Felder E, Franzini-Armstrong C. Type 3 ryanodine receptors of skeletal muscle are segregated in a parajunctional position. Proc Natl Sci USA. 2002;99:1695–1700.
13. Fleischer S, Inui M. Biochemistry and biophysics of excitation-contraction coupling. Annu Rev Biophys Biophys Chem. 1989;18:333–364.
14. Franzini-Armstrong C, Jorgensen AO. Structure and development of E-C coupling units in skeletal muscle. Annu Rev Physiol. 1994;56:509–534.
15. Franzini-Armstrong C, Peachey LD. Striated muscle-contractile and control mechanisms. J Cell Biol. 1981;91:166s–186s.
16. Franzini-Armstrong C, Protasi F. Ryanodine receptors of striated muscles: a complex channel capable of multiple interactions. Physiol Rev. 1997;77:699–729.
17. Gillespie D, Xu L, Wang Y, Meissner G. (De)constructing the ryanodine receptor: modeling ion permeation and selectivity of the calcium release channel. J Phys Chem B. 2005;109:15598–15610.
18. González-Serratos H. Inward spread of activation in vertebrate muscle fibres. J Physiol. 1971;212:777–799.
19. Hodgkin AL, Horowicz P. Effects of K and Cl on the membrane potential of isolated muscle fibres. J Physiol. 1957;137:30P.
20. Jong DS, Pape PC, Geibel J, Chandler WK. Sarcoplasmic reticulum calcium release in frog cut muscle fibres in the presence of a large concentration of EGTA. Soc Gen Physiol Ser. 1996;51:255–268.
21. Klein MG, Schneider MF. Ca2+ sparks in skeletal muscle. Prog Biophys Mol Biol. 2006;92:308–332.
22. Kushnir A, Betzenhauser MJ, Marks AR. Ryanodine receptor studies using genetically engineered mice. FEBS Lett. 2010;584:1956–1965.
23. Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol, 2010;:2.
24. Leong P, MacLennan DH. Complex interactions between skeletal muscle ryanodine receptor and dihydropyridine receptor proteins. Biochem Cell Biol. 1998;76:681–694.
25. Lu X, Xu L, Meissner G. Phosphorylation of dihydropyridine receptor II-III loop peptide regulates skeletal muscle calcium release channel function Evidence for an essential role of the beta-OH group of Ser687. J Biol Chem. 1995;270:18459–18464.
26. Meissner G. Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium. 2004;35:621–628.
27. Meissner G. Adenine nucleotide stimulation of Ca2+-induced Ca2+ release in sarcoplasmic reticulum. J Biol Chem. 1984;259:2365–2374.
28. Meissner G, Lu X. Dihydropyridine receptor-ryanodine receptor interactions in skeletal muscle excitation-contraction coupling. Biosci Rep. 1995;15:399–408.
29. Melzer W, Herrmann-Frank A, Luttgau HC. The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim Biophys Acta. 1995;1241:59–116.
30. Monnier N, Procaccio V, Stieglitz P, Lunardi J. Malignant-hyperthermia susceptibility is associated with a mutation of the alpha 1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am J Hum Genet. 1997;60:1316–1325.
31. Nakai J, Dirksen RT, Nguyen HT, Pessah IN, Beam KG, Allen PD. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 1996;380:72–75.
32. Nakai J, Ogura T, Protasi F, Franzini-Armstrong C, Allen PD, Beam KG. Functional nonequality of the cardiac and skeletal ryanodine receptors. Proc Natl Acad Sci USA. 1997;94:1019–1022.
33. Obermair GJ, Tuluc P, Flucher BE. Auxiliary Ca(2+) channel subunits: lessons learned from muscle. Curr Opin Pharmacol. 2008;8:311–318.
34. O’Brien J, Meissner G, Block BA. The fastest contracting muscles of nonmammalian vertebrates express only one isoform of the ryanodine receptor. Biophys J. 2019;Brien et al., 1993;65:2418–2427.
35. Paolini C, Fessenden JD, Pessah IN, Franzini-Armstrong C. Evidence for conformational coupling between two calcium channels. Proc Natl Acad Sci USA. 2004;101:12748–12752.
36. Peachey LD. The sarcoplasmic reticulum and transverse tubules of the frog’s sartorius. J Cell Biol. 1965;25(Suppl):209–231.
37. Peachey LD, Eisenberg BR. Helicoids in the T system and striations of frog skeletal muscle fibers seen by high voltage electron microscopy. Biophys J. 1978;22:145–154.
38. Pritchard TJ, Kranias EG. Junctin and the histidine-rich Ca2+ binding protein: potential roles in heart failure and arrhythmogenesis. J Physiol. 2009;587:3125–3133.
39. Rios E, Pizarro G. Voltage sensor of excitation-contraction coupling in skeletal muscle. Physiol Rev. 1991;71:849–908.
40. Samsó M, Wagenknecht T. Contributions of electron microscopy and single-particle techniques to the determination of the ryanodine receptor three-dimensional structure. J Struct Biol. 1998;121:172–180.
41. Schneider MF. Control of calcium release in functioning skeletal muscle fibers [Review]. Annu Rev Physiol. 1994;56:463–484.
42. Schneider MF, Chandler WK. Voltage dependent charge movement of skeletal muscle: a possible step in excitation-contraction coupling. Nature. 1973;242:244–246.
43. Sheridan DC, Takekura H, Franzini-Armstrong C, Beam KG, Allen PD, Perez CF. Bidirectional signaling between calcium channels of skeletal muscle requires multiple direct and indirect interactions. Proc Natl Acad Sci USA. 2006;103:19760–19765.
44. Suh-Kim H, Wei X, Klos A, et al. Reconstitution of the skeletal muscle dihydropyridine receptor Functional interaction among alpha 1, beta, gamma and alpha 2 delta subunits. Receptors Channels. 1996;4:217–225.
45. Sutko JL, Airey JA. Ryanodine receptor Ca2+ release channels: does diversity in form equal diversity in function?. Physiol Rev. 1996;76:1027–1071.
46. Tanabe T, Beam KG, Adams B, Niidome T, Numa S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature. 1990;346:567–569.
47. Tanabe T, Beam KG, Powell J, Numa S. Restoration of excitation-contraction coupling and slow calcium current in dysgenic muscle by dihydropyridine receptor complementary DNA. Nature. 1988;336:134–139.
48. Tanabe T, Takeshima H, Mikami A, et al. Primary structure of the receptor for calcium channel blockers from skeletal muscle. Nature. 1987;328:313–318.
49. Tinker A, Lindsay AR, Williams AJ. A model for ionic conduction in the ryanodine receptor channel of sheep cardiac muscle sarcoplasmic reticulum. J Gen Physiol. 1992;100:495–517.
50. Xu L, Wang Y, Gillespie D, Meissner G. Two rings of negative charges in the cytosolic vestibule of type-1 ryanodine receptor modulate ion fluxes. Biophys J. 2006;90:443–453.
51. Yamaguchi N, Takahashi N, Xu L, Smithies O, Meissner G. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca release channel. J Clin Invest. 2007;117:1344–1353.
52. Yu FH, Yarov-Yarovoy V, Gutman GA, Catterall WA. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol Rev. 2005;57:387–395.
53. Vergara J, Delay M. A transmission delay and the effect of temperature at the triadic junction of skeletal muscle. Proc R Soc Lond B Biol Sci. 1986;229:97–110.
54. Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367–385.
55. Zhou J, Brum G, Gonzalez A, Launikonis BS, Stern MD, Rios E. Ca2+ sparks and embers of mammalian muscle Properties of the sources. J Gen Physiol. 2003;122:95–114.