
1.1 Microscopic Anatomy of the Nervous System
Introduction
Neurologic diseases can only be understood on the basis of the anatomy and physiology of the nervous system. Genetic abnormalities underlie many of them. Modern methods such as electron microscopy, electrophysiologic testing, and biochemical and molecular-biological analysis have yielded new insights into neural structure and function that are playing an increasingly important role in the classification and diagnosis of nervous diseases, as well as in their treatment.
It follows that a basic knowledge of neuroanatomy (both gross and microscopic), neurophysiology, and neurogenetics is indispensable for contemporary medical practice. In this chapter, we briefly recapitulate the essential facts in these three areas.
For the student, the important questions are:
What are the microscopic building blocks of which the nervous system is composed?
What are the fundamental processes in neurophysiology?
What role do genetic factors play in the pathogenesis of disease?
The last question is becoming ever more important. Many neurologic diseases are hereditary, that is, partly or entirely due to genetic abnormalities; they will come to the reader’s attention again and again in the pages of this book. At present, in the era of molecular biology, the genetic defects underlying many of them have already been identified (with still more to come). For these diseases, diagnosis by deoxyribonucleic acid (DNA) testing is now possible even before the patient develops any overt symptoms. Such testing should be done only after the patient has been thoroughly informed of the potential consequences.
Key Point
Neurons are the structural and functional building blocks of the nervous system. They are specialized for the reception, integration, and transmission of electric impulses.
The neuronal cell body (soma) is enclosed by the cell membrane and contains the cell nucleus, mitochondria, endoplasmic reticulum, neurotubules, and neurofilaments ( ▶ Fig. 1.1). Dendrites are short, more or less extensively branched cellular processes that conduct afferent impulses toward the soma. They provide the cell with a much larger surface area than the soma alone, thereby increasing the area available for intercellular contact and for the deployment of cell membrane receptors. Each type of neuron has its own characteristic dendritic structure: the dendritic tree of a cerebellar Purkinje cell, for example, resembles a deer’s antlers ( ▶ Fig. 1.2). The axon is a single, elongated cell process that emerges from the soma at the axon hillock. It conducts efferent impulses away from the soma to another neuron or to an effector organ.
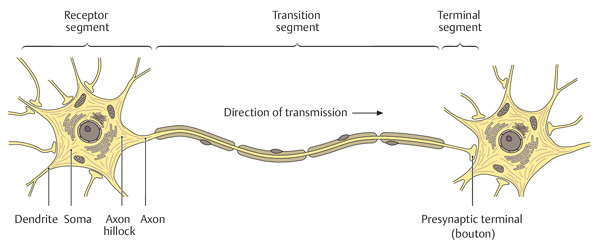
Fig. 1.1 Fine structure of a neuron. (Adapted from Schuenke et al. Thieme Atlas of Anatomy. Head and Neuroanatomy. New York, NY: Thieme Medical Publishers; 2011. Illustration by Markus Voll.)

Fig. 1.2 Cerebellar Purkinje cell (microphotograph). Note the numerous synapses on the dendrites. (Image provided courtesy of Dr. Marco Vecellio, Histological Institute of the University of Fribourg, Switzerland.)
In general, every neuron has a soma, an axon, and one or more dendrites. The structure and configuration of the neuronal processes (especially the dendrites) vary depending on the function of the neuron. Thus, neurons can be classified into several morphologic subtypes ( ▶ Fig. 1.3).
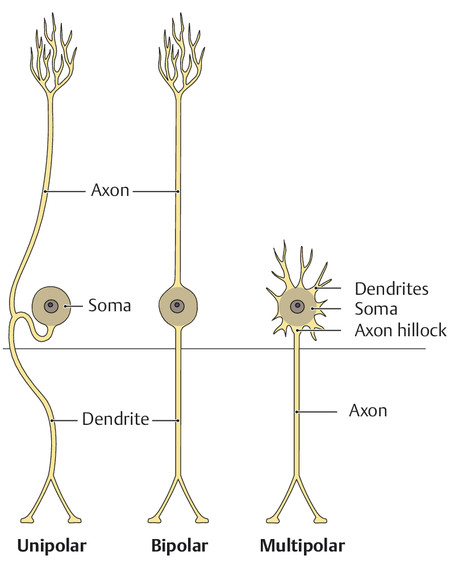
Fig. 1.3 Three types of neurons. (Adapted from Schuenke et al. Thieme Atlas of Anatomy. Head and Neuroanatomy. New York, NY: Thieme Medical Publishers; 2011. Illustration by Markus Voll.)
The neurons are traditionally thought to constitute the important functional part of the nervous system; they are surrounded by supportive cells, which are collectively called neuroglia. Astrocytes are neuroglial cells with a starlike structure. They make contact with nonsynaptic sites on the neuronal surface and also have perivascular foot processes that make contact with 85% of the capillaries of the nervous system. The astrocytes supply nutrients to the neurons and are an important constituent of the blood–brain barrier. Other types of supportive cell in the central nervous system are the oligodendrocytes, microglia, ependymal cells, and choroid plexus cells.
Axons less than 1 µm in diameter are usually unmyelinated; thicker ones are sheathed in myelin. The myelin sheath is generated when an axon “sinks” into an oligodendrocyte, giving rise to a mesaxon, that is, a double sheet of oligodendrocyte membrane. (In the peripheral nervous system, Schwann cells play the role of oligodendrocytes.) The mesaxon wraps around the axon multiple times to create the myelin sheath, a thick coat of electrically insulating material. Individual myelin segments (up to 1 mm long) are separated by segments of “naked” axon called nodes of Ranvier, which play an important role in impulse propagation (see section ▶ 1.1.4). The nodes are 1 to 4µm wide and are only partly covered by processes of the neighboring Schwann cells. They are thus separated from the endoneural interstitium by little more than the neuronal cell membrane (called the neurilemma or axolemma). The nodal axolemma mainly contains voltage-dependent sodium channels; the axonal segments between the nodes mainly contain potassium channels.
The sites at which neurons transmit impulses to each other are called synapses. The structures making up a synapse include: a bulblike expansion at the end of an axon, called an axon terminal (or bouton); the synaptic cleft; and the postsynaptic membrane of the receiving neuron or effector organ ( ▶ Fig. 1.4). Myelinated axons lose their myelin sheath just proximal to the axon terminal. A single neuron can receive synaptic input from one or more axons, and the impulses it receives can be either excitatory or inhibitory. An axon can form a synapse onto a cell body, a dendrite, or another axon. Ongoing structural and functional changes at the synapses give the nervous system functional adaptability (“plasticity”) even after the organism has reached maturity. Neural impulses are transmitted across synapses by chemical substances called neurotransmitters: some of the more important ones in the central nervous system are dopamine, serotonin, acetylcholine, and γ-aminobutyric acid (GABA). Specialized synapses connect the axons of the peripheral nervous system to effector organs such as muscle cells (motor end plates, see section ▶ 15.1.3) or secretory cells in glands.
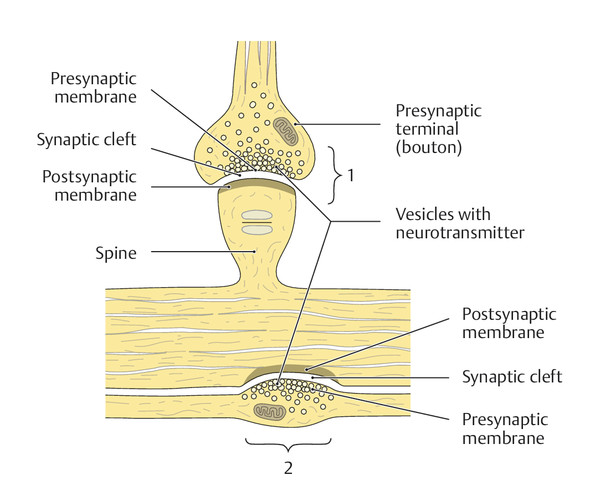
Fig. 1.4 Fine structure of a synapse. The two most common types of synapse are shown: 1 a spiny synapse and 2 a parallel contact or bouton en passage. (Reproduced from Schuenke et al. Thieme Atlas of Anatomy. Head and Neuroanatomy. New York, NY: Thieme Medical Publishers; 2011. Illustration by Markus Voll.)
Key Point
The resting membrane potential of a neuron or myocyte can undergo a rapid, transient change, called an action potential, in response to an incoming stimulus or impulse. The action potential is generated by transient changes of ion permeability across the cell membrane. Action potentials along neuronal processes and chemical impulse transmission between neurons at synapses are the mechanisms used by the nervous system for information transfer.
Neurons are enclosed by a double-layered cell membrane with an inner phospholipid layer and an outer glycoprotein layer. Specialized protein molecules within the cell membrane form channels that are selectively permeable to sodium, potassium, or chloride ions. Some ion channels (e.g., in synapses) open only when a specific ligand binds to them, for example, a neurotransmitter molecule. These channels are called ligand-dependent ion channels. Voltage-dependent ion channels, on the other hand, are found mainly on axons. They open and close depending on the transmembrane electric potential.
An electric potential difference arises across the neuronal membrane because of the unequal concentrations of ions in the intracellular and extracellular spaces (ICS, ECS), combined with the varying electric conductivity of the membrane to different types of ion. This resting potential is mainly determined by the ratio of intra- to extracellular potassium concentration. Its origin can be explained as follows. At rest, the membrane is highly permeable to potassium ions and relatively impermeable to sodium ions. The potassium concentration in the ICS is roughly 35 times higher than in the ECS. Thus, potassium ions tend to diffuse out of the cell. A buildup of negative charge on the inner surface of the membrane results; this, in turn, generates a difference of electric potential across the membrane that opposes further potassium ion outflow. An equilibrium is reached at which the potential difference exactly cancels out the force arising from the difference in potassium ion concentration. As there is no further net transfer of potassium ions across the membrane, the resting membrane potential remains stable, with a value ranging from –60 to –90 mV.
Because the sodium ion concentration is roughly 20 times higher in the ECS than in the ICS, the neurotransmitter-induced opening of ligand-sensitive postsynaptic sodium channels is followed by a rapid influx of sodium ions into the cell. The inner surface of the cell membrane becomes positively charged, and an action potential is generated whose amplitude and time course are independent of the nature and intensity of the depolarizing impulse (this is the all-or-nothing law of cellular excitation). The transmembrane potential difference reaches a peak ranging from +20 to +50 mV. Then, after a brief delay, the cell membrane becomes more permeable to potassium, and a net outflow of potassium ions results. This compensates for the preceding sodium influx and causes membrane repolarization. An active sodium pump also participates in this process. Until repolarization is complete, the membrane cannot conduct any further impulses; there is an initial absolute refractory period, followed by a relative refractory period.
The axon potential begins at the axon hillock and is conducted along the axonal membrane by the successive opening of voltage-dependent sodium channels. This wave of excitation (local depolarization) travels down the axon at a speed depending on the thickness of the axon and the thickness of its myelin sheath. The nodes of Ranvier play a major role in this process: the myelin sheaths lower the capacitance of the axonal membrane and raise its electric resistance. The action potentials are therefore initiated only at the nodes of Ranvier, “jumping over” the internodal segments (so-called saltatory conduction). This mechanism enables myelinated nerve fibers to conduct action potentials much more rapidly than unmyelinated fibers. The normal motor and sensory conduction velocity of peripheral nerves is 50 to 60 m/s.
Key Point
Many neurologic diseases are caused by a genetic defect or favored by a genetic predisposition. In this section, we present the basics of both “classic” (mendelian) inheritance and molecular genetics, which are prerequisites for the understanding of these diseases and the counseling of affected patients and their families.
Note
The physical characteristics of an organism in health and in disease, its phenotype, are determined by its genetic makeup, or genotype, along with environmental factors. DNA is the carrier of genetic information.
DNA molecules are located in the cell nucleus and mitochondria. A segment of DNA containing the information needed for the synthesis of a protein molecule is called a gene, and the totality of the organism’s genes is called the genome. Human nuclear genes are contained in 23 pairs of chromosomes: 22 pairs of autosomes and 1 pair of sex chromosomes (gonosomes), which are either XX (in females) or XY (in males).
Growth requires many cell divisions (mitosis). In each mitosis, the nuclear genetic material doubles in amount (replicates) and is distributed to the two daughter cells, so that each one, like the original cell, contains a complete (diploid) set of chromosomes. For sexual reproduction, a reductive cell division (meiosis) occurs, producing germ cells with a haploid set of chromosomes—that is, only one of each chromosome. The union of an egg cell and a sperm cell yields a zygote with a full (diploid) complement of chromosomes, half derived from the maternal genome and half from the paternal genome.
By the rules of mendelian inheritance, maternally derived and paternally derived properties (genes) are assorted randomly and independently to the germ cells, and thereby to the offspring. Yet, the independence of gene transmission is not total, because genes that lie on the same chromosome are usually transmitted together to the daughter cells. There is a phase of meiosis in which corresponding DNA segments on homologous chromatids can be exchanged with each other to produce a new arrangement of genetic material; this is called crossing-over or genetic recombination. The further apart two genes are on a chromosome, the more often recombination will occur between them.
Alongside these physiologic mechanisms of genetic change and reassortment (random mixing of maternal and paternal chromosomes in meiosis and fertilization, recombination of genes on homologous chromosomes), genes can also undergo spontaneous change, that is, mutation. Mutations in the germ line are passed on to the offspring (see later).
Unlike nuclear DNA, mitochondrial DNA is passed on only from the mother to her offspring, by way of the egg cell.
An allele (version of a gene) that markedly influences or completely determines the phenotype of the individual in the heterozygous state (i.e., even if it is present in only one copy) is called dominant. If either parent is heterozygous for a dominant allele, then the child has a 50% chance of being affected in both genotype and phenotype.
An allele of an autosomal gene that has an overt effect only in the heterozygous state (i.e., only if present in two copies) is called recessive. If both parents are heterozygous for such an allele, then 50% of their children will be asymptomatic heterozygotes (carriers) and 25% will be symptomatic homozygotes, and 25% will not inherit the recessive allele from either parent and will thus be both genetically and phenotypically normal.
Sons receive an X-chromosome from their mother and a Y-chromosome from their father; daughters receive X-chromosomes from both parents. A mother who is heterozygous for an abnormal X-chromosomal allele will pass it on to half of her children (of either sex), while a father bearing such an allele will pass it on to all of his daughters, but none of his sons. Dominantly inherited X-chromosomal diseases affect both sexes; recessively inherited X-chromosomal diseases mainly affect males, with females affected only if they have received the abnormal allele from both parents. Any affected male must have received the allele from his mother; as long as his mate is not a carrier, all of his daughters will be carriers. Female carriers with unaffected mates will pass on the disease to 50% of their sons; all of their daughters will be phenotypically normal, but half will be carriers.
Mitochondrial DNA is inherited exclusively in the maternal line. Thus, mitochondrial genetic diseases are passed on only by mothers to their children (both male and female), but never by fathers. Mitochondria with mutated DNA can coexist in the same cell with other mitochondria whose DNA is normal. This phenomenon, called heteroplasmy, has no counterpart in the nuclear genome, which is the same in all cells of the body. In mitochondrial genetic diseases, the extent of abnormality of the cells and tissues (the phenotype) depends on the number of mutated mitochondria and on the ratio of mutated to normal mitochondrial DNA.
Mutations are necessary for evolution; without them, no species (including man) could exist. Unfortunately, adverse mutations can also cause deformity and disease. Mutations are classified as either genomic or intragenic.
These are of two types, called numerical and structural chromosomal aberrations. In the former, the number of chromosomes is abnormal (e.g., monosomy, trisomy). In the latter, a chromosome has an abnormal structure—caused by a deletion, translocation, or inversion of a chromosomal segment.
Intragenic mutations involve alterations of the DNA. Within each chromosome, DNA is arranged linearly. The DNA segments (genes) that code for amino acid sequences (proteins), which are called exons, alternate with noncoding sequences called introns. Exons make up only ~5% of human chromosomal DNA. When the DNA is transcribed into RNA, the primary RNA transcript still contains a copy of the introns, which are then removed by “splicing” to yield the mature transcript, messenger RNA (mRNA).
Each trinucleotide sequence in the mRNA molecule (called a triplet or codon) encodes an amino acid in the protein being synthesized. “Stop codons” between exons signal the beginning and end of the gene and thereby determine the length of the protein. The replacement of a DNA nucleotide by a different one can alter the sense of the codon to which it belongs (missense mutations), causing the “wrong” amino acid to be inserted into the gene product. This can affect its function in a variety of ways. If a nucleotide replacement happens to generate or destroy a stop codon, then either a truncated protein or an excessively long one will be produced (nonsense mutations). Mutations involving either the insertion of an extra nucleotide into the DNA or the deletion of a nucleotide will alter the rhythm of nucleotide triplets and are therefore called frameshift mutations: these tend to cause marked abnormalities of protein structure and function (e.g., Duchenne muscular dystrophy; see section ▶ 15.3.1).
Another type of mutation of special importance in neurology changes the number of trinucleotides (triplets) in a gene. Normal human DNA contains many repetitive trinucleotide sequences that affect gene function and expression. An important group of neurodegenerative diseases is caused by mutations involving abnormally long (expanded) triplet repeat sequences; these are called trinucleotide or triplet repeat diseases. A normal repeat sequence might contain only a few triplets; diseased sequences contain dozens or even hundreds. The longer the expansion, the earlier the age of onset of disease and the more severe its manifestations. The abnormal repeat sequences tend to lengthen from one generation to the next, so that the disease tends to appear ever earlier (“anticipation”), and in ever greater severity, as it is passed down through the generations.
These impair oxidative metabolism in the mitochondria, causing various kinds of disease, including mitochondrial encephalomyopathies (see section ▶ 15.5.2).
Note
The triplet diseases are of special relevance in neurology.
Triplet repeat diseases The neurodegenerative diseases caused by expanded triplet repeats are listed in ▶ Table 1.1; their common features are as follows:
Autosomal dominant or X-chromosomal inheritance.
Onset usually between the ages of 25 and 45 years.
Gradual progression of disease.
Symmetric neuronal loss and gliosis in the brain.
Anticipation (earlier disease onset in successive generations).
The diagnosis can be established by DNA analysis.
The number of triplet repeats is correlated with the age of onset and the severity of the disease.
|
Disease |
Major clinical manifestations |
Triplet |
Chromosomal localization |
|
Fragile X-chromosome |
Diminished intelligence, sometimes facial dysmorphism, connective tissue dysplasia |
CGG |
Xq27.3 |
|
Fragile X tremor/ataxia syndrome (FXTAS) |
Progressive intention tremor, extrapyramidal hypokinesia, impotence, and cognitive impairment in old age |
CGG |
Xq27.3 |
|
Myotonic dystrophy of Steinert (DM1) |
Progressive, mainly distal muscular dystrophy and myotonia |
CTG |
19q13.3 |
|
Myotonic dystrophy type 2 (proximal myotonic myopathy, PROMM, DM2) |
Mainly proximal muscular dystrophy and myotonia |
CCTG |
3q13.3-q24 |
|
Friedreich ataxia |
Ataxia, areflexia, pyramidal tract signs, dysarthria |
GAA |
9q13-q21.1 |
|
Spinobulbar muscle atrophy (Kennedy syndrome) |
Muscle atrophy, dysarthria, fasciculations, gynecomastia |
CAG |
Xq13-q21 |
|
Huntington disease |
Chorea, rarely spasticity or rigidity, cognitive and behavioral disturbances |
CAG |
4p16.3 |
|
Dentate-rubro-pallido-Luysian atrophy (DRPLA) |
Ataxia, myoclonus, epilepsy, choreoathetosis, dementia |
CAG |
12p13.31 |
|
Spinocerebellar ataxia type 1 (SCA1) |
cerebellar ataxia, sometimes chorea or dystonia, polyneuropathy, often pyramidal tract signs, sometimes dementia |
CAG |
6p22.3 |
|
Spinocerebellar ataxia type 2 (SCA2) |
Cerebellar ataxia, sometimes chorea or dystonia, myoclonus, polyneuropathy, sometimes pyramidal tract signs and dementia |
CAG |
12q24.12 |
|
Spinocerebellar ataxia type 3 (SCA3); Machado–Joseph disease |
Cerebellar ataxia, sometimes chorea or dystonia, polyneuropathy, sometimes pyramidal tract signs and dementia |
CAG |
14q32.12 |
|
Spinocerebellar ataxia type 6 (SCA6) |
Cerebellar ataxia, sometimes polyneuropathy and pyramidal tract signs |
CAG |
19p13.2 |
|
Spinocerebellar ataxia type 7 (SCA7) |
Cerebellar ataxia, sometimes chorea or dystonia, retinal degeneration, polyneuropathy, sometimes pyramidal tract signs |
CAG |
3p14.1 |
|
Spinocerebellar ataxia type 8 (SCA8) |
cerebellar ataxia, spasticity, impaired vibration sense |
CTG |
13q21.33 |
|
Spinocerebellar ataxia type 10 (SCA10) |
cerebellar ataxia, epileptic seizures, sometimes polyneuropathy |
ATTCT |
22q13.31 |
|
Spinocerebellar ataxia type 12 (SCA12) |
Cerebellar ataxia, extrapyramidal hypokinesia, later dementia |
CAG |
5q32 |
|
Spinocerebellar ataxia type 17 (SCA17) |
Cerebellar ataxia, spasticity, cognitive impairment, psychosis, epileptic seizures |
CAG |
6q27 |
Most common inherited mitochondrial diseases
Progressive external ophthalmopathy.
Kearns–Sayre syndrome.
Leber’s hereditary optic neuropathy.
Mitochondrial encephalomyopathy with lactic acidosis and stroke.
Leigh disease.
Neuropathy, ataxia, and retinitis pigmentosa syndrome.
Myoclonus epilepsy with ragged red fibers.
Myoneurogastrointestinal encephalopathy.
Practical Tip
Ever more genetic defects are being identified. Rapid access to current knowledge is best obtained via the Internet. Two useful sites are Online Mendelian Inheritance in Man (OMIM) and Medline/Pubmed.
Many genetic mutations can be detected directly by DNA analysis. The results are highly specific. Thus, many diseases can be diagnosed even before they become symptomatic. Sadly, these diseases are generally untreatable and inexorably progressive.
Note
The findings of DNA analysis can be emotionally devastating for the patient. Before DNA analysis, the patient must be thoroughly informed and counseled by a physician.
Before obtaining a DNA analysis, the treating physician should:
Perform a meticulous clinical examination.
Obtain a detailed family history and personally examine the patient’s relatives, if possible.
Inform the patient and his or her relatives in detail about the suspected disease.
Explain the consequences of the proposed DNA analysis to them in a readily understandable manner.
A negative DNA analysis can provide relief and freedom from anxiety. A positive result, on the other hand, can propel the patient into a severe depression, as he or she will then face the certainty of developing a hereditary disease, often with a grim prognosis, and may not be able to cope with this knowledge. A known genetic defect can also severely strain a marriage or other relationship. Social problems of other kinds can arise as well, because, unfortunately, persons with inherited diseases can easily become pariahs in our postindustrial society. They may have difficulties finding or keeping a job, not least because they may become uninsurable. For all these reasons, genetic testing generally causes fewer problems if it is performed after the disease has become symptomatic. Asymptomatic children should not have their DNA tested even if their parents ask for it. They should decide for themselves whether to be tested once they are mature enough to do so and have attained legal majority.
Many patients and their relatives decide not to be tested after being fully informed about their potential genetic disease and the consequences of DNA analysis. In particular, pre- and asymptomatic persons often prefer not to know whether they are going to develop the disease in the future. A positive test result would destroy their hopes for good health in later life.
If the patient does decide to undergo DNA analysis and then tests positive, the physician should inform the patient and his or her relatives in a personal discussion, with ample time to address all of the implications. Test results should never be imparted over the telephone or in writing. Patients who have tested positive often need long-term psychotherapy. Nor does the physician–patient relationship end when the test results are given: many persons with hereditary diseases are greatly helped by ongoing psychological support and symptomatic treatment.