 GENERAL PRINCIPLES GENERAL PRINCIPLES
GENERAL PRINCIPLES GENERAL PRINCIPLESThe evaluation of patients with suspected neurologic disease remains first and foremost a bedside exercise. Accurate diagnosis requires consideration of individual patient and disease differences. Despite the benefits of evidence-based medicine, conclusions are more relevant to populations than to individuals. Confounding variables that are part of the human experience may be overlooked or overemphasized by testing algorithms. This textbook will repeatedly emphasize the strongly held philosophy of its authors, that is, patient management flows from an accurate diagnosis. An accurate diagnosis is most likely to be obtained based on a differential diagnosis driven by clinical assessment and hypotheses. These hypotheses should be formulated on the basis of the principles of neurologic localization, the correlation of the chronologic course of symptom development with the behaviors of differing disease conditions, and the application of risk factor analysis. Ideally, the tests described in the subsequent two chapters and throughout the text would be utilized with the primary intent of resolving a clinically established differential diagnosis ideally to prove a working diagnosis. As all tests are potentially fallible, the credibility of their results diminishes when they are used as screening procedures. A laboratory abnormality, occurring without the context of clinical correlation, fails to establish the desired confidence in a cause and effect relationship with the patient’s complaint(s). Metaphorically, laboratory tests are analogous to a carpenter’s tools. They are of great value when placed in the hands of a skillful artisan, but are potentially damaging if used injudiciously.
In this book, a neuromuscular disorder will refer to any condition that affects the structure and/or function of any component of the neuromuscular system, beginning and working centrifugally from the cell bodies of the anterior horn and dorsal root ganglion. This will include disorders of nerve root, plexus, nerve, neuromuscular junction and muscle. In essence, with the exception of disorders affecting small, poorly, or unmyelinated nerve fibers such as the small fiber or pure autonomic neuropathies, a neuromuscular disorder may alternatively be defined as one that can be potentially detected by electromyography and nerve conduction studies. Disorders affecting the peripheral autonomic system or cranial nerves will be discussed only as necessary to better understand diseases affecting their somatic and spinal counterparts.
Many neuromuscular disorders are the result of or are influenced by single gene or complex genetic mutations. Many of these patients will not recognize the hereditary nature of their disease. This may be due to a recessive inheritance pattern, spontaneous mutation, false paternity, or incomplete or delayed penetrance. Frequently, it is due to a lack of familiarity with the medical issues of other family members. In suspected hereditary disease, acquisition of family history, particularly if done in a cursory fashion, may be insufficient. Examination of other family members, even if only briefly, is strongly recommended when heritable diseases are considered.
The differential diagnosis of disorders of the neuromuscular system is in part age-dependent. The differential diagnosis of neuromuscular conditions in infants, children, and adolescents is both overlapping and unique in comparison to their adult counterparts (Tables 1-1 to 1-3).1,2 The applied diagnostic principles are similar although both the examination and review of symptoms may be hampered in infants. In the pediatric population, parents must be questioned with great care and sensitivity. The heightened concern of the parents may cause them to unconsciously omit important details of the patient’s status or assume a benign attribution as the cause of the symptom. Parents may also bring a considerable amount of guilt to the examination, which may limit their willingness to share information. The parents’ fears and associated guilt should be addressed. If necessary, professional counseling should be offered in addition to treating the patient. Often, when a child is ill, the entire family is affected, which can in turn have profound repercussions on the entire family from both a physical and a psychological standpoint.
 TABLE 1-1. DIFFERENTIAL DIAGNOSIS OF THE FLOPPY INFANT
TABLE 1-1. DIFFERENTIAL DIAGNOSIS OF THE FLOPPY INFANT
Central nervous system disorders (most common etiology)
Anterior horn cell
Spinal muscular atrophy types I and II
Peripheral neuropathy
CMT III (Dejerine–Sottas, congenital hypomyelinating/amyelinating neuropathy)
CMT I and CMT II—rare
Giant axonal neuropathy
Neuromuscular junction
Infantile botulism
Transient neonatal myasthenia gravis
Congenital myasthenic syndromes
Myopathy
Congenital myopathies (all of them can present in infancy)
Muscular dystrophies
Congenital muscular dystrophies
Dystrophinopathy/sarcoglycanopathy (rare)
Congenital myotonic dystrophy
Metabolic myopathies
Glycogen storage defects
Acid maltase deficiency
Debrancher deficiency
Branching enzyme deficiency
Myophosphorylase deficiency (rare)
Disorders of lipid metabolism
Carnitine deficiency
Other fatty acid/acyl-CoA dehydrogenase deficiencies
Mitochondrial myopathies
Benign and fatal infantile myopathy
Leigh’s syndrome
Endocrine myopathies (e.g., hypothyroidism)
Modified with permission from Dumitru D, Amato AA. Introduction to myopathies and muscle tissue’s reaction to injury. In: Dumitru D, Amato AA, Swartz MJ, eds. Electrodiagnostic Medicine. 2nd ed. Philadelphia, PA: Hanley & Belfus; 2002.
TABLE 1-2. NEUROMUSCULAR CAUSES OF WEAKNESS PRESENTING IN CHILDHOOD OR EARLY ADULTHOOD
Anterior horn cell
Spinal muscular atrophy type III
Poliomyelitis
Amyotrophic lateral sclerosis
Peripheral neuropathy
Acute or chronic inflammatory demyelinating polyneuropathy
Hereditary neuropathies
Neuromuscular junction
Botulism
Myasthenia gravis
Congenital myasthenic syndromes
Lambert–Eaton syndrome
Myopathy
Congenital myopathies
Central core
Multicore
Centronuclear
Nemaline
Muscular dystrophies
Dystrophinopathy (Duchenne or Becker)
Limb-girdle muscular dystrophies
Myofibrillar myopathy
Myotonic dystrophy
Other dystrophies (e.g., FSHD and EDMD)
Metabolic myopathies
Glycogen storage defects
Acid maltase deficiency
Debrancher and branching enzyme deficiency
Disorders of lipid metabolism
Carnitine deficiency
Other fatty acid/acyl-CoA dehydrogenase deficiencies
Mitochondrial myopathies
Periodic paralysis
Electrolyte imbalance
Hyperkalemia
Hypokalemia
Hypophosphatemia
Hypercalcemia
Endocrine myopathies
Toxic myopathies
Inflammatory myopathies
Dermatomyositis
Polymyositis (after the age of 20 years)
Infectious myositis
FSHD, facioscapulohumeral muscular dystrophy; EDMD, Emery–Dreifuss muscular dystrophy.
Modified with permission from Dumitru D, Amato AA. Introduction to myopathies and muscle tissue’s reaction to injury. In Dumitru D, Amato AA, Swartz MJ. eds. Electrodiagnostic Medicine. 2nd ed. Philadelphia, PA: Hanley & Belfus; 2002.
TABLE 1-3. NEUROMUSCULAR CAUSES OF WEAKNESS PRESENTING IN MIDDLE TO LATE ADULTHOOD
Anterior horn cell
Spinal muscular atrophy type III
Kennedy disease
Poliomyelitis
Amyotrophic lateral sclerosis
Peripheral neuropathy
Hereditary neuropathies
Acute or chronic inflammatory demyelinating polyneuropathy
Drug-induced or toxic neuropathies
Diabetic neuropathy
Amyloid
Vasculitis
Neuromuscular junction
Botulism
Myasthenia gravis
Lambert–Eaton syndrome
Myopathy
Muscular dystrophies
Dystrophinopathy (Becker)
Limb-girdle muscular dystrophies
Myofibrillar myopathy
Oculopharyngeal dystrophy
Bent spine/dropped head syndrome
Metabolic myopathies
Glycogen storage defects
Acid maltase deficiency
Debrancher deficiency
Disorders of lipid metabolism (rare)
Congenital myopathies
Sporadic late onset nemaline myopathy
Mitochondrial myopathies
Periodic paralysis
Familial hypo-KPP manifest within the first three decades
Familial hyper-KPP usually manifests in the first decade
Electrolyte imbalance
Hyperkalemia
Hypokalemia
Hypophosphatemia
Hypercalcemia
Endocrine myopathies
Toxic myopathies
Myopathy associated with systemic disease (e.g., cancer), poor nutrition, and disuse
Amyloid myopathy
Inflammatory myopathies
Inclusion body myositis (most common inflammatory myopathy after the age of 50 years)
Dermatomyositis
Polymyositis (after the age of 20 years)
Infectious myositis
hypo-KPP, hypokalemic periodic paralysis; hyper-KPP, hyperkalemic periodic paralysis.
Modified with permission from Dumitru D, Amato AA. Introduction to myopathies and muscle tissue’s reaction to injury. In: Dumitru D, Amato AA, Swartz MJ, eds. Electrodiagnostic Medicine. 2nd ed. Philadelphia, PA: Hanley & Belfus; 2002.
The nature of neurologic practice is such that many patients evaluated by a neurologist will have complaints that are attributable neither to a specific neuromuscular disorder nor to the nervous system in general. Confidence in the ability to exclude neuromuscular disorders from consideration is enhanced by a thorough knowledge of how these conditions behave. The strategies outlined in this chapter are based on the general principle that diagnostic accuracy is enhanced by correlation of the patient’s signs and symptoms, with knowledge of the natural history and behavior of the ever-expanding menu of neuromuscular diseases. In our opinion, adherence to these principles will improve diagnostic accuracy. This chapter will attempt to focus on information that is important to elicit, and also on an organizational framework to allow accurate interpretation.
DOES THE PATIENT HAVE A NEUROMUSCULAR PROBLEM?Neuromuscular diseases manifest themselves through some symptoms or combination of symptoms attributable directly or indirectly to the dysfunction of peripheral motor, sensory and autonomic nerves, neuromuscular junction or muscle. Motor symptoms are typically expressed in a “negative” fashion (weakness or atrophy). Occasionally, “positive” symptoms referable to overactivity [e.g., muscle cramps and fasciculations with LMN involvement and stiffness or flexor spasms in upper motor neuron (UMN) involvement] may dominate the clinical presentation. Sensory symptoms may also manifest with either a positive (e.g., paresthesia) or a negative (e.g., numbness or sensory ataxia) manner. Although pain may be considered a positive sensory symptom, it will be considered as an independent symptom in this text as it is neither a common or dominant feature in many neuromuscular conditions.
Neuromuscular disorders which manifest themselves solely within the domain of the motor system typically originate from anterior horn cells, the neuromuscular junction, muscle or rarely motor nerve fibers. Sensory symptoms typically imply a disorder of nerve root, dorsal root ganglion, plexus, or one or more peripheral nerve trunks. During history acquisition, there is considerable value in identifying both the location and the nature of the initial symptom(s), including the context in which that symptom developed. The subsequent evolution of symptoms should then be developed in a chronologic fashion with particular attention to the topographical distribution. The value of this approach may be illustrated with the example of multifocal neuropathy. At the time of their initial neurologic assessment, the patient’s deficits may have become confluent and indistinguishable from a length-dependent neuropathy and its far more extensive differential diagnosis. Identifying that the initial symptom occurred in a focal nerve distribution limits the differential diagnosis and improves diagnostic accuracy. The benefit of defining the chronologic course is that the differential diagnosis of acute neuromuscular disorders is notably disparate from that of its chronic counterparts (Tables 1-4 to 1-6).
TABLE 1-4. NEUROMUSCULAR DISORDERS PRESENTING WITH ACUTE OR SUBACUTE PROXIMAL OR GENERALIZED WEAKNESS
Anterior horn cell
Poliomyelitis
Peripheral neuropathy
Guillain–Barré syndrome
Porphyria
Diphtheria
Tick paralysis
Toxic neuropathies
Diabetic amyotrophy
Vasculitis
Carcinomatous infiltration (e.g., leukemia and lymphoma)
Paraneoplastic neuropathy
Neuromuscular junction
Botulism
Lambert–Eaton syndrome
Myasthenia gravis
Myopathy
Periodic paralysis
Electrolyte imbalance
Endocrinopathies
Inflammatory myopathies
Dermatomyositis
Polymyositis
Infectious myositis
Immune mediated necrotizing myopathy
Toxic myopathies
Metabolic myopathies
Glycogen and lipid disorders
Neuromyopathy
Critical illness neuromyopathy
Reroduced with permission from Dumitru D, Amato AA. Introduction to myopathies and muscle tissue’s reaction to injury. In: Dumitru D, Amato AA, Swartz MJ, eds. Electrodiagnostic Medicine. 2nd ed. Philadelphia, PA: Hanley & Belfus; 2002.
TABLE 1-5. DIFFERENTIAL DIAGNOSIS OF CHRONIC PROGRESSIVE PROXIMAL WEAKNESS
Anterior horn cell
Amyotrophic lateral sclerosis
Spinal muscular atrophy type III
Kennedy disease
Peripheral neuropathy
Chronic inflammatory demyelinating polyneuropathy
Multifocal motor neuropathy
Toxic neuropathies
Neuropathy associated with systemic disorders
Connective tissue disease (e.g., vasculitis)
Diabetes mellitus
Amyloidosis
Paraneoplastic
Carcinomatous infiltration (e.g., leukemia and lymphoma)
Neuromuscular junction
Lambert–Eaton syndrome
Myasthenia gravis
Myopathy
Muscular dystrophy
Periodic paralysis
Electrolyte imbalance
Endocrinopathies
Inflammatory myopathies
Dermatomyositis
Polymyositis
Infectious myositis
Toxic myopathies
Metabolic myopathies
Glycogen and lipid disorders
Miscellaneous: Tick paralysis, hypophosphatemia; hypokalemia
Reproduced with permission from Dumitru D, Amato AA. Introduction to myopathies and muscle tissue’s reaction to injury. In: Dumitru D, Amato AA, Swartz MJ, eds. Electrodiagnostic Medicine. 2nd ed. Philadelphia, PA: Hanley & Belfus; 2002.
TABLE 1-6. NEUROMUSCULAR CAUSES OF CHRONIC DISTAL WEAKNESS CAUSING BILATERAL FOOT AND/OR HEEL DROP
Anterior horn cell
ALSa
Distal spinal muscular atrophy
Polio and other enterovirusa
Conus medullaris syndrome—e.g., myelodysplasia, ependymoma, syringomyelia
Scapuloperoneal form of SMA
Nerve
Charcot–Maries–Tooth disease
Multifocal neuropathiesa—infiltrative (neoplastic, amyloid, sarcoid, neurofibromatosis), vasculitic, immune mediated (MADSAM, MMN)
NMJ
Autoimmune myasthenia gravis (rare)
Congenital myasthenia
Muscle
Distal myopathies—Nonaka, Miyoshi, Udd, Welander, Laing
Muscular dystrophies—scapuloperoneal, fascioscapuloperoneal, Emery–Dreifuss, oculopharyngeal distal myopathy, calveolinopathy
Congenital myopathies—nemaline, central core, nemaline
Glycogen storage diseases—brancher, debrancher/polyglucosan body disease, Pompe, phosphorylase B kinase deficiency
Lipid storage disorders—neutral lipid storage myopathy, multiple acyl-coA dehydrogenase deficiency
Myofibrillar myopathy
Inflammatory—inclusion body myositisa
aUsual notable asymmetries.
In the history acquisition, it is imperative not to accept words at face value and to explore what that word means to a patient. For example, it is not uncommon for patients to say numb when they mean weak, and weak when they mean numb. The mechanism of impaired function should be explored. For example, questions should be formulated to determine whether a fall is due to proximal weakness resulting in failure of antigravity muscles, tripping due to a foot drop, or loss of balance due to impaired proprioception, vestibular function, or disordered postural reflexes originating at the central nervous system level. Detailed questioning may be required to determine whether the inability to get out of the chair is due to proximal weakness or impaired central nervous initiation.
It is important to identify symptoms not only referable to the peripheral neuromuscular system but to symptoms relating to impairment of higher cortical or cranial nerve function. In addition, a major discriminator in the development of a differential diagnosis is the presence or absence of symptoms referable to involvement of other organ systems. A careful system review is important in an attempt not only to achieve a diagnosis but also to fully anticipate the scope of its potential morbidity. For example, the recognition of orthostasis either by history or examination can provide insight that an evolving, otherwise nonspecific neuropathy pattern may be attributable to amyloidosis. Symptoms referable to cardiomyopathy or cardiac conduction defects, impaired GI motility, cutaneous change, and contractures may clarify the differential diagnosis in the heritable myopathies.
As muscle weakness is usually the most objective manifestation of neuromuscular disease, emphasis is placed not only on its existence but on its characteristics (e.g., upper or lower motor neuron) and on the pattern of involvement (Tables 1-4 to 1-7). The existence of weakness may be apparent either through history taking or, more commonly, by examination. Even though muscle weakness is the hallmark of neuromuscular disease, patients frequently identify weakness by its functional consequences. Patients with proximal upper extremity weakness commonly complain of activities of daily living (ADLs) that involve use of the arms at or above shoulder level. Shaving or drying hair, obtaining objects off shelves, or getting arms in coat sleeves are notable examples. Distal upper extremity weakness interferes with a wide variety of activities such as diminished grip strength, difficulty with opening flip tops on beverage cans, buttoning or using nail clippers. Patients with hip flexor weakness have trouble going up stairs or getting their legs into vehicles. Patients with hip or knee extensor weakness have troubles with stairs in either direction, getting up from a squat or a deep chair. Patients with foot dorsiflexion weakness may trip whereas patients with plantar flexion weakness cannot run or walk as fast and cannot reach for objects as effectively.
TABLE 1-7. PATTERNS OF MUSCLE WEAKNESS AND CORRELATIONS WITH NEUROMUSCULAR LOCALIZATION
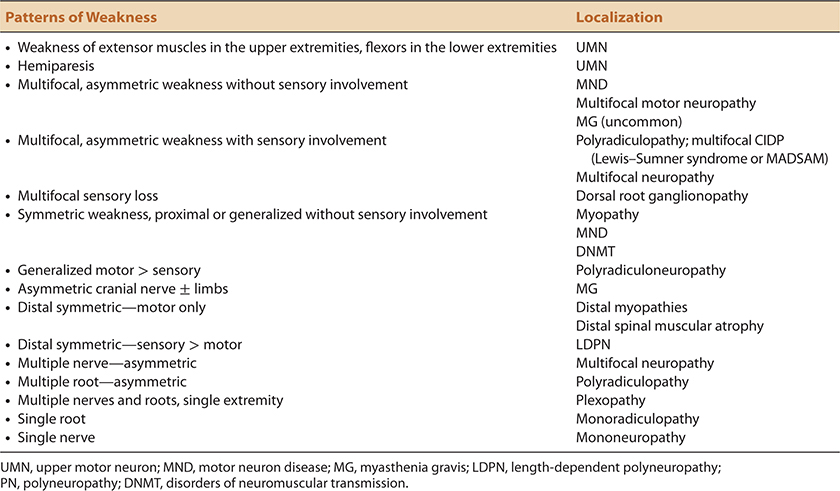
Conversely, the complaint of weakness is more commonly used by patients as a synonym for asthenia—a more pervasive, generalized complaint due to a number of different conditions. History taking pertaining to muscle weakness should focus on the identification of specific functions or activities that the patient finds difficult. If a patient who claims to be weak cannot describe a specific activity that is problematic for them, the existence of true muscle weakness remains suspect unless subsequently corroborated by the physical examination. Conversely, it is not rare for a disorder such as Lambert–Eaton myasthenic syndrome where credible functional impairments due to muscle weakness appear disproportionate to actual weakness found on bedside examination.
At times, weakness may present with pain rather than with symptoms directly attributable to weakness. For example, patients with trapezius weakness commonly present with shoulder pain, presumably due to traction on pain-sensitive structures resulting from their “shoulder drop.” Pain originating from strain on joints or soft tissues, as a secondary consequence of neuromuscular disease and the weakness it produces, is not uncommon.
UMN involvement needs to be considered in patients with potential neuromuscular disease, either as an alternative explanation for symptoms, or as a component of their neuromuscular condition. UMN pathology interferes with the synergistic functions of multiple muscle groups. As a result, functional activities highly dependent on coordinated muscle actions are commonly impaired early in the disease course. Impaired running and hand dexterity are notable examples. In addition, positive motor symptoms that occur commonly in UMN disease such as limb stiffness or spasms are readily recognized. They may complain of a tendency to drag one or both lower extremities. If the corticobulbar tracts are affected, swallowing and articulation are affected early and prominently, as these functions are dependent on the coordinated interplay of multiple muscle groups. The speech pattern that results is often halting, effortful, and “strangled” in its characteristics. Patients may lose their ability to effectively sniff or blow their nose. Patients with corticobulbar tract involvement may also develop lability of affect known as pseudobulbar palsy or forced yawning.
In contrast, as the final common pathway, lower motor neuron disorders express themselves in a limited number of ways, typically as a direct effect of functional loss due to weakness. Depending on a patient’s handedness, vocation or hobbies, this may not be noticed until the weakness is substantial. Less commonly, the patient’s initial complaints pertaining to lower motor neuron loss may reflect awareness of atrophy, fasciculations, or cramps.
Patients with weakness of hip flexion will have difficulty getting in and out of a car without manually lifting their thighs. Unless there is concomitant knee extensor weakness, patients will have more difficulty going upstairs than down as the former requires active hip flexion against gravity. Patients with weakness of hip abductors will waddle as a compensatory maneuver to maintain their center of gravity and balance. Patients with chronic weakness of hip extension will have difficulty rising from a chair and a tendency to have exaggerated lumbar lordosis as well, the latter resulting from posterior displacement of the shoulders for the same compensatory reasons. Knee extension weakness will result in difficulty getting up from a squat or out of deep chairs and commonly results in falls due to buckling of one or both knees. These patients may hyperextend their knees in order to prevent this while standing or walking (i.e., genu recurvatum). Ankle dorsiflexion weakness often results in tripping. Ankle plantar flexion weakness affects the efficiency of walking and deprives individuals from the ability to stand on their toes and run effectively.
In the upper extremity, people with weakness of the shoulder girdle will have difficulty with antigravity movements such as washing their hair, lifting heavy pans, inserting arms into coat sleeves, or retrieving objects from shelves. Weakness of elbow flexion and extension often goes unnoticed until fairly severe but may be recognized while attempting to open doors that require pull and push, respectively. Wrist and digit weaknesses interfere with grip and dexterity, which may impair multiple ADLs, including opening of bottles and cans, grasping zipper tabs, turning ignition keys, or buttoning buttons.
Neuromuscular disorders often affect the motor and to a lesser extent sensory functions of cranial nerves. Extraocular muscle involvement is a key discriminating factor in working through the differential diagnosis of neuromuscular disorders. For example, the extraocular muscles are rarely affected in motor neuron disease (MND), the majority of polyneuropathies or acquired inflammatory myopathies. Conversely, they may represent prominent manifestations of the inflammatory demyelinating polyneuropathies, disorders of neuromuscular transmission, and a finite list of muscle diseases, typically heritable in nature.
Patients typically become aware of ptosis by personal or family observation (Table 1-8). Occasionally, they first become aware when their vision is impaired by the drooping eyelid. Extraocular muscle involvement is typically expressed as diplopia, although patients with slowly progressive, symmetric involvement of the extraocular muscles such as in chronic progressive external ophthalmoplegia may have limited awareness of their deficit.
TABLE 1-8. NEUROMUSCULAR CAUSES OF PTOSIS OR OPHTHALMOPLEGIA
Peripheral neuropathy
Guillain–Barré syndrome
Miller–Fisher syndrome
CANOMAD
Mitochondrial (SANDO)
Neuromuscular junction
Botulism
Lambert–Eaton syndrome (ptosis only)
Myasthenia gravis (pupil sparing)
Congenital myasthenia
Myopathy
Mitochondrial myopathies
Kearn–Sayres syndrome
Progressive external ophthalmoplegia
Oculopharyngeal and oculopharyngodistal muscular dystrophy
Myotonic dystrophy (ptosis only)
Congenital myopathy
Myotubular
Nemaline (ptosis only)
Congenital fiber type disproportion
Multiminicore disease
Hyperthyroidism/Graves disease (ophthalmoplegia without ptosis)
Hereditary inclusion body myopathy type III
Notable exceptions: anterior horn cell diseases; acquired inflammatory myopathies
CANOMAD, chronic ataxic neuropathy ophthalmology IgM paraprotein cold agglutinins disialosyl antibodies; SANDO, sensory ataxic neuropathy, dysarthria, ophthalmoplegia.
Modified with permission from Dumitru D, Amato AA. Introduction to myopathies and muscle tissue’s reaction to injury. In: Dumitru D, Amato AA, Swartz MJ, eds. Electrodiagnostic Medicine. 2nd edn. Philadelphia, PA: Hanley & Belfus; 2002.
Patients with acute onset of unilateral facial weakness are usually very aware of the existence and nature of their problem. In many neuromuscular disorders, facial weakness is often chronic and symmetric, and as a result, the patient may not be aware of their deficit (Table 1-9). It is not rare for chronic bifacial weakness to be recognized for the first time on a routine neurologic examination. Questions pertaining to a tendency to sleep with eyes incompletely closed, the ability to blow up balloons or whistle may help to estimate the duration of a problem in situations such as these.
TABLE 1-9. NEUROMUSCULAR DISORDERS ASSOCIATED WITH FACIAL WEAKNESS29
Anterior horn cell
Amyotrophic lateral sclerosis
Spinal muscular atrophy
Kennedy disease
Polycranialradiculoneuropathy
Lyme
Sarcoidosis
Neoplastic meningitis
Chronic meningitis
GBS
CIDP
Neuromuscular junction
Autoimmune myasthenia gravis
Congenital myasthenia gravis
Lambert–Eaton myasthenia gravis
Botulism
Muscle
Facioscapulohumeral muscular dystrophy
Congenital myopathies
Myotonic muscular dystrophy
Inclusion body myositis
Oculopharyngeal distal myopathy
Symptomatic jaw weakness is an infrequent neuromuscular complaint. When present, it is often overshadowed by symptoms referable to muscles concomitantly affecting speech, swallowing, and breathing. Difficulty with chewing should nonetheless be inquired about, as it may sometimes be the initial or key symptom in a limited number of disorders such as myasthenia or Kennedy disease.
Symptoms referable to tongue weakness are common in many neuromuscular disorders. Patients typically become aware of tongue weakness as a result of dysarthria. Other issues may include the inability to manipulate food properly within their mouth. This kind of detail is uncommonly volunteered by the patient and is more frequently elucidated by detailed questioning.
Weakness of the neck muscles may be noticed by patients or their families when the neck extensors can no longer support the weight of the head and head drop develops by the development of head drop (Table 1-10). This is often accompanied by nuchal discomfort, presumably due to the constant and unaccustomed traction on posterior cervical ligamentous structures. Neck discomfort from head drop may be distinguished from other, more common causes of neck pain, by the relief allowed by neck support. Head drop may contribute to dysphagia as well. Trapezius weakness is most commonly symptomatic when acute and unilateral and is usually a result of a mononeuropathy of the accessory nerve. As discussed above, trapezius weakness is usually presents with shoulder pain as the index symptoms. Shoulder drop can be easily missed unless the patient is viewed from the rear, with the back exposed.
TABLE 1-10. NEUROMUSCULAR DISORDERS ASSOCIATED WITH HEAD DROP7-24
Anterior horn cell
Amyotrophic lateral sclerosis
Radiation myelopathy
Syringomyelia
Neuromuscular junction
Autoimmune myasthenia gravis
Neuropathy
Guillain-Barré syndrome
CIDP
Muscle
Polymyositis
Inclusion body myositis
Focal myositis
Sporadic late onset nemaline myopathy (SLONM)
Hereditary inclusion body myopathy
Laminopathy
Selenoproteinopathy
Isolated neck extensor myopathy
Proximal myotonic myopathy
Carnitine deficiency
Facioscapulohumeral muscular dystrophy
Mitochondrial myopathy
Hyperparathyroidism
Hypokalemia
Myofibrillar myopathy (desmin)
Weakness of the scapula can result from weakness of either the trapezius or serratus anterior muscles (Table 1-11). Scapular winging interferes with both shoulder-girdle strength and mobility. Patients may note either difficulty in raising an arm above the head or an inability to push with the accustomed force, for example, while doing pushups.
TABLE 1-11. NEUROMUSCULAR DISORDERS ASSOCIATED WITH SCAPULAR WINGING25
Anterior horn cell
Scapuloperoneal spinal muscular atrophy
Nerve
Accessory nerve palsy
Long thoracic nerve palsy
Davidenkow’s syndrome
Muscle
Facioscapulohumeral muscular dystrophy
Scapuloperoneal muscular dystrophy
Limb-girdle muscular dystrophy (e.g., calpainopathy)
Acid maltase deficiency
Neuromuscular diseases where scapular winging occurs uncommonly
Myotonic muscular dystrophy
Emery–Dreifuss muscular dystrophy
Myotubular myopathy
Nemaline rod myopathy
Central core myopathy
Phosphofructokinase deficiency
The symptoms of ventilatory muscle weakness represent an ominous, occasionally initial manifestation of a selective group of neuromuscular disorders (Table 1-12).3 In this text, ventilation will refer to the mechanical act of air exchange from atmosphere to alveoli as opposed to respiration, the act of gas exchange between alveoli and the circulation. Dyspnea on exertion is the typical symptom of hypoventilation but may not become evident in this population due to the limited ability of patients to exert themselves. Diaphragmatic weakness is more symptomatic in the supine position leading to orthopnea. Symptomatic hypoventilation in the neuromuscular disorders often presents in a protean fashion with nonspecific, frequently nocturnal and unrecognized symptoms.4 The nocturnal predilection may be multifactorial. In addition to orthopnea from diaphragmatic weakness, the supine position also places more of the surface area of the chest wall against surfaces that add further resistance to chest wall expansion. Weakness of pharyngeal musculature may diminish the support of the upper airway further compromising the upper airway integrity during inspiration. Patients who are dependent on accessory muscles, paralyzed during REM sleep, will experience further compromise of ventilation during this stage of sleep. Resulting nocturnal hypercarbia may interrupt normal sleep cycling and promote nocturnal restlessness and diurnal fatigue. Early morning headache and confusion due to carbon dioxide retention are usually late symptoms that clearly warrant the provision of positive pressure airway support.
TABLE 1-12. NEUROMUSCULAR DISORDERS ASSOCIATED WITH VENTILATORY MUSCLE WEAKNESS26–28,32
Anterior horn cell
Motor neuron disease/amyotrophic lateral sclerosis
Poliomyelitis
West Nile virus
Nerve
Bilateral phrenic neuropathies (brachial plexus neuritis)
Critical illness neuropathy
Guillain–Barré syndrome
CIDP (consider POEMS)
CMT2 C
Multifocal motor neuropathy with phrenic neuropathy (rare)
Amyloidosis
Porphyria
Toxins (thallium, lead, arsenic, organophosphates, vincristine)
Neuromuscular junction
Autoimmune myasthenia gravis
Congenital myasthenia
Botulism
Lambert–Eaton myasthenic syndrome
Envenomations (reptile, insect, marine)
Tick paralysis
Muscle
Myotonic muscular dystrophy
Dystrophinopathies
Limb-girdle muscular dystrophy (2C–F, 2I)
Emery–Dreifuss muscular dystrophy
Acid maltase deficiency
Phosphofructokinase deficiency (rare)
Carnitine deficiency
Poly/dermatomyositis (rare)
Myotubular myopathy
Multiminicore disease with rigid spine (SEPN-1)
Carnitine palmitoyl transferase deficiency and rhabdomyolysis
Nemaline rod myopathy
Congenital fiber type disproportion
Critical illness myopathy
Mitochondrial myopathy (rare)
Myofibrillar myopathy (desmin)
Necrotizing myopathy
Myopathy associated with signal recognizing protein (SRP) antibodies
Metabolic (hypokalemia, hypophosphatemia)
With the sensory history, there is great value in allowing the patient to identify the topographic area of involvement which is frequently more accurately identified by the patient than by the examining physician. For example, paresthesia confined to one or two contiguous digits would, in the vast majority of cases, indicate a disorder of the neuromuscular system that may be difficult to corroborate even by a detailed sensory examination conducted by an experienced physician. With the sensory history, it is also important to identify any associated morbidity, for example, loss of balance or ability to identify a coin in a pocket due to proprioceptive loss.
Disorders that affect sensory neurons may lead to a variety of perceived sensations that may in part be related to the size of the sensory axons affected and the duration of the illness. Paresthesias (a positive or abnormal spontaneous sensation) may be described as tingling, prickly, burning, shooting or electrical sensations, often with an unpleasant or painful characteristic. The latter three sensations are thought to indicate preferential involvement of small unmyelinated sensory nerve endings. Other abnormal although probably less specific perceptions include coldness as well as itching. If large myelinated sensory fibers are affected, the patient may describe a band-like, wrapped, swollen, “pad-like,” or wooden sensation. They may feel as though they have cotton stuffed between their toes or that their body parts are encased in plastic, dried glue, or that their skin is foreign to them. Pain associated with large diameter nerve fibers is often deep, dull, and aching in characteristic.
Numbness can be conceptualized as a loss of sensation, that is, a negative sensory symptom. In actuality, it is really a sign in that it may not be recognized by the patient until the affected body region is touched. It is largely held that unrecognized numbness unaccompanied by paresthesia is indicative of a very chronic, slowly progressive process. As an example, unrecognized sensory loss without paresthesia is one of the characteristic features of Charcot–Marie–Tooth disease.
As with the motor history, it is important to explore the functional consequences of sensory loss although these may be less specific. In the authors’ experience, the complaint of “dropping things” from the hands has poor discriminating value in the separation of definable from nondefinable neurologic disease. Conversely, impaired balance from large fiber sensory loss, that is, sensory ataxia, is an important symptom associated with significant morbidity. Inquiries should be made regarding nocturnal balance, the use of a night-light, and balance in the shower while hair washing.
Impaired autonomic system function occurs in certain causes of peripheral neuropathy as well as in presynaptic disorders of neuromuscular transmission. Identification of dysautonomia may aid greatly in focusing the differential diagnosis. Common symptoms include orthostatic intolerance with faintness and nuchal discomfort, constipation, diarrhea, or early satiety, urinary retention, incontinence, erectile dysfunction, sweating abnormalities including dry cracked feet, blurred vision, dry eyes, or dry mouth.
Perception of pain is dependent on nerve function but results from injury to other tissues. Pain caused by nerve injury or dysfunction, is referred to as neuropathic pain. Neuropathic pain is recognized by its characteristics or by its association with objective evidence of relevant nerve injury. It is often linear in its orientation and often, but not always has burning, lancinating, deep boring or electrical characteristics. Allodynia, or cutaneous pain triggered by a normally innocuous stimulus, for example, the touch of bed clothes may occur in patterns not typically recognized as typical nerve or nerve root distributions. The truncal neuropathy of diabetes is a notable example of this. Muscle pain is also a common complaint brought to the attention of the neuromuscular clinician. Along similar lines, myalgia without a definable trigger, associated weakness, or some other objective finding is unlikely to be of neuromuscular causation. As mentioned previously, pain commonly occurs as a consequence of neuromuscular disease, frequently mechanical in nature and related to imbalanced forces on joints and other connective tissues promoted by muscle weakness or impaired sensation.
Time constraints are a medical reality. Examining clothed patients represents an understandable but unfortunate response to this inconvenience. In neuromuscular medicine, this short cut is not a viable option. As emphasized later in this section, there are numerous observations that can be made only by direct observation of exposed body parts that provide clues integral to accurate diagnosis.
The strategy of the neuromuscular examination is to identify patterns of weakness and sensory loss and correlate them with typical patterns of specific disorders. In certain cases, such as a multifocal neuropathy, the patterns are more readily identifiable early in the disease, whereas in others, for example, ALS, some degree of disease evolution may be required for the diagnosis to become apparent. Either by history or examination but ideally by both, involvement of motor, sensory and/or autonomic systems should be sought. Recognized patterns such as distal symmetric, that is length-dependent, proximal symmetric, UMN, single or multiple peripheral nerve patterns, and single or multiple nerve root patterns should be sought for and ideally recognized. In an analogous manner, sensory loss should be characterized as small fiber, large fiber or both. If possible, the recognition of length-dependent, multifocal, single nerve and single nerve root distribution of sensory signs, and symptoms will provide an invaluable diagnostic clue.
The motor examination of cranial nerves begins with observation. In childhood spinal muscular atrophies the upper lip may have a tented configuration. A number of myopathies will produce “myopathic facies” with a transverse smile with little or no elevation of the corners of the mouth. With severe weakness of muscles of mastication, the jaw may be slack and hang open. Patients with facial weakness affecting the obicularis oculi may have ptosis of the lower lid resulting in visible sclera between the lower limbus of the cornea and the margin of the lower eye lid. These same patients may be observed not to oppose their eyelids completely while blinking. More subtle facial weakness may be noticeable when the eyelids are not completely “buried” when the patient is asked to squeeze their eyes shut as hard as they can.
Atrophy in muscles innervated by cranial nerves may be evident in the temporalis, sternocleidomastoid, and particularly in the tongue. The former two are common features of myotonic muscular dystrophy. Tongue atrophy can be seen in a number of neuromuscular disorders most notably the MNDs. Fasciculations of the face and tongue are key diagnostic features, particularly in the evaluation of bulbar syndromes, and should be actively sought for in suspected amyotrophic lateral sclerosis (ALS) and the spinal muscular atrophies. As with any other muscle, it is important to examine the muscle in a relaxed rather than partially contracted state as muscle movement in the latter situation may be readily misinterpreted as fasciculations. It is also important to distinguish a generalized tremulousness of the tongue, which occurs frequently in normal patients from the random twitching of individual motor units that represent fasciculations.
Manual muscle testing in cranial innervated muscle is an integral part of the neuromuscular examination. Facial weakness can be assessed by attempting to pry the tightly closed eyes and/or lips apart. We grade facial weakness on a mild, moderate, and severe scale. Mild weakness means that the eyelids oppose and generate some but inadequate strength with an attempt to open them. Moderate weakness means that the eyelids oppose but offer minimal resistant whereas severe weakness means that the eyelids cannot completely oppose. With the lips, mild weakness is determined by the ability to blow up the cheeks with air but the inability to prevent air leakage when the cheeks are compressed. Moderate weakness is the ability to oppose the lips but not puff out the cheeks whereas severe weakness is the inability to oppose the lips.
Jaw strength can be tested by looking for lateral chin deviation upon opening or by trying to pry open the fully closed jaw by placing the fingers on the back of the neck and applying downward pressure with the thumbs. Attempting to assess jaw opening strength should be done with caution as inadvertent trauma to the teeth may occur if the jaw snaps shut inadvertently.
Tongue strength is best tested by having the patient “pocket” each cheek with manual pressure being placed on the cheek and indirectly on the tongue attempting to force it back to the midline. Again we use a mild, moderate, and severe scale. Mild weakness is a retained ability to pocket but an inability to resist pressure. Moderate weakness is the ability to pocket the cheek in a limited fashion with little or no resistance to pressure. Severe weakness refers to little or no tongue movement. Neck flexion and extension strength is tested in the customary isometric manner by having the patient resist full neck flexion and extension, respectively, with or without the use of a dynamometer.
Myotonia and paramyotonia of eyelid opening and closing, as well as in limb muscles, may be sought for in the appropriate context, particularly in suspected paramyotonia myotonia and myotonia congenita. In assessing for eyelid myotonia or paramyotonia, the patient is asked to repetitively close their eyes tightly and open them quickly. With myotonia, the delay in opening is most apparent with the first attempt whereas in paramyotonia, it gets worse with subsequent efforts. The examiner can also ask the patient to look up for several seconds and then rapidly look back down to the primary position. If the eyelid does not return to the primary position as fast as the globe, myotonia of the eyelid elevators may be considered along with other causes of lid lag. Myotonia can also be sought for by percussing the tongue with the assistance of gauze and two tongue blades but this is cumbersome procedure that and probably adds little to the assessment of myotonia through grip or percussion of limb muscles. An additional eyelid sign of potential use in neuromuscular disease is the Cogan eyelid twitch. The patient is asked to look down, and then rapidly saccade to mid-position. A positive result is identified by an overshoot of the upper lid followed by a few oscillatory movements of the upper lid until it settles back to its normal relationship with the globe.
Relevant to this is our belief that ptosis, proptosis, and to a certain extent facial weakness are best recognized by understanding the normal anatomic relationship between the eyelids and the globe. Typically, the lower margin of the upper lid covers the upper 2 to 3 mm of the limbus whereas the upper margin of the lower lid typically intersects the lower limbus. The observation of sclera between the upper lid and the limbus indicates eyelid retraction or proptosis. The observation of sclera between the lower lid and the limbus represents obicularis oculi weakness or proptosis. A narrowed palpebral fissure represents squinting, blepharospasm, atrophy, or retraction of the globe.
Observation of the eyebrow position is also helpful in the interpretation of abnormal eyelid positioning. If the lower margin of the upper lid is lower than it should be due to ptosis, the eyebrow is typically elevated in a compensatory attempt of the frontalis muscle to elevate it unless the frontalis is weak as well, for example, myasthenia. Conversely, if the upper lid position is lowered by squinting from blepharospasm, the eyebrow is usually lower than the opposite side if uninvolved.
The pupils should be examined, preferably, at least initially, in a dimly lit room to assess for the possibility of Horner’s syndrome. The lack of pupillary reactivity may represent an autonomic component of the patient’s disorder. Perhaps, the greatest value of the pupil examination in neuromuscular disease is to distinguish neuromuscular disorders causing ophthalmoparesis that spare the pupil from those that do not. Myasthenia, most diabetic third nerve palsies, and myopathic causes of ophthalmoparesis fit into the former category. Ophthalmoparesis with pupillary involvement may occur as a consequence of Guillain–Barré syndrome and its variants and also due to presynaptic disorders of neuromuscular transmission such as botulism.
Examination of limb and trunk muscles also begins with observation. Again, it is our strongly held belief that although the patient should be gowned with appropriate undergarments, that every part of the body should be available to direct observation. There are many potential clues that can be obtained in this manner. Muscle atrophy, focal or generalized, and muscle hypertrophy should be sought for. Viewing the shoulder girdles from the back may identify shoulder drop from trapezius weakness or overt scapular winging. Viewing the chest in males may disclose gynecomastia. Viewing the shoulder girdles from the front may disclose a crease in the pectoralis, an elevated scapula producing a pseudohypertrophic appearance of the trapezius or a horizontally oriented clavicle all resulting from weakness of periscapular muscles. In a similar vein, abnormal scapular positioning may affect the positioning of the arms which may be internally rotated so that back of the hand rather than the thumb is anteriorly oriented producing a simian posture. Arm movement during conversation, that is, gesticulation may identify diminished spontaneous movements of one or both upper extremities due to proximal weakness, limitation of joint movement, or central nervous system disease. Conversely, the physician may notice completely normal spontaneous movement under these conditions which is subsequently found to be incongruous with the patient’s inability (or unwillingness) to use the limb properly during the examination, implicating decreased effort from pain, apraxia, or psychogenic etiology.
Muscles should be closely observed for adventitious movements such as tremor, fasciculations, myokymia, or rippling. In our experience, benign fasciculations tend to be felt by the patient more frequently than they are seen, are most commonly seen in the calves and feet, and occur briefly and repetitively in a single spot before disappearing. Postural tremor is not rare in neuromuscular disease and may be a notable feature of Charcot–Marie–Tooth disease, CIDP, or spinal muscular atrophy. Fasciculations that occur in multiple locations in multiple muscles simultaneously are more ominous and suggest excessive cholinesterase inhibitor effect, a nerve hyperexcitability disorder, or most commonly, a motor system disease.
Muscle contractures (nonphysiologic) or other dysmorphic features may be noted either by observation or during passive movement of limbs. Contractures may be seen in a number of neuromuscular conditions as listed in Table 1-13 and may provide key diagnostic clues. Dysmorphic features such as long thin facies, high-arched palates, kyphoscoliosis, exaggerated lumbar lordosis, cavus foot deformities, and hammer toes are also key diagnostic clues. Cavus foot deformities are usually indicative of long-standing disorders dating to childhood and are frequent accompaniments of Charcot–Marie–Tooth disease, distal forms of spinal muscular atrophy, hereditary spastic paraparesis, and Friedreich ataxia. There are many neuromuscular conditions with accompanying dermal or epidermal changes. These include the ecchymoses of Cushing disease, the angiokeratomas of Fabry disease, the skin changes of POEMS syndrome, the skin and nail bed changes of dermatomyositis, Mee’s lines in finger and toe nails representing growth arrest in response to arsenic or lead intoxication among others.
TABLE 1-13. NEUROMUSCULAR DISORDERS ASSOCIATED WITH EARLY JOINT CONTRACTURES
Anterior horn cell
Arthrogryposis multiplex congenita
SMA
Nerve
CMT3
Neuromuscular junction
Congenital myasthenia gravis—rapsyn
Muscle
Central core disease
Nemaline myopathy
Congenital fiber type disproportion
Bethlem myopathy
Ullrich congenital muscular dystrophy
Dystrophinopathy
Emery–Dreifuss muscular dystrophy types I–III
Dominant myopathy with ankle contractures and high CK
Juvenile dermatomyositis
The identification of scapular winging may require provocative posturing as well as observation. It is an important and easily overlooked diagnostic clue in the assessment of neuromuscular disease. Affected patients will be unable to raise their hand over their head effectively. Scapular winging may be evident by simply looking at the patient from the rear. It may be accentuated by a number of maneuvers depending on which muscles are weak. Scapular winging due to weakness of the serratus anterior results in the inferior-medial angle of the scapula being elevated more off the ribcage and migrating further away from the midline than its superior-medial counterpart. It can be accentuated by having the patient push against a wall or by putting downward pressure on the humerus when the arm is flexed at the shoulder. With scapular winging resulting from trapezius weakness, the entire medial border is elevated. Winging is accentuated by attempted external rotation, or abduction of the arm at the shoulder against resistance. The dynamics of scapular winging resulting from the more diffuse myopathic and motor neuron disorders are more complex.
Provocative muscle testing should also be performed when relevant. Percussion myotonia is most commonly tested in the extensor digitorum communis (EDC) and thenar eminence. In the former, the forearm is supported by the examiner in a pronated position, allowing the wrist and fingers to hang limply. The EDC is percussed just distal to the head of the radius. A normal response is no movement or a minimal brief flicker of digit extension. The presence of myotonia is suggested when one or more of the digits extends at the metacarpal phalangeal joints and sustains this posture for a second or so. Percussion of the thenar eminence is performed in a similar manner with the wrist and forearm supported while the forearm is fully supinated. The thumb should be maintained limply in the same plane as the palm. Myotonia is identified when the thumb abducts notably in response to a brief percussive strike to the abductor pollicis brevis muscle. Grip myotonia is sought for by having the patient tightly grip an object, for example the examiners index and middle finger for a few seconds, then rapidly release the grip. A slow and deliberate extension of the fingers indicates myotonia. Typical myotonia improves with repeated trials. Paradoxical myotonia worsens with repeated trials. Myoedema refers to a mounding of muscle in response to percussion of a muscle belly that represents an uncommon finding in some muscle diseases.
The foundation of the neuromuscular examination is the assessment for the presence and pattern of muscle weakness. Two strategies are typically employed: isometric manual muscle resistance and functional testing. Ideally, suspected weakness identified by the first method, for example, reduced resistance of foot dorsiflexors, would be confirmed by the latter, that is, the inability to walk on the heels. There is an art to manual muscle testing, which is undoubtedly improved upon by experience, particularly in the distinction of true weakness as opposed to that due to impaired effort. Muscles are typically tested in an isometric fashion that is a contracted position with the patient asked to resist the force applied by the examiner. For example, elbow flexors are tested with the patient’s fist resting against his or her shoulder. The patient is held by the examiner in such a way that the muscle(s) tested are isolated to the extent possible. Again, in the case of the elbow flexors, the examiner would place the hand that delivers the force just proximal to the wrist to produce the greatest mechanical advantage, while at the same time removing wrist movement from consideration. The other hand, which serves to stabilize, is placed on the biceps just proximal to the elbow.
In order to obtain full patient effort, the patient has to have confidence that the examiner will not harm them. The examiner should sustain full effort long enough to detect either true weakness with its smooth characteristics or “give way” weakness with its ratchety and inconsistent character. It is important however, to relinquish effort before the full range of motion is exhausted so as to avoid injury. Along similar lines, great caution should be exercised to avoid pathologic fracture in any patient with cancer potentially metastatic to bone.
Mild degrees of weakness may easily go unrecognized by both patient and examiner alike. This is particularly true in strong muscles like the quadriceps and the gastrocnemius, or when the strength or effort of examiner is limited. It is imperative that the examiner place themselves at the greatest mechanical advantage and gives an appropriate effort so as to avoid a false-negative result. For example, ideal examination of neck flexion, elbow flexion, knee extension, and trunk flexion, the patient should be tested in the supine position, where the patient has to move against gravity and resistance. Testing a patient on their side is ideal for testing hip abduction and the prone position optimal for elbow, hip and neck extension, and knee flexion.
It is in these same strong muscles where functional testing is particularly useful. For example, hip and knee extension strength can be assessed by the patient’s ability to get up from a deep chair or their ability to perform a partial squat or hop on one leg. Foot plantar flexion strength can be assessed by having the patient elevate their heel while standing on one leg.
Once weakness is recognized, two characteristics are of paramount importance: its pattern and its severity. The primary importance of the pattern of weakness is in the formulation of the initial diagnosis. Pattern recognition as a diagnostic tool is addressed in Tables 1-4 to 1-7 and will be elaborated on repeatedly in this and subsequent chapters. The importance of the degree of weakness may also contribute to the diagnosis, for example, demonstrating progression both within and between different muscle groups is a key in the diagnosis of ALS. In addition, and perhaps more importantly, establishing the degree of weakness is also key in establishing treatment responsiveness.
To this end, accurate quantitative measurements of strength are paramount. Historically, the Medical Research Council (MRC) scale has been used by most institutions for this purpose. This is a 0–5 scale, with 5 representing normal strength and 0 representing no discernible muscle movement. By definition, the MRC scale requires muscles be examined against gravity. An MRC grade of 3 preserves ability to move the joint through a full range of motion against gravity but with negligible resistance to the examiner. An MRC grade of 2 represents movement through a complete range of motion with gravity eliminated. An MRC grade of 1 represents observed muscle contraction with little or no limb or digit movement. With the MRC scale, the majority of weak muscles will fall into the four (modest weakness) range. For this reason, the MRC scale has been modified to include a 4– and 4+ category to expand this largest group of weak muscles.
The MRC scale is problematic, as it may be insensitive, qualitative, and subjective.5 The potential exists for considerable inter-examiner variability. It has been documented that patients may lose 80% or more of their motor units in a given muscle before they receive a 3 or less MRC rating.5 In the opinion of the authors’, it is a poor tool to measure motor deficits in UMN disease where functional impairment may be more on the basis of altered coordination and tone rather than loss of strength. Increasingly in clinical trials, and to some extent in clinical practice, tools such as hand-held dynamometry are used in an attempt to measure strength in a more objective, linear, and reproducible manner. As an example, in the experience of the authors’ most men can generate 40 or more kilograms of force in the majority of upper extremity muscles. An MRC grade of 3 approximates a force of 10 kg, implying that a modified MRC grade between 4- and 4+ represents approximately 75% of the weakness spectrum in these muscle groups.
Ventilation can be assessed at the bedside by a number of techniques. There is value in asking the patient to generate a forceful sniff or cough. Use of accessory muscles should be noted as well as a tendency for the patient to interrupt sentences to catch their breath. Shallow breathing can be detected by auscultation. The vital capacity can be roughly estimated in the cooperative patients by having them inspire fully and then count out loud at the rate of 1 per second until that single breath is exhausted. That number multiplied by a hundred will estimate their vital capacity measured in cubic centimeters. There may be value as well in examining the patient in the supine position to assess for paradoxical abdominal movements (outward abdominal movement in response to inspiration) as an indicator of diaphragmatic weakness.
UMN signs in the cranial nerve distribution are limited in number and in specificity. An enhanced jaw jerk or gag reflex, the presence of a snout reflex, forced yawning and a pseudobulbar affect are all accepted UMN signs. Reduction in the speed in which a patient is able to repetitively blink or wiggle their tongue back and forth, in the absence of weakness or mechanical restriction of the respective muscles probably represents central nervous system dysfunction but is unlikely to specify corticobulbar tract pathology. The same is likely true for synkinesis of two muscles innervated by different cranial nerves, for example the inability to keep the jaw from moving side to side when the requested task is wiggling the tongue back and forth in the horizontal plane.
Impaired motor function of corticospinal tract origin may include weakness, particularly if acute in onset, but tends to be dominated by impaired coordination and function. Clumsiness disproportionate to the degree of weakness is a sensitive, albeit nonspecific indicator of UMN disease. UMN weakness may also be suspected on the basis of topographic pattern of involvement. A hemiparetic pattern, even in ALS (also known as the Mill’s variant) is rarely LMN in nature. A paraparetic or quadriparetic pattern often occurs as a result of corticospinal involvement of the spinal cord but may just as easily occur in a neuromuscular disorder as well. UMN weakness when limited in distribution is often more distal than proximal, particularly in the upper extremity. Often, UMN weakness can be implicated when flexors are stronger than extensors in the upper limbs and the opposite in the lower extremities. For example, weakness of hip flexion, knee flexion, and foot dorsiflexion in combination strongly suggests UMN disease. Impaired motor function of central nervous system origin can often be deduced by observation, that is, the reduced spontaneous use of a body part such as diminished gesturing of an arm during talking.
UMN disease is also implicated when deep tendon reflexes are exaggerated, or with the existence of pathologic reflexes or spastic tone. The detection of hyperactive deep tendon reflexes can be somewhat subjective. Sustained clonus is undoubtedly pathologic in all cases. Deep tendon reflexes that persist in a limb that is weak and atrophic, unsustained clonus, and reflex spread are all suggestive of UMN pathology but are probably not pathognomonic. Babinski signs are universally accepted as a marker of UMN pathology but bilateral Hoffman’s signs and absent abdominal reflexes need to be interpreted with some caution.
Like its motor counterpart, the results of the sensory examination are most credible when they are concordant with both the history and available functional tests of sensation. There are many sensory examination strategies. In the authors’ experience, the application of sensory stimuli in a random fashion with subsequent attempts to identify the boundaries of the sensory loss is often difficult to interpret and may produce false-positive results. An alternative technique is a hypothesis-driven approach in which the examiner attempts to prove or disprove a specified pattern of sensory loss, for example, a length-dependent pattern in a patient with numb feet. As examiners can apply stimuli with different intensities inadvertently and as patients have different thresholds for what they consider reduced (or increased), it is important to perform sensory testing in a reproducible and as unbiased manner as is possible. For this reason, there is a benefit from testing with the patient’s eyes closed and with the addition of random null stimuli. This is particularly true with vibration where patients commonly confuse the touch of the tuning fork with vibration as the sensation in question. Using the tip of the examiner’s finger as a random substitute for the tuning fork is a means to ensure that the patient is responding positively to vibration and not simply to pressure.
There are a few important points to recognize in performing the sensory examination. As already emphasized, it is not uncommon to be unable to convincingly demonstrate sensory loss in a symptomatic region in a person with credible sensory complaints. Conversely and somewhat paradoxically, it is not uncommon to find patients in the setting of a partial nerve injury who claim to react to a stimulus in a hypersensitive manner in an area that they claim to be numb. Finally, it is important to realize that the topographical area where sensory symptoms are perceived and sensory loss is found is often far smaller than published anatomical charts would suggest for any nerve or dermatomal distribution. Presumably, this is the result of the considerable overlap between contiguous nerve territories.
There are a limited number of functional sensory tests to corroborate the findings on the direct sensory examination. The best known of these is the Romberg test, which assesses proprioceptive (or less likely bilateral vestibular) dysfunction in the lower extremities arising from either the peripheral or the central nervous system. The finger–nose test, also done with the eyes closed, is an analogous test for proprioceptive loss in the upper extremities. Stereognosis testing can be helpful at times. Even with severe nerve injuries, absolute anesthesia is rare. Patients who claim to feel absolutely nothing in the hands yet can readily manipulate an object in that hand with their eyes closed are unlikely to have the degree of sensory loss that is claimed.
Common bedside screening tests of autonomic function include observation of pupillary responses as described above. The feet should be observed for the presence of dry, cracked skin suggesting the possibility of anhydrosis. Pulse variation in response to deep breathing is a test of parasympathetic function. Arguably, the most commonly performed and valuable bedside autonomic test is orthostatic blood pressure and pulse measurements. They should be done after a few minutes in the supine position. Both blood pressure and pulse should be measured immediately on standing (or sitting) and at 1-minute intervals for at least 3 minutes, depending on the index of suspicion and the result.
Examination of young children, particularly infants, can be a challenge. Infants can be placed in a prone position to observe if they are capable of extending their head. An inability to do so suggests weakness of the neck extensor muscles. Most infants have considerable subcutaneous fat that makes muscle palpation quite difficult. Palpating neck extensor muscles is a good place to attempt this evaluation as little subcutaneous fat overlies this muscle group. Neck flexion strength can be assessed as the child is pulled by the arms from a supine to a sitting position. Crying during the examination allows the opportunity to assess the child’s vocalization (e.g., presence of a weak cry) and fatigability to the physical examination. Muscle weakness in infants is usually characterized by an overall decrease in muscle tone and many children with profound weakness are characterized as “floppy.” This terminology does not necessarily imply a neuromuscular disorder. In fact, most floppy infants result from a central nervous system problem. In view of prominent subcutaneous tissue, fasciculations may be visible only in the tongue. Observation of tremor is important as it may be a feature of spinal muscular atrophy and some hereditary neuropathies. It is important to examine the parents of floppy infants for the possibility of a neuromuscular disorder. This is particularly important in children suspected of having myotonic dystrophy. In addition, weakness can transiently develop in infants born to mothers with myasthenia gravis.
WHAT IS NEUROMUSCULAR PROBLEM?The following section will attempt to summarize the patterns of motor and sensory involvement that typify the diseases described in this text, in an attempt to facilitate the localization process (Table 1-6). Further formulation of the differential diagnosis will require knowledge of the behaviors and natural histories of the disorders that are addressed in Tables 1-1 to 1-3 and described in detail in subsequent chapters of this book.
The hallmark of the MND, also known as anterior horn cell diseases or motor neuronopathies, is painless weakness and atrophy frequently accompanied by the positive symptoms of cramps and fasciculations. Although cramps and fasciculations may occur in apparent absence of disease, and can be seen with any peripheral nerve disorder, they are far more prevalent in disorders of the anterior horn. As mentioned above, benign fasciculations are commonly evanescent and confined to a singular area at any given time. Conversely, fasciculations seen in numerous locations on a continuous or near continuous basis is almost always the result of a motor neuron disorder. The absence of fasciculations does not preclude a motor neuron localization, particularly where there is considerable subcutaneous tissue that may obscure their observation, infants and those with an elevated body mass index being the most notable examples. Sensory symptoms and sensory loss do not typically occur in MND except in Kennedy disease. Nonetheless, it may occasionally occur in ALS due to other unrelated problems or potentially as a consequence of the occasional multisystem variants of this disorder.6
Most motor neuron disorders are hereditary/degenerative in nature and as a result have an insidiously progressive course. The rate of progression varies both with and between different MND, ALS, and spinal muscular atrophy type I having the most virulent courses. The pattern of weakness varies with the disorder. With ALS, onset is typically focal and asymmetric, for example, foot drop. Even early in the course however, weakness can be recognized as being multisegmental and outside of a single nerve or nerve root distribution. Poliomyelitis and other neurotropic viruses may present focally as well with marked asymmetry or with a more generalized presentation. The spinal muscular atrophies tend to have a symmetric presentation that is generalized or proximally predominant in both the X-linked bulbospinal (Kennedy disease) and infantile forms. The more uncommon distal spinal muscular atrophies have a distal, symmetric pattern of weakness that may mimic neuropathies or distal myopathies. Juvenile segmental spinal muscular atrophy (Hirayama disease) presents focally in the distal aspect of first one and at times the other upper extremity.
The recognition of MND is also aided by the identification of functions that are spared. Most notably, patients with MND virtually never experience ptosis or ophthalmoparesis except in the rare cases of ALS that behave more like a multisystem disorder. Impaired bulbar function (i.e., speech and swallowing) is common in many MND. Facial and jaw weakness may occur but are typically less prominent than the weakness of the tongue and throat muscles. Deep tendon reflexes tend to be lost unless there is concomitant UMN disease such as in ALS.
These disorders, also known as sensory neuronopathies, are characterized by non–length-dependent, multi-focal sensory signs and symptoms. Like many nerve diseases, distal aspects of limbs tend to be more afflicted than proximal, thus potentially mimicking the far more common length-dependent polyneuropathy pattern. Careful history taking may be required to identify the non–length-dependent or asymmetric features. Both the resulting chronologic course and the presence or absence of pain is variable and in large part dependent on etiology. Electrodiagnosis is useful to demonstrate that sensory fibers alone are affected. In polyneuropathies, there is almost always some indication of motor involvement, even when not apparent clinically, particularly if fibrillation potentials within intrinsic foot muscles are sought for. Sensory ataxia is a common manifestation of these disorders. Dorsal root ganglionopathies may be autoimmune, toxic, infectious, or at times degenerative in etiology.
Monoradiculopathies are among the most common neurologic problems, commonly due to some mechanism associated with degenerative spine and disc disease. Their phenotype is in turn dependent on the mechanism and acuity of nerve root compression. The prototypical symptom of an acute monoradiculopathy, usually related to disc herniation is pain, limited to one extremity, and following the course of the involved dermatome. The pain may not affect the entire dermatome simultaneously, for example, buttock and anterolateral leg pain sparing the thigh in an L5 radiculopathy. Contrary to common belief, the pain usually begins in the scapular and the buttock area rather than the neck or back. Sensory and motor deficits are not universal, but when present, should be confined to a single segment. Weakness should be confined to a single myotome but involve more than one peripheral nerve distribution. For example, in a C7 monoradiculopathy, both elbow extension (C7/radial) and wrist flexion (C7/median) are often involved. Conversely, weakness may not be detectable in all muscles innervated by that particular myotome. For example, demonstrating weakness only in the extensor hallicus longus is not uncommon in an L5 monoradiculopathy. A helpful caveat is the recognition that a given muscle is virtually never completely paralyzed from a single nerve root lesion as virtually all muscles have multiple segmental innervation.
In a similar fashion, sensory symptoms and deficits virtually always involve a smaller region than is predicted from dermatomal maps due to overlap of territories from contiguous dermatomes. For example, patients with C6 radiculopathies describe their numbness or paresthesias as affecting only the tip of their thumb.
A deep tendon reflex(s) may be diminished if appropriate to the involved root. The pain of a monoradiculopathy in the lower extremity may be reproduced by the straight-leg or reverse straight-leg raising signs or by lateral bending toward the affected extremity. In the cervical region, it may be reproduced by extending and laterally bending the head and neck toward the symptomatic side in an attempt to promote foraminal compression.
In chronic radiculopathies, pain may be intermittent and position/activity dependent such as in lumbar spinal stenosis, or may be minimal or nonexistent. Chronic radiculopathies typically occur from some component of spondylotic spine disease resulting from bone spurs or hypertrophied ligaments. Multiple rather than single nerve roots are more commonly affected by this process and motor and sensory deficits may be less dramatic in their manifestations.
The typical phenotype of polyradiculopathy is the sequential development of motor and sensory signs and symptoms involving more than one spinal segment in one or more extremities. These disorders are typically painful, and with certain etiologies, involve cranial nerves as well.
The etiologies are heterogeneous and in many cases involve diseases with a predilection for cerebrospinal fluid, the meninges, or neural foramen nerve roots and cranial nerves pass through on their journey from spinal cord to limbs, head, and trunk. The most common cause of polyradiculopathy is lumbosacral spinal stenosis typically presenting with back and lower extremity pain provoked by standing and walking. Diabetic radiculoplexopathy can be another common cause of what may be considered a polyradiculopathy. This typically presents as an acute painful disorder affecting the L2–L4 innervated muscles in one leg. Some patients will have their other leg affected on a delayed basis. Other causes of polyradiculopathy are relatively uncommon and are typically related to inflammatory, infectious, or neoplastic disorders that produce a chronic meningitis. Cranial nerves, both motor and sensory, are commonly affected in these disorders.
A plexopathy is suspected when sensory and motor deficits are restricted to a single limb, the deficits being more widespread than can be explained on the basis of a single nerve or nerve root dysfunction. Pain is the rule rather than the exception, as the causes of plexopathy are most commonly traumatic, inflammatory, or neoplastic which either compress, infiltrate or inflame nerve. Occasionally, most notably with acute brachial plexus neuritis, or diabetic radiculoplexopathy, sensory signs and symptoms may be modest or nonexistent. The reasons for this may be multifactorial. Acute brachial plexus neuritis has a predilection affecting purely motor nerves, for example, the long thoracic or anterior interosseous nerves. In fact, it is this multifocal nerve pattern confined to one upper extremity or adjacent cranial or upper cervical nerves that often serves as a diagnostic clue. The motor predominant nature of acute brachial plexopathy may be related to a demyelinating pathophysiology that may preferentially affect motor function in a manner similar to the Guillain–Barré syndrome.
Mononeuropathy syndromes are usually readily recognizable due to their frequency and relative homogeneity of presentation for a particular compression or entrapment syndrome. They most commonly result from the anatomic vulnerability to compression (external forces—e.g., Saturday night palsy) or entrapment (internal forces—e.g., carpal tunnel syndrome) of particular nerves at specific locations. The mode of presentation between different mononeuropathies is variable, in part due to the constituency of the nerve, for example, pure sensory nerves such as the lateral femoral cutaneous nerve. More commonly, the mode of presentation varies due to pathophysiology which may be primarily axonal or due to differing mechanisms of demyelination. In the case of carpal tunnel syndrome and ulnar neuropathies at the elbow, sensory symptoms tend to initially predominate. Common peroneal or radial neuropathies at the spiral groove tend to have more of a motor predominance. Pain may or may not be an issue. Pain without motor, sensory, or reflex signs or symptoms is uncommonly due to a definable mononeuropathy despite descriptions of alleged mononeuropathy syndromes such as the piriformis and pronator syndromes.
In any event, signs and symptoms should be restricted to the distribution of a single peripheral nerve, distal to the site of nerve injury. The converse is not always true. For example, it may be very difficult to demonstrate weakness of ulnar forearm muscles, which are at risk from ulnar neuropathies at the elbow. This phenomenon has been attributed to selective fascicular involvement. As nerve fibers destined for the same muscle tend to sequester themselves in the same fascicle even in proximal locations, these fascicles may be relatively spared from a compression or entrapment process that may affect certain fascicles more than others. Alternatively, weakness of ulnar wrist flexion may be obscured by the preservation of median wrist flexion.
Length-dependent polyneuropathy is one of the most common neuromuscular problems encountered both by neurologists and other physicians. Long, narrow axons are presumably vulnerable to the axonal transport mechanisms on which they are dependent, and the 200 or more etiologies that can adversely affect them. Despite a phenotype that usually begins with symmetric motor and/or sensory involvement of the toes and feet, there is considerable heterogeneity in clinical expression. Conceptually, the majority of these disorders result from toxic, metabolic, or hereditary disturbances of cell body metabolism or myelin growth resulting in impaired axon transport or nerve impulse transmission. This provides a cogent explanation for preferential involvement of most distal aspects of the longest nerves in the body affected in a symmetric, “length-dependent” fashion. Usually, sensory, motor, and reflex functions are all impaired in this length-dependent pattern although sensory signs and symptoms typically predominate. The best explanation for this phenomenon is that sensory nerve endings of the feet have no backup system. Denervation of intrinsic foot muscles that flex and extend the toes however, is clinically masked by leg muscles providing the same function. The inability to spread the toes provides a nonspecific but sensitive means of clinically suspecting motor involvement in length-dependent neuropathies. Identifying fibrillation potentials or low amplitude compound muscle action potentials in intrinsic foot muscles may be the only reliable way to detect early motor involvement in many length-dependent polyneuropathies. Again, it is important to plot the evolution of sensory symptoms to ensure that they are not asymmetric or non–length-dependent in pattern suggesting a different anatomic localization such as multifocal neuropathy, polyradiculopathy, or sensory neuronopathy.
Polyradiculoneuropathy refers to a disorder that affects multiple nerves both at the nerve and nerve root level. The most commonly encountered polyradiculoneuropathies are acquired, inflammatory, and demyelinating in nature, for example, the Guillain–Barré syndrome and chronic inflammatory demyelinating polyneuropathy (CIDP). Uncommonly, this pattern may occur as an axon loss process secondary to a disorder like acute intermittent porphyria or Lyme disease.
Polyradiculoneuropathies are usually readily distinguished from length-dependent polyneuropathy. They tend to be motor rather than sensory predominant. The pattern of involvement is typically symmetric but is usually more generalized and non–length-dependent. There may be cranial nerve involvement, which would be an extremely rare occurrence in most causes of length-dependent polyneuropathy. Reflex loss is typically generalized rather than length dependent. This is a consequence of the demyelinating pathophysiology with differential slowing in different fibers within the same nerve that desynchronizes impulse transmission rendering functions dependent on synchronous impulse transmission such as deep tendon reflexes and vibration perception impaired.
MULTIFOCAL NEUROPATHYMultifocal neuropathy has been historically referred to as mononeuritis multiplex or multiple mononeuropathies. It is not a universally accepted term but will be the preferred term in this chapter for the following reasons. Multiple mononeuropathy is an equally accurate descriptor but may imply to some a more benign multifocal compressive syndrome in contrast to many causes of multifocal neuropathy which tend to be systemic in nature. Mononeuritis multiplex is a frequently used designation but is unsatisfactory to us in that it implies an inflammatory pathology that may not exist or may go unproven. For this reason, we have chosen to avoid it.
The deficits of multifocal neuropathy are often abrupt and painful, occurring haphazardly (although usually distally) and asymmetrically, with weakness and sensory loss being mapped to individual peripheral nerve distributions in more than one extremity. As described above, clinical recognition may depend on examination of the patient early in the disease, or obtaining an accurate history of early disease evolution, prior to the inevitable confluence of deficits.
Multifocal neuropathy is often the result of disorders that infiltrate (sarcoidosis, lymphoma, amyloidosis) or infarct (vasculitis, diabetes) nerve, or provide susceptibility to compressive and/or demyelinating nerve injury (multifocal motor neuropathy, multifocal acquired demyelinating sensory and motor neuropathy, hereditary liability to pressure palsy).
NEUROMUSCULAR TRANSMISSION DISORDERSThese disorders are difficult to lump together from a clinical perspective, as there is phenotypic variability. Like MND and myopathy, the signs and symptoms are attributable exclusively to the motor domain. As the neuromuscular junction is a more physiologically dynamic structure than nerve or muscle, fluctuations in strength and stamina are hallmarks of these disorders. In acquired disorders of neuromuscular transmission, muscle atrophy is notable for its absence.
In postsynaptic disorders of neuromuscular transmission like myasthenia, for reasons not clearly understood, there is a predilection for cranial innervated musculature. Ptosis, diplopia, dysarthria, dysphagia, and chewing difficulties are common complaints. The deficits can be quite asymmetric and at times remarkably focal. Rarely, myasthenia may present with limb weakness with little, if any, oculobulbar involvement. This can also be either symmetric in nature mimicking a myopathy or focal such as a finger or foot drop, thus potentially mimicking an MND. Postsynaptic disorders of neuromuscular transmission do not affect the cholinergic receptors of the autonomic nervous system. Pupils should be spared even with complete ophthalmoparesis. Deep tendon reflexes are commonly spared in myasthenia gravis unless involved muscles are significantly weak.
Signs and symptoms of cholinergic dysautonomia are commonplace in presynaptic disorders of neuromuscular transmission such as botulism and the Lambert–Eaton myasthenic syndrome. Weakness in these two disorders tends to be symmetric and is often proximally predominant and generalized. Cranial nerve involvement is very common in botulism. It does occur in the Lambert–Eaton myasthenic syndrome, although not as prominently as in botulism or myasthenia gravis. Deep tendon reflexes are commonly lost in a generalized pattern in any presynaptic disorder of neuromuscular transmission.
MYOPATHIESMyopathy is suspected in three different clinical settings, fixed, typically painless and symmetric weakness, periodic weakness due to disorders of ion channels, and exercise-induced muscle pain, fatigue, and stiffness due to disorders of muscle energy metabolism. With fixed weakness, symmetry is a relative term. Minor asymmetries are common in disorders such as facioscapulohumeral muscular dystrophy and inclusion body myositis (IBM). The distribution of weakness is often proximal, but there are many notable exceptions. Myopathies presenting with symmetric, distally predominant weakness are not rare. These usually begin in the lower extremities but may begin in the hands as well. Myopathies, particularly those of a hereditary nature (e.g., facioscapulohumeral or oculopharyngeal dystrophy) and IBM, may also be recognized by regional patterns of weakness. Weakness in neck flexors and extensors should be sought in all neuromuscular disease but are particularly common in myopathy in addition to disorders of neuromuscular transmission and anterior horn cells. Cranial muscle involvement is variable. Dysphagia, ptosis, ophthalmoparesis, facial, jaw, and tongue weakness may occur and aid in the differential diagnosis of myopathic disorders. Reflexes may be lost or preserved, depending on the pattern and severity of muscle involvement.
Attention to other elements of the examination may aid in the identification of the existence, type, and potential complications of muscle disease. Percussion, grip, or electrical myotonia will serve to identify a select group of myopathies (Table 2-1, Chapter 2). A number of myopathies may associate with joint contractures or skeletal abnormalities. Muscle hypertrophy is a constant feature of the dystrophinopathies and may occur with certain limb-girdle dystrophy phenotypes as well as infiltrative disorders of muscle such as amyloid myopathy. Involvement of ventilatory and cardiac muscle as well as other organ systems may aid in diagnosis and allow anticipation of future morbidity (Table 1-12).
The clinical phenotypes related to ion channel disorders and metabolic muscle diseases that may differ from the fixed weakness that typify most muscle disease will be discussed in the relevant chapters that follow. As rhabdomyolysis and myoglobinuria may result from numerous causes, it will be more convenient to discuss the topic here. Rhabdomyolysis and myoglobinuria, although conceptually different, are terms that are often used interchangeably. In this book, they will be discussed as a singular clinical and laboratory entity (RHB/MGU).33 Although discussed later in this book in the context of individual diseases, it is addressed here in order to provide an overview and strategic approach to the problem. Rhabdomyolysis refers to an acute, large scale breakdown of striated muscle fibers whereas myoglobinuria implicates the urinary excretion of the pigment myoglobin released into the bloodstream as a consequence. Attempts have been made to define these terms quantitatively. Myoglobin is visible in the urine when its concentration exceeds 100 μg/dL or when plasma levels exceed 1.5 mg/dL but is an insensitive means to detect small or chronic CK elevations and usually becomes undetectable within hours, long before serum CK normalizes. Rhabdomyolysis has been defined by serum CK levels exceeding five times the upper limits of normal. As there are many patients who may carry CK levels in excess of this chronically and even asymptomatically, this quantitative definition fails to conceptually capture the acute and potentially catastrophic nature of the RHB/MGU syndrome. The RHB/MGU syndrome is most commonly associated with serum CK levels in tens of thousands.
There are numerous potential causes of RHB/MGU that are typically monophasic and result from toxic, traumatic, or infectious insults (Table 1-14). In addition, a number of heritable metabolic myopathies pose a risk for recurrent episodes. There is some evidence that there may be genetic susceptibility underlying some individuals who experience RHB/MGU in apparent response to an environmental provocation.34 Unfortunately, many cases remained undiagnosed despite intensive evaluation.
TABLE 1-14. CAUSES OF RHABDOMYOLYSIS/MYOGLOBINURIA30,31,38
Toxic
Envenomation—specific species of snake, bees, spiders
Ingestion—(1) animals who have ingested toxins—e.g., Haff disease (fish), quail that have eaten hemlock, (2) certain mushroom species
Recreational drugs—cocaine, heroin, alcohol, amphetamines, LSD, loxapine, hemlock, mercuric chloride, phencyclidine, strychnine, terbutaline
Environmental exposure—monensin, chlorophenoxy herbicides, toluene, pentaborane
Medications—amiodarone, emetine, cholesterol lowering agents, epsilon amino caproic acid, isoniazid, lamotrigine, pentamidine, propofol, proton pump inhibitors, valproate, zidovudine, neuroleptic malignant syndrome
Metabolic
Hypokalemia
Hyperthermia
Hyperosmolality (hyperglycemia)
Hypophosphatemia
Infectious (more common in children)
Bacteria (tetanus, salmonella, Legionella, group A beta hemolytic strep)
Virus (influenza, EBV, CMV, HIV)
Parasites (malaria)
Rickettsial
Ischemic—acute peripheral arterial occlusion
Traumatic
Crush injury
Compartment syndrome
Miscellaneous
Status epilepticus
Delirium tremens
High voltage electrical shock
Excessive exercise in the deconditioned
Heritable
Mitochondrial—cytochrome c oxidase deficiency, complex I deficiency, primary coenzyme Q10 deficiency, cytochrome b deficiency, lipoamide dehydrogenase, succinate dehydrogenase and aconitase
Glycogen storage disease—myophosphorylase, phosphofructokinase, phosphoglycerate mutase and kinase, LDH deficiency, phosphorylase B kinase deficiency
Lipid storage disease—carnitine palmitoyl transferase deficiency, carnitine translocase, acyl-CoA dehydrogenase deficiency
Channelopathies—malignant hyperthermia types I—VI, hypokalemic periodic paralysis
Congenital myopathies—King–Denborough syndrome, central core disease
Muscular dystrophy—DM1 (rare), dystrophinopathy (rare) LGMI 2I
Inflammatory—dermatomyositis (rare)
The symptoms of RHB/MGU are often nonspecific. There is often a nonspecific fever and malaise, usually accompanied by the more specific myalgias, muscle swelling, tenderness, and a generalized sense of weakness (asthenia). In the author’s experience, it can be difficult to discern the cause of diminished patient movement. Both muscle pain and actual weakness may play a role, the former seemingly being the primary mechanism in most cases. The release of myofiber contents into the blood stream can lead to nausea and vomiting, cardiac arrhythmia (hyperkalemia), and even CNS side effects such as confusion and coma. Rhabdomyolysis, if severe enough, can lead to enough movement of fluid into muscle to cause intravascular volume depletion and hypotension. Rhabdomyolysis can trigger the coagulation cascade and lead to disseminated intravascular coagulation. The most notorious complication is the development of acute tubular necrosis secondary accumulation of myoglobin and case formation within renal tubules. Ventilatory failure due to involvement of diaphragm and chest wall muscles is a rare complication of rhabdomyolysis secondary to carnitine palmitoyl transferase deficiency.32
The pathophysiology of RHB/MGU is not fully understood. The intracellular migration of sodium and water may lead to a secondary exchange of intracellular sodium with extracellular calcium in muscle fibers that remain viable. This in turn may lead to persistent activation of the myofiber contractile apparatus that may perpetuate muscle injury. The need to pump sodium out of cells may deplete ATP as an additional adverse effect of this cascade. Furthermore, injury may occur as a consequence of the ischemia created by increased compartmental pressure, even in the absence of crush injury, by the fluid migration described above. The biochemical milieu created by muscle breakdown may in itself be toxic either through the release of the normally sequestered contents of the muscle fibers, from calcium-dependent proteases and phospholipases, or from the cytokines release by the customary inflammatory response to injury.
Recognition of RHB/MGU may or may not be obvious. A high index of suspicion should be maintained with predisposing conditions such as crush injury, prolonged immobilization, or introduction of potentially myotoxic drugs. Myoglobinuria and acute, diffuse myalgias are obvious clues but are not always readily evident, particularly when detailed history is not readily available. Patients typically have a leukocytosis. The measurement of serum CK is the most efficient and direct diagnostic tool. A positive urine dipstick for (which detects both myoglobin and hemoglobin) in the absence of red blood cells on microscopic examination of the urine strongly suggests myoglobinuria.
Diagnosis of the underlying cause may also be obvious or remain enigmatic despite extensive evaluation. In most series, drugs and metabolic disturbances are the most common identified cause of RHB/MGU.35 It is estimated that approximately a third to a half of patients who do not have an obvious cause of their RHB/MGU, will be found to have an identifiable enzymatic deficiency or underlying muscle disease as a cause of their syndrome.36 The yield will be predictably higher in patients who have had recurrent episodes.37
In RHB/MGU, CK levels typically peak in 12–24 hours and remain at peak levels for a few days. In response to minor muscle injury such as EMG examinations, CK levels typically normalize in a week. In more protracted causes of muscle injury such as infection or with very high CK elevations, it would be prudent to wait a few weeks to determine if CK has normalized. Once the CK peaks, it can be anticipated that it will drop by 50% every 48 hours. This is a potentially valuable diagnostic tool as normalization of CK should suggest a monophasic cause of RHB/MGU or carnitine palmitoyl transferase deficiency. Persistent elevations, although not universally found, would be more in keeping with glycogen storage disease or other pre-existing and persistent myopathic disorders.
The forearm exercise test is a valuable screening tool for those glycogen storage disorders characterized by exercise-induced symptomatology and the potential for RHB/MGU. Its yield will be greatest in those individuals whose RHB/MGU is provoked by brief periods of intense exercise. Its yield will be less in individuals whose RHB/MGU appears to occur sporadically, on a delayed basis after more protracted exertion, or following fasting. In these circumstances, CPT deficiency will be the most common hereditary etiology. Forearm exercise testing will be described in detail in the section on glycogen storage disease. In summary, baseline determinations of venous lactate and ammonia obtained. Forearm muscles are then repetitively and forcefully exercised, typically with a grip dynamometer. Using a blood pressure cuff to render the forearm ischemic is no longer done by the majority of neuromuscular specialists as the risk of patient injury is felt to exceed any additional diagnostic yield. Serial measurements of venous lactate and ammonia are obtained from the ipsilateral basilic or cephalic veins in the antecubital fossa. In patients with glycogen storage disease, these measurements are often superfluous as the affected patient will develop a sustained painful “contracture” of the forearm muscles often leading to a dystonic posture with forearm pronation, wrist flexion with ulnar deviation, and finger flexion. A three-fold elevation or more above baseline of either lactate or ammonia signifies adequate effort. With glycogen storage disease, ammonia will elevate but lactate will not. The converse will occur with myoadenylate deaminase deficiency, the clinical significance of which remains controversial. In young adults experiencing RHB/MGU during or after protracted exercise, particularly if recurrent or with normalization of CK following the acute episode, we commonly obtain genetic testing for CPT deficiency.
It is difficult to provide dogmatic advice regarding the role of muscle biopsy in the evaluation of RHB/MGU. Muscle biopsy in the acute setting is to be avoided. Random myofiber necrosis in similar stages of degeneration, affecting both type I and type II fibers, without any specific diagnostic features is the anticipated outcome. Whether muscle biopsy should be performed subsequent to the RHB/MGU episode is a more difficult question to answer. The yield in identifying the exact cause of RHB/MGU is extremely low, and the cost of providing a comprehensive analysis of the muscle biopsy can be substantial. In our opinion, muscle biopsy in RHB/MGU should be reserved for those individuals with recurrent episodes of RHB/MGU of indeterminate cause with negative exercise forearm testing and genetic analysis for CPT deficiency.
Hydration is the most important therapeutic intervention. Six to twelve liters of intravenous fluids in the first 24 hours are recommended in the absence of comorbidities. Urine output should ideally be in the 200–300 mL/h range. The fluid should not be hypotonic. Mannitol and sodium bicarbonate are frequently added although there is limited evidence to support their efficacy. Vigorous diuresis with furosemide represents a mainstay of treatment as well. A high index of suspicion for compartment syndrome should be maintained and fasciotomy considered when clinically appropriate. Hypokalemia, hypocalcemia, and hypophosphatemia should be screened for and treated if necessary. Awareness of adult respiratory distress syndrome, ischemic bowel, and the possibility of hemorrhage secondary to DIC should be maintained. Alkalinization should be utilized where appropriate.
SUMMARYDiagnostic accuracy is in large part dependent on a clinician’s ability to elicit and formulate pertinent information from three domains of the patient’s history and examination, these being anatomic localization, definition of the disease course, and relevant risk factors. The astute clinician will learn to discard information that is not germane to the patient’s current problem, and formulate a differential diagnosis by accurately matching the information from the three domains mentioned above to the known behaviors of different diseases. An accurate diagnosis may be evident solely from this clinical process, or may require further testing to confirm or refute the differential diagnosis generated by this process. This chapter has addressed the strategies that serve as the foundation for this problem-solving approach. Subsequent chapters will discuss the features of the neuromuscular diseases in an attempt to complete this diagnostic and hopefully therapeutic endeavor.
1. David WS, Jones HR Jr. Electromyography and biopsy correlation with suggested protocol for evaluation of the floppy infant. Muscle Nerve. 1994;17(4):424–430.
2. Dumitru D, Amato AA. Introduction to myopathies and muscle tissue’s reaction to injury. In Dumitru D, Amato AA, Swartz MJ, eds. Electrodiagnostic Medicine. 2nd ed. Philadelphia, PA: Hanley & Belfus; 2002:1229–1264.
3. Shoesmith CL, Findlater K, Rowe A, Strong MJ. Prognosis of amyotrophic lateral sclerosis with respiratory onset. J Neurol Neurosurg Psychiatry. 2007;78:629–631.
4. Chokroverty S. Sleep-disordered breathing in neuromuscular disorders: A condition in search of recognition. Muscle Nerve. 2001;24(4):451–455.
5. Cudkowicz ME, Qureshi M, Shefner J. Measure and markers in amyotrophic lateral sclerosis. NeuroRx. 2004;1:273–283.
6. Isaacs JD, Dean AF, Shaw CE, Al-Chalabi A, Mills KR, Leigh PN. Amyotrophic lateral sclerosis with sensory neuropathy: Part of a multisystem disorder? J Neurol Neurosurg Psychiatry. 2007;78:750–753.
7. Wolfe GI, Bank WJ. Pseudokyphosis in motor neuron disease corrected by the pocket sign [abstract]. Muscle Nerve. 1994; 17:1091.
8. Van Gerpen JA. Camptocormia secondary to early amyotrophic lateral sclerosis. Mov Disord. 2001;16:358–360.
9. Hoffman D, Gutmann L. The dropped head syndrome with chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 1994;17:808–810.
10. Keating JM, Yapundich RA, Claussen GC, Oh SJ. Head drop syndrome as a presenting feature of polymyositis [abstract]. Muscle Nerve. 1996;9:1190.
11. Hund E, Heckl R, Goebel HH, Meinck UM. Inclusion body myositis presenting with isolated erector spinae paresis. Neurology. 1995;45:993–994.
12. Biran I, Cohen O, Diment J, Peyser A, Bahnof R, Steiner I. Focal, steroid responsive myositis causing dropped head syndrome. Muscle Nerve. 1999;22:769–771.
13. Lomen-Hoerth C, Simmons ML, Dearmond SJ, Layzer RB. Adult-onset nemaline myopathy: another cause of dropped head. Muscle Nerve. 1999;22:1146–1150.
14. Chanin N, Selcen D, Engel AG. Sporadic late onset nemaline myopathy. Neurology. 2005;65:1158–1164.
15. Katirji B, Hachwi R, Al-Shekhlee A, Cohen ML, Bohlmann HH. Isolated head drop due to adult- onset nemaline myopathy treated by posterior fusion. Neurology. 2005;65:1504–1505.
16. Luque FA, Rosenkilde C, Valsamis M, Danon MJ. Inclusion body myositis (IBM) presenting as the “dropped head syndrome” (DHS) [abstract]. Brain Pathol. 1994;4:568.
17. Evidente VG, Cook A. Floppy head syndrome resulting from proximal myotonic dystrophy [abstract]. Ann Neurol. 1997;42:417.
18. Serratrice J, Weiller PJ, Pouget J, Serratrice G. [An unrecognized cause of camptocormia: proximal myotonic myopathy]. Presse Med. 2000;29(20):1121–1123. French.
19. Karpati G, Carpenter S, Engel AG, et al. The syndrome of systemic carnitine deficiency: clinical, morphologic, biochemical, and pathophysiologic features. Neurology. 1975;25:16–24.
20. Umapathi T, Chaudhry V, Cornblath D, Drachman D, Griffin J, Kuncl R. Head drop and camptocormia. J Neurol Neurosurg Psychiatry. 2002;73:1–7.
21. Baquis GD, Moral L, Sorrell M. Neck extensor myopathy: a mitochondrial disease [abstract]. Neurology. 1997;48(suppl 2):A443.
22. Berenbaum F, Rajzbaum G, Bonnichon P, Amor B. Une hyperparathyroidie revelee par une chute de la tete. Rev Rheum Ed Fr. 1993;60:467–469.
23. Beekman R, Tijssen CC, Visser LH, Schellens RL. Dropped head as the presenting symptom of primary hyperparathyroidism. J Neurol. 2002;249(12):1738–1739.
24. Yoshida S, Takayama Y. Licorice-induced hypokalemia as a treatable cause of dropped head syndrome. Clin Neurol Neurosurg. 2003;105(4):286–287.
25. Barohn RL, McVey AL, DiMauro S. Adult acid maltase deficiency. Muscle Nerve. 1993;16:672–676.
26. Serrano MC, Rabinstein AA. Cause and outcomes of acute neuromuscular respiratory failure. Arch Neurol. 2010;67(9):1089–1094.
27. Hughes RA, Bihari D. Acute neuromuscular respiratory paralysis. J Neurol Neurosurg Psych. 1993;56(4):334–343.
28. Nicolle MW, Stewart DJ, Remtulla H, Chen R, Bolton CF. Lambert-Eaton myasthenic syndrome presenting with severe respiratory failure. Muscle Nerve. 1996;19:1328–1333.
29. Durmus H, Laval SH, Deymeer F, et al. Oculopharyngodistal myopathy is a distinct entity: clinical and genetic features of 47 patients. Neurology. 2011;76:227–235.
30. Huerta-Alardίn AL, Varon J, Marik PE. Bench-to-bedside review: rhabdomyolysis- an overview for clinicians. Crit Care. 2005;9(2):158–169.
31. Mathews KD, Stephan CM, Laubenthal K, et al. Myoglobinuria and muscle pain are common in patients with limb-girdle muscular dystrophy 2I. Neurology. 2011;76:194–195.
32. Berardo A, DiMauro S, Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neuroscience Rep. 2010; 10:118–126.
33. Warren JD, Blumbergs PC, Thompson PD. Rhabdomyolysis: a review. Muscle Nerve. 2002;25:332–347.
34. Vladutiu GD, Simmons Z, Isackson PJ, et al. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve. 2006;34(2):153–162.
35. Melli G, Chaudhry V, Cornblath DR. Rhabdomyolysis: an evaluation of 475 hospitalized patients. Medicine (Baltimore). 2005 84:377–385.
36. Gabow PA, Kaehny WD, Kelleher SP. The spectrum of rhabdomyolysis. Medicine. 1982;61:141–152.
37. Lofberg M, Jankala H, Paetau A, Harkonen M, Sormer H. Metabolic causes of recurrent rhabdomyolysis. Acta Neurol Scand. 1998;98:268–275.
38. Rose MR, Kissel JT, Bickley LS, Griggs RC. Sustained myoglobinuria: the presenting manifestation of dermatomyositis. Neurology. 1996;47:119–123.