 INTRODUCTION INTRODUCTION
INTRODUCTION INTRODUCTIONThe ideal of every patient and physician is to identify a diagnosis whose natural history is self-limited, or if not, a diagnosis for which an effective treatment can be administered. Autoimmunity is believed to be the contributing, if not causal, mechanism of a significant number of neuromuscular disorders.1Accordingly, patients with proven or suspected autoimmune neuromuscular disorders become candidates for treatments that modulate or suppress immune-mediated nerve, neuromuscular junction, or muscle dysfunction or injury. Familiarity with drugs or other interventions that suppress or modulate the patient’s immune system is therefore a prerequisite for anyone practicing neuromuscular medicine.
In this book, we will define immunomodulation as any therapy that affects in any way the native activities of a patient’s immune system in an attempt to mitigate disease. We will define immunosuppression as a subcategory of immunomodulation in which a patient’s immunologic response is impaired by one of the three recognized mechanisms.2,3 One mechanism, as occurs with drugs such as azathioprine, cyclophosphamide, mycophenolate, and methotrexate, curtails B-cell and T-cell proliferation by cell cycle interruption. Another mechanism, as exemplified by drugs such as the calcineurin inhibitors (e.g., cyclosporine and tacrolimus) and corticosteroids, is impairment of T-cell activation. A final mechanism of immunosuppression is accomplished by monoclonal antibodies–directed cell surface antigens, rituximab being the most notable example. Conversely, we will consider interventions such as intravenous immunoglobulin (IVIg) or plasma exchange (PLEX) to be immunomodulating, not immunosuppressive.
The authors strongly endorse the concept of evidence-based medicine. At the same time, we recognize that evidence-based medicine applies to populations and that strict adherence to evidence guidance is not always in the best interests of the individual patients we are responsible for. In neuromuscular medicine, there are numerous examples of treatments that are universally considered to be efficacious yet remain of unproven benefit by “evidence-based” standards.4 Corticosteroids in myasthenia gravis (MG) is one notable example, discovered by innovative effort by individual clinicians. Because of the accepted efficacy of this and other historically identified empiric treatments, it is unlikely that a number of currently accepted treatments will ever be validated by large prospective studies.
Our position is also supported by personal witness of unequivocal benefit to individual patients, who respond to treatments demonstrated to be ineffective to larger populations with the same disease. Rituximab in MuSK-positive MG is such an example.5 Accordingly, this chapter will describe, and in some cases endorse, the off-label uses of immunomodulating treatments for various neuromuscular diseases even in the absence of evidence-based support. We do so cautiously as we recognize that these idiosyncratic responses may be harmful as well as helpful. Ultimately, each physician needs, along with their patient, to determine whether the potential benefits of immunomodulating treatment, of proven or unproven benefit, exceed the probability and magnitude of potential risk.
This chapter will approach immunomodulating treatment of presumed immune-mediated disorders by focusing on the treatments, rather than the disorders themselves which will be the subject matter of subsequent chapters. A summary of these agents, and the disorders for which they may or may not be effective are summarized in Table 4-1. Details regarding dosing, side effect profiles, and recommended screening procedures are summarized in Table 4-2.
 TABLE 4-1. IMMUNOMODULATING DRUGS IN NM DISORDERS—CURRENT USAGE
TABLE 4-1. IMMUNOMODULATING DRUGS IN NM DISORDERS—CURRENT USAGE
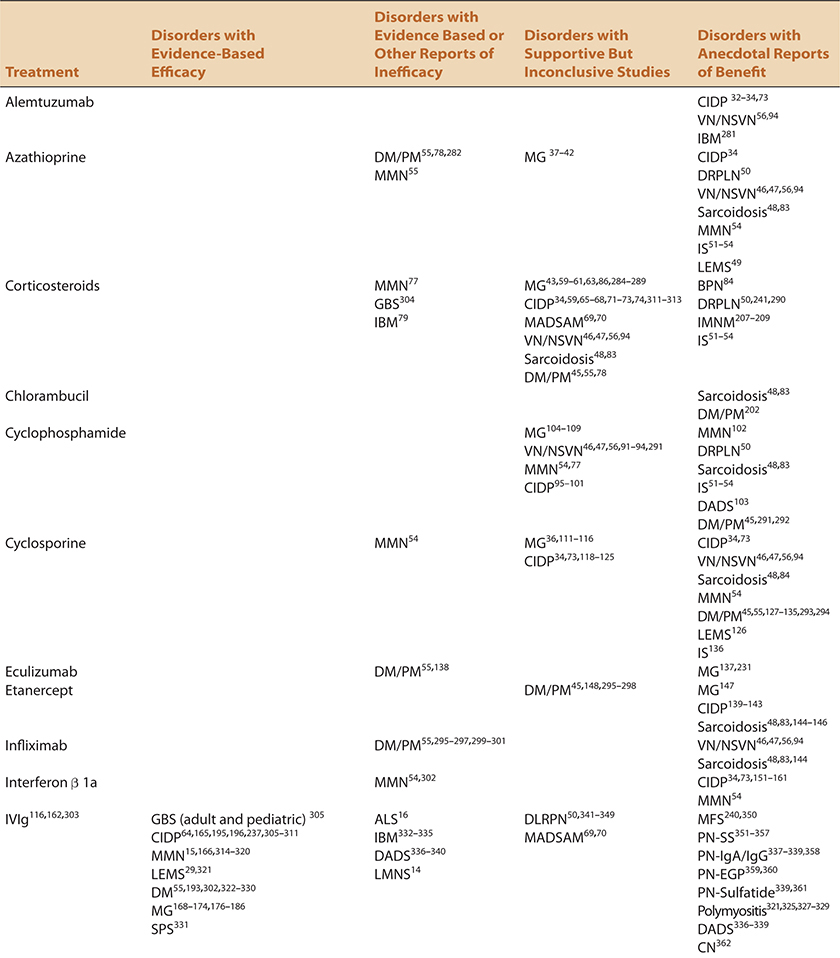

TABLE 4-2. IMMUNOSUPPRESSIVE THERAPY FOR NEUROMUSCULAR DISORDERS

GENERAL CONSIDERATIONSBefore initiating immunomodulating therapy, it is critical to consider the probability and magnitude of both the potential risk to an individual patient, as well as the potential benefit. There is a consensus that patients who receive immunomodulating treatment are at an increased risk for both infection and malignancy.6 There is also a consensus that the risk is probably dependent on numerous variables including the genetics and comorbidities of the individual patient, the agent or agents used, as well as cumulative dose and duration of treatment. The following section will review some of these considerations facing clinicians and their patients who are contemplating immunomodulating treatment. On discussing risk with patients, we find it useful to utilize the World Health Organization guidelines which define risk as very common >1/10, common >1/100, uncommon >1/1,000, rare >1/10,000, and very rare >1/100,000.7
In consideration of immunomodulating treatment, it is important to be armed with knowledge relevant to a number of key issues in order to make rational treatment decisions. Of primary importance is the identification of an objective parameter to measure. A pretreatment baseline should be established in order to determine whether treatment is effective or not in the future. The ideal parameter(s) chosen should be not only quantifiable and reproducible, it (they) should correlate with meaningful improvements in patient comfort and function.
In neuromuscular disease, measurements of strength are the most commonly utilized. We have found manual muscle strength testing and handheld dynamometry (e.g., microFET2®) to be helpful in this regard, along with quantitative bedside assessments of sensation (e.g., timed vibration or the Rydel-Seiffer® tuning fork). There are many functional or symptomatic scales that have been developed for specific diseases, e.g. ALS, myasthenia, and peripheral neuropathy, that also facilitate determination of treatment response.8–13 Unfortunately in neuromuscular disease, other biomarkers such as imaging, electromyography and nerve conduction study data, and measurement of serologic markers are not always accurate or practical means of monitoring treatment response.
A master clinician understands the natural history of the disease they are treating as well as the properties of the agents they are using. The latency between treatment and response is dependent on at least two parameters, the pharmacology of the immunomodulating agents used and the pathophysiology of the disease. For example, morbidity created by disorders that impede ion channel function, demyelinate axons without otherwise injuring them, or that injure relatively easily repairable components of the neuromuscular system such as ACh receptors may be expected to respond to an effective treatment relatively rapidly, within days to weeks in many cases. Conversely, disorders that require axon regrowth may require months before return of function becomes evident depending on the number of axons injured, the distance between the site of injury and the muscle(s) that require(s) reinnervation. Lastly, disorders that lead to significant destruction of motor or sensory cell bodies are limited in their ability to recover as their regeneration is unlikely, even if an effective treatment is initiated.
Furthermore, it is important to be familiar with the duration of treatment benefit as well as the latency between therapeutic intervention and clinical response. Without an appreciation of both, at least three potential risks may be encountered. A clinician may give up on a treatment before it has had a chance to work, and by doing so, initiate a second, potentially unnecessary and harmful agent. Conversely, a clinician may be overly optimistic, waiting too long for an ineffective agent to work, thus delaying exposure to an additional, potentially beneficial treatment. In addition, a clinician may unnecessarily procrastinate by waiting too long to initiate subsequent maintenance doses, allowing potentially avoidable relapses to occur and by doing so, eroding a patient’s confidence in their physician.
A particularly vexing problem in this age of evidence-based medicine is the patient with a suspected or proven autoimmune disorder for which no known proven treatments exist. In these cases, a diagnostic and potentially therapeutic trial may be undertaken. IVIg is frequently used for this purpose both for its relatively rapid onset of action, its efficacy in many autoimmune diseases, and in consideration of its relative safety. For example, although current evidence does not support the routine use of IVIg in lower motor neuron syndromes, some of these patients are thought to represent cases of multifocal motor neuropathy (MMN) without demonstrable biomarkers such as elevated GM1 autoantibody titres or conduction block.14,15 In this and other comparable situations, we follow the lead of others by typically providing a 3-month therapeutic trial of IVIg.14,16 Although this practice may be considered somewhat arbitrary in its duration, it represents in our mind a reasonable compromise between a sufficient interval to detect benefit in this demyelinating neuropathy and the waste of an expensive resource. In the case of an MMN suspect, unequivocal stabilization would provide sufficient proof of treatment efficacy as this would differ from the inexorable progression of motor neuron disease which is the primary differential diagnostic consideration.14
Immunomodulating treatment strategies vary in consideration with the treatment modality employed, individual disease characteristics, and individual patient context. There are general principles however, that include the recognition of maximal achievable benefit with the goal of avoiding excessive treatment. If disease remission can be achieved, with the potential in that particular disorder for a durable, treatment-free response, an attempt should be made to wean by reducing the amount and/or frequency of administration. For example, it is not uncommon for patients with vasculitic neuropathies who respond to immunosuppression to eventually be successfully weaned from treatment after 2–3 years and enjoy years of subsequent treatment-free stability. The goal with any patient, is to ensure, through clinical or when relevant other means, continued patient improvement or stabilization. At the same time, the goal is to also limit potential adverse effects and costs of chronic treatment while at the same time achieve and sustain the best potential outcome.
These differing treatment strategies are illustrated in the following examples. Corticosteroids in MG or inflammatory myopathies or IVIg in MMN are often initiated at high “induction” doses and then gradually weaned in an attempt to identify the smallest dose or longest interval between treatments that will achieve remission or maintain an acceptable level of morbidity. Conversely, with IVIg in Guillain–Barré syndrome (GBS) or rituximab in MG, a singular prescribed course is initially delivered regardless of initial response and repeated in the future only with initial response and subsequent relapse.
RISK CONSIDERATIONS WITH IMMUNOMODULATING TREATMENTSPneumocystis jirovecii, formerly known as Pneumocystis carinii (PCP), is a fungal interstitial pneumonia that occurs predominantly in individuals who are immunosuppressed as a result of their disease or its treatment.17 It is widely accepted that 70–90% of patients who acquire PCP have received corticosteroid treatment.18 PCP prophylaxis advocates justify its use in immunosuppressed individuals due to the potential morbidity and mortality associated infection. The mortality risk is estimated to be 30–50%, even if recognized and treated.18
The onset of PCP may be subacute or indolent. Typical symptoms are dyspnea on exertion, nonproductive cough, fever, and tachycardia. Diagnosis is supported by imaging evidence of bilateral pulmonary infiltrates extending outward from the perihilar regions and an elevated serum LDH. Confirmation typically requires bronchoalveolar lavage.
Prophylaxis is preferentially achieved with trimethoprim–sulfamethoxazole, 160–800 mg/day or double strength three times a week.19 Either regimen is felt to be 90% successful in preventing PCP. In those intolerant of sulfa, atovaquone, dapsone, and pentamidine represent alternatives. For those who favor PCP prophylaxis, current recommendations suggest it should be introduced if prednisone is utilized at a dose greater than 20 mg/day for a duration that exceeds 4 weeks.
PCP prophylaxis in neuromuscular patients treated with immunomodulating agents is not practiced universally. The evidence basis for PCP prophylaxis is largely derived from cancer and pulmonary patient populations.18 In addition, there is a paucity of information to guide clinicians regarding adjusted risk based on the number and types of agents utilized, their doses, and the duration of exposure. Although the severity of PCP infection is unquestioned, its frequency in neuromuscular patients treated with immunomodulating agents is less well known. Although many disciplines such as rheumatology and infectious disease seem to favor its use, neurologists appear to be in general less sanguine about prophylactic necessity. For example, a recent poll neuromuscular specialists posted on Rick’s Real Neuromuscular Friends indicated that 53% of 45 respondents do not provide routine PCP prophylaxis in this population.20 Many have never seen a case of PCP despite having treated many patients chronically with one or more immunomodulating agents generating a more conservative perspective than suggested above.
The risk of tuberculosis reactivation is considered to be approximately three to six times greater in patients receiving tumor necrosis factor (TNF) inhibitors and corticosteroids.21 It becomes prudent therefore to ascertain the risk of latent tuberculosis infection (LTBI) in any individual in whom immunomodulating treatment is considered. Screening would include determining risk of prior, chest x-ray, and in those who are at risk, either tuberculin skin testing (TST) or an interferon (IFN) gamma release assay (IGRA). TST is thought to be 98% sensitive in detecting LTBI. Its limitations are that it may not detect infection in the first 8 weeks following exposure, may be falsely negative in individuals already immunocompromised, or may be falsely positive in individuals who have received prior BCG vaccination. IGRAs are complementary diagnostic tools for LTBI. They are in vitro blood tests of cell-mediated immune response to Mycobacterium tuberculosis and measure T-cell release of IFN-gamma following stimulation by antigens specific to M. tuberculosis. The two available IGRAs are QuantiFERON-TB Gold In-Tube and the T-SPOT.TB. They are the preferred means to confirm LTBI in individuals with prior BCG exposure. The IGRAs appear to be somewhat more specific and less sensitive for predicting future active TB than the tuberculin skin test but the differences are modest. Like TST, IGRA false negatives are more likely to occur in immunosuppressed individuals. Both IGRAs and the TST have high negative predictive values for development of future infection.21
Like all clinical decisions, prophylactic treatment of patients suspected of LBTI needs to consider the relative benefits and risks. Currently, someone with suspected LBTI who is going to receive immunosuppressant treatment with corticosteroids, TNF-α inhibitors and probably other agents, is recommended to receive prophylactic treatment with isoniazid (with pyridoxine), rifampin, or a combination of both. Isoniazid is typically prescribed as 300 mg daily for 9 months or 900 mg twice a week for 6 months. Rifampin alone is dosed at 600 mg/day for 4 months. When both drugs are used together, 600 mg of rifampin and 300 mg of isoniazid are given daily for 3 months.21 Other considerations are the risk of isoniazid hepatotoxicity which increases with age and exposure to other hepatotoxic agents and the numerous drug interactions that occur with rifampin use.
Progressive multifocal leukoencephalopathy (PML) is a demyelinating central nervous system disorder caused by infection with the John Cunningham (JC) virus. The JC virus is ubiquitous, found in 50–60% of the normal population and is typically sequestered in peripheral organs.22 PML is a disorder seen almost exclusively in the immunosuppressed. Even in this population, the virus rarely gains access to the central nervous system.
To date, PML has been associated with two agents employed in immune-mediated neuromuscular disorders, rituximab and mycophenolate mofetil (MMF).23 Infliximab, etanercept, and alemtuzumab, have also been reported as PML risk factors as have other currently available monoclonal antibodies that may be employed in the treatment of neuromuscular disease in the future.24 Undoubtedly, it will be described in association with other immunomodulating agents relevant to neuromuscular disease in the future. The risk for PML appears to be predominantly in those who harbor the JC virus prior to the introduction of immunosuppressant treatment.
Surveillance and treatment paradigms have been developed for PML in multiple sclerosis patients exposed to natalizumab.22 Presumably as the risk of developing PML with the immunosuppressant drugs described in this chapter is thought to be very rare, we are unaware of any recommendations for JC virus surveillance and prophylaxis for any agents described in this chapter. As anticipated, recognition of PML in any patient receiving immunosuppressant drugs warrants drug discontinuation in virtually any clinical context.
Stongyloidiasis is a parasite that is endemic in warm moist tropical and subtropical climates such as Eastern Europe, South and Southeast Asia, Central America, South America, and sub-Saharan Africa.25–27 It is transmitted via skin penetration by infective filariform larvae following exposure to water or soil contaminated by human or canine fecal material. The larvae are hematogenously carried to the lungs, regurgitated, and then swallowed where they mature into adults in the intestines.25 The reproductive cycle of the nematode and resultant reinfection may continue indefinitely. The autoinfected human host frequently remains asymptomatic or experiences mild nonspecific skin and gastrointestinal symptoms.25,26 This equilibrium may persist indefinitely.
Immunosuppression however, particularly with corticosteroids, may result in multiorgan dissemination and hyperinfection.25,26 Control of parasitic infections requires Th2 cytokine, eosinophilic, and IgE influence, all of which are suppressed by steroids and other immunomodulating agents.25,26 With hyperinfection, mortality is estimated to be 60–85%.27 For this reason, parasitic and serologic surveillance is recommended for anyone at increased risk prior to immunomodulating treatment. The absence of hypereosinophilia occurs frequently in infected individuals and does not represent a sensitive screening test. Ideally, patients at risk should undergo three negative surveillance stool specimens and an ELISA screening test for IgG Strongyloides stercoralis antibodies available through the Centers for Disease Control before treatment is begun.25,26 The ELISA test is thought to be 80–100% sensitive and highly specific in immunocompetent individuals. Its sensitivity drops significantly in immunosuppressed individuals. A negative test needs to be interpreted cautiously in individuals already exposed to immunomodulating treatment.25 If infected, ivermectin, thiabendazole, and albendazole are the most commonly used therapeutic agents.26,27 One suggested regimen for strongyloidiasis prophylaxis would be ivermectin 200 μg/kg/day for 2 days, repeated within 2 weeks.26
Questions regarding vaccination of patients in whom immunomodulating treatment is being considered or is already being received are very relevant to the practice of neuromuscular medicine. Current recommendations hold that ideally, patients should be vaccinated against influenza, pneumococcus, tetanus, hepatitis A and perhaps B prior to initiation of immunosuppression.28 Once immunosuppression has commenced, there appears to be a consensus that vaccines containing dead virus can be utilized without undue risk but that live-virus vaccines should be avoided.6
Management of immunomodulating treatment in women of child-bearing age is difficult. Current evidence holds that use of corticosteroids or IVIg provides no additional risk for mother or child during pregnancy.7,29 We are very reticent to use other immunomodulating agents in childhood unless patient morbidity provides no other options. If corticosteroids are used before full growth is achieved, it is recommended that linear growth be tested regularly and growth hormone treatment considered if necessary.7
Patients who receive immunosuppressive therapy are believed to be at increased risk of developing malignancy.28 This risk is attributed to oncogenic infectious agents, reduced immune surveillance of cells having undergone mutation in relationship to age or environmental factors, or direct effects on oncogenes.30 Notable oncogenic organisms whose proliferation may be aided by immunosuppression include Epstein–Barr virus, human herpesvirus 8, human papillomavirus, hepatitis B and C, and helicobacter. The malignancies most commonly associated with immunosuppression include lymphoproliferative disorders, Kaposi’s sarcoma, as well as anogenital, liver, and stomach cancer.28 Data pertaining to the relative risk of developing these malignancies, indexed to the numbers, types, and length of exposure to immunosuppressant medications are lacking although it is widely accepted that both increased dose and duration of exposure are relevant.30 Of interest, available data suggests that cancer risk rapidly dissipates following discontinuation of immunomodulating treatment.28 The pragmatic benefit of this knowledge is uncertain given the presumption that discontinuation of an effective immunosuppressive agent is unlikely unless cancer develops. Recommendations regarding rational, evidence-based cancer surveillance protocols for patients on immunomodulating treatments are elusive and are beyond the scope of this text. Discussion of this risk should nonetheless be part of the informed consent process. Consideration of dose reduction and potentially discontinuation is recommended in those who achieve complete disease remission.
There also appears to be an increased risk of skin cancers in patients receiving immunosuppressant drugs. The incidence of squamous cell carcinoma is believed to be increased by 14–82 fold and malignant melanoma increased by a factor of 2.4 in the solid organ transplant population.31 We routinely advise patients on immunosuppressant drugs of the increased skin cancer risk and recommend limiting sun exposure, ample use of sun-blocking agents, and routine skin surveillance.
INDIVIDUAL TREATMENTS MODALITIESIn the following section, individual immunomodulating treatments commonly used in neuromuscular disease will be discussed. Consideration of mechanisms of action, specific disorders in which individual agents are often used, adverse effects and management strategies will be addressed for each modality. For more detailed management strategies, the reader is referred to the relevant chapter on the disease in question.
Alemtuzumab is a monoclonal antibody directed against the CD52 antigen found on the cell surface of mature lymphocytes. It is used primarily in the treatment of chronic lymphocytic leukemia, cutaneous and other T-cell lymphomas. There has been limited experience with its use in neuromuscular disorders.
Seven chronic inflammatory demyelinating polyneuropathy (CIDP) patients who have received alemtuzumab have been reported to demonstrate some degree of efficacy.32,33 An open-label multicenter trial of alemtuzumab in CIDP is underway.34
Some patients receiving alemtuzumab have been reported to develop autoimmune disease, most notably Graves disease and hemolytic anemia.34
One reported protocol consists of five daily intravenous infusions of 30 mg with repeated courses as required.34
Azathioprine is a purine analog that acts as a cytotoxic immunosuppressive agent.35 Its main active metabolite, 6-mercaptopurine, is a purine antagonist.36 It is a cell-cycle–specific inhibitor, exerting its actions mainly in the resting (G1) and DNA synthesis (S) phases of the cell cycle through suppression of GTPase Rac1 activation.2 Although its primary effects are directed at T cells, it is efficacious in T-cell–dependent antibody-mediated disorders such as MG.1 In addition to reducing numbers of circulating T cells, it also reduces levels of B-cell–derived immunoglobulins and interleukin-2 (IL-2).
Azathioprine is a commonly used maintenance, “steroid-sparing” therapy in MG. Several trials have demonstrated the efficacy of azathioprine alone or in combination with prednisone.37–42 Improvement is noted in 70–90% of patients with myasthenia treated with azathioprine, including some patients who are steroid resistant.43 We commonly initiate azathioprine (or other steroid-sparing agent) along with corticosteroids in any myasthenic patient with generalized disease in whom we anticipate the need for long-term immunomodulating treatment. We do so in the hope of facilitating steroid weaning, thereby limiting risk of long-term steroid side effects. By starting early, we take advantage of the short-term benefits of corticosteroids, recognizing the delayed therapeutic latency (3–15 months) of azathioprine which may require up to 2 years to achieve full effect.1,44
Azathioprine is also used a second-line agent in a number of presumed immune-mediated neuromuscular diseases, either as an adjunct to steroids, as a long-term maintenance agent. On occasion, it may become a first-line agent when more typical first-line agents (e.g., corticosteroids, IVIg, or PLEX) have failed. For the most part, the use of azathioprine in nonmyasthenic NM diseases is based on expert opinion or small case series. It has been used in dermatomyositis, polymyositis, CIDP, MMN, Lambert–Eaton myasthenic syndrome (LEMS), distal acquired demyelinating sensory neuropathy (DADS) vasculitic neuropathy, diabetic lumbosacral radiculoplexus neuropathy, Isaac’s syndrome, and sarcoidosis.34,35,45–56 We would advocate for its use in dermatomyositis, polymyositis, CIDP, LEMS, and sarcoid neuropathy in a patient with significant morbidity, who responds to first-line treatment, but who experiences unacceptable side effects or other impediments to long-term treatment with more conventional first-line treatments for these disorders.
Side effects have been reported in 35–42% of individuals. Fortunately, they are often mild and tolerable. They typically, although not invariably, develop within days to weeks of drug exposure. Many individuals tolerate the drug without any apparent side effects for protracted periods of time, which along with its potential effectiveness, make it an attractive agent.35,40 A systemic reaction characterized by fever, abdominal pain, nausea, vomiting, and anorexia occurs in 12% of patients requiring discontinuation of the drug. As mentioned, this reaction generally occurs within the first few weeks of therapy and resolves within a few days of discontinuing the azathioprine. Rechallenge with azathioprine may be successful but usually results in the recurrence of the systemic reaction. Other uncommon but major complications of azathioprine are bone marrow suppression, hepatic toxicity, pancreatitis, teratogenicity, risk of opportunistic infection and oncogenicity including increased risk of skin cancer.35,40
Azathioprine is available in 50 mg tablets without a parenteral analog. We typically begin with one tablet a day and escalate slowly to a maintenance dose of 2–3 mg/kg/day, typically 2.5 mg/kg/day. Prior to beginning azathioprine, we typically screen for thiopurine methyltransferase (TPMT) deficiency. Patients who are heterozygous for the TPMT mutation may be able to tolerate azathioprine at lower dosages but those who are homozygous should not receive drug. They cannot metabolize it and may experience severe bone marrow toxicity. Fortunately, the majority of patients who develop adverse hematologic responses in response to azathioprine recover fully once the drug is discontinued.
In patients receiving azathioprine, complete blood count (CBC) and liver function tests are monitored every 2 weeks until the patient is on a stable dose of azathioprine and then every 3–6 months for 2 years. After that, with stable blood counts, yearly surveillance is likely to be sufficient. If the white blood count falls below 4,000/mm3, the dose should be decreased. Azathioprine is held if the white blood count declines to 2,500/mm3 or the absolute neutrophil count falls to 1,000/mm3. Leukopenia can develop as early as 1 week or as late as 2 years after initiating azathioprine. As in most drugs with potential hepatotoxic effects, azathioprine should be discontinued if transaminases increase more than two to three times the baseline values. In the treatment of patients with myositis, it is important to determine whether transaminase elevation is due to liver damage from drug or muscle injury from disease. Accordingly, in these situations, we follow glutamyl transpeptidase levels (GGT), an enzyme present in liver but not muscle, in addition to AST and ALT for this reason. Liver toxicity from azathioprine generally develops within the first several months of treatment or increase in dosage. Leukopenia generally reverses in 1 month and hepatotoxicity can take several months to resolve.
An elevated mean corpuscular volume is an anticipated effect of azathioprine therapy and is used by some clinicians as an indicator of a biologic response. Allopurinol should be avoided in patients who require azathioprine because it interferes with azathioprine metabolism, increasing drug levels, and increasing the risk of bone marrow and liver toxicity.
Glucocorticoid effects are mediated through both genomic, nuclear glucocorticoid receptors as well as nongenomic cell surface receptors.57,58 They are one of the most versatile immunomodulating agents available in that they affect both cell-mediated and antibody-mediated autoimmunity.1 Corticosteroids largely affect T-cell function by producing T-cell apoptosis, suppressing the transcription of proinflammatory cytokines and impairing dendritic cell maturation.36 Specifically, glucocorticoids increase the rate of lipocortin synthesis which promotes anti-inflammatory effects by inhibition of phospholipase A2 as well as the proinflammatory cytokines IL-1, IL-2, the IL-2 receptor, INF gamma, and TNF.
The use of corticosteroids in autoimmune MG deserves special consideration. The recognition that corticosteroids were beneficial to patients with MG was historically delayed by the initial disease worsening that occurs in approximately 30% of patients receiving high-dose steroids, typically beginning between week 1–3 and lasting approximately 1 week.43,59–62 The mechanism of the worsening appears to be unique to disorders of neuromuscular transmission, apparently secondary to weak neuromuscular blocking properties of the drug supported by the demonstration of decremental responses to slow repetitive stimulation.61
Fortunately, the benefits of corticosteroids in MG became subsequently recognized. They remain a mainstay of MG treatment despite the lack of evidence-based support for its use. Corticosteroids, typically prednisone, are the first-line drug in anyone whose disease severity requires immunomodulating therapy unless other confounding clinical variables coexist.43 Its efficacy in MG appears to be universally accepted, making it unlikely that enrollment in a placebo-controlled trial would ever succeed.
Seventy-five percent of myasthenics are estimated to improve with corticosteroid use, 30% achieving remission and 45% marked improvement.36 Improvement may become evident within 2 weeks of initiation of high dose (0.75–1.5 mg/kg) daily dosing and is typically well established by 6–8 weeks. Absence of a significant response within 4 weeks suggests treatment failure and should prompt consideration of alternative or additional treatments. There have been reports of myasthenics who appear resistant to both the adverse and beneficial effects of prednisone who respond well to prednisolone.36
In patients in crisis who are intubated, parenteral methylprednisolone may be prescribed at doses of up to 1 g/day for up to 7 days before tapering to a 60–100 mg/day prednisone equivalent. In these patients, the risk of crisis provoked by steroids becomes largely irrelevant.63 In someone with significant morbidity from generalized disease, who we do not feel is in imminent danger of crisis, and in whom we are confident that adequate monitoring can occur, we may initiate high-dose prednisone, typically at 1–1.5 mg/kg up to 100 mg/day. In others whose morbidity warrants immunomodulating treatment, but where neither disease severity or risk of crisis warrants initial high-dose treatment, we utilize the so-called “start low, go slow” approach beginning at 10 or 20 mg of prednisone per day and gradually increasing by 5–10 mg/day every week or two until the target dose of 50–100 mg/day is reached.
Corticosteroids are used in numerous other presumed immune-mediated neuromuscular disorders including a number of neuropathy syndromes. The best evidence for efficacy exists in classic CIDP with a phenotype of generalized symmetric weakness, sensory signs and symptoms, and areflexia.34,59,64–68 Steroids also appear to be effective for the presumed CIDP variant, multifocal acquired demyelinating sensory and motor neuropathy (MADSAM), (a.k.a. Lewis–Sumner syndrome).65,69,70 Steroids have less apparent efficacy with other presumed CIDP variants, particularly when they are pure sensory or pure motor.65 Although prednisone is the most commonly used glucocorticoid for CIDP, successful intravenous methylprednisolone and oral dexamethasone regimens have been reported as well.71,72
The weight of existing evidence suggests that steroids provide no benefit and may be harmful in the aggregate in GBS, MMN, and DADS associated with IgM monoclonal proteins, or other neuropathies associated with monoclonal gammopathy of unknown significance.65,73–77
Corticosteroids are also considered to be effective in some but not all inflammatory myopathies.45,59 They are commonly used as a first-line treatment based on expert opinion in dermatomyositis, polymyositis, and immune-mediated necrotizing myopathy/myositis but are considered ineffective in inclusion body myositis.56,78,79
There is little doubt that immunomodulating treatment favorably alters the natural history of the systemic vasculitides. Corticosteroids, often with concomitant cyclophosphamide or rituximab are the backbones of treatment for these disorders.47,56,80–82 There is considerable support for the use of corticosteroids in the treatment of sarcoidosis and sarcoid neuropathy but no evidence-based confirmation. 83,84 There is a dearth of evidence in support of corticosteroid use in brachial plexus neuritis.84 We are of the opinion that steroids may benefit the painful aspects of this disorder if prescribed early but they appear to have a limited, if any, benefit in altering the natural history of the disease. Corticosteroids have been used with anecdotal reports or reports based on expert opinion of benefit in stiff person syndrome and diabetic lumbosacral radiculoplexus neuropathy.85
It is estimated that at least 30% of patients will experience corticosteroid-induced side effects, dependent upon dose and duration of therapy.36 Once a desired therapeutic effect is achieved, an attempt is made to wean to the lowest effective maintenance dose with 20 mg/day considered an acceptable balance between the benefits and drawbacks of long-term steroid side effects.36 Adverse effects of corticosteroids are largely dose-dependent and include diabetes, hypertension, peptic ulcer disease, osteopenia, cataracts, glaucoma, opportunistic infections, dyslipidemia, hypokalemia, increased appetite and weight gain, insomnia, and myopathy.59,62 Steroid psychosis and aseptic necrosis appear to be adverse effects that are idiosyncratic in nature. Interventions intended to prophylax against these complications will be addressed in the management considerations section below.
Corticosteroids are frequently administered in a single daily morning dose to parallel the normal circadian peak of endogenous cortisol production.1,59 Therapeutically, this strategy has been demonstrated to have some advantage in patients with rheumatoid arthritis although interpretation may be somewhat confounded by relief of morning stiffness, a notable source of morbidity in this disease.7
Once maximal efficacy has been achieved, we attempt to wean to the smallest effective maintenance dose and typically do so in an every other day format.1,59,86 We have utilized two different strategies. One is to begin the weaning process by initially doubling the induction dose on odd days and alternating this with zero on even days while beginning to reduce the aggregate dose. For example, a patient with an induction dose of 60 mg/day would be switched to 110 mg alternating with zero on any every other day basis. The alternative strategy is to initiate the weaning process by subtracting from the odd day dosage while maintaining the even dosage, for example, 60 mg on even days, 50 mg on odd days.
The speed of weaning proceeds based on individual patient context. For example, development of significant steroid side effects such as myopathy will accelerate the weaning pace whereas any indication of disease exacerbation may put the weaning process on hold. As a general guideline, we reduce the dose by 10 mg every 2 weeks. With the first regimen, this would mean 100 mg alternating with zero. With the second regimen this would mean 60 mg alternating with 40 mg. Once a dose of 20 mg every other day is reached, we taper more slowly, typically in increments of 2 mg every 2–4 weeks based on clinical response and the potential development of signs and symptoms of potential adrenal insufficiency.
Tuberculosis, strongyloidiasis, and herpes zoster are the three infectious agents we are aware of where corticosteroids may fulminantly exacerbate pre-existing, indolent infection. Consideration may be given to shingles vaccination prior to steroid initiation in individuals. It is estimated that it is safe to administer steroids or other immunomodulating agents 2 weeks or more after administration of this or any other live virus.28 We routinely question patients for potential exposure to tuberculosis and when in doubt perform PPD, chest x-ray, and IGRA testing. If there is suggestion for indolent TB infection, and immunomodulating treatment is medically necessary, we initiate isoniazid and pyridoxine treatment concomitantly unless otherwise contraindicated. If the patient comes from an area where strongyloidiasis is endemic, we consider baseline serologic testing and stool analysis.
Patients on long-term corticosteroids should receive baseline screening and periodic monitoring of intraocular pressure, blood pressure, blood sugar, lipids, and bone density. In particular, glucocorticoids facilitate osteopenia by interfering with bone formation through apoptosis of osteocytes and enhancing bone resorption through inhibition of osteoprotegerin, an endogenous antiresorptive cytokine.87 In any patient who will be receiving corticosteroids for more than 3 months, it is prudent to obtain a bone density and initiate daily treatment with 2,000 IU of vitamin D3 and 1,000 mg of calcium, promote exercise and suggest no more than modest alcohol intake.87–89 In men >50, women who are postmenopausal, or anyone with a T score of -1.5 or below, initiation of a bisphosphonate such as alendronate at a dose of 10 mg daily or 70 mg weekly, or 35 mg three times a week is recommended.87,90
We do not routinely recommend gastric protection in patients using chronic corticosteroids unless they are symptomatic or at increased risk for gastritis because of concomitant use of nonsteroidal anti-inflammatory agents. In these situations, prophylactic treatment with a proton pump inhibitor, misoprostol, or a cyclooxygenase 2 inhibitor is utilized.7 In addition, patients are instructed to start a low-sodium, low-carbohydrate, high-protein diet to prevent excessive weight gain and in the case of a high-protein diet, to theoretically reduce the risk of steroid myopathy. Patients are also encouraged to slowly begin an aerobic exercise program as it is hypothesized that both osteopenia and steroid myopathy are enhanced by immobility. Lastly, augmentation of corticosteroid dosing should be considered perioperatively in order to avoid risk of adrenal insufficiency in any patient who has been receiving these drugs for more than a month.7
Cyclophosphamide is a DNA-alkylating drug and nonspecific cell cycle inhibitor, with more pronounced effects on B cells than on T cells.2
Within the realm of neuromuscular diseases, cyclophosphamide use is probably best established in the treatment of neuropathy. It is commonly recognized as a mainstay of treatment in systemic vasculitic neuropathy, usually in conjunction with corticosteroids and has been reported to be of benefit in nonsystemic vasculitic neuropathy as well.45,46,91–94 In our experience and that of others, it can be an effective treatment in refractory CIDP patients.95–101 There are anecdotal reports of beneficial effect in DADS neuropathy and MMN as well.102,103
Cyclophosphamide may benefit patients with MG including those with MuSK autoantibodies.104–109 In view of its risk profile, it is typically used in the MG population only when other less-toxic agents have proven unsatisfactory.2 A protocol that utilizes high-dose cyclophosphamide to “reboot” the bone marrow has been suggested in individuals who have failed attempts at other less-toxic regimens.107 Because of the significant side effects described below, most clinicians avoid cyclophosphamide for MG if at all possible. Cyclophosphamide may be considered in patients with severe generalized MG refractory to other modes of immunotherapy. We have used it in refractory cases of polymyositis and dermatomyositis as well.
Cyclophosphamide has a substantial side effect profile that includes frequent nausea and vomiting with administration. Serious side effects include opportunistic infection, hemorrhagic cystitis, bone marrow depression, sterility, teratogenicity, alopecia, and late development of malignancy, particularly lymphoma and bladder cancer. The incidence of malignancy associated with cyclophosphamide use appears to increase when the cumulative dose exceeds 85 g.1
Cyclophosphamide may be administered either orally or intravenously. The oral dose is 2–2.5 mg/kg/day typically administered as single dose each morning. The intravenous dose is 1 g/m2 monthly for 6 consecutive months. The latter is preferred as it is thought to be less toxic and as the adverse hematologic effects are easier to time and monitor. The therapeutic latency between administration and manifest benefit is relatively short and is estimated to be 2–6 months, dependent on both drug effect and end organ healing.44
Patients receiving intravenous cyclophosphamide should have a baseline CBC and platelet count and urinalysis done and at minimum, a repeat CBC prior to each subsequent infusion to ensure a safe neutrophil level. Like azathioprine, repeat cyclosporine infusions are held if the WBC count and absolute neutrophil count which typically nadirs between 1–2 weeks post infusion does not reestablish itself to >2,500/mm3 or >1,000/mm3 respectively prior to the next dose. It is frequently recommended that cyclophosphamide recipients have yearly urine cytologic surveillance.
In the cytoplasm, cyclosporine binds to its immunophilin, cyclophilin. The cyclosporine–cyclophilin complex binds to and blocks the function of the enzyme calcineurin, eventually inhibiting T-cell activation and reducing production of the proinflammatory cytokine IL-2.2,110
Cyclosporine has been demonstrated to be effective in the treatment of patients with MG.111–116 The therapeutic latency is typically 1–3 months following treatment initiation.44 In psoriasis, the time of onset of action has been reported to be 6 weeks.117 Mean time to maximum improvement is approximately 7 months. Cyclosporine also appears to have a steroid-sparing effect. As many as 95% of patients are able to discontinue or decrease their corticosteroid dose.
There are a number of reports that describe cyclosporine as beneficial in CIDP patients.34,73,118–125 Like many reports of immunomodulating therapy in neuromuscular disorders, interpretation is confounded by its use in patients who have used other agents either concomitantly or previously. Cyclosporine may benefit patients with vasculitic neuropathy, both systemic and nonsystemic, sarcoid neuropathy, LEMS, dermatomyositis, and polymyositis.45,48,56,94,126–136 Its effectiveness in MMN is suspect.55 It is used by most clinicians as a third-line drug in patients unresponsive to other modalities.
Renal toxicity occurs in approximately a quarter of patients. The need to monitor creatinine and trough cyclosporine levels frequently has limited the enthusiasm of some clinicians for its use. Patients receiving cyclosporine or any calcineurin inhibitor may also develop a calcineurin inhibition syndrome that includes prominent and at times debilitating tremor requiring dose reduction. Cyclosporine is used in MG primarily in patients who are refractory to prednisone and azathioprine. There is a general perception that brand name drugs, for example, Neoral® or Sandimmune®, are preferable to their generic counterparts.
Initially a total dose of 3.0–4.0 mg/kg/day in two divided doses is used and gradually increased to a maximum dose of 6.0 mg/kg/day as necessary. The cyclosporine dose is initially being titrated to maintain trough serum cyclosporine levels of 100–200 ng/mL. Blood pressure, electrolytes and renal function, and trough cyclosporine levels need to be monitored on a monthly basis. The dose is lowered as necessary to keep the creatinine level stable while maintaining the trough level within therapeutic range. Any upward trend of creatinine levels should promote a dose reduction. After patients achieve maximum improvement, the dose is reduced over several months to the minimum dose necessary to maintain the therapeutic response. Patients need to be informed of the numerous drugs that can aggravate renal toxicity including nonsteroidal anti-inflammatory agents and drugs that may raise blood pressure or affect serum potassium levels.
Eculizumab is a humanized monoclonal antibody which acts by blocking the formation of terminal complement complex by specifically preventing the enzymatic cleavage of complement 5 (C5).137
Its labeled indications include paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome.
Eculizumab has been used infrequently in the treatment of autoimmune neuromuscular disease. Dermatomyositis, polymyositis, and myasthenia are the most notable examples.55,137,138 In a prospective, placebo-controlled trial of myasthenic patients refractory to other agents, eculizumab demonstrated clinically meaningful improvements in the treatment group and a larger phase 3 clinical trial is underway.137
The most common adverse effects reported in the prospective study of eculizumab and MG were nausea, back pain, nasopharyngitis, and headache.137 Renal insufficiency, anemia, leukopenia, dyspepsia, diarrhea, tachycardia, peripheral edema, fatigue, and both hypo- and hypertension have been reported. There is also an increased risk of meningococcal meningitis.
In MG, one described protocol is a 600-mg eculizumab infusion weekly for 4 weeks followed by a 900 mg maintenance infusion every 2 weeks for an additional six doses.137 Patients should receive meningococcal vaccination prior to its administration.
As the inflammatory response is dependent on inflammatory cytokines, and as TNF-α represents one of the major proinflammatory cytokines, it stands to reason that etanercept and other TNF-α inhibitors would be studied in the treatment of autoimmune neuromuscular disorders. Its labeled uses include rheumatoid arthritis, inflammatory spondyloarthropathy, and plaque psoriasis.
There is one report of etanercept benefitting patients with CIDP.139 Conversely, there are reports of patients developing neuropathy with features characteristic of CIDP in association with the use of TNF antagonists.140–143 Etanercept has also been suggested as a potential treatment in sarcoidosis but it has been reported to potentially cause sarcoidosis as well.48,83,144–146 A clinical trial of etanercept in MG reported modest benefits in five out of eight patients.147 Two patients experienced worsening however, and at least one patient has been reported to develop MG while receiving etanercept for rheumatoid arthritis.146 A small randomized trial suggested a steroid-sparing benefit for etanercept in dermatomyositis.148 It is our current perspective that etanercept should be used cautiously if at all in CIDP, MG, and sarcoid neuropathy.
In the etanercept study in refractory dermatomyositis patients, the six serious adverse events occurred in three participants comprising pregnancy and miscarriage in a partner; hospitalization for a urinary tract infection and fever of unknown origin; postherpetic neuralgia and two admissions for psychosis. Five participants in the etanercept-treated group compared to one in the placebo group had worsening of their skin disease.148
Other adverse effects other than the development of sarcoidosis that have been reported include anaphylaxis and other hypersensitivity reactions, positive antinuclear antibody titers with rare lupus-like syndromes, autoimmune hepatitis, rare central and peripheral nervous system demyelinating diseases including optic neuritis, transverse myelitis, multiple sclerosis, GBS and other demyelinating neuropathies, seizures or seizure exacerbation, heart failure or decreased left ventricular function, rare cases of pancytopenia and aplastic anemia, reactivation of infections notably hepatitis B, fungus, and tuberculosis. It is believed that the risk of these opportunistic infections is increased with concomitant use of corticosteroids or other agents. In addition, lymphoma has been reported in children and adolescent patients receiving TNF-blocking agents, including etanercept. Skin cancers, notably melanoma, nonmelanoma skin cancer, and Merkel cell carcinoma may develop in adults.
Etanercept is typically administered subcutaneously in a dose of 25 or 50 mg once or twice weekly. Etanercept’s therapeutic latency in psoriasis is estimated to be 6.6 and 9.5 weeks for high-dose and low-dose regimens respectively and is estimated at 2–6 months in MG.44,117 Surveillance for the numerous potential adverse effects listed above should be undertaken.
Infliximab is another TNF-α inhibitor that has been used sparingly in the treatment of immune-mediated neuromuscular disorders. Again, its potential mechanism of action is to suppress the effects of the proinflammatory cytokine TNF. Infliximab is approved for the treatment of inflammatory bowel disease psoriatic and rheumatoid arthritis, plaque psoriasis, and inflammatory spondyloarthropathy.
Infliximab has been reported to have a modest, suggested benefit in dermatomyositis and associated interstitial lung disease.149 Its use has been reported in both sarcoid and vasculitic neuropathy with uncertain benefit.48,56 We are aware of a single report describing infliximab’s use in myasthenia.150 Infliximab is one of the fastest acting immunomodulating agents. Its time of action in psoriasis is estimated to be 3.5 weeks.117
The most frequently occurring adverse effects have been reported to be headache, nausea, diarrhea, abdominal pain, increased transaminases, development or increased titres of antinuclear and double stranded DNA antibodies, abscess particularly in those with Crohn’s disease, upper respiratory tract infections, and an infusion-related reaction. Warnings concerning risk of malignancy, myelosuppression, and opportunistic infection with agents such as hepatitis B and tuberculosis are identical to etanercept. Like etanercept, the development of CIDP has been described to occur in concert with infliximab use.143
A typical regimen for infliximab infusion is 5 mg/kg at 0, 2, and 6 weeks, followed by 5 mg/kg every 8 weeks thereafter. In patients who initially respond but appear to develop tolerance, an increase to 10 mg/kg is permitted. In the absence of therapeutic effect, treatment is discontinued by 14 weeks.
INF β is a naturally occurring cytokine that downregulates inflammatory responses. Its biologic actions are copied by the recombinant protein INF β1a. INF α, bioengineered as INF α2a, is also a naturally occurring cytokine which upregulates the inflammatory response and it has been used primarily for the treatment of hepatitis C. Not surprisingly, it has been reported to cause autoimmune disease.151,152 Paradoxically, it has also been used to treat patients with autoimmune diseases, notably CIDP.125,153,154
In neuromuscular disease, INF β1 a has been utilized primarily in the treatment of CIDP.34,73,155–160 Although there are occasional case reports suggesting a beneficial effect, the weight of experience would not favor its use. In addition, in at least one case of concomitant multiple sclerosis and CIDP, the neuropathy worsened in response to INF β1 a.161 With INF α, beneficial effects in CIDP are once again difficult to ascertain because of confounding considerations such as monoclonal proteins, use of other agents concomitantly, or the tendency to study patients who have been historically resistant to other treatments. Although benefit has been reported in occasional patients, at times dramatically, the use of either INF cannot be currently endorsed in CIDP.125,153,154
INF β1 a is well tolerated. Potential side effects include minor alterations of liver function and white cell counts. With subcutaneous preparations, skin reactions occur but serious side effects are uncommon. With INF α2 a, minor side effects such as fatigue, fever, malaise and myalgia, and arthralgia occur frequently.34
INF β1 a is typically given at a dose of 22–44 ug via subcutaneous injection three times a week. INF α2 a is also delivered subcutaneously three times a week, typically at a dose of 3 million IU.34
IVIg is a blood product collected from thousands of donors composed of 95% IgG and less than 2.5% IgA. In addition, CD4, CD8, HLA molecules, and other plasma components are typically included.16 IVIg’s half-life is estimated at 18–33 days but some of its beneficial effects seem to extend beyond this period.16,162 There is heterogeneity in different IVIg products pertaining to IgA and sodium content, type of sugar, pH and osmolality, and viral inactivation strategies employed.162
There have been multiple proposed IVIg mechanisms of action, one or more of which may be relevant depending on the pathophysiology of the treated disease.163,164 Anti-idiotype antibodies contained within IVIg may react with the Fc or antigen-binding regions of pathologic autoantibodies and neutralize their effects. IVIg including nonimmunoglobulin components may beneficially interfere with T-cell function in a number of ways. It may restore the balance between T cells releasing proinflammatory Th1 and anti-inflammatory Th2 cytokines. It may cause T-cell apoptosis, interfere with T-cell interaction with antigen-presenting cells, and interrupt T-cell migration through the blood–nerve barrier. Interference with B-cell function including production of autoantibodies, activation of complement, formation of membrane attack complex, and macrophage function are other proposed mechanisms of action.16,162
In GBS, IVIg is typically administered as five daily doses of 0.4 mg/kg of ideal body weight, initiated as soon as possible, and preferably within the first 2 weeks of symptoms. There is currently no evidence-based support for repeat administration although we would consider this if a patient demonstrated unequivocal improvement in response to the initial 2 g/kg regimen, and then subsequently relapsed. Some neuromuscular clinicians advocate waiting for a specified threshold of morbidity such as compromised ambulation to occur before initiating treatment. Our practice is to be more aggressive, as we are not confident in our ability to predict the natural history in any individual patient while at the same time adhering to the premise that nerve injury is easier to prevent than heal.
In CIDP, we frequently use IVIg as our first-line treatment beginning with single 5-day infusion totaling 2 g/kg of IBW. If there is no response after 1 month, we will prescribe two additional monthly IVIg courses and then disband if no significant benefit occurs. If there is a meaningful response, we will continue to observe until the benefit plateaus. As rare CIDP patients have a monophasic course, we offer no further treatment unless relapse occurs in a patient who normalizes after a single 5-day infusion.165 If the patient improves partially but incompletely, we then initiate a maintenance program. Three commonly used protocols are 0.5 g/kg every 2 weeks, 1 g/kg every 3 weeks, or 2 g/kg every month. In CIDP patients requiring maintenance IVIg treatment, we attempt to minimize the dose, and maximize the interval between doses attempting to sustain the maximal degree of achievable improvement. As the symptoms in relapsing CIDP may return insidiously allowing time for successful intervention to take place, we often discontinue treatment in individuals in whom we can achieve a drug-free remission.
Our use of IVIg in MMN differs somewhat from CIDP. The monophasic course seen in some CIDP patients does not seem to occur with the same frequency in MMN. Although spontaneous remission in individual nerve injury has been reported, we are unaware of reported self-limited cases with protracted remission. In our experience, relapses may be abrupt and severe. Reversal of deficits may be difficult if treatment is delayed, presumably due to transition to axon loss that appears to occur frequently in untreated patients.166,167,15 The long-term natural history of untreated cases appears unfavorable.15 As a result, we typically maintain patients on chronic treatment indefinitely in the hope of preventing these unpredictable events.15
We use IVIg in myasthenia predominantly in two situations, in crisis or in preparation for thymectomy in patients who we feel that improvement in their strength will reduce risk of postoperative complications.168 In general, our experience, like others, suggests that PLEX is somewhat superior to IVIg in the management of myasthenic crises.169 We acknowledge however, that the preponderance of literature views IVIg and PLEX to be of equivalent efficacy in this context.168–192
IVIg has also been demonstrated to be efficacious in the treatment of dermatomyositis. A double-blinded, placebo-controlled study demonstrated significant efficacy in patients who were already utilizing prednisone. All eight of the patients initially randomized to IVIg demonstrated significant improvement in strength and neuromuscular symptoms as did 9 out of /12 patients subsequent to crossover.193 The tendency to use IVIg preferably is in consideration of the relative ease of use comparatively and reduced side effect profile for IVIg.172 In general, we attempt to avoid both IVIg and PLEX in chronic disorders like myasthenia, dermatomyositis, and polymyositis unless a satisfactory response cannot be achieved by oral agents. Our position is in consideration of patient lifestyle, cost, and safety. If we use these agents as maintenance therapy in these diseases, we ascribe to a regimen similar to that described in CIDP above. We rarely employ IVIg for those disorders listed in the disorders with anecdotal reports of benefit column in Table 4-1.
Minor infusion-related symptoms such as chills, nausea, myalgia, headache, and vasomotor disturbances are fairly common with IVIg infusion. In an attempt to avoid this reaction, we typically pretreat our patients with 650 mg of acetaminophen and 25 mg of diphenhydramine orally. If an allergic reaction is experienced, we add 100 mg of intravenous hydrocortisone to the pretreatment regimen prior to every infusion.
Serious side effects from IVIg are rare and include thromboembolic disease such as stroke and myocardial infarction, renal failure, aseptic meningitis, congestive heart failure, and anaphylactic reactions.16,162 The incidence of serious side effects has been reported at 4.5% although this has not been our experience.194 Thromboembolic risk may be higher in those with atherosclerotic cardiovascular risk factors or in those with monoclonal proteins or other reasons for increased blood viscosity. Aseptic meningitis has been reported to occur in up to 10% of patients although this also has not been our experience.16 It does not appear to be related to a particular IVIg formulation(s), occurs more frequently in migraineurs, and does not appear to be preventable with steroid pretreatment. Rechallenging patients who have previously experienced IVIg-induced aseptic meningitis should probably be avoided unless there are particularly strong indications for its use. Rechallenge appears to have a greater success in patients without a history of migraine.16 As IVIg represents a pooled blood product, a theoretical risk of infection continues to exist. Historically, one case of transmitted hepatitis C was reported in 1994. Subsequent exposure of all IVIg products to solvent detergent of 10% caprylate have seemingly eliminated this and other viral risks.16
Anaphylaxis has been reported in patients deficient in IgA. The initial suggestion that this risk exists from IgG autoantibodies directed at IgA autoantibodies has been refuted by safe infusion in IgA deficient, IgG versus IgA autoantibody-carrying patients. Recent evidence suggests that IgE versus IgA is the more likely causative pathophysiology of this reaction. As the incidence of anaphylaxis in patients receiving IVIg is extremely uncommon, screening for IgA deficiency is not recommended as a routine practice prior to IVIg exposure.16,162
Other consequences of IVIg treatment of note include elevation of serum inflammatory markers such as erythrocyte sedimentation rates which may exceed 100 mm/h which may confound patient management. Following IVIg infusion, determination of a patient’s actual immunity toward certain diseases may also be confounded.16 Antibodies detected in a patient’s serum may represent those passively transferred rather than one’s generated by the patient’s own immune system.
The standard induction dose of IVIg is 2 g/kg typically delivered in 1 g/kg doses on two consecutive days in stable outpatients or 0.4 g/kg doses on 5 consecutive days on intolerant or fragile patients or those who will be hospitalized for that period of time. Increasingly, recommended dosing is based on IBW. For men greater than or equal to 60 in in height, this figure is calculated by the formula 50 kg + (2.3 × height in inches -60). If the height is less than 60 in, 50 kg is used as the IBW. For women the formula is identical other than 46 kg is substituted for 50 kg, both as the initial number of the formula, and as the fallback IBW if patient height is less than 5 ft.
The therapeutic onset for IVIg effect in MG is estimated to be 1–2 weeks.162 As suggested above, the onset latency for IVIg may be equally fast if the damaged structure is an ion channel but may be more protracted if myelin, myofiber, or particularly axonal regeneration is required. The duration of effect for IVIg is variable between both disease and patient. As a general rule, IVIg half-life predicts a 4–6 weeks’ benefit with responses of up to 60 days reported.170 In consideration of the half-life of the agent wishing to neither overtreat nor allow relapse, we begin maintenance treatment at 4-week intervals. IVIg may also be successfully delivered subcutaneously, notably in CIDP.195,196
Subsequent dosing, as described above, is dependent on clinical context, taking into consideration the specific disease and its natural history as well as disease pathophysiology. Knowing what the likely outcome is in any given disease and the time it takes to for that improvement to become evident is critical to rational decision making. For example, the majority of outcome improvements in GBS have been measured at a month or more after treatment. Initiating IVIg after PLEX, or a second course of IVIg within the first month due to the perception that the patient has not improved sufficiently, cannot be routinely supported, based either on clinical experience or clinical trials.197
Methotrexate, which also mainly affects the G1 and S phases of the cell cycle, is a folate antagonist that can inhibit de novo synthesis of both purines and pyrimidines.2 It is a selective inhibitor of both dihydrofolate reductase and lymphocyte proliferation. As a structural analog of folic acid, it affects adenosine-mediated inflammatory mediators resulting in apoptosis and clonal deletion of T-cell lines. It also acts to decreases the production of proinflammatory cytokines IL-1 and IL-6.
In neuromuscular disease, methotrexate is probably used with the greatest frequency in the treatment of dermato- and polymyositis.1,45,55,78,127,198–203 It is our suspicion that this is based on the frequency with which these disorders are treated by rheumatologists who are perhaps more comfortable with its use than neurologists, rheumatologists not as frequently involved in the treatment of other neuromuscular conditions.
There are reports of methotrexate being utilized as a second or third treatment in CIDP, systemic and nonsystemic vasculitic neuropathy, sarcoidosis, MMN, myasthenia, and immune-mediated necrotizing myopathy.2,34,46–48,54,56,94,125,204–210 One advantage of methotrexate is its relatively short onset latency in comparison to other immunomodulating agents. Although reported in reference to the treatment of psoriasis, its beneficial effects have been measured to occur with a mean time (in 25% of treated individuals) of 3.2 weeks with high doses and 9.9 weeks with low doses.117
Potential side effects of methotrexate therapy include hepatotoxicity which is relatively uncommon, interstitial pneumonitis, which presents with dyspnea, fever, and dry cough and potentially leads to pulmonary fibrosis, infection, neoplasia, infertility, bone marrow suppression with leukopenia, alopecia, gastric irritation with nausea, vomiting and diarrhea, fatigue, rash, dizziness, ulcerative stomatitis, and teratogenicity.211 Like most immunosuppressant medications, the exact incidence of neoplasia and its causal relationship to methotrexate is difficult to quantitate.2
Methotrexate is typically initiated orally at 7.5 mg/week or lower in the context of renal insufficiency. It can be given in a single dose or in divided doses. One regimen is three divided doses administered 12 hours apart, often Saturday morning and evening and Sunday morning so that any side effects might have a chance to dissipate before the work week begins. The total dose may be gradually escalated dependent on the development of beneficial or adverse effect to a maximal dose of 25 mg/week. All patients are concomitantly treated with folate, at least 5 mg/week. Methotrexate can also be delivered subcutaneously once weekly at a dose of 20–50 mg.
Hepatic enzymes, platelet, and white blood cell counts should be monitored closely. In patients with pulmonary symptoms, methotrexate should be held until an infectious cause and pulmonary fibrosis can be excluded. Chest imaging, pulmonary function testing, and pulmonary consultation are recommended in symptomatic patients.
MMF (Cellcept) inhibits lymphocytic purine synthesis by selectively inhibiting the enzyme inosine-5′-monophosphate dehydrogenase. Like azathioprine, it acts predominantly on the G1 and S phases of the cell cycle.2 Its use results in T-cell and B-cell depletion.212
Within the spectrum of neuromuscular disorders, mycophenolate has been most diligently studied in MG.213–223 Initial open-label studies suggested notable benefit in an estimated 75% of treated individuals, primarily as an adjunctive therapy.216,218,221 Subsequent prospective studies have not been as supportive.213,214 Mycophenolate remains a frequently utilized “steroid-sparing” agent in the treatment of MG as many neuromuscular clinicians feel that it is an effective agent, an impression supported by a study reported in a retrospective 2010 analysis of 102 patients. There is also the perception that the negative studies were flawed due to their relatively short periods of observation.222
There have been eight studies suggesting a benefit for mycophenolate in the treatment of CIDP.221,224–229 Interpretation of these studies is confounded by the numerous other treatment variables that many of these patients were exposed to. There have been case reports or case series suggesting benefit of mycophenolate treatment in systemic and nonsystemic vasculitic neuropathy, sarcoid neuropathy and myositis, immune-mediated necrotizing myopathy, and Isaac’s syndrome.45–48,51–54,56,94,207,208
MMF is usually well tolerated and has little or no renal or liver toxicity. The major side effect is diarrhea. Starting slowly and increasing the dose slowly may diminish the risk and severity of this troublesome side effect. Less common side effects include abdominal discomfort, nausea, peripheral edema, fever, and leukopenia. Measurement of drug levels is not done routinely. Adverse hematologic effects may be more common in doses of greater than 2,000 mg/day. In view of the relatively short experience with this agent, long-term safety for mycophenolate is still in question. Malignancy rates do not appear higher in the transplant population; however, there are rare reports of lymphoma or lymphoproliferative disorders developing in MG patients.219,230 Progressive multifocal encephalopathy has been reported in a patient receiving mycophenolate for systemic lupus erythematosus and organ transplantation.231
MMF, available in 500 mg tablets, is typically dosed at 1,000 mg twice a day. Starting at 500 mg daily or twice a day and gradually increasing the dose may diminish the incidence and severity of diarrhea. The maximal recommended dose is 1,500 mg BID. Patients who do not respond to a daily dose of 2,000 mg but respond to higher doses are relatively uncommon in our experience. Improvement has been noted in MG with a mean therapeutic latency of 9–11 weeks with a maximal effect noted by 6 months in most patients. Occasionally, the full effect may not become manifest before a year.231 Mycophenolate is excreted through the kidneys; therefore, the dose should be decreased (no more than 1 g/day total dose) in patients with renal insufficiency.
Mycophenolic acid or mycophenolate sodium (Myfortic®) is an alternative preparation that comes in 180 and 360 mg enteric-coated tablets. As a result, it has the potential for less gastrointestinal side effects. Unlike MMF, it is not available for parenteral use. The daily dose is 720–1,440 mg/day in two divided doses.
Therapeutic PLEX reduces the titer of circulating antibodies within the blood stream through filtration.232
The most established uses for PLEX in NM disease is in GBS, CIDP, and MG.169,170,172,175,183,184,187–192,197,233–236 Once again, because of cost, safety, and logistical considerations, PLEX is used primarily in initial induction treatment, not in chronic maintenance therapy. In MG, it is used primarily in patients in crisis or in those with moderate weakness prior to thymectomy in order to maximize their perioperative strength and minimize risk of postoperative complications. The American Academy of Neurology was unable to endorse the use of PLEX as treatment for MG in its evidence-based guideline.233 In the mind of many neuromuscular clinicians however, including ourselves, it is as effective if not more effective than IVIg in the treatment of MG crisis.4,231
There are reports that provide convincing support for the use of PLEX in association with IgA and IgG monoclonal protein of uncertain significance.233,239 There are individual or small case series that suggest potential benefit in diabetic radiculoplexus neuropathy, systemic and nonsystemic vasculitic neuropathy, Miller Fisher, stiff person and Isaac’s syndrome.46,47,51–54,56,232,240,241
There are reports suggesting that PLEX is both effective and ineffective for LEMS and inflammatory myopathies.49,55,232 PLEX appears to have no role in the treatment of MMN and DADS neuropathy.15,55,77,233
PLEX has several limitations. It has limited availability and like IVIg, cost is an issue. Many of the risks related to PLEX are related to the need for large bore catheters that need to be placed in central veins in a significant portion of individuals. Potential complications include symptoms related to alkalosis, pneumothorax, hypotension particularly in GBS patients prone to dysautonomia, sepsis, and pulmonary embolism.233
The standard PLEX protocol is to exchange a volume of 200–250 mL/kg/day (2–3 L) for 5 days typically spread out over a 7–10-day period of time. In MG, improvement is noticeable after two to four exchanges.232 The durability of the effect is usually a few weeks.
Rituximab is a monoclonal antibody directed against CD20 B cells, developed for the treatment of B-cell lymphomas. It produces B-cell depletion, postulated to occur via complement-mediated cytotoxicity, antibody-dependent cell-mediated cytotoxicity, and induction of apoptosis.2 It also reduces T-lymphocyte activation and decreases cytokine production.36
There are an increasing number of reports suggesting that both AChR and MuSK MG respond favorably.242–248 Other reports are somewhat more circumspect.249–251
In consideration of rituximab’s efficacy in lymphoproliferative disorders, it is rational to consider its use in neuromuscular conditions associated with monoclonal proteins. DADS, an acquired, demyelinating sensory predominant neuropathy is one such disorder. It is frequently associated with an IgM kappa monoclonal protein, and frequently associated with myelin-associated glycoprotein autoantibodies. Initial reports were optimistic but subsequent, randomized, placebo-controlled trials found no benefit.252 Others have reported disease worsening following rituximab exposure.253 Our experience is that most patients do not respond, with rare patients responding dramatically in a manner that contrasts significantly with the natural history of this disorder. Whether the response is durable in these uncommon patients is uncertain.
There are a handful of case reports and small case series describing a benefit of rituximab in CIDP.125,254–265 Again these are difficult to interpret as the majority of the cases are confounded by the use of other agents, the presence of monoclonal proteins or concurrent diseases like lymphoma.
There were also initial optimistic reports of rituximab in MMN but our experience and that of others has been disappointing.55 The use of rituximab has also been reported in vasculitic neuropathy and stiff person syndrome but convincing evidence of its efficacy in these and other immune-mediated neuromuscular diseases remains lacking.34,46,47,56,207,208,265–268
Although a large randomized, placebo-controlled trial of rituximab showed no benefit in adults and children with polymyositis and dermatomyositis, there were many methodologic issues in regard to trial design that may have impeded the detection of efficacy.269 We and others have used rituximab in myositis patients (non-IBM) and have found it particularly effective in patients with immune-mediated necrotizing myopathy.270
Rituximab is well tolerated by the majority of patients. Pruritus, nausea, vomiting, dizziness, headache, angina, cardiac dysrhythmia, anemia, leukopenia, and thrombocytopenia have been reported. Myelosuppression, PML, and lymphoma are uncommon but potential risks.23
There are a number of dosing regimens utilized for rituximab. Our experience has been with 375 mg/m2 infused weekly for 4 consecutive weeks. Pretreatment with 650 mg of acetaminophen and 25 mg of diphenhydramine by mouth is suggested although the drug is well tolerated by most individuals. The therapeutic latency for rituximab in myasthenics appears to be about a month but is undoubtedly longer for some disorders.44 We have seen patients respond for 12–24 months before reinfusion is required. Our approach has been to wait at least 6 months before reinfusing and then only if initial improvement and then subsequent clinical deterioration takes place.
Tacrolimus binds to the FK506-binding protein (FKB), forming the tacrolimus–FKB complex, which also binds to and blocks calcineurin. Although cyclosporine and tacrolimus bind to different target molecules, both drugs inhibit T-cell activation in the same manner and as calcineurin inhibitors, reduce production of the proinflammatory cytokine IL-2.2,110 As is the case with cyclosporine, these are two immunomodulating drugs developed for and used by the organ transplant community.
Tacrolimus, like cyclosporine, has been undoubtedly used most frequently in MG within the spectrum of neuromuscular disease.110,116,223,271–279 It has been used either as monotherapy, or more commonly in conjunction with thymectomy or other therapies. Both favorable and equivocal results are reported.271,273 We are aware of reported benefit in dermatomyositis/polymyositis and case reports of suggesting benefit in CIDP and vasculitic neuropathy.56,280 Its use has been suggested in both Isaac’s syndrome and LEMS without convincing literature support to our knowledge.136
Tacrolimus has similar toxicities to cyclosporine. It tends to be less nephrotoxic but more inclined to cause hyperglycemia.44 Sirolimus has less renal toxicity than tacrolimus or cyclosporine. In view of this, our anecdotal experience with it in couple of patients with refractory MG has demonstrated no apparent benefit.
Tacrolimus has been prescribed for myasthenic patients at a dosage of 0.1 mg/kg/day in two divided doses, and subsequently adjusted for plasma drug concentrations between 7 and 8 mg/mL.116 Tacrolimus has been reported to benefit some who have failed to respond or become refractory to the effects of cyclosporine.
1. Drachman DB. Immunotherapy in neuromuscular disorders: Current and future strategies. Muscle Nerve. 1996;19:1239–1251.
2. Sathasivam S. Steroids and immunosuppressant drugs in myasthenia gravis. Nat Clin Pract Neurol. 2008;4(6):317–327.
3. Taylor AL, Christopher CJ, Bradley JA. Immunosuppressive agents in solid organ transplantation: Mechanisms of action and therapeutic efficacy. Crit Rev Oncol Hematol. 2005;56:23–46.
4. Khatri BO, McQuillen MP, Kaminski M, et al. Evidence-based guideline update: Plasmapheresis in neurologic disorders. Neurology. 2011;77:e101–e103.
5. Keung B, Robeson KR, DiCapua DB, et al. Long-term benefit of rituximab in MuSK autoantibody myasthenia gravis patients. J Neurol Neurosurg Psychiatry. 2013;84(12):1407–1409.
6. Rahier KF, Moutschen M, Van Gompel A, et al. Vaccinations in patients with immune-mediated inflammatory diseases. Rheumatology. 2010;49:1815–1827.
7. Hoes JN, Hacobs JW, Boers M, et al. EULAR evidence-based recommendations on the management of systemic glucocorticoid therapy in rheumatic diseases. Ann Rheum Dis. 2007;66:1560–1567.
8. Sharshar T, Chevret S, Mazighi M, et al. Validity and reliability of two muscle strength scores commonly used as endpoints in assessing treatment of myasthenia gravis. J Neurol. 2000;247:286–290.
9. Cederbaum J, Stambler N, Malta E, et al. The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. J Neurol Sci. 1999;169:13–21.
10. Dyck PJ, Davies JL, Litchy WJ, O’Brien PC. Longitudinal assessment of diabetic polyneuropathy using a composite score in the Rochester Diabetic Neuropathy Study Cohort. Neurology. 1997;49:229–239.
11. Gajdos P, Sharshar T, Chevret S. Standards of measurements in myasthenia gravis. Ann N Y Acad Sci. 2003;998:445–452.
12. Barohn RJ, McIntire D, Herbelin L, Wolfe GI, Nations S, Bryan WW. Reliability testing of the quantitative myasthenia gravis score. Ann N Y Acad Sci. 1998;841:769–772.
13. Bedlack RS, Simel DL, Bosworth H, Samsa G, Tucker-Lipscomb B, Sanders DB. Quantitative myasthenia gravis score: Assessment of responsiveness and longitudinal validity. Neurology. 2005;64(11):1968–1970.
14. Simon NG, Ayer G, Lomen-Hoerth C. Is IVIg therapy warranted in progressive lower motor neuron syndromes without conduction block? Neurology. 2013;81(24):2116–2020.
15. Nobile-Orazio E, Cappellari A, Priori A. Multifocal motor neuropathy: Current concepts and controversies. Muscle Nerve. 2005;31:663–680.
16. Ruzhansky K, Brannagan TH III. Intravenous immunoglobulin for treatment of neuromuscular disease. Neurol Clin Pract. 2013;440–446.
17. Thomas CF, Limper AH. Pneumocystis pneumonia. N Engl J Med. 2004;350:2487–2498.
18. Kelly DM, Cronin S. PCP prophylaxis with use of corticosteroids by neurologists. Pract Neurol. 2014;14:74–76.
19. Neumann S, Krause SW, Maschmeyer G, Schiel X, von Lilienfeld-Toal M. Primary prophylaxis of bacterial infections and Pneumocystis jirovecii pneumonia in patients with hematological malignancies and solid tumors. Ann Hematol. 2013;92:433–442.
20. www.rrnmf.com. Accessed on March 24, 2012.
21. Horsburgh CR Jr, Rubin EJ. Latent tuberculosis infection in the United States. N Engl J Med. 2011;364:1441–1448.
22. Wingerchuk DM, Carter JL. Multiple sclerosis: Current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc. 2014;89(2):225–240.
23. Zaheer F, Berger JR. Treatment-related progressive multifocal leukoencephalopathy: Current understanding and future steps. Ther Adv Drug Safe. 2012;3(5):227–239.
24. Takao M. Targeted therapy and progressive multifocal leukoencephalopathy (PML): PML in the era of monoclonal antibody therapies. Brain Nerve. 2013;65(11):1363–1374.
25. Basile A, Simzar S, Bentow J, et al. Disseminated Strongyloides stercoralis: Hyperinfection during medical immunosuppression. J Am Acad Derm. 2010;63(5):896–902.
26. Santiago M, Leitão B. Prevention of strongyloides hyperinfection syndrome: A rheumatological point of view. Eur J Intern Med. 2009;20(8):744–788.
27. Iriemenam NC, Sanyaolu AO, Oyibo WA, Fagbenro-Beyioku AF. Strongyloides stercoralis and the immune response. Parasitol Int. 2010;59:9–14.
28. Riminton DS, Hartung HP, Reddel SW. Managing the risks of immunosuppression. Curr Opin Neurol. 2011;24:217–223.
29. Ringel I, Zettl UK. Intravenous immunoglobulin therapy in neurological disease during pregnancy. J Neurol. 2006;253 (suppl 5):v70–v74.
30. Gutierrez-Dalmau A, Campistol JM. Immunosuppressive therapy and malignancy in organ transplant recipients: A systematic review. Drugs. 2007;67(8):1167–1198.
31. Dahlke E, Murray CA, Kitchen J, Chan AW. Systematic review of melanoma incidence and prognosis in solid organ transplant recipients. Transplant Res. 2014;3:10.
32. Hirst C, Raasch S, Llewelyn G, Robertson N. Remission of chronic inflammatory demyelinating polyneuropathy after alemtuzumab. J Neurol Neurosurg Psychiatry. 2006;77(6):800–802.
33. Marsh EA, Hirst CL, Llewelyn JG, et al. Alemtuzumab in the treatment of IVIG-dependent chronic inflammatory demyelinating polyneuropathy. J Neurol. 2010;257(6):913–919.
34. Mahdi-Rogers M, van Doorn PA, Hughes RA. Immunomodulatory treatment other than corticosteroids, immunoglobulin and plasma exchange for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 2013;6:CD003280.
35. Kissel JT, Levy RJ, Mendell JR, Griggs RC. Azathioprine toxicity in neuromuscular disease. Neurology. 1986;36:35–39.
36. Sanders DB, Evoli A. Immunosuppressive therapies in myasthenia gravis. Autoimmunity. 2010;43(5–6):428–435.
37. Gajdos P, Elkharrat D, Chevret S, Chastang C. A randomized clinical trial comparing prednisone and azathioprine in myasthenia gravis. Results of the second interim analysis. J Neurol Neurosurg Psychiatry. 1993;56:1157–1163.
38. Bromberg MB, Wald JJ, Forshew DA, Feldman EL, Albers JW. Randomized trial of azathioprine or prednisone for initial immunosuppressive treatment of myasthenia. J Neurol Sci. 1997;150:59–62.
39. Palace J, Newsom-Davis J, Lecky B. A randomized double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Neurology. 1998;50:1778–1783.
40. Hohlfeld R, Michels M, Heininger K, Besinger U, Toyka KV. Azathioprine toxicity during long-term immunosuppression of generalized myasthenia gravis. Neurology. 1988;38:258–261.
41. Myasthenia Gravis Clinical Study Group. A randomized clinical trial comparing prednisone and azathioprine in myasthenia gravis. Results of the second interim analysis. J Neurol Neurosurg Psychiatry. 1993;56:1157–1163.
42. Herrllinger U, Weller M, Dichgans J, Melms A. Association of primary central nervous system lymphoma with long-term azathioprine therapy for myasthenia gravis. Ann Neurol. 2000;47:682–683.
43. Sanders DB, Scoppetta C. The treatment of patients with myasthenia gravis. Neurol Clin. 1994;12:342–368.
44. Silvestri NJ, Wolfe GI. Treatment-refractory myasthenia gravis. J Clin Neuromuscul Dis. 2014;15(4):167–178.
45. Amato AA, Greenberg SA. Inflammatory myopathies. Continuum (Minneap Minn). 2013;19(6 Muscle Disease):1615–3163.
46. Collins MP, Periquet MI, Mendell JR, Sahenk Z, Nagaraja HN, Kissel JT. Nonsystemic vasculitic neuropathy: Insights from a clinical cohort. Neurology. 2003;61:623–630.
47. Collins MP, Dyck PJ, Gronseth GS, et al. Peripheral Nerve Society: Guideline on the classification, diagnosis, investigation, and immunosuppressive therapy of non-systemic vasculitic neuropathy: Executive summary. J Periph Nerv Syst. 2010; 15:176–184.
48. Lacomis D. Neurosarcoidosis. Curr Neuropharmacol. 2011;9:429–436.
49. Maddison P. Treatment in Lambert-Eaton myasthenic syndrome. Ann N Y Acad Sci. 2012;1275:78–84.
50. Krendel DA, Costigan DA, Hopkins LC. Successful treatment of neuropathies in patients with diabetes mellitus. Arch Neurol. 1995;52:1053–1061.
51. Alessi G, De Reuck J, De Bleecker J, Vancayzeele S. Successful immunoglobulin treatment in a patient with neuromyotonia. Clin Neurol Neurosurg. 2000;102:173–175.
52. Ishii A, Hayashi A, Ohkoshi N, et al. Clinical evaluation of plasma exchange and high dose intravenous immunoglobulin in a patient with Isaacs’ syndrome. J Neurol Neurosurg Psychiatry. 1994;57:840–842.
53. Van den Berg JS, van Engelen BG, Boerman RH, de Baets MH. Acquired neuromyotonia: Superiority of plasma exchange over high-dose intravenous immunoglobulin [letter]. J Neurol. 1999;246:623–625.
54. Jinka M, Chaudhry V. Treatment of multifocal motor neuropathy. Curr Treat Opinions Neurol. 2014;16(2):269.
55. Gordon PA, Winer JB, Hoogendijk JE, Choy EH. Immunosuppressant and immunomodulatory treatment for dermatomyositis and polymyositis. Cochrane Database Syst Rev. 2012;8:CD003643.
56. Gwathney KG, Burns TM, Collins MP, Dyck PJ. Vasculitic neuropathy. Lancet Neurol. 2014;13:67–82.
57. Gomes JA, Stevens RD, Lewin JJ III, Mirski MA, Bhardwaj A. Glucocorticoid therapy in neurologic critical care. Crit Care Med. 2005;33:1214–1224.
58. McDaneld LM, Fields JD, Bourdette DN, Bhardwaj A. Immunomodulatory therapies in neurologic critical care. Neurocrit Care. 2010;12(1):132–143.
59. Bromberg MB, Carter O. Corticosteroid use in the treatment of neuromuscular disorders: Empirical and evidence-based data. Muscle Nerve. 2004;30:20–37.
60. Johns TR. Long-term corticosteroid treatment of myasthenia gravis. Ann N Y Acad Sci. 1987;505:568–583.
61. Miller RG, Milner-Brown HS, Mirka A. Prednisone-induced worsening of neuromuscular function in myasthenia gravis. Neurology. 1986;36:729–732.
62. Panegyres PK, Squire M, Newsom-Davis J. Acute myopathy associated with large parenteral dose of corticosteroid in myasthenia gravis. J Neurol Neurosurg Psychiatry. 1993;56:702–704.
63. Lindberg C, Andersen O, Lefvert AK. Treatment of myasthenia gravis with methylprednisolone pulse: A double-blind study. Acta Neurol Scand. 1998;97:370–373.
64. Eftimov F, Winer JB, Vermeulen M, de Haan R, van Schaik IN. Intravenous immunoglobulin for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev 2013;12:CD001797.
65. Nobile-Orazio E. Chronic inflammatory demyelinating polyradiculoneuropathy and variants: Where we are and where we should go. J Peripher Nerve Syst. 2014;19(1):2–13.
66. Hughes RA, Mehndiratta MM. Corticosteroids for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 2012;8:CD002062.
67. Dyck PJ, Lais AC, Ohta M, Bastron JA, Okazaki H, Groover RV. Chronic inflammatory polyradiculoneuropathy. Mayo Clin Proc. 1975;50:621–637.
68. Dyck PJ, O’Brien P, Swanson C, Low P, Daube J. Combined azathioprine and prednisone in chronic inflammatory demyelinating polyneuropathy. Neurology. 1985;35:1173–1176.
69. Lewis RA, Sumner AJ, Brown MJ, Asbury AK. Multifocal demyelinating neuropathy with persistent conduction block. Neurology. 1982;32:958–964.
70. Viala K, Renie´ L, Maisonobe T, et al. Follow-up study and response to treatment in 23 patients with Lewis-Sumner syndrome. Brain. 2004;127:2010–2017.
71. Börü ÜT, Erdoğan H, Alp R, et al. Treatment of chronic inflammatory demyelinating polyneuropathy with high dose intravenous methylprednisolone monthly for five years: 10-year follow up. Clin Neurol Neurosurg. 2014;118:89–93.
72. van Schaik IN, Eftimov F, van Doorn PA, et al. Pulsed high-dose dexamethasone versus standard prednisolone treatment for chronic inflammatory demyelinating polyradiculoneuropathy (PREDICT study): A double-blind, randomised, controlled trial. Lancet Neurol. 2010;9(3):245–253.
73. Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society – First Revision. J Peripher Nerv Syst. 2010;15:1–9.
74. Katz JS, Saperstein DS, Gronseth G, Amato AA, Barohn RJ. Distal acquired demyelinating symmetric neuropathy. Neurology. 2000;54:615–620.
75. Hughes RA, Newsom-Davis JM, Perkin GD, Pierce JM. Controlled trial of prednisolone in acute polyneuropathy. Lancet. 1978;2:750–753.
76. Hughes RA. Ineffectiveness of high-dose intravenous methylprednisolone in Guillain-Barré syndrome. Lancet. 1991;338:1142.
77. Feldman EL, Bromberg MB, Albers JW, Pestronk A. Immunosuppressive treatment in multifocal motor neuropathy. Ann Neurol. 1991;30:397–401.
78. Joffe MM, Love LA, Leff RL, et al. Drug therapy of the idiopathic inflammatory myopathies: Predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med. 1993;94(4):379–387.
79. Lotz BP, Engel AG, Nishino H, Stevens JC, Litchy WJ. Inclusion body myositis. Observations in 40 patients. Brain. 1989;112:727–747.
80. Miloslavsky EM, Specks U, Merkel PA, et al. Clinical outcomes of remission induction therapy for severe antineutrophil cytoplasmic antibody-associated vasculitis. Arth Rheum. 2013;65(9):2441–2449.
81. Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363:211–220.
82. Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–232.
83. Burns TM, Dyck PJ, Aksamit AJ, Dyck PJ. The natural history and long-term outcome of 57 limb sarcoidosis neuropathy cases. J Neurol Sci. 2006;244(1–2):77–87.
84. van Eijk JJ, van Alfen N, Berrevoets M, van der Wilt GJ, Pillen S, van Engelen BG. Evaluation of prednisolone treatment in the acute phase of neuralgic amyotrophy: An observational study. J Neurol Neurosurg Psychiatry. 2009;80(10):1120–1124.
85. Dalakas MC. Advances in the pathogenesis and treatment of patients with stiff person syndrome. Curr Neurol Neurosci Rep. 2008;8(1):48–55.
86. Howard FM, Duane DD, Lambert EH, Daube JR. Alternate-day prednisone: Preliminary report of a double-blind controlled study. Ann N Y Acad Sci. 1976;274:596–697.
87. Rothman MS, West SG, McDermott MT. Osteoporosis for the practicing neurologists. Neurol Clin Pract. 2014;4:34–43.
88. Adachi JD, Bensen WG, Brown J, et al. Intermittent etidronate therapy to prevent corticosteroid-induced osteoporosis. N Engl J Med. 1997;337:382–387.
89. Pereira RM, Carvalho JF, Paula AP, et al. Guidelines for the prevention and treatment of glucocorticoid-induced osteoporosis. Rev Bras Rheumatol. 2012;52(4):580–593.
90. Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. N Engl J Med. 1998;339:292–292.
91. Adu D, Pall A, Luqmani RA, et al. Controlled trial of pulse versus continuous prednisolone and cyclophosphamide in the treatment of vasculitis. Q J Med. 1997;90:401–409.
92. Guillevin L, Cordier JF, Lhote F, et al. A prospective, multicenter, randomized trial comparing steroids and pulse cyclophosphamide versus steroids and oral cyclophosphamide in the treatment of generalized Wegener’s granulomatosis. Arthritis Rheum. 1997;40:2187–2198.
93. Gorson KC. Therapy for vasculitic neuropathies. Curr Opinion Treat Neurology. 2006;8(2):105–117.
94. Vrancken AF, Hughes RA, Said G, Wokke JH, Notermans NC. Immunosuppressive treatment for non-systemic vasculitic neuropathy. Cochrane Database Syst Rev. 2007;(1):CD006050.
95. Dalakas MC, Engel WK. Chronic relapsing (dysimmune) polyneuropathy: Pathogenesis and treatment. Ann Neurol. 1981;9(suppl):134–145.
96. McCombe PA, Pollard JD, McLeod JG. Chronic inflammatory demyelinating polyradiculoneuropathy. A clinical and electro-physiological study of 92 cases. Brain. 1987;110(6):1617–1630.
97. Bouchard C, Lacroix C, Planté V, et al. Clinicopathologic findings and prognosis of chronic inflammatory demyelinating polyneuropathy. Neurology. 1999;52(3):498–503.
98. Brannagan TH, Pradhan A, Heiman-Patterson T, et al. High-dose cyclophosphamide without stem-cell rescue for refractory CIDP. Neurology. 2002;58(12):1856–1858.
99. Good JL, Chehrenama M, Mayer RF, Koski CL. Pulse cyclophosphamide therapy in chronic inflammatory demyelinating polyneuropathy. Neurology. 1998;51(6):1735–1738.
100. Gladstone DE, Prestrud AA, Brannagan TH 3rd. High-dose cyclophosphamide results in long-term disease remission with restoration of a normal quality of life in patients with severe refractory chronic inflammatory demyelinating polyneuropathy. J Peripher Nerv Syst. 2005;10(1):11–6.
101. Prineas JW, McLeod JG. Chronic relapsing polyneuritis. J Neurol Sci. 1976;27(4):427–458.
102. Pestronk A, Cornblath DR, Ilyas A, et al. A treatable multifocal motor neuropathy with antibodies to GM1 ganglioside. Ann Neurol. 1988;24:73–78.
103. Leitch MM, Sherman WH, Brannagan TH 3rd. Distal acquired demyelinating symmetric polyneuropathy progressing to classic chronic inflammatory demyelinating polyneuropathy and response to fludarabine and cyclophosphamide. Muscle Nerve. 2013;47(2):292–296.
104. Lewis RA, Lisak RP. “Rebooting” the immune system with cyclophosphamide: Taking risks for a “cure”? Ann Neurol. 2003;53:7–9.
105. Lin PT, Martin BA, Winacker AB, So YT. High-dose cyclophosphamide in refractory myasthenia with MuSK antibodies. Muscle Nerve. 2006;33:433–435.
106. DeFeo LG, Schottlender J, Martelli NA, Molfino NA. Use of intravenous pulsed cyclophosphamide in severe, generalized myasthenia gravis. Muscle Nerve. 2002;26:32–36.
107. Drachman DB, Jones RJ, Brodsky RA. Treatment of refractory myasthenia: “Rebooting” with high-dose cyclophosphamide. Ann Neurol. 2003;53:29–34.
108. Nagappa M, Netravathi M, Taly AB, Sinha S, Bindu PS, Mahadevan A. Long-term efficacy and limitations of cyclophosphamide in myasthenia gravis. J Clin Neurosci. 2014;21(11):1909–1914.
109. El-Salem K, Yassin A, Al-Hayk K, Yahya S, Al-Shorafat D, Dahbour SS. Treatment of MuSK-associated myasthenia gravis. Curr Treat Options Neurol. 2014;16(4):283.
110. Benatar M, Sanders D. The importance of studying history: Lessons learnt from a trial of tacrolimus in myasthenia gravis. J Neurol Neurosurg Psychiatry. 2011;82:945.
111. Ciafaloni E, Nikhar NK, Massey JM, Sanders DB. Retrospective analysis of the use of cyclosporine in myasthenia gravis. Neurology. 2000;55:448–450.
112. Sanders DB, Ciafaloni E, Nikhar NK, Massey JM. Retrospective analysis of the use of cyclosporine in myasthenia [abstract]. Neurology. 2000;54(suppl 3):A394.
113. Tindall RS, Rollins JA, Phillips JT, Greenlee RG, Wells L, Belendiuk G. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med. 1987;316:719–724.
114. Tindall RS, Phillips JT, Rollins JA, Wells L, Hall K. A clinical therapeutic trial of cyclosporine in myasthenia gravis. Ann N Y Acad Sci. 1993;681:539–551.
115. Kurokawa T, Nishiyama T, Yamamoto R, Kishida H, Hakii Y, Kuroiwa Y. Anti-MuSK antibody positive myasthenia gravis with HIV infection successfully treated with cyclosporin: A case report. Rinsho Shinkeigaku. 2008;48(9):666–669.
116. Ponseti JM, Azem J, Fort JM, et al. Long-term results of tacrolimus in cyclosporine- and prednisone-dependent myasthenia gravis. Neurology. 2005;64:1641–1643.
117. Nast A, Sporbeck B, Rosumeck S, et al. Which antipsoriatic drug has the fastest onset of action? Systematic review on the rapidity of the onset of action. J Invest Dermatol. 2013;133:1963–1970.
118. Hefter H, Sprenger KB, Arendt G, Hafner D. Treatment of chronic relapsing inflammatory demyelinating polyneuropathy by cyclosporin A and plasma exchange. A case report. J Neurol. 1990;237(5):320–233.
119. Kolkin S, Nahman NS Jr, Mendell JR. Chronic nephrotoxicity complicating cyclosporine treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 1987;37(1):147–149.
120. Hodgkinson SJ, Pollard JD, McLeod JG. Cyclosporin A in the treatment of chronic demyelinating polyradiculoneuropathy. J Neurol Neurosurg Psychiatry. 1990;53(4):327–330.
121. Barnett MH, Pollard JD, Davies L, McLeod JG. Cyclosporin A in resistant chronic inflammatory demyelinating polyradiculoneuropathy. Muscle Nerve. 1998;21(4):454–460.
122. Mahattanakul W, Crawford TO, Griffin JW, Goldstein JM, Cornblath DR. Treatment of chronic inflammatory demyelinating polyneuropathy with cyclosporin-A. J Neurol Neurosurg Psychiatry. 1996;60(2):185–187.
123. Matsuda M, Hoshi K, Gono T, Morita H, Ikeda S. Cyclosporin A in treatment of refractory patients with chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol Sci. 2004;224(1–2):29–35.
124. Odaka M, Tatsumoto M, Susuki K, Hirata K, Yuki N. Intractable chronic inflammatory demyelinating polyneuropathy treated successfully with ciclosporin. J Neurol Neurosurg Psychiatry. 2005;76(8):1115–1120.
125. Cocito D, Grimaldi S, Paolasso I, et al. Italian Network for CIDP Register. Immunosuppressive treatment in refractory chronic inflammatory demyelinating polyradiculoneuropathy. A nation-wide retrospective analysis. Eur J Neurol. 2011;18(12):1417–1421.
126. Sanders DB. Lambert-Eaton myasthenic syndrome: Diagnosis and treatment. Ann N Y Acad Sci. 2003;998:500–508.
127. Vencovsky J, Jarosova K, Machacek S, et al. Cyclosporine A versus methotrexate in the treatment of polymyositis and dermatomyositis. Scand J Rheumatol. 2000;29(2):95–102.
128. Borleffs JC. Cyclosporine as monotherapy for polymyositis? Transplant Proc. 1988;20(3 suppl 4):333–334.
129. Correia O, Polonia J, Nunes JP, Resende C, Delgado L. Severe acute form of adult dermatomyositis treated with cyclosporine. Int J Dermatol. 1992;31(7):517–519.
130. Girardin E, Dayer JM, Paunier L. Cyclosporine for juvenile dermatomyositis. J Pediatr. 1988;112(1):165–166.
131. Heckmatt J, Hasson N, Saunders C, et al. Cyclosporin in juvenile dermatomyositis. Lancet. 1989;1(8646):1063–1066.
132. Jones DW, Snaith ML, Isenberg DA. Cyclosporine treatment for intractable polymyositis. Arthritis Rheum. 1987;30(8):959–960.
133. Lueck CJ, Trend P, Swash M. Cyclosporin in the management of polymyositis and dermatomyositis. J Neurol Neurosurg Psychiatry. 1991;54(11):1007–1008.
134. Mehregan DR, Su WP. Cyclosporine treatment for dermatomyositis/polymyositis. Cutis. 1993;51(1):59–61.
135. Pistoia V, Buoncompagni A, Scribanis R, et al. Cyclosporin A in the treatment of juvenile chronic arthritis and childhood polymyositis-dermatomyositis. Results of a preliminary study. Clin Exp Rheumatol. 1993;11(2):203–208.
136. van Sonderen A, Wirtz PW, Verschuuren JJ, Titulaer MJ. Paraneoplastic syndromes of the neuromuscular junction: Therapeutic options in myasthenia gravis, Lambert-Eaton myasthenic syndrome, and neuromyotonia. Curr Treat Options Neurol. 2013;15(2):224–239.
137. Howard JF Jr, Barohn RJ, Cutter GR, et al. A randomized, double-blind, placebo-controlled phase II study of eculizumab in patients with refractory generalized myasthenia gravis. Muscle Nerve. 2013;48(1):76–84.
138. Takada K, Bookbinder S, Furie R, et al. A pilot study of eculizumab in patients with dermatomyositis. Arthritis Rheum. 2002;46(suppl):S489.
139. Chin RL, Sherman WH, Sander HW, Hays AP, Latov N. Etanercept (Enbrel) therapy for chronic inflammatory demyelinating polyneuropathy. J Neurol Sci. 2003;210(1–2):19–21.
140. Alshekhlee A, Basiri K, Miles JD, Ahmad SA, Katirji B. Chronic inflammatory demyelinating polyneuropathy associated with tumor necrosis factor-alpha antagonists. Muscle Nerve. 2010;41(5):723–727.
141. Hamon MA, Nicolas G, Deviere F, Letournel F, Dubas F. Demyelinating neuropathy during anti-TNF alpha treatment with a review of the literature. Rev Neurol. 2007;163(12):1232–1235.
142. Lozeron P, Denier C, Lacroix C, Adams D. Long-term course of demyelinating neuropathies occurring during tumor necrosis factor-blocker therapy. Arch Neurol. 2009;66(4):490–497.
143. Richez C, Blanco P, Lagueny A, Schaeverbeke T, Dehais J. Neuropathy resembling CIDP in patients receiving tumor necrosis factor-alpha blockers. Neurology. 2005;64(8):1468–1470.
144. Moravan M, Segal BM. Treatment of CNS sarcoidosis with infliximab and mycophenolate mofetil. Neurology. 2009;72(4):337–340.
145. Baughman RP, Lower EE, Bradley DA, Raymond LA, Kaufman A. Etanercept for refractory ocular sarcoidosis: Results of a double-blind randomized trial. Chest. 2005;128(2):1062.
146. Thongpooswan S, Abrudescu A. Lung sarcoidosis in etanercept treated rheumatoid arthritis patient: A case report and review of the literature. Case Rep Rheumatol. 2014;2014:358567.
147. Rowin J, Meriggioli MN, Tüzün E, Leurgans S, Christadoss P. Etanercept treatment in corticosteroid-dependent myasthenia gravis. Neurology. 2004;63:2390–2392.
148. The Muscle Study Group. A randomized, pilot trial of etanercept in dermatomyositis. Ann Neurol. 2011;70:427–436.
149. Chen D, Wang XB, Zhou Y, Zhu XC. Efficacy of infliximab in the treatment for dermatomyositis with acute interstitial pneumonia: A study of fourteen cases and literature review. Rheumatol Int. 2013;33(10):2455–2458.
150. Kakoulidou M, Bjelak S, Pirskanen R, Lefvert AK. A clinical and immunological study of a myasthenia gravis patient treated with infliximab. Acta Neurol Scand. 2007;115(4):279–283.
151. Marzo ME, Tintoré M, Fabregues O, Montalbán X, Codina A. Chronic inflammatory demyelinating polyradiculoneuropathy during treatment with interferon alpha. J Neurol Neurosurg Psychiatry. 1998;65(4):604.
152. Meriggioli MN, Rowin J. Chronic inflammatory demyelinating polyneuropathy after treatment with interferon-alpha. Muscle Nerve. 2000;23(3):433–435.
153. Gorson KC, Allam G, Simovic D, Ropper AH. Improvement following interferon-alpha 2A in chronic inflammatory demyelinating polyneuropathy. Neurology. 1997;48(3):777–780.
154. Pavesi G, Cattaneo L, Marbini A, Gemignani F, Mancia D. Long-term efficacy of interferon-alpha in chronic inflammatory demyelinating polyneuropathy. J Neurol. 2002;249(6):777–779.
155. Choudhary PP, Thompson N, Hughes RA. Improvement following interferon beta in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 1995;242(4):252–253.
156. Radziwill AJ, Kuntzer T, Fuhr P, Steck AJ. Long term follow up in chronic inflammatory demyelinating polyneuropathy (CIDP) under interferon beta-1a (IFN-b1 a) and intravenous immunoglobulin (IVIg). J Neurol Neurosurg Psychiatry. 2001;70:273.
157. Kuntzer T, Radziwill AJ, Lettry-Trouillat R, et al. Interferon-beta 1a in chronic inflammatory demyelinating polyneuropathy. Neurology. 1999;53(6):1364–1365.
158. Martina IS, van Doorn PA, Schmitz PI, Meulstee J, van der Meché FG. Chronic motor neuropathies: Response to interferon-beta 1a after failure of conventional therapies. J Neurol Neurosurg Psychiatry. 1999;66(2):197–201.
159. Vallat JM, Hahn AF, Léger JM, et al. Interferon beta-1a as an investigational treatment for CIDP. Neurology. 2003;60(8 suppl 3):S23–S28.
160. Hadden RD, Sharrack B, Bensa S, Soudain SE, Hughes RA. Randomized trial of interferon beta-1a in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 1999;53(1):57–61.
161. Matsuse D, Ochi H, Tashiro K, et al. Exacerbation of chronic inflammatory demyelinating polyradiculoneuropathy during interferon beta-1b therapy in a patient with childhood-onset multiple sclerosis. Intern Med. 2005;44(1):68–72.
162. Donofrio PD, Berger A, Brannagan TH III, et al. Consensus statement: The use of intravenous immunoglobulin in the treatment of neuromuscular conditions, report of the ad hoc committee of the AANEM. Muscle Nerve. 2009;40:890–900.
163. Dwyer JM. Manipulating the immune system with immune globulin. N Engl J Med. 1992;326(2):107–116.
164. Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med. 2012;367:2015–2025.
165. Querol L, Rojas R, Casasnovas C, et al. Long-term outcome in chronic inflammatory demyelinating polyneuropathy patients treated with intravenous immunoglobulin: A retrospective study. Muscle Nerve. 2013;48:879–876.
166. Nobile-Orazio E, Meucci N, Barbieri S, Carpo M, Scarlato G. High dose intravenous immunoglobulin therapy in multifocal motor neuropathy. Neurology. 1993;43:537–544.
167. O’Leary CP, Mann AC, Lough J, Willison HJ. Muscle hypertrophy in multifocal motor neuropathy is associated with continuous motor unit activity. Muscle Nerve. 1997;20:479–485.
168. Huang CS, Hsu HS, Kao KP, Huang MH, Huang BS. Intravenous immunoglobulin in the preparation of thymectomy for myasthenia gravis. Acta Neurol Scand. 2003;108:136–138.
169. Stricker RB, Kwiatkowski BJ, Habis JA, Kiprov DD. Myasthenic crisis: Response to plasmapheresis following failure of intravenous gamma-globulin. Arch Neurol. 1993;50:837–840.
170. Barth D, Nabavi NM, Ng E, New P, Bril V. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology. 2011;76:2017–2023.
171. Zinman L, Ny E, Bril V. IV Immunoglobulin in patients with myasthenia gravis: A randomized controlled trial. Neurology. 2007;68:837–841.
172. Gajdos P, Chevret S, Clair B, Tranchant C, Chastang C. Clinical trial of plasma exchange and high-dose intravenous immunoglobulin in myasthenia gravis. Myasthenia Gravis Clinical Study Group. Ann Neurol. 1997;41:789–796.
173. Gajdos P, Tranchant C, Clair B, et al. Treatment of myasthenia gravis exacerbation with intravenous immunoglobulin: A randomized double-blind clinical trial. Arch Neurol. 2005;62:1689–1693.
174. Gajdos P, Chevret S, Toyka K. Intravenous immunoglobulin for myasthenia gravis. Cochrane Database Syst Rev. 2006;19: CD002277.
175. Gajdos P, Chevret S, Toyka K. Plasma exchange for myasthenia gravis. Cochrane Database Syst Rev. 2002;4:CD002275.
176. Achiron A, Barak Y, Miron S, Sarova-Pinhas I. Immunoglobulin treatment in refractory myasthenia gravis. Muscle Nerve. 2000;23:551–555.
177. Cosi V, Lombardi M, Piccolo G, Erbetta A. Treatment of myasthenia gravis with high-dose intravenous immunoglobulin. Acta Neurol Scand. 1991;84:81–84.
178. Jongen JL, van Doorn PA, van der Meche FG. High-dose intravenous immunoglobulin therapy for myasthenia gravis. J Neurol. 1998;245:26–31.
179. Howard JF. Intravenous immunoglobulin for the treatment of acquired myasthenia gravis. Neurol. 1998;51(suppl 5):S30–S36.
180. Qureshi AI, Choudhry MA, Akbar MS, et al. Plasma exchange versus intravenous immunoglobulin treatment in myasthenic crisis. Neurology. 1999;52:629–632.
181. Shibata-Hamaguchi A, Samuraki M, Furui E, et al. Long-term effect of intravenous immunoglobulin on anti-MuSK antibody-positive myasthenia gravis. Acta Neurol Scand. 2007; 116:406–408.
182. Takahashi H, Kawaguchi N, Nemoto Y, Hattori T. High-dose intravenous immunoglobulin for the treatment of MuSK antibody-positive seronegative myasthenia gravis. J Neurol Sci. 2006;247:239.
183. Ronager J, Ravnborg M, Hermansen I, Vorstrup S. Immunoglobulin treatment versus plasma exchange in patients with chronic moderate to severe myasthenia gravis. Artif Organs. 2001;25:967–973.
184. Bril V, Barnett-Tapia C, Barth D, Katzberg HD. IVIG and PLEX in the treatment of myasthenia gravis. Ann N Y Acad Sci. 2012;1275(1):1–6.
185. Herrmann DN, Carney PR, Wald JJ. Juvenile myasthenia gravis: Treatment with immune globulin and thymectomy. Pediatr Neurol. 1998;18:63–66.
186. Selcen D, Dabrowski ER, Michon AM, Nigro MA. High-dose immunoglobulin therapy in juvenile myasthenia gravis. Pediatr Neurol. 2000;22:40–43.
187. Pinching AJ, Peters DK, Newsom-Davis J. Remission of myasthenia gravis following plasma-exchange. Lancet. 1976; 2:1373–1376.
188. Antozzi C, Gemma M, Regi B, et al. A short plasma exchange protocol is effective in severe myasthenia gravis. J Neurol. 1991;238:103–107.
189. Mandawat A, Mandawat A, Kaminski H, Shaker Z, Alawi AA, Alshekhlee A. Outcome of plasmapheresis in myasthenia gravis: Delayed therapy is not favorable. Muscle Nerve. 2011;43;578–584.
190. Howard JF. The treatment of myasthenia gravis with plasma exchange. Semin Neurol. 1982;2:273–279.
191. Guptill JT, Oakley D, Kuchibhatla M, et al. A retrospective study of complications of therapeutic plasma exchange in myasthenia. Muscle Nerve. 2013;47:170–176.
192. Dau PC, Lindstrom JM, Cassel CK, Denys EH, Shev EE, Spitler LE. Plasmapheresis and immunosuppressive drug therapy in myasthenia gravis. N Engl J Med. 1977;297:1134–1140.
193. Dalakas M, Illa I, Dambrosia J, et al. A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med. 1993;329:1993–2000.
194. Brannagan TH 3rd, Nagle KJ, Lange DJ, Rowland LP. Complications of intravenous immune globulin treatment in neurologic disease. Neurology. 1996;47:674–677.
195. Lazzaro C, Lopiano L, Cocito D. Subcutaneous vs intravenous administration of immunoglobulin in chronic inflammatory demyelinating polyneuropathy: An Italian cost-minimization analysis. Neurol Sci. 2014;35(7):1023–1034.
196. Markvardsen LH, Harbo T, Sindrup SH, Christiansen I, Andersen H, Jakobsen J; The Danish CIDP and MMN Study Group. Subcutaneous immunoglobulin preserves muscle strength in chronic inflammatory demyelinating polyneuropathy. Eur J Neurol. 2014;21(12):1465–1470.
197. Plasma Exchange/Sandoglobulin Guillain-Barre Syndrome Trial Group. Randomized trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barré syndrome. Lancet. 1997;349:225–230.
198. Metzger AL, Bohan A, Goldberg LS, Bluestone R, Pearson CM. Polymyositis and dermatomyositis: Combined methotrexate and corticosteroid therapy. Ann Intern Med. 1974;81(2):182–189.
199. Sokoloff MC, Goldberg LS, Pearson CM. Treatment of corticosteroid-resistant polymyositis with methotrexate. Lancet. 1971;1(7688):14–16.
200. Miller LC, Sisson BA, Tucker LB, DeNardo BA, Schaller JG. Methotrexate treatment of recalcitrant childhood dermatomyositis. Arthritis Rheum. 1992; 35(10):1143–1149.
201. Villalba L, Hicks JE, Adams EM, et al. Treatment of refractory myositis: A randomized crossover study of two new cytotoxic regimens. Arthritis Rheum. 1998;41(3):392–399.
202. Cagnoli M, Marchesoni A, Tosi S. Combined steroid, methotrexate and chlorambucil therapy for steroid-resistant dermatomyositis. Clin Exp Rheumatol. 1991;9(6):658–659.
203. Giannini M, Callen JP. Treatment of dermatomyositis with methotrexate and prednisone. Arch Dermatol. 1979; 115(10):1251–1252.
204. Mahdi-Rogers M; For the Randomised Methotrexate Chronic Inflammatory Demyelinating polyradiculoneuropathy (RMC) Trial Group. A pilot randomised controlled trial of methotrexate for CIDP: Lessons for future trials. J Peripher Nerv Syst. 2008;13(2):176.
205. Fialho D, Chan YC, Allen DC, Reilly MM, Hughes RA. Treatment of chronic inflammatory demyelinating polyradiculoneuropathy with methotrexate. J Neurol Neurosurg Psychiatry. 2006;77(4):544–547.
206. Díaz-Manera J, Rojas-García R, Gallardo E, Illa I. Response to methotrexate in a chronic inflammatory demyelinating polyradiculoneuropathy patient. Muscle Nerve. 2009;39(3):386–388.
207. Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arth Rheum. 2010; 62(9):2757–2766.
208. Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve. 2010;41:185–190.
209. Mohassel P, Mammen AL. Statin-associated autoimmune myopathy and anti-HMGCR autoantibodies. Muscle Nerve. 2013;48:477–483.
210. Pasnoor M, He J, Herbelin L, Dimachkie M, Barohn RJ; Muscle Study Group. Phase II trial of methotrexate in myasthenia gravis. Ann N Y Acad Sci. 2012;1275(1):23–28.
211. Visser K, van der Heijde DM. Risk and management of liver toxicity during methotrexate treatment in rheumatoid and psoriatic arthritis: A systematic review of the literature. Clin Exp Rheumatol. 2009;27:1017–1025.
212. Graves D, Vernino S. Immunotherapies in neurologic disease. Med Clin North Am. 2012;96(3):497–523.
213. The Muscle Study Group. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology. 2008;71:394–399.
214. Sanders DB, Hart IK, Mantegazza R, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology. 2008;71:400–406.
215. Meriggioli MN, Rowin J, Richman JG, Leurgans S. Mycophenolate mofetil for myasthenia gravis: A double-blind, placebo-controlled pilot study. Ann N Y Acad Sci. 2003;998:494–499.
216. Meriggioli MN, Ciafaloni E, Al-Hayk KA, et al. Mycophenolate mofetil for myasthenia gravis: An analysis of efficacy, safety, and tolerability. Neurology. 2003;61:1438–1440.
217. Sanders D, McDermott M, Thornton C, Tawil A, Barohn R; The Muscle Study Group. A trial of mycophenolate mofetil (MMF) with prednisone as initial immunotherapy in myasthenia gravis (MG) [abstract]. Neurology. 2007;68:A107.
218. Ciafaloni E, Massey JM, Tucker-Lipscomb B, Sanders DB. Mycophenolate mofetil for myasthenia gravis: An open-label pilot study. Neurology. 2001;56:97–99.
219. Vernino S, Salomao DR, Habermann TM, O’Neill BP. Primary CNS lymphoma complicating treatment of myasthenia gravis with mycophenolate mofetil. Neurology. 2005;65:639–641.
220. Hauser RA, Malek AR, Rosen R. Successful treatment of a patient with severe refractory myasthenia gravis using mycophenolate mofetil. Neurology. 1998;51:912–913.
221. Chaudhry V, Cornblath DR, Griffin JW, O’Brien R, Drachman DB. Mycophenolate mofetil: A safe and promising immunosuppressant in neuromuscular diseases. Neurology. 2001;56:94–96.
222. Hehir MK, Burns TM, Alpers J, Conaway MR, Sawa M, Sanders DB. Mycophenolate mofetil in AChR-antibody-positive myasthenia gravis: Outcomes in 102 patients. Muscle Nerve. 2010; 41(5):593–598.
223. Schneider-Gold C, Hartung HP, Gold R. Mycophenolate mofetil and tacrolimus: New therapeutic options in neuroimmunological diseases. Muscle Nerve. 2006;34(3):284–291.
224. Gorson KC, Amato AA, Ropper AH. Efficacy of mycophenolate mofetil in patients with chronic immune demyelinating polyneuropathy. Neurology. 2004;63(4):715–717.
225. Radziwill AJ, Schweikert K, Kuntzer T, Fuhr P, Steck AJ. Mycophenolate mofetil for chronic inflammatory demyelinating polyradiculoneuropathy: An open label study. Eur J Neurol. 2006;56(1):37–38.
226. Mowzoon N, Sussman A, Bradley WG. Mycophenolate (Cell-Cept) treatment of myasthenia gravis, chronic inflammatory polyneuropathy and inclusion body myositis. J Neurol Sci. 2001;185(2):119–122.
227. Benedetti L, Grandis M, Nobbio L, et al. Mycophenolate mofetil in dysimmune neuropathies: A preliminary study. Muscle Nerve. 2004;29(5):748–749.
228. Bedi G, Brown A, Tong T, Sharma KR. Chronic inflammatory demyelinating polyneuropathy responsive to mycophenolate mofetil therapy. J Neurol Neurosurg Psychiatry. 2010; 81(6):634–636.
229. Umapathi T, Hughes R. Mycophenolate in treatment resistant inflammatory neuropathies. Eur J Neurol. 2002;9(6):683–685.
230. Dubal DB, Mueller S, Ruben BS, Engstrom JW, Josephson SA. T-cell lymphoproliferative disorder following mycophenolate treatment for myasthenia gravis. Muscle Nerve. 2009; 39(6):849–850.
231. Silvestri NJ, Wolfe GI. Myasthenia gravis. Semin Neurol. 2012; 32(3):215–226.
232. Sorgun MH, Erdogan S, Bay M, et al. Therapeutic plasma exchange in treatment of neuroimmunologic disorders: Review of 92 cases. Transfus Apher Sci. 2013;49(2):174–180.
233. Cortese I, Chaudhry V, So YT, Cantor F, Cornblath DR, Rae-Grant A. Evidence-based guideline update: Plasmapheresis in neurologic disorders. Neurology. 2011;76:294–300.
234. NIH Consensus DevelopmentConference. The utility of therapeutic plasmapheresis for neurological disorders. NIH Consensus Development. JAMA. 1986;256:1333–1337.
235. Yeh JH, Chiu HC. Plasmapheresis in myasthenia gravis. A comparative study of daily versus alternately daily schedule. Acta Neurol Scand. 1999;99:147–151.
236. Mahalati K, Dawson RB, Collins JO, Mayer RF. Predictable recovery for myasthenia gravis crisis with plasma exchange: 36 cases and review of current management. J Clin Apher. 1999;14:1–8.
237. Dyck PJ, Litchy WJ, Kratz KM, et al. Plasma exchange versus immune globulin infusion trial in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol. 1994; 36:838–845.
238. Mehndiratta MM, Hughes RA. Plasma exchange for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 2012;9:CD003906.
239. Dyck PJ, Low PA, Windebank AJ, et al. Plasma exchange in polyneuropathy associated with monoclonal gammopathy of undetermined significance. N Engl J Med. 1991;325(21):1482–1486.
240. Bai HX, Wang ZL, Tan LM, Xiao B, Goldstein JM, Yang L. The effectiveness of immunomodulating treatment on Miller Fisher syndrome: A retrospective analysis of 65 Chinese patients. J Peripher Nerv Syst. 2013;18(2):195–196.
241. Thaisetthawatkul P, Dyck PJ. Treatment of diabetic and non-diabetic lumbosacral radiculoplexus neuropathy. Curr Treat Options Neurol. 2010;12(2):95–99.
242. Baek WS, Bashey A, Sheean GL. Complete remission induced by rituximab in refractory, seronegative, muscle-specific kinase positive myasthenia gravis. J Neurol Neurosurg Psychiatry. 2007;78:771.
243. Díaz-Manera J, Martínez-Hernandez E, Querol L, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology. 2012;78:189–193.
244. Hain B, Jordan K, Deschauer M, Zierz S. Successful treatment of MuSK antibody-positive myasthenia gravis with rituximab. Muscle Nerve. 2006;33(4):575–580.
245. Nowak RJ, Dicapua DB, Zebardast N, Goldstein JM. Response of patients with refractory myasthenia gravis to rituximab: A retrospective study. Ther Adv Neurol Disord. 2011;4:259–266.
246. Collongues N, Casez O, Lacour A, et al. Rituximab in refractory and non-refractory myasthenia: A retrospective multicenter study. Muscle Nerve. 2012;46:687–691.
247. Steiglbauer K, Topakian R, Schäffer G, Aichner FT. Rituximab for myasthenia gravis: Three case reports and review of the literature. J Neurol Sci. 2009;280:120–122.
248. Blum S, Gillis D, Brown H, et al. Use and monitoring of low-dose rituximab in myasthenia gravis. J Neurol Neurosurg Psychiatry. 2011;82:659–663.
249. Kuntzer T, Carota A, Novy J, Cavassini M, Du Pasquier RA. Rituximab is successful in an HIV positive patient with MuSK myasthenia. Neurology. 2011;76:757–758.
250. Maddison P, McConville J, Farrugia ME, et al. The use of rituximab in myasthenia gravis and Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 2011;82:671–673.
251. Gajra A, Vajpayee N, Grethlein SJ. Response of myasthenia gravis to rituximab in a patient with non-Hodgkin lymphoma. Am J Hematol. 2004;77(2):196–197.
252. Souayah N, Noopur R, Tick-Chong PS. Beneficial effects of Rituximab in patients with anti-MAG (myelin-associated glycoprotein) neuropathy: Case reports. Immunopharmacol Immunotoxicol. 2013;35(5):622–624.
253. Stork AC, Notermans NC, Vrancken AF, Cornblath DR, van der Pol WL. Rapid worsening of IgM anti-MAG demyelinating polyneuropathy during rituximab treatment. J Peripher Nerv Syst. 2013;18(2):189–191.
254. Gorson KC, Natarajan N, Ropper AH, Weinstein R. Rituximab treatment in patients with IVIg-dependent immune polyneuropathy: A prospective pilot trial. Muscle Nerve. 2007;35(1):66–69.
255. D’Amico A, Catteruccia M, De Benedetti F, et al. Rituximab in a childhood-onset idiopathic refractory chronic inflammatory demyelinating polyneuropathy. Eur J Paediatr Neurol. 2012;16(3):301–303.
256. Knecht H, Baumberger M, Tobòn A, Steck A. Sustained remission of CIDP associated with Evans syndrome. Neurology. 2004; 63(4):730–732.
257. Sadnicka A, Reilly MM, Mummery C, Brandner S, Hirsch N, Lunn MP. Rituximab in the treatment of three coexistent neurological autoimmune diseases: Chronic inflammatory demyelinating polyradiculoneuropathy, Morvan syndrome and myasthenia gravis. J Neurol Neurosurg Psychiatry. 2011;82(2):230–232.
258. Münch C, Anagnostou P, Meyer R, Haas J. Rituximab in chronic inflammatory demyelinating polyneuropathy associated with diabetes mellitus. J Neurol Sci. 2007;256(1–2):100–102.
259. Kilidireas C, Anagnostopoulos A, Karandreas N, Mouselimi L, Dimopoulos MA. Rituximab therapy in monoclonal IgM-related neuropathies. Leuk Lymphoma. 2006;47(5):859–864.
260. Bodley-Scott DD. Chronic inflammatory demyelinating polyradiculoneuropathy responding to rituximab. Pract Neurol. 2005;5(4):242–245.
261. Briani C, Zara G, Zambello R, Trentin L, Rana M, Zaja F. Rituximab-responsive CIDP. Eur J Neurol. 2004;11(11):788.
262. Benedetti L, Briani C, Franciotta D, et al. Rituximab in patients with chronic inflammatory demyelinating polyradiculoneuropathy: A report of 13 cases and review of the literature. J Neurol Neurosurg Psychiatry. 2011;82(3):306–308.
263. Benedetti L, Franciotta D, Beronio A, Cadenotti L, Gobbi M, Mancardi GL, et al. Rituximab efficacy in CIDP associated with idiopathic thrombocytopenic purpura. Muscle Nerve. 2008;38(2):1076–1077.
264. Lunn MP. Rituximab usage in CIDP: A retrospective e-mail based data collection. J Peripher Nerv Syst. 2009;(14):92.
265. Kasamon YL, Nguyen TN, Chan JA, Nascimento AF. EBV-associated lymphoma and chronic inflammatory demyelinating polyneuropathy in an adult without overt immunodeficiency. Am J Hematol. 2002;69(4):289–293.
266. Baker MR, Das M, Isaacs J, Fawcdett PR, Bates D. Treatment of stiff person syndrome with rituximab. J Neurol Neurosurg Psychiatry. 2005;76:999–1001.
267. Dupond JL, Essalmi L, Gil H, Meaux-Ruault N, Hafsaoui C. Rituximab treatment of stiff-person syndrome in a patient with thymoma, diabetes mellitus and autoimmune thyroiditis. J Clin Neurosci. 2010;17(3):389–391.
268. Katoh N, Matsuda M, Ishii W, Morita H, Ikeda S. Successful treatment with rituximab in a patient with stiff-person syndrome complicated by dysthyroid ophthalmopathy. Intern Med. 2010;49(3):237–241.
269. Oddis CV, Reed AM, Aggarwal R, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: A randomized, placebo-phase trial. Arthritis Rheum. 2013;65:314–324.
270. Valiyil R, Casciola-Rosen L, Hong G, Mammen A, Christopher-Stine L. Rituximab therapy for myopathy associated with anti-signal recognition particle antibodies: A case series. Arthritis Care Res (Hoboken). 2010;62(9):1328–1334.
271. Yoshikawa H, Kiuchi T, Saida T, Takamori M. Randomised double blind, placebo controlled study of tacrolimus in myasthenia gravis. J Neurol Neurosurg Psychiatry. 2011;82:970–977.
272. Ponseti JM, Gamez J, Azem J, et al. Post-thymectomy combined treatment of prednisone and tacrolimus versus prednisone alone for the consolidation of complete stable remission in patients with myasthenia gravis: A non-randomized, non-controlled study. Curr Med Res Opin. 2007;23:1269–1278.
273. Ponseti JM, Gamez J, Azem J, López-Cano M, Vilallonga R, Armengol M. Tacrolimus for myasthenia gravis: A clinical study of 212 patients. Ann N Y Acad Sci. 2008;1132:254–263.
274. Nagane Y, Utsugisawa K, Obara D, Kondoh R, Terayama Y. Efficacy of low-dose FK506 in the treatment of myasthenia gravis–a randomized pilot study. Eur Neurol. 2005;53:146–150.
275. Evoli A, Di Schino C, Marsili F, Punzi C. Successful treatment of myasthenia gravis with tacrolimus. Muscle Nerve. 2002;25(1):111–114.
276. Konishi T, Yoshiyama Y, Takamori M, Yagi K, Mukai E, Saida T. Japanese FK506 MG Study Group. Clinical study of FK506 in patients with myasthenia gravis. Muscle Nerve. 2003;28(5):570–574.
277. Sanders DB, Aarli JA, Cutter GR, Jaretzki A III, Kaminski HJ, Phillips LH II. Long-term results of tacrolimus in cyclosporine- and prednisone-dependent myasthenia gravis [comment]. Neurology. 2006; 66(6):954–955.
278. Tada M, Shimohata T, Tada M, et al. Long-term therapeutic efficacy and safety of low-dose tacrolimus (FK506) for myasthenia gravis. J Neurol Sci. 2006;247(1):17–20.
279. Wakata N, Saito T, Tanaka S, Hirano T, Oka K. Tacrolimus hydrate (FK506): Therapeutic effects and selection of responders in the treatment of myasthenia gravis. Clin Neurol Neurosurg. 2003;106(1):5–8.
280. Ahlmén J, Andersen O, Hallgren G, Peilot B. Positive effects of tacrolimus in a case of CIDP. Transplant Proc. 1998;30(8):4194.
281. Dalakas MC, Rakocevic G, Schmidt J, et al. Effect of Alemtuzumab (CAMPATH 1-H) in patients with inclusion-body myositis. Brain. 2009;132(Pt 6):1536–1544.
282. Bunch TW, Worthington JW, Combs JJ, Ilstrup DM, Engel AG. Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Ann Intern Med. 1980;92(3):365–369.
283. Heaney D, Geddes JF, Nagendren K, Swash M. Sarcoid polyneuropathy responsive to intravenous immunoglobulin. Muscle Nerve. 2004;29:447–450.
284. Agius MA. Treatment of ocular myasthenia gravis with corticosteroids: Yes. Arch Neurol. 2000;57:750–751.
285. Pascuzzi RM, Coslett HB, Johns TR. Long-term corticosteroid treatment of myasthenia gravis: Report of 116 patients. Ann Neurol. 1984;15:291–298.
286. Schneider-Gold C, Gajdos P, Toyka KV, Hohlfeld RR. Corticosteroids for myasthenia gravis. Cochrane Database Syst Rev. 2005;2:CD002828.
287. Bae JS, Go SM, Kin BJ. Clinical predictors of steroid-induced exacerbation in myasthenia gravis. J Clin Neurosci. 2006;13:1006–2010.
288. Arsura EL, Brunner NG, Namba T, Grob D. High-dose intravenous methylprednisolone in myasthenia gravis. Arch Neurol. 1985;42:1149–1153.
289. Mann JD, Johns TR, Campa JF. Long-term administration of corticosteroids in myasthenia gravis. Neurology. 1976;26:729–740.
290. Dyck PJ, O’Brien P, Bosch P, et al. The multi-center double-blind controlled trial of IV methylprednisolone in diabetic lumbosacral radiculoplexus neuropathy. Neurology. 2006;66(suppl 2):A191.
291. Kono DH, Klashman DJ, Gilbert RC. Successful IV pulse cyclophosphamide in refractory PM in 3 patients with SLE. J Rheumatol. 1990;17(7):982–983.
292. Leroy JP, Drosos AA, Yiannopoulos DI, Youinou P, Moutsopoulos HM. Intravenous pulse cyclophosphamide therapy in myositis and Sjogren’s syndrome. Arthritis Rheum. 1990;33(10):1579–1581.
293. Daniel MG, Malcangi G, Palmieri C, et al. Cyclosporin A and intravenous immunoglobulin treatment in polymyositis/dermatomyositis. Ann Rheum Dis. 2002;61:37–41.
294. Goei The HS, Jacobs P, et al. Cyclosporine in the treatment of intractable polymyositis. Arthritis Rheum. 1985;28(12):1436–1437.
295. Hengstman GJ, van den Hoogen FH, Barrera P, et al. Successful treatment of dermatomyositis and polymyositis with anti-tumor-necrosis-factor-alpha: Preliminary observations. Eur Neurol. 2003;50(1):10–15.
296. Hengstman GJ, van den Hoogen FH, van Engelen BG. Treatment of dermatomyositis and polymyositis with anti-tumor necrosis factor-alpha: Long-term follow-up. Eur Neurol. 2004;52(1):61–63.
297. Efthimiou P, Schwartzman S, Kagen LJ. Possible role for tumour necrosis factor inhibitors in the treatment of resistant dermatomyositis and polymyositis: A retrospective study of eight patients. Ann Rheum Dis. 2006;65(9):1233–1236.
298. Sprott H, Glatzel M, Michel BA. Treatment of myositis with etanercept (Enbrel), a recombinant human soluble fusion protein of TNF-alpha type II receptor and IgG1. Rheumatology (Oxford). 2004;43(4):524–526.
299. Coyle K, Pokrovnichka A, French K, Joe G, Shrader J, Swan L, et al. A randomized, double-blind, placebo controlled trial of infliximab in patients with polymyositis and dermatomyositis. Arth Rheum 2008;58(suppl); Abstract No:2058.
300. Riley P, McCann LJ, Maillard SM, Woo P, Murray KJ, Pilkington CA. Effectiveness of infliximab in the treatment of refractory juvenile dermatomyositis with calcinosis. Rheumatology (Oxford). 2008;47(6):877–880.
301. Labioche I, Liozon E, Weschler B, Loustaud-Ratti V, Soria P, Vidal E. Refractory polymyositis responding to infliximab: Extended follow-up. Rheumatology (Oxford). 2004;43(4):531–532.
302. Radziwill AJ, Botez SA, Novy J, Kuntzer T. Interferon beta-1a as adjunctive treatment for multifocal motor neuropathy: An open label trial. J Peripher Nerv Syst. 2009;14(3):201–202.
303. Patwa HS, Chaudhry H. Katzberg H, Rae-Grant AD, So YT. Evidence-based guideline: Intravenous immunoglobulin in the treatment of neuromuscular disorders: Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2012;78:1009–1015.
304. Hughes RA, Raphael JC, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2006(1):CD002063.
305. Hughes RA, Donofrio PD, Bril V, et al.; ICE Group. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): A randomized placebo-controlled trial. Lancet Neurol. 2008;7:115–116.
306. Hahn AF, Bolton CF, Zochodne DW, Feasby TE. Intravenous immunoglobulin treatment in chronic inflammatory demyelinating polyneuropathy: A double blind, placebo controlled, cross over study. Brain. 1996;119:1067–1077.
307. Hughes RA, Bensa S, Willison HJ, et al. Randomized controlled trial of intravenous gammaglobulin vs oral prednisolone in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol. 2001;50:195–201.
308. Mendell JR, Barohn RJ, Freimer ML, et al. Randomized controlled trial of IVIG in untreated chronic inflammatory demyelinating polyradiculoneuropathy. Neurology. 2001;56:445–449.
309. Vermulen M, van Doorn PA, Brand A, Strengers PF, Jennekens FG, Bussch HF. Intravenous immunoglobulin treatment in patients with chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry. 1993;56:36–39.
310. Van Schaik I, Winer JB, De Haan R, Vermeulen M. Intravenous immunoglobulins for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 2002;(2):CD001797.
311. Viala K, Maisonobe T, Stojkovic T, et al. A current view of the diagnosis, clinical variants, response to treatment and prognosis of chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. 2010;15(1):50–56.
312. Mehndiratta MM, Hughes RA. Corticosteroids for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev. 2012;8:CD002062.
313. Eftimov F, Vermeulen M, van Doorn PA, Brusse E, van Schaik IN; PREDICT Study Group. Long-term remission of CIDP after pulsed high-dose dexamethasone or short term prednisolone treatment. Neurology. 2012;78:1079–1084.
314. Azulay JP, Blin O, Pouget J, et al. Intravenous immunoglobulin treatment in patients with motor neuron syndromes associated with anti-GM1 antibodies: A double-blind, placebo-controlled study. Neurology. 1994;44:429–432.
315. Federico P, Zochodne DW, Hahn AF, Brown WF, Feasby TE. Multifocal motor neuropathy improved by IVIG: Randomized, double-blind, placebo-controlled study. Neurology. 2000;55:1256–1262.
316. Leger JM, Chassande B, Musset L, Meininger V, Bouche P, Baumann N. Intravenous immunoglobulin therapy in multifocal motor neuropathy: A double-blind, placebo-controlled study. Brain. 2001;124:145–153.
317. van Schaik IN, van den Berg LH, de Haan R, Vermeulen M. Intravenous immunoglobulin for multifocal motor neuropathy. Cochrane Database Syst Rev. 2005;18:CD004429.
318. Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. J Peripher Nerv Syst. 2006;11:1–8.
319. Van den Berg LH, Franssen H, Wokke JH. Improvement of multifocal motor neuropathy during long-term weekly treatment with human immunoglobulin. Neurology. 1995;45:987–988.
320. Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of multifocal motor neuropathy. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society – first revision. J Peripher Nerv Syst. 2010;15:295–301.
321. Bain PG, Motomura M, Newsom-Davis J, et al. Effects of intravenous immunoglobulin on muscle weakness and calcium-channel autoantibodies in the Lambert-Eaton myasthenic syndrome. Neurology. 1996;47:678–683.
322. Cherin P, Herson S, Weschsler B, et al. Efficacy of intravenous gammaglobulin therapy in chronic refractory polymyositis and dermatomyositis; an open study with 20 adult patients. Am J Med. 1991;91:162–168.
323. Dalakas MC. Intravenous immune globulin for dermatomyositis. N Engl J Med. 1994;330:1392–1393.
324. Kuwano Y, Ihn H, Yazawa N, et al. Successful treatment of dermatomyositis with high-dose intravenous immunoglobulin. Acta Derm Venereol. 2006;86:158–159.
325. Mastaglia FL, Phillips BA, Zilko PJ. Immunoglobulin therapy in inflammatory myopathies. J Neurol Neurosurg Psychiatry. 1998;63:107–110.
326. Williams L, Chang PY, Park E, Gorson KC, Bayer-Zwirello L. Successful treatment of dermatomyositis during pregnancy with intravenous immunoglobulin monotherapy. Obstet Gynecol. 2007;109(Pt 2):561–563.
327. Cherin P, Piette JC, Wechsler B, et al. Intravenous gamma globulin as first line therapy in polymyositis and dermatomyositis: An open study in 11 adult patients. J Rheumatol. 1994;21:1092–1097.
328. Cherin P, Pelletier S, Teixeira A, et al. Results and longterm followup of intravenous immunoglobulin infusions in chronic, refractory polymyositis: An open study with thirty five adult patients. Arthritis Rheum. 2002;46:467–474.
329. Genevay S, Saudan-Kister A, Guerne PA. Intravenous gammaglobulins in refractory polymyositis: Lower dose for maintenance treatment is effective. Ann Rheum Dis. 2001;60:635–636.
330. Wang DX, Shu XM, Tian XL, et al. Intravenous immunoglobulin therapy in adult patients with polymyositis/dermatomyositis: A systematic literature review. Clin Rheumatol. 2012;31(5):801–806.
331. Dalakas MC, Fujii M, Li M, Lufti B, Kyhos J, McElroy B. High-dose intravenous immune globulin for stiff-person syndrome. N Engl J Med. 2001;345:1870–1876.
332. Dalakas M, Koffman B, Fujii M, Spector S, Sivakumar K, Cupler E. A controlled study of intravenous immunoglobulin combined with prednisone in the treatment of IBM. Neurology. 2001;56:323–327.
333. Dalakas MC, Sonies B, Dambrosia J, Sekul E, Cupler E, Sivakumar K. Treatment of inclusion body myositis with IVIG: A double-blind placebo controlled study. Neurology. 1997;48:712–716.
334. Walter MC, Lochmuller H, Toepfer M, et al. High-dose immunoglobulin therapy in sporadic inclusion body myositis: A double blind placebo-controlled study. J Neurol. 2000;247:22–28.
335. Mukunda BN, Kumar PD, Smith HR. Long-lasting effectiveness of intravenous immunoglobulin in a patient with inclusion-body myositis. Ann Intern Med. 2001;134:1156.
336. Dalakas MC, Quarles RH, Farrer RG, et al. A controlled study of intravenous immunoglobulin in demyelinating neuropathy with IgM gammopathy. Ann Neurol. 1996;40:792–795.
337. Comi G, Roveri L, Swan A, et al. A randomised controlled trial of intravenous immunoglobulin in IgM paraprotein associated demyelinating neuropathy. J Neurol. 2002;249:1370–1377.
338. Gorson KC, Ropper AH, Weinberg DH, Weinstein R. Efficacy of intravenous immunoglobulin in patients with IgG monoclonal gammopathy and polyneuropathy. Arch Neurol. 2002;59:766–772.
339. Kornberg AJ, Pestronk A. Antibody-associated polyneuropathy syndromes: Principles and treatment. Semin Neurol. 2003;23:181–189.
340. Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies. Report of a Joint Task Force of the European Federation of Neurological Societies and the Peripheral Nerve Society—first revision. J Peripher Nerv Syst. 2010;15:185–195.
341. Courtney AE, McDonnell GV, Patterson VH. Human immunoglobulin for diabetic amyotrophy—a promising prospect? Postgrad Med. 2001;77:326–328.
342. Fernandes Filho JA, Nathan BM, Palmert MR, Katieji B. Diabetic amyotrophy in an adolescent responsive to intravenous immunoglobulin. Muscle Nerve. 2005;32:818–820.
343. Morlii T, Fujita H, Toyoshima I, Sageshima M, Ito S. Efficacy of immunoglobulin and prednisolone in diabetic amyotrophy. Endocr J. 2003;50:831–832.
344. Ogawa T, Taguchi T, Tanaka Y, Ikeguchi K, Nakano I. Intravenous immunoglobulin therapy for diabetic amyotrophy. Intern Med. 2001;40:349–352.
345. Said G, Goulon-Goeau C, Lacroix C, Moulonguet A. Nerve biopsy findings in different patterns of proximal diabetic neuropathy. Ann Neurol. 1994;35:559–569.
346. Pascoe MK, Low PA, Windebank AJ, Litchy WJ. Subacute diabetic proximal neuropathy. Mayo Clin Proc. 1997;72:1123–1132.
347. Romdenne P, Mukendi R, Stasse P, Indekeu P, Buysschaert M, Colin IM. An unusual neuropathy in a diabetic patient: Evidence for intravenous immunoglobulin induced effective therapy. Diabetes Metab. 2001;27:155–158.
348. Wada Y, Yanagihara C, Nishimura Y, Oka N. A case of diabetic amyotrophy with severe atrophy and weakness of shoulder girdle muscles showing good response to intravenous immune globulin. Diabetes Res Clin Pract. 2007;75:107–110.
349. Zochodne DW, Isaac D, Jones C. Failure of immunotherapy to prevent, arrest or reverse lumbosacral plexopathy. Acta Neurol Scand. 2003;107:299.
350. Mori M, Kuwabara S, Fukutake T, Hattori T. Intravenous immunoglobulin therapy for Miller Fisher syndrome. Neurology. 2007;68:1144–1146.
351. Kizawa M, Mori K, Iijima M, Koike H, Hattori N, Sobue G. Intravenous immunoglobulin treatment in painful sensory neuropathy without sensory ataxia associated with Sjogren’s syndrome. J Neurol Neurosurg Psychiatry. 2006;77:967–969.
352. Molina JA, BenitoLeon J, Bermejo F, Jimenez FJ, Olivan J. Intravenous immunoglobulin therapy in sensory neuropathy associated with Sjogren’s syndrome. J Neurol Neurosurg Psychiatry. 1996;60:699.
353. Pascual J, Cid C, Berciano J. High-dose i.v. immunoglobulin for peripheral neuropathy associated with Sjogren’s syndrome. Neurology. 1998;51:650–651.
354. Taguchi Y, Takashima S, Takata M, Dougu N, Asaoka E, Inoue H. High-dose intravenous immunoglobulin in the treatment of sensory ataxic neuropathy with Sjogren’s syndrome: A case report. No To Shinkei. 2004;56:421–424.
355. Takahashi Y, Takata T, Hoshino M, Sakurai M, Kanazawa I. Benefit of IVIG for long-standing ataxic sensory neuronopathy with Sjögren’s syndrome. Neurology. 2003;60:503–505.
356. Wolfe GI, Nations SP, Burns DK, Herbelin LL, Barohn RJ. Benefit of IVIG for long-standing ataxic sensory neuronopathy with Sjögren’s syndrome. Neurology. 2003;61:873.
357. Burns TM, Quijano-Roy S, Jones HR. Benefit of IVIG for long-standing ataxic sensory neuronopathy with Sjögren’s syndrome. Neurology. 2003;61:873.
358. Ponsford S, Willison H, Veitch J, Morris R, Thomas PK. Long-term clinical and neurophysiological follow-up of patients with peripheral neuropathy associated with benign monoclonal gammopathy. Muscle Nerve. 2000;23:164–174.
359. Levy Y, Uziel Y, Zandman GG, et al. Intravenous immunoglobulin in peripheral neuropathy associated with vasculitis. Ann Rheum Dis. 2003;62:1221–1223.
360. Tsurikisawa N, Taniguchi M, Saito H, et al. Treatment of Churg-Strauss syndrome with high-dose intravenous immunoglobulin. Ann Allergy Asthma Immunol. 2004;92:80–87.
361. Carpo M, Meucci N, Allaria S, et al. Anti-sulfatide IgM antibodies in peripheral neuropathy. J Neurol Sci. 2000;176:144–150.
362. Kuhl V, Vogt T, Anghelescu I. Intravenous immunoglobulin and prednisolone treatment of cryoglobulinemic polyneuropathy. Nervenarzt. 2001;72:445–448.
363. Gondim FA, Brannagan TH III, Sander HW, Chin RL, Latov N. Peripheral neuropathy in patients with inflammatory bowel disease. Brain. 2005;128:867–879.
364. Nobuhara Y, Saito M, Goto R, et al. Chronic progressive sensory ataxic neuropathy associated with limited systemic sclerosis. J Neurol Sci. 2006;241:103–106.
365. Lesprit P, Mouloud F, Bierling P, et al. Prolonged remission of SLE-associated polyradiculoneuropathy after a single course of intravenous immunoglobulin. Scand J Rheumatol. 1996;25:177–179.
366. Nakajima M, Fujioka S, Ohno H, Iwamoto K. Partial but rapid recovery from paralysis after immunomodulation during early stage of neuralgic amyotrophy. Eur Neurol. 2006;55:227–229.
367. Takakura, Y, Murai H, Furaya H, Ochi H, Kira J. A case of brachial diplegia accompanied with Sjögren’s syndrome presenting good response to immunotherapies in the early course of the disease. Rinsho Shinkeigaku. 2005;45:346–350.
368. Tsao BE, Avery R, Shield RW. Neuralgic amyotrophy precipitated by Epstein-Barr virus. Neurology. 2004;62:1234–1235.
369. Léger JM, Viala K, Nicolas G, et al. Placebo-controlled trial of rituximab in IgM anti-myelin associated glycoprotein neuropathy. Neurology. 2013;80:2217–2225.