TABLE 17-1. INFECTIOUS AGENTS ASSOCIATED WITH NEUROPATHIES
TABLE 17-1. INFECTIOUS AGENTS ASSOCIATED WITH NEUROPATHIESNeuropathies can result directly from various bacterial and viral infections, as well as from an indirect or parainfectious autoimmune response to the infection (Table 17-1). Parainfectious neuropathies (e.g., Guillain–Barré syndrome associated with various infections and vasculitis associated with hepatitis) are discussed in detail in other chapters.
TABLE 17-1. INFECTIOUS AGENTS ASSOCIATED WITH NEUROPATHIES
Bacterial:
Mycobacterium leprae (Leprosy)
Borrelia burdorferi (Lyme disease)
Corynebacterium diphtheriae (Diphtheria)
Viral:
Human immunodeficiency virus (HIV)
Distal symmetric polyneuropathy
Acute inflammatory demyelinating polyradiculoneuropathy
Chronic inflammatory demyelinating polyradiculoneuropathy
Other polyradiculoneuropathy
Mononeuropathy multiplex
Autonomic neuropathy
Sensory ganglionopathy
Human T-lymphocytic type 1 (HTLV-1)
Cytomegalovirus (CMV)
Hepatitis B and C
Herpes varicella-zoster (HVZ)
 LEPROSY (HANSEN DISEASE)
LEPROSY (HANSEN DISEASE)Leprosy is caused by the acid-fast bacteria Mycobacterium leprae. Leprosy is the most common cause of peripheral neuropathy in Southeast Asia, Africa, and South America. The main route of transmission is felt to be from person-to-person spread via nasal droplets. The bacteria is very slow growing with an incubation period that can vary between 2 and 40 years, customarily between 5 and 7 years.1
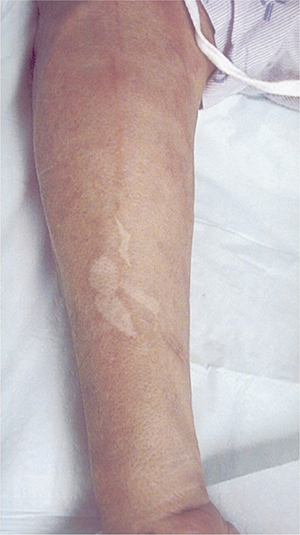
There is a spectrum of clinical manifestations ranging from tuberculoid leprosy at one end to lepromatous leprosy on the other end of the spectrum, with borderline leprosy in between based upon the Ridley–Joplin classification (Table 17-2).1–5 The World Health Organization (WHO) introduced a simpler classification based on the number of skin lesions to help guide treatment: multibacillary (six more skin lesions) and paucibacillary (fewer than six skin lesions) leprosy.1 In general, lepromatous leprosy is always multibacillary, and tuberculoid leprosy is usually paucibacillary; borderline leprosy can be either multibacillary or paucibacillary. The clinical manifestations of the disease are determined by the immunological response of the host to the infection. In tuberculoid leprosy, the cell-mediated immune response is intact.1–5 Thus, there are focal, circumscribed inflammatory responses to the bacteria within the affected areas of skin and nerves. The resulting skin lesions appear as well-defined, scattered hypopigmented patches and plaques with raised, erythematous borders (Figs. 17-1 and 17-2). Cutaneous nerves are often affected, resulting in a loss of sensation in the center of these skin lesions. Cooler regions of the body (e.g., face and limbs) are more susceptible than warmer regions such as the groin or axilla. In addition, the ulnar nerve at the medial epicondyle, the median nerve at the distal forearm, the peroneal nerve at the fibular head, the sural nerve, the greater auricular nerve, and the superficial radial nerve at the wrist are common sites of involvement and become encased with granulomas, leading to mononeuropathy or mononeuropathy multiplex. These nerves are thickened and often palpable.
TABLE 17-2. CLINICAL, LABORATORY, IMMUNOLOGICAL, AND HISTOPATHOLOGICAL FEATURES OF LEPROSY

Figure 17-1. Tuberculoid leprosy. Hypopigmented skin lesions are evident on lateral aspect of forearm in a patient with tuberculoid leprosy.
Figure 17-2. Borderline leprosy. A patient with borderline leprosy has multiple skin lesions with hypopigmented center with raised erythematous borders on the back (A) and on the leg (B). (Reproduced with permission from Amato AA, Dumitru D. Acquired neuropathies. In: Dumitru D, Amato AA, Swartz MJ, eds. Electrodiagnostic Medicine, 2nd ed. Philadelphia, PA: Hanley & Belfus; 2002.)
In lepromatous leprosy, cell-mediated immunity is severely impaired, leading to extensive infiltration of the bacilli and hematogenous dissemination, producing confluent and symmetrical areas of rash, anesthesia, and anhidrosis.1–5 Neuropathies tend to be more severe in the lepromatous subtype. As in the tuberculoid form, there is a predilection for the involvement of cooler regions of the body. Infiltration of the organism in the face leads to the loss of eyebrows and eyelashes and exaggeration of the natural skin folds, leading to the so-called “leonine facies.” Superficial cutaneous nerves of the ears and distal limbs are also commonly affected. A slowly progressive symmetric sensorimotor polyneuropathy gradually develops due to widespread invasion of the bacilli into the epi-, peri-, and endoneurium. Distal extremity weakness may be seen, but large fiber sensory modalities and muscle stretch reflexes are relatively spared. Involvement of nerve trunks leads to superimposed mononeuropathies, including facial neuropathy.
Neuropathies are most common in patients with borderline leprosy.2,3,5 Patients can develop generalized symmetric sensorimotor polyneuropathies, mononeuropathies, and mononeuropathy multiplex, including multiple mononeuropathies in atypical locations, such as the brachial plexus. Borderline leprosy is associated with clinical and histological features of both the lepromatous and the tuberculoid forms of leprosy (Table 17-2 and Fig. 17-3). There is partial impairment in cellular immunity in patients with borderline leprosy, such that there is some degree of mycobacterial spread as well as an inflammatory response. The immunological state is considered unstable in patients with borderline leprosy in that the immune response and clinical manifestations can shift up and down the spectrum.
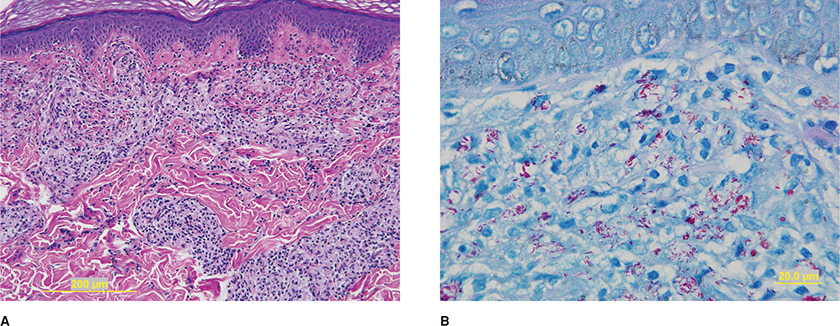
Figure 17-3. Borderline leprosy. Skin biopsy demonstrates marked inflammatory cell infiltrate, H&E (A). Red staining bacilli are evident on higher power with a Fite stain (B).
Patients with leprosy may present with isolated peripheral neuropathy without skin lesions, particularly in endemic areas.6,7 Most cases of the so-called pure neuritic leprosy have the tuberculoid or borderline tuberculoid subtypes of the disease.
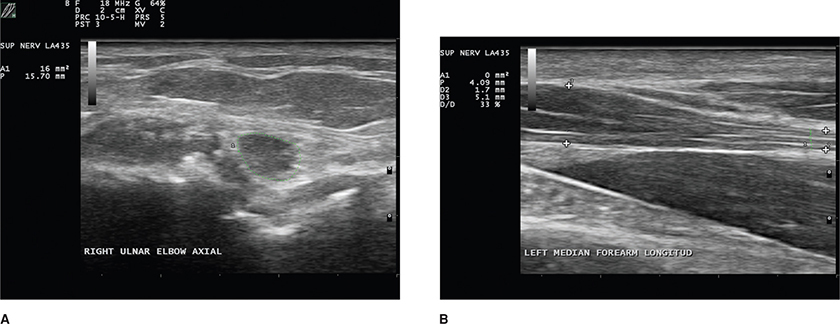
Sensory nerve conduction studies (NCS) are usually absent in the lower limb and are reduced in amplitude in the arms.1,6,7 Motor NCS may demonstrate reduced amplitudes in affected nerves.8,9 Motor conduction velocities are normal or slightly reduced; however, a few patients may demonstrate values less than 20 m/s in both the upper and the lower limb. Electromyography (EMG) reveals mild-to-moderate degrees of active denervation. The pattern of involvement on the EMG and NCS can be generalized as symmetric or reflective of a mononeuropathy or multiple mononeuropathies, as apparent from the clinical features. Ultrasound can demonstrate enlarged axons, particularly of the median nerve in distal forearm near the carpal tunnel and of the ulnar nerve just proximal to the medial epicondyle (Fig. 17-4).10
Figure 17-4. Ultrasound in patient with lepromatous leprosy demonstrates swelling of the ulnar nerve at that elbow just proximal to the medial epicondyle with an area of 16 mm-squared (normal <10 mm2) (A). There was also swelling of the median nerve in the distal forearm as it approached the carpel tunnel as the diameter 1.4 mm at D2 to 5.1 mm (B).
Leprosy is usually diagnosed with skin lesion biopsy and using the Fite method to stain the acid-fast bacilli red (Fig. 17-3).3 The morphological index (MI) is the ratio of viable to nonviable organisms on skin smears. The bacteriological index (BI) is a logarithmically scaled measure of the density of bacilli in the dermis. Both the MI and BI have been used to measure treatment response.
The host’s immune response to the bacilli determines the histopathology (Table 17-2).1–5 Nerve biopsies can also be diagnostic, particularly when there are no apparent skin lesions. The tuberculoid form is characterized by granulomas formed by macrophages and T lymphocytes (CD4 T lymphocytes greater than CD8). Caseatiing granulomas may or may not be present. Importantly, bacilli are not seen. In contrast, with lepromatous leprosy, large number of infiltrating bacilli, CD8 greater than CD4 lymphocytes, and organism-laden, foamy macrophages with minimal granulomatous infiltration are evident (Fig. 17-5A). The bacilli are best appreciated using the Fite stain, where they can be seen as red staining rods in clusters within the endoneurium, within macrophages, or within Schwann cells (Fig. 17-5B). On electron microscopy, the bacilli appear as dense osmiophilic rods surrounded by a clear halo (Figs. 17-5C and D). Borderline leprosy can have histological features of both tuberculoid and lepromatous leprosy.
Figure 17-5. Borderline leprosy. Sural nerve biopsy perivascular and diffuse endoneurial inflammation consisting of lymphocytes and macrophages, paraffin section stained with trichrome (A). Fite stain reveals red staining bacilli (the so-called “red snappers”) sometimes in clusters in the endoneurium and within Schwann cells (B). Electron microscopy reveals electron-dense bacilli with surrounding clear halos within the cytoplasm of a Schwann cell surrounding a myelinated axon (C) and on higher power within a Schwann cell surrounding unmyelinated axons (D).
The clinical and pathological spectrum of the disease is dependent on the host’s immune response to M. leprae and reflects the relative balance between the Th1 and Th2 response (Table 17-2).1–5 The tuberculoid form defines one end of the spectrum, in which the CD4 T cells predominate. These CD4 T cells produce interleukin-2 and gamma-interferon which in turn lead to activation of macrophages. On the other extreme, the lepromatous form is dominated by CD8 cells, which produce interleukin-4, interleukin-5, and interleukin-10, thereby downregulating cell-mediated immunity and inhibiting macrophages. The borderline subtypes exhibit immune responses spanning the spectrum between the tuberculoid and lepromatous forms.
Patients are treated with multiple drugs: dapsone, rifampicin, and clofazimine depending on the form of leprosy they have (Table 17-2).1–4 The current WHO recommendations for adults with multibacillary leprosy are as follows: rifampicin (600 mg once a month), dapsone (100 mg daily), and clofazimine (300 mg once a month and 50 mg daily) for 1 to 2 years.11 For adults with paucibacillary leprosy, the WHO recommends rifampicin (600 mg once a month) and dapsone (100 mg daily) for 6 months.11 For adults with single skin lesion paucibacillary leprosy the recommended WHO regimen is a single dose of rifampicin: 600 mg, ofloxacin: 400 mg, and minocycline: 100 mg. Proven relapses are re-treated with multidrug regimen.
The WHO recommendations are controversial as there have been no clinical trials to support their efficacy and the relapse rate of multibacillary leprosy is high. Thus, some advocate for more aggressive treatment based in part on Ridley–Joplin classification: tuberculoid leprosy to be treated with dapsone 100 mg daily for 5 years and lepromatous leprosy to be treated with rifampin 600 mg daily for 3 years and dapsone 100 mg daily for life.1
Patients should be instructed on the side effects of these medications before starting treatment. Rifampicin may make the urine turn a slightly reddish color for a few hours after its intake. Clofazimine causes brownish-black discoloration and dryness of skin. However, this disappears within few months after stopping treatment. The main side effect of dapsone is allergic reaction, causing itchy skin rashes and exfoliative dermatitis. Patients known to be allergic to drugs containing sulfa should not be given dapsone.
Treatment is sometimes complicated by the so-called reversal reaction, particularly in borderline leprosy.1–3 The reversal reaction can occur at any time during treatment and develops because of a shift to the tuberculoid end of the spectrum, as the result of an increase in cellular immunity during treatment. The cellular response is upregulated as evidenced by an increased release of tumor necrosis factor-alpha, gamma-interferon, and interleukin-2 with new granuloma formation. This can result in an exacerbation of the skin lesions and the neuropathy. High-dose corticosteroids blunt this adverse reaction and is often used prophylactically in high-risk patients (i.e., those with borderline leprosy) at treatment onset.
Erythema nodosum leprosum is another adverse reaction that usually occurs during treatment of patients with lepromatous leprosy.1–3,12 Multiple erythematous, sometimes painful, subcutaneous nodules appear, and may be associated with worsening of the neuropathy. Erythema nodosum leprosum probably results from slow degradation of bacilli and release of new antigens. Subsequently, antigen–antibody complexes form and complement is activated in affected tissue. Erythema nodosum leprosum is commonly treated with corticosteroids, clofazimine, or thalidomide.
A recent review looked at 13 studies involving 445 participants treated for erythema nodosum leprosum with corticosteroids, clofazimine, thalidomide, and other agents (i.e., pentoxifylline, indomethacin, and levamisole).12 The quality of the trials was generally poor and results could not be pooled due to the treatments being so heterogeneous. That said, clofazimine treatment was felt to be superior to prednisolone and thalidomide.12
Prevention of leprosy is of primary importance. It is recommended that children exposed to leprosy in the household be prophylactically treated with rifampin daily for 6 months.2,3 Various vaccinations are available, including BCG, killed leprae, and chemically modified organisms.
LYME DISEASELyme disease is caused by infection with Borrelia burgdorferi, a spirochete, transmitted by ticks. The deer tick, Ixodes dammini, is responsible for the disease in most cases. Ticks acquire the spirochetes by feeding on an infected host (e.g., deer) and then transmit the spirochetes to the next host (e.g., humans) at a later feed. It takes approximately 12–24 hours of tick attachment to transfer the spirochetes.
There are three recognized stages of Lyme disease: (1) early infection with localized erythema migrans, (2) disseminated infection, and (3) late-stage infection. The localized response occurs within 1 month of a tick bite. It consists of an erythematous circular region centered around the area of the original tick bite. The erythematous area gradually expands and the center of the lesion becoming clear creating a bull’s eye appearance. The rash resolves spontaneously after approximately a month. Importantly, not all patients with Lyme disease develop erythema migrans. The second stage of the illness is marked by dissemination of the spirochetes throughout the body. Patients develop systemic symptoms including fever, chills, localized adenopathy, fatigue, myalgias, headache, neck and back pain, and additional skin lesions about the body. Cardiac involvement may lead to pericarditis and heart block. Inflammatory arthritis of large and small joints may also occur.
Neurological complications may develop during the second and third stages of infection (Table 17-3).13–22 Facial neuropathy is the most common neurological manifestation of Lyme disease and is bilateral in about half of cases, which is rare for idiopathic Bell palsy. Involvement of nerves is frequently asymmetric. Patients with Lyme disease may also manifest with multiple mononeuropathies or, more commonly in our experience, with radiculopathy or polyradiculopathy. Although often considered in the differential diagnosis of GBS, it usually does not resemble cases of GBS given the asymmetric nature and electrophysiological features (see below). Rarely, affected patients develop an inflammatory myopathy as opposed to neuropathy.22
TABLE 17-3. NEUROLOGICAL DISORDERS ASSOCIATED WITH LYME DISEASE
Encephalitis/meningitis
Myelitis
Cranial neuropathies (e.g., facial nerve palsy)
Peripheral neuropathy
Mononeuropathies
Multiple mononeuropathies
Radiculopathy
Plexopathy
Inflammatory myopathy
The late stage of infection is characterized by further destructive inflammatory changes in the joints. The distal extremities develop a bluish discoloration of the skin (acrodermatitis chronica atrophicans). Spirochetes may be readily cultured from biopsies of these sites. Approximately 50% of patients have numbness, paresthesia, weakness, and cramps in the distal extremities, and proprioception and vibration are reduced as are muscle stretch reflexes.
Examination of the cerebrospinal fluid (CSF) should demonstrate lymphocytic pleocytosis and increased protein in patients with polyradiculitis, cranial neuropathies, and central nervous system involvement. Immunofluorescent or enzyme-linked immunosorbent assay may detect antibodies directed against the spirochete in the serum and CSF. False-positive reactions are not uncommon and, therefore, Western blot analysis should be performed to confirm a positive enzyme-linked immunosorbent assay.
Electrodiagnostic studies are suggestive of a primary axonopathy. In a patient with a mononeuropathy or multiple mononeuropathies, NCS typically reveal reduced compound muscle action potential (CMAP) and sensory nerve action potential (SNAP) amplitudes.13,17–22 Those with facial nerve palsies have reduced facial nerve CMAPs and abnormal blink reflexes.15 The electrophysiological abnormalities are often asymmetric.23,24 Needle EMG reveals increased insertional and spontaneous activity in the form of fibrillation potentials and positive sharp waves and decreased recruitment of neurogenic-appearing motor unit action potentials (MUAPs). Patients presenting with a radiculopathy may have normal motor and sensory NCS, but the EMG is abnormal as above.
Nerve biopsies are not typically performed in patients with Lyme disease and symptoms of neuropathy, but can reveal perivascular infiltration of plasma cells and lymphocytes around small endoneurial, perineurial, and epineurial blood vessels without clear necrotizing vasculitis. Axonal degeneration and secondary demyelination can be seen.
Peripheral nerve involvement may be the result of an indirect immunological response and/or some form of vasculopathy.
Recommended treatment of facial nerve palsies in adults is the combination of amoxicillin 500 mg p.o. q.i.d. plus probenecid 500 mg q.i.d. for 2–4 weeks. Patients who are allergic to penicillin can be treated with doxycycline 100 mg p.o. b.i.d. for 2–4 weeks. Children less than 4 years of age can be treated with amoxicillin 20–40 mg/kg/d in four divided doses for 2–4 weeks. If allergic to penicillin, children can be treated with erythromycin 30 mg/kg/d in four divided doses for 2–4 weeks.
Adult patients with other types of peripheral neuropathy are treated with intravenous (IV) penicillin 20–24 million units/d for 10–14 days or ceftriaxone 2 g IV q.d. for 2–4 weeks. Those allergic to penicillin should receive doxycycline 100 mg p.o. b.i.d. for 30 days. Children with Lyme neuropathy can receive IV penicillin G 250,000 U/kg/d in divided doses for 10–14 days or ceftriaxone 50–80 mg/kg/d IV for 2–4 weeks.
DIPHTHERITIC NEUROPATHYDiphtheria is caused by the bacteria Corynebacterium diphtheriae. Individuals who are infected present with “flu-like” symptoms of generalized myalgias, headache, fatigue, low-grade fever, and irritability within a week to 10 days of the exposure. A whitish membranous exudate may be appreciated in the pharynx with or without swollen or tender cervical lymph nodes. Cardiovascular involvement can manifest as cardiac arrhythmias and hypotension. About 20–70% of patients develop a peripheral neuropathy caused by a toxin released by the bacteria.25–28 Three to four weeks after infection, patients may note decreased sensation in their throat and begin to develop dysphagia, dysarthria, or hoarseness. Around the same time, patients develop blurred vision, particularly when looking at near objects. Pupils react to light but fail to accommodate. Additional cranial nerves may also become involved. Ventilatory muscle weakness can develop due to phrenic nerve involvement. A generalized polyneuropathy may manifest 2 or 3 months following the initial infection characterized by numbness, paresthesia, and weakness of the arms and legs. Neurological examination reveals a reduction in perception of all sensory modalities. Distal greater than proximal weakness is seen. Weakness may progress over a period of weeks, such that patients are unable to ambulate. Muscle stretch reflexes are diminished or absent throughout in keeping with the demyelinating nature of the neuropathy. Rarely, bowel and bladder function are affected.
CSF protein can be elevated with or without a lymphocytic pleocytosis.29 Sensory NCS often reveal absent SNAPs.27 Motor NCS demonstrate markedly reduced conduction velocities (<50% of mean values) in the arms and legs.26,27,29,30 The distal motor latencies are only mildly to moderately prolonged. The NCS become abnormal by 2 weeks following onset of neuropathic symptoms and reach their nadir by 5–8 weeks. Subsequently, there is slow and steady improvement in the NCS that lag behind clinical recovery.
Segmental demyelination and axonal degeneration have been appreciated in the nerve roots and the more distal segments of the peripheral nerve.25 Degeneration of dorsal root ganglia may be observed as well.
The bacteria release diphtheria exotoxin which binds to Schwann cells and inhibits synthesis of myelin proteins.31
Antitoxin and antibiotics should be given within 48 hours of symptom onset. Although early treatment reduces the incidence and severity of some complications (i.e., cardiomyopathy), it does not appear to alter the natural history of the associated peripheral neuropathy. The neuropathy usually resolves after several months. Patients need to be managed with supportive care (e.g., mechanical ventilation, PE prophylaxis, physical therapy) as discussed in the chapter on GBS (Chapter 13).
HUMAN IMMUNODEFICIENCY VIRUSHuman immunodeficiency virus (HIV) infection can result in a variety of neuromuscular complications (Table 17-4), including peripheral neuropathies. Approximately 20% of individuals infected with HIV develop a neuropathy which may be as a direct result of the virus itself, other associated viral infections [e.g., cytomegalovirus (CMV) infection], or neurotoxicity secondary to antiviral medications.32–35 The neuropathy associated with antiviral medications are discussed in Chapter 20. The major presentations of peripheral neuropathy associated with HIV infection include (1) distal symmetric polyneuropathy (DSP), (2) inflammatory demyelinating polyneuropathy (including both AIDP and CIDP), (3) multiple mononeuropathies (e.g., vasculitis, CMV related), (4) polyradiculopathy (usually CMV related), (5) autonomic neuropathy, and (6) sensory ganglionitis.36–43
TABLE 17-4. NEUROLOGICAL COMPLICATIONS ASSOCIATED WITH HIV INFECTION
Central nervous system
Opportunistic infections
Progressive multifocal leukoencephalopathy
HIV-associated encephalopathy (AIDS dementia)
Lymphomas and other malignancies
Subacute combined degeneration (B12 deficiency)
Vacuolar myelopathy
Peripheral nervous system disorders
Distal symmetric polyneuropathy
Motor neuronopathy
Acute and chronic inflammatory demyelinating polyradiculoneuropathy
Polyradiculoneuropathy/multiple mononeuropathies caused by other infections (e.g., cytomegalovirus, hepatitis B or C, and herpes zoster)
Autonomic neuropathy
Sensory ganglionopathy
Toxic neuropathy (antiretroviral medications)
Myopathy
Toxic myopathy (antiretroviral medications)
Inflammatory (polymyositis or inclusion body myositis)
Infectious (opportunistic infections)
Myopathy secondary to wasting/cachexia
HIV-RELATED DISTAL SYMMETRIC POLYNEUROPATHYDSP is the most common form of peripheral neuropathy associated with HIV infection and usually is seen in patients with AIDS.32,33,35,44,45 It is characterized by numbness and painful paresthesia involving the distal legs and arms. Some patients are asymptomatic but have reduced sensation to all modalities on examination. Mild distal muscle weakness may be appreciated. Proximal leg and distal arm weakness may develop late in the course of the disease. Muscle stretch reflexes are reduced at the ankles but are relatively preserved at the knees and in the arms.
CSF examination may demonstrate an increased protein and mild lymphocytic pleocytosis in patients with HIV infection regardless of the stage of the infection and the presence or absence of peripheral neuropathy.46,47 Vitamin B12 deficiency is noted in some48,49 but not all patients.50–52 NCS and EMG reveal abnormalities suggestive of a symmetric, axonal sensory greater than motor polyneuropathy.32,34,53–56
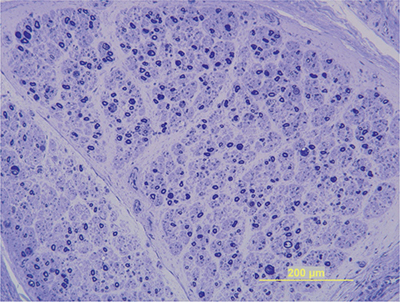
Nerve biopsies are not routinely performed in HIV patients with DSP but typically reveal axonal degeneration and a loss in the total number of both myelinated and demyelinated axons (Fig. 17-6).41,53,56–59 A reduction of cell bodies in the dorsal root ganglia may be appreciated, as well as secondary degeneration of the dorsal columns. Mild perivascular inflammation consisting of macrophages and T lymphocytes is seen along with the evidence of increased cytokine expression. Reduced density of small myelinated epidermal nerve fibers is appreciated in the epidermis with skin biopsy.60
Figure 17-6. HIV neuropathy. Sural nerve biopsy in a patient with distal symmetric sensory neuropathy demonstrates a mild reduction in myelinated nerve fibers. Epoxy embedded, toluidine blue stain.
The pathogenic basis for DSP is unknown but is not due to actual infection of the peripheral nerves. Viral coat proteins may mediate nerve fiber damage and hyperalgesia through direct and indirect mechanisms.45 These proteins may be directly toxic to axons. In addition, the glial and macrophage response activated by virus may indirectly damage neurons by the release of cytokines from surrounding inflammatory cells. Vitamin B12 deficiency may contribute to some cases but is not a major cause of most cases of DSP. Various antiretroviral agents (e.g., dideoxycytidine, dideoxyinosine, and stavudine) are also neurotoxic and may cause a painful sensory neuropathy.55,61–63
The neuropathy is not responsive to treatment with antiretroviral medications and therapy is largely symptomatic. We usually initiate treatment with neuropathic pain medications (see Chapter 22, Table 22-3).
HIV-RELATED INFLAMMATORY DEMYELINATING POLYRADICULONEUROPATHYBoth AIDP and CIDP can occur as a complication of HIV infection.35,45,64 AIDP usually develops at the time of seroconversion, whereas CIDP can occur anytime in the course of the infection. Clinical features are indistinguishable from idiopathic AIDP or CIDP.
In addition to elevated protein levels, lymphocytic pleocytosis is evident in the CSF—a finding that helps distinguish HIV-associated polyradiculoneuropathy from idiopathic AIDP/CIDP. Motor and sensory NCS are similar to that seen in idiopathic AIDP and CIDP (Chapters 13 and 14).30,64–66 Motor and sensory NCS may demonstrate slow conduction velocities, prolonged distal latencies and F waves, conduction block, and/or temporal dispersion.
Nerve biopsies are not be performed routinely, but can show features identical to those found in idiopathic AIDP and CIDP.30,57
We usually treat patients with HIV-associated AIDP or CIDP with intravenous immunoglobulin (IVIg) or plasmapheresis (PE).30,65 Prednisone can be used in CIDP, but we try to avoid steroids and other second-line immunosuppressive agents because of the long-term implications of immunosuppression in patients with HIV.
HIV-RELATED PROGRESSIVE POLYRADICULOPATHY (SECONDARY CMV INFECTION)An acute, progressive lumbosacral polyradiculoneuropathy secondary to CMV infection can develop in patients with AIDS.35,45 Patients usually present with severe radicular pain, numbness, and weakness in the legs, which is usually asymmetric. Loss of perineal sensation with bowel and bladder incontinence is common. The arms and cranial nerves may also be affected. Reduced or absent muscle stretch reflexes are appreciated on examination. Plantar responses are usually flexor but can be extensor if a superimposed CMV myelitis is also present. Patients usually have evidence of CMV infection in other parts of the body (i.e., CMV retinitis).
CSF is abnormal, demonstrating an increased protein level along with a reduced glucose concentration and, notably, a neutrophilic pleocytosis. CMV can be cultured from the CSF, blood, and urine. NCS often demonstrate an asymmetric reduction of amplitudes of the SNAPs and CMAPs with active denervation changes on EMG in muscles innervated by affected nerve roots and nerves including the paraspinals.67,68 The axonal nature and distribution of these abnormalities are quite distinct from those found in both CIDP and DSP respectively, helping to differentiate these various disorders.
With postmortem examination, inflammatory infiltrates associated with varying degrees of axonal loss are evident in the ventral and dorsal roots, particularly in the lumbar regions. Occasionally, the cranial nerves exiting from the brainstem may be involved in association with the myelitis. CMV inclusions may be found in endothelial cells and macrophages on nerve biopsy specimens when obtained.69
The polyradiculoneuropathy may be caused by the direct infection of neurons by CMV or ischemia secondary to associated vasculitis.
The polyradiculoneuropathy may improve with ganciclovir or foscarnet, if treatment is started early.68,70 However, the prognosis is poor, and most patients die within several weeks or months.
HIV-RELATED MULTIPLE MONONEUROPATHIESMultiple mononeuropathies can also develop in patients with HIV infection usually in the context of AIDS.35,45 Weakness, numbness, paresthesia, and pain are evident in the distribution of affected nerves.
Elevated CSF protein and mononuclear pleocytosis may be seen. EMG and NCS demonstrate features of axon loss as seen with other forms of multiple mononeuropathies caused by vasculitis (see Chapter 15).71
Nerve biopsies can reveal axonal degeneration with necrotizing vasculitis or perivascular inflammation.72 CMV inclusions may be seen in endothelial cells and macrophages on electron microscopy.69
The pathogenic basis for this disorder is likely multifactorial. The neuropathy may be caused by vasculitis related to deposition of HIV antigen–antibody complexes in the walls of blood vessel, concomitant hepatitis B or C infection, or CMV infection.
Corticosteroid treatment is indicated in vasculitis directly due to HIV infection. Multiple mononeuropathies secondary to concurrent hepatitis B or C infection can be treated with plasma exchange, antiviral agents (e.g., vidarabine), or α-interferon. Short courses of prednisone and cyclophosphamide may be necessary. If CMV is suspected, treatment with ganciclovir or foscarnet should be initiated.
HIV-RELATED AUTONOMIC NEUROPATHYAn autonomic neuropathy characterized by orthostatic hypotension, impaired sweating, diarrhea, impotence, and bladder dysfunction can develop acutely or insidiously in patients with HIV infection.73–75 Clinical features are similar to those seen with idiopathic autonomic neuropathy.
CSF can reveal pleocytosis and increased protein. Most patients have electrodiagnostic features similar to that noted in DSP. In addition, autonomic function testing is usually abnormal.74
An immune-mediated mechanism similar to that suspected in idiopathic autonomic neuropathy is likely.
A trial of corticosteroids, IVIg, or PE may be tried. Symptoms of autonomic neuropathy are treated symptomatically.
HIV-RELATED SENSORY NEURONOPATHY/GANGLIONOPATHYDorsal root ganglionitis is a very rare complication of HIV infection, but neuronopathy can be the presenting manifestation.76 Patients develop sensory ataxia similar to idiopathic sensory neuronopathy/ganglionopathy. Autopsies have demonstrated inflammatory cell infiltrate in the dorsal root ganglia along with the loss of cell bodies and degeneration of myelinated nerve fibers in the peripheral nerves. NCS reveal amplitudes or absence of SNAPs. Again, a trial of corticosteroids, IVIg, or PE may be considered.
HUMAN T-LYMPHOCYTE TYPE 1 INFECTIONBesides the more common myelopathy (tropical spastic paraparesis), human T-lymphocyte type 1 (HTLV-1) infection is also associated with an axonal sensorimotor polyneuropathy.77–79 The neuropathy can be seen even in patients without a myelopathy. HTLV-1 infection has also been associated with myositis. NCS demonstrate abnormalities suggestive of an axonal, sensory greater than motor, length-dependent neuropathy.79 Sural nerve biopsy when performed can reveal axonal degeneration with secondary demyelination and inflammatory cell infiltrates. There is one report of CIDP occurring in the setting of HTLV-1 infection.80
CYTOMEGALOVIRUSCMV can cause an acute lumbosacral polyradiculopathy and multiple mononeuropathies in patients with HIV infection or other causes of severe immunosuppression as previously noted.
EPSTEIN–BARR VIRUSEpstein–Barr virus infection has been associated with AIDP, cranial neuropathies, mononeuropathy multiplex, brachial plexopathy, lumbosacral radiculoplexopathy, and sensory neuronopathies.81
HEPATITIS VIRUSESHepatitis B and C can cause multiple mononeuropathies related to vasculitis, AIDP, or CIDP, as previously discussed (Chapters 13 and 14).
HERPES VARICELLA-ZOSTER VIRUSPeripheral neuropathy from herpes varicella-zoster (HVZ) infection is the result of reactivation of latent virus or a primary infection. Primary infection is the cause of “chicken pox.” Reactivation of the virus later in life leads to dermal zoster. In patients who are immunocompromised, HVZ infection can be associated with severe disseminated zoster. Two-thirds of infections in adults are characterized by dermal zoster, in which severe pain and paresthesias develop in a dermatomal region, followed within a week or two by a vesicular rash in the same distribution. The vesicular skin lesions clear by 2 weeks. Approximately 25% of patients who are affected have continued pain (postherpetic neuralgia). In a large series of patients, zoster developed in thoracic dermatomes in nearly 50%, lumbosacral region in 18%, trigeminal distribution in the head in an additional 18%, and the cervical dermatomes in the remainder.
Weakness in muscles innervated by roots corresponding to the dermatomal distribution of skin lesions occurs in 5–30% of patients.82–84 The weakness usually develops within the first 2 weeks of the skin eruption but can vary between several hours and a month. Unilateral phrenic nerve involvement can lead to hemidiaphragmatic paralysis.85 When the thoracic myotomes are involved, hernias can occur through weakened abdominal wall musculature.86,87 Muscle strength usually improves over time. Rarely, patients develop AIDP following HVZ infection.88,89 Additional neurological manifestations of herpes zoster infection include encephalitis and angiitis leading to vascular events.
CSF protein may be elevated with or without pleocytosis. The virus is difficult to culture from the CSF, but polymerase chain reaction can be used to confirm the presence of the virus in the CSF. Sensory NCS reveal reduced or absent SNAPs in affected nerves.90–92 Motor NCS demonstrate normal reduced CMAP amplitudes.83,84,90,92 Positive sharp waves and fibrillation potentials and neurogenic-appearing MUAPs and recruitment can be observed on needle EMG in muscles of affected myotomes.83,84,90,92
The basic pathological neural reaction is that of axonal degeneration with some degree of secondary segmental demyelination. With respect to the sensory system, severe infections can result in the destruction of dorsal root ganglion cells with the secondary loss of posterior column fibers.
Following initial infection, the HVZ migrates up the sensory nerves and takes residence in the sensory ganglia, where the virus appears to be insulated from the host’s immune defense mechanisms. When the host becomes immunosuppressed, the virus can reactivate and replicate. HVZ travels down the sensory nerves including cutaneous nerves and result in the typical cutaneous zoster lesions. The inflammatory response in the spinal nerve may involve motor axons resulting in muscle weakness.
Acyclovir helps improve the rate of healing of the skin lesions, but acyclovir neither alone nor in combination with corticosteroids reduces the frequency or severity of postherpetic neuralgia. Intravenous acyclovir should be administered in immunocompromised patients with severe infections. The treatment of postherpetic neuralgia is symptomatic. Our first-line treatment of choice is lidoderm patches applied over the regions with neuralgic pain. Gabapentin,93 carbamazepine, topical capsaicin ointment, and tricyclic antidepressants94 may also reduce the pain in some patients. Opioids are warranted as well in patients with refractory pain.95
SUMMARYNeuropathies associated with infection are not uncommon. In fact, lepromatous neuropathy may be the most common form of neuropathy, particularly in nonindustrialized nations. Further, neuropathies related to HIV infection have increased, owing to the spread of this infection and longer life span of treated individuals rendering them susceptible to the neurotoxic effects of the infection and antiretroviral infections. Lyme disease likewise needs to be considered in endemic regions. Notably, many of these neuropathies are treatable; thus, diagnosis is essential.
1. Rodrigues LC, Lockwood DN. Leprosy now: epidemiology, progress, challenges, and research gaps. Lancet Infect Dis. 2011;11:464–470.
2. Altman D, Amato A. Lepromatous neuropathy. J Clin Neuromuscul Dis. 1999;1:68–73.
3. Nations SP, Katz JS, Lyde CB, Barohn RJ. Leprous neuropathy: an American perspective. Semin Neurol. 1998;18(1):113–124.
4. Ooi WW, Srinivasan J. Leprosy and the peripheral nervous system: basic and clinical aspects. Muscle Nerve. 2004;30(4): 393–409.
5. Ridley DS, Jopling WH. Classification of leprosy according to immunity. A five-group system. Int J Lepr Other Mycobact Dis. 1966;34:255–276.
6. Rodriguez G, Sanchez W, Chaleta JG, Soto J. Primary neuritic leprosy. J Am Acad Dermatol. 1993;29:1050–1052.
7. Jardim MR, Chimelli L, Faria SC, et al. Clinical, electroneuromyographic and morphological studies of pure neural leprosy in a Brazilian referral centre. Lepr Rev. 2004;75(3):242–253.
8. McLeod JG, Hargrave JC, Walsh JC, Booth GC, Gye RS, Barron A. Nerve conduction studies in leprosy. Int J Lepr Other Mycobact Dis. 1975;43:21–31.
9. Amato AA, Dumitru D. Acquired neuropathies. In: Dumitru D, Amato AA, Swartz MJ, eds. Electrodiagnostic Medicine, 2nd ed. Philadelphia, PA: Hanley & Belfus; 2002:937–1041.
10. Bathala L, Kumar K, Pathapati R, Jain S, Visser LH. Ulnar neuropathy in hansen disease: clinical, high-resolution ultrasound and electrophysiologic correlations. J Clin Neurophysiol. 2012;29:190–193.
11. World Health Organization (WHO) recommended MDT regimens. http://www.who.int/lep/mdt/regimens/en
12. Van Veen NH, Lockwood DN, Van Brakel WH, Ramirez J Jr, Richardus JH. Interventions for erythema nodosum leprosum. A Cochrane review. Lepr Rev. 2009;80:355–372.
13. Halperin JJ, Little BW, Coyle PK, Dattwyler RJ. Lyme disease: cause of a treatable peripheral neuropathy. Neurology. 1987;37:1700–1706.
14. Halperin J, Luft BJ, Volkman DJ, Dattwyler RJ. Lyme neuroborreliosis. Brain. 1990;113:1207–1221.
15. Krishnamurthy KB, Liu GT, Logigian EL. Acute Lyme neuropathy presenting with polyradicular pain, abdominal protrusion, and cranial neuropathy. Muscle Nerve. 1993; 16:1261–1264.
16. Logigian EL, Steere AC. Clinical and electrophysiologic findings in chronic neuropathy of Lyme disease. Neurology. 1992; 42:303–311.
17. Oey PL, Franssen H, Bersen RA, Wokke JH. Multifocal conduction block in a patient with Borrelia burgdorferi infection. Muscle Nerve. 1991;14:375–377.
18. Scelsa SN, Hershkovitz S, Berger AR. A predominantly motor polyradiculopathy of Lyme disease. Muscle Nerve. 1996; 19:780–783.
19. Pachner AR, Steere AC. The triad of neurologic manifestations of Lyme disease: meningitis, cranial neuritis, and radiculoneuritis. Neurology. 1985;35:47–53.
20. Vallat JM, Hugon J, Lubeau M, Leboutet MJ, Dumas M, Desproges-Gotteron R. Tick-bite meningoradiculoneuritis: clinical, electrophysiologic, and histologic findings in 10 cases. Neurology. 1987;37:749–753.
21. Wulff CH, Hansen K, Strange P, Trojaborg W. Multiple mononeuritis and radiculitis with erythema, pain, elevated CSF protein and pleocytosis (Bannwarth’s syndrome). J Neurol Neurosurg Psychiatry. 1983;46:485–490.
22. Schoenen J, Sianard-Gainko J, Carpentier M, Reznik M. Myositis during Borrelia burgdorferi infection (Lyme disease). J Neurol Neurosurg Psychiatry. 1989;52:1002–1005.
23. Halperin JJ, Volkman DJ, Luft BJ, Dattwyler RJ. Carpal tunnel syndrome in Lyme borrelliosis. Muscle Nerve. 1989;12: 397–400.
24. Halperin JJ, Pass HL, Anand AK, Luft BJ, Volkman DJ, Dattwyler RJ. Nervous system abnormalities in Lyme disease. Ann N Y Acad Sci. 1988;539:24–34.
25. Fisher CM, Adams RD. Diphtheritic polyneuritis; a pathological study. J Neuropathol Exp Neurol. 1956;15:243–268.
26. Kazemi B, Tahjernia AC, Zandian K. Motor nerve conduction in diphtheria and diphtheritic myocarditis. Arch Neurol. 1973;29:104–106.
27. Kurdi A, Abdul-Kader M. Clinical and electrophysiological studies of diphtheritic neuritis in Jordan. J Neurol Sci. 1979;42:243–250.
28. Solders G, Nennesmo I, Persson A. Diphtheritic neuropathy, an analysis based on muscle and nerve biopsy and repeated neurophysiological and autonomic function tests. J Neurol Neurosurg Psychiatry. 1989;52:876–880.
29. Créange A, Meyrignac C, Roualdes B, Degos JD, Gherardi RK. Diphtheritic neuropathy. Muscle Nerve. 1995;18:1460–1463.
30. Cornblath DR, McArthur JC, Kennedy PG, Witte AS, Griffin JW. Inflammatory demyelinating peripheral neuropathies associated with human T-cell lymphotropic virus type III infection. Ann Neurol. 1987;21:32–40.
31. Pleasure DE, Feldmann B, Prokop DJ. Diphtheria toxin inhibits the synthesis of myelin proteolipid and basic proteins by peripheral nerve in vivo. J Neurochem. 1973;20:81–90.
32. Floeter MK, Civetello LA, Everett CR, Dambrosia J, Luciano CA. Peripheral neuropathy in children with HIV infection. Neurology. 1997;49:207–212.
33. Marra CM, Boutin P, Collier AC. Screening for distal sensory peripheral neuropathy in HIV-infected persons in research and clinical settings. Neurology. 1998;51:1678–1681.
34. Tagliati M, Grinnell J, Godbold J, Simpson DM. Peripheral nerve function in HIV infection: clinical, electrophysiologic, and laboratory findings. Arch Neurol. 1999;56:84–89.
35. Robinson-Papp J, Simpson DM. Neuromuscular diseases associated with HIV-1 infection. Muscle Nerve. 2009;40:1043–1053.
36. Barohn RJ, Gronseth GS, LeForce BR, et al. Peripheral nervous system involvement in a large cohort of human immunodeficiency virus-infected individuals. Arch Neurol. 1993;50:167–171.
37. Cornblath DR, McArthur JC. Predominantly sensory neuropathy in patients with AIDS and AIDS-related complex. Neurology. 1988;38:794–796.
38. Dalakas MC, Pezeshkpour GH. Neuromuscular diseases associated with human immunodeficiency virus infection. Ann Neurol. 1988;23:S38–S48.
39. de la Monte SM, Gabuzda DH, Ho DD, et al. Peripheral neuropathy in the acquired immunodeficiency syndrome. Ann Neurol. 1988;23:485–492.
40. Fuller GN, Jacobs JM, Guiloff RJ. Nature and incidence of peripheral nerve syndrome in HIV infection. J Neurol Neurosurg Psychiatry. 1993;56:372–381.
41. Hall CD, Snyder CR, Messenheimer JA, et al. Peripheral neuropathy in a cohort of human immunodeficiency virus infected patients. Incidence and relationship to other nervous system dysfunction. Arch Neurol. 1991;48:1273–1274.
42. Lange DJ. AAEM minimonograph #41: neuromuscular diseases associated with HIV-1 infection. Muscle Nerve. 1994;17:16–30.
43. McArthur JC, Cohen BA, Selnes OA, et al. Low prevalence of neurological and neuropsychological abnormalities in otherwise healthy HIV-1-infected individuals: results from the multicenter AIDS Cohort Study. Ann Neurol. 1989;26:601–611.
44. Kamerman PR, Moss PJ, Weber J, Wallace VC, Rice AS, Huang W. Pathogenesis of HIV-associated sensory neuropathy: evidence from in vivo and in vitro experimental models. J Peripher Nerv Syst. 2012;17:19–31.
45. Simpson DM, Olney RK. Peripheral neuropathies associated with human immunodeficiency virus infection. Neurol Clin. 1992;10:685–711.
46. Barohn RJ, Gronseth GS, Amato AA, et al. Is there any relationship between cerebral spinal fluid and nerve conduction abnormalities in HIV positive individuals? J Neurol Sci. 1996;136:81–85.
47. Marshall DW, Brey RL, Butzin CA, et al. CSF changes in a longitudinal study of 124 neurologically normal HIV-1-infected U.S. Air Force personnel. J Acquir Immune Defic Syndr. 1991;4:777–781.
48. Beach RS, Morgan R, Wilkie F, et al. Plasma vitamin B12 level as a potential cofactor in studies of human immunodeficiency virus type 1-related cognitive changes. Arch Neurol. 1992;49:501–506.
49. Kieburtz KD, Giang DW, Schiffer RB, Vakil N. Abnormal vitamin B12 metabolism in human immunodeficiency virus infection: association with neurological dysfunction. Arch Neurol. 1991;48:312–314.
50. Dal Pan GJ, Allen RH, Glass JD, et al. Cobalamin (vitamin B12)-dependent metabolism is not altered in HIV-1-associated vacuolar myelopathy. Ann Neurol. 1993;34:281–282.
51. Robertson KR, Stern RA, Hall CD, et al. Vitamin B12 deficiency and nervous system disease in HIV infection. Arch Neurol. 1993;50:807–811.
52. Veilleux M, Paltiel O, Falutz J. Sensorimotor neuropathy and abnormal vitamin B12 metabolism in early HIV infection. Can J Neurol Sci. 1995;22:43–46.
53. Bailey RO, Baltch AL, Venkatesh R, Singh JK, Bishop MB. Sensory motor neuropathy associated with AIDS. Neurology. 1988;38:886–891.
54. Chavanet P, Solary E, Giroud M, et al. Infraclinical neuropathies related to immunodeficiency virus infection associated with higher T-helper cell count. J Acquir Immune Defic Syndr. 1989;2:564–569.
55. Dubinsky RM, Yarchoan R, Dalakas M, Broder S. Reversible axonal neuropathy from treatment of AIDS and related disorders with 2′,3′-dideoxycytidine (ddC). Muscle Nerve. 1989;12:856–860.
56. Fuller GN, Jacobs JM, Guiloff RJ. Subclinical peripheral nerve involvement in AIDS: an electrophysiological and pathological study. J Neurol Neurosurg Psychiatry. 1991;54:318–324.
57. Chaunu MP, Ratinahirana H, Raphael M, et al. The spectrum of the changes on 20 nerve biopsies in patients with HIV infection. Muscle Nerve. 1989;12:452–459.
58. Fuller GN, Jacobs JM, Guiloff RJ. Axonal atrophy in painful peripheral neuropathy in AIDS. Acta Neuropathol. 1990;81:198–203.
59. Rance NE, McArthur JC, Cornblath DR, Landstrom DL, Griffin JW, Price DL. Gracile tract degeneration in patients with sensory neuropathy and AIDS. Neurology. 1998;38:265–271.
60. Herrman DN, Griffin JW, Hauer P. Intraepidermal nerve fiber density, sural nerve morphometry and electrodiagnosis in peripheral neuropathies. Neurology. 1999;53:1634–1640.
61. Berger AR, Arezzo JC, Schaumburg HH, et al. 2′,3′-dideoxycytidine (ddC) toxic neuropathy: a study of 52 patients. Neurology. 1993;43:358–362.
62. Kieburtz KD, Seidlin M, Lambert JS, Dolin R, Reichman R, Valentine F. Extended follow-up of peripheral neuropathy in patients with AIDS and AIDS-related complex treated with dideoxyinosine. J Acquir Immune Defic Syndr. 1992;5:60–64.
63. Leung GP. Iatrogenic mitochondriopathies: a recent lesson from nucleoside/nucleotide reverse transcriptase inhibitors. Adv Exp Med Biol. 2012;942:347–369.
64. Brannagan TH III, Zhou Y. HIV-associated Guillain–Barre syndrome. J Neurol Sci. 2003;208(1–2):39–42.
65. Leger JM, Bouche P, Bolgert F, et al. The spectrum of polyneuropathies in patients infected with HIV. J Neurol Neurosurg Psychiatry. 1989;52:1369–1374.
66. Przedbroski S, Liesnard C, Voordecker P, et al. Inflammatory demyelinating polyradiculoneuropathy associated with human immunodeficiency virus infection. J Neurol. 1988; 235:359–361.
67. Dalakas MC, Yarchoan R, Spitzer R, Elder G, Sever JL. Treatment of human immunodeficiency virus-related polyneuropathy with 3′-azido-2′,3′-dideoxythymidine. Ann Neurol. 1988;23:S92–S94.
68. Miller RG, Storey JR, Greco CM. Ganciclovir in the treatment of progressive AIDS-related polyradiculopathy. Neurology. 1990;40:569–574.
69. Roullet E, Assuerus V, Gozlan J, et al. Cytomegalovirus multifocal neuropathy in AIDS: analysis of 15 consecutive cases. Neurology. 1994:44:2174–2182.
70. Kim YS, Hollander H. Polyradiculopathy due to cytomegalovirus: report of two cases in which improvement occurred after prolonged therapy and review of the literature. Clin Infect Dis. 1993;17:32–37.
71. Lipkin WI, Parry G, Kiprov D, Abrams D. Inflammatory neuropathy in homosexual men with lymphadenopathy. Neurology. 1985;35:1479–1483.
72. Said G, Lacroix-Ciaudo C, Fujimura H, Blas C, Faux N. The peripheral neuropathy of necrotizing arteritis: a clinicopathological study. Ann Neurol. 1988;23:461–465.
73. Cohen JA, Laudenslager M. Autonomic nervous system involvement in patients with human immunodeficiency virus infection. Neurology. 1989;39:1111–1112.
74. Craddock C, Pasvol G, Bull R, Protheroe A, Hopkin J. Cardiorespiratory arrest and autonomic neuropathy in AIDS. Lancet. 1987;2:16–18.
75. Lin-Greenberger A, Taneja-Uppal N. Dysautonomia and infection with the human immunodeficiency virus. Ann Intern Med. 1987;106:167.
76. Elder G, Dalakas M, Pezeshkpour G, Sever J. Ataxic neuropathy due to ganglioneuronitis after probable acute human immunodeficiency virus infection. Lancet. 1986;2:1275–1276.
77. Kiwaki T, Umehara F, Arimura Y, et al. The clinical and pathological features of peripheral neuropathy accompanied with HTLV-I associated myelopathy. J Neurol Sci. 2003;206(1):17–21.
78. Leite AC, Silva MT, Alamy AH, et al. Peripheral neuropathy in HTLV-I infected individuals without tropical spastic paraparesis/HTLV-I-associated myelopathy. J Neurol. 2004; 251(7):877–881.
79. Saeidi M, Sasannejad P, Foroughipour M, Shahami S, Shoeibi A. Prevalence of peripheral neuropathy in patients with HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP). Acta Neurol Belg. 2011;111:41–44.
80. Ali A, Char G, Hanchard B. Chronic inflammatory demyelinating polyneuropathy in a patient infected with human T lymphotropic virus type I. BMJ Case Rep. 2009; doi: 10.1136/bcr03.2009.1680.
81. Rubin DI, Daube JR. Subacute sensory neuropathy associated with Epstein–Barr virus. Muscle Nerve. 1999;22:1607–1610.
82. Greenberg MK, McVey AL, Hayes T. Segmental motor involvement in herpes zoster: an EMG study. Neurology. 1992;42: 1122–1123.
83. Haanpää M, Häkkinen V, Nurmikko T. Motor involvement in acute herpes zoster. Muscle Nerve. 1997;20:1433–1438.
84. Modelli M, Scarpini C, Malandrini A, Romano C. Painful neuropathy after diffuse herpes zoster. Muscle Nerve. 1997; 20:229–231.
85. Dutt AK. Diaphragmatic paralysis caused by herpes zoster. Am Rev Respir Dis. 1970;101:755–758.
86. Glantz RH, Ristanovic RK. Abdominal muscle paralysis from herpes zoster. J Neurol Neurosurg Psychiatry. 1988;51: 885–886.
87. Gottschau P, Trojaborg W. Abdominal muscle paralysis associated with herpes zoster. Acta Neurol Scand. 1991;84:344–347.
88. Dayan AD, Ogul E, Graveson GS. Polyneuritis and herpes zoster. J Neurol Neurosurg Psychiatry. 1972;35:170–175.
89. Sander EA, Peters AC, Gratana JW, Hughes RA, Guillain–Barré syndrome after varicella zoster infection. J Neurol. 1987;234:437–439.
90. Gardner-Thorpe C, Foster JB, Barwick DD. Unusual manifestations of herpes zoster. A clinical and electrophysiological study. J Neurol Sci. 1976;28:427–447.
91. Rosenfeld T, Price MA. Paralysis in herpes zoster. Aust N Z J Med. 1985;15:712–716.
92. Sachs GM. Segmental zoster paresis: An electrophysiological study. Muscle Nerve. 1996;19:784–786.
93. Segal AZ, Rordorf G. Gabapentin as novel treatment for postherpetic neuralgia. Neurology. 1996;46:1175–1176.
94. Max MB. Treatment of post-herpetic neuralgia: antidepressants. Ann Neurol. 1994;35:S50–S53.
95. Rowbotham MC. Managing post-herpetic neuralgia with opioids and local anesthetics. Ann Neurol. 1994;35:S46–S49.