TABLE 18-1. NUTRITIONAL DEFICIENCY ASSOCIATED WITH PERIPHERAL NEUROPATHY
TABLE 18-1. NUTRITIONAL DEFICIENCY ASSOCIATED WITH PERIPHERAL NEUROPATHYPatients can develop neuropathies due to inadequate nutrition and subsequent vitamin deficiency (Table 18-1). Nutritional deficiency-related polyneuropathies are currently uncommon, especially in developed countries. However, these neuropathies do occur and are important because they are potentially treatable. Malnutrition may occur in chronic alcoholics and in patients with chronic illness, unusual diets, and obesity surgery. Some vitamin deficiencies (e.g., vitamins B12 and E) often occur because of impaired gastrointestinal absorption rather than poor dietary intake. In other cases, neuropathy may develop secondary to the effects of medications (e.g., isoniazid causing vitamin B6 deficiency). The clinical and laboratory features of most nutritional polyneuropathies are similar to those of the more common polyneuropathies. Timely and accurate diagnosis is important because patients can improve with replacement therapy.
TABLE 18-1. NUTRITIONAL DEFICIENCY ASSOCIATED WITH PERIPHERAL NEUROPATHY
Thiamine (vitamin B1) deficiency
Pyridoxine (vitamin B6) deficiency
Cobalamin (vitamin B12) deficiency
Folate deficiency
Vitamin E deficiency
Copper deficiency
Hypophosphatemia
 THIAMINE (VITAMIN B1) DEFICIENCY
THIAMINE (VITAMIN B1) DEFICIENCYThiamine deficiency or beriberi is uncommon nowadays and primarily occurs as a consequence of chronic alcohol abuse, recurrent vomiting, total parenteral nutrition, inappropriately restrictive diets, and perhaps bariatric surgery.1 The symptoms arising from insufficient dietary intake of thiamine are known as beriberi and may present in two forms: dry beriberi and wet beriberi. The difference between these two types of beriberi is simply the presence (wet beriberi) or absence (dry beriberi) of congestive heart failure and lower limb edema. Affected individuals usually present with numbness, tingling, and burning in the distal lower extremities, which subsequently spread to involve the proximal legs and upper extremities.2 On examination, a mild-to-moderate reduction in all sensory modalities is noted in a stocking distribution along with diminished muscle stretch reflexes. Mild, predominantly distal weakness may be appreciated. Congestive heart failure with edema of the lower legs is seen in the so-called wet beriberi.
Measuring thiamine concentration in serum and urine is not very reliable.3 Assay of erythrocyte transketolase activity and the increase in activity after adding thiamine pyrophosphate (TPP) appears to be more accurate and reliable.4–7 Sensory nerve conduction studies (NCS) reveal reduced or absent sensory nerve action potentials (SNAPs) amplitudes with relative preservation of distal sensory latencies and conduction velocities.2 The motor NCS may be normal or demonstrate slightly reduced amplitudes.
Sural nerve biopsies reveal loss of primarily large myelinated axons.1,8 Necropsy studies have demonstrated chromatolysis of the anterior horn cells and dorsal root ganglia cells along with axonal degeneration and secondary demyelination of the posterior columns.
Most meats and vegetables contain adequate amounts of thiamine, in particular unrefined cereal grains, wheat germ, yeast, soybean flour, and pork.3 It is absorbed in the small intestine by both passive diffusion and active transport. Here, thiamine is converted to TPP.3 Because stores of thiamine in the body are limited and its half-life is only 10–14 days, 1–1.5 mg daily of thiamine should be part of any routine diet else deficiency can arise.3
Thiamine and TPP catalyze the decarboxylation of alpha-ketoacids to coenzyme A moieties, an important process in ATP synthesis in mitochondria.3 TPP plays a role in the formation of myelin.9 Thiamine may also affect neuronal conduction by altering membrane sodium channel function.10,11
Thiamine 100 mg/d should be given intravenously or intramuscularly in deficient patients. In patients with thiamine deficiency secondary to alcohol use, discontinuation of alcohol is imperative. In addition to the likely direct toxic influences on Schwann cells and peripheral nerves, ethanol is likely to impair thiamine utilization even when blood levels are normal.12 Cardiomyopathy usually is quite responsive to thiamine replacement, although improvement in neurologic function is more variable and less dramatic.13 Motor deficits appear to improve more so than sensory.14 Some improvement is expected in most patients, but this typically occurs slowly over 6–12 months. In patients with severe neuropathy permanent deficits are typical.2
PYRIDOXINE (VITAMIN B6 DEFICIENCY)Pyridoxine not only is neurotoxic when taken in large dosages (see Chapter 20),15–18 but can also be associated with a sensorimotor polyneuropathy when deficient. Pyridoxine deficiency is usually associated with isoniazid and hydralazine treatment.19–21 Pyridoxine deficiency may also result from malnutrition (e.g., chronic alcoholism) or in patients receiving chronic peritoneal dialysis.22 The symptoms of vitamin B6 deficiency are nonspecific. Affected individuals manifest with a sensory greater than motor polyneuropathy similar to most idiopathic neuropathies. The electrophysiology studies reflect an axonal sensorimotor polyneuropathy.19,20 Vitamin B6 levels can be measured in blood. Deficient patients should be treated with 50–100 mg/d of vitamin B6.23,24 This should also be given prophylactically in patients being treated with isoniazid or hydralazine.25
COBALAMIN (VITAMIN B12) DEFICIENCYPatients with vitamin B12 deficiency can present with central nervous system (CNS) or peripheral nervous system (PNS) abnormalities with or without hematologic findings (megaloblastic anemia).26–34 Those affected may manifest with numbness and sensory ataxia due to posterior column dysfunction and spastic weakness due to pyramidal tract insult (subacute combined degeneration). In addition, they may have altered mental status. Most patients have signs and symptoms of both CNS and PNS involvement, with reduction of vibratory perception and proprioception, positive Romberg sign, sensory ataxia, decreased or absent reflexes at the ankles, and brisk reflexes elsewhere. Plantar responses can be either extensor or flexor. Because of the myelopathy, patients may present with numbness restricted to the hands potentially mimicking carpal tunnel syndrome. A subacute onset and constant, rather than intermittent numbness would favor vitamin B12 deficiency. A positive Lhermitte’s sign may be present owing to swelling in the cervical spinal cord.
Serum vitamin B12 assays are not sensitive, as many symptomatic patients may have serum vitamin B12 levels that are within the normal range.35,36 Serum levels of the vitamin B12 metabolites, methylmalonic acid (MMA) and homocysteine (Hcy), are much more sensitive in detecting deficiency of B12.36,37 MMA and Hcy levels are increased (i.e., evidence of B12 deficiency) in 5–10% of patients with serum vitamin B12 levels less than 300 pg/mL and in 0.1–1% of those with levels greater than 300 pg/mL.37 We measure MMA and Hcy levels in patients with polyneuropathy who are suspected of having vitamin B12 deficiency (e.g., those with a sudden onset of symptoms, symptoms beginning in the hands, findings suggestive of myelopathy, or risk factors for vitamin B12 malabsorption). In addition, we routinely measure copper, ceruloplasmin, and zinc levels in the same group of patients as copper deficiency manifests in a virtually identical manner.
In the absence of symptomatic gastrointestinal disease, it probably is not necessary to seek a diagnosis of pernicious anemia in a patient with vitamin B12 deficiency because this information will not alter management.38 A Schilling test can be done to diagnose pernicious anemia.39 It is a multistep and therefore inconvenient test which is now uncommonly utilized. Anti-intrinsic factor antibodies are specific for pernicious anemia but are found in only 50% of patients.40 The combination of elevated gastrin and antiparietal cell antibodies is more sensitive and specific for pernicious anemia.41
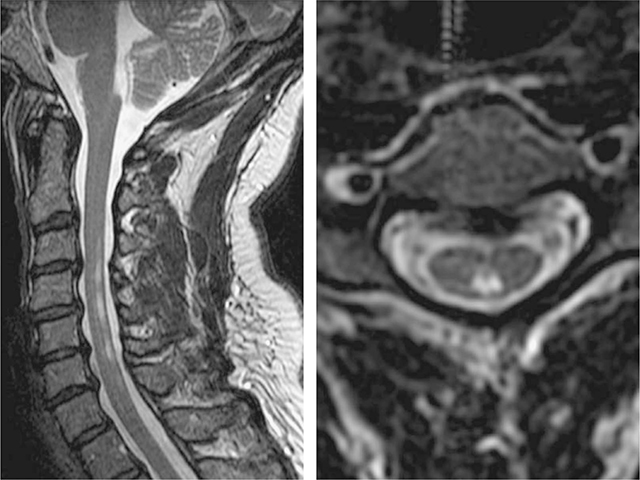
NCS reveal absent or reduced SNAP amplitudes with CMAPs amplitudes that are normal or slightly reduced. Motor and sensory distal latencies and conduction velocities are essentially normal or only mildly abnormal.26–30,34,42 Somatosensory-evoked potentials and magnetic stimulation studies may reveal prolongation of central conduction time.29,32 Magnetic resonance imaging (MRI) scans of the cervical cord can reveal increased signal on T2 images in the posterior columns (Fig. 18-1).43
Figure 18-1. Vitamin B12 deficiency. Sagittal (left image) and axial (right image) T2 MRI in subacute combined degeneration (SCD) showing abnormal hyperintensity in the posterior columns. The patient had markedly reduced vibration and position sense and a Romberg sign; the tendon reflexes were preserved and there were no corticospinal tract or peripheral nerve signs. (Reproduced with permission from Ropper AH, Samuels MA, Klein JP, eds. Adams and Victor’s Principles of Neurology, 10th ed. New York, NY: McGraw-Hill; 2014.)
Degeneration of the posterior columns and corticospinal tracts has been found at autopsies. Nerve biopsies reveal loss of large myelinated fibers, axonal degeneration, and secondary segmental demyelination.31,34,44
Cobalamin is found in meat, fish, and dairy products but is not present in fruits, vegetables, and grains. Vitamin B12 requires a transport molecule, intrinsic factor, which is synthesized and secreted by gastric parietal cells. Vitamin B12 deficiency can result from lack of dietary intake (strict vegetarian diet), lack of intrinsic factor (pernicious anemia with autoimmune destruction of parietal cells or gastrectomy), malabsorption syndromes (sprue or lower ileum resection), genetic defects in methionine synthetase, and bacteria (blind-loop syndrome) or bacterial or parasitic consumption prior to its absorption. Cobalamin functions as an enzyme necessary for demethylation of methyltetrahydrofolate.45 Tetrahydrofolate, in turn, is required for the production of folate coenzymes that are necessary for DNA synthesis. The pathogenic mechanism for the neuropathy/myelopathy associated with cobalamin deficiency is not known but may be related to impairment in DNA synthesis, decreased methylation of myelin phospholipids, or buildup of methylmalonic and propionic acids that serve as abnormal substrates for fatty acid synthesis, leading to aberrant myelination.45
We generally treat deficient patients with B12 1,000 μg IM/week for 1 month, followed by 1,000 μg IM/month thereafter. It may be possible to treat vitamin B12 deficiency with oral replacement. A randomized trial comparing treatment with 2,000-mg oral vitamin B12/day to 1,000-mg intramuscular vitamin B12/month showed similar improvements in hematologic indices, serum MMA and Hcy, and neurologic symptoms.46 However, a minority of subjects had neurologic symptoms, and the methods by which clinical efficacy was assessed were lacking.
Approximately 2% of patients experience worsening sensory symptoms for unclear reasons during the first month of treatment.47 The response to treatment of vitamin B12 deficiency polyneuropathy, separate from other neurologic complications of vitamin B12, has not been well studied. Patients with vitamin B12 deficiency polyneuropathy/myelopathy probably do not show an immediate response to treatment and may not respond at all.34,48 The duration of symptoms is an important determinant of treatment response.47,49,50
VITAMIN B12 DEFICIENCY SECONDARY TO NITROUS OXIDE INHALATIONNitrous oxide can inactivate methylcobalamin, leading to neuropathy and subacute combine degeneration in individuals with low or borderline vitamin B12 levels, euphemistically referred to as “anesthetica paresthetica.”51–54 Physical examination, electrodiagnostic findings, and nerve biopsies are similar to that seen in B12 deficiency, as described in the previous section.
FOLATE DEFICIENCYFolate deficiency is associated with neurologic abnormalities similar to those complicating B12 deficiency.55,56 Subacute combined degeneration of the posterior columns and corticospinal tracts, sensorimotor peripheral neuropathy, and altered mental status can develop.
Serum folate levels should be reduced. It is necessary to measure both serum folate and vitamin B12 levels to define a pure folic acid deficiency. Megaloblastic anemia may be evident on a complete blood count and smear. Sensory and motor NCS are similar to those seen with B12 deficiency.
Folate is found in fruit and vegetables and in liver. It is primarily absorbed in the proximal jejunum. Isolated folic acid deficiencies are extremely rare but can occur in the elderly on poor diets, alcoholics, young persons’ consuming only snack foods, partial gastrectomies, duodenojejunal resections, celiac disease, and disorders of the jejunal mucosa.55,56 Several drugs (e.g., phenytoin, phenobarbital, sulfasalazine, and colchicine) can also interfere with the optimal utilization of folic acid. The mechanism by which folic acid deficiency results in a polyneuropathy is not known; however, folic acid is required in DNA synthesis.
Administration of folic acid usually results in good clinical recovery.
VITAMIN E DEFICIENCYVitamin E or alpha-tocopherol is a lipid-soluble antioxidant vitamin that is present in the lipid bilayer constituting the cell membrane.57,58 There is a close relationship between the metabolism of lipids and that of vitamin E. There are three major mechanisms associated with vitamin E deficiency: (1) deficient fat absorption (e.g., cystic fibrosis, chronic cholestasis, short-bowel syndrome, and intestinal lymphangiectasia), (2) deficient fat transport (abetalipoproteinemia, hypobetalipoproteinemia, normotriglyceridemic abetalipoproteinemia, and chylomicron retention disease), and (3) a genetically based abnormality of vitamin E metabolism. Patients with vitamin E deficiency usually present with progressive difficulty ambulating and impaired coordination of the hands.59–62 Some individuals complain of weakness and sensory loss. Dysarthria can also occur.
Physical examination is remarkable for ataxia of the trunk and upper and lower extremities.59–62 There is prominent loss of proprioception and vibratory perception. Muscle stretch reflexes are reduced or absent. Manual muscle testing can be difficult secondary to the ataxia, but there can be proximal muscle weakness, suggesting a superimposed myopathic process. Ocular examination may reveal ophthalmoplegia and retinopathy.
Vitamin E (alpha-tocopherol) levels in the serum are low. With hyperlipidemia, the vitamin E level may be normal. In such cases, the ratio of total serum vitamin E to the total serum lipid concentration is a more sensitive indicator of vitamin E deficiency.63
NCS reveal reduced amplitudes or absent SNAPs.57–64 The sensory nerve conduction velocities are normal or only slightly reduced. Somatosensory-evoked potentials demonstrate normal peripheral nerve potentials with marked slowing and attenuation of central responses consistent with slowing of central conduction with loss of posterior column fibers.65 Motor NCS are normal.
Autopsy studies demonstrate swelling and degeneration of axons in the posterior columns and spinocerebellar tracts along with neuronal loss and lipofuscin accumulation in the gracile and cuneate nuclei.60,66,67 Changes within the basal ganglia may be seen. Sural nerve biopsies show nonspecific abnormalities including the loss of large myelinated fibers, axonal degeneration, regenerating sprouts, occasional vacuoles in the myelin sheath, and breakup of the Schmidt–Lanterman incisures, but little in the way of primary demyelination.68,69
There are four main types of vitamin E, the most active of which is alpha-tocopherol. Vitamin E is lipid soluble and absorbed in the small intestine. Vitamin E is incorporated into chylomicrons and is transported to the liver. Here, vitamin E is incorporated into very low-density lipoproteins in a step requiring alpha-tocopherol transfer protein. Deficiency of this transfer protein is associated with hereditary vitamin E deficiency (discussed in Chapter 12). Vitamin E may serve to eliminate free radicals and stabilize cell membrane structure.70
Vitamin E deficiency is usually due to factors other than insufficient intake.71 As mentioned, deficiency can result secondary to disorders of lipid malabsorption or transport. Abetalipoproteinemia is a rare autosomal-dominant disorder characterized by steatorrhea, pigmentary retinopathy, acanthocytosis, and progressive ataxia that is associated with vitamin E deficiency.72 Cystic fibrosis can also be complicated by vitamin E deficiency secondary to malabsorption. There are also genetic forms of isolated vitamin E.73,74 Mutations in the alpha-tocopherol transfer protein gene, TTPA, located on chromosome 8q13 result in loss of vitamin E.75,76 Vitamin E deficiency may also complicate various liver and biliary disorders as well as surgical removal of parts of the intestines leading to short bowel or dumping syndrome.64,77,78
Therapy is aimed at preventing progression, but improvement in neurologic function may occur. The specific dose of vitamin E is dependent upon the cause of deficiency.71 In cases of isolated vitamin E deficiency, patients are treated with 1,500–6,000 international units (IU)/day in divided doses. Patients with chronic cholestasis are initially treated with 50 IU/kg/d and the dose is increased in 50 IU/kg increments up to a 200 IU/kg/d as required to obtain a normal serum tocopherol to lipid ratio. Patients with cystic fibrosis, who are receiving oral pancreatic enzyme therapy, require doses of 5–10 IU/kg/d. Those with short bowel syndrome are given 300–5,400 IU/d. Abetalipoproteinemia is treated with vitamin E 150–300 IU/kg/d and vitamin A 15,000–20,000 IU/d.
POSTGASTRECTOMY/BARIATRIC SURGERY DEFICIENCIESPolyneuropathy may complicate gastric/bariatric surgery for gastric ulcers, cancer, or morbid obesity.14,79–82 The clinical picture is variable and may include acute or subacute sensory loss, burning feet, generalized weakness that can resemble Guillain–Barré syndrome, mononeuropathies, and radiculoplexus neuropathy.82–86 Some cases are complicated by CNS dysfunction resembling Wernicke–Korsakoff syndrome. In the largest retrospective series, 71 out of 435 (16%) of patients who underwent bariatric surgery developed some type of peripheral neuropathy. The neuropathy is associated with malnutrition and the rapidity of weight loss and usually develops within the first 1½ years following weight loss surgery.79,82,86 The latency between surgery and symptoms ranges from a few months to years in patients following total or partial gastrectomy for ulcer or cancer.14,87
Weight reduction surgical procedures include gastrojejunostomy, gastric stapling, vertical banded gastroplasty, and gastrectomy with Roux-en-Y anastomosis. Although thiamine deficiency seems to be a factor (given the frequent co-occurrence of the Wernicke–Korsakoff syndrome), there is not good documentation of thiamine deficiency in the reported cases. In some cases, one or more vitamin deficiencies are identified.88 In many cases, no specific deficiency is identified. Electrodiagnostic studies most commonly reveal evidence of a length-dependent, axonal, sensory greater than motor polyneuropathy.
Sural nerve biopsies when obtained may reveal active axonal degeneration and mild perivascular, endoneurial, and epineurial infiltrate.
The basis of the neuropathies is unclear but likely to result from multiple nutritional deficiencies.
Patients should be treated with parenteral vitamin supplementation and, on occasion, reversal of the surgical bypass.82,88,89 Patients with protracted vomiting after weight reduction surgery should receive total parenteral nutrition and vitamins. Patients can recover if started on treatment early, though some will have persistent sensory loss and weakness. The duration and severity of deficits before identification and treatment of neuropathy are important predictors of final outcome.
COPPER DEFICIENCYCopper deficiency is associated with an unusual myeloneuropathy, neutropenia, and sometimes pancytopenia.82,90–100 The clinical phenotype is similar to vitamin B12 deficiency. Most patients manifest with numbness and tingling in the legs, weakness, spasticity, and gait difficulties. Large-fiber sensory function is impaired, reflexes are brisk, and plantar responses are extensor. In some cases, light touch and pinprick sensation are affected and NCS indicate sensorimotor axonal polyneuropathy in addition to myelopathy.91,93 A severe motor axonopathy can also be seen.94 The weakness and sensory loss in some cases is primarily due to a myelopathy.95 Demyelinating lesions may be appreciated on brain MRI, and some patients have ocular dysmetria indicating brain involvement.95
Besides low–serum-copper levels, some cases are associated with high levels of zinc. Microcytic anemia and neutropenia90,93–98,101,102 and occasionally pancytopenia91 are also seen. Bone marrow biopsy may reveal abnormalities of a myelodysplastic syndrome. Cerebrospinal fluid may be normal or show mildly elevated protein or immunoglobulin synthesis rate.91,94,95,97 MRI may demonstrate abnormal T2-weighted signal in the dorsal columns.90,94,95,97,98
NCS may reveal features of a sensorimotor axonal polyneuropathy.91,93,97,98 Somatosensory-evoked potentials demonstrate impaired conduction in the central pathway in those with myelopathy.97,98
Sural nerve biopsies may show evidence of axonal degeneration.97,98
Copper is absorbed in the stomach and proximal jejunum accounting for why deficiency may complicate gastric surgery.90,93,103 The reason that zinc can lead to copper deficiency is that it upregulates the production of metallothionein in the gut, which in turn reduces copper absorption.104,105 Interestingly, some denture creams contain a large amount of zinc and can lead to hypocupremia and neurologic disease.96 Copper deficiency may also result from malnutrition, prematurity, total parenteral nutrition, and copper chelating agents.101,103
The myeloneuropathy may improve with oral or intravenous copper replacement quickly90,92,93 but benefits may not be seen for months or years.91,95,97 and some patients do not improve at all.94 In contrast to the variable clinical improvement, the pancytopenia usually normalizes with copper replacement therapy.97
Hyperalimentation with inadequate phosphate supplementation can lead to hypophosphatemia and the development of a subacute and severe sensorimotor peripheral neuropathy, which can clinically resemble Guillain–Barré syndrome.106,107 Typically, serum phosphate levels need to be below 1 mg/dL for this to occur. Paresthesias are initially noted in the feet and ascend to involve the upper limbs and remainder of the body. Impaired ambulation secondary to both weakness and sensory ataxia occurs over the course of hours to days. Generalized weakness, ataxia, depressed muscle stretch reflexes, and reduced perception of all sensory modalities are appreciated on examination. Weakness may also involve the ventilatory muscles requiring mechanical assistance. NCS reveal an absence of SNAPs, reduced CMAP amplitudes, and slow conduction velocities. Correction of the hypophosphatemia results in clinical and electrophysiologic improvement.
ALCOHOLIC NEUROPATHYAlcoholics can develop a generalized axonal sensorimotor polyneuropathy.12,108–112 Usually the neuropathy is slowly progressive, although some cases with acute or subacute presentation resembling Guillain–Barré syndrome have been reported.113,114 Unlike Guillain–Barré syndrome, CSF protein in alcohol-related acute axonal polyneuropathy is usually normal or only slightly elevated. Most cases are preceded by prominent weight loss for 2–3 months. Most patients manifest with an insidious onset of numbness, paresthesia, and burning pain suggestive of a small fiber polyneuropathy. It is estimated that the equivalent of 10 oz of 86 proof-distilled spirits were 3 L of beer a day for 3 or more years, which is the threshold for alcoholic neuropathy. It has also been hypothesized that the lead content of wine may also contribute to the pathogenesis of alcoholic neuropathy.12
Examination demonstrates a reduction of all sensory modalities in a glove and stocking distribution, worse in the lower compared to upper limbs. Muscle stretches are reduced or absent. Mild distal leg weakness may be appreciated, but proximal leg and arm strength is usually normal. An occasional patient presents with symptoms and signs suggestive of a myopathy as opposed to neuropathy. NCS reveal features suggestive of a generalized axonal sensory or sensorimotor polyneuropathy.12,108–110,112
Nerve biopsies may reveal loss of large- and small-caliber myelinated fibers along with Wallerian degeneration and secondary segmental demyelination.112,114
The exact etiology of peripheral nerve insult in alcoholism is unknown but may in part be related to both a nutritional deficiency (e.g., vitamin B group and folate) and a direct toxic effect of alcohol on peripheral nerves.12
Abstaining from alcohol and consuming an optimal diet can result in an improvement of the peripheral neuropathy.114
SUMMARYNutritional neuropathies are not particularly common. However, because they can be treatable with correction of the deficit, it is important to be vigilant for signs and symptoms that would suggest a nutritional deficiency. In particular, those patients with gastrointestinal disease or history of gastric bypass may be particularly vulnerable.
1. Ohnishi A, Tsuji S, Igisu H, et al. Beriberi neuropathy. Morphometric study of sural nerve. J Neurol Sci. 1980;45:177–190.
2. Hong CZ. Electrodiagnostic findings of persisting polyneuropathies due to previous nutritional deficiency in former prisoners of war. Electromyogr Clin Neurophysiol. 1986;26:351–363.
3. McCormick DB, Greene HL. Vitamins. In: Burtis CA, Ash-wood ER, eds. Tierz Textbook of Clinical Chemistry. Philadelphia, PA: Saunders; 1999:999–1028.
4. Brin M, Tai M, Ostashever AS, Kalinsky H. The effect of thiamine deficiency on the activity of erythrocyte hemolysate transketolase. J Nutr. 1960;71:273–281.
5. Brin M. Erythrocyte transketolase in early thiamine deficiency. Ann N Y Acad Sci. 1962;98:528–541.
6. Jeyasingham MD, Pratt OE, Burns A, Shaw GK, Thomson AD, Marsh A. The activation of red blood cell transketolase in groups of patients especially at risk from thiamin deficiency. Psychol Med. 1987;17:311–318.
7. Jeyasingham MD, Pratt OE, Shaw GK, Thomson AD. Changes in the activation of red blood cell transketolase of alcoholic patients during treatment. Alcohol. 1987;22:259–365.
8. Takahashi K, Nakamura H. Axonal degeneration in beriberi neuropathy. Arch Neurol. 1976;33:836–841.
9. Collins RC, Lonergan ET. Transketolase and myelin. N Engl J Med. 1971;285:751–752.
10. Cooper JR, Pincus JH. The role of thiamin in nervous tissue. Neurochem Res. 1979;4:223–229.
11. Schoffeniels E. Thiamine phosphorylated derivatives and bioelectrogenesis. Arch Int Physiol Biochim. 1983;91:233–242.
12. Mellion M, Gilchrist JM, de la Monte S. Alcohol- related peripheral neuropathy: nutritional, toxic, or both? Muscle Nerve. 2011;43:309–316.
13. Jolliffe N. The diagnosis, treatment and prevention of vitamin B1 deficiency. Bull N Y Acad Med. 1939;15:469–478.
14. Koike H, Misu K, Hattori N, et al. Postgastrectomy polyneuropathy with thiamine deficiency. J Neurol Neurosurg Psychiatry. 2001;71:357–362.
15. Albin RL, Albers JW, Greenberg HS, et al. Acute sensory neuropathy-neuronopathy from pyridoxine overdose. Neurology. 1987;37:1729–1732.
16. Dalton K, Dalton MJ. Characteristics of pyridoxine overdose neuropathy syndrome. Acta Neurol Scand. 1987;76:8–11.
17. Parry GJ, Bredesen DE. Sensory neuropathy with low dose pyridoxine. Neurology. 1985;35:1466–1468.
18. Schaumburg H, Kaplan J, Windsbank A, et al. Sensory neuropathy from pyridoxine abuse. A new megavitamin syndrome. N Engl J Med. 1983;309:445–448.
19. Gammon GD, Burge FW, King G. Neural toxicity in tuberculous patients treated with isoniazid (isonicotinic acid hydrazide). AMA Arch Neurol Psychiatry. 1953;70:64–69.
20. Lubing HN. Peripheral neuropathy in tuberculosis patients treated with isoniazid. Am Rev Tuberc. 1953;68:458–461.
21. Selikoff IJ, Robitzek EH, Ornstein CG. Treatment of pulmonary tuberculosis with hydrazide derivatives of nicotinic acid. J Am Med Assoc. 1952;150(10):973–980.
22. Moriwaki K, Kanno Y, Nakamoto H, Okada H, Suzuki H. Vitamin B6 deficiency in elderly patients on chronic peritoneal dialysis. Adv Perit Dial. 2000;16:308–312.
23. Ruffin JM, Smith DT. Treatment of pellagra with special reference to the use of nicotinic acid. South Med J. 1939;32:40–47.
24. Sebrel WH, Butler RE. Riboflavin deficiency in man (ariboflavinosis). Public Health Rep. 1939;54:2121–2131.
25. Marcus R, Coulston AN. Water-soluble vitamins. In: Gilman AG, Goodman LS, Rall TW, Murad F, eds. Goodman and Gilman’s the Pharmacological Basis of Therapeutics, 7th ed. New York, NY: Macmillan Publishing Company; 1985:1551–1572.
26. Fine EJ, Hallett M. Neurophysiological study of subacute combined degeneration. J Neurol Sci. 1980;45:331–336.
27. Fine EJ, Soria E, Paroski MW, Petryk D, Thomasula L. The neurophysiological profile of vitamin B12 deficiency. Muscle Nerve. 1990;13:158–164.
28. Hahn AF, Gilbert JJ, Brown WF. A study of the sural nerve in pernicious anemia. Can J Neurol Sci. 1976;3:217.
29. Hemmer B, Glocker FX, Schumacher M, Deuschl G, Lucking CH. Subacute combined degeneration: clinical, electrophysiological, and magnetic resonance imaging findings. J Neurol Neurosurg Psychiatry. 1998;65:822–827.
30. Kayser-Gatchalian MC, Neundorfer B. Peripheral neuropathy with vitamin B12 deficiency. J Neurol. 1977;214:183–193.
31. Kosik KS, Mullins TF, Bradley WG, Tempelis LD, Cretella AJ. Coma and axonal degeneration in vitamin B12 deficiency. Arch Neurol. 1980;37:590–592.
32. Krumholz A, Weiss HD, Goldstein PJ, Harris KC. Evoked responses in vitamin B12 deficiency. Ann Neurol. 1981;9:407–409.
33. Lockner D, Reizenstein P, Wennberg A, Widén L. Peripheral nerve function in pernicious anemia before and after treatment. Acta Haematol. 1969;41:257–263.
34. McCombe PA, McLeod JG. The peripheral neuropathy of vitamin B12 deficiency. J Neurol Sci. 1984;66:117–126.
35. Carmel R. Current concepts in cobalamin deficiency. Annu Rev Med. 2000;51:357–375.
36. Savage DG, Lindenbaum J, Stabler SP, Allen RH. Sensitivity of serum methylmalonic acid and total homocysteine determinations for diagnosing cobalamin and folate deficiencies. Am J Med. 1994;96:239–246.
37. Lindenbaum J, Savage DG, Stabler SP, Allen RH. Diagnosis of cobalamin deficiency: II. Relative sensitivities of serum cobalamin, methylmalonic acid, and total homocysteine concentrations. Am J Hematol. 1990;34:99–107.
38. Stabler SP. Screening the older population for cobalamin (vitamin B12) deficiency. J Am Geriatr Soc. 1995;43:1290–1297.
39. Swain R. An update of vitamin B12 metabolism and deficiency states. J Fam Pract. 1995;41:595–600.
40. Chanarin I. The Megaloblastic Anemias. 2nd ed. Oxford, England: Blackwell Scientific Publications; 1979.
41. Metz J, Bell AH, Flicker L, et al. The significance of subnormal serum vitamin B12 concentration in older people: a case control study. J Am Geriatr Soc. 1996;44:1355–1361.
42. Saperstein DS, Wolfe GI, Nations SP, Herbelin LL, Barohn RJ. Electrodiagnostic features of cobalamin deficiency polyneuropathy. Muscle Nerve. 2002;26:574.
43. Bou-Haidar P, Peduto AJ, Karunaratne N. Differential diagnosis of T2 hyperintense spinal cord lesions: part B. J Med Imaging Radiat Oncol. 2009;53:152–159.
44. Abarbanel JM, Frishers S, Osimani A. Vitamin B12 deficiency neuropathy: sural nerve biopsy study. Isr J Med Sci. 1986;22: 909–911.
45. Green R, Kinsella LJ. Current concepts in the diagnosis of cobalamin deficiency. Neurology. 1995;45:1435–1440.
46. Kuzminski AM, Del Giacco EJ, Allen RH, Stabler SP, Lindenbaum J. Effective treatment of cobalamin deficiency with oral cobalamin. Blood. 1998;92:1191–1198.
47. Healton EB, Savage DG, Brust JC, Garrett TJ, Lindenbaum J. Neurologic aspects of cobalamin deficiency. Medicine (Baltimore). 1991;70:229–245.
48. Saperstein DS, Wolfe GI, Gronseth GS, et al. Challenges in the identification of cobalamin-deficiency polyneuropathy. Arch Neurol. 2003;60:1296–1301.
49. Hyland HH, Farquharson RF. Subacute combined degeneration of the spinal cord in pernicious anemia. Arch Neurol Psychiatry. 1936;36:1166–1205.
50. Ungley CC. Subacute combined degeneration of the cord: I. Response to liver extracts. II. Trials with vitamin B12. Brain. 1949;72:382–427.
51. Heyer EJ, Simpson DM, Bodis-Wollner I, Diamond SP. Nitrous oxide: clinical and electrophysiologic investigation of neurologic complications. Neurology. 1986;36:1618–1622.
52. Layzer RB, Fishman RA, Schafer JA. Neuropathy following abuse of nitrous oxide. Neurology. 1978;28:504–506.
53. Sahenk Z, Mendell JR, Couri D, Nachtman J. Polyneuropathy from inhalation of N2O cartridges through a whipped-cream dispenser. Neurology. 1978;28:485–487.
54. Vishnubhakat SM, Beresford HR. Reversible myeloneuropathy of nitrous oxide abuse: serial electrophysiological studies. Muscle Nerve. 1991;14:22–26.
55. Enk C, Hougaard K, Hippe E. Reversible dementia and neuropathy associated with folate deficiency 16 years after partial gastrectomy. Scand J Haematol. 1980;25:63–66.
56. Fehling C, Jagerstad M, Linstrand K, et al. Folate deficiency and neurological disease. Arch Neurol. 1974;30:263–265.
57. Guggenheim MA, Ringel SP, Silverman A, Grabert BE. Progressive neuromuscular disease in children with chronic cholestasis and vitamin E deficiency: diagnosis and treatment with alpha tocopherol. J Pediatr. 1982;100:51–58.
58. Harding AE. Vitamin E and the nervous system. Crit Rev Neurobiol. 1987;3(1):89–103.
59. Bertoni JM, Abraham FA, Falls HF, Itabashi HH. Small bowel resection with vitamin E deficiency and progressive spinocerebellar syndrome. Neurology. 1984;34:1046–1052.
60. Rosenblum JL, Keating JP, Prensky AL, Nelson JS. A progressive neurologic syndrome in children with chronic liver disease. N Engl J Med. 1981;304:503–508.
61. Ko HY, Park-Ko I. Electrophysiologic recovery after vitamin E-deficient neuropathy. Arch Phys Med Rehab. 1999;80:964–967.
62. Brin M, Pedley TA, Lovelace RE, et al. Electrophysiologic features of abetalipoproteinemia: functional consequences of vitamin E deficiency. Neurology. 1986;36:669–673.
63. Sokol RJ, Heubi JE, Iannaccone ST, Bove KE, Balistreri WF. Vitamin E deficiency with normal serum vitamin E concentrations in children with chronic cholestasis. N Engl J Med. 1984;310:1209–1212.
64. Satya-Murti S, Howard L, Krohel G, Wolf B. The spectrum of neurologic disorder from vitamin E deficiency. Neurology. 1986;36:917–921.
65. Kaplan PW, Rawal K, Erwin CW, D’Souza BJ, Spock A. Visual and somatosensory evoked potentials in vitamin E deficiency with cystic fibrosis. Electroencephalogr Clin Neurophysiol. 1988; 71:266–272.
66. Jeffrey GP, Muller DPR, Burroughs AK, et al. Vitamin E deficiency and its clinical significance in adults with primary biliary cirrhosis and other forms of chronic liver disease. J Hepatol. 1987;4:307–317.
67. Sung JH, Stadlan EM. Neuroaxonal dystrophy in congenital biliary atresia. J Neuropathol Exp Neurol. 1966;25:341–361.
68. Traber MG, Sokol RJ, Ringel SP, Neville HE, Thellman CA, Kayden HJ. Lack of tocopherol in peripheral nerves of vitamin E-deficient patients with peripheral neuropathy. N Engl J Med. 1987;317:262–265.
69. Yokota T, Wada Y, Furukawa T, Tsukagoshi H, Uchihara T, Watabiki S. Adult-onset spinocerebellar syndrome with idiopathic vitamin E deficiency. Ann Neurol. 1987;22:84–87.
70. Tappel AL. Vitamin E and free radical peroxiadation of lipids. Ann NY Acad Sci. 1972;203:12–28.
71. Sokol RJ. Vitamin E and neurologic deficits. Adv Pediatr. 1990;37:119–148.
72. Muller DP, Harries JT, Lloyd JK. The relative importance of the factors involved in the absorption of vitamin E in children. Gut. 1974;15:966–971.
73. Harding AE, Matthews S, Jones S, Ellis CJ, Booth IW, Muller DP. Spinocerebellar degeneration associated with a selective defect of vitamin E absorption. N Engl J Med. 1985;313:32–35.
74. Sokol RJ, Kayden HJ, Bettis DB, et al. Isolated vitamin E deficiency in the absence of fat malabsorption–familial and sporadic cases: characterization and investigation of causes. J Lab Clin Med. 1988;111:548–559.
75. Ouahchi K, Arita M, Kayden H, et al. Ataxia with isolated vitamin E deficiency is caused by mutations in the α-tocopherol transfer protein. Nat Genet. 1995;9:141–145.
76. Gotoda T, Arita M, Arai H, et al. Adult-onset spinocerebellar dysfunction caused by a mutation in the gene for α-tocopherol transfer protein. N Engl J Med. 1995;333:1313–1318.
77. Harding AE, Muller DP, Thomas PK, Willison HJ. Spinocerebellar degeneration secondary to chronic intestinal malabsorption: a vitamin E deficiency syndrome. Ann Neurol. 1982; 12:419–424.
78. Howard L, Ovensen L, Satya-Murti S, Chu R. Reversible neurological symptoms caused by vitamin E deficiency in a patient with short bowel syndrome. Am J Clin Nutr. 1982;36:1243–1249.
79. Cirignotta F, Manconi M, Mondini S, Buzzi G, Ambrosetto P. Wernicke–Korsakoff encephalopathy and polyneuropathy after gastroplasty for morbid obesity. Arch Neurol. 2000;57:1356–1359.
80. Harwood SC, Chodoroff G, Ellenberg MR. Gastric partitioning complicated by peripheral neuropathy with lumbosacral plexopathy. Arch Phys Med Rehab. 1987;68:310–312.
81. Somer H, Bergstrom L, Mustajoki P, Rovamo L. Morbid obesity, gastric application and a severe neurological deficit. Acta Med Scand. 1985;217:575–576.
82. Koffman BM, Greenfield LJ, Ali II, Pirzada NA. Neurologic complications after surgery for obesity. Muscle Nerve. 2006;33(2):166–176.
83. Feit H, Glasberg M, Ireton C, Rosenberg RN, Thal E. Peripheral neuropathy and starvation after gastric partitioning for morbid obesity. Ann Intern Med. 1982;96:453–455.
84. Williams JA, Hall GS, Thompson AG, Cooke WT. Neurological disease after partial gastrectomy. Br Med J. 1969;3:210–212.
85. Abarbanel JM, Berginer VM, Osimani A, Solomon H, Charuzi I. Neurologic complications after gastric restriction surgery for morbid obesity. Neurology. 1987;37:196–200.
86. Thaisetthawatkul P, Collazo-Clavell ML, Sarr MG, Noreel JE, Dyck PJ. A controlled study of peripheral neuropathy after bariatric surgery. Neurology. 2004;63:1462–1470.
87. Hoffman PM, Brody JA. Neurological disorders in patients following surgery for peptic ulcer. Neurology. 1972;22:450.
88. Rudnicki SA. Prevention and treatment of peripheral neuropathy after bariatric surgery. Curr Treat Options Neurol. 2010; 12:29–36.
89. Thaisetthawatkul P, Collazo-Clavell ML, Sarr MG, Norell JE, Dyck PJ. Good nutritional control may prevent polyneuropathy after bariatric surgery. Muscle Nerve. 2010;42:709–714.
90. Schleper B, Stuerenburg HJ. Copper deficiency-associated myelopathy in a 46-year-old woman. J Neurol. 2001;248:705–706.
91. Hedera P, Fink JK, Bockenstedt PL, Brewer GJ. Myelopolyneuropathy and pancytopenia due to copper deficiency and high zinc levels of unknown origin: further support for existence of a new zinc overload syndrome. Arch Neurol. 2003;60:1303–1306.
92. Kumar N, Gross JB Jr, Ahlskog JE. Myelopathy due to copper deficiency. Neurology. 2003;61:273–274.
93. Kumar N, McEvoy KM, Ahlskog JE. Myelopathy due to copper deficiency following gastrointestinal surgery. Arch Neurol. 2003;60:1782–1785.
94. Greenberg SA, Briemberg HR. A neurological and hematological syndrome associated with zinc and excess and copper deficiency. J Neurol. 2004; 251:111–114.
95. Prodan CI, Holland NR, Wisdom PJ, Burstein SA, Bottomley SS. CNS demyelination associated with copper deficiency and hyperzincemia. Neurology. 2002;59:1453–1456.
96. Nations SP, Boyer PJ, Love LA, et al. Denture cream: An unusual source of excess zinc, leading to hypocupremia and neurologic disease. Neurology. 2008;71:639–643.
97. Kumar N, Gross JB Jr, Ahlskog JE. Copper deficiency myelopathy produces a clinical picture like subacute combined degeneration. Neurology. 2004;63:33–39.
98. Kumar N. Copper deficiency myelopathy (human sway-back). Mayo Clin Proc. 2006;81(10):1371–1384.
99. Rowin J, Lewis SL. Copper deficiency myeloneuropathy and pancytopenia secondary to overuse of zinc supplementation. J Neurol Neurosurg Psychiatry. 2005;76(5):750–751.
100. Prodan CI, Bottomley SS, Holland NR, Lind SE. Relapsing hypocupraemic myelopathy requiring high-dose oral copper replacement. J Neurol Neurosurg Psychiatry. 2006;77(9):1092–1093.
101. Bottomley SS. Sideroblastic anemias. In: Lee GR, Foerster J, Lukens J, et al., eds. Wintrobe’s Clinical Hematology. 10th ed. Baltimore, MD: Lippincott Williams & Wilkins; 1999:1022–1045.
102. Gregg XT, Reddy V, Prchal JT. Copper deficiency masquerading as myelodysplastic syndrome. Blood. 2002;100:1493–1495.
103. Solomons NW. Biochemical, metabolic, and clinical role of copper in human nutrition. J Am Coll Clin Nutr. 1985;4:83–105.
104. Irving JA, Mattman A, Lockitch G, Farrell K, Wadsworth LD. Element of caution: a case of reversible cytopenias associated with excessive zinc supplementation. CMAJ. 2003;169:129–131.
105. Fiske DN, McCoy HE III, Kitchens CS. Zinc-induced sideroblastic anemia: report of a case, review of the literature, and description of the hematologic syndrome. Am J Hematol. 1994; 46:147–150.
106. Weintraub MI. Hypophosphatemia mimicking acute Guillain–Barré–Strohl syndrome: a complication of hyperalimentation. JAMA. 1976;235:1040–1041.
107. Yagnik P, Singh N, Burns R. Peripheral neuropathy with hypophosphatemia in patient receiving intravenous hyperalimentation. Muscle Nerve. 1982;5:562.
108. Casey EB, Le Quesne PM. Electrophysiological evidence for a distal lesion in alcoholic neuropathy. J Neurol Neurosurg Psychiatry. 1972;35:624–630.
109. Mawdsley C, Mayer RF. Nerve conduction in alcoholic polyneuropathy. Brain. 1985;88:335–356.
110. Shankar K, Maloney FP, Thompson C. An electrodiagnostic study in chronic alcoholic subjects. Arch Phys Med Rehab. 1987;68:803–805.
111. Shields RW Jr. Alcoholic polyneuropathy. Muscle Nerve. 1985;8:183–187.
112. Walsh JC, McLeod JG. Alcoholic neuropathy: an electrophysiological and histological study. J Neurol Sci. 1970;10:457–469.
113. Tabaraud F, Vallat JM, Hugon J, Ramiandrisoa H, Dumas M, Signoret JL. Acute or subacute alcoholic neuropathy mimicking Guillain–Barré syndrome. J Neurol Sci. 1990;97:195–205.
114. Wöhrle JC, Spengos K, Steinke W, Goebel HH, Hennerici M. Alcohol-related acute axonal polyneuropathy. A differential diagnosis of Guillain–Barré syndrome. Arch Neurol. 1998;55:1329–1334.