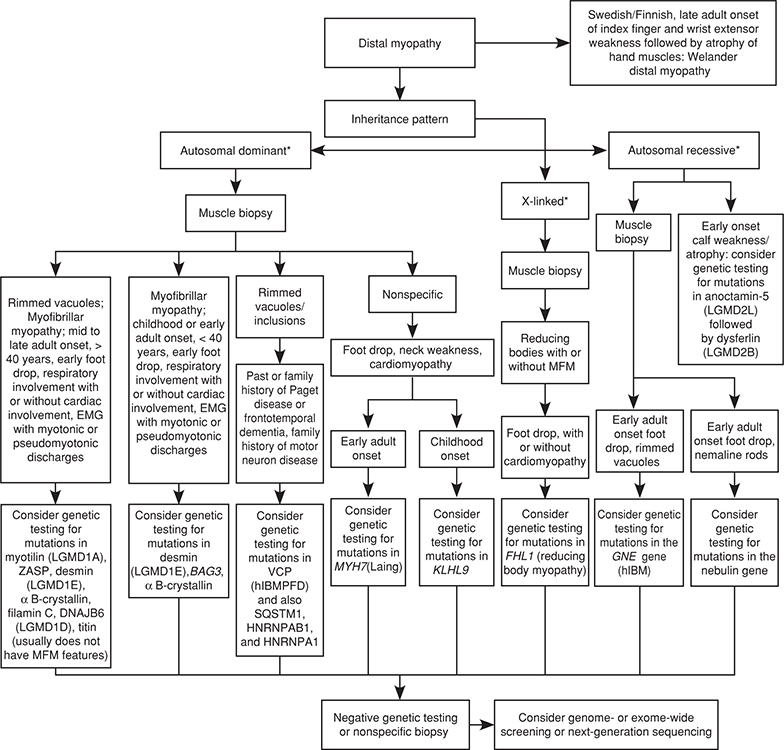
Although distal weakness is often presumed to be neuropathic in etiology, a variety of neuromuscular disorders, including myopathies, are associated with distal extremity weakness (Table 27-1, Fig. 27-27).2,378,379 The distal myopathies are characterized clinically by progressive atrophy and weakness of distal arm or leg muscles and histologically by nonspecific myopathic features on muscle biopsy. We consider the distal myopathies to be forms of muscular dystrophy. Advances in the molecular genetics of these disorders support this notion, as some types of distal myopathy have been found to be allelic with specific types of LGMD (tibial myopathy and LGMD 2 J caused by titin mutations and Miyoshi myopathy and LGMD 2B caused by dysferlin mutations). Furthermore, there is a clear overlap of some distal myopathies with some forms of h-IBM and MFM. The distal myopathies can be subdivided, based on the clinical features, age of onset, CK levels, muscle histology, and mode of inheritance.
Figure 27-27. Diagnostic approach to patients with a distal pattern of weakness and suspected muscular dystrophy. *Autosomal dominant, autosomal recessive, or X-linked inheritance may be responsible in sporadic cases. h-IBM, hereditary inclusion body myopathy; h-IBMPFD, hereditary inclusion body myopathy with Paget Disease and Frontotemporal Dementia. (Reproduced with permission from Narayanaswami P, Weiss M, Selcen D, et al: Evidence-based guideline summary: diagnosis and treatment of limb-girdle and distal dystrophies: report of the guideline development subcommittee of the American Academy of Neurology and the practice issues review panel of the American Association of Neuromuscular & Electrodiagnostic Medicine, Neurology. 2014;83(16):1453–1463.)
Welander originally described the features of this autosomal dominant myopathy in a report of 249 cases from 72 Scandinavian families.380 Onset of weakness usually begins in the fifth decade of life, with rare cases beginning before the age of 30 years (mean age of onset 47 years, range 20–77 years). Weakness is usually first noted in the wrist and finger extensors and slowly progresses to involve the distal lower limbs—ankle dorsiflexors more than the plantar flexors.2,380–382 However, in approximately 10% of cases, weakness is initially appreciated in the legs or there is simultaneous involvement of the distal arms and legs. Although the extensor muscle groups are more severely affected, the flexor groups are involved in over 40% of cases. Rarely, proximal muscles become weak. Sensation is usually normal. Muscle stretch reflexes are initially preserved, but the brachioradialis and Achilles’ reflexes diminish or disappear over time.
Serum CK levels are usually normal or only minimally abnormal.2 Motor and sensory nerve conduction studies are usually normal for age. Diminished temperature and vibratory perception quantitative sensory testing has been demonstrated in some patients.381–383 Needle EMG demonstrates early recruitment of small-amplitude, short-duration MUAPs.2,381–383 Quantitative EMG further suggests a myopathic process.381
Muscle biopsies demonstrate variability in fiber size, increased central nuclei, split fibers, and increased endomysial connective tissue and adipose cells in longstanding disease.2,381–386 Furthermore, rimmed vacuoles typical of IBM, h-IBM, and OPMD are seen in scattered muscle fibers. EM also reveals 15–18-nm cytoplasmic and nuclear filaments similar to those observed in IBM, h-IBM, and OPMD. In addition, disruption of myofibrils and accumulation of Z-disc-derived material similar to that found in MFM can also be demonstrated. Nerve biopsies may reveal a moderate reduction of mainly small-diameter, myelinated fibers.383
Welander myopathy is caused by mutations in the TIA1 gene that encodes for T-cell restricted intracellular antigen, which is a RNA-binding protein.387 The TIA1 protein appears to aggregate in granules in muscle biopsy in patients with Welander myopathy. The mutations in TIA1 may make the transcribed protein more prone to self-aggregation and aggregate with other proteins.387,388
As previously discussed, this autosomal dominant distal myopathy is associated with mutations in the TTN. Udd distal myopathy usually presents after the age of 35 years (usually in the fifth to seventh decade), with weakness of the anterior compartment of the lower legs resulting in unilateral or bilateral foot drop.2,209,210,211–220,389,390 The disorder is slowly progressive, beginning in the toe extensors and gradually involving anterior tibial muscles. Occasionally, the proximal legs and distal upper limbs (predominately the hand intrinsics and wrist extensors) are affected. Rarely, the arms are affected more than the legs, posterior calves are involved with sparing of the anterior tibial muscles, or patients have a limb-girdle distribution of weakness.209 Facial muscles are usually spared, although bulbar weakness has been reported. Sensation is normal. Achilles’ tendon reflexes are usually reduced. Unlike other forms of titinopathy, cardiac and ventilatory muscles are usually spared in Udd distal myopathy.
Serum CK is normal or only slightly elevated.2,209,210–213,215–220,389,390 Motor and sensory NCS are normal. EMG of affected muscles reveals fibrillation potentials and positive sharp waves as well as small-amplitude, brief-duration MUAPs that recruit early.2,212,390 Imaging scans of muscle revealed fatty infiltration in the anterior tibial and extensor digitorum longus more than the gastrocnemius muscles; the proximal pelvic muscles, gluteus medius and minimus may be affected later.210,215
Muscle biopsies reveal dystrophic features fibers with rimmed vacuoles.2,212,388,390 Other features of MFM are usually not seen in titinopathy manifesting with the Udd distal myopathy, but may be seen in those with the HMERF presentation as discussed in LGMD2J section.
Udd distal myopathy is caused by mutations in the TTN gene on chromosome 2q31–33 encoding for titin.2,209,391 As previously discussed, the disorder is allelic to autosomal recessive LGMDJ and autosomal dominant HMERF. Why dominant mutations in the TTN typically lead to such different phenotypes and inheritance pattern is not entirely clear. A confounding factor is the variability of clinical phenotype sometimes seen even within families. The giant protein titin (also known as connectin) is attached to the Z-disc and spans from the M- to the Z-line of the sarcomere. Titin serves to connect the myosin filaments to the Z-disc and probably plays an important role in myofibrillogenesis.
This is another late-onset, autosomal dominant distal myopathy, typically beginning in the anterior compartment of the legs with onset in third to eighth decade of life.2,392,393 Some patients develop proximal leg and distal arm weakness (wrist and finger extensors) as well. A dilated cardiomyopathy is common.
Serum CK is normal or only mildly elevated. Motor and sensory nerve conduction studies are usually normal. Serum CK is usually mildly elevated, and EMG reveals features of a myopathy with muscle membrane irritability.
Muscle biopsies demonstrate rimmed vacuoles and features of MFM.
Markesbery–Griggs distal myopathy is caused by mutations in LIM domain binding 3 gene (LDB3) that encodes for Z-band alternatively spliced PDZ-motif containing protein or ZASP.2,393
This autosomal recessive myopathy was initially reported in Japan,395–398 but it occurs worldwide, and is allelic to autosomal recessive inclusion body myopathy (h-IBM type 1).2,399–402 The preferred term nowadays is “GNE myopathy.” Affected individuals usually develop weakness of the anterior compartment of the distal lower limb, leading to foot drop in the second or third decade of life. The posterior compartment of the legs and distal upper limb muscles are also affected early, but to a lesser degree. The proximal arm and leg muscles as well as the neck flexors may become weak over time. The quadriceps may become affected but usually remain relatively spared compared to other muscle groups, as are ocular and bulbar muscles. Sensation is normal. Muscle stretch reflexes can be normal or absent.
Serum CK is normal or only mildly elevated. Motor and sensory NCS are usually normal. EMG reveals positive sharp waves and early recruitment of small-amplitude, brief-duration MUAPs in weak muscles.
Muscle biopsies demonstrate rimmed vacuoles with muscle fibers as well as other nonspecific myopathic features, as described in the other forms of distal myopathy.395–402 Because of the frequent rimmed vacuoles, the biopsy can be erroneously interpreted as sporadic inclusion body myositis (s-IBM). However, inflammation cell infiltrate and major histochemistry antigen 1 expression on muscle fibers are usually absent. EM can demonstrate 15–18-nm intranuclear and cytoplasmic tubulofilaments similar to that found in sporadic IBM.
Nonaka myopathy and autosomal-recessive h-IBM are allelic disorders caused by mutations in the GNE gene on chromosome 9p1-q1 that encodes for UDP-N-acetylglucosamine 2-epimerase/n-acetylmannosamine kinase.399,401,403 This may be involved in the post-translational glycosylation of proteins to form glycoproteins and in the production of sialic acid.
This recessively inherited myopathy is associated with early adult onset of calf atrophy and weakness and markedly elevated serum CKs. It is caused by mutations in the dysferlin gene and was discussed in greater detail in the section on LGMD 2B.
Laing and colleagues initially described an Australian family with dominant inheritance (nine affected members over four generations) with weakness beginning in the anterior compartment of the distal lower limbs and neck flexors between the ages of 4 and 25 years.404 Subsequently, this myopathy has been widely reported.2,405–409 Over time, there is involvement of finger extensors and later, to a lesser extent, the shoulder and hip girdle muscles. Finger flexors and hand intrinsic muscles are spared. Scapular winging, scoliosis, pes cavus, ankle contractures, and/or lumbar hyperlordosis are seen in approximately 50% of patients. Hand tremor may occur.
Serum CK is normal or slightly elevated.2,404,407,408 Motor and sensory NCS are normal. EMG reveals occasional fibrillation potentials and positive sharp waves and small-amplitude, short-duration, polyphasic MUAPs in distal more than proximal muscles.
Muscle biopsies demonstrate nonspecific myopathic features. Rimmed vacuoles are not seen. Large deposits of MyHC in the subsarcolemmal region of type 1 muscle fibers led some authorities to label this as a form of myosin storage myopathy.19,410
Laing distal myopathy is caused by mutations in the slow/beta cardiac MyHC 1 gene, MYH7, located on chromosome 14q.2,411–414 MyHC is the major myosin isoform expressed in type 1 muscle fibers. Of note, mutations have also been identified in the MYH7 in hyaline body myopathy (discussed in Chapter 28). Mutations in MYH7 are also a common cause of familial hypertrophic and dilated cardiomyopathy, although patients with the cardiomyopathy usually do not have much symptomatic skeletal muscle involvement and vice versa.19 That said, we have followed patients with Laing myopathy, who also had a severe cardiomyopathy requiring transplantation, so cardiac evaluation in all patients is important.
Williams distal myopathy is an autosomal dominant disorder that manifests as progressive, predominantly lower extremity weakness that can affect proximal or distal muscles either in the arms or legs with an onset in the teens to fifth decade of life.2,415–418 Some patients develop a cardiomyopathy.
Serum CK is usually mildly elevated and EMG is myopathic.
Muscle biopsies may demonstrate features of MFM as will be discussed in a later section.
William distal myopathy is caused by mutations in the FLN-C gene that encodes for filamin C, an actin-binding protein felt to be important in cytoskeletal formation.
 OTHER DISTAL MYOPATHIES
OTHER DISTAL MYOPATHIESMutations in the nebulin gene (NEB), although usually associated with nemaline myopathy with a congenital onset, can cause a later onset distal myopathy with nemaline rods.419,420 Such affected individuals develop slowly progressive weakness with foot drop along with finger extensor and neck flexor weakness later in childhood or in the teens. CK levels are normal or only slightly elevated. PFTs may reveal a reduced FVC.419 Fatty degeneration in the anterior compartment of the lower legs may be apparent on skeletal muscle MRI.419,420 The diagnosis is made by demonstration of nemaline bodies on muscle biopsy in patients with the characteristic phenotype and with confirmatory genetic testing.
Mutations in KLHL9 that encodes for KELCH-Like Homologue 9 is also associated with progressive foot drop followed by intrinsic hand weakness with onset in the first or second decade of life.421 Inheritance is autosomal dominant. Weakness is slowly progressive such that affected individuals retained the ability to walk until the seventh decade. CK levels are normal or mildly elevated. Muscle biopsy reveals nonspecific dystrophic changes without rimmed vacuoles.
Late-onset, autosomal dominant vocal cord and pharyngeal distal myopathy (VCPDM) is a rare and controversial disorder, in regard to the localization of the lesion (i.e., motor neuron disease versus myopathy).422–425 Weakness usually begins in the anterior tibial muscles in the fourth to sixth decade. Weakness is asymmetric in some. Vocal cord and pharyngeal involvement develops after the limb weakness manifested. Ventillatory weakness can ensue.
The initial description of a large family in America was that of a vacuolar a myopathy.422.423 Subsequently, some affected individuals developed progressive ventilatory failure resulting in death with 15 years of onset and examinations showed hyperreflexia of the lower limbs, indicative of upper motor neuron involvement as well as tongue fasciculations.424 These clinical findings led to the reclassification of this disorder in this family by one group of authors as a form of slowly progressive familial amyotrophic lateral sclerosis (ALS21) rather than a myopathy.424 However, a recent paper involving 6 other families with the same MTR3 mutation reported a clearly progressive degenerative myopathy without any evidence of lower motor neuron defects by clinical examination, EMG, and histopathology.425 This again calls into question the site of the localization, but we feel most cases are really a myopathy.
Serum CK levels are normal to moderately elevated. Motor and sensory NCS are usually normal but may reveal mild slowing of conduction velocities. EMG may reveal either myopathic or neurogenic features, but the description of these features was limited. Fibrillations and positive sharp waves have been described, but not fasciculation potentials.
Muscle biopsies demonstrated nonspecific myopathic features along with numerous rimmed vacuoles. Notably, fiber-type grouping and grouped angulated atrophic fibers have not been reported.
This disorder is caused by mutations in MTR3 that encodes for matrin-3.423–425 Matrin-3 is a component of the nuclear matrix and appears to have roles in DNA replication, transcription, and RNA splicing. As mentioned, the localization of the site of pathology (motor neuron vs. muscle) is of debate. Perhaps, this may be similar to other disorders such as seen with mutations affecting similar genes such as VCP, SQTM1, HNRNPA2B1, and HNRNPA1 that can all be associated with familial ALS or a h-IBM (also associated with rimmed vacuoles in muscle—see later section on h-IBMPFD).
TREATMENT OF THE DISTAL MYOPATHIESThere is no specific medical treatment currently available for distal myopathies. Braces for lower limb weakness and other orthotic devices may be of benefit in improving gait and functional abilities.
OTHER MUSCULAR DYSTROPHIESMFM is a clinically and genetically heterogeneous group of disorders, characterized by the pathologic finding of myofibrillar disruption on EM and excessive desmin accumulation in muscle fibers.2,20,21,426–438 Because desmin is not the only protein that accumulates, the term “MFM” was suggested to be a more accurate description of the spectrum of the histologic abnormalities.435 This myopathy has been reported as desmin storage myopathy, desmin myopathy, familial desminopathy, spheroid body myopathy, cytoplasmic body myopathy, Mallory body myopathy, familial cardiomyopathy with subsarcolemmal vermiform deposits, myopathy with intrasarcoplasmic accumulation of dense granulofilamentous material, and h-IBM with early respiratory failure.426 In addition, some cases previously diagnosed with other forms of distal myopathy (Markesbery–Griggs distal myopathy) have MFM histopathology.392 MFM has been classified by some in the past as congenital myopathies, but are probably best considered a form of muscular dystrophy.
As mentioned, MFM is associated with a wide spectrum of clinical phenotypes.2,426,429,431–438 Most affected individuals develop weakness between 25 and 45 years of age, although weakness may be noticeable in infancy or may present later in adulthood. Weakness can be predominantly proximal, distal, or generalized. In addition, some patients have a facioscapulohumeral or scapuloperoneal distribution of weakness. Facial and pharyngeal muscles can also be affected in some individuals. Rigidity of the spine can also be seen.
In addition to skeletal muscle, the heart can also be affected and cardiac arrhythmias and CHF may be the predominant features of the disease. In severe cardiomyopathies, pacemaker insertion or cardiac transplantation may be required. In addition, severe ventilatory muscle involvement can develop in MFM. Also, smooth muscle involvement may lead to intestinal pseudo-obstructions.
Serum CK is normal or usually only slightly increased in MFM.2 EKGs may demonstrate conduction defects or arrhythmia, while echocardiograms may reveal a dilated or hypertrophic cardiomyopathy. NCS are usually normal, although low CMAP and SNAP amplitudes and slowing of conduction velocities can be seen. EMG reveals increased insertional and spontaneous activity with fibrillation potentials, positive sharp waves, pseudomyotonic potentials, complex repetitive discharges, and early recruitment of short-duration, small-amplitude, polyphasic MUAPs.2 Long-duration, large-amplitude MUAPs may also be seen, owing to the chronicity of the disorder.
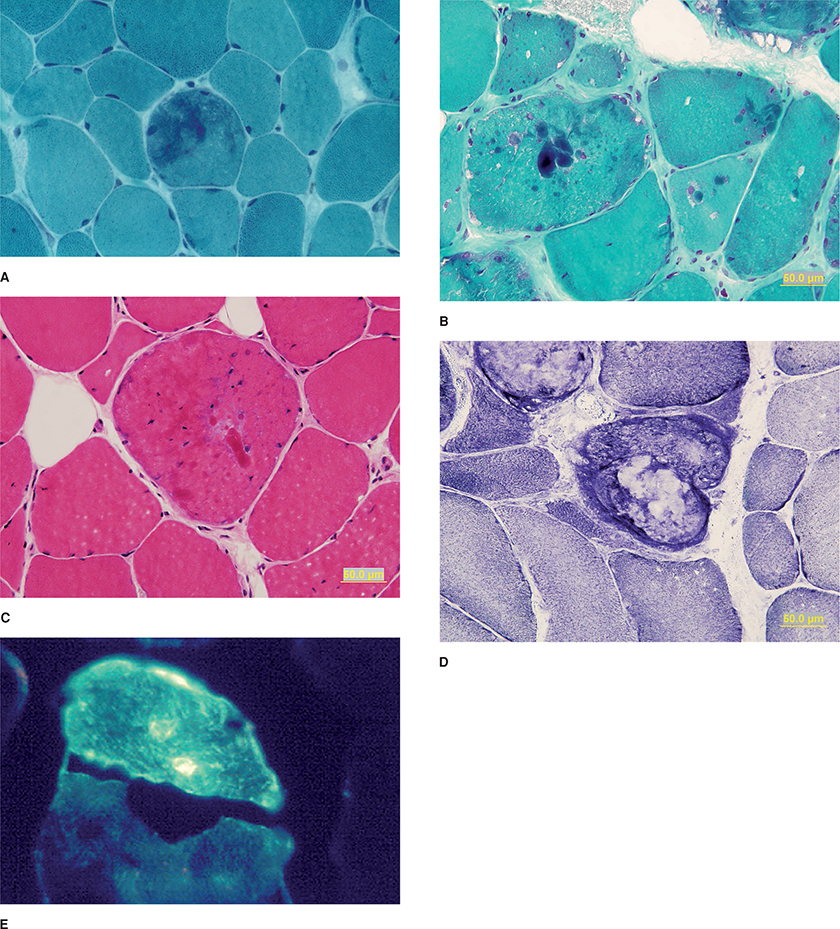
Muscle biopsies reveal variability in fiber size, increased internalized nuclei, occasionally type 1 fiber predominance, and in some cases, scattered fibers with rimmed vacuoles.2 In addition, Nakano et al. defined two major types of lesions on light and EM that characterize MFM: hyaline structures and nonhyaline lesions (Fig. 27-28).434 The hyaline structures are cytoplasmic granular inclusions, which are typically eosinophilic on H&E and dark blue-green or occasionally red on modified Gomori trichrome stains. They do not stain for NADH. On EM, the hyaline lesions resemble cytoplasmic, spheroid, or Mallory bodies. The nonhyaline lesions appear as dark green areas of amorphous material on Gomori trichrome stains. On EM, these nonhyaline lesions correspond to foci of myofibrillar destruction and consist of disrupted myofilaments, Z-disc-derived bodies, dappled dense structures of Z-disc origin, and streaming of the Z-disc.434,436 In addition, larger-size tubulofilaments (14–20 nm) typical of the inclusion body myopathies may accumulate.
Figure 27-28. Myofibrillar myopathy. Nonhyaline lesions appear as amorphous accumulation of reddish-purple or dark green material (A), while the hyaline lesions are denser and can have the appearance of cytoplasmic or spheroid bodies (B) on Trichrome stain. The hyaline lesions are eosinophilic on H&E but less well seen than on the trichrome stains (C). The hyaline and nonhyaline lesions do not stain with NADH-TR (D). Immunostaining reveals that the lesions are immunoreactive to desmin (E).
Immunohistochemistry reveals that both the hyaline and the nonhyaline lesions contain desmin and numerous other proteins.2,22,426–434,436,438 Abnormal accumulation of desmin is not specific for MFM and can be seen in a variety of neuromuscular conditions, including X-linked myotubular myopathy, congenital myotonic dystrophy, spinal muscular atrophy, nemaline rod myopathy, fetal myotubes, IBM, and in regenerating muscle fibers of any etiology. Abnormal accumulation of desmin has been demonstrated in cardiac muscles in MFM patients with cardiomyopathy. Immunohistochemistry also reveals that the nonhyaline lesions react strongly not only for desmin, but also for dystrophin, gelsolin, N terminus of β-amyloid precursor protein, and NCAM in addition to desmin. In addition, the nonhyaline lesions are depleted of actin, α-actinin, myosin, and, less consistently, titin and nebulin. In contrast, the hyaline structures are composed of compacted and degraded remnants of thick and thin filaments and react to actin, α-actinin, and myosin, in addition to dystrophin, gelsolin, filamin c, and the N terminus of β-amyloid precursor protein; they do not react to NCAM and react variably to desmin. Both types of lesions also react for αB-crystallin, α-1 antichymotrypsin, and ubiquitin and can be congophilic. The abnormal muscle fibers also abnormally express several cyclin-dependent kinases (CDC/CDK) in the cytoplasm, including CDC2, CDK2, CDK4, and CDK7.426,438
Nerve and intramuscular nerve biopsies have demonstrated enlarged axons with accumulation of intermediate-sized neurofilaments and formation of axonal spheroids in some patients.439,440
The pathogenesis of MFM is likely related to disruption of the Z-disc.2,20,21,426,435,436 Mutations have been identified in the genes that encode for desmin, αB-crystallin, myotilin, filamin c, BCL2-associated athanogene 3 (BAG3), FHL-1, ZASP (Z-band alternatively spliced PDX motif-containing protein), titin, selenoprotein N, DNAJB6, and transportin 3. Most of these proteins are Z-disc-related proteins. Most familial cases demonstrate autosomal-dominant inheritance, although autosomal-recessive (some desmin) and X-linked inheritance (FHL1) occur.
Mutations in the desmin gene, DES, located on chromosome 2q35 is associated with autosomal-dominant LGMD1E and recessive LGMD2R that have MFM histopathology as previously discussed.2,129–138,428,432,436,441 Mutations in DES were demonstrated in the initial family reported with scapuloperoneal myopathy. Desmin is an intermediate filament protein of skeletal, cardiac, and some smooth muscle cells. This cytoskeletal protein links Z-bands with the sarcolemma and the nucleus. The intermediate filament network is important in the stability of the muscle fiber and during mitosis/regeneration of muscle cells. These abnormal desmin filaments form insoluble aggregates, which prevent the genesis of the normal filamentous network.442
Mutations in the αB-crystallin gene, CRYAB, on chromosome 11q21–23 have been demonstrated in some autosomal- dominant kinships.435–437 Alpha B-crystallin possesses “molecular chaperone” activity and is felt to interact with desmin in the assembly of the intermediate filament network.
Missense mutations in the myotilin gene, MYOT, located on chromosome 5q22–31 cause late-onset MFM and are allelic to LGMD 1 A as previously discussed.2,20,90–99 These patients had late onset of distal greater than proximal weakness, polyneuropathy, and cardiopathy. Myotilin is a component of the Z-disc where it interacts with α-actinin, actin, and filamin c and probably plays a fundamental role in myofibrillar assembly.
In addition, missense mutations in the LDP3 gene located on chromosome 10q22.3–10q23.2 that encodes for ZASP are responsible for Markesbury–Griggs distal myopathy, which has MFM histopathology as previously discussed.2,21,393 ZASP is expressed in skeletal and cardiac muscle and it binds to α-actinin, a component of the Z-disc that in turn cross-links thin filaments of adjacent sarcomeres.21
Mutations in BAG3 located on chromosome 10q26.11 are associated with proximal and/or distal weakness, hypernasal speech, ventilatory weakness, and dilated cardiomyopathy with onset in the first or second decade.394 BAG3 interacts with heat shock proteins and may have a role in cellular response to environmental stress.
Mutations in FLN-C located on chromosome 7q32 that encodes for filamin-C cause Williams distal myopathy, as previously discussed.2,22,415–418 Filamin-C binds actin and is involved in the formation of the Z-disc. In addition, filamin-c also binds γ- and δ-sarcoglycan at the sarcolemmal membrane and may also play a role in signaling pathways from the sarcolemma to the myofibril.22
Mutations in FHL1 on Xq26.3 encoding for four and a half LIM protein some cases of scapuloperoneal dystrophy with reducing bodies and features of MFM on muscle biopsy.2,329
Mutations in the selenoprotein N gene (SEPN1) located on chromosome 1p36 were identified in autosomal-recessive MFM associated with Mallory-body-like inclusions.301 These patients have an early onset of axial muscle weakness, ventilatory weakness, and rigidity of the spine. Of note, mutations in SEPN1 gene can cause MDC with rigid spine and multi/minicore myopathy, but can also have histopathology resembling MFM.
Finally, LGMD1D and LGMD1E caused by mutations in the genes encoding for DNAJB6 and transportin 3, respectively, can have MFM-like histopathology.140
There is no proven medical therapy to improve skeletal muscle weakness. Antiarrhythmic and cardiotropic medications are sometimes necessary in patients with cardiopathy. Cardiac transplantation can be lifesaving in patients with severe cardiomyopathy.
There are autosomal-dominant and autosomal-recessive forms of h-IBM.2
The most common form of autosomal recessive h-IBM2 is allelic Nonaka distal myopathy being caused by mutations in GNE that encodes for UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase.2,395–402 As previously discussed, the preferred term now is “GNE myopathy.” This myopathy was initially reported in Iranian Jews and other Middle Eastern Karaites and Arab Muslims of Palestinian and Bedouin origin as a form of hIBM, while Nonaka distal myopathy was the name given for similar patients reported in Japanese, Korean, and Chinese families. The age of onset and pattern of weakness in GNE myopathy are different from that of s-IBM. Most patients with s-IBM present over the age of 50 years with weakness of quadriceps and wrist and finger flexors. In contrast, most patients with GNE myopathy present in the late teens to early 40s with anterior tibial involvement leading to foot drop. There is insidious progression with gradual involvement of the iliopsoas, thigh adductors, and to a lesser extent the glutei muscles. Importantly, in differentiating from s-IBM, the quadriceps are usually normal or relatively spared.399–402 However, rarely the quadriceps can be affected. The proximal arms and neck flexors can also become affected. There can be asymmetry of muscle weakness. Progression is variable, with some patients becoming wheelchair dependent within a few years of onset, while others are ambulatory several decades later.
There are a few reports of familial cases of s-IBM in which siblings, even twins, had the characteristic clinical phenotype and histological features of s-IBM.402 These cases do not represent h-IBM. Rather, there may be a familial predisposition for development of s-IBM, similar to that described for other autoimmune disorders.
Serum CK levels are normal or only mildly elevated. Motor and sensory nerve conduction studies are usually normal. EMG demonstrates fibrillation potentials, positive sharp waves, and complex repetitive discharges. There is a mixture of small-amplitude, short-duration polyphasic MUAPs with large-amplitude, long-duration polyphasic MUAPs.
Muscle biopsies are similar to s-IBM, except for the lack of significant endomysial inflammation and invasion of non-necrotic muscle fibers.2,399–402 Fiber size variability, split fibers, increased central nuclei, and fibers with rimmed vacuoles are evident. Amyloid and other “Alzheimer characteristic proteins” are seen in vacuolated muscle fibers, although they are much less frequent compared to s-IBM. As in s-IBM, EM demonstrates the abnormal accumulation of 15–18-nm tubulofilaments in the cytoplasm and nuclei of muscle fibers.
Nonaka-type distal myopathy and autosomal-recessive h-IBM are allelic disorders caused by mutations in GNE on chromosome 9p1-q1 encoding UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase.2,399–402 GNE is involved in the post-translational glycosylation of proteins to form glycoproteins. Disturbed glycosylation is therefore now recognized as a newly identified molecular genetic defect for muscular dystrophies. However, other mechanisms may be involved in the pathogenesis of this myopathy. GNE is an important enzyme in the sialic acid biosynthesis pathway.
There is no medical treatment available for GNE myopathy. Patients with distal lower limb weakness may benefit from bracing.
This myopathy usually presents with congenital arthrogryposis and ophthalmoparesis with mild proximal weakness beginning in adulthood. Face and distal extremity weakness may occur. Some patients complain of muscle pain. As muscle biopsies may demonstrate rimmed vacuoles and tubulofilamentous inclusions, this disorder has also been called h-IBM type 3.2,19,443,444
Serum CK levels are usually.
Muscle biopsies reveal small and infrequent type II fibers, particularly type 2 A fibers, focal disorganization of the myofibrils, and rimmed vacuoles and inclusion consisting of 15- to 20-nm tubulofilaments.2 Lobulated fibers and minicores have also been reported.
Mutations have been identified in the MYH2 gene located on chromosome 17p13.1, which encodes for MyHC IIa.2,9,443,444 The MyHC IIa isoform of MyHCs is expressed in type 2 A muscle fibers.
This autosomal-recessive h-IBM is associated with an infantile onset of progressive proximal greater than distal weakness, legs worse than arms, and marked cerebral white matter abnormalities on CT and MRI.445 Despite the apparent leukoencephalopathy on radiological imaging, intellectual function was normal in all the cases. Motor nerve conduction studies were mildly slow, suggesting dysmyelination of peripheral nerves as well.
h-IBM associated with Paget disease of the bone and frontotemporal dementia, or IBMPFD, is a rare autosomal-dominant disorder is usually caused by mutations in VCP that encodes for valosin-containing protein (VCP).2,446–463 It is characterized by adult onset (range late first to ninth decade, with mean in the 40s) of limb-girdle, distal, or scapuloperoneal weakness. There also appears to be a mild asymmetry and variability in the patterns of muscle weakness. Frontotemporal dementia is seen in approximately 30–50% with onset approximately 10 years after weakness (average age 54 years). Paget disease of the bone (PDB) tends to occur earlier than in the more common sporadic forms of PDB and is seen with variable frequency. The complete triad of h-IBM, PDB, and frontotemporal dementia occurs in only about one-third of cases. In addition, mutations in the same gene cause a form of familial amyotrophic lateral sclerosis (fALS) with or without frontotemporal dementia.464,465 A dilated cardiomyopathy may be seen in a quarter of patients.459 Ultimately, the cause of death is through progressive muscle weakness and ventilatory failure. There is significant heterogeneity in clinical phenotype and severity both between and within families.
Serum CK levels are normal to slightly elevated. Serum alkaline phosphatase levels can be a screening test but may not be elevated in those without PDB. EMG shows myopathic changes with muscle membrane irritability.
Muscle biopsy reveal fibers with rimmed vacuoles and inclusions that immunostain with ubiquitin, TDP-43, and VCP.2,455,456 Neurogenic features of type grouping and angulated fibers, which is notable VCP mutations can also be associated with motor neuron disease.455,456 EM may show paired helical filaments in muscle and in PDB osteoclasts.
h-IBMPFD is usually caused by mutations in the gene encoding VCP, a member of the AAA-ATPase superfamily.2,446,447 VCP is associated with a variety of cellular activities, including cell-cycle control, membrane fusion, and the ubiquitin–proteasome degradation pathway. VCP normally localizes to nuclei and specifically near nucleoli. Mutations in VCP gene may disrupt in nuclear structure or normal translation of mRNA. In addition, mutations in SQTM1, HNRPA2B1, and HNRNPA1, have been noted to cause hIBM or fALS, while mutations in SQTM1 can also cause PDB. Some have termed these disorders as “multisystem proteinopathies.”466,467.
SUMMARYWith so many different types of muscular dystrophies and the variability of clinical phenotypes associated with specific forms of dystrophy, even within individual families, the evaluation of patients presenting with weakness can be quite daunting. However, rather than ordering every genetic test possible or doing a muscle biopsy initially on every patient, an approach to ordering tests based on clinical phenotype (inheritance pattern, age of onset, pattern of weakness, and associated manifestations—early contractures and cardiac or ventilatory involvement) should be useful (Figs. 27-8, 27-10, 27-16, 27-19, 27-23, 27-27).2,250 However, as next generation, whole genome, and whole exome sequencing become more widely available and less expensive, such large scale genetic testing will become a more accessible tool due to cost considerations. Nonetheless, accurate clinical assessment will remain a prerequisite in order to distinguish pathological mutations from the benign polymorphisms that this technology will uncover.
Unfortunately, there are limited beneficial medical treatments, other than corticosteroids for children with DMD. Still with supportive treatments (physical and occupational therapy, bracing, respiratory, and cardiac), quality of life can be improved in patients. More work needs to be done to further understand the pathogenesis of these disorders and discover targeted and better treatments.
1. Wicklund MP. The muscular dystrophies. Continuum (Minneap Minn). 2013;19(6 Muscle Disease):1535–1570.
2. Narayanaswami P, Weiss M, Selcen D, et al. Summary of evidence-based guideline: Diagnosis and treatment of limb-girdle and distal muscular dystrophies. Neurology. 2014;83 (16):1453–1463.
3. Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928.
4. Hoffman EP, Fischbeck KH, Brown RH, et al. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne’s or Becker’s muscular dystrophy. N Engl J Med. 1988;318(21):1363–1368.
5. Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509–517.
6. Cohn RD, Campbell KP. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23(10):1456–1471.
7. Michele DE, Barresi R, Kanagawa M, et al. Post-translational disruption of dystroglycan–ligand interactions in congenital muscular dystrophies. Nature. 2002;418(6896):417–422.
8. Wewer UM, Engvall E. Merosin/laminin-2 and muscular dystrophy. Neuromuscul Disord. 1996;6(6):409–418.
9. Matsumura K, Yamada H, Saito F, Sunada Y, Shimizu T. Peripheral nerve involvement in merosin-deficient congenital muscular dystrophy and dy mouse. Neuromuscul Disord. 1997;7(1):7–12.
10. Vachon PH, Xu H, Liu L, et al. Integrins (alpha7beta1) in muscle function and survival. Disrupted expression in merosin-deficient congenital muscular dystrophy. J Clin Invest. 1997; 100(7):1870–1881.
11. Bansal D, Miyake K, Vogel SS, et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003; 423(6936):168–172.
12. Cenacchi G, Fanin M, De Giorgi LB, Angelini C. Ultrastructural changes in dysferlinopathy support defective membrane repair mechanism. J Clin Pathol. 2005;58(2):190–195.
13. Bashir R, Britton S, Strachan T, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet. 1998;20 (1):37–42.
14. Matsuda C, Aoki M, Hayashi YK, Ho MF, Arahata K, Brown RH Jr. Dysferlin is a surface membrane-associated protein that is absent in Miyoshi myopathy. Neurology. 1999;53(5):1119–1122.
15. Carbone I, Bruno C, Sotgia F, et al. Mutation in the CAV3 gene causes partial caveolin-3 deficiency and hyperCKemia. Neurology. 2000;54(6):1373–1376.
16. Minetti C, Sotgia F, Bruno C, et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet. 1998;18(4):365–368.
17. Sotgia F, Woodman SE, Bonuccelli G, et al. Phenotypic behavior of caveolin-3 R26Q, a mutant associated with hyperCKemia, distal myopathy, and rippling muscle disease. Am J Physiol Cell Physiol. 2003;285(5):C1150–C1160.
18. Woodman SE, Sotgia F, Galbiati F, Minetti C, Lisanti MP. Caveolinopathies: Mutations in caveolin-3 cause four distinct autosomal dominant muscle diseases. Neurology. 2004;62(4):538–543.
19. Oldfors A, Tajsharghi H, Darin N, Lindberg C. Myopathies associated with myosin heavy chain mutations. Acta Myol. 2004;23(2):90–96.
20. Selcen D, Engel AG. Mutations in myotilin cause myofibrillar myopathy. Neurology. 2004;62(8):1363–1371.
21. Selcen D, Engel AG. Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann Neurol. 2005;57(2):269–276.
22. Vorgerd M, van der Ven PF, Bruchertseifer V, et al. A mutation in the dimerization domain of filamin c causes a novel type of autosomal dominant myofibrillar myopathy. Am J Hum Genet. 2005;77(2):297–304.
23. Nagano A, Arahata K. Nuclear envelope proteins and associated diseases. Curr Opin Neurol. 2000;13(5):533–539.
24. Nagano A, Koga R, Ogawa M, et al. Emerin deficiency at the nuclear membrane in patients with Emery–Dreifuss muscular dystrophy. Nat Genet. 1996;12(3):254–259.
25. Ognibene A, Sabatelli P, Petrini S, et al. Nuclear changes in a case of X-linked Emery–Dreifuss muscular dystrophy. Muscle Nerve. 1999;22(7):864–869.
26. Sabatelli P, Lattanzi G, Ognibene A, et al. Nuclear alterations in autosomal-dominant Emery–Dreifuss muscular dystrophy. Muscle Nerve. 2001;24(6):826–829.
27. Bonne G, Mercuri E, Muchir A, et al. Clinical and molecular genetic spectrum of autosomal dominant Emery–Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. 2000;48(2):170–180.
28. Felice KJ, Schwartz RC, Brown CA, Leicher CR, Grunnet ML. Autosomal dominant Emery–Dreifuss dystrophy due to mutations in rod domain of the lamin A/C gene. Neurology. 2000;55(2):275–280.
29. Frosk P, Weiler T, Nylen E, et al. Limb-girdle muscular dystrophy type 2 H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am J Hum Genet. 2002;70(3):663–672.
30. Brockington M, Blake DJ, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet. 2001;69(6):1198–1209.
31. Brockington M, Yuva Y, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1 C. Hum Mol Genet. 2001;10(25):2851–2859.
32. Brown SC, Torelli S, Brockington M, et al. Abnormalities in alpha-dystroglycan expression in MDC1 C and LGMD2I muscular dystrophies. Am J Pathol. 2004;164(2):727–737.
33. Beltran-Valero de Bernabe D, Voit T, Longman C, et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet. 2004;41(5):e61.
34. Brockington M, Torelli S, Prandini P, et al. Localization and functional analysis of the LARGE family of glycosyltransferases: Significance for muscular dystrophy. Hum Mol Genet. 2005;14(5):657–665.
35. Longman C, Brockington M, Torelli S, et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet. 2003;12(21):2853–2861.
36. Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9(1):77–93.
37. Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurol. 2010;9(2):177–189.
38. Flanigan KM. Duchenne and Becker Muscular Dystrophies. Neurol Clin. 2014;32(3):671–688.
39. Emery AE. Population frequencies of inherited neuromuscular diseases-a world survey. Neuromuscul Disord. 1991;1(1):19–29.
40. Brooke MH, Fenichel GM, Griggs RC, et al. Duchenne muscular dystrophy: Patterns of clinical progression and effects of supportive therapy. Neurology. 1989;39(4):475–481.
41. Brooke MH, Griggs RC, Mendell JR, Fenichel GM, Shumate JB. The natural history of Duchenne muscular dystrophy: A caveat for therapeutic trials. Trans Am Neurol Assoc. 1981;106:195–199.
42. Farah MG, Evans EB, Vignos PJ Jr. Echocardiographic evaluation of left ventricular function in Duchenne’s muscular dystrophy. Am J Med. 1980;69(2):248–254.
43. Perloff JK, Roberts WC, de Leon AC Jr, O’Doherty D. The distinctive electrocardiogram of Duchenne’s progressive muscular dystrophy. An electrocardiographic-pathologic correlative study. Am J Med. 1967;42(2):179–188.
44. Sanyal SK, Johnson WW. Cardiac conduction abnormalities in children with Duchenne’s progressive muscular dystrophy: Electrocardiographic features and morphologic correlates. Circulation. 1982;66(4):853–863.
45. Leibowitz D, Dubowitz V. Intellect and behaviour in Duchenne muscular dystrophy. Dev Med Child Neurol. 1981;23(5):577–590.
46. Arahata K, Engel AG. Monoclonal antibody analysis of mononuclear cells in myopathies. I: Quantitation of subsets according to diagnosis and sites of accumulation and demonstration and counts of muscle fibers invaded by T cells. Ann Neurol. 1984;16(2):193–208.
47. Bushby KM, Thambyayah M, Gardner-Medwin D. Prevalence and incidence of Becker muscular dystrophy. Lancet. 1991;337(8748):1022–1024.
48. Bushby KM, Gardner-Medwin D. The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. I. Natural history. J Neurol. 1993;240(2):98–104.
49. Sunohara N, Arahata K, Hoffman EP, et al. Quadriceps myopathy: Forme fruste of Becker muscular dystrophy. Ann Neurol. 1990;28(5):634–639.
50. Comi GP, Prelle A, Bresolin N, et al. Clinical variability in Becker muscular dystrophy. Genetic, biochemical and immunohistochemical correlates. Brain. 1994;117(Pt 1):1–14.
51. Gospe SM, Jr., Lazaro RP, Lava NS, Grootscholten PM, Scott MO, Fischbeck KH. Familial X-linked myalgia and cramps: A nonprogressive myopathy associated with a deletion in the dystrophin gene. Neurology. 1989;39(10):1277–1280.
52. Doriguzzi C, Palmucci L, Mongini T, Chiadò-Piat L, Restagno G, Ferrone M. Exercise intolerance and recurrent myoglobinuria as the only expression of Xp21 Becker type muscular dystrophy. J Neurol. 1993;240(5):269–271.
53. Muntoni F, Cau M, Ganau A, et al. Brief report: Deletion of the dystrophin muscle-promoter region associated with X-linked dilated cardiomyopathy. N Engl J Med. 1993;329(13):921–925.
54. Towbin JA, Hejtmancik JF, Brink P, et al. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation. 1993;87(6):1854–1865.
55. de Visser M, de Voogt WG, la Rivière GV. The heart in Becker muscular dystrophy, facioscapulohumeral dystrophy, and Bethlem myopathy. Muscle Nerve. 1992;15(5):591–596.
56. Kaido M, Arahata K, Hoffman EP, Nonaka I, Sugita H. Muscle histology in Becker muscular dystrophy. Muscle Nerve. 1991;14(11):1067–1073.
57. Hoffman EP, Kunkel LM, Angelini C, Clarke A, Johnson M, Harris JB. Improved diagnosis of Becker muscular dystrophy by dystrophin testing. Neurology. 1989;39(8):1011–1017.
58. Hoffman EP, Arahata K, Minetti C, Bonilla E, Rowland LP. Dystrophinopathy in isolated cases of myopathy in females. Neurology. 1992;42(5):967–975.
59. Arahata K, Ishihara T, Kamakura K, et al. Mosaic expression of dystrophin in symptomatic carriers of Duchenne’s muscular dystrophy. N Engl J Med. 1989;320(3):138–142.
60. Clerk A, Rodillo E, Heckmatt JZ, Dubowitz V, Strong PN, Sewry CA. Characterisation of dystrophin in carriers of Duchenne muscular dystrophy. J Neurol Sci. 1991;102(2):197–205.
61. Minetti C, Chang HW, Medori R, et al. Dystrophin deficiency in young girls with sporadic myopathy and normal karyotype. Neurology. 1991;41(8):1288–1292.
62. Hyser CL, Doherty RA, Griggs RC, et al. Carrier assessment for mothers and sisters of isolated Duchenne dystrophy cases: The importance of serum enzyme determinations. Neurology. 1987;37(9):1476–1480.
63. Hyser CL, Griggs RC, Mendell JR, et al. Use of serum creatine kinase, pyruvate kinase, and genetic linkage for carrier detection in Duchenne and Becker dystrophy. Neurology. 1987;37(1):4–10.
64. Bakker E, Veenema H, Den Dunnen JT, et al. Germinal mosaicism increases the recurrence risk for ‘new’ Duchenne muscular dystrophy mutations. J Med Genet. 1989;26(9):553–559.
65. Prior TW, Bartolo C, Pearl DK, et al. Spectrum of small mutations in the dystrophin coding region. Am J Hum Genet. 1995;57(1):22–33.
66. Brooke MH, Fenichel GM, Griggs RC, et al. Clinical investigation of Duchenne muscular dystrophy. Interesting results in a trial of prednisone. Arch Neurol. 1987;44(8):812–817.
67. Fenichel GM, Florence JM, Pestronk A, et al. Long-term benefit from prednisone therapy in Duchenne muscular dystrophy. Neurology. 1991;41(12):1874–1877.
68. Griggs RC, Moxley RT 3rd, Mendell JR, et al. Prednisone in Duchenne dystrophy. A randomized, controlled trial defining the time course and dose response. Clinical Investigation of Duchenne Dystrophy Group. Arch Neurol. 1991;48(4):383–388.
69. Griggs RC, Moxley RT 3rd, Mendell JR, et al. Duchenne dystrophy: Randomized, controlled trial of prednisone (18 months) and azathioprine (12 months). Neurology. 1993;43 (3 Pt 1):520–527.
70. Mendell JR, Moxley RT, Griggs RC, et al. Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophy. N Engl J Med. 1989;320(24):1592–1597.
71. Moxley RT 3rd, Ashwal S, Pandya S, et al. Practice parameter: Corticosteroid treatment of Duchenne dystrophy: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2005;64(1):13–20.
72. Backman E, Henriksson KG. Low-dose prednisolone treatment in Duchenne and Becker muscular dystrophy. Neuromuscul Disord. 1995;5(3):233–241.
73. Connolly AM, Schierbecker J, Renna R, Florence J. High dose weekly oral prednisone improves strength in boys with Duchenne muscular dystrophy. Neuromuscul Disord. 2002;12(10):917–925.
74. Angelini C, Pegoraro E, Turella E, Intino MT, Pini A, Costa C. Deflazacort in Duchenne dystrophy: Study of long-term effect. Muscle Nerve. 1994;17(4):386–391.
75. Bonifati MD, Ruzza G, Bonometto P, et al. A multicenter, double-blind, randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve. 2000;23(9):1344–1347.
76. Fenichel GM, Griggs RC, Kissel J, et al. A randomized efficacy and safety trial of oxandrolone in the treatment of Duchenne dystrophy. Neurology. 2001;56(8):1075–1079.
77. Tarnopolsky M, Martin J. Creatine monohydrate increases strength in patients with neuromuscular disease. Neurology. 1999;52(4):854–857.
78. Walter MC, Lochmuller H, Reilich P, et al. Creatine monohydrate in muscular dystrophies: A double-blind, placebo-controlled clinical study. Neurology. 2000;54(9):1848–1850.
79. Mendell JR, Kissel JT, Amato AA, et al. Myoblast transfer in the treatment of Duchenne’s muscular dystrophy. N Engl J Med. 1995;333(13):832–838.
80. Fairclough RJ, Wood MJ, Davies KE. Therapy for Duchenne muscular dystrophy: Renewed optimism from genetic approaches. Nat Rev Genet. 2013;14:373–378.
81. Cirak S, Arechavala-Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet. 2011;378(9791):595–605.
82. Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74(5):637–647.
83. Goemans NM, Tulinius M, van den Akker JT, et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N Engl J Med. 2011;364(16):1513–1522.
84. Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014;50(4):477–487.
85. Darras BT, Francke U. Myopathy in complex glycerol kinase deficiency patients is due to 3 deletions of the dystrophin gene. Am J Hum Genet. 1988;43(2):126–130.
86. Guggenheim MA, McCabe ER, Roig M, et al. Glycerol kinase deficiency with neuromuscular, skeletal, and adrenal abnormalities. Ann Neurol. 1980;7(5):441–449.
87. Seltzer WK, Angelini C, Dhariwal G, Ringel SP, McCabe ER. Muscle glycerol kinase in Duchenne dystrophy and glycerol kinase deficiency. Muscle Nerve. 1989;12(4):307–313.
88. Gilchrist JM, Pericak-Vance M, Silverman L, Roses AD. Clinical and genetic investigation in autosomal dominant limb-girdle muscular dystrophy. Neurology. 1988;38(1):5–9.
89. Chutkow JG, Heffner RR Jr, Kramer AA, Edwards JA. Adult-onset autosomal dominant limb-girdle muscular dystrophy. Ann Neurol. 1986;20(2):240–248.
90. Hauser MA, Horrigan SK, Salmikangas P, et al. Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum Mol Genet. 2000;9(14):2141–2147.
91. Olivé M, Goldfarb LG, Shatunov A, Fischer D, Ferrer I. Myotilinopathy: Refining the clinical and myopathological phenotype. Brain. 2005;128:2315–2326.
92. Pénisson-Besnier I, Talvinen K, Dumez C, et al. Myotilinopathy in a family with late onset myopathy. Neuromuscul Disord. 2006;16:427–431.
93. Schramm N, Born C, Weckbach S, Reilich P, Walter MC, Reiser MF. Involvement patterns in myotilinopathy and desminopathy detected by a novel neuromuscular whole-body MRI protocol. Eur Radiol. 2008;18:2922–2936.
94. Olivé M, Odgerel Z, Martínez A, et al. Clinical and myopathological evaluation of early- and late-onset subtypes of myofibrillar myopathy. Neuromuscul Disord. 2011;21:533–542.
95. Gilchrist JM, Pericak-Vance M, Silverman L, Roses AD. Clinical and genetic investigation in autosomal dominant limb-girdle muscular dystrophy. Neurology. 1988;38:5–9.
96. Marconi G, Pizzi A, Arimondi CG, Vannelli B. Limb girdle muscular dystrophy with autosomal dominant inheritance. Acta Neurol Scand. 1991;83(4):234–238.
97. Reilich P, Krause S, Schramm N, et al. A novel mutation in the myotilin gene (MYOT) causes a severe form of limb girdle muscular dystrophy 1 A (LGMD1 A). J Neurol. 2011;258(8):1437–1444.
98. McNeill A, Birchall D, Straub V, et al. Lower limb radiology of distal myopathy due to the S60 F myotilin mutation. Eur Neurol. 2009;62(3):161–166.
99. Schessl J, Bach E, Rost S, et al. Novel recessive myotilin mutation causes severe myofibrillar myopathy. Neurogenetics. 2014; 15(3):151–156.
100. Fang W, Huang CC, Chu NS, Chen CJ, Lu CS, Wang CC. Childhood-onset autosomal-dominant limb-girdle muscular dystrophy with cardiac conduction block. Muscle Nerve. 1997;20(3):286–292.
101. van der Kooi AJ, Ledderhof TM, de Voogt WG, et al. A newly recognized autosomal dominant limb girdle muscular dystrophy with cardiac involvement. Ann Neurol. 1996;39(5):636–642.
102. Bonne G, Di Barletta MR, Varnous S, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery–Dreifuss muscular dystrophy. Nat Genet. 1999;21(3):285–288.
103. Bonne G, Mercuri E, Muchir A, et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. 2000;48:170–180.
104. Jimenez-Escrig A, Gobernado I, Garcia-Villanueva M, Sanchez-Herranz A. Autosomal recessive Emery-Dreifuss muscular dystrophy caused by a novel mutation (R225Q) in the lamin A/C gene identified by exome sequencing. Muscle Nerve. 2012;45:605–610.
105. Rankin J, Auer-Grumbach M, Bagg W, et al. Extreme phenotypic diversity and nonpenetrance in families with the LMNA gene mutation R644 C. Am J Med Genet A. 2008;146A:1530–1542.
106. van der Kooi AJ, Bonne G, Eymard B, et al. Lamin A/C mutations with lipodystrophy, cardiac abnormalities, and muscular dystrophy. Neurology. 2002;59:620–623.
107. van der Kooi AJ, van Meegen M, Ledderhof TM, McNally EM, de Visser M, Bolhuis PA. Genetic localization of a newly recognized autosomal dominant limb-girdle muscular dystrophy with cardiac involvement (LGMD1B) to chromosome 1q11–21. Am J Hum Genet. 1997;60:891–895.
108. Vantyghem M, Pigny P, Maurage CA, et al. Patients with familial partial lipodystrophy of the Dunnigan type due to a LMNA R482 W mutation show muscular and cardiac abnormalities. J Clin Endocrinol Metab. 2004;89:5337–5346.
109. Carboni N, Mura M, Marrosu G, et al. Muscle imaging analogies in a cohort of patients with different clinical phenotypes caused by LMNA gene mutations. Muscle Nerve. 2010;41:458–463.
110. Deconinck N, Dion E, Ben Yaou R, et al. Differentiating Emery-Dreifuss muscular dystrophy and collagen VI-related myopathies using a specific CT scanner pattern. Neuromuscul Disord. 2010;20:517–523.
111. Mercuri E, Counsell S, Allsop J, et al. Selective muscle involvement on magnetic resonance imaging in autosomal dominant Emery-Dreifuss muscular dystrophy. Neuropediatrics. 2002;33:10–14.
112. Mercuri E, Clements E, Offiah A, et al. Muscle magnetic resonance imaging involvement in muscular dystrophies with rigidity of the spine. Ann Neurol. 2010;67:201–208.
113. Minetti C, Sotgia F, Bruno C, et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet. 1998;18(4):365–368.
114. Aboumousa A, Hoogendijk J, Charlton R, et al. Caveolinopathy–new mutations and additional symptoms. Neuromuscul Disord. 2008;18(7):572–578.
115. González-Pérez P, Gallano P, González-Quereda L, et al. Phenotypic variability in a Spanish family with a Caveolin-3 mutation. J Neurol Sci. 2009;276:95–98.
116. Cagliani R, Bresolin N, Prelle A, et al. A CAV3 microdeletion differentially affects skeletal muscle and myocardium. Neurology. 2003;61:1513–1519.
117. Catteruccia, M, Sanna T, Santorelli FM, et al. Rippling muscle disease and cardiomyopathy associated with a mutation in the CAV3 gene. Neuromuscul Disord. 2009;19:779–783.
118. Dotti MT, Malandrini A, Gambelli S, Salvadori C, De Stefano N, Federico A. A new missense mutation in caveolin-3 gene causes rippling muscle disease. J Neurol Sci. 2006;243:61–64.
119. Fischer D, Schroers A, Blümcke I, et al. Consequences of a novel caveolin-3 mutation in a large German family. Ann Neurol. 2003;53:233–241.
120. Fulizio L, Nascimbeni AC, Fanin M, et al. Molecular and muscle pathology in a series of caveolinopathy patients. Hum Mutat. 2005;25:82–89.
121. Jacobi C, Ruscheweyh R, Vorgerd M, Weber MA, Storch-Hagenlocher B, Meinck HM. Rippling muscle disease: Variable phenotype in a family with five afflicted members. Muscle Nerve. 2010;41:128–132.
122. Ricker K, Moxley RT, Rohkamm R. Rippling muscle disease. Arch Neurol. 1989;46:405–408.
123. Sundblom J, Stålberg E, Osterdahl M, et al. Bedside diagnosis of rippling muscle disease in CAV3 p.A46 T mutation carriers. Muscle Nerve. 2010;41:751–757.
124. Vorgerd M, Bolz H, Patzold T, Kubisch C, Malin JP, Mortier W. Phenotypic variability in rippling muscle disease. Neurology. 1999;52:1453–1459.
125. Yabe I, Kawashima A, Kikuchi S, et al. Caveolin-3 gene mutation in Japanese with rippling muscle disease. Acta Neurol Scand. 2003;108:47–51.
126. Hackman P, Sandell S, Sarparanta J, et al. Four new Finnish families with LGMD1D; refinement of the clinical phenotype and the linked 7q36 locus. Neuromuscul Disord. 2011;21:338–344.
127. Sarparanta J, Jonson PH, Golzio C, et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet. 2012;44:450–455, S1–S2.
128. Harms MB, Sommerville RB, Allred P, et al. Exome sequencing reveals DNAJB6 mutations in dominantly-inherited myopathy. Ann Neurol. 2012;71:407–416.
129. Goldfarb LG, Park KY, Cervenáková L, et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. 1998;19:402–403.
130. Bergman JE, Veenstra-Knol HE, van Essen AJ, et al. Two related Dutch families with a clinically variable presentation of cardioskeletal myopathy caused by a novel S13 F mutation in the desmin gene. Eur J Med Genet. 2007;50:355–366.
131. Dalakas MC, Dagvadorj A, Goudeau B, et al. Progressive skeletal myopathy, a phenotypic variant of desmin myopathy associated with desmin mutations. Neuromuscul Disord. 2003; 13:252–258.
132. Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. 2000;342:770–780.
133. Walter MC, Reilich P, Huebner A, et al. Scapuloperoneal syndrome type Kaeser and a wide phenotypic spectrum of adult-onset, dominant myopathies are associated with the desmin mutation R350P. Brain. 2007;130:1485–1496.
134. Goudeau B, Rodrigues-Lima F, Fischer D, et al. Variable pathogenic potentials of mutations located in the desmin alpha-helical domain. Hum Mutat. 2006;27:906–913.
135. Bär H, Goudeau B, Wälde S, et al. Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum Mutat. 2007;28:374–376.
136. Dagvadorj A, Olivé M, Urtizberea JA, et al. A series of West European patients with severe cardiac and skeletal myopathy associated with a de novo R406 W mutation in desmin. J Neurol. 2004;251:143–149.
137. Olivé M, Armstrong J, Miralles F, et al. Phenotypic patterns of desminopathy associated with three novel mutations in the desmin gene. Neuromuscul Disord. 2007;17:443–450.
138. Greenberg SA, Salajegheh M, Judge DP, et al. Etiology of limb girdle muscular dystrophy 1D/1E determined by laser capture microdissection proteomics. Ann Neurol. 2012;71:141–145.
139. Palenzuela L, Andreu AL, Gàmez J, et al. A novel autosomal dominant limb-girdle muscular dystrophy (LGMD 1 F) maps to 7q32.1–32.2. Neurology. 2003;61(3):404–406.
140. Melià MJ, Kubota A, Ortolano S, et al. Limb-girdle muscular dystrophy 1 F is caused by a microdeletion in the transportin 3 gene. Brain. 2013;136:1508–1517.
141. Fardeau M, Eymard B, Mignard C, Tomé FM, Richard I, Beckmann JS. Chromosome 15-linked limb-girdle muscular dystrophy: Clinical phenotypes in Reunion Island and French metropolitan communities. Neuromuscul Disord. 1996; 6(6):447–453.
142. Fardeau M, Hillaire D, Mignard C, et al. Juvenile limb-girdle muscular dystrophy. Clinical, histopathological and genetic data from a small community living in the Reunion Island. Brain. 1996;119(Pt 1):295–308.
143. Kawai H, Akaike M, Kunishige M, et al. Clinical, pathological, and genetic features of limb-girdle muscular dystrophy type 2 A with new calpain 3 gene mutations in seven patients from three Japanese families. Muscle Nerve. 1998;21(11):1493–1501.
144. Penisson-Besnier I, Richard I, Dubas F, Beckmann JS, Fardeau M. Pseudometabolic expression and phenotypic variability of calpain deficiency in two siblings. Muscle Nerve. 1998;21(8):1078–1080.
145. Spencer MJ, Tidball JG, Anderson LV, et al. Absence of calpain 3 in a form of limb-girdle muscular dystrophy (LGMD2 A). J Neurol Sci. 1997;146(2):173–178.
146. Guglieri M, Magri F, D’Angelo MG, et al. Clinical, molecular, and protein correlations in a large sample of genetically diagnosed Italian limb girdle muscular dystrophy patients. Hum Mutat. 2008;29:258–266.
147. Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: In-depth analysis of a muscle clinic population. Brain. 2009;132(Pt 11):3175–3186.
148. van der Kooi AJ, Frankhuizen WS, Barth PG, et al. Limb-girdle muscular dystrophy in the Netherlands: Gene defect identified in half the families. Neurology. 2007;68:2125–2128.
149. Krahn M, Lopez De Munain A, Streichenberger N, et al. CAPN3 mutations in patients with idiopathic eosinophilic myositis. Ann Neurol. 2006;59(6):905–911.
150. Amato AA. Adults with eosinophilic myositis and calpain-3 mutations. Neurology. 2008;70:730–731.
151. Jaka O, Azpitarte M, Paisán-Ruiz C, et al. Entire CAPN3 gene deletion in a patient with limb-girdle muscular dystrophy type 2 A. Muscle Nerve. 2014;50(3):448–453.
152. Degardin A, Morillon D, Lacour A, Cotten A, Vermersch P, Stojkovic T. Morphologic imaging in muscular dystrophies and inflammatory myopathies. Skeletal Radiol. 2010;39(12):1219–1227.
153. Mercuri E, Bushby K, Ricci E, et al. Muscle MRI findings in patients with limb girdle muscular dystrophy with calpain 3 deficiency (LGMD2 A) and early contractures. Neuromuscul Disord. 2005;15:164–171.
154. Beckmann JS, Richard I, Broux O, et al. Identification of muscle-specific calpain and beta-sarcoglycan genes in progressive autosomal recessive muscular dystrophies. Neuromuscul Disord. 1996;6(6):455–462.
155. Fanin M, Fulizio L, Nascimbeni AC, et al. Molecular diagnosis in LGMD2 A: Mutation analysis or protein testing? Hum Mutat. 2004;24(1):52–62.
156. Richard I, Broux O, Allamand V, et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2 A. Cell. 1995;81(1):27–40.
157. Richard I, Roudaut C, Saenz A, et al. Calpainopathy-a survey of mutations and polymorphisms. Am J Hum Genet. 1999; 64(6):1524–1540.
158. Luo SS, Xi JY, Zhu WH, et al. Genetic variability and clinical spectrum of Chinese patients with limb-girdle muscular dystrophy type 2 A. Muscle Nerve. 2012;46(5):723–729.
159. Fanin M, Pegoraro E, Matsuda-Asada C, Brown RH Jr, Angelini C. Calpain-3 and dysferlin protein screening in patients with limb-girdle dystrophy and myopathy. Neurology. 2001; 56(5):660–665.
160. Brown RH, Jr., Amato A. Calpainopathy and eosinophilic myositis. Ann Neurol. 2006;59(6):875–877.
161. Illa I, Serrano-Munuera C, Gallardo E, et al. Distal anterior compartment myopathy: A dysferlin mutation causing a new muscular dystrophy phenotype. Ann Neurol. 2001;49(1):130–134.
162. Illarioshkin SN, Ivanova-Smolenskaya IA, Tanaka H, et al. Clinical and molecular analysis of a large family with three distinct phenotypes of progressive muscular dystrophy. Brain. 1996;119(Pt 6):1895–1909.
163. Mahjneh I, Marconi G, Bushby K, Anderson LV, Tolvanen-Mahjneh H, Somer H. Dysferlinopathy (LGMD2B): A 23-year follow-up study of 10 patients homozygous for the same frameshifting dysferlin mutations. Neuromuscul Disord. 2001;11(1):20–26.
164. Mahjneh I, Passos-Bueno MR, Zatz M, et al. The phenotype of chromosome 2p-linked limb-girdle muscular dystrophy. Neuromuscul Disord. 1996;6(6):483–490.
165. Suzuki N, Aoki M, Takahashi T, et al. Novel dysferlin mutations and characteristic muscle atrophy in late-onset Miyoshi myopathy. Muscle Nerve. 2004;29(5):721–723.
166. Takahashi T, Aoki M, Tateyama M, et al. Dysferlin mutations in Japanese Miyoshi myopathy: Relationship to phenotype. Neurology. 2003;60(11):1799–1804.
167. Vilchez JJ, Gallano P, Gallardo E, et al. Identification of a novel founder mutation in the DYSF gene causing clinical variability in the Spanish population. Arch Neurol. 2005;62:1256–1259.
168. Ho M, Gallardo E, McKenna-Yasek D, De Luna N, Illa I, Brown RH Jr. A novel, blood-based diagnostic assay for limb girdle muscular dystrophy 2B and Miyoshi myopathy. Ann Neurol. 2002;51(1):129–133.
169. Paradas C, Llauger J, Diaz-Manera J, et al. Redefining dysferlinopathy phenotypes based on clinical findings and muscle imaging studies. Neurology. 2010;75:316–323.
170. Kesper K, Kornblum C, Reimann J, Lutterbey G, Schröder R, Wattjes MP. Pattern of skeletal muscle involvement in primary dysferlinopathies: A whole-body 3.0-T magnetic resonance imaging study. Acta Neurol Scand. 2009;120:111–118.
171. Brummer D, Walter MC, Palmbach M, et al. Long-term MRI and clinical follow-up of symptomatic and presymptomatic carriers of dysferlin gene mutations. Acta Myol. 2005;24:6–16.
172. Piccolo F, Moore SA, Ford GC, Campbell KP. Intracellular accumulation and reduced sarcolemmal expression of dysferlin in limb-girdle muscular dystrophies. Ann Neurol. 2000;48(6):902–912.
173. Gallardo E, Rojas-Garcia R, de Luna N, Pou A, Brown RH Jr, Illa I. Inflammation in dysferlin myopathy: Immunohistochemical characterization of 13 patients. Neurology. 2001;57(11):2136–2138.
174. Selcen D, Stilling G, Engel AG. The earliest pathologic alterations in dysferlinopathy. Neurology. 2001;56(11):1472–1481.
175. Spuler S, Carl M, Zabojszcza J, et al. Dysferlin-deficient muscular dystrophy features amyloidosis. Ann Neurol. 2008;63:323–328.
176. Liu J, Aoki M, Illa I, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20(1):31–36.
177. Angelini C, Fanin M, Freda MP, Duggan DJ, Siciliano G, Hoffman EP. The clinical spectrum of sarcoglycanopathies. Neurology. 1999;52(1):176–179.
178. Duggan DJ, Gorospe JR, Fanin M, Hoffman EP, Angelini C. Mutations in the sarcoglycan genes in patients with myopathy. N Engl J Med. 1997;336(9):618–624.
179. Melacini P, Fanin M, Duggan DJ, et al. Heart involvement in muscular dystrophies due to sarcoglycan gene mutations. Muscle Nerve. 1999;22(4):473–479.
180. Angelini C, Fanin M, Menegazzo E, Freda MP, Duggan DJ, Hoffman EP. Homozygous alpha-sarcoglycan mutation in two siblings: One asymptomatic and one steroid-responsive mild limb-girdle muscular dystrophy patient. Muscle Nerve. 1998;21(6):769–775.
181. Bonnemann CG, Modi R, Noguchi S, et al. Beta-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat Genet. 1995; 11(3):266–273.
182. Campbell KP. Adhalin gene mutations and autosomal recessive limb-girdle muscular dystrophy. Ann Neurol. 1995;38(3):353–354.
183. Duggan DJ, Fanin M, Pegoraro E, Angelini C, Hoffman EP. Alpha-Sarcoglycan (adhalin) deficiency: Complete deficiency patients are 5% of childhood-onset dystrophin-normal muscular dystrophy and most partial deficiency patients do not have gene mutations. J Neurol Sci. 1996;140(1-2):30–39.
184. Duggan DJ, Manchester D, Stears KP, Mathews DJ, Hart C, Hoffman EP. Mutations in the delta-sarcoglycan gene are a rare cause of autosomal recessive limb-girdle muscular dystrophy (LGMD2). Neurogenetics. 1997;1(1):49–58.
185. Ljunggren A, Duggan D, McNally E, et al. Primary adhalin deficiency as a cause of muscular dystrophy in patients with normal dystrophin. Ann Neurol. 1995;38(3):367–372.
186. McNally EM, Duggan D, Gorospe JR, et al. Mutations that disrupt the carboxyl-terminus of gamma-sarcoglycan cause muscular dystrophy. Hum Mol Genet. 1996;5(11):1841–1847.
187. Diniz G, Tosun Yildirim H, Akinci G, et al. Sarcolemmal alpha and gamma sarcoglycan protein deficiencies in Turkish siblings with a novel missense mutation in the alpha sarcoglycan gene. Pediatr Neurol. 2014;50:640–647.
188. Moreira ES, Vainzof M, Marie SK, Sertié AL, Zatz M, Passos-Bueno MR. The seventh form of autosomal recessive limb-girdle muscular dystrophy is mapped to 17q11–12. Am J Hum Genet. 1997;61(1):151–159.
189. Moreira ES, Wiltshire TJ, Faulkner G, et al. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat Genet. 2000;24(2):163–166.
190. Mues A, van der Ven PF, Young P, Fürst DO, Gautel M. Two immunoglobulin-like domains of the Z-disc portion of titin interact in a conformation-dependent way with telethonin. FEBS Lett. 1998;428(1-2):111–114.
191. Mayans O, van der Ven PF, Wilm M, et al. Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature. 1998;395(6705):863–869.
192. Weiler T, Greenberg CR, Zelinski T, et al. A gene for autosomal recessive limb-girdle muscular dystrophy in Manitoba Hutterites maps to chromosome region 9q31–q33: Evidence for another limb-girdle muscular dystrophy locus. Am J Hum Genet. 1998;63(1):140–147.
193. Schoser BG, Frosk P, Engel AG, Klutzny U, Lochmüller H, Wrogemann K. Commonality of TRIM32 mutation in causing sarcotubular myopathy and LGMD2 H. Ann Neurol. 2005;57(4):591–595.
194. Borg K, Stucka R, Locke M, et al. Intragenic deletion of TRIM32 in compound heterozygotes with sarcotubular myopathy/LGMD2 H. Hum Mutat. 2009;30(9):E831–E844.
195. Saccone V, Palmieri M, Passamano L, et al. Mutations that impair interaction properties of TRIM32 associated with limb-girdle muscular dystrophy 2 H. Hum Mutat. 2008;29:240–247.
196. Driss A, Amouri R, Ben Hamida C, et al. A new locus for autosomal recessive limb-girdle muscular dystrophy in a large consanguineous Tunisian family maps to chromosome 19q13.3. Neuromuscul Disord. 2000;10(4-5):240–246.
197. Mercuri E, Brockington M, Straub V, et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol. 2003;53(4):537–542.
198. Poppe M, Bourke J, Eagle M, et al. Cardiac and respiratory failure in limb-girdle muscular dystrophy 2I. Ann Neurol. 2004;56(5):738–741.
199. Poppe M, Cree L, Bourke J, et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology. 2003;60(8):1246–1251.
200. Boito CA, Melacini P, Vianello A, et al. Clinical and molecular characterization of patients with limb-girdle muscular dystrophy type 2I. Arch Neurol. 2005;62:1894–1899.
201. Brockington M, Yuva Y, Prandini P, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet. 2001;10(25):2851–2859.
202. Schwartz M, Hertz JM, Sveen ML, Vissing J. LGMD2I presenting with a characteristic Duchenne or Becker muscular dystrophy phenotype. Neurology. 2005;64(9):1635–1637.
203. Lindberg C, Sixt C, Oldfors A. Episodes of exercise-induced dark urine and myalgia in LGMD 2I. Acta Neurol Scand. 2012;125(4):285–287.
204. Mathews KD, Stephan CM, Laubenthal K, et al. Myoglobinuria and muscle pain are common in patients with limb-girdle muscular dystrophy 2I. Neurology. 2011;76(2):194–195.
205. Müller T, Krasnianski M, Witthaut R, Deschauer M, Zierz S. Dilated cardiomyopathy may be an early sign of the C826 A Fukutin-related protein mutation. Neuromuscul Disord. 2005;15(5):372–376.
206. Palmieri A, Manara R, Bello L, et al. Cognitive profile and MRI findings in limb-girdle muscular dystrophy 2I. J Neurol. 2011;258(7):1312–1320.
207. Matsumoto H, Noguchi S, Sugie K. Subcellular localization of fukutin and fukutin-related protein in muscle cells. J Biochem (Tokyo). 2004;135(6):709–712.
208. Esapa CT, McIlhinney RA, Blake DJ. Fukutin-related protein mutations that cause congenital muscular dystrophy result in ER-retention of the mutant protein in cultured cells. Hum Mol Genet. 2005;14(2):295–305.
209. Udd B, Vihola A, Sarparanta J, Richard I, Hackman P. Titinopathies and extension of the M-line mutation phenotype beyond distal myopathy and LGMD2 J. Neurology. 2005;64(4):636–642.
210. Udd B. Limb-girdle type muscular dystrophy in a large family with distal myopathy: Homozygous manifestation of a dominant gene? J Med Genet. 1992;29(6):383–389.
211. Udd B, Rapola J, Nokelainen P, Arikawa E, Somer H. Nonvacuolar myopathy in a large family with both late adult onset distal myopathy and severe proximal muscular dystrophy. J Neurol Sci. 1992;113(2):214–221.
212. Udd B, Partanen J, Halonen P, et al. Tibial muscular dystrophy. Late adult-onset distal myopathy in 66 Finnish patients. Arch Neurol. 1993;50(6):604–608.
213. Udd B, Haravuori H, Kalimo H, et al. Tibial muscular dystrophy–from clinical description to linkage on chromosome 2q31. Neuromuscul Disord. 1998;8(5):327–332.
214. Van den Bergh PY, Bouquiaux O, Verellen C, et al. Tibial muscular dystrophy in a Belgian family. Ann Neurol. 2003;54(2):248–251.
215. Mahjneh I, Lamminen AE, Udd B, et al. Muscle magnetic resonance imaging shows distinct diagnostic patterns in Welander and tibial muscular dystrophy. Acta Neurol Scand. 2004;110(2):87–93.
216. Pénisson-Besnier I, Hackman P, Suominen T, et al. Myopathies caused by homozygous titin mutations: Limb-girdle muscular dystrophy 2 J and variations of phenotype. J Neurol Neurosurg Psychiatry. 2010;81(11):1200–1202.
217. Ohlsson M, Hedberg C, Brådvik B, et al. Hereditary myopathy with early respiratory failure associated with a mutation in A-band titin. Brain. 2012;135(Pt 6):1682–1694.
218. Pfeffer G, Elliott HR, Griffin H, et al. Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain. 2012;135(Pt 6):1695–1713.
219. Carmignac V, Salih MA, Quijano-Roy S, et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol. 2007;61(4):340–351.
220. Udd B, Kääriänen H, Somer H. Muscular dystrophy with separate clinical phenotypes in a large family. Muscle Nerve. 1991;14(11):1050–1058.
221. Godfrey C, Clement E, Mein R, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130(Pt 10):2725–2735.
222. Hicks D, Sarkozy A, Muelas N, et al. A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy. Brain. 2011;134(Pt 1):171–182.
223. Penttilä S, Palmio J, Suominen T, et al. Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5. Neurology. 2012;78(12):897–903.
224. Bolduc V, Marlow G, Boycott KM, et al. Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2 L and distal MMD3 muscular dystrophies. Am J Hum Genet. 2010;86(2):213–221.
225. Mahjneh I, Jaiswal J, Lamminen A, et al. A new distal myopathy with mutation in anoctamin 5. Neuromuscul Disord. 2010; 20(12):791–795.
226. Schessl J, Kress W, Schoser B. Novel ANO5 mutations causing hyper-CK-emia, limb girdle muscular weakness and Miyoshi type of muscular dystrophy. Muscle Nerve. 2012;45(5):740–742.
227. Sarkozy A, Hicks D, Hudson J, et al. ANO5 gene analysis in a large cohort of patients with anoctaminopathy: Confirmation of male prevalence and high occurrence of the common exon 5 gene mutation. Hum Mutat. 2013;34(8):1111–1118.
228. Jimenez-Mallebrera C, Torelli S, Feng L, et al. A comparative study of alpha-dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of alpha-dystroglycan does not consistently correlate with clinical severity. Brain Pathol. 2009;19(4):596–611.
229. Godfrey C, Escolar D, Brockington M, et al. Fukutin gene mutations in steroid-responsive limb girdle muscular dystrophy. Ann Neurol. 2006;60(5):603–610.
230. Biancheri R, Falace A, Tessa A, et al. POMT2 gene mutation in limb-girdle muscular dystrophy with inflammatory changes. Biochem Biophys Res Commun. 2007;363(4):1033–1037.
231. Clement EM, Godfrey C, Tan J, et al. Mild POMGnT1 mutations underlie a novel limb-girdle muscular dystrophy variant. Arch Neurol. 2008;65(1):137–141.
232. Hara Y, Balci-Hayta B, Yoshida-Moriguchi T, et al. A dystroglycan mutation associated with limb-girdle muscular dystrophy. N Eng J Med. 2011;364(10):939–946.
233. Banwell BL, Russel J, Fukudome T, Shen XM, Stilling G, Engel AG. Myopathy, myasthenic syndrome, and epidermolysis bullosa simplex due to plectin deficiency. J Neuropathol Exp Neurol. 1999;58(8):832–846.
234. Charlesworth A, Chiaverini C, Chevrant-Breton J, et al. Epidermolysis bullosa simplex with PLEC mutations: New phenotypes and new mutations. Br J Dermatol. 2013;168(4):808–814.
235. Chavanas S, Pulkkinen L, Gache Y, et al. A homozygous nonsense mutation in the PLEC1 gene in patients with epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest. 1996;98(10):2196–2200.
236. Forrest K, Mellerio JE, Robb S, et al. Congenital muscular dystrophy, myasthenic symptoms and epidermolysis bullosa simplex (EBS) associated with mutations in the PLEC1 gene encoding plectin. Neuromuscul Disord. 2010;20(11):709–711.
237. Gache Y, Chavanas S, Lacour JP, et al. Defective expression of plectin/HD1 in epidermolysis bullosa simplex with muscular dystrophy. J Clin Invest. 1996;97(10):2289–2298.
238. Pulkkinen L, Smith FJ, Shimizu H, et al. Homozygous deletion mutations in the plectin gene (PLEC1) in patients with epidermolysis bullosa simplex associated with late-onset muscular dystrophy. Hum Mol Genet. 1996;5(10):1539–1546.
239. Smith FJ, Eady RA, Leigh IM, et al. Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nat Genet. 1996;13:450–457.
240. Yiu EM, Klausegger A, Waddell LB, et al. Epidermolysis bullosa with late-onset muscular dystrophy and plectin deficiency. Muscle Nerve. 2011;44:135–141.
241. Selcen D, Juel VC, Hobson-Webb LD, et al. Myasthenic syndrome caused by plectinopathy. Neurology. 2011;76(4):327–336.
242. Maselli RA, Arredondo J, Cagney O, et al. Congenital myasthenic syndrome associated with epidermolysis bullosa caused by homozygous mutations in PLEC1 and CHRNE. Clin Genet. 2011;80(5):444–451.
243. McMillan JR, Akiyama M, Rouan F, et al. Plectin defects in epidermolysis bullosa simplex with muscular dystrophy. Muscle Nerve. 2007;35(1):24–35.
244. Fine JD, Stenn J, Johnson L, Wright T, Bock HG, Horiguchi Y. Autosomal recessive epidermolysis bullosa simplex. Generalized phenotypic features suggestive of junctional or dystrophic epidermolysis bullosa, and association with neuromuscular diseases. Arch Dermatol. 1989;125:931–938.
245. Bolling MC, Pas HH, de Visser M, et al. PLEC1 mutations underlie adult-onset dilated cardiomyopathy in epidermolysis bullosa simplex with muscular dystrophy. J Invest Dermatol. 2010;130(4):1178–1181.
246. Gundesli H, Talim B, Korkusuz P, et al. Mutation in exon 1f of PLEC, leading to disruption of plectin isoform 1f, causes autosomal-recessive limb girdle muscular dystrophy. Am J Hum Genet. 2010;87(6):834–841.
247. Cetin N, Balci-Hayta B, Gundesli H, et al. A novel desmin mutation leading to autosomal recessive limb-girdle muscular dystrophy: Distinct histopathological outcomes compared with desminopathies. J Med Genet. 2013;50(7):437–443.
248. Henderson M, De Waele L, Hudson J, et al. Recessive desmin-null muscular dystrophy with central nuclei and mitochondrial abnormalities. Acta Neuropathol. 2013;125(6):917–919.
249. Bögershausen N, Shahrzad N, Chong JX, et al. Recessive TRAPPC11 mutations cause a disease spectrum of limb girdle muscular dystrophy and myopathy with movement disorder and intellectual disability. Am J Hum Genet. 2013;93(1):181–90.
250. Bönnemann CG, Wang CH, Quijano-Roy S, et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord. 2014;24(4):289–311.
251. Muntoni F, Voit T. The congenital muscular dystrophies in 2004: A century of exciting progress. Neuromuscul Disord. 2004;14(10):635–649.
252. Muntoni F, Voit T. 133rd ENMC International Workshop on Congenital Muscular Dystrophy (IXth International CMD Workshop) 21–23 January 2005, Naarden, the Netherlands. Neuromuscul Disord. 2005;15(11):794–801.
253. Philpot J, Cowan F, Pennock J, et al. Merosin-deficient congenital muscular dystrophy: The spectrum of brain involvement on magnetic resonance imaging. Neuromuscul Disord. 1999;9(2):81–85.
254. Prandini P, Berardinelli A, Fanin M, et al. LAMA2 loss-of-function mutation in a girl with a mild congenital muscular dystrophy. Neurology. 2004;63(6):1118–1121.
255. Sewry CA, Naom I, D’Alessandro M, et al. Variable clinical phenotype in merosin-deficient congenital muscular dystrophy associated with differential immunolabeling of two fragments of the laminin alpha 2 chain. Neuromuscul Disord. 1997;7(3):169–175.
256. Shorer Z, Philpot J, et al. Demyelinating peripheral neuropathy in merosin-deficient congenital muscular dystrophy. J Child Neurol. 1995;10(6):472–475.
257. Cohn RD, Herrmann R, Muntoni F, Sewry C, Dubowitz V, et al. Laminin alpha2 chain-deficient congenital muscular dystrophy: Variable epitope expression in severe and mild cases. Neurology. 1998;51(1):94–100.
258. Mora M, Moroni I, Uziel G, et al. Mild clinical phenotype in a 12-year-old boy with partial merosin deficiency and central and peripheral nervous system abnormalities. Neuromuscul Disord. 1996;6(5):377–381.
259. Morandi L, Di Blasi C, Farina L, et al. Clinical correlations in 16 patients with total or partial laminin alpha2 deficiency characterized using antibodies against 2 fragments of the protein. Arch Neurol. 1999;56(2):209–215.
260. Naom I, Sewry C, D’Alessandro M, et al. Prenatal diagnosis in merosin-deficient congenital muscular dystrophy. Neuromuscul Disord. 1997;7(3):176–179.
261. Pegoraro E, Marks H, Garcia CA, et al. Laminin alpha2 muscular dystrophy: Genotype/phenotype studies of 22 patients. Neurology. 1998;51(1):101–110.
262. Tan E, Topaloglu H, Sewry C, et al. Late onset muscular dystrophy with cerebral white matter changes due to partial merosin deficiency. Neuromuscul Disord. 1997;7(2):85–89.
263. Bushby K, Anderson LV, Pollitt C, Naom I, Muntoni F, Bindoff L. Abnormal merosin in adults. A new form of late onset muscular dystrophy not linked to chromosome 6 q2. Brain. 1998;121(Pt 4):581–588.
264. Mercuri E, Pennock J, Goodwin F, et al. Sequential study of central and peripheral nervous system involvement in an infant with merosin-deficient congenital muscular dystrophy. Neuromuscul Disord. 1996;6(6):425–429.
265. Sewry CA, D’Alessandro M, Wilson LA, et al. Expression of laminin chains in skin in merosin-deficient congenital muscular dystrophy. Neuropediatrics. 1997;28(4):217–222.
266. Guicheney P, Vignier N, Helbling-Leclerc A, et al. Genetics of laminin alpha 2 chain (or merosin) deficient congenital muscular dystrophy: From identification of mutations to prenatal diagnosis. Neuromuscul Disord. 1997;7(3):180–186.
267. Hayashi YK, Chou FL, Engvall E, et al. Mutations in the integrin alpha7 gene cause congenital myopathy. Nat Genet. 1998;19(1):94–97.
268. Yonekawa T, Nishino I. Ullrich congenital muscular dystrophy: Clinicopathological features, natural history and pathomechanism(s). J Neurol Neurosurg Psychiatry. 2015;86(3):280–287.
269. Ishikawa H, Sugie K, Murayama K, et al. Ullrich disease due to deficiency of collagen VI in the sarcolemma. Neurology. 2004;62(4):620–623.
270. Baker NL, Morgelin M, Peat R, et al. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet. 2005;14(2):279–293.
271. Demir E, Ferreiro A, Sabatelli P, et al. Collagen VI status and clinical severity in Ullrich congenital muscular dystrophy: Phenotype analysis of 11 families linked to the COL6 loci. Neuropediatrics. 2004;35(2):103–112.
272. Meilleur KG, Zukosky K, Medne L, et al. Clinical, pathologic, and mutational spectrum of dystroglycanopathy caused by LARGE mutations. J Neuropathol Exp Neurol. 2014;73(5):425–441.
273. Manzani MC, Tambunan DE, Hill RS, et al. Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am J Hum Genet. 2012;91(3):541–547.
274. Buysse K, Riemersma M, Powell G, et al. Missense mutations in [beta]-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1) cause Walker-Warburg syndrome. Hum Mol Genet. 2013;22(9):1746–1754.
275. Stevens E, Carss KJ, Cirak S, et al. Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of [alpha]-dystroglycan. Am J Hum Genet. 2013;92(3):354–365.
276. Hedberg C, Oldfors A, Darin N. B3GALNT2 is a gene associated with congenital muscular dystrophy with brain malformations. Eur J Hum Genet. 2014;22(5):707–710.
277. Vuillaumier-Barrot S, Bouchet-Séraphin C, Chelbi M, et al. Identification of mutations in TMEM5 and ISPD as a cause of severe cobblestone lissencephaly. Am J Hum Genet. 2012;91(6):1135–1143.
278. Carss KJ, Stevens E, Foley AR, et al. Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of [alpha]-dystroglycan. Am J Hum Genet. 2013;93(1):29–41.
279. Kim DS, Hayashi YK, et al. POMT1 mutation results in defective glycosylation and loss of laminin-binding activity in alpha-DG. Neurology. 2004;62(6):1009–1011.
280. Beltran-Valero de Bernabe D, Currier S, Steinbrecher A, et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker–Warburg syndrome. Am J Hum Genet. 2002;71(5):1033–1043.
281. Cormand B, Pihko H, Bayés M, et al. Clinical and genetic distinction between Walker–Warburg syndrome and muscle–eye–brain disease. Neurology. 2001;56(8):1059–1069.
282. Diesen C, Saarinen A, Pihko H, et al. POMGnT1 mutation and phenotypic spectrum in muscle–eye–brain disease. J Med Genet. 2004;41(10):e115.
283. Wewer UM, Durkin ME, Zhang X, et al. Laminin beta 2 chain and adhalin deficiency in the skeletal muscle of Walker–Warburg syndrome (cerebro-ocular dysplasia-muscular dystrophy). Neurology. 1995;45(11):2099–2101.
284. Toda T, Kobayashi K, Kondo-Iida E, et al. The Fukuyama congenital muscular dystrophy story. Neuromuscul Disord. 2000;10(3):153–159.
285. Toda T, Yoshioka M, Nakahori Y, et al. Genetic identity of Fukuyama-type congenital muscular dystrophy and Walker–Warburg syndrome. Ann Neurol. 1995;37(1):99–101.
286. Kobayashi K, Nakahori Y, Miyake M, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394(6691):388–392.
287. Yoshida A, Kobayashi K, Manya H, et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. 2001;1(5):717–724.
288. van Reeuwijk J, Janssen M, van den Elzen C, et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker Warburg syndrome. J Med Genet. 2005;42(12):907–912.
289. Balci B, Uyanik G, Dincer P, et al. An autosomal recessive limb girdle muscular dystrophy (LGMD2) with mild mental retardation is allelic to Walker–Warburg syndrome (WWS) caused by a mutation in the POMT1 gene. Neuromuscul Disord. 2005;15(4):271–275.
290. Haltia M, Leivo I, Somer H, et al. Muscle–eye–brain disease: A neuropathological study. Ann Neurol. 1997;41(2):173–180.
291. Santavuori P, Somer H, Sainio K, et al. Muscle–eye–brain disease (MEB). Brain Dev. 1989;11(3):147–153.
292. Santavuori P, Valanne L, Autti T, Haltia M, Pihko H, Sainio K. Muscle–eye–brain disease: Clinical features, visual evoked potentials and brain imaging in 20 patients. Eur J Paediatr Neurol. 1998;2(1):41–47.
293. Louhichi N, Triki C, Quijano-Roy S, et al. New FKRP mutations causing congenital muscular dystrophy associated with mental retardation and central nervous system abnormalities. Identification of a founder mutation in Tunisian families. Neurogenetics. 2004;5(1):27–34.
294. Lotz BP, Stubgen JP. The rigid spine syndrome: A vacuolar variant. Muscle Nerve. 1993;16(5):530–536.
295. Mercuri E, Talim B, Moghadaszadeh B, et al. Clinical and imaging findings in six cases of congenital muscular dystrophy with rigid spine syndrome linked to chromosome 1p (RSMD1). Neuromuscul Disord. 2002;12(7–8):631–638.
296. Merlini L, Granata C, Ballestrazzi A, Marini ML. Rigid spine syndrome and rigid spine sign in myopathies. J Child Neurol. 1989;4(4):274–282.
297. Moghadaszadeh B, Topaloglu H, Merlini L, et al. Genetic heterogeneity of congenital muscular dystrophy with rigid spine syndrome. Neuromuscul Disord. 1999;9(6–7):376–382.
298. Reichmann H, Goebel HH, Schneider C, Toyka KV. Familial mixed congenital myopathy with rigid spine phenotype. Muscle Nerve. 1997;20(4):411–417.
299. Taylor J, Muntoni F, Robb S, Dubowitz V, Sewry C. Early onset autosomal dominant myopathy with rigidity of the spine: A possible role for laminin beta 1? Neuromuscul Disord. 1997;7(4):211–216.
300. Flanigan KM, Kerr L, Bromberg MB, et al. Congenital muscular dystrophy with rigid spine syndrome: A clinical, pathological, radiological, and genetic study. Ann Neurol. 2000;47(2):152–161.
301. Ferreiro A, Ceuterick-de Groote C, Marks JJ, et al. Desmin-related myopathy with Mallory body-like inclusions is caused by mutations of the selenoprotein N gene. Ann Neurol. 2004;55 (5):676–686.
302. Tawil R, Van Der Maarel SM. Facioscapulohumeral muscular dystrophy. Muscle Nerve. 2006;34(1):1–15.
303. Personius KE, Pandya S, King WM, Tawil R, McDermott MP. Facioscapulohumeral dystrophy natural history study: Standardization of testing procedures and reliability of measurements. The FSH DY Group. Phys Ther. 1994;74(3):253–263.
304. Tawil R, McDermott MP, Mendell JR, Kissel J, Griggs RC. Facioscapulohumeral muscular dystrophy (FSHD): Design of natural history study and results of baseline testing. FSH-DY Group. Neurology. 1994;44(3 Pt 1):442–446.
305. Tawil R, McDermott MP, Pandya S, et al. A pilot trial of prednisone in facioscapulohumeral muscular dystrophy. FSH-DY Group. Neurology. 1997;48(1):46–49.
306. van der Kooi AJ, Visser MC, Rosenberg N, et al. Extension of the clinical range of facioscapulohumeral dystrophy: Report of six cases. J Neurol Neurosurg Psychiatry. 2000;69(1):114–116.
307. Felice KJ, North WA, Moore SA, Mathews KD. FSH dystrophy 4q35 deletion in patients presenting with facial-sparing scapular myopathy. Neurology. 2000;54(10):1927–1931.
308. Wohlgemuth M, van der Kooi EL, van Kesteren RG, van der Maarel SM, Padberg GW. Ventilatory support in facioscapulohumeral muscular dystrophy. Neurology. 2004;63(1):176–178.
309. Laforet P, de Toma C, Eymard B, et al. Cardiac involvement in genetically confirmed facioscapulohumeral muscular dystrophy. Neurology. 1998;51(5):1454–1456.
310. Felice KJ, Jones JM, Conway SR. Facioscapulohumeral dystrophy presenting as infantile facial diplegia and late-onset limbgirdle myopathy in members of the same family. Muscle Nerve. 2005;32(3):368–372.
311. Munsat TL, Piper D, Cancilla P, Mednick J. Inflammatory myopathy with facioscapulohumeral distribution. Neurology. 1972;22(4):335–347.
312. Spuler S, Engel AG. Unexpected sarcolemmal complement membrane attack complex deposits on nonnecrotic muscle fibers in muscular dystrophies. Neurology. 1998;50(1):41–46.
313. Wijmenga C, Hewitt JE, Sandkuijl LA, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet. 1992;2(1):26–30.
314. Tawil R, Forrester J, Griggs RC, et al. Evidence for anticipation and association of deletion size with severity in facioscapulohumeral muscular dystrophy. The FSH-DY Group. Ann Neurol. 1996;39(6):744–748.
315. Lemmers RJ, Tawil R, Petek LM, et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. 2012;44(12), 1370–1374.
316. Sacconi S, Lemmers RJ, Balog J, et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am J Hum Genet. 2013;93(4),744–751.
317. Kissel JT, McDermott MP, Natarajan R, et al. Pilot trial of albuterol in facioscapulohumeral muscular dystrophy. FSHDY Group. Neurology. 1998;50(5):1402–1406.
318. Kissel JT, McDermott MP, Mendell JR, et al. Randomized, double-blind, placebo-controlled trial of albuterol in facioscapulohumeral dystrophy. Neurology. 2001;57(8):1434–1440.
319. Andrews CT, Taylor TC, Patterson VH. Scapulothoracic arthrodesis for patients with facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 1998;8(8):580–584.
320. Letournel E, Fardeau M, Lytle JO, Serrault M, Gosselin RA. Scapulothoracic arthrodesis for patients who have fascioscapulohumeral muscular dystrophy. J Bone Joint Surg Am. 1990;72(1):78–84.
321. Chakrabarti A, Pearce JM. Scapuloperoneal syndrome with cardiomyopathy: Report of a family with autosomal dominant inheritance and unusual features. J Neurol Neurosurg Psychiatry. 1981;44(12):1146–1152.
322. Feigenbaum JA, Munsat TL. A neuromuscular syndrome of scapuloperoneal distribution. Bull Los Angeles Neurol Soc. 1970;35(2):47–57.
323. Kaeser HE. Scapuloperoneal muscular atrophy. Brain. 1965; 88(2):407–418.
324. Takahashi K, Nakamura H, Nakashima R. Scapuloperoneal dystrophy associated with neurogenic changes. J Neurol Sci. 1974;23(4):575–583.
325. Thomas PK, Schott GD, Morgan-Hughes JA. Adult onset scapuloperoneal myopathy. J Neurol Neurosurg Psychiatry. 1975;38(10):1008–1015.
326. Wilhelmsen KC, Blake DM, Lynch T, et al. Chromosome 12-linked autosomal dominant scapuloperoneal muscular dystrophy. Ann Neurol. 1996;39(4):507–520.
327. Tawil R, Myers GJ, Weiffenbach B, Griggs RC. Scapuloperoneal syndromes. Absence of linkage to the 4q35 FSHD locus. Arch Neurol. 1995;52(11):1069–1072.
328. Probst A, Ulrich J, Kaeser HE, Heitz P. Scapulo-peroneal muscular atrophy. Full autopsy report. Unusual findings in the anterior horn of the spinal cord. Lipid storage in muscle. Eur Neurol. 1977;16(1–6):181–196.
329. Quinzi CM, Vu TH, Min KC, et al. X-linked dominant scapuloperoneal myopathy is due to mutation in the gene encoding four-and-a-half-LIM protein 1. Am J Hum Genet. 2008;82:208–213.
330. Thomas PK, Calne DB, Elliott CF. X-linked scapuloperoneal syndrome. J Neurol Neurosurg Psychiatry. 1972;35(2):208–215.
331. Marques J, Duarte ST, Costa S, et al. Atypical phenotype in two patients with LAMA2 mutations. Neuromuscul Disord. 2014;24(5):419–424.
332. Emery AE, Dreifuss FE. Unusual type of benign x-linked muscular dystrophy. J Neurol Neurosurg Psychiatry. 1966;29(4):338–342.
333. Hopkins LC, Jackson JA, Elsas LJ. Emery–Dreifuss humeroperoneal muscular dystrophy: An x-linked myopathy with unusual contractures and bradycardia. Ann Neurol. 1981;10(3):230–237.
334. Kubo S, Tsukahara T, Takemitsu M, et al. Presence of emerinopathy in cases of rigid spine syndrome. Neuromuscul Disord. 1998;8(7):502–507.
335. Emery AE. Emery–Dreifuss muscular dystrophy—a 40 year retrospective. Neuromuscul Disord. 2000;10(4–5):228–232.
336. Schessl J, Columbus A, Hu Y, et al. Familial reducing body myopathy with cytoplasmic bodies and rigid spine revisited: Identification of a second LIM domain mutation in FHL1. Neuropediatrics. 2010;41(1):43–46.
337. Schoser B, Goebel HH, Janisch I, et al. Consequences of mutations within the C terminus of the FHL1 gene. Neurology. 2009;73(7):543–551.
338. Shalaby S, Hayashi YK, Nonaka I, Noguchi S, Nishino I. Novel FHL1 mutations in fatal and benign reducing body myopathy. Neurology. 2009;72(4):375–376.
339. Chen DH, Raskind WH, Parson WW, et al. A novel mutation in FHL1 in a family with X-linked scapuloperoneal myopathy: Phenotypic spectrum and structural study of FHL1 mutations. J Neurol Sci. 2010;296(1–2):22–29.
340. Selcen D, Bromberg MB, Chin SS, Engel AG. Reducing bodies and myofibrillar myopathy features in FHL1 muscular dystrophy. Neurology. 2011;77(22):1951–1959.
341. Shalaby S, Hayashi YK, Goto K, et al. Rigid spine syndrome caused by a novel mutation in four-and-a-half LIM domain 1 gene (FHL1). Neuromuscul Disord. 2008;18:959–961.
342. Bione S, Maestrini E, Rivella S, et al. Identification of a novel X-linked gene responsible for Emery–Dreifuss muscular dystrophy. Nat Genet. 1994;8(4):323–327.
343. van der Kooi AJ, van Meegen M, Ledderhof TM, McNally EM, de Visser M, Bolhuis PA. Genetic localization of a newly recognized autosomal dominant limb-girdle muscular dystrophy with cardiac involvement (LGMD1B) to chromosome 1q11–21. Am J Hum Genet. 1997;60(4):891–895.
344. Fatkin D, MacRae C, Sasaki T, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341(23):1715–1724.
345. Taylor J, Sewry CA, Dubowitz V, Muntoni F. Early onset, autosomal recessive muscular dystrophy with Emery–Dreifuss phenotype and normal emerin expression. Neurology. 1998;51(4):1116–1120.
346. Zhang Q, Bethmann C, Worth NF, et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. 2007;16(23):2816–2833.
347. Liang WC, Mitsuhashi H, Keduka E, et al. TMEM43 mutations in Emery-Dreifuss muscular dystrophy-related myopathy. Ann Neurol. 2011;69(6):1005–1013.
348. Arts WF, Bethlem J, Volkers WS. Further investigations on benign myopathy with autosomal dominant inheritance. J Neurol. 1978;217(3):201–206.
349. Bertini E, Pepe G. Collagen type VI and related disorders: Bethlem myopathy and Ullrich scleroatonic muscular dystrophy. Eur J Paediatr Neurol. 2002;6(4):193–198.
350. Bethlem J, Wijngaarden GK. Benign myopathy, with autosomal dominant inheritance. A report on three pedigrees. Brain. 1976;99(1):91–100.
351. Haq RU, Speer MC, Chu ML, Tandan R. Respiratory muscle involvement in Bethlem myopathy. Neurology. 1999;52(1):174–176.
352. Jobsis GJ, Boers JM, Barth PG, de Visser M. Bethlem myopathy: A slowly progressive congenital muscular dystrophy with contractures. Brain. 1999;122(Pt 4):649–655.
353. Jobsis GJ, Keizers H, Vreijling JP, et al. Type VI collagen mutations in Bethlem myopathy, an autosomal dominant myopathy with contractures. Nat Genet. 1996;14(1):113–115.
354. Pepe G, Bertini E, Bonaldo P, et al. Bethlem myopathy (BETHLEM) and Ullrich scleroatonic muscular dystrophy: 100th ENMC international workshop, 23–24 November 2001, Naarden, the Netherlands. Neuromuscul Disord. 2002;12(10):984–993.
355. Pepe G, de Visser M, Bertini E, et al. Bethlem myopathy (BETHLEM) 86th ENMC international workshop, 10–11 November 2000, Naarden, the Netherlands. Neuromuscul Disord. 2002;12(3):296–305.
356. Mercuri E, Lampe A, Allsop J, et al., Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord. 2005;15(4), 303–310.
357. ten Dam L, van der Kooi AJ, van Wattingen M, de Haan RJ, de Visser M. Reliability and accuracy of skeletal muscle imaging in limb-girdle muscular dystrophies. Neurology. 2012;79(16), 2276–2277.
358. Pan TC, Zhang RZ, Pericak-Vance MA, et al. Missense mutation in a von Wille-brand factor type A domain of the alpha 3(VI) collagen gene (COL6A3) in a family with Bethlem myopathy. Hum Mol Genet. 1998;7(5):807–812.
359. Katz JS, Wolfe GI, Burns DK, et al. Isolated neck extensor myopathy: A common cause of dropped head syndrome. Neurology. 1996;46(4):917–921.
360. Rose MR, Levin KH, Griggs RC. The dropped head plus syndrome: Quantitation of response to corticosteroids. Muscle Nerve. 1999;22(1):115–118.
361. Serratrice G, Pouget J, Pellissier JF. Bent spine syndrome. J Neurol Neurosurg Psychiatry. 1996;60(1):51–54.
362. Suarez GA, Kelly JJ, Jr. The dropped head syndrome. Neurology. 1992;42(8):1625–1627.
363. Chahin N, Selcen D, Engel AG. Sporadic late onset nemaline myopathy. Neurology. 2005;65(8):1158–1164.
364. Biran I, Cohen O, Diment J, Peyser A, Bahnof R, Steiner I. Focal, steroid responsive myositis causing dropped head syndrome. Muscle Nerve. 1999;22(6):769–771.
365. Dominick J, Sheean G, Schleimer J, Wixom C. Response of the dropped head/bent spine syndrome to treatment with intravenous immunoglobulin. Muscle Nerve. 2006;33(6):824–826.
366. Duarte S, Oliveira J, Santos R, et al. Dominant and recessive RYR1 mutations in adults with core lesions and mild muscle symptoms. Muscle Nerve. 2011;44(1):102–108.
367. Løseth S, Voermans NC, Torbergsen T, et al. A novel late-onset axial myopathy associated with mutations in the skeletal muscle ryanodine receptor (RYR1) gene. J Neurol. 2013;260(6):1504–1510.
368. Brais B, Rouleau GA, Bouchard JP, Fardeau M, Tomé FM. Oculopharyngeal muscular dystrophy. Semin Neurol. 1999;19(1):59–66.
369. Hill ME, Creed GA, Bouchard JP, Fardeau M, Tomé FM. Oculopharyngeal muscular dystrophy: Phenotypic and genotypic studies in a UK population. Brain. 2001;124(Pt 3):522–526.
370. Blumen SC, Brais B, Korczyn AD, et al. Homozygotes for oculopharyngeal muscular dystrophy have a severe form of the disease. Ann Neurol. 1999;46(1):115–118.
371. Brais B, Bouchard JP, Xie YG, et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18(2):164–167.
372. Calado A, Tome FM, Brais B, et al. Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Hum Mol Genet. 2000;9(15):2321–2328.
373. Shanmugam V, Dion P, Rochefort D, Laganière J, Brais B, Rouleau GA. PABP2 polyalanine tract expansion causes intranuclear inclusions in oculopharyngeal muscular dystrophy. Ann Neurol. 2000;48(5):798–802.
374. Fukuhara N, Kumamoto T, Tsubaki T, Mayuzumi T, Nitta H. Oculopharyngeal muscular dystrophy and distal myopathy. Intrafamilial difference in the onset and distribution of muscular involvement. Acta Neurol Scand. 1982;65(5):458–467.
375. Goto I, Hayakawa T, Miyoshi T, Ino K, Kusunoki R. Case of oculo-pharyngo-distal myopathy with cardiopathy. Rinsho Shinkeigaku. 1973;13(9):529–536.
376. Amato AA, Jackson CE, Ridings LW, Barohn RJ. Childhood-onset oculopharyngodistal myopathy with chronic intestinal pseudo-obstruction. Muscle Nerve. 1995;18(8):842–847.
377. Vita G, Dattola R, Santoro M, Messina C. Familial oculopharyngeal muscular dystrophy with distal spread. J Neurol. 1983;230(1):57–64.
378. Dimachkie MM, Barohn RJ. Distal myopathies. Neurol Clin. 2014;32(3):817–842.
379. Barohn RJ, Amato AA, Griggs RC. Overview of distal myopathies: From the clinical to the molecular. Neuromuscul Disord. 1998;8(5):309–316.
380. Welander, L. Myopathia distalis tarda hereditaria; 249 examined cases in 72 pedigrees. Acta Med Scand Suppl. 1951;265:1–124.
381. Borg K, Ahlberg G, Borg J, Edström L. Welander’s distal myopathy: Clinical, neurophysiological and muscle biopsy observations in young and middle aged adults with early symptoms. J Neurol Neurosurg Psychiatry. 1991;54(6):494–498.
382. Lindberg C, Borg K, Edström L, Hedström A, Oldfors A. Inclusion body myositis and We-lander distal myopathy: A clinical, neurophysiological and morphological comparison. J Neurol Sci. 1991;103(1):76–81.
383. Borg K, Solders G, Borg J, Edström L, Kristensson K. Neurogenic involvement in distal myopathy (Welander). Histochemical and morphological observations on muscle and nerve biopsies. J Neurol Sci. 1989;91(1–2):53–70.
384. Borg K, Tome FM, Edström L. Intranuclear and cytoplasmic filamentous inclusions in distal myopathy (Welander). Acta Neuropathol (Berl). 1991;82(2):102–106.
385. Edstrom, L. Histochemical and histopathological changes in skeletal muscle in late-onset hereditary distal myopathy (Welander). J Neurol Sci. 1975;26(2):147–157.
386. Markesbery WR, Griggs RC, Herr B. Distal myopathy: Electron microscopic and histochemical studies. Neurology. 1977; 27(8):727–735.
387. Hackman P, Sarparanta J, Lehtinen S, et al. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann Neurol. 2013;73(4):500–9.
388. Klar J, Sobol M, Melberg A, et al. Welander distal myopathy caused by an ancient founder mutation in TIA1 associated with perturbed splicing. Hum Mutat. 2013;34(4):572–577.
389. Udd B, Bushby K, Nonaka I, Griggs R. 104th European Neuromuscular Centre (ENMC) International Workshop: Distal myopathies, 8–10th March 2002 in Naarden, the Netherlands. Neuromuscul Disord. 2002;12(9):897–904.
390. Udd B, Haravuori H, Kalimo H, et al. Tibial muscular dystrophy—from clinical description to linkage on chromosome 2q31. Neuromuscul Disord. 1998;8(5):327–332.
391. Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71(3):492–500.
392. Markesbery WR, Griggs RC, Leach RP, Lapham LW. Late onset hereditary distal myopathy. Neurology. 1974;24(2):127–134.
393. Griggs R, Vihola A, Hackman P, et al. Zaspopathy in a large classic late-onset distal myopathy family. Brain. 2007;130:1477–1484.
394. Lin X, Ruiz J, Bajraktari I, et al. Z-disc-associated, alternatively spliced, PDZ motif-containing protein (ZASP) mutations in the actin-binding domain cause disruption of skeletal muscle actin filaments in myofibrillar myopathy. J Biol Chem. 2014;289(19):13615–13626.
395. Mizusawa H, Kurisaki H, Takatsu M, et al. Rimmed vacuolar distal myopathy: A clinical, electrophysiological, histopathological and computed tomographic study of seven cases. J Neurol. 1987;234(3):129–136.
396. Nonaka I, Sunohara N, Ishiura S, Satoyoshi E. Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J Neurol Sci. 1981;51(1):141–155.
397. Nonaka I, Sunohara N, Satoyoshi E, Terasawa K, Yonemoto K. Autosomal recessive distal muscular dystrophy: A comparative study with distal myopathy with rimmed vacuole formation. Ann Neurol 1985;17(1):51–59.
398. Sunohara N, Nonaka I, Kamei N, Satoyoshi E. Distal myopathy with rimmed vacuole formation. A follow-up study. Brain. 1989;112(Pt 1):65–83.
399. Argov Z, Eisenberg I, Grabov-Nardini G, et al. Hereditary inclusion body myopathy: The Middle Eastern genetic cluster. Neurology. 2003;60(9):1519–1523.
400. Argov Z, Yarom R. “Rimmed vacuole myopathy” sparing the quadriceps. A unique disorder in Iranian Jews. J Neurol Sci. 1984;64(1):33–43.
401. Eisenberg I, Grabov-Nardini G, Hochner H, et al. Mutations spectrum of GNE in hereditary inclusion body myopathy sparing the quadriceps. Hum Mutat. 2003;21(1):99.
402. Sivakumar K, Dalakas MC. The spectrum of familial inclusion body myopathies in 13 families and a description of a quadriceps-sparing phenotype in non-Iranian Jews. Neurology. 1996;47(4):977–984.
403. Eisenberg I, Avidan N, Potikha T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29(1):83–87.
404. Laing NG, Laing BA, Meredith C, et al. Autosomal dominant distal myopathy: Linkage to chromosome 14. Am J Hum Genet. 1995;56(2):422–427.
405. Mastaglia FL, Phillips BA, Cala LA, et al. Early onset chromosome 14-linked distal myopathy (Laing). Neuromuscul Disord. 2002;12:350–357.
406. Meredith C, Herrmann R, Parry C, et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1). Am J Hum Genet. 2004;75:703–708.
407. Muelas N, Hackman P, Lugue H, et al. MYH7 gene tail mutation causing myopathic profiles beyond Laing distal myopathy. Neurology. 2010;75:732–741.
408. Overeem S, Schelhaas HJ, Blijham PJ, et al. Symptomatic distal myopathy with cardiomyopathy due to a MYH7 mutation. Neuromuscul Disord. 2007;17:490–493.
409. Tasca G, Ricci E, Penttilä S, et al. New phenotype and pathology features in MYH7-related distal myopathy. Neuromuscul Disord. 2012;22:640–647.
410. Lamont PJ, Udd B, Mastaglia FL, et al. Laing early onset distal myopathy: Slow myosin defect with variable abnormalities on muscle biopsy. J Neurol Neurosurg Psychiatry. 2006;77(2):208–215.
411. Tajsharghi H, Thornell LE, Lindberg C, Lindvall B, Henriksson KG, Oldfors A. Myosin storage myopathy associated with a heterozygous missense mutation in MYH7. Ann Neurol. 2003;54(4):494–500.
412. Tajsharghi H, Oldfors A, Macleod DP, Swash M. Homozygous mutation in MYH7 in myosin storage myopathy and cardiomyopathy. Neurology. 2007;68:962.
413. Masuzugawa S, Kuzuhara S, Narita Y, Naito Y, Taniguchi A, Ibi T. Autosomal dominant hyaline body myopathy presenting as scapuloperoneal syndrome: Clinical features and muscle pathology. Neurology. 1997;48:253–257.
414. Bohlega S, Abu-Amero SN, Wakil SM, et al. Mutation of the slow myosin heavy chain rod domain underlies hyaline body myopathy. Neurology. 2004;62:518–521.
415. Williams DR, Reardon K, Roberts L, et al. A new dominant distal myopathy affecting posterior leg and anterior upper limb muscles. Neurology. 2005;64:1245–1254.
416. Duff RM, Tay V, Hackman P, et al. Mutations in the N-terminal actin-binding domain of filamin C cause a distal myopathy. Am J Hum Genet. 2011;88:729–740.
417. Guergueltcheva V, Peeters K, Baets J, et al. Distal myopathy with upper limb predominance caused by filamin C haploinsufficiency. Neurology. 2011;77:2105–2114.
418. Luan X, Hong D, Zhang W, Wang Z, Yuan Y. A novel heterozygous deletion-insertion mutation (2695–2712 del/GTTTGT ins) in exon 18 of the filamin C gene causes filaminopathy in a large Chinese family. Neuromuscul Disord. 2010;20:390–396.
419. Wallgren-Pettersson C, Lehtokari VL, Kalimo H, et al. Distal myopathy caused by homozygous missense mutations in the nebulin gene. Brain. 2007;130:1465–1476.
420. Lehtokari VL, Pelin K, Herczegfalvi A, et al. Nemaline myopathy caused by mutations in the nebulin gene may present as a distal myopathy. Neuromuscul Disord. 2011;21:556–562.
421. Cirak S, von Deimling F, Sachdev S, et al. Kelch-like homologue 9 mutation is associated with an early onset autosomal dominant distal myopathy. Brain. 2010;133:2123–2135.
422. Feit H, Silbergleit A, Schneider LB, et al. Vocal cord and pharyngeal weakness with autosomal dominant distal myopathy: Clinical description and gene localization to 5q31. Am J Hum Genet. 1998;63(6):1732–1742.
423. Senderek J, Garvey SM, Krieger M, et al. Autosomal-dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am J Hum Genet. 2009;84:511–518.
424 Johnson JO, Pioro EP, Boehringer A, et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci. 2014;17(5):664–666.
425. Muller TJ, Kraya T, Stoltenburg-Didinger G, et al. Phenotype of the Matrin-3-distal myopathy in 16 German patients. Ann Neurol. 2014;76:669–680.
426. Amato AA, Kagan-Hallet K, Jackson CE, et al. The wide spectrum of myofibrillar myopathy suggests a multifactorial etiology and pathogenesis. Neurology. 1998;51(6):1646–1655.
424. Dalakas MC, Dagvadorj A, Goudeau B, et al. Progressive skeletal myopathy, a phenotypic variant of desmin myopathy associated with desmin mutations. Neuromuscul Disord. 2003;13(3):252–258.
428. Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. 2000;342(11):770–780.
429. De Bleecker JL, Engel AG, Ertl BB. Myofibrillar myopathy with abnormal foci of desmin positivity. II. Immunocytochemical analysis reveals accumulation of multiple other proteins. J Neuropathol Exp Neurol. 1996;55(5):563–577.
430. Engel AG. Myofibrillar myopathy. Ann Neurol. 1999;46(5):681–683.
431. Goebel HH. Desmin-related neuromuscular disorders. Muscle Nerve. 1995;18(11):1306–1320.
432. Goldfarb LG, Park KY, Cervenáková L, et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. 1998;19(4):402–403.
433. Goldfarb LG, Vicart P, Goebel HH, Dalakas MC. Desmin myopathy. Brain. 2004;127(Pt 4):723–734.
434. Nakano S, Engel AG, Waclawik AJ, Emslie-Smith AM, Busis NA. Myofibrillar myopathy with abnormal foci of desmin positivity. I. Light and electron microscopy analysis of 10 cases. J Neuropathol Exp Neurol. 1996;55(5):549–562.
435. Selcen D, Engel AG. Myofibrillar myopathy caused by novel dominant negative alpha B-crystallin mutations. Ann Neurol. 2003;54(6):804–810.
436. Selcen D, Ohno K, Engel AG. Myofibrillar myopathy: Clinical, morphological and genetic studies in 63 patients. Brain. 2004;127(Pt 2):439–451.
437. Vicart P, Caron A, Guicheney P, et al. A missense mutation in the alphaBcrystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20(1):92–95.
438. Nakano S, Engel AG, Akiguchi I, Kimura J. Myofibrillar myopathy. III. Abnormal expression of cyclin-dependent kinases and nuclear proteins. J Neuropathol Exp Neurol. 1997;56(8):850–856.
439. Bertini E, Biancalana V, Bolino A, et al. 118th ENMC International Workshop on Advances in Myotubular Myopathy, 26–28 September 2003, Naarden, the Netherlands (5th Workshop of the International Consortium on Myotubular Myopathy). Neuromuscul Disord. 2004;14(6):387–396.
440. Sabatelli M, Bertini E, Ricci E, et al. Peripheral neuropathy with giant axons and cardiomyopathy associated with desmin type intermediate filaments in skeletal muscle. J Neurol Sci. 1992;109(1):1–10.
441. Munoz-Marmol AM, Strasser G, Isamat M, et al. A dysfunctional desmin mutation in a patient with severe generalized myopathy. Proc Natl Acad Sci U S A. 1998;95(19):11312–11317.
442. Li M, Dalakas MC. Abnormal desmin protein in myofibrillar myopathies caused by desmin gene mutations. Ann Neurol. 2001;49(4):532–536.
443. Tajsharghi H, Darin N, Rekabdar E, et al. Mutations and sequence variation in the human myosin heavy chain IIa gene (MYH2). Eur J Hum Genet. 2005;13(5):617–622.
444. Martinsson T, Oldfors A, Darin N, et al. Autosomal dominant myopathy: Missense mutation (Glu-706 –> Lys) in the myosin heavy chain IIa gene. Proc Natl Acad Sci U S A. 2000;97: 14614–14619.
445. Cole AJ, Kuzniecky R, Karpati G, Carpenter S, Andermann E, Andermann F. Familial myopathy with changes resembling inclusion body myositis and periventricular leucoencephalopathy. A new syndrome. Brain. 1988;111(Pt 5):1025–1037.
446. Watts GD, Thorne M, Kovach MJ, Pestronk A, Kimonis VE. Clinical and genetic heterogeneity in chromosome 9p associated hereditary inclusion body myopathy: Exclusion of GNE and three other candidate genes. Neuromuscul Disord. 2003;13(7-8):559–567.
447. Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36(4):377–381.
448. Watts GD, Thomasova D, Ramdeen SK, et al. Novel VCP mutations in inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Clin Genet. 2007;72:420–426.
449. Watts GD. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381.
450. Kimonis VE, Kovach MJ, Waggoner B, et al. Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet Med. 2000;2:232–241.
451. Kimonis VE, Mehta SG, Fulchiero EC, et al. Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia. Am J Med Genet A. 2008;146A:745–757.
452. Kimonis VE, Watts GD. Autosomal dominant inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Alzheimer Dis Assoc Disord. 2005;19(Suppl 1):S44–S47.
453. Guyant-Maréchal L, Laquierrière A, Duyckaerts C, et al. Valosin-containing protein gene mutations: Clinical and neuropathologic features. Neurology. 2006;67:644–651.
454. Haubenberger D, Bittner RE, Rauch-Shorny S, et al. Inclusion body myopathy and Paget disease is linked to a novel mutation in the VCP gene. Neurology. 2005;65:1304–1305.
455. Miller TD, Jackson AP, Barresi R, et al. Inclusion body myopathy with Paget disease and frontotemporal dementia (IBMPFD): Clinical features including sphincter disturbance in a large pedigree. J Neurol Neurosurg Psychiatry. 2009;80:583–584.
456. Kim EJ, Park YE, Kim DS, et al. Inclusion body myopathy with Paget disease of bone and frontotemporal dementia linked to VCP p.Arg155Cys in a Korean family. Arch Neurol. 2011;68:787–796.
457. Kumar KR, Needham M, Mina K, et al. Two Australian families with inclusion-body myopathy, Paget’s disease of bone and frontotemporal dementia: Novel clinical and genetic findings. Neuromuscul Disord. 2010;20:330–334.
458. Stojkovic T, Hammouda el H, Richard P, et al. Clinical outcome in 19 French and Spanish patients with valosin-containing protein myopathy associated with Paget’s disease of bone and frontotemporal dementia. Neuromuscul Disord. 2009;19:316–323.
459. Viassolo V, Previtali SC, Schiatti E, et al. Inclusion body myopathy, Paget’s disease of the bone and frontotemporal dementia: Recurrence of the VCP R155 H mutation in an Italian family and implications for genetic counselling. Clin Genet. 2008;74:54–60.
460. Waggoner B, Kovach MJ, Winkelman M, et al. Heterogeneity in familial dominant Paget disease of bone and muscular dystrophy. Am J Med Genet. 2002;108:187–191.
461. Shi Z, Hayashi YK, Mitsuhashi S, et al. Characterization of the Asian myopathy patients with VCP mutations. Eur J Neurol. 2012;19:501–509.
462. van der Zee J, Pirici D, Van Langenhove T, et al. Clinical heterogeneity in 3 unrelated families linked to VCP p.Arg159His. Neurology. 2009;73:626–632.
463. Palmio J, Sandell S, Suominen T, et al. Distinct distal myopathy phenotype caused by VCP gene mutation in a Finnish family. Neuromuscul Disord. 2011;21:551–555.
464. Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68(5):857–864.
465. González-Pérez P, Cirulli ET, Drory VE, et al. Novel mutation in VCP gene causes atypical amyotrophic lateral sclerosis. Neurology. 2012;79(22):2201–2208.
466. Kim HJ, Kim NC, Wang YD, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473.
467. Bucelli RC, Arhzaouy K, Pestronk A, et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology. 2015. (in-press)