TABLE 30-1. CLASSIFICATION OF THE MITOCHONDRIAL MYOPATHIES BY METABOLIC FUNCTION AFFECTED
TABLE 30-1. CLASSIFICATION OF THE MITOCHONDRIAL MYOPATHIES BY METABOLIC FUNCTION AFFECTEDMitochondrial myopathies and neuropathies or neuromyopathies refer to a heterogeneous group of disorders caused by dysfunction of mitochondria.1–10 Mitochondrial disorders can be classified according to the associated biochemical, genetic defects, or clinical phenotype (Tables 30-1 to 30-3). One difficulty in classifying patients by any particular scheme is the clinical-phenotypic heterogeneity associated with specific mitochondrial mutations and the genetic heterogeneity in well-defined clinical phenotypes that are seen with mitochondrial disorders.
TABLE 30-1. CLASSIFICATION OF THE MITOCHONDRIAL MYOPATHIES BY METABOLIC FUNCTION AFFECTED
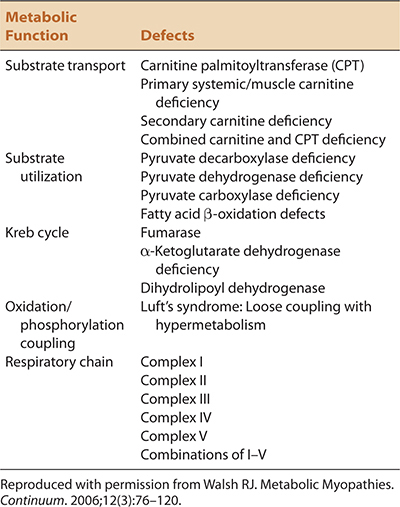
TABLE 30-2. CLASSIFICATION OF MITOCHONDRIAL DISORDERS BY GENETIC MUTATIONS
I. Mitochondrial DNA mutations
A. Large scale deletions
1. Kearns–Sayre syndrome
2. PEO
B. Mutations in mtDNA protein–coding genes
1. ATP6 is associated with Leigh syndrome and NARP
2. Cytochrome b is associated with exercise intolerance and recurrent myoglobinuria
3. Cytochrome c oxidase is associated with fatal and benign infantile myopathies, Leigh syndrome, MELAS, recurrent rhabdomyolysis
C. Mutations in mitochondrial tRNA and rRNA genes
1. MERRF is usually associated with mutations in tRNALys gene. MERRF has also been associated with mutations in tRNALeu and tRNASer
2. MELAS is usually associated with mutations in tRNALeu gene. MELAS also occurs with mutations in tRNAVal, tRNACys, ND5 of complex 1, and in cytochrome b
II. Nuclear gene mutations
A. Nuclear mutations associated with mtDNA maintenance and replication (multiple mtDNA deletions and mtDNA depletion)
1. Thymidine phosphorylase gene (TYMP) is associated with autosomal recessive MNGIE
2. Adenine nucleotide translocator 1 (ANT1) is associated with autosomal dominant PEO
3. Twinkle (C10orf2) is associated with autosomal dominant PEO and SANDO
4. Polymerase gamma (POLG1 and POLG2) is associated with autosomal recessive and dominant PEO, SANDO, MIRAS
5. Ribonucleotidase reductase (RRM2B) is associated with MNGIE and PEO
6. Thymidine kinase 2 (TK2) is associated with severe myopathy
7. DNA replication helicase/nuclease 2 (DNA2) is associated with PEO
8. Deoxyguanosine kinase (DGUOK) is associated with PEO, recurrent rhabdomyolysis, and mtDNA depletion myopathy
9. Paraplegin (SGP7) is usually associated with hereditary spastic paraglegia but can be associated with PEO and spasticity
B. Nuclear mutations associated with abnormal mitochondrial fusion/fission
1. Mitofusin 2 (MFN2) associated with CMT2A
2. Ganglioside-induced differentiation associated-protein 1 (GDAP1) associated with CMT2K and CMT4A
3. Optic atrophy 1 (OPA1) associated with optic atrophy 1 syndrome/CMT with optic atrophy
4. Mitochondrial inner protein 17 (MPV17) associated with Navajo neurohepatopathy and PEO
C. Nuclear DNA mutations directly affecting components of mitochondrial respiratory chain
1. Leigh syndrome may be caused by mutations in SURF1 and several different subunits of Complexes I, II, IV of the respiratory chain encoded by nuclear genes
2. NARP caused by mutations in MTATP6
3. SDH mutations can be associated with exercise intolerance
TABLE 30-3. CLASSIFICATION OF MITOCHONDRIAL MYOPATHIES BY CLINICAL FEATURES AND GENOTYPE

The mitochondria are responsible for converting fuels (carbohydrates, lipids, and proteins) into energy for the cells. Fatty acids are metabolized into molecules of acetyl-CoA within the mitochondria. Amino acids are converted to pyruvate in the mitochondria. Carbohydrates are metabolized to pyruvate in the cytoplasm and then transported into the mitochondria. Pyruvate from either source is converted into acetyl-CoA. Acetyl-CoA, then enters into the Krebs cycle from which electrons are generated. Electrons derived from the Krebs cycle are shuttled to the respiratory chain and processed through complexes I–V to generate ATP molecules. Thus, mitochondrial disorders can be classified according to the metabolic defect present (1) transport, (2) substrate utilization, (3) Krebs cycle, (4) oxidation/phosphorylation coupling, and (5) respiratory chain (Table 30-1).
Some of the biochemical abnormalities seen in various mitochondrial disorders are nonspecific and the result of primary “upstream” defects in metabolic pathways. For example, cytochrome oxidase (COX) deficiency is seen in many types of mitochondrial myopathy and does not imply that the primary mutation lies in one of the genes encoding for subunits of COX. The rapid advances of molecular genetics may provide a better classification scheme. The mitochondrial disorders may be classified by their genetic defect (1) mitochondrial DNA (mtDNA), (2) nuclear DNA (nDNA) mutations directly or indirectly affecting the mitochondrial respiratory chain complex, or (3) nDNA mutations that are involved in mtDNA maintenance or mitochondrial dynamics (Table 30-2; Fig. 30-1).1–6 Notably though, there is significant phenotypic variability even in patients with the same genetic mutation. Therefore, a combined classification scheme is currently favored because of phenotypical variability and problems inherent in current genotyping capabilities (Table 30-3). Prior to discussing specific disorders, we will review a few basic principles regarding the mitochondrial genome and different inheritance patterns of mitochondrial disorders.
Figure 30-1. Genes associated with mitochondrial disease. Mutations have been identified in genes encoding CI, CII, CIII, CIV, and CV subunits and assembly factors; genes involved in mitochondrial DNA (mtDNA) maintenance (via nucleotide metabolism), mtDNA replication, and mtDNA expression; genes affecting the electron carriers coenzyme Q (CoQ) and cytochrome c (CytC); genes affecting FeS assembly; and genes involved in protein import, toxicity/apoptosis, and membrane function. Recently identified genes described in this review are highlighted in red. Genes affecting oxidative phosphorylation for which mutations are not reported to cause neuropathology are underlined. rRNA, ribosomal RNA; tRNA, transfer RNA. Currently, most cases of mitochondrial encephalopathy are untreatable, other than by relieving certain symptoms. Therefore, there is a great need to better understand the genetics of mitochondrial disease, which will enable prenatal diagnoses and will deliver the deeper understanding of mitochondrial function needed for the development of effective therapies. Recent advances in sequencing technology indicate that we may be on the cusp of a revolution in the way genetic diseases, such as mitochondrial encephalopathy, are diagnosed. (Reproduced with permission from Tucker EJ, Compton AG, Thorburn DR: recent advances in the genetics of mitochondrial encephalopathies. Curr Neurol Neurosci Rep. 2010;10(4):277–285.)
 COMPOSITION OF MITOCHONDRIAL DNA AND PROTEINS
COMPOSITION OF MITOCHONDRIAL DNA AND PROTEINSThe mitochondrial genome comprises 16.5-kB circular double-stranded DNA that contains no noncoding regions (i.e., introns). In fact, contiguous mitochondrial genes overlap in some areas. There is a single promoter site, and transcription is polycistronic such that mitochondrial genes are transcribed as two large RNAs. These are subsequently cleaved into 13 respective messenger RNAs (mRNA), 2 ribosomal RNAs (rRNA), and 22 transfer RNAs (tRNA). Interestingly, the genetic code for translation of human mitochondrial genes differs from the standard code which governs the translation of human nuclear genes.
The 13 mRNAs are translated into 13 polypeptides that are subunits of the respiratory chain complexes. Also note that any mutation in a mitochondrial tRNA gene can impair the proper translation of the 13 mitochondrial mRNAs. Importantly, the 13 proteins encoded by the mitochondrial genome account for less than 5% of all mitochondrial proteins. Thus, the majority of mitochondrial proteins are encoded by the nuclear genome that are translated in the cytoplasm and subsequently are transported into the mitochondria. Furthermore, the nucleus appears to regulate replication of the mitochondrial genome.
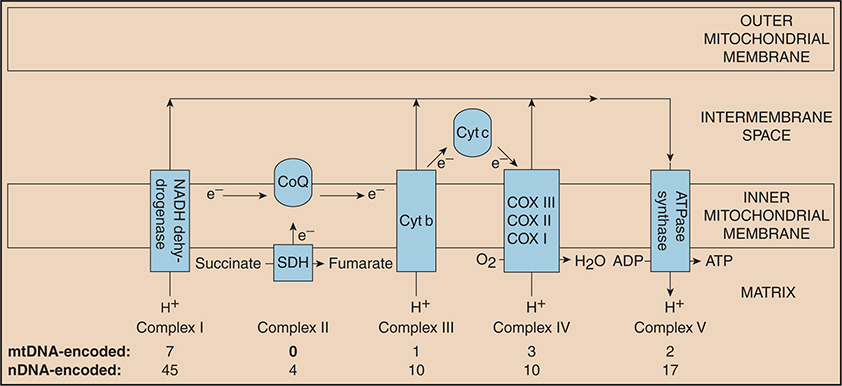
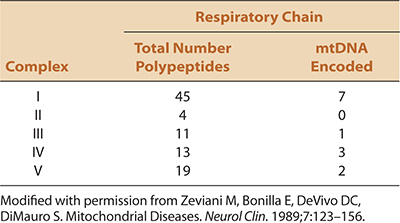
The respiratory chain comprises five multienzyme complexes (complexes I–V) (Fig. 30-2). Complex I (NADH-CoQ reductase) contains 45 subunits, seven encoded by mtDNA; complex II [succinate dehydrogenase (SDH) CoQ reductase] comprises four subunits, each encoded by nuclear genes; complex III (CoQH2-cytochrome c reductase) comprises 11 polypeptide units, one of which is encoded by mtDNA; complex IV (cytochrome c oxidase) has 13 subunits, three encoded by mtDNA; and complex V (ATPase synthetase) comprises 19 subunits, two encoded by mtDNA (Table 30-4).
Figure 30-2. Schematic view of the respiratory chain. This diagram shows the number of subunits encoded by mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) for each complex. All of the subunits for complex II are encoded by nDNA. Electrons (e–) flow down the respiratory chain and protons (H+) are pumped from the matrix to the intermembranous space through complexes I, III, and IV, then back into the matrix through complex V (ATPase synthase). Cytochrome c (Cyt c) and coenzyme Q (CoQ) are electron carriers. This process generates adenosine triphosphate (ATP). (Reproduced with permission from Walsh RJ. Metabolic Myopathies. Continuum. 2006;12(3):76–120.)
TABLE 30-4. COMPOSITION AND GENETIC CONTROL OF THE MITOCHONDRIAL RESPIRATORY CHAIN
GENETICS OF MITOCHONDRIAL DISORDERSA population-based study in Northern England found that 6.57 per 100,000 adults have a mitochondrial disease and 12.48 per 100,000 of children and adults are at risk for developing a mitochondrial disorder on the basis of identifiable mtDNA mutations.11 Because this study included only disorders caused by mtDNA mutations and not nDNA mutations affecting mitochondria, the prevalence of mitochondrial disorders is certainly higher. Remember that during fertilization, all the mitochondria are contributed by the mother. Hundreds of mitochondria are present in most cells in the body and every mitochondrion has several copies of mtDNA. Mutations involving mtDNA are more common and more likely to manifest clinically than mutations in nuclear genes because of the lack of introns and decreased DNA repair mechanisms in the mitochondrial genome. mtDNA mutations are randomly distributed in subsequent generations of somatic cells during mitosis and germ cells during meiosis. Therefore, some cells will have few or no mutant genomes (normal homoplasty), some will have a mixture of mutant and normal or wild-type mtDNA (heteroplasty), and some will have predominantly mutant genomes (mutant homoplasty). Phenotypic expression depends on the relative proportion of mutant and wild-type mitochondria within each cell within a given organ system. When the number of mitochondria bearing sufficient mutated mtDNA exceed a certain threshold, mitochondrial function becomes impaired and patients manifest clinical symptoms and signs of disease (threshold affect).
During mitosis and meiosis, the proportion of mutant mitochondria in daughter cells can shift, thus changing the genotype and possibly the phenotype (mitotic/meiotic segregation). In addition, mutant mitochondria may utilize the mitochondrial-encoded mRNAs and tRNAs from neighboring normal mitochondria in a process called complementation. Thus, there can be some degree of normal translation of mtDNA-encoded proteins even in mitochondria harboring large DNA deletions.
Different organs have differing susceptibility for mitochondrial abnormalities depending on their energy requirements. Because the central nervous system (CNS) is in constant demand for energy, small decreases in energy production can lead to severe abnormalities. In contrast, skeletal muscle has low energy demands at rest, but these demands drastically increase with exercise. This is the basis for exercise-intolerance in many patients with mitochondrial myopathies.
Primary mutations of mtDNA can only be inherited from the mother. Unlike X-linked disorders that are also passed on only from the mother, women and men are equally affected in inherited mitochondrial diseases, while men are generally more severely affected with an X-linked inheritance pattern. Further, based on the degree of mitochondrial segregation and heteroplasty, all the children of an affected mother may be affected to a variable degree, which is different from autosomal dominant and recessive inheritance patterns.
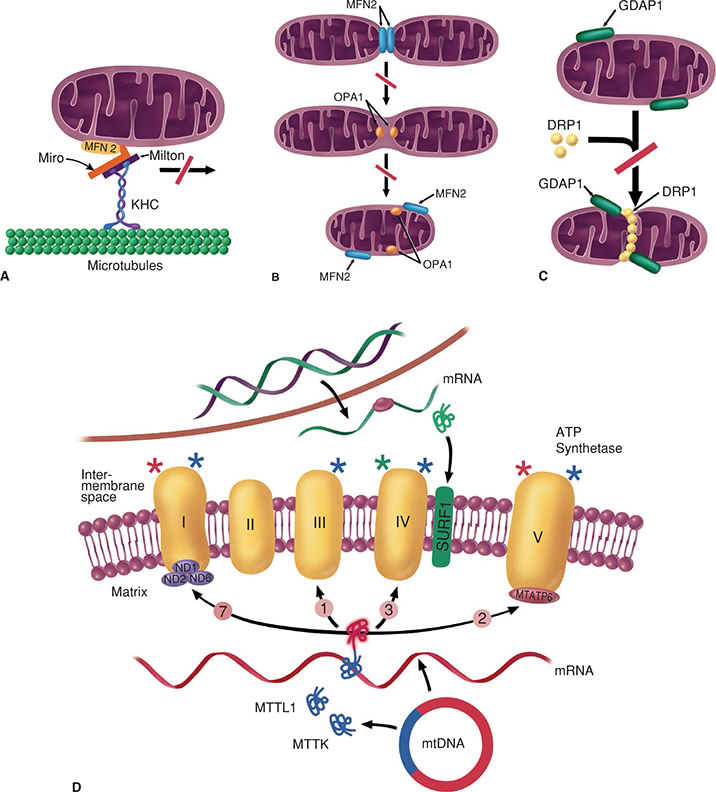
Mitochondrial disorders are not strictly inherited from an affected mother. Because over 95% of mitochondrial proteins are encoded from nuclear genes, mitochondrial disorders can be inherited in an autosomal dominant [e.g., some forms of progressive external ophthalmoplegia (PEO)], autosomal recessive [e.g., mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) syndrome], and even X-linked (e.g., some forms of Leigh syndrome) fashion. In addition, the presence of mutations involving mtDNA does not imply a maternal/mitochondrial inheritance pattern. In this regard, Kearns–Sayre syndrome (KSS) is associated with large mtDNA deletions, but is sporadic in nature. In addition, as noted earlier there appears to be some nuclear control over the replication and/or maintenance mitochondrial genome. Thus, mutations in some nuclear genes result in syndromes associated with depletion or multiple deletions of mtDNA. These disorders can demonstrate autosomal recessive or dominant inheritance patterns and are caused by defects in enzymes required for mtDNA replication and for maintaining the proper balance within mitochondria of deoxynucleoside triphosphates (dNTPs) that serve as the building blocks for mtDNA (Figs. 30-1 and 30-3). Replication of mtDNA requires the catalytic subunit of polymerase (encoded by the POLG1 gene), the accessory subunit (encoded by POLG2), and the replicative helicases, twinkle (encoded by PEO1), and DNA replication helicase/nuclease 2 (encoded by DNA2). Mutations in these genes result in mtDNA depletion and/or multiple mtDNA deletions that are found in various mitochondrial disorders [e.g., PEO, MNGIE, sensory ataxia neuropathy dysarthria/dysphagia ophthalmoplegia (SANDO)], which are discussed later.1–6 A delicate balance of the 4 dNTPs (dATP, dGTP, dCTP, and dTTP) are also necessary for mtDNA replication. Several nuclear-encoded enzymes are key in preserving this balance (i.e., TK2, DGUOK SUCLA2, SUCLG1, RRM2B, TYMP, and MPV17). Mutations in these genes also lead to mtDNA depletion and cause various mitochondrial disorders (e.g., mitochondrial depletion myopathy, MNGIE, Navajo neurohepatopathy).1–6 Furthermore, some nuclear genes are responsible for normal mitochondrial dynamics (Figs. 30-1 and 30-3). For example, fusion of mitochondria is dependent on nuclear-encoded profusion GTPases that are located in the mitochondrial membranes and mitofusin 1 and 2 (MFN1 and MFN2).1–6

Figure 30-3. Overview of mechanisms underlying the main mitochondrial peripheral neuropathies. (A) MFN2 is located in the outer mitochondrial membrane and interacts with Miro and Milton proteins, which belong to the molecular complex that links mitochondria to kinesin (KHC) motors. (B) MFN2 participates in bringing the outer membranes of two mitochondria into close proximity. Thus, MFN2 mutations can lead to defects in mitochondrial motility along the cytoskeletal microtubular tracks, and to dysfunction in fusion of the outer mitochondrial membranes of opposing mitochondria. Similarly, mutations in OPA1, which is located in the inner mitochondrial membrane, lead to dysfunction in the fusion process of the inner mitochondrial membrane. (C) Loss-of-function mutations in GDAP1, located in the outer mitochondrial membrane, can lead to dysfunction in the mitochondrial fission process, since GDAP1 might be a positive effector of assembly of the fission mediator DRP1. Dysfunction is shown by red oblique bars. (D) Dysfunctions in the respiratory chain can be due to the following: Direct mutations in mitochondrial protein-coding genes (red segment of circular mtDNA) ND1, ND4, and ND6, which encode for subunits of complex I (NADH dehydrogenase), and MTATP6, which encodes for a subunit of complex V (ATP-synthase); mutations in mitochondrial tRNA-coding genes (light blue segment) MTTL1 and MTTK, which lead to dysfunction in transcription of mitochondrial protein-coding genes; or direct mutations in the nuclear gene SURF1 (in green), which encodes an ancillary protein involved in complex IV (cytochrome c oxidase) assembly. Dysfunction in the corresponding respiratory chain complexes are shown by red, light blue, and green asterisks, respectively. Encircled numbers show the number of subunits encoded by mtDNA. (E) Dysfunctions of the respiratory chain can also occur as a result of changes in mtDNA synthesis (red oblique bar), which lead to mtDNA depletion or multiple mtDNA deletions. mtDNA synthesis might be disturbed by the following: Mutations in nuclear genes encoding components of the mitochondrial replisome—that is, polymerase gamma POLG and the helicase C10orf2 genes; loss-of-function mutations (red oblique bar) in genes involved in the synthesis of nucleotides—that is, RRM2B and TYMP—which lead to nucleotide depletion (red lines show inhibition); or mutations in MPV17, a nuclear gene encoding an inner mitochondrial membrane protein of unknown function. Finally, mitochondrial peripheral neuropathies might result from changes in intermediary metabolism that eventually lead to decreased ATP synthesis. Mutations in SLC25A19 inhibit the passage of TPP from the cytosol to the mitochondrial matrix, thus leading to lactate accumulation and an increase in αKG (an intermediate of the Krebs cycle), since TPP is an essential cofactor of PDH and αKGD (green lines show stimulation); similarly, a gain-of-function mutation (green tick mark) in PDK3 might lock PDH in an inactive state, limiting glucose oxidation and favoring a switch toward anaerobic lactate production. mtDNA, mitochondrial DNA; NDPs, nucleoside diphosphates; dNDPs, deoxynucleoside diphosphates; dCTD, deoxycytidine; dCMP, deoxycytosine monophosphate; dCTP, deoxycytosine triphosphate; α-KG, α-ketoglutarate; α-KGD, α-ketoglutarate dehydrogenase; SCA, succinyl-CoA; TPP, thiamine pyrophosphate; PDH, pyruvate dehydrogenase.
There can be significant genetic heterogeneity even within well-defined clinical syndromes. For example, PEO can be associated with multiple mtDNA deletions, point mutations in various mitochondrial tRNA genes, or have no mtDNA mutations. In addition, specific mutations of mitochondrial-encoded genes can manifest with heterogeneous clinical phenotypes. For example, point mutations in the mitochondrial tRNALeu can result in mitochondrial myopathy lactic acidosis and strokes (MELAS), PEO, encephalomyopathy, or a generalized myopathy with exercise intolerance. Variability in clinical phenotype can also be apparent within families with identical mtDNA mutations. The vast clinical and genetic heterogeneity of the various mitochondrial disorders can be explained by the different segregation patterns of mutant mitochondria, the degree of mutant heteroplasty, tissue-specific thresholds, and the severity of the biochemical impairment related to the specific mutations.
LABORATORY FEATURESSerum creatine kinase (CK), lactic acid, and pyruvate levels can be normal or elevated. In addition, lactic acid levels may also be elevated in cerebrospinal fluid (CSF). Some mitochondrial disorders (e.g., mtDNA depletion) can be associated with renal tubular defects characterized by glycosuria, proteinuria, and aminoaciduria.
Bicycle ergometry can sometimes be a useful test. Low levels of workload lead to an excessive rise in pulse rate and oxygen consumption. The degree of exercise intolerance correlates directly with the severity of impaired muscle oxidative phosphorylation as indicated by the peak capacity for muscle oxygen extraction and mitochondrial mutation load.12,13 The diagnostic value of a constant workload protocol may be superior to an incremental cycle test, but the test is less sensitive for mitochondrial myopathies than simple testing of resting lactate and muscle morphology.14
A forearm exercise test can be performed where bicycle ergometry testing is not available.15 The patient is instructed to open and close their hand (about once every 2 seconds at 40% of maximal voluntary contraction for 3 minutes). A butterfly needle can be placed in the antecubital fossa and venous oxygen and lactate levels can be measured at baseline and each minute during and immediately following exercise. Patients with mitochondrial myopathies and exercise intolerance often demonstrate excessive and prolonged lactate production and paradoxically increased venous oxygen saturation.15 The range of elevated venous PO2 during forearm exercise in mitochondrial myopathy patients (32–82 mm Hg) correlates closely with the severity of oxidative impairment as assessed during cycle exercise.16 Thus, the measurement of venous PO2 during aerobic forearm exercise provides an easily performed screening test that sensitively detects impaired oxygen use and accurately assesses the severity of oxidative impairment in patients with mitochondrial myopathy and exercise intolerance.
Nerve conduction studies (NCS) may be normal or abnormal. Some mitochondrial disorders are associated with a myopathy and/or a neuropathy. The neuropathy can be an axonal [i.e., neuropathy ataxia and retinitis pigmentosa (NARP)] or demyelinating in nature (e.g., MNGIE). Electromyography (EMG) is usually normal, although some myopathies are associated with increased insertional and spontaneous activity as well as early recruitment of small motor unit action potentials (MUAPs), while neurogenic disorders may be associated with decreased recruitment and large MUAPs. Conduction defects may be apparent on electrocardiograms (EKG).
Magnetic resonance imaging (MRI) and CT of the brain as well as electroencephalography (EEG) are typically abnormal in patients with a mitochondrial encephalomyopathy. MRI of skeletal muscle can reveal morphologic changes that resemble muscular dystrophies.17 Magnetic resonance spectroscopy (MRS) with 31P and 1H compounds permits the analysis of ATP, creatine phosphate, inorganic phosphate, and pH in muscle and brain.18,19 In mitochondrial disorders, there is a rapid fall in levels of creatine phosphate and an abnormal accumulation of inorganic phosphates in tissues with exercise. In addition, there is a delay in the recovery of phosphocreatine levels to normal after exercise. These techniques may also be potentially valuable in evaluating efficacy of various treatments.20
If a mitochondrial disorder is suspected from the clinical history and laboratory results, a muscle biopsy may be useful to confirm the diagnosis. Mutational analysis may be done on white blood cells. However, in certain syndromes, this is not as sensitive in finding mitochondrial mutations as in muscle tissue, particularly in those with large mtDNA deletions.
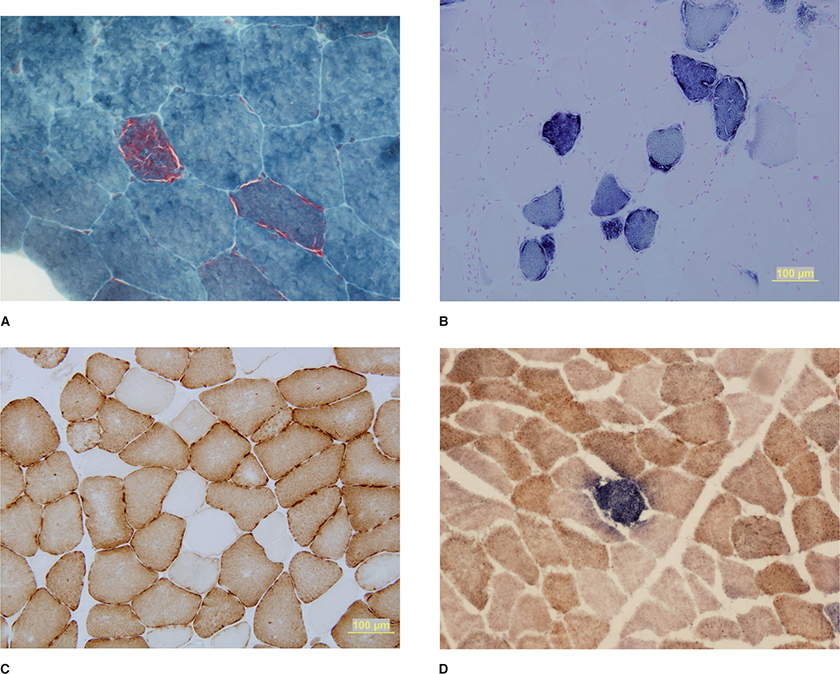
HISTOPATHOLOGYThe histopathological abnormalities in nerve biopsies of the various mitochondrial disorders are nonspecific and generally not helpful. However, muscle biopsies are often useful in diagnosing a mitochondrial disorder, particularly if there is significant muscle involvement. The characteristic histological features are the presence of ragged red fibers on the modified-Gomori trichrome stain (Fig. 30-4). An increased number of lipid droplets are also often evident within these abnormal muscle fibers. Oxidative enzyme stains nicotinamide adenine dinucleotide dehydrogenase (NADH), SDH, and cytochrome c oxidase (COX) are also invaluable. The aggregated mitochondria intensely react to NADH and SDH stains forming ragged blue fibers (Fig. 30-4). Some patients with mitochondrial myopathies (in particular disorders not associated with mt-tRNA mutations) may have no ragged red fibers and normal NADH and SDH staining. COX stain (directed against one of the subunits encoded by mtDNA) appears to be the most sensitive stain and can demonstrate scattered muscle fibers with reduced or absent stain (Fig. 30-4). In addition, COX can highlight the subsarcolemmal accumulations of mitochondria. Reduced COX staining can be seen in both ragged red and otherwise normal-appearing muscle fibers. The variability of COX staining in combination with intense SDH staining is characteristic of disorders with mtDNA mutations. Remember, the SDH component of complex II is entirely encoded by nDNA, while 3 of 13 subunits of complex IV (COX) are encoded by mtDNA. Mutations of mtDNA often lead to a proliferation of mitochondria, perhaps in a compensatory response. Because SDH is encoded by nDNA, its transcription is generally increased in disorders caused by mtDNA mutations. The variability of COX staining reflects the heteroplasmic population of mutant and wild-type mitochondria. COX staining is not always abnormal in mitochondrial myopathies. Some patients with MELAS, point mutations in either ND genes or cytochrome b, or multiple mtDNA deletions (e.g., due to POLG1 and other nDNA mutations) can have normal muscle histochemistry, including COX staining.9
Figure 30-4. Muscle biopsy demonstrates ragged red fibers resulting from the accumulation of abnormal mitochondrial below the sarcolemma of muscle fibers on modified Gomori-trichrome stain (A). Mitochondrial myopathies associated with mtDNA mutations often spare SDH, which is entirely encoded by the nuclear genome. Therefore, muscle fibers with proliferating mitochondria stain intensely with SDH stain—so-called ragged blue fibers (B). The most sensitive stain is for cytochrome c oxidase (COX). Scattered COX-negative fibers are often appreciated in mitochondrial myopathies (C). Combining the COX and SDH stains is very helpful as well. The presence of COX negativity in an SDH-positive fiber (blue staining) is suggestive of an mtDNA mutation, though this may be secondary to mutations in nuclear genes regulating mtDNA (D).
Ultrastructural alterations in mitochondria are usually apparent on EM. These abnormalities include an increased number of normal-appearing mitochondria, enlarged mitochondria with abnormal cristae, and mitochondria with paracrystalline inclusions (Fig. 30-5). The paracrystalline inclusions are accumulations of dimeric mitochondrial creatine kinase (mtCK). This enzyme exists in both a dimeric and octamer form, but the increased radical generation in patients with mitochondrial disorders favors the production and crystallization of dimeric mtCK.21
Figure 30-5. Electron microscopy. EM reveals increased numbers of mitochondria with abnormal paracrystalline inclusions.
BIOCHEMICAL ANALYSIS OF MITOCHONDRIAL FUNCTIONMitochondrial enzyme activities can be assayed in muscle biopsy specimens. This can be useful when the routine muscle histochemistry is unrevealing, but the diagnosis of a mitochondrial myopathy is still suspected because of the clinical phenotype. It can also be used to target genes for mutation screening. There is no standard method for performing mitochondrial metabolic analysis. Some centers prefer to assay only fresh muscle biopsy specimens (this is necessary for measurement of substrate oxidation). Rates of flux, substrate oxidation, and ATP production can be measured by polarography or using 14 C-labeled substrates. More commonly, measurement of enzyme activity of each of the individual respiratory complexes is performed on frozen muscle tissue.
MOLECULAR GENETIC ANALYSISMutation analysis of mtDNA and nuclear genes traditionally have been guided by the clinical phenotype, laboratory features, histochemistry, and biochemistry (Fig. 30-6).1–10 In patients with classic clinical syndromes (e.g., MERRF, MELAS), one can proceed directly toward mutation screening for the most common mutations associated with these disorders. As will be discussed, however, there is wide genetic heterogeneity even within well-defined clinical phenotypes. If a mutation is not found in white blood cells, then a muscle biopsy and mitochondrial enzyme analysis can be performed. Then, screening for mutations can be done based on clinical phenotype aided by histochemical features and biochemical analysis. That said, recent advances in molecular genetics have allowed for less expensive and extensive screening of mtDNA and nDNA by next-generation sequencing and other techniques.
Figure 30-6. Flowchart illustrating routes of investigation in cases of suspected mitochondrial muscle disease. (Reproduced with permission from Taylor RW, Schaefer AM, Baron MJ, et al: the diagnosis of mitochondrial muscle disease. Neuromuscul Disord. 2004;14(4):237–245.)
SPECIFIC MITOCHONDRIAL DISORDERSMyoclonic epilepsy and ragged red fibers (MERRF) is characterized by myoclonus, generalized seizures (myoclonic and tonic–clonic), ataxia, dementia, sensorineural hearing loss, optic atrophy, and progressive muscular weakness developing in childhood or adult life.1–3,22–31 The clinical spectrum is variable, which may be a reflection of the percentage of abnormal mitochondria that segregate into the respective tissues. Age of onset, spectrum and severity of involvement, and the course can vary, even within families. Muscle weakness and atrophy can be generalized, but there is a predilection for involvement of proximal arm and leg muscles. In addition, a generalized sensorimotor polyneuropathy and pes cavus deformities may be appreciated. The myoclonus is stimulus sensitive, but can be present at rest. The seizures may be photosensitive. Patients are often misdiagnosed as having juvenile myoclonic epilepsy,32 until other signs or symptoms (e.g., weakness, ataxia) manifest. Unlike KSS and PEO, individuals with MERRF do not usually ptosis, ophthalmoparesis, and pigmentary retinopathy. However, cardiomyopathy with conduction block or heart failure may also be seen in MERRF, particularly those cases presenting early.28 MERRF can also be complicated by ventilatory muscle weakness and associated with life-threatening hypoventilation in the setting of surgery, sedation, or intercurrent infection.28,33 Some patients also manifest with multiple symmetric lipomatosis.34,35
Serum CK can be normal or mildly elevated. Serum lactate can be normal or elevated as well. Generalized slowing of the background activity and bursts of spikes and slow waves may be apparent on EEG. MRI or CT scan of the brain often reveals cerebral and cerebellar atrophy. NCS may demonstrate reduced decreased amplitudes of sensory nerve action potentials consistent with a superimposed axonopathy in some patients.23,36,37 EMG is usually normal, although early recruitment of small MUAPs might be evident in weak muscles.
Muscle histopathology is abnormal as noted previously. Many ragged red fibers and COX-negative fibers are evident as well as fibers with increased SDH staining. Neuronal loss and gliosis of the dentate nuclei, globus pallidus, red nuclei, substantia nigra, inferior olivary nuclei, optic nerves, and cerebellar cortex are apparent on autopsy.24 In addition, demyelination and gliosis are evident in the corticospinal and spinothalamic tracts, and posterior columns.
There is non-Mendelian maternal inheritance of MERRF. Approximately 80% of MERRF causes are caused by a point mutation at nucleotide position 8344 of the mitochondrial genome that results in an A to G transition in the tRNALys gene (MTTK).30,38–40 Of note, there is clinical heterogeneity with this specific mutation as patients can present with PEO, Leigh syndrome, or multiple symmetric lipomatosis.24,35 MERRF has also been described with mutations at other locations in the tRNALys gene (positions 8356 and 8366) and with mutations in the tRNALeu (MTTL1) that is most commonly mutated in MELAS. Other tRNA mutations associated with MERRF include tRNAHis (MTTH), tRNAPhe (MTTF), and tRNASer (MTTS1). In addition, an MERRF clinical phenotype can also be found in patients with multiple mtDNA deletions caused by mutations in the polymerase gamma 1 gene (POLG1) and in ND5 (MTND5). Mutations can be demonstrated by polymerase chain reaction of mtDNA in leukocytes or muscle specimens, but the frequency of abnormal mtDNA is greater in muscle.
As described previously, the mitochondrial tRNA gene mutations impair the translation of mitochondrial-DNA–encoded respiratory chain proteins. Assays of mitochondrial enzyme activity in biopsied muscle tissue reveals diminished activity of complex I and IV. At least 90% of the mitochondria must harbor mutations in order for clinical abnormalities to appear.34
There is no specific therapy for MERRF other than treating the myoclonus (e.g., clonazepam) and the seizures with antiepileptic medications. A slight benefit was reported in a few patients with MERRF treated with creatine monohydrate (5–10 g/day).41,42 Special care must be taken as patients with mitochondrial myopathies can develop marked alveolar hypoventilation in response to sedating medications and anesthetic agents.43,44
MELAS is characterized by muscle weakness, high lactate levels in the serum or CSF, and stroke-like episodes.1–3,44–48 Onset occurs in the first year of life in fewer than 10% with 60–80% developing symptoms and signs of the illness by the age of 15 years.44,47 Rarely, MELAS can present as late as the eighth decade.45 Most affected individuals have recurrent stroke-like episodes manifesting as migraine-type headaches with nausea and vomiting, hemiparesis, hemianopsia, or cortical blindness. These stroke-like attacks may be provoked by exercise or intercurrent infection. Progressive dementia may ensue. Most patients exhibit proximal muscle weakness and complain of easy fatigue and myalgias with exercise.
As with other mitochondrial disorders, many patients with MELAS are short-statured. Some affected individuals develop myoclonus, seizures, or ataxia and thus overlap clinically with MERRF. Ptosis, ophthalmoparesis, pigmentary retinopathy, and/or cardiomyopathy occur in less than 10% of patients. Rare individuals with the most common 3243 mutation in the MTTL1 gene manifest with only diabetes mellitus and/or deafness.
Serum CK can be normal or elevated. Lactate levels are elevated in the serum and CSF in the majority of patients. EEG may demonstrate epileptiform activity. NCS are normal, but EMG may reveal early recruitment of myopathic-appearing MUAPs.
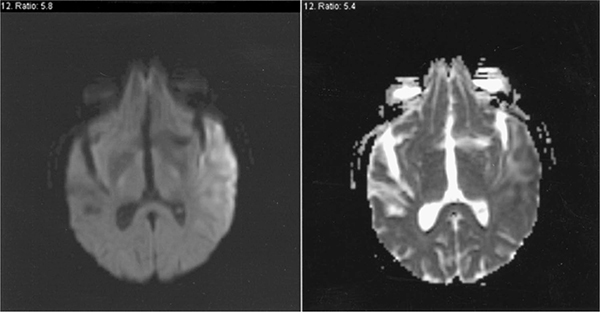
MRI scans of the brain reveal cortical atrophy and high T(2) signal and FLAIR abnormalities in the cerebral cortex, basal ganglia, and thalamus (Fig. 30-7). The apparent diffusion coefficient (ADC) of the lesions may be increased or decreased.49 MRS of acute cortical lesions reveal severely elevated lactate levels and reduced concentrations of N-acetylaspartyl compounds, glutamate, and myo-inositol.19 In addition, MRS of skeletal muscle may demonstrate a reduced phosphocreatine level, elevated concentrations of inorganic phosphate and free adenosine 5′-diphosphate, and an abnormally low phosphorylation potential.19
Figure 30-7. MRI in MELAS. The MRI brain imaging, during a stroke-like episode in a 61-year-old patient with genetically proven MELAS, shows a DWI high-intensity lesion in the left temporal lobe in the first image and absence of corresponding hypointensity on the corresponding ADC image. (Reproduced with permission from Walsh RJ. Metabolic Myopathies. Continuum. 2006;12(3):76–120.)
Muscle biopsies are indistinguishable from other mitochondrial myopathies as described previously. There are many ragged red fibers that have variable COX staining, ranging from increased reactivity to absent staining. This variability in COX staining is more prominent than that seen in MERRF. The COX-negative fibers intensely stain with SDH. Arterioles are also strongly SDH-reactive and an increased number of mitochondria are evident in the muscular walls of small blood vessels. Mitochondrial enzyme analysis of muscle tissue reveals reduced activities of complexes I, III, IV, and V.
MELAS is inherited maternally in a non-Mendelian pattern. Over 70% of cases are caused by a mtDNA mutation, an A to G substitution, at nucleotide position 3243 in the gene (MTTL1) encoding for tRNALeu.44 There is genetic heterogeneity of MELAS as mutations have also been identified at positions 3252, 3260, 3271, 3291 in the tRNALeu gene as well as in the genes for tRNAVal (MTTV), tRNALys (MTTK), tRNAPhe (MTTF), tRNASer (MTTS1), the dehydrogenase-ubiquinone oxidoreductase (ND) subunits of complex I: ND1 (MTND1), ND4 (MTND4), ND5 (MTND5), and cytochrome b of complex III (MTCYB).50 In addition there is phenotypic heterogeneity even within different family members who carry the common 3243 mutation within the tRNALeu gene. Abnormal mitochondria in cerebral blood vessels or the neurons both may be responsible for the stroke-like episodes secondary to impaired energy production in metabolically active regions of the brain.
No specific medical therapy is available other than treatment for seizures and myoclonus. Coenzyme Q does not appear to be of any significant benefit. A small study reported some improvement with dichloroacetate (DCA),51 but a double-blind, placebo-controlled, randomized, 3-year crossover trial of DCA (25 mg/kg/day) in 30 patients demonstrated no efficacy with peripheral nerve toxicity.52 Creatine monohydrate (5–10 g/day) modestly improved strength in a few patients with MELAS,41,42,53 but again, blinded controlled studies are lacking.
KSS is characterized by the clinical triad of PEO, pigmentary retinopathy, and cardiomyopathy with onset usually before the age of 20 years.1–3,54–60 Other clinical features include short stature, proximal muscle weakness, sensorineural hearing loss, dementia, ataxia, depressed ventilatory drive, and multiple endocrinopathies (e.g., diabetes mellitus, hypothyroidism, hypoparathyroidism, delayed secondary sexual characteristics). Affected individuals are very sensitive to sedatives and anesthetic agents can provoke ventilatory failure.61,62
Serum CK level is typically normal; however, lactate and pyruvate levels may be elevated. CSF protein is usually increased. The EKG often reveals conduction defects. NCS are usually normal, although diminished amplitudes suggestive of an axonal sensory or sensorimotor polyneuropathy may be seen. EMG usually demonstrates normal insertional and spontaneous activity, but may reveal early recruitment of small polyphasic MUAPs in weak muscles.
Muscle biopsies demonstrate ragged red fibers; however, unlike MERRF and MELAS, there is little variability of COX staining with most of the ragged red fibers lacking COX reactivity.44,63 The number of ragged red fibers and COX-negative fibers correlate with the percentage of mitochondria harboring large deletions. Autopsy may reveal spongy degeneration of the cerebral white matter.
Single large mtDNA deletions (ranging from 1.3 to 8.8 kB) can be demonstrated in most patients with KSS.56,58,60,63 As many as 43% of patients have a characteristic 4.9-kB deletion, suggesting there may be “hot spots” in the mitochondrial genome for these large deletions. One is more likely to find mtDNA mutations in muscle tissue than in peripheral white blood cells with the percentage of affected mitochondrial genomes in muscle biopsies ranging from 20% to 90%.58 These large deletions most likely arise during oogenesis.2 Mitochondrial disorders with single large deletions need to be differentiated from disorders with multiple deletions—see later). The large deletions usually involve several tRNA genes, thus impairing the adequate translation of mtDNA-encoded proteins. The single deletion mutations are usually sporadic in nature, although rare cases with familial occurrences have been reported.28,56
The clinical phenotype of individuals harboring single large mtDNA is again heterogeneous. Some patients develop migraines and stroke-like episodes with or without PEO, some have PEO with or without limb weakness, retinopathy, or deafness, others have an encephalopathy without PEO (including Leigh syndrome), while rare patients manifest only with diabetes mellitus, deafness, or Pearson syndrome.
Some patients with KSS treated with creatine supplementation (0.08–0.35 g/kg body weight/day) have improved exercise capacity measured with bicycle ergometry.53 Patients with cardiac conduction defects may require pacemaker insertion. Ptosis may be treated with eyelid surgery provided there is sufficient facial strength to allow full eye closure. Tarsorraphy should be undertaken judiciously, however, as there is risk for corneal injury due to exposure/trauma if the eyelids cannot completely close, and risk of ptosis recurrence. Patients and their physicians need to be made aware of the extreme sensitivity to CNS depressants and potential for decreased respiratory drive.61,62
Patients with PEO have ptosis and ophthalmoparesis (Fig. 30-8) with or without extremity weakness, but they lack pigmentary retinopathy, cardiac conduction defects, or other systemic manifestations (e.g., endocrinopathies).1–3,56–58,63 Some cases that are sporadic in nature and associated with single large deletions of mtDNA probably represent a partial clinical expression of KSS. There are autosomal dominant and recessive forms as well as maternally (mitochondrial) inherited forms of PEO.
Figure 30-8. Progressive external ophthalmoplegia. A patient has ptosis and inability to move the eyes.
Serum CK, serum lactate, and CSF lactate can be normal or elevated. CSF protein may be increased. In contrast to classic KSS, the EKG does not demonstrate cardiac conduction defects. NCS are normal. EMG is also usually normal, although myopathic MUAPs may be found in weak extremity muscles.
Muscle pathology is indistinguishable from KSS.
Approximately 40–70% of patients with PEO have single large mtDNA deletions similar to KSS.1–3,56–58,60,63 Such cases are generally sporadic in nature and PEO is not passed on to subsequent generations. Point mutations have been identified within various mitochondrial tRNAs (Leu, Ile, Asn, Trp) genes in several kinships with maternal inheritance of PEO.64
Autosomal dominant PEO is usually associated with multiple mtDNA deletions and is genetically heterogeneous (Table 30-2).1–3,55,57,64–68 Several genes have been identified in autosomal dominant PEO (PEOA): PEOA1 due to mutations in the polymerase gamma 1 gene (POLG1); PEOA2 is caused by mutations in the ANT1 gene that encodes for adenine nucleotide translocator (ANT); and PEOA3 caused by mutation in the twinkle gene (C10orf2). ANT is the most abundant mitochondrial protein and is responsible for transporting adenosine triphosphate across the inner mitochondrial membrane, while twinkle and POLG are involved in mtDNA replication. Less common, PEO has been associated with mutations in POLG2, TK2, OPA1, DGOUK, and RRM2B.
Autosomal recessive progressive external ophthalmoplegia (PEOB) is also most commonly caused by mutations in the POLG1 gene. Mutations in this gene also have been implicated in Alper syndrome, which causes a clinical triad of psychomotor retardation, intractable epilepsy, and liver failure in infants and young children. Less common mutations have been identified in the TK2, DGOUK, RRM2B, MPV17, DNA2, and SPG7 genes.1–3,60,67,69
Surgery to correct ptosis may help. As with other mitochondrial disorders, individuals with PEO can develop hypoventilation in with infections and in response to sedatives or anesthetic agents.61,62 Improvement in ventilatory muscle weakness has been reported with vitamin E treatment.61,62
Only a few patients have been reported with this rare syndrome characterized by childhood-onset PEO, facial and proximal limb weakness, and severe cardiomyopathy.71,72 Affected individuals frequently complained of chest pain, dyspnea, and palpitations secondary to severe dilated cardiomyopathy and some require heart transplantation. The severe cardiomyopathy and autosomal recessive inheritance pattern help to distinguish this myopathy from autosomal dominant and maternally inherited PEO. The reported patients had no evidence of pigmentary retinopathy, hearing loss, ataxia, or peripheral neuropathy, although deep tendon reflexes were reduced.
Serum CK levels are mildly elevated. Serum lactate can be normal at rest but increases excessively during exercise. The EKG reveals cardiac conduction defects, while echocardiogram usually demonstrates dilated ventricles and a reduced ejection fraction. NCS are normal but EMG may reveal myopathic-appearing MUAPs.
Muscle biopsies reveal ragged red fibers which are strongly reactive to SDH but COX-negative.71,72 However, there are also many COX-negative fibers which do not co-localize to ragged red and SDH-positive fibers. Biochemical assay have demonstrated decreased activity of respiratory chain enzymes containing mtDNA-encoded subunits, sparing the entirely nuclear-encoded SDH and citrate synthetase.
Multiple mtDNA deletions may be found and the genetic defect is suspected to lie in nuclear genes involved in regulating the mitochondrial genome.
Many patients will die from the severe cardiomyopathy within the first two decades of life, unless they receive cardiac transplantation.
The mtDNA depletion syndromes (MDS) are a heterogeneous group of autosomal recessive disorders characterized by decreased mtDNA copy number in affected tissues (Table 30-2).1–7,61,73–98 The MDS are associated with a severe myopathy that usually presents in infancy or early childhood, but milder cases manifesting in adult life have been reported. There is a predilection for proximal muscle involvement but ptosis and ophthalmoplegia are also common. Some also have a superimposed polyneuropathy. Muscle stretch reflexes are diminished or absent. In addition, some affected individuals can develop a cardiomyopathy, De Toni–Fanconi–Debré syndrome (a renal tubular defect), seizures, or liver failure. When the onset is in infancy, the muscle weakness is typically severe and progressive leading to feeding difficulties, respiratory failure, and death usually within the first year of life. Some forms of PEO (previously discussed) and MNGIE, mitochondria recessive ataxic syndrome (MIRAS), optic atrophy 1 (OA1), and Navajo neurohepatopathy (discussed in subsequent sections) are other mitochondrial disorders associated with MDS.
Serum CK can be normal or elevated as can serum lactate levels. The associated renal tubular defect results in glycosuria, proteinuria, and aminoaciduria. Cerebral atrophy and patchy areas of hypomyelination of subcortical white matter may be apparent on MRI scans.80 Unlike most other mitochondrial myopathies, EMG may demonstrate numerous fibrillation potentials and positive sharp waves in those myopathies with mtDNA depletion. Motor units can have a mixed myopathic and neuropathic appearance. NCS can be normal or reveal features of an axonal or demyelinating sensorimotor neuropathy.
Muscle biopsies demonstrate many COX-negative fibers, although ragged red fibers may not be apparent.74,80 EM shows enlarged mitochondria, some with concentric or whorled cristae, dense bodies, or paracrystalline inclusions. Biochemical assay of COX activity in skeletal muscle tissue of affected patients is greatly diminished or absent.
MDS is associated with mutations in nuclear genes that control and maintain mtDNA.1–7 At least half the cases are sporadic in nature but some are inherited in an autosomal recessive fashion. A depletion of mtDNA was first reported by Moraes et al. in 199181 and subsequently confirmed by others.74,77–98 The quantity of mtDNA indirectly correlates with the clinical severity of the disorder. As much as a 99% reduction in mtDNA is present in the fatal infantile myopathy form of the disease, while the more benign myopathy has been demonstrated to have lesser depletions (36–88%) of mtDNA. Mitochondrial depletion myopathy is usually caused by mutations in the gene that encodes for thymidine kinase 2 (TK2),77,86–97 but rare cases are caused by mutations in the deoxyguanosine kinase gene (DGOUK).67,97,98
Mitochondrial depletion can be seen by mutations in other genes that are associated with other clinical syndromes: thymidine phosphorylase (TYMP), POLG1 and 2, twinkle (C10orf2), mitochondrial inner membrane protein 17 (MPV17), ribonucleotidase reductase (RRM2B), optic atrophy 1 (OPA1), succinate-CoA ligase alpha subunit (SUCLG1), and succinate-CoA ligase ADP-forming, beta subunit (SUCLA2). Disorders associated with mutations in these genes are discussed in other sections.
No specific medical therapy has been demonstrated to be effective.
MNGIE, also referred to as POLIP syndrome, (Polyneuropathy, Ophthalmoplegia, Leukoencephalopathy, and Intestinal Pseudo-obstruction) is an autosomal recessive mitochondrial disorder.1–7,55,99–101 As the acronyms imply, the disorder is associated with a sensorimotor polyneuropathy, leukoencephalopathy on MRI of the brain, ragged red fibers on muscle biopsy, and chronic intestinal pseudo-obstruction. The disorder usually manifests before the age of 20 years (mean 13.9 years, range 2.5–32 years).65 The course is progressive with severe disability or death by the third or fourth decade of life. The earliest symptoms are often caused by gastrointestinal dysmotility (i.e., dyspepsia, bloating, eructations, cramps, intolerance of large meals, and episodic nausea, vomiting, and diarrhea). Affected individuals gradually develop distal greater than proximal muscle weakness and atrophy, a stocking–glove distribution of sensory loss, and reduced muscle stretch reflexes throughout. Most patients have ptosis and extraocular muscle weakness. Despite the leukoencephalopathy apparent on MRI and on autopsies, most affected patients have little in the way of CNS symptoms or signs. However, rare patients have mental retardation. Other clinical features include pigmentary retinopathy, sensorineural hearing loss, facial weakness, hoarseness, or dysarthria.
Serum CK can be normal or mildly elevated. Lactate, pyruvate, and CSF protein levels are typically elevated. Thymidine phosphorylase activity is decreased in leukocytes and platelets, thymidine levels are increased in the plasma, and deoxyuridine is increased in serum and plasma in those cases caused by mutation in the TYMP gene that encodes for thymidine phosphorylase.7
Leukoencephalopathy of the cerebral and cerebellar white matter is apparent on MRI scans. Radiological studies also demonstrate dilatation and dysmotility of the esophagus, stomach, and small intestine. EKG has shown conduction defects in some patients, although they remained asymptomatic from a cardiac standpoint. Motor and sensory nerve conduction velocities may be slow to within the demyelinating range, while F-wave latencies are usually markedly prolonged.65,101,102 Other cases are more suggestive of a primary axonopathy with reduced SNAP and CMAP amplitudes. EMG may reveal fibrillation potentials and positive sharp waves.65 Recruitment of MUAPs can be decreased, suggestive of denervation in distal muscles. However, quantitative electromyography of proximal muscles may reveal small duration MUAPs suggesting a superimposed myopathic process. The generalized weakness coupled with the demyelinating polyneuropathy can lead to misdiagnosis as chronic inflammatory demyelinating polyneuropathy.101
Muscle biopsies may demonstrate ragged red fibers, ragged blue fibers with NADH and SDH staining, and COX-negative fibers.65 Neurogenic atrophy may be apparent biopsies of distal muscles. Nerve biopsies have shown loss of myelinated nerve fibers, demyelination/remyelination, and rare onion-bulb formation, in addition to features of axonal degeneration. Abnormal mitochondria with paracrystalline inclusions occur in both muscle fibers and Schwann cells. Diminished COX and other respiratory complex activities can be demonstrated on enzymatic assays of muscle tissue.65
Autopsies have revealed widespread endoneurial fibrosis and demyelination in the peripheral nervous system and poorly defined white matter changes in the cerebral and cerebellar white matter.101 Cranial nerves and spinal roots are less severely involved. Neurons of the brainstem and spinal cord appeared relatively intact. Interestingly, a loss of neurons and fibrotic changes of the autonomic ganglia and of the celiac and myenteric plexuses has been noted, which likely explains the associated gastrointestinal dysmotility.101
Multiple mtDNA deletions similar to those found in some cases of autosomal dominant PEO have been demonstrated in some patients with autosomal recessive MNGIE.1–7,64,100,103 Some cases of MNGIE are caused by mutations in the thymidine phosphorylase (TYPM or ECGG1) gene located on chromosome 22q13.32-qter.100,103,104 Thymidine phosphorylase converts thymidine to 2-deoxy D-ribose 1-phosphate and may regulate thymidine availability for DNA synthesis. Interestingly, thymidine phosphorylase is not normally expressed in muscle tissue, so how it leads to multiple mtDNA deletions is unclear. It may lead to a reduction in the nucleotide pool within the mitochondria. Rare cases of MNGIE have been shown to be caused by mutations in POLG1, RRM2B, and the tRNALys (MTTK) gene.7,105–107 Neuropathy is demyelinating in patients carrying TYPM or RRM2B mutations, whereas patients with POLG1 mutations mainly have axonal electrodiagnostic (EDX) features.7
No specific medical therapy is available. PEG tube or parenteral feedings for nutritional support are required in the majority of cases. Ankle foot orthotics may be beneficial in patients with foot drop.
SANDO (sensory ataxic neuropathy, dysarthria, ophthalmoplegia) is a disorder that may occur sporadically or in either a dominantly or recessively inherited fashion.5–7,108–112 The onset of sensory ataxia is typically in early adulthood. Not all patients have each element of the SANDO syndrome. In addition to the phenotypic features described in the acronym, cerebellar ataxia, facial weakness, mild proximal weakness, exercise intolerance, Parkinsonism, myoclonus epilepsy, and hepatic failure have been described.108,110
NCS may reveal amplitude reduction and/or slowing of sensory SNAPs.5–7,108–112
Muscle biopsies may reveal ragged red and COX-negative muscle fibers. Sural nerve biopsy is not routinely performed but has revealed loss of large and small myelinated fibers and demonstrated onion-bulb formations.5,6 Autopsy studies have shown axonal and dorsal column degeneration, suggesting involvement of dorsal root ganglia.7
SANDO is usually autosomal recessively inherited and caused by mutations in POLG1 that results in multiple mtDNA deletions and mtDNA depletion as mentioned previously.109,111,112 SANDO can also result from mutations in C10orf2, which encodes twinkle helicase that also is involved in mtDNA replication.113 Mutations in this gene can cause an autosomal dominant or recessive syndrome.
No specific medical therapy is available.
MIRAS is very similar to SANDO and typically presents as a juvenile or adult onset ataxic neuropathy. Associated phenotypic features may include CPEO, dysarthria, seizures, nystagmus, cognitive impairment, involuntary movements, and psychiatric symptoms.114–119 Like SANDO, it is usually caused by mutations in POLG1.
NARP is associated with a childhood or adult onset axonal neuropathy, cerebellar ataxia, and retinitis pigmentosa. There may be proximal or distal weakness as well as pes cavus on examination.5–7,120–123
NCS reveal features suggestive of an axonal sensory neuropathy.
Muscle biopsy does not generally reveal ragged red or COX-negative fibers as the causal gene (see below) is involved only in the last step of ATP production. Nerve biopsy reveals loss of myelinated nerve fibers.5,6
NARP is caused by mutations in mitochondrial-encoded ATPase 6 (MTATP6), which is also a cause of Leigh syndrome.
No specific medical therapy is available.
This manifests in infancy or early childhood with failure to thrive, diarrhea, vomiting, and signs of liver failure, including recurrent metabolic acidosis. In addition, infants have generalized hypotonia and weakness. Other neurological signs included microcephaly, seizures, ataxia, and dystonia. Those individuals who survive typically develop a severe sensorimotor polyneuropathy.5–7,124–127 Loss of sensation leads to acromutilation and corneal ulcerations.
Serum transaminases and bilirubin are elevated while albumin is low. There may be evidence of a coagulopathy. Aminoaciduria is event on urine screen. MRI scans reveal increase signal in cortex and subcortical white matter as well as the dentate nuclei and cerebellar white matter.5,6 NCS reveal slow of conduction velocities suggestive of a demyelinating sensorimotor polyneuropathy.5–7,124,125
Nerve biopsies have demonstrated loss of myelinated and unmyelinated nerve fibers.124,127
This is caused by homozygous mutations (Arg50Trp) in the mitochondrial inner membrane protein gene (MPV17).124,125
No specific medical therapy is available.
OA1 usually manifests with progressive optic atrophy and is inherited in an autosomal dominant pattern.7,128–132 It is allelic to what has also been called HMSN VI (CMT with optic atrophy). Approximately 20% of affective individuals develop neurological abnormalities, such as progressive external ophthalmoplegia, peripheral neuropathy, proximal myopathy, and hearing loss. The neuropathy is usually mild, and mostly sensory, but mild distal muscle atrophy and weakness along with pes cavus may be seen. Some patients have a sensory or cerebellar ataxia.
NCS are most suggestive of an axonal sensory greater than motor polyneuropathy.129,132
Muscle biopsy often shows multiple mtDNA deletions and sometimes depletion, ragged red fibers, and COX-negative fibers. Literature on nerve biopsy findings are lacking.
This is caused my mutations in the OPA1 gene, encoding a dynamin-related GTPase, which is important in mitochondrial fusion, fission, and cristae organization.5,6,131,132
There is no specific medical treatment for OPA1.
Leigh syndrome, or subacute necrotizing encephalomyopathy, usually presents in infancy or early childhood, but can rarely develop in adult life.133,134 Affected individuals can manifest with recurrent vomiting, psychomotor retardation, hypotonia, generalized weakness and atrophy, ptosis, ophthalmoplegia, poor suck, respiratory failure, nystagmus, optic atrophy, hearing loss, involuntary movements, seizures, spasticity, ataxia, and peripheral neuropathy. The rate of progression varies, but the disorder is generally fatal.
Serum and CSF lactate levels are elevated as can be the lactate:pyruvate ratio. The syndrome is biochemically heterogeneous. Defects in activity of the pyruvate dehydrogenase (PDH), pyruvate decarboxylase (PDC), COX, and complex I have been described in some patients with Leigh syndrome. MRI demonstrates symmetric lesions in the thalamus, brainstem, cerebellum, and spinal cord reflecting the underlying pathology.
Muscle biopsy can demonstrate reduced or absent COX staining (mitochondrial and nuclear-encoded COX subunits) of muscle fibers, although ragged red fibers are usually not seen. Unlike fatal infantile myopathy, COX staining is also deficient in muscle spindles and in the smooth muscle of intramuscular blood vessels. Autopsy studies of the brain and spinal cord demonstrate symmetric cystic necrosis, spongioform changes, demyelination, and vascular proliferation in the thalamus, basal ganglia, brainstem, cerebellar white matter, dentate nuclei, and posterior columns.
Leigh syndrome is genetically heterogeneous. Mutations have been identified in both nuclear- and mitochondrial-encoded genes. These genes are all involved in energy metabolism, including the generation of ATP, components of the PDH complex and mitochondrial respiratory chain complexes I, II, III, IV, and V, which are involved in oxidative phosphorylation.
Complex I comprises at least 45 subunits, of which seven are encoded by the mitochondrial genome (ND1–6, ND4 L) and the others are encoded by nuclear genes. Multiple complex I genes have been implicated in Leigh syndrome including mitochondrial-encoded MTND3, MTND5, and MTND6, and nuclear-encoded NDUFV1, NDUFS1, NDUFS3, NDUFS4, NDUFS7, and NDUFS8 genes.135,136
From complex II, a mutation has been found in the nDNA gene flavoprotein subunit A (SDHA).137 In complex III, a mutation has been found in the nDNA gene BCS1 L, which is involved in the assembly of complex III.
Complex IV mutated genes include mitochondrial-encoded cytochrome c oxidase subunit 3 (MTCO3) and nuclear-encoded cytochrome c oxidase assembly proteins 10 (COX10), and 15 (COX15). Two other nuclear-encoded genes with mutations are: (1) synthesis of cytochrome c oxidase 2 (SCO2), and (2) surfeit 1(SURF1). Surfeit1 is involved in the assembly of complex IV.138 A mutation has also been found in a complex V gene, the mitochondrial-encoded ATPase 6 (MTATP6).139 Of note, mutations in this gene are also responsible for the mitochondrial disorder termed NARP (neuropathy, ataxia, and retinitis pigmentosa) as previously discussed. When the proportion of mutated mtDNA is high (>90%), Leigh syndrome occurs; but NARP develops when the burden of mtDNA mutations is lower.
Mutations in multiple genes encoding mitochondrial tRNA proteins have also been identified in patients with maternally inherited Leigh syndrome: TRNAVal (MTTV), tRNALys (MTTK), tRNATrp (MTTW), and tRNALeu (MTTL1).133,140 Single large deletions of mtDNA have also been demonstrated.141
Leigh syndrome may also be caused by mutations in components of the PDH complex. The gene DLD encodes for dihydrolipoamide dehydrogenase, which is a component not only of the PDH complex, but also of the alpha-ketoglutarate dehydrogenase complex, and the branched-chain alpha-keto acid dehydrogenase complex. Compound heterozygous mutations in DLD have been implicated in Leigh syndrome. X-linked Leigh syndrome is caused by mutation in the gene encoding the E1-alpha subunit of the PDH complex (PDHA1).142–144
The French-Canadian (or Saguenay-Lac Saint Jean) type of Leigh syndrome with COX deficiency (LSFC) is caused by mutation in the leucine-rich PPR motif-containing protein gene (LRPPRC). This gene encodes for an mRNA-binding protein involved in the processing and trafficking of mtDNA-encoded transcripts, but how this causes COX deficiency is not yet clear.
There is no specific medical therapy available.
This disorder has been described in only a few patients.145–146 A sister and brother presented in the second decade of life with exertional muscle pain and fatigue, myoglobinuria, and mild proximal weakness.145 Their father of this pair was asymptomatic but had an elevated serum CK. Congenital weakness, hypotonia, delayed motor milestones, and mental retardation has also been reported.146
Serum CK levels are mild to moderately elevated and serum lactate levels are normal. A decreased selenium level has been described in one patient.146 NCS are normal, but EMG may reveal myopathic MUAPs.
The most striking histologic feature, for which this disorder is named, is focal depletion of mitochondria in the center of the sarcoplasm in type 2 muscle fibers. At the periphery of muscle fibers, the mitochondria are enlarged. Scattered degenerating and regenerating fibers can be appreciated.
This myopathy is presumably autosomal dominant. No molecular or quantitative defects of mtDNA have been reported in patients with this syndrome. Similar histological findings have been demonstrated in patients with myopathy felt to be related to selenium deficiency.147
There is no specific medical therapy. A trial of selenium replacement should be considered in patients who are deficient in selenium.
Some patients with mitochondrial myopathy manifest only with exercise-induced myalgias beginning in infancy or early adulthood.1–3,148–159 They are typically short-statured and have generalized reduction in muscle bulk. Muscle strength may be normal or there can be mild proximal weakness. Recurrent episodes of myoglobinuria can also occur and be provoked by exercise and alcohol intake. However, provocative factors often are not present. We have seen patients with progressive deafness as well.
Serum lactate and pyruvate may be normal or slightly at rest but become significantly elevated with aerobic exercise. Serum CK can be normal or mildly elevated between episodes of myoglobinuria. EMG and NCS are typically normal.
Muscle biopsies may reveal scattered ragged red fibers, increased SDH and NADH stains (ragged blue fibers), as well as COX-negative fibers. However, COX stain can be normal, particularly in patients with mutations in MTCO1, MTCO2, MTCO3, ND4, and MTCYB (see below). Decreased COX activity has been found on enzyme analysis of muscle tissue in some,148 but not all cases.149 Abnormal mitochondria with paracrystalline inclusions can be detected on EM.
This is a genetically heterogeneous group of disorders. Multiple mtDNA deletions were demonstrated in two brothers with presumed autosomal recessive inheritance.138 In addition, point mutations in tRNAPhe have been found in kinships with and without recurrent myoglobinuria. Mutations within the gene encoding for subunits of cytochrome c oxidase (MTCO1, MTCO2, MTCO3) have been reported in sporadic cases.2,142,150,151 Other cases of exercise intolerance and recurrent myoglobinuria have be ascribed to mutations in the mtDNA genes encoding for tRNAGly143 subunit 4 of NADH dehydrogenase (ND4),154 and cytochrome b (MTCYB).76,77,155,156 Mutations in ND4, may also produce Leber hereditary optic neuropathy157 or Wolfram syndrome.158 In addition, mutations in the gene encoding the iron-sulphur cluster assembly protein (ISCU) have been associated with exercise intolerance and myoglobinuria and muscle biopsies demonstrating succinate dehydrogenase deficiency and accumulation of iron in muscle fibers.159
Attenuation of free-radical production and paracrystalline inclusions in muscle biopsies has been reported following a 5-week trial of creatine supplementation in a patient with a novel cytochrome b (MTCYB) mutation.21 However, the patient did not feel any subjective improvement and there was no effect on his maximal oxygen consumption. There is no specific medical therapy other than treatment of myoglobinuria and avoidance of strenuous activity and alcohol.
There are at least three proteins involved in mitochondrial dynamics that cause forms of CMT: CMT2 A caused by mutations in the mitofusin 2 gene (MFN2). CMT2 K and CMT4 A are associated with mutations in ganglioside-induced differentiation associated-protein 1 (GDAP1), and OPA1 mutations are associated with CMT associated with optic atrophy as previously discussed (Fig. 30-3).5–7 CMT caused by mutations involving MFN2 and GDAP1 are discussed in more detail in Chapter 11 (Charcot–Marie Tooth Disease and Related Disorders). MFN and GDAP1 are involved in the fusion and fission of mitochondria which are essential in controlling the shape, size, number, and transport of mitochondria within cells. Dynamin-like GTPases located in the outer membrane (e.g., MFN2) and inner membrane (e.g., OPA1) control mitochondrial fusion. MFN2 helps tether mitochondria during fusion, while OPA1 is important for fusion of the inner membrane and formation of cristae. GDAP1, located in the outer membrane, is important in mitochondrial fission.
SUMMARYThere is a wide range of phenotypic and genotypic variability in patients with mitochondrial disorders. Due to high energy requirements, many of these disorders are associated with disorders of peripheral nerve and/or muscle. This phenotypic and genotypic heterogeneity makes definitive diagnosis (i.e., identifying specific genetic mutation) an often long and expensive process. Although there is a greater understanding regarding the molecular pathogenesis of the different forms of mitochondrial encephalomyopathies, these advances have not as yet led to easy diagnosis in many cases or effective medical treatments, other than supportive measures.
1. DiMauro S, Schon EA, Carelli V, Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. 2013;9(8):429–444.
2. Milone M, Wong LJ. Diagnosis of mitochondrial myopathies. Mol Genet Metab. 2013;110(1–2):35–41.
3. Pfeffer G, Chinnery PF. Diagnosis and treatment of mitochondrial myopathies. Ann Med. 2013;45(1):4–16.
4. Tucker EJ, Compton AG, Thorburn DR. Recent advances in the genetics of mitochondrial encephalopathies. Curr Neurol Neurosci Rep. 2010;10(4):277–285.
5. Finsterer J, Ahting U. Mitochondrial depletion syndromes in children and adults. Can J Neurol Sci. 2013;40:635–644.
6. Finsterer J. Inherited mitochondrial neuropathies. J Neurol Sci. 2011;304(1–2):9–16.
7. Pareyson D, Piscosquito G, Moroni I, Salsano E, Zeviani M. Peripheral neuropathy in mitochondrial disorders. Lancet Neurol. 2013;12(10):1011–1024.
8. Schmiedel J, Jackson S, Schafer J, Reichmann H. Mitochondrial cytopathies. J Neurol. 2003;250(3):267–277.
9. Taylor RW, Schaefer AM, Baron MJ, McFarland R, Turnbull DM. The diagnosis of mitochondrial muscle disease. Neuromuscul Disord. 2004;14:237–245.
10. Vu TH, Hirano M, DiMauro S. Mitochondrial diseases. Neurol Clin. 2002;20(3):809–839.
11. Chinnery PF, Johnson MA, Wardell TM, et al. The epidemiology of pathogenic mitochondrial DNA mutations. Ann Neurol. 2000;48:188–193.
12. Jeppesen TD, Schwartz M, Olsen DB, Vissing J. Oxidative capacity correlates with muscle mutation load in mitochondrial myopathy. Ann Neurol. 2003;54(1):86–92.
13. Taivassalo T, Jensen TD, Kennaway N, DiMauro S, Vissing J, Haller RG. The spectrum of exercise tolerance in mitochondrial myopathies: a study of 40 patients. Brain. 2003;126:413–423.
14. Jeppesen TD, Olsen D, Vissing J. Cycle ergometry is not a sensitive diagnostic test for mitochondrial myopathy. J Neurol. 2003;250(3):293–299.
15. Jensen TD, Kazemi-Esfarjani P, Skomorowska E, Vissing J. A forearm exercise screening test for mitochondrial myopathy. Neurology. 2002;58(10):1533–1538.
16. Taivassalo T, Abbott A, Wyrick P, Haller RG. Venous oxygen levels during aerobic forearm exercise: an index of impaired oxidative metabolism in mitochondrial myopathy. Ann Neurol. 2002;51(1):38–44.
17. Olsen DB, Langkilde AR, Orngreen MC, Rostrup E, Schwartz M, Vissing J. Muscle structural changes in mitochondrial myopathy relate to genotype. J Neurol. 2003;250(11):1328–1334.
18. Laforet P, Wary C, Duteil S, et al. [Exploration of exercise intolerance by 31P NMR spectroscopy of calf muscles coupled with MRI and ergometry]. Rev Neurol. 2003;159(1):56–67.
19. Moller HE, Wiedermann D, Kurlemann G, Hilbich T, Schuierer G. Application of NMR spectroscopy to monitoring MELAS treatment: a case report. Muscle Nerve. 2002;25(4):593–600.
20. Bendahan D, Mattei JP, Kozak-Ribbens G, Cozzone PJ. Non invasive investigation of muscle diseases using 31P magnetic resonance spectroscopy: potential in clinical applications. Rev Neurol. 2002;158(5 pt 1):527–540.
21. Tarnopolsky MA, Simon DK, Roy BD, et al. Attenuation of free radical production and paracrystalline inclusions by creatine supplementation in a patient with a novel cytochrome b mutation. Muscle Nerve. 2004;29:537–547.
22. Blumenthal DT, Shanske S, Schochet SS, et al. Myoclonus epilepsy with ragged red fibers and multiple mtDNA deletions. Neurology. 1998;50:524–525.
23. Fang W, Huang CC, Chu NS, et al. Myoclonic epilepsy with ragged-red fibers (MERRF) syndrome: report of a Chinese family with mitochondrial DNA point mutation in the tRNALys gene. Muscle Nerve. 1994;17:52–57.
24. Fukuhara N. Clinicopathological features of MERRF. Muscle Nerve. 1995;(suppl 3):S90–S94.
25. Fukuhara N. MERRF: a clinicopathological study. Relationships between myoclonus epilepsy and mitochondrial myopathies. Rev Neurol. 1991;147:476–479.
26. Fukuhara N, Tokiguchi S, Shirakawa K, Tsubaki T. Myoclonus epilepsy associated with ragged-red fibers (mitochondrial abnormalities): disease entity or a syndrome? J Neurol Sci. 1980;47:117–133.
27. Lombes A, Mendell JR, Nakase H, et al. Myoclonic epilepsy and ragged red fibers with cytochrome oxidase deficiency: neuropathology, biochemistry, and molecular genetics. Ann Neurol. 1989;26:20–33.
28. Ozawa M, Goto Y, Sakuta R, Tanno Y, Tsuji S, Nonaka I. The 8,344 mutation in mitochondrial DNA: a comparison between the proportion of mutant DNA and clinical pathologic findings. Neuromuscul Disord. 1995;5:483–488.
29. Rosing HS, Hopkins LC, Wallace DC, Epstein CM, Weidenheim K. Maternally inherited mitochondrial myopathy and myoclonic epilepsy. Ann Neurol. 1985;17:228–237.
30. Silvestri G, Ciafoni E, Santorelli FM, et al. Clinical features associated with the A→G transition at nucleotide 8344 of mtDNA (“MERRF mutation”). Neurology. 1993;43:1200–1206.
31. Tsairis P, Engel WK, Kark P. Familial myoclonic epilepsy syndrome associated with skeletal muscle mitochondrial abnormalities [abstract]. Neurology. 1973;23:408.
32. Greenberg DA, Durner M, Keddache M, et al. Reproducibility and complications in gene searches: linkage on chromosome 6, heterogeneity, association, and maternal inheritance in juvenile myoclonic epilepsy. Am J Hum Genet. 2000;66:508–516.
33. Bryne E, Dennet X, Trounce I. Burdon J. Mitochondrial myoneuropathy with respiratory failure and myoclonic epilepsy. J Neurol Sci. 1985;71:273–281.
34. Larsson NG, Tulinius MH, Holme E, Oldfors A. Pathologenetic aspects of the A8344G mutation of mitochondrial DNA associated with MERRF syndrome and multiple symmetric lipomas. Muscle Nerve. 1995;(suppl 3):S102–S106.
35. Muñoz-Málaga A, Bautista J, Salazar JA, et al. Lipomatosis, proximal myopathy, and the mitochondrial 8344 mutation. A lipid storage myopathy? Muscle Nerve. 2000;23:538–542.
36. Mizusawa H, Watanabe M, Kanazawa I, et al. Familial mitochondrial myopathy associated with peripheral neuropathy: partial deficiencies of complex I and complex IV. J Neurol Sci. 1988;86:171–184.
37. Pezeshkpour G, Krarup C, Buchthal F, DiMauro S, Bresolin N, McBurney J. Peripheral neuropathy in mitochondrial disease. J Neurol Sci. 1987;77:285–304.
38. Hammans SR, Sweeny MG, Brockington M, et al. The mitochondrial DNA transfer RNALys A → G(8344) mutation and the syndrome of myoclonic epilepsy with ragged red fibres (MERRF). Relationship of the clinical phenotype to proportion of mutant mitochondrial DNA. Brain. 1993;116: 617–632.
39. Shoffner JM, Lott MT, Lezza AMS, Seibel P, Ballinger SW, Wallace DC. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell. 1990;61:931–937.
40. Yoneda M, Miyatake T, Attardi G. Heteroplasmic mitochondrial tRNALys mutation and its complementation in MERRF patient-derived mitochondrial transformants. Muscle Nerve Suppl. 1995;(suppl 3):S95–S101.
41. Tarnopolsky M, Martin J. Creatine monohydrate increases strength in patients with neuromuscular disease. Neurology. 1999;52:854–857.
42. Tarnopolsky MA, Roy BD, MacDonald JR. A randomized, controlled trial of creatine monohydrate in patients with mitochondrial cytopathies. Muscle Nerve. 1997;20:1502–1509.
43. Feit H, Kirkpatrick J, VanWoert MH, Pandian G. Myoclonus, ataxia, and hypoventilation: response to L-5-hydroxytrptophan. Neurology. 1983;33:109–112.
44. Ciafaloni E, Ricci E, Shanske S, et al. MELAS. Clinical features, biochemistry, and molecular genetics. Ann Neurol. 1992;31: 391–398.
45. Crimmins D, Morris JGL, Walker GL, et al. Mitochondrial encephalomyopathy: variable clinical expression within a single kindred. J Neurol Neurosurg Psychiatry. 1993;56:900–905.
46. Goto YI. Clinical features of MELAS and mitochondrial DNA mutations. Muscle Nerve. 1995;(suppl 3):A107–S112.
47. Goto Y, Horai S, Matsuoka T, et al. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS): correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42:545–550.
48. Pavlakis SG, Phillips PC, DiMauro S, DeVivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode: a distinctive clinical syndrome. Ann Neurol. 1984;16:481–488.
49. Iizuka T, Sakai F, Kan S, Suzuki N. Slowly progressive spread of the stroke-like lesions in MELAS. Neurology. 2003;61(9): 1238–1244.
50. Servidei S. Mitochondrial encephalomyopathies: gene mutation. Neuromuscul Disord. 2000;10:10–15.
51. Saitoh S, Momoi MY, Yamagata T, Mori Y, Imai M. Effects of dochorpacetate in three patients with MELAS. Neurology. 1998;50:531–534.
52. Kaufmann P, Engelstad K, Wei Y, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66(3):324–330.
53. Komura K, Hobbiebrunken E, Wilichowski EK, Hanefeld FA. Effectiveness of creatine monohydrate in mitochondrial encephalomyopathies. Pediatr Neurol. 2003;28(1):53–58.
54. Berenberg RA, Pellock JM, DiMauro S, et al. Lumping or splitting? “Ophthalmoplegia-plus” or Kearns-Sayre syndrome? Ann Neurol. 1977;1:37–54.
55. DiMauro S, Bonilla E, Lombes A, Shanske S, Minneti C, Moraes CT. Mitochondrial encephalomyopathies. Neurol Clin. 1990;8:483–506.
56. Holt IJ, Harding AE, Cooper JM, et al. Mitochondrial myopathies: clinical and biochemical features of 30 patients with major deletions of muscle mitochondrial DNA. Ann Neurol. 1989;26:699–708.
57. Laforêt P, Lombes A, Eymard B, et al. Chronic progressive external ophthalmoplegia with ragged-red fibers: clinical, morphological, and genetic investigations in 43 patients. Neuromuscul Disord. 1995;5:399–413.
58. Moraes CT, DiMauro S, Zeviani M, et al. Mitochondrial deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med. 1989;320:1293–1299.
59. Rowland LP. Progressive external ophthalmoplegia and ocular myopathies. In: Rowland LP, DiMauro S, eds. Handbook of Clinical Neurology. Vol 18(62). Amsterdam: Elsevier Science Publishers BV; 1992:287–329.
60. Zeviani M, Moraes CT, DiMauro S, et al. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology. 1988;38:1339–1346.
61. Barohn RJ, Clanton T, Sahenk Z, Mendell JR. Recurrent respiratory insufficiency and depressed ventilatory drive complicating mitochondrial myopathies. Neurology. 1990;40:103–106.
62. Carroll JE, Zwillich C, Weil JV, Brooke MH. Depressed ventilatory response in oculocraniosomatic neuromuscular disease. Neurology. 1976;26:140–146.
63. Goto Y, Koga S, Horai S, Nonaka I. Chronic progressive external ophthalmoplegia: a correlative study of mitochondrial DNA deletions and their phenotypic expression in muscle biopsies. J Neurol Sci. 1990;100:63–69.
64. Servidei S, Zeviani M, Manfredi G, et al. Dominantly inherited mitochondrial myopathy with multiple deletions of mitochondrial DNA: clinical, morphologic, and biochemical studies. Neurology. 1991;41:1053–1059.
65. Hirano M, Silvestri G, Blake DM, et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): clinical, biochemical, and genetic features of an autosomal recessive mitochondrial disorder. Neurology. 1994;44:721–727.
66. Kaukonen J, Juselius JK, Tiranti V, et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science. 2000;289:782–785.
67. Ronchi D, Garone C, Bordoni A. Next-generation sequencing reveals DGUOK mutations in adult patients with mitochondrial DNA multiple deletions. Brain. 2012:135:3404–3415.
68. Bakker HD, Scholte HR, Van den Bogert C, et al. Adenine nucleotide translocator deficiency in muscle: potential therapeutic value of vitamin E. J Inherit Metab Dis. 1993;16:548–552.
69. Ronchi D, Di Fonzo A, Lin W, et al. Mutations in DNA2 link progressive myopathy to mitochondrial DNA instability. Am J Hum Genet. 2013;92:293–300.
70. Pfeffer G, Gorman GS, Griffin H, et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain. 2014:137;1323–1336.
71. Bohlega S, Tanji K, Santorelli FM, Hirano M, al-Jishi A, DiMauro S. Multiple mitochondrial DNA deletions associated with autosomal recessive ophthalmoplegia and severe cardiomyopathy. Neurology. 1996;46:1329–1334.
72. Carrozzo R, Hirano M, Fromenty B, et al. Multiple mtDNA deletions in autosomal dominant and recessive diseases suggests distinct pathogeneses. Neurology. 1998;50:99–106.
73. Minchum PE, Dormer RL, Hughs IA, et al. Fatal infantile myopathy due to cytochrome c oxidase deficiency. J Neurol Sci. 1983;60:453–463.
74. Tritschler H-J, Andreetta F, Moraes CT, et al. Mitochondrial myopathy of childhood associated with depletion of mitochondrial DNA. Neurology. 1992;42:209–217.
75. Tritschler HJ, Bonilla E, Lombes A, et al. Differential diagnosis of fatal and benign cytochrome c oxidase deficient myopathies of infancy: an immunohistochemical approach. Neurology. 1991;41:300–305.
76. Zeviani M, Peterson P, Servidei S, et al. Benign reversible muscle cytochrome c oxidase deficiency: a second case. Neurology. 1987;37:64–67.
77. Mancuso M, Filosto M, Bonilla E, et al. Mitochondrial myopathy of childhood associated with mitochondrial DNA depletion and a homozygous mutation (T77M) in the TK2 gene. Arch Neurol. 2003;60(7):1007–1009.
78. Mancuso M, Filosto M, Stevens JC, et al. Mitochondrial myopathy and complex III deficiency in a patient with a new stop-codon mutation (G339X) in the cytochrome b gene. J Neurol Sci. 2003;209(1–2):61–63.
79. Campos Y, Martin MA, Garcia-Silva T, et al. Clinical heterogeneity associated with mitochondrial DNA depletion in muscle. Neuromuscul Disord. 1998;8:568–573.
80. Vu TH, Sciacco M, Tanji K, et al. Clinical manifestations of mitochondrial DNA depletion. Neurology. 1998;50:1783–1790.
81. Moraes CT, Shanske S, Tritschler HJ, et al. Mitochondrial DNA depletion with variable tissue specificity: a novel genetic abnormality in mitochondrial diseases. Am J Hum Genet. 1991;48:492–501.
82. Durham SE, Bonilla E, Samuels DC, DiMauro S, Chinnery PF. Mitochondrial DNA copy number threshold in mtDNA depletion myopathy. Neurology. 2005;65:453–455.
83. Figarella-Branger D, Pelssier JF, Scheiner C, Wernert F, Desnuelle C. Defects of the mitochondrial respiratory chain complexes in three pediatric cases with hypotonia and cardiac involvement. J Neurol Sci. 1992;108:105–113.
84. Mazziotta MR, Ricci E, Bertini E, et al. Fatal infantile liver failure associated with mitochondrial DNA depletion. J Pediatr. 1992;121:896–901.
85. Telerman-Toppet N, Biarent D, Bouton JM, et al. Fatal cytochrome c oxidase-deficient myopathy of infancy associated with mtDNA depletion: differential involvement of skeletal muscle and cultured fibroblasts. J Inherit Metab Dis. 1992;15:323–326.
86. Saada A, Ben-Shalom E, Zyslin R, Miller C, Mandel H, Elpeleg O. Mitochondrial deoxyribonucleoside triphosphate pools in thymidine kinase 2 deficiency. Biochem Biophys Res Commun. 2003;310(3):963–966.
87. Saada A, Shaag A, Elpeleg O. Mtdna depletion myopathy: elucidation of the tissue specificity in the mitochondrial thymidine kinase (TK2) deficiency. Mol Genet Metab. 2003;79(1):1–5.
88. Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutation mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet. 2001;29:342–344.
89. Vila MR, Segovia-Silvestre T, Gamez J, et al. Reversion of mtDNA depletion in a patient with TK2 deficiency. Neurology. 2003;60(7):1203–1205.
90. Chanprasert S, Wang J, Weng SW, et al. Molecular and clinical characterization of the myopathic form of mitochondrial DNA depletion syndrome caused by mutations in the thymidine kinase (TK2) gene. Mol Genet Metab. 2013;110(1–2):153–161.
91. Paradas C, Gutiérrez Ríos P, Rivas E, Carbonell P, Hirano M, DiMauro S. TK2 mutation presenting as indolent myopathy. Neurology. 2013;80(5):504–506.
92. Béhin A, Jardel C, Claeys KG, et al. Adult cases of mitochondrial DNA depletion due to TK2 defect: an expanding spectrum. Neurology. 2012;78(9):644–648.
93. Lesko N, Naess K, Wibom R, et al. Two novel mutations in thymidine kinase-2 cause early onset fatal encephalomyopathy and severe mtDNA depletion. Neuromuscul Disord. 2010;20(3):198–203.
94. Collins J, Bove KE, Dimmock D, Morehart P, Wong LJ, Wong B. Progressive myofiber loss with extensive fibro-fatty replacement in a child with mitochondrial DNA depletion syndrome and novel thymidine kinase 2 gene mutations. Neuromuscul Disord. 2009;19(11):784–787.
95. Blakely E, He L, Gardner JL, et al. Novel mutations in the TK2 gene associated with fatal mitochondrial DNA depletion myopathy. Neuromuscul Disord. 2008;18(7):557–560.
96. Oskoui M, Davidzon G, Pascual J, et al. Clinical spectrum of mitochondrial DNA depletion due to mutations in the thymidine kinase 2 gene. Arch Neurol. 2006;63(8):1122–1126.
97. Wang L, Limongelli A, Vila MR, Carrara F, Zeviani M, Eriksson S. Molecular insight into mitochondrial DNA depletion syndrome in two patients with novel mutations in the deoxyguanosine kinase and thymidine kinase 2 genes. Mol Genet Metab. 2005;84(1):75–82.
98. Buchaklian AH, Helbling D, Ware SM, Dimmock DP. Recessive deoxyguanosine kinase deficiency causes juvenile onset mitochondrial myopathy. Mol Genet Metab. 2012;107(1–2):92–94.
99. Bardosi A, Creutzfeldt W, DiMauro S, et al. Myo-neuro-gastrointestinal encephalopathy (MNGIE syndrome) due to partial deficiency of cytochrome C oxidase: a new mitochondrial multisystem disorder. Acta Neuropathol (Berl). 1987;74:248–258.
100. Nishino I, Spinazzola A, Papadimitriou A, et al. Mitochondral neurogastrointestinal encephalomyopathy: an autosomal recessive disorder due to thymidine phosphorylase mutations. Ann Neurol. 2000;47:729–800.
101. Simon LT, Horoupian DS, Dorfman LJ, et al. Polyneuropathy, ophthalmoplegia, leukoencephalopathy, and intestinal pseudo-obstruction: POLIP syndrome. Ann Neurol. 1990;28:349–360.
102. Bedlack RS, Vu T, Hammans S, et al. MNGIE neuropathy: five cases mimicking chronic inflammatory demyelinating polyneuropathy. Muscle Nerve. 2004;29(3):364–368.
103. Nishino I, Spinazzola A, Hirano M. Thymidine phosphorylase gene mutations in MNGIE: a human mitochondrial disorder. Science. 1999;283:689–692.
104. Hirano M, Garcia-de-Yebenes J, Jones AC, et al. Mitochondrial neurogastrointestinal encephalomyopathy syndrome maps to chromosome 22q13.32-qter. Am J Hum Genet. 1998;63:526–533.
105. Van Goethem G, Schwartz M, Lofgren A, Dermaut B, Van Broeckhoven C, Vissing J. Novel POLG mutations in progressive external ophthalmoplegia mimicking mitochondrial neurogastrointestinal encephalomyopathy. Eur J Hum Genet. 2003;11:547–549.
106. Tang S, Dimberg EL, Milone M, Wong LJ. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)-like phenotype: an expanded clinical spectrum of POLG1 mutations. J Neurol. 2012;259:862–868.
107. Shaibani A, Shchelochkov OA, Zhang S, et al. Mitochondrial neurogastrointestinal encephalopathy due to mutations in RRM2B. Arch Neurol. 2009;66:1028–1032.
108. Fadic R, Russell JA, Russell JA. Sensory ataxic neuropathy as the presenting feature of a novel mitochondrial disease. Neurology. 1997;49:239–245.
109. Mancuso M, Filosto M, Bellan M, et al. POLG mutations causing ophthalmoplegia, sensorimotor polyneuropathy, ataxic and deafness. Neurology. 2004;62:316–318.
110. van Domburg PH, Gabreels-Festen AA, ter Laak H, et al. Mitochondrial cytopathy presenting as hereditary sensory neuropathy with progressive external ophthalmoplegia, ataxia and fatal myoclonic epileptic status. Brain. 1996;119:997–1010.
111. Milone M, Brunetti-Pierri N, Tang LY, et al. Sensory ataxic neuropathy with ophthalmoparesis caused by POLG mutations. Neuromuscul Disord. 2008;18;626–632.
112. Wong LJ, Naviaux RK, Brunetti-Pierri N, et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum Mutat. 2008;29(9):E150–E172.
113. Martin-Negrier ML, Sole G, Jardel C, Vital C, Ferrer X, Vital A. TWINKLE gene mutation: report of a French family with an autosomal dominant progressive external ophthalmoplegia and literature review. Eur J Neurol. 2011;18:436–441.
114. Hakonen AH, Heiskanen S, Juvonen V, et al. Mitochondrial DNA polymerase W748 S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am J Hum Genet. 2005;77:430–441.
115. Wintgerthun S, Ferrari G, He L, et al. Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase gamma mutations. Neurology. 2005;64:1204–1208.
116. Rantamaki MT, Soini HK, Finnila SM, Majamaa K, Udd B. Adult-onset ataxia and polyneuropathy caused by mitochondrial 8993 T>C mutation. Ann Neurol. 2005;58:337–340.
117. Van Goethem G, Luoma P, Rantamaki M, et al. POLG mutations in neurodegenerative disorders with ataxia but no muscle involvement. Neurology. 2004;63:1251–1257.
118. Luoma PT, Luo N, Loscher WN, et al. Functional defects due to spacer region mutations of human mitochondrial DNA polymerase in a family with an ataxia-myopathy syndrome. Hum Mol Genet. 2005;14:1907–1920.
119. Tzoulis C, Engelsen BA, Telstad W, et al. The spectrum of clinical disease caused by the A467 T and W748 S POLG mutations: a study of 26 cases. Brain. 2006;129:1685–1692.
120. Holt IJ, Harding AE, Petty RK, Morgan-Hughes JA. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am J Hum Genet. 1990;46:428–433.
121. Santorelli FM, Tanji K, Shanske S, DiMauro S. Heterogeneous clinical presentation of the mtDNA NARP/T8993G mutation. Neurology. 1997;49:270–273.
122. Childs AM, Hutchin T, Pysden K, et al. Variable phenotype including Leigh syndrome with a 9185TNC mutation in the MTATP6 gene. Neuropediatrics. 2007;38:313–316.
123. Gelfand JM, Duncan JL, Racine CA, et al. Heterogeneous patterns of tissue injury in NARP syndrome. J Neurol. 2011;258:440–448.
124. Karadimas CL, Vu TH, Holve SA, et al. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am J Hum Genet. 2006;79:544–548.
125. Spinazzola A, Viscomi C, Fernandez-Vizarra E, et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38:570–575.
126. Lawlor MW, Holve S, Stubbs EB Jr. Assessment of serum-mediated neurotoxicity in Navajo neuropathy. Electromyogr Clin Neurophysiol. 2000;40:211–214.
127. Appenzeller O, Kornfeld M, Snyder R. Acromutilating, paralyzing neuropathy with corneal ulceration in Navajo children. Arch Neurol. 1976;33:733–738.
128. Yu-Wai-Man P, Griffiths PG, Gorman GS, et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain. 2010;133:771–786.
129. Amati-Bonneau P, Valentino ML, Reynier P, et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain. 2008;131:338–351.
130. Hudson G, Amati-Bonneau P, Blakely EL, et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. 2008;131:329–337.
131. Liguori M, La Russa A, Manna I, et al. A phenotypic variation of dominant optic atrophy and deafness (ADOAD) due to a novel OPA1 mutation. J Neurol. 2008;255:127–129.
132. Voo I, Allf BE, Udar N, Silva-Garcia R, Vance J, Small KW. Hereditary motor and sensory neuropathy type VI with optic atrophy. Am J Ophthalmol. 2003;136:670–677.
133. Chalmers RM, Lamont PJ, Nelson I, et al. A mitochondrial DNA tRNAVal point mutation associated with adult-onset Leigh syndrome. Neurology. 1997;49:589–592.
134. Lombes A, Nakase H, Tritschler HJ, et al. Biochemical and molecular analysis of cytochrome c oxidase deficiency in Leigh’s syndrome. Neurology. 1991;41:491–498.
135. Loeffen J, Smeitink J, Triepels R, et al. The first nuclear-encoded complex 1 mutation in a patient with Leigh syndrome. Am J Hum Genet. 1998;63:1598–1604.
136. Triepels RH, Vanden Heuven L, Loeffen JL, et al. Leigh syndrome associated with a mutation in the NDUFS7 (PSST) nuclear encoded subunit of complex I. Ann Neurol. 1999;45:787–790.
137. Bougeron T, Roustin P, Chretien D, et al. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat Genet. 1995;11:144–149.
138. Ahu Z, Yao J, Johns T, et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet. 1998;20:337–343.
139. Shoffner JM, Fernhoff PM, Krawiecki NS, et al. Subacute necrotizing encephalopathy: oxidative phosphorylation defects and the ATPase 6 point mutation. Neurology. 1992;42:2168–2174.
140. Sweeney MG, Hammans SR, Duchen LW, et al. Mitochondrial DNA mutation underlying Leigh’s syndrome: clinical, pathological, biochemical, and genetic studies of a patient presenting with progressive myoclonic epilepsy. J Neurol Sci. 1994;121:57–65.
141. Yamamoto M, Clemens PR, Engel AG. Mitochondrial DNA deletions in mitochondrial cytopathies: observations in 19 patients. Neurology. 1991;41:1822–1828.
142. DeVivo D. Complexities of the pyruvate dehydrogenase complex. Neurology. 1998;51:1247–1249.
143. Lissens W, Desguerre I, Benelli C, et al. Pyruvate dehydrogenase deficiency in a female due to a 4 base pair deletion in exon 10 of the E1 gene. Hum Mol Genet. 1995;4:307–308.
144. Matthews PM, Marchington DR, Squire M, Land J, Brown RM, Brown GK. Molecular genetic characterization of an X-linked form of Leigh’s syndrome. Ann Neurol. 1993;33:652–655.
145. Genge A, Karpati G, Arnold D, Shoubridge EA, Carpenter S. Familial myopathy with conspicuous depletion of mitochondria in muscle fibers: a morphologically distinct disease. Neuromuscul Disord. 1995;5:139–144.
146. Nishino I, Kobayshi O, Goto Y-I, et al. A new distinct muscular dystrophy with mitochondrial structural abnormalities. Muscle Nerve. 1998;21:40–47.
147. Osaki Y, Nishino I, Murakami N, et al. Mitochondrial abnormalities in selenium-deficient myopathy. Muscle Nerve. 1998; 21:637–639.
148. Saunier P, Chretien D, Wood C, et al. Cytochrome c oxidase deficiency presenting as recurrent neonatal myoglobinuria. Neuromuscul Disord. 1995;5:285–289.
149. Ohno K, Tanaka M, Sahashi K, et al. Mitochondrial DNA deletions in inherited recurrent myoglobinuria. Ann Neurol. 1991;29:364–369.
150. Chinnery PF, Johnson MA, Taylor RW, Durward WF, Turnbull DM. A novel mitochondrial tRNA isoleucine gene mutation causing chronic progressive external ophthalmoplegia. Neurology. 1997;49:1166–1168.
151. Moslemi AR, Lindberg C, Toft J, Holme E, Kollberg G, Oldfors A. A novel mutation in the mitochondrial tRNA(Phe) gene associated with mitochondrial myopathy. Neuromuscul Disord. 2004;14:46–50.
152. Keightley JA, Hoffbuhr KC, Burton MD, et al. A microdeletion in cytochrome c oxidase (COX) subunit III associated with COX deficiency and recurrent myoglobinuria. Nat Genet. 1996;12(4):410–416.
153. Nishigaki Y, Bonilla E, Shanske S, Gaskin DA, DiMauro S, Hirano M. Exercise-induced muscle “burning,” fatigue, and hyper-CKemia: MtDNA T10010 C mutation in tRNA(Gly). Neurology. 2002;8(8):1282–1285.
154. Andreu AL, Tanji K, Bruno C, et al. Exercise intolerance due to a nonsense mutation in the mtDNA ND4 gene. Ann Neurol. 1999;45:820–823.
155. Andreu AL, Bruno C, Dunne TC, et al. A nonsense mutation (G15059 A) in the cytochrome b gene in a patient with exercise intolerance and myoglobinuria. Ann Neurol. 1999;45: 127–130.
156. Andreu AL, Bruno C, Shanske S, et al. Missense mutation in the mtDNA cytochrome b gene in a patient with myopathy. Neurology. 1998;51:1444–1447.
157. Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–1430.
158. Pilz D, Quarrell OW, Jones EW. Mitochondrial mutation commonly associated with Leber’s hereditary optic neuropathy observed in a patient with Wolfram syndrome (DIDMOAD). J Med Genet. 1994;31:328–330.
159. Kollberg G, Melberg A, Holme E, Oldfors A. Transient restoration of succinate dehydrogenase activity after rhabdomyolysis in iron-sulphur cluster deficiency myopathy. Neuromuscul Disord. 2011;21:115–120.