TABLE 31-1. MYOTONIC DISORDERS
TABLE 31-1. MYOTONIC DISORDERSMyotonic dystrophy is the most common myotonic disorder (Table 31-1). There are at least two genetically distinct forms of myotonic dystrophy: Dystrophica myotonia type 1 (DM1) and dystrophica myotonia type 2 (DM2), the later of which is also known as proximal myotonic myopathy (PROMM).
TABLE 31-1. MYOTONIC DISORDERS
Myotonic dystrophy type 1
Myotonic dystrophy type 2/proximal myotonic myopathy
Myotonia congenita
Paramyotonia congenita
Potassium-aggravated myotonia
Hyperkalemic periodic paralysis
Chondrodystrophic myotonia (Schwartz–Jampel syndrome)
Drug induced
Cholesterol-lowering agents (statin medications, fibrates)
Cyclosporine
Chloroquine
 MYOTONIC DYSTROPHY (DM1)
MYOTONIC DYSTROPHY (DM1)DM1 in an autosomal dominant manner with a prevalence of 13.5 per 100,000.1–4 DM1 can present at any age, including infancy. Limb weakness begins distally in the extremities and can progress slowly to affect proximal muscles. Wrist flexors are often weaker than wrist extensors. The neck flexors, including the sternocleidomastoids, are also affected early. Atrophy and weakness of temporalis and other facial muscles as well as the jaw muscles giving rise to the characteristic “hatchet face” appearance (Fig. 31-1). Ptosis is often evident. Some patients develop dysarthria and dysphagia due to pharyngeal and lingual muscles involvement.
Figure 31-1. Myotonic dystrophy type 1. Note the typical myotonic facies of a DM 1 patient with frontal balding and temporal, jaw, and facial muscle atrophy, and weakness.
Many patients do not complain or are not aware of their myotonia, although it is usually readily apparent on examination, particularly in the hands. Delayed relaxation of the fingers is seen following a forceful hand grip (action myotonia). The myotonia is lessened with repeated muscle contractions, a so-called warm-up phenomenon. Percussion of muscle groups, in particular of the thenar eminence or finger extensors also gives rise to delayed relaxation (percussion myotonia). Muscle reflexes are diminished, but sensory testing is normal.5 Adult patients with DM1 may have a mild reduction in cognitive abilities, while severe mental retardation is associated with congenital myotonic dystrophy.6,7
Congenital myotonic dystrophy is much more severe than adult-onset DM1. Affected infants are invariably born to mothers with myotonic dystrophy.8,9 It is important to examine mothers of floppy infants, as they may not even be aware that they have the disorder. Pregnancy may be complicated by polyhydramnios and diminished fetal movements. Infants with congenital myotonic dystrophy have severe generalized weakness and hypotonia and may also have arthrogryposis. Clinical myotonia is not apparent in the neonatal period and may not be noticeable until about 5 years of age. However, myotonic discharges can be appreciated on electromyography (EMG) before the appearance of clinical myotonia. Many infants require ventilatory assistance due to ventilatory insufficiency. The mortality rate in infancy is approximately 25%. Severe psychomotor abnormalities affect 75% of surviving children. Most will have some degree of mental retardation. Life expectancy is greatly reduced in DM1 patients, particularly those with early onset of the disease and significant proximal, in addition to distal, weakness.10,11
DM1 is a systemic disorder affecting the gastrointestinal tract, the uterus, ventilatory muscles, cardiac muscle, the lens, and the endocrine system.12 In addition to dysphagia, reduced gastrointestinal motility can lead to chronic pseudo-obstruction.13,14 Alveolar hypoventilation can arise from involvement of the diaphragm and intercostal muscles. It is more severe in congenital myotonic dystrophy and may lead to ventilatory failure, but this certainly occurs in later onset cases as well.9 It is unclear if decreased central drive contributes to hypoventilation.15,16 Nonetheless, many patients develop symptoms suggestive of sleep apnea: frequent nocturnal arousals, excessive daytime hypersomnolence, and morning headaches. Pulmonary hypertension can develop and may lead to cor pulmonale.
Cardiac abnormalities are common with approximately 90% of patients having conduction defects on electrocardiograms (EKGs).17 Sudden cardiac death secondary to arrhythmia is well documented. However, the severity of the cardiomyopathy does not necessarily correlate with the severity of skeletal muscle weakness. The size of the mutation (discussed in Pathogenesis section) and the severity of the skeletal muscle weakness do not correlate with the occurrence of cardiac conduction abnormalities or sudden death.18 It seems that risk of sudden death increases with duration of disease and age, and that risk is higher in male patients.18
Neurobehavioral abnormalities are common in patients with DM1.19,20 Neuropsychological testing demonstrates elements of obsessive–compulsive, passive–aggressive, dependent, and avoidant personality traits in many patients. Apathy and depression are also frequent. Cognitive impairment, particularly in memory and spatial orientation, may be demonstrated. The neuropsychological deficits appear to correlate with brain single photon emission computed tomography, which shows frontal and parieto-occipital hypoperfusion.20
Other systemic manifestations include posterior subscapular cataracts, frontal balding, testicular atrophy and impotence in men, and a high rate of fetal loss and complications of pregnancy in women. Hyperinsulinemia is common following glucose tolerance tests, however, the frequency of overt diabetes mellitus is not increased.21
Some epidemiological studies have reported an increased risk of cancer in patients with DM1. In a study of Swedish and Danish populations, the risk of malignancy was double that of the general population.22 Specifically, they observed an increased risk of endometrial, ovarian, colon, and brain cancer. In a study from the Mayo Clinic, there was an increased risk of thyroid cancer and choroidal melanoma, as well as perhaps testicular and prostate cancer.23 However, they found no increased risk of endometrial, ovarian, breast, colorectal, lung, renal, bladder, or brain cancers.
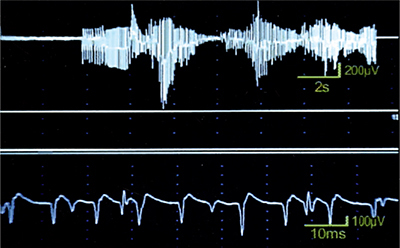
Serum creatine kinase (CK) may be normal or mildly increased. Motor and sensory nerve conduction studies (NCS) are usually normal. EMG demonstrates myotonic discharges (Fig. 31-2). It is important to sample multiple muscles as myotonic discharges are not necessarily appreciated in every muscle studied.24 Facial and intrinsic hand muscles are the most commonly affected. In congenital myotonic dystrophy, electrical myotonia may be observed as early as 5 days to 3 weeks following birth and increases with age.25,26 Fibrillation potentials, positive sharp waves, and myopathic motor unit action potentials (MUAPs) may also be seen but they can be obscured by the myotonic discharges.
Figure 31-2. Myotonic dystrophy. Electromyography reveals myotonic discharges which wax and wane in frequency and amplitude.
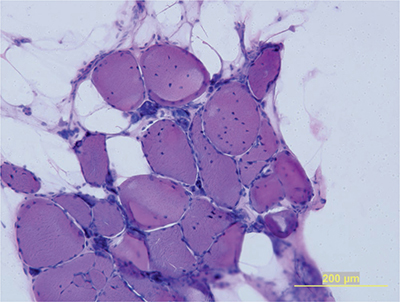
Muscle biopsies demonstrate an increased number of internalized nuclei in the muscle fibers (Fig. 31-3). Type 1 predominance and atrophy are very common. In addition, hypertrophic type 2 fibers, ring fibers, small angulated fibers, atrophic fibers with pyknotic nuclear clumps, and sarcoplasmic masses are also frequently observed. In contrast to other muscular dystrophies, necrotic fibers and increased connective tissue are less conspicuous. Autopsy studies of the brain demonstrate neurofibrillary degeneration with abnormal tau expression.27
Figure 31-3. Myotonic dystrophy type 1. Muscle biopsies reveal adipose tissue and remaining muscle fibers with numerous internalized nuclei and atrophic fibers with pyknotic nuclear clumps.
DM1 is caused by an expansion of unstable polymorphic cytosine–thymine–guanine (CTG) trinucleotide repeats in the 3′ untranslated region of the myotonin protein kinase gene, (DMPK), that is located on chromosome 19q13.2.12,28–35 This CTG repeat is copied in the gene up to 27 times in normals, but 50 to more than 4,000 copies are found in DM1 patients. The severity of the myopathy directly correlates with the size of the CTG repeat, which is unstable. The mutation size usually expands from one generation to the next, which accounts for the anticipation phenomena (i.e., the earlier presentation and/or more severe disease in each generation). More marked expansion of the CTG repeat usually occurs in children of mothers with DM1, which explains the severe phenotype of congenital myotonic dystrophy.
It is not the abnormal expression of myotonin protein kinase itself that is responsible for the disorder. Rather, DM1 seems to be a consequence of nuclear retention of mutant mRNA containing expanded CTG repeats, rather than a specific lack or gain of function of the DMPK protein. Indeed, the myopathy and other systemic features appear to be due to a toxic gain of function of the mutant mRNA.36
The transcribed mRNA with expanded CTG (DM1) accumulates as abnormal focal collections in the nucleus that cannot be transported to the cytoplasm, where RNA translation into protein takes place.37–40 Aggregates of mutated mRNA are directly toxic to cells by sequestering RNA-binding proteins (such as muscleblind proteins), which in turn, lead to abnormal splicing of pre-mRNA from various target genes (e.g., chloride ion channel, insulin receptor, tau protein, cardiac troponin, ryanodine receptor, and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase).37–39,41–45 Therefore, there is abnormal translation of the RNAs into functional proteins, and this explains the multiple organ/systemic manifestations of DM1. Other studies have shown that mutant RNA binds and sequesters transcription factors with up to 90% depletion of selected transcription factors from active chromatin.46 This leads to reduced expression of a variety of genes, including the chloride ion channel (CIC-1), which is also mutated in myotonia congenita and is the likely origin of the myotonic discharges that occur in both disorders.
There are no medical therapies that clearly improve muscle strength. A small pilot study of dehydroepiandrosterone sulfate (DHEAS) in 11 patients with DM1 seemed to be beneficial in a few patients, but larger controlled trials are necessary before commenting on the possible efficacy.47 Small trials of creatine monohydrate in DM1 failed to demonstrate efficacy.48 A recent study of recombinant human insulin-like growth factor 1 (rhIGF-1) complexed with IGF-binding protein 3 (rhIGF-1/rhIGFBP-3) in patients with DM1 reported that drug was associated with increased lean body mass and improvement in metabolism, but not increased muscle strength or function.49
Patients are usually not so bothered by the myotonia to warrant treatment. Further, some drugs that may improve myotonia, such as quinine, procainamide, and tocainide, can also potentiate cardiac arrhythmias and should be avoided. A recent study demonstrated that mexiletine was helpful in reducing myotonia.50 We initiate treatment with mexiletine 150 mg daily and gradually increase as tolerated and as necessary to control the symptoms, up to a maximum of 300 mg tid. We assess baseline EKG and with each increment of dosage. In addition, aerobic training is safe and may improve fitness effectively in patients with myotonic dystrophy.51
We obtain yearly EKGs to monitor for evidence of conduction defects/arrhythmias. If abnormalities are detected, we obtain a cardiology consultation, 24-hour Holter monitoring, and echocardiograms because some patients may require antiarrhythmic medication or pacemaker insertion. Pulmonary function tests are routinely performed. Patients with DM1 are at risk for pulmonary and cardiac complications from general anesthesia and neuromuscular blocking medications.52–55 These agents should be used with extreme caution.
We obtain overnight polysomnography in patients with symptoms and signs of sleep apnea. Patients with significant hypoventilation or sleep apnea may benefit from noninvasive ventilatory assistance with BiPAP. Modafinil 200–400 mg per day is also effective in reducing the excessive daytime somnolence that is commonly associated with DM1.56–58
Some patients require excision of their cataracts. Occasionally for bothersome ptosis, we refer patients for blepharoplasty. However, it is important to discuss with patients the associated risk of inadvertent exposure keratitis. Physical and occupational therapy are important. Orthotic devices such as ankle braces are indicated in patients with foot drop to assist their gait.
Genetic counseling is of utmost importance. Patients need to know that the risk of passing the disease on to their children is 50% with each pregnancy. Further, the disease severity is generally worse from one generation to the next, particularly when the mother has DM1. Prenatal diagnosis is possible via amniocentesis or chorionic villus sampling.
MYOTONIC DYSTROPHY TYPE 2 OR PROXIMAL MYOTONIC MYOPATHYMyotonic dystrophy type 2 (DM2) is a multisystem, autosomal dominant disorder that resembles DM1 with myotonia, weakness, cataracts, testicular failure, glucose intolerance, hypogammaglobulinemia, and cardiac conduction defects.2–4,12,21,40,59–69 In a study of 234 individuals with DM2, 90% had electrical myotonia, 82% weakness, 61% cataracts, 23% diabetes, and 19% cardiac involvement.59 Most patients with DM2 become symptomatic between the ages of 20 and 60 years, although onset can occur in childhood. The initial symptoms are usually intermittent stiffness and pain of the thigh muscles in one or both legs. Myotonia may be evident in proximal and distal extremity muscles as well as facial muscles, however it is variable and not always present. Myotonia can initially manifest or worsen during pregnancy.70,71 There is an associated “warm-up” phenomenon with decreased myotonia following repeated muscle contractions. The clinical myotonia does not exacerbate with cold temperature, although a few affected individuals have described worsening of symptoms with warm temperatures.72
Patients often describe pain that is episodic and disabling, with burning, tearing, or jabbing characteristics. This pain typically affects the thighs, shoulders, and upper arms and is not necessarily related to the myotonic stiffness of the muscles. They may complain of peculiar chest pains as well, leading to cardiac evaluations to rule out coronary artery disease.
Slowly progressive proximal and distal weakness develops in the majority of patients. The characteristic pattern of muscle weakness involves the neck flexors, elbow extensors, thumb and deep finger flexors, and hip flexors and extensors in the legs. In general, the proximal muscle is often affected earlier than one sees in DM1, thus the name “proximal myotonic myopathy.” Some patients describe fluctuations of their weakness with episodes of increased weakness lasting hours or weeks.64 During these periods of increased weakness, repeated activity can lead to transient improvement in strength. Significant loss of muscle bulk is not apparent early, however, approximately 9% of patients develop considerable atrophy of proximal muscles late in life.59 Calf hypertrophy occurs in some patients, which can be asymmetric. Rarely, myoglobinuria can occur as a complication of DM2.
Symptoms and severity can vary within families. Studies have demonstrated an earlier onset of symptoms among offspring of affected individuals, suggesting that anticipation is also a feature of DM2.59,64,67 However, in contrast to DM1, anticipation in DM2 is much milder and a congenital form has never been described.59,67
Cataracts that are indistinguishable from those seen in DM1 are common in DM2.12,59 These cataracts usually appear before the age of 50 years and have even developed in patients in their late childhood. Cardiac abnormalities may also develop.12,59,73 Syncope, near-syncopal spells, or symptomatic tachycardia occur in 8%, cardiac conduction defects in 20%, and a potentially life-threatening cardiomyopathy occur in as many as 7% of individuals who are affected.59 Unlike DM1, most series have not reported an increased incidence of alveolar hypoventilation in patients with DM2, however, some patients develop sleep apnea and excessive daytime somnolence.72
Also, in contrast to DM1, mental retardation is not a prominent feature. However, white matter abnormalities may be appreciated on magnetic resonance imaging (MRI) of the brain.13 In addition, some affected individuals have stroke-like symptoms, seizures, parkinsonian features, and hypersomnia. Further, neuropsychological testing reveals lower scores on tests of frontal lobe function compared to normal along with avoidant personality traits; brain single photon emission and computed tomography can show frontal and parieto-occipital hypoperfusion similar to DM1.20 Frontal balding has been reported in as many as 20–50% of men aged 21–34 years. Testicular atrophy with gonadal insufficiency can occur. Gastrointestinal hypomotility has not been described. Late-onset deafness was reported in one kinship with atypical DM2.
Serum CK levels are often mildly elevated. Low testosterone levels may be seen in as many as 29% of affected males and insulin insensitivity in 75% of patients.59 A high GGT was demonstrated in 64%, low IgG in 65%, and low IgM in 11%.59 Abnormalities are common in EKG as described earlier.
Motor and sensory NCS are normal. EMG reveals myotonic discharges even in patients without clinical myotonia, although these discharges can be difficult to detect in some patients. Despite the prominent proximal muscle involvement clinically, electrical myotonia is often more easily detected in distal muscles.
Muscle biopsy reveals nonspecific myopathic features including a mild to moderate increase in internalized nuclei, variability of fiber size with atrophy of type 2 fibers, small angular fibers, and atrophic fibers with pyknotic nuclear clumps.59,74,75 In contrast to that seen in DM1, selective type 1 fiber atrophy, sarcoplasmic masses, and ringed fibers are not usually appreciated on DM2 muscle biopsies. Autopsy studies demonstrate neurofibrillary degeneration with abnormal tau expression as in DM1.27
DM2 and PROMM are allelic disorders caused by CCTG repeat expansions in intron 1 of the zinc finger protein 9 gene, (ZNF9), located on chromosome 3.40,59 The transcribed mRNA with expanded CCTG repeats accumulates as abnormal focal collections in the nucleus similar to expanded CTG repeats seen in DM1.37–40 As with DM1, the aggregates of mutated mRNA appear to exert their toxic effect on cells by sequestering RNA-binding proteins that leads to abnormal splicing of pre-mRNA from various target genes (e.g., chloride ion channel, insulin receptor, tau protein, cardiac troponin).37–39,42,45 The subsequent abnormal translation of the RNAs into functional proteins explains the multiple organ/systemic manifestations of both DM1 and DM2.
There is no specific treatment for DM2. A small randomized controlled trial of creatine monohydrate in DM2 was ineffective.76 There is insufficient information regarding the efficacy of various antimyotonia agents, but mexiletine or carbamazepine, or phenytoin can be tried if the myotonia or muscle pain is bothersome to the patient.72,77 Cataracts may need surgical excision. It seems prudent to carefully monitor patients during surgery and the postoperative period. One patient with PROMM developed increased muscle pain, myoglobinuria, and transient renal insufficiency after minor surgery.64
SUMMARYDM1 and DM2 are multi systemic disorders caused by expanded repeats in the noncoding regions of the DMPK and ZNF9 genes, respectively. The novel pathogenic consequence of these mutations is not due to a loss of function created by loss of DMPK and ZNF9 protein products, but rather a toxic effect on the cells by the accumulation of abnormal mRNA. The mutant mRNA sequesters necessary RNA-binding proteins and this results in abnormal splicing of pre-mRNA from various target genes (e.g., chloride ion channel, insulin receptor, tau protein, cardiac troponin), thus explaining the multisystemic manifestations of DM1 and DM2. There may be additional forms of myotonic dystrophy not localized to the DM1 and DM2 loci. Treatment of these disorders at this time is largely symptomatic.
1. Emery AE. Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul Disord. 1991;1:19–29.
2. Machuca-Tzili L, Brook D, Hilton-Jones D. Clinical and molecular aspects of the myotonic dystrophies: A review. Muscle Nerve. 2005;32(1):1–18.
3. Tramonte JJ, Burns TM. Myotonic dystrophy. Arch Neurol. 2005;62(8):1316–1319.
4. van Engelen BG, Eymard B, Wilcox D. 123rd ENMC International Workshop: Management and therapy in myotonic dystrophy, 6-8 February 2004, Naarden, The Netherlands. Neuromuscul Disord. 2005;15(5):389–394.
5. Messina C, Tonali P, Scoppetta C. The lack of deep reflexes in myotonic dystrophy: A neurophysiologic study. J Neurol Sci. 1976;30:303–311.
6. Bird TD, Follett C, Griep E. Cognitive and personality function in myotonic dystrophy. J Neurol Neurosurg Psychiatry. 1983;46:971–980.
7. Portwood MM, Wicks JJ, Lieberman JS, Duveneck MJ. Intellectual and cognitive function in adults with myotonic dystrophy. Arch Phys Med Rehabil. 1986;67:299–303.
8. Hageman AT, Gabreels FJ, Liem KD, Renkawek K, Boon JM. Congenital myotonic dystrophy; a report on thirteen cases and a review of the literature. J Neurol Sci. 1993;115:95–101.
9. Reardon W, Newcombe R, Fenton I, Sibert J, Hfarper PS. The natural history f congenital myotonic dystrophy: Mortality and long term clinical aspects. Arch Dis Child. 1993;68:177–181.
10. de Die-Smulders CE, Howeler CJ, Thijs C, et al. Age and causes of death in adult-onset myotonic dystrophy. Brain. 1998;121: 1557–1563.
11. Mathieu J, Allard P, Potvin L, Prevost C, Begin P. A 10 year study of mortality in a cohort of patients with myotonic dystrophy. Neurology. 1999;52:1658–1662.
12. Meola G. Genetic and clinical heterogeneity in myotonic dystrophies. Muscle Nerve. 2000;13:1789–1799.
13. Hund E, Jansen O, Koch MC, et al. Proximal myotonic myopathy with white matter abnormalities of the brain. Neurology. 1997;48:33–37.
14. Nowak TV, Anuras S, Brown BP, Ionasescu V, Green JB. Small intestine motility in myotonic dystrophy patients. Gastroenterology. 1984;86:808–813.
15. Begin R, Bureau MA, Lupien L, Lemiex B. Control and modulation of respiration in Steinert’s myotonic dystrophy. Am Rev Respir Dis. 1980;121:281–289.
16. Hansotia P, Frens D. Hypersomnia associated with alveolar hypoventilation in myotonic dystrophy. Neurology. 1981;31: 1336–1337.
17. Motta J, Guilleminault C, Billingham M, Barry W, Mason J. Cardiac abnormalities in myotonic dystrophy. Electrophysiologic and histologic studies. Am J Med. 1979;67:467–473.
18. Sabovic M, Medica I, Logar N, Mandic E, Zidar J, Peterlin B. Relation of CTG expansion and clinical variables to electrocardiogram conduction abnormalities and sudden death in patients with myotonic dystrophy. Neuromuscul Disord. 2003;13(10):822–826.
19. Delaporte C. Personality patterns in patients with myotonic dystrophy. Arch Neurol. 1998;55:635–640.
20. Meola G, Sansone V, Perani D, et al. Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM-1) and in proximal myotonic myopathy (PROMM/DM-2). Neuromuscul Disord. 2003;13(10):813–821.
21. Moxley RT 3rd, Griggs RC, Goldblatt D. VanGelder V, Herr BE, Thiel R. Decreased insulin sensitivity of forearm muscle in myotonic dystrophy. J Clin Invest. 1978;62:857–867.
22. Gadalla SM, Lund M, Pfeiffer RM, et al. Cancer risk among patients with myotonic muscular dystrophy. JAMA. 2011;306: 2480–2486.
23. Win AK, Perattur PG, Pulido JS, Pulido CM, Lindor NM. Increased cancer risks in myotonic dystrophy. Mayo Clin Proc. 2012;87(2):130–135.
24. Streib EW, Sun SF. Distribution of electrical myotonia in myotonic muscular dystrophy. Ann Neurol. 1983;14:80–82.
25. Dodge PR, Gamstrop I, Byers RK, Russell P. Myotonic dystrophy in infancy and childhood. Pediatrics. 1965;35:3–19.
26. Swift TR, Ignacio OJ, Dyken PR. Neonatal dystrophica myotonica. Electrophysiological studies. Am J Dis Child. 1975;129: 734–737.
27. Maurage CA, Udd B, Ruchoux MM, et al. Similar brain tau pathology in DM2/PROMM and DM1/Steinert disease. Neurology. 2005;65:1636–1638.
28. Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of transcript encoding protein kinase family member. Cell. 1992;68:799–808.
29. Fischbeck KH. The mechanism of myotonic dystrophy. Ann Neurol. 1994;35:255–256.
30. Fu YH, Friedman DL, Richards S, et al. Decreased expression of myotonin-protein kinase messenger RNA and protein in adult form of myotonic dystrophy. Science. 1993;260:235–238.
31. Fu YH, Pizzuti A, Fenwick R Jr, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258.
32. Harper PS, Harley HG, Reardon W, Shaw DJ. Review article: Anticipation in myotonic dystrophy: New light on an old problem. Am J Hum Genet. 1992;51:10–16.
33. Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255.
34. Ptacek LJ, Johnson KJ, Griggs RC. Genetics and physiology of the myotonic disorders. N Engl J Med. 1993;328:482–489.
35. Shelbourne P, Davies J, Buxton J, et al. Direct diagnosis of myotonic dystrophy with a disease-specific DNA marker. N Engl J Med. 1993;328:471–475.
36. Tian B, White RJ, Xia T, et al. Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA. 2000;6:79–87.
37. Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of CIC-1 chloride channel prem-RNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44.
38. Mankodi A, Teng-Umnauay P, Krym M, Hendierson D, Swanson M, Thornton CA. Ribonuclear inclusions in skeletal myotonic dystrophy types 1 and 2. Ann Neurol. 2003;54:760–768.
39. Mankodi A, Thornton CA. Myotonic syndromes. Curr Opin Neurol. 2002;15(5):545–552.
40. Udd B, Meola G, Krahe R, et al. Myotonic dystrophy type 2 (DM2) and related disorders report of the 180th ENMC workshop including guidelines on diagnostics and management 3–5 December 2010, Naarden, The Netherlands. Neuromuscul Disord. 2011;21:443–450.
41. Berg J, Jiang H, Thornton CA, Cannon SC. Truncated ClC-1 mRNA in myotonic dystrophy exerts a dominant-negative effect on the Cl current. Neurology. 2004;63(12):2371–2375.
42. Day JW, Ranum LP. RNA pathogenesis of the myotonic dystrophies. Neuromuscul Disord. 2005;15(1):5–16.
43. Kimura T, Nakamori M, Lueck JD, et al. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1. Hum Mol Genet. 2005;14(15):2189–2200.
44. Pascual M, Vicente M, Monferrer L, Artero R. The Muscleblind family of proteins: An emerging class of regulators of developmentally programmed alternative splicing. Differentiation. 2006;74(2–3):65–80.
45. Kanadia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302: 1978–1980.
46. Ebralidze A, Wang Y, Petkova V, Ebralidse K, Junghans RP. RNA leaching of transcription factors disrupts transcription in myotonic dystrophy. Science. 2004;303(5656):383–387.
47. Sugino M, Ohsawa N, Ito T, et al. A pilot study of dehydroepiandrosterone sulfate in myotonic dystrophy. Neurology. 1998;51(2):586–589.
48. Tarnopolsky M, Mahoney D, Thompson T, Naylor H, Doherty TJ. Creatine monohydrate supplementation does not increase muscle strength, lean body mass, or muscle phosphocreatine in patients with myotonic dystrophy type 1. Muscle Nerve. 2004;29(1):51–58.
49. Heatwole CR, Eichinger KJ, Friedman DI, et al. Open-label trial of recombinant human insulin-like growth factor 1/recombinant human insulin-like growth factor binding protein 3 in myotonic dystrophy type 1. Arch Neurol. 2011;68:37–44.
50. Logigian EL, Martens WB, Moxley RT 4th, et al. Mexiletine is an effective antimyotonia treatment in myotonic dystrophy type 1. Neurology. 2010;74:1441–1448.
51. Orngreen MC, Olsen DB, Vissing J. Aerobic training in patients with myotonic dystrophy type 1. Ann Neurol. 2005;57(5): 754–757.
52. Aldridge LM. Anesthetic problems in myotonic dystrophy. A case report and review of the Aberdeen experience comprising 48 general anaesthetics in a further 16 patients. Br J Anaesth. 1985;57:1119–1130.
53. Brahams D. Postoperative monitoring in patients with muscular dystrophy. Lancet. 1989;2:1053–1054.
54. Harper PS. Postoperative complications in myotonic dystrophy. Lancet. 1989;2:1269.
55. Mathieu J, Allard P, Gobeil G, Girard M, De Braekeleer M, Begin P. Anesthetic and surgical complications in 219 cases of myotonic dystrophy. Neurology. 1997;49:1646–1650.
56. Damian MS, Gerlach A, Schmidt F, Lehman E, Reichmann H. Modafinil for excessive daytime sleepiness in myotonic dystrophy. Neurology. 2001;56:794–796.
57. MacDonald JR, Hill JD, Tarnopolsky MA. Modafinil reduces excessive somnolence and enhances mood in patients with myotonic dystrophy. Neurology. 2002;59(12):1876–1880.
58. Talbot K, Stradling J, Crosby J, Hilton-Jones D. Reduction in excess daytime sleepiness by modafinil in patients with myotonic dystrophy. Neuromuscul Disord. 2003;13(5):357–364.
59. Day JW, Ricker K, Jacobsen JF, et al. Myotonic dystrophy type 2: Molecular, diagnostic, and clinical spectrum. Neurology. 2003;60:657–664.
60. Meola G, Sansone V, Radice S, Skradski S, Ptacek L. A family with an unusual myotonic and myopathic phenotype and no CTG expansion (Proximal myotonic myopathic syndrome): A challenge for future molecular studies. Neuromuscul Disord. 1996;6:143–150.
61. Moxley RT 3rd. Proximal myotonic myopathy: Mini-review of a recently delineated clinical disorder. Neuromuscul Disord. 1996;6:87–93.
62. Ricker K, Grimm T, Koch MC, et al. Linkage of proximal myotonic myopathy to chromosome 3q. Neurology. 1999;52:170–171.
63. Ricker K, Koch MC, Lehmann-Horn F, et al. Proximal myotonic myopathy: A new dominant disorder with myotonia, muscle weakness, and cataracts. Neurology. 1994;44:1448–1452.
64. Ricker K, Koch MC, Lehmann-Horn F, et al. Proximal myotonic myopathy. Clinical features of a multisystemic disorder similar to myotonic dystrophy. Arch Neurol. 1995;52:25–31.
65. Ricker K, Moxley RT 3rd, Heine R, Lehmann-Horn F. Myotonia fluctuans. A third type of muscle sodium channel disease. Arch Neurol. 1994;44:500–1503.
66. Ricker K, Moxley RT 3rd, Heine R, Lehmann-Horne F. Myotonia fluctuans. A third type of muscle sodium channel disease. Arch Neurol. 1994;51:1095–1102.
67. Schneider C, Ziegler A, Ricker K, et al. Proximal myotonic myopathy. Evidence for anticipation in families with linkage to chromosome 3q. Neurology. 2000;55:383–388.
68. Thornton CA, Ashizawa T. Getting a grip on the myotonic dystrophies. Neurology. 1999;52:12–13.
69. Thornton CA, Griggs RC, Moxley RT 3rd. Myotonic dystrophy with no trinucleotide repeat expansion. Ann Neurol. 1994;35:269–272.
70. Newman B, Meola G, O’Donovan DG, Schapira AH, Kingston H. Proximal myotonic myopathy (PROMM) presenting as myotonia during pregnancy. Neuromuscul Disord. 1999;9:144–149.
71. Rudnik-Schoneborn S, Schneider-Gold C, Raabe U, Kress W, Zerres K, Schoser BG. Outcome and effect of pregnancy in myotonic dystrophy type 2. Neurology. 2006;66(4):579–580.
72. Sander HW, Tavoulareas G, Chokroverty S. Heat sensitive myotonia in proximal myotonic myopathy. Neurology. 1996;47:956–962.
73. Schoser BG, Ricker K, Schneider-Gold C, et al. Sudden cardiac death in myotonic dystrophy type 2. Neurology. 2004;63 (12):2402–2404.
74. Schoser BG, Schneider-Gold C, Kress W, et al. Muscle pathology in 57 patients with myotonic dystrophy type 2. Muscle Nerve. 2004;29(2):275–281.
75. Vihola A, Bassez G, Meola G, et al. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology. 2003;60:1854–1857.
76. Schneider-Gold C, Beck M, Wessig C, et al. Creatine monohydrate in DM2/PROMM: A double-blind placebo-controlled clinical study. Proximal myotonic myopathy. Neurology. 2003;60(3):500–502.
77. Moxley RT 3rd. Myotonic disorders in childhood. Diagnosis and treatment. J Child Neurol. 1997;12:116–129.