TABLE 35-1. TOXIC MYOPATHIES
TABLE 35-1. TOXIC MYOPATHIESMany drugs can cause a myopathy.1–10 The pathophysiological mechanisms are diverse and, in many instances, unclear. Medications can have either a direct or an indirect adverse effect on muscle. The direct effect can be focal, as might occur secondary to a drug being injected into tissue, or more commonly generalized. Indirect toxic effects may result from the agent creating an electrolyte imbalance or inducing an immunological reaction. Muscle fibers may undergo necrosis as a result of the drug directly disrupting the sarcolemma, nuclear or mitochondria function, or that of other organelles. In this chapter, we classify the toxic myopathies according to their presumed pathogenic mechanisms (Table 35-1).

 NECROTIZING MYOPATHIES
NECROTIZING MYOPATHIESA number of drugs can cause a generalized necrotizing myopathy. Affected individuals may complain of myalgias or weakness, or they might just have asymptomatic elevations of their serum creatine kinase (CK) levels. Severe necrotizing myopathy may be complicated by myoglobinuria and renal failure. The degree of serum CK elevation is proportionate to the amount of muscle damaged.
Cholesterol-lowering medications including 3-hydroxy-3-methylglutaryl-coenzyme A (3-HMG-CoA) reductase inhibitors,11–19 fibric acid derivatives,16,20–30 niacin,31,32 and ezetimibe33–36 may cause a toxic myopathy. Most patients just have mild elevations in serum CK without any symptoms. Others have myalgias and less frequently weakness. Myoglobinuria is a rare event but can be complicated by death. With discontinuation of the offending agent, the myalgias, weakness, and elevated serum CK levels tend to completely resolve, but it may take several days to months. However, rarely these agents may trigger an immune-mediated inflammatory myopathy, usually necrotizing, that requires treatment with immunosuppressive medications.
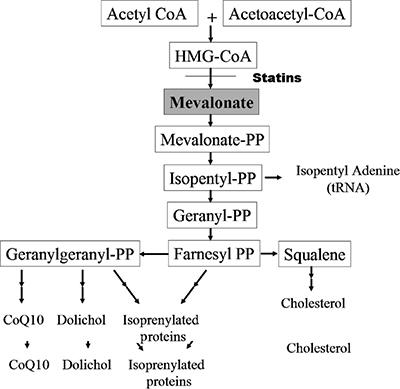
Statin agents inhibit 3-HMG-CoA reductase, the rate controlling enzyme in cholesterol synthesis (Fig. 35-1). Adverse side effects including asymptomatic hyper-CK-emia, myalgias, proximal weakness, and, less commonly, myoglobinuria occur with all of the major HMG-CoA reductase inhibitors: lovastatin,13,16,18,19,32,37,38 simvastatin,12,14,15,38,39 provastatin,17,38 atorvastatin,11,38,40 fluvastatin,38 and cerivastatin.38,41,42 The nomenclature regarding statin-induced toxic myopathies in the published literature is unfortunately quite unsatisfactory, listing “myalgias,” “myositis,” and “myopathy” as three independent types of muscle disorders caused by statin use, when these three subtypes may just reflect the spectrum of severity of the myopathy.43–45
Figure 35-1. Hydroxy-3methyl-glutaryl-coenzyme A pathway. (Reproduced with permission from Greenberg SA, Amato AA. Statin myopathies. Continuum. 2006;12(3):169–184.)
Reviews discussing statin myopathies cite a 2–7% incidence of myalgias, 0.1–1.0% incidence of weakness or elevated CK, and myoglobinuria developing in <0.5% of patients.1,43,46–49 The National Heart Lung and Blood Institute advisory panel estimated the incidence of severe myopathy to be approximately 0.08% for lovastatin, simvastatin, and pravastatin.44 The risk of toxic myopathy increases with the concomitant use of fibric acids,18,19,27,31,32,40 niacin,31,32 cyclosporine,18,19 ezetimibe,33–36 triazole antibiotics,50 rapamycin,51and sirolimus48 as well as in the presence of renal insufficiency or hepatobiliary dysfunction. In this regard, 5% of patients taking both lovastatin and gemfibrozil developed a severe myopathy,27 while a severe myopathy complicated as many as 30% of patients receiving both lovastatin and cyclosporine.13,18,19
Most patients with a statin myopathy improve within a few weeks of stopping the agent. One dilemma we face is if patients who exhibited symptoms or signs of statin-intolerance might be rechallenged once the muscle symptoms have resolved. In one study of 51 patients, who previously experienced myalgias or elevated transaminase levels on a variety of different statins, 37 (72.5%) were able to tolerate every other day rosuvastatin.52
Although the term “myositis” has been used to denote cases associated with markedly elevated serum CK levels, histopathological confirmation is lacking in most cases. “Myositis” denotes an autoimmune attack on muscle. True cases of myositis, particularly dermatomyositis, have been described in association with statin use.17,53–60 More recently, an immune-mediated necrotizing myopathy has been reported to occur in the setting of statin use (discussed in greater detail in Chapter 33).49,61–64 In these cases weakness continues to progress despite discontinuation of the statin, and only improves if the patients are treated with immunosuppressive agents. Furthermore, disease activity often flares, if the immunosuppressive medications are discontinued.
Asymptomatic elevation of serum CK is common in patients taking statin medications. Marked elevations of CK occur in patients with severe weakness and myoglobinuria. Routine motor and sensory nerve conduction studies (NCS) are normal. Fibrillation potentials, positive sharp waves, and myotonic discharges with early recruitment of small-duration motor unit action potentials (MUAPs) are apparent in weak muscles.65 Electromyography (EMG) in patients with asymptomatic serum CK elevations is often normal.
Interestingly, autoantibodies directed against HMG-CoA reductase have been reported in cases of statin-associated immune-mediated necrotizing myopathies.61,62 These antibodies are not usually seen in patients who just take statin medications or those that have the more typical statin myotoxicity that resolves upon discontinuation of the offending medication.
Muscle biopsies reveal muscle fiber necrosis with phagocytosis and small regenerating fibers in patients with elevated serum CKs and weakness or myalgias (Fig. 35-2). Lipid-filled vacuoles within myofibers and cytochrome oxidase–negative myofibers may be appreciated, but these are not consistent findings.66 Patients with statin-induced necrotizing myopathy often have increased expression of major histocompatibility antigen 1 (MHC1) and membrane attack complex deposition on the sarcolemma of non-necrotic muscle fibers.61–64
Figure 35-2. Statin myopathy. Muscle biopsy demonstrates scattered necrotic muscle fibers. Modified Gomori trichrome.
The pathogenesis of the myopathy secondary to HMG-CoA reductase inhibitors is unclear, as several pathways may potentially be interrupted downstream (Fig. 35-1).1,49,67 Mevalonate is the immediate product of HMG-CoA reductase metabolism. Subsequently, mevalonate is metabolized to farnesol, which is converted to either squalene or geranylgeraniol. Squalene is the first metabolite committed to the synthesis of cholesterol. In contrast, geranylgeraniol is important in the biosynthesis of coenzyme Q10 [a mitochondrial enzyme important in the production of adenosine triphosphate (ATP)], dolichol (important in glycoprotein synthesis), and isopentylamine (a component of tRNA), and in the activation of regulatory proteins (G-proteins). It is possible that statins could diminish cholesterol within muscle membranes, thereby predisposing the muscle fibers to rhabdomyolysis. However, the depletion of metabolites of geranylgeraniol, and not the inhibition of cholesterol synthesis, may be the primary cause of myotoxicity. In this regard, HMG-CoA reductase inhibitors decrease the levels of coenzyme Q, which could impair energy production.
A couple genome-wide association study conducted in patients with suspected statin-induced toxic myopathy revealed a strong association of myopathy with a single nucleotide polymorphism (SNP), rs4363657, located within the SLCO1B1 gene on chromosome 12.68 This gene encodes a protein that regulates the hepatic uptake of statins. Of note, more than 60% statin myopathy patients carried this SNP. No SNPs in any other region were clearly associated with myopathy, including those genes associated with metabolic myopathies.
There are several reports of patients treated with statins who developed dermatomyositis55–57,59,60 or polymyositis.17,53,54,58 The most common myositis that we and others have seen in patients on a statin medication is a necrotizing myopathy.61–64 Unlike polymyositis, muscle biopsies in these cases many necrotic fibers without much in the way of endomysial inflammation, except around and within the necrotic fibers. In many instances, the myositis does not improve following discontinuation of the statin medication (after 6 months or more) and only improves after treatment with an immunosuppressant medication. In addition, the myopathy may worsen after discontinuation of the immunosuppressant agent and improve once again upon reinstituting immunotherapy. In addition, as noted previously, autoantibodies directed against the HMG-CoA reductase have been reported in these cases of statin-associated necrotizing myopathies.61,62 These features suggest that these rare cases of necrotizing myopathy do not represent a “toxic” myopathy per se, but a distinct immune-mediated process.
Clofibrate and gemfibrozil are branched-chain fatty acid esters, which are used to treat hyperlipidemia. These fibric acid derivatives can cause a myopathy that typically presents within 2 or 3 months after starting the drug.16,20–30,40 However, the toxic myopathy may develop up to 2 years following initiation of treatment. Affected individuals complain of generalized weakness, myalgias, and cramps. Myoglobinuria is also a rare complication. Patients with renal insufficiency, those taking both clofibrate and gemfibrozil, and especially also those receiving an HMG-CoA inhibitor, are particularly at increased risk of developing myotoxicity.
Elevated serum CK levels are usually noted. Motor and sensory NCS are normal.22,24,28 Needle EMG may demonstrate fibrillation potentials, positive sharp waves, complex repetitive discharges, myotonic discharges, and short-duration, small-amplitude polyphasic MUAPs in affected muscle groups.20,21,25,29,69
Muscle biopsies demonstrate scattered necrotic muscle fibers. In animal models, clofibrate is also known to result in noninflammatory necrosis of muscle tissue with fiber size variation and groups of small atrophic muscle fibers.70
The pathogenic mechanism of the myopathy associated with fibric acid derivatives is not known. These medications might somehow destabilize the lipophilic muscle membrane leading to muscle fiber degeneration.27
Rarely, niacin use is complicated by myalgias and cramps.31 Serum CK levels can be elevated as much as 10-fold. The symptoms improve and CK levels normalize after discontinuation of niacin. Electrodiagnostic studies and muscle biopsies have not been reported in detail. In most cases, rhabdomyolysis occurred in patients who were also taking a statin agent.31 Of note, niacin can inhibit HMG-CoA reductase; therefore, the pathogenic mechanism of the myopathy is likely similar to that of the statins.
Ezetimibe selectively inhibits the absorption of intestinal cholesterol. There are a few reports of ezetimibe-induced myopathy.33,36,71 Similar to other cholesterol-lowering agents, patients may develop hyper-CK-emia with or without myalgias or weakness. Most cases occur in patients who are already on a statin agent, but some occur with Ezetimibe monotherapy. The toxic myopathy usually resolves within a few weeks after the medication is discontinued. However, we have also seen rare cases of what appear to be an immune-mediated necrotizing myopathy as discussed in the statin section in which the myopathy improved only after the affected patients were treated with immunosuppressive agents.
A common question posed to neuromuscular specialists is if patients with known myopathies (e.g., muscular dystrophy, metabolic myopathies, polymyositis) can be treated with cholesterol-lowering agents to control their hypercholesterolemia. There is very little evidence that there is increased risk of statin-induced myotoxicity in patients with an underlying myopathy (aside from those that have the rare statin-induced immune-mediated necrotizing myopathy). Furthermore, there is no strong evidence that statin medications (or other lipid-lowering agents) can exacerbate any underlying myopathy. Given the well-known benefits of statins in patients at risk for cardiovascular disease and lack of any strong evidence of increased risk of these medications in patients with underlying myopathy, we again see no contraindication to their use in most patients. We do tend to follow them closer and periodically check their CK levels.
The immunophilins (i.e., cyclosporine and tacrolimus) are commonly used as immunosuppressive agents, especially in patients requiring transplantation.72 Rarely, generalized myalgias and proximal muscle weakness develop within months after starting these medications.72–77 Myoglobinuria can also occur, particularly in patients receiving cyclosporine or tacrolimus concurrent with cholesterol-lowering agents or colchicine.18,19,78–81 Tacrolimus has also been associated with hypertrophic cardiomyopathy and congestive heart failure.82 Myalgias, muscle strength, and cardiac function improve with reduction or discontinuation of the offending cyclophilin.
Serum CK is usually elevated. NCS are normal. EMG often reveals increased muscle membrane instability with fibrillation potentials, positive sharp waves, and myotonic potentials.74 Early recruitment of small-amplitude, short-duration MUAPs may be demonstrated in weak muscle groups.
Muscle biopsies demonstrate necrosis, vacuoles, and type 2 muscle fiber atrophy.
The pathogenic basis of immunophilin-induced myopathy and cardiomyopathy is not known. Perhaps, the agents destabilize the lipophilic muscle membrane leading to muscle fiber degeneration, similar to the cholesterol-lowering agents. In this regard, cyclosporine itself has a cholesterol-lowering effect. This may explain the increased risk of myopathy in patients receiving cyclosporine and the more classic lipid-lowering agents (e.g., fibric acid derivatives and statins).
There are a few reports of necrotizing myopathy associated with the use of the antihypertensive agent, labetalol.83,84 Patients can develop acute or insidious onset of proximal weakness or myalgias, which resolve following discontinuation of the medication.
Serum CK can be markedly elevated. EMG may demonstrate increased insertional and spontaneous activity with fibrillation potentials and positive sharp waves. Short-duration, small-amplitude, polyphasic MUAPs, which recruit early, are evident.
Routine light microscopy can be normal83 or can reveal necrotic and regenerating fibers.84 Electron microscopy (EM) revealed subsarcolemmal vacuoles in one case.83
The pathogenic etiology for the muscle necrosis seen is not known.
Propofol is an anesthetic agent that is frequently used for sedating patients who are mechanically ventilated and sometimes for the treatment of status epilepticus. Myoglobinuria, metabolic acidosis, hypoxia, and myocardial arrest are rare adverse events associated with the use of propofol.85–87 Propofol does not appear to be associated with malignant hyperthermia. Acute quadriplegic myopathy (AQM) in the intensive care unit (ICU) has also developed in patients treated with propofol in combination with high-dose intravenous corticosteroids.88 The myopathy in these individuals could be explained by the high-dose corticosteroids rather than the use of propofol. It remains to be determined if propofol is an independent risk factor for the development of AQM.
Serum CK levels are markedly elevated. Electrophysiologic studies have not been performed or were not reported in the cases associated with rhabdomyolysis in children. The adult patients with AQM have low-amplitude compound muscle action potentials (CMAPs), profuse fibrillation potentials, positive sharp waves, and early recruitment of short-duration, small-amplitude polyphasic MUAPs.88
Muscle biopsies reveal necrosis of skeletal and cardiac muscle.85–87 Patients with AQM, may have prominent necrosis and loss of thick filaments.88
The mechanism for muscle destruction is unknown.
Propofol should be discontinued and supportive therapy instituted for myoglobinuria, metabolic acidosis, hyperkalemia, and renal failure.
Amphiphilic drugs contain separate hydrophobic and hydrophilic regions, which allow the drugs to interact with the anionic phospholipids of cell membranes and organelles. In addition to a myopathy, these agents can also cause a toxic neuropathy that is even more severe than the direct toxicity on muscle.
Chloroquine, a quinoline derivative, is used to treat malaria, sarcoidosis, systemic lupus erythematosus (SLE), and other connective tissue diseases.7,8,89–92 Some patients develop slowly progressive, painless, proximal weakness and atrophy, which affect the legs more than the arms. A cardiomyopathy can also occur. Sensation is often reduced as are muscle stretch reflexes, particularly at the ankles, secondary to a concomitant neuropathy. This “neuromyopathy” usually does not occur unless patients take 500 mg a day for a year or more but has been reported with doses as low as 200 mg/day. The neuromyopathy improves after chloroquine discontinuation.
Serum CK levels are usually elevated. Motor and sensory NCS reveal mild-to-moderate reduction in the amplitudes with slightly slow velocities in patients with a superimposed neuropathy.90,92 Individuals with only the myopathy usually have normal motor and sensory studies.89 Increased insertional activity in the form of positive sharp waves, fibrillation potentials, and myotonic discharges are seen primarily, but not exclusively, in the proximal limb muscles.89,90,92 Early recruitment of small-amplitude, short-duration polyphasic MUAPs are appreciated in weak proximal muscles. Neurogenic appearing units and reduced recruitment may be seen in distal muscles that are more affected by the toxic neuropathy.
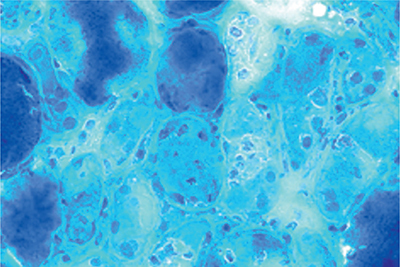
Autophagic vacuoles are evident in as many as 50% of skeletal and cardiac muscle fibers (Fig. 35-3).7,8,89,91,92 Type 1 fibers appear to be preferentially affected. The vacuoles stain positive for acid phosphatase, suggesting lysosomal origin. On EM, the vacuoles are noted to contain concentric lamellar myeloid debris and curvilinear structures. Autophagic vacuoles are also evident in nerve biopsies.
Figure 35-3. Chloroquine myopathy. Chloroquine can cause a vacuolar myopathy (A), hematoxylin and eosin (H&E). Electron microscopy reveals a bundle of dilated tubules (B). (Reproduced with permission from Wasay M, Wolfe GI, Herrold JM, Burns DK, Barohn RJ. Chloroquine myopathy and neuropathy with elevated CSF protein. Neurology. 1998;51(4):1226–1227.)
Chloroquine is believed to interact with lipid membranes, forming drug–lipid complexes that are resistant to digestion by lysosomal enzymes. This results in the formation of the autophagic vacuoles filled with myeloid debris.
Hydroxychloroquine is structurally similar to chloroquine and can cause a neuromyopathy.90 The myopathy is usually not as severe as seen in chloroquine. Vacuoles are less appreciated on routine light microscopy, but EM still usually demonstrates the abnormal accumulation of myeloid and curvilinear bodies.
Amiodarone is an antiarrhythmic medication that can also cause a neuromyopathy.93–97 The neuromyopathy is characterized by severe proximal and distal weakness along with distal sensory loss and reduced muscle stretch reflexes. The legs are more affected than the arms. Some patients develop tremor or ataxia. Amiodarone can also cause hypothyroidism, which may also contribute to proximal weakness. Patients with renal insufficiency are predisposed to developing the toxic neuromyopathy. Muscle strength gradually improves following discontinuation of amiodarone.
Serum CK levels are elevated. Motor and sensory NCS reveal reduced amplitudes and slow conduction velocities, particularly in the lower extremities.96,97 EMG demonstrates fibrillation potentials and positive sharp waves in proximal and distal muscles. In proximal muscles, MUAPs are typically polyphasic, short in duration, small in amplitude, and recruit early. Distal muscles are more likely to have large-amplitude, long-duration polyphasic MUAPs with decreased recruitment.
Muscle biopsies demonstrate scattered fibers with autophagic vacuoles. In addition, neurogenic atrophy can also be appreciated, particularly in distal muscles. EM reveals myofibrillar disorganization and autophagic vacuoles filled with myeloid debris. Myeloid inclusions are also apparent on nerve biopsies. These lipid membrane inclusions may be evident in muscle and nerve biopsies as long as 2 years following discontinuation of amiodarone.
The pathogenesis is presumably similar to other amphiphilic medications (e.g., chloroquine).
ANTIMICROTUBULAR MYOPATHIESColchicine is commonly prescribed for individuals with gout. Colchicine can also cause a generalized toxic neuromyopathy. It is weakly amphiphilic, but its toxic effect is believed to arise secondary to its binding with tubulin and prevention of tubulin’s polymerization into microtubular structures.5,8,9 The neuromyopathy usually develops after chronic administration, but it can also develop secondary to acute intoxication.5,98–100 Chronic renal failure and age over 50 years are risk factors for the development of neuromyopathy. Patients usually manifest with progressive proximal muscle weakness over several months. Clinical myotonia has been described.101 A superimposed toxic neuropathy leads to distal sensory loss as well as diminished reflexes. The neuromyopathy weakness typically resolves within 4–6 months after discontinuing the colchicine.
Serum CK level is elevated up to 50-fold in symptomatic patients. Serum CK may also be mildly elevated in asymptomatic patients taking colchicine. NCS reveal reduced amplitudes, slightly prolonged latencies, and mildly slow conduction velocities of motor and sensory nerves in the arms and legs.98–100,102 Needle EMG demonstrates positive sharp waves, fibrillation potentials, and complex repetitive discharges, which are detected with ease in all muscle regions. Myotonic discharges may also be seen.101 The myopathic MUAP abnormalities can be masked in the distal limb muscles secondary to the superimposed peripheral neuropathy.
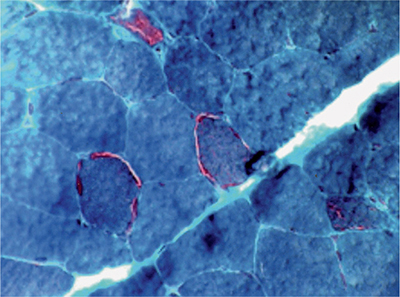
Muscle biopsy demonstrates acid phosphatase–positive autophagic vacuoles containing membranous debris (Fig. 35-4). In addition, nerve biopsies can reveal evidence suggestive of a mild axonal neuropathy.
Figure 35-4. Colchicine myopathy. Colchicine can cause a vacuolar myopathy as evident on modified Gomori trichrome stain (A) and hematoxylin and eosin stain (B).
The abnormal assembly of microtubules most likely disrupts intracellular movement or localization of lysosomes, leading to the accumulation of autophagic vacuoles.99
Vincristine is a chemotherapeutic agent, which disrupts gene transcription and also promotes the polymerization of tubulin into microtubules.8 The dose limiting side effect of vincristine is a toxic axonal sensorimotor polyneuropathy that is associated with distal muscle weakness and sensory loss. Proximal muscle weakness and myalgias are less common.103
Serum CK levels have not been reported in patients suspected of having a superimposed myopathy. NCS demonstrate markedly reduced amplitudes of SNAPs and CMAPs, while the distal latencies are slightly prolonged, and conduction velocities are mildly slow.103 Needle EMG demonstrates positive sharp waves, fibrillation potentials, and neurogenic appearing MUAPs in the distally located muscles of the upper and lower extremities.
Biopsies of distal muscles demonstrate evidence of neurogenic atrophy and, occasionally, the accumulation of lipofuscin granules. Proximal muscle biopsies reveal scattered necrotic fibers.103 On EM, there is prominent myofibrillar disarray and subsarcolemmal accumulation of osmiophilic material. In addition, some myonuclei contain membrane-bound inclusions. Autophagic vacuoles with spheromembranous debris have been noted in research animals104,105 but have not been appreciated in humans.103
The pathogenic basis of the neuromyopathy is presumably similar to that of colchicine.
DRUG-INDUCED MITOCHONDRIAL MYOPATHYPatients with azidothymidine (AZT) myopathy usually present with an insidious onset of progressive proximal muscle weakness and myalgias.106–116 However, these clinical features do not help in distinguishing AZT myopathy from other HIV-related myopathies. Other myopathies related to HIV infection are heterogeneous and include inflammatory myopathy, microvasculitis, noninflammatory necrotizing myopathy, and type 2 muscle fiber atrophy secondary to disuse or wasting due to their chronic debilitated state.107,110–112,114–124 Furthermore, weakness in an HIV-infected patient can also be secondary to peripheral neuropathy (e.g., chronic inflammatory demyelinating polyneuropathy) or myasthenia gravis. Clinically, AZT myopathy and the other myopathic disorders associated with HIV infection are indistinguishable, compounding the diagnostic difficulty. Regardless of etiology of the myopathy, patients manifest with progressive proximal muscle weakness and myalgias. In addition, muscle weakness may be multifactorial.
Serum CK levels are normal or only mildly elevated in AZT myopathy. However, similar elevations are evident in other forms of HIV-related myopathy. An elevated serum CK (e.g., greater than five times the upper limited of normal) is more suggestive of an HIV-associated myositis. Motor and sensory NCS are normal, unless there is a concomitant peripheral neuropathy. Needle EMG may demonstrate positive sharp waves and fibrillation potentials and early recruitment of short-duration, small-amplitude polyphasic MUAPs.114,119,122,125 In addition, small polyphasic MUAPs with early recruitment but no abnormal spontaneous activity was reported in patients with AIDS, along with ultrastructural mitochondrial abnormalities but no inflammation or nemaline rods on biopsy.112
Muscle biopsies are remarkable for the presence of ragged red fibers, suggesting mitochondrial abnormalities in AZT myopathy (Fig. 35-5). The number of ragged red fibers correlates with the cumulative dose of AZT.110,111 In addition, necrotic fibers, cytoplasmic bodies, nemaline rods, and fibers with microvacuolation may be seen in addition to ragged red fibers.107,110,111 In contrast to HIV-associated inflammatory myopathy, significant endomysial inflammation with or without invasion of non-necrotic fibers should not be present in cases of pure AZT myopathy. EM reveals abnormalities of the mitochondria and myofilaments.
Figure 35-5. Azidothymidine myopathy. Ragged red fibers suggestive of abnormal mitochondria are evident on modified Gomori trichrome stain.
AZT acts as a false substitute for the viral reverse transcriptase, thereby inhibiting its enzymatic activity and replication of the HIV virus. However, AZT also inhibits the activity of mitochondrial DNA polymerase, which probably accounts for the mitochondrial abnormalities. When treated with AZT, patients with HIV have a decrease in quantity of mitochondrial DNA and decline in respiratory chain enzymatic activity, compared to untreated infected patients.121,126 The histological and molecular abnormalities on repeated muscle biopsies resolve coinciding with clinical improvement following discontinuation of AZT.127 Although, AZT is responsible for at least some of the mitochondrial abnormalities evident on muscle biopsy, the contribution of these mitochondrial abnormalities to the muscle weakness remains controversial.
In the past, anywhere from 18% to 100% of patients with “AZT” myopathy improved following discontinuation of the medication.107,110,112,114,116,119,120,128 AZT is not used as much anymore as other antiviral agents are typical used nowadays in the treatment of HIV (see below).
The risk of mitochondrial myopathy with other nucleoside reverse transcriptase inhibitors (e.g., lamivudine), zalcitabine, didanosine is probably less than that of AZT.129,130 However, these agents are clearly associated with mitochondrial toxicity, and patients may develop associated hyperlactemia and hepatic steatosis on these medications. The AIDS Clinical Trial group randomized 2,467 patients to receive one of four single or combination regimens with AZT, didanosine, zalcitabine, and their respective placebo.123 Approximately 10% of patients had myalgias prior to treatment and 7% developed myalgia during treatment. There was no significant difference between treatment arms and the rate of myalgia or muscle weakness in any group. Five patients (0.5%) had elevated serum CK (>4× normal) prior to treatment, and 52 (5%) developed increased CK during treatment. Serum CK levels were significantly higher in the AZT-zalcitabine group, but this did not correlate with symptoms of myopathy. Unfortunately, there was no comment on muscle biopsies, and thus it is unclear if the myopathies were secondary to mitochondrial toxicity or myositis.
The main treatment of HIV infection currently is with highly active antiretroviral therapy (HAART) consisting of a combination of nucleoside reverse transcriptase inhibitors and protease inhibitors. Rare cases of rhabdomyolysis and myoglobinuria occur in patients taking other HAART medications including tenofovir131 and ritonavir.132 A review of 563 patients between 1995 and 1998 demonstrated a prevalence of “HIV-associated myopathy” in 1.5% of patients treated with HAART.91 It was not clearly stated how the myopathy was defined (e.g., clinical symptoms or signs, elevated serum CK, EMG, or biopsy). Further, it is unclear if the myopathy was felt to be due to mitochondrial toxicity, myositis, or wasting syndrome.
DRUG-INDUCED INFLAMMATORY MYOPATHIESAs discussed in the Necrotizing Myopathies section, dermatomyositis, polymyositis, and in particular, an immune-mediated necrotizing myopathy rarely occur in patients taking statin medications and occasionally the other cholesterol lowering agents.61–64 These inflammatory myopathies do not improve with the discontinuation of the cholesterol-lowering agent. Rather, patients need to be treated with immunotherapy.
Eosinophilia–myalgia syndrome was described in the late 1980s and early 1990s and was found to be caused by a contaminant used in the production of L-tryptophan.133–139 The clinical, laboratory, electrophysiological, and histopathological features were similar to that seen in diffuse fasciitis with eosinophilia (Shulman syndrome).140 Patients developed a subacute onset of generalized muscle pain and tenderness with variable degrees of weakness. Onset could have been within a few weeks or after several years of starting tryptophan. Numbness, paresthesias, arthralgias, lymphadenopathy, dyspnea, abdominal pain, mucocutaneous ulcers, and an erythematous rash were also common. Some patients developed a severe generalized sensorimotor polyneuropathy mimicking Guillain–Barré syndrome138,141 or multiple mononeuropathies suggestive of a vasculitis.142
Serum CK level are normal or elevated. Autoantibodies are absent and ESR is usually normal. The absolute eosinophil count was elevated (>1 × 109 cells/L). Decreased amplitudes of compound muscle and sensory nerve action potentials (SNAPs) with normal or mildly reduced conduction velocities are evident in patients with a polyneuropathy.138,143 A few patients with severe Guillain–Barré syndrome had electrophysiologic studies showing multifocal conduction block and slowing of conduction velocities.141 Needle EMG revealed muscle membrane instability in the form of fibrillation potentials, positive sharp waves, and complex repetitive discharges.136,138,141 Small and large polyphasic MUAPs with early recruitment are seen as a result of the chronic myopathy.136,138 Large polyphasic MUAPs with decreased recruitment are seen in patients with severe neuropathy.138 The electrophysiological abnormalities improve with discontinuation of tryptophan.
Muscle biopsies demonstrated diffuse or perivascular inflammatory infiltrate in the fascia, perimysium, and, to a lesser extent, in the endomysium.138 The majority of inflammatory cells are CD8+ T cells and macrophages, while eosinophils and B cells comprised <3% of the infiltrating cells. Unlike DM, there is no deposition of membrane attack complex on small blood vessels. Nerve biopsies show predominately perivascular inflammatory infiltrates, mainly mononuclear, with occasional eosinophils in the epineurium, endoneurium, and/or perineurium with axonal degeneration.136,138,141,143,144
The disorder was caused by a contaminant(s) in the manufacture of tryptophan. Two trace adulterants have been identified as the possible toxins: 3-phenylaminoalanine and 1,1′-ethylidenebis tryptophan.145 The mechanism by which this contaminant resulted in the disorder is unknown, but the eosinophilia and eosinophilic infiltrate in tissues suggest some form of allergic reaction.
Discontinuation of L-tryptophan and treatment with high-dose corticosteroids were usually effective in the prior epidemic. Some patients experienced relapses upon withdrawal of steroids.
The toxic oil syndrome occurred as a single epidemic in Spain and has not recurred since 1981. It was quite similar to the eosinophilia–myalgia syndrome associated with tryptophan.146 The disorder was linked to the ingestion of illegally marked, denatured rapeseed oil as a cooking substitute for olive oil. Interestingly, the toxic contaminant in the rapeseed oil, 3-phenylamino-1, 2-propanediol, is chemically similar to 3-phenylaminoalanine, the presumed adulterant in tryptophan causing the eosinophilia–myalgia syndrome.145
D-Penicillamine is rarely used nowadays to treat Wilson disease, rheumatoid arthritis (RA), and other connective tissue disorders. Approximately 0.2–1.4% of patients treated with D-penicillamine developed an inflammatory myopathy reminiscent of polymyositis or dermatomyositis.147–150 It has also been associated with myasthenia. Discontinuation of the drug results in resolution of the symptoms. The medication may be restarted at a lower dosage without recurrence of the inflammatory myopathy.
Rare cases of inflammatory myopathy have been reported with cimetidine, a histamine H2 receptor antagonist.151 One patient developed generalized weakness and myalgias associated with CK elevations up to 40,000 U/L and interstitial nephritis. The muscle biopsy revealed perivascular inflammation, predominantly consisting of CD8+ lymphocytes. No deposition of immunoglobulin or complement on small blood vessels was noted, nor did the patients have a cutaneous rash to suggest dermatomyositis. However, cases of cutaneous vasculitis have been described with cimetidine use.152
Proximal muscle weakness and myalgias rarely occur with procainamide usage.41,153 Serum CK levels are elevated, and EMG has been reported as being consistent with a “patchy” myopathy. Muscle biopsies demonstrate perivascular inflammation and rare necrotic muscle fibers. The pathogenesis may be related to lupus-like vasculitis, which can occur in patients treated with procainamide. The myopathy resolves following withdrawal of procainamide.
A single case of proximal muscle weakness and myalgias has been reported in a patient with Parkinson disease treated with L-Dopa.154 The patient developed the muscle symptoms after treatment with L-Dopa for over 4 years. The serum CK was elevated 10-fold. Gastrocnemius and quadriceps muscle biopsies revealed perivascular inflammation and rare necrotic fibers. The authors suggested that the patient developed a hypersensitivity vasculitis to the L-Dopa; however, it is more likely that the myositis occurred incidentally.
Myalgias and weakness may develop in patients treated with phenytoin due to hypersensitivity reactions.155 Serum CK levels can be elevated, and muscle biopsies show scattered necrotic and regenerating muscle fibers. EMG can reveal increased spontaneous activity with fibrillation potentials and positive sharp waves. Small-amplitude, short-duration, polyphasic MUAPs, which recruit early may be observed. The myopathy improves with discontinuation of the phenytoin and a short course of corticosteroids.
We have seen a case of severe myoglobinuria and renal failure associated with a generalized rash, anemia, leukopenia, and thrombocytopenia shortly after the patient was started on lamotrigine. The clinical and laboratory features resembled thrombocytic thrombocytopenic purpura. The patient improved with plasmapheresis and discontinuation of lamotrigine.
Alpha-interferon is used in the treatment of viral hepatitis and certain malignancies [e.g., chronic myelogenous leukemia (CML) and melanoma]. A rare side effect of alpha-interferon is the occurrence of autoimmune disorders including myasthenia gravis and myositis.156–158 Further, as discussed in Chapter 33, the overproduction of type 1 interferons, such as alpha-interferon, have been implicated in the pathogenesis of dermatomyositis.
Tumor necrosis factor alpha (TNF-α) blockers are used to treat RA, ankylosing spondylitis, and psoriatic arthritis. Side effects of TNF-α blockers include induction of other autoimmune disorders such as SLE and autoimmune neuropathies. Some patients with myositis patients treated with various TNF-α blockers improve, while others worsen.159 In addition, there are reports of patients with previous history of myositis, who develop an inflammatory myopathy while being treated with a TNF-α blocker.160,161
Imatinib mesylate is a tyrosine kinase inhibitor used to treat patients with CML. Imatinib inhibits the tyrosine kinase activity of the BCR-ABL oncoprotein in CML. Imatinib is well tolerated, but myalgias occur in 21–52% of patients. We reported a patient with CML who developed polymyositis while taking imatinib.162 CML28 antibodies were detected in the patient’s serum. CML28 is identical to hRrp46p, a component of the human exosome, a multiprotein complex involved in processing of RNA. Antibodies directed against hRrp46p and other components of the human exosome (e.g., PM-Scl 100 and PMScl 75) have been noted in patients with polymyositis (see Chapter 33). The patient’s strength and serum CK normalized with discontinuation of the imatinib and a course of corticosteroids.
Tyrosine kinases are involved in signal transduction, cell growth, and differentiation. The mechanism by which imatinib therapy could cause myositis is unclear. The previous use of an immunomodulatory agent (i.e., alpha-interferon) followed by imatinib leads to rapid apoptosis of leukemic cells. The subsequent release of a large bolus of leukemia antigens may have cross-reactivity with muscle antigens and generate an autoimmune response.
MYOPATHIES SECONDARY TO IMPAIRED PROTEIN SYNTHESIS OR INCREASED CATABOLISMSteroid myopathy manifests as proximal muscle weakness and atrophy affecting the legs more than the arms.163–170 The distal extremities, oculobulbar, and facial muscles are normal as are sensation and muscle stretch reflexes. Most patients exhibit a Cushingoid appearance with facial edema and increased truncal adipose tissue. Prednisone at doses of 30 mg/day or more (or equivalent doses of other corticosteroids) is associated with an increased risk of myopathy.167 Any synthetic glucocorticoid can cause the myopathy, but those that are fluorinated (triamcinolone > betamethasone > dexamethasone) are more likely to result in muscle weakness than the nonflourinated compounds.171 Women appear to be more at risk than men (approximately 2:1) of developing a steroid myopathy. Alternate-day dosing may reduce the risk of corticosteroid-induced weakness.
Muscle weakness can develop within several weeks following the administration of corticosteroids; however, it more commonly occurs as a complication of chronic administration of oral high–dose corticosteroids. Acute onset of severe generalized weakness can occur in patients receiving high dosages of intravenous corticosteroids with or without concomitant administration of neuromuscular blocking agents or sepsis (see section regarding Acute Quadriplegic Myopathy).
Serum CK is normal. Serum potassium can be low as a result of glucocorticoid excess and can cause some degree of weakness. Motor and sensory nerve conductions are normal in steroid myopathy.172 Repetitive stimulation studies should not demonstrate a significant decrement or increment. Needle EMG is normal as well.
The paucity of abnormalities is understandable, as corticosteroids preferentially affect type 2 muscle fibers. The first recruited motor units are comprised of type 1 muscle fibers. Because these are not affected as severely as type 2 fibers, there is little in the way of electrophysiological abnormality to observe.
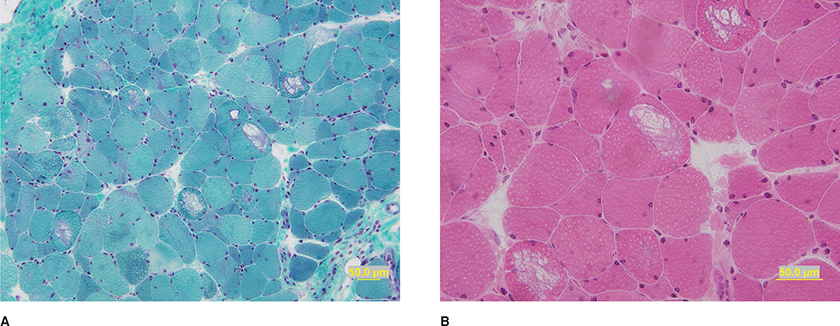
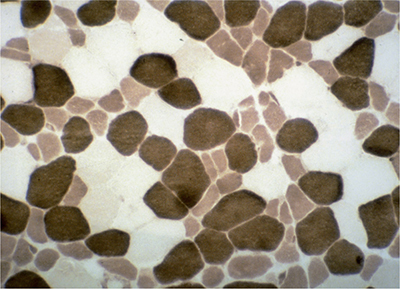
Muscle biopsies reveal atrophy of type 2 fibers, especially the fast-twitch, glycolytic-type 2B fibers (Fig. 35-6).166,167,173,174 There may also be a lesser degree of atrophy of type 1 muscle fibers. Lipid droplets are commonly noted in type 1 fibers, and rare mitochondrial abnormalities have been seen on EM.
Figure 35-6. Steroid myopathy. Selective atrophy of the intermediately staining type 2B fibers is evident. ATPase pH 4.5.
Corticosteroids bind to receptors on target cells and are subsequently internalized into the nuclei, where these regulate the transcription of specific genes. How corticosteroids cause a myopathy is not known, but could be the result of decreased protein synthesis, increased protein degradation, alterations in carbohydrate metabolism, mitochondrial alterations, or reduced sarcolemmal excitability.166,167
Reduction in the dose, tapering to an alternate-day regimen, or switching to a nonflourinated steroid along with a low carbohydrate diet and exercise to prevent concomitant disuse atrophy are major modes of therapy.167,171,175
Of particular concern is to distinguish steroid myopathy from an exacerbation of underlying immune-mediated neuromuscular disorder (e.g., inflammatory myopathy, myasthenia gravis, and chronic inflammatory demyelinating polyneuropathy) in a patient being treated with corticosteroids.169,175–177 If the weakness occurs while the patient is tapering the corticosteroid, relapse of the underlying disease process would be most likely. In contrast, if weakness developed while the patient was on chronic high doses of steroids, a steroid myopathy should be considered. In the case of an inflammatory myopathy, an increasing serum CK and an EMG with prominent increase in insertional and spontaneous activity would point to an exacerbation of the myositis.175 In some cases, it is impossible to state with certainty whether the new weakness is related to a relapse of the underlying disease or secondary to the corticosteroid treatment. In such cases, we taper the corticosteroid medication and closely observe the patient. If muscle strength improves presumably, the patient had a steroid myopathy. If patient’s strength declines then more likely the weakness is caused by an exacerbation of the underlying autoimmune disease and requires increased doses of corticosteroids or other immunosuppressive medication.
Finasteride is used to treat benign prostatic hypertrophy. It is a 4-azasteroid that inhibits 5α-reductase, and thus blocks dihydrotestosterone production and androgen action in the prostate and skin. One patient developed severe proximal greater than distal weakness and atrophy while being treated with finasteride (5 mg qd).178 Sensation and muscle stretch reflexes were normal.
Serum CK levels were normal.
NCS were normal, while the EMG demonstrated showed small polyphasic MUAPs.
Muscle biopsy revealed only mild variability in fiber size, type 2 muscle fiber atrophy, and increased central nuclei.
The pathophysiologic mechanism for the myopathy is not known. Finasteride is one of the 4-azasteroids and its parent compound, as well as the metabolites, has structural similarity to corticosteroids. Thus, the pathogenic mechanism may be similar to that seen of steroid myopathy.
Discontinuation of finasteride was associated with normalization of strength and improvement in EMG abnormalities.
Emetine hydrochloride is an emetic agent that has been abused, particularly in patients with anorexia nervosa and bulimia. A severe proximal myopathy and cardiomyopathy can occur with overuse of emetine (500–600 mg/day for over 10 days).179–181 Patients also complain of muscle pain, tenderness, and stiffness. Muscle stretch reflexes are usually diminished, but the sensory examination is completely normal. The myopathy is reversible following discontinuation of the medication.
The serum CK levels may be mildly to moderately elevated. Sensory and motor NCS are normal.179–181 Needle EMG examination can be normal although, positive sharp waves and fibrillation potentials are usually apparent. There is early recruitment of small-amplitude, short-duration MUAPs.
Muscle biopsies reveal scattered necrotic fibers, small atrophic and regenerating fibers as well as many fibers containing cytoplasmic bodies. Oxidative enzyme stains demonstrate targetoid or moth-eaten structures. On EM, there is evidence of myofibrillar degeneration in addition to compacted myofibrillar debris (cytoplasmic bodies). The histological appearance of light microscopy and EM is similar to myofibrillar myopathy (see Chapter 27).182,183
The exact pathogenic basis for the disorder is not known, but it is postulated that emetine might inhibit the synthesis of important muscle proteins.
The myopathy resolves following discontinued use of emetine.
TOXIC MYOPATHIES WITH MULTIFACTORIAL OR UNKNOWN PATHOGENIC MECHANISMSPatients in the ICU may develop generalized weakness due to critical illness polyneuropathy,184,185 prolonged neuromuscular blockage,186,187 or secondary to a myopathic process. The myopathic disorder has been termed AQM, acute illness myopathy, critical illness myopathy, and myopathy associated with thick filament (myosin loss).88,187–201 It can be difficult to differentiate AQM from critical illness neuropathy or prolonged neuromuscular blockade, and patients can potentially have a combination of the above. Some series of ICU weakness report critical illness neuropathy to be more frequent than AQM,202,203 while others found the myopathy to be more prevalent.194,198,204 In the largest series involving 88 patients who developed weakness while in an ICU, AQM was three times as common as critical illness neuropathy (42% vs. 13%); prolonged neuromuscular blockade occurred in only one patient who also had AQM.194 In our experience, AQM is much more common than critical illness neuropathy.
The first reported case of AQM was a 24-year-old woman with status asthmaticus who developed severe generalized weakness following treatment with high doses of intravenous corticosteroids and neuromuscular blockade.196 Subsequently, there have been numerous reports of AQM usually developing in patients who received high-dose intravenous corticosteroids and/or nondepolarizing neuromuscular blockers.88,187–189,192,193–195,197,198–201,205–207 The myopathy can also develop in patients with sepsis or multiorgan failure who never received either corticosteroids or nondepolarizing neuromuscular blocking agents.190,191,200 Recent organ transplantation appears to be at increased risk factor for the development of AQM, perhaps related to high doses of intravenous corticosteroids used for prevention of rejection and neuromuscular blocking agents in the perioperative period. Because of their immunosuppressed state, patients undergoing transplant are also prone to infection and sepsis, which also predisposes to AQM.
The incidence of AQM is uncertain because there have been only a few prospective series published on the subject.205,208 In a study of 25 consecutive patients requiring mechanical ventilation for severe asthma, myopathy developed in 9/25 (36%) and elevated serum CK levels in 19/22 (76%) of patients tested.208 The patients were treated with dexamethasone 10 mg every 8 hours or hydrocortisone 250 mg every 6 hours; 22 of the 25 patients also received vecuronium. Mechanical ventilation lasted an average of 3.1 +/– 3.1 days in patients without myopathy and 12.9 +/– 6.6 days in those with myopathy. In a prospective study of 100 consecutive adult patients undergoing liver transplantation, 7 patients developed AQM.205 Patients were treated with nondepolarizing neuromuscular blocking agents and high-dose steroids in the perioperative period. Three of six patients tested had elevated serum CK levels, as high as 10 times the upper limit of normal by the twenty-fifth postoperative day. Four patients had muscle biopsies demonstrating necrosis and selected loss of myosin (see below). Three patients later died from sepsis and multiorgan failure. The remaining patients slowly regained strength and the ability to ambulate over 1–3 months.
Patients with AQM usually exhibit severe generalized weakness of the trunk, extremities, and respiratory muscles which can rarely involve the extraocular muscles.201,207 The myopathy is usually initially recognized by the inability to wean the patient from the ventilator. Sensory examination is usually normal, but this of course can be difficult to determine in an intubated patient with altered mental status. Muscle reflexes are decreased or absent. The mortality is high, approximately 30% in one large series, secondary to multiple organ failure and sepsis rather than the myopathy.194 The morbidity and mortality in AQM and critical illness neuropathy appear to be similar.194 In patients who survive, muscle strength recovers slowly over several months.
Serum CK levels can be normal but are moderately elevated in about 50% of patients. NCS reveal marked CMAP amplitude reduction with normal distal latencies and conduction velocities. In contrast, SNAP amplitudes should be normal or mildly reduced (>80% of the lower limit of normal) in comparison. Markedly reduced amplitudes of SNAPs should lead to the consideration of critical illness myopathy. However, the SNAPs may be affected, if the patient has a baseline (unrelated neuropathy), and many of these patients have illnesses that are associated with neuropathy (diabetes mellitus and renal or liver failure). SNAPs in the lower extremities may be obscured by edema as well. Thus, reduced SNAP amplitudes in and of itself does not exclude AQM.
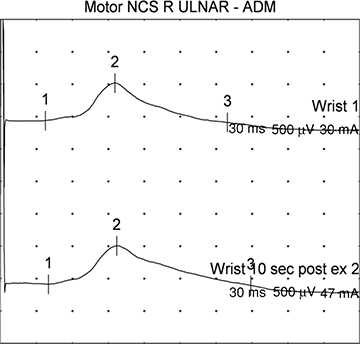
Direct muscle stimulation may help to distinguish AQM from critical illness myopathy, but these studies are fraught with the possibility of technical error.198,199,209 Direct muscle stimulation bypasses the distal motor nerve and neuromuscular junction. In critical illness neuropathy or prolonged neuromuscular blockade, the muscle membranes should retain their excitability, and direct muscle stimulation CMAP (dmCMAP) should be near normal despite a low or absent nerve stimulation-evoked CMAP (neCMAP). In contrast, if the muscle membrane excitability is reduced as seen in AQM, both the neCMAP and the dmCMAP should be very low. Theoretically, the ratio of neCMAP to dmCMAP should be close to 1:1 in a myopathy and should approach zero in a neuropathy or neuromuscular junction disorder. In this regard, absent or reduced amplitudes of the dmCMAP with neCMAP/dmCMAP ratios >0.9 were demonstrated in 11 patients with AQM, while neCMAP/dmCMAP ratios were 0.5 or less in patients with severe neuropathy.198,199 Significant slowing of muscle-fiber conduction velocity (MFCV) and muscle-fiber conduction block during the acute phase of AQM can be demonstrated. This correlates with prolonged CMAP duration, clinical severity, and course (Fig. 35-7).210
Figure 35-7. Synchronous dispersion causing a reduced amplitude and a long tail of the negative phase of the CMAP resulting in a prolonged duration of the ulnar CMAP (15 ms) from a patient with critical illness myopathy.
EMG usually demonstrates prominent fibrillation potentials and positive sharp waves; however, abnormal spontaneous activity is not always evident. Early recruitment of short-duration, small-amplitude, polyphasic MUAPs may be seen if the patient has sufficient strength to generate any MUAPs. Patients with severe weakness may be unable to volitionally recruit any MUAP. The inability to quantitate MUAP morphology and recruitment can make it difficult to distinguish AQM from critical illness neuropathy in patients who may have incidentally abnormal sensory conduction studies. Sequential EMG studies in AQM have reported profuse spontaneous activity and inability to actively recruit MUAPs early, followed by the appearance of small polyphasic MUAPs with early recruitment during the recovery period.206
Muscle biopsies reveal a wide spectrum of histological abnormalities (Fig. 35-8). Type 2 muscle fiber atrophy with or without type 1 fiber atrophy is common.187,188,191,192,194,200,201,207 Scattered necrotic muscle fibers may be seen.194,197,200,205,207 Focal or diffuse loss of reactivity for myosin ATPase activity in type 1 fibers more than type 2 fibers, corresponding to the loss of thick filaments (myosin) apparent on EM, is typically observed (Fig. 32-6).187,189,190,192–194,200,205 Other structural proteins (actin, titin, and nebulin) are relatively spared.200
Figure 35-8. Critical illness/acute quadriplegic myopathy. Muscle biopsy demonstrates marked degeneration and atrophy of muscle fibers on modified Gomori trichrome (A). Electron microscopy demonstrates a muscle fiber with a preserved sarcomeres adjacent to a degenerating muscle fiber (B). Higher power view on EM reveals the preserved Z-disc and thin filaments but the loss of the myosin thick filaments (C).
The variable laboratory, histological, and electrophysiological features suggest that the pathogenesis is multifactorial. Some biopsies demonstrate widespread necrosis, which certainly can account for the muscle weakness observed in patients. The mechanism of muscle fiber necrosis is not known, and, importantly, not all patients have this feature on biopsy. Myosin is selectively lost in some but not all patients. Calcium-activated proteases (calpains) may be responsible for proteolysis of myosin.200 Perhaps, glucocorticoids, nondepolarizing neuromuscular agents, or the milieu of critical illness induces the expression of calpains. In addition, the enhanced expression of cytokines during sepsis may, in turn, lead to a catabolic state in muscle with breakdown of proteins, glycogen, and lipid.
The reduced muscle membrane excitability may be the result of a combination of several factors: (1) partial depolarization of the resting membrane potential, (2) reduced muscle membrane resistance, and (3) decreased sodium currents.198,199,209,211 Denervation and neuromuscular blockade normally decrease the resting membrane muscle potential, increase membrane resistance secondary to decreased chloride conductance, and increase the number of sodium channels on the muscle membrane. In denervated rats treated with corticosteroids, the resting membrane potential does not significantly decrease but muscle membrane resistance decreases (rather than increase) as a result of increased chloride conductance. The reduced membrane resistance decreases the depolarization caused by the opening of sodium channels. In addition, there is diminished sodium current secondary to a reduction in the number of sodium channels, decreased sodium channel conductance, or impaired voltage-dependent gating.
Supportive care and treating underlying systemic abnormalities (e.g., antibiotics in sepsis and dialysis in renal failure) are the only modes of therapy. Corticosteroids or nondepolarizing neuromuscular blockers should be discontinued if possible. Patients require extensive physical and occupational therapy to prevent contractures and help regain muscle strength and functional abilities.
Omeprazole inhibits the H+/K+ ATPase enzyme system (the proton pump) at the secretory surface of the gastric parietal cell and is used for the treatment of gastric and duodenal ulcers and reflux. Rare cases of neuromyopathy have been reported with the use of omeprazole.212,213 Patients develop proximal weakness and myalgias along with paresthesias and a stocking distribution of sensory loss, predominantly in the legs. Muscle reflexes are diminished or absent.
Serum CK levels are normal or mildly elevated. NCS may be normal or reveal an axonal sensorimotor polyneuropathy.212 EMG can be normal or show small polyphasic MUAPs.213
Muscle biopsies in the two reported patients revealed only type 2 muscle fiber atrophy.212,213 Superficial peroneal nerve biopsy in one patient demonstrated axonal degeneration.212
The pathogenic mechanism for the neuromyopathy is unknown.
Muscle strength and sensation improve and serum CK levels normalize following discontinuation of omeprazole. Symptoms may recur if omeprazole is restarted.
Isotretinoin (Accutane) is used for treatment of severe acne. Exercise-induced myalgias and proximal weakness can occur.214–216
Serum CK levels can be normal or elevated 100-fold.214,215 Decreased serum carnitine levels may be seen. EMG can demonstrate small polyphasic MUAPs.
Muscle biopsy in a single reported patient demonstrated only atrophy of muscle fibers.
The basis for the myopathy is not clear. The diminished carnitine levels and response to L-carnitine in some patients suggest that perturbation of lipid metabolism may be contributory.
The myalgias, weakness, and CK elevations improve with discontinuation of isotretinoin.
Hypokalemia can be a complication of a variety of medications (e.g., diuretics, laxatives, mineralocorticoids, amphotericin, and lithium). Further, excessive eating of licorice may have an aldosterone-like effect and cause hypokalemia. Hypokalemic myopathy has also been associated with alcohol abuse and inhalation of toluene. The clinical, laboratory, histopathological, and electrophysiologic features of hypokalemic myopathy are similar, regardless of the etiology of the hypokalemia. Affected individuals develop acute or subacute generalized weakness that can resemble Guillain–Barré syndrome. Weakness usually does not occur unless the serum potassium levels are less than 2 mEq/L. The serum CK levels are elevated. EMGs can be normal or demonstrate mild irritability in the form of fibrillation potentials and positive sharp waves in severely weakened muscles. Muscle biopsies are not typically performed as the diagnosis is apparent with the appropriate laboratory testing. However, muscle biopsies may demonstrate scattered necrotic and regenerating muscle fibers as well as vacuoles that arise from T-tubules. The weakness improves with correction of the hypokalemia.
MYOPATHIES ASSOCIATED WITH ANESTHETIC AGENTS AND CENTRALLY ACTING MEDICATIONSMalignant hyperthermia is a genetically heterogeneous group of disorders characterized by severe muscle rigidity, myoglobinuria, fever, tachycardia, cyanosis, and cardiac arrhythmias precipitated by depolarizing muscle relaxants (e.g., succinylcholine) and inhalational anesthetic agents (e.g., halothane).217 The incidence of malignant hyperthermia ranges from 0.5% to 0.0005%.217 At least 50% of patients have had previous anesthesia without any problems.4 The signs of malignant hyperthermia usually appear during surgery but can develop in the postoperative period. Rarely, attacks of malignant hyperthermia have been triggered by exercise, ingestion of caffeine, and stress.218
Serum CK can be normal or mildly elevated between attacks in patients susceptible to malignant hyperthermia. During attacks of malignant hyperthermia, serum CK levels are markedly elevated and myoglobinuria can develop. Hyperkalemia is also usually present. Metabolic and respiratory acidosis is evident with lactic acidosis, hypoxia, and hypercarbia. NCS and EMG are usually normal in the interictal periods. However, EMG performed shortly after an attack of malignant hyperthermia may demonstrate increased spontaneous activity and, perhaps, small polyphasic MUAPs that recruit early.
The in vitro muscle contracture test can be performed to assess the susceptibility of malignant hyperthermia in individuals who may be at risk (i.e., family members with history of malignant hyperthermia).217 However, the test is not routinely available and false-positive and false-negative tests occur. Varying concentrations of halothane and caffeine are applied to strips of muscle that are stimulated at 0.1–0.2 Hz for 1–5 seconds, while tension is measured by a stain gauge. In patients susceptible to malignant hyperthermia, much lower concentrations of caffeine and halothane produce muscle contractions than are required to produce a similar in normal muscle tissue.
Muscle biopsies demonstrate nonspecific myopathic features including fiber size variability, increased internal nuclei, moth-eaten fibers, and necrotic fibers after an attack of malignant hyperthermia.
At least some cases of malignant hyperthermia probably arise secondary to excessive calcium release by the sarcoplasmic reticulum calcium channels. Increased intracytoplasmic calcium leads to excessive muscle contraction, increased use of oxygen and ATP, and overproduction of heat. Why various anesthetic agents and depolarizing muscle relaxants trigger this exaggerated release of calcium from the sarcoplasmic reticulum in predisposed individuals is not known.
Malignant hyperthermia susceptibility is genetically very heterogeneous, as families have been linked to different chromosomes and genes. The first mutations were discovered in the ryanodine receptor gene located on chromosome 19q13.1 (MHS1).142,219,220 The ryanodine receptor bridges the gap between the sarcoplasmic reticulum and the T tubule. Mutations in the ryanodine receptor may result in a functional alteration of the associated calcium channel such that there is an excessive release of calcium into the cytoplasm upon activation. Of note, mutations in this gene also cause the congenital myopathy, central core disease. Mutations in the ryanodine receptor gene account for only a minority of patients with malignant hyperthermia; other genetic loci have been identified. MHS2 localizes to chromosome 17q11.2–q24 (possibly the gene for the subunit of the sodium channel).221 Thus, MHS2 may be allelic to potassium-sensitive periodic paralysis, paramyotonia congenita, and related disorders. MHS3 links to chromosome 7q21–q22 (possibly to a gene CACNA2D1, encoding a subunit of the calcium channel).222 MHS4 localizes to chromosome 3q13.1, but the gene has yet to be identified.223 Mutations in the dihydropteridine receptor gene on chromosome 1q31 (allelic to hypokalemic periodic paralysis type 1) cause MHS5.224 Linkage to chromosome 5p has been demonstrated in still other families (MHS6).225 In addition, patients with muscular dystrophies, myotonic dystrophies, mitochondrial myopathies, and other channelopathies are susceptible to developing malignant hyperthermia.226 Thus, it appears that malignant hyperthermia may occur in various myopathic disorders, affecting the structural proteins of the muscle membrane or ion channels.
Individuals at risk of malignant hyperthermia should not be given known triggering anesthetic agents if possible. Malignant hyperthermia is a medical emergency, requiring several therapeutic steps.217 The anesthetic agent must be discontinued, while 100% oxygen is delivered. Dantrolene 2–3 mg/kg every 5 minutes for a total of 10 mg/kg should be administered. The stomach, bladder, and lower gastrointestinal tract are lavaged with iced saline solution, and cooling blankets are applied. Acidosis and hyperkalemia are treated with sodium bicarbonate, hyperventilation, dextrose, insulin, and occasionally calcium chloride. Urinary output must be maintained with hydration, furosemide, or mannitol. The patient must be monitored and treated for cardiac arrhythmias.
MYOPATHIES SECONDARY TO DRUGS OF ABUSEChronic alcohol abuse is more often attributed to causing neuropathy than myopathy.9 However, several forms of a toxic myopathy due to alcohol have been described: (1) acute necrotizing myopathy, (2) acute hypokalemic myopathy, (3) chronic alcoholic myopathy, (4) asymptomatic alcoholic myopathy, and (5) alcoholic cardiomyopathy.227–231
An acute necrotizing myopathy manifests as acute muscle pain, tenderness to palpation, cramping, swelling, and weakness following or during a recent particularly intense binge. The severity of the myopathy is highly variable. Severe cases can be associated with myoglobinuria and acute renal failure. The muscle cramps resolve over the course of several days, while the remainder of symptoms may last several weeks. Serum CK levels are markedly elevated during these attacks. Muscle biopsies reveal widespread muscle fiber necrosis and occasionally fibers with tubular aggregates. Disorganization of the sarcomeres and degeneration of mitochondria may be appreciated on EM. Patents require appropriate supportive medical care and nutritional supplementation as many are malnourished.
Alcohol abuse can lead to acute hypokalemia, which can cause generalized weakness. Muscle weakness evolves over the time period of 1 or 2 days. Serum potassium is very low, <2 mEq/L, and the CK levels are elevated. Muscle biopsy performed in the acute time frame may reveal vacuoles with the muscle fibers. The myopathy resolves with correction of the serum potassium.
Some alcoholics develop the insidious onset of primarily proximal limb-girdle weakness, especially of the lower limbs, which has been attributed to a chronic alcoholic myopathy. Muscle biopsy may reveal scattered muscle fiber atrophy, necrosis, and regeneration. Whether the muscle weakness is caused by a toxic influence of alcohol on muscle, a toxic peripheral neuropathy, or malnutrition is unclear.
An asymptomatic alcoholic myopathy has been suggested in some patients on the basis of an elevated serum CK levels found coincidentally. There is no complaint of weakness, and the physical examination does not reveal striking evidence of a myopathic disorder. Histological findings are not available for this class of patients, and the true nature of this presumed form of alcoholic myopathy is questionable. The elevated serum CK may be related to subclinical necrotizing myopathy, hypokalemia, or muscle trauma.
Serum CK levels may be normal or slightly elevated and potassium levels may be reduced or normal. Reduced amplitudes of the sensory and, occasionally, motor nerve conductions studies may be seen, if patients have a concomitant alcoholic neuropathy. Needle EMG may reveal positive sharp waves, fibrillation potentials, and early recruitment of short-duration, low-amplitude MUAPs firing at high rates with minimal force production in weak muscles in patients with a necrotizing alcoholic myopathy.227–231
The pathogenic basis for the various forms of alcoholic myopathies is not known. The metabolism of alcohol may lead to the accumulation of toxic metabolites (e.g., acetaldehyde) or free radicals that may be toxic to lipid membranes.
MYOPATHIES SECONDARY TO ILLICIT DRUGSIllicit drugs including opioids (e.g., heroin, meperidine, cocaine, pentazocine, piritramide, amphetamines, etc.) may be myotoxic.232–235 Muscle injury can be related to direct muscle trauma (e.g., needle injury), rhabdomyolysis secondary to pressure and ischemic necrosis related to prolonged loss of consciousness, ischemia due to vasoconstriction, rhabdomyolysis caused by generalized status epilepticus, or the direct toxic effects of the drugs (or adulterants) on muscle tissue. Serum CK levels should be markedly elevated, and muscle biopsies reveal widespread necrosis in such cases.
Inhalation of volatile agents (e.g., toluene) can also cause generalized muscle weakness and, occasionally, myoglobinuria. Toluene causes distal renal tubular acidosis with associated severe hypokalemia, hypophosphatemia, and mild hypocalcemia. Muscle strength returns after correction of the electrolyte abnormalities and abstinence from further exposure.
SUMMARYVarious drugs can cause muscle damage and from a variety of different mechanisms. It is imperative to take a good medical history including current and previous medication history (as well as history of illicit drug use and alcohol abuse), as stopping the offending agent usually leads to improvement of the myopathy. However, continued use can be associated with significant morbidity and even death (e.g., from myoglobinuria). The most common toxic myopathy is associated with statin use in keeping with how frequently these medications are prescribed. That said, most individuals treated with statin medications and other medications known to cause toxic myopathy have no complications.
1. Amato AA, Dumitru D. Acquired myopathies. In: Dumitru D, Amato AA, Zwarts MJ, eds. Electrodiagnostic Medicine. 2nd ed. Philadelphia, PA: Hanley & Belfus; 2002:1371–1432.
2. Argov Z, Mastaglia FL. Drug-induced neuromuscular disorders in man. In: Walton JN, ed. Disorders of Voluntary Muscle. 5th ed. Edinburgh: Churchill-Livingstone; 1988:981–1014.
3. Baker PC. Drug-induced and toxic myopathies. Semin Neurol. 1983;3:265–273.
4. Griggs RC, Mendell JR, Miller RG. Myopathies of systemic disease. In: Evaluation and Treatment of Myopathies. Philadelphia, PA: FA Davis; 1995:355–385.
5. Kuncl RW, Wiggins WW. Toxic myopathies. Neurol Clin. 1988;6:593–619.
6. Lane RJM, Mastaglia FL. Drug-induced myopathies in man. Lancet. 1978;2:562–565.
7. Mastaglia FL. Adverse effects of drugs on muscle. Drugs. 1982;24:304–321.
8. Mastaglia FL. Toxic myopathies. In: Rowland LP, DiMauro S, eds. Handbook of Clinical Neurology. Vol 18, No. 62: Myopathies. Amsterdam: Elsevier Science Publishers BV; 1992:595–622.
9. Victor M, Sieb JP. Myopathies due to drugs, toxins, and nutritional deficiency. In: Engel AG, Franzini-Armstrong C, eds. Myology. 2nd ed. New York, NY: McGraw-Hill; 1994:1697–1725.
10. Mastaglia FL. Iatrogenic myopathies. Curr Opin Neurol. 2010;23:445–449.
11. Bakker-Arema RG, Best J, Fayyad R, et al. A brief review paper on the efficacy and safety of atorvastatin in early clinical trials. Atherosclerosis. 1997;131:17–23.
12. Berland Y, Coponat H, Durand C, Baz M, Laugier R, Musso JL. Rhabdomyolysis and simvastatin use. Nephron. 1991;57:365–366.
13. Corpier C, Jones P, Suki W, et al. Rhabdomyolysis and renal injury with lovastatin use. JAMA. 1988;260:239–241.
14. Davidson MH, Stein EA, Dujoven CA, et al. The efficacy and six week tolerability of simvastatin 80 and 160 mg/day. Am J Cardiol. 1997;79:38–42.
15. Deslypere J, Vermuelen A. Rhabdomyolysis and simvastatin. Ann Intern Med. 1991;114:342.
16. Marais GE, Larson KK. Rhabdomyolysis and acute renal failure induced by combination lovastatin and gemfibrozil therapy. Ann Intern Med. 1990;112:228–230.
17. Schalke BB, Schmidt B, Toyka K, Hartung HP. Pravastatin-associated inflammatory myopathy. N Engl J Med. 1992;327:649–650.
18. Tobert J. Efficacy and long-term adverse effect pattern of lovastatin. Am J Cardiol. 1988;62:28J–33J.
19. East C, Alivizatos PA, Grundy SM, Jones PH, Farmer JA. Rhabdomyolysis in patients receiving lovastatin after cardiac transplantation. N Engl J Med. 1988;318:48.
20. Abourizk N, Khalil BA, Bahuth N, Afifi AK. Clofibrate induced muscular syndrome. J Neurol Sci. 1979;42:1–9.
21. Denizot M, Fabre J, Pometa D, Wildi E. Clofibrate, nephrotic syndrome, and histological changes in muscle. Lancet. 1973;1:1326.
22. Gabriel R, Pearce JM. Clofibrate induced myopathy and neuropathy. Lancet. 1976;2:906.
23. Kwiecinski H. Myotonia induced with clofibrate in rats. J Neurol. 1978;219:107–116.
24. Langer T, Levy RI. Acute muscular syndrome associated with administration of clofibrate. N Engl J Med. 1968;279:856–858.
25. London F, Gross KF, Ringel SP. Cholesterol-lowering agent myopathy (CLAM). Neurology. 1991;41:1159–1160.
26. Magarian GJ, Lucas LM. Gemfibrozil-induced myopathy. Arch Intern Med. 1991;151:1873–1874.
27. Pierce LR, Wysowski DK, Gross TP. Myopathy and rhabdomyolysis associated with lovastatin-gemfibrozil combination therapy. JAMA. 1990;264:71–75.
28. Pierides AM, Alvarez-Ude F, Kerr DN. Clofibrate induced muscle damage in patients with chronic renal failure. Lancet. 1975;2:1279–1282.
29. Rush P, Baron M, Kapusta M. Clofibrate myopathy: a case report and a review of the literature. Semin Arthritis Rheum. 1986;15:226–229.
30. Shepherd J. Fibrates and statins in the treatment of hyperlipidemia: an appraisal of their efficacy and safety. Eur Heart J. 1995;16:5–13.
31. Litin SC, Andersone CF. Nicotinic acid-associated myopathy: a report of three cases. Am J Med. 1989;86:481–483.
32. Reaven P, Witzum J. Lovastatin, nicotinic acid and rhabdomyolysis [letter]. Ann Intern Med. 1988;109:597–598.
33. Fux R, Morike K, Gundel UF, Hartmann R, Gleiter CH. Ezetimibe and statin-associated myopathy. Ann Intern Med. 2004;140(8):671–672.
34. Havranek JM, Wolfsen AR, Warnke GA, Phillips PS. Monotherapy with ezetimibe causing myopathy. Am J Med. 2006;119 (3):285–286.
35. Simard C, Poirier P. Ezetimibe-associated myopathy in monotherapy and in combination with a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Can J Cardiol. 2006;22(2):141–144.
36. Perez-Calvo J, Civeira-Murillo F, Cabello A. Worsening myopathy associated with ezetimibe in a patient with McArdle disease. QJM. 2005;98(6):461–462.
37. Dujovne CA, Chremos AN, Pool JL, et al. Expanded Clinical Evaluation of Lovastatin (EXCEL) study results. IV. Additional perspectives on the tolerability of lovastatin. Am J Med. 1991;91(suppl 1B):25–30.
38. Jones P, Kafonek S, Laurora I, Hunningshake D. Comparative dose efficacy study of atorvastatin versus simvastatin, provastatin, lovastatin, and fluvastatin in patients with hypercholesterolemia (the CURVES Study). Am J Cardiol. 1998;81:582–587.
39. Galper JB. Increase incidence of myositis in patients treated with high dose simvastatin. Am J Cardiol. 1998;81:259.
40. Duell PB, Connor WE, Illingsworth DR. Rhabdomyolysis after taking atorvastatin with gemfibrozil. Am J Cardiol. 1998;81:368–369.
41. Furberg CD, Pitt B. Commentary: withdrawal of cervistatin from the world market. Curr Control Trials Cardiovasc Med. 2001;2:205–207.
42. von Keutz E, Schluter G. Preclinical safety evaluation of cerivastation, a novel HMG-CoA reductase inhibitor. Am J Cardiol. 1998;82(4B):11J–17J.
43. Hamilton-Craig I. Statin-associated myopathy. Med J Aust. 2001;175:486–489.
44. Pasternak RC, Smith SC, Bairey-Merz CN, Grundy S, Cleeman J, Lenfant C. ACC/AHA/NHLBI clinical advisory on the use and safety of statins. J Am Coll Cardiol. 2002;40:556–572.
45. Ucar M, Mjorndal T, Dahlqvist R. HMG-CoA reductase inhibitors and myotoxicity. Drug Saf. 2000;22:441–457.
46. Hodel C. Myopathy and rhabdomyolysis with lipid-lowering drugs. Toxicol Lett. 2002;128:159–168.
47. Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA. 2003;289:1681–1690.
48. dos Santos AG, Guardia AC, Pereira TS, et al. Rhabdomyolysis as a clinical manifestation of association with ciprofibrate, sirolimus, cyclosporine, and pegylated interferon-α in liver-transplanted patients: A case report and literature review. Transplant Proc. 2014;46:1887–1888.
49. Mammen AL, Amato AA. Statin myopathy: a review of recent progress. Curr Opin Rheumatol. 2010;22:644–650.
50. Shanmugam VK, Matsumoto C, Pien E, et al. Voriconazole associated myositis. J Clin Rheumatol. 2009;15:350–353.
51. Basic-Jukic N, Kes P, Bubic-Filipi L, Vranjican Z. Rhabdomyolysis and acute kidney injury secondary to concomitant use of fluvastatin and rapamycin in a renal transplant recipient. Nephrol Dial Transplant. 2010;25:2036.
52. Backes JM, Venero CV, Gibson CA, et al. Effectiveness and tolerability of every-other-day rosuvastatin dosing in patients with prior statin intolerance. Ann Pharmacother. 2008;42:341–346.
53. Fauchais AL, Iba Ba J, Maurage P, et al. Polymyositis induced or associated with lipid-lowering drugs: five cases. Rev Med Interne. 2004;25(4):294–298.
54. Giordano N, Senesi M, Mattii G, Battisti E, Villanova M, Gennari C. Polymyositis associated with simvastatin. Lancet. 1997;349(9065):1600–1601.
55. Hill C, Zeitz C, Kirkham B. Dermatomyositis with lung involvement in a patient treated with simvastatin. Aust N Z J Med. 1995;25(6):745–746.
56. Khattak FH, Morris IM, Branford WA. Simvastatin-associated dermatomyositis. Br J Rheumatol. 1994;33(2):199.
57. Noel B, Cerottini JP, Panizzon RG. Atorvastatin-induced dermatomyositis. Am J Med. 2001;110(8):670–671.
58. Riesco-Eizaguirre G, Arpa-Gutierrez FJ, Gutierrez M, Toribio E. Severe polymyositis with simvastatin use. Rev Neurol. 2003;37(10):934–936.
59. Rodriguez-Garcia JL, Serrano Commino M. Lovastatin-associated dermatomyositis. Postgrad Med J. 1996;72(853):694.
60. Vasconcelos OM, Campbell WW. Dermatomyositis-like syndrome and HMG-CoA reductase inhibitor (statin) in-take. Muscle Nerve. 2004;30(6):803–807.
61. Christopher-Stine L, Casciola Rosen L, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200 and 100 kDa proteins is associated with an immune mediated necrotizing myopathy. Arthritis Rheum. 2010;62:2757–2766.
62. Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum. 2011;63:713–721.
63. Needham M, Fabian V, Knezevic W, Panegyres P, Zilko P, Mastaglia FL. Progressive myopathy with up-regulation of MHC-I associated with statin therapy. Neuromuscul Disord. 2007;17(2):194–200.
64. Grable-Esposito P, Katzberg HD, Greenberg SA, Srinivasan J, Katz J, Amato AA. Immune-mediated necrotizing myopathy associated with statins. Muscle Nerve. 2010;41:185–190.
65. Meriggioli MN, Barboi A, Rowin J, Cochran EJ. HMGCoA reductase inhibitor myopathy: clinical, electrophysiologic, and pathologic data in five patients. J Clin Neuromuscul Dis. 2001;2:129–134.
66. Phillips PS, Haas RH, Bannykh S, et al. Statin-associated myopathy with normal creatine kinase levels. Ann Intern Med. 2002;137:581–585.
67. Greenberg SA, Amato AA. Statin myopathies. In: Amato AA, ed. Continuum: Muscle Diseases. Vol 12. 2006, pp. 169–184.
68. SEARCH Collaborative Group; Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy: a genomewide study. N Engl J Med. 2008;359:789–799.
69. Kra SJ. Muscle syndrome with clofibrate usage. Conn Med. 1974;38:348–349.
70. Afifi AK, Hajj SS, Tekian A, et al. Clofibrate-induced myotoxicity in rats. Eur Neurol. 1984;23:182–197.
71. Slim H, Thompson PD. Ezetimibe-related myopathy: a systematic review. J Clin Lipidol. 2008;2:328–234.
72. Amato AA, Barohn RJ. Neurological complications of transplantations. In: Harati Y, Rolack LA, eds. Practical Neuroimmunology. Boston, MA: Butterworth-Heineman; 1997:341–375.
73. Arellano F, Krup P. Muscular disorders associated with cyclosporine [letter]. Lancet. 1991;337:915.
74. Costigan DA. Acquired myotonia, weakness and vacuolar myopathy secondary to cyclosporine [abstract]. Muscle Nerve. 1989;12:761.
75. Goy JJ, Stauffer JC, Deruaz JP, et al. Myopathy as a possible side effect cyclosporine. Lancet. 1989;1:1446–1449.
76. Grezard O, Lebranchu Y, Birmele B, Sharobeem R, Nivet H, Bagros P. Cyclosporine-induced muscular toxicity. Lancet. 1990;1:177.
77. Noppen D, Verlkeriers B, Dierckx R, Bruyland M, Vanhaelst L. Cyclosporine and myopathy. Ann Intern Med. 1987;107:945–946.
78. Hibi S, Hisawa A, Tamai M, et al. Severe rhabdomyolysis with tacrolimus [letter]. Lancet. 1995;346:702.
79. Norman D, Illingworth DR, Munson J, Hosenpud J. Myolysis and acute renal failure in heart transplant recipient receiving lovastatin. N Engl J Med. 1988;318:46–47.
80. Rieger EH, Halasz NA, Wahlstrom HE. Colchicine neuromyopathy after renal transplantation. Transplantation. 1990;49:1196–1198.
81. Volin L, Jarventie G, Ruut U. Fatal rhabdomyolysis as a complication of bone marrow transplantation. Bone Marrow Transplant. 1990;6:59–60.
82. Atkinson P, Joubert G, Barron A, et al. Hypertrophic cardiomyopathy with tacrolimus in paediatric transplant patient. Lancet. 1995;345:894–896.
83. Teicher A, Rosenthal T, Kissen E, Sarova I. Labetalol-induced toxic myopathy. Br Med J. 1981;282:1824–1825.
84. Willis JK, Tilton AH, Harkin JC, Boineau FG. Reversible myopathy due to labatolol. Pediatr Neurol. 1990;6:275–276.
85. Hanna JP, Ramundo ML. Rhabdomyolysis and hypoxia associated with prolonged propofol infusion in children. Neurology. 1988;50:301–303.
86. Parke TJ, Steven JE, Rice AS, et al. Metabolic acidosis and myocardial failure after propofol infusion in children: five case reports. Br Med J. 1992;305:613–616.
87. Strickland RA, Murray MJ. Fatal metabolic acidosis in pediatric patient receiving and infusion of propofol in the intensive care unit: is there a relationship? Crit Care Med. 1995;23:405–409.
88. Hanson P, Dive A, Brucher JM, Bisteau M, Dangoisse M, Deltombe T. Acute corticosteroid myopathy in intensive care patients. Muscle Nerve. 1997;20:1371–1380.
89. Eadie MJ, Ferrier TM. Chloroquine myopathy. J Neurol Neurosurg Psychiatry. 1966;29:331–337.
90. Estes ML, Ewing-Wilson D, Chou SM, et al. Chloroquine neuromyotoxicity. Clinical and pathological perspective. Am J Med. 1987;82:447–455.
91. Maschke M, Kastrup O, Esser S, Ross B, Hengge U, Hufnagel A. Incidence and prevalence of neurological disorders associated with HIV since the introduction of highly active antiretroviral therapy (HAART). J Neurol Sci. 2000;69:376–380.
92. Mastaglia FL, Papadimitriou JM, Dawkins RL, Beveridge B. Vacuolar myopathy associated with chloroquine, lupus erythematosus and thymoma. J Neurol Sci. 1977;34:315–328.
93. Alderson K, Griffin JW, Cornblath DR, Levine JH, Kuncl RW, Griffin LSC. Neuromuscular complications of amiodarone therapy. Neurology. 1987;37(suppl):355.
94. Costa-Jussa FR, Jacobs JM. The pathology of amiodarone neurotoxicity. I. Experimental changes with reference to changes in other tissues. Brain. 1985;108:735–752.
95. Jacobs JM, Costa-Jussa FR. The pathology of amiodarone neurotoxicity. II. Peripheral neuropathy in man. Brain. 1985;108:753–769.
96. Meier C, Kauer B, Muller U, Ludin HP. Neuromyopathy during amiodarone treatment: a case report. J Neurol. 1979;220:231–239.
97. Roth R, Itabashi H, Louie J, Anderson T, Narahara KA. Amiodarone toxicity: myopathy and neuropathy. Am Heart J. 1990;119:1223–1225.
98. Kuncl RW, Duncan G, Watson D, Alderson K, Rogawski MA, Peper M. Colchicine myopathy and neuropathy. N Engl J Med. 1987;316:1562–1568.
99. Kuncl RW, Cornblath DR, Avila O, Duncan G. Electrodiagnosis of human colchicine myoneuropathy. Muscle Nerve. 1989;12:360–364.
100. Riggs JE, Schochet SS, Gutmann L, Crosby TW, DiBartolomeo AG. Chronic colchicine neuropathy and myopathy. Arch Neurol. 1986;43:521–523.
101. Rutkove SB, De Girolami U, Preston DC, et al. Myotonia in colchicine myoneuropathy. Muscle Nerve. 1996;19:870–875.
102. Kontos HA. Myopathy associated with chronic colchicine toxicity. N Engl J Med. 1962;266:38–39.
103. Bradley WG, Lassman LP, Pearce GW, Walton JN. The neuromyopathy of Vincristine in man: clinical, electrophysiological and pathological studies. J Neurol Sci. 1970;10:107–131.
104. Anderson P, Song S, Slotwiner P. The fine structure of spheromembranous degeneration of skeletal muscle induced by vincristine. J Neuropathol Exp Neurol. 1967;26:15–24.
105. Slotwiner P, Song S, Andersone P. Spheromembranous degeneration of muscle induced by vincristine. Arch Neurol. 1966; 15:172–176.
106. Dalakas M. HIV or zidovudine myopathy? [letter]. Neurology. 1994;44:360–361.
107. Dalakas MC, Illa I, Pezeshkpour GH, Laukaitis JP, Cohen B, Griffin JL. Mitochondrial myopathy caused by long-term zidovudine therapy. N Engl J Med. 1990;322:1098–1105.
108. Gherardi R, Chariot P. HIV or zidovudine myopathy [letter]? Neurology. 1994;44:361–362.
109. Grau JM, Casademont J. HIV or zidovudine myopathy? [letter]. Neurology. 1994;44:361.
110. Grau JM, Masanes F, Pedreo E, Casdemont J, FernandezSola J, Urbano-Marquez A. Human immunodeficiency virus type 1 infection and myopathy: clinical relevance of zidovidine therapy. Ann Neurol. 1993;34:206–211.
111. Mhiri C, Baudrimont M, Bonne G, et al. Zidovudine myopathy: a distinctive disorder associated with mitochondrial dysfunction. Ann Neurol. 1991;29:606–614.
112. Peters BS, Winer J, Landon DN, Stoffer A, Pinching AJ. Mitochondrial myopathy associated with chronic zidovudine therapy in AIDS. Q J Med. 1993;86:5–15.
113. Richman DD, Fischl MA, Grieco HM, et al. The toxicity of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. N Engl J Med. 1987;317:192–197.
114. Simpson DM, Bender AN. Human immunodeficiency virus-associated myopathy: analysis of 11 patients. Ann Neurol. 1988;24:79–84.
115. Simpson DM, Bender AN, Farraye J, Mendelson SG, Wolfe DE. Human immunodeficiency virus wasting syndrome represents a treatable myopathy. Neurology. 1990;40:535–538.
116. Simpson DM, Slasor P, Dafni U, Berger J, Fischl MA, Hall C. Analysis of myopathy in a placebo-controlled zidovudine trial. Muscle Nerve. 1997;20:382–385.
117. Bailey RO, Turok DI, Jaufmann BP, Singh JK. Myositis and acquired immunodeficiency syndrome. Hum Pathol. 1987;18:749–751.
118. Bessen LJ, Green JB, Louie E, Seitzman P, Weinberg H. Severe polymyositis-like syndrome associated with zidovudine therapy of AIDS and ARC. N Engl J Med. 1988;318:708.
119. Chalmers AC, Greco CM, Miller RG. Prognosis in AZT myopathy. Neurology. 1991;41:1181–1184.
120. Manji H, Harrison MJ, Round JM, et al. Muscle disease, HIV and zidovudine: the spectrum of muscle disease in HIV-infected individuals treated with zidovudine. J Neurol. 1993;240:479–488.
121. Reyes MG, Casanova J, Varricchio F, Sequeira W, Fresco K. Zidovudine myopathy. Neurology. 1992;42:1252.
122. Simpson DM, Citak KA, Godfrey E, Godbold J, Wolfe D. Myopathies associated with human immunodeficiency virus and zidovudine: can their effects be distinguished? Neurology. 1993;43:971–976.
123. Simpson DA, Katzenstein DA, Hughes MD, et al. Neuromuscular function in HIV infection: analysis of a placebo-controlled combination antiviral trial. AIDS Clinical Group 175/801 Study Team. AIDS. 1998;12:2425–2432.
124. Till M, McDonnel KB. Myopathy with human immunodeficiency virus type 1 (HIV) infection. HIV-1 or zidovudine? Ann Intern Med. 1990;113:492–494.
125. Panegyres PK, Papadimitriou JM, Hollingsworth PN, Armstrong JA, Kakulas BA. Vesicular changes in the myopathies of AIDS. Ultrastructural observations and their relationship to zidovudine treatment. J Neurol Neurosurg Psychiatry. 1990;53:649–655.
126. Arnaudo E, Dalakas M, Shanske S, Moraes CT, DiMauro S, Schon EN. Depletion of mitochondrial DNA in AIDS patients with zidovudine-induced myopathy. Lancet. 1991;1:508–510.
127. Masanes F, Barrientos A, Cebrian M, et al. Clinical, histological, and molecular reversibility of zidovudine myopathy. J Neurol Sci. 1998;159:225–228.
128. Jay CA, Hench K, Ropka M. Improvement of AZT myopathy after change to dideoxyinosine (ddI) or dideoxycytosine (ddC) [abstract]. Neurology. 1993;43(suppl 2):A373–A374.
129. Benbrik E, Chariot P, Boanvaud S, et al. Cellular and mitochondrial toxicity of zidovudine (AZT), didanosione (ddI), and zacitabine (ddC) on cultured human muscle cells. J Neurol Sci. 1997;149:19–25.
130. Pedrol E, Masanes F, Fernandez-Sola J, et al. Lack of myotoxicity with didanosine (dI). Clinical and experimental studies. J Neurol Sci. 1996;138:42–48.
131. Callens S, De Roo A. Fanconi-like syndrome and rhabdomyolysis in a person with HIV infection on highly active antiretroviral treatment occluding tenofovir [letter]. J Infect Dis. 2003;47:262–263.
132. Mah Ming JB, Gill MJ. Drug-induced rhabdomyolysis after concomitant use of clarithermycine, atorvastatin, lopinivir/ritanovir in a patient with HIV. AIDS Patient Care STDS. 2003;17:207–210.
133. Belongia EA, Hedberg CW, Gleich GJ, et al. An investigation of the case of the eosinophilia myalgia syndrome associated with tryptophan use. N Engl J Med. 1990;323:357–365.
134. Donofrio PD, Stanton C, Miller VS, et al. Demyelinating polyneuropathy in eosinophilia-myalgia syndrome. Muscle Nerve. 1992;15:796–805.
135. Hertzman PA, Blevins WL, Mayer J, Greenfield B, Ting M, Gleich GJ. Association of the eosinophilia-myalgia syndrome with the ingestion of tryptophan. N Engl J Med. 1990;322:869–873.
136. Sagman DL, Melamed JC. L-Tryptophan induced eosinophilia-myalgia syndrome and myopathy. Neurology. 1990;40:1629–1630.
137. Sakimoto K. The cause of the eosinophilia-myalgia syndrome associated with tryptophan use. N Engl J Med. 1990;323:992.
138. Smith BE, Dyck PJ. Peripheral neuropathy in the eosinophilic-myalgia syndrome associated with Ltryptophan ingestion. Neurology. 1990;40:1035–1040.
139. Tanhehco JL, Wiechers DO, Golbus J, Neely SE. Eosinophiliamyalgia syndrome: myopathic electrodiagnostic characteristics. Muscle Nerve. 1992;15:561–567.
140. Shulman LE. Diffuse fasciitis with eosinophilia: a new syndrome? Trans Assoc Am Physicians. 1975;88:70–86.
141. Heiman-Patterseon TD, Bird SJ, Parry GJ, et al. Peripheral neuropathy associated with eosinophilia-myalgia syndrome. Ann Neurol. 1990;28:522–528.
142. Quane KA, Keating KE, Manning BM, et al. Detection of a common mutation in the ryanodine receptor gene in malignant hyperthermia: implication for diagnosis and heterogenetic studies. Hum Mol Genet. 1994;3:471–476.
143. Selwa JF, Feldman EL, Blaivas M. Mononeuropathy multiplex in tryptophan-associated eosinophilic-myalgia syndrome. Neurology. 1990;40:1632–1633.
144. Turi GK, Solitaire GB, James N, Dicker R. Eosinophiliamyalgia syndrome (L-tryptophan-associated neuromyopathy). Neurology. 1990;40:1793–1796.
145. Mayeno AN, Belongia EA, Lin F, Lundy SK, Gleich GJ. 3-(Phenylamino) alanine—a novel aniline-derived aminoacid associated with the eosinophilic-myalgia syndrome: a link to the toxic oil syndrome. Mayo Clin Proc. 1992;67:1134.
146. Kilbourne EM, Rigau-Perez JG, Heath CW, et al. Clinical epidemiology of toxic-oil syndrome. N Engl J Med. 1983;309:1408–1414.
147. Dawkins RL, Zilko PJ, Carrano J, Garlepp MJ, McDonald BL. Immunobiology of D-penicillamine. J Rheumatol. 1981;8(suppl):56–61.
148. Hall JT, Fallahi S, Koopman WJ. Penicillamine-induced myositis: observations and unique features in two patients and review of the literature. Am J Med. 1984;77:719.
149. Takahashi K, Ogita T, Okudaira H, Yoshinoya S, Yoshizawa H, Miyamoto T. D-penicillamine induced polymyositis in patients with rheumatoid arthritis. Arthritis Rheum. 1986;29:560–564.
150. Taneja V, Mehra N, Singh YN, Kumar A, Malaviya A, Singh RR. HLA-D region genes and susceptibility to D-penicillamine-induced polymyositis. Arthritis Rheum. 1990;33:1445–1447.
151. Watson AJ, Dalbow MH, Stachura I, et al. Immunologic studies in cimetidine-induced nephropathy and polymyositis. N Engl J Med. 1983;308:142–145.
152. Mitchell CG, Magnussen AR, Weiler JM. Cimetidine-induced cutaneous vasculitis. Am J Med. 1983;75:875.
153. Lewis CA, Boheimer N, Rose P, Jackson G. Myopathy after short term administration of procainamide. Br Med J. 1986;292:593–597.
154. Wolf S, Goldberg LS, Verity MA. Neuromyopathy and periarteriolitis in a patient receiving levodopa. Arch Intern Med. 1976;136:1055–1057.
155. Harney J, Glasberg MR. Myopathy and hypersensitivity to phenytoin. Neurology. 1983;33:790–791.
156. Cirigliano G, Della RA, Tavoni A, Tavoni A, Vicava P, Bombardieri S. Polymyositis occurring during alpha-interferon treatment for malignant melanoma: a case report and review of the literature. Rheumatol Int. 1999;19:65–67.
157. Dietrich L, Bridges AJ, Albertini MR. DM after interferon alpha treatment. Med Oncol. 2000;17:64–69.
158. Hengstman GJ, Vogels OJ, ter Laak HJ, de Witte T, van Engelen BG. Myositis during ling-term interferon-alpha treatment. Neurology. 2000;54:2186.
159. The Muscle Study Group. A randomized, pilot trial of etanercept in dermatomyositis. Ann Neurol. 2011;70(3):427–436.
160. Klein R, Rosenbach M, Kim EJ, Kim B, Werth VP, Dunham J. Tumor necrosis factor inhibitor-associated dermatomyositis. Arch Dermatol. 2010;146:780–784.
161. Ishikawa Y, Yukawa N, Ohmura K, et al. Etanercept-induced anti-Jo-1-antibody-positive polymyositis in a patient with rheumatoid arthritis: a case report and review of the literature. Clin Rheumatol. 2010;29:563–566.
162. Srinivasan J, Wu C, Amato AA. Inflammatory myopathy associated with imitinab therapy. J Clin Neuromuscul Dis. 2004;5:119–121.
163. Coomes EN. The rate of recovery of reversible myopathies and the effects of anabolic agents in steroid myopathy. Neurology. 1965;18:523–530.
164. Engel AG. Metabolic and endocrine myopathies. In: Walton JN, ed. Disorders of Voluntary Muscle. 5th ed. Edinburgh: Churchill-Livingstone; 1988:811–868.
165. Golding DN, Murray SM, Pearce GW, Thompson M. Corticosteroid myopathy. Ann Phys Med. 1962;6:171–177.
166. Kaminski HJ, Ruff RL. Endocrine myopathies (hyper- and hypofunction of adrenal, thyroid, pituitary, and parathyroid glands and iatrogenic corticosteroid myopathy). In: Engel AG, Franzini-Armstrong C, eds. Myology. 2nd ed. New York, NY: McGraw-Hill; 1994:1726–1753.
167. Kissel JT, Mendell JR. The endocrine myopathies. In: Rowland LP, DiMauro S, eds. Handbook of Clinical Neurology. Vol 18. No. 62: Myopathies. Amsterdam: Elsevier Science Publishers BV; 1992:527–551.
168. Muller R, Kugelberg E. Myopathy in Cushing’s syndrome. J Neurol Neurosurg Psychiatry. 1959;22:314–319.
169. Williams RS. Triamcinolone myopathy. Lancet. 1959;516:698–700.
170. Yates DAH. Steroid myopathy. In: Walton JN, Canal N, Scarlato G, eds. Muscle Diseases: Proceedings of an International Congress Milan. 19–21 May, 1969. Amsterdam: Excerpta Medica; 1970:482–488.
171. Faludi G, Gotlieb J, Meyers J. Factors influencing the development of steroid-induced myopathy. Ann NY Acad Sci. 1967; 138:61–72.
172. Buchthal F. Electrophysiological abnormalities in metabolic myopathies and neuropathies. Acta Neurol Scand. 1970;46(suppl 43):129–176.
173. Hudgson P, Kendall-Taylor P. Endocrine myopathies. In: Mastaglia FL, Walton JN, eds. Skeletal Muscle Pathology. Edinburgh: Churchill Livingstone; 1992:493–509.
174. Pleasure DE, Walsh GO, Engel WK. Atrophy of skeletal muscle in patients with Cushing’s syndrome. Arch Neurol. 1970;22:118–125.
175. Amato AA, Barohn RJ. Idiopathic Inflammatory myopathies. Neurol Clin. 1997;15:615–648.
176. Afifi AK, Bergman RA, Harvey JC. Steroid myopathy. Johns Hopkins Med J. 1968;123:158–174.
177. MacLean D, Schurr PH. Reversible amyotrophy complicating treatment with fluodrocortisone. Lancet. 1959;516:701–702.
178. Haan J, Hollander JM, van Duinin SG, Sacena PR, Wintzen AR. Reversible severe myopathy during treatment with finasteride. Muscle Nerve. 1997;20:502–504.
179. Bennett HS, Spiro AJ, Pollack MA, Zucker P. Ipecac-induced myopathy simulating dermatomyositis. Neurology. 1982;32:91–94.
180. Mateer JE, Farrell BJ, Chou SS, Gutmann L. Reversible ipecac myopathy. Arch Neurol. 1985;42:188–190.
181. Palmer EP, Guay AT. Reversible myopathy secondary to abuse of ipecac in patients with major eating disorders. N Engl J Med. 1985;313:1457–1459.
182. Amato AA, Jackson CE, Lampin S, Kaggan-Hallet K. Myofibrillar myopathy: no evidence of apoptosis by TUNEL. Neurology. 1999;52(4):861–863.
183. Amato AA, Kagan-Hallet K, Jackson CE, et al. The wide spectrum of myofibrillar myopathy suggests a multifactorial etiology and pathogenesis. Neurology. 1998;51(6):1646–1655.
184. Bolton CF, Gilbert JJ, Hahn AF, Sibbald WJ. Polyneuropathy in critically ill patients. J Neurol Neurosurg Psychiatry. 1984;47:1223–1231.
185. Zochodne DW, Bolton CF, Wells GF, et al. Critical illness polyneuropathy. A complication of sepsis and multiorgan failure. Brain. 1987;110:819–842.
186. Barohn RJ, Jackson CE, Rogers SJ, Ridings LW, McVey AL. Prolonged paralysis due to nondepolarizing neuromuscular blocking agents and corticosteroids. Muscle Nerve. 1994; 17:647–654.
187. Gooch JL. AAEM case report #29: prolonged paralysis after neuromuscular blockade. Muscle Nerve. 1995;18:937–942.
188. Al-Lozi MT, Pestronk A, Yee WC, Flaris N, Cooper J. Rapidly evolving myopathy with myosin-deficient fibers. Ann Neurol. 1994;35:273–279.
189. Danon MJ, Carpenter S. Myopathy with thick filament (myosin) loss following prolonged paralysis with vecuronium during steroid treatment. Muscle Nerve. 1991;14:1131–1139.
190. Deconinck N, Van Parijs V, Beckers-Bleukx G, Van den Bergh P. Critical illness myopathy unrelated to corticosteroids or neuromuscular blocking agents. Neuromuscul Disord. 1998;8:186–192.
191. Gutmann L, Blumenthal D, Schochet SS. Acute type II myofiber atrophy in critical illness. Neurology. 1996;46:819–821.
192. Hirano M, Ott BR, Rapps EC, et al. Acute quadriplegic myopathy: a complication of treatment with steroids, nondepolarizing blocking agents, or both. Neurology. 1992;42:2082–2087.
193. Lacomis D, Giuliani MJ, Van Cott A, Kramer DJ. Acute myopathy of the intensive care: clinical, electromyographic, and pathological aspects. Ann Neurol. 1996;40:645–654.
194. Lacomis D, Petrella JT, Giuliani MJ. Causes of neuromuscular weakness in the intensive care unit: a study of ninety-two patients. Muscle Nerve. 1998;21:610–617.
195. Lacomis D, Smith TW, Chad DA. Acute myopathy and neuropathy in status asthmaticus: Case report and literature review. Muscle Nerve. 1993;16:84–90.
196. McFarlane IA, Rosenthal FD. Severe myopathy after status asthmaticus. Lancet. 1977;2:615.
197. Ramsay DA, Zochodne DW, Robertson DM, Nag S, Ludwin SK. A syndrome of acute severe muscle necrosis in intensive care unit patients. J Neuropathol Exp Neurol. 1993;52:387–398.
198. Rich MM, Bird SJ, Raps EC, McClaskey LF, Teener JW. Direct muscle stimulation in acute quadriplegic myopathy. Muscle Nerve. 1997;20:665–673.
199. Rich MM, Teener JW, Raps EC, Schotland DL, Bird SJ. Muscle is electrically inexcitable in acute quadriplegic myopathy. Neurology. 1996;46:731–736.
200. Showalter CJ, Engel AG. Acute quadriplegic myopathy: analysis of myosin isoforms and evidence for calpain-mediated proteolysis. Muscle Nerve. 1997;20:316–322.
201. Sitwell LD, Weishenker BG, Monipetit V, Reid D. Complete ophthalmoplegia as a complication of acute corticosteroid- and pancuronium-associated myopathy. Neurology. 1991;41:921–922.
202. Op de Coul AA, Lambregts PC, Koeman J, van Puyenbroek MJ, Ter Laak HJ, Gabreels-Festen AA. Neuromuscular complications in patients given Pavulon (pancuronium bromide) during artificial ventilation. Clin Neurol Neurosurg. 1985;87:17–22.
203. Witt NJ, Zochodne DW, Bolton CF, et al. Peripheral nerve function in sepsis and multiorgan failure. Chest. 1991;99:176–184.
204. Latronico N, Fenzi F, Recupero D, et al. Critical illness myopathy and neuropathy. Lancet. 1996;347:1579–1582.
205. Campellone JV, Lacomis D, Kramer DJ, Van Cott AC, Giuliani MJ. Acute myopathy after liver transplantation. Neurology. 1998;50:46–53.
206. Road J, Mackie G, Jiang TX, Stewart H, Eisen A. Reversible paralysis with status asthmaticus, steroids, and pancuronium: clinical electrophysiological correlates. Muscle Nerve. 1997;20:1587–1590.
207. Zochodne DW, Ramsey DA, Saly V, Shelley S, Moffatt S. Acute necrotizing myopathy of the intensive care: electrophysiological studies. Muscle Nerve. 1994;17:285–292.
208. Douglass JA, Tuxen DV, Horne M, et al. Myopathy in severe asthma. Am Rev Respir Dis. 1992;146:517–519.
209. Rich MM, Pinter MJ, Kraner SD, Barchi RL. Loss of electrical excitability in an animal model of acute quadriplegic myopathy. Ann Neurol. 1998;43:171–179.
210. Allen DC, Arunachalam R, Mills KR. Critical illness myopathy: further evidence from muscle-fiber excitability studies of an acquired channelopathy. Muscle Nerve. 2008;37:14–22.
211. Ruff RL. Why do ICU patients become paralyzed? Ann Neurol. 1998;43:154–155.
212. Faucheux JM, Tourneize P, Viguier A, Arne-Bes MC, Larrue M, Geraud G. Neuromyopathy secondary to omeprazole treatment. Muscle Nerve. 1998;21:261–262.
213. Garrot FJ, Lacambrac D, Del Sert T, García Díaz B, Obeso G, Solís J. Subacute myopathy during omeprazole therapy [letter]. Lancet. 1994;340:672.
214. Sarifakioglu E, Onur O, Kart H, Yilmaz AE. Acute myopathy and acne fulminans triggered by isotretinoin therapy. Eur J Dermatol. 2011;21(5):794–795.
215. Chroni E, Monastirli A, Tsambaos D. Neuromuscular adverse effects associated with systemic retinoid dermatotherapy: monitoring and treatment algorithm for clinicians. Drug Saf. 2010;33:25–34.
216. Kaymak Y. Creatine phosphokinase values during isotretinoin treatment for acne. Int J Dermatol. 2008;47:398–340.
217. Bertorini TE. Myoglobinuria, malignant hyperthermia, neuroleptic malignant syndrome and serotonin syndrome. Neurol Clin. 1997;15:649–671.
218. Nelson TE, Flewellen EH. Current concepts: the malignant hyperthermia syndrome. N Engl J Med. 1983;309:416–418.
219. MacLennan DH, Phillips MS. Malignant hyperthermia. Science. 1992;256:789–794.
220. McCarthy TV, Healy JM, Heffron JJ, et al. Localization of the malignant hyperthermia susceptibility locus to human chromosome 19q12–13.2. Nature. 1990;343:562–564.
221. Moslehi R, Lanlois S, Yam I, Fiedman J. Linkage of malignant hyperthermia and hyperkalemic periodic paralysis to the adult skeletal muscle sodium channel (SCNa4) gene in a large pedigree. Am J Hum Genet. 1998;76:21–27.
222. Iles DE, Lehman-Horn F, Scherer SW, et al. Localization of the gene encoding the d2/d-subunits of the L-type voltage dependent calcium channel to chromosome 7q and analysis of the segregation of flanking markers in malignant hyperthermia susceptible families. Hum Mol Genet. 1994;3:969–975.
223. Subrak R, Procaccio V, Klasusnitzer M, et al. Mapping of a further malignant hyperthermia susceptibility locus to chromosome 3q13.1. Am J Hum Genet. 1995;56:684–691.
224. Monnier N, Procaccio V, Stieglitz P, Lunardi J. Malignant hyperthermia susceptibility is associated with a mutation of the a1-subunit of the human dihydropyridine-sensitive L-type voltage -dependent calcium-channel receptor in skeletal muscle. Am J Hum Genet. 1997;60:1316–1325.
225. Robinson RL, Monnier N, Jung M, et al. A genome wide search for susceptibility loci in three European malignant hyperthermia pedigrees. Hum Mol Genet. 1997;6:953–961.
226. Sethna NF, Rockoff MA, Worthen HM, Rosnow JM. Anesthesia-related complications of children with Duchenne muscular dystrophy. Anesthesiology. 1988;68:462–465.
227. Ekbom K, Hed R, Kirstein L, Astrom KE. Muscular affections in chronic alcoholism. Arch Neurol. 1964;10:449–458.
228. Mayer RF, Garcia-Mullin R, Eckholdt JW. Acute alcoholic myopathy. Neurology. 1968;18:275.
229. Oh SJ. Chronic alcoholic myopathy: an entity difficult to diagnose. South Med J. 1972;65:449–452.
230. Perkoff GT, Dioso MM, Bleisch V, Klinkerfuss G. A spectrum of myopathy associated with alcoholism. I. Clinical and laboratory features. Ann Intern Med. 1967;67:481–492.
231. Rubenstein AE, Wainapel SF. Acute hypokalemic myopathy in alcoholism: a clinical entity. Arch Neurol. 1977;34:553–555.
232. Cogen FC, Rigg G, Simmons JL, Domino EF. Phencyclidine-associated acute rhabdomyolysis. Ann Intern Med. 1978;88:210–212.
233. Richter RW, Challenor YB, Pearson J, Kagen LJ, Hamilton LL, Ramsey WH. Acute myoglobinuria associated with heroin addiction. J Am Med Assoc. 1971;216:1172–1176.
234. Richter RW, Pearson J, Bruun B, Challenor YB, Brust JC, Baden MM. Neurological complications of heroin addiction. Bull N Y Acad Med. 1973;49:1–21.
235. Van den Bergh PY, Guettat L, Vande Berg BC, Martine JJ. Focal myopathy associated with chronic intramuscular injection of piritramide. Muscle Nerve. 1997;20(12):1598–1600.