Chapter 5
Carbocations, Carbanions, Free Radicals, Carbenes, and Nitrenes
There are four types of organic species in which a carbon atom has a valence of only 2 or 3.1 They are usually very short-lived, and most exist only as intermediates that are quickly converted to more stable molecules. However, some are more stable than others and fairly stable examples have been prepared for three of the four types. The four types of species are carbocations (A), carbon radicals (B), carbanions (C), and carbenes (D). Of the four, only carbanions have a complete octet around the carbon. There are many other organic ions and radicals with charges and unpaired electrons on atoms other than carbon, but only nitrenes (E), the nitrogen analogues of carbenes, will be discussed. Each of these five types is discussed in a separate section, which in each case includes brief summaries of the ways in which the species form and react. These summaries are short and schematic. The generation and fate of the five types are more fully treated for the appropriate specific reactions in Part II.
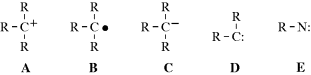
5.A. Carbocations2
5.A.i. Nomenclature
First, the nomenclature of carbocations (A) is discussed. For many years, these species were called “carbonium ions,” although it was suggested3 as long ago as 1902 that this was inappropriate because “-onium” usually refers to a covalency higher than that of the neutral atom. Nevertheless, the name “carbonium ion” was well established and created few problems4 until some years ago, when Olah and co-workers2,5 found evidence for another type of intermediate in which there is a positive charge at a carbon atom, but in which the formal covalency of the carbon atom is five rather than three. The simplest example is the methanonium ion (CH5+; see Reaction 12-01). Olah5 proposed that the name “carbonium ion” be henceforth reserved for pentacoordinated positive ions, and that A be called a “carbenium ions.” He also proposed the term “carbocation” to encompass both types. IUPAC has accepted these definitions.6 For the most part, intermediates such as A are called carbenium ions or carbocations, but the latter term will be used more often in this book.
5.A.ii. Stability and Structure of Carbocations
[Reprinted with permission from Kato, T.; Reed, C.A. Angew. Chem. Int. Ed. 2004, 43, 2908, Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. Copyright © 2004 by Wiley–VCH Verlag]
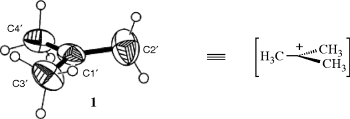
Carbocations are intermediates in several kinds of reactions.7 The more stable ones have been prepared in solution and in some cases even as solid salts. X-ray crystallographic structures also have been obtained in some cases.8 The X-ray of the tert-butyl cation complexed with dichloromethane was reported,9 for example, and is presented as 1 with the solvent molecules removed for clarity. The IR spectrum of the tert-butyl cation has been recorded in the gas phase.10 An isolable dioxa-stabilized pentadienylium ion was isolated and its structure was determined by  NMR,
NMR,  NMR, mass spectrometry, and IR.11 A β-fluoro substituted 4-methoxyphenethyl cation has been observed directly by laser flash photolysis.12 In solution, the carbocation may be free (this is more likely in polar solvents, in which it is solvated) or it may exist as an ion pair,13 which means that it is closely associated with a negative ion, called a counterion or gegenion. Ion pairs are more likely in nonpolar solvents.
NMR, mass spectrometry, and IR.11 A β-fluoro substituted 4-methoxyphenethyl cation has been observed directly by laser flash photolysis.12 In solution, the carbocation may be free (this is more likely in polar solvents, in which it is solvated) or it may exist as an ion pair,13 which means that it is closely associated with a negative ion, called a counterion or gegenion. Ion pairs are more likely in nonpolar solvents.
Among simple alkyl carbocations,14 the order of stability is tertiary > secondary > primary. There are many known examples of rearrangements of primary or secondary carbocations to tertiary, both in solution and in the gas phase (see Sec. 18.A.ii). Since simple alkyl cations are unstable in ordinary strong-acid solutions (e.g., H2SO4), the study of these species was greatly facilitated by the discovery that many of them could be kept indefinitely as stable solutions in mixtures of fluorosulfuric acid and antimony pentafluoride. Such mixtures, usually dissolved in SO2 or SO2ClF, are among the strongest acidic solutions known and are often called superacids.15 The original experiments involved the addition of alkyl fluorides to SbF5.16
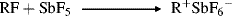
Subsequently, it was found that the same carbocations could also be generated from alcohols in superacid–SO2 at −60 °C17 and from alkenes by the addition of a proton from superacid or HF–SbF5 in SO2 or SO2ClF at low temperatures.18 Even alkanes give carbocations in superacid by loss of H−. For example,19 2-methylpropane gives the tert-butyl cation.
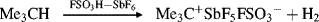
No matter how they are generated, study of the simple alkyl carbocations has provided dramatic evidence for the stability order.20 Both propyl fluorides gave the isopropyl cation; all four butyl fluorides21 gave the tert-butyl cation, and all seven of the pentyl fluorides examined gave the tert-pentyl cation. n-Butane, in superacid, gave only the tert-butyl cation. To date, no primary cation has survived long enough for detection. Neither methyl nor ethyl fluoride gave the corresponding carbocations when treated with SbF5. At low temperatures, methyl fluoride gave chiefly the methylated sulfur dioxide salt [(CH3OSO)+SbF6−],22 while ethyl fluoride rapidly formed the tert-butyl and tert-hexyl cations by addition of the initially formed ethyl cation to ethylene molecules also formed.23 At room temperature, methyl fluoride also gave the tert-butyl cation.24 In accord with the stability order, hydride ion is abstracted from alkanes by superacid most readily from tertiary and least readily from primary positions.
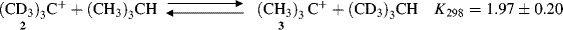
The stability order can be explained by the polar effect and by hyperconjugation (Sec. 2.M). In the polar effect, nonconjugated substituents exert an influence on stability through bonds (inductive effect) or through space (field effect). Since a tertiary carbocation has more carbon substituents on the positively charged carbon, relative to a primary, there is a greater polar effect that leads to great stability. In the hyperconjugation explanation,25 a primary carbocation is compared with a tertiary, and “the hyperconjugation concept arises from model-building procedures (see Sec. 2.M). In general, this means that the model must be corrected by including some delocalization in order to get a good enough description.”26 Evidence used to support the hyperconjugation explanation is that the equilibrium constant for this reaction involving 2 and 3 is 1.97, showing that 3 is more stable than 2.27 Due to a β secondary isotope effect, there is less hyperconjugation in 2 than in 3 (see Sec. 6.J.V.ii for isotope effects).28 The field effect explanation is that the electron-donating effect of alkyl groups increases the electron density at the charge-bearing carbon, reducing the net charge on the carbon, and in effect spreading the charge over the α carbons. It is a general rule that the more concentrated any charge is, the less stable the species bearing it will be. There are several structural types of delocalization, as summarized in Table 5.1.29
Table 5.1 Structural Types of Delocalizationa
| Valence Structures | Abbreviation | Name |
 |
ππ | Simple conjugation |
 |
σπ | Hyperconjugation |
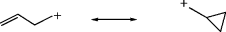 |
πσ | Homoconjugation |
 |
σσ | Homohyperconjugation |
 |
σπ/ππ | Hyperconjugation/conjugation |
 |
σπ/σπ | Double hyperconjugation |
| [Reprinted with permission from Radom, L.; Poppinger, D.; Haddon, R.C. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 5, Wiley, NY, 1976, pp. 2303–2426, Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. Copyright © 1976 by Wiley–VCH Verlag]. | ||
| a. See Ref. 25. | ||
The most stable of the simple alkyl cations is the tert-butyl cation. Even the relatively stable tert-pentyl and tert-hexyl cations fragment at higher temperatures to produce the tert-butyl cation, as do all other alkyl cations with four or more carbons to date studied.30 Methane,31 ethane, and propane, in superacid, also yield tert-butyl cations as the main product (see Reaction 12-20). Even paraffin wax and polyethylene give a tert-butyl cation. Solid salts of tert-butyl and tert-pentyl cations, [e.g., Me3C+ SbF6−], have been prepared from superacid solutions and are stable below −20 °C.32
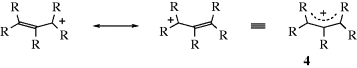
In carbocations, where the positive carbon is in conjugation with a double bond, as in allylic cations (the allyl cation is 4, R = H), the stability is greater because of increased delocalization due to resonance33 where the positive charge is spread over several atoms instead of being concentrated on one (see the molecular orbital picture of 4 in Sec. 2.C, category 2). Each of the terminal atoms in 4 has a charge of 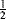 (the charge is exactly
(the charge is exactly 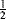 if all of the R groups are the same). Stable cyclic and acyclic allylic-type carbocations34 have been prepared by dissolving conjugated dienes in concentrated sulfuric acid; the cyclopentadienyl cation, (5) is an example.35
if all of the R groups are the same). Stable cyclic and acyclic allylic-type carbocations34 have been prepared by dissolving conjugated dienes in concentrated sulfuric acid; the cyclopentadienyl cation, (5) is an example.35
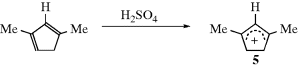
Stable allylic carbocations have also been obtained by the reaction between alkyl halides, alcohols, or alkenes (by hydride extraction) and SbF5 in SO2 or SO2ClF.36 Bis(allylic) cations37 are more stable than the simple allylic type, and some of these have been prepared in concentrated sulfuric acid.38 Arenium ions (Sec. 11.A.i) are familiar examples of this type. Propargyl cations (RC CC+R2) have also been prepared.39
CC+R2) have also been prepared.39
Canonical forms can be drawn for benzylic carbocations, as shown,40 and they are similar to those shown above for allylic cations.
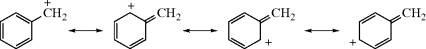
A number of benzylic carbocations have been obtained in solution as SbF6− salts.41 Diarylmethyl and triarylmethyl cations are even more stable because more canonical forms are possible (i.e., there is more extensive delocalization, hence greater stability). Chlorotriphenylmethane ionizes in polar solvents to give the stable triphenylmethyl cation (trityl cation, see 18), for example, because the solvent does not react with the ion,

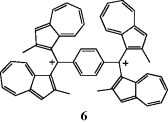
whereas water does react with the ion. In liquid SO2, for example, the ion remains stable for many years. Both triphenylmethyl and diphenylmethyl cations have been isolated as solid salts.42 In fact, Ph3C+ BF4− and related salts are available commercially. Arylmethyl cations are further stabilized if they have electron-donating substituents in ortho or para positions.43 Dications44 and trications are also possible, including the particularly stable dication (6), where each positively charged benzylic carbon is stabilized by two azulene rings.45 A related trication is known where two azulene rings stabilize each benzylic cationic center.46
Cyclopropylmethyl carbocations47 are even more stable than benzylic carbocations. Carbocations 7, 8, and similar ions have been prepared by dissolution of the alcohols in FSO3H–SO2–SbF5,48 and 9 has been prepared from the corresponding alcohol in 96% H2SO4.49 This special stability, which increases with each additional cyclopropyl group, is
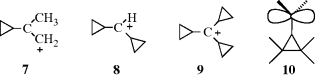
a result of conjugation between the bent orbitals of the cyclopropyl rings (Sec. 4.Q.i) and the vacant p orbital of the cationic carbon (see 10). Nuclear magnetic resonance and other studies have shown that the vacant p orbital lies parallel to the C-2–C-3 bond of the cyclopropane ring and not perpendicular to it.50 In this respect, the geometry is similar to that of a cyclopropane ring conjugated with a double bond (Sec. 4.Q.i). Cyclopropylmethyl cations are further discussed in Section 10.C.i, category 4. The stabilizing effect just discussed is unique to cyclopropyl groups. Cyclobutyl and larger cyclic groups are about as effective at stabilizing a carbocation as ordinary alkyl groups.51
Another structural feature that increases carbocation stability is the presence, adjacent to the cationic center, of a heteroatom bearing an unshared pair52 (e.g., oxygen,53 nitrogen,54 or halogen).55 Such ions are stabilized by resonance, as with the oxocarbenium ion (R2C=O+Me).

This methoxymethyl cation can be obtained as a stable solid, MeOCH2+ SbF6−.56 Carbocations containing either α, β, or γ silicon atom are also stabilized,57 relative to similar ions without the silicon atom. γ-Silyl cyclobutylcarbocations are known.58 In superacid solution, ions [e.g., CX3+ (X = Cl, Br, I)] have been prepared.59 Vinyl-stabilized halonium ions are also known.60
Simple acyl cations (RCO+) have been prepared61 in solution and the solid state.62 The acetyl cation (CH3CO+) is about as stable as the tert-butyl cation (see Table 5.1). The 2,4,6-trimethylbenzoyl and 2,3,4,5,6-pentamethylbenzoyl cations are especially stable (for steric reasons) and are easily formed in 96% H2SO4.63 These ions, often referred to as acylium ions, are stabilized by a canonical form containing a triple bond (12), although the positive charge is principally located on the carbon,64 so that 11 contributes more than 12.

The stabilities of many other stable carbocations can also be attributed to resonance. Among these are the tropylium, cyclopropenium,65 and other aromatic cations discussed in Chapter 2. Where resonance stability is completely lacking, as in the phenyl (C6H5+) or vinyl cations,66 the ion, if formed at all, is usually very short lived.67 Neither a vinyl68 nor a phenyl cation has as yet been prepared as a stable species in solution.69 However, stable alkenyl carbocations have been generated on Zeolite Y,70 and the phenyl cation has been observed in cryogenic argon matrices.71
Various quantitative methods have been developed to express the relative stabilities of carbocations.72 One of the most common of these, although useful only for relatively stable carbocations that are formed by ionization of alcohols in acidic solutions, is based on the equation73
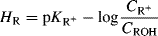
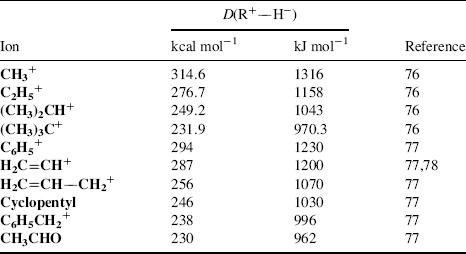
pKR+ is the pK value for the reaction 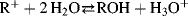 and is a measure of the stability of the carbocation. The HR parameter is an easily obtainable measurement of the stability of a solvent (Sec. 8.C) and approaches pH at low concentrations of acid. In order to obtain pKR+, for a cation R+, one dissolves the alcohol ROH in an acidic solution of known HR. Then, the concentration of R+ and ROH are obtained, generally from spectra, and pKR+ is easily calculated.74 A measure of carbocation stability that applies to less stable ions is the dissociation energy [D(R+–H−)] for the cleavage reaction R–H → R+ + H−, which can be obtained from PES (Sec. 1.E) and other measurements. Some values of D(R+–H−) are shown in Table 5.2.75–7875767778 Within a given class of ion, (primary, secondary, allylic, aryl, etc.), D(R+–H−) has been shown to be a linear function of the logarithm of the number of atoms in R+, with larger ions being more stable.77
and is a measure of the stability of the carbocation. The HR parameter is an easily obtainable measurement of the stability of a solvent (Sec. 8.C) and approaches pH at low concentrations of acid. In order to obtain pKR+, for a cation R+, one dissolves the alcohol ROH in an acidic solution of known HR. Then, the concentration of R+ and ROH are obtained, generally from spectra, and pKR+ is easily calculated.74 A measure of carbocation stability that applies to less stable ions is the dissociation energy [D(R+–H−)] for the cleavage reaction R–H → R+ + H−, which can be obtained from PES (Sec. 1.E) and other measurements. Some values of D(R+–H−) are shown in Table 5.2.75–7875767778 Within a given class of ion, (primary, secondary, allylic, aryl, etc.), D(R+–H−) has been shown to be a linear function of the logarithm of the number of atoms in R+, with larger ions being more stable.77
Table 5.2 R–H → R+ + H− Dissociation Energies in the Gas Phase.
Since the central carbon of tricoordinated carbocations has only three bonds and no other valence electrons, the bonds are sp2 and should be planar.79 Raman, IR, and NMR spectroscopic data on simple alkyl cations show this to be so.80 In methylcycohexyl cations, there are two chair conformations where the carbon bearing the positive charge is planar (13 and 14), and there is evidence that 14 is more stable due to a difference in hyperconjugation.81 Arenonium ions (15) are also known, and are relatively stable.82 Other evidence is that carbocations are difficult to form at bridgehead atoms in [2.2.1] systems,83 where they cannot be planar (see Sec. 10.A.ii).84 Bridgehead carbocations are known, however, as in [2.1.1]hexanes85 and cubyl carbocations.86 However, larger bridgehead ions can exist. For example, the adamantyl cation (15) has been synthesized, as the SF6− salt.87 The relative stability of 1-adamantyl cations is influenced by the number and nature of substituents. For example, the stability of the 1-adamantyl cation increases with the number of isopropyl substituents at C-3, C-5, and C-7.88 Among other bridgehead carbocations that have been prepared in superacid solution at –78 °C are the dodecahydryl cation (16)89 and the 1-trishomobarrelyl cation (17).90 In the latter case, the instability of the bridgehead position is balanced by the extra stability gained from the conjugation with the three cyclopropyl groups.
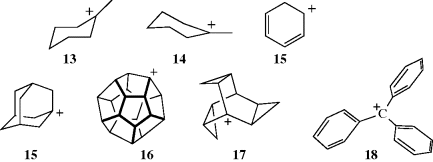
Triarylmethyl cations (e.g., the triphenylmethyl carbocation, 18),91 are propeller-shaped, although the central carbon and the three ring carbons connected to it are in a plane:92 The three benzene rings cannot be all in the same plane due to steric hindrance, although increased resonance energy would be gained if they could.
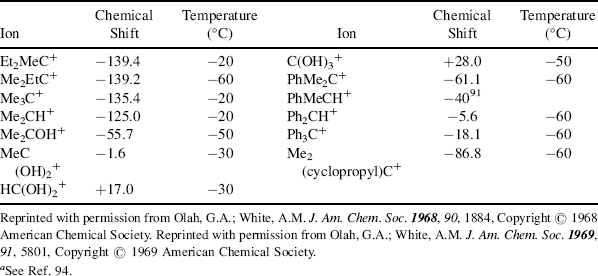
An important tool for the investigation of carbocation structure is measurement of the 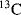 NMR chemical shift of the carbon atom bearing the positive charge.93 This shift approximately correlates with electron density on the carbon. The
NMR chemical shift of the carbon atom bearing the positive charge.93 This shift approximately correlates with electron density on the carbon. The 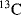 chemical shifts for a number of ions are given in Table 5.3.94 As shown in Table 5.3, the substitution of an ethyl for a methyl or a methyl for a hydrogen atom causes a downfield shift, indicating that the central carbon becomes somewhat more positive. On the other hand, the presence of hydroxy or phenyl groups decreases the positive character of the central carbon. The
chemical shifts for a number of ions are given in Table 5.3.94 As shown in Table 5.3, the substitution of an ethyl for a methyl or a methyl for a hydrogen atom causes a downfield shift, indicating that the central carbon becomes somewhat more positive. On the other hand, the presence of hydroxy or phenyl groups decreases the positive character of the central carbon. The 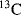 chemical shifts are not always in exact order of carbocation stabilities, as determined in other ways. Thus the chemical shift shows that the triphenylmethyl cation has a more positive central carbon than diphenylmethyl cation, although the former is more stable. Also, the 2-cyclopropylpropyl and 2-phenylpropyl cations have shifts of −86.8 and −61.1, respectively, although we have seen that according to other criteria a cyclopropyl group is better than a phenyl group at stabilizing a carbocation.95 The reasons for this discrepancy are not fully understood.88,96
chemical shifts are not always in exact order of carbocation stabilities, as determined in other ways. Thus the chemical shift shows that the triphenylmethyl cation has a more positive central carbon than diphenylmethyl cation, although the former is more stable. Also, the 2-cyclopropylpropyl and 2-phenylpropyl cations have shifts of −86.8 and −61.1, respectively, although we have seen that according to other criteria a cyclopropyl group is better than a phenyl group at stabilizing a carbocation.95 The reasons for this discrepancy are not fully understood.88,96
Table 5.3 The NMRChemical-Shift Values, in Parts per Million from S2, for the Charged Carbon Atom of Some Carbocations in SO2ClF–SbF5, SO2–FSO3H–SbF6, or SO2–SbF5a
Nonclassical carbocations are discussed in section 10.C.i.
5.A.iii. The Generation and Fate of Carbocations
A number of methods are available to generate carbocations, stable or unstable.
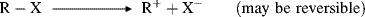
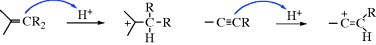
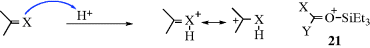
There are two principal pathways by which carbocations react to give stable products that are effectively the reverse of the two pathways just described.
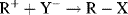
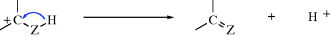
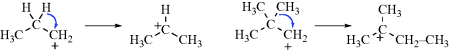
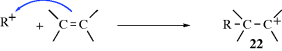
5.B. Carbanions
5.B.i. Stability and Structure105
Formally, a carbanion is a trivalent carbon atom with an unshared electron pair, and a formal charge of −1. In fact, there are few carbanions that do not have an anion-stabilizing group attached to the carbon atom. Stabilization may be by resonance delocalization or by orbital participation of an atom with d orbitals or orbitals associated withy a metal.
By definition, every carbanion possesses an unshared pair of electrons and is formally a base. When a carbanion donates an electron to a proton, it is converted to its conjugate acid (an acid–base reaction, see Chapter 8). If the carbanion (R3C:−) were available, reaction with an acid generates the conjugate acid (R3C–H), an alkane. The stability of the carbanion is directly related to the strength of the conjugate acid. The weaker that conjugate acid, the greater the base strength of the carbanion, and the lower the stability of the carbanion.106 Stability here is judged by diminished reactivity (lower electron-donating ability) with a proton. The greater the stability, the lower the electron-donating ability (lower reactivity) for reaction of the carbanion with a proton (any acid that is sufficiently strong), and hence the longer lived the carbanion. Thus the determination of the order of stability of a series of carbanions is equivalent to a determination of the inverse order of strengths of the conjugate acids, and one can obtain information about relative carbanion stability from a table of acid strengths (e.g., Table 8.1).
While formation of simple carbanions (e.g, CH3−) is rare, formation of a carbon–metal bond often generates a molecule (e.g., R3C–M, where M = a metal atom) that has a polarized bond in which the carbon is electron rich (δ−). An organic molecule that contains a carbon–metal bond is called an organometallic compound. Organometallic compounds where the metal is Mg, Li, or other metals are carbanion surrogates, and in much of their chemistry they react as if they were carbanions (see Reactions 12-22–12.39). Many such compounds are known, and organometallic chemistry is a very large area, occupying a borderline region between organic and inorganic chemistry. This section will discuss carbanions with little reference to a metal. Section 5.B.ii will discuss the structures of organometallic compounds, which are often carbanion surrogates.
Carbanions are very strong bases, and the conjugate acids of simple unsubstituted carbanions are very weak acids, with very few exceptions. Unfortunately, it is not easy to measure acid strengths of very weak acids. There is little doubt that carbanions are very unstable in solution, and in contrast to the situation with carbocations, efforts to prepare solutions in which carbanions (e.g., ethyl or isopropyl) exist in a relatively free state have not yet been successful. It has also not been possible to form these carbanions in the gas phase. Indeed, there is evidence that simple carbanions (e.g., ethyl and isopropyl) are unstable, losing an electron, which converts them to radicals.107 Nevertheless, there have been several approaches to the problem. Applequist and O'Brien108 studied the position of equilibrium for the reaction:
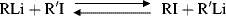
This reaction was done in ether or an ether–pentane mixture. The reasoning in these experiments was that the R group that forms the more stable carbanion would be more likely to be bonded to lithium than to iodine. Carbanion stability was found to be in the order: vinyl > phenyl > cyclopropyl > ethyl > n-propyl > isobutyl > neopentyl > cyclobutyl > cyclopentyl. In a somewhat similar approach, Dessy et al.109 treated a number of alkylmagnesium compounds with a number of alkylmercury compounds in THF, setting up the equilibrium:
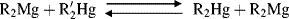
where the group of greater carbanion stability is linked to magnesium. The carbanion stability determined this way was phenyl > vinyl > cyclopropyl > methyl > ethyl > isopropyl. The two stability orders are in fairly good agreement, and they show that stability of simple carbanions decreases in the order methyl > primary > secondary. It was not possible to determine the position of tert-butyl by the experiments reported by Dessy et al.109, but there seems little doubt that it is still less stable. This stability order can be interpreted as solely a consequence of the field effect since resonance is absent. The electron-donating alkyl groups of isopropyl result in a greater negative charge density at the central carbon atom (compared with methyl), thus decreasing its stability. The results of Applequist and O'Brien108 show that β branching also decreases carbanion stability. Cyclopropyl occupies an apparently anomalous position, but this is probably due to the large amount of s character in the carbanionic carbon (see Sec. 5.B.i, category 2). Strongly electron-withdrawing groups (e.g., trifluoromethylsulfonyl) provide exceptional stability to carbanions.110
A different approach to the problem of hydrocarbon acidity, and hence carbanion stability, is that of Shatenshtein and Shapiro, who treated hydrocarbons with deuterated potassium amide and measured the rates of hydrogen exchange.111 The experiments did not measure thermodynamic acidity, since rates were measured, not positions of equilibria. They measured kinetic acidity; that is, which compounds gave up protons most rapidly (see Sec. 6.F for the distinction between thermodynamic and kinetic control of product). Measurements of rates of hydrogen exchange enable one to compare acidities of a series of acids against a given base even where the positions of the equilibria cannot be measured because they lie too far to the side of the starting materials; that is, where the acids are too weak to be converted to their conjugate bases in measurable amounts. Although the correlation between thermodynamic and kinetic acidity is far from perfect,112 the results of the rate measurements, too, indicated that the order of carbanion stability is methyl > primary > secondary > tertiary.111
Experiments described above were done in solution, and experiments in the gas phase gave different results. In reactions of −OH with alkyltrimethylsilanes, it is possible to cleave either R or Me. Since the R or Me come off as a carbanion or incipient carbanion, the product ratio RH/MeH can be used to establish the relative stabilities of various R groups. From these experiments a stability order of neopentyl > cyclopropyl > tert-butyl > n-propyl > methyl > isopropyl > ethyl was found.113 On the other hand, in a different kind of gas-phase experiment, Graul and Squires114 were able to observe CH3− ions, but not the ethyl, isopropyl, or tert-butyl ions.
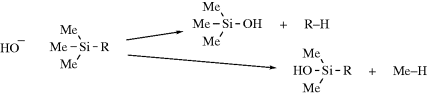
As mentioned above, carbanion-stabilizing groups can increase the stability of carbanions, which influences their ease of formation. Six structural features that lead to improved stability are listed:

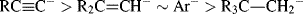

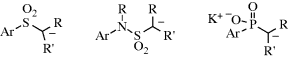
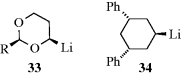
5.B.ii. The Structure of Organometallic Compounds152
Whether a carbon–metal bond is ionic or polar-covalent is determined chiefly by the electronegativity of the metal and the structure of the organic part of the molecule. Ionic bonds become more likely as the negative charge on the metal-bearing carbon is decreased by resonance or field effects. Thus the sodium salt of acetoacetic ester has a more ionic carbon–sodium bond than methylsodium.
Most organometallic bonds are polar-covalent. Only the alkali metals have electronegativities low enough to form ionic bonds with carbon, and even here the behavior of lithium alkyls shows considerable covalent character. The simple alkyls and aryls of Na, K, Rb, and Cs153 are nonvolatile solids154 insoluble in benzene or other organic solvents, while alkyllithium reagents are soluble, although they too are generally nonvolatile solids. Organolithium reagents with alkyl units (alkyllithium reagents) do not exist as monomeric species in hydrocarbon solvents or ether.155 In benzene and cyclohexane, freezing-point-depression studies have shown that alkyllithium reagents are normally hexameric unless steric interactions favor tetrameric aggregates.156 Nuclear magnetic resonance studies, especially measurements of 13C–6Li coupling, have also shown aggregation in hydrocarbon solvents.157 Boiling-point-elevation studies have been performed in ether solutions, where alkyllithium reagents exist in two- to fivefold aggregates.158 Even in the gas phase159 and in the solid state,160 alkyllithium reagents exist as aggregates. X-ray crystallography has shown that methyllithium has the same tetrahedral structure in the solid state as in ether solution.160 However, tert-butyllithium is monomeric in THF, although dimeric in ether and tetrameric in hydrocarbon solvents.161 Neopentyllithium exists as a mixture of monomers and dimers in THF.162
The C–Mg bond in Grignard reagents is covalent and not ionic. The actual structure of Grignard reagents in solution has been a matter of much controversy over the years.163 In 1929, it was discovered164 that the addition of dioxane to an ethereal Grignard solution precipitates all the magnesium halide and leaves a solution of R2Mg in ether; (i.e., there can be no RMgX in the solution since there is no halide). The following equilibrium, now called the Schlenk equilibrium, was proposed as the composition of the Grignard solution:
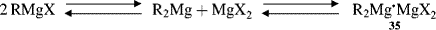
in which 35 is a complex. Much work has demonstrated that the Schlenk equilibrium actually exists and that the position of the equilibrium depends on the identity of R, X, the solvent, the concentration, and the temperature.165 It has been known for many years that the magnesium in a solution of a Grignard reagent, no matter whether it is RMgX, R2Mg, or MgX2, can coordinate with two molecules of ether in addition to the two covalent bonds to generate the solvent-coordinated species shown.
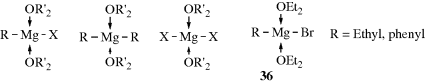
Rundle and Guggenberger166 performed X-ray diffraction studies on solid phenylmagnesium bromide dietherate and on ethylmagnesium bromide dietherate, which they obtained by cooling ordinary ethereal Grignard solutions until the solids crystallized. They found that the structures were magnesium bromides (e.g., 36). These solids still contained ether. When ordinary ethereal Grignard solutions167 prepared from bromomethane, chloromethane, bromoethane, and chloroethane were evaporated at ~100 °C under vacuum so that the solid remaining contained no ether, X-ray diffraction showed no RMgX, but a mixture of R2Mg and MgX2.168 These results indicate that in the presence of ether, RMgX√2Et2O is the preferred structure, while the loss of ether drives the Schlenk equilibrium to R2Mg + MgX2. However, conclusions drawn from a study of the solid materials do not necessarily apply to the structures in solution.
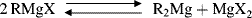
Boiling-point-elevation and freezing-point-depression measurements have demonstrated that in THF at all concentrations and in ether at low concentrations (up to ~0.1 M) Grignard reagents prepared from alkyl bromides and iodides are monomeric, (i.e., there are few or no molecules with two Mg atoms).169 Thus, part of the Schlenk equilibrium is operating but not the other part (i.e., 35 is not present in measurable amounts). This was substantiated by  NMR spectra of the ethyl Grignard reagent in THF, which showed the presence of three peaks, corresponding to EtMgBr, Et2Mg, and MgBr2.170 That the equilibrium between RMgX and R2Mg lies far to the left for “ethylmagnesium bromide” in ether was shown by Smith and Becker,171 who mixed 0.1 M ethereal solutions of Et2Mg and MgBr2 and found that a reaction occurred with a heat evolution of 3.6 kcal mol−1 (15 kJ mol−1) of Et2Mg, and that the product was monomeric (by boiling-point elevation measurements). When either solution was added little by little to the other, there was a linear output of heat until almost a 1 : 1 molar ratio was reached. Addition of an excess of either reagent gave no further heat output. These results show that at least under some conditions the Grignard reagent is largely RMgX (coordinated with solvent), but that the equilibrium can be driven to R2Mg by evaporation of all the ether or by addition of dioxane.
NMR spectra of the ethyl Grignard reagent in THF, which showed the presence of three peaks, corresponding to EtMgBr, Et2Mg, and MgBr2.170 That the equilibrium between RMgX and R2Mg lies far to the left for “ethylmagnesium bromide” in ether was shown by Smith and Becker,171 who mixed 0.1 M ethereal solutions of Et2Mg and MgBr2 and found that a reaction occurred with a heat evolution of 3.6 kcal mol−1 (15 kJ mol−1) of Et2Mg, and that the product was monomeric (by boiling-point elevation measurements). When either solution was added little by little to the other, there was a linear output of heat until almost a 1 : 1 molar ratio was reached. Addition of an excess of either reagent gave no further heat output. These results show that at least under some conditions the Grignard reagent is largely RMgX (coordinated with solvent), but that the equilibrium can be driven to R2Mg by evaporation of all the ether or by addition of dioxane.
For some aryl Grignard reagents, it is possible to distinguish separate NMR chemical shifts for ArMgX and Ar2Mg.172 From the area under the peaks, it is possible to calculate the concentrations of the two species, and from them, equilibrium constants for the Schlenk equilibrium. These data show172 that the position of the equilibrium depends very markedly on the aryl group and the solvent, but that conventional aryl Grignard reagents in ether are largely ArMgX. In THF the predominance of ArMgX is less, and with some aryl groups there is actually more Ar2Mg present. Separate NMR chemical shifts have also been found for alkyl RMgBr and R2Mg in HMPA173 and in ether at low temperatures.174 When Grignard reagents from alkyl bromides or chlorides are prepared in triethylamine the predominant species is RMgX.175 Thus the most important factor determining the position of the Schlenk equilibrium is the solvent. For primary alkyl groups the equilibrium constant for the reaction as written above is lowest in Et3N, higher in ether, and still higher in THF.176
However, Grignard reagents prepared from alkyl bromides or iodides in ether at higher concentrations (0.5–1 M) contain dimers, trimers, and higher polymers, and those prepared from alkyl chlorides in ether at all concentrations are dimeric,177 so that 35 is in solution, probably in equilibrium with RMgX and R2Mg (i.e., the complete Schlenk equilibrium seems to be present).
The Grignard reagent prepared from 1-chloro-3,3-dimethylpentane in ether undergoes rapid inversion of configuration at the Mg containing carbon (demonstrated by NMR; this compound is not chiral).178 The mechanism of this inversion is not completely known. Despite the mechanistic ambiguity, in almost all cases, it is not possible to retain the configuration of a stereogenic carbon while forming a Grignard reagent.
Organolithium reagents (RLi) are very important reagents in organic chemistry. In recent years, a great deal has been learned about their structure179 in both the solid state and in solution. X-ray analysis of complexes of n-butyllithium with tetramethylethylenediamine (TMEDA), THF, and 1,2-dimethoxyethane (DME) shows them to be dimers and tetramers [e.g., (BuLi·DME)4];180 they are aggregates.181 X-ray analysis of isopropyllithium shows it to be a hexamer [(iPrLi)6],182 and unsolvated lithium aryls are tetramers.183 α-Ethoxyvinyllithium [CH2=C(OEt)Li] shows a polymeric structure with tetrameric subunits.184 Aminomethyl aryllithium reagents have been shown to be chelated and dimeric in solvents (e.g., THF).185 There are several functionalized organolithium reagents.186
The dimeric, tetrameric, and hexameric structures of organolithium reagents187 in the solid state is often retained in solution, but this is dependent on the solvent and complexing additives, if any. A tetrahedral organolithium compound is known,188 and the X-ray of an α,α-dilithio hydrocarbon has been reported.189 Phenyllithium is a mixture of tetramers and dimers in diethyl ether, but stoichiometric addition of THF, DME, or TMEDA leads to the dimer.190 The solution structures of mixed aggregates of butyllithium and amino-alkaloids has been determined191 as well as the solution structure of sulfur-stabilized allyllithium compounds.192 Vinyllithium is an 8:1 mixture of tetramer/dimer in THF at −90 °C, but addition of TMEDA changes the ratio of tetramer/dimer to 1 : 13 at −80 °C.193 Internally solvated allylic lithium compounds have been studied, showing the coordinated lithium to be closer to one of the terminal allyl carbons.194 A relative scale of organolithium stability has been established,195 and the issue of configurational stability of enantioenriched organolithium reagents has been examined.196
Enolate anions are an important class of carbanions that appear in a variety of important reactions, including alkylation α to a carbonyl group and the aldol (16-34) and Claisen condensation (16-85) reactions. Metal enolate anions of aldehydes, ketones, esters, and other acid derivatives exist as aggregates in ether solvents.197 There is evidence that the lithium enolate of isobutyrophenone is a tetramer in THF,198 but a dimer in DME.199 X-ray crystallography of ketone enolate anions have shown that they can exist as tetramers and hexamers.200 There is also evidence that the aggregate structure is preserved in solution and is probably the actual reactive species. Lithium enolate anions derived from esters are as dimers in the solid state201 that contain four THF molecules. It has also been established that the reactivity of enolate anions in alkylation and condensation reactions is influenced by the aggregate state of the enolate. It is also true that the relative proportions of (E) and (Z) enolate anions are influenced by the extent of solvation and the aggregation state. Addition of LiBr to a lithium enolate anion in THF suppresses the concentration of monomeric enolate.202Ab initio studies confirm the aggregate state of acetaldehyde.203 It is also known that α-Li benzonitrile [PhCH(Li)CN] exists as a dimer in ether and with TMEDA.204 Mixed aggregates of tert-butyllithium and lithium tert-butoxide are known to be hexameric.205
It might be mentioned that matters are much simpler for organometallic compounds with less-polar bonds. Thus Et2Hg and EtHgCl are both definite compounds, the former is a liquid and the latter is a solid. Organocalcium reagents are also known, and are formed from alkyl halides via a single electron transfer (SET) mechanism with free radical intermediates.206
5.B.iii. The Generation and Fate of Carbanions
There are two principal ways in which most carbanions are generated.

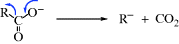
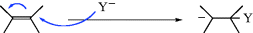
The most common reaction of carbanions is to donate electrons to a positive species, often a proton, or with another species that has an empty orbital in its outer shell (a Lewis acid–base reaction):

This means that carbanions react with electrophilic atoms (those functionalized so there is a δ+ carbon atom); see Chapter 16.
Carbanions may also form a bond with a carbon that already has four bonds, by displacing one of the four groups (SN2 reaction, see Chapter 10):
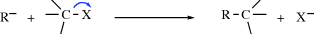
Like carbocations, carbanions can also react in ways in which they are converted to species that are still charged. They can add to double bonds (usually C=O double bonds; see Chapters 10 and 16),

or rearrange, although this is rare (see Chapter 18),

or they are oxidized to free radicals.208 A system in which a carbocation [Ph(p-Me2NC6H4)2C+] oxidizes a carbanion [(p-NO2C6H4)3C−] to give two free radicals, reversibly, so that all four species are present in equilibrium, has been demonstrated.209,210
Organometallic compounds that are not ionic but polar–covalent behave very much as if they were ionic and give similar reactions.
5.C. Free Radicals
5.C.i. Stability and Structure211
A free radical (usually just called a radical) may be defined as a species that contains one or more unpaired electrons. Note that this definition includes certain stable inorganic molecules (e.g., NO and NO2), as well as many individual atoms (e.g., Na and Cl). As with carbocations and carbanions, simple alkyl radicals are very reactive and are usually transient species. For the most part, their lifetimes are extremely short in solution, but they can be kept frozen for relatively long periods of time within the crystal lattices of other molecules.212 There are, however, many stable radicals,213 some of which will be noted below. Many spectral214 measurements have been made on radicals trapped in this manner. Even under these conditions the methyl radical decomposes with a half-life of 10–15 min in a methanol lattice at 77 K.215 Since the lifetime of a radical depends not only on its inherent stability, but also on the conditions under which it is generated, the terms persistent and stable are usually used for the different senses. A stable radical is inherently stable; a persistent radical has a relatively long lifetime under the conditions at which it is generated, although it may not be very stable.
Radicals can be characterized by several techniques (e.g., mass spectrometry216 or the characterization of alkoxycarbonyl radicals by Step–Scan Time-Resolved Infrared Spectroscopy).217 Another technique makes use of the magnetic moment that is associated with the spin of an electron, which can be expressed by a quantum number of 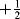 or
or 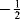 . According to the Pauli principle, any two electrons occupying the same orbital must have opposite spins, so the total magnetic moment is zero for any species in which all the electrons are paired. In radicals, however, one or more electrons are unpaired, so there is a net magnetic moment and the species is paramagnetic. Radicals can therefore be detected by magnetic-susceptibility measurements, but for this technique a relatively high concentration of radicals is required.
. According to the Pauli principle, any two electrons occupying the same orbital must have opposite spins, so the total magnetic moment is zero for any species in which all the electrons are paired. In radicals, however, one or more electrons are unpaired, so there is a net magnetic moment and the species is paramagnetic. Radicals can therefore be detected by magnetic-susceptibility measurements, but for this technique a relatively high concentration of radicals is required.
A much more important technique is electron spin resonance (ESR), also called electron paramagnetic resonance (EPR).218 The principle of ESR is similar to that of NMR, except that electron spin is involved rather than nuclear spin. The two electron spin states (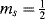 and
and 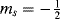 ) are ordinarily of equal energy, but in a magnetic field the energies are different. As in NMR, a strong external field is applied and electrons are caused to flip from the lower state to the higher by the application of an appropriate radio frequency signal. Inasmuch as two electrons paired in one orbital must have opposite spins that cancel, an ESR spectrum arises only from species that have one or more unpaired electrons (i.e., free radicals).
) are ordinarily of equal energy, but in a magnetic field the energies are different. As in NMR, a strong external field is applied and electrons are caused to flip from the lower state to the higher by the application of an appropriate radio frequency signal. Inasmuch as two electrons paired in one orbital must have opposite spins that cancel, an ESR spectrum arises only from species that have one or more unpaired electrons (i.e., free radicals).
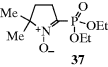
Since only free radicals give an ESR spectrum, the method can be used to detect the presence of radicals and to determine their concentration.219 Furthermore, information concerning the electron distribution (and hence the structure) of free radicals can be obtained from the splitting pattern of the ESR spectrum (ESR peaks are split by nearby protons).220 Fortunately (for the existence of most free radicals is very short), it is not necessary for a radical to be persistent for an ESR spectrum to be obtained. Electron spin resonance spectra have been observed for radicals with lifetimes considerably <1 s. Failure to observe an ESR spectrum does not prove that radicals are not involved, since the concentration may be too low for direct observation. In such cases, the spin-trapping technique can be used.221 In this technique, a compound is added that is able to combine with very reactive radicals to produce more persistent radicals; the new radicals can be observed by ESR. Azulenyl nitrones have been developed as chromotropic spin-trapping agents.222 An important class of spin-trapping compounds are nitroso compounds, which react with radicals to give stable nitroxide radicals:223 RN=O + R′√ → RR′N–O√. An N-oxide spin trap has been developed [37; 2(diethylphosphino)-5,5-dimethyl-1-pyrroline-N-oxide], and upon trapping a reactive free radical, 31P NMR can be used to identify it.224 This technique is effective, and short-lived species (e.g., the oxiranylmethyl radical) have been detected by spin trapping.225 Other molecules have been used to probe the intermediacy of radicals via SET processes. They are called SET probes.226
Because there is an equal probability that a given unpaired electron will have a quantum number of 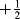 or
or 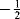 , radicals are observed as a single line in an ESR spectrum unless they interact with other electronic or nuclear spins or possess magnetic anisotropy, in which case two or more lines may appear in the spectrum.227
, radicals are observed as a single line in an ESR spectrum unless they interact with other electronic or nuclear spins or possess magnetic anisotropy, in which case two or more lines may appear in the spectrum.227
Another magnetic technique for the detection of free radicals uses an ordinary NMR instrument. It was discovered228 that if an NMR spectrum is taken during the course of a reaction, certain signals might be enhanced, either in a positive or negative direction; others may be reduced. When this type of behavior, called chemically induced dynamic nuclear polarization229 (CIDNP), is found in the NMR spectrum of the product of a reaction, it means that at least a portion of that product was formed via the intermediacy of a free radical.230 For example, the question was raised whether radicals were intermediates in the exchange reaction between ethyl iodide and ethyllithium (Reaction 12-39):
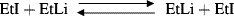
Curve a in Fig. 5.1231 shows an NMR spectrum taken during the course of the reaction. Curve b is a reference spectrum of ethyl iodide (CH3 protons at δ = 1.85; CH2 protons at δ = 3.2). Note that in curve a some of the ethyl iodide signals are enhanced; others go below the base line (negative enhancement; also called emission). Thus the ethyl iodide formed in the exchange shows CIDNP and so was formed via a free radical intermediate. Chemically induced dynamic nuclear polarization results when protons in a reacting molecule become dynamically coupled to an unpaired electron while traversing the path from reactants to products. Although the presence of CIDNP almost always means that a free radical is involved,232 its absence does not prove that a free radical intermediate is necessarily absent, since reactions involving free radical intermediates can also take place without observable CIDNP. Also, the presence of CIDNP does not prove that all of a product was formed via a free radical intermediate, only that some of it was. Note that dynamic nuclear polarization (DNP) enhances signal intensities in the NMR spectra of solids and liquids. In a contemporary DNP experiment, a diamagnetic sample is doped with a paramagnet and the large polarization of the electron spins is transferred to the nuclei via microwave irradiation of the EPR spectrum.233 Dynamic nuclear polarization has been used to examine biradicals.234
Fig. 5.1231 (a) The NMR spectrum taken during reaction between EtI and EtLi in benzene (the region between 0.5 and 3.5 δ was scanned with an amplitude twice that of the remainder of the spectrum). The signals at 1.0–1.6 δ are due to butane, some of which is also formed in the reaction. (b) Reference spectrum of EtI. [Reprinted with permission from Ward, H.R.; Lawler, R.G.; Cooper, R.A. J. Am. Chem. Soc. 1969, 91, 746. Copyright © 1969 American Chemical Society.]

As with carbocations, the stability order of free radicals is tertiary > secondary > primary, explainable by field effects and hyperconjugation, analogous to that in carbocations (Sec. 5.A.ii).235
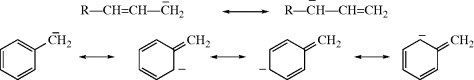
With resonance possibilities, the stability of free radicals increases;236 some can be kept indefinitely.237 Benzylic and allylic238 radicals for which canonical forms can be drawn similar to those shown for the corresponding cations (Sec. 5.A.ii) and anions (Sec. 5.B.i, category 1) are more stable than simple alkyl radicals, but still have only a transient existence under ordinary conditions. Note that 2-phenylethyl radicals have been shown to exhibit bridging of the phenyl group.239
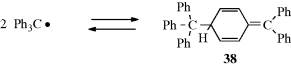
The triphenylmethyl and similar radicals240 are stable enough to exist in solution at room temperature, although they are in equilibrium with a dimeric form. The concentration of triphenylmethyl radical in benzene solution is ~2% at room temperature. For many years, it was assumed that Ph3C√, the first stable free radical known,241 dimerized to hexaphenylethane (Ph3C–CPh3),242 but UV and NMR investigations have shown that the true structure is 38.243 Although triphenylmethyl-type radicals are stabilized by resonance:
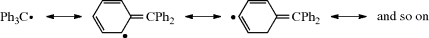
steric hindrance to dimerization and not resonance is the major cause of their stability.244 This was demonstrated by the preparation of the radicals 39 and 40.245 These radicals are electronically very similar, but 39, being planar, has much less steric hindrance to dimerization than Ph3C√, while 40, with six groups in ortho positions, has much more. On the other hand, the planarity of 35 means that it has a maximum amount of
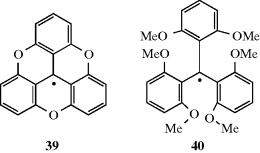
resonance stabilization, while 40 must have much less, since its degree of planarity should be even less than Ph3C√, which itself is propeller shaped and not planar. Thus if resonance is the chief cause of the stability of Ph3C√, 40 should dimerize and 39 should not, but if steric hindrance is the major cause, the reverse should happen. It was found233 that 40 gave no evidence of dimerization, even in the solid state, while 39 existed primarily in the dimeric form, which is dissociated to only a small extent in solution.246 This result indicates that steric hindrance to dimerization is the major cause for the stability of triarylmethyl radicals. A similar conclusion was reached in the case of (NC)3C√, which dimerizes readily although it is considerably stabilized by resonance.247 Nevertheless, that resonance is still an important contributing factor to the stability of radicals is shown by the facts that (1) the radical t-Bu(Ph)2C√ dimerizes more than Ph3C√, while p-PhCOC6H4(Ph2)C√ dimerizes less.248 The latter has more canonical forms than Ph3C√, but steric hindrance should be about the same (for attack at one of the two rings). (2) A number of radicals (p-XC6H4)3C√, with X = F, Cl, O2N, CN, and so on, do not dimerize, but are kinetically stable.249 Completely chlorinated triarylmethyl radicals are more stable than the unsubstituted kind, probably for steric reasons, and many are quite inert in solution and in the solid state.250
Allylic radical are relatively stable, and the pentadienyl radical is particularly stable, but (E,E)-, (E,Z)-, and (Z,Z)-stereoisomers can form. It has been calculated that the (Z,Z)-pentadienyl radical is 5.6 kcal mol−1 less stable than the (E,E)-pentadienyl radical.251 Note that vinyl radicals have (E)- and (Z)-forms and the inversion barrier from one to the other increases as the electronegativity of substituents increase.252 Conjugated propargylic radicals are calculated to have diminished stability as the conjugation increases, in contrast to the behavior of alkenes.253 Cyclopropyl alkynes have been used as mechanistic probes to distinguish between vinyl radicals and ionic intermediates.254 Enolate radicals are also known.255
It has been postulated that the stability of free radicals is enhanced by the presence at the radical center of both an electron-donating and an electron-withdrawing group.256 This finding is called the push–pull or captodative effect (see also, Sec. 4.K.i). The effect arises from increased resonance, as in 41.
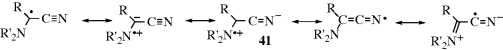
There is some evidence in favor257 of the captodative effect, some of it from ESR studies.258 However, there is also experimental259 and theoretical260 evidence against it. There is evidence that while FCH2√ and F2CH√ are more stable than CH3√, the radical CF3√ is less stable; that is, the presence of the third F destabilizes the radical.261
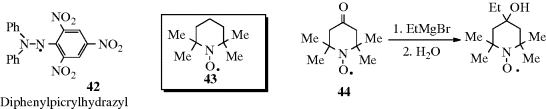
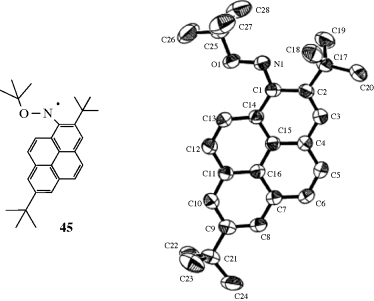
Certain radicals with unpaired electron on a carbon are also very stable.262 Radicals can be stabilized by intramolecular hydrogen bonding.263 Diphenylpicrylhydrazyl (42) is a solid that can be kept for years, and stable neutral azine radicals have been prepared.264 Nitroxide radicals were mentioned previously,265 and the commercially available TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) free radical (43) is a stable nitroxyl radical used in chemical reactions (e.g., oxidations),266 or as a spin trap.267 Nitroxyl radical 44 is a nitroxide radical so stable that reactions can be performed on it (e.g., the Grignard reaction shown with 44; see Reaction 16-24) without affecting the unpaired electron268 (the same is true for some of the chlorinated triarylmethyl radicals mentioned above269). Several nitrogen-containing groups are known to stabilize radicals, and the most effective radical stabilization is via spin delocalization.270 A number of persistent N-tert-butoxy-1-aminopyrenyl radicals (e.g., 45) have been isolated as monomeric radical crystals (see 46, the X-ray crystal
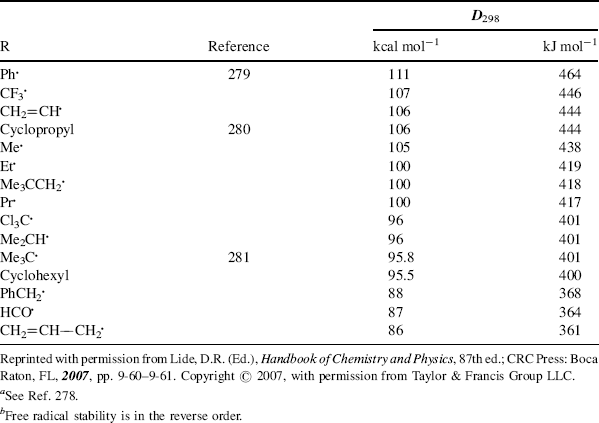
structure of 45),271 and monomeric N-alkoxyarylaminyls have been isolated.272 α-Trichloromethylbenzyl(tert-butyl)aminoxyl (47) is extremely stable.273 In aqueous media it is stable for > 30 days, and in solution in an aromatic hydrocarbon solvent it has survived for >90 days.273 Although the stable nitroxide radicals have the α-carbon blocked to prevent radical formation there, stable nitroxide radicals are also known with hydrogen at the α-carbon,274 and long-lived vinyl nitroxide radicals are known.275 A stable organic radical lacking resonance stabilization has been prepared (48), and its X-ray crystal structure was obtained.276 Dissociation energies (D values) of R–H bonds provide a measure of the relative inherent stability of free radicals R.277 Table 5.4 lists such values. 278279280281 The higher the D value, the less stable the radical. Bond-dissociation energies also have been reported for the C–H bond of alkenes and dienes282 and for the C–H bond in radical precursors XYC–H, where X,Y can be H, alkyl, COOR, COR, SR, CN, NO2, and so on.283 Bond dissociation energies for the C–O bond in hydroperoxide radicals (ROO√) have also been reported.284 However, note that basing radical stabilization energy on the difference between the bond dissociation energy (BDE) of CH3–H, as a reference point, and of R–H has been observed to have shortcomings.285 The problem is that these values are only applicable to carbon-centered radicals, and the stabilization energies are not transferable and cannot be used to estimate BDE of R–R′, R–R, or any R–X compounds.282
Table 5.4 The D298 Values for Some R–H Bondsa,b
There are two possible structures for simple alkyl radicals.286 They might have sp2 bonding, in which case the structure would be planar, with the odd electron in a p orbital, or the bonding might be sp3, which would make the structure pyramidal and place the odd electron in an sp3 orbital. The ESR spectra of √CH3 and other simple alkyl radicals as well as other evidence indicate that these radicals have planar structures.287 This finding is in accord with the known loss of optical activity when a free radical is generated at a stereogenic carbon.288 In addition, electronic spectra of the CH3 and CD3 radicals (generated by flash photolysis) in the gas phase have definitely established that under these conditions the radicals are planar or near planar.289 The IR spectra of √CH3 trapped in solid argon led to a similar conclusion.290 Despite the usual loss of optical activity noted above, asymmetric radicals can be prepared in some cases. For example, asymmetric nitroxide radicals are known.291 An anomeric effect was observed in alkoxy radical 49, where the ratio of 49a/49b was 1:1.78.292
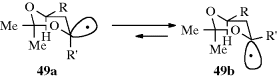
Evidence from studies on bridgehead compounds shows that although a planar configuration is more stable, pyramidal structures are not impossible. In contrast to the situation with carbocations, free radicals have often been generated at bridgeheads, although studies have shown that bridgehead free radicals are less rapidly formed than the corresponding open-chain radicals.293 In sum, the available evidence indicates that although simple alkyl free radicals prefer a planar, or near-planar shape, the energy difference between a planar and a pyramidal free radical is not great. However, free radicals in which the carbon is connected to atoms of high electronegativity (e.g., √CF3), prefer a pyramidal shape;294 increasing the electronegativity increases the deviation from planarity.295 Cyclopropyl radicals are also pyramidal.296 Free radicals with resonance are definitely planar, although triphenylmethyl-type radicals are propeller shaped,297 like the analogous carbocations (Sec. 5.A.i). Radicals possessing simple alkyl substituents attached to the radical carbon (C√) that have Csp3–Csp3 bonds, and rotation about those bonds is possible. The internal rotation barrier for the tert-butyl radical (Me3C√), for example, was estimated to be ~1.4 kcal mol−1 6 kJ mol−1.298
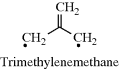
A number of diradicals (also called biradicals) are known,299 and the thermodynamic stability of diradicals has been examined.300 Orbital phase theory has been applied to the development of a theoretical model of localized 1,3-diradicals, and used to predict the substitution effects on the spin preference and S–T gaps, and to design stable localized carbon-centered 1,3-diradicals.301 When the unpaired electrons of a diradical are widely separated, for example, as in √CH2CH2CH2CH2√, the species behaves spectrally like two doublets. When they are close enough for interaction or can interact through an unsaturated system (as in trimethylenemethane),302 they can have total spin numbers of +1, 0, or -1, since each electron could be either 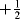 or
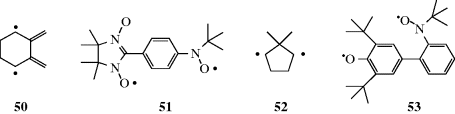
or 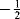 . Spectroscopically they are called triplets,303 since each of the three possibilities is represented among the molecules and gives rise to its own spectral peak. In triplet molecules, the two unpaired electrons have the same spin. Not all diradicals have a triplet ground state. In 2,3-dimethylelecycohexane-1,4-diyl (50), the singlet and triplet states were found to be almost degenerate.304 Diradicals (e.g., 51) are very stable with a triplet ground state.305 Diradicals are generally short-lived species. The lifetime of 52 was measured to be <0.1 ns and other diradicals were found to have lifetimes in the 4–316-ns range.306 Diradical 53 [3,5-di-tert-butyl-3′-(N-tert-butyl-N-aminoxy)-4-oxybiphenyl] was found to have a lifetime of weeks even in the presence of oxygen, and survived brief heating in toluene up to ~60 °C.307 Radicals with both unpaired electrons on the same carbon are discussed under carbenes. 1,4-Biradicals are known, and α-carbonyl substituents increase the lifetime of the radical, and negative α-hyperconjugation (see Sec. 2.M) has been suggested as the cause.308
. Spectroscopically they are called triplets,303 since each of the three possibilities is represented among the molecules and gives rise to its own spectral peak. In triplet molecules, the two unpaired electrons have the same spin. Not all diradicals have a triplet ground state. In 2,3-dimethylelecycohexane-1,4-diyl (50), the singlet and triplet states were found to be almost degenerate.304 Diradicals (e.g., 51) are very stable with a triplet ground state.305 Diradicals are generally short-lived species. The lifetime of 52 was measured to be <0.1 ns and other diradicals were found to have lifetimes in the 4–316-ns range.306 Diradical 53 [3,5-di-tert-butyl-3′-(N-tert-butyl-N-aminoxy)-4-oxybiphenyl] was found to have a lifetime of weeks even in the presence of oxygen, and survived brief heating in toluene up to ~60 °C.307 Radicals with both unpaired electrons on the same carbon are discussed under carbenes. 1,4-Biradicals are known, and α-carbonyl substituents increase the lifetime of the radical, and negative α-hyperconjugation (see Sec. 2.M) has been suggested as the cause.308
5.C.ii. The Generation and Fate of Free Radicals309
Free radicals are formed from molecules by breaking a bond so that each fragment keeps one electron.310,311 The energy necessary to break the bond is supplied in one of two ways.
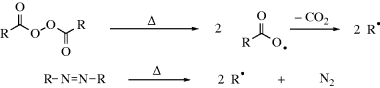
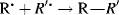
A propagation reaction is one in which a radical reacts to give at least one radical product, which continues the radical reaction sequence. There are four principal propagation reactions, of which the first two are most common:
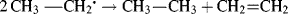
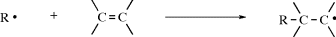

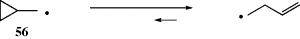
5.C.iii. Radical Ions333
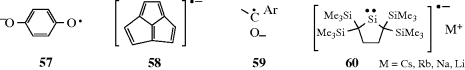
Several types of radical anions are known with the unpaired electron or the charge or both on atoms other than carbon. Examples include semiquinones334 (57), acepentalenes (58),335 ketyls336 (59) and the radical anion of the isolable dialkylsilylene (60).337 Radical anions are formed by the reaction of carbene anions with chloromethanes.338 Reactions in which alkali metals are reducing agents often involve radical anion intermediates (Birch reduction, e.g., Reaction 15-13) that proceed via radical anion 61.
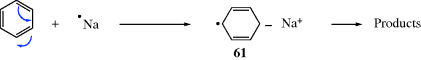
Several types of radical cation are also known.339 Typical examples include alkyl azulene cation radicals (62),340 trialkyl amine radical cations,341 1,2-bis(dialkylamino)benzenes radical cations (e.g., 63),342 dimethylsulfonium cation radicals (Me2S√+),343N-alkyl substituted imine cation radicals (Ph2C=NEt√+),344 dibenzo[a,e]cyclooctene (64, a nonplanar cation radical),345 and [n.n]paracyclophane cation radicals.346 A twisted radical cation derived from bicyclo[2.2.2]oct-2-ene has been reported.347
5.D. Carbenes
5.D.i. Stability and Structure348
Carbenes are highly reactive species, and practically all have lifetimes considerably < 1 s. With exceptions noted below (Sec. 5.D.ii), carbenes have been isolated only by entrapment in matrices at low temperatures (77 K or less).349 The parent species (CH2) is usually called methylene, although derivatives are more often named by the carbene nomenclature. Thus CCl2 is generally known as dichlorocarbene, although it can also be called dichloromethylene.
The two nonbonded electrons of a carbene can be either paired or unpaired. If they are paired, the species is spectrally a singlet, while, as seen above (Sec. 5.C.i), two unpaired electrons appear as a triplet. An ingenious

method of distinguishing between the two possibilities was developed by Skell,350 based on the common reaction of addition of carbenes to double bonds to form cyclopropane derivatives (Reaction 15-51). If the singlet species adds to cis-2-butene, the resulting cyclopropane should be the cis isomer since the movements of the two pairs of electrons
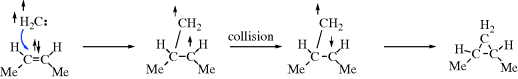
should occur either simultaneously or with one rapidly succeeding another. However, if the attack is by a triplet species, the two unpaired electrons cannot both go into a new covalent bond, since by Hund's rule they have parallel spins. So one of the unpaired electrons will form a bond with the electron from the double bond that has the opposite spin, leaving two unpaired electrons that have the same spin and therefore cannot form a bond at once, but must wait until, by some collision process, one of the electrons can reverse its spin. During this time, there is free rotation about the C–C bond and a mixture of cis- and trans-1,2-dimethylcyclopropanes should result.351
The results of this type of experiment show that CH2 itself is usually formed as a singlet species, which can decay to the triplet state, which consequently has a lower energy (MO calculations352 and experimental determinations show that the difference in energy between singlet and triplet CH2 is ~8–10 kcal mol−1 or 33–42 kJ mol−1)353. However, it is possible to prepare triplet CH2 directly by a photosensitized decomposition of diazomethane.354 The CH2 group is so reactive355 that it generally reacts as the singlet before it has a chance to decay to the triplet state.356 As to other carbenes, some react as triplets, some as singlets, and others as singlets or triplets, depending on how they are generated. There are, however, molecules that generate persistent triplet carbenes.357 Indeed, remarkably stable diaryl triplet carbenes have been prepared,358 and protected diphenylcarbenes are particularly stable.359 There are also persistent singlet carbenes, although radical fragmentation is a problem.360
There is a limitation to the use of stereospecificity of addition as a diagnostic test for singlet or triplet carbenes.361 When carbenes are generated by photolytic methods, they are often in a highly excited singlet state. When they add to the double bond, the addition is stereospecific; but the cyclopropane formed carries excess energy (i.e., it is in an excited state). It has been shown that under certain conditions (low pressures in the gas phase) the excited cyclopropane may undergo cis–trans isomerization after it is formed, so that triplet carbene may seem to be involved although in reality the singlet was present.362
Studies of the IR spectrum of CCl2 trapped at low temperatures in solid argon indicate that the ground state for this species is the singlet.363 The geometrical structure of triplet methylene can be investigated by ESR measurements,364 since triplet species are diradicals. Such measurements made on triplet CH2 trapped in matrices at very low temperatures (4 K) show that triplet CH2 is a bent molecule, with an angle of ~136°.365 The EPR measurements cannot be made on singlet species, but from electronic spectra of CH2 formed in flash photolysis366 of diazomethane it was concluded that singlet CH2 is also bent, with an angle of ~103°.367 Singlet CCl2300 and CBr2368 are also bent, with angles of 100 and 114°, respectively. It has long been known that triplet aryl carbenes are bent.369
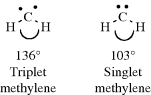
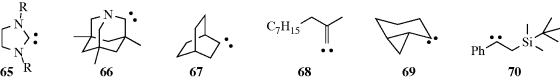
The most common carbenes are :CH2 and :CCl2,370 but many others have been reported,371 including heterocyclic carbenes372 diboron carbenes,373 65 (stabilized by the steric constraints of the ring geometry),374 66 (an aminocarbene without π conjugation),375 bicyclo[2.2.2]octylidene, (67),376 alkylidene carbenes (e.g., 68),377 conformationally restricted cyclopropylcarbenes, (e.g., 69),378 β-silylcarbenes (e.g., 70),379 α-keto carbenes,380 vinyl carbenes,381 and chiral carbenoids.382 Fluoro(phenoxy)carbene is stable for several days if it is generated within the cavity of a hemicarcerand (see Sec. 3.C.iii).383 In the case of 65 (R = Ph),384 the precursor is a tetraaminoethylene, and when potassium hydride is present to preclude electrophilic catalysis, starting tetraaminoethylenes are recovered unchanged.
Flash photolysis of CHBr3 produced the intermediate CBr,385 which is a carbyne.
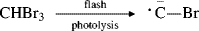
The intermediates CF and CCl were generated similarly from CHFBr2 and CHClBr2, respectively. Triplet acetylenes have been reported as equivalents for 1,2-bicarbenes.386
5.D.ii. The Generation and Fate of Carbenes387
There are two primary methods to form carbenes, although other pathways are also known.
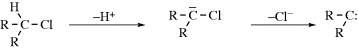
 |
Ref. 390 |
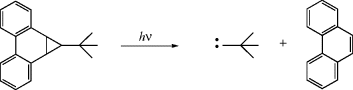 |
Ref. 391 |
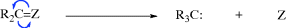
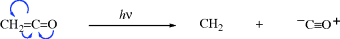
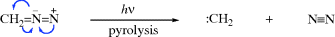

The reactions of carbenes are more varied than those of the species previously discussed in this chapter.400 Solvent effects have been observed in carbene reactions. The selectivity of certain carbenes is influenced by the nature of the solvent.401 The distribution of rearrangement products (see below) from tert-butylcarbene402 are influenced by changes in solvent.403 It is known that singlet methylene forms a charge-transfer complex with benzene.404 Solvent interactions for chlorophenylcarbene and fluorophenylcarbene, however, are weak.405
 |
Ref. 416 |
 |
Ref. 417 |
 |
Ref. 418 |
 |
Ref. 419 |
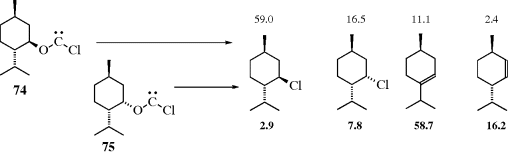
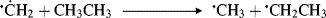
5.E. Nitrenes
Nitrenes (R–N),427 are the nitrogen analogues of carbenes, and most of the comments about carbenes also applies to them. Nitrenes are too reactive for isolation under ordinary conditions,428 although ab initio calculations show that nitrenes are more stable than carbenes with an enthalpy difference of 25–26 kcal mol−1 (104.7–108.8 kJ mol−1).429

Alkyl nitrenes have been isolated by trapping in matrices at 4 K,430 while aryl nitrenes, which are less reactive, can be trapped at 77 K.431 The ground state of NH, and probably of most nitrenes,432 is a triplet, although nitrenes can be generated in both triplet433 and singlet states. A quartet ground-state nitreno radical has been reported.434 In additions of EtOOC–N to C=C double bonds two species are involved, one of which adds in a stereospecific manner and the other not. By analogy with Skell's proposal involving carbenes (Sec. 5.D.i) these are taken to be the singlet and triplet species, respectively.435
The two principal means of generating nitrenes are analogous to those used to form carbenes.
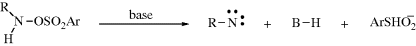
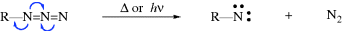
The reactions of nitrenes are also similar to those of carbenes.437 As in that case, many reactions in which nitrene intermediates are suspected probably do not involve free nitrenes. It is often very difficult to obtain proof in any given case that a free nitrene is or is not an intermediate.


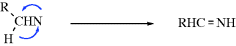

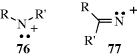
Notes
1. For general references, see Isaacs, N.S. Reactive Intermediates in Organic Chemistry, Wiley, NY, 1974; McManus, S.P. Organic Reactive Intermediates, Academic Press, NY, 1973. Two serial publications devoted to review articles on this subject are Reactive Intermediates (Wiley) and Reactive Intermediates (Plenum).
2. See Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, 5 Vols., Wiley, NY, 1968–1976; Vogel, P. Carbocation Chemistry, Elsevier, NY, 1985. See Saunders, M.; Jiménez-Vázquez, H.A. Chem. Rev. 1991, 91, 375; Arnett, E.M.; Hofelich, T.C.; Schriver, G.W. React. Intermed. (Wiley) 1987, 3, 189. For reviews of dicarbocations, see Lammertsma, K.; Schleyer, P.v.R.; Schwarz, H. Angew. Chem. Int. Ed. 1989, 28, 1321. See also, the series Advances in Carbocation Chemistry.
3. Gomberg, M. Ber. 1902, 35, 2397.
4. For a history of the term “carbonium ion”, see Traynham, J.G. J. Chem. Educ. 1986, 63, 930.
5. Olah, G.A. CHEMTECH 1971, 1, 566; J. Am. Chem. Soc. 1972, 94, 808.
6. Gold, V.; Loening, K.L.; McNaught, A.D.; Sehmi, P. Compendium of Chemical Terminology, IUPAC Recommendations, Blackwell Scientific Publications, Oxford, 1987.
7. Olah, G.A. J. Org. Chem. 2001, 66, 5943. See Olah, G.A.; Prakash, G.K.S. (Eds.), Carbocation Chemistry, Wiley Intersience, Hoboken, NJ, 2004.
8. See Laube, T. J. Am. Chem. 2004, 126, 10904 and references therein. For the X-ray of a vinyl carbocation see Müller, T.; Juhasz, M.; Reed, C.A. Angew. Chem. Int. Ed. 2004, 43, 1543.
9. Kato, T.; Reed, C.A. Angew. Chem. Int. Ed. 2004, 43, 2908.
10. Douberly, G.E.; Ricks, A.M.; Ticknor, B.W.; Schleyer, P.v.R.; Duncan, M.A. J. Am. Chem. Soc. 2007, 129, 13782.
11. Lüning, U.; Baumstark, R. Tetrahedron Lett. 1993, 34, 5059.
12. McClelland, R.A.; Cozens, F.L.; Steenken, S.; Amyes, T.L.; Richard, J.P. J. Chem. Soc. Perkin Trans. 2 1993, 1717.
13. For a treatise, see Szwarc, M. Ions and Ion Pairs in Organic Reactions, 2 Vols., Wiley, NY, 1972–1974.
14. For a review, see Olah, G.A.; Olah, J.A. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2, WIley, NY, 1969, pp. 715–782. Also see, Farcasiu, D.; Norton, S.H. J. Org. Chem. 1997, 62, 5374.
15. See Olah, G.A.; Prakash, G.K.S.; Sommer, J. in Superacids, Wiley, NY, 1985, pp. 65–175.
16. Olah, G.A.; Baker, E.B.; Evans, J.C.; Tolgyesi, W.S.; McIntyre, J.S.; Bastien, I.J. J. Am. Chem. Soc. 1964, 86, 1360; Kramer, G.M. J. Am. Chem. Soc. 1969, 91, 4819.
17. Olah, G.A.; Sommer, J.; Namanworth, E. J. Am. Chem. Soc. 1967, 89, 3576.
18. Olah, G.A.; Halpern, Y. J. Org. Chem. 1971, 36, 2354. See also, Herlem, M. Pure Appl. Chem. 1977, 49, 107.
19. Olah, G.A.; Lukas, J. J. Am. Chem. Soc. 1967, 89, 4739.
20. See Amyes, T.L.; Stevens, I.W.; Richard, J.P. J. Org. Chem. 1993, 58, 6057 for a recent study.
21. See Saunders, M.; Hagen, E.L.; Rosenfeld, J. J. Am. Chem. Soc. 1968, 90, 6882; Saunders, M.; Cox, D.; Lloyd, J.R. J. Am. Chem. Soc. 1979, 101, 6656; Myhre, P.C.; Yannoni, C.S. J. Am. Chem. Soc. 1981, 103, 230.
22. Olah, G.A.; Donovan, D.J. J. Am. Chem. Soc. 1978, 100, 5163.
23. Olah, G.A.; Olah, J.A. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2, Wiley, NY, 1969, p. 722.
24. Bacon, J.; Gillespie, R.J. J. Am. Chem. Soc. 1971, 91, 6914.
25. See Radom, L.; Poppinger, D.; Haddon, R.C. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 5, Wiley, NY, 1976, pp. 2303–2426.
26. Lowry, T.H.; Richardson, K.S. Mechanism and Theory in Organic Chemistry, 3rd ed., HarperCollins, NY, 1987, p. 68.
27. Meot-Ner, M. J. Am. Chem. Soc. 1987, 109, 7947.
28. If only the field effect were operating, 2 would be more stable than 3, since deuterium is electron-donating with respect to hydrogen (Sec. 1.J), assuming that the field effect of deuterium could be felt two bonds away.
29. Lambert, J.B.; Ciro, S.M. J. Org. Chem. 1996, 61, 1940.
30. Olah, G.A.; Lukas, J. J. Am. Chem. Soc. 1967, 89, 4739; Olah, G.A.; Olah, J.A. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2, Wiley, NY, 1969, pp. 750–764.
31. Olah, G.A.; Klopman, G.; Schlosberg, R.H. J. Am. Chem. Soc. 1969, 91, 3261. See also, Hogeveen, H.; Gaasbeek, C.J. Recl. Trav. Chim. Pays-Bas 1968, 87, 319.
32. Olah, G.A.; Svoboda, J.J.; Ku, A.T. Synthesis 1973, 492; Olah, G.A.; Lukas, J. J. Am. Chem. Soc. 1967, 89, 4739.
33. See Barbour, J.B.; Karty, J.M. J. Org. Chem. 2004, 69, 648; Mo, Y. J. Org. Chem. 2004, 69, 5563 and references cited therein.
34. For reviews, see Deno, N.C. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2 Wiley, NY, 1970, pp. 783–806; Richey, Jr., H.G. in Zabicky, J. The Chemistry of Alkenes, Vol. 2, Wiley, NY, 1970, pp. 39–114.
35. Deno, N.C.; Richey, Jr., H.G.; Friedman, N.; Hodge, J.D.; Houser, J.J.; Pittman, Jr., C.U. J. Am. Chem. Soc. 1963, 85, 2991.
36. Olah, G.A.; Spear, R.J. J. Am. Chem. Soc. 1975, 97, 1539 and references cited therein.
37. For a review of divinylmethyl and trivinylmethyl cations, see Sorensen, T.S. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2, Wiley, NY, 1970, pp. 807–835.
38. Deno, N.C.; Pittman, Jr., C.U. J. Am. Chem. Soc. 1964, 86, 1871.
39. Olah, G.A.; Spear, R.J.; Westerman, P.W.; Denis, J. J. Am. Chem. Soc. 1974, 96, 5855.
40. For a review of benzylic, diarylmethyl, and triarymethyl cations, see Freedman, H.H. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 4, Wiley, NY, 1971, pp. 1501–1578.
41. Olah, G.A.; Porter, R.D.; Jeuell, C.L.; White, A.M. J. Am. Chem. Soc. 1972, 94, 2044.
42. Volz, H.; Schnell, H.W. Angew. Chem. Int. Ed. 1965, 4, 873.
43. Deno, N.C.; Schriesheim, A. J. Am. Chem. Soc. 1955, 77, 3051.
44. Prakash, G.K.S. Pure Appl. Chem. 1998, 70, 2001.
45. Ito, S.; Morita, N.; Asao, T. Tetrahedron Lett. 1992, 33, 3773.
46. Ito, S.; Morita, N.; Asao, T. Tetrahedron Lett. 1994, 35, 751.
47. For reviews, see in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 3, Wiley, NY, 1972: Richey, Jr., H.G. pp. 1201–294; Wiberg, K.B.; Hess, Jr., B.A.; Ashe, III, A.H. pp. 1295–1345.
48. Pittman, Jr., C.U.; Olah, G.A. J. Am. Chem. Soc. 1965, 87, 2998; Deno, N.C.; Liu, J.S.; Turner, J.O.; Lincoln, D.N.; Fruit, Jr., R.E. J. Am. Chem. Soc. 1965, 87, 3000.
49. Deno, N.C.; Richey, Jr., H.G.; Liu, J.S.; Hodge, J.D.; Houser, H.J.; Wisotsky, M.J. J. Am. Chem. Soc. 1962, 84, 2016.
50. See Poulter, C.D.; Spillner, C.J. J. Am. Chem. Soc. 1974, 96, 7591; Childs, R.F.; Kostyk, M.D.; Lock, C.J.L.; Mahendran, M. J. Am. Chem. Soc. 1990, 112, 8912.
51. Sorensen, T.S.; Miller, I.J.; Ranganayakulu, K. Aust. J. Chem. 1973, 26, 311.
52. See Hevesi, L. Bull. Soc. Chim. Fr. 1990, 697; Olah, G.A.; Liang, G.; Mo, Y.M. J. Org. Chem. 1974, 39, 2394; Borch, R.F. J. Am. Chem. Soc. 1968, 90, 5303; Rabinovitz, M.; Bruck, D. Tetrahedron Lett. 1971, 245.
53. For a review of ions of the form R2C+–OR', see Rakhmankulov, D.L.; Akhmatdinov, R.T.; Kantor, E.A. Russ. Chem. Rev. 1984, 53, 888. For a review of ions of the form R'C+(OR)2 and C+(OR)3, see Pindur, U.; Müller, J.; Flo, C.; Witzel, H. Chem. Soc. Rev. 1987, 16, 75.
54. For a review of such ions where nitrogen is the heteroatom, see Scott, F.L.; Butler, R.N. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 4, Wiley, NY, 1974, pp. 1643–1696.
55. See Allen, A.D.; Tidwell, T.T. Adv. Carbocation Chem. 1989, 1, 1. See also, Teberekidis, V.I.; Sigalas, M.P. Tetrahedron 2003, 59, 4749.
56. Olah, G.A.; Svoboda, J.J. Synthesis 1973, 52.
57. See Lambert, J.B. Tetrahedron 1990, 46, 2677; Lambert, J.B.; Zhao, Y.; Emblidge, R.W.; Salvador, L.A.,;Liu, X.; So, J.-H.; Chelius, E.C. Acc. Chem. Res.1999, 32, 183. See also, Lambert, J.B.; Chelius, E.C. J. Am. Chem. Soc. 1990, 112, 8120.
58. Creary, X.; Kochly, E.D. J. Org. Chem. 2009, 74, 9044.
59. Olah, G.A.; Heiliger, L.; Prakash, G.K.S. J. Am. Chem. Soc. 1989, 111, 8020.
60. Haubenstock, H.; Sauers, R.R. Tetrahedron 2004, 60, 1191.
61. see Al-Talib, M.; Tashtoush, H. Org. Prep. Proced. Int. 1990, 22, 1; Olah, G.A.; Germain, A.; White, A.M. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 5, Wiley, NY, 1976, pp. 2049–2133; Lindner, E. Angew. Chem. Int. Ed. 1970, 9, 114.
62. See Olah, G.A.; Dunne, K.; Mo, Y.K.; Szilagyi, P. J. Am. Chem. Soc. 1972, 94, 4200; Olah, G.A.; Svoboda, J.J. Synthesis 1972, 306.
63. Hammett, L.P.; Deyrup, A.J. J. Am. Chem. Soc. 1933, 55, 1900; Newman, M.S.; Deno, N.C. J. Am. Chem. Soc. 1951, 73, 3651.
64. Boer, F.P. J. Am. Chem. Soc. 1968, 90, 6706; Le Carpentier, J.; Weiss, R. Acta Crystallogr. Sect. B, 1972, 1430. See also, Olah, G.A.; Westerman, P.W. J. Am. Chem. Soc. 1973, 95, 3706.
65. See Komatsu, K.; Kitagawa, T. Chem. Rev. 2003, 103, 1371. Also see, Gilbertson, R.D.; Weakley, T.J.R.; Haley, M.M. J. Org. Chem. 2000, 65, 1422.
66. See Gronheid, R.; Lodder, G.; Okuyama, T. J. Org. Chem. 2002, 67, 693. For a discussion of aryl substituted vinyl cations, see Müller, T.; Margraf, D.; Syha, Y. J. Am. Chem. Soc. 2005, 127, 10852.
67. For a review of destabilized carbocations, see Tidwell, T.T. Angew. Chem. Int. Ed. 1984, 23, 20.
68. See Abram, T.S.; Watts, W.E. J. Chem. Soc. Chem. Commun,. 1974, 857; Siehl, H.; Carnahan, Jr., J.C.; Eckes, L.; Hanack, M. Angew. Chem. Int. Ed. 1974, 13, 675. Also see Franke, W.; Schwarz, H.; Stahl, D. J. Org. Chem. 1980, 45, 3493. See also, Siehl, H.; Koch, E. J. Org. Chem. 1984, 49, 575.
69. See Stang, P.J.; Rappoport, Z.; Hanack, M.; Subramanian, L.R. Vinyl Cations, Academic Press, NY, 1979; Hanack, M. Pure Appl. Chem. 1984, 56, 1819, Acc. Chem. Res. 1976, 9, 364; Ambroz, H.B.; Kemp, T.J. Chem. Soc. Rev. 1979, 8, 353; Richey, Jr., H.G.; Richey, J.M. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2, Wiley, NY, 1970, pp. 899–957; Richey, Jr., H.G. in Zabicky, J. The Chemistry of Alkenes, Vol. 2, Wiley, NY, 1970, pp. 42–49; Stang, P.J. Prog. Phys. Org. Chem. 1973, 10, 205. See also, Charton, M. Mol. Struct. Energ. 1987, 4, 271. For a computational study, see Glaser, R.; Horan, C. J.; Lewis, M.; Zollinger, H. J. Org. Chem. 1999, 64, 902.
70. Yang, S.; Kondo, J.N.; Domen, K. Chem. Commun. 2001, 2008.
71. Winkler, M.; Sander, W. J. Org. Chem. 2006, 71, 6357.
72. For reviews, see Bagno, A.; Scorrano, G.; More O'Ferrall, R.A. Rev. Chem. Intermed. 1987, 7, 313; Bethell, D.; Gold, V. Carbonium Ionds, Academic Press, NY, 1967, pp. 59–87.
73. Deno, N.C.; Berkheimer, H.E.; Evans, W.L.; Peterson, H.J. J. Am. Chem. Soc. 1959, 81, 2344.
74. For a list of stabilities of 39 typical carbocations, see Arnett, E.M.; Hofelich, T.C. J. Am. Chem. Soc. 1983, 105, 2889. See also, Schade, C.; Mayr, H.; Arnett, E.M. J. Am. Chem. Soc. 1988, 110, 567; Schade, C.; Mayr, H. Tetrahedron 1988, 44, 5761.
75. Hammett, L.P.; Deyrup, A.J. J. Am. Chem. Soc. 1933, 55, 1900; Newman, M.S.; Deno, N.C. J. Am. Chem. Soc. 1951, 73, 3651; Boer, F.P. J. Am. Chem. Soc. 1968, 90, 6706; Le Carpentier, J.; Weiss, R. Acta Crystallogr. Sect. B, 1972, 1430. See also, Arnett, E.M.; Petro, C. J. Am. Chem. Soc. 1978, 100, 5408; Arnett, E.M.; Pienta, N.J. J. Am. Chem. Soc. 1980, 102, 3329.
76. Schultz, J.C.; Houle, F.A.; Beauchamp, J.L. J. Am. Chem. Soc. 1984, 106, 3917.
77. Lossing. F.P.; Holmes, J.L. J. Am. Chem. Soc. 1984, 106, 6917.
78. Vinyl cations are generated by photolysis of vinyl iodonium salts. See Slegt, M.; Gronheid, R.; van der Vlugt, D.; Ochiai, M.; Okuyama, T.; Zuilhof, H.; Overkleeft, H.S.; Lodder, G. J. Org. Chem. 2006, 71, 2227.
79. See Schleyer, P.v.R. in Chiurdoglu, G. Conformational Analysis, Academic Press, NY, 1971, p. 241; Hehre, W.J. Acc. Chem. Res. 1975, 8, 369; Freedman, H.H. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 4, Wiley, NY, 1974, pp. 1561–574.
80. Olah, G.A.; DeMember, J.R.; Commeyras, A.; Bribes, J.L. J. Am. Chem. Soc. 1971, 93, 459; Yannoni, C.S.; Kendrick, R.D.; Myhre, P.C.; Bebout, D.C.; Petersen, B.L. J. Am. Chem. Soc. 1989, 111, 6440.
81. Rauk, A.; Sorensen, T.S.; Maerker, C.; de M. Carneiro, J.W.; Sieber, S.; Schleyer, P.v.R. J. Am. Chem. Soc. 1996, 118, 3761.
82. Lawlor, D.A.; More O'Ferrall, R.A.; Rao, S.N. J. Am. Chem. Soc. 2008, 130, 17997.
83. For a review of bridgehead carbocations, see Fort, Jr., R.C. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 4, Wiley, NY, 1974, pp. 1783–1835.
84. Della, E.W.; Schiesser, C.H. J. Chem. Soc. Chem. Commun. 1994, 417.
85. Åhman, J.; Somfai, P.; Tanner, D. J. Chem. Soc. Chem. Commun. 1994, 2785.
86. Della, E.W.; Head, N.J.; Janowski, W.K.; Schiesser, C.H. J. Org. Chem. 1993, 58, 7876.
87. Olah, G.A.; Prakash, G.K.S.; Shih, J.G.; Krishnamurthy, V.V.; Mateescu, G.D.; Liang, G.; Sipos, G.; Buss, V.; Gund, T.M.; Schleyer, P.v.R. J. Am. Chem. Soc. 1985, 107, 2764. See also, Kruppa, G.H.; Beauchamp, J.L. J. Am. Chem. Soc. 1986, 108, 2162; Laube, T. Angew. Chem. Int. Ed. 1986, 25, 349.
88. Takeuchi, K.; Okazaki, T.; Kitagawa, T.; Ushino, T.; Ueda, K.; Endo, T.; Notario, R. J. Org. Chem. 2001, 66, 2034.
89. Olah, G.A.; Prakash, G.K.S.; Fessner, W.; Kobayashi, T.; Paquette, L.A. J. Am. Chem. Soc. 1988, 110, 8599.
90. de Meijere, A.; Schallner, O. Angew. Chem. Int. Ed. 1973, 12, 399.
91. See Sundaralingam, M.; Chwang, A.K. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 5, Wiley, NY, 1976, pp. 2427–2476.
92. Schuster, I.I.; Colter, A.K.; Kurland, R.J. J. Am. Chem. Soc. 1968, 90, 4679.
93. For reviews of the NMR spectra of carbocations, see Young, R.N. Prog. Nucl. Magn. Reson. Spectrosc., 1979, 12, 261; Farnum, D.G. Adv. Phys. Org. Chem. 1975, 11, 123.
94. Olah, G.A.; White, A.M. J. Am. Chem. Soc. 1968, 90, 1884; 1969, 91, 5801. For 13C NMR data for additional ions, see Olah, G.A.; Donovan, D.J. J. Am. Chem. Soc. 1977, 99, 5026; Olah, G.A.; Prakash, G.K.S.; Liang, G. J. Org. Chem. 1977, 42, 2666.
95. Olah, G.A.; Porter, R.D.; Kelly, D.P. J. Am. Chem. Soc. 1971, 93, 464.
96. See Brown, H.C.; Peters, E.N. J. Am. Chem. Soc. 1977, 99, 1712; Kitching, W.; Adcock, W.; Aldous, G. J. Org. Chem. 1979, 44, 2652. See also, Larsen, J.W.; Bouis, P.A. J. Am. Chem. Soc. 1975, 97, 4418; Volz, H.; Shin, J.; Streicher, H. Tetrahedron Lett. 1975, 1297; Larsen, J.W. J. Am. Chem. Soc. 1978, 100, 330.
97. Peterson, P.E.; Slama, F.J. J. Am. Chem. Soc., 1968, 90, 6516.
98. Carlier, P.R.; Deora, N.; Crawford, T.D. J. Org. Chem. 2006, 71, 1592.
99. Mascal, M.; Hafezi, N.; Meher N.K.; Fettinger, J.C. J. Am. Chem. Soc. 2008, 130, 13532.
100. Prakash, G.K.S.; Bae, C.; Rasul, G.; Olah, G.A. J. Org. Chem. 2002, 67, 1297.
101. Richard, J.P.; Amyes, T.L.; Williams, K.B. Pure. Appl. Chem. 1998, 70, 2007.
102. Toteva, M.M.; Richard, J.P. J. Am. Chem. Soc. 1996, 118, 11434.
103. Vrcek, V.; Saunders, M.; Kronja, O. J. Am. Chem. Soc. 2004, 126, 13703.
104. Kronja, O.; Kohli, T.-P.; Mayr, H.; Saunders, M. J. Am. Chem. Soc. 2000, 122, 8067.
105. See Buncel, E.; Durst, T. Comprehensive Carbanion Chemistry, pts. A, B, and C; Elsevier, NY, 1980, 1984, 1987; Bates, R.B.; Ogle, C.A. Carbanion Chemistry, Springer, NY, 1983; Stowell, J.C. Carbanions in Organic Synthesis, Wiley, NY, 1979; Cram, D.J. Fundamentals of Carbanion Chemistry, Academic Press, NY, 1965; Staley, S.W. React. Intermed. (Wiley) 1985, 3, 19; Staley, S.W.; Dustman, C.K. React. Intermed. (Wiley) 1981, 2, 15. For reviews of NMR spectra of carbanions, see Young, R.N. Prog. Nucl. Magn. Reson. Spectrosc. 1979, 12, 261. For a review of dicarbanions, see Thompson, C.M.; Green, D.L.C. Tetrahedron 1991, 47, 4223.
106. See Reutov, O.A.; Beletskaya, I.P.; Butin, K.P. CH-Acids, Pergamon, Elmsford, NY, 1978; Fischer, H.; Rewicki, D. Prog. Org. Chem. 1968, 7, 116.
107. See Graul, S.T.; Squires, R.R. J. Am. Chem. Soc. 1988, 110, 607.
108. Applequist, D.E.; O'Brien, D.F. J. Am. Chem. Soc. 1963, 85, 743.
109. Dessy, R.E.; Kitching, W.; Psarras, T.; Salinger, R.; Chen, A.; Chivers, T. J. Am. Chem. Soc. 1966, 88, 460.
110. Terrier, F.; Magnier, E.; Kizilian, E.; Wakselman, C.; Buncel, E. J. Am. Chem. Soc. 2005, 127, 5563.
111. For reviews, see Jones, J.R. Surv. Prog. Chem. 1973, 6, 83; Shatenshtein, A.I.; Shapiro, I.O. Russ. Chem. Rev. 1968, 37, 845.
112. See Bordwell, F.G.; Matthews, W.S.; Vanier, N.R. J. Am. Chem. Soc. 1975, 97, 442.
113. DePuy, C.H.; Gronert, S.; Barlow, S.E.; Bierbaum, V.M.; Damrauer, R. J. Am. Chem. Soc. 1989, 111, 1968. The same order (for t-Bu, Me, iPr, and Et) was found in gas-phase cleavages of alkoxides (Reaction 12-41): Tumas, W.; Foster, R.F.; Brauman, J.I. J. Am. Chem. Soc. 1984, 106, 4053.
114. Graul, S.T.; Squires, R.R. J. Am. Chem. Soc. 1988, 110, 607.
115. See Richey, Jr., H.G. in Zabicky, J. The Chemistry of Alkenes, Vol. 2, Wiley, NY, 1970, pp. 67–77.
116. See Bockrath, B.; Dorfman, L.M. J. Am. Chem. Soc. 1974, 96, 5708.
117. See Buncel, E.; Menon, B. in Buncel, E.; Durst, T. Comprehensive Carbanion Chemistry, pts. A, B, and C, Elsevier, NY, 1980, 1984, 1987, pp. 97–124.
118. Olmstead, M.M.; Power, P.P. J. Am. Chem. Soc. 1985, 107, 2174.
119. Laferriere, M.; Sanrame, C. N.; Scaiano, J. C. Org. Lett. 2004, 6, 873.
120. Kinoshita, T.; Fujita, M.; Kaneko, H.; Takeuchi, K-i.; Yoshizawa, K.; Yamabe, T. Bull. Chem. Soc. Jpn. 1998, 71, 1145.
121. Eldin, S.; Whalen, D.L.; Pollack, R.M. J. Org. Chem. 1993, 58, 3490.
122. Abbotto, A.; Bradamante, S.; Pagani, G.A. J. Org. Chem. 1993, 58, 449.
123. Perkins, M.J.; Peynircioglu, N.B. Tetrahedron 1985, 41, 225.
124. Okamoto, K.; Kitagawa, T.; Takeuchi, K.; Komatsu, K.; Kinoshita, T.; Aonuma, S.; Nagai, M.; Miyabo, A. J. Org. Chem. 1990, 55, 996. See also, Okamoto, K.; Kitagawa, T.; Takeuchi, K.; Komatsu, K.; Miyabo, A. J. Chem. Soc. Chem. Commun. 1988, 923.
125. See Richey, Jr., H.G. in Zabicky, J. The Chemistry of Alkenes, Vol. 2, Wiley, NY, 1970, pp. 49–56.
126. See Oae, S.; Uchida, Y. in Patai, S.; Rappoport, Z.; Stirling, C. The Chemistry of Sulphones and Sulphoxides, Wiley, NY, 1988, pp. 583–664; Wolfe, S. in Bernardi, F.; Csizmadia, I.G.; Mangini. A. Organic Sulfur Chemistry, Elsevier, NY, 1985, pp. 133–190; Block, E. Reactions of Organosulfur Compounds, Academic Press, NY, 1978, pp. 42–56; Durst, T.; Viau, R. Intra-Sci. Chem. Rep. 1973, 7 (3), 63. Also see, Reich, H.J. in Liotta, DC. Organoselenium Chemistry, Wiley, NY, 1987, pp. 243–276.
127. See Wolfe, S.; LaJohn, L.A.; Bernardi, F.; Mangini, A.; Tonachini, G. Tetrahedron Lett. 1983, 24, 3789; Wolfe, S.; Stolow, A.; LaJohn, L.A. Tetrahedron Lett. 1983, 24, 4071.
128. See Borden, W.T.; Davidson, E.R.; Andersen, N.H.; Denniston, A.D.; Epiotis, N.D. J. Am. Chem. Soc. 1978, 100, 1604; Bernardi, F.; Bottoni, A.; Venturini, A.; Mangini, A. J. Am. Chem. Soc. 1986, 108, 8171.
129. Bernasconi, C.F.; Kittredge, K.W. J. Org. Chem. 1998, 63, 1944.
130. Wetzel, D.M.; Brauman, J.I. J. Am. Chem. Soc. 1988, 110, 8333.
131. For a review of such carbanions, see Beak, P.; Reitz, D.B. Chem. Rev. 1978, 78, 275. See also, Rondan, N.G.; Houk, K.N.; Beak, P.; Zajdel, W.J.; Chandrasekhar, J.; Schleyer, P.v.R. J. Org. Chem. 1981, 46, 4108.
132. See Werstiuk, N.H. Tetrahedron 1983, 39, 205; Hunter, D.H.; Stothers, J.B.; Warnhoff, E.W. in de Mayo, P. Rearrangements in Ground and Excited States, Vol. 1, Academic Press, NY, 1980, pp. 410–437.
133. See Werstiuk, N.H.; Yeroushalmi, S.; Timmins, G. Can. J. Chem. 1983, 61, 1945; Lee, R.E.; Squires, R.R. J. Am. Chem. Soc. 1986, 108, 5078; Peiris, S.; Ragauskas, A.J.; Stothers, J.B. Can. J. Chem. 1987, 65, 789; Shiner, C.S.; Berks, A.H.; Fisher, A.M. J. Am. Chem. Soc. 1988, 110, 957.
134. For reviews of carbanion pairs, see Hogen-Esch, T.E. Adv. Phys. Org. Chem. 1977, 15, 153; Jackman, L.M.; Lange, B.C. Tetrahedron 1977, 33, 2737. See also, Laube, T. Acc. Chem. Res. 1995, 28, 399.
135. Zook, H.D.; Gumby, W.L. J. Am. Chem. Soc. 1960, 82, 1386.
136. Solov'yanov, A.A.; Karpyuk, A.D.; Beletskaya, I.P.; Reutov, O.A. J. Org. Chem. USSR 1981, 17, 381. See also, Solov'yanov, A.A.; Beletskaya, I.P.; Reutov, O.A. J. Org. Chem. USSR 1983, 19, 1964.
137. See DePalma, V.M.; Arnett, E.M. J. Am. Chem. Soc. 1978, 100, 3514; Buncel, E.; Menon, B. J. Org. Chem. 1979, 44, 317; O'Brien, D.H.; Russell, C.R.; Hart, A.J. J. Am. Chem. Soc. 1979, 101, 633; Streitwieser, Jr., A.; Shen, C.C.C. Tetrahedron Lett. 1979, 327; Streitwieser, Jr., A. Acc. Chem. Res. 1984, 17, 353.
138. See Schade, C.; Schleyer, P.v.R.; Geissler, M.; Weiss, E. Angew. Chem. Int. Ed. 1986, 21, 902.
139. Ellison, G.B.; Engelking, P.C.; Lineberger, W.C. J. Am. Chem. Soc. 1978, 100, 2556.
140. Retention of configuration has never been observed with simple carbanions. Cram has obtained retention with carbanions stabilized by resonance. However, these carbanions are known to be planar or nearly planar, and retention was caused by asymmetric solvation of the planar carbanions (see Sec. 12.A.ii).
141. See Peoples, P.R.; Grutzner, J.B. J. Am. Chem. Soc. 1980, 102, 4709.
142. See Feit, B.; Melamed, U.; Speer, H.; Schmidt, R.R. J. Chem. Soc. Perkin Trans. 1 1984, 775; Chou, P.K.; Kass, S.R. J. Am. Chem. Soc. 1991, 113, 4357.
143. Boche, G.; Harms, K.; Marsch, M. J. Am. Chem. Soc. 1988, 110, 6925; Boche, G.; Walborsky, H.M. Cyclopropane Derived Reactive Intermediates, Wiley, NY, 1990. For a review, see Boche, G.; Walborsky, H.M. in Rappoport, Z. The Chemistry of the Cyclopropyl Group, pt. 1, Wiley, NY, 1987, pp. 701–808.
144. See Cram, D.J. Fundamentals of Carbanion Chemistry, Academic Press, NY, 1965, pp. 85–105.
145. Bordwell, F.G.; Phillips, D.D.; Williams, Jr., J.M. J. Am. Chem. Soc. 1968, 90, 426; Annunziata, R.; Cinquini, M.; Colonna, S.; Cozzi, F. J. Chem. Soc. Chem. Commun. 1981, 1005; Chassaing, G.; Marquet, A.; Corset, J.; Froment, F. J. Organomet. Chem. 1982, 232, 293; Cram, D.J. Fundamentals of Carbanion Chemistry, Academic Press, NY, 1965, pp. 105–113; Hirsch, R.; Hoffmann, R.W. Chem. Ber. 1992, 125, 975.
146. Hoffmann, R.W.; Rühl, T.; Chemla, F.; Zahneisen, T. Liebigs Ann. Chem. 1992, 719.
147. Rychnovsky, S.D.; Plzak, K.; Pickering, D. Tetrahedron Lett. 1994, 35, 6799.
148. Reich, H.J.; Medina, M.A.; Bowe, M.D. J. Am. Chem. Soc. 1992, 114, 11003.
149. Jenkins, P.R.; Symons, M.C.R.; Booth, S.E.; Swain, C.J. Tetrahedron Lett. 1992, 33, 3543.
150. Gais, H.; Müller, J.; Vollhardt, J.; Lindner, H.J. J. Am. Chem. Soc. 1991, 113, 4002. For a contrary view, see Trost, B.M.; Schmuff, N.R. J. Am. Chem. Soc. 1985, 107, 396.
151. Grossert, J.S.; Hoyle, J.; Cameron, T.S.; Roe, S.P.; Vincent, B.R. Can. J. Chem. 1987, 65, 1407.
152. See Elschenbroich, C.; Salzer, A. Organometallics, VCH, NY, 1989; Oliver, J.P. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 2, Wiley, NY, 1985, pp. 789–826; Coates, G.E.; Green, M.L.H.; Wade, K. Organometallic Compounds, 3rd ed., Vol. 1, Methuen: London, 1967; Grovenstein, Jr., E. in Buncel, E.; Durst, T. Comprehensive Carbanion Chemistry, pt. C, Elsevier, NY, 1987, pp. 175–221.
153. See Schade, C.; Schleyer, P.v.R. Adv. Organomet. Chem. 1987, 27, 169.
154. For X-ray crystallography studies, see Weiss, E.; Sauermann, G. Chem. Ber. 1970, 103, 265; Weiss, E.; Köster, H. Chem. Ber. 1977, 110, 717.
155. See Setzer, W.N.; Schleyer, P.v.R. Adv. Organomet. Chem. 1985, 24, 353; Schleyer, P.v.R. Pure Appl. Chem. 1984, 56, 151; Brown, T.L. Pure Appl. Chem. 1970, 23, 447, Adv. Organomet. Chem. 1965, 3, 365; Kovrizhnykh, E.A.; Shatenshtein, A.I. Russ. Chem. Rev. 1969, 38, 840. For reviews of the structures of lithium enolate anions and related compounds, see Boche, G. Angew. Chem. Int. Ed. 1989, 28, 277; Seebach, D. Angew. Chem. Int. Ed. 1988, 27, 1624. Also see Günther, H.; Moskau, D.; Bast, P.; Schmalz, D. Angew. Chem. Int. Ed. 1987, 26, 1212; Wakefield, B.J. Organolithium Methods, Academic Press, NY, 1988, The Chemistry of Organolithium Compounds, Pergamon, Elmsford, NY, 1974.
156. Lewis, H.L.; Brown, T.L. J. Am. Chem. Soc. 1970, 92, 4664; Brown, T.L.; Rogers, M.T. J. Am. Chem. Soc. 1957, 79, 1859; Weiner, M.A.; Vogel, G.; West, R. Inorg. Chem. 1962, 1, 654.
157. Thomas, R.D.; Jensen, R.M.; Young, T.C. Organometallics 1987, 6, 565. See also, Kaufman, M.J.; Gronert, S.; Streitwieser, Jr., A. J. Am. Chem. Soc. 1988, 110, 2829.
158. Wittig, G.; Meyer, F.J.; Lange, G. Liebigs Ann. Chem. 1951, 571, 167. See also, Bates, T.F.; Clarke, M.T.; Thomas, R.D. J. Am. Chem. Soc. 1988, 110, 5109.
159. Plavš i , D.; Srzi, D.; Klasinc, L. J. Phys. Chem. 1986, 90, 2075.
, D.; Srzi, D.; Klasinc, L. J. Phys. Chem. 1986, 90, 2075.
160. Weiss, E.; Sauermann, G.; Thirase, G. Chem. Ber. 1983, 116, 74.
161. Bauer, W.; Winchester, W.R.; Schleyer, P.v.R. Organometallics 1987, 6, 2371.
162. Fraenkel, G.; Chow, A.; Winchester, W.R. J. Am. Chem. Soc. 1990, 112, 6190.
163. For reviews, see Ashby, E.C. Bull. Soc. Chim. Fr. 1972, 2133; Q. Rev. Chem. Soc. 1967, 21, 259; Wakefield, B.J. Organomet. Chem. Rev. 1966, 1, 131; Bell, N.A. Educ. Chem. 1973, 143.
164. Schlenk, W.; Schlenk, Jr., W. Ber. 1929, 62B, 920.
165. See Parris, G.; Ashby, E.C. J. Am. Chem. Soc. 1971, 93, 1206; Salinger, R.M.; Mosher, H.S. J. Am. Chem. Soc. 1964, 86, 1782.
166. Guggenberger, L.J.; Rundle, R.E. J. Am. Chem. Soc. 1968, 90, 5375.
167. See Sakamoto, S.; Imamoto, T.; Yamaguchi, K. Org. Lett. 2001, 3, 1793.
168. Weiss, E. Chem. Ber. 1965, 98, 2805.
169. Ashby, E.C.; Smith, M.B. J. Am. Chem. Soc. 1964, 86, 4363; Vreugdenhil, A.D.; Blomberg, C. Recl. Trav. Chim. Pays-Bas 1963, 82, 453, 461.
170. Benn, R.; Lehmkuhl, H.; Mehler, K.; Rufi ska, A. Angew. Chem. Int. Ed. 1984, 23, 534.
ska, A. Angew. Chem. Int. Ed. 1984, 23, 534.
171. Smith, M.B.; Becker, W.E. Tetrahedron 1966, 22, 3027.
172. Evans, D.F.; Fazakerley, V. Chem. Commun. 1968, 974.
173. Ducom, J. Bull. Chem. Soc. Fr. 1971, 3518, 3523, 3529.
174. See Parris, G.; Ashby, E.C. J. Am. Chem. Soc. 1971, 93, 1206.
175. Ashby, E.C.; Walker, F. J. Org. Chem. 1968, 33, 3821.
176. Parris, G.; Ashby, E.C. J. Am. Chem. Soc. 1971, 93, 1206.
177. Ashby, E.C.; Smith, M.B. J. Am. Chem. Soc. 1964, 86, 4363.
178. Fraenkel, G.; Cottrell, C.E.; Dix, D.T. J. Am. Chem. Soc. 1971, 93, 1704; Pechhold, E.; Adams, D.G.; Fraenkel, G. J. Org. Chem. 1971, 36, 1368; Maercker, A.; Geuss, R. Angew. Chem. Int. Ed. 1971, 10, 270.
179. See Pratt, L.M.; Kass, S.R. J. Org. Chem. 2004, 69, 2123.
180. Nichols, M.A.; Williard, P.G. J. Am. Chem. Soc. 1993, 115, 1568.
181. See Jones, A.C.; Sanders, A.W.; Bevan, M.J.; Reich, H.J. J. Am. Chem. Soc. 2007, 129, 3492.
182. Siemeling, U.; Redecker, T.; Neumann, B.; Stammler, H.-G. J. Am. Chem. Soc. 1994, 116, 5507.
183. Ruhlandt-Senge, K.; Ellison, J.J.; Wehmschulte, R.J.; Pauer, F.; Power, P.P. J. Am. Chem. Soc. 1993, 115, 11353. Also see Betz, J.; Hampel, F.; Bauer, W. Org. Lett. 2000, 2, 3805.
184. Sorger, K.; Bauer, W.; Schleyer, P.v.R.; Stalke, D. Angew. Chem. Int. Ed., 1995, 34, 1594.
185. Reich, H.J.; Gudmundsson, B.O.; Goldenberg, W.S.; Sanders, A.W.; Kulicke, K.J.; Simon, K.; Guzei, I. A. J. Am. Chem. Soc. 2001, 123, 8067.
186. Nájera, C.; Yus, M. Tetrahedron 2005, 61, 3137.
187. See Parisel, O.; Fressigne, C.; Maddaluno, J.; Giessner-Prettre, C. J. Org. Chem. 2003, 68, 1290.
188. Sekiguchi, A.; Tanaka, M. J. Am. Chem. Soc. 2003, 125, 12684.
189. Linti, G.; Rodig, A.; Pritzkow, H. Angew. Chem. Int. Ed. 2002, 41, 4503.
190. Reich, H.J.; Green, D.P.; Medina, M.A.; Goldenberg, W.S.; Gudmundsson, B.Ö.; Dykstra, R.R.; Phillips. N.H. J. Am. Chem. Soc. 1998, 120, 7201.
191. Sun, X.; Winemiller, M.D.; Xiang, B.; Collum, D.B. J. Am. Chem. Soc. 2001, 123, 8039. See also, Rutherford, J.L.; Hoffmann, D.; Collum, D.B. J. Am. Chem. Soc. 2002, 124, 264.
192. Piffl, M.; Weston, J.; Günther, W.; Anders, E. J. Org. Chem. 2000, 65, 5942.
193. Bauer, W.; Griesinger, C. J. Am. Chem. Soc. 1993, 115, 10871.
194. Fraenkel, G.; Chow, A.; Fleischer, R.; Liu, H. J. Am. Chem. Soc. 2004, 126, 3983.
195. Graña, P.; Paleo, M.R.; Sardina, F.J. J. Am. Chem. Soc. 2002, 124, 12511.
196. Basu, A.; Thayumanavan, S. Angew. Chem. Int. Ed. 2002, 41, 717. See also, Fraenkel, G.; Duncan, J.H.; Martin, K.; Wang, J. J. Am. Chem. Soc. 1999, 121, 10538.
197. Stork, G.; Hudrlik, P.F. J. Am. Chem. Soc. 1968, 90, 4464; Bernstein, M.P.; Collum, D.B. J. Am. Chem. Soc. 1993, 115, 789; Collum, D.B. Acc. Chem. Res. 1992, 25, 448.
198. Jackman, L.M.; Lange, B.C. J. Am. Chem. Soc. 1981, 103, 4494.
199. Jackman, L.M.; Lange, B.C. Tetrahedron 1977, 33, 2737.
200. Williard, P.G.; Carpenter, G.B. J. Am. Chem. Soc. 1986, 108, 462; Williard, P.G.; Carpenter, G.B. J. Am. Chem. Soc. 1985, 107, 3345 and references cited therein.
201. Seebach, D.; Amstutz, R.; Laube, T.; Schweizer, W.B.; Dunitz, J.D. J. Am. Chem. Soc. 1985, 107, 5403.
202. Abu-Hasanayn, F.; Streitwieser, A. J. Am. Chem. Soc. 1996, 118, 8136.
203. Abbotto, A.; Streitwieser, A.; Schleyer, P.v.R. J. Am. Chem. Soc. 1997, 119, 11255.
204. Carlier, P.R.; Lucht, B.L.; Collum, D.B. J. Am. Chem. Soc. 1994, 116, 11602.
205. DeLong, G.T.; Pannell, D.K.; Clarke, M.T.; Thomas, R.D. J. Am. Chem. Soc. 1993, 115, 7013.
206. Walborsky, H.M.; Hamdouchi, C. J. Org. Chem. 1993, 58, 1187.
207. For a review of such reactions, see Durst, T. in Buncel, E.; Durst, T. Comprehensive Carbanion Chemistry, pt. B, Elsevier, NY, 1984, pp. 239–291.
208. For a review, see Guthrie, R.D. in Buncel, E.; Durst, T. Comprehensive Carbanion Chemistry, pt. A, Elsevier, NY, 1980, pp. 197–269.
209. Arnett, E.M.; Molter, K.E.; Marchot, E.C.; Donovan, W.H.; Smith, P. J. Am. Chem. Soc. 1987, 109, 3788.
210. Okamoto, K.; Kitagawa, T.; Takeuchi, K.; Komatsu, K.; Kinoshita, T.; Aonuma, S.; Nagai, M.; Miyabo, A. J. Org. Chem. 1990, 55, 996. See also, Okamoto, K.; Kitagawa, T.; Takeuchi, K.; Komatsu, K.; Miyabo, A. J. Chem. Soc. Chem. Commun. 1988, 923.
211. See Alfassi, Z.B. N-Centered Radicals, Wiley, Chichester, 1998; Alfassi, Z.B. Peroxyl Radicals, Wiley, Chichester, 1997; Alfassi, Z.B. Chemical Kinetics of Small Organic Radicals, 4 Vols., CRC Press: Boca Raton, FL, 1988; Nonhebel, D.C.; Tedder, J.M.; Walton, J.C. Radicals, Cambridge University Press, Cambridge, 1979; Nonhebel, D.C.; Walton, J.C. Free-Radical Chemistry, Cambridge University Press, Cambridge, 1974; Kochi, J.K. Free Radicals, 2 Vols., Wiley, NY, 1973; Hay, J.M. Reactive Free Radicals, Academic Press, NY, 1974;. For reviews, see Kaplan, L. React. Intermed. (Wiley) 1985, 3, 227; Griller, D.; Ingold, K.U. Acc. Chem. Res. 1976, 9, 13.
212. See Dunkin, I.R. Chem. Soc. Rev. 1980, 9, 1; Jacox, M.E. Rev. Chem. Intermed. 1978, 2, 1. For a review of the study of radicals at low temperatures, see Mile, B. Angew. Chem. Int. Ed. 1968, 7, 507.
213. See. Hicks, R.G. Org. Biomol. Chem. 2007, 5, 1321. See also, Hioe, J.; Zipse, H. Org. Biomol. Chem. 2010, 8, 3609.
214. See Andrews, L. Annu. Rev. Phys. Chem. 1971, 22, 109.
215. Sullivan, P.J.; Koski, W.S. J. Am. Chem. Soc. 1963, 85, 384.
216. Sablier, M.; Fujii, T. Chem. Rev. 2002, 102, 2855.
217. Bucher, G.; Halupka, M.; Kolano, C.; Schade, O.; Sander, W. Eur. J. Org. Chem. 2001, 545.
218. See Wertz, J.E.; Bolton, J.R. Electron Spin Resonance, McGraw-Hill, NY, 1972 [reprinted by Chapman and Hall, NY, and Methuen: London, 1986]; Assenheim, H.M. Introduction to Electron Spin Resonance, Plenum, NY, 1967; Bersohn, R.; Baird, J.C. An Introduction to Electron Paramagnetic Resonance, W.A. Benjamin, NY, 1966. For reviews, see Bunce, N.J. J. Chem. Educ. 1987, 64, 907; Hirota, N.; Ohya-Nishiguchi, H. in Bernasconi, C.F. Investigation of Rates and Mechanisms of Reactions, 4th ed., pt. 2, Wiley, NY, 1986, pp. 605–655; Griller, D.; Ingold, K.U. Acc. Chem. Res. 1980, 13, 193; Norman, R.O.C. Chem. Soc. Rev. 1980, 8, 1; Fischer, H. in Kochi, J.K. Free Radicals, Vol. 2, Wiley, NY, 1973, pp. 435–491; Turro, N.J.; Kleinman, M.H.; Karatekin, E. Angew. Chem. Int. Ed. 2000, 39, 4437; Kurreck, H.; Kirste, B.; Lubitz, W. Angew. Chem. Int. Ed. 1984, 23, 173. See also, Poole, Jr., C.P. Electron Spin Resonance. A Comprehensive Treatise on Experimental Techniques, 2nd ed., Wiley, NY, 1983.
219. Davies, A.G. Chem. Soc. Rev. 1993, 22, 299.
220. See Walton, J.C. Rev. Chem. Intermed. 1984, 5, 249; Kochi, J.K. Adv. Free-Radical Chem. 1975, 5, 189; Bielski, B.H.J.; Gebicki, J.M. Atlas of Electron Spin Resonance Spectra, Academic Press, NY, 1967.
221. See Janzen, E.G.; Haire, D.L. Adv. Free Radical Chem. (Greenwich, Conn.) 1990, 1, 253; Perkins, M.J. Adv. Phys. Org. Chem. 1980, 17, 1; Zubarev, V.E.; Belevskii, V.N.; Bugaenko, L.T. Russ. Chem. Rev. 1979, 48, 729; Evans, C.A. Aldrichimica Acta 1979, 12, 23; Janzen, E.G. Acc. Chem. Res. 1971, 4, 31. See also, the collection of papers on this subject in Can. J. Chem. 1982, 60, 1379.
222. Becker, D.A.; Natero, R.; Echegoyen, L.; Lawson, R.C. J. Chem. Soc. Perkin Trans. 2 1998, 1289. Also see, Klivenyi, P.; Matthews, R.T.; Wermer, M.; Yang, L.; MacGarvey, U.; Becker, D.A.; Natero, R.; Beal, M.F. Experimental Neurobiology 1998, 152, 163.
223. For a series of papers on nitroxide radicals, see Pure Appl. Chem. 1990, 62, 177.
224. Janzen, E.G.; Zhang, Y.-K. J. Org. Chem. 1995, 60, 5441. For the preparation of a new but structurally related spin trap see Karoui, H.; Nsanzumuhire, C.; Le Moigne, F.; Tordo, P. J. Org. Chem. 1999, 64, 1471.
225. Grossi, L.; Strazzari, S. Chem. Commun. 1997, 917.
226. Timberlake, J.W.; Chen, T. Tetrahedron Lett. 1994, 35, 6043; Tanko, J.M.; Brammer, Jr., L.E.; Hervas', M.; Campos, K. J. Chem. Soc. Perkin Trans. 2 1994, 1407.
227. Harry Frank, University of Connecticut, Storrs, CT., Personal Communication.
228. Ward, H.R.; Lawler, R.G.; Cooper, R.A. J. Am. Chem. Soc. 1969, 91, 746; Lepley, A.R. J. Am. Chem. Soc. 1969, 91, 749; Lepley, A.R.; Landau, R.L. J. Am. Chem. Soc. 1969, 91, 748.
229. See Lepley, R.L.; Closs, G.L. Chemically Induced Magnetic Polarization, Wiley, NY, 1973. Bargon, J. Helv. Chim. Acta 2006, 89, 2082. For reviews, see Adrian, F.J. Rev. Chem. Intermed. 1986, 7, 173; Closs, G.L.; Miller, R.J.; Redwine, O.D. Acc. Chem. Res. 1985, 18, 196; Closs, G.L. Adv. Magn. Reson. 1974, 7, 157; Lawler, R.G. Acc. Chem. Res. 1972, 5, 25; Kaptein, R. Adv. Free-Radical Chem. 1975, 5, 319.
230. A related technique is called CIDEP. For a review, see Hore, P.J.; Joslin, C.G.; McLauchlan, K.A. Chem. Soc. Rev. 1979, 8, 29.
231. Ward, H.R.; Lawler, R.G.; Cooper, R.A. J. Am. Chem. Soc. 1969, 91, 746.
232. It has been shown that CIDNP can also arise in cases where para hydrogen (H2 in which the nuclear spins are opposite) is present: Eisenschmid, T.C.; Kirss, R.U.; Deutsch, P.P.; Hommeltoft, S.I.; Eisenberg, R.; Bargon, J.; Lawler, R.G.; Balch, A.L. J. Am. Chem. Soc. 1987, 109, 8089.
233. Wind, R.A.; Duijvestijn, M.J.; van der Lugt, C.; Manenschijn, A.; Vriend, J. Prog. Nucl. Magn. Reson. Spectrosc. 1985, 17, 33.
234. Hu, K.-N.; Yu, H.-h.; Swager, T. M.; Griffin, R. G. J. Am. Chem. Soc. 2004, 126, 10844. A discussion of electronic effects is found in Wagner, P.J.; Wang, L. Org. Lett. 2006, 8, 645.
235. For a discussion of the role of alkyl substitution with respect to radical stabilization, see Gronert, S. J. Org. Chem. 2006, 71, 7045. For a discussion concerning data that hyperconguation stabilizes alkyl radicals, see Gronert, S. Org. Lett. 2007, 9, 2211.
236. For a discussion, see Robaugh, D.A.; Stein, S.E. J. Am. Chem. Soc. 1986, 108, 3224.
237. See Forrester, A.R.; Hay, J.M.; Thomson, R.H. Organic Chemistry of Stable Free Radicals, Academic Press, NY, 1968.
238. For an electron diffraction study of the allyl radical, see Vajda, E.; Tremmel, J.; Rozsondai, B.; Hargittai, I.; Maltsev, A.K.; Kagramanov, N.D.; Nefedov, O.M. J. Am. Chem. Soc. 1986, 108, 4352.
239. Asensio, A.; Dannenberg, J. J. J. Org. Chem. 2001, 66, 5996.
240. For a review, see Sholle, V.D.; Rozantsev, E.G. Russ. Chem. Rev. 1973, 42, 1011.
241. Gomberg, M. J. Am. Chem. Soc. 1900, 22, 757, Ber. 1900, 33, 3150.
242. For hexaphenylethane derivatives, see Stein, M.; Winter, W.; Rieker, A. Angew. Chem. Int. Ed. 1978, 17, 692; Yannoni, N.; Kahr, B.; Mislow, K. J. Am. Chem. Soc. 1988, 110, 6670.
243. Volz, H.; Lotsch, W.; Schnell, H. Tetrahedron 1970, 26, 5343; McBride, J. Tetrahedron 1974, 30, 2009. See Guthrie, R.D.; Weisman, G.R. Chem. Commun. 1969, 1316; Takeuchi, H.; Nagai, T.; Tokura, N. Bull. Chem. Soc. Jpn. 1971, 44, 753; Peyman, A.; Peters, K.; von Schnering, H.G.; Rüchardt, C. Chem. Ber. 1990, 123, 1899.
244. For a review of steric effects in free radical chemistry, see Rüchardt, C. Top. Curr. Chem. 1980, 88, 1.
245. Sabacky, M.J.; Johnson, Jr., C.S.; Smith, R.G.; Gutowsky, H.S.; Martin, J.C. J. Am. Chem. Soc. 1967, 89, 2054.
246. Müller, E.; Moosmayer, A.; Rieker, A.; Scheffler, K. Tetrahedron Lett. 1967, 3877. See also, Neugebauer, F.A.; Hellwinkel, D.; Aulmich, G. Tetrahedron Lett. 1978, 4871.
247. Kaba, R.A.; Ingold, K.U. J. Am. Chem. Soc. 1976, 98, 523.
248. Zarkadis, A.K.; Neumann, W.P.; Marx, R.; Uzick, W. Chem. Ber. 1985, 118, 450; Zarkadis, A.K.; Neumann, W.P.; Uzick, W. Chem. Ber. 1985, 118, 1183.
249. Dünnebacke, D.; Neumann, W.P.; Penenory, A.; Stewen, U. Chem. Ber. 1989, 122, 533.
250. For reviews, see Ballester, M. Adv. Phys. Org. Chem. 1989, 25, 267, pp. 354–405; Acc. Chem. Res. 1985, 18, 380. See also, Hegarty, A.F.; O'Neill, P. Tetrahedron Lett. 1987, 28, 901.
251. Fort, Jr., R.C.; Hrovat, D.A.; Borden, W.T. J. Org. Chem. 1993, 58, 211.
252. Galli, C.; Guarnieri, A.; Koch, H.; Mencarelli, P.; Rappoport, Z. J. Org. Chem. 1997, 62, 4072.
253. Rogers, D.W.; Matsunaga, N.; Zavitsas, A.A. J. Org. Chem. 2006, 71, 2214.
254. Gottschling, S.E.; Grant, T.N.; Milnes, K.K.; Jennings, M.C.; Baines, K.M. J. Org. Chem. 2005, 70, 2686.
255. Giese, B.; Damm, W.; Wetterich, F.; Zeltz, H.-G.; Rancourt, J.; Guindon, Y. Tetrahedron Lett. 1993, 34, 5885.
256. For reviews, see Sustmann, R.; Korth, H. Adv. Phys. Org. Chem. 1990, 26, 131; Viehe, H.G.; Janousek, Z.; Merényi, R.; Stella, L. Acc. Chem. Res. 1985, 18, 148.
257. See Pasto, D.J. J. Am. Chem. Soc. 1988, 110, 8164. See also, Ashby, E.C. Bull. Soc. Chim. Fr. 1972, 2133; Bell, N.A. Educ. Chem. 1973, 143.
258. See Sakurai, H.; Kyushin, S.; Nakadaira, Y.; Kira, M. J. Phys. Org. Chem. 1988, 1, 197; Rhodes, C.J.; Roduner, E. Tetrahedron Lett. 1988, 29, 1437; Viehe, H.G.; Merényi, R.; Janousek, Z. Pure Appl. Chem. 1988, 60, 1635; Bordwell, F.G.; Lynch, T. J. Am. Chem. Soc. 1989, 111, 7558.
259. See Bordwell, F.G.; Bausch, M.J.; Cheng, J.P.; Cripe, T.H.; Lynch, T.-Y.; Mueller, M.E. J. Org. Chem. 1990, 55, 58; Bordwell, F.G.; Harrelson, Jr., J.A. Can. J. Chem. 1990, 68, 1714.
260. See Pasto, D.J. J. Am. Chem. Soc. 1988, 110, 8164.
261. Jiang, X.; Li, X.; Wang, K. J. Org. Chem. 1989, 54, 5648.
262. For reviews of radicals with the unpaired electron on atoms other than carbon, see, in Kochi, J.K. Free Radicals, Vol. 2, Wiley, NY, 1973, the reviews by Nelson, S.F. pp. 527–593 (N-centered); Bentrude, W.G. pp. 595–663 (P-centered); Kochi, J.K. pp. 665–710 (O-centered); Kice, J.L. pp. 711–740 (S-centered); Sakurai, H. pp. 741–807 (Si, Ge, Sn, and Pb centered).
263. Maki, T.; Araki, Y.; Ishida, Y.; Onomura, O.; Matsumura, Y. J. Am. Chem. Soc. 2001, 123, 3371.
264. Jeromin, G.E. Tetrahedron Lett. 2001, 42, 1863.
265. See Novak, I.; Harrison, L.J.; Kova , B.; Pratt, L.M. J. Org. Chem. 2004, 69, 7628.
, B.; Pratt, L.M. J. Org. Chem. 2004, 69, 7628.
266. See Anelli, P.L.; Montanari, F.; Quici, S. Org. Synth. 1990, 69, 212; Fritz-Langhals, E. Org. Process Res. Dev. 2005, 9, 577. See also, Rychnovsky, S.D.; Vaidyanathan, R.; Beauchamp, T.; Lin, R.; Farmer, P.J. J. Org. Chem. 1999, 64, 6745.
267. Volodarsky, L.B.; Reznikov, V.A.; Ovcharenko, V.I. Synthetic Chemistry of Stable Nitroxides, CRC Press, Boca Raton, FL, 1994; Keana, J.F.W. Chem. Rev. 1978, 78, 37; Aurich, H.G. Nitroxides. In Nitrones, Nitronates, Nitroxides, Patai, S.; Rappoport, Z., Eds., Wiley, NY, 1989; Chap. 4.
268. Neiman, M.B.; Rozantsev, E.G.; Mamedova, Yu.G. Nature (London) 1963, 200, 256. See Breuer, E.; Aurich, H.G.; Nielsen, A. Nitrones, Nitronates, and Nitroxides, Wiley, NY, 1989, pp. 313–399; Rozantsev, E.G.; Sholle, V.D. Synthesis 1971, 190, 401.
269. See Ballester, M.; Veciana, J.; Riera, J.; Castañer, J.; Armet, O.; Rovira, C. J. Chem. Soc. Chem. Commun. 1983, 982.
270. Adam, W.; Ortega Schulte, C. M. J. Org. Chem. 2002, 67, 4569.
271. Miura, Y.; Matsuba, N.; Tanaka, R.; Teki, Y.; Takui, T. J. Org. Chem. 2002, 67, 8764. For another stable nitroxide radical, see Huang, W.-l.; Chiarelli, R.; Rassat, A. Tetrahedron Lett. 2000, 41, 8787.
272. Miura, Y.; Tomimura, T.; Matsuba, N.; Tanaka, R.; Nakatsuji, M.; Teki, Y. J. Org. Chem. 2001, 66, 7456. See also, Miura, Y.; Muranaka, Y.; Teki, Y. J. Org. Chem. 2006, 71, 4786; Miura, Y.; Mu, Y. Chem. Lett. 2005, 34, 48
273. Janzen, E.G.; Chen, G.; Bray, T.M.; Reinke, L.A.; Poyer, J.L.; McCay, P.B. J. Chem. Soc. Perkin Trans. 2. 1993, 1983.
274. Reznikov, V.A.; Volodarsky, L.B. Tetrahedron Lett. 1994, 35, 2239.
275. Reznikov, V.A.; Pervukhina, N.V.; Ikorskii, V.N.; Ovcharenko, V.I..; Grand, A. Chem. Commun. 1999, 539.
276. Apeloig, Y.; Bravo-Zhivotovskii, D.; Bendikov, M.; Danovich, D.; Botoshansky, M.; Vakulrskaya, T.; Voronkov, M.; Samoilova, R.; Zdravkova, M.; Igonin, V.; Shklover, V.; Struchkov, Y. J. Am. Chem. Soc. 1999, 121, 8118.
277. It has been claimed that relative D values do not provide such a measure: Nicholas, A.M. de P.; Arnold, D.R. Can. J. Chem. 1984, 62, 1850, 1860.
278. Except where noted, these values are from Lide, D.R. (Ed.), Handbook of Chemistry and Physics, 87th ed.; CRC Press: Boca Raton, FL, 2007, pp. 9-60–9-61. For another list of D values, see McMillen, D.F.; Golden, D.M. Annu. Rev. Phys. Chem. 1982, 33, 493. See also, Holmes, J.L.; Lossing, F.P.; Maccoll, A. J. Am. Chem. Soc. 1988, 110, 7339; Holmes, J.L.; Lossing, F.P. J. Am. Chem. Soc. 1988, 110, 7343; Roginskii, V.A. J. Org. Chem. USSR 1989, 25, 403.
279. For the IR of a matrix-isolated phenyl radical, see Friderichsen, A. V.; Radziszewski, J. G.; Nimlos, M. R.; Winter, P. R.; Dayton, D. C.; David, D. E.; Ellison, G. B. J. Am. Chem. Soc. 2001, 123, 1977.
280. For a review of cyclopropyl radicals, see Walborsky, H.M. Tetrahedron 1981, 37, 1625. See also, Boche, G.; Walborsky, H.M. Cyclopropane Derived Reactive Intermediates, Wiley, NY, 1990.
281. This value is from Gutman, D. Acc. Chem. Res. 1990, 23, 375.
282. Zhang, X.-M. J. Org. Chem. 1998, 63, 1872.
283. Brocks, J.J.; Beckhaus, H.-D.; Beckwith, A.L.J.; Rüchardt, C. J. Org. Chem. 1998, 63, 1935.
284. Pratt, D.A.; Porter, N.A. Org. Lett. 2003, 5, 387.
285. Zavitsas, A.A.; Rogers, D.W.; Matsunaga, N. J. Org. Chem. 2010, 75, 5697.
286. For a review, see Kaplan, L. in Kochi, J.K. Free Radicals, Vol. 2, Wiley, NY, 1973, pp. 361–434.
287. See Giese, B.; Beckhaus, H. Angew. Chem. Int. Ed. 1978, 17, 594; Ellison, G.B.; Engelking, P.C.; Lineberger, W.C. J. Am. Chem. Soc. 1978, 100, 2556. See, however, Paddon-Row, M.N.; Houk, K.N. J. Am. Chem. Soc. 1981, 103, 5047.
288. There are a few exceptions. See Section 14.A.iv.
289. Herzberg, G. Proc. R. Soc. London, Ser. A 1961, 262, 291. See also, Tan, L.Y.; Winer, A.M.; Pimentel, G.C. J. Chem. Phys. 1972, 57, 4028; Yamada, C.; Hirota, E.; Kawaguchi, K. J. Chem. Phys. 1981, 75, 5256.
290. Andrews, L.; Pimentel, G.C. J. Chem. Phys. 1967, 47, 3637; Milligan, D.E.; Jacox, M.E. J. Chem. Phys. 1967, 47, 5146.
291. Tamura, R.; Susuki, S.; Azuma, N.; Matsumoto, A.; Todda, F.; Ishii, Y. J. Org. Chem. 1995, 60, 6820.
292. Rychnovsky, S.D.; Powers, J.P.; LePage, T.J. J. Am. Chem. Soc. 1992, 114, 8375.
293. Danen, W.C.; Tipton, T.J.; Saunders, D.G. J. Am. Chem. Soc. 1971, 93, 5186; Fort, Jr., R.C.; Hiti, J. J. Org. Chem. 1977, 42, 3968; Lomas, J.S. J. Org. Chem. 1987, 52, 2627.
294. Fessenden, R.W.; Schuler, R.H. J. Chem. Phys. 1965, 43, 2704; Rogers, M.T.; Kispert, L.D. J. Chem. Phys. 1967, 46, 3193; Pauling, L. J. Chem. Phys. 1969, 51, 2767.
295. See Chen, K.S.; Tang, D.Y.H.; Montgomery, L.K.; Kochi, J.K. J. Am. Chem. Soc. 1974, 96, 2201. For a discussion, see Krusic, P.J.; Bingham, R.C. J. Am. Chem. Soc. 1976, 98, 230.
296. See Deycard, S.; Hughes, L.; Lusztyk, J.; Ingold, K.U. J. Am. Chem. Soc. 1987, 109, 4954.
297. Adrian, F.J. J. Chem. Phys. 1958, 28, 608; Andersen, P. Acta Chem. Scand. 1965, 19, 629.
298. Kubota, S.; Matsushita, M.; Shida, T.; Abu-Raqabah, A.; Symons, M.C.R.; Wyatt, J.L. Bull. Chem. Soc. Jpn. 1995, 68, 140.
299. See Borden, W.T. Diradicals, Wiley, NY, 1982; Johnston, L.J.; Scaiano, J.C. Chem. Rev. 1989, 89, 521; Doubleday Jr., C.; Turro, N.J.; Wang, J. Acc. Chem. Res. 1989, 22, 199; Scheffer, J.R.; Trotter, J. Rev. Chem. Intermed. 1988, 9, 271; Wilson, R.M. Org. Photochem. 1985, 7, 339; Borden, W.T. React. Intermed. (Wiley) 1985, 3, 151; 1981, 2, 175; Borden, W.T.; Davidson, E.R. Acc. Chem. Res. 1981, 14, 69. See also, Döhnert, D.; Koutecky, J. J. Am. Chem. Soc. 1980, 102, 1789. For a series of papers on diradicals, see Tetrahedron 1982, 38, 735. For a stable hydrocarbon diradical, see Rajca, A.; Shiraishi, K.; Vale, M.; Han, H.; Rajca, S. J. Am. Chem. Soc. 2005, 127, 9014.
300. Zhang, D. Y.; Borden, W. T. J. Org. Chem. 2002, 67, 3989.
301. Ma, J.; Ding, Y.; Hattori, K.; Inagaki, S. J. Org. Chem. 2004, 69, 4245
302. For reviews of trimethylenemethane, see Borden, W.T.; Davidson, E.R. Ann. Rev. Phys. Chem. 1979, 30, 125; Bergman, R.G. in Kochi, J.K. Free Radicals, Vol. 1, Wiley, NY, 1973, pp. 141–149.
303. See Turro, N.J. J. Chem. Educ. 1969, 46, 2; Wasserman, E.; Hutton, R.S. Acc. Chem. Res. 1977, 10, 27; Ichinose, N.; Mizuno, K.; Otsuji, Y.; Caldwell, R.A.; Helms, A.M. J. Org. Chem. 1998, 63, 3176.
304. Matsuda, K.; Iwamura, H. J. Chem. Soc. Perkin Trans. 2 1998, 1023. Also see, Roth, W.R.; Wollweber, D.; Offerhaus, R.; Rekowski, V.; Lenmartz, H.-W.; Sustmann, R.; Müller, W. Chem. Ber. 1993, 126, 2701.
305. Inoue, K.; Iwamura, H. Angew. Chem. Int. Ed. 1995, 34, 927. Also see, Ulrich, G.; Ziessel, R.; Luneau, D.; Rey, P. Tetrahedron Lett. 1994, 35, 1211.
306. Engel, P.S.; Lowe, K.L. Tetrahedron Lett. 1994, 35, 2267.
307. Liao, Y.; Xie, C.; Lahti, P.M.; Weber, R.T.; Jiang, J.; Barr, D.P. J. Org. Chem. 1999, 64, 5176.
308. Cai, X.; Cygon, P.; Goldfuss, B.; Griesbeck, A.G.; Heckroth, H.; Fujitsuka, M.; Majima, T. Chemistry: European J. 2006, 12, 4662.
309. See Giese, B. Radicals in Organic Synthesis: Formation of Carbon–Carbon Bonds, Pergamon, Elmsford, NY, 1986, pp. 267–281; Brown, R.F.C. Pyrolytic Methods in Organic Chemistry, Academic Press, NY, 1980, pp. 44–61.
310. See Harmony, J.A.K. Methods Free-Radical Chem. 1974, 5, 101.
311. See Barker, P.J.; Winter, J.N. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 2, Wiley, NY, 1985, pp. 151–218.
312. Matsuyama, K.; Sugiura, T.; Minoshima, Y. J. Org. Chem. 1995, 60, 5520; Ryzhkov, L.R. J. Org. Chem. 1996, 61, 2801. See Howard, J.A. in Patai, S. The Chemistry of Peroxides, Wiley, NY, 1983, pp. 235–258; Batt, L.; Liu, M.T.H. in the same volume, pp. 685–710.
313. See Engel, P.S. Chem. Rev. 1980, 80, 99; Adams, J.S.; Burton, K.A.; Andrews, B.K.; Weisman, R.B.; Engel, P.S. J. Am. Chem. Soc. 1986, 108, 7935; Schmittel, M.; Rüchardt, C. J. Am. Chem. Soc. 1987, 109, 2750.
314. Cossy, J.; Ranaivosata, J.-L.; Bellosta, V. Tetrahedron Lett. 1994, 35, 8161.
315. Courtneidge, J.L. Tetrahedron Lett. 1992, 33, 3053.
316. Pasto, D.J.; Cottard, F. Tetrahedron Lett. 1994, 35, 4303.
317. Halliwell, B.; Gutteridge, J.M.C. in Free Radicals in Biology and Medicine, Oxford University Press, Oxford, 1999, pp 246–350; DeMatteo, M.P.; Poole, J.S.; Shi, X.; Sachdeva, R.; Hatcher, P.G.; Hadad, C.M.; Platz, M.S. J. Am. Chem. Soc. 2005, 127, 7094.
318. Johnston, L.J. Chem. Rev. 1993, 93, 251.
319. See Costentin, C.; Robert, M.; Saveant, J.-M. J. Am. Chem. Soc. 2003, 125, 105.
320. See Pilling, M.J. Int. J. Chem. Kinet. 1989, 21, 267; Khudyakov, I.V.; Levin, P.P.; Kuz'min, V.A. Russ. Chem. Rev. 1980, 49, 982; Gibian, M.J.; Corley, R.C. Chem. Rev. 1973, 73, 441.
321. Cuerva, J.M.; Campaña, A.G.; Justicia, J.; Rosales, A.; Oller-López, J.L.; Robles, R.; Cárdenas, D.J.; Buñuel, E.; Oltra, J.E. Angew. Chem. Int. Ed. 2006, 45, 5522.
322. Hammerum, S. J. Am. Chem. Soc. 2009, 131, 8627.
323. Dolenc, D.; Plesniar, B. J. Org. Chem. 2006, 71, 8028.
324. Bietti, M.; Salamone, M. Org. Lett. 2010, 12, 3654.
325. See Stevenson, J. P.; Jackson, W. F.; Tanko, J. M. J. Am. Chem. Soc. 2002, 124, 4271.
326. LeTadic-Biadatti, M.-H.; Newcomb, M. J. Chem. Soc. Perkin Trans. 2 1996, 1467. See also, Choi, S.-Y.; Horner, J. H.; Newcomb, M. J. Org. Chem. 2000, 65, 4447; Cooksy, A. L.; King, H. F.; Richardson, W. H. J. Org. Chem. 2003, 68, 9441; Tian, F.; Dolbier, Jr., W.R. Org. Lett. 2000, 2, 835.
327. Halgren, T. A.; Roberts, J. D.; Horner, J. H.; Martinez, F. N.; Tronche, C.; Newcomb, M. J. Am. Chem. Soc. 2000, 122, 2988.
328. Newcomb, M.; Choi, S.-Y.; Toy, P. H. Can. J. Chem. 1999, 77, 1123; Nevill, S. M.; Pincock, J. A. Can. J. Chem. 1997, 75, 232.
329. See Barton, D.H.R.; Jacob, M.; Peralez, E. Tetrahedron Lett. 1999, 40, 9201.
330. Choi, S.-Y.; Horner, J.H.; Newcomb, M. J. Org. Chem. 2000, 65, 4447; Engel, P.S.; He, S.-L.; Banks, J.T.; Ingold, K.U.; Lusztyk, J. J. Org. Chem. 1997, 62, 1210.
331. See Leardini, R.; Lucarini, M.; Pedulli, G.F.; Valgimigli, L. J. Org. Chem. 1999, 64, 3726; Roschek, Jr., B.; Tallman, K.A.; Rector, C.L.; Gillmore, J.G.; Pratt, D.A.; Punta, C.; Porter, N.A. J. Org. Chem. 2006, 71, 3527.
332. See Khudyakov, I.V.; Kuz'min, V.A. Russ. Chem. Rev. 1978, 47, 22.
333. See Kaiser, E.T.; Kevan, L. Radical Ions, Wiley, NY, 1968; Gerson, F.; Huber, W. Acc. Chem. Res. 1987, 20, 85; Todres, Z.V. Tetrahedron 1985, 41, 2771; Holy, N.L.; Marcum, J.D. Angew. Chem. Int. Ed. 1971, 10, 115. See Chanon, M.; Rajzmann, M.; Chanon, F. Tetrahedron 1990, 46, 6193. For a series of papers on this subject, see Tetrahedron 1986, 42, 6097.
334. See Depew, M.C.; Wan, J.K.S. in Patai, S.; Rappoport, Z. The Chemistry of the Quinonoid Compounds, Vol. 2, pt. 2, Wiley, NY, 1988, pp. 963–1018; Huh, C.; Kang, C.H.; Lee, H.W.; Nakamura, H.; Mishima, M.; Tsuno, Y.; Yamataka, H. Bull. Chem. Soc. Jpn. 1999, 72, 1083.
335. de Meijere, A.; Gerson, F.; Schreiner, P.R.; Merstetter, P.; Schüngel, F.-M. Chem. Commun. 1999, 2189.
336. See Russell, G.A. in Patai, S.; Rappoport, Z. The Chemistry of Enones, pt. 1, Wiley, NY, 1989, pp. 471–512. See Davies, A.G.; Neville, A.G. J. Chem. Soc. Perkin Trans. 2 1992, 163, 171.
337. Ishida, S.; Iwamoto, T.; Kira, M. J. Am. Chem. Soc. 2003, 125, 3212; Sekiguchi, A.; Tanaka, T.; Ichinohe, M.; Akiyama, K.; Tero-Kubota, S. J. Am. Chem. Soc. 2003, 125, 4962; Inoue, S.; Ichinohe, M.; Sekiguchi, A. J. Am. Chem. Soc. 2007, 129, 6096.
338. Villano, S.M.; Eyet, N.; Lineberger, W.C.; Bierbaum, V.M. J. Am. Chem. Soc. 2008, 130, 7214.
339. See Roth, H.D. Acc. Chem. Res. 1987, 20, 343; Courtneidge, J.L.; Davies, A.G. Acc. Chem. Res. 1987, 20, 90; Symons, M.C.R. Chem. Soc. Rev. 1984, 13, 393; Marchetti, F.; Pinzino, C.; Zacchini. S.; Guido, G. Angew. Chem. Int. Ed. 2010, 49, 5268.
340. Gerson, F.; Scholz, M.; Hansen, H.-J.; Uebelhart, P. J. Chem. Soc. Perkin Trans. 2 1995, 215.
341. de Meijere, A.; Chaplinski, V.; Gerson, F.; Merstetter, P.; Haselbach, E. J. Org. Chem. 1999, 64, 6951.
342. Neugebauer, F.A.; Funk, B.; Staab, H.A. Tetrahedron Lett. 1994, 35, 4755. See Stickley, K.R.; Blackstock, S.C. Tetrahedron Lett. 1995, 36, 1585.
343. Dauben, W.G.; Cogen, J.M.; Behar, V.; Schultz, A.G.; Geiss, W.; Taveras, A.G. Tetrahedron Lett. 1992, 33, 1713.
344. Rhodes, C.J.; AgirBas H. J. Chem. Soc. Perkin Trans. 2 1992, 397.
345. Gerson, F.; Felder, P.; Schmidlin, R.; Wong, H.N.C. J. Chem. Soc. Chem. Commun. 1994, 1659.
346. Wartini, A.R.; Valenzuela, J.; Staab, H.A.; Neugebauer, F.A. Eur. J. Org. Chem. 1998, 139.
347. Nelson, S.F.; Reinhardt, L.A.; Tran, H.Q.; Clark, T.; Chen, G.-F.; Pappas, R.S.; Williams, F. Chem. Eur. J. 2002, 8, 1074.
348. See Jones, Jr., M.; Moss, R.A. Carbenes, 2 Vols., Wiley, NY, 1973–1975; Rees, C.W.; Gilchrist, T.L. Carbenes, Nitrenes, and Arynes, Nelson, London, 1969; Minkin, V.I.; Simkin, B.Ya.; Glukhovtsev, M.N. Russ. Chem. Rev. 1989, 58, 622; Moss, R.A.; Jones, Jr., M. React. Intermed. (Wiley) 1985, 3, 45; Liebman, J.F.; Simons, J. Mol. Struct. Energ. 1986, 1, 51.
349. See Nefedov, O.M.; Maltsev, A.K.; Mikaelyan, R.G. Tetrahedron Lett. 1971, 4125; Wright, B.B. Tetrahedron 1985, 41, 1517. For reviews, see Zuev, P.S.; Nefedov, O.M. Russ. Chem. Rev. 1989, 58, 636; Sheridan, R.S. Org. Photochem. 1987, 8, 159, pp. 196–216; Trozzolo, A.M. Acc. Chem. Res. 1968, 1, 329.
350. Skell, P.S. Tetrahedron 1985, 41, 1427.
351. See Closs, G.L. Top. Stereochem. 1968, 3, 193, pp. 203–210; Bethell, D. Adv. Phys. Org. Chem. 1969, 7, 153, p. 194; Hoffmann, R. J. Am. Chem. Soc. 1968, 90, 1475.
352. Richards, Jr., C.A.; Kim, S.-J.; Yamaguchi, Y.; Schaefer, III, H.F. J. Am. Chem. Soc. 1995, 117, 10104.
353. See Lengel, R.K.; Zare, R.N. J. Am. Chem. Soc. 1978, 100, 7495; Borden, W.T.; Davidson, E.R. Ann. Rev. Phys. Chem. 1979, 30, 125, see pp. 128–134; Leopold, D.G.; Murray, K.K.; Lineberger, W.C. J. Chem. Phys. 1984, 81, 1048.
354. Kopecky, K.R.; Hammond, G.S.; Leermakers, P.A. J. Am. Chem. Soc. 1961, 83, 2397; 1962, 84, 1015; Duncan, F.J.; Cvetanovi, R.J. J. Am. Chem. Soc. 1962, 84, 3593.
355. For a review of the kinetics of CH2 reactions, see Laufer, A.H. Rev. Chem. Intermed. 1981, 4, 225.
356. See Turro, N.J.; Cha, Y.; Gould, I.R. J. Am. Chem. Soc. 1987, 109, 2101.
357. Tomioka, H. Acc. Chem. Res. 1997, 30, 315; Kirmse, W. Angew. Chem. Int. Ed. 2003, 42, 2117; Hirai, K.; Itoh, T.; Tomioka, H. Chem. Rev. 2009, 109, 3275.
358. Woodcock, H.L.; Moran, D.; Schleyer, P.v.R.; Schaefer, III, H.F. J. Am. Chem. Soc. 2001, 123, 4331.
359. Itoh, T.; Nakata, Y.; Hirai, K.; Tomioka, H. J. Am. Chem. Soc. 2006, 128, 957.
360. Cattoën, X.; Miqueu, K.; Gornitzka, H.; Bourissou, D.; Bertrand, G. J. Am. Chem. Soc. 2005, 127, 3292.
361. For other methods of distinguishing singlet from triplet carbenes, see Hendrick, M.E.; Jones, Jr., M. Tetrahedron Lett. 1978, 4249; Creary, X. J. Am. Chem. Soc. 1980, 102, 1611.
362. Rabinovitch, B.S.; Tschuikow-Roux, E.; Schlag, E.W. J. Am. Chem. Soc. 1959, 81, 1081; Frey, H.M. Proc. R. Soc. London, Ser. A 1959, 251, 575; Lambert, J.B.; Larson, E.G.; Bosch, R.J. Tetrahedron Lett. 1983, 24, 3799.
363. Andrews, L. J. Chem. Phys. 1968, 48, 979.
364. The technique of spin trapping (Sec. 5.C.i) has been applied to the detection of transient triplet carbenes: Forrester, A.R.; Sadd, J.S. J. Chem. Soc. Perkin Trans. 2 1982, 1273.
365. Wasserman, E.; Kuck, V.J.; Hutton, R.S.; Anderson, E.D.; Yager, W.A. J. Chem. Phys. 1971, 54, 4120; Bernheim, R.A.; Bernard, H.W.; Wang, P.S.; Wood, L.S.; Skell, P.S. J. Chem. Phys. 1971, 54, 3223.
366. Hahn, F.E. Angew. Chem. Int. Ed. 2006, 45, 1348. For imidazopyridine carbenes, see Moss, R.A.; Tian, J.; Sauers, R.R.; Krogh-Jespersen, K. J. Am. Chem. Soc. 2007, 129, 10019.
367. Herzberg, G.; Johns, J.W.C. J. Chem. Phys. 1971, 54, 2276 and cited references.
368. Ivey, R.C.; Schulze, P.D.; Leggett, T.L.; Kohl, D.A. J. Chem. Phys. 1974, 60, 3174.
369. Senthilnathan, V.P.; Platz, M.S. J. Am. Chem. Soc. 1981, 103, 5503; Gilbert, B.C.; Griller, D.; Nazran, A.S. J. Org. Chem. 1985, 50, 4738.
370. For reviews of halocarbenes, see Burton, D.J.; Hahnfeld, J.L. Fluorine Chem. Rev. 1977, 8, 119; Margrave, J.L.; Sharp, K.G.; Wilson, P.W. Fort. Chem. Forsch. 1972, 26, 1, pp. 3–13.
371. See Stang, P.J. Acc. Chem. Res. 1982, 15, 348; Chem. Rev. 1978, 78, 383; Marchand, A.P.; Brockway, N.M. Chem. Rev. 1974, 74, 431; Schuster, G.B. Adv. Phys. Org. Chem. 1986, 22, 311. For a review of carbenes with neighboring hetero atoms, see Taylor, K.G. Tetrahedron 1982, 38, 2751.
372. Alcarazo, M.; Roseblade, S.J.; Cowley, A.R.; Fernández, R.; Brown, J.M.; Lassaletta, J.M. J Am. Chem. Soc. 2005, 127, 3290. See also, Kassaee, M.Z.; Shakib, F.A.; Momeni, M.R.; Ghambarian, M.; Musavi, S.M. J. Org. Chem. 2010, 75, 2539.
373. Krahulic, K.E.; Enright, G.D.; Parvez, M.; Roesler, R. J. Am. Chem. Soc. 2005, 127, 4142.
374. Herrmann, W.A. Angew. Chem. Int. Ed. 2002, 41, 1290.
375. Ye, Q.; Komarov, I. V.; Kirby, A. J.; Jones, Jr., M. J. Org. Chem. 2002, 67, 9288.
376. Ye, Q.; Jones Jr., M.; Chen, T.; Shevlin, P.B. Tetrahedron Lett. 2001, 42, 6979.
377. Ohira, S.; Yamasaki, K.; Nozaki, H.; Yamato, M.; Nakayama, M. Tetrahedron Lett. 1995, 36, 8843. For dimethylvinylidene carbene see Reed, S.C.; Capitosti, G.J.; Zhu, Z.; Modarelli, D.A. J. Org. Chem. 2001, 66, 287. For a review of akylidenecarbenes, see Knorr, R. Chem. Rev. 2004, 104, 3795.
378. Fernamberg, K.; Snoonian, J.R.; Platz, M.S. Tetrahedron Lett. 2001, 42, 8761.
379. Creary, X.; Butchko, M.A. J. Org. Chem. 2002, 67, 112.
380. Bonnichon, F.; Richard, C.; Grabner, G. Chem. Commun. 2001, 73.
381. Zuev, P. S.; Sheridan, R. S. J. Am. Chem. Soc. 2004, 126, 12220.
382. Topolski, M.; Duraisamy, M.; Racho, J.; Gawronski, J.; Gawronska, K.; Goedken, V.; Walborsky, H.M. J. Org. Chem. 1993, 58, 546.
383. Kirmse, W. Angew. Chem. Int. Ed. 2005, 44, 2476.
384. See Wanzlick, H.-W.; Schikora, E. Angew. Chem. 1960, 72, 494.
385. Ruzsicska, B.P.; Jodhan, A.; Choi, H.K.J.; Strausz, O.P. J. Am. Chem. Soc. 1983, 105, 2489.
386. Zeidan, T.A.; Kovalenko, S.V.; Manoharan, M.; Clark, R.J.; Ghiviriga, I.; Alabugin, I.V. J. Am. Chem. Soc. 2005, 127, 4270.
387. See Jones, Jr., M. Acc. Chem. Res. 1974, 7, 415; Kirmse, W. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 9; Elsevier, NY, 1973, pp. 373–415; Ref. 348; Petrosyan, V.E.; Niyazymbetov, M.E. Russ. Chem. Rev. 1989, 58, 644.
388. For a review of formation of carbenes in this manner, see Kirmse, W. Angew. Chem. Int. Ed. 1965, 4, 1.
389. Ashby, E.C.; Deshpande, A.K.; Doctorovich, F. J. Org. Chem. 1993, 58, 4205. For a preparation from diclorodiazirine, see Chu, G.; Moss, R.A.; Sauers, R.R. J. Am. Chem. Soc. 2005, 127, 14206. Also see Moss, R.A.; Tian, J.; Sauers, R.R.; Ess, D.H.; Houk, K.N.; Krogh-Jespersen, K. J. Am. Chem. Soc. 2007, 129, 5167.
390. Wagner, W.M. Proc. Chem. Soc. 1959, 229.
391. Glick, H.C.; Likhotvovik, I.R.; Jones, Jr., M. Tetrahedron Lett. 1995, 36, 5715; Stang, P.J. Acc. Chem. Res. 1982, 15, 348; Chem. Rev. 1978, 78, 383.
392. For a review, see Regitz, M.; Maas, G. Diazo Compounds, Academic Press, NY, 1986, pp. 170–184.
393. For example, see Mieusset, J.-L.; Brinker, U.H. J. Org. Chem. 2006, 71, 6975.
394. See Martinu, T.; Dailey, W.P. J. Org. Chem. 2004, 69, 7359.
395. Liu, M.T.H. Chemistry of Diazirines, 2 Vols, CRC Press, Boca Raton, FL, 1987. For reviews, see Moss, R.A. Acc. Chem. Res. 2006, 39, 267; Liu, M.T.H. Chem. Soc. Rev. 1982, 11, 127.
396. Moss, R.A.; Fu, X. Org. Lett. 2004, 6, 3353.
397. Fede, J.-M.; Jockusch, S.; Lin, N.; Moss, R.A.; Turro, N.J. Org. Lett. 2003, 5, 5027.
398. Toscano, J.P.; Platz, M.S.; Nikolaev, V.; Cao, Y.; Zimmt, M.B. J. Am. Chem. Soc. 1996, 118, 3527.
399. For a review, see Nefedov, O.M.; D'yachenko, A.I.; Prokof'ev, A.K. Russ. Chem. Rev. 1977, 46, 941.
400. For a discussion of the nucleophilcity of dichlorocarbene, see Moss, R.A.; Zhang, M.; Krogh-Jespersen, K. Org. Lett. 2009, 11, 1947.
401. Tomioka, H.; Ozaki, Y.; Izawa, Y. Tetrahedron 1985, 41, 4987.
402. Krogh-Jespersen, K.; Yan, S.; Moss, R.A. J. Am. Chem. Soc. 1999, 121, 6269.
403. Ruck, R. T.; Jones, Jr., M. Tetrahedron Lett. 1998, 39, 2277.
404. Khan, M. I.; Goodman, J. L. J. Am. Chem. Soc. 1995, 117, 6635.
405. Sun, Y.; Tippmann, E. M.; Platz, M. S. Org. Lett. 2003, 5, 1305.
406. Ruck, R.T.; Jones, Jr., M. Tetrahedron Lett. 1998, 39, 2277.
407. See Halberstadt, M.L.; McNesby, J.R. J. Am. Chem. Soc. 1967, 89, 3417.
408. See Buncel, E.; Wilson, H. J. Chem. Educ. 1987, 64, 475; Johnson, C.D. Tetrahedron 1980, 36, 3461; Chem. Rev. 1975, 75, 755; Giese, B. Angew. Chem. Int. Ed. 1977, 16, 125; Pross, A. Adv. Phys. Org. Chem. 1977, 14, 69. See also, Srinivasan, C.; Shunmugasundaram, A.; Arumugam, N. J. Chem. Soc. Perkin Trans. 2 1985, 17; Bordwell, F.G.; Branca, J.C.; Cripe, T.A. Isr. J. Chem. 1985, 26, 357; Formosinho, S.J. J. Chem. Soc. Perkin Trans. 2 1988, 839; Johnson, C.D.; Stratton, B. J. Chem. Soc. Perkin Trans. 2 1988, 1903. For a group of papers on this subject, see Isr. J. Chem. 1985, 26, 303.
409. Closs, G.L.; Coyle, J.J. J. Am. Chem. Soc. 1965, 87, 4270.
410. See Tomioka, H.; Ozaki, Y.; Izawa, Y. Tetrahedron 1985, 41, 4987; Frey, H.M.; Walsh, R.; Watts, I.M. J. Chem. Soc. Chem. Commun. 1989, 284.
411. For a discussion, see Regitz, M. Angew. Chem. Int. Ed. 1991, 30, 674.
412. Arduengo, III, A.J.; Harlow, R.L.; Kline, M. J. Am. Chem. Soc. 1991, 113, 361.
413. See Locatelli, F.; Candy, J.-P.; Didillon, B.; Niccolai, G.P.; Uzio, D.; Basset, J.-M. J. Am. Chem. Soc. 2001, 123, 1658; Brown, R.F.C. Pyrolytic Methods in Organic Chemistry, Academic Press, NY, 1980, pp. 115–163; Wentrup, C. Adv. Heterocycl. Chem,. 1981, 28, 231; Jones, W.M. in de Mayo, P. Rearrangements in Ground and Excited States, Vol. 1, Academic Press, NY, 1980, pp. 95–160; Schaefer, III, H.F. Acc. Chem. Res. 1979, 12, 288; Kirmse, W. Carbene Chemistry, 2nd ed., Academic Press, NY, 1971, pp. 457–496.
414. The activation energy for the 1,2-hydrogen shift has been estimated at 1.1 kcal mol–1 (4.5 kJ mol–1), an exceedingly low value: Stevens, I.D.R.; Liu, M.T.H.; Soundararajan, N.; Paike, N. Tetrahedron Lett. 1989, 30, 481. Also see, Pezacki, J. P.; Couture, P.; Dunn, J. A.; Warkentin, J.; Wood, P. D.; Lusztyk, J.; Ford, F.; Platz, M. S. J. Org. Chem. 1999, 64, 4456.
415. Bettinger, H.F.; Rienstra-Kiracofe, J.C.; Hoffman, B.C.; Schaefer, III, H.F.; Baldwin, J.E.; Schleyer, P.v.R. Chem. Commun. 1999, 1515.
416. Liu, M.T.H.; Bonneau, R. J. Am. Chem. Soc. 1989, 111, 6873; Jackson, J.E.; Soundararajan, N.; White, W.; Liu, M.T.H.; Bonneau, R.; Platz, M.S. J. Am. Chem. Soc. 1989, 111, 6874; Ho, G.; Krogh-Jespersen, K.; Moss, R.A.; Shen, S.; Sheridan, R.S.; Subramanian, R. J. Am. Chem. Soc. 1989, 111, 6875; LaVilla, J.A.; Goodman, J.L. J. Am. Chem. Soc. 1989, 111, 6877.
417. Friedman, L.; Shechter, H. J. Am. Chem. Soc. 1960, 82, 1002.
418. McMahon, R.J.; Chapman, O.L. J. Am. Chem. Soc. 1987, 109, 683.
419. Friedman, L.; Berger, J.G. J. Am. Chem. Soc. 1961, 83, 492, 500.
420. For a review, see Jones, W.M. Acc. Chem. Res. 1977, 10, 353.
421. Moss, R.A.; Johnson, L.A.; Kacprzynski, M.; Sauers, R.R. J. Org. Chem. 2003, 68, 5114.
422. See Yao, G.; Rempala, P.; Bashore, C.; Sheridan, R.S. Tetrahedron Lett. 1999, 40, 17.
423. Moss, R. A.; Ma, Y.; Sauers, R. R.; Madni, M. J. Org. Chem. 2004, 69, 3628.
424. Mekley, N.; El-Saidi, M.; Warkentin, J. Can. J. Chem. 2000, 78, 356.
425. Vignolle, J.; Catton, X.; Bourissou, D. Chem. Rev. 2009, 109, 3333.
426. Roth, H.D. J. Am. Chem. Soc. 1971, 93, 1527, 4935, Acc. Chem. Res. 1977, 10, 85.
427. See Scriven, E.F.V. Azides and Nitrenes, Academic Press, NY, 1984; Lwowski, W. React. Intermed. (Wiley) 1985, 3, 305; 1981, 2, 315; 1978, 1, 197; Abramovitch, R.A. in McManus, S.P. Organic Reactive Intermediates, Academic Press, NY, 1973, pp. 127–192; Kuznetsov, M.A.; Ioffe, B.V. Russ. Chem. Rev. 1989, 58, 732 (N- and O-nitrenes); Meth-Cohn, O. Acc. Chem. Res. 1987, 20, 18 (oxycarbonylnitrenes); Abramovitch, R.A.; Sutherland, R.G. Fortsch. Chem. Forsch. 1970, 16, 1 (sulfonyl nitrenes); Ioffe, B.V.; Kuznetsov, M.A. Russ. Chem. Rev. 1972, 41, 131 (N-nitrenes).
428. McClelland, R.A. Tetrahedron 1996, 52, 6823.
429. Kemnitz, C.R.; Karney, W.L.; Borden, W.T. J. Am. Chem. Soc. 1998, 120, 3499.
430. Wasserman, E.; Smolinsky, G.; Yager, W.A. J. Am. Chem. Soc. 1964, 86, 3166. See Carrick, P.G.; Brazier, C.R.; Bernath, P.F.; Engelking, P.C. J. Am. Chem. Soc. 1987, 109, 5100.
431. Smolinsky, G.; Wasserman, E.; Yager, W.A. J. Am. Chem. Soc. 1962, 84, 3220. For a review, see Sheridan, R.S. Org. Photochem. 1987, 8, 159, pp. 159–248.
432. See Sigman, M.E.; Autrey, T.; Schuster, G.B. J. Am. Chem. Soc. 1988, 110, 4297.
433. See Singh, P.N.D.; Mandel, S.M.; Robinson, R.M.; Zhu, Z.; Franz, R.; Ault, B.S.; Gudmundsdottir, A.D. J. Org. Chem. 2003, 68, 7951.
434. Sander, W.; Grote, D.; Kossmann, S.; Neese, F. J. Am. Chem. Soc. 2008, 130, 4396.
435. McConaghy, Jr., J.S.; Lwowski, W. J. Am. Chem. Soc. 1967, 89, 2357, 4450; Mishra, A.; Rice, S.N.; Lwowski, W. J. Org. Chem. 1968, 33, 481.
436. See Dyall, L.K. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement D, pt. 1, Wiley, NY, 1983, pp. 287–320; Dürr, H.; Kober, H. Top. Curr. Chem. 1976, 66, 89; L'Abbé, G. Chem. Rev. 1969, 69, 345.
437. See Subbaraj, A.; Subba Rao, O.; Lwowski, W. J. Org. Chem. 1989, 54, 3945.
438. See Abramovitch, R.A.; Kyba, E.P. J. Am. Chem. Soc. 1971, 93, 1537.
439. Maltsev, A.; Bally, T.; Tsao, M.-L.; Platz, M. S.; Kuhn, A.; Vosswinkel, M.; Wentrup, C. J. Am. Chem. Soc. 2004, 126, 237.
440. See, for example, Leyva, E.; Platz, M.S.; Persy, G.; Wirz, J. J. Am. Chem. Soc. 1986, 108, 3783.
441. Novak, M.; Rajagopal, S. Adv. Phys. Org. Chem. 2001, 36, 167; Falvey, D. E. in Moss, R. A.; Platz, M. S.; Jones, Jr., M. Reactve Intermediate Chemistry, Wiley–Interscience, Hoboken, NJ, 2004, Vol. 1, pp 593–650.
442. Winter, A.H.; Falvey, D.E.; Cramer, C.J. J. Am. Chem. Soc., 2004, 126, 9661.
443. See Abramovitch, R.A.; Jeyaraman, R. in Scriven, E.F.V. Azides and Nitrenes, Acaademic Press, NY, 1984, pp. 297–357; Gassman, P.G. Acc. Chem. Res. 1970, 3, 26; Lansbury, P.T. in Lwowski, W. Nitrenes, Wiley, NY, 1970, pp. 405–419.
444. Gassman, P.G.; Cryberg, R.L. J. Am. Chem. Soc. 1969, 91, 5176.