
• Interim Guidelines for Collecting, Handling, and Testing Clinical Specimens
• Interim Laboratory Biosafety Guidelines
• Laboratory Biosafety Frequently Asked Questions
Interim Guidance for Laboratories
• Interim Guidelines for Collecting, Handling, and Testing Clinical Specimens
• Interim Laboratory Biosafety Guidelines
• Laboratory Biosafety Frequently Asked Questions
Real-Time RT-PCR Resources
• Processing of Sputum Specimens for Nucleic Acid Extraction [1 page]
• CDC 2019-nCoV Real-Time RT-PCR Diagnostic Panel Instructions for Use
• FAQ for Diagnostic Tools and Virus
• Information about COVID-19 diagnostic tests under FDA Emergency Use Authorization
• To request rRT-PCR kits, CDC’s International Reagent Resource (IRR)
• To request 2019-nCoV grown in cell culture, NIH’s BEI Resources Repository
Requests for COVID-19 Diagnostic Tools and Virus
CDC COVID-19 Lab Work
• CDC Tests for COVID-19
• CDC Grows SARS-CoV-2 in Cell Culture
• Serology Test for COVID-19
Fact Sheets to Accompany 2019-nCoV Real-Time RT-PCR Diagnostic Panel Test Results
• Fact Sheet for Healthcare Providers
Frequently Asked Questions on COVID-19 Testing at Laboratories
Q. Where do public health laboratories get access to testing supplies to detect the virus that causes Coronavirus Disease 2019 (COVID-19)?
a. CDC provides the test reagents for public health laboratories (PHLs) to perform real-time RT-polymerase chain reaction (rRT-PCR) detection of the SARS-CoV-2 virus (the virus that causes COVID-19) in respiratory specimens. CDC received authorization from the Food and Drug Administration (FDA) on February 4, 2020 for use of this rRT-PCR test to detect the virus in upper and lower respiratory specimens. These test reagents are available through the International Reagent Resource (IRR). For over ten years, CDC has provided test reagents to PHLs through the IRR. This resource was established to support state and local public health laboratories, Department of Defense laboratories, and other qualified laboratories participating in public health surveillance and studies. Clinical and commercial laboratories conducting COVID-19 testing access test reagents from commercial reagent manufacturers which have received authorization from the Food and Drug Administration (FDA).
Q. What is the CDC’s International Reagent Resource (IRR)?
a. The International Reagent Resource (IRR) was established by the Centers for Disease Control and Prevention (CDC) to provide registered users with reagents, tools and information for studying and detection of Influenza and other pathogens, including the SARS-CoV-2 virus (the virus that causes COVID-19). The IRR acquires, authenticates, and produces reagents that scientists need to carry out basic research and develop improved diagnostic tests, vaccines, and detection methods. By centralizing these functions within the IRR, access to and use of these materials in the scientific and public health community is monitored and quality control of the reagents is assured. The International Reagent Resource is managed under a CDC contract by American Type Culture Collection (ATCC).
Q. What reagents do public health laboratories need to perform testing to detect the virus that causes Coronavirus Disease 2019 (COVID-19)?
a. Public health laboratories in the U.S. performing COVID-19 testing of respiratory specimens are provided the following reagents from the CDC’s International Reagent Resource (IRR):
i. Equipment and Extraction Kits – These kits are used in the preparation of specimens
1. QIAGEN with QIAmp DSP Viral RNA Mini Kit (obtained from IRR)
2. QIAGEN EZ1 Advanced XL with EZ1 DSP Virus Kit (obtained from IRR)
3. QIAGEN QIAcube with QIAmp DSP Viral RNA Mini Kit (obtained from IRR))
4. Roche MagNA Pure LC with Total Nucleic Acid Kit
5. Roche MagNA Pure Compact with Nucleic Acid Isolation Kit I
6. Roche MagNA Pure 96 with DNA and Viral NA Small Volume Kit
ii. rRT-PCR Test Kits (CDC 2019-nCoV Real-Time RT-PCR Diagnostic Panel) – These kits include vials of test reagents that detect the virus that causes COVID-19 in respiratory specimens (obtained from IRR)
iii. Reagents –
1. Master Mix Kits (rRT-PCR Enzyme Mastermix (TaqPath™ 1-Step RT-qPCR Master Mix, CG) – These kits contain the enzymes and other components needed to run the PCR test. (obtained from IRR)
2. Human Specimen Control (HSC) (obtained from IRR)
3. EUA Positive Control (obtained from IRR)
Q. How do clinicians get access to COVID-19 testing?
a. As availability of diagnostic testing for COVID-19 increases, clinicians will be able to access laboratory tests for diagnosing COVID-19 through clinical laboratories performing tests authorized by FDA under an Emergency Use Authorization (EUA). Clinicians should consult with the laboratories that routinely perform their diagnostic services to see how best to access testing for COVID-19.
b. A list of Coronavirus Disease 2019 (COVID-19) Emergency Use Authorizations provided by FDA are available at https://www.fda.gov/medical-devices/emergency-situations-medical-devices/emergency-use-authorizations#coronavirus2019
c. Clinicians are also able to access laboratory testing through public health laboratories in their A list of available public health laboratory testing locations is provided by the Association of Public Health Laboratories (APHL). Questions about testing can be directed to a clinician’s state health department.
Q. Where can additional information about laboratory testing guidance from CDC be found?
a. CDC has published the following interim guidelines, but this is a very dynamic response so please check CDC’s website for the most up to date information:
i. Frequently Asked Questions about Biosafety and COVID-19
ii. 2019-nCoV Real Time RT-PCR Diagnostic Panel Instructions for Use
iii. Interim Guidelines for Collecting, Handling, and Testing Clinical Specimens from Patients Under Investigation for 2019-nCoV
iv. Interim Laboratory Biosafety Guidelines for Handling and Processing Specimens Associated with 2019-nCoV
On February 11, 2020, the International Committee on Taxonomy of Viruses, charged with naming new viruses, named the virus causing coronavirus disease 2019 (COVID-19), severe acute respiratory syndrome coronavirus 2, shortened to SARS-CoV-2.
As the name indicates, the virus is related to the sars-associated coronavirus (SARS-CoV) that caused an outbreak of severe acute respiratory syndrome (SARS) in 2002-2003, however it is not the same virus.
CDC Grows SARS-CoV-2 Virus in Cell Culture
CDC has developed a new laboratory test kit for use in testing patient specimens for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus that causes COVID-19. The test kit is called the “Centers for Disease Control and Prevention (CDC) 2019-Novel Coronavirus (2019-nCoV) Real-Time Reverse Transcriptase (RT)-PCR Diagnostic Panel.” It is intended for use with the Applied Biosystems 7500 Fast DX Real-Time PCR Instrument with SDS 1.4 software. This test is intended for use with upper and lower respiratory specimens collected from persons who meet CDC criteria for COVID-19 testing. CDC’s test kit is intended for use by laboratories designated by CDC as qualified, and in the United States, certified under the Clinical Laboratory Improvement Amendments (CLIA) to perform high complexity tests. The test kits also will be shipped to qualified international laboratories, such as World Health Organization (WHO) Global Influenza Surveillance Response System (GISRS) laboratories. The test will not be available in U.S. hospitals or other primary care settings. The kits will be distributed through the International Reagent Resource (IRR)
On Monday, February 3, 2020, CDC submitted an Emergency Use Authorization (EUA) package to the U.S. Food and Drug Administration (FDA) in order to expedite FDA permitted use of the CDC diagnostic panel in the United States. The EUA process enables FDA to consider and authorize the use of unapproved, but potentially life-saving medical or diagnostic products during a public health emergency. The U.S. Secretary of Health and Human Services declared the SARS-CoV-2 virus a U.S. public health emergency on Friday, January 31, 2020. FDA issued the EUA on February 4, 2020. IRR began distribution of the test kits to states, but shortly thereafter performance issues were identified related to a problem in the manufacturing of one of the reagents which led to laboratories not being able to verify the test performance. CDC is remanufacturing the reagents with more robust quality control measures. New tests will be distributed once this issue has been addressed. CDC continues to perform initial and confirmatory testing.

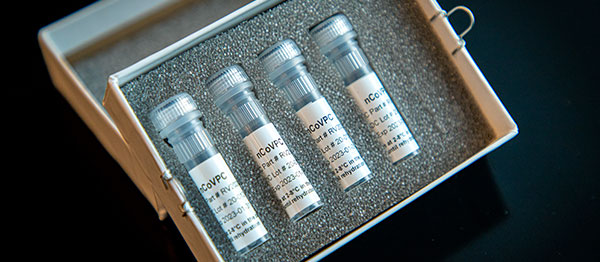
This is a picture of CDC’s laboratory test kit for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). CDC is shipping the test kits to laboratories CDC has designated as qualified, including U.S. state and local public health laboratories, Department of Defense (DOD) laboratories and select international laboratories. The test kits are bolstering global laboratory capacity for detecting SARS-CoV-2.
CDC is working to develop a new laboratory test to assist with efforts to determine how much of the U.S. population has been exposed to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus that causes COVID-19.
The serology test will look for the presence of antibodies, which are specific proteins made in response to infections. Antibodies can be found in the blood and in other tissues of those who are tested after infection. The antibodies detected by this test indicate that a person had an immune response to SARS-CoV-2, whether symptoms developed from infection or the infection was asymptomatic. Antibody test results are important in detecting infections with few or no symptoms.
Initial work to develop a serology test for SARS-CoV-2 is underway at CDC. In order to develop the test, CDC needs blood samples from people who had COVID-19 at least 21 days after their symptoms first started. Researchers are currently working to develop the basic parameters for the test, which will be refined as more samples become available. Once the test is developed, CDC will need additional samples to evaluate whether the test works as intended.
Interim Guidelines for Collecting, Handling, and Testing Clinical Specimens from Persons Under Investigation (PUIs) for Coronavirus Disease 2019 (COVID-19)
Summary of Recent Changes
Revisions were made on March 9, 2020. to reflect the following:
Updates to all specimen storage guidelines for consistency with EUA IFU.
Recommendation to include combined NP/OP specimens as an option for upper respiratory specimen collection.
March 9, 2020
Health care providers should contact their local/state health department immediately to notify them of patients with fever and lower respiratory illness who live in or have recently traveled to an affected area with sustained transmission or have been in close contact with a confirmed COVID-19 patient. Local and state public health staff will determine if the patient meets the criteria for a person under investigation (PUI) for COVID-19. Clinical specimens should be collected from PUIs for routine testing of respiratory pathogens at either clinical or public health labs. Note that clinical laboratories should NOT attempt viral isolation from specimens collected from COVID-19 PUIs.
Now that the CDC’s diagnostic test has been authorized by FDA under the EUA, the International Reagent Resource (IRR) has begun to distribute the test to requesting laboratories.
Clinicians who have identified a potential PUI should immediately notify their state or local health department. Local and state public health staff will determine if the person is a PUI and whether testing for COVID-19 is indicated. The state and local health department will then assist clinicians to collect, store, and ship specimens appropriately, including during afterhours or on weekends/holidays.
Testing for other pathogens by the provider should be done as part of the initial evaluation and should not delay specimen shipping.
If a PUI tests positive for another respiratory pathogen, after clinical evaluation and consultation with public health authorities, they may no longer be considered a PUI. This may evolve as more information becomes available on possible COVID-19 co-infections.
For initial diagnostic testing for COVID-19, CDC recommends collecting and testing upper respiratory (nasopharyngeal AND oropharyngeal swabs), and lower respiratory (sputum, if possible) for those patients with productive coughs. Induction of sputum is not recommended. Specimens should be collected as soon as possible once a PUI is identified, regardless of the time of symptom onset. Maintain proper infection control when collecting specimens.
Store specimens at 2-8°C and ship overnight to CDC on ice pack. Label each specimen container with the patient’s ID number (e.g., medical record number), unique specimen ID (e.g., laboratory requisition number), specimen type (e.g., serum) and the date the sample was collected. Complete a CDC Form 50.34 for each specimen submitted. In the upper left box of the form, 1) for test requested select “Respiratory virus molecular detection (non-influenza) CDC-10401” and 2) for At CDC, bring to the attention of enter “Stephen Lindstrom: 2019-nCoV PUI”.
A. Lower respiratory tract
Bronchoalveolar lavage, tracheal aspirate
Collect 2-3 mL into a sterile, leak-proof, screw-cap sputum collection cup or sterile dry container.
Sputum
Have the patient rinse the mouth with water and then expectorate deep cough sputum directly into a sterile, leak-proof, screw-cap sputum collection cup or sterile dry container.
B. Upper respiratory tract
Nasopharyngeal swab AND oropharyngeal swab (NP/OP swab)
Use only synthetic fiber swabs with plastic shafts. Do not use calcium alginate swabs or swabs with wooden shafts, as they may contain substances that inactivate some viruses and inhibit PCR testing. Place swabs immediately into sterile tubes containing 2-3 ml of viral transport media. NP and OP specimens may be kept in separate vials or combined at collection into a single vial.
Nasopharyngeal swab: Insert a swab into the nostril parallel to the palate. Leave the swab in place for a few seconds to absorb secretions.
Oropharyngeal swab (e.g., throat swab): Swab the posterior pharynx, avoiding the tongue.
Nasopharyngeal wash/aspirate or nasal aspirate
Collect 2-3 mL into a sterile, leak-proof, screw-cap sputum collection cup or sterile dry container.
Store specimens at 2-8°C for up to 72 hours after collection. If a delay in testing or shipping is expected, store specimens at -70°C or below.
Specimens PUI’s must be packaged, shipped, and transported according to the current edition of the International Air Transport Association (IATA) Dangerous Goods Regulations. Store specimens at 2-8°C and ship overnight to CDC on ice pack. If a specimen is frozen at -70°C ship overnight to CDC on dry ice. Additional useful and detailed information on packing, shipping, and transporting specimens can be found at Interim Laboratory Biosafety Guidelines for Handling and Processing Specimens Associated with Coronavirus Disease 2019 (COVID-19).
Interim Laboratory Biosafety Guidelines for Handling and Processing Specimens Associated with Coronavirus Disease 2019 (COVID-19)
Summary of Recent Changes
Revisions were made on February 10, 2020, to reflect the following:
• The term “certified” was added to clarify that a certified Class II Biological Safety Cabinet (BSC) should be used for any laboratory procedure with the potential to generate aerosols or droplets.
• Additional details were provided for any laboratory procedures that are performed outside of a BSC.
• Additional details were provided about the use of EPA-registered hospital disinfectants.
• Additional details were provided for handling COVID-19 laboratory waste.
• The need for both site- and activity-specific risk assessments was added to determine if additional laboratory biosafety control measures are necessary.
February 10, 2020
Until more information becomes available, precautions should be taken in collecting and handling specimens that may contain SARS-CoV-2. Timely communication between clinical and laboratory staff is essential to minimize the risk incurred in handling specimens from patients with possible SARS-CoV-2 infection. Such specimens should be labeled accordingly, and the laboratory should be alerted to ensure proper specimen handling. General and specific biosafety guidelines for handling SARS-CoV-2 specimens are provided below.
For additional detailed instructions please refer to the following:
• Biosafety in Microbiological and Biomedical Laboratories (BMBL) – Fifth Edition
• Laboratory Biosafety Manual – Third Edition
General Guidelines (for working with potentially infectious materials)
Laboratory workers should wear appropriate personal protective equipment (PPE) which includes disposable gloves, laboratory coat/gown and eye protection when handling potentially infectious specimens.
Any procedure with the potential to generate aerosols or droplets (e.g., vortexing) should be performed in a certified Class II Biological Safety Cabinet (BSC). Appropriate physical containment devices (e.g., centrifuge safety buckets; sealed rotors) should be used for centrifugation. Ideally, rotors and buckets should be loaded and unloaded in a BSC. For any procedures outside of a BSC, eye and face protection (e.g. goggles, mask, face shield) or other physical barriers (e.g. splash shield) should be used to minimize the risk of exposure to laboratory staff.
After specimens are processed, decontaminate work surfaces and equipment with appropriate disinfectants. Use EPA-registered hospital disinfectants with label claims to be effective against other respiratory pathogens, such as seasonal influenza and other human coronaviruses. Follow manufacturer’s recommendations for use – dilution (i.e., concentration), contact time, and care in handling.
For SARS-CoV-2 laboratory waste, follow standard procedures associated with other respiratory pathogens, such as seasonal influenza and other human coronaviruses.
Virus isolation in cell culture and initial characterization of viral agents recovered in cultures of SARS-CoV-2 specimens are NOT recommended at this time, except in a BSL3 laboratory using BSL3 work practices.
The following activities may be performed in BSL-2 facilities using standard BSL-2 work practices:
• Pathologic examination and processing of formalin-fixed or otherwise inactivated tissues
• Molecular analysis of extracted nucleic acid preparations
• Electron microscopic studies with glutaraldehyde-fixed grids
• Routine examination of bacterial and mycotic cultures
• Routine staining and microscopic analysis of fixed smears
• Final packaging of specimens for transport to diagnostic laboratories for additional testing. Specimens should already be in a sealed, decontaminated primary container.
• Inactivated specimens (e.g., specimens in nucleic acid extraction buffer)
The following activities involving manipulation of potentially infected specimens should be, at a minimum, performed as above and in a certified Class II BSC in a BSL-2 facility. Site- and activity-specific risk assessments should be performed to determine if enhanced biosafety precautions are warranted based on situational needs (e.g. high testing volumes):
• Aliquoting and/or diluting specimens
• Inoculating bacterial or mycological culture media
• Performing diagnostic tests that do not involve propagation of viral agents in vitro or in vivo
• Nucleic acid extraction procedures involving potentially infected specimens
• Preparation and chemical- or heat-fixing of smears for microscopic analysis
Clinical laboratories performing routine hematology, urinalysis, and clinical chemistry studies, and microbiology laboratories performing diagnostic tests on serum, blood, or urine specimens should follow standard laboratory practices, including Standard Precautions, when handling potential COVID-19 patient specimens. For additional information, see Biosafety in Microbiological and Biomedical Laboratories (BMBL) – Fifth Edition (page 225).
Packing, Shipping and Transport
Packaging, shipping, and transport of specimens from suspect cases or PUI’s of COVID-19 must follow the current edition of the International Air Transport Association (IATA) Dangerous Goods Regulations.
Follow shipping regulations for UN 3373 Biological Substance, Category B when sending potential COVID-19 patient specimens.
• Packaging Checklist, see Category B Saf-T-Pak
• Packing Instructions 650 for UN 3373
○ Click on “Infectious substances” and there is an option to download the packing instructions.
• Labels for UN 3373
○ When using cold pack – Include the name and telephone number of the person who will be available during normal business hours who knows the content of the shipment (can be someone at CDC). Place the label on one side of the box and cover the label completely with clear tape (do not tape just the edges of the label).
○ When using dry ice – Include the name and telephone number of the person who will be available during normal business hours who knows the content of the shipment (can be someone at CDC). Place the label on one side of the box and cover the label completely with clear tape (do not tape just the edges of the label).
• Schematic for packaging, UN 3373 Category B
FAQ for Diagnostic Tools and Virus
How do I learn more about the CDC 2019-nCoV Real-Time RT-PCR Diagnostic Panel?
• The diagnostic panel’s FDA-authorized instructions for use contain information about the test, its intended use, test procedure and performance characteristics. FDA’s EUA website has published the FDA Letter of Authorization for the diagnostic panel, which defines the authorized use and the conditions of authorization that apply to CDC and to testing laboratories.
How do I order a reagent diagnostic panel?
The International Reagent Resource (IRR) will distribute the diagnostic panel and most of the associated reagents.
Which labs will receive the diagnostic panel?
The Food and Drug Administration (FDA) authorized the Emergency Use Authorization (EUA) on February 4, 2020.
• CDC’s diagnostic panel is intended for use by laboratories designated by CDC as qualified, and in the United States, certified under the Clinical Laboratory Improvement Amendments (CLIA) to perform high complexity tests. This includes U.S. state and local public health laboratories and Department of Defense (DoD) laboratories. The test kits also will be shipped to qualified international laboratories, such as World Health Organization (WHO) Global Influenza Surveillance Response System (GISRS) laboratories For more information, see the section labeled CDC Laboratory Test Kit Distribution (general audiences).
• Each laboratory that places an order will receive one diagnostic panel initially, and each panel can test 700-800 patient specimens.
This is a picture of CDC’s laboratory test kit for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). CDC is shipping the test kits to laboratories CDC has designated as qualified, including U.S. state and local public health laboratories, Department of Defense (DOD) laboratories and select international laboratories. The test kits are bolstering global laboratory capacity for detecting SARS-CoV-2.
What does the diagnostic panel include?
The CDC 2019-nCoV Real-Time RT-PCR Diagnostic Panel contains the following:
• 2019-nCoV_N1, 2019-nCoV_N2 and 2019-nCoV_N3 primers and probes that target the nucleocapsid (N) gene and are designed for both universal detection of SARS-like coronavirus as well as specific detection of the 2019-nCoV;
• RP primers and probes that target the Human RNase P gene; and
• nCoVPC, the 2019-nCoV positive control used in the assay.
What other equipment will labs need to perform tests using the diagnostic panel?
The CDC 2019-nCoV Real-Time RT-PCR Diagnostic Panel also requires the use of additional authorized materials and authorized ancillary reagents that are not included with the test but are commonly used in clinical laboratories and are described in the authorized CDC 2019-nCoV Real-Time RT-PCR Diagnostic Panel Instructions for Use Package Insert.
The CDC 2019-nCoV Real-Time RT-PCR Diagnostic Panel requires the following control materials, or other authorized control materials; all controls listed below must generate expected results for a test to be considered valid, as outlined in the CDC 2019-nCoV Real-Time RT-PCR Diagnostic Panel Instructions for Use:
• Human Specimen Control (HSC): A human cell culture preparation used as an extraction control and positive control for the RNase P primer and probe set that is extracted and tested concurrently with each specimen extraction run.
• Positive Control for COVID-19 (nCoVPC): Run with each batch of specimens. Monitors for failures of rRT-PCR reagents and reaction conditions.
• No Template Control (NTC): Nuclease-free water included in each run. Monitors for reagent and system contamination.
Should I be testing all patients for COVID-19?
At this time, CDC only recommends diagnostic testing of patients who meet the clinical criteria for a COVID-19 person under investigation (PUI), per Interim Guidelines for Collecting, Handling and Testing Clinical Specimens from Persons Under Investigation (PUIs) for Coronavirus Disease 2019 (COVID-19).
• People who have been in close contact with a confirmed COVID-19 patient, and people who live in or recently traveled from an area with sustained transmission, are at elevated risk of COVID-19.
• For more information about interpreting test results, see section labeled “Interpretation of test results from CDC 2019 novel coronavirus (2019-nCoV) Real-Time RT-PCR Diagnostic Panel.”
What safety equipment should labs use when using the diagnostic tool?
Use appropriate personal protective equipment when collecting and handling specimens from individuals suspected of being infected with COVID-19 as outlined in the CDC Interim Guidelines for Collecting, Handling and Testing Clinical Specimens from Persons Under Investigation (PUIs) for Coronavirus Disease 2019 (COVID-19).
How can my lab get the virus?
The National Institute of Health (NIH)’s Biodefense and Emerging Infections Research Resources Repository (BEI Resources) will provide laboratories with COVID-19.
I believe that I have found a treatment or vaccine for COVID-19. Is CDC the best place to submit my idea?
BARDA is providing a portal to support U.S. government medical countermeasure research and development. Interested stakeholders can learn more here.
Research Use Only Real-Time RT-PCR Protocol for Identification of 2019-nCoV
NOT FOR DIAGNOSTIC USE
For CDC diagnostic testing guidance and information about CDC’s FDA-authorized diagnostic testing panel, please visit Information for Laboratories.
Table of Contents
• Introduction
• Specimens
• Reagents, Supplies and Equipment Requirements
• Nucleic Acid Extraction
• Quality Control
• rRT-PCR Assays
• Interpreting Test Results
• Assay Limitations
• Contact Information
Purpose: This document describes the use of real-time RT PCR (rRT-PCR) assays for the in vitro qualitative detection of 2019-Novel Coronavirus (2019-nCoV) in respiratory specimens and sera. The 2019-nCoV primer and probe sets are designed for the universal detection of SARS-like coronaviruses (N3 assay) and for specific detection of 2019-nCoV (N1 and N2 assays).
Protocol Use Limitations: The rRT-PCR assays described here have not been validated for platforms or chemistries other than those described in this document.
Biosafety Precautions
Wear appropriate personal protective equipment (e.g. gowns, gloves, eye protection) when working with clinical specimens. Specimen processing should be performed in a certified class II biological safety cabinet following biosafety level 2 or higher guidelines. For more information, refer to:
• Interim Guidelines for Collecting, Handling, and Testing Clinical Specimens from Persons Under Investigation (PUIs) for 2019 Novel Coronavirus (2019-nCoV) https://www.cdc.gov/coronavirus/2019-nCoV/lab/guidelines-clinical-specimens.html
• Biosafety in Microbiological and Biomedical Laboratories 5th edition available at http://www.cdc.gov/biosafety/publications/.
Acceptable Specimens
• Respiratory specimens including: nasopharyngeal or oropharyngeal aspirates or washes, nasopharyngeal or oropharyngeal swabs, broncheoalveolar lavage, tracheal aspirates, and sputum.
○ Swab specimens should be collected only on swabs with a synthetic tip (such as polyester or Dacron®) with aluminum or plastic shafts. Swabs with calcium alginate or cotton tips with wooden shafts are not acceptable.
Specimen Handling and Storage
• Specimens can be stored at 4oC for up to 72 hours after collection.
• If a delay in extraction is expected, store specimens at -70oC or lower.
• Extracted nucleic acids should be stored at -70oC or lower.
Specimen Rejection criteria:
• Specimens not kept at 2-4°C (≤4 days) or frozen at -70°C or below.
• Incomplete specimen labeling or documentation.
• Inappropriate specimen type.
• Insufficient specimen volume.
Reagents, Supplies and Equipment Requirements
Disclaimer: Names of vendors or manufacturers are provided as examples of suitable product sources. Inclusion does not imply endorsement by the Centers for Disease Control and Prevention.
Reagents and Supplies
• rRT-PCR primer/probe sets
• Positive template control
• TaqPath™ 1-Step RT-qPCR Master Mix, CG (ThermoFisher; cat # A15299 or A15300)
• Molecular grade water, nuclease-free
• Disposable powder-free gloves
• P2/P10, P200, and P1000 aerosol barrier tips
• Sterile, nuclease-free 1.5 mL microcentrifuge tubes
• 0.2 mL PCR reaction tube strips or 96-well real-time PCR reaction plates and optical 8-cap strips
• Laboratory marking pen
• Cooler racks for 1.5 microcentrifuge tubes and 96-well 0.2 mL PCR reaction tubes
• Racks for 1.5 ml microcentrifuge tubes
• Acceptable surface decontaminants
○ DNAZapTM (Life Technologies, cat. #AM9890)
○ DNA AwayTM (Fisher Scientific; cat. #21-236-28)
○ RNAse AwayTM (Fisher Scientific; cat. #21-236-21
○ 10% bleach (1:10 dilution of commercial 5.25-6.0% sodium hypochlorite)
Equipment
• PCR Work Station [UV lamp; Laminar flow (Class 100 HEPA filtered)]
• Vortex mixer
• Microcentrifuge
• Micropipettes (2 or 10 µl, 200 µl and 1000 µl)
• Multichannel micropipettes (5-50 µl)
• 2 x 96-well cold blocks
• -20oC (nonfrost-free) and -70oC freezers; 4oC refrigerator
• Real-time PCR detection system
• Nucleic acid extraction system
• Performance of rRT-PCR amplification based assays depends on the amount and quality of sample template RNA. RNA extraction procedures should be qualified and validated for recovery and purity before testing specimens.
• Commercially available extraction procedures that have been shown to generate highly purified RNA when following manufacturer’s recommended procedures for sample extraction include: bioMérieux NucliSens® systems, QIAamp® Viral RNA Mini Kit, QIAamp® MinElute Virus Spin Kit or RNeasy® Mini Kit (QIAGEN), EZ1 DSP Virus Kit (QIAGEN), Roche MagNA Pure Compact RNA Isolation Kit, Roche MagNA Pure Compact Nucleic Acid Isolation Kit, and Roche MagNA Pure 96 DNA and Viral NA Small Volume Kit, and Invitrogen ChargeSwitch® Total RNA Cell Kit.
• Retain residual specimen and nucleic extract and store immediately at -70oC
• Only thaw the number of specimen extracts that will be tested in a single day. Do not freeze/thaw extracts more than once before testing.
Due to the sensitivity of rRT-PCR, these assays should be conducted using strict quality control and quality assurance procedures. Following these guidelines will help minimize chance of false-positive amplification.
General Considerations
• Personnel must be familiar with the protocol and instruments used.
• Maintain separate areas and dedicated equipment (e.g., pipettes, microcentrifuges) and supplies (e.g., microcentrifuge tubes, pipette tips, gowns and gloves) for assay reagent setup and handling of extracted nucleic acids.
• Work flow must always be from the clean area to the dirty area.
• Wear clean disposable gowns and new, previously unworn, powder-free gloves during assay reagent setup and handling of extracted nucleic acids. Change gloves whenever contamination is suspected.
• Store primer/probes and enzyme master mix at appropriate temperatures (see package inserts). Do not use reagents beyond their expiry dates.
• Keep reagent tubes and reactions capped as much as possible.
• Clean and decontaminate surfaces.
• Do not bring extracted nucleic acid or PCR products into the assay setup area.
• Use aerosol barrier (filter) pipette tips only.
• Use PCR plate strip caps only. Do not use PCR plate sealing film.
Assay Controls
• Assay controls should be run concurrently with all test samples.
• PTC – positive template control with an expected Ct value range
• NTC – negative template control added during rRT-PCR reaction set-up
• HSC – human specimen extraction control extracted concurrently with the test samples; provides a nucleic acid extraction procedural control and a secondary negative control that validates the nucleic extraction procedure and reagent integrity
• RP – all clinical samples should be tested for human RNAse P (RNP) gene to assess specimen quality
• Note: Keep running logs of PTC performance. After each rRT-PCR run of clinical samples, the control Ct values should be recorded.
Stock Reagent Preparation
rRT-PCR Primers/Probe Sets (see package insert if provided by CDC)
• Precautions: These reagents should only be handled in a clean area and stored at appropriate temperatures (see below) in the dark. Freeze-thaw cycles should be avoided. Maintain cold when thawed.
• Using aseptic technique, suspend dried reagents in 1.5 mL nuclease-free water and allow to rehydrate for 15 min at room temperature in the dark.
• Mix gently and aliquot primers/probe in 300 μL volumes into 5 pre-labeled tubes. Store a single aliquot of primers/probe at 2-8oC in the dark. Do not refreeze (stable for up to 4 months). Store remaining aliquots at ≤-20oC in a non-frost-free freezer.
Positive Template Control (PTC) (if using CDC positive control, nCoVPC, see package insert provided by CDC)
• Precautions: This reagent should be handled with caution in a dedicated nucleic acid handling area to prevent possible contamination. Freeze-thaw cycles should be avoided. Maintain on ice when thawed.
• Used to assess performance of rRT-PCR assays. Resuspend dried reagent in each tube in 1 mL of nuclease-free water to achieve the proper concentration. Make single use aliquots (approximately 30 μL) and store at ≤ -70°C.
• Thaw a single aliquot of diluted positive control for each experiment and hold on ice until adding to plate. Discard any unused portion of the aliquot.
• The nCoVPC also contains human DNA that serves as the positive control for the RP assay.
Equipment preparation
• Clean and decontaminate all work surfaces, pipets, centrifuges and other equipment prior to use using RNase Away® or 10% freshly prepared bleach.
• Turn on AB 7500 Fast DX and allow block to reach optimal temperature.
• Perform plate set up and select cycling protocol on the instrument.
• Instrument Settings: Detector (FAM); Quencher (None); Passive Reference: (None); Run Mode: (Standard); Sample Volume (20 µL)
|
Step |
Cycles |
Temp |
Time |
|
UNG Incubation |
1 |
25°C |
2 min |
|
RT incubation |
1 |
50°C |
15 min |
|
Enzyme activation |
1 |
95°C |
2 min |
|
Amplification |
45 |
95°C |
3 sec |
|
55°C |
30 sec |
||
|
* Fluorescence data (FAM) should be collected during the 55°C incubation step. |
|||
Reaction Master Mix and Plate Set-Up
Note: Plate set-up configuration can vary with the number of specimens and work day organization. NTCs and nCoVPCs must be included in each run.
1. In the reagent set-up room clean hood, place r4X Master Mix and primer/probes on ice or cold-block. Keep cold during preparation and use.
2. Thaw 4X Reaction Mix prior to use.
3. Mix 4X Master Mix and primer/probes by inversion 5 times.
4. Briefly centrifuge 4X Master Mix and primers/probes and return to cold block.
5. Label one 1.5 mL microcentrifuge tube for each primer/probe set.
6. Determine the number of reactions (N) to set up per assay. It is necessary to make excess reaction mix for the NTC, nCoVPC, and RP reactions and for pipetting error. Use the following guide to determine N:
○ If number of samples (n) including controls equals 1 through 14, then N = n + 1
○ If number of samples (n) including controls is 15 or greater, then N = n + 2
7. For each primer/probe set, calculate the amount of each reagent to be added for each reaction mixture (N = # of reactions).
TaqPath™ 1-Step RT-qPCR Master Mix
|
Step # |
Reagent |
Vol. of Reagent Added per Reaction |
|---|---|---|
|
1 |
Nuclease-free Water |
N x 8.5 µL |
|
2 |
Combined Primer/Probe Mix |
N x 1.5 µL |
|
3 |
TaqPathTM 1-Step RT-qPCR Master Mix (4x) |
N x 5.0 µL |
|
|
Total Volume |
N x 15.0 µL |
8. Dispense reagents into each respective labeled 1.5 mL microcentrifuge tube. After addition of the reagents, mix reaction mixtures by pipetting up and down. Do not vortex.
9. Centrifuge for 5 seconds to collect contents at the bottom of the tube, and then place the tube in a cold rack.
10. Set up reaction strip tubes or plates in a 96-well cooler rack.
11. Dispense 15 µL of each master mix into the appropriate wells going across the row as shown below (Figure 1):
Figure 1: Example of Reaction Master Mix Plate Set-Up
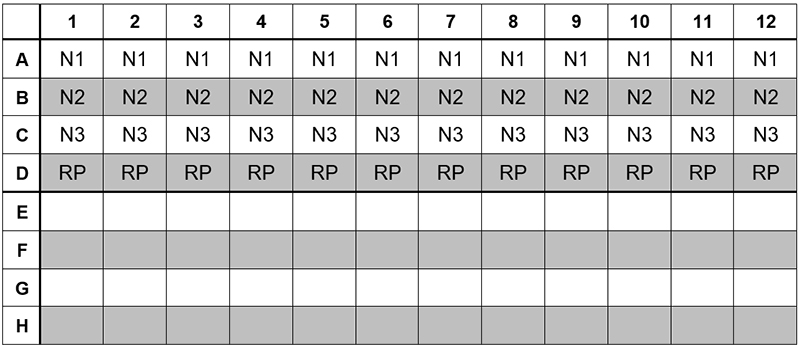
12. Prior to moving to the nucleic acid handling area, prepare the No Template Control (NTC) reactions for column #1 in the assay preparation area.
13. Pipette 5 µL of nuclease-free water into the NTC sample wells. Securely cap NTC wells before proceeding.
14. Cover the entire reaction plate and move the reaction plate to the specimen nucleic acid handling area.
1. Gently vortex nucleic acid sample tubes for approximately 5 seconds.
2. After centrifugation, place extracted nucleic acid sample tubes in the cold rack.
3. Samples should be added to column 2-11 (column 1 and 12 are for controls) to the specific assay that is being tested as illustrated in Figure 2. Carefully pipette 5.0 µL of the first sample into all the wells labeled for that sample (i.e. Sample “S1” down column #2). Keep other sample wells covered during addition. Change tips after each addition.
4. Securely cap the column to which the sample has been added to prevent cross contamination and to ensure sample tracking.
5. Change gloves often and when necessary to avoid contamination.
6. Repeat steps #3 and #4 for the remaining samples.
7. If necessary, add 5 µL of Human Specimen Control (HSC) extracted sample to the HSC wells (Figure 2, column 11). Securely cap wells after addition
8. Cover the entire reaction plate and move the reaction plate to the positive template control handling area.
1. Pipette 5 µL of nCoVPC RNA to the sample wells of column 12 (Figure 2). Securely cap wells after addition of the control RNA.
NOTE:If using 8-tube strips, label the TAB of each strip to indicate sample position. DO NOT LABEL THE TOPS OF THE REACTION TUBES!
2. Briefly centrifuge reaction tube strips for 10-15 seconds. After centrifugation return to cold rack.
NOTE: If using 96-well plates, centrifuge plates for 30 seconds at 500 x g, 4°C.
Figure 2. 2019-nCoV rRT-PCR Diagnotic Panel: Example of Sample and Control Set-up
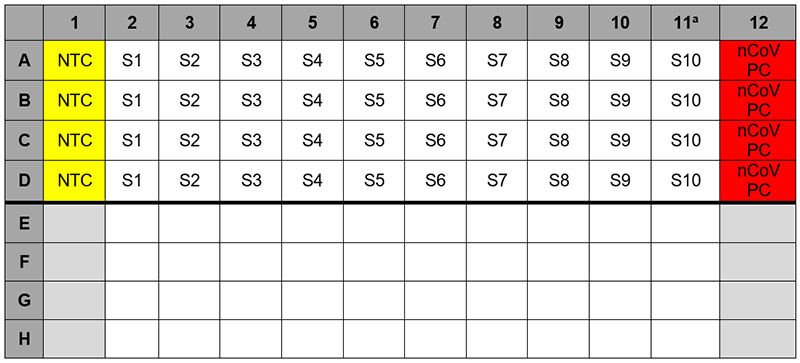
a Replace the sample in this column with extracted HSC if necessary
Data Analysis
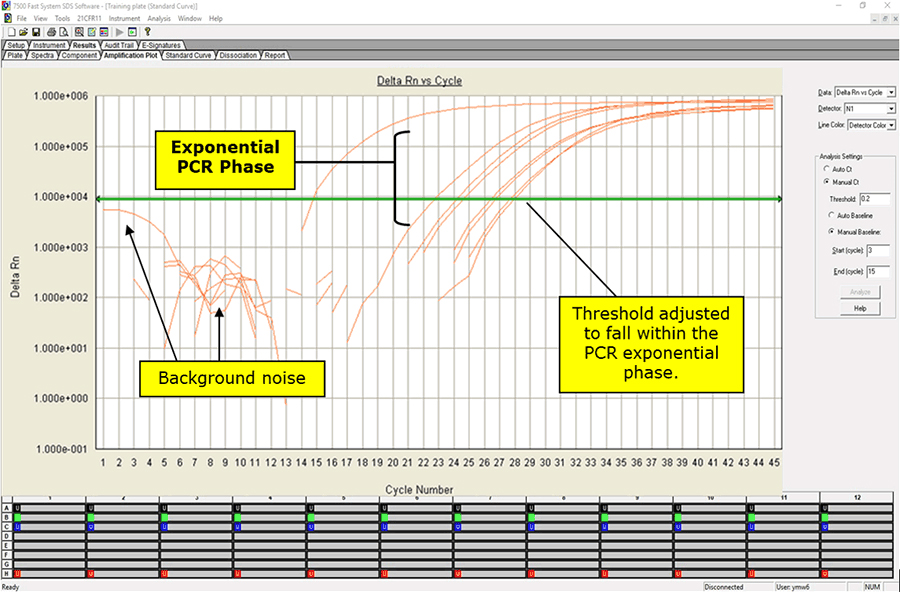
1. After completion of the run, save and analyze the data following the instrument manufacturer’s instructions. Analyses should be performed separately for each target using a manual threshold setting. Thresholds should be adjusted to fall within exponential phase of the fluorescence curves and above any background signal (refer to Figure 3). The procedure chosen for setting the threshold should be used consistently.
Figure 3. Amplification Plot Window
Interpreting Test Results
• NTCs should be negative and not exhibit fluorescence growth curves that cross the threshold line.
○ If a false positive occurs with one or more of the primer and probe NTC reactions, sample contamination may have occurred.
■ Invalidate the run and repeat the assay with stricter adherence to the procedure guidelines.
• PTC reaction should produce a positive result with an expected Ct value for each target included in the test.
○ If expected positive reactivity is not achieved, invalidate the run and repeat the assay with stricter adherence to procedure guidelines.
○ Determine the cause of failed PTC reactivity, implement corrective actions, and document results of the investigation and corrective actions.
○ Do not use PTC reagents that do not generate expected result.
• RP should be positive at or before 35 cycles for all clinical samples and HSC, thus indicating the presence of sufficient nucleic acid from human RNase P gene and that the specimen is of acceptable quality.
○ Failure to detect RNase P in HSC may indicate:
■ Improper assay set up and execution
■ Reagent or equipment malfunction
○ Detection of RNase P in HSC but failure to detect RNase P in any of the clinical samples may indicate:
■ Improper extraction of nucleic acid from clinical materials resulting in loss of nucleic acid or carry-over of PCR inhibitors from clinical specimens
■ Absence of sufficient human cellular material in sample to enable detection
• HSC should be negative for 2019-nCoV specific primer/probe sets.
○ If any 2019-nCoV specific primer/probes exhibit a growth curve that crosses the threshold line, interpret as follows:
■ Contamination of nucleic acid extraction reagents may have occurred. Invalidate the run and confirm reagent integrity of nucleic acid extraction reagents prior to further testing.
○ Cross contamination of samples occurred during nucleic acid extraction procedures or assay setup. Invalidate the run and repeat the assay with stricter adherence to procedure guidelines.
• When all controls exhibit the expected performance, a specimen is considered negative if all 2019-nCoV markers (N1, N2, N3) cycle threshold growth curves DO NOT cross the threshold AND the RNase P growth curve DOES cross the threshold line.
• When all controls exhibit the expected performance, a specimen is considered positive for 2019-nCoV if all markers (N1, N2, N3) cycle threshold growth curve crosses the threshold line. The RNase P may or may not be positive as described above, but the 2019-nCoV result is still valid.
• When all controls exhibit the expected performance and the growth curves for the 2019-nCoV markers (N1, N2, N3) AND the RNase P marker DO NOT cross the cycle threshold growth curve, the result is invalid. The extracted RNA from the specimen should be re-tested. If residual RNA is not available, re-extract RNA from residual specimen and re-test. If the re-tested sample is negative for all markers and all controls exhibit the expected performance, the result is “Invalid.”
• When all controls exhibit the expected performance and the cycle threshold growth curve for any one or two markers, (N1, N2, N3) but not all three crosses the threshold line the result is inconclusive for 2019-nCoV. Re-extract RNA from residual specimen and re-test.
2019-nCoV rRT-PCR Diagnostic Panel Results Interpretation
|
2019 nCoV_N1 |
2019 nCoV_N2 |
2019 nCoV_N3 |
RP |
Result Interpretationa |
|---|---|---|---|---|
|
+ |
+ |
+ |
± |
2019-nCoV detected |
|
If only one, or two, of three targets is positive |
± |
Inconclusive Result |
||
|
– |
– |
– |
+ |
2019-nCoV not detected |
|
– |
– |
– |
– |
Invalid Result |
• Analysts should be trained and familiar with testing procedures and interpretation of results prior to performing the assay.
• A false negative result may occur if inadequate numbers of organisms are present in the specimen due to improper collection, transport or handling.
• RNA viruses in particular show substantial genetic variability. Although efforts were made to design rRT-PCR assays to conserved regions of the viral genomes, variability resulting in mis-matches between the primers and probes and the target sequences can result in diminished assay performance and possible false negative results.
Suggestions and questions concerning this procedure may be sent to: respvirus@cdc.gov.
Research Use Only 2019-Novel Coronavirus (2019-nCoV) Real-time RT-PCR Primer and Probe Information
NOT FOR DIAGNOSTIC USE
Reagents manufactured from these sequences may not be used for diagnostic testing under FDA’s authorization of the CDC 2019-nCoV Real-Time RT-PCR Diagnostic Panel.
Only primers and probes labeled for EUA use and distributed by the International Reagent Resource may be used for diagnostic testing with the CDC 2019-nCoV Real-Time RT-PCR Diagnostic Panel.
DISCLAIMER
These sequences are intended to be used for the purposes of respiratory virus surveillance and research. The recipient agrees to use them in compliance with all applicable laws and regulations. Every effort has been made to assure the accuracy of the sequences, but CDC cannot provide any warranty regarding their accuracy. The recipient can acknowledge the source of sequences in any oral presentations or written publications concerning the research project by referring to the Division of Viral Diseases, National Center for Immunization and Respiratory Diseases, Centers for Disease Control and Prevention, Atlanta, GA, USA.
|
Name |
Description |
Oligonucleotide Sequence (5’>3’) |
Label1 |
Working Conc. |
|---|---|---|---|---|
|
2019-nCoV_N1-F |
2019-nCoV_N1 Forward Primer |
5’-GAC CCC AAA ATC AGC GAA AT-3’ |
None |
20 µM |
|
2019-nCoV_N1-R |
2019-nCoV_N1 Reverse Primer |
5’-TCT GGT TAC TGC CAG TTG AAT CTG-3’ |
None |
20 µM |
|
2019-nCoV_N1-P |
2019-nCoV_N1 Probe |
5’-FAM-ACC CCG CAT TAC GTT TGG TGG ACC-BHQ1-3’ |
FAM, BHQ-1 |
5 µM |
|
2019-nCoV_N2-F |
2019-nCoV_N2 Forward Primer |
5’-TTA CAA ACA TTG GCC GCA AA-3’ |
None |
20 µM |
|
2019-nCoV_N2-R |
2019-nCoV_N2 Reverse Primer |
5’-GCG CGA CAT TCC GAA GAA-3’ |
None |
20 µM |
|
2019-nCoV_N2-P |
2019-nCoV_N2 Probe |
5’-FAM-ACA ATT TGC CCC CAG CGC TTC AG-BHQ1-3’ |
FAM, BHQ-1 |
5 µM |
|
2019-nCoV_N3-F |
2019-nCoV_N3 Forward Primer |
5’-GGG AGC CTT GAA TAC ACC AAA A-3’ |
None |
20 µM |
|
2019-nCoV_N3-R |
2019-nCoV_N3 Reverse Primer |
5’-TGT AGC ACG ATT GCA GCA TTG-3’ |
None |
20 µM |
|
2019-nCoV_N3-P |
2019-nCoV_N3 Probe |
5’-FAM-AYC ACA TTG GCA CCC GCA ATC CTG-BHQ1-3’ |
FAM, BHQ-1 |
5 µM |
|
RP-F |
RNAse P Forward Primer |
5’-AGA TTT GGA CCT GCG AGC G-3’ |
None |
20 µM |
|
RP-R |
RNAse P Reverse Primer |
5’-GAG CGG CTG TCT CCA CAA GT-3’ |
None |
20 µM |
|
RP-P |
RNAse P Probe |
5’-FAM – TTC TGA CCT GAA GGC TCT GCG CG – BHQ-1-3’ |
FAM, BHQ-1 |
5 µM |
|
1 TaqMan® probes are labeled at the 5′-end with the reporter molecule 6-carboxyfluorescein (FAM) and with the quencher, Black Hole Quencher 1 (BHQ-1) (Biosearch Technologies, Inc., Novato, CA) at the 3′-end. Note: Oligonucleotide sequences are subject to future changes as the 2019-Novel Coronavirus evolves. |
||||