A mother brings her 12-month-old child, a new patient for your clinic, for a well-child visit. You immediately note the child to be small for her age. Her weight is below the 5th percentile on standardized growth curves (50th percentile for an 8-month-old), her length is at the 25th percentile, and her head circumference is at the 50th percentile. Her vital signs and her examination otherwise are normal.
![]() What is the next step in the management of this patient?
What is the next step in the management of this patient?
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the next step in the evaluation?
What is the next step in the evaluation?
ANSWERS TO CASE 1: Failure to Thrive
Summary: A 12-month-old girl has poor weight gain, but no etiology is suggested on examination.
• Next step: Gather more information, including birth, past medical, family, social, and developmental histories. A dietary history is especially important.
• Most likely diagnosis: Failure to thrive (FTT), most likely “nonorganic” in etiology.
• Next step in evaluation: Limited screening laboratory testing to identify organic causes of FTT, dietary counseling, and frequent office visits to assess weight gain.
1. Know the historical clues necessary to recognize organic and nonorganic FTT.
2. Understand the appropriate use of the laboratory in an otherwise healthy child with FTT.
3. Appreciate the treatment and follow-up of a child with nonorganic FTT.
This patient’s growth pattern (inadequate weight gain, potentially modest length retardation, and head circumference sparing) suggests FTT, most likely nonorganic given that the examination is normal. A nonorganic FTT diagnosis is made after organic etiologies are excluded, and, after adequate nutrition and an adequate environment is assured, growth resumes normally after catch-up growth is demonstrated. Diagnostic and therapeutic maneuvers aimed at organic causes are appropriate when supported by the history (prematurity, maternal infection) or examination (enlarged spleen, significant developmental delay). Although organic and nonorganic FTT can occur simultaneously, attempts to differentiate the two forms are helpful because the evaluation, treatment, and follow-up may be different.
Note: Had the same practitioner followed this patient since birth or had records from the previous health-care provider, earlier detection of FTT and its potential etiology might have occurred, thus allowing more rapid intervention. For instance, patients with poor caloric intake usually fail to gain weight but maintain length and head circumference. As nutrition remains poor, length becomes affected next and then ultimately head circumference.
Failure to Thrive
FAILURE TO THRIVE (FTT): A physical sign, not a final diagnosis. It is suspected when a child’s growth is below the 3rd or 5th percentile, in a child less than 6 months old who does not gain weight for 2 to 3 months, or in a child whose growth crosses more than two major growth percentiles in a short time frame. Usually seen in children younger than 5 years whose physical growth is significantly less than that of their peers.
NONORGANIC (PSYCHOSOCIAL) FTT: Poor growth without a medical etiology. Nonorganic FTT often is related to poverty or poor caregiver–child interaction. It constitutes one-third to one-half of FTT cases identified in tertiary care settings and nearly all cases in primary care settings.
ORGANIC FTT: Poor growth caused by an underlying medical condition, such as inflammatory bowel disease, renal disease, or congenital heart conditions.
The goals of the history, physical examination, and laboratory testing are to establish whether the child’s caregiver is supplying enough calories, whether the child is consuming enough calories, and whether the child is able to use the calories for growth. Identification of which factor is the likely source of the problem helps guide management.
The history and physical examination are the most important tools in an FTT evaluation. A dietary history can offer important clues to identify an etiology. The type of milk (breast or bottle) and frequency and quality of feeding, voiding, vomiting, and stooling should be recorded. The milk used (commercial or homemade formula) and the mixing process (to ensure appropriate dilution) should be reviewed (adding too much water to powdered formula results in inadequate nutrition). The amount and type of juices and solid foods should be noted for older children. Significant food aversions might suggest gastric distress of malabsorption. A 2-week food diary (the parent notes all foods offered and taken by the child) and any associated symptoms of sweating, choking, cyanosis, difficulty sucking, and the like can be useful.
Pregnancy and early neonatal histories may reveal maternal infection, depression, drug use, intrauterine growth retardation, prematurity, or other chronic neonatal conditions. When children suspected of having FTT are seen in families whose members are genetically small or with a slow growth history (constitutional delay), affected children are usually normal and do not require an exhaustive evaluation. In contrast, a family history of inheritable disease associated with poor growth (cystic fibrosis) should be evaluated more extensively. Because nonorganic FTT is more commonly associated with poverty, a social history is often useful. The child’s living arrangements, including primary and secondary caregivers, housing type, caregiver’s financial and employment status, the family’s social supports, and unusual stresses (such as spousal abuse) should be reviewed. While gathering the history, the clinician can observe for unusual caregiver–child interactions.
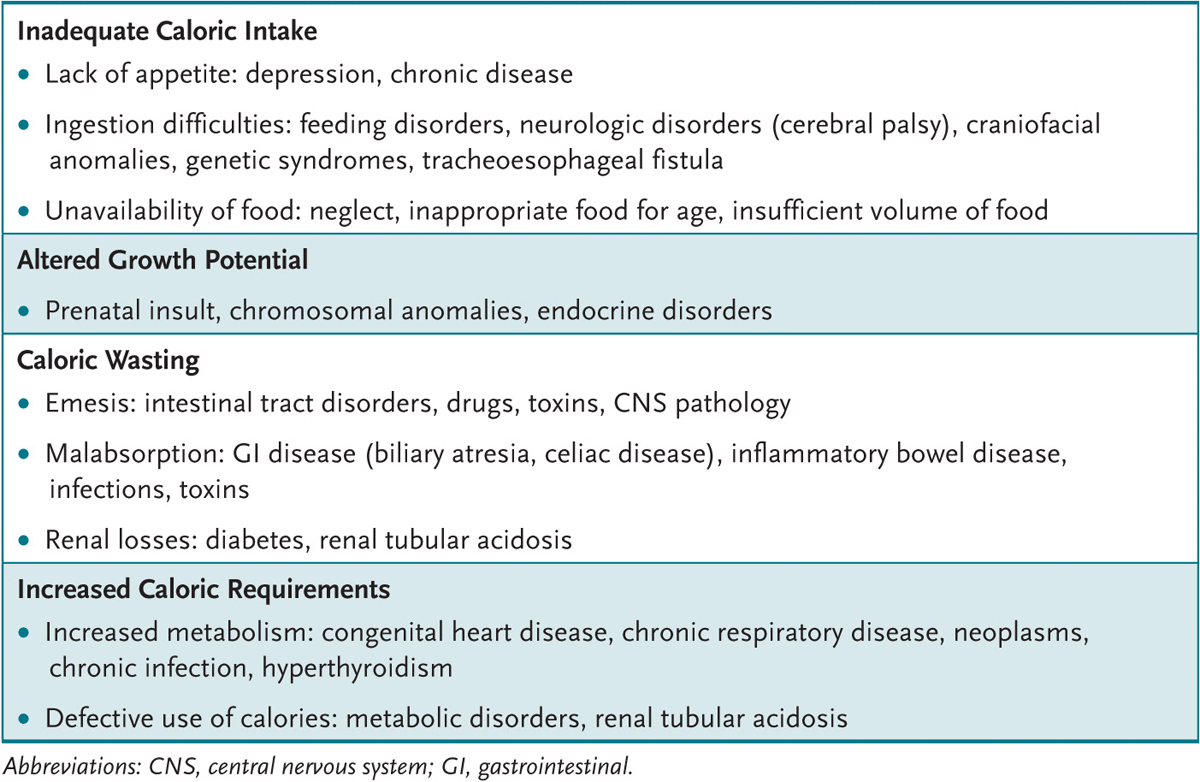
All body organ systems potentially harbor a cause for organic FTT (Table 1-1). The developmental status (possibly delayed in organic and nonorganic FTT) needs evaluation. Children with nonorganic FTT may demonstrate an occipital bald spot from lying in a bed and failure to attain appropriate developmental milestones resulting from lack of parental stimulation; may be disinterested in their environment; may avoid eye contact, smiling, or vocalization; and may not respond well to maternal attempts of comforting. Children with some types of organic FTT (renal tubular acidosis) and most nonorganic FTT show “catch-up” in developmental milestones with successful therapy. During the examination (especially of younger infants) the clinician can observe a feeding, which may give clues to maternal-child interaction bonding issues or to physical problems (cerebral palsy, oral motor or swallowing difficulties, velum cleft palate).
Table 1-1 • MAJOR CAUSES OF INADEQUATE WEIGHT GAIN
The history or examination suggestive of organic FTT directs the laboratory and radiologic evaluation. In most cases, results of the newborn state screen are critical. A child with cystic fibrosis in the family requires sweat chloride or genetic testing, especially if this testing is not included on the newborn state screen. A child with a loud, harsh systolic murmur and bounding pulses deserves a chest radiograph, an electrocardiogram (ECG), and perhaps an echocardiogram and cardiology consult.
Most FTT children have few or no signs. Thus, laboratory evaluation is usually limited to a few screening tests: a complete blood count (CBC), lead level (especially for patients in lower socioeconomic classes or in cities with a high lead prevalence), thyroid and liver function tests, urinalysis and culture, and serum electrolyte levels (including calcium, blood urea nitrogen [BUN], and creatinine). A tuberculosis skin test and human immunodeficiency virus testing may also be indicated. Abnormalities in screening tests are pursued more extensively.
The treatment and follow-up for organic FTT are disease specific. Patients with nonorganic FTT are managed with improved dietary intake, close follow-up, and attention to psychosocial issues.
Healthy infants in the first year of life require approximately 120 kcal/kg/d of nutrition and about 100 kcal/kg/d thereafter; FTT children require an additional 50% to 100% to ensure adequate catch-up growth. A mealtime routine is important. Families should eat together in a nondistracting environment (television off!), with meals lasting between 20 and 30 minutes. Solid foods are offered before liquids; children are not force-fed. Low-calorie drinks, juices, and water are limited; age-appropriate high-calorie foods (whole milk, cheese, dried fruits, peanut butter) are encouraged. Formulas containing more than the standard 20 cal/oz may be necessary for smaller children, and high-calorie supplementation (PediaSure or Ensure) may be required for larger children. Frequent office or home health visits are indicated to ensure weight gain. In some instances, hospitalization of an FTT child is required; such infants often have rapid weight gain, supporting the diagnosis of nonorganic FTT.
Nonorganic FTT treatment requires not only the provision of increased calories but also attention to contributing psychosocial issues. Referral to community services (Women, Infants, and Children [WIC] Program, Food Stamp Program, and local food banks) may be required. Caregiver help in the form of job training, substance and physical abuse prevention, parenting classes, and psychotherapy may be available through community programs. Older children and their families may benefit from early childhood intervention and Head Start programs.
Some children with organic FTT also have nonorganic FTT. For instance, a poorly growing special-needs premature infant is at increased risk for superimposed nonorganic FTT because of psychosocial issues, such as poor bonding with the family during a prolonged hospital stay. In such cases, care for the organic causes is coordinated with attempts to preclude nonorganic FTT.
1.1 Parents bring their 6-month-old son to see you. He is symmetrically less than the 5th percentile for height, weight, and head circumference on routine growth curves. He was born at 30 weeks’ gestation and weighed 1000 g. He was a planned pregnancy, and his mother’s prenatal course was uneventful until an automobile accident initiated the labor. He was ventilated for 3 days in the intensive care unit (ICU) but otherwise did well without ongoing problems. He was discharged at 8 weeks of life. Which of the following is the mostly likely explanation for his small size?
A. Chromosomal abnormality
B. Protein-calorie malnutrition
C. Normal ex-premie infant growth
D. Malabsorption secondary to short gut syndrome
E. Congenital hypothyroidism
1.2 A 13-month-old child is noted to be at the 25th percentile for weight, the 10th percentile for height, and less than the 5th percentile for head circumference. She was born at term. She was noted to have a small head at birth, to be developmentally delayed throughout her life, and to have required cataract surgery shortly after birth. She currently takes phenobarbital for seizures. Which of the following would most likely explain this child’s small size?
A. Congenital cytomegalovirus (CMV) infection
B. Down syndrome
C. Glycogen storage disease type II
D. Congenital hypothyroidism
E. Craniopharyngioma
1.3 A 2-year-old boy had been slightly less than the 50th percentile for weight, height, and head circumference, but in the last 6 months he has fallen to slightly less than the 25th percentile for weight. The pregnancy was normal, his development is as expected, and the family reports no psychosocial problems. The mother says that he is now a finicky eater (wants only macaroni and cheese at all meals), but she insists that he eat a variety of foods. The meals are marked by much frustration for everyone. His examination is normal. Which of the following is the best next step in his care?
A. Sweat chloride testing
B. Ophthalmologic examination for retinal hemorrhages
C. Reassurance and counseling for family about childhood normal developmental stage
D. Testing of stool for parasites
E. Magnetic resonance imaging (MRI) of the brain
1.4 A 4-month-old child has poor weight gain. Her current weight is less than the 5th percentile, height about the 10th percentile, and head circumference at the 50th percentile. The planned pregnancy resulted in a normal, spontaneous, vaginal delivery; mother and child were discharged after a 48-hour hospitalization. Feeding is via breast and bottle; the quantity seems sufficient. The child has had no illness. The examination is unremarkable except for the child’s small size. Screening laboratory shows the hemoglobin and hematocrit are 11 mg/dL and 33%, respectively, with a platelet count of 198,000/mm3. Serum electrolyte levels are sodium 140, chloride 105, potassium 3.5, bicarbonate 17, blood urea nitrogen 15, and creatinine 0.3. Liver function tests are normal. Urinalysis reveals a pH of 8 with occasional epithelial cells but no white blood cells, bacteria, protein, ketones, or reducing substances. Which of the following is the most appropriate therapy for this child?
A. Transfusion with packed red blood cells (PRBCs)
B. Intravenous (IV) infusion of potassium chloride
C. Sweat chloride analysis
D. Growth hormone determination
E. Oral supplementation with bicarbonate
1.1 C. The expected weight versus age must be modified for a preterm infant. Similarly, growth for children with Down or Turner syndrome varies from that for other children. Thus, use of an appropriate growth curve is paramount. For the child in the question, weight gain should follow or exceed that of term infants. For this premature infant, when his parameters are plotted on a “premie growth chart,” normal growth is revealed.
1.2 A. The developmental delay, intrauterine growth retardation (including microcephaly), cataracts, seizures, hepatosplenomegaly, prolonged neonatal jaundice, and purpura at birth are consistent with a congenital cytomegalovirus (CMV) or toxoplasmosis infection. Calcified brain densities of CMV typically are found in a periventricular pattern; in toxoplasmosis, they are found scattered throughout the cortex.
1.3 C. Between 18 and 30 months of age children often become “picky eaters.” Their growth rate can plateau, and the period can be distressing for families. Calm counseling of parents to provide nutrition, avoid “force-feeding,” and avoid providing snacks is usually effective. Close follow-up is required.
1.4 E. The patient has evidence of renal tubular acidosis (probably distal tubular), a well-described cause of FTT. Upon confirmation of the findings, oral bicarbonate supplementation would be expected to correct the elevated chloride level, the low bicarbonate and potassium levels (although potassium supplements may be required), and poor growth.
![]() In the United States, psychosocial failure to thrive is more common than organic failure to thrive; it often is associated with poverty or poor parent-child interaction.
In the United States, psychosocial failure to thrive is more common than organic failure to thrive; it often is associated with poverty or poor parent-child interaction.
![]() Inexpensive laboratory screening tests, dietary counseling, and close observation of weight changes are appropriate first steps for most healthy-appearing infants with failure to thrive.
Inexpensive laboratory screening tests, dietary counseling, and close observation of weight changes are appropriate first steps for most healthy-appearing infants with failure to thrive.
![]() Organic failure to thrive can be associated with abnormalities of any organ system. Clues in history, examination, or screening laboratory tests help identify affected organ systems.
Organic failure to thrive can be associated with abnormalities of any organ system. Clues in history, examination, or screening laboratory tests help identify affected organ systems.
![]() Up to one-third of patients with psychosocial failure to thrive have developmental delay as well as social and emotional problems.
Up to one-third of patients with psychosocial failure to thrive have developmental delay as well as social and emotional problems.
![]() Patients with renal tubular acidosis, a common cause of organic failure to thrive, can have proximal tubule defects (type 2) caused by impaired tubular bicarbonate reabsorption or distal tubule defects (type 1) caused by impaired hydrogen ion secretion. Type 4 is also a distal tubule problem associated with impaired ammoniagenesis.
Patients with renal tubular acidosis, a common cause of organic failure to thrive, can have proximal tubule defects (type 2) caused by impaired tubular bicarbonate reabsorption or distal tubule defects (type 1) caused by impaired hydrogen ion secretion. Type 4 is also a distal tubule problem associated with impaired ammoniagenesis.
Bunik M, Brayden RM, Fox D. Ambulatory & office pediatrics. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:238-239.
Chiang ML, Hill LL. Renal tubular acidosis. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1886-1892.
Chiesa A, Sirotnak AP. Child abuse & neglect. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:216-217.
Kirkland RT. Failure to thrive. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006: 900-906.
Lum GM. Kidney & urinary tract. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:690-692.
McLean HS, Price DT. Failure to thrive. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1147-1149.
McLeod R. Toxoplasmosis (Toxoplasma gondii). In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011: 1208-1216.
Noel RJ. Approach to the infant and child with feeding difficulty. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:117-123.
Raszka WV. Neonatal toxoplasmosis. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006: 530-532.
Sanchez PJ, Siegel JD. Cytomegalovirus. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:511-516.
Shaw JS, Palfrey JS. Health maintenance issues. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:27-34.
Sreedharan R, Avner ED. Renal tubular acidosis. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011: 1808-1811.
Stagno S. Cytomegalovirus. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1115-1117.
A healthy 16-year-old adolescent arrives at your office with his parents, who are concerned about his several months’ history of erratic behavior. At times he has a great deal more energy, seems to be in a terrific mood, and has unusually high self-esteem; during these episodes he has difficulty concentrating, remembering things, and often has headaches. At other times he seems to be his “normal” self. He had previously been a good student, but his grades have fallen this year. Last evening he appeared flushed and agitated, he had dilated pupils and a rapid heart rate, and he complained “people were out to get him.” The family reluctantly reports that he was arrested for burglary 2 weeks previously. You know him to be in otherwise good health. Today he appears normal.
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the next step in the evaluation?
What is the next step in the evaluation?
![]() What is the long-term evaluation and therapy?
What is the long-term evaluation and therapy?
ANSWERS TO CASE 2: Adolescent Substance Abuse
Summary: A 16-year-old previously healthy adolescent with recent behavior changes and declining school performance.
• Most likely diagnosis: Drug abuse (probably MDMA [ecstasy] or possibly cocaine or amphetamines).
• Next steps in evaluation: History, examination, urine drug screen, and screening for other commonly associated drug abuse consequences (sexually transmitted infections [STIs], hepatitis).
• Long-term evaluation and therapy: Threefold approach: (1) detoxification program, (2) follow-up with developmentally appropriate psychosocial support systems, and (3) possible long-term assistance with a professional trained in substance abuse management.
1. Learn the pattern of behavior found among drug-abusing adolescents.
2. Know the signs and symptoms of the drugs most commonly abused by adolescents.
3. Understand the general approach to therapy for an adolescent abusing drugs.
Rarely, a brain tumor could explain an adolescent with new onset of behavior changes. In general, however, an adolescent’s new-onset truant behavior, depression or euphoria, or declining grades is more commonly associated with substance abuse. A previously undiagnosed psychiatric history (mania or bipolar disease), too, must be considered. A history, family history, physical examination (especially the neurologic and psychological portions), and screening laboratory will help provide clarity. Information can come from the patient, his family, or other interested parties (teachers, coaches, and friends). Direct questioning of the adolescent alone about substance abuse is appropriate during routine health visits or when signs and symptoms are suggestive of abuse.
The Substance-Abusing Adolescent
SUBSTANCE ABUSE: Alcohol or other drug use leading to impairment or distress, causing failure of school or work obligations, physical harm, substance-related legal problems, or continued use despite social or interpersonal consequences resulting from the drug’s effects.
SUBSTANCE DEPENDENCE: Alcohol and other drug use, causing loss of control with continued use (tolerance requiring higher doses or withdrawal when terminated), compulsion to obtain and use the drug, and continued use despite persistent or recurrent negative consequences.
Experimentation with alcohol and other drugs is common among adolescents; some consider this experimentation “normal.” Others argue it is to be avoided because substance abuse is often a cause of adolescent morbidity and mortality (homicide, suicide, and unintentional injuries). In all cases, a health-care provider is responsible for discussing facts about alcohol and drugs in an attempt to reduce the adolescent’s risk of harm and for identifying those requiring intervention.
Children at risk for drug use include those with significant behavior problems, learning difficulties, and impaired family functioning. Cigarettes and alcohol are the most commonly used drugs; marijuana is the most commonly used illicit drug. Some adolescents abuse common household products (inhalation of glue or aerosols); others abuse a sibling’s medications (methylphenidate, which is often snorted with cocaine).
The American Academy of Pediatrics (AAP) recommends pediatricians ask about alcohol or drug use during the adolescent’s annual health examination or when an adolescent presents with evidence of substance abuse. Direct questions can identify drug or alcohol use and their effect on school performance, family relations, and peer interactions. Should problems be identified, an interview to determine the degree of drug use (experimentation, abuse, or dependency) is warranted.
Historical clues to drug abuse include significant behavioral changes at home, a decline in school or work performance, or involvement with the law. An increased incidence of intentional or accidental injuries may be alcohol or drug related. Risk-taking activities (trading sex for drugs, driving while impaired) can be particularly serious and may suggest serious drug problems. Alcohol or other drug users usually have a normal examination, especially if the use was not recent. Needle marks and nasal mucosal injuries are rarely found.
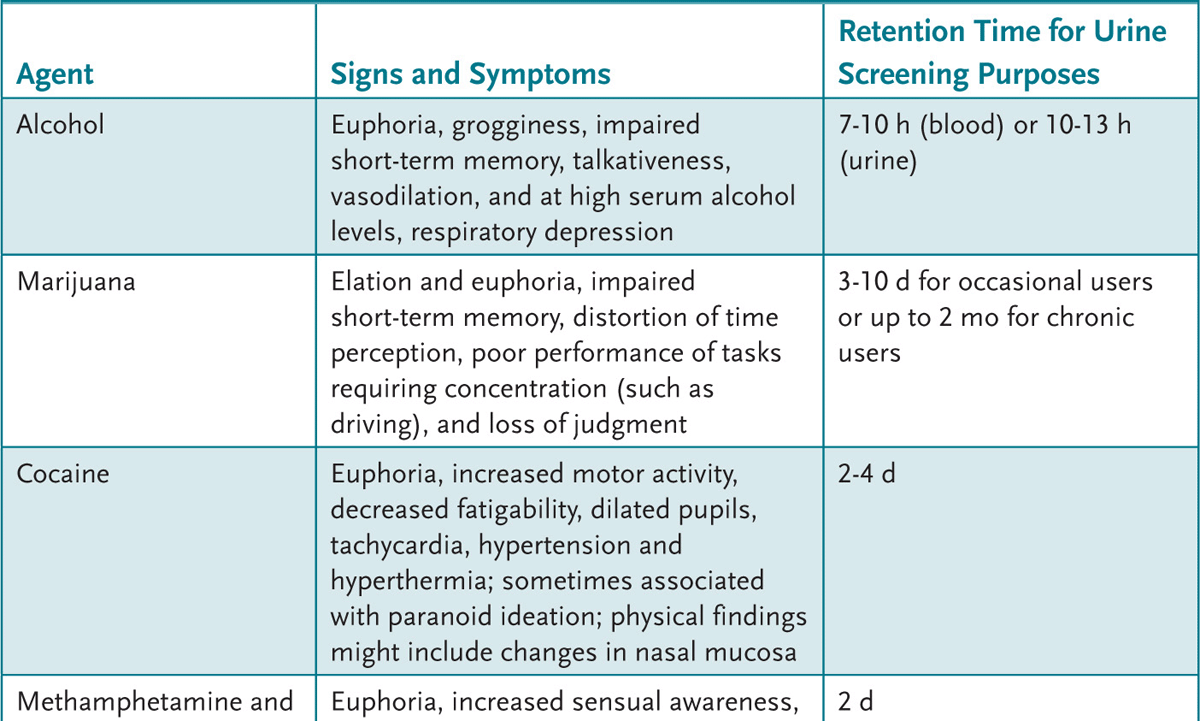
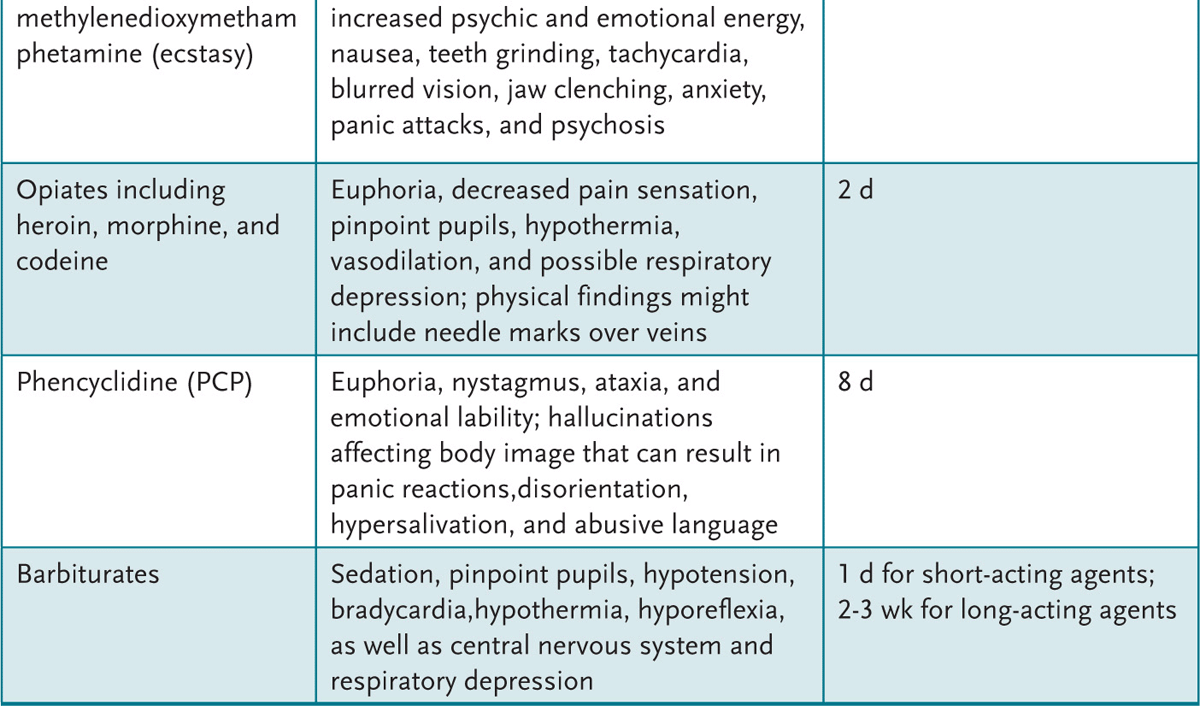
An adolescent with recent alcohol or drug use can present with a variety of findings (Table 2-1). A urine drug screen (UDS) can be helpful to evaluate the adolescent who: (1) presents with psychiatric symptoms, (2) has signs and symptoms commonly attributed to drugs or alcohol, (3) is in a serious accident, or (4) is part of a recovery monitoring program. An attempt to obtain the adolescent’s permission and maintain confidentiality is paramount.
Table 2-1 • CLINICAL FEATURES OF SUBSTANCE ABUSE
Treatment of life-threatening acute problems related to alcohol or drug use follows the ABCs of emergency care: manage the Airway, control Breathing, and assess the Circulation. Treatment then is directed at the offending agent (if known). After stabilization, a treatment plan is devised. For some, inpatient programs that disrupt drug use allow for continued outpatient therapy. For others, an intensive outpatient therapy program can be initiated to help develop a drug-free lifestyle. The expertise necessary to assist an adolescent through these changes is often beyond a general pediatrician’s expertise. Assistance with this chronic problem by qualified health professionals in a developmentally appropriate setting can maximize outcome. Primary care providers can, however, assist families to find suitable community resources.
2.1 A 14-year-old boy has ataxia. He is brought to the local emergency department, where he appears euphoric, emotionally labile, and a bit disoriented. He has nystagmus and hypersalivation. Many notice his abusive language. Which of the following agents is most likely responsible for his condition?
A. Alcohol
B. Amphetamines
C. Barbiturates
D. Cocaine
E. Phencyclidine (PCP)
2.2 Parents bring their 16-year-old daughter for a “well-child” checkup. She looks normal on examination. As part of your routine care you plan a urinalysis. The father pulls you aside and asks you to secretly run a urine drug screen (UDS) on his daughter. Which of the following is the most appropriate course of action?
A. Explore the reasons for the request with the parents and the adolescent, and perform a UDS with the adolescent’s permission if the history warrants.
B. Perform the UDS as requested, but have the family and the girl return for the results.
C. Perform the UDS in the manner requested.
D. Refer the adolescent to a psychiatrist for further evaluation.
E. Tell the family to bring the adolescent back for a UDS when she is exhibiting signs or symptoms such as euphoria or ataxia.
2.3 A previously healthy adolescent male has a 3-month history of increasing headaches, blurred vision, and personality changes. Previously he admitted to marijuana experimentation more than a year ago. On examination he is a healthy, athletic-appearing 17-year-old with decreased extraocular range of motion and left eye visual acuity. Which of the following is the best next step in his management?
A. Acetaminophen (APAP) and ophthalmology referral
B. Glucose measurement
C. Neuroimaging
D. Trial of methysergide (Sansert) for migraine
E. Urine drug screen
2.4 An 11-year-old girl has dizziness, pupillary dilatation, nausea, fever, tachycardia, and facial flushing. She says she can “see” sound and “hear” colors. Which of the following agents is responsible for this?
A. Alcohol
B. Amphetamines
C. Ecstasy
D. Lysergic acid diethylamide (LSD)
E. PCP
2.1 E. PCP is associated with hyperactivity, hallucinations, abusive language, and nystagmus.
2.2 A. The adolescent’s permission should be obtained before drug testing. Testing “secretly” in this situation destroys the doctor-patient relationship.
2.3 C. Despite previous drug experimentation, his current neurologic symptoms and physical findings make drug use a less likely etiology. Evaluation for possible brain tumor is warranted.
2.4 D. LSD is associated with symptoms that begin 30 to 60 minutes after ingestion, peak 2 to 4 hours later, and resolve by 10 to 12 hours, including delusional ideation, body distortion, and paranoia. “Bad trips” result in the user becoming terrified or panicked; treatment usually is reassurance of the user in a controlled, safe environment.
![]() Cigarettes and alcohol are the most commonly used drugs in adolescence.
Cigarettes and alcohol are the most commonly used drugs in adolescence.
![]() Marijuana is the most common illicit drug used in adolescence.
Marijuana is the most common illicit drug used in adolescence.
![]() Substance abuse behaviors include drug dealing, prostitution, burglary, unprotected sex, automobile accidents, and physical violence.
Substance abuse behaviors include drug dealing, prostitution, burglary, unprotected sex, automobile accidents, and physical violence.
![]() Children at risk for drug use include those with significant behavior problems, learning difficulties, and impaired family functioning.
Children at risk for drug use include those with significant behavior problems, learning difficulties, and impaired family functioning.
Crewe SN, Marcell AV. Substance use and abuse. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:278-282.
Heyman RB. Adolescent substance abuse and other high-risk behaviors. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:579-584.
Kaul P. Adolescent substance abuse. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:145-158.
Kulig JW. The Role of the pediatrician in prevention, identification, and management of substance abuse. Pediatrics. 2005:115; 816-821.
Stager MM. Substance abuse. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:671-685.
A 36-year-old woman with little prenatal care delivers a 3900 g girl. The infant has decreased tone, upslanting palpebral fissures, epicanthal folds, redundant nuchal skin, fifth finger clinodactyly and brachydactyly, and a single transverse palmar crease.
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the next step in the evaluation?
What is the next step in the evaluation?
ANSWERS TO CASE 3: Down Syndrome
Summary: A newborn with dysmorphic features is born to a woman of advanced maternal age.
• Most likely diagnosis: Down syndrome (trisomy 21).
• Next step in evaluation: Infant chromosomal evaluation to confirm diagnosis, evaluation for other features of the syndrome, counseling, and family support.
1. Know the physical features and problems associated with Down syndrome (DS) and other common trisomy conditions.
2. Understand the evaluation of a child with dysmorphic features consistent with DS.
3. Appreciate the counseling and support required by a family with a special-needs child.
This newborn has many DS features; confirmation is made with a chromosome evaluation. Upon identification of a child with possible DS, the health-care provider attempts to identify potentially life-threatening features, including cardiac or gastrointestinal (GI) anomalies. A thorough evaluation of the family’s psychosocial environment is warranted; these children can be physically, emotionally, and financially challenging.
Note: This woman of advanced maternal age had limited prenatal care but was at high risk for pregnancy complications. Adequate care may have included a serum triple screen between the 15th and 20th weeks of pregnancy or a “genetic” ultrasound, which could have demonstrated a DS pattern. Further evaluation (amniocentesis for chromosomal analysis) then could have been offered.
APPROACH TO:
The Dysmorphic Child
ADVANCED MATERNAL AGE: The incidence of DS increases each year beyond the age of 35 years. At 35 years, the incidence is 1 in 378 live born infants, increasing to 1 in 106 by the age of 40 and to 1 in 11 by the age of 49 years.
BRACHYDACTYLY: Excessive shortening of hand and foot tubular bones resulting in a boxlike appearance.
CLINODACTYLY: Incurving of one of the digits (in DS the fifth digit curves toward the fourth digit due to midphalanx dysplasia).
DYSMORPHIC CHILD: A child with problems of generalized growth or body structure formation. These children can have a syndrome (a constellation of features from a common cause; ie, DS features caused by extra chromosome 21 material), an association (two or more features of unknown cause occurring together more commonly than expected; ie, VATER [Vertebral problems, Anal anomalies, Trachea problems, Esophageal abnormalities, and Radius or renal anomalies]), or a sequence (a single defect that leads to subsequent abnormalities; ie, Potter disease’s lack of normal infant kidney function, causing reduced urine output, oligohydramnios, and constraint deformities; common facial features include wide-set eyes, flattened palpebral fissures, prominent epicanthus, flattened nasal bridge, mandibular micrognathia, and large, low-set, cartilage-deficient ears).
SERUM TRISOMY SCREENING: Measurements of α-fetoprotein (AFP), human chorionic gonadotropin (hCG), inhibin A, and estriol levels, usually performed at 15 to 20 weeks’ gestation. These tests screen for a variety of genetic problems. Approximately 75% of DS babies and 80% to 90% of babies with neural tube defects will be identified by this testing.
The first newborn evaluation occurs in the delivery room where attempts are made to successfully transition the infant from an intrauterine to an extrauterine environment; it focuses primarily on the ABCs of medicine—Airway, Breathing, and Circulation. The infant is then evaluated for possible abnormalities, including those that might fit into a pattern such as DS.
The prenatal history and course provide some important clues in the evaluation of a dysmorphic child. The parents’ age (increased chromosomal abnormalities with increased maternal and sometimes paternal age), degree of fetal movement, maternal drug or teratogen exposure, family history of dysmorphia, and prenatal testing results, including triple screening and chorioamnionic or chorionic villus testing, may prove helpful. For instance, an older mother with a low AFP on her triple screen is at higher risk for having a DS child.
The physical examination is critical to the diagnosis of a dysmorphic child. For DS, a distinctive pattern can lead to a presumptive diagnosis; more than 90% of such children have features, including upslanting palpebral fissures, Brushfield spots (white or grey spots in the periphery of the iris), flat facial profile, small and rounded ears, excess nuchal skin, widespread nipples, pelvic dysplasia, joint hyperflexibility, fifth finger clinodactyly, a single transverse palmar (simian) crease, hypotonia, and a poor Moro reflex. Other features include brachycephaly (disproportionate shortness of the head), epicanthal folds, brachydactyly, wide spacing between first and second toes, and short stature.
In newborns with suspected DS, at least two potentially life-threatening conditions must be addressed. Approximately 50% of DS infants have cardiac defects—most commonly an endocardial cushion defect (60%), ventricular septal defect (VSD, 32%), and tetralogy of Fallot (6%). A cardiology consultation and echocardiogram usually are indicated. Approximately 12% of DS infants have intestinal (usually duodenal) atresia, some presenting with a history of polyhydramnios. All DS infants have hypotonia and sometimes slower feeding. Should an infant with presumed DS develop persistent vomiting after feeds (especially if bilious), an upper GI study likely will reveal the characteristic “double-bubble” pattern of duodenal atresia; surgical intervention is warranted.
Confirmation of DS requires chromosomal analysis. A complete, extra chromosome 21 (nondisjunction, ie, failure to segregate during meiosis) occurs in almost 95% of cases. Two percent of cases are caused by translocations (breakage and removal of a large DNA segment from one chromosome and attachment to a different one), and 3% are mosaics (more than one cell type; usually described as an abnormal cell percentage). Parents of a child with translocation-caused DS are evaluated for chromosomal aberrations; the recurrence risk can approach 100% in some cases.
Other newborn conditions associated with DS include hearing loss, strabismus, cataracts, nystagmus, and congenital hypothyroidism. Hearing is evaluated by the age of 3 months. An ophthalmologist evaluates the eyes by the age of 6 months, and thyroid function is assessed as part of the routine newborn screening program. Longer-term DS consequences include obesity, a higher leukemia risk, acquired hypothyroidism, atlantoaxial (cervical spine) instability, and premature aging with an increased risk of Alzheimer disease. All DS children are mentally retarded, but the intelligence quotients vary widely (mosaics can exhibit near-normal intelligence).
“Well-child care” takes on special meaning for DS children. In addition to providing routine care based on the American Academy of Pediatrics (AAP) guidelines for health supervision that apply to all children, the AAP has promulgated DS-specific guidelines (see www.aap.org). Periodic objective thyroid, hearing, and vision screenings are focal points of concern. Equally important in successful DS management is appropriate psychosocial intervention. Proper home or environmental, educational, and vocational interventions can improve the DS child’s functioning level, facilitating his or her transition to adulthood. Providing family support and assisting with financial and medical support program applications are within the pediatrician’s realm.
3.1 A small-for-gestational age infant is born to a 35-year-old woman. He has low-set and malformed ears, microcephaly, rocker-bottom feet, inguinal hernias, cleft lip and palate, and micrognathia. Chromosomal analysis is likely to reveal which of the following?
A. Down syndrome (trisomy 21)
B. Edwards syndrome (trisomy 18)
C. Holt-Oram syndrome
D. Patau syndrome (trisomy 13)
E. Turner syndrome
3.2 A 15-day-old infant has respiratory distress. A quick observation suggests she has slight cyanosis, hepatosplenomegaly, and features consistent with DS. The cardiac examination demonstrates a loud first heart sound, a wide and fixed split second heart sound, a low-pitched, mid-diastolic murmur at the lower left sternal border, and a harsh apical holosystolic murmur in the mitral area. An echocardiogram is likely to demonstrate which of the following?
A. Complete atrioventricular (AV) canal (endocardial cushion defect)
B. Hypoplastic left heart
C. Total anomalous venous return
D. Transposition of the great vessels
E. Tricuspid atresia
3.3 A small-for-gestational age, dysmorphic newborn infant has microcephaly and sloping forehead, cutis aplasia (missing portion of the skin and hair) of the scalp, polydactyly, microphthalmia, and omphalocele. Which of the following is the most likely diagnosis?
A. Down syndrome (trisomy 21)
B. Edwards syndrome (trisomy 18)
C. Holt-Oram syndrome
D. Patau syndrome (trisomy 13)
E. Turner syndrome
3.4 The parents of an 8-year-old DS boy arrive for his annual well-child visit. He wants to participate in sports, including the Special Olympics. Until further evaluation can be completed, which of the following sports would you suggest as being safe?
A. Diving
B. Football
C. Tennis
D. Tumbling
E. Wrestling
3.1 B. The child has trisomy 18. Other features include clenched hands with overlapping digits, small palpebral fissures, prominent occiput, short sternum, and cardiac defects (ventricular septal defect [VSD], atrial septal defect [ASD], patent ductus arteriosus [PDA], or coarctation of the aorta).
3.2 A. Although VSDs are common in DS, the most characteristic lesion is endocardial cushion defect (or atrioventricular [AV] canal defect). Slight cyanosis occurs because of the mixing of deoxygenated with oxygenated blood. In the AV canal, a range of defects involving the atrial septum, the ventricular septum, and one or both of the AV valves can be seen. A complete AV canal includes ASDs and VSDs with a common AV valve. A partial AV canal includes defects of the atrial septum and separate mitral and tricuspid valve orifices.
3.3 D. The appearance of cutis aplasia and polydactyly suggests trisomy 13. Other common features include holoprosencephaly (failure of growth of the forebrain), cleft lip or palate, postaxial polydactyly, flexed and overlapping fingers, coloboma, and cardiac defects (VSD, ASD, PDA, dextrocardia).
3.4 C. Until lateral cervical flexion–extension films confirm normal anatomy, contact sports and other activities that may result in forceful flexion of the neck should be avoided.
![]() Down syndrome is the most common autosomal chromosome abnormality in live born infants, increasing in incidence with advanced maternal age.
Down syndrome is the most common autosomal chromosome abnormality in live born infants, increasing in incidence with advanced maternal age.
![]() The most common neonatal Down syndrome features are hypotonia with poor Moro reflex, flat faces, slanted palpebral fissures, laxity of joints, and excessive skin on the back of the neck.
The most common neonatal Down syndrome features are hypotonia with poor Moro reflex, flat faces, slanted palpebral fissures, laxity of joints, and excessive skin on the back of the neck.
![]() Common problems associated with Down syndrome include cardiac defects and duodenal atresia.
Common problems associated with Down syndrome include cardiac defects and duodenal atresia.
![]() Common features of trisomy 18 (Edwards) syndrome include weak cry, single umbilical artery, micrognathia with small mouth and high arched palate, clenched hand with overlapping of index finger over the third finger, simian crease, rocker-bottom feet, small pelvis, and short sternum.
Common features of trisomy 18 (Edwards) syndrome include weak cry, single umbilical artery, micrognathia with small mouth and high arched palate, clenched hand with overlapping of index finger over the third finger, simian crease, rocker-bottom feet, small pelvis, and short sternum.
![]() Common features of trisomy 13 (Patau) syndrome include microcephaly and sloping forehead, deafness, scalp cutis aplasia, microphthalmia, coloboma, cardiac defect (especially ventricular septal defect), omphalocele, single umbilical artery, and hypersensitivity to agents containing atropine and pilocarpine.
Common features of trisomy 13 (Patau) syndrome include microcephaly and sloping forehead, deafness, scalp cutis aplasia, microphthalmia, coloboma, cardiac defect (especially ventricular septal defect), omphalocele, single umbilical artery, and hypersensitivity to agents containing atropine and pilocarpine.
American Academy of Pediatrics. Health supervision for children with Down syndrome. Pediatrics. 2001: 107; 442-449.
Bacino CA, Lee B. Cytogenetics. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:394-415.
Bernstein D. Atrioventricular septal defects (ostium primum and atrioventricular canal or endocardial cushion defects). In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1554-1556.
Carey JC. Chromosome disorders. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:691-697.
Lewanda AF, Boyadjiev SA, Jabs EW. Dysmorphology: genetic syndromes and associations. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:2629-2630.
South ST, Carey JC. Human cytogenetics. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:688-691.
Sponseller PD. Cervical spine. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:2491.
Tsai AC-H, Manchester DK, Elias ER. Genetics & dysmorphology. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:1037-1038.
Vick GW, Bezoild LI. Defects of the atrial septum, including the atrioventricular canal. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1565-1574.
An 8-year-old boy presents to your clinic with a 3-day history of a “white coating” in his mouth. He denies having a sore throat, upper respiratory infection symptoms, gastrointestinal distress, change in appetite, or fever. His immunizations are current, he has no significant past medical history, and he has been developing normally per his mother. His weight, however, has fallen from the 25th percentile to the 5th percentile, and he has been hospitalized on three occasions in the last year with pneumonia or dehydration. His family history is remarkable only for maternal hepatitis C infection related to past intravenous (IV) drug use. The patient is afebrile today, but his examination is notable for severe gingivitis, bilateral cervical and axillary lymphadenopathy, exudates on his buccal mucosa, and hepatomegaly.
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the next step in evaluation?
What is the next step in evaluation?
ANSWERS TO CASE 4: Immunodeficiency
Summary: A child with lymphadenopathy, organomegaly, weight loss, recurring infection, and oral lesions consistent with candidiasis.
• Most likely diagnosis: Immunodeficiency.
• Next step in evaluation: Gather additional history, including birth history, details of hospitalizations, dietary history, and patient and family histories of recurring or atypical infection. Consider testing for human immunodeficiency virus type 1 (HIV) and obtaining a complete blood count and comprehensive metabolic panel to assess cell counts, organ function, and nutritional status.
1. Differentiate between primary and secondary immunodeficiency.
2. Understand selected etiologies of pediatric immunodeficiency.
3. Identify and manage pediatric HIV disease.
Recurring infections in this patient presenting with oral lesions, weight loss, and lymphadenopathy are concerning for immune system dysfunction. He may have a primary immunodeficiency due to an inheritable defect or an acquired (secondary) immunodeficiency related to HIV infection, malignancy, malnutrition, or other disorder. The maternal history of IV drug use makes pediatric HIV infection a strong likelihood, probably due to vertical transmission. Additional patient and family histories and selected initial laboratory tests will aid in diagnosis and help guide management.
APPROACH TO:
The Child with Immunodeficiency
HIV DNA POLYMERASE CHAIN REACTION (PCR): Primary assay to diagnose HIV infection in children under 18 months of age; detects HIV DNA in white blood cells; sensitivity and specificity greater than 95%; definitive exclusion of HIV with two negative assays after 1 month of age, assuming other immunologic studies are negative.
HIV ANTIBODY ELISA: Enzyme-linked immunosorbent assay (ELISA) screening for HIV immunoglobulin G (IgG); initially detectable 2 weeks to 6 months after exposure; sensitivity and specificity greater than 99%; false-positive rate less than 5 in 100,000 assays; false-negative results may occur after immunization or in hepatic disease, autoimmune disease, or advanced acquired immunodeficiency syndrome (AIDS).
WESTERN BLOT: Direct visualization of antibodies to virion proteins; can be used to confirm screening antibody assay; results can be indeterminate and require repeat testing.
CD4 (T HELPER) CELL: Essential for humoral (B-cell) and cellular (T-cell) immunity; binds to antigens presented by B cells, prompting antibody production, and to antigens presented by phagocytes, prompting lymphokine release; rendered dysfunctional in HIV infection.
Evaluation of patients with recurring or atypical infection starts with a comprehensive history and systems review. Clinicians should inquire about perinatal history, growth and development, and past illnesses. Immunosuppression is suggested by failure to thrive (FTT) or atypical or difficult-to-eradicate infections (recurring otitis refractory to multiple antimicrobials). Family history includes parental health concerns (unexplained weight loss, growth failure, or developmental delay in siblings) and recurring or atypical infection in immediate family members. A focused physical examination should then be performed to identify signs consistent with immunosuppression (wasting, generalized lymphadenopathy, and organomegaly).
Primary (syndromic) immunodeficiency is due to a genetic defect, either inherited or related to gene mutation; most are humoral in origin or characterized by both humoral and cellular dysfunction (severe combined immunodeficiency). Other primary immunodeficiencies include phagocytic cell deficiency (chronic granulomatous disease due to defective macrophages) and complement deficiency (autoimmune disease or serious bacterial infection due to C2 deficiency). Patients with secondary immunodeficiency have normal immune function at birth, but subsequently develop an illness or metabolic abnormality that disrupts immune cell production or function. Conditions adversely affecting a patient’s immune status include HIV infection, diabetes mellitus, malnutrition, hepatic disease, autoimmune disease (scleroderma), aging, and stress.
HIV is a global epidemic, with over 30 million people presumably infected worldwide. Unprotected sexual intercourse and needle sharing with IV drug use are known means of transmission. Prior to the mid-1980s, blood transfusion was also a risk factor. In the pediatric population, HIV is typically acquired through vertical transmission. Approximately 80% of pediatric cases involve intrapartum transfer, but HIV can also be acquired from infected secretions at delivery and from breast milk. It is important to know the HIV status of the pregnant female, so that antiretroviral therapy can be administered during pregnancy to decrease viral replication and diminish the potential for transfer to the neonate. An infected mother has a 25% chance of transmitting the virus to her newborn if antiretroviral therapy is not received during pregnancy. Zidovudine, when started by the mother during the second trimester and given to the baby through the age of 6 weeks, reduces the risk of HIV transmission to less than 10%.
HIV infection gives rise to dysfunctional CD4 cells resulting in overall immune system compromise and eventual opportunistic infection. Approximately 75% of pediatric patients who acquire HIV vertically follow a course similar to adults, with an extended period of disease inactivity; a patient will often remain asymptomatic for a decade or more until the CD4 count falls to a critical level. The remainder of patients progress rapidly during the first several months of life. Therefore, early determination of maternal HIV status and measures to decrease transmission are critical (avoiding breast-feeding, aggressive and appropriate neonatal HIV testing, early antiretroviral therapy).
Verification of HIV infection is made in the patient older than 18 months by performing an HIV antibody ELISA and subsequent Western blot for confirmation. Because of placental transfer of maternal antibodies, diagnosis in younger patients is made by HIV DNA PCR testing. Two assays are performed on separate occasions to confirm the diagnosis. Subsequently, HIV RNA activity, CD4 cell count, and clinical findings are used to determine disease status. Centers for Disease Control and Prevention (CDC) classification of HIV status is based on the presence and severity of signs or symptoms and degree of immunosuppression. For example, a patient with Pneumocystis jiroveci (carinii) pneumonia (PCP), an AIDS-defining opportunistic infection, is classified “severe” disease (category C). Degree of immunosuppression is based on an age-adjusted CD4 count. For the patient in this case, a normal CD4 count would be more than or equal to 500 or 25%. Severe suppression is denoted by a CD4 count less than 200 or 15%.
Neonates born to HIV-positive women are tested at birth and at selected intervals through approximately 6 months of age. Traditionally, the exposed neonate receives 6 weeks of antiretroviral therapy in the form of zidovudine starting in the first few hours of life. PCP prophylaxis in the form of trimethoprim (TMP)-sulfamethoxazole (SMX) commences at approximately 6 weeks of age for HIV-positive infants. CD4 levels are followed in quarterly intervals in the patient who becomes HIV-positive. HIV RNA activity is followed and typically correlates with disease progression; RNA activity of more than 100,000 copies/mL has been associated with advanced progression and early death.
Treatment for HIV-positive patients is started early to diminish viral replication before mutation and antiretroviral resistance occur. The three major classes of anti-retrovirals are nucleoside reverse transcriptase inhibitors (didanosine, stavudine, zidovudine),nonnucleoside reverse transcriptase inhibitors (efavirenz, nevirapine), and protease inhibitors (indinavir, nelfinavir). Combination retroviral therapy in children has led to a marked decline in child mortality. Common adverse effects for all include headache, emesis, abdominal pain, and diarrhea. Osteopenia and drug rash can also be seen. Possible other abnormalities include anemia, neutropenia, elevated transaminases, hyperglycemia, and hyperlipidemia.
The current pediatric antiretroviral therapy recommendation consists of three drugs: two nucleoside reverse transcriptase inhibitors and one protease inhibitor. An existing treatment regimen is altered when toxicity becomes an issue or disease progression occurs. Ultimately, HIV treatment requires a multidisciplinary approach with input from nutritionists, social workers, and pediatric HIV and mental health specialists. In addition to periodic monitoring of viral activity and prophylaxis against opportunistic infection, close monitoring of growth, development, and emotional health is important in pediatric HIV disease management. Immunizations should be kept current, with all vaccines administered per the recommended pediatric schedule, excluding live vaccines such as measles-mumps-rubella (MMR) and varicella for symptomatic HIV-infected children with a CD4 count less than 15%.
4.1 A 15-year-old adolescent girl has a 1-month history of urinary frequency without dysuria and the complaint of a recent onset of an itchy rash beneath both breasts. She has been gaining weight over the past year and regularly complains of fatigue. She is a febrile with a weight greater than the 99th percentile and has an erythematous, macular rash beneath both breasts characterized by satellite lesions. Urinalysis is significant for 2+ glucosuria, but no pyuria. Which of the following is the best next step in your evaluation?
A. HIV RNA level
B. Hemoglobin A1c
C. CD4 cell count
D. Herpes simplex virus-1 IgG
E. Thyroid stimulating hormone
4.2 A mother notes her 6-week-old son’s umbilical cord is still attached. His activity and intake are normal; he has had no illness or fever. Delivery was at term without problems. His examination is notable for a cord without evidence of separation and a shallow, 0.5-cm ulceration at the occiput without discharge or surrounding erythema. Mother declares that the “sore,” caused by a scalp probe, has been slowly healing since birth and was deemed unremarkable at his 2-week checkup. Which of the following is consistent with this child’s likely diagnosis?
A. Defective humoral response
B. Functional leukocyte adherence glycoproteins
C. Marked neutrophilia
D. Normal wound healing
E. Purulent abscess formation
4.3 A 6-month-old girl is seen after an emergency room visit for decreased intake, emesis, and watery diarrhea for the past 3 days. She was diagnosed yesterday with “stomach flu” and given IV fluids. She is doing better today with improved intake and resolution of her emesis and diarrhea. The father is concerned about her thrush since birth (despite multiple courses of an oral antifungal), and that she has been hospitalized twice for pneumonia over the past 4 months. Her weight has dropped from the 50th percentile on her 4-month visit to the 5th percentile today. She has no findings consistent with dehydration, but she does appear to have some extremity muscle wasting. Her examination is remarkable for buccal mucosal exudates and hyperactive bowel sounds. Vital signs and the remainder of her examination are normal. You suspect severe combined immunodeficiency (SCID). Which of the following is consistent with the diagnosis?
A. Autosomal dominant inheritance
B. Persistent lymphocytosis
C. Defective cellular immunity
D. Normal vaccine immune response
E. No curative therapy
4.4 You are called urgently to examine a term, 2-hour-old newborn with temperature instability, difficulty with feeding, and a suspected seizure. He has atypical facies (wide-set eyes, a prominent nose, and a small mandible), a cleft palate, and a holosystolic murmur. A chest radiograph reveals a boot-shaped heart. Which of the following is consistent with this infant’s likely diagnosis?
A. Hypercalcemia
B. Chromosomal duplication
C. Parathyroid hyperplasia
D. Hypophosphatemia
E. Thymic aplasia
4.1 B. The obese adolescent in this case has findings of diabetes mellitus. An elevated hemoglobin A1c (glycosylated hemoglobin) is a good diagnostic tool for diabetes. This patient’s cutaneous candidiasis is likely an indication of secondary immunosuppression related to hyperglycemia. In diabetes, hyperglycemia promotes neutrophil dysfunction, and circulatory insufficiency contributes to ineffective neutrophil chemotaxis during infection. HIV infection is possible and testing might be reasonable, but this scenario is most consistent with hyperglycemia.
4.2 C. You suspect leukocyte adhesion deficiency (LAD) as the etiology of this child’s problem. LAD is an inheritable disorder of leukocyte chemotaxis and adherence characterized by recurring sinopulmonary, oropharyngeal, and cutaneous infections with delayed wound healing. Neutrophilia is common with WBC counts typically more than 50,000 cells/mm3. Severe, life-threatening infection is possible with Staphylococcus species, Enterobacteriaceae, and Candida species. Good skin and oral hygiene are important; broad-spectrum antimicrobials and surgical debridement are early considerations with infection.
4.3 C. Severe combined immunodeficiency (SCID) is an autosomal recessive or X-linked disorder of both humoral and cellular immunity. Serum immunoglobulins and T cells are often markedly diminished or absent. Thymic dysgenesis is also seen. Recurring cutaneous, gastrointestinal, or pulmonary infections occur with opportunistic organisms such as cytomegalovirus (CMV) and Pneumocystis pneumonia (PCP). Death typically occurs in the first 12 to 24 months of life unless bone marrow transplantation is performed.
4.4 E. The child in the question has typical features of DiGeorge syndrome, caused by a 22q11 microdeletion. This syndromic immunodeficiency is characterized by decreased T-cell production and recurring infection. Findings include characteristic facies and velocardiofacial defects such as ventricular septal defect and tetralogy of Fallot. Thymic or parathyroid dysgenesis can occur, accompanied by hypocalcemia and seizures. Developmental and speech delay are common in older patients.
![]() Primary immunodeficiency is an inheritable disorder characterized by weakened immunity and recurring, serious infection early in life.
Primary immunodeficiency is an inheritable disorder characterized by weakened immunity and recurring, serious infection early in life.
![]() A variety of illnesses can provoke secondary immunodeficiency; malignancy, malnutrition, hepatic disease, and HIV infection are known to adversely influence both humoral and cellular immunity.
A variety of illnesses can provoke secondary immunodeficiency; malignancy, malnutrition, hepatic disease, and HIV infection are known to adversely influence both humoral and cellular immunity.
![]() Pediatric HIV disease can be deterred by appropriate testing and treatment of pregnant females and judicious antiretroviral prophylaxis in the exposed neonate. Exposed patients should be closely followed by clinicians and a team approach used in the management of active disease.
Pediatric HIV disease can be deterred by appropriate testing and treatment of pregnant females and judicious antiretroviral prophylaxis in the exposed neonate. Exposed patients should be closely followed by clinicians and a team approach used in the management of active disease.
American Academy of Pediatrics. Human immunodeficiency virus infection. In: Pickering LK, ed. 2009 Red Book: Report of the Committee on Infectious Diseases. 28th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2009:380-400.
Borkowsky W. Acquired immunodeficiency syndrome and human immunodeficiency virus. In: Katz SL, Hotez PJ, Gerson AA, eds. Krugman’s Infectious Diseases of Children. 11th ed. Philadelphia, PA: Mosby; 2004:1-26.
Buckley RH. Evaluation of suspected immunodeficiency. In: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:715-722.
Church JA. Human immunodeficiency virus infection. In: Osborn LM, DeWitt TG, First LR, Zenel JA, eds. Pediatrics. 1st ed. Philadelphia, PA: Elsevier-Mosby; 2005:1132-1139.
Yogev R, Chadwick EG. Acquired immunodeficiency syndrome (human immunodeficiency virus). In: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1157-1177.
A 13-year-old boy arrives for routine care. His mother reports that he seems to be much more immature and insecure than her older son was at the same age. His school performance is below average, and this year he has begun to receive special education for language-based classes. On physical examination you note that he is at the 95th percentile for height-age, his extremities are longer than expected, and he is embarrassed by his gynecomastia. His physical examination shows that he has Tanner stage 1 sexual development with small gonads.
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the best test to diagnose this condition?
What is the best test to diagnose this condition?
ANSWERS TO CASE 5: Klinefelter Syndrome
Summary: A tall, immature, and insecure 13-year-old boy with hypogonadism, long limbs, gynecomastia, and developmental delay.
• Most likely diagnosis: Klinefelter syndrome, a trisomy syndrome of nondisjunction affecting approximately 1 in 600 to 800 male infants.
• Best diagnostic test: Chromosomal analysis.
1. Understand the signs and symptoms of Klinefelter syndrome.
2. Appreciate the variety of causes of childhood mental retardation (MR).
3. Learn the signs and symptoms of syndromes involving missing or duplicate sex chromosomes.
This child’s mother has identified this adolescent’s development and behavior to be different from her other children. The school recently has identified his need for special education, especially in the language-based classes. A thorough history (including all school performance and behavioral problems) and physical examination can provide diagnostic clues. The etiology of his condition impacts his psychosocial outcome, his future medical therapy, and his parents, family planning decisions.
APPROACH TO:
Klinefelter Syndrome
KLINEFELTER SYNDROME: A specific syndrome associated with behavioral problems (immaturity, insecurity), developmental delay (speech, language, lower IQ), and physical findings (gynecomastia, hypogonadism, long limbs) caused by an extra X chromosome in boys and men.
MENTAL RETARDATION (MR): A clinically and socially important impairment of measured intelligence and adaptive behavior that is diagnosed before 18 years of age.
Causes of MR include preconceptual and early embryonic disruptions (teratogens, chromosomal abnormalities, placental dysfunction, congenital central nervous system [CNS] malformations); fetal brain insults (infections, toxins, placental problems); perinatal difficulties (prematurity, metabolic disorders, infections); postnatal brain injuries (infections, trauma, metabolic disorders, toxins, poor nutrition); and miscellaneous postnatal family difficulties (poverty, poor caregiver-child interaction, parental mental illness). A category of “unknown etiology” includes children with MR who do not fit into the above categories.
The history of a child with possible MR includes an evaluation of the child’s psychosocial skills and a review of school reports. The ultimate diagnosis may require formal testing to determine if the IQ falls below some set point, such as 80. A determination of whether formal testing should be performed is based on physical examination findings, developmental and school histories, and concerns of the family and teachers. Males with Klinefelter syndrome often have developmental delay, especially in verbal cognitive areas where they underachieve in reading, spelling, and mathematics; their full IQ may be normal, but their verbal IQ usually is somewhat decreased. In variants with multiple X chromosomes, the incidence and severity of MR increases. Boys with Klinefelter syndrome often go unidentified until puberty because of the subtleness of the clinical findings. The diagnosis should be considered for all boys (regardless of age) who have been identified as having mental retardation, or psychosocial, school, or adjustment problems.
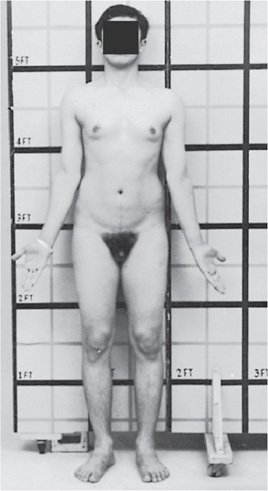
Physical findings to be considered in patients with nonspecific MR include the size of the occiput, unusual hair color or distribution, distinctive eyes, malformed ears or nose, and abnormalities in jaw size, mouth shape, or palate height. The hands and feet may have short metacarpals or metatarsals, overlapping or supernumerary digits, and abnormal creases or nails. The skin may have café au lait spots or depigmented nevi, and the genitalia may be abnormally sized or ambiguous. Patients with MR caused by Klinefelter syndrome typically are tall, slim, and thin with long extremities (see Figure 5-1). Their testes and sometimes the phallus are small for age, but these latter findings may not become apparent until puberty. As adults, males with Klinefelter syndrome develop gynecomastia, sparse facial hair, and azoospermia. The incidence of breast cancer (as well as some hematologic cancers) is elevated in Klinefelter syndrome.
Figure 5-1. Klinefelter syndrome (XXY) in a 20-year-old man. Note relatively increased lower/upper body segment ratio, gynecomastia, small penis, and sparse body hair with a female pubic hair pattern. (Reproduced, with permission, from Gardner DG, Shoback D. Greenspan’s Basic & Clinical Endocrinology, 9th ed., New York: McGraw-Hill, 2011. Figure 12-7.)
Laboratory testing of a child with MR is based on the clinical findings and developmental milestones. A chromosomal analysis is often included in the evaluation of a child with mental retardation; for Klinefelter syndrome such an analysis will demonstrate the extra X-chromosome material. Other MR testing may include urine and serum amino and organic acids, serum levels of various compounds including ammonia, lead, zinc, and copper, and serum titers for congenital infections. Radiologic evaluation may include cranial computed tomography (CT), magnetic resonance imaging (MRI), or electroencephalogram (EEG).
Management of children with MR includes specialized educational services, early childhood interventions, social services, vocational training, and psychiatric interventions. Further interventions for children with specific underlying etiologies may include diet modification, genetic counseling, or reviewing the natural disease course with the family.
5.1 An institutionalized male juvenile delinquent upon close examination has severe nodulocystic acne, mild pectus excavatum, large teeth, prominent glabella, and relatively long face and fingers. His family says he has poor fine motor skills (such as penmanship), an explosive temper, and a low–normal IQ. What is the most likely diagnosis?
A. Fragile X syndrome
B. Klinefelter syndrome (XXY)
C. Turner syndrome (XO)
D. XXX syndrome
E. XYY male
5.2 A tall, thin 14-year-old adolescent male has no signs of puberty. He was delayed in his speech development and always has done less well in school than his siblings. He is shy, and teachers report that his activity is immature. Physical examination reveals breast development and long limbs with a decreased upper segment–lower segment ratio. He has small testes and phallus. What is the most likely diagnosis?
A. Fragile X syndrome
B. Klinefelter syndrome (XXY)
C. Turner syndrome (XO)
D. XXX syndrome
E. XYY male
5.3 A 15-year-old adolescent girl with primary amenorrhea is noted to be well below the fifth percentile for height. She has hypertension, a low posterior hairline, prominent and low-set ears, and excessive nuchal skin. What is the most likely diagnosis?
A. Fragile X syndrome
B. Klinefelter syndrome (XXY)
C. Turner syndrome (XO)
D. XXX syndrome
E. XYY phenotypic female
5.4 A 7-year-old boy with MR was born at home at 26 weeks’ gestation to a 28-year-old mother who had received no prenatal care. An evaluation is likely to suggest his MR is related to which of the following?
A. Brain tumor
B. Chromosomal aberration
C. Complications of prematurity
D. Congenital infection with cytomegalovirus
E. Elevated serum lead levels
5.1 E. XYY-affected males often have explosive tempers. Other findings include long and asymmetrical ears, increased length versus breadth for the hands, feet, and cranium, and mild pectus excavatum. By the age of 5 to 6 years, they tend to be taller than their peers and begin displaying aggressive or defiant behavior.
5.2 B. With Klinefelter syndrome, testosterone replacement allows for more normal adolescent male development, although azoospermia is the rule; the breast cancer incidence approaches that of women.
5.3 C. Turner syndrome also includes widely spaced nipples and broad chest; cubitus valgus (increased carrying angle of arms); edema of the hands and feet in the newborn period; congenital heart disease (coarctation of the aorta or bicuspid aortic valve); horseshoe kidney; short fourth metacarpal and metatarsal; hypothyroidism; and decreased hearing. Mental development usually is normal.
5.4 C. Prematurity, especially when earlier than 28 weeks’ gestation, is associated with complications (such as intraventricular hemorrhage) that can result in developmental delay and low IQ.
![]() Males with Klinefelter syndrome (XXY) have mild mental delay, eunuchoid habitus, gynecomastia, long arms and legs, and hypogonadism.
Males with Klinefelter syndrome (XXY) have mild mental delay, eunuchoid habitus, gynecomastia, long arms and legs, and hypogonadism.
![]() XYY males have explosive (often antisocial) behavior, weakness with poor fine motor control, accelerated growth in mid-childhood, large teeth, prominent glabella and asymmetrical ears, and severe acne at puberty.
XYY males have explosive (often antisocial) behavior, weakness with poor fine motor control, accelerated growth in mid-childhood, large teeth, prominent glabella and asymmetrical ears, and severe acne at puberty.
![]() Girls with Turner syndrome (45, XO) have short stature, amenorrhea, excessive nuchal skin, low posterior hairline, broad chests with widely spaced nipples, cubitus valgus, and coarctation of the aorta. Hypertension is common, possibly due to renal abnormalities (horseshoe kidney).
Girls with Turner syndrome (45, XO) have short stature, amenorrhea, excessive nuchal skin, low posterior hairline, broad chests with widely spaced nipples, cubitus valgus, and coarctation of the aorta. Hypertension is common, possibly due to renal abnormalities (horseshoe kidney).
![]() Fragile X syndrome, the most common form of inherited mental retardation, is seen primarily in boys and can be diagnosed in patients with mental retardation (particularly boys) who have macrocephaly, long face, high arched palate, large ears, and macroorchidism after puberty.
Fragile X syndrome, the most common form of inherited mental retardation, is seen primarily in boys and can be diagnosed in patients with mental retardation (particularly boys) who have macrocephaly, long face, high arched palate, large ears, and macroorchidism after puberty.
Accardo PJ, Accardo JA, Capute AJ. Mental retardation. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:608-614.
Ali O, Donohoue PA. Hypofunction of the testes. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1943-1951.
American Academy of Pediatrics: Committee on Genetics. Health supervision for children with fragile X syndrome. Pediatrics. 2011:127; 994-1006.
Bacino CA, Lee B. Cytogenetics. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:394-415.
Carey JC. Chromosome disorders. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:691-697.
Goldson E, Reynolds A. Child development & behavior. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:64-103.
Lewanda AF, Boyadjiev SA, Jaabs EW. Dysmorphology: genetic syndromes and associations. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:2629-2670.
Shapiro BK, Batshaw ML. Intellectual disability. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:122-129.
South ST Carey JC. Human cytogenetics. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:688-691.
Tsai AC-H, Manchester DK, Elias ER. Genetics & dysmorphology. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:1038-1039.
A 6-month-old child arrives for a well-child examination. His family recently moved to the United States from Turkey. His medical and family histories are unremarkable except that his sole source of nutrition is goat’s milk. He appears to be healthy on examination.
![]() What hematologic problem is most likely to develop?
What hematologic problem is most likely to develop?
![]() What nonhematologic concerns are considered in an infant fed on goat’s milk?
What nonhematologic concerns are considered in an infant fed on goat’s milk?
ANSWERS TO CASE 6: Megaloblastic Anemia
Summary: This is a 6-month-old child exclusively fed on goat’s milk.
• Likely complication: Megaloblastic anemia from folate or B12 deficiency.
• Other concerns: Brucellosis if milk is unpasteurized.
1. Appreciate the benefits of breast-feeding.
2. Know the nutritional supplements recommended for breast-feeding mothers.
3. Understand the special needs of infants and toddlers fed on goat’s milk or vegan diets.
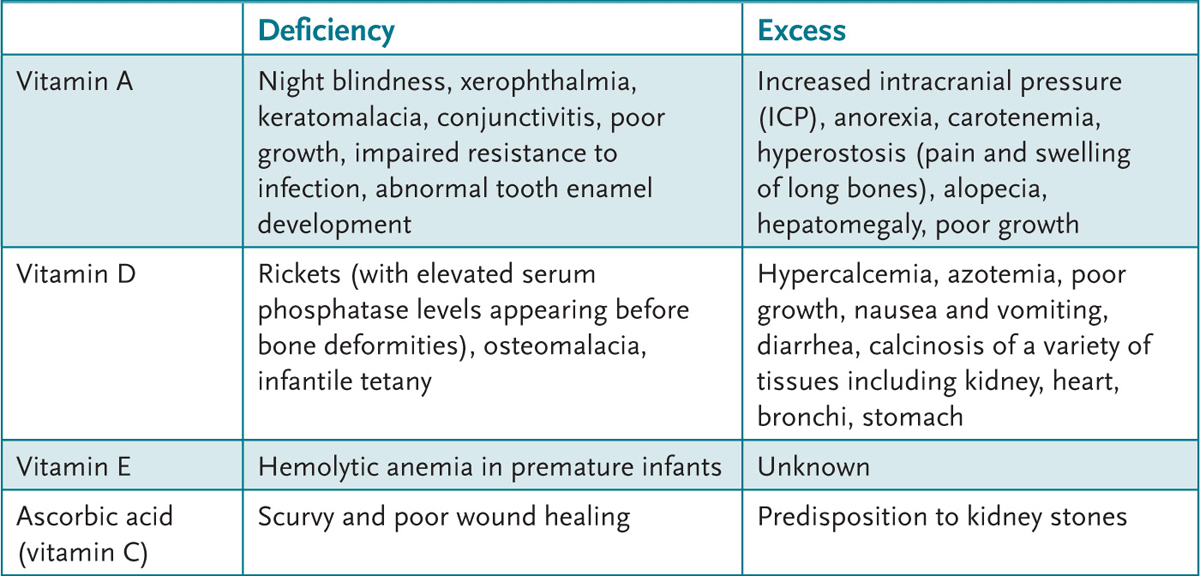
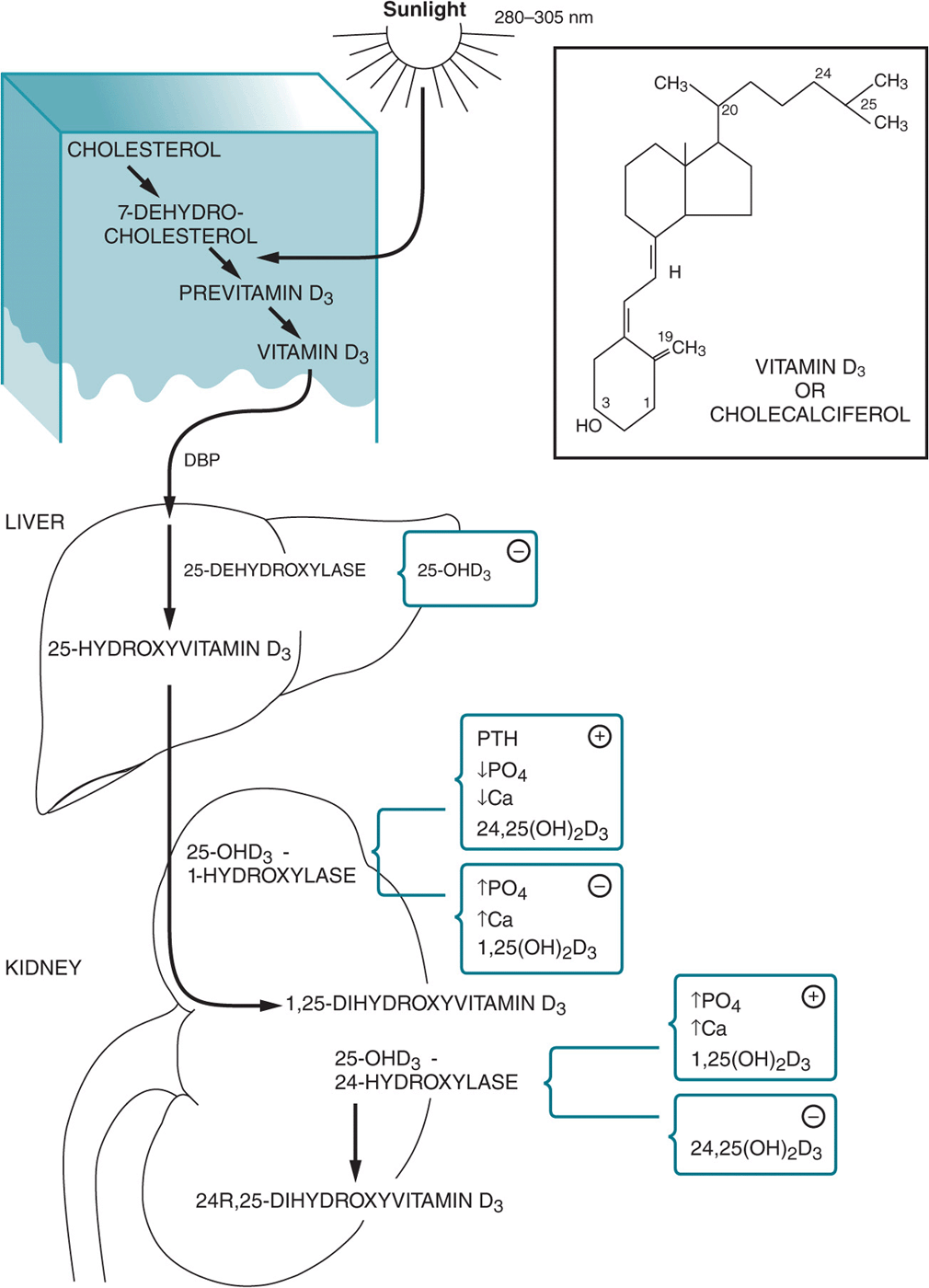
4. Appreciate the clinical syndromes resulting from vitamin excesses and deficiencies.
A variety of feeding regimens exist for infants and toddlers—breast-feeding, goat’s milk, other types of nonformula milk, and commercial or handmade foods. Health-care providers can educate parents about the benefits and potential dangers of various diet choices.
APPROACH TO:
Infant Nutrition
LACTOVEGETARIAN: Diet devoid of animal products but includes milk.
OMNIVORE: Diet includes both animal and vegetable products.
OVOVEGETARIAN: Diet devoid of animal products but includes eggs.
VEGAN: Vegetarian diet devoid of all animal products.
Infant formulas containing goat’s milk are not routinely available in the United States, but they are available elsewhere. Goat’s milk has lower sodium levels but more potassium, chloride, linoleic acid, and arachidonic acid than does cow’s milk. It is low in vitamin D, iron, folate, and vitamin B12; infants receiving goat’s milk as a primary nutrition source are given folate and vitamin B12 (to prevent megaloblastic anemia) and iron (to prevent iron deficiency anemia). Goat’s milk is boiled before ingestion; goats are particularly susceptible to brucellosis.
Breast milk is considered the ideal human infant food because it contains complete nutrition (with the possible exception of vitamin D and fluoride); iron levels are low but highly bioavailable and do not require supplementation until 4 to 6 months of age. In addition, it has antimicrobial properties and offers psychological advantages to mothers and infants. In developing countries, it is associated with lower infant morbidity and mortality, not only due to a reduction in diarrhea associated with contaminated water used in formula preparation but also because it contains high concentrations of immunoglobulin A (IgA), which reduces viruses and bacteria intestinal wall adherence, and macrophages, which inhibit Escherichia coli growth. Disadvantages include potential HIV (and other virus) transmission, occasional jaundice exacerbation due to increased unconjugated bilirubinemia levels (resolved with a 12- to 24-hour breast-feeding interruption), and its association with low vitamin K levels, contributing to hemorrhagic disease of the newborn (prevented by vitamin K administration at birth).
Formula feeding is substituted for breast-feeding for a variety of reasons. Commercial formula manufacturers strive to provide products similar to human milk. Infant growth rates with cow’s milk formula are similar to those in infants receiving breast milk. Improved sterilization procedures and refrigeration in developed and developing countries have reduced to some degree the gastrointestinal (GI) infections noted with formula feedings.
Formulas are available for special-needs infants. Infants with phenylketonuria require formulas low in phenylalanine, and those unable to digest protein require nitrogen in the form of amino acid mixtures.
Vegan diets supply all necessary nutrients if a variety of vegetables is selected. Some evidence suggests that high-fiber vegetarian diets lead to faster gastrointestinal transit time, resulting in reduced serum cholesterol levels, less diverticulitis, and a lower appendicitis incidence. Breast-feeding vegan mothers are given vitamin B12 to prevent the infant’s developing methylmalonic acidemia (an amino acid metabolism disorder involving a defect in the conversion of methylmalonyl-coenzyme A [CoA] to succinyl-CoA); patients can present with failure to thrive, seizure, encephalopathy, stroke, or other neurologic manifestations. Toddlers on a vegan diet are given vitamin B12 and, because of the high fiber content and rapid gastrointestinal transit time, are given trace minerals that can be depleted.
Vitamin deficiencies and excesses can result in a variety of clinical syndromes. Although rare, these syndromes usually can be averted with appropriate nutrition (Table 6-1).
Table 6-1 • EFFECTS OF VITAMIN AND MINERAL DEFICIENCY OR EXCESS
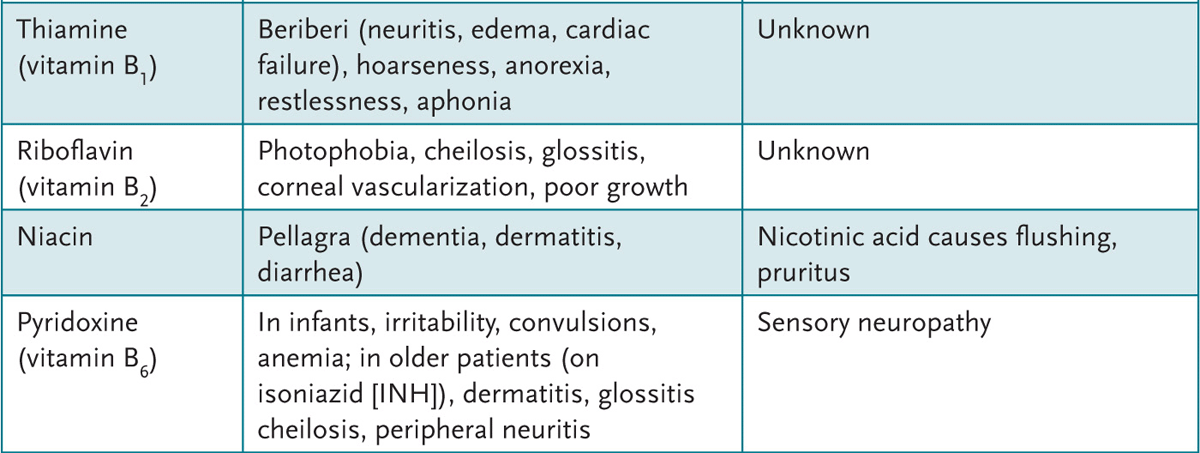
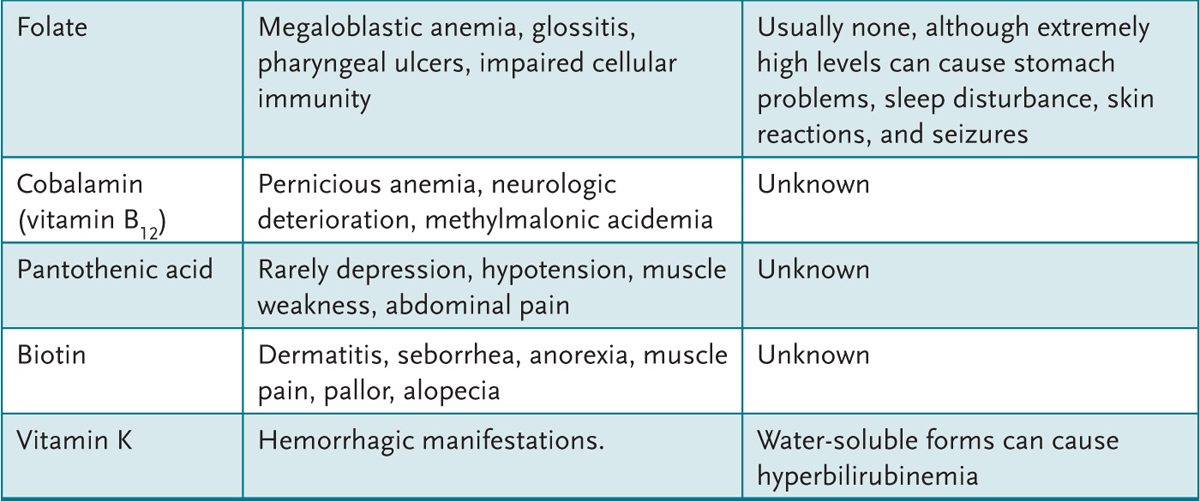
6.1 A 2-day-old infant has significant nasal and rectal bleeding. The pregnancy was without complications; he was delivered by a midwife at home. His Apgar scores were 9 at 1 minute and 9 at 5 minutes. He has breast-fed well and has not required a health-care professional visit since birth. Which of the following vitamin deficiencies might explain his condition?
A. Vitamin A
B. Vitamin B1
C. Vitamin C
D. Vitamin D
E. Vitamin K
6.2 A 6-month-old infant has been growing poorly. His parents have changed his formula three times without success. His examination is remarkable for a pale, emaciated child with little subcutaneous fat and anterior fontanelle fullness. His laboratory test results are notable for a hemolytic anemia and prolonged bleeding times. Which of the following is the most appropriate next step?
A. Gather urine for pH and electrolytes.
B. Measure serum factor IX levels.
C. Measure serum immunoglobulins.
D. Obtain a sweat chloride concentration.
E. Perform a hemoglobin electrophoresis.
6.3 An exclusively breast-fed infant with poor routine care is switched at 6 months of age to whole milk and table foods. Screening laboratories at 9 months of age demonstrate the hemoglobin and hematocrit to be 8 mg/dL and 25%, respectively, and the lead level to be less than 2 µg/dL. A follow-up complete blood count (CBC) 2 weeks later shows the hemoglobin to be at 7.8 mg/dL, the hematocrit 25%, the mean corpuscular volume (MCV) 62%, the platelet count to be 750,000/mm3, and a reticulocyte count of 1%. Which of the following would be the next step in the management of this child?
A. Order a hemoglobin electrophoresis.
B. Obtain a bone marrow aspiration.
C. Initiate iron supplementation.
D. Refer to a pediatric hematologist.
E. Initiate soybean-based formula.
6.4 A 3-week-old is admitted for failure to thrive, diarrhea, and a septic appearance. He does well on intravenous fluids; when begun on routine infant formula with iron, his symptoms return. It is Saturday and the state health department laboratory is closed. You should begin feeds with which of the following?
A. Amino acid-based formula (Nutramigen or Pregestimil)
B. Low-phenylalanine formula (Lofenalac or Phenex-1)
C. Low-iron, routine infant formula (Similac with low iron or Enfamil with low iron)
D. Low-isoleucine, low-leucine, low-valine infant formula (Ketonex-1 or MSUD 1)
E. Soy-based formula (Isomil or ProSobee)
6.1 E. Newborn infants have a relative vitamin K deficiency, especially if they are breast-fed; most infants are given vitamin K at birth to prevent deficiency-related bleeding complications.
6.2 D. The patient appears to have failure to thrive, with deficiencies of vitamin K (bleeding problems), vitamin A (fontanelle fullness), and vitamin E (hemolytic anemia). Cystic fibrosis (associated with vitamin malabsorption) would explain the condition.
6.3 C. The child in the question most likely did not get iron (or vitamin D) supplementation in the first 6 months of life while exclusively breast-feeding, and was switched to whole milk (low in iron) and to table foods (not supplemented with iron as are baby foods) at too young an age. All of the laboratory data are consistent with iron deficiency anemia; iron supplementation in this child with a resultant brisk erythrocyte response is both diagnostic and therapeutic. Failure of the child to respond to the iron therapy would require further evaluation.
6.4 E. This patient appears to have galactosemia; uridyl transferase deficiency is the cause, and the condition results in features of jaundice, hepatosplenomegaly, vomiting, hypoglycemia, seizures, lethargy, irritability, poor feeding and failure to thrive, aminoaciduria, liver failure, mental retardation, and an increased risk of E coli sepsis. Children with galactosemia are managed with a lactose-free formula. The low-phenylalanine formulas are for infants with phenylketonuria; low-iron formulas serve no purpose other than causing iron deficiency anemia; the low-isoleucine, low-leucine, low-valine infant formulas are useful for patients with maple syrup urine disease (MSUD); and the amino acid-based formulas are excellent for children with malabsorption syndromes.
![]() Breast-feeding is associated with lower morbidity and mortality (especially in developing countries) mostly because of a reduction in enteric pathogens and diarrhea associated with contaminated water used in formula preparation.
Breast-feeding is associated with lower morbidity and mortality (especially in developing countries) mostly because of a reduction in enteric pathogens and diarrhea associated with contaminated water used in formula preparation.
![]() Breast-feeding provides all the nutrients necessary for infant growth with the possible exceptions of vitamin D and fluoride, which usually are supplemented.
Breast-feeding provides all the nutrients necessary for infant growth with the possible exceptions of vitamin D and fluoride, which usually are supplemented.
![]() A breast-feeding vegan should supplement her infant’s or toddler’s diet with vitamin B 12 to prevent methylmalonic acidemia and trace minerals.
A breast-feeding vegan should supplement her infant’s or toddler’s diet with vitamin B 12 to prevent methylmalonic acidemia and trace minerals.
American Academy of Pediatrics. Section on breastfeeding. Breastfeeding and the use of human milk. Pediatrics. 2005:115; 496-506.
Baker RD, Greer FR. Diagnosis and prevention of iron deficiency and iron-deficiency anemia in infants and young children (0-3 years of age). Pediatrics. 2010:126; 1040-1050.
Egan M. Cystic fibrosis. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1481-1497.
Federico MJ, Kerby GS, Deterding RR, et al. Cystic fibrosis. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:501-502.
Finberg L. Feeding the healthy child. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:109-118.
Greenbaum LA. Rickets and hypervitaminosis D. In: Kleigman RM, Stanton BF, St. Geme JW, Schnor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:200-209.
Greenbaum LA. Vitamin E deficiency. In: Kleigman RM, Stanton BF, St. Geme JW, Schnor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:209-211.
Kirby M. Infant formula and complementary foods. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:99-105.
Kishnani PS, Chen Y-T. Defects in galactose metabolism. In: Kleigman RM, Stanton BF, St. Geme JW, Schnor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:502-503.
Krebs NF, Primak LE, Haemer M. Normal childhood nutrition & its disorders. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:277-278.
Lerner NB, Sills R. Iron-deficiency anemia. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1655-1658.
Martin PL. Nutritional anemias. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1692-1696.
Orenstein DM. Cystic fibrosis. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:1977-1986.
Rosenstein BJ. Cystic fibrosis. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1425-1438.
Sachdev HPS, Shah D. Vitamin B complex deficiency and excess. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:191-198.
Shah D, Sachdev HPS. Vitamin C (ascorbic acid). In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:198-200.
Stettler N, Bhatia J, Parish A, Stallings V. The feeding of healthy infants, children, and adolescents. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:160-170.
Suchy FJ. Disorders of carbohydrate metabolism. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:1503-1504.
Wappner RS. Disorders of carbohydrate metabolism. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:2181-2192.
Zile M. Vitamin A deficiencies and excess. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:188-191.
An 8-month-old child has a 24-hour history of increased crying when she moves her right leg. She has a prominent bulge over the mid-right thigh where she had received an immunization the previous day. She has not had fever or change in appetite, and she seems upset only when the leg is disturbed. The child underwent a failed Kasai procedure for biliary atresia and is awaiting a liver transplant. A radiograph of the leg demonstrates a mid-shaft fracture and poor mineralization.
![]() What is the mechanism for this condition?
What is the mechanism for this condition?
![]() What are the best diagnostic tests to diagnose this condition?
What are the best diagnostic tests to diagnose this condition?
Summary: An 8-month-old child with a chronic medical condition, including biliary atresia, poor bone mineralization, and a fracture.
• Mechanism: Malabsorption of vitamin D (among other fat-soluble vitamins) due to lack of intestinal secretion of bile salts, resulting in rickets.
• Best diagnostic tests: Serum 25(OH)D, calcium, phosphorus, and alkaline phosphatase levels. Radiographs demonstrate poor bone mineralization.
1. Become familiar with the clinical presentation of rickets.
2. Understand the pathophysiology behind nutritional and nonnutritional rickets.
3. Appreciate some of the other causes of childhood fractures.
This child has biliary atresia and underwent a failed Kasai procedure. Metabolic aberrations are expected while this child awaits liver transplantation. A review of her medications and compliance in receiving them is warranted. Because of the brittle nature of her bones, her leg was fractured while receiving immunizations.
APPROACH TO:
The Child with Possible Rickets
BILIARY ATRESIA: A congenital condition affecting approximately 1 in 16,000 live births in which the liver’s bile ducts become blocked and fibrotic, resulting in reduced bile flow into the bowel.
GENU VALGUM: “Knock” knees.
GENU VARUM: “Bowed” legs.
KASAI PROCEDURE: An operative procedure in which a bowel loop forms a duct to allow bile to drain from a liver with biliary atresia.
RICKETS: Poor mineralization of growing bone or of osteoid tissue.
A patient with liver failure has poor bile salt secretion, resulting in poor fat-soluble vitamin absorption, including vitamin D. The poor vitamin D absorption causes low serum 25(OH)D, occasionally reduced serum calcium levels, markedly elevated serum alkaline phosphatase, poor bone mineralization, and an increased risk of fractures. Children with liver failure and ascites are treated with loop diuretics, which often cause urinary calcium losses. Treatment, aimed at restoring normal bone mineralization, consists of high vitamin D doses and calcium supplementation.
Nutritional rickets, resulting from inadequate dietary vitamin D or a lack of sunlight exposure (Figure 7-1), is rare in industrialized countries in healthy children. It is occasionally seen in dark-skinned infants who do not receive vitamin D supplementation or in breast-fed infants not exposed to sunlight. More common causes of rickets are liver or renal failure and a variety of biochemical abnormalities in calcium or phosphorus metabolism (Table 7-1).
Figure 7-1. Vitamin D metabolism.
Table 7-1 • COMMON CAUSES OF ABNORMAL METABOLISM OF CALCIUM AND PHOSPHORUS
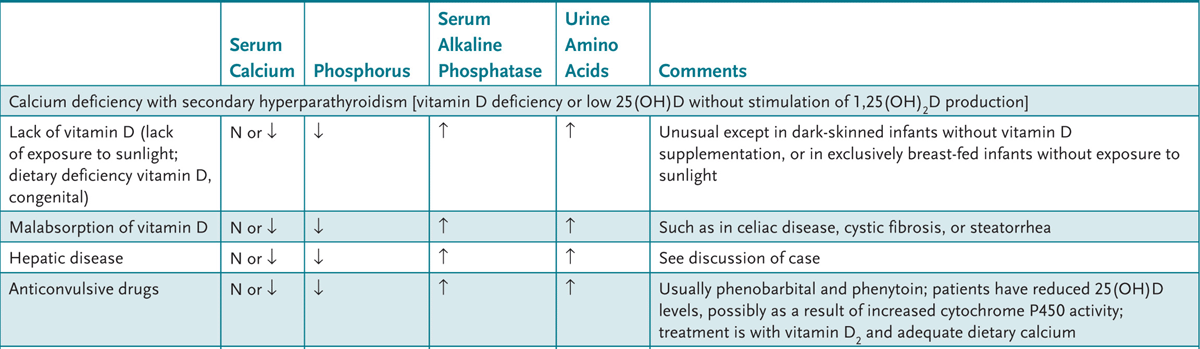
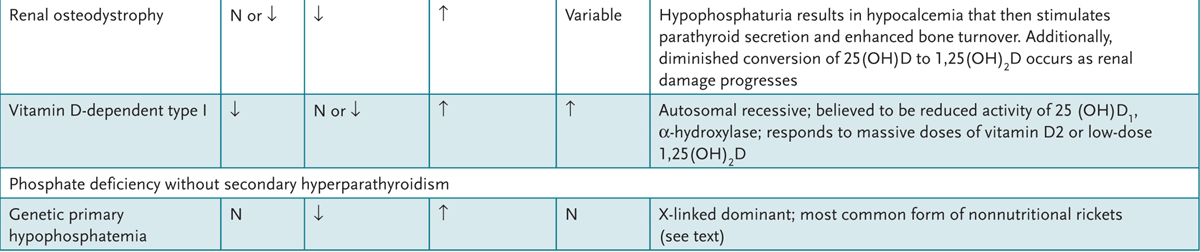
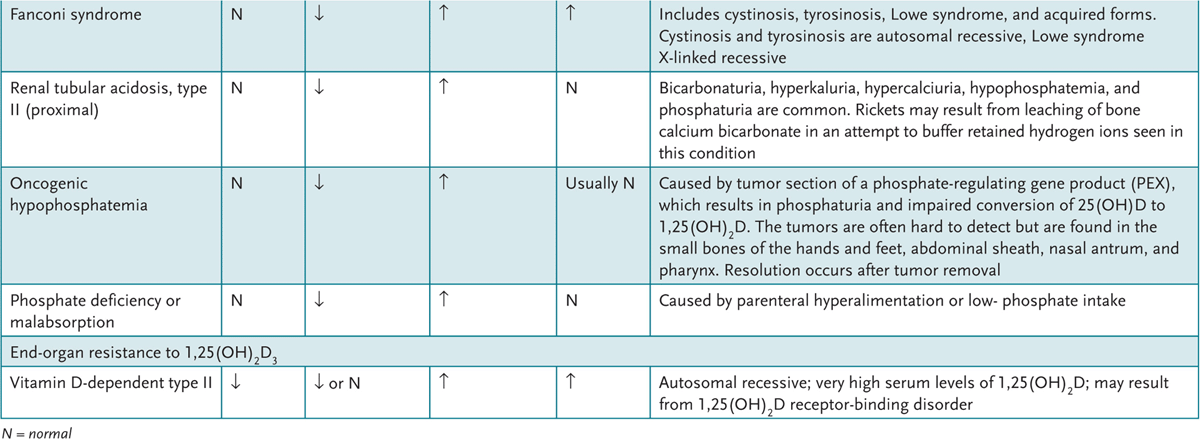
The most common form of nonnutritional rickets is familial, primary hypophosphatemia (X-linked dominant) in which phosphate reabsorption is defective, and conversion of 25(OH)D to 1,25(OH)2D in the proximal tubules of the kidneys is abnormal. This results in low serum 1,25(OH)2D, low–normal serum calcium, moderately low serum phosphate, and elevated serum alkaline phosphatase levels. Additionally hyperphosphaturia without evidence of hyperparathyroidism is present. Children at the age of walking present with smooth lower-extremity bowing (as compared to angular bowing of calcium-deficient rickets), a waddling gait, genu varum, genu valgum, coxa vara, and short stature. Other findings of calcium-deficient rickets (myopathy, rachitic rosary, pectus deformities, tetany) usually are not seen. Familial hypophosphatemia can cause intraglobular dentin deformities, whereas calcium-deficient rickets causes enamel defects. Radiologic findings include coarse-appearing trabecular bone and widening, fraying, and cupping of the metaphysis of the proximal and distal tibia, distal femur radius, and ulna.
7.1 A 14-month-old child has lower-extremity bowing, a waddling gait, and genu varum, and is at the 5th percentile for height. Laboratory data include low–normal serum calcium, moderately low serum phosphate, and elevated serum alkaline phosphatase levels, hyperphosphaturia, and normal parathyroid levels. Which of the following is the most likely diagnosis?
A. Fanconi syndrome
B. Genetic primary hypophosphatemia
C. Malabsorption of vitamin D
D. Phosphate malabsorption
E. Renal osteodystrophy
7.2 An 8-month-old African-American baby arrives to the emergency department with his mother with the complaint of decreased left arm movement. He is born after a normal term pregnancy, has had no medical problems, and was in good health when his mother dropped him off at the day care center. Upper arm radiographs show a left humerus spiral fracture. Which of the following is the most appropriate next step in management?
A. Admit the child and call child protective services.
B. Obtain serum 1,25(OH)2D levels.
C. Order serum alkaline phosphatase levels.
D. Obtain stool for analysis for fat-soluble vitamins.
E. Send chromosome sample for osteogenesis imperfecta analysis.
7.3 The diet of a 3-year-old child with cystic fibrosis should be supplemented with which of the following?
A. Folate
B. Sodium
C. Vitamin C
D. Vitamin B12
E. Vitamin D
7.4 A 5-year-old girl is somewhat short and has mild leg bowing. Her medical history is significant only for well-controlled seizure disorder. Serum calcium, phosphorus, and alkaline phosphatase levels and urinary amino acid concentration are normal. A bone age is notable for abnormal distal radius and ulna mineralization. Which of the following is the most likely diagnosis?
A. Cystic fibrosis
B. Fanconi syndrome
C. Genetic primary hypophosphatemia
D. Rickets associated with anticonvulsive drug use
E. Schmid metaphyseal dysplasia
7.1 B. Lower-extremity bowing, low–normal calcium and phosphate levels, and normal parathyroid hormone levels suggest familial primary hypophosphatemia.
7.2 A. A spiral fracture of the humerus is suspicious but not diagnostic for child abuse. While further laboratory testing is appropriate, the next step in the management of this child is to provide a safe environment until more data are available.
7.3 E. In addition to pancreatic enzyme replacement therapy, supplementation with fat-soluble vitamins (A, D, E, and K), often iron, and sometimes zinc is recommended.
7.4 E. All of the rickets syndromes present with elevated alkaline phosphatase levels. Schmid metaphyseal dysplasia, an autosomal dominant condition, presents in a similar way with short stature, leg bowing, and waddling gait. Radiographs show irregular long bone mineralization. Biochemically, Schmid-type metaphyseal dysostosis present with normal serum calcium, phosphorus, and alkaline phosphatase activity and normal urinary amino acid levels.
![]() Nutritional rickets (inadequate dietary vitamin D or sunlight exposure) is rare in healthy children in industrialized countries. Medical conditions (liver or renal failure) or abnormalities in calcium and phosphorus metabolism usually are responsible.
Nutritional rickets (inadequate dietary vitamin D or sunlight exposure) is rare in healthy children in industrialized countries. Medical conditions (liver or renal failure) or abnormalities in calcium and phosphorus metabolism usually are responsible.
![]() Primary hypophosphatemia (X-linked dominant) is the most common cause of nonnutritional rickets; proximal kidney tubule defects in phosphate reabsorption and conversion of 25(OH)D to 1,25(OH)2D are seen. Findings include low–normal serum calcium, moderately low serum phosphate, elevated serum alkaline phosphatase, and low serum 1,25(OH)2D levels, hyperphosphaturia, and no evidence of hyperparathyroidism.
Primary hypophosphatemia (X-linked dominant) is the most common cause of nonnutritional rickets; proximal kidney tubule defects in phosphate reabsorption and conversion of 25(OH)D to 1,25(OH)2D are seen. Findings include low–normal serum calcium, moderately low serum phosphate, elevated serum alkaline phosphatase, and low serum 1,25(OH)2D levels, hyperphosphaturia, and no evidence of hyperparathyroidism.
Brewer ED. Pan-proximal tubular dysfunction (Fanconi syndrome). In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1892-1897.
Chesney RW. Metabolic bone disease. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:2446-2447.
Chiang ML. Disorders of renal phosphate transport. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1898-1901.
Egan M. Cystic fibrosis. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1481-1497.
Federico MJ, Kerby GS, Deterding RR, et al. Cystic fibrosis. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:501-502.
Geary DF. Chronic kidney disease. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:1749-1755.
Greenbaum LA. Rickets and hypervitaminosis D. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:200-209.
Hill LL, Chiang ML. Renal tubular acidosis. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1886-1892.
Kohaut EC. Chronic renal failure. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1841-1844.
Lum GM. Chronic renal failure. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:686-688.
Orenstein DM. Cystic fibrosis. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:1977-1986.
Porter CC, Avner ED. Toxic nephropathies-renal failure. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1816-1818.
Root AW. Rickets and osteomalacia. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:2097-2101.
Rosenstein BJ. Cystic fibrosis. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1425-1438.
Sokol RJ, Narkewicz MR. Biliary atresia. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:639-640.
Zeitler PS, Travers SH, Nadeau K, et al. Disorders of calcium & phosphorus metabolism. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:959-963.
CASE 8
A family reports that their 5-year-old son has been increasingly confused over the last several hours. His emergency department vital signs show a heart rate of 180 beats/min, a blood pressure of 80/50 mm Hg, a temperature of 36.1 °C (97°F), and slow, deep respirations. His capillary refill is 5 seconds, and he has skin tenting as well as altered mental status. His mother reports that he has had a several pounds of weight loss over the last few weeks, has been increasingly tired for several days, and that she has been concerned about his 2- or 3-day history of thirst, frequent daytime urination, and new onset of nocturnal enuresis.
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the best therapy?
What is the best therapy?
ANSWERS TO CASE 8: Diabetic Ketoacidosis
Summary: A 5-year-old with weight loss, polydipsia, and polyuria who presents with dehydration and Kussmaul breathing.
• Most likely diagnosis: Diabetic ketoacidosis (DKA).
• Best therapy: Fluid rehydration, insulin, and close monitoring of serum glucose level and acidemia.
1. Understand the presentation of patients in DKA.
2. Appreciate the initial treatment strategies in the management of DKA.
3. Become familiar with pitfalls in the treatment of DKA.
This patient is in extremis. He is tachycardic, hypotensive, hypothermic, and has delayed capillary refill with tenting of the skin. The ABCs of medicine apply. He is confused but not obtunded; he probably requires neither his Airway controlled nor his Breathing regulated. His examination suggests at least 10% dehydration; his Circulatory status is marginal and requires rapid volume restoration. His history and physical examination suggest diabetes; a finger-stick glucose test confirms the diagnosis. The therapy for DKA rests on: (1) aggressive volume repletion, (2) glucose control with insulin, and (3) correction of metabolic abnormalities.
APPROACH TO:
Diabetic Ketoacidosis
KETOACIDOSIS: A condition resulting from deficient insulin availability, leading to lipid oxidation and metabolism rather than glucose metabolism. The insulin absence results in free fatty acid (FFA) released from adipose tissue and in unregulated hepatic FFA oxidation and ketogenesis.
TYPE I DIABETES: Known by a variety of names, it is caused by a severe endogenous insulin deficiency and a requirement for exogenous insulin to prevent ketoacidosis.
TYPE II DIABETES: Known by a variety of names, it usually consists of tissue-level insulin resistance (although exogenous insulin is often required) and rarely leads to ketoacidosis.
KUSSMAUL BREATHING: Deep, rapid respirations associated with acidosis.
Patients with DKA represent a medical emergency. Such patients may require intubation, but usually this is seen later in the disease course. Children more commonly present signs and symptoms of severe dehydration and acidosis. The history often is positive for polyuria, polydipsia, nausea, vomiting, and abdominal complaints. Hypothermia, hypotension, Kussmaul respirations, and acetone on the breath are common. As these signs and symptoms may be nonspecific, especially in younger children, a high index of suspicion is required to make the diagnosis.
Laboratory data demonstrate an elevated glucose level (often 400-800 mg/dL), metabolic acidosis (with anion gap, ie, excess endogenous anion production such as from lactic acid), and hyperketonemia. Serum electrolyte levels usually show hyponatremia and normal or slightly elevated potassium (despite potentially serious intracellular potassium depletion). Elevated blood urea nitrogen and creatinine levels are commonly seen, reflecting the dehydration. White blood cell counts (WBCs) often are elevated, especially if a bacterial infection is exacerbating the condition.
Treating DKA includes initial vascular volume expansion (often with normal saline) and then correction of the hyperglycemia and hyperketonemia. Intravenous (IV) fluid boluses sufficient to stabilize the heart rate and blood pressure are often required, and then a slower IV rate (usually a saline solution with or without some glucose) to replace fluid losses and to ensure adequate urine flow is initiated. Potassium is added to IV fluids after urine output is established to counteract the patient’s total body potassium depletion (treatment of the hyperglycemia and acidosis drives potassium intracellularly; hypokalemia is an avoidable life-threatening complication). A continuous insulin infusion at a rate of approximately 0.1 U/kg/h is also started (a bolus of 0.1 U/kg is often given initially), with the IV rate adjusted based on the results of hourly glucose measurements. Glucose is added to IV fluids when the serum glucose level drops to approximately 250 or 300 mg/dL, and additional insulin rate adjustments are made based on serum glucose levels. The low plasma pH and elevated serum ketone levels will correct significantly in the first 8 to 10 hours; the serum bicarbonate level may remain low for 24 hours or more. Improvement is characterized by a decrease in IV insulin doses and resolution of the ketonuria; then, the patient can take oral feedings, and insulin is converted from the IV to subcutaneous route.
Several pitfalls should be avoided during the treatment of DKA. Intravenous fluids with insulin and improvement in acidosis levels often are associated with a fall in serum potassium levels; addition of potassium to the IV fluids usually is indicated to prevent serious hypokalemia. Bicarbonate infusion usually is avoided except in extreme situations, because it may: (1) precipitate hypokalemia, (2) shift the oxygen dissociation curve to the left, worsening organ oxygen delivery, (3) overcorrect the acidosis, and (4) result in worsening cerebral acidosis while the plasma pH is being corrected (transfer into the cerebrum of CO2 formed when the bicarbonate is infused in an acid serum). Cerebral edema (etiology unknown) sometimes occurs, manifesting as headache, personality changes, vomiting, and decreased reflexes. Treatment of cerebral edema consists of reduction in IV fluid, administration of IV mannitol, and hyperventilation. Episodes of DKA (especially in the known diabetic) can be precipitated by bacterial infection. An evaluation for infection sources with institution of antibiotics (if appropriate) is required.
8.1 A 14-year-old adolescent girl from another state was monitored for 7 years for a history of insulin-dependent diabetes mellitus. At your clinic her hemoglobin A1C is 14.9%. This laboratory test indicates which of the following?
A. Her glucose control is poor.
B. She does not have insulin-dependent diabetes.
C. She has entered the “honeymoon phase” of her diabetes.
D. She has an underlying infection.
E. She is demonstrating the Somogyi phenomenon.
8.2 Six months after being diagnosed with what appears to be insulin-dependent diabetes, the 5-year-old in the case presentation has a significant decrease in his insulin requirement. Which of the following is the most likely explanation?
A. His diagnosis of insulin-dependent diabetes was incorrect.
B. He had a chronic infection that is now under control.
C. He has followed his diabetes diet so well that he requires less insulin.
D. He is demonstrating the Somogyi phenomenon.
E. He has entered the “honeymoon phase” of his diabetes.
8.3 A 15-year-old adolescent girl has experienced abdominal pain, vomiting, and lethargy for 3 days. Her chest and throat examinations are clear, but her abdominal examination is significant for right lower quadrant pain. Rectal examination is equivocal for pain, and her pelvic examination is remarkable for pain upon movement of her cervix. Laboratory data include a white blood cell count of 18,000/mm3, serum glucose level of 145 mg/dL, and serum bicarbonate level of 21 mEq/dL. Her urinalysis is remarkable for 1+ white blood cells, 1+ glucose, and 1+ ketones. Which of the following is the most likely diagnosis?
A. Appendicitis
B. Diabetic ketoacidosis (DKA)
C. Gastroenteritis
D. Pelvic inflammatory disease (PID)
E. Right lower lobe pneumonia
8.4 A 16-year-old adolescent girl has enuresis, frequent urination, a white vaginal discharge, and a dark rash around her neck. She is greater than the 95th percentile for her age. Her serum glucose level is 250 mg/dL, and her urinalysis is positive for 2+ glucose but is otherwise negative. Which of the following is the most likely diagnosis?
A. Chemical vaginitis
B. Chlamydia cervicitis
C. Psoriasis
D. Type II diabetes
E. Urinary tract infection (UTI)
8.1 A. The patient most likely has poor diabetes control. The hemoglobin A1C test, commonly used to follow glucose control, reflects the average glucose levels over the previous 2 or 3 months. The hemoglobin A1C goal for most diabetics is 6% to 9%. Levels greater than 12% suggest poor control, and levels of 9% to 12% represent fair control. In the Somogyi phenomenon, a patient has nocturnal hypoglycemic episodes manifested as night terrors, headaches, or early morning sweating and then presents a few hours later with hyperglycemia, ketonuria, and glucosuria. Counter-regulatory hormones, in response to the hypoglycemia, cause the hyperglycemia.
8.2 E. Up to 75% of newly diagnosed diabetics have a progressive decrease in the daily insulin requirement in the months after their diabetes diagnosis; a few patients temporarily require no insulin. This “honeymoon” period usually lasts a few months, and then an insulin requirement returns. Patients are told that the “honeymoon” period is not a cure and that they should expect a return to insulin requirement.
8.3 D. The patient likely has PID; glucosuria is a stress response to the infection and does not represent glucose metabolism problems. All of the options in the question can cause abdominal pain. Although diabetes mellitus is in the differential, DKA more likely presents with ketoacidosis (significantly decreased serum bicarbonate levels) and high serum glucose levels.
8.4 D. The description is of an obese adolescent female with candida vaginitis (the vaginal discharge) and acanthosis nigricans (the nuchal dark rash) consistent with type II diabetes. This condition is far more common in overweight children, especially those with a family history of the condition.
![]() Diabetic ketoacidosis (DKA) is a medical emergency that can present with nonspecific signs of dehydration, polyuria, nausea, vomiting, and abdominal complaints. Hypothermia, hypotension, Kussmaul respirations, and acetone on the breath are also seen. A high index of suspicion is required to make the diagnosis, especially in the younger child.
Diabetic ketoacidosis (DKA) is a medical emergency that can present with nonspecific signs of dehydration, polyuria, nausea, vomiting, and abdominal complaints. Hypothermia, hypotension, Kussmaul respirations, and acetone on the breath are also seen. A high index of suspicion is required to make the diagnosis, especially in the younger child.
![]() Cerebral edema is a potentially life-threatening complication in diabetic ketoacidosis treatment presenting as headache, personality changes, vomiting, and decreased reflexes.
Cerebral edema is a potentially life-threatening complication in diabetic ketoacidosis treatment presenting as headache, personality changes, vomiting, and decreased reflexes.
![]() Electrolyte disturbances are common in diabetic ketoacidosis. Hypokalemia can occur during treatment if adequate sources are not provided. Bicarbonate administration usually is avoided except in extreme situations for a variety of physiologic reasons.
Electrolyte disturbances are common in diabetic ketoacidosis. Hypokalemia can occur during treatment if adequate sources are not provided. Bicarbonate administration usually is avoided except in extreme situations for a variety of physiologic reasons.
Alemzadeh R, Ali O. Diabetes mellitus. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1068-1997.
Chase HP, Eisenbarth GS. Diabetes mellitus. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:984-991.
Cooke DW. Type 2 diabetes mellitus. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:2115-2122.
Plotnick LP. Type 1 (insulin-dependent) diabetes mellitus. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:2103-2115.
Rosenbloom AL. Diabetes mellitus. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:2104-2125.
An 8-year-old child presents to the emergency department with the complaint of right-sided weakness. The child is one of your well-known sickle cell disease (SCD) patients having been followed by your practice since birth. The child’s previous history has been relatively benign with only two previous hospitalizations, once at 6 months for fever and another at 12 months for a swollen, painful left wrist.
![]() What is the next step in the care of this patient?
What is the next step in the care of this patient?
![]() What long-term strategies might be employed to prevent recurrence?
What long-term strategies might be employed to prevent recurrence?
ANSWER TO CASE 9: Sickle Cell Disease with Probable Stroke
Summary: A healthy 8-year-old-child known to have SCD presents with acute onset of weakness.
• Next step: Admit to the hospital (probably the intensive care unit) and arrange for a simple or partial exchange transfusion to reduce the amount of circulating sickle cells and thus reduce the chances for further neurologic damage.
• Long-term strategy: This child’s chance of a second stroke in the upcoming 2 years is approximately 70% to 80%. Thus, chronic transfusion therapy is indicated to reduce the risk of such neurologic events.
1. Become familiar with the goals of the routine well-child (or health supervision) session for a patient with SCD.
2. Learn the common complications and treatment strategies for a child with SCD.
Well-child care for healthy children typically is uncomplicated. For children with special needs, such as SCD or Down syndrome, guidelines outline their specific considerations. For children with multiple handicaps, such as those resulting from extreme prematurity, no specific guidelines exist; the providers adapt national “well-child care” guidelines as appropriate.
APPROACH TO:
Sickle Cell Disease
Goals of a health supervision visit for all children including those with sickle cell and other disease incorporate evaluating a child’s physical, developmental, psychosocial, and educational status to identify problems early; prompt intervention then can be instituted. Anticipatory guidance aims to foster good health habits, prevent illness, and assist in family communication. For the child with a diagnosis such as SCD, additional strategies are also employed including ensuring the child is linked to a comprehensive SCD program.
Hemoglobinopathies such as SCD are often diagnosed at birth as part of each state’s newborn screening program. Routine care for the child with SCD then can be implemented, which typically includes such things as initiation of daily penicillin therapy by 2 months of age and folate by 6 months of age. Special vaccinations for these children include administration at 2 years of age—meningococcal and the 23-valent polysaccharide pneumococcal vaccines; additional doses of these vaccines may be required. Additional laboratory, radiologic, and other testing also may be indicated.
Children with SCD are at high risk for sepsis; those who present with temperatures greater than approximately 38.5°C require evaluation and initiation of antibiotic therapy. Hospital admission is indicated for febrile young children, all children who have evidence of toxicity, or children whose laboratory evaluation is of concern.
Pain crises are not uncommon among children with SCD. Children whose pain is inadequately controlled with home medications regimens must be evaluated. Additional pain medications, such as morphine or hydromorphone, along with hydration may be attempted in the outpatient setting. If more than one or two doses of these additional pain medications are required, inpatient hospitalization is required.
Children with SCD who have significant respiratory symptoms such as severe cough, shortness of breath, or chest pain may be exhibiting symptoms of acute chest syndrome. Should these children with lower respiratory symptoms have hypoxemia and a new infiltrate on chest radiograph, hospital admission is warranted. Therapies might include oxygen, hydration, blood transfusion, pain control, and antibiotics. Close observation of respiratory failure is warranted.
Parents of the child with SCD are taught to palpate the abdomen of their younger children to observe for splenic enlargement. A child who has abdominal pain, distension, or acute enlargement of the spleen likely has acute splenic sequestration and requires hospitalization, possibly in the intensive care unit, to observe for cardiovascular collapse. Blood transfusions, perhaps even emergently, may be required and will be life-saving. As the child ages the spleen usually auto-infarcts, eliminating the SCD complication of splenic sequestration but increasing the odds of an encapsulated organism infection.
About 10% of children with SCD have acute stroke. Symptoms might include paresis, aphasia, seizures, cranial nerve palsy, headache, or coma; all such children are admitted to the hospital. Emergency neuroimaging is warranted, repeated neurologic examinations are conducted, and partial or simple transfusions are performed to reduce the percentage of sickle cells. Physical therapy and rehabilitation are provided as the patient recovers. Chronic transfusions are instituted to reduce the risk of recurrence. As part of the routine well-child care of an SCD patient, transcranial Doppler (TCD) ultrasonography is often recommended to identify those with increased flow velocity in the large cerebral blood vessels and thus at high-risk for developing a first stroke. Chronic transfusion among these high-risk children has resulted in reduced risk of first stroke.
A child with SCD who presents with a significant increase in pallor, fatigue, or lethargy may be exhibiting signs of aplastic crisis. These children will have a hemoglobin level below their normal baseline and a low reticulocyte count. These children require hospitalization to observe for evidence of cardiovascular collapse. Blood transfusions may be required.
A boy with SCD who has a priapism episode persisting for more than 3 to 4 hours must be evaluated by a urologist. Intravenous fluid hydration and pain control are provided; ice is not to be used. The urologist may be required to aspirate and irrigate the corpora cavernosa to achieve detumescence. Failure of 3 or 4 aspirations in the outpatient setting requires more extensive inpatient management including exchange blood transfusions, further pain control, and additional surgical interventions.
Significant vomiting or diarrhea in the patient with SCD puts the patient at risk for dehydration and a vaso-occlusive crisis. Intravenous fluids until the patient is able to tolerate liquids orally may be required.
9.1 A 14-year-old girl is known to have SCD. Over the previous 2 or 3 months, she has been having increasingly frequent episodes of right upper quadrant, cramping pain. Which of the following strategies is likely to identify her medical condition?
A. Measure hepatitis B surface antigen and antibody levels.
B. Obtain urine for routine analysis and culture.
C. Obtain an ultrasound of her gallbladder.
D. Order a chest radiograph for new infiltrates.
E. Measure via echocardiogram her cardiac output.
9.2 Appropriate advice for a mother of a 2-week-old child identified on newborn state screening to have SCD includes which of the following?
A. Initiation of iron therapy.
B. Emergent genetic testing of both parents for hemoglobinopathy status.
C. Initiation of hydroxyurea therapy.
D. Purchase of an apnea monitor.
E. Enrollment in a comprehensive sickle cell program.
9.3 During the triage of a “well-child” visit, the staff record that the parents of a previously healthy 5-month-old offer a great amount of information. Which of the following bits of information is of most concern?
A. A diet that includes baby cereal, five different baby vegetables, and one baby fruit.
B. Consuming 32 oz of infant formula per day.
C. Intermittent tugging on the ears.
D. The child appears to be more pale than usual.
E. Rolling from front to back but not back to front.
9.4 Which of the following statements about “routine” procedures for an SCD patient is accurate?
A. All SCD children have baseline and then periodic CBC and reticulocyte measurement screenings beginning at about 2 months.
B. To reduce the risk of sepsis, polysaccharide pneumococcal 23 vaccines are administered at 2, 4, and 6 months of age.
C. To identify new infiltrates, chest radiographs are obtained at all routine visits beginning at about 12 months of age.
D. Yearly gallbladder ultrasounds are indicated beginning at adolescence to identify the presence of stones.
E. Human papilloma virus vaccines are contraindicated in the SCD population.
9.1 C. The child presented has pain referred to the right upper quadrant; she is at high risk for developing gallstones because of her SCD. The one test likely to identify the stones is the ultrasound. Part of her routine childhood immunizations should have been vaccination for hepatitis B, thus making this type of hepatitis unlikely. She may need periodic chest radiographs or echocardiograms, especially if she has evidence of acute chest syndrome or chronic cardiac/pulmonary disease, but the case as presented does not suggest such findings.
9.2 E. This child must be enrolled in a comprehensive SCD program to ensure the best possible outcome. At 2 weeks of age, the child has no reason to be iron deficient, and combined with future blood transfusions that may be required, iron therapy could result in iron overload. The newborn state screen has shown the child to have SCD and that both parents have at least a single sickle cell gene; further testing of the family may be warranted but not as an emergency. Hydroxyurea is used to increase the levels of fetal hemoglobin; this child already has significant quantities of that hemoglobin. SCD is not an indication for an apnea monitor.
9.3 D. All of the items presented are normal for the child’s age except increasing pallor, which may be due to splenic sequestration of aplastic anemia.
9.4 A. SCD patients require baseline and periodic blood counts as described. The 23 valent polysaccharide pneumococcal vaccine is initiated at 2 years of age, while the conjugate pneumococcal vaccine is administered at the younger ages outlined. Chest radiographs typically are obtained at approximately 2 years of age and periodically thereafter for screening purposes, for recent acute chest syndrome, or if the child has chronic cardiac or pulmonary disease. Ultrasounds of the gallbladder are reserved for patients with symptoms referable to that area.
![]() Children with SCD who have fever (risk of sepsis), pallor (aplastic crisis), abdominal pain or distension (splenic sequestration), pain crisis, evidence of lower respiratory disease (acute chest syndrome), priapism, new neurologic findings (stroke), or dehydration must be evaluated urgently.
Children with SCD who have fever (risk of sepsis), pallor (aplastic crisis), abdominal pain or distension (splenic sequestration), pain crisis, evidence of lower respiratory disease (acute chest syndrome), priapism, new neurologic findings (stroke), or dehydration must be evaluated urgently.
![]() Additions to routine care required for all children include initiation of penicillin and folate therapies as well as initiation of meningococcal and polysaccharide vaccines at early-than-typical ages.
Additions to routine care required for all children include initiation of penicillin and folate therapies as well as initiation of meningococcal and polysaccharide vaccines at early-than-typical ages.
![]() A variety of screening tests, such as routine CBC and reticulocyte measurements, begin at 2 months of age or at diagnosis.
A variety of screening tests, such as routine CBC and reticulocyte measurements, begin at 2 months of age or at diagnosis.
Ambruso DR, Hays T, Goldenberg NA. Sickle cell disease. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:846-848.
American Academy of Pediatrics. Recommendations for preventive pediatric health care. Available at: http://www.aap.org. Accessed on April 18, 2012.
Debaun MR, Frei-Jones M, Vichinsky E. Sickle cell disease. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1663-1670.
Lane PA, Buchanan GR, Hutter JJ, et al. Sickle cell disease in children and adolescents: diagnosis, guidelines for comprehensive care, and care paths and protocols for management of acute and chronic complications. Sickle Cell Disease Care Consortium. Accessed April 18, 2012.
Martin PL. Sickle cell disease and trait. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1696-1698.
Quinn CT. Hemoglobinopathies. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:1556-1561.
A 4-year-old boy has a 2-day history of runny nose, productive cough, and wheezing. Subjective fever and decreased appetite also were noted today. He has no known cardiorespiratory disease, and his immunizations are current. His two younger siblings are recovering from “chest colds.” On examination, he is febrile to 103.2°F (39.6°C), with a respiratory rate of 22 breaths/min. His examination is remarkable for congested nares, clear rhinorrhea, coarse breaths sounds in all lung fields, and bibasilar end-expiratory wheezes.
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the next step in evaluation?
What is the next step in evaluation?
Summary: A toddler presents with cough, fever, and an abnormal chest examination.
• Most likely diagnosis: Pneumonia.
• Next step in evaluation: A chest x-ray (CXR) often is indicated to ascertain if radiographic changes support clinical findings. In addition to chest radiography, pulse oximetry and selected laboratory tests (complete blood count [CBC], blood culture, and nasal wash for selected viral antigens) may help elucidate the etiology and extent of infection, as well as direct possible antimicrobial therapy.
1. Describe the etiologies of pneumonia and their age predilections.
2. Describe various clinical and radiographic findings in pneumonia.
3. Describe the evaluation and treatment of pneumonia.
The most important initial goal in managing this patient is to ensure adequacy of the ABCs (maintaining the Airway, controlling the Breathing, and ensuring adequate Circulation). A patient with pneumonia may present with varying degrees of respiratory compromise. Oxygen may be required, and in severe cases respiratory failure may be imminent, necessitating intubation and mechanical ventilation. The patient with pneumonia and sepsis also may have evidence of circulatory failure (septic shock) and require vigorous fluid resuscitation. After the basics of resuscitation have been achieved, further evaluation and management can be initiated.
APPROACH TO:
The Child with Pneumonia
RALES: Wet or “crackly” inspiratory breath sounds due to alveolar fluid or debris; usually heard in pneumonia or congestive heart failure (CHF).
PLEURAL RUB: Inspiratory and expiratory “rubbing” or scratching breath sounds heard when inflamed visceral and parietal pleurae come together.
STACCATO COUGH: Coughing spells with quiet intervals, often heard in pertussis and chlamydial pneumonia.
PLEURAL EFFUSION: Fluid accumulation in the pleural space; may be associated with chest pain or dyspnea; can be transudate or exudate depending on results of fluid analysis for protein and lactate dehydrogenase; origins include cardiovascular (congestive heart failure), infectious (mycobacterial pneumonia), and malignant (lymphoma).
EMPYEMA: Purulent infection in the pleural space; may be associated with chest pain, dyspnea, or fever; usually seen in conjunction with bacterial pneumonia or pulmonary abscess.
PULSE OXIMETRY:Noninvasive estimation of arterial oxyhemoglobin concentration (SPO2) using select wavelengths of light.
Pneumonia or lower respiratory tract infection (LRTI) is a diagnosis made clinically and radiographically. The typical pediatric patient with pneumonia may have traditional findings (fever, cough, tachypnea, and toxicity) or very few signs, depending on the organism involved and the patient’s age and health status.
Lower respiratory tract infection (LRTI) typically begins with organism acquisition via inhalation of infected droplets or contact with a contaminated surface. Depending on the organism, spread to distal airways occurs over varying intervals. Bacterial infection typically progresses rapidly over a few days; viral pneumonia may develop more gradually. With infection progression, an inflammatory cascade ensues with airways affected by humoral and cellular mediators. The resulting milieu adversely affects ventilation–perfusion, and respiratory symptoms develop.
The pneumonia process may produce few findings or may present with increased work of breathing manifested as nasal flaring, accessory muscle use, or tachypnea, the latter being a relatively sensitive indicator of pneumonia. Associated symptoms may include malaise, headache, abdominal pain, nausea, or emesis. Toxicity can develop, especially in bacterial pneumonia. Fever is not a constant finding. Subtle temperature instability may be noted in neonatal pneumonia. Clinically, pneumonia can be associated with decreased or abnormal breathing (rales or wheezing). Chest examination may be equivocal, especially in the neonate. Hypoxia can be seen. Pneumonia complications (pleural effusion) may be identified by finding localized decreased breath sounds or rubs.
Radiographic findings in LRTI may be limited, nonexistent, or lag the clinical symptoms, especially in the dehydrated patient. Findings may include single or multilobar consolidation (pneumococcal or staphylococcal pneumonia), air trapping with a flattened diaphragm (viral pneumonia with bronchospasm), or perihilar lymphadenopathy (mycobacterial pneumonia). Alternatively, an interstitial pattern may predominate (mycoplasmal pneumonia). Finally, pleural effusion and abscess formation are more consistent with bacterial infection.
LRTI occurs more frequently in the fall and winter and with greater frequency in younger patients, especially those in group environments (large households, day care facilities, and elementary schools). When all age groups are considered, approximately 60% of pediatric pneumonias are bacterial in origin, with pneumococcus topping the list. Viruses (respiratory syncytial virus [RSV], adenovirus, influenza, parainfluenza, enteric cytopathic human orphan [ECHO] virus, and coxsackie virus) run a close second.
Identifying an organism in pediatric pneumonia may prove difficult; causative organisms are identified in only 40% to 80% of cases. Routine culturing of the nasopharynx (poor sensitivity/specificity) or sputum (difficulty obtaining specimens in young patients) usually is not performed. Thus, diagnosis and treatment usually are directed by a patient’s symptoms, physical and radiographic findings, and age.
In the first few days of life, Enterobacteriaceae and group B Streptococcus (GBS) are the primary bacterial etiologies; other possibilities include Staphylococcus aureus, Streptococcus pneumoniae (pneumococcus), and Listeria monocytogenes. In the newborn with pneumonia, broad-spectrum antimicrobials (ampicillin with either gentamicin or cefotaxime) are customarily prescribed. During the first few months of life, Chlamydia trachomatis is a possibility, particularly in the infant with staccato cough and tachypnea, with or without conjunctivitis or known maternal chlamydia history. These infants also may have eosinophilia, and bilateral infiltrates with hyperinflation on chest radiograph; treatment is erythromycin. Viral etiologies include herpes simplex virus (HSV), enterovirus, influenza, and RSV; of these, HSV is the most concerning and prevalent viral pneumonia in the first few days of life. Intravenous acyclovir is an important consideration if HSV is suspected.
Beyond the newborn period and through approximately 5 years of age, viral pneumonia is common; adenovirus, rhinovirus, RSV, influenza, and parainfluenza are possibilities. Bacterial etiologies include pneumococcus and nontypeable Haemophilus influenzae. Patients with nasal and chest congestion with increased work of breathing, wheezing, and hypoxemia regularly present to the emergency room during the winter months and are admitted for observation, oxygen, and bronchodilator therapies. The diagnosis of a viral process may be made clinically or with CXR findings (perihilar interstitial infiltrates). Nucleic acid polymerase chain reaction (PCR) amplification of secretions from a nasal swab or wash often is performed to confirm a viral etiology. A mixed viral and bacterial pneumonia can be present in approximately 20% of patients. Antibacterial coverage should be considered if the clinical scenario, examination, or x-ray findings suggest bacterial infection.
The pediatric patient older than approximately 5 years of age with LRTI typically has Mycoplasma. However, most of the viral and bacterial etiologies previously listed are possible, except GBS and Listeria. Antibiotics in this age group are directed toward Mycoplasma and typical bacteria (pneumococcus). Treatment options include macrolides (azithromycin) or cephalosporins (ceftriaxone or cefuroxime).
Pneumonia in the intubated intensive care patient with central lines may be related to Pseudomonas aeruginosa or fungal species (Candida). Pseudomonas and Aspergillus are possibilities in the patient with chronic lung disease (cystic fibrosis). Varicella-zoster virus should be considered in the patient with typical skin findings and pneumonia; cytomegalovirus (CMV) if concomitant retinitis is present; Legionella pneumophila if the patient has been exposed to stagnant water; and Aspergillus if a patient has refractory asthma or a classic “fungal ball” on chest radiograph. Travel to the southwestern United States may expose patients to Coccidioides immitis, infected sheep or cattle to Coxiella brunetti, and spelunking or working on a farm east of the Rocky Mountains to Histoplasma capsulatum.
One important subset of LRTI is tuberculosis (TB). Mycobacterium tuberculosishas become more problematic over the past decade; multidrug resistance is increasingly seen. Patients may present with symptoms ranging from a traditional cough, bloody sputum, fever, and weight loss to subtle or nonspecific symptoms. A positive purified protein derivative (PPD) is defined by induration diameter in the context of a patient’s exposure history, radiographic findings, and immune status. For instance, 5-mm induration may be considered a “positive PPD” at 48 to 72 hours in a patient with confirmed exposure, abnormal chest radiograph, or immunodeficiency. This same measurement in an otherwise healthy child without exposures would not be considered positive. Possible sources for acid-fast bacilli for stain and culture (depending on the age of the patient) include sputum samples, first-morning gastric aspirates, cerebrospinal fluid, bronchial washes or biopsy obtained through bronchoscopy, and empyema fluid analysis or pleural biopsy if surgical intervention is required. Standard antituberculous therapy, while awaiting culture and sensitivities, includes isoniazid, rifampin, and pyrazinamide. For possible drug-resistant organisms, ethambutol can be added temporarily as long as visual acuity can be followed. The typical antibiotic course consists of an initial phase of approximately 2 months’ duration on three or four medications, followed by a continuation phase of 4 to 10 months on isoniazid and rifampin. Therapy for 9 to 12 months is recommended for CNS or disseminated TB. Ultimately, total therapy duration is dependent upon the extent of imaging abnormalities, resistance patterns, and results of follow-up sputum samples in the age-appropriate patient. Directly observed therapy should be routinely advised.
10.1 A 6-week-old boy, born by vaginal delivery after an uncomplicated term gestation, has experienced cough and “fast breathing” for 2 days. His mother relates that he has a 1-week history of nasal congestion and watery eye discharge, but no fever or change in appetite. He has a temperature of 99.4°F (37.4°C) and a respiratory rate of 44 breaths/min. He has nasal congestion, clear rhinorrhea, erythematous conjunctivae bilaterally, and watery, right eye discharge. His lungs demonstrate scattered crackles without wheezes. Which of the following is the most likely pathogen?
A. C trachomatis
B.L monocytogenes
C. Respiratory syncytial virus
D. Rhinovirus
E.S pneumoniae
10.2 A 2-year-old girl has increased work of breathing. Her father notes she has had cough and subjective fever over the past 3 days. She has been complaining that her “belly hurts” and has experienced one episode of posttussive emesis, but no diarrhea. Her immunizations are current, and she is otherwise healthy. Her temperature is 102°F (38.9°C). She is somnolent but easily aroused. Respirations are 28 breaths/min, and her examination is remarkable for decreased breath sounds at the left base posteriorly with prominent crackles. Which of the following acute interventions is the next best step in your evaluation?
A. Blood culture
B. Chest radiography
C. Pulse oximetry
D. Sputum culture
E. Viral nasal swab
10.3 You are evaluating a previously healthy 8-year-old boy with subjective fever, sore throat, and cough over the past week. There has been no rhinorrhea, emesis, or diarrhea, and his appetite is unchanged. According to your clinic records, his immunizations are current and his weight was at the 25th percentile on his examination 6 months ago. Today, he is noted at the 10th percentile for weight. He is a febrile, with clear nares and posterior oropharynx, and a normal respiratory effort. He has bilateral cervical and right supraclavicular lymphadenopathy. Chest auscultation is notable for diminished breath sounds at the left base. Beyond obtaining a chest radiograph, which of the following is the best next step in your evaluation?
A. Rapid strep throat swab
B. Viral nasal swab
C. PPD placement
D. Lymph node biopsy
E. Bordetella pertussis direct fluorescent antibody testing
10.4 A 13-year-old adolescent female complains of dry cough, slight fever, and fatigue over the past 2 weeks. She noted increased chest congestion and coughing yesterday when walking outside in the cold air. She denies nasal congestion, rhinorrhea, emesis, or diarrhea. Her mother declares her daughter is generally healthy with a history of only summertime allergies. Her vital signs, respiratory effort, and chest examination are normal. Which of the following is the most likely pathogen?
A. H influenzae
B. M pneumoniae
C. Respiratory syncytial virus
D. S aureus
E. S pneumoniae
10.1 A. Cough and increased respiratory effort in an afebrile infant with eye discharge are consistent with Chlamydia. Transmission typically occurs during vaginal delivery. Approximately 25% of infants born to mothers with Chlamydia develop conjunctivitis; about half of these develop pneumonia. Most infants present with respiratory infection in the second month of life, but symptoms can be seen as early as the second week. Inner eyelid swabs are sent for PCR, and oral erythromycin or sulfisoxazole (latter only in infants older than 2 months of age) is given for 2 weeks for either conjunctivitis or pneumonia.
10.2 C. Tachypnea and lethargy are prominent in this patient with clinical pneumonia. Pulse oximetry should urgently be performed to ascertain whether oxygen is required. Sputum culturing is reasonable for an older patient who can produce sputum, but an adequate and diagnostically useful specimen can only be obtained from a 2-year-old by endotracheal aspirate or bronchoscopy. In this otherwise healthy toddler for whom concerns for atypical pneumonia are high, invasive maneuvers are not indicated. Viruses (RSV and adenovirus) are prominent at this age; one might consider performing a nasal swab for viral antigens. Abdominal pain, as noted in this question, can be seen as a presenting symptom in pneumonia, probably as a result of irritation of the diaphragm by pulmonary infection.
10.3 C. The scenario is typical for pediatric tuberculosis. Neck and perihilar or mediastinal lymphadenopathy and pulmonary or extrapulmonary manifestations can occur, with miliary disease and meningitis more common in infants and younger children. Fever, weight loss, and lower respiratory tract signs and symptoms (possible left pleural effusion in this patient) are archetypal TB findings. A purified protein derivative (PPD) (usually with control) should be placed, and consideration given to hospitalizing this patient in negative pressure isolation for further evaluation beyond PPD placement (pleurocentesis, bronchoalveolar lavage, gastric aspirates) and possible antituberculous treatment.
10.4 B. All of these findings are consistent with mycoplasmal infection (“walking pneumonia”). The incubation period for Mycoplasma is 5 to 7 days, and most symptoms are noted during the second to third week of infection. Hemolysis occurs as antibodies attach to red blood cells, prompting reticulocyte production. If necessary, nasopharyngeal aspirate for PCR or measurement of cold agglutinins may help aid in the diagnosis. Auscultatory and radiographic findings vary in this infection; a normal CXR or one with an interstitial pattern, effusion, or atelectasis could be seen.
![]() The etiology of pneumonia varies according to the patient’s age. Neonates have the greatest risk of group B Streptococcus, toddlers are more likely to have respiratory syncytial virus, and adolescents usually contract Mycoplasma.
The etiology of pneumonia varies according to the patient’s age. Neonates have the greatest risk of group B Streptococcus, toddlers are more likely to have respiratory syncytial virus, and adolescents usually contract Mycoplasma.
![]() Efforts in tuberculosis management should be directed toward isolating an organism and obtaining sensitivities, thus allowing selection of the optimal antituberculous regimen.
Efforts in tuberculosis management should be directed toward isolating an organism and obtaining sensitivities, thus allowing selection of the optimal antituberculous regimen.
American Academy of Pediatrics. Tuberculosis. In: Pickering LK, ed. 2009 Red Book: Report of the Committee on Infectious Diseases. 28th ed. Elk Grove Village, IL: American Academy of Pediatrics; 2009:680-701.
Kennedy WA. Disorders of the lungs and pleura. In: Osborn LM, DeWitt TG, First LR, Zenel JA, eds. Pediatrics. 1st ed. Philadelphia, PA: Elsevier-Mosby; 2005:803-818.
Moscona A, Murrell MT, Horga M, Burroughs M. Respiratory infections. In: Katz SL, Hotez PJ, Gerson AA, eds. Krugman’s Infectious Diseases of Children. 11th ed. Philadelphia, PA: Mosby; 2005:493-524.
Roosevelt GE. Acute inflammatory upper airway obstruction. In: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1445-1449.
Sandora TJ, Sectish TC. Pneumonia. In: Kliegman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1474-1479.
You are moonlighting in a rural emergency room when a father rushes his 3-year-old daughter into the waiting area. You quickly determine that the child had been playing with her chihuahua at a relative’s farm where they were spraying for insects in a field. While at the farm she developed abdominal cramping, cough, drooling, and tearing. While in route the child seems to be having increased respiratory difficulty, and the dad notes she soiled and urinated upon herself.
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() How is the diagnosis made?
How is the diagnosis made?
![]() What is the best therapy?
What is the best therapy?
ANSWERS TO CASE 11: Organophosphate Poisoning
Summary: A 3-year-old, previously healthy child who, while playing where spraying for insects was ongoing, develops salivation, lacrimation, respiratory distress, and gastrointestinal (GI) symptoms.
• Most likely diagnosis: Organophosphate poisoning.
• Making the diagnosis:High index of suspicion so therapy is not delayed; confirmation via decreased serum pseudocholinesterase and erythrocyte cholinesterase levels.
• Best therapy: Decontamination of the child, supportive care, administration of atropine or pralidoxime.
1. Understand the signs, symptoms, and treatment of organophosphate poisoning.
2. Be familiar with the treatment options of various commonly ingested agents.
This child is demonstrating evidence of organophosphate poisoning, the leading cause of nonpharmaceutical ingestion fatality in children. She was exposed during the spraying for insects in a field, and is at risk for ongoing absorption of toxin until decontamination of her clothing is achieved.
Note:For some children exposed to a toxic substance, parents are able to provide a container of the toxic agent. For others either the container is not available or the symptoms are not obviously related to a toxic exposure. In all cases a thorough history and physical examination, along with a high index of suspicion in younger children, is required to ensure the diagnosis of accidental toxic exposure.
APPROACH TO:
Organophosphate Poisoning
NICOTINIC SYMPTOMS:Cardiac (hypertension, tachycardia, arrhythmia); muscle (fasciculations, weakness, tremors); respiratory failure due to diaphragm paralysis; hypertension.
MUSCARINIC SYMPTOMS: Gastrointestinal (emesis, urinary and fecal incontinence); respiratory (bronchorrhea, bronchospasm); cardiac (hypotension, bradycardia); tearing and drooling; miosis.
Millions of children are poisoned each year with about 90% of exposures occurring in the home. About half of all accidental poisonings occur in children younger than 5 years of age. Children aged from about 6 to 12 years are much less likely to be exposed, and those with toxic exposures beyond 12 years of age often do so intentionally. Death due to accidental poisonings has become unusual since a variety of measures have been implemented, including poison prevention as part of all well-child visits, development of regional poison control centers, child-resistant packaging, and improved medical management.
Organophosphate poisoning can occur across skin or mucous membranes, by inhalation, or by ingestion. Commonly found in such pesticides as parathion, malathion, and diazinon, organophosphates bind irreversibly to cholinesterase of neurons and erythrocytes, as well as to liver pseudocholinesterase. The common finding is failure to terminate the effects of acetylcholine at the receptor sites.
Signs and symptoms of cholinergic excess are often remembered with the mnemonic “dumb bells,” which includes:

In addition to these muscarinic and nicotinic effects, central effects including obtundation, seizures, and apnea are also seen.
Confirmation of the exposure can be confirmed by finding decreased serum pseudocholinesterase and erythrocyte cholinesterase levels, but the correlation of these levels to the magnitude of exposure or the symptoms observed is poor. Thus, a high index of suspicion must be maintained to quickly and accurately diagnose organo-phosphate exposure.
Treatment of the patient exposed to organophosphate consists of rapid decontamination by removing all clothing and washing of all skin surfaces. For ingestions, gastric lavage or activated charcoal may be attempted, but the organophosphate compounds are rapidly absorbed and the benefits somewhat limited. The ABCs of medicine apply: preserve the Airway (intubation may be required); maintain Breathing (excessive secretions may require frequent suctioning); and ensure appropriate Circulation.
Two specific therapies to counter the effects of organophosphate poisonings include atropine and pralidoxime. Atropine works by antagonizing the muscarinic receptor; large, repeated, and sometimes continuous doses may be required. Often the amount and number of atropine doses required correlates to the degree of exposure, and may assist in the prediction of course duration. Pralidoxime is a cholinesterase-reactivating oxime, often used for patients with significant muscle weakness, especially if mechanical ventilation is required owing to muscle failure.
Careful attention to a child’s environment can help prevent countless instances of toxic ingestion. Counseling parents to “poison proof” their home is a first step toward prevention. Written and video materials are readily available through the American Academy of Pediatrics, local and state health departments, and poison control centers. All families are taught to become familiar with the national network of poison control centers, reached toll-free at 1-800-222-1222.
11.1 Students attending a school built in 1951 are at risk for which of the following?
A. Arsenic
B. Asbestos
C. Dichlorodiphenyltrichloroethane (DDT)
D. Mercury
E. Polychlorinated biphenyls (PCBs)
11.2 An 8-year-old, mentally delayed child ingests the contents of a mercury thermometer. Which of the following symptoms are most likely to be seen?
A. Ataxia, dysarthria, and paresthesias
B. Chest pain and dyspnea
C. Gingivostomatitis, tremor, and neuropsychiatric disturbances
D. No symptoms
E. Pulmonary fibrosis
11.3 A 4-year-old child is found with a bottle of insecticide that contains arsenic. Which of the following symptoms is most likely to occur?
A. Bradycardia with third-degree heart block
B. Constipation
C. Hemorrhagic gastroenteritis with third spacing of fluids
D. Hyperreflexia
E. Hypothermia
11.4 Exposure to environmental toxins can occur in a number of ways. Which of the following is the most likely mechanism of exposure?
A. Asbestos exposure from hazardous arts and crafts materials
B. Exposure of a child to beryllium from the child’s parents’ clothing
C. Iron intoxication from vehicular emissions
D. Lead toxicity from ingesting pieces of a pencil
E. Transplacental exposure to benzene
11.1 B. Between 1947 and 1973 asbestos was commonly sprayed on school ceilings as a fire retardant. Deterioration results in release of microscopic fibers into the air. Drop ceilings or placement of barriers usually is sufficient protection against this carcinogen.
11.2 D. The child in the question is unlikely to develop symptoms (the quantity of mercury is small); a larger acute elemental ingestion might result in a variety of gastrointestinal (GI) complaints. If the elemental mercury were in vapor form, GI complaints would be seen, along with fever, chills, headaches, visual changes, cough, chest pain, and possibly pneumonitis and pulmonary edema. Exposure to inorganic mercury salts (pesticides, disinfectants, explosives, dry batteries) can cause gastroesophageal burns, nausea, vomiting, abdominal pain, hematemesis, hematochezia, cardiovascular collapse, or death. Ataxia, dysarthria, and paresthesias are seen in methyl mercury intoxication (contaminated fish exposure). Gingivostomatitis, tremor, and neuropsychiatric disturbances are seen with chronic inorganic mercury intoxication; indeed, the term “mad as a hatter” originates from the occupational hazard of workers’ exposure in the early industrial period to mercury-containing vapors during the process of felt hat making.
11.3 C. Acute arsenic ingestions can cause nausea, vomiting, abdominal pain, and diarrhea. The third spacing and hemorrhage in the gut can lead to hypovolemic shock. Cardiac symptoms include ventricular tachycardia (QT prolongation) and congestive heart failure (CHF). These patients can develop seizures, cerebral edema, encephalopathy, and coma. Early on, patients develop loss of deep tendon reflexes, paralysis, painful dysesthesias, and respiratory failure similar to Guillain-Barré syndrome. Fever, anemia, alopecia, hepatitis, and renal failure also can be seen.
11.4 B. Fat-soluble compounds can be transmitted transplacentally (but benzene would be unusual). Parents’ work clothes can transmit potentially hazardous compounds. Arts and crafts supplies likely do not contain asbestos. Vehicular emissions are responsible for a number of pollutants including lead prior to lead-free gasoline, many of which are carcinogens, but iron intoxication would be unusual. Pencil “lead” is actually graphite (carbon) and not elemental lead.
![]() Organophosphate poisoning is the leading cause of nonpharmacologic ingestion fatality in the United States.
Organophosphate poisoning is the leading cause of nonpharmacologic ingestion fatality in the United States.
![]() The mnemonic “dumb bells” outlines the signs and symptoms of cholinergic excess.
The mnemonic “dumb bells” outlines the signs and symptoms of cholinergic excess.
![]() Therapy for organophosphate toxicity includes supportive care and use of either atropine or pralidoxime.
Therapy for organophosphate toxicity includes supportive care and use of either atropine or pralidoxime.
Feng S-Y, Goto CS, Baker MD. Toxic ingestions and exposures. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:455-469.
Fortenberry JD, Mariscalco MM. General principles of poisoning management. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:747-754.
O’Donnell KA, Ewald MB. Poisonings. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:250-270.
Rumack BH, Dart RC. Poisoning. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:321-347.
A mother brings her 2-year-old son to your clinic because she has seen over the last 2 days drops of bright red blood in his diaper with each stool and with wiping. He is eating his regular diet of grilled cheese sandwiches with 20 ounces of milk each day. He has been afebrile with no vomiting or diarrhea. Mom has noted he cries with stooling. Your examination of abdomen is normal. On inspection of the rectal area you find a 7-mm linear split in the posterior midline traversing from the anocutaneous junction to the dentate line. There is also a small skin appendage in the same area. His parents are the only caregivers.
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the best management for this condition?
What is the best management for this condition?
![]() What is your next step in the evaluation?
What is your next step in the evaluation?
ANSWERS TO CASE 12: Rectal Bleeding
Summary: A 2-year-old boy presents with rectal bleeding and pain on defecation.
• Most likely diagnosis:Anal fissure
• Best management: Confirm that the red substance is blood by performing a fecal occult blood test. Red-pigmented foods such as gelatin, breakfast cereals, or beets can mimic blood. When the reagent of the fecal occult blood test combines with hemoglobin, a blue color appears. This test is very sensitive. The concomitant rectal exam can assess for impacted or hard stool.
• Next step: Quantify the amount of blood loss and review vital signs; tachycardia is the initial sign of rapid blood loss. Hypotension is a late finding. Begin dietary changes and stool softeners; oral polyethylene glycolate is most commonly used and may be required for a few months to break the cycle of constipation. Parents should minimize foods known to be constipating (such as dairy products), increase water intake, and avoid bulking agents (such as fiber, psyllium).
1. Know the differential diagnosis for rectal bleeding at various ages.
2. Know how to manage rectal bleeding.
3. Be familiar with methods of investigating the cause of bleeding.
The presentation of gastrointestinal (GI) tract bleeding will often depend on the site of bleeding and the rate of hemorrhage. Hematochezia usually indicates the site is in the large intestine, but if there is massive hemorrhage in the small intestine, it may present similarly. Otherwise, bleeding in the small intestine tends to cause melena.
APPROACH TO:
Rectal Bleeding
HEMATOCHEZIA: Blood in the stool that is red or maroon-colored.
MELENA:Black tarry stools; color is produced when heme is oxidized by intestinal flora.
While gastric and duodenal bleeding can cause nausea, vomiting, or diarrhea, other sites of hemorrhage in the intestinal tract rarely cause GI symptoms. Tachycardia and hypotension may be the first symptoms before hematochezia or melena follow. If there is any change in vital signs, immediate stabilization is necessary. Patients are admitted to the hospital for monitoring, and intravascular volume is initially restored with isotonic saline, then packed red blood cells may be needed. Frequent measurement of hemoglobin or hematocrit is indicated, and bleeding can be monitored in each stool via guaiac testing if blood is not grossly visible.
Laboratory evaluation for contributing coagulopathic conditions should be performed, which includes measurement of platelets, prothrombin time (PT), activated partial thromboplastin time (APTT), liver enzymes, and creatinine. Blood urea nitrogen (BUN) levels may be elevated due to urea being produced from hemoglobin breakdown in the GI tract. Investigation for an infectious cause, such as colitis or enteritis, should be undertaken if there is a history of fever or diarrhea.
X-rays are frequently used in neonates to look for signs of necrotizing enterocolitis (NEC), such as intramural air entering the portal venous system. In infants or children, a dilated portion of proximal bowel with air distally that outlines a telescoped portion signals intussusception. An obstructive pattern can also be seen with volvulus. Special imaging techniques include Meckel scan if Meckel diverticulum is suspected, ultrasound or air-contrast enema for intussusception, angiography which also allows for embolization by an interventional radiologist, or a tagged red blood cell scan for low flow bleeds.
Pediatric gastroenterologists may be needed to identify the site or cause of the bleeding. The duodenum, stomach, and esophagus are evaluated with esophago-gastroduodenoscopy (EGD). Capsule endoscopy is a new tool that can be used to evaluate the small intestine. For lower GI bleeding, colonoscopy is performed once the patient is stabilized.
12.1 A 2-month-old boy presents with his mom after having 3 days of stools with blood streaks intermixed in them. Over the past week, he has been stooling more often with an increase from 4 stools to 8 stools per day. He has been happy and afebrile, and no blood is found when she wipes him. He continues to take 3 ounces of standard infant formula at each feed. Which of the following statements about his condition is most accurate?
A. He needs to be changed to soy formula.
B. Treatment will consist of changing him to an elemental formula, or if mom is breastfeeding, have her eliminate milk products from her diet.
C. Broad-spectrum antibiotics should be given for 10 days.
D. Provide reassurance that the condition is benign and transient and will resolve without any intervention.
E. There is usually a positive family history of lactose intolerance.
12.2 A 2-year-old girl presents with her second episode of bloody stool. Mom brings a diaper that is filled with brick-colored stool. The first episode had occurred 6 months ago and had improved over the course of a day, so no workup had been done. The child has had less appetite than usual for the day but no fever, vomiting, or complaints of pain. What would be the next steps in management?
A. Prescribe a laxative and anti-hemorrhoid cream for 1 week and then follow-up if symptoms recur.
B. Instruct mom to remove milk and milk products from her daughter’s diet for 1 year, and then slowly re-introduce it.
C. Inquire more about the amount of ibuprofen the girl has been taking and prescribe omeprazole.
D. Admit the patient to the hospital for observation and Meckel scan.
E. Ask mom about any family history of Crohn disease or ulcerative colitis, and send an erythrocyte sedimentation rate (ESR).
12.3 A 3-day-old girl is brought in to the emergency room (ER) by her parents after they noted blood-streaked stools. She has been feeding well; parents deny emesis and diarrhea. Which of the following components of the history would be leastuseful in determining the cause of the hematochezia?
A. Ask the parents whether the baby was born at home or in the hospital.
B. Review mom’s records for the color of her amniotic fluid.
C. Ask about the length of time it required for the patient to stool after birth and her specific number of stools.
D. Ask if the baby was premature.
E. Ask mom if she is breastfeeding and, if so, if she has any bleeding from the nipples.
12.4 A 15-month-old boy presents to the ER with two episodes of nonbilious vomiting. His dad reports that his son would not eat breakfast, was very fussy and irritable before the episodes began, appeared to have abdominal pain, but after the emesis, became calm and fell asleep. Two hours later, he awoke screaming and was inconsolable for about 30 minutes. He then fell back asleep. On your examination, the child awakens but lies quietly in his dad’s arms. The abdominal and genitourinary examinations are normal. Guaiac of stool from the rectal exam is positive. What is the next best step in management?
A. Consult a pediatric gastroenterologist and type and cross 5 cc/kg of packed red blood cells.
B. Administer a dose of ceftriaxone and have patient follow-up with his pediatrician the day after for results of stool studies.
C. Consult a pediatric surgeon and order an air-contrast enema.
D. Measure the levels of Helicobacter pylori antibodies and administer omeprazole.
E. Reassure dad that night terrors are common in this age group and may be provoked by an illness.
12.1 B. Allergic proctocolitis is induced by allergy to the protein in cow’s milk. Standard infant formulas are composed of this protein. Soy protein is similar in structure so cross-allergy often exists. The protein can cross over into breast milk. Elemental formulas are made of amino acids rather than complete proteins. If the inciting protein is not removed from the diet, the infant can progress to enterocolitis with resulting severe diarrhea, malabsorption, vomiting, and dehydration. The condition usually presents before 3 months of age and is more common in boys. There may be a family history of atopy.
12.2 D. Meckel diverticulum is a pouch off the ileum due to a remnant of the omphalomesenteric duct. It is usually 3 to 6 cm in size and located 50 to 75 cm from the ileocecal valve. It is often lined with endothelium that has undergone meta-plastic change that simulates gastric mucosa; the acid that is secreted causes ulceration of the adjacent ileal mucosa. Symptoms of intermittent painless rectal bleeding usually appear at the age of 2 years. It produces 50% of all lower GI bleeds in children under the age of 2 years. A Meckel radionuclide scan is needed to confirm the diagnosis, but it has a high false-negative rate, so a diagnostic laparoscopy may be needed. Even if the bleeding stops, surgical excision of the mucosa is often done to prevent re-bleeding, obstruction, or diverticulitis.
12.3 D. Necrotizing enterocolitis (NEC) occurs predominantly in preterm infants, with term infants accounting for less than 25% of the cases. Swallowed blood syndrome occurs on the 2nd to 3rd day of life. The blood may be from delivery or from the mother’s nipple. The Apt test involves differentiating the fetal hemoglobin from maternal hemoglobin based on the infant’s blood being alkali-resistant. Hemorrhagic disease of the newborn occurs in infants who do not receive vitamin K. The condition is usually not seen in newborns that are born in the hospital since intramuscular vitamin K is routinely given shortly after birth. Hirschsprung disease presents with a delay in passing meconium after birth; it can progress to toxic megacolon and enterocolitis, which will present with bloody stools and even diarrhea.
12.4 C. Intussusception is the most common cause of intestinal obstruction in children under the age of 2 years. In most cases, a lead point is not identifiable, but the condition occurs when part of the small bowel telescopes into the lumen of a distal portion of bowel. The lumen of the inserted portion collapses and causes abdominal obstruction. Peristalsis is still active and attempts to propel contents past the obstruction; this creates episodes of severe colicky intermittent pain that on subsidence leave the patient calm or lethargic. This is a hallmark symptom of abdominal obstruction and requires a pediatric surgery consult. If the bowel wall becomes ischemic with resultant areas of necrosis, blood may appear, and this will usually be in the first 12 hours of the obstruction. Only 60% of infants will have the classic “currant jelly” stool composed of blood and mucus. An air-contrast enema can be diagnostic as well as therapeutic, but a pediatric surgeon should be available in case perforation occurs or if reduction is unsuccessful.
![]() Anal fissures are the most common cause of hematochezia in infants, children, and adolescents.
Anal fissures are the most common cause of hematochezia in infants, children, and adolescents.
![]() The differential diagnosis of hematochezia varies by age.
The differential diagnosis of hematochezia varies by age.
![]() In newborns, the cause of hematochezia is most often a life-threatening condition, whereas the etiology in infants and children is more often benign.
In newborns, the cause of hematochezia is most often a life-threatening condition, whereas the etiology in infants and children is more often benign.
![]() If the patient is ill-appearing and there is no visible anal fissure, further investigation with stool studies, laboratory, or imaging is indicated.
If the patient is ill-appearing and there is no visible anal fissure, further investigation with stool studies, laboratory, or imaging is indicated.
![]() Tachycardia is the first indication that the rate or volume of bleeding is significant and warrants admission to the hospital.
Tachycardia is the first indication that the rate or volume of bleeding is significant and warrants admission to the hospital.
Densmore JC, Lal DR. Intussusception. In: Rudolph CD, Rudolph AM, Lister GE, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:1428-1429.
Kennedy M, Liacouras CA. Intussusception. In: Kliegman RM, Stanton BF, St. Geme III J, Schor N, Behrman R, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1287-1289.
Kennedy M, Liacouras CA. Meckel diverticulum and other remnants of the omphalomesenteric duct. In: Kliegman RM, Stanton BF, St. Geme III J, Schor N, Behrman R, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1281-1282.
Klinker DB, Gourlay DM. Omphalo-mesenteric duct remnants. In: Rudolph CD, Rudolph AM, Lister GE, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:1425-1427.
Maheshwari A, Carlo WA. Hemorrhage in the newborn infant. In: Kliegman RM, Stanton BF, St. Geme III J, Schor N, Behrman R, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:621.
Noel RJ. Upper and lower gastrointestinal bleeding. In: Rudolph CD, Rudolph AM, Lister GE, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:1389-1391.
Sampson HA, Leung DYM. Adverse reactions to foods. In: Kliegman RM, BF, St. Geme III J, Schor N, Behrman R, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:821.
Sreedharan R, Liacouras CA. Major symptoms and signs of digestive tract disorders. In: Kliegman RM, Stanton BF, St. Geme III J, Schor N, Behrman R, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1248-1249.
Stafford SJ, Klein MD. Anal fissure. In: Kliegman RM, Stanton BF, St. Geme III J, Schor N, Behrman R, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:1359.
Warner B. Necrotizing enterocolitis. In: Rudolph CD, Rudolph AM, Lister GE, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:246-248.
A 4-year-old child complains of ear pain. He has a temperature of 102.1°F (38.9°C) and has had a cold for several days, but he has been eating well and his activity has been essentially normal.
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the best therapy?
What is the best therapy?
ANSWERS TO CASE 13: Acute Otitis Media
Summary: A preschool child presents with ear pain and fever.
• Most likely diagnosis: Acute otitis media (AOM)
• Best therapy: Oral antibiotics
1. Be familiar with the epidemiology of otitis media (OM) in children.
2. Understand the treatment of this condition.
3. Learn the consequences of severe infection.
Otitis media is high on the differential diagnosis for this child with upper respiratory infection (URI) and ear pain. The diagnosis can be confirmed by pneumatic otoscopy and treatment started. A “telephone diagnosis” should be avoided. Figure 13-1 illustrates the anatomy of the middle ear.
Figure 13-1. Anatomy of the middle ear. (Redrawn, with permission, from Rudolph CD, Rudolph AM, Hostetter MK, Lister G, Siegel NJ, eds. Rudolph’s Pediatrics. 21st ed. New York, NY: McGraw-Hill; 2003:1240.)
Acute Otitis Media
ACUTE OTITIS MEDIA (AOM): A condition of otalgia (ear pain), fever, and other symptoms along with findings of a red, opaque, poorly moving, bulging tympanic membrane (TM).
MYRINGOTOMY AND PLACEMENT OF PRESSURE EQUALIZATION TUBES:A surgical procedure involving TM incision and placement of pressure equalization (PE) tubes (tiny plastic or metal tubes anchored into the TM) to ventilate the middle ear and help prevent reaccumulation of middle ear fluid.
OTITIS MEDIA WITH EFFUSION: A condition in which fluid collects behind the TM but without signs and symptoms of AOM. Sometimes also called serous OM.
PNEUMATIC OTOSCOPY:The process of obtaining a tight ear canal seal with a speculum and then applying slight positive and negative pressure with a rubber bulb to verify TM mobility.
TYMPANOCENTESIS: A minor surgical procedure in which a small incision is made into the TM to drain pus and fluid from the middle ear space. This procedure is rarely done in the primary care office, but rather is done by the specialist.
Otitis media is a common childhood diagnosis. Common bacterial pathogens include Streptococcus pneumoniae, nontypeable Haemophilus influenzae,and Moraxella catarrhalis. Other organisms, Staphylococcus aureus, Escherichia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa, are seen in neonates and patients with immune deficiencies. Viruses can cause AOM, and in many cases the etiology is unknown. Acute OM is diagnosed in a child with fever (usually <104°F [40°C]), ear pain (often nocturnal, awakening child from sleep), and generalized malaise. Systemic symptoms may include anorexia, nausea, vomiting, diarrhea, and headache. Examination findings include a red, bulging TM that does not move well with pneumatic otoscopy. The TM may be opaque with pus behind it, the middle ear landmarks may be obscured, and, if the TM has ruptured, pus may be seen in the ear canal. Normal landmarks are shown in Figure 13-2.
Figure 13-2. The tympanic membrane. (Reproduced, with permission, from Rudolph CD, Rudolph AM, Hostetter MK, Lister G, Siegel NJ, eds. Rudolph’s Pediatrics. 21st ed. New York, NY: McGraw-Hill; 2003:1240.)
In some situations and in a child with few symptoms, a “watchful waiting” period of a few days may be indicated since many AOM cases self-resolve. Should antibiotics be deemed necessary and depending on a community’s bacterial resistance patterns, amoxicillin at doses up to 80 to 90 mg/kg/d for 7 to 10 days is often the initial treatment. If clinical failure is noted after 3 treatment days, a change to amoxicillin-clavulanate, cefuroxime axetil, azithromycin, cefixime, ceftriaxone, or tympanocentesis is considered. Adjuvant therapies (analgesics or antipyretics) are often indicated, but other measures (antihistamines, decongestants, and corticosteroids) are ineffective.
After an AOM episode, middle ear fluid can persist for up to several months. If hearing is normal, middle ear effusion often is treated with observation; some practitioners treat with antibiotics. When the fluid does not resolve or recurrent episodes of suppurative OM occur, especially if hearing loss is noted, myringotomy with PE tubes is often implemented.
Rare but serious OM complications include mastoiditis, temporal bone osteomyelitis, facial nerve paralysis, epidural and subdural abscess formation, meningitis, lateral sinus thrombosis, and otitic hydrocephalus (evidence of increased intracranial pressure with OM). An AOM patient whose clinical course is unusual or prolonged is evaluated for one of these conditions.
13.1 An 8-year-old boy has severe pain with ear movement. He has no fever, nausea, vomiting, or other symptoms. He has been in good health, having just returned from summer camp where he swam, rode horses, and water-skied. Ear examination reveals a somewhat red pinna that is extremely tender with movement, a very red and swollen ear canal, but an essentially normal TM. Which of the following is the most appropriate next course of therapy?
A. Administration of topical mixture of polymyxin and corticosteroids
B. High-dose oral amoxicillin
C. Intramuscular ceftriaxone
D. Intravenous vancomycin
E. Tympanocentesis and culture
13.2 Three days after beginning oral amoxicillin therapy for OM, a 4-year-old boy has continued fever, ear pain, and swelling with redness behind his ear. His ear lobe is pushed superiorly and laterally. He seems to be doing well otherwise. Which of the following is the most appropriate course of action?
A. Change to oral amoxicillin-clavulanate
B. Myringotomy and parenteral antibiotics
C. Nuclear scan of the head
D. Topical steroids
E. Tympanocentesis
13.3 A 5-year-old girl developed high fever, ear pain, and vomiting a week ago. She was diagnosed with OM and started on amoxicillin-clavulanate. On the third day of this medication she continued with findings of OM, fever, and pain. She received ceftriaxone intramuscularly and switched to oral cefuroxime. Now, 48 hours later, she has fever, pain, and no improvement in her OM; otherwise she is doing well. Which of the following is the most logical next step in her management?
A. Addition of intranasal topical steroids to the oral cefuroxime
B. Adenoidectomy
C. High-dose oral amoxicillin
D. Oral trimethoprim-sulfamethoxazole
E. Tympanocentesis and culture of middle ear fluid
13.4 A 1-month-old boy has a fever to 102.7°F (39.3°C), is irritable, has diarrhea, and has not been eating well. On examination he has an immobile red TM that has pus behind it. Which of the following is the most appropriate course of action?
A. Admission to the hospital with complete sepsis evaluation
B. Intramuscular ceftriaxone and close outpatient follow-up
C. Oral amoxicillin-clavulanate
D. Oral cefuroxime
E. High-dose oral amoxicillin
13.1 A. The patient likely has an otitis externa that was caused by his swimming (also known as swimmer’s ear). Treatment is the application of a topical agent as described. Insertion of a wick may assist in excess fluid absorption in the macerated, swollen, and occluded ear canal. Causative organisms include Pseudomonas species (or other gram-negative organisms), S aureus, and occasionally fungus (Candida or Aspergillus species).
13.2 B. The child has mastoiditis, a clinical diagnosis that can require computed tomography scan confirmation. Treatment includes myringotomy, fluid culture, and parenteral antibiotics. Surgical drainage of the mastoid air cells may be needed if improvement is not seen in 24 to 48 hours.
13.3 E. After failing several antibiotic regimens, tympanocentesis and culture of the middle ear fluid are indicated.
13.4 A. Very young children with OM (especially if irritable or lethargic) are at higher risk for bacteremia or other serious infection. Hospitalization and parenteral antibiotics often are needed.
![]() The most common bacterial pathogens causing otitis media (OM) are S pneumoniae, nontypeable H influenzae, and M catarrhalis.
The most common bacterial pathogens causing otitis media (OM) are S pneumoniae, nontypeable H influenzae, and M catarrhalis.
![]() Examination findings of OM include a red, bulging tympanic membrane that does not move well with pneumatic otoscopy, an opaque tympanic membrane with pus behind it, obscured middle-ear landmarks, and, if the tympanic membrane has ruptured, pus in the ear canal.
Examination findings of OM include a red, bulging tympanic membrane that does not move well with pneumatic otoscopy, an opaque tympanic membrane with pus behind it, obscured middle-ear landmarks, and, if the tympanic membrane has ruptured, pus in the ear canal.
![]() Initial treatment of OM often includes amoxicillin (depending on local bacterial resistance patterns). If a clinical failure is seen on day 3, a change to amoxicillin-clavulanate, cefuroxime axetil, ceftriaxone, or a tympanocentesis is indicated.
Initial treatment of OM often includes amoxicillin (depending on local bacterial resistance patterns). If a clinical failure is seen on day 3, a change to amoxicillin-clavulanate, cefuroxime axetil, ceftriaxone, or a tympanocentesis is indicated.
![]() Complications are rare but include mastoiditis, temporal bone osteomyelitis, facial nerve palsy, epidural and subdural abscess formation, meningitis, lateral sinus thrombosis, and otitic hydrocephalus.
Complications are rare but include mastoiditis, temporal bone osteomyelitis, facial nerve palsy, epidural and subdural abscess formation, meningitis, lateral sinus thrombosis, and otitic hydrocephalus.
Haddad J. External otitis (otitis externa). In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:2196-2199.
Kerschner JE. Otitis media. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:2199-2213.
Klein JO. Otitis media. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:973-979.
Kline MW. Otitis externa. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1496-1497.
Kline MW. Mastoiditis. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1501-1502.
Rudolph C. Otitis externa. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:979.
Schwarzwald H, Kline MW. Otitis media. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:1497-1500.
Yoon PJ, Kelley PE, Friedman NR. Acute otitis media. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:453-464.
Yoon PJ, Kelley PE, Friedman NR. Mastoiditis. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:464-465.
Yoon PJ, Kelley PE, Friedman NR. Otitis externa. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:452-453.
You are called to the delivery room because a now 2-minute-old male was born floppy and blue; his Apgar scores were 4 and 5. He has not responded well to stimulation and blow-by oxygen. The obstetrician who is resuscitating the infant informs you that the child was born by a spontaneous vaginal delivery to a 24-year-old primagravida parity 1 woman. Her pregnancy was uncomplicated. Fetal heart tones were stable throughout the labor. Spinal epidural anesthesia was administered but was only partially effective; the obstetrician supplemented her labor analgesia with intravenous meperidine (Demerol) and promethazine (Phenergan). The amniotic fluid was not meconium stained, and the mother had no evidence of intraamniotic infection.
![]() What is the next step?
What is the next step?
ANSWER TO CASE 14: Neonatal Resuscitation
Summary: A newborn is born floppy, blue, and has responded poorly to initial resuscitation efforts of warming, drying, and stimulation.
• Next step: Evaluate heart rate (HR) and respirations. If no respirations are found or if HR is less than100 bpm (beats/min), initiate positive-pressure ventilation (PPV) by bag and mask. Because this mother received meperidine during the labor process, naloxone (Narcan) administration is an important step in resuscitation.
1. Understand the steps of newborn delivery room resuscitation.
2. Become familiar with use of the Apgar score.
3. Become familiar with conditions causing newborn transition problems.
This depressed infant was born to a healthy mother without prenatal or delivery complications other than the partially effective epidural anesthesia, which was supplemented with meperidine and promethazine. PPV was initiated and naloxone administered. The provider must appreciate the timing of maternal meperidine administration and its continued effects on the neonate.
APPROACH TO:
Neonatal Resuscitation
NARCOSIS:The condition of deep stupor or unconsciousness produced by a chemical substance such as a drug or anesthesia.
PERINATAL HYPOXIA: Inadequate oxygenation of a neonate that, if severe, can lead to brainstem depression and secondary apnea unresponsive to stimulation.
POSITIVE-PRESSURE VENTILATION (PPV): Mechanically breathing using a bag and mask.
Delivery room resuscitation follows the ABC rules of resuscitation for patients of all ages: establish and maintain the Airway, control the Breathing, and maintain the Circulation with medications and chest compressions (if necessary).
In this case, the meperidine given during labor probably is responsible for the infant’s apnea and poor respiratory effort. Neonates with narcosis usually have a good HR response but poor respiratory effort in response to bag-and-mask ventilation. The therapy for narcotic-related depression is intravenous (IV), intramuscular (IM), subcutaneous (SQ), or endotracheal administration of naloxone (Narcan); repeated doses may be required should respiratory depression recur.
The Apgar score (Table 14-1) is widely used to evaluate a neonate’s transition from the intra- to extrauterine environment. Scores of 0, 1, or 2 are given at 1 and 5 minutes of life for the listed signs. The 1-minute score helps to determine an infant’s well-being in the period just prior to delivery, and scores less than 3 historically have been used to indicate the need for immediate resuscitation. In current practice, HR, color, and respiratory rate (RR) rather than the 1-minute Apgar score are used to determine this need. The 5-minute score is one indicator of how successful the resuscitation efforts were. Some continue to measure Apgar scores beyond the 5-minute period to determine the continued response to resuscitation efforts. The Apgar score alone cannot determine neonatal morbidity or mortality.
Table 14-1 • APGAR EVALUATION OF A NEWBORN
14.1 A female infant is born through emergency cesarean section to a 34-year-old mother whose pregnancy was complicated by hypertension and abnormal fetal heart monitoring. At delivery she is covered in thick, green meconium and is limp, apneic, and bradycardic. Which of the following is the best first step in her resuscitation?
A. Administer IV bicarbonate.
B. Administer IV naloxone.
C. Initiate bag-and-mask ventilation.
D. Initiate chest compressions immediately.
E. Intubate with an endotracheal tube and suction meconium from the trachea.
14.2 A term male is delivered vaginally to a 22-year-old mother. Immediately after birth he is noted to have a scaphoid abdomen, cyanosis, and respiratory distress. Heart sounds are heard on the right side of the chest, and the breath sounds seem to be diminished on the left side. Which of the following is the most appropriate next step in his resuscitation?
A. Administer IV bicarbonate.
B. Administer IV naloxone.
C. Initiate bag-and-mask intubation.
D. Initiate chest compressions immediately.
E. Intubate with an endotracheal tube.
14.3 A 37-week-gestation male is born after an uncomplicated pregnancy to a 33-year-old mother. At birth he was lethargic and had an HR of 40. Oxygen was administered via bag and mask, and he was intubated; his HR remained at 40 bpm. Which of the following is the most appropriate next step?
A. Administer IV bicarbonate.
B. Administer IV atropine.
C. Administer IV epinephrine.
D. Administer IV calcium chloride.
E. Begin chest compressions.
14.4 A term female infant is born vaginally after an uncomplicated pregnancy. She appears normal but has respiratory distress when she stops crying. When crying she is pink; when not she makes vigorous respiratory efforts but becomes dusky. Which of the following is the likely explanation for her symptoms?
A. Choanal atresia
B. Diaphragmatic hernia
C. Meconium aspiration
D. Neonatal narcosis
E. Pneumothorax
14.1 E. An attempt is made to remove the meconium from the oropharynx and the airway prior to initiation of respirations. Ideally, the obstetrician will begin suctioning the meconium upon delivery of the head, and the pediatrician will further remove meconium with an aspirator or through endotracheal intubation with suction. Ventilation is initiated after meconium is removed. The goal is to remove airway meconium and to prevent its aspiration into the small airways where ventilation-perfusion mismatch may occur with deleterious effects.
14.2 E. The case describes diaphragmatic hernia. As a result of herniated bowel in the chest, these children often have pulmonary hypoplasia. Bag-and-mask ventilation will cause accumulation of bowel gas (which is located in the chest) and further respiratory compromise. Therefore, endotracheal intubation is the best course of action.
14.3 E. If the HR is still less than 60 bpm despite PPV with 100% oxygen, then chest compressions are given for 30 seconds. If the HR is still less than 60 bpm, then drug therapy (usually epinephrine) is indicated.
14.4 A. Infants are obligate nose breathers until about 4 months of age. When crying they can breathe through their mouth, but they must have a patent nose when quiet. Choanal atresia is identified by passing a feeding tube through each nostril or by identification of clouding on cold metal held under the infant’s nose. Should choanal atresia be diagnosed, endotracheal intubation bypasses the airway obstruction until surgical repair can be completed.
![]() An infant with slow heart rate, poor color, and inadequate respiratory effort requires immediate resuscitation.
An infant with slow heart rate, poor color, and inadequate respiratory effort requires immediate resuscitation.
![]() The therapy for narcosis (newborn respiratory depression because of maternal pain control) is intravenous, intramuscular, subcutaneous, or endotracheal administration of naloxone (Narcan).
The therapy for narcosis (newborn respiratory depression because of maternal pain control) is intravenous, intramuscular, subcutaneous, or endotracheal administration of naloxone (Narcan).
![]() A child with diaphragmatic hernia often presents with immediate respiratory distress, scaphoid abdomen, cyanosis, and heart sounds displaced to the right side of the chest.
A child with diaphragmatic hernia often presents with immediate respiratory distress, scaphoid abdomen, cyanosis, and heart sounds displaced to the right side of the chest.
![]() Choanal atresia results in respiratory distress when a child stops crying; immediate treatment is intubation until surgical correction can be completed.
Choanal atresia results in respiratory distress when a child stops crying; immediate treatment is intubation until surgical correction can be completed.
Carlo WA. Delivery room emergencies. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:575-579.
Carlo WA. Routine delivery room and initial care. In: Kleigman RM, Stanton BF, St. Geme JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: WB Saunders; 2011:536-538.
Ekrenkranz RA. Newborn resuscitation. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:207-213.
Thilo EH, Rosenberg AA. Diaphragmatic hernia. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:46-47.
Thilo EH, Rosenberg AA. Perinatal resuscitations. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:25-30.
Wyckoff MH. Delivery room resuscitation. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:164-170.
Yoon PJ, Kelley PE, Friedman NR. Choanal atresia. In: Hay WW, Levin MJ, Sondheimer JM, Deterding RR. Current Diagnosis & Treatment: Pediatrics. 20th ed. New York, NY: McGraw-Hill; 2011:473.
Zenel JA. Nose. In: Rudolph CD, Rudolph AM, Lister G, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:177.
A 12-month-old boy whom you have followed since birth arrives for a well-child visit. The mother is concerned that the baby’s manner of crawling, where he drags his legs rather than using a four-limbed movement, is abnormal. She says that the child only recently began crawling and he does not pull to a stand. You noted at his 6-month visit that he was not yet rolling over or sitting; previous visits were unremarkable as was the mother’s pregnancy and vaginal delivery. On examination today, you note that he positions his legs in a “scissoring” posture (ie, legs extended and crossed) when held by the axillae.
![]() What is the initial step in the evaluation of this child?
What is the initial step in the evaluation of this child?
![]() What is the most likely diagnosis?
What is the most likely diagnosis?
![]() What is the next step in the evaluation?
What is the next step in the evaluation?
ANSWERS TO CASE 15: Cerebral Palsy
Summary: A 12-month-old boy crawls using primarily his upper extremities, and holds his legs in a “scissoring” posture when suspended.
• Initial step: Gather detailed history, focusing on the age at which developmental milestones were achieved; obtain thorough pregnancy, birth, social, and family histories; and perform a detailed neurologic examination.
• Most likely diagnosis: Cerebral palsy (CP).
• Next step: Vision and hearing testing, consider a brain magnetic resonance imaging (MRI) scan, and arrange for therapy with a developmental specialist.
1. Know the definition of CP.
2. Recognize the classifications of CP.
3. Know the basic therapeutic approach to CP.
The spasticity of the baby’s lower extremities described is abnormal and is suggestive of CP. He has gross motor delay. A complete developmental and neurologic assessment is crucial for initiating therapies that will help him achieve maximal functional outcome. Although often of low yield, an attempt should be made to identify the etiology of the child’s CP. Knowing the etiology can aid in developing a treatment plan, subsequent family planning (especially if the etiology is inherited), and assuaging parental guilt for this child’s condition.
APPROACH TO:
Cerebral Palsy
CEREBRAL PALSY (CP): A disorder of nonprogressive movement and posture that results from an insult to or anomaly of the developing central nervous system (CNS). This definition recognizes the central origin of the dysfunction, thus distinguishing it from neuropathies and myopathies.
DEVELOPMENTAL DELAY: Failure of a child to reach developmental milestones of gross motor, fine motor, language, or social-adaptive skills at anticipated ages.
NEUROLOGIC DEFICIT: Abnormal functioning or lack of function of a part of the nervous system.
With a prevalence of 3 to 4 cases per 1000 live births, CP is the most common childhood movement disorder. Approximately one-third of CP patients also have seizures, and approximately 60% are mentally retarded. Deafness, visual impairments, swallowing difficulty with concomitant aspiration, limb and sensory impairments, and behavioral disturbances are common comorbidities.
Most children with CP have no identifiable risk factors. Current research indicates that CP most likely is the result of antenatal insults. Subsequent difficulties during the pregnancy, delivery, and perinatal period are thought to reflect these insults and are probably not the primary cause of CP.
Cerebral palsy, or “static” encephalopathy, is the result of a one-time CNS insult. In contrast, progressive encephalopathies destroy brain function with time. The term static is misleading, however, because the manifestations of CP may change with age. Contractures and postural deformities may become more severe with time or may improve with therapy. Also, a child’s changing developmental stages early in life can alter the expression of his or her neurologic deficits.
Immaturity of the CNS at birth makes diagnosis of CP nearly impossible in a neonate. If a CNS insult is suspected, head imaging (by ultrasound or MRI) can be helpful in recognizing CP early. Possible imaging findings include periventricular leukomalacia, atrophy, or focal infarctions. Beyond infancy, CP is suspected when a child fails to meet anticipated developmental milestones.
Examples of concerning findings are:
• A stepping response after the age of 3 months
• A Moro reflex beyond 6 months
• An asymmetrical tonic neck reflex beyond 6 months
Cerebral palsy can be classified in terms of physiologic, topographic, or functional categories. Physiologic descriptors identify the major motor abnormality and are divided into pyramidal (spastic) and extrapyramidal (nonspastic) categories. Extrapyramidal types can be subdivided further into choreoathetoid, ataxic, dys-tonic, or rigid types.
The topographic classification categorizes CP types according to limb involvement. Hemiplegia refers to involvement of a single lateral side of the body, with greater impairment of the upper extremities than the lower extremities. Diplegia describes four-limb involvement, with greater impairment of the lower extremities. Spastic quadriplegia is four-limb involvement with significant impairment of all extremities, although the upper limbs may be less impaired than lower limbs. (The term paraplegia is reserved for spinal and lower motor neuron disorders.)
The functional classification of CP relies on the “motor quotient” to place patients into minimal, mild, moderate, and severe (profound) categories. The motor quotient is derived by dividing the child’s “motor age” (ie, motor skills developmental age) with the chronologic age. A motor quotient of 75 to 100 represents minimal impairment, 55 to 70 mild impairment, 40 to 55 moderate impairment, and lesser quotients severe impairment. These categories help clinicians identify children with less obvious impairments so that early treatment can be provided.
The evaluation of CP is based on the history and physical examination. The yield of diagnostic findings with brain imaging and metabolic or genetic testing is low but can be helpful in managing the patient, in future family planning, and in reassuring the parents. Identification of comorbid conditions includes cognitive testing for mental retardation and electroencephalography (EEG) for seizures.
Treatment goals include maximizing motor function and preventing secondary handicaps. During the preschool years, the child’s communication ability is important. School performance and peer acceptance become important issues for older children. Physical therapy for motor deficits may be supplemented with pharmacologic and surgical interventions. Occupational therapy improves positioning and allows for better interaction with the environment and eases care as the child grows. The family’s psychological and social needs should not be overlooked; children may require extensive physical and emotional support.
15.1 A term infant requires resuscitation after a spontaneous vaginal delivery. The Apgar scores at 1, 5, and 10 minutes were 2, 7, and 9, respectively. The mother’s medical records show that she received routine prenatal care with normal prenatal ultrasonogram, triple screen, and glucose tolerance tests. The nurse tells you that the father seemed very agitated and mentioned “suing the obstetrician if the baby does not turn out normal.” Your examination of the baby reveals no abnormalities. In counseling the family, which of the following is most appropriate?
A. Inform them that your examination findings indicate that everything is fine.
B. Tell them that the low Apgar scores at 1 and 5 minutes indicate that the baby suffered perinatal asphyxia.
C. Inform them that because the pregnancy was uncomplicated, any neurologic deficit that the baby may develop likely can be attributed to events occurring at delivery.
D. Tell them that your examination findings are reassuring, and that you will perform a careful developmental assessment at every well-child visit.
E. Avoid speaking to the parents until you have had a chance to speak with the obstetrician and to see the cord blood gas results.
15.2 A 4-year-old child with CP comes to your clinic for the first time for a routine visit. He walks with the help of leg braces and a walker, and his speech is slurred and limited to short phrases. He has never been hospitalized, and he does not have swallowing problems. He began walking at the age of 2.5 years, and he is unable to take off his clothes and use the toilet without help. On examination you find that the boy has only minimally increased tone in the upper extremities but good fine motor coordination; he has significantly increased tone and deep tendon reflexes in the lower extremities. How would you categorize this child’s CP?
A. Mild, diplegic
B. Mild, hemiplegic
C. Moderate, diplegic
D. Moderate, quadriplegic
E. Severe, diplegic
15.3 A female is born through spontaneous vaginal delivery at 28-week gestation because of an incompetent cervix. Which of the following features of her clinical course in the neonatal intensive care unit (ICU) is most likely to correlate with her clinical outcome 5 years from now?
A. Administration of surfactant
B. Apnea of prematurity
C. Grade IV intraventricular hemorrhage
D. Retinopathy of prematurity stage 1 on initial ophthalmologic examination
E. Umbilical artery catheterization
15.4 The parents of a 2-year-old girl, recent immigrants from Guatemala, bring their child to you for the first time. The child was born at term after an uncomplicated pregnancy and delivery, and her neonatal course was uneventful. She sat without support at 6 months of age, pulled to a stand at 10 months, and walked at 14 months. She has a 10-word vocabulary, is able to drink from a cup, and feeds herself with a spoon. A previous child in the family died at the age of 5 years from “heart trouble.” On physical examination, you note lower extremity contractures, hand stiffness, somewhat coarse facial features, and hepatosplenomegaly. The child’s growth is within normal limits, and her examination is otherwise normal. Which of the following is the most appropriate next step to diagnose this child’s condition?
A. Abdominal computerized tomography (CT)
B. Brain magnetic resonance imaging (MRI)
C. Chromosomal analysis
D. Tests for a storage disorder
E. Thyroid function studies
15.1 D. The Apgar score at 1 minute reflects the neonatal environment immediately prior to birth; the 5-minute score correlates the infant’s response to resuscitation. The Apgar scores are not an accurate reflection of morbidity. An examination is a better indicator of the child’s outcome, but CP cannot be ruled out on the basis of a normal neonatal physical examination. A discussion of the events of delivery is best left to the obstetrician; the majority of difficult deliveries are the result of a previously unidentified antenatal insult. However, avoidance of the parents will likely only further their anxiety and may impede your efforts to provide care for the child.
15.2 C. In diplegia all four extremities are affected, with greater impairment of the lower extremities. As most children walk by the age of 14 months, this child’s motor quotient is 14 months/30 months = 0.47, which classifies him as moderately impaired.
15.3 C. Intraventricular hemorrhage is a complication in preterm infants. It is associated with seizures, hydrocephalus, and periventricular leukomalacia. A grade IV bleed involves the brain parenchyma, putting this child at higher risk for neurodevelopmental handicap.
15.4 D. The enlarged liver and spleen, the coarse facies, and the history of a previous child’s death from “heart trouble” point to a storage disorder. Her joint contractures and hand stiffness may be explained by an abnormal metabolism rather than a CNS deficit as in CP.
![]() Cerebral palsy is a disorder of movement or posture resulting from an insult to, or an anomaly of, the central nervous system.
Cerebral palsy is a disorder of movement or posture resulting from an insult to, or an anomaly of, the central nervous system.
![]() Most children with cerebral palsy have no identifiable risk factors for the disorder.
Most children with cerebral palsy have no identifiable risk factors for the disorder.
![]() Optimal treatment plans for cerebral palsy use a multidisciplinary approach.
Optimal treatment plans for cerebral palsy use a multidisciplinary approach.
American Academy of Pediatrics. Use and abuse of the Apgar score. Available at: http://www.aap.org. Accessed April 23, 2012.
Cooley WC and Committee on Children with Disabilities. Providing a primary care medical home for children and youth with cerebral palsy. Pediatrics. 2004; 114:1106-1113.
Johnston MV. Cerebral palsy. In: Kliegman RM, Stanton BF, St. Geme III JW, Schor NF, Behrman RE, eds. Nelson Textbook of Pediatrics. 19th ed. Philadelphia, PA: Elsevier WB Saunders; 2011:2061-2065.
Rust RS, Urion DK. Cerebral palsy and static encephalopathies. In: Rudolph CD, Rudolph AM, Lister GE, First LR, Gershon AA, eds. Rudolph’s Pediatrics. 22nd ed. New York, NY: McGraw-Hill; 2011:2178- 2181.
Shapiro BK, Capute AJ. Cerebral palsy. In: McMillan JA, Feigin RD, DeAngelis CD, Jones MD, eds. Oski’s Pediatrics: Principles and Practice. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:2251-2258.