Cutaneous Appendageal Tumors
TUMORS WITH HAIR FOLLICLE DIFFERENTIATION
Benign Nongerminative Follicular Neoplasms
Dilated Pore of Winer and Pilar Sheath Acanthoma
Tumor of the Follicular Infundibulum
Proliferating Trichilemmal Cyst (Benign Proliferating Trichilemmal Tumor)
Benign Follicular Neoplasms with Germinative-Type Differentiation
Neoplasms with Differentiation Toward Follicular Mesenchyme
Pseudoneoplastic Sebaceous Proliferations
Tumors with Hair Follicle Differentiation
The hair follicle is a microscopically complex structure, and, correspondingly, it is not surprising that classification schemes for lesions showing follicular differentiation are numerous and similarly complex—particularly to students who are approaching the subject for the first time. For the purposes of this chapter, the author will categorize these lesions according to their predominant lines of differentiation: outer root sheath epithelium (nongerminative), germinative epithelium, follicular mesenchyme with or without an epithelial component, and malignant pilar neoplasms. Several nevoid lesions of hair follicles will also be discussed. It will also be seen that varying degrees of differentiation toward follicular epithelium can be observed in the nonmalignant pilar lesions. Thus, the epithelial elements of hair follicle nevi bear a close resemblance to normal follicles, and the same can be said of dilated pore of Winer, trichofolliculoma, and trichogenic trichoblastoma. On the other hand, the epithelial structures in proliferating trichilemmal tumor, pilomatricoma, and trichogerminoma (trichoblastoma) may be barely recognizable as having any association with hair follicles whatsoever. In addition to the better delineated adnexal tumors, there are also benign and malignant neoplasms with mixed differentiation that may show as one element a degree of follicular differentiation. Such tumors are difficult to classify but appear from time to time, especially in a busy consultation practice.
Nevoid Follicular Lesions
Clinical Features: Hair follicle nevi, or congenital vellus hamartomas, are considered malformations rather than neoplasms. They are found most often in young individuals and tend to arise in the head and neck regions.1–4 There are multiple flesh-colored papules that sometimes coalesce, ranging from 1 to 5 mm in diameter. They can be associated with either hypertrichosis or alopecia. Linear lesions are sometimes noted, being distributed along Blaschko lines.5 One example is associated with frontonasal dysplasia.6 Note that the so-called “wooly hair nevus” of the scalp is composed of structurally abnormal hair shafts but otherwise lacks significant histopathologic abnormalities in the underlying skin.7
Microscopic Findings: There are collections of small follicular structures, usually displayed in cross-sectional profiles. They have the usual structural characteristics of vellus follicles, although some may have a slightly immature appearance.1–4 Basement membranes are inconspicuous, and the follicles are surrounded by ordinary-appearing connective tissue.
Differential Diagnosis: Facial biopsies sectioned tangentially, particularly when obtained from younger individuals, may give the impression of increased numbers of small vellus follicles but still be within the range of normal. The observation that some of the follicles in a lesion are developmentally immature should lead to the consideration of a hair follicle nevus. Hamartomas with a hair follicle component, such as the common accessory tragus and the uncommon rhabdomyomatous mesenchymal hamartoma, have some of the features of hair follicle nevus and may represent part of a spectrum of lesions. However, these also have distinctive connective tissue components. Goldenhar syndrome is an uncommon condition that combines accessory tragi with malar, maxillary, or mandibular hypoplasia; epibulbar dermoids; and numerous other anomalies.8 The epibulbar dermoid is regarded as an example of choristoma—a benign congenital overgrowth of abnormally located tissue. In this case, the tissue composing the epibulbar dermoid is almost a perfect mimic of skin. The microscopic findings of epibulbar dermoid and accessory tragus in the same individual can lead to a reasonably confident diagnosis of Goldenhar syndrome.
Unlike the hair follicle nevus, trichofolliculoma features a central dilated follicle from which branch numerous secondary follicles in a radial arrangement. Nevus comedonicus contains dilated follicles filled with keratin and showing irregular proliferations of their walls; connections to the surface epidermis are readily identified. In earlier texts, pigmented hair nevus, also known as Becker nevus, was sometimes included as a type of hair follicle nevus. Although this lesion features prominent hypertrichosis, the follicles themselves have a mature appearance and are not clustered but widely dispersed within these radially extensive lesions. Furthermore, the epidermis features acanthosis with pointed rete ridges and basilar hypermelanosis, and an association with connective tissue nevus or smooth muscle hamartoma is sometimes demonstrable.
Basaloid Follicular Hamartoma
Clinical Features: Basaloid follicular hamartoma is a pilar-related tumor that has been variously interpreted as a malformation or as a neoplasm. It may be seen in one of four clinical settings: as a sporadic solitary papule, as a plaque on the scalp with associated alopecia, as a group of linear papules, and as a group of generalized papules in patients with autoimmune conditions such as myasthenia gravis and lupus erythematosus.9 Lesions range from 1 to 5 mm in diameter, are flesh colored, and can be encountered in individuals of all ages; multiple lesions in children are usually associated with autosomal dominant inheritance.10,11
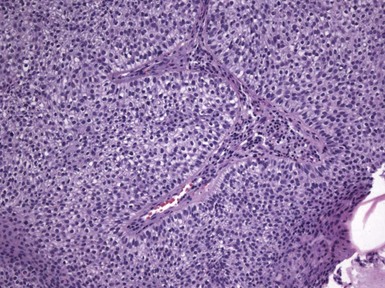
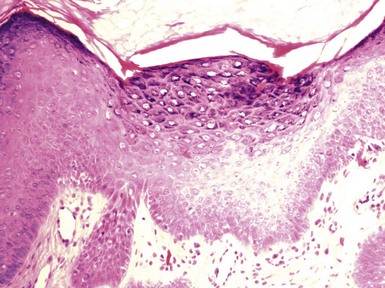
Microscopic Findings: Basaloid follicular hamartomas show anastomosing cords and strands of cytologically bland basaloid or polygonal cells, attached to basilar epidermis or hair follicles12,13 (Fig. 19-1). Apoptosis is uncommon, and mitotic figures are usually absent. Clefting artifact or myxoid stromal changes, as seen in basal cell carcinomas, are not evident. Structures suggesting follicular bulbs and papillary mesenchymal bodies can be observed.14
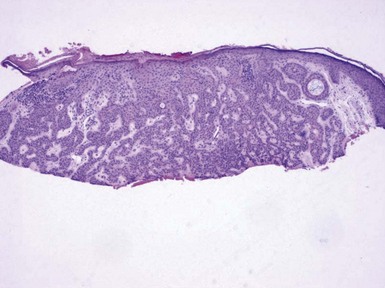
Differential Diagnosis: The chief differential diagnostic considerations are superficial basal carcinoma with a fronded or reticulated configuration and the infundibulocystic variant of basal cell carcinoma. The latter can also be multifocal and occur as a genodermatosis.15 However, as mentioned previously, mitotic activity, stromal retraction, and mucinous connective tissue are features of basal cell carcinoma that are not seen in basaloid follicular hamartoma. In a recent report, the authors were able to differentiate nevoid basal cell carcinoma syndrome from basaloid follicular hamartoma genetically, confirming the former by finding a novel PTCH1 germline mutation.16 The view has been expressed that basaloid follicular hamartoma may be a variant of trichoepithelioma.17 The classic form of the latter lesion has a more nodular configuration, and papillary mesenchymal bodies are more readily identified. However, a recent study of CD34, BCL2, and CD10 expression showed similarities among trichoepithelioma, the lesions of generalized basaloid follicular hamartoma syndrome, and two other benign lesions—vellus hair hamartoma and neurofollicular hamartoma.18 There is also some resemblance to tumor of the follicular infundibulum. However, that tumor has a more platelike growth beneath the epidermis, although with connections to the epidermal surface, and the constituent cells are more squamoid in appearance.19 With regard to immunohistochemical staining, basaloid follicular hamartomas show CD34 positivity around the epithelial strands and have a low proliferative index via Ki-67 staining, in contrast to basal cell carcinomas.
Benign Nongerminative Follicular Neoplasms
Dilated Pore of Winer and Pilar Sheath Acanthoma
Clinical Features: The dilated pore of Winer presents as a large open comedo on the face or trunk, particularly in elderly individuals.20 These lesions are only of cosmetic concern. An association with nevus comedonicus and other follicular tumors has been reported.21 The pilar sheath acanthoma is usually found on the face, particularly above the upper lip, and presents as a nodule with a central keratin plug. Both lesions are effectively removed by simple excision.
Microscopic Findings: The dilated pore consists of a keratin-filled, epithelial-lined cyst or sinus surrounded by mildly proliferative, budding epithelium. The resemblance to a follicular unit is supported by the occasional presence of sebaceous glands or vellus hairs (Fig. 19-2). Pilar sheath acanthoma shows a cystically dilated, infundibulum-like pore surrounded by markedly proliferative, lobulated epithelium (Fig. 19-3). The constituent cells are either polyhedral, with pale eosinophilic to clear cytoplasm, or basaloid, thereby having a resemblance to outer root sheath epithelium. The findings in these two lesions strongly suggest that they are closely related, probably representing points within the same pathologic spectrum.
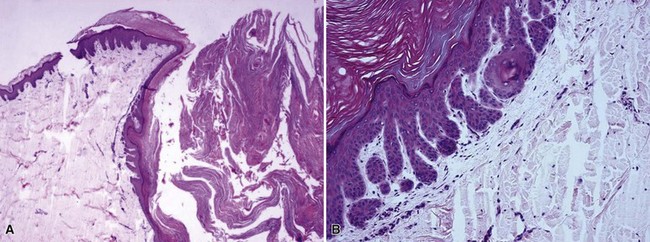

Differential Diagnosis: The clinical appearance of pilar sheath acanthoma may suggest a small version of keratoacanthoma, but that is not borne out by the microscopic features. In particular, the epithelium lacks the degree of proliferation, glassy cytoplasm, or the intraepithelial neutrophilic microabscesses characteristic of keratoacanthoma. As indicated previously, the differences between dilated pore and pilar sheath acanthoma are largely ones of degree, with dilated pore displaying a closer resemblance to a mature hair follicle.
Trichoadenoma
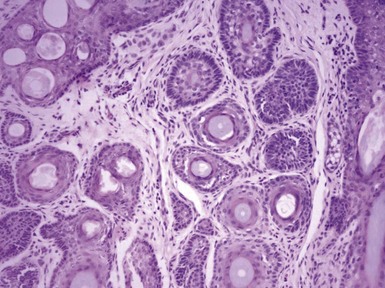
Clinical Features: Trichoadenoma was once considered by some to represent a variant of trichoepithelioma—presumably one consisting of mostly horn cysts—but appears to be unique enough to merit a separate designation. It sometimes bears the eponymic name trichoadenoma of Nikolowski.22 It is typically a solitary nodule that develops on the head and neck or occasionally the trunk. Simple excision is effective treatment.
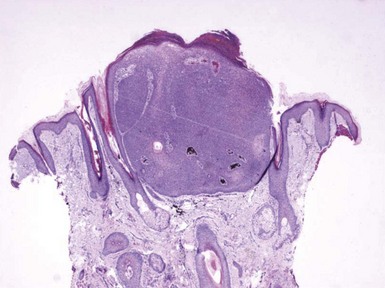
Microscopic Findings: This lesion consists of numerous keratin-filled cysts composed of polyhedral cells with eosinophilic to clear cytoplasm; some of the lining cells display keratohyaline granules. Solid buds of epithelium are also noted, although some of these exhibit cystic spaces on serial sectioning. Basaloid elements are not evident. The surrounding stroma is fibroblastic; attachments to the epidermis are not observed, and hair shafts are not identified in the cystic spaces (Fig. 19-4). Staining for carcinoembryonic antigen (CEA), epithelial membrane antigen (EMA), chromogranin A, and CD15 is negative.
Differential Diagnosis: The chief differential considerations include two lesions with similarly appearing horn cysts: trichoepithelioma and microcystic adnexal carcinoma.23,24 However, both of these lesions also feature narrow cords of epithelial cells, with evidence of lumen formation in those of microcystic adnexal carcinoma. In a sufficiently large biopsy/excisional specimen, trichoepitheliomas (particularly the desmoplastic variants) have a sharply demarcated base, whereas microcystic adnexal carcinomas show deep infiltration within the subcutis.24 Calcification within the horn cysts is a typical feature of trichoepithelioma. In contrast to trichoadenomas, desmoplastic trichoepitheliomas are usually positive for EMA and chromogranin, whereas microcystic adnexal carcinomas are positive for CEA and CD15. The findings can also be almost identical in chloracne, a distinctly different clinical picture but one in which horn cysts are prominent and sebaceous lobules inconspicuous.
Trichilemmoma
Clinical Features: Trichilemmoma is primarily a tumor of the head and neck region that is most commonly seen in adults. In most instances, this tumor arises sporadically, with no particular disease association. However, multiple lesions are observed in Cowden syndrome, an autosomal dominant disorder that combines various skin tumors with hamartomatous intestinal polyps and carcinomas of the breast, thyroid gland, and other sites. This syndrome has been linked to a mutation in a tumor suppressor gene known as PTEN on chromosome 10.25 The trichilemmoma is also one of the adnexal tumors that can arise in association with a nevus sebaceus. Simple excision is sufficient for removal.
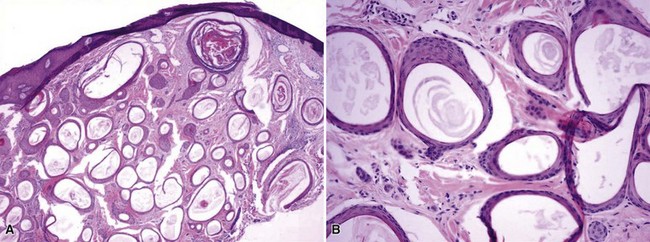
Microscopic Findings: Trichilemmomas are characterized by lobulated, follicle-centered proliferations of bland, uniform polyhedral cells with clear glycogenated cytoplasm (Fig. 19-5). The peripheral basal cells of the lobular proliferations demonstrate striking nuclear palisading, and the lobules are surrounded by a closely applied, cuticular basement membrane (Fig. 19-6). The surface frequently shows papillomatosis, parakeratosis, and scale crusting, with a configuration mimicking that of verruca. The lobules of an ordinary trichilemmoma have rounded contours with “pushing” borders, but the variant of this tumor called desmoplastic trichilemmoma shows a pseudoinfiltrative base, with islands of epithelial cells with somewhat fusiform appearance irregularly interdigitating between fibromyxoid connective tissue (Fig. 19-7). Preservation of the cuticular basement membrane is a useful diagnostic clue, and this structure can be highlighted with periodic acid–Schiff (PAS), collagen IV, or laminin stains. One study demonstrated CD34 reactivity in all trichilemmoma variants.26

Differential Diagnosis: The differential diagnosis of trichilemmoma includes other tumors with clear cell variants, including basal cell carcinoma and squamous cell carcinoma. The greater degrees of atypia in those two lesions should ordinarily allow distinction, and the CD34 positivity of trichilemmoma may also constitute a distinguishing feature. However, a variant of basal cell carcinoma has been described with thickened basement membrane, capable of mimicking trichilemmoma and other benign tumors.27 In addition, basal cell carcinoma has been reported to arise in association with desmoplastic trichilemmomas.28 Inverted follicular keratosis has an endophytic, lobulated growth arrangement similar to that of trichilemmoma, and in fact the two are often discussed together as benign, lobulated verruciform lesions. However, the inverted follicular keratosis has a closer resemblance to the irritated seborrheic keratosis and is often considered an endophytic variant of that lesion. Accordingly, there are usually horn cysts and some degree of intercellular edema (spongiosis), while the distinctive peripheral palisading and cuticular basement membrane of trichilemmoma are not apparent. Although the distinction between the two benign lesions ordinarily would not be crucial, recognizing that a lesion represents trichilemmoma could be important because of the condition’s known association with Cowden disease. Neither trichilemmoma nor inverted follicular keratosis has been shown to contain the DNA of human papillomavirus.29,30
Tumor of the Follicular Infundibulum
Clinical Features: Tumor of the follicular infundibulum is an uncommon lesion with a predilection for middle-aged or elderly women. It typically presents as a solitary papule or nodule measuring less than 1 cm in diameter. Multiple or eruptive lesions have been reported but are less common. As is the case for trichilemmoma, these lesions have been reported in Cowden syndrome,19 although they have also been identified in the Schöpf-Schulz-Passarge syndrome, a rare autosomal dominant disorder that also includes keratoderma, apocrine hidrocystomas, hypotrichosis, and nail and tooth abnormalities.31
Microscopic Findings: There is a subepidermal epithelial growth with a platelike configuration and multiple connections with the surface epidermis. Exaggerated nuclear palisading of peripheral basal cells is evident, together with a prominent surrounding basement membrane, and the constituent keratinocytes have a glycogenated appearance, resembling that of the outer root sheath (trichilemmal sheath)19,32,33 (Fig. 19-8). Cytologic atypia and mitotic activity are not observed in the lesion. These are not hair-producing tumors, although follicles, sebaceous glands, or sweat glands may be present within the lesion. Deposits of elastic tissue have been found in the dermis immediately beneath the lesion.19
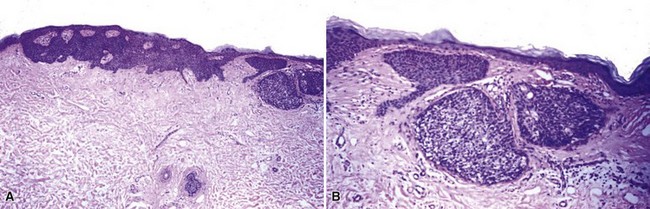
Differential Diagnosis: These lesions have a rather unique configuration. Some microscopic details—glycogenated cells, distinctly palisaded basilar cells, prominent basement membrane—are also seen in trichilemmoma, which is a closely related tumor and shares an association with Cowden syndrome. There is some resemblance to islands of superficial basal cell carcinoma, which can occasionally display cells with clear cytoplasm. However, in contrast to basal cell carcinoma, tumor of the follicular infundibulum is negative for BerEp4.
Proliferating Trichilemmal Cyst (Benign Proliferating Trichilemmal Tumor)
See Chapter 18, Epidermal Cysts and Tumors.
Benign Follicular Neoplasms with Germinative-Type Differentiation
In contrast to the previously discussed tumors, this group of neoplasms features one or more of the germinative components of the follicular unit, including matrical cells, cortical cells, and cells of the inner root sheath. They can show evidence of hair formation to varying degrees, ranging from the keratinization and “shadow cell” formation of pilomatricomas to the actual formation of hair shafts seen in trichofolliculoma.
Trichofolliculoma
Clinical Features: This lesion presents as a solitary, flesh-colored nodule developing in the head and neck region.34–36 A unique feature is the tuft of (often) white, vellus hairs emerging from the central portion of the lesion. Trichofolliculomas are removed by simple excision.
Microscopic Findings: There is a central, dilated primary follicle opening to the skin surface and lined by epithelium characteristic of the follicular infundibulum or isthmus. Secondary follicles bud from this primary follicle in a centrifugal configuration (Fig. 19-9), and depending on sectioning display either germinative epithelial differentiation or formation of mature hairs, and their cells possess isthmic or outer root sheath characteristics (Fig. 19-10). Sebaceous differentiation within the secondary follicles has been termed sebaceous trichofolliculoma.37 These epithelial features are present within a densely collagenous matrix.

Differential Diagnosis: Trichofolliculoma is occasionally confused with trichoepithelioma, but this is mainly related to the similarity of the terms. The tumor of trichoepithelioma appears more primitive and consists mainly of nests and cords of basaloid cells; horn cysts are present, but hair production is not observed. Similar considerations help distinguish trichofolliculoma from basal cell carcinoma with pilar differentiation; in addition, the stroma of basal cell carcinomas most often is of fibromyxoid type. Another lesion that bears a close resemblance to trichofolliculoma is the folliculosebaceous cystic hamartoma (see “Sebaceous Tumors”), which typically has a more cystic configuration and a stroma that may include neural, vascular, and adipocytic components. It has been proposed that these two lesions represent part of a spectrum,38 or that folliculosebaceous cystic hamartoma represents the late stage of a trichofolliculoma,39 but not all authors agree that these tumors have a pathogenetic relationship.40
Trichoepithelioma
Clinical Features: There are two major variants of this follicular tumor: classic trichoepithelioma and desmoplastic trichoepithelioma. Classic trichoepithelioma usually appears as a single, slowly growing, flesh-colored nodule on facial skin. It often appears in children41,42 but occasionally is first noted in older adults. It can reach a large size and appear on other body sites, although some experts would categorize the latter lesions among the trichoblastic tumors (see later discussion). Multiple trichoepitheliomas, arising usually in the head and neck region but occasionally elsewhere, are inherited as an autosomal dominant trait and are termed epithelioma adenoides cysticum or Brooke tumors. Multiple trichoepitheliomas sometimes coexist with multiple cylindromas (“turban tumors”), spiradenomas, salivary gland dermal analog tumors, trichilemmomas, or basal cell carcinomas.43,44 Associations of trichoepitheliomas with more generalized or systemic disorders are seen in Rombo syndrome (including milia, hypotrichosis, atrophoderma, basal cell carcinoma, and vasodilatation with cyanosis) or with systemic lupus erythematosus or myasthenia gravis.45
Molecular studies have shown that, when presenting in the context of a syndrome, trichoepithelioma is linked to genes with tumor suppressor characteristics that reside on chromosomes 9p21 and 16q12q13.46–48 In fact, multiple germline mutations in the CYLD gene (mapped to chromosome 16q12q13) have been found in families with multiple trichoepitheliomas, multiple cylindromas, or the Brooke-Spiegler syndrome (multiple trichoepitheliomas, cylindromas, or spiradenomas).49 On the other hand, sporadic trichoepithelioma and basal cell carcinoma share a loss of heterozygosity and chromosomal deletions at 9q22.3, the patched gene,50 as well as overexpression of the Gli1 transcription factor.51 These findings suggest a close relationship between these two tumors and raise the possibility that sporadic trichoepithelioma might be a variant form of basal cell carcinoma.
Desmoplastic trichoepithelioma often appears as a flesh-colored plaque in the central facial region of young or middle-aged women.52 It is typical for these lesions to have an elevated border and a somewhat depressed, sclerotic center. Although patients usually present with solitary lesions,17 the author has seen desmoplastic trichoepitheliomas in the setting of multiple tumors, some of which were of the classic type. Both lesions show indolent biologic behavior and are cured by simple excision; even partial excision may be sufficient in many cases.
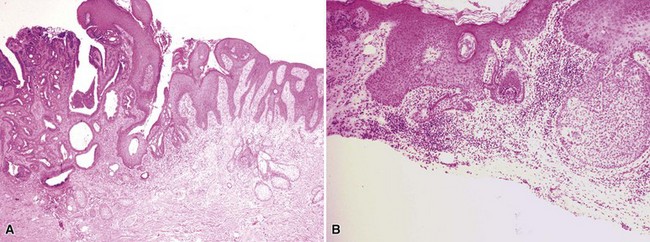
Microscopic Findings: Classic trichoepitheliomas show lobular dermal aggregates of uniform basaloid cells, separated by a mature, collagenized and sometimes hypercellular matrix. The lobules often display a fronted configuration and may interconnect by narrow cords of cells. Some contain keratinous cysts that are composed of cells with infundibular or isthmic differentiation (Fig. 19-11). Calcification within these cysts is occasionally seen. Another demonstration of the close association between epithelium and stroma in these tumors is the formation of papillary mesenchymal bodies—aggregates of fibroblast-like cells that are believed to represent attempts to form the dermal hair papillae responsible for induction of normal follicular units.53 These can be seen in close approximation to epithelial islands and sometimes appear within an epithelial invagination or in proximity to structures resembling hair bulbs (Fig. 19-12). Recognition of papillary mesenchymal bodies can be of substantial diagnostic importance (see subsequent discussion).
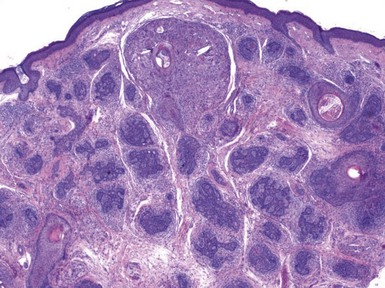
Figure 19-11 Trichoepithelioma. This is an example of a “classic” lesion, showing lobular aggregates of fronded basaloid cells within a cellular fibrotic matrix. Horn cysts can be identified, one of which has a surrounding granulomatous infiltrate.
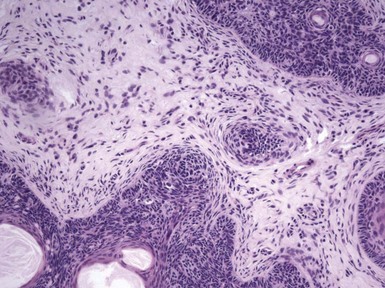
Figure 19-12 Trichoepithelioma: papillary mesenchymal body. A cluster of mesenchymal cells invaginates an island of basaloid cells.
In desmoplastic trichoepithelioma, low-power microscopic examination shows a disk-shaped dermal lesion (wider than it is deep) with sharply demarcated lateral margins. Narrow cords of compact polygonal epithelial cells are identified, without a palisaded basaloid component. Keratinous microcysts are evident, and these may display calcification or, rarely, metaplastic ossification (Fig. 19-13). The epithelial elements are again found within a dense and collagenous stroma, with variable cellularity. When biopsies are sufficiently deep, it is apparent that the base of the lesion is also well demarcated, the depth often limited to the midreticular dermis (Fig. 19-14). Deep subcutaneous infiltration is not observed, and perineural invasion is not a feature. An association with intradermal nevi has been reported on several occasions; when this occurs, nevus cell aggregates are found to reside within cords of epithelial cells in an intimate admixture that would not be expected in a “collision” tumor.54–56
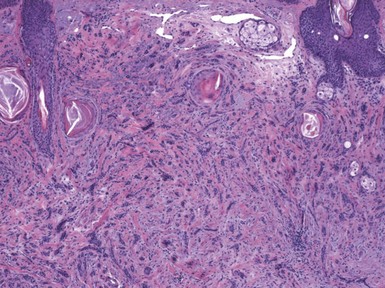
Figure 19-13 Desmoplastic trichoepithelioma. Cords of epithelial cells and microcysts are identified within a collagenous, cellular stroma.

Special stains, including immunohistochemical methods, are chiefly used to distinguish trichoepitheliomas from basal cell carcinoma and other adnexal tumors. Therefore, these are considered in “Differential Diagnosis.”
Differential Diagnosis: The chief differential diagnostic consideration when evaluating for possible trichoepithelioma is basal cell carcinoma. In contrast to trichoepithelioma, basal cell carcinoma is more apt to feature a fibromyxoid stroma, clefting artifact separating tumor islands from this adjacent stroma, and apoptotic foci in the presence of melanin deposition. In contrast, trichoepitheliomas more often feature papillary mesenchymal bodies,53 although these can be seen occasionally in basal cell carcinomas with follicular differentiation (“keratotic” basal cell carcinomas). Numerous studies have endeavored to find ways to distinguish trichoepithelioma from basal cell carcinomas using special stains and immunohistochemical methods. Thus, basal cell carcinomas are said to contain more elastic fibers than trichoepithelioma.57 CD34 positivity in peritumoral stroma is a feature of trichoepithelioma.58 Basal cell carcinomas typically stain strongly and uniformly for BerEp4 and BCL2, whereas trichoepitheliomas may show peripheral or more variable staining of tumor islands with these markers.59,60 Other markers that have been tried include the matrix metalloproteinase, stromelysin-3 (positive in the fibroblastic cells surrounding islands of morpheaform basal cell carcinoma but not in desmoplastic trichoepithelioma),61 CD10 (positive mainly in peritumoral stroma of trichoepitheliomas, in basaloid cells of basal cell carcinoma),62 and keratin 15 (peripheral localization is most typical for trichoepithelioma).57 A panel of stains consisting of keratin 20 (positive in desmoplastic trichoepithelioma but not in morpheaform basal cell carcinoma) and androgen receptor (less frequent in desmoplastic trichoepithelioma than in morpheaform basal cell carcinoma) has been recommended by Katona and colleagues.63 However, none of these stains, either in isolation or in combination, has proven to be completely reliable in distinguishing between these two lesions.60,64 This fact, combined with the often close morphologic resemblances and genetic similarities described earlier, leads to the conclusion that trichoepithelioma and basal cell carcinoma are part of a continuum. From a practical standpoint, trichoepitheliomas should be removed completely wherever possible, or at least subjected to close clinical surveillance.
Two other adnexal tumors that bear a passing resemblance to classic trichoepithelioma include cylindroma and spiradenoma, both thought to display sweat gland differentiation. Both of these lesions show much more intercellular basement membrane material. Cylindroma features mucin-filled cylinders of cells, whereas spiradenoma shows internal dispersion of mature lymphocytes and prominent lymphatic spaces. Trichoblastic tumors also bear a close resemblance to trichoepithelioma, and in fact, some authorities regard the former lesions as variants of trichoepithelioma. See “Trichoblastic Tumors” for a more detailed discussion of these tumors. In contrast to morpheaform basal cell carcinoma, the desmoplastic trichoepithelioma is better circumscribed both laterally and at its base, lacks a fibromucinous stroma or retraction artifact, and is not composed of truly basaloid elements.65 The distinction of desmoplastic trichoepithelioma from microcystic adnexal carcinoma can be quite difficult or impossible when superficial shave biopsies are submitted for evaluation. However, the latter consists of more tubular cell nests, and deeper biopsy shows that infiltrating cellular islands permeate deeply into the subcutaneous tissue and are prone to perineural infiltration.
Pilomatricoma
Clinical Features: Pilomatricoma, also termed calcifying epithelioma of Malherbe, is a tumor that displays differentiation toward the follicular matrix. It has a bimodal age distribution, with the greatest peak in children and young adults and a second peak in individuals older than 50 years of age. It presents as a deep, slow-growing, cystic nodule that is firm and sometimes rock hard on palpation. It most often arises on the head and neck but is sometimes found in other locations.66–69 Surface erosion is occasionally evident. Multiple pilomatricomas may be seen in myotonic dystrophy,70 and, as mentioned elsewhere, cysts showing features of pilomatricoma are identified in patients with Gardner syndrome.71 A possible association with Turner syndrome is less clear.72
Changes suggesting an intermediate-grade lesion (atypical or proliferating pilomatricoma) can be seen (see microscopic description). There is also a pilomatrix carcinoma, which is discussed in a later section. Occasionally, the deep location of a pilomatricoma may create a resemblance to an enlarged lymph node, prompting the use of fine needle aspiration biopsy. Diagnosis can be made with this technique,73 although the cytologic atypia often seen in specimens obtained by this method may lead to an erroneous diagnosis of metastatic carcinoma. Local excision is the recommended therapy for these lesions.
Microscopic Findings: It has been proposed that pilomatricoma begins as a cystic lesion lined by infundibular epithelium and basaloid, germinative matrical cells.74 This idea is supported by the finding of cysts with pilomatrical components in patients with Gardner syndrome71,75 (Fig. 19-15). However, in actual practice, a cystic configuration is only occasionally identified in biopsies of pilomatricoma—and then only in a portion of the lesion. Instead, there are two major components of these lesions organized with respect to one another, although sometimes appearing to be distributed through the lesion in a random arrangement: (1) aggregates of basaloid cells with monotonous round nuclei, dark chromatin, small nucleoli, and scattered mitotic figures, and (2) eosinophilic, keratinous material (Fig. 19-16). The basaloid cells tend to be aggregated at the periphery and show transition into an intermediate zone in which nuclei take on a washed-out appearance and are found within an amorphous, eosinophilic matrix; these are the so-called shadow cells (Fig. 19-17). Shadow cells exclusively occupy the central portion of these formations. Dystrophic calcification may be found in these keratinized zones; hence the other name for this tumor (calcifying epithelioma of Malherbe) (Fig. 19-18). Over time, the basaloid cells are replaced by keratinized shadow cells, associated with calcification and occasionally metaplastic ossification. The exposed keratinous material often elicits a vigorous, partly granulomatous inflammatory reaction.74 Foci of ossification occasionally show intratumoral extramedullary hematopoiesis.76
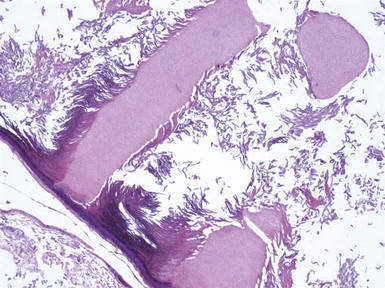
Figure 19-15 Pilomatricoma changes in cyst of Gardner syndrome. Arising in the wall of an epidermal cyst are columns of shadow cells. A few basaloid cells are seen between the two columns; these are elements of pilomatricoma.
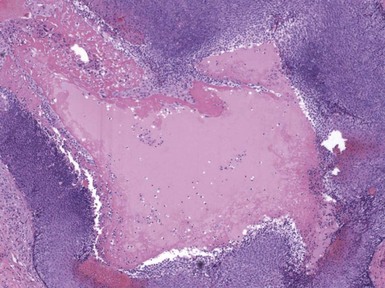
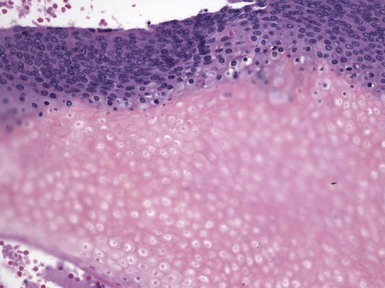
Figure 19-17 Pilomatricoma. The transition from basaloid cells to shadow cells can be seen in this image.
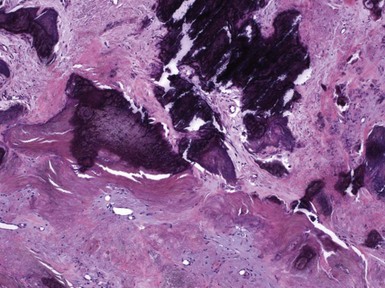
Microscopic variants of pilomatricoma include pilomatricoma with anetoderma, probably the result of elastophagocytosis by the granulomatous reaction that is frequently present77; perforating pilomatricoma, with extrusion of keratinous material through a disrupted epithelial surface78,79; and pigmented pilomatricoma.80 In another form of pilomatricoma, budding groups of tumor cells infiltrate into the surrounding tissue and elicit a desmoplastic reaction. These have been designated atypical, invasive, proliferating, or aggressive pilomatricomas, and unlike ordinary pilomatricomas, they are prone to local recurrence.81,82
Differential Diagnosis: Samples that show predominantly basaloid cells can be confused with conventional basal cell carcinoma, but the latter tumor usually shows peripheral palisading and clefting artifact that separates tumor islands from adjacent fibromucinous matrix, features not seen in pilomatricoma. Basal cell carcinomas with pilar differentiation may rarely show matrical differentiation, but other more typical features of basal cell carcinoma usually allow distinction. Pilomatricomas subjected to fine needle aspiration cytology can be confused with metastatic carcinoma; recognition of shadow cells in aspirates can then be a key to making the correct diagnosis.83 Shadow cells in general have been considered to be a clue to follicular differentiation; in addition to pilomatricoma and pilar basal cell carcinoma with matrical differentiation, they have been seen in hair shafts in some alopecias,84 the horn cysts of conventional and desmoplastic trichoepithelioma as well as microcystic adnexal carcinoma,84,85 mixed tumor of skin,84 proliferating trichilemmal tumor,86 and even intracranial dermoid cyst.87 Other features of these lesions should permit ready differentiation from pilomatricoma.
Trichoblastic Tumors
Clinical Features: In 1976, Headington proposed a grouping of hair follicle tumors that show features comparable to the odontogenic tumors; he called these trichoblastic tumors.88,89 They are classified microscopically according to degrees of differentiation and proportions of epithelial and mesenchymal components. Lesions present as nodules that range from 1 to 2 cm in diameter, may be deep-seated, and can be seen in virtually any location with the exception of the distal extremities. These generally benign lesions are managed by simple excision. One exception, a trichogerminoma reported by Sau and associates, metastasized.90
Microscopic Findings: The most primitive tumor in this group is known as trichoblastoma or trichogerminoma. It is a well-demarcated lesion, centered in the deep dermis, composed of relatively uniform basaloid cells with dispersed chromatin, small nucleoli, and mitotic activity. Tumor islands are separated from one another by a cellular, collagenous to fibromucinous stroma. The characteristic feature is the formation of cellular “balls” within the larger tumor aggregates: these are small nodular groupings that have a concentric arrangement suggestive of hair bulbs (Fig. 19-19). Other features include nuclear palisading, PAS-positive membranes surrounding the larger tumor islands, microcysts, and apoptotic bodies. The trichoblastic fibroma is often regarded as the most common of the trichoblastic tumors. It consists of interconnecting cords of basaloid cells, two to three cells in thickness, separated by a cellular stroma91 (Fig. 19-20). A hyaline membrane surrounds the cords of cells. A rippled pattern trichoblastoma has been described.92 Small cystlike structures can be seen, and there are often basilar buds invaginated by stromal cells, recapitulating the hair bulb and primitive dermal hair papilla. Pigmented93 and multinodular94 subtypes have been reported. Lesions with a cellular, mucinous stroma have been termed trichoblastic myxoma, a tumor presumably analogous to the odontogenic myxoma88,92 (Fig. 19-21). In the uncommon trichogenic trichoblastoma, there is advanced follicular differentiation. There tends to be a centrifugal organization of the tumor, in which hair bulbs are arranged at the periphery of tumor islands and hairs are arranged centrally, found within small keratinizing cysts89 (Fig. 19-22). In the case of Requena and coworkers, advanced differentiation was demonstrated by dermal papilla-like structures and inner root sheath differentiation, although hair shaft formation could not be demonstrated.95
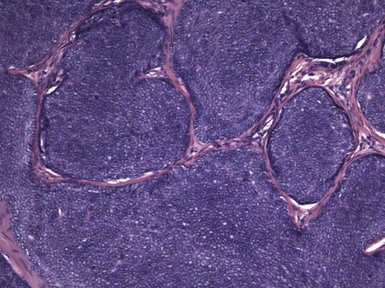
Figure 19-19 Trichoblastoma (trichogerminoma). Basaloid tumor islands show cellular “balls” with a vague suggestion of hair bulbs.
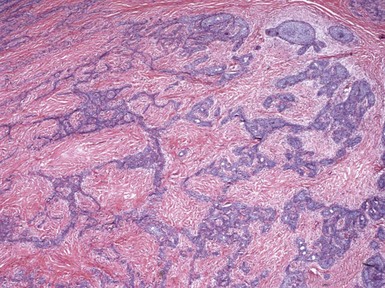
Figure 19-20 Trichoblastic fibroma. There are interconnecting cords of basaloid cells within a cellular stroma.
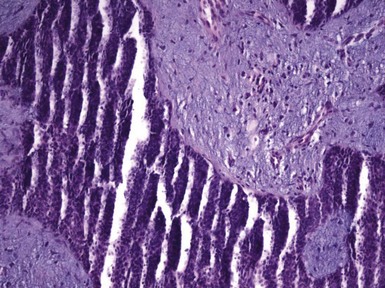
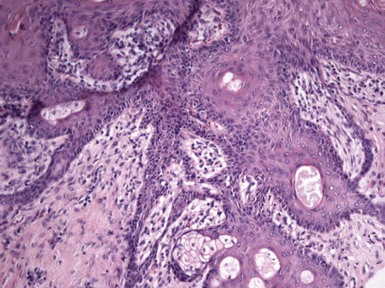
Figure 19-22 Trichogenic trichoblastoma. Advanced follicular differentiation is present. This view shows follicular units oriented toward a central point.
A feature often seen in trichoblastic tumors, particularly the trichoblastic fibroma, is adamantinoid change, resembling the odontogenic tumor ameloblastoma. This consists of tumor islands with palisades of basaloid cells surrounding loosely arranged cells in an edematous background, mimicking the stellate reticulum of the enamel organ. A lesion called cutaneous lymphadenoma probably represents an adamantinoid trichoblastoma, with the additional finding of prominent lymphocytic infiltration of the tumor islands.96,97 This relationship is further supported by a recent study by McNiff and colleagues, in which staining for CK20 (Merkel cells), BCL2, CD34, S-100, and CD1a produced similar results for lymphadenomas and trichoblastomas.98
Differential Diagnosis: The cellular “balls” in trichoblastoma (trichogerminoma) are probably sufficiently distinctive to distinguish this tumor from basal cell carcinoma or trichoepithelioma. Differentiation of trichoblastic fibroma from basaloid follicular hamartoma can be challenging because the latter also shows single-file cords of cells and a close association of epithelium and stroma. However, embryonic hair follicle–like structures are not as commonly seen in basaloid follicular hamartoma as they are in trichoblastoma. Trichogenic trichoblastomas that produce hair could have some features in common with trichofolliculomas, but the former are larger tumors that are less differentiated and do not show the organization of small but mature secondary follicles entering on a central, cystically dilated follicle. Some have suggested that trichoblastic tumors are simply variants of trichoepithelioma. However, the former often appear quite different clinically (larger lesions, often in locations other than the head and neck) and have much more variable histopathology with, in some cases, more advanced follicular differentiation. In several immunohistochemical studies of small nodular and rippled pattern trichoblastomas, Yamamoto and associates found similar keratin profiles for the two trichoblastic tumors, including expression of CK 7, which is not found in trichoepithelioma.92,99 CD10 may have some use in separating basal cell carcinoma from trichoblastoma; trichoblastomas show only peritumoral stromal staining for CD10, whereas basal cell carcinomas typically show intraepithelial staining. A combination of both epithelial and peritumoral stromal staining is seen in basal cell carcinomas with follicular differentiation.100
Neoplasms with Differentiation toward Follicular Mesenchyme
Several lesions have been described that have as a distinctive feature perifollicular stromal differentiation. Lesions with features of trichodiscoma, fibrofolliculoma, and perifollicular fibroma have been associated with the Birt-Hogg-Dube syndrome. It now appears that the findings these terms describe may actually represent variations on the same theme and that apparent differences among them are, in fact, effects of tissue sectioning.101 For that reason, these three findings will be discussed together.
Trichodiscoma, Fibrofolliculoma, and Perifollicular Fibroma
Clinical Features: The trichodiscoma has been proposed to represent differentiation toward the hair disc or haarscheibe.102 The hair disc is a receptor complex, including nerve ends and a fibrovascular stroma, associated with overlying epidermal Merkel cells.103,104 Trichodiscomas often present as a group of small, painless papules or small nodules in the head and neck region. They may be associated with other follicular neoplasms, particularly fibrofolliculomas. Simple excision is reported to be curative. The clinical presentation of fibrofolliculoma is similar. These lesions can be multiple, “associated” with trichodiscomas as part of the Birt-Hogg-Dube syndrome, but they can also be solitary and nonhereditary. Lesions described as perifollicular fibroma are sometimes grouped with fibrous papule. As such, they can also occur as solitary or multiple lesions.105 It has been suggested that “perifollicular fibroma” is actually a microscopic feature rather than a clinicopathologic entity and that it can be seen in a variety of guises—as a component of the Birt-Hogg-Dube syndrome, as a fibrous papule, in an angiofibroma, or as a kind of connective tissue nevus.
Microscopic Findings: In trichodiscoma, there is a superficial, ill-defined “disc” composed of hypocellular fibrovascular tissue that contains nerve twigs, the latter requiring special staining techniques for identification. Mucin and elastin are present in the stroma. Follicular units are seen at the periphery of these connective tissue changes, and it sometimes appears that follicles are “pushed aside” by the stroma102 (Fig. 19-23). Fibrofolliculoma shows a distorted, dilated, centrally located follicle with branching cords of epithelium that penetrate a surrounding fibrous mantle106 (Fig. 19-24). This feature has been termed an epithelial net,107 although it may imply a more complex network of epithelium than is usually observed. In perifollicular fibroma, there is lamellar fibrosis concentrated around hair follicles (Fig. 19-25). Similar changes can be seen around dermal vessels,108 an image closely resembling fibrous papule105 and angiofibroma.109 In fact, multiple lesions with features of angiofibroma can occur in the Birt-Hogg-Dube syndrome as well as in tuberous sclerosis and multiple endocrine neoplasia type I.109
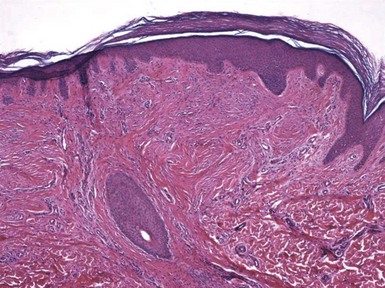
Figure 19-23 Trichodiscoma. A “disc” composed of fibrovascular tissue appears to “push aside” a follicular unit.
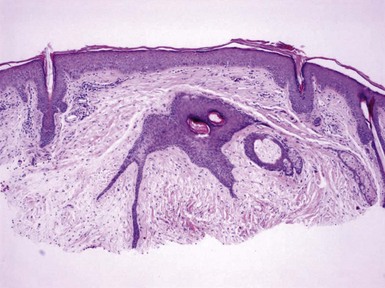
Figure 19-24 Fibrofolliculoma. There is a centrally located, distorted follicular unit with branching cords of epithelium that penetrate the surrounding connective tissue.
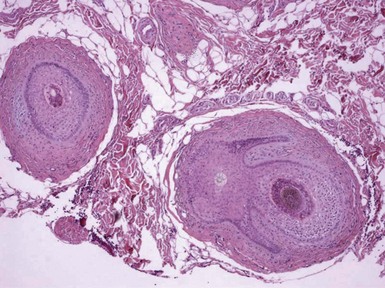
Figure 19-25 Perifollicular fibroma. Lamellar fibrosis surrounds two cross-sectional profiles of hair follicles. One of these also shows small branching epithelial cords.
The Birt-Hogg-Dube syndrome is an autosomal dominant disorder that links the cutaneous lesions described above with other abnormalities, including renal tumors (oncocytoma, papillary renal cell carcinoma, chromophobe renal carcinoma),110 pulmonary cysts with spontaneous pneumothorax,111,112 multiple lipomas, angiolipomas, parathyroid adenomas,113 and flecked chorioretinopathy.114 The association of multiple skin lesions with features of fibrofolliculoma, familial polyposis, and colorectal carcinoma had been designated the Hornstein-Knickenberg syndrome, but these features are now considered part of the spectrum of the Birt-Hogg-Dube syndrome.115 Much genetic research has linked the Birt-Hogg-Dube syndrome to abnormalities in chromosome 17p12-q11.2,112,116 in an area known as the BHD locus, or FLCN. This region codes for a protein called folliculin, which may have a tumor suppressor function. Multiple different mutations have been identified in this coding region.117
The skin lesions most closely associated with this syndrome have been traditionally designated trichodiscoma, fibrofolliculomas, and acrochordons. However, the acrochordons of Birt-Hogg-Dube syndrome actually have microscopic features consistent with the trichodiscoma-fibrofolliculoma spectrum,118 and careful studies suggest that all of these lesions represent fibrofolliculomas.101,119
Differential Diagnosis: It is apparent that careful orientation and multiple levels are needed to correctly classify these lesions, because many or most of them may show branching cords of follicular epithelium or laminated perifollicular fibrosis. Fibromyxoid changes resembling those of trichodiscoma are seen in superficial angiomyxoma120 and were also reported in fibromyxoid tumors of the adventitial dermis in tissues surrounding a meningomyelocele scar.121 These lesions appear quite different clinically from the usual trichodiscoma of facial skin, but an argument could be made that they are histogenetically related lesions. The microscopic features of fibrofolliculoma are rather unique, whereas, as noted above, changes of perifollicular fibroma may be found in forms of angiofibroma or connective tissue nevi.
Malignant Pilar Neoplasms
There are several well-delineated and specific malignant pilar neoplasms, which will be described below. In addition, carcinomas with pilar differentiation that are otherwise difficult to classify are encountered not infrequently in consultation material. Many of these show several lines of differentiation, including sweat gland and sebaceous as well as pilar. The author’s approach is to designate such lesions as adnexal carcinomas with divergent differentiation rather than to devise unnecessarily complex diagnostic terminology.122,123
Trichilemmal Carcinoma
Clinical Features: Trichilemmal carcinoma was first described by Headington in 1976.88 Since then, several published series support its designation as a distinct entity.124,125 This lesion commonly occurs in older adults, and frequent locations include the face, scalp, and ears. It presents as a single, hyperkeratotic, smooth-surfaced or ulcerated nodule or plaque, although multiple concurrent lesions have also been reported.126 The initial clinical diagnosis is often actinic keratosis, basal cell carcinoma, or squamous cell carcinoma. A linkage with Cowden syndrome has not been reported.127
Microscopic Findings: Trichilemmal carcinoma is a lobulated, follicular-based lesion that appears to replace the sheath of one or several follicular units. Tumor cells replace both the involved follicles and the perifollicular epidermis, separated by uninvolved adjacent epithelium. There is irregular infiltration of the dermis, associated with surrounding fibroplasia and chronic inflammation. Peripheral palisading may be demonstrated in tumor lobules. Some tumor cells possess clear cytoplasm due to the presence of glycogen (PAS-positive, diastase-sensitive cells) (Fig. 19-26). Foci of trichilemmal keratinization can be seen, and tumors may be accompanied by pagetoid intraepidermal proliferation and intravascular or perineural invasion.125
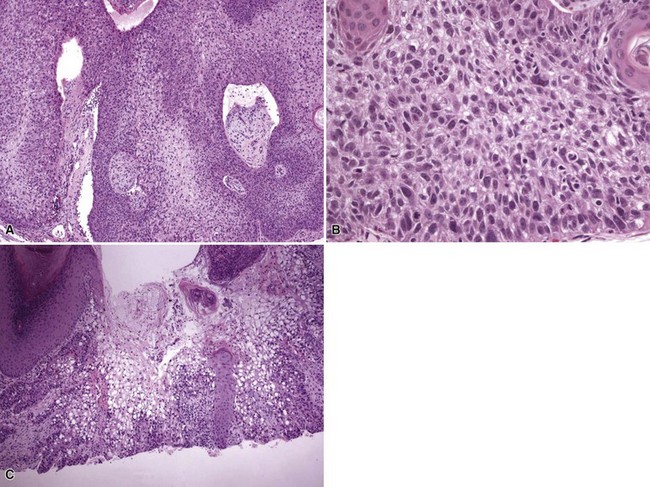
Differential Diagnosis: The differential diagnosis chiefly includes those tumors characterized by clear cell change, including clear cell variants of squamous cell, basal cell, sebaceous, and eccrine carcinomas. Also included are metastatic clear cell adenocarcinoma and melanomas with clear cell change. Knowledge of the clinical features and close attention to the microscopic findings, especially to the relationship of tumor with follicular units, should permit an accurate diagnosis of trichilemmal carcinoma. PAS staining is useful in establishing the presence of glycogen in clear cells, and immunostaining for selected keratins (such as the pilar-related keratins AE13 and AE14, or keratins 1, 10, 14 and 17) can provide further diagnostic support.125,128
Malignant Proliferating Trichilemmal Tumor
Clinical Features: The clinical setting of malignant proliferating trichilemmal tumor is quite similar to that of benign proliferating trichilemmal tumor (see Chapter 18). In fact, some of these tumors arise through transformation of proliferating trichilemmal tumors, in which case signs of malignancy include rapid enlargement or ulceration of existing lesions.129,130
Microscopic Findings: There are two forms of malignant proliferating trichilemmal tumor. The first, a low-grade variant, closely resembles the benign lesion but shows irregularly shaped cords or “buds” of neoplastic cells that invade adjacent connective tissue. The second, a high-grade lesion, lacks the lobulated configuration of the benign variant and shows geographic necrosis, marked nuclear pleomorphism, atypical mitotic figures, and sometimes spindle cell features129,131 (Fig. 19-27). Low-grade tumors tend to recur locally, but high-grade tumors are capable of distant metastasis.129
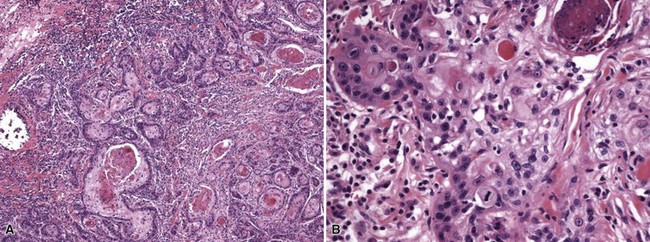
Differential Diagnosis: The chief consideration in the differential diagnosis is primary cutaneous squamous cell carcinoma, but in contrast to the usual examples of the former lesion, malignant proliferating trichilemmal tumors lack connections with the epidermis and show foci of trichilemmal keratinization. Anaplastic ductal eccrine adenocarcinoma shows some evidence of gland formation. Metastatic carcinoma with squamous characteristics does not show areas of trichilemmal keratinization, and the clinical presentation is generally quite different, because there are often multifocal and rapidly evolving lesions.
Pilomatrix Carcinoma
Clinical Features: The pilomatrix carcinoma is a rare tumor that most commonly arises in older adults132 but has been reported during the first two decades of life.81 Although pilomatricomas are more common in females, pilomatrix carcinomas arise more commonly in males.133 They are often reported to develop from preexisting pilomatricomas.81,134 Most commonly noted in the head and neck region,135 they have been reported on the trunk and extremities.134 Clinically they may resemble calcified cysts133 and can be quite large and ulcerated.136 Treatment is surgical excision,81,137 and there is a report of a case managed with Mohs surgery.138 Metastases to lymph nodes or viscera have been reported in 10% of cases.134,139
Microscopic Findings: As noted previously, and analogous to the low-grade malignant proliferating trichilemmal tumors, there is an intermediate pilomatrical lesion, termed proliferating pilomatricoma, that is mainly characterized by irregular cellular growth into adjacent tissue. In contrast to true pilomatrix carcinoma, these lesions do not appear to have metastatic potential, although they can recur locally. Pilomatrix carcinoma shows zones of geographic necrosis, invasive growth, and marked cytologic atypia, consisting of cells with high nuclear-to-cytoplasmic ratios, vesicular chromatin, and prominent nucleoli.132,136,140,141 The finding of shadow cells is the distinctive feature that marks the tumor as matrical in nature (Fig. 19-28). Immunohistochemistry has generally not been helpful in distinguishing between benign and malignant pilomatrical tumors, and therefore morphologic assessment is the principal means of establishing the diagnosis of pilomatrix carcinoma.140
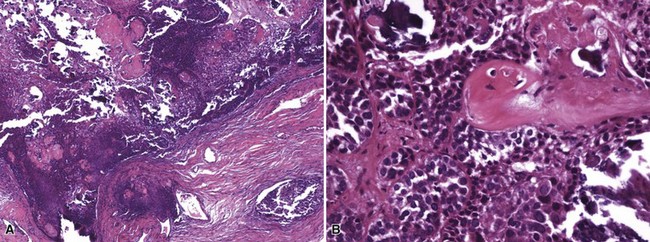
Differential Diagnosis: β-Catenin, a protein involved in signal transduction, is recognized as an oncogene in colon cancer and melanoma. Several recent studies have investigated the expression of β-catenin in the basaloid cells of pilomatricoma, because its expression has been found to correlate with mutations in its encoding gene CTNNB1. Both pilomatricomas and pilomatrix carcinomas show nuclear and cytoplasmic expression of β-catenin, indicating two things: (1) its expression does not explain the differences in biologic behavior of these two tumors, and (2) staining for β-catenin cannot be exploited in differentiating between them.142,143 However, the findings lend support to the idea that pilomatrix carcinomas from time to time may indeed arise from preexisting pilomatricomas.143 Small samples of pilomatrix carcinomas may create confusion with lymphoepithelioma-like carcinoma144 or undifferentiated metastatic carcinoma. At present, the best means for ruling out these two neoplasms are through clinical history and the demonstration of matrical differentiation by identifying shadow cells in pilomatrix carcinoma.
Sebaceous Tumors
Benign sebaceous tumors are relatively common, and they therefore frequently come into play when contemplating the diagnosis of a nonmelanocytic cutaneous lesion. On the other hand, it is perhaps a fortunate circumstance that the number and variety of sebaceous tumors is limited. This section will consider the classification and diagnostic approach to this group of lesions. Several other general considerations regarding sebaceous tumors should be mentioned here. First, it should be recalled that sebaceous glands constitute a component of the primary epithelial germ, and therefore it should not be surprising that there are composite adnexal tumors, difficult to classify, that may have follicular and/or apocrine as well as sebaceous elements. Second, a diagnostic dilemma is often encountered when confronting tumors composed of clear cells, especially in the case of carcinomas, because well-defined sebaceous cells with compact nuclei and multivacuolated cytoplasm may not be easily recognized in some sebaceous carcinomas. Third, the existence of sebaceous adenomas, sebaceomas, or sebaceous carcinomas may raise the possibility of Muir-Torre syndrome, prompting the use of immunohistochemistry for assessment of the products of mismatch repair genes.
Pseudoneoplastic Sebaceous Proliferations
Clinical Features: Hyperplasia is the most common sebaceous proliferation. It is most commonly encountered in middle-aged to elderly individuals. Sebaceous hyperplasia most often presents as umbilicated yellow-orange, telangiectatic papules of the face and eyelids, although it may also be found in the areola of the breasts, lips, or genitalia. Coalescence of lesions to form plaques is not uncommon.145 The umbilication and telangiectasias associated with this lesion can create the appearance of basal cell carcinoma, which is frequently considered in the clinical differential diagnosis. A causal relationship has been suggested between sebaceous hyperplasia and some immunosuppressive medications.146–148 Familial sebaceous hyperplasia is an uncommonly encountered, autosomal dominant condition that begins early in life.149,150
Microscopic Findings: Several sebaceous lobules are grouped around a common, dilated duct that opens on the epidermal surface. A vellus hair can sometimes be found in the vicinity. The lobules are composed of mature sebocytes and lined by a thin layer, or several layers, of basaloid cells151 (Fig. 19-29). Plaquelike lesions show several such structures immediately adjacent to one another.
Differential Diagnosis: The major differential diagnostic consideration is sebaceous adenoma, which structurally can appear to be quite similar, but has a more prominent component of basaloid cells. The clustering of sebaceous lobules around ducts and overall sharp lesional demarcation of sebaceous hyperplasia differ from rhinophyma or other manifestations of glandular hyperplastic rosacea, in which sebaceous lobules are not as well organized. Nevus sebaceus lesions tend to lack the sharp demarcation and grouping of lobules about a common dilated duct, while they show verruciform surface changes and/or an underlying apocrine nevus (especially in adults).
Nevus Sebaceus (Organoid Nevus)
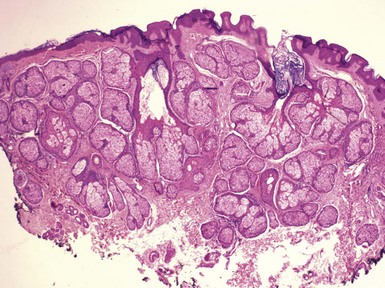
Clinical Features: Nevus sebaceus is a hamartoma that is most often found on the head and neck; most cases involve the scalp.152 It is occasionally inherited as an autosomal dominant trait. The condition is sometimes regarded as a phakomatosis (neurocutaneous syndrome), because extensive linear lesions have been associated with seizures, mental retardation, skeletal abnormalities, pigmentary changes, ocular lesions, and renal hamartomas.153 The lesions themselves most often present in early life as hairless, yellow or tan plaques of up to several centimeters in diameter. At puberty, these lesions can become distinctly verrucoid. This is a feature shared with epidermal nevus. Recently, epidermodysplasia verruciformis-associated and genital mucosal human papillomavirus (HPV; especially HPV 16) DNA has been found with high frequency in lesions of nevus sebaceus; the author suggests that this finding implies maternal transmission of HPV and infection of ectodermal stem cells, resulting in altered skin development along Blaschko lines154 (Fig. 19-30). Importantly, secondary neoplasms may develop in these lesions, including basal cell carcinomas (some of which in reality may be basaloid hyperplasias155 and a variety of other epithelial and appendageal tumors, including trichilemmoma, syringocystadenoma papilliferum, and trichoblastic tumors156,157 (Fig. 19-31). Adnexal carcinomas can also arise in nevus sebaceus; apocrine, pilar, and complex carcinomas have been described, some of which are capable of regional or generalized metastases.158 It has recently been shown that tissues from lesions of nevus sebaceus show deletions in the PTCH gene on chromosome 9q22.3.159 The same locus is aberrant in basal cell carcinomas, possibly explaining the frequent development of basal cell carcinomas (and perhaps other tumors) in nevus sebaceus.
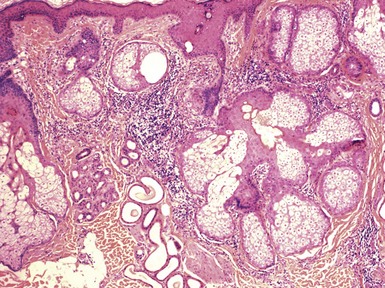
Microscopic Findings: The microscopic appearance of nevus sebaceus varies depending largely on the patient’s age.156 In infants and younger children, there appear to be immature and malformed follicular units with small sebaceous lobules (the latter being found especially after the first 3 months of life, with the diminished influence of maternal androgens), mainly occupying the superficial dermis (Fig. 19-32). After puberty, there is usually marked acanthosis with papillomatosis, having at times a distinctly verruciform configuration (Fig. 19-33). The sebaceous glands become enlarged, and there are proliferations of apocrine elements, dilatation of apocrine ducts, and, sometimes, increased numbers of eccrine sweat glands (Fig. 19-34). Sebaceous glands appear to attach directly to the overlying epidermis via a dilated duct or incompletely formed follicular unit (see Fig. 19-33) with an occasional vellus hair. There may also be disorder in the orientation of dermal collagen bundles, hypercellularity, and alteration in the density of neurovascular structures. This constellation of findings explains why many prefer the term organoid nevus for these lesions.
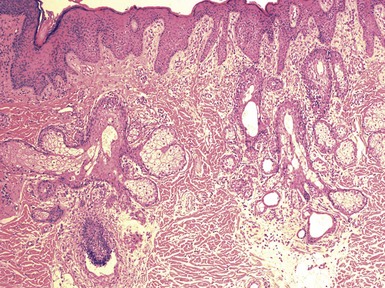
Figure 19-32 Nevus sebaceus in a 9-month-old child. Note immature, malformed follicular units with small sebaceous lobules in the superficial dermis.

Differential Diagnosis: Small biopsies, submitted without a clinical history, may generate a rather broad differential diagnosis. The surface changes can resemble those of epidermal nevus (without the other features of nevus sebaceus), verruca vulgaris, seborrheic keratosis, or even well-differentiated squamous cell carcinoma. The enlarged sebaceous glands encountered following puberty may suggest sebaceous hyperplasia, the sebaceous gland enlargement often seen in adult facial skin (particularly in individuals with glandular hyperplastic rosacea), or the sebaceous lobules sometimes seen overlying dermatofibromas as a manifestation of “stromal induction.” Biopsies that are focused on a secondary neoplasm developing in a nevus sebaceus (e.g., a basal cell carcinoma), may not show sufficient surrounding changes to indicate the lesion of origin. Knowing that one of these adnexal tumors has arisen on the scalp of a relatively young individual, within a preexisting lifelong lesion, can prompt consideration of the diagnosis and lead to a determination of the most appropriate therapy.
Folliculosebaceous Cystic Hamartoma
Clinical Features: The folliculosebaceous cystic hamartoma, a recently described condition,160 presents most often as a papule or nodule located in the central part of the face. Rarely, the lesion has been found elsewhere on the body surface,161 and a giant lesion has been reported.162 Rarely diagnosed clinically, it is a slow-growing lesion with a benign clinical course.
Microscopic Findings: There is an infundibulocystic structure in the dermis, to which are attached sebaceous lobules in radial array (Fig. 19-35). A characteristic stroma surrounding this epithelial structure appears to be sharply demarcated from the surrounding normal connective tissue; the stroma contains fibrous and adipose tissues, along with vessels and nerve bundles (Fig. 19-36). Immunohistochemistry demonstrates CD34 and factor XIIIa positivity within this surrounding tissue.163 In their immunohistochemical study, Toyoda and Morohashi found that nerve bundles were reactive for PGP 9.5 (a general neuronal marker protein gene product) but not for a variety of neuropeptides that are contained in normal cutaneous nerves. This finding was interpreted as supporting the concept that folliculosebaceous cystic hamartoma is a tumor-like malformation with overgrowth of normal skin components.164
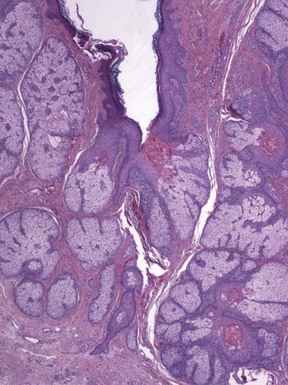
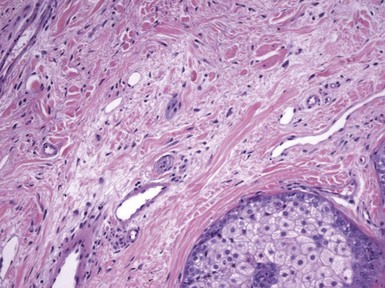
Differential Diagnosis: The unique constellation of features usually allows distinction of folliculosebaceous cystic hamartoma from other sebaceous neoplasms. However, as noted earlier, there is some resemblance between this lesion and trichofolliculoma, particularly the sebaceous variant. Although it has been proposed that these two lesions represent part of a spectrum,38 or that folliculosebaceous cystic hamartoma represents the late stage of a trichofolliculoma,39 not all authors agree.40 Misago and coworkers suggest that the up-regulation of nestin expression in lesions of folliculosebaceous cystic hamartoma may explain the unique mesenchymal changes in these lesions that are not appreciated in trichofolliculomas.165 Nestin is an intermediate filament protein expressed in follicle stem cells in the bulge region of the hair follicle, and its actions may result in the production of various mesenchymal and neural tissues.
Benign Sebaceous Neoplasms
Clinical Features: Sebaceous adenoma is a slowly enlarging facial nodule with a yellowish appearance, seen most often in individuals older than 50 years of age. Occasionally, this tumor may arise in unusual sites such as the oral cavity, ear canal, or salivary gland.145,166 It measures up to several centimeters in diameter. Clinical confusion of sebaceous adenoma with basal cell carcinoma is common. This lesion and other sebaceous tumors, including sebaceomas, sebaceous carcinomas, and keratoacanthomas (some of which may contain sebaceous elements) have been associated with the Muir-Torre syndrome, together with visceral malignancies.167,168 This complex of clinical features is associated with microsatellite instability, resulting from inactivating germline mutations in various DNA mismatch repair genes, particularly MSH-2 and MLH-1.169 Microsatellite instability is found in hereditary nonpolyposis colon cancer syndromes, including the Muir-Torre syndrome. The internal neoplasms in patients with Muir-Torre syndrome are usually laryngeal, mammary, or gastrointestinal carcinomas, but other types may be seen as well, including malignant lymphoma.168,170
Microscopic Findings: In sebaceous adenoma, there is a circumscribed proliferation of enlarged sebaceous lobules, composed of mature sebocytes. The tumor often appears to compress or thin the overlying epidermis and is surrounded by a fibrous pseudocapsule in some cases. The latter feature, as well as an occasional appendageal collarette, reflects the slow growth of sebaceous adenoma. The tumor lobules may show focal ductlike differentiation and holocrine secretion, which potentially results in the formation of microcysts; lesions with this feature have been termed sebocrine adenomas.171,172 There is a prominent basaloid component, representing germinative cells, although mature sebaceous cells predominate (Fig. 19-37). Despite the presence of small nucleoli, significant nuclear atypia or mitotic activity is absent. Forms of sebaceous adenoma in patients with Muir-Torre syndrome are not as well demarcated from the adjacent dermis, and some tumors include larger numbers of basaloid, germinative epithelial cells at their periphery. Rutten and colleagues have also found that cystic change in cutaneous sebaceous tumors is more frequent in patients with Muir-Torre syndrome, and this is compatible with the author’s experience.173
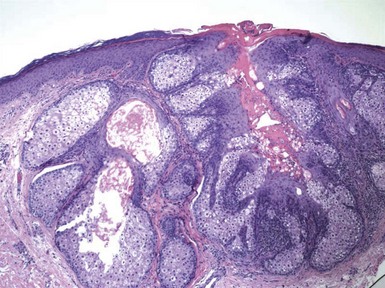
Figure 19-37 Sebaceous adenoma. This is a lobulated tumor having some architectural similarities to sebaceous hyperplasia but with a more prominent basaloid component.
With regard to special staining, Hassanein and associates have suggested that staining for Thomsen-Friedenreich antigen, a glycoprotein found on epithelial cells, can separate (trifluoroacetic acid [TFA]-positive) sebaceous carcinoma from other tumors with sebaceous differentiation.174 The author does not have personal experience with the use of this antibody; however, another paper suggests that TFA staining is also positive in a variety of other skin cancers.175 The other useful procedure is immunohistochemical staining for mismatch repair proteins, especially MSH-2 and MLH-1. A lack of nuclear staining for one of these markers within the tumor is evidence of a germline mutation of the corresponding gene, and in the particular context of a sebaceous tumor supports the diagnosis of Muir-Torre syndrome. The value of this procedure has been documented in a number of studies169,176,177 (Fig. 19-38). Recently, it has been reported that mutation of MSH-6 can also be found in patients with Muir-Torre syndrome, and in fact was the mismatch repair protein most commonly lost in one study.178 This determination can be important in prompting surveillance for internal neoplasms as well as in genetic counseling.
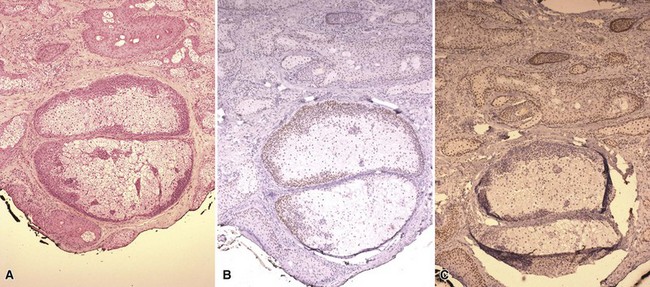
Figure 19-38 Sebaceous adenoma with immunohistochemical staining for mismatch repair proteins. This sebaceous adenoma has been identified at the base of a keratoacanthoma-like lesion. A, Findings on H&E-stained sections. B, Positive staining of nuclei for MLH-1. C, Negative staining of nuclei for MSH-2, in the face of positive staining of nuclei in adjacent epithelium and normal sebaceous lobules.
Differential Diagnosis: The differential diagnosis of sebaceous adenoma includes other sebaceous tumors and tumor-like proliferations, basal cell carcinoma with sebaceous differentiation, and sebaceous carcinoma. Sebaceous hyperplasia can have a low-power configuration that is quite similar to that of sebaceous adenoma, but the latter has a more prominent basal cell population, and lesions associated with the Muir-Torre syndrome show a degree of disorganization not encountered in sebaceous hyperplasia. Sebaceoma (see subsequent discussion) has a greater proportion of basaloid cells and in addition may show some features in common with trichoepithelioma, dermal duct tumor, cylindroma, and trichilemmoma. Basal cell carcinoma with sebaceous differentiation can show considerable histopathologic overlap with sebaceous adenoma, but shows a higher proportion of basaloid cells (>50% of the total cell population), a fibromyxoid stroma, and clefting artifact separating tumor lobules from the adjacent stroma. Sebaceous adenomas lack the degree of cytologic atypia and infiltrative growth pattern frequently observed in sebaceous carcinomas (although there may be circumscription and even peripheral palisading in tumor islands of the basaloid variant of sebaceous carcinoma).
Superficial Epithelioma with Sebaceous Differentiation
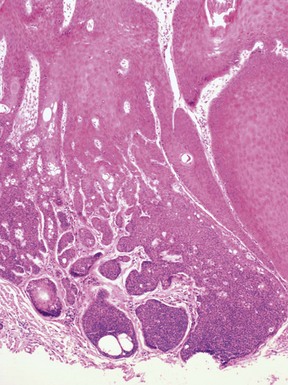
This is a rarely encountered neoplasm with sebaceous cells, described as a slow-growing papule or verrucoid nodule on the face, trunk, or proximal extremities; multiple lesions have been reported. Microscopic descriptions in various reports consistently describe a multilobular, platelike proliferation of tumor cells in the upper dermis with multiple broad connections to the overlying epidermis. The tumor cells are basaloid and feature single or aggregated sebocytes, with more lobulated arrangements in lower portions of the tumor179–183 (Fig. 19-39). Ductlike structures with eosinophilic luminal cuticles are frequently present, and squamous eddies can be observed. The image has features particularly reminiscent of tumor of the follicular infundibulum with sebaceous elements. Thus far, there has been no reported association with the Muir-Torre syndrome.183
Sebaceoma
Sebaceoma, a term proposed by Troy and Ackerman,184 has largely supplanted the older term sebaceous epithelioma, and this newer name probably encompasses a wider variety of microscopic changes. This condition presents as a circumscribed nodular lesion that is rarely diagnosed clinically. Microscopically, these lesions present as lobulated tumors in the upper to mid-dermis, composed predominantly of basaloid cells with a minority of sebocytes, singly dispersed and in clusters. Ductlike structures and areas of trichilemmal keratinization can also be observed184,185 (Fig. 19-40). Examples of sebaceoma with rippled,186 cribriform-reticulated,187 or carcinoid-like188 configurations have been reported. Sebaceoma can usually be distinguished from the more organized sebaceous adenoma, which usually has a less prominent basaloid component. However, overlapping features of the two can occur, particularly in some of the sebaceous neoplasms of patients with the Muir-Torre syndrome. A “rippled” configuration can occur in a variety of other tumors, including trichoblastoma.188 Such tumors can usually be distinguished from sebaceoma, but there could be confusion with one variant form that has been termed rippled-pattern sebaceous trichoblastoma.188

Malignant Sebaceous Neoplasms
Clinical Features: Although sebaceous carcinomas have traditionally been grouped into two categories, ocular and extraocular, partly because ocular lesions were said to be associated with a worse prognosis, a review of the literature shows that ocular and extraocular tumors actually have similar risks for local recurrence, distant metastases, and tumor-related mortality.189–191 However, ocular involvement (eyelids or adnexa) is more common than extraocular involvement. Sebaceous carcinoma usually arises in middle-aged or elderly patients. Tumors of the eyelids also occur in relation to radiation therapy or secondarily following treatment of a primary malignancy of another type.192 Ocular lesions present as painless masses of lid margins or conjunctiva, and commonly resemble blepharitis, chalazion, or conjunctivitis.193 Multifocal involvement can occur. Lesions can also develop elsewhere in the head and neck region or on the trunk or extremities, where they may be typified by rapid growth, pain, or ulceration. As previously noted, sebaceous carcinoma can be a manifestation of the Muir-Torre syndrome.194 Overall, these tumors have shown a high rate of recurrence and metastasis.195
Microscopic Findings: Sebaceous carcinomas are graded based on growth patterns rather than cytologic features.196 Basically, grade I lesions are comprised of rounded, well-demarcated cellular aggregates (Fig. 19-41A), grade III lesions show infiltrative cords of tumor cells or sheetlike arrangements (see Fig. 19-41B), and grade II lesions show combinations of these growth patterns. In general, most examples consist of lobulated groups of atypical, polygonal cells within a fibrovascular stroma (Fig. 19-42A). Central areas of the cellular islands frequently show necrosis with the configuration of “comedo necrosis.” As might be expected, well-differentiated variants show reasonably numerous cells with vacuolated cytoplasm and oval, vesicular nuclei, whereas more atypical lesions show greater nuclear pleomorphism, high nuclear-to-cytoplasmic ratios, more numerous (and atypical) mitoses, and cytoplasm that is not as obviously vacuolated (see Fig. 19-42B).
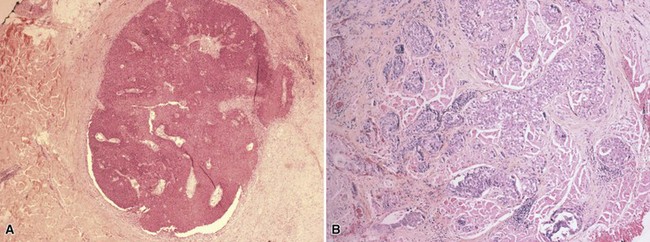
Figure 19-41 Sebaceous carcinoma. A, A grade I lesion shows a well-demarcated cellular aggregate. B, A grade III lesion has infiltrative cords of tumor cells.
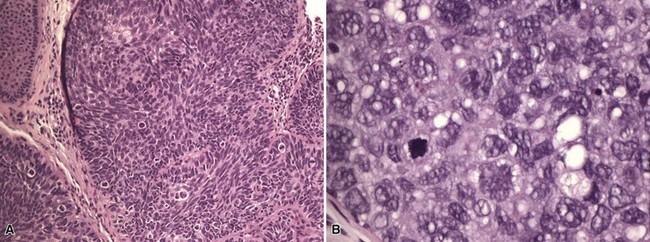
Figure 19-42 Sebaceous carcinoma. A, This lobulated tumor has nuclear pleomorphism. A few sebaceous cells can be identified. B, This atypical tumor shows substantial nuclear pleomorphism and mitotic activity.
Several microscopic features or subtypes are important in diagnosing sebaceous carcinoma. An association with intraepithelial Paget disease–like change sometimes occurs,197 and examples of intraepithelial sebaceous carcinoma without underlying tumor have been reported198 (Fig. 19-43). Basaloid sebaceous carcinomas are composed of small cells with relatively high nuclear-to-cytoplasmic ratios and peripheral palisading, but there may be significant stromal infiltration, marked nuclear atypia, and scattered clear to vacuolated cells that only vaguely suggest sebaceous differentiation. Other variants include sebaceous carcinomas with squamous metaplasia and a sarcomatoid form composed of spindled cells.
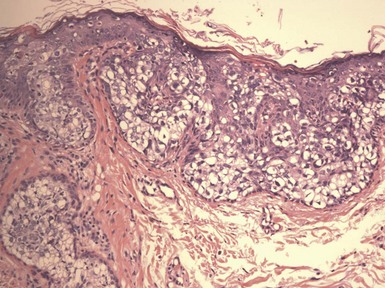
Figure 19-43 Pagetoid sebaceous carcinoma in situ. This is an intraepithelial tumor without invasion of the underlying connective tissue.
Special staining can be useful in recognizing that a particular tumor shows sebaceous differentiation. Lipid stains, including Oil Red O or Sudan IV, can be used, but they require frozen sections that may be formalin-fixed but not paraffin-embedded. Among immunohistochemical stains, neoplastic sebaceous cells are often positive for EMA,199 and they may label for androgen receptor protein,200 BRST-1,199 and cellular adhesion molecule 5.2.199 Adipophilin is a monoclonal antibody directed against adipose differentiation-related protein, which is located on the surface of intracellular lipid droplets. This stain is positive in most sebaceous tumors, including sebaceous carcinomas.201 As is also the case for sebaceous adenoma and sebaceoma, staining for DNA mismatch repair gene products can be useful in establishing a relationship to the Muir-Torre syndrome.202
Differential Diagnosis: The histopathologic differential diagnosis of sebaceous carcinoma is broad and particularly includes other malignant (and some benign) clear cell tumors of skin.203 Particular entities that should be considered are squamous cell carcinoma with clear cell features; clear cell basal cell carcinoma; trichilemmal carcinoma; balloon cell melanoma; clear cell sarcoma extending into the dermis; and metastatic clear cell carcinomas from other sites, particularly metastatic renal cell carcinoma (one of the metastatic tumors that can appear in skin before detection of the primary lesion).
Among these tumors, only balloon cell melanoma has the distinctive vacuolization expected in a sebaceous tumor; however, as previously noted, poorly differentiated sebaceous carcinoma may display only rudimentary sebaceous differentiation, and cytoplasmic vacuolization may therefore be difficult to discern. EMA can be helpful in identifying intracytoplasmic vesicles in some cases. Sebaceous carcinomas also lack several protein products that are identified in sweat gland tumors or metastatic carcinomas, including carcinoembryonic antigen, S-100 protein, gross cystic disease fluid protein-15 (GCDFP-15), cancer antigen 125 (CA-125), CA-19-9, or the renal cell carcinoma marker. A possible pitfall in diagnosis can result from the use of adipophilin, which stains cells of renal cell carcinoma as well as sebaceous carcinoma.201,204 However, the staining pattern in nonsebaceous tumors tends to be granular rather than membranous-vesicular.201 Basal cell carcinoma could potentially be confused with basaloid sebaceous carcinoma. However, basal cell carcinoma with sebaceous differentiation tends to show a higher degree of sebaceous differentiation than is the case in sebaceous carcinoma. Other basal cell carcinomas may also show clear cell change. However, the nuclei of basal cell carcinomas are not as vesicular as those in sebaceous carcinomas. In contrast to sebaceous carcinoma, the stroma of basal cell carcinoma is fibromyxoid in type. Both tumors can express BerEp4, but EMA staining is negative in basal cell carcinoma except for any areas that might show clear-cut sebaceous differentiation. Another problem that arises is the distinction between sebaceous carcinoma with squamoid features and conventional squamous cell carcinoma. Here, BerEp4 can be helpful, in that sebaceous carcinoma is positive for this marker, whereas squamous cell carcinoma is negative. Sarcomatoid sebaceous carcinoma can be distinguished from true sarcoma by its keratin positivity, but it may be difficult to recognize the tumor as specifically sebaceous if sebocytic differentiation cannot be recognized. Sebaceous differentiation can sometimes be encountered in other malignancies, including microcystic adnexal carcinoma,205 lymphoepithelioma-like carcinoma,206 and the difficult-to-classify adnexal carcinoma with divergent differentiation,122 but the mere finding of sebaceous cells in these circumstances should not lead to a diagnosis of sebaceous carcinoma in the absence of other attributes of that tumor.
Sweat Gland Tumors
The following section will consider the sweat gland tumors of both eccrine and apocrine differentiation. These are organized according to degrees of differentiation. Pseudoneoplastic lesions of sweat glands are either hamartomas or reactive sweat gland changes, and these conditions include eccrine and apocrine nevi, eccrine and apocrine hidrocystomas, apocrine cystadenomas, and syringometaplasias. The well-known (to dermatologists and dermatopathologists) benign adnexal tumors include eccrine lesions (syringoma, cylindroma, spiradenoma, poroma and variants, syringofibroadenoma, papillary eccrine adenoma, acrospiroma) and apocrine lesions (syringocystadenoma papilliferum, tubular apocrine adenoma, and hidradenoma papilliferum). Mixed tumors of skin also show mixed forms of differentiation, although the majority of these appear to be apocrine. The reader should note that the classification of these tumors as either eccrine or apocrine is controversial and seemingly ever changing. Currently, apocrine derivation is in the ascendancy; thus, most hidrocystomas are believed to be apocrine, acrospiromas (with few exceptions) are now widely considered to show apocrine differentiation (although they are grouped with the eccrine tumors here), and there is believed to be an apocrine variant of poroma. The precise categorization of these tumors may be the subject of debate, but fortunately, this controversy is not critical in the recognition of these tumors or their management.
Malignant eccrine sweat gland tumors include porocarcinoma, mucinous eccrine carcinoma, adenoid cystic carcinoma, digital papillary adenocarcinoma, microcystic adnexal carcinoma, mucoepidermoid carcinoma of the skin, and carcinoma ex cylindroma and spiradenoma. Malignant apocrine tumors include ductopapillary apocrine carcinoma and extramammary Paget disease with or without invasive carcinoma. Carcinomas of both differentiation lines can clearly mimic metastatic adenocarcinomas; examples of particular importance in this regard include ductal and clear cell eccrine carcinoma, ductopapillary apocrine carcinoma, and signet-ring adenocarcinoma. Finally, it should be recognized that there exists a group of poorly defined adnexal carcinomas with mixed differentiation; such tumors can show varying combinations of follicular, sebaceous, and apocrine (in other words, primary epithelial germ) or eccrine elements.
Nevoid and Malformations of Sweat Glands
Clinical Features: These lesions can present as isolated lesions (sometimes nodules), often but not invariably associated with sweat production,207 or as linear papules. The latter arrangement can be seen, particularly over the palms and soles, in porokeratotic eccrine ostial and dermal duct nevus, which combines the clinical features of verrucous epidermal nevus and nevus comedonicus (see Chapter 18).208,209 An additional lesion, the eccrine angiomatous hamartoma, presents as a red-tan papule or plaque on the extremities, associated with localized sweating and pain on palpation.210
Microscopic Findings: In examples composed mainly of glandular elements, there are well-defined sweat gland coils, more numerous than in normal skin and with perhaps a slight degree of disorganization.211 Nevertheless, the coils are composed of compact polygonal cells with cuticular lumina and surrounded by a basement membrane (Fig. 19-44). Sometimes, ductal hyperplasia predominates. In the porokeratotic eccrine ostial and dermal duct nevus, cornoid lamellae involve dilated ostia of anomalous eccrine ducts.212 In eccrine angiomatous hamartoma, nevoid sweat gland coils are associated with clusters of blood vessels and small nerve twigs.210
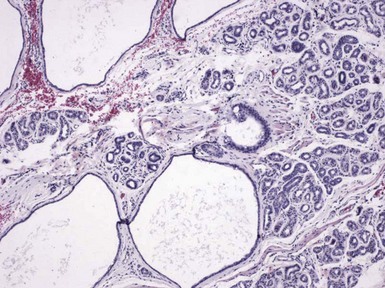
Differential Diagnosis: Eccrine nevi that are predominately glandular differ from normal sweat glands mainly in terms of numbers of glands per unit area. Their essentially normal appearance in other respects distinguishes them from other sweat gland tumors, such as syringoma or acrospiroma. The porokeratotic eccrine ostial and dermal duct nevus resembles true porokeratosis because of the formation of cornoid lamellae (see Chapter 18). However, in the nevus these structures occupy dilated eccrine ostia, and there is a lack of the cytologic atypia usually seen in the invaginated underlying epidermis of lesions of porokeratosis. The admixture of eccrine coils with vessels and nerve twigs forms a unique configuration not shared with other sweat gland lesions.
Apocrine Nevus
Clinical Features: Isolated apocrine nevi are rare, presenting as papules or isolated small nodules. They may manifest with localized tenderness and swelling; when this occurs in an axillary location, hidradenitis suppurativa may be the suspected clinical diagnosis.213,214 Most apocrine nevi are associated with nevus sebaceus or syringocystadenoma papilliferum.
Microscopic Findings: Increased numbers of mature apocrine glands with “apical snouts” (decapitation secretion) or their ducts are identified within the reticular dermis or subcutis; some of the ducts may be dilated (Fig. 19-45). Immunohistochemically, glandular epithelium demonstrates positivity for EMA, GCDFP, and low-molecular-weight keratin.213

Differential Diagnosis: Diagnosis depends in part on the recognition that aggregates of apocrine glands form a discrete lesion. This may be difficult to appreciate in small punch biopsies, where there may be overlap with the appearance of normal apocrine glands, especially in locations characterized by prominence of apocrine tissue, such as the axilla or anogenital region. Assessment of overlying epidermal changes can help exclude the possibility of coexistent nevus sebaceus or syringocystadenoma papilliferum (see later discussion).
Eccrine Hidrocystoma
Clinical Features: These were originally described as small translucent nodules in the periorbital regions. They may be bilateral and symmetrical. The nodules have a propensity to become more prominent in hot environments and decrease in size (and even become unnoticeable) in cold climates. Now considered uncommon, most of these hidrocystomas are currently believed to be of apocrine origin.215,216
Microscopic Findings: There is a cystic cavity lined by two layers of cuboidal epithelial cells. Proximity to an eccrine coil or duct is considered a clue to the diagnosis (Fig. 19-46). The cyst itself is indistinguishable from a “pure” apocrine hidrocystoma, because the ductal components (double layer of cuboidal epithelial cells) of these two glands are the same.

Apocrine Hidrocystoma and Apocrine Cystadenoma
Clinical Features: Traditionally, these were considered mostly solitary lesions of periorbital tissues, but they can be multiple and bilateral. Eyelid lesions are known as cysts of Moll gland.217 Hidrocystomas and cystadenomas are occasionally found in other anatomic locations and may be associated with complex apocrine tumors or nevus sebaceus (organoid nevus).
Microscopic Findings: True apocrine hidrocystomas are lined by two layers of flattened cuboidal cells218 (Fig. 19-47). The lining cells of apocrine cystadenomas show decapitation secretion and often demonstrate papillary projections within the cyst lumen that may be quite extensive219 (Fig. 19-48).
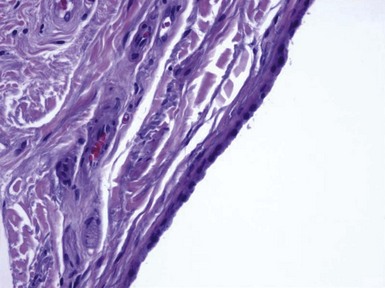
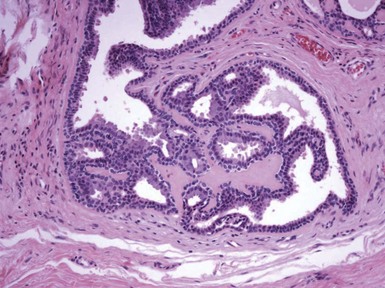
Differential Diagnosis: Structurally, apocrine and eccrine hidrocystomas are identical, both being lined by two layers of cuboidal epithelium. Proximity to other apocrine structures with characteristic decapitation secretion enables a correct diagnosis, although it is currently believed that most hidrocystomas are apocrine. Markedly papillary variants of apocrine cystadenoma can have overlapping features with tubular apocrine adenoma.
Syringometaplasia
Clinical Features: There are several forms of syringometaplasia. Mucinous syringometaplasia presents as a verruciform lesion typically in an acral location or as a plaque in other locations.220 Squamous metaplasia can be seen in a number of circumstances: accompanying mucinous syringometaplasia; associated with neutrophilic eccrine hidradenitis (which can present as erythematous papules and plaques in a variety of anatomic locations); as a response to certain drugs, particularly chemotherapeutic agents but also nonsteroidal anti-inflammatory drugs and some antimicrobials; and accompanying certain tumors, particularly squamous cell carcinoma.221,222 Changes precisely mimicking syringofibroadenoma (see later discussion) can be seen at the periphery of squamous cell carcinomas, other primary malignant tumors of the skin, or chronic inflammatory or fibrosing conditions.223,224
Microscopic Findings: Mucinous syringometaplasia shows an epidermal invagination, into which enter eccrine ducts with mucin-containing goblet cells220 (Fig. 19-49A). The squamous metaplasia accompanying neutrophilic eccrine hidradenitis, associated with chemotherapeutic agents (with or without changes of neutrophilic eccrine hidradenitis) or other drugs, shows prominent eccrine ducts composed of increased numbers of large keratinocytes, which sometimes show nuclear pleomorphism. Apoptotic keratinocytes can also be observed (see Fig. 19-49B). Eccrine coils underlying squamous cell carcinomas often show squamous metaplasia that can mimic invasive tumor islands. In syringofibroadenomatous change, elongated cords of epithelium extend from the epidermal surface into the dermis surrounding a central neoplastic, inflammatory, or fibrotic process (see Fig. 19-49C). Some of these epithelial cords can be shown to contain small ducts, and they are surrounded by a fibrovascular stroma.
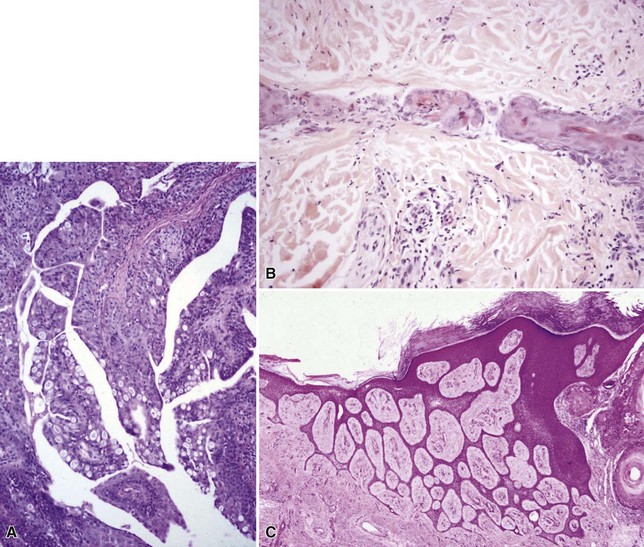
Figure 19-49 Forms of syringometaplasia. A, This is an example of mucinous metaplasia. Beneath an epidermal invagination, there are eccrine ducts with mucin-containing goblet cells. B, Squamous syringometaplasia with an eccrine duct that shows increased numbers of large keratinocytes, with nuclear pleomorphism and apoptosis. C, syringofibroadenoma-like changes: elongated, thin cords of epithelium extend into the dermis. This change was found adjacent to a squamous cell carcinoma.
Benign Sweat Gland Tumors
Clinical Features: Syringomas are benign eccrine ductal proliferations that most often present as multiple flesh-colored papules, typically on the face, and usually in an infraorbital location. Solitary and linear lesions have been described.225,226 The author has also seen these lesions above the upper lip227 and in a genital location.228 Eruptive syringomas of the trunk are seen in young individuals. Syringomas may be familial and have been associated with Down syndrome.229 An association between clear cell syringomas and diabetes mellitus has been described.230,231
Microscopic Findings: Within the dermis, there are well-demarcated aggregates of ducts lined by two rows of flattened epithelial cells; some are associated with comma-like epithelial cords232 (Fig. 19-50). Occasionally, the cells composing these structures possess clear cytoplasm233 (Fig. 19-51). Keratin cysts resembling milia are also seen. The surrounding stroma is often sclerotic. Syringomas lack nuclear atypia or a permeative growth pattern.
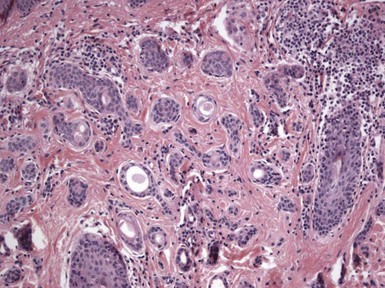
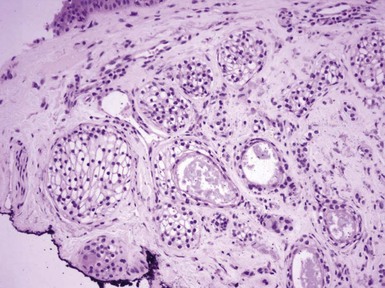
Differential Diagnosis: A small superficial biopsy of a syringoma can display features resembling microcystic adnexal carcinoma and sometimes desmoplastic trichoepithelioma. Each of these can have small keratin cysts resembling milia. A lack of ductal differentiation would favor desmoplastic trichoepithelioma. However, a deeper biopsy, sufficient to show changes in the subcutis, is the preferred method to exclude microcystic adnexal carcinoma, because the latter is not well demarcated, shows permeative growth into the subcutis, and is likely to display perineural infiltration.
Cylindroma
Clinical Features: Cylindroma is one of the sweat gland tumors that is consistently found in the face, neck, and scalp. Solitary, sporadic nodules occur, or there may be multiple nodules involving the scalp, often called “turban tumors” (the Ancell-Spiegler syndrome).44 These are inherited in an autosomal dominant fashion, sometimes together with multiple trichoepitheliomas. The mutated gene in familial cylindromatosis, CYLD, is localized to chromosome 16q.46,49 The link with trichoepithelioma and other immunohistochemical evidence suggests that cylindromas demonstrate primary epithelial germ (apocrine) differentiation, although other evidence suggests that it may be an eccrine tumor.
Microscopic Findings: Cylindromas are composed of compact basaloid tumor islands, molded to one another forming a “jigsaw puzzle” configuration. The tumor islands are composed of two cell populations: compact basaloid cells and polygonal cells with amphophilic cytoplasm. The cellular islands are surrounded by a hyaline basement membrane, and droplets of this material can be identified within the cell aggregates (Fig. 19-52). Mitotic activity is often, but not invariably, sparse. Buds of epithelium, seemingly detached from the main tumor mass, can be seen at the periphery of the lesion, but this does not imply biologic aggressiveness. Malignant lesions (carcinoma ex cylindroma) are rare, and they feature obviously anaplastic tumor cells and a less apparent organoid configuration.
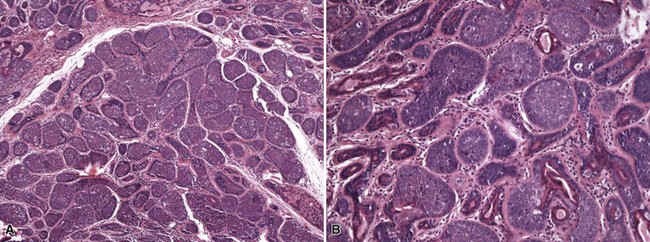
Differential Diagnosis: Occasionally, the configuration of cylindroma can be mimicked by basal cell carcinoma. However, that tumor lacks the closely applied hyaline basement membrane or hyaline droplets, and instead shows a fibromucinous stroma with clefting artifact. Perhaps the tumor most closely resembling cylindroma is the spiradenoma, and in fact these tumors can occur together, either in the same lesion or as separate lesions, or as components of the Ancell-Spiegler syndrome.234 Spiradenoma tends to consist of larger basophilic nodules, contains ductlike structures with two cell types (cells with hyperchromatic nuclei and other, larger cells with pale nuclei), and is associated with an edematous stroma with dilated vessels. Nevertheless, the occasional association with cylindroma and overlapping histopathologic features suggest a close relationship between these two lesions.
Spiradenoma
Clinical Features: This tumor usually presents as a solitary nodule and may occur in a variety of locations, most often on the upper (usually ventral) half of the body surface. Multiple spiradenomas have also been reported, typically in a linear distribution.235,236 A number of instances of “giant” spiradenomas have been reported in various locations, their size being related not only to the epithelial component but to the prominent vascular stroma.237 Despite the generally nonspecific clinical features of this tumor, it is noteworthy for its association with tenderness and pain.
Microscopic Findings: There are one or more well-demarcated lobules composed of interconnecting cords of basaloid cells (Fig. 19-53A). Two types of cells can be identified: cells with small dark nuclei at the periphery of cellular aggregates and cells with larger, pale nuclei in center of aggregates, sometimes with a suggestion of lumen formation238 (see Fig. 19-53B). The entire lesion is often permeated by lymphocytes. There are dilated intratumoral spaces that can be edematous or contain erythrocytes or lymph fluid; at times, this change can be sufficiently prominent to suggest a vascular tumor and accounts for those lesions designated “giant (vascular) spiradenoma239 (see Fig. 19-53C). As noted earlier, a resemblance to or association with cylindroma has been frequently observed (Fig. 19-54). In the author’s experience, it is not always possible to recognize the two cell types in spiradenomas, so the diagnosis is often based on a constellation of other characteristic findings.
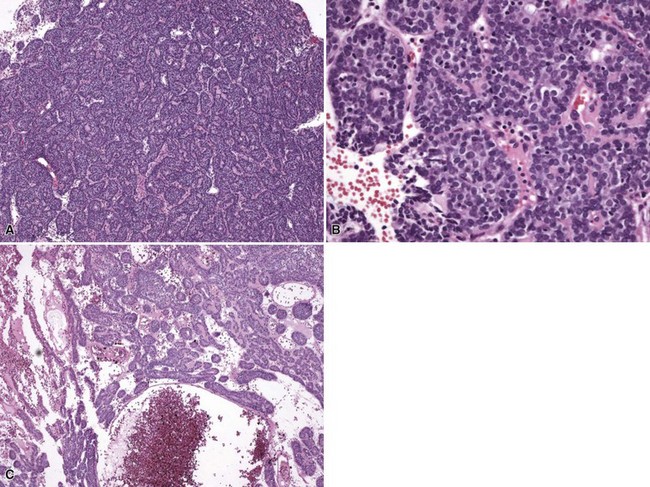
Figure 19-53 Spiradenoma. A, There is a large lobule composed of interconnecting cords of basaloid cells. B, In this higher magnification view, two cell types may be identified: a peripheral layer of cells with smaller, darker nuclei and a central zone of cells with larger, paler nuclei. Numerous lymphocytes are also identified. C, In this giant (vascular) spiradenoma, tumor islands are separated by edema, and there are dilated intratumoral spaces that contain erythrocytes.
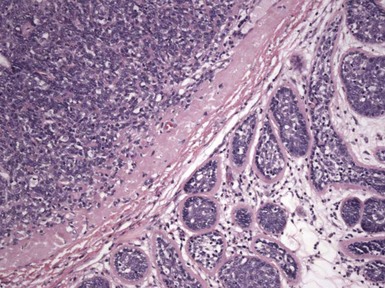
Differential Diagnosis: The resemblance to cylindroma can be striking; as noted previously the two can coexist, sometimes in the same lesion. However, basaloid tumor islands in spiradenoma tend to be larger, molding of tumor islands in a “jigsaw puzzle” arrangement is usually not observed, and hyaline basement membranes surrounding the tumor islands tend to be absent or not well developed. In addition, an edematous, vascular stroma is more typical of spiradenoma. Carcinoma ex spiradenoma shows elements of the benign tumor but also undifferentiated foci that indicate neoplastic transformation.
Poroma, Dermal Duct Tumor, and Hidroacanthoma Simplex
Clinical Features: Most poromas are believed to show differentiation toward the acrosyringium and are therefore considered “eccrine” lesions. However, a less common variant shows apocrine features and/or associated primary epithelial germ (follicular or sebaceous) differentiation.240 Classic poromas present as isolated, exophytic papules or small nodules, often (but not invariably) on distal extremities. The occurrence of numerous, widely scattered small papules, termed poromatosis, is quite uncommon241 and has been associated with hidrotic ectodermal dysplasia.242 The variant called dermal duct tumor can also have a wide geographic distribution. Some authorities include this entity under the term poroid hidradenoma—essentially a poroid variant of acrospiroma.243 Hidroacanthoma simplex is considered an intraepidermal variant of poroma. These lesions have been reported to occur on the trunk or extremities.244,245 A clinical as well as microscopic resemblance to seborrheic keratosis has been reported—potential sources of diagnostic difficulty, as will be discussed (see “Differential Diagnosis”). All three types of poroid tumors have been identified in a single lesion.246
Microscopic Findings: In classic poroma, broad, anastomosing bands of small cuboidal cells show multiple connections to the epidermis (Fig. 19-55). Tumor cells are often sharply demarcated from the adjacent keratinocytes of normal epidermis. Duct formation can be identified. Nuclei are bland in appearance; nucleoli are absent and mitoses difficult to identify, and the cytoplasm is amphophilic.247,248 An association with other pilosebaceous elements or evidence of apocrine differentiation leads to a diagnosis of apocrine poroma.249,250 The dermal duct tumor shows predominant dermal involvement, but the cytology is similar to that of classic poroma, and ductlike spaces are identified within tumor islands251,252 (Fig. 19-56). In hidroacanthoma simplex, discrete nests of cells are found within the epidermis, an example of the Borst-Jadassohn phenomenon; ductal lumina can occasionally be encountered within the nests (Fig. 19-57). Pigmented variants of both poroma and hidroacanthoma simplex have been described.253,254 Two interesting observations are that (1) it is difficult to find reports of pigmented dermal duct tumors and (2) a high proportion of reported cases of pigmented hidroacanthoma simplex have been associated with porocarcinoma.255,256
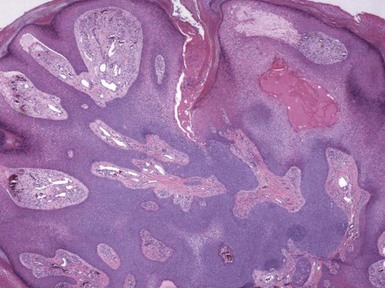
Figure 19-55 Poroma. There are broad, anastomosing bands of small cuboidal cells with multiple connections to the overlying epidermis.
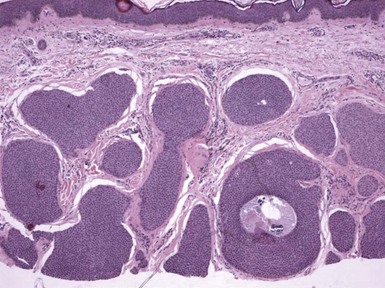
Figure 19-56 Dermal duct tumor. There are intradermal tumor islands with cytology similar to that of classic poroma. Ductlike spaces can be identified within some tumor islands.
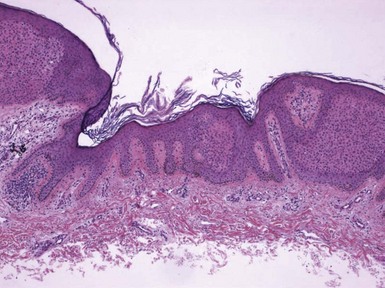
Figure 19-57 Hidroacanthoma simplex. Discrete nests of cells are identified within the epidermis in this intraepidermal poroma variant.
Immunohistochemistry has often been disappointing in the evaluation of these lesions, particularly hidroacanthoma simplex. In one study, neither CEA nor EMA were helpful in identifying the cells within the intraepidermal nests.257 CEA and EMA have been detected by some investigators within ductal structures and intracytoplasmic lumina.255,258 Electron microscopy has shown that the nested cells do not resemble luminal acrosyringeal cells.257 A study of keratin expression showed that all poroid tumors expressed CK14 and showed islands of CK10-positive and CK77-negative large cells; CK77 (a keratin specific for luminal cells of the eccrine dermal sweat duct) stained only luminal cells of intact ductal structures. The authors postulated that the common histogenesis of these tumors is derivation from basal keratinocytes of the sweat duct ridge and lower acrosyringium.259
Differential Diagnosis: A distinction of dermal duct tumor from poroid hidradenoma may be difficult or impossible. Finding epidermal connections with characteristic features of poroma would be one way, but then such tumors would probably be classified either as poromas or as combined tumors with features of both poroma and dermal duct tumor. The most important differential consideration is porocarcinoma. Although portions of porocarcinomas may bear a close resemblance to their benign counterparts, true porocarcinomas show infiltrative growth and areas of spontaneous necrosis. Some poromas show focal nuclear enlargement or nucleoli, sometimes with limited mitotic activity.260 Nevertheless, if the overall configuration is that of a conventional poroma, a diagnosis of malignancy should not be rendered.
Syringofibroadenoma
Clinical Features: This lesion was first described by Mascaro in 1963.261 Initially reported as a solitary nodule or plaque on the extremities, several varieties of lesions have since been reported and have arisen in many anatomic locations.262 These include solitary; multiple, in association with several syndromes; multiple without other cutaneous manifestations; unilateral linear; and reactive, in association with other skin lesions. The syndromes associated with multiple syringofibroadenomas include hidrotic ectodermal dysplasia (Clouston syndrome, combining poromas and palmoplantar syringofibroadenomas with alopecia, nail dystrophy, eye changes, and palmoplantar keratoderma) and the Schöpf-Schulz-Passarge syndrome (multiple palmoplantar syringofibroadenomas and keratoderma, associated with multiple apocrine hidrocystomas, hypodontia, hypotrichosis, and nail dystrophy263). HPV 10 DNA has been detected in the eccrine syringofibroadenomas of Clouston syndrome.264 In the author’s experience, the most common setting for syringofibroadenomatous change, by far, is as secondary change adjacent to another cutaneous lesion, typically squamous cell carcinoma, a chronic ulcer, or chronic infectious disease.223,265–267
Microscopic Findings: Thin, anastomosing cords of epithelium extend from the surface epidermis, sometimes with formation of small lumina,268 within a fibrovascular matrix (Fig. 19-58). The overlying epidermis is often acanthotic and occasionally pseudoepitheliomatous. The lesion is usually sharply circumscribed, with minimal if any nuclear atypia or mitotic activity. The “pure” form of syringofibroadenoma is much less common than the secondary syringofibroadenomatous change that often accompanies squamous cell carcinoma or other lesions. These changes are essentially identical to those of the primary tumor, and they may range from mild epithelial proliferation that is somewhat disorganized, to bilaterally symmetrical epithelial cords surrounding a tumor (often squamous cell carcinoma), ulcer, or chronic inflammatory or infectious process.
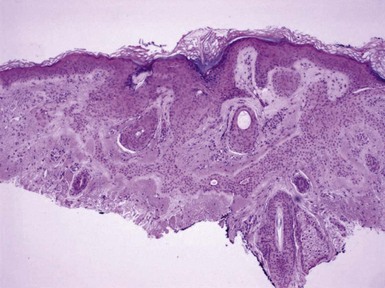
Differential Diagnosis: The overall configuration of the lesion bears a resemblance to fibroadenoma of the breast or fibroepithelioma of Pinkus but can be differentiated by the clinical setting or, in the latter case, by the absence of buds of basilar epithelium with the characteristics of basal cell carcinoma. The absence of an associated chronic inflammatory or neoplastic lesion argues against secondary syringofibroadenomatous change, which (in contrast to primary syringofibroadenoma) the author regards as an extremely common histopathologic finding.
Papillary Eccrine Adenoma
Clinical Features: Papillary eccrine adenoma can be regarded as the eccrine equivalent of the tubular apocrine adenoma. This lesion typically presents as a solitary dermal nodule, located on the extremities, without clinically distinctive features. A common presentation is on the lower extremity of African-American women.269
Microscopic Findings: There is a well-demarcated dermal aggregate of numerous ductlike structures within a fibrovascular matrix (Fig. 19-59A). Verruciform surface changes or cutaneous horn formation can be identified, but in at least one study, a workup for HPV infection was negative.269 The ductlike structures are composed of two or more layers of polyhedral cells that form papillary projections within their lumina (see Fig. 19-59B). Tubular configurations are also seen in some examples.270,271 The constituent cells lack significant pleomorphism, nucleoli, or mitotic activity. However, recurrences are reported.
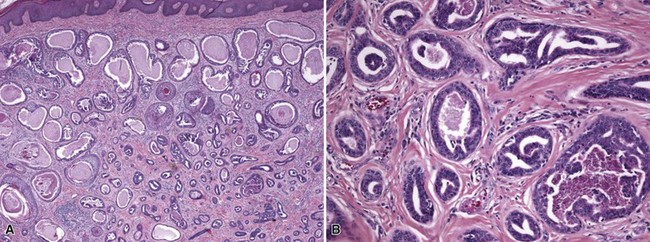
Differential Diagnosis: Tubular apocrine adenoma may bear a close resemblance to this tumor. However, there is evidence of apocrine-type (decapitation) secretion and other differences based on ultrastructural findings and enzyme histochemistry. In addition, tubular apocrine adenomas occur most commonly about the head and neck, particularly on the scalp. The (usually) good circumscription of these lesions and lack of significant nuclear pleomorphism or mitotic activity argue against a sweat gland carcinoma or metastatic adenocarcinoma.
(Eccrine) Acrospiroma
Clinical Features: Other names applied to these tumors are hidradenoma, clear cell hidradenoma, nodular hidradenoma, and solid-cystic hidradenoma, creating great confusion for novices and experienced pathologists alike. These lesions were once routinely grouped with the eccrine tumors, and at least some of them were thought to be closely related to the poroma. The current trend has been to consider most acrospiromas to be of apocrine differentiation, the one exception being the poromatous variant of acrospiroma (poroid hidradenoma). In the author’s opinion, this newer classification is still subject to debate, and this discussion retains the eccrine designation for these lesions. Acrospiromas present as dermal nodules that can arise in virtually any cutaneous site. Ulceration has been reported.272 Some examples are quite cystic; therefore, they can be clinically confused with other types of cystic or vascular lesions. However, they generally lack sufficiently distinctive clinical features to allow diagnosis without histopathologic study.273
Microscopic Findings: Acrospiromas typically show a nodular dermal growth pattern (occasionally with epidermal connections) composed of monotonous polygonal cells (Fig. 19-60). Clear cell change may predominate or form a limited component of the lesion (Fig. 19-61). Cystic changes are often observed and may be prominent (see Fig. 19-60). Micropapillary and ductal features are also present272 (Fig. 19-62), and foci of squamous metaplasia are common.274 Mitotic figures can be frequent, but this feature by itself does not equate with malignancy.275 The stroma sometimes has a hyalinized appearance (see Fig. 19-62). Most examples have rounded, “pushing” borders, but in some cases, cords of tumor cells appear to infiltrate into the adjacent dermis. This feature is of concern in that recurrences are frequent; moreover, recurrent tumors may develop progressively greater degrees of nuclear anaplasia and geographic necrosis with the biologic characteristics of malignancy (hidradenocarcinomas). Because of the sometimes surprisingly aggressive biologic behavior of even bland-appearing acrospiromas, the author’s general recommendation has been assurance of complete removal of all of these lesions.
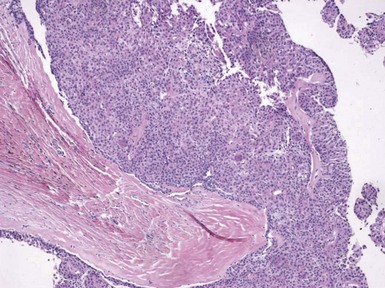
Figure 19-60 Acrospiroma. There is a nodular intradermal growth pattern composed of numerous polygonal cells. Cystic changes can also be observed.
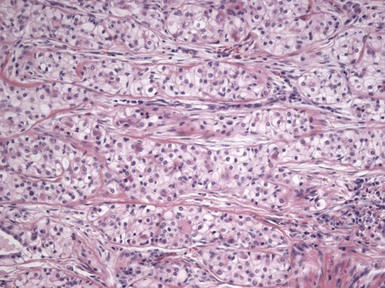
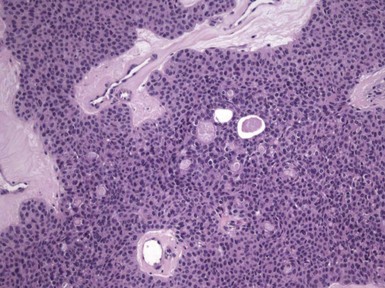
Differential Diagnosis: There is considerable microscopic overlap among poroid hidradenoma, poroma, and dermal duct tumor; in fact, they probably have a close histogenetic relationship (hence the concept of “acrospiroma, poroma type”). A distinction among these may be of some academic interest but makes little difference in terms of the prognosis and management of these benign lesions. Clear cell variants of acrospiroma can be confused with other clear cell tumors, including trichilemmoma and metastatic renal cell carcinoma. However, trichilemmomas show distinct peripheral palisading of their nuclei and a surrounding cuticular basement membrane, whereas acrospiromas tend to show large cystic spaces as well as micropapular and ductal features. Renal cell carcinomas of clear cell type often show a tubular arrangement of cells and tend to have a highly vascular stroma with hemorrhage; furthermore, immunostaining for renal cell carcinoma is positive in the majority of cases. As is the case with a number of appendageal tumors, examples of acrospiroma with cords of cells either budding or infiltrating into the adjacent dermis should probably be considered low-grade carcinomas (hidradenocarcinomas), and complete excision should be assured in such cases.
Syringocystadenoma Papilliferum
Clinical Features: Syringocystadenoma papilliferum usually manifests as a verruciform, friable nodule, most often located in the head and neck, but about one fourth of lesions occur in other anatomic locations, and there are some case reports of linear lesions.276 It most often represents a sporadic tumor277 but can be associated with nevus sebaceus of Jadassohn.278 Simple excision is sufficient for the removal of these lesions.
Microscopic Findings: There is a cystic invagination of epidermis lined by cuboidal or low columnar epithelial cells that display decapitation secretion, typical of apocrine epithelium. Papillary structures lined by the same type of epithelium protrude into the invagination (Fig. 19-63A); these are comprised of fibrin cores containing numerous plasma cells278 (see Fig. 19-63B). Tubular structures may be seen in the subjacent dermis. Atypical features are uncommon except in those lesions that have been subjected to trauma. In the author’s opinion, a well-documented example of malignant transformation in syringocystadenoma papilliferum has not yet been reported.
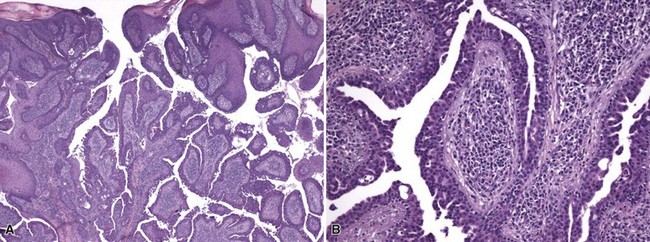
Figure 19-63 Syringocystadenoma papilliferum. A, Within a cystic invagination of the epidermis, there are papillary structures lined by cuboidal or low columnar epithelial cells that focally display decapitation secretion. B, The papillary structures are composed of fibrin cores and contain numerous plasma cells.
Differential Diagnosis: The microscopic findings in syringocystadenoma papilliferum are rather unique and not generally confused with those of other entities. The one lesion that can be confused with this lesion is the tubular apocrine adenoma, and in fact lesions with overlapping features have been described.279,280 However, the classic tubular apocrine adenoma has a configuration more reminiscent of papillary eccrine adenoma, the papillary projections lack stroma, and there are fewer inflammatory cells (including plasma cells). Both lesions are benign entities and can be associated with nevus sebaceus (organoid nevus)279; therefore, differentiation may be largely of academic interest.
Tubular Apocrine Adenoma
Clinical Features: First described by Landry and Winkelmann in 1972,281 the tubular apocrine adenoma presents most often as a smooth surfaced nodule, usually occurring on the scalp. As might be suspected, it is rarely if ever diagnosed without histopathologic confirmation.
Microscopic Findings: There is a collection of tubular structures in the dermis (Fig. 19-64A). These structures possess myoepithelial cells, an outer layer of flattened cuboidal cells, and a luminal layer of columnar cells that show decapitation secretion. Papillary projections are frequently observed (see Fig. 19-64B). Connection to the overlying epidermis via a ductlike structure is often identified,282 and overlying changes resembling syringocystadenoma papilliferum have been reported on numerous occasions.283,284
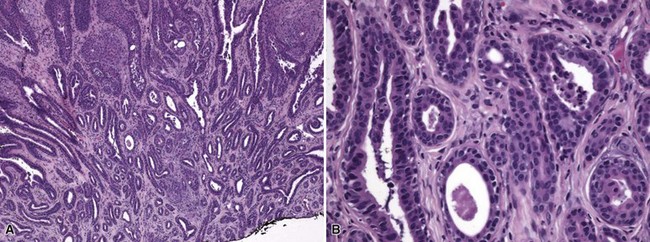
Differential Diagnosis: The location on the scalp and presence of decapitation secretion in luminal cells favors tubular apocrine adenoma over papillary eccrine adenoma, which usually arises on the extremities (particularly the lower legs) and lacks apocrine-type secretion. The distinction from syringocystadenoma papilliferum has already been discussed, although as noted the two lesions can coexist. Apocrine cystadenomas are frequently present in periorbital tissues; they have similar papillary features, but a large cystlike conformation predominates over collections of smaller tubular structures. The presence of myoepithelial cells, absence of significant cytologic atypia, and lack of stromal infiltration argue against a form of apocrine carcinoma.
Hidradenoma Papilliferum
Clinical Features: Hidradenoma papilliferum is a benign tumor with apocrine differentiation that presents as a reddish-blue dermal tumor, most often in women. The lesion usually occurs in the perineum, although uncommon locations include the eyelids and axillae. It has been rarely reported in men.285,286
Microscopic Findings: This is a well-circumscribed dermal tumor without epidermal connections (Fig. 19-65A). It consists in part of a cystic space with numerous micropapillary projections that demonstrate complex branching and interconnecting papillae, forming tubular profiles. The lumina are lined either by a single row of columnar cells showing decapitation secretion or by both luminal columnar cells and an outer layer of cuboidal myoepithelial cells287 (see Fig. 19-65B).
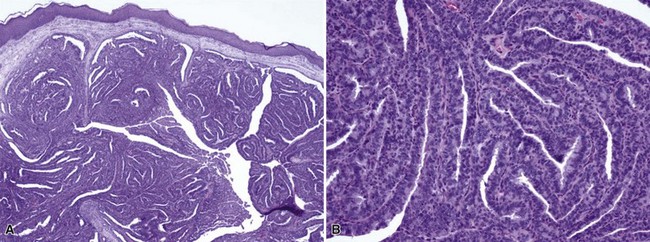
Differential Diagnosis: The exuberant proliferation of tubules can raise concerns about malignancy, particularly if the lesion has been traumatized. However, the characteristic sharply circumscribed profile of these lesions is a reliable clue to the correct diagnosis. Case reports of malignancy arising in hidradenoma papilliferum are elusive; one recent report described microscopic changes within a hidradenoma papilliferum that were considered diagnostic of ductal carcinoma in situ.288 These tumors do not closely resemble the other lesions referred to as hidradenoma—so-called nodular hidradenoma or solid-cystic hidradenoma. The latter are considered variants of acrospiroma, and in contrast to hidradenoma papilliferum lack the overall sharp circumscription, may show epidermal connections, lack complex interconnecting papillae, and often have solid areas comprised of monotonous (and sometimes clear) cuboidal cells.
Mixed Tumor of Skin
Clinical Features: The cutaneous mixed tumor most often manifests as a nondescript dermal or subcutaneous nodule in the head and neck region. The lesions may be firm due to the presence of chondroid and/or bone. The alternate term chondroid syringoma derives from the frequent association of the former component with these lesions289; however, chondroid is not always present. Most mixed tumors are now believed to show apocrine differentiation, but one variant featuring small tubular lumina is regarded by some as an eccrine lesion and may more closely fit the “chondroid syringoma” designation. In any event, mixed tumor of skin has become the umbrella term applying to all of these lesions.
Microscopic Features: These tumors feature both an epithelial and mesenchymal component, probably representing divergent differentiation from a common stem cell.290 The epithelial component is usually composed of either branching tubules (Fig. 19-66A) or small tubules and clusters of epithelial cells, often but not invariably showing evidence of apocrine-type secretion291 (see Fig. 19-66B). The mesenchymal component can consist of mucin, chondroid material (Fig. 19-67A), and occasionally bone (one example of a cutaneous tumor in which bone develops through enchondral ossification) (see Fig. 19-67B). Oncocytic change may be a focal or predominant feature. Lesions with dense spindle cell growth or clear cell change are termed myoepithelioma of skin; the latter cells stain for low-molecular-weight keratins, S-100 protein, and variably for smooth muscle actin.292
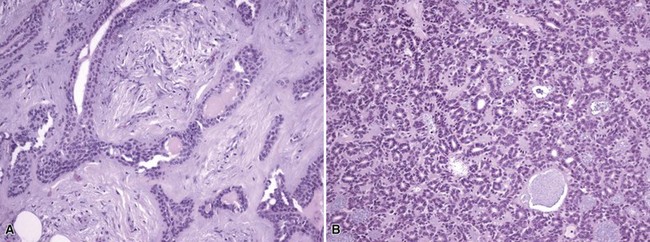
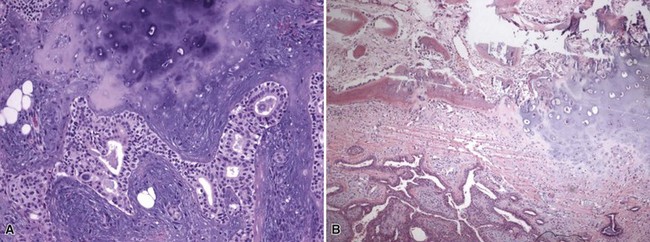
Differential Diagnosis: The combination of epithelial and mesenchymal elements is unique among cutaneous tumors. However, mixed tumors of salivary gland can have a close microscopic resemblance, and some deeply located mixed tumors obtained from the facial region may in fact represent salivary mixed tumors.293 Chondroma, osteoma, or mesenchymal hamartomas of skin might also be considered, but these lack the tubular structures of mixed tumor.
Malignant Sweat Gland Tumors
Clinical Features: Porocarcinoma usually presents as a dermal nodule, sometimes ulcerated, occurring on acral sites in adults. The tumor has also developed in children.294 Considered as a whole, the biologic aggressiveness of poromas is intermediate, with metastases most commonly to regional lymph nodes.
Microscopic Findings: Porocarcinoma differs from poroma by infiltrative growth, often within a desmoplastic stroma. Tumor necrosis, anaplasia, clear cell change, and vascular or perineural invasion may be observed.295,296 This tumor may represent another example of an adnexal carcinoma in which there are both an intermediate-grade lesion, characterized mainly by stromal infiltration and prone to local recurrence (Fig. 19-68), and a cytologically malignant lesion with a definite metastatic risk (Fig. 19-69). An intraepidermal variant is termed malignant hidroacanthoma simplex, analogous to the benign variant of intraepidermal poroma.297 This lesion has a “clonal,” micronodular configuration composed of atypical polygonal cells nested within the epidermis. CEA and S-100 staining can help in identifying this tumor and distinguishing it from other intraepidermal nested tumors.298
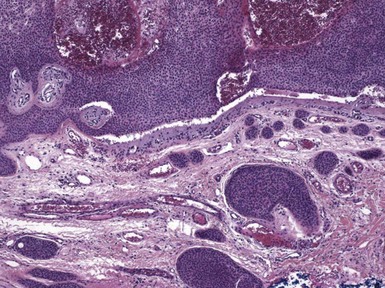
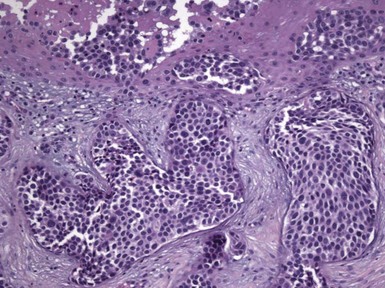
Differential Diagnosis: Porocarcinoma can be distinguished from poroma by its infiltrative growth and/or cytologic atypia. Metastatic carcinoma is sometimes considered in the differential diagnosis, especially in the uncommon situation where intraepidermal growth occurs.299 This phenomenon has been reported, for example, in metastatic prostate carcinoma. Differential staining may help in these circumstances.300 However, prostate-specific antigen has been identified in cells of extramammary Paget disease without associated adenocarcinoma of the prostate.301 The variant of porocarcinoma termed malignant hidroacanthoma simplex should be distinguished from other lesions characterized by intraepidermal nesting, such as Bowen disease and seborrheic keratosis. These lesions are positive for keratin, as is porocarcinoma, but only porocarcinoma would express CEA and/or S-100 protein. Pagetoid melanoma in situ could also resemble intraepidermal porocarcinoma, but whereas melanoma also expresses S-100 protein, the cells in question would be negative for keratin.
Mucinous Eccrine Carcinoma
Clinical Features: Mucinous eccrine carcinoma is an example of sweat gland carcinoma that has no benign counterpart. It may take the clinical form of a reddish nodule, sometimes ulcerated, and it occurs most commonly in the head and neck region, particularly in the periorbital area. Local recurrences develop in about one third of cases, but the incidence of metastatic disease is low.302
Microscopic Findings: There are large extracellular pools of mucin of epithelial origin (sialomucin), within which are rounded, irregular nests or branching cords of polyhedral tumor cells303,304 (Fig. 19-70A). These display amphophilic to slightly eosinophilic cytoplasm and nuclei with dispersed chromatin. Mitotic figures are uncommon. The mucin stains with a variety of methods, including PAS (diastase-resistant), Alcian blue, and colloidal iron; it is hyaluronidase-resistant and does not stain metachromatically with toluidine blue (see Fig. 19-70B).
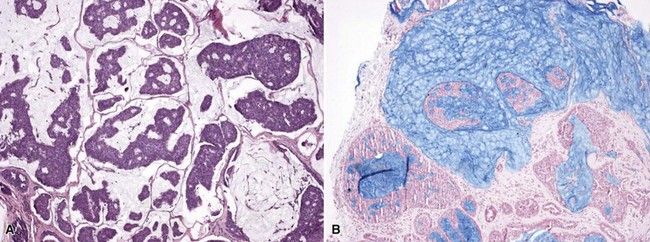
Differential Diagnosis: Mucinous eccrine carcinoma can resemble mucinous carcinoma of breast or gastrointestinal tract, but clinical history is often decisive, because metastases from these tumors do not involve the skin except in the case of obvious disseminated disease.305 This particular sweat gland carcinoma does not express p63, which is frequently a means of distinguishing primary adnexal carcinomas (p63-positive) from metastatic carcinomas (p63-negative); however, p63 staining of the myoepithelial cells of sweat glands occupied by carcinoma cells may be a clue that the mucinous carcinoma is of primary cutaneous origin.306 A mixed tumor with abundant mucin could be confused with mucinous eccrine carcinoma, but the mesenchymal component of the former often contains fusiform stromal cells, whereas acellular mucin pools are characteristic of mucinous eccrine carcinoma.
Adenoid Cystic Carcinoma of Skin
Clinical Features: Tumors with this designation arise in a number of organs (especially salivary glands), including skin, where they are noted for local aggressiveness (recurrence rate about 50%) but late, infrequent metastasis. Such tumors, when found in skin, are almost always primarily cutaneous in origin, but the author has seen a case of recurrent adenoid cystic carcinoma of salivary origin that involved the overlying dermis of facial skin. Adenoid cystic carcinomas of skin usually affect older patients and have a male-to-female ratio of 4 : 1. The scalp is the most common site of involvement.307 Surgical excision is the treatment of choice and should be sufficiently broad to minimize the risk of recurrence.
Microscopic Findings: Microscopically, these tumors consist of monomorphic basaloid cells arranged in tubules, nests, and cords, often with an infiltrative configuration (Fig. 19-71A). Duct formation may be present. Some cell islands may show a cribriform growth pattern, a fairly characteristic feature in these tumors (see Fig. 19-71B). Linear or globular basement membrane material may also be observed, and perineural invasion is relatively common.308–310 The constituent cells show scant amphophilic cytoplasm and nuclei with dispersed chromatin and indistinct nucleoli; mitotic figures are rare. The tumor cells of adenoid cystic carcinoma have demonstrated coexpression of keratins of simple epithelia (keratins 7, 8, 18, and 19) and stratified epithelia (keratins 1, 5, 10, and 14); this coexpression is apparently observed in fetal but not in adult sweat glands.311 The luminal surfaces of tubular and ductlike structures stain for both EMA and CEA.312 Myoepithelial cells at the periphery of tumor islands express appropriate antigens, presumably including p63, smooth muscle actin,313 and CD10.314
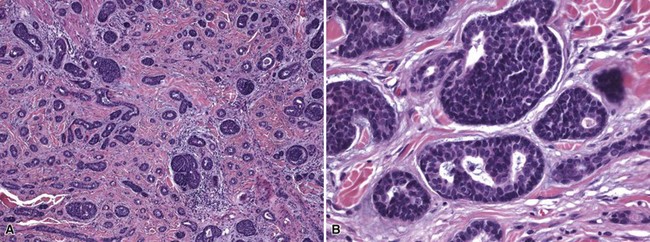
Differential Diagnosis: There is a close resemblance between adenoid cystic carcinomas of cutaneous and salivary gland origin, and comparative immunohistochemical studies show similar staining patterns for many of the markers listed above.312 The adenoid basal cell carcinoma has a number of overlapping microscopic features. However, in contrast to adenoid cystic carcinoma, basal cell carcinoma shows apoptotic cells in the central portions of tumor islands, foci of necrosis en masse, and sometimes evidence of follicular differentiation or melanin pigment. Basal cell carcinomas are negative for EMA, whereas adenoid cystic carcinomas are positive for EMA. It remains to be seen whether the CD117 expression reported in adenoid cystic carcinoma315 may allow distinction from adenoid basal cell carcinoma.
Digital Papillary Adenocarcinoma
Clinical Features: Eccrine tumors with this designation are unique to the distal extremities, especially the digits. Initially, these tumors were subdivided into two types: aggressive digital papillary adenoma and (aggressive) digital papillary adenocarcinoma.316 However, it was soon realized that these are parts of a spectrum of malignant neoplasms.317 These tumors commonly present as a solitary cystic nodule of distal digits, especially between the distal interphalangeal joint and the nail. Ulceration and bleeding may be present, and lesions may be initially interpreted as an infection.318 Recurrences are frequent, and metastasis occurs in about 15% of cases,319 most often to the lungs.
Microscopic Findings: The hallmark of digital papillary adenocarcinoma is the formation of papillae, some of which qualify as macropapillae, projecting into large cystic spaces (Fig. 19-72A). Solid nests and tubules of polygonal cells (sometimes displaying clear cell change) can also be found to infiltrate the surrounding connective tissue, and underlying bone can be involved (see Fig. 19-72B). Lower grade lesions display nuclei with dispersed chromatin, indistinct nucleoli, and low mitotic activity,316,320 whereas higher grade lesions feature nuclei with vesicular chromatin, prominent nucleoli, and numerous mitoses. Squamous metaplasia is most often identified in lower grade lesions, whereas geographic necrosis is common in higher grade lesions. Tumor cells are S-100 positive, and cyst contents express CEA and EMA.321
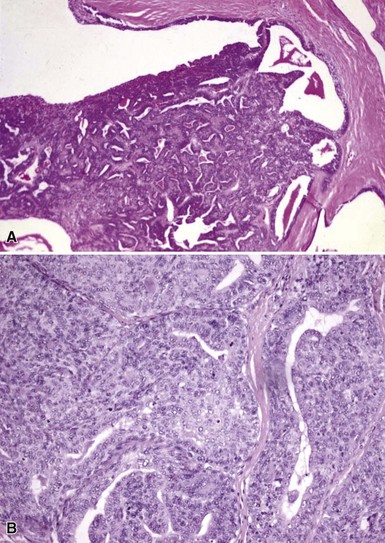
Differential Diagnosis: The image of digital papillary adenocarcinoma is similar to that of papillary carcinoma of the breast, but the latter only involves the skin in the setting of widespread metastatic disease. Hidradenoma papilliferum can have some resemblance to digital papillary adenocarcinoma, especially the low-grade variant, but the former has not been reported in the digits, and an unequivocal malignant variant has yet to be described. Papillary eccrine adenoma has a similar name but consists of micropapillae projecting into numerous small tubular lumina, organized in an overall bland and well-demarcated configuration.
Microcystic Adnexal Carcinoma
Clinical Features: This tumor typically presents as a slow-growing plaque on the central portion of face, especially the upper lip. It is most common among middle-aged women. Although it is particularly noteworthy for local recurrence rather than metastasis, rare instances of metastases have been reported.322–325 Immunosuppression or incomplete removal (with persistence and transformation to a higher grade neoplasm) may play a role in some of these cases. Metastases have been reported to regional lymph nodes, bone, and liver, and intracranial perineural spread has been described.322
Microscopic Findings: Microcystic adnexal carcinoma has a syringoma-like profile. The presence of keratin-forming microcysts suggests an element of pilar differentiation and explains the designation as microcystic adnexal carcinoma (Fig. 19-73). Despite a relative absence of cytologic atypia (nucleoli and mitotic activity are typically absent), small aggregates and cords of cells extend deeply into the dermis and subcutis (Fig. 19-74A). Some of these epithelial cords show luminal secretory material. Rarely, sebaceous or pilar elements are observed.85 However, there are examples lacking horn cysts, and these lesions are often called sclerosing sweat duct carcinoma. Variant forms show more solid nests and cords or clear cell changes.326 Whorled arrangements of cellular cords are often seen in deeper portions of the lesion, and perineural infiltration is particularly common (see Fig. 19-74B), as is involvement of vascular adventitia or skeletal muscle.


Differential Diagnosis: The differential diagnosis centers on other primary cutaneous lesions rather than metastatic tumors. These include the familiar triad of syringoma, desmoplastic trichoepithelioma, and infiltrative (morpheaform) basal cell carcinoma. On clinical grounds, microcystic adnexal carcinoma is plaquelike and ill defined, whereas syringoma and desmoplastic trichoepithelioma are small and well circumscribed. Microscopically, both syringoma and desmoplastic trichoepithelioma are sharply demarcated, laterally and at their bases, usually extending no deeper than mid-dermal, whereas microcystic adnexal carcinoma extends deeply into the subcutis. For this reason, an accurate diagnosis requires a deep biopsy. Morpheaform basal cell carcinoma displays more branching of epithelial structures than is the case for microcystic adnexal carcinoma, and clefting between tumor and stroma is a feature not observed in the latter tumor. Luminal CEA staining may help distinguish microcystic adnexal carcinoma from non–duct-forming tumors, but sometimes this marker also stains keratinized foci nonspecifically. By report, CK15 positivity separates microcystic adnexal carcinoma from infiltrative basal cell carcinoma and squamous cell carcinoma with ductal differentiation; however, CK15 and BerEp4 do not reliably separate microcystic adnexal carcinoma from desmoplastic trichoepithelioma, and therefore other methods need to be explored.327 A panel including hard keratins, EMA, and CEA may be useful in distinguishing microcystic adnexal carcinoma from desmoplastic trichoepithelioma,328 and EMA positivity of microcystic adnexal carcinoma may facilitate its distinction from morpheaform basal cell carcinoma, because the latter tumor is negative for EMA. Microcystic adnexal carcinoma with numerous horn cysts can resemble trichoadenoma of Nikolowski, but infiltrative cellular cords with syringoma-like profiles are not seen in the latter tumor. Although immunohistochemistry can be helpful, careful morphologic evaluation is still the gold standard for diagnosing this tumor.
Other Sweat Gland Carcinomas
Mucoepidermoid carcinoma of the skin is a rare lesion that may ulcerate and can be found in a variety of anatomic locations. It usually presents as a single nodule with indistinct borders. The tumor is composed of polyhedral squamous cells admixed with cuboidal cells and mucin-forming cuboidal or low columnar cells; mucin-filled microcysts and clear cell change may be identified.329,330 Constituent nuclei are bland in appearance, with small nucleoli and dispersed chromatin; mitoses are rare. Dermal infiltration with perineural or lymphatic invasion can be seen, associated with admixed chronic inflammatory cells. Cutaneous metastasis from a mucoepidermoid carcinoma arising from salivary glands or the respiratory system would be a rare event, generally occurring only after the primary tumor has been identified and treated. In contrast to primary cutaneous lesions, secondary mucoepidermoid carcinomas are multinodular, are sharply demarcated from the surrounding connective tissue, and lack the admixture of inflammatory cells. The expression of p63 in primary cutaneous mucoepidermoid carcinomas also allows distinction from mucoepidermoid carcinomas arising from other sites.
Carcinoma ex cylindroma and spiradenoma is a type of malignant tumor that arises in “parent” cylindromas or spiradenomas. This event is suggested clinically by rapid expansion of a previously indolent cutaneous nodule. Ulceration and pain may be noted. Microscopically, remnants of conventional cylindroma or spiradenoma are present. The malignant component consists of aggregates of markedly atypical epithelioid, stellate, or fusiform cells with brisk mitotic activity (Fig. 19-75), producing an image resembling either carcinosarcoma (with divergent mesenchymal differentiation) or squamous cell carcinoma/adenocarcinoma. Despite some high-grade neoplastic elements, carcinoma ex cylindroma may show indolent biologic behavior.331,332
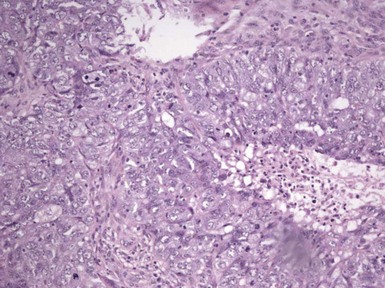
Figure 19-75 Carcinoma ex spiradenoma. Markedly atypical epithelioid to slightly fusiform cells are identified.
Ductopapillary apocrine carcinomas usually arise in the eyelids, axillae, and genital-perineal skin. Biopsy findings include nests of cells with internal papillations that show deep and irregular infiltration of the dermis (Fig. 19-76). Tumor cells have granular eosinophilic cytoplasm and sometimes show evidence of decapitation secretion.333 Iron staining is positive (a feature of apocrine differentiation), as is immunohistochemical staining for GDCFP-15.
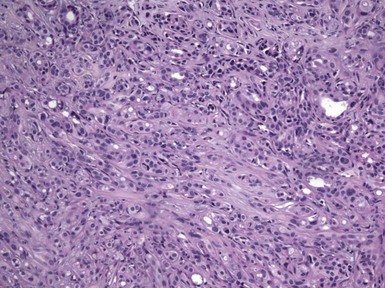
Figure 19-76 Ductopapillary apocrine carcinoma. Nests of cells, some with ductal features and others with internal papillations, extensively infiltrate the dermis.
Several sweat gland carcinomas particularly simulate metastases. Ductal eccrine adenocarcinoma is considered by some to be the most common eccrine carcinoma.334 This tumor presents as a solitary, slow-growing nodule arising on the head and neck or proximal extremities. The metastatic rate for all variants is about 40%. Histopathologic features include cords or tubules of polygonal cells that permeate the dermis. Ductal lumina are focally present. The stroma may be desmoplastic with varying degrees of chronic inflammation (Fig. 19-77). The tumor cells possess amphophilic cytoplasm, sometimes with vacuolization, and their nuclei display dense chromatin with prominent nucleoli. Squamoid differentiation may be present. Among the described variants are (1) ductal eccrine adenocarcinoma with fibromyxoid stroma, which arises on distal extremities, may contact the epidermis, and shows basaloid features, and (2) a fusiform or sarcomatoid type, whose features suggest myoepithelial differentiation. Clear cell eccrine carcinoma most often presents as a solitary nodule, usually on the skin of the head and neck. The clinical presentation contrasts with that of clear cell metastatic carcinoma, which arises primarily on the trunk and is more likely to be multifocal and of rapid onset.

Figure 19-77 Ductal eccrine adenocarcinoma. Tubules of polygonal cells, some containing ductal lumina, infiltrate within a desmoplastic stroma.
The clear cell eccrine carcinoma is closely related to malignant acrospiroma. It is multilobulated and lacks epidermal connections. Broad sheets or clusters of polyhedral cells with clear cytoplasm are separated by a fibrovascular stroma. The nuclei are either compact, with evenly dispersed chromatin and small nucleoli, or vesicular, with prominent nucleoli. Tumor necrosis and perineural or perivascular invasion can be seen. Mitotic rates vary.335 With regard to immunohistochemistry, CEA positivity is helpful, in that renal cell and urogenital clear cell carcinomas are CEA negative; furthermore, CA-125, a marker for renal cell carcinoma and female genital tract carcinoma, is absent in clear cell eccrine carcinoma. Signet-ring cell adenocarcinoma manifests as a nodular or plaquelike lesion of the eyelids and is most often found in adults. In contrast, signet-ring cell adenocarcinomas originating in other organs tend to metastasize to the skin of the trunk. Microscopically, there are small epithelioid cells in the dermis or subcutis, arranged in cords or small clusters; perineural invasion is infrequent, but when present supports cutaneous origin for these lesions. Rounded tumor cell nuclei with dispersed chromatin and small nucleoli are displaced to the periphery of each cell by single cytoplasmic vacuoles (primitive lumina) that sometimes contain secretory material. Tumor necrosis is rare, and mitoses are scant.336
Mammary and Extramammary Paget Disease
Clinical Features: Views differ about the origin of the intraepidermal cells of these lesions, it being a long-standing belief that mammary Paget disease represents migration of tumor cells into the epidermis from an underlying carcinoma of the breast, whereas extramammary Paget disease has been thought to arise within the epidermis, representing a primitive cell with a capacity for glandular differentiation. On the other hand, a number of us believe that both forms of the disease arise from pluripotential intraepidermal stem cells. In fact, about 3% to 5% of cases of mammary Paget disease are apparently not associated with an underlying carcinoma despite extensive research. In both instances, lesions present as erythematous plaques or eczematoid patches, either involving the nipple and areola or the anogenital region, particularly the penis, scrotum, vulva, or perianal region; axillary involvement has also been reported in extramammary cases. Most examples of mammary Paget disease are associated with an underlying invasive or intraductal carcinoma. Figures vary for extramammary Paget disease, but there may be an associated underlying adnexal carcinoma in more than 20% of cases, and another 10% or more can have malignancies of other tissues, including prostate, cervix, rectum, breast, bladder, and skin.337,338 Excision or Mohs surgery has been used to treat the lesions of Paget disease, along with excision of any associated malignancy. Recurrences and metastases have been reported.
Microscopic Findings: Epithelioid tumor cells populate the epidermis, arranged singly or in small clusters (Fig. 19-78). The underlying basilar layer may be compressed by aggregates of these cells, which have pale, sometimes vacuolated amphophilic cytoplasm and nuclei with dispersed chromatin and small nucleoli (Fig. 19-79); mitotic figures are sparse.339,340 There is an anaplastic variant of Paget disease, characterized by marked variation in nuclear size and shape.341 Pigmented examples of Paget disease have been reported, in which Paget cells contain melanin. Tumor cells contain mucin that is PAS (Fig. 19-80), mucicarmine, Alcian blue, and/or colloidal iron positive. Colloidal iron is the most sensitive of the mucin stains, whereas aldehyde fuchsin at pH 1.7 is the most specific. Positive staining for mucin tends to be more common in extramammary than in mammary Paget disease. The neoplastic cells are reactive for CEA, GCDFP-15, or CK7 (Fig. 19-81). In the author’s experience, polyclonal CEA is usually positive in these cases, whereas monoclonal CEA is frequently negative. Positivity for CA-19-9 or keratin 20 in extramammary disease raises the possibility of associated adenocarcinoma of the rectum, urinary bladder, or endocervix, because these particular markers indicate an enteric immunophenotype.342 Alpha-methylacyl-coenzyme A racemase, a marker associated with some prostate carcinomas, is frequently positive in cases of extramammary Paget disease.343
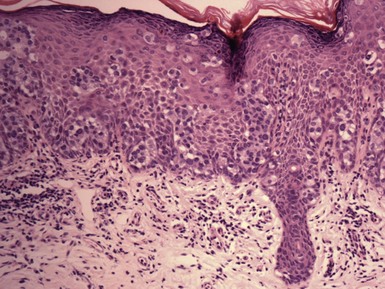
Figure 19-78 Mammary and extramammary Paget disease. Cells with pale or vacuolated cytoplasm populate the epidermis, arranged singly and in small clusters.
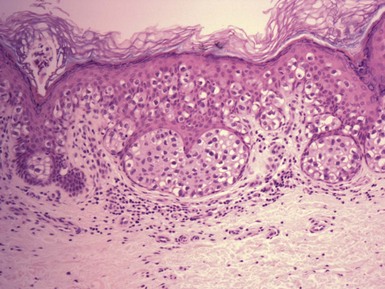
Figure 19-79 Mammary and extramammary Paget disease. The epidermal basilar layer is focally compressed by aggregates of Paget cells.

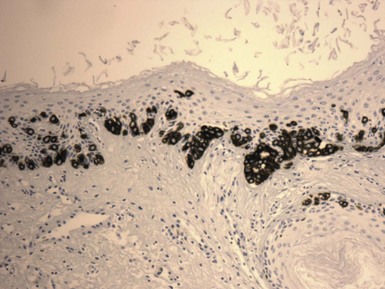
Differential Diagnosis: The differential diagnosis includes superficial spreading (pagetoid) melanoma and pagetoid Bowen disease. Neither melanoma cells nor the cells of squamous cell carcinoma produce mucin. The immunohistochemical profile for Paget disease is also distinctive. Paget cells do not express S-100 or human melanoma black-45 (HMB-45). Differential keratin staining can be used to distinguish Paget disease from Bowen disease: the neoplastic cells of Bowen disease express 57-kD and 66-kD keratins and not 54-kD keratin, whereas those of Paget disease are variable for 57-kD keratin, negative for 66-kD keratin, and positive for 54-kD keratin.344 Other markers that may help in the distinction of Paget disease from pagetoid Bowen disease include syndecan-1 (membranous in cells of pagetoid Bowen disease, cytoplasmic in extramammary Paget disease),345 cystic fibrosis transmembrane conductance regulator (CFTR) (positive staining favors extramammary Paget disease),346 and p63 (negative in primary vulvar Paget disease; positive in Bowen disease and in vulvar Paget disease associated with urothelial carcinoma).347,348 Pagetoid change also occurs in sebaceous carcinoma; most often the underlying invasive component substantiates this diagnosis, but in rare examples of primary sebaceous carcinoma confined to the epidermis, differentiation can be achieved by correlation with the clinical features and differential staining using mucins, sebaceous markers, and the characteristic markers for antigens associated with Paget disease. Clear cell papulosis contains intraepidermal cells similar to those of Paget disease that are believed by some to possibly represent precursors of mammary or extramammary Paget disease. Mucin and immunohistochemical stains are similar. However, these are whitish papules that develop along the milk lines of children. Pagetoid dyskeratosis is an incidental finding, frequently seen in polypoid lesions (skin tags) but also on the lips, hand, and in hemorrhoidal disease. These pale cells are few in number, are sometimes seen in loosely organized foci, and contain high-molecular-weight keratin.
References
1. Pippione, M, Aloi, F, Depaoli, MA. Hair-follicle nevus. Am J Dermatopathol. 1984;6(3):245–247.
2. Labandeira, J, Peteiro, C, Toribio, J. Hair follicle nevus: case report and review. Am J Dermatopathol. 1996;18(1):90–93.
3. Davis, DA, Cohen, PR. Hair follicle nevus: case report and review of the literature. Pediatr Dermatol. 1996;13(2):135–138.
4. Choi, EH, Ahn, SK, Lee, SH, et al. Hair follicle nevus. Int J Dermatol. 1992;31(8):578–581.
5. Germain, M, Smith, KJ. Hair follicle nevus in a distribution following Blaskho’s lines. J Am Acad Dermatol. 2002;46(5 Suppl):S125–S127.
6. Kuwahara, H, Lao, LM, Kiyohara, T, et al. Hair follicle nevus occurring in frontonasal dysplasia: an electron microscopic observation. J Dermatol. 2001;28(6):324–328.
7. Reda, AM, Rogers, RS, III., Peters, MS. Woolly hair nevus. J Am Acad Dermatol. 1990;22(2 Pt 2):377–380.
8. Sohi, AS, Sohi, BK. Oculo-auriculo-vertebral syndrome (Goldenhar’s syndrome). Int J Dermatol. 1978;17(4):339–341.
9. Ricks, M, Elston, DM, Sartori, CR. Multiple basaloid follicular hamartomas associated with acrochordons, seborrhoeic keratoses and chondrosarcoma. Br J Dermatol. 2002;146(6):1068–1070.
10. Brownstein, MH. Basaloid follicular hamartoma: solitary and multiple types. J Am Acad Dermatol. 1992;27(2 Pt 1):237–240.
11. Girardi, M, Federman, GL, McNiff, JM. Familial multiple basaloid follicular hamartomas: a report of two affected sisters. Pediatr Dermatol. 1999;16(4):281–284.
12. Nelson, BR, Johnson, TM, Waldinger, T, et al. Basaloid follicular hamartoma: a histologic diagnosis with diverse clinical presentations. Arch Dermatol. 1993;129(7):915–917.
13. Mehregan, AH, Baker, S. Basaloid follicular hamartoma: three cases with localized and systematized unilateral lesions. J Cutan Pathol. 1985;12(1):55–65.
14. Toyoda, M, Kagoura, M, Morohashi, M. Solitary basaloid follicular hamartoma. J Dermatol. 1998;25(7):434–437.
15. Requena, L, Farina, MC, Robledo, M, et al. Multiple hereditary infundibulocystic basal cell carcinomas: a genodermatosis different from nevoid basal cell carcinoma syndrome. Arch Dermatol. 1999;135(10):1227–1235.
16. Hellani, A, Baghdadi, H, Dabbour, N, et al. A novel PTCH1 germline mutation distinguishes basal cell carcinoma from basaloid follicular hamartoma: a case report. J Med Case Reports. 2009;3:52.
17. Starink, TM, Lane, EB, Meijer, CJ. Generalized trichoepitheliomas with alopecia and myasthenia gravis: clinicopathologic and immunohistochemical study and comparison with classic and desmoplastic trichoepithelioma. J Am Acad Dermatol. 1986;15(5 Pt 2):1104–1112.
18. Ramos-Ceballos, FI, Pashaei, S, Kincannon, JM, et al. Bcl-2, CD34 and CD10 expression in basaloid follicular hamartoma, vellus hair hamartoma and neurofollicular hamartoma demonstrate full follicular differentiation. J Cutan Pathol. 2008;35(5):477–483.
19. Cribier, B, Grosshans, E. Tumor of the follicular infundibulum: a clinicopathologic study. J Am Acad Dermatol. 1995;33(6):979–984.
20. Steffen, C. Winer’s dilated pore: the infundibuloma. Am J Dermatopathol. 2001;23(3):246–253.
21. Dudley, K, Barr, WG, Armin, A. Nevus comedonicus in association with widespread, well-differentiated follicular tumors. J Am Acad Dermatol. 1986;15(5 Pt 2):1123–1127.
22. Sieron, J, Thein, T, Pirsig, W. [The Nikolowski trichoadenoma: a rare tumor of the ENT area]. Laryngorhinootologie. 1993;72(3):140–142.
23. Cooper, PH, Mills, SE, Leonard, DD, et al. Sclerosing sweat duct (syringomatous) carcinoma. Am J Surg Pathol. 1985;9(6):422–433.
24. Kumar, K, McGregor, JC, Watson, JD. Microcystic adnexal carcinoma: a report of three cases. J R Coll Surg Edinb. 1998;43(6):412–414.
25. Kubo, Y, Urano, Y, Hida, Y, et al. A novel PTEN mutation in a Japanese patient with Cowden disease. Br J Dermatol. 2000;142(6):1100–1105.
26. Illueca, C, Monteagudo, C, Revert, A, et al. Diagnostic value of CD34 immunostaining in desmoplastic trichilemmoma. J Cutan Pathol. 1998;25(8):435–439.
27. El-Shabrawi, L, LeBoit, PE. Basal cell carcinoma with thickened basement membrane: a variant that resembles some benign adnexal neoplasms. Am J Dermatopathol. 1997;19(6):568–574.
28. Crowson, AN, Magro, CM. Basal cell carcinoma arising in association with desmoplastic trichilemmoma. Am J Dermatopathol. 1996;18(1):43–48.
29. Penneys, NS, Mogollon, RJ, Nadji, M, et al. Papillomavirus common antigens. Papillomavirus antigen in verruca, benign papillomatous lesions, trichilemmoma, and bowenoid papulosis: an immunoperoxidase study. Arch Dermatol. 1984;120(7):859–861.
30. Leonardi, CL, Zhu, WY, Kinsey, WH, et al. Trichilemmomas are not associated with human papillomavirus DNA. J Cutan Pathol. 1991;18(3):193–197.
31. Verplancke, P, Driessen, L, Wynants, P, et al. The Schopf-Schulz-Passarge syndrome. Dermatology. 1998;196(4):463–466.
32. Koch, B, Rufli, T. Tumor of follicular infundibulum. Dermatologica. 1991;183(1):68–69.
33. Mehregan, AH. Tumor of follicular infundibulum. Dermatologica. 1971;142(3):177–183.
34. Macmillan, A, Roberts, SO. Trichofolliculoma. Br J Dermatol. 1971;85(5):491–492.
35. Schulz, T, Hartschuh, W. The trichofolliculoma undergoes changes corresponding to the regressing normal hair follicle in its cycle. J Cutan Pathol. 1998;25(7):341–353.
36. Mizutani, H, Senga, K, Ueda, M. Trichofolliculoma of the upper lip: report of a case. Int J Oral Maxillofac Surg. 1999;28(2):135–136.
37. Plewig, G. Sebaceous trichofolliculoma. J Cutan Pathol. 1980;7(6):394–403.
38. Wu, YH. Folliculosebaceous cystic hamartoma or trichofolliculoma? A spectrum of hamartomatous changes inducted by perifollicular stroma in the follicular epithelium. J Cutan Pathol. 2008;35(9):843–848.
39. Schulz, T, Hartschuh, W. Folliculo-sebaceous cystic hamartoma is a trichofolliculoma at its very late stage. J Cutan Pathol. 1998;25(7):354–364.
40. Cole, P, Kaufman, Y, Dishop, M, et al. Giant, congenital folliculosebaceous cystic hamartoma: a case against a pathogenetic relationship with trichofolliculoma. Am J Dermatopathol. 2008;30(5):500–503.
41. Simpson, W, Garner, A, Collin, JR. Benign hair-follicle derived tumours in the differential diagnosis of basal-cell carcinoma of the eyelids: a clinicopathological comparison. Br J Ophthalmol. 1989;73(5):347–353.
42. Marrogi, AJ, Wick, MR, Dehner, LP. Benign cutaneous adnexal tumors in childhood and young adults, excluding pilomatrixoma: review of 28 cases and literature. J Cutan Pathol. 1991;18(1):20–27.
43. Szepietowski, JC, Wasik, F, Szybejko-Machaj, G, et al. Brooke-Spiegler syndrome. J Eur Acad Dermatol Venereol. 2001;15(4):346–349.
44. Cecchi, R, Crudeli, F, Fedi, E, et al. [Multiple trichoepithelioma, cylindroma, eccrine spiradenoma present in the same family. Histologic and histopathogenetic considerations]. G Ital Dermatol Venereol. 1985;120(2):149–152.
45. Akasaka, T, Kon, S, Mihm, MC, Jr. Multiple basaloid cell hamartoma with alopecia and autoimmune disease (systemic lupus erythematosus). J Dermatol. 1996;23(11):821–824.
46. Zhang, XJ, Liang, YH, He, PP, et al. Identification of the cylindromatosis tumor-suppressor gene responsible for multiple familial trichoepithelioma. J Invest Dermatol. 2004;122(3):658–664.
47. Salhi, A, Bornholdt, D, Oeffner, F, et al. Multiple familial trichoepithelioma caused by mutations in the cylindromatosis tumor suppressor gene. Cancer Res. 2004;64(15):5113–5117.
48. Harada, H, Hashimoto, K, Ko, MS. The gene for multiple familial trichoepithelioma maps to chromosome 9p21. J Invest Dermatol. 1996;107(1):41–43.
49. Bowen, S, Gill, M, Lee, DA, et al. Mutations in the CYLD gene in Brooke-Spiegler syndrome, familial cylindromatosis, and multiple familial trichoepithelioma: lack of genotype-phenotype correlation. J Invest Dermatol. 2005;124(5):919–920.
50. Matt, D, Xin, H, Vortmeyer, AO, et al. Sporadic trichoepithelioma demonstrates deletions at 9q22.3. Arch Dermatol. 2000;136(5):657–660.
51. Hatta, N, Hirano, T, Kimura, T, et al. Molecular diagnosis of basal cell carcinoma and other basaloid cell neoplasms of the skin by the quantification of Gli1 transcript levels. J Cutan Pathol. 2005;32(2):131–136.
52. Brownstein, MH, Shapiro, L. Desmoplastic trichoepithelioma. Cancer. 1977;40(6):2979–2986.
53. Brooke, JD, Fitzpatrick, JE, Golitz, LE. Papillary mesenchymal bodies: a histologic finding useful in differentiating trichoepitheliomas from basal cell carcinomas. J Am Acad Dermatol. 1989;21(3 Pt 1):523–528.
54. Brownstein, MH, Starink, TM. Desmoplastic trichoepithelioma and intradermal nevus: a combined malformation. J Am Acad Dermatol. 1987;17(3):489–492.
55. Rahbari, H, Mehregan, AH. Trichoepithelioma and pigmented nevus. A combined malformation. J Cutan Pathol. 1975;2(5):225–231.
56. Fukui, Y, Ono, H, Umemura, T, et al. A combined case of desmoplastic trichoepithelioma and nevus cell nevus. J Dermatol. 1990;17(8):506–509.
57. Choi, CW, Park, HS, Kim, YK, et al. Elastic fiber staining and cytokeratin 15 expression pattern in trichoepithelioma and basal cell carcinoma. J Dermatol. 2008;35(8):499–502.
58. Kirchmann, TT, Prieto, VG, Smoller, BR. CD34 staining pattern distinguishes basal cell carcinoma from trichoepithelioma. Arch Dermatol. 1994;130(5):589–592.
59. Krahl, D, Sellheyer, K. Monoclonal antibody Ber-EP4 reliably discriminates between microcystic adnexal carcinoma and basal cell carcinoma. J Cutan Pathol. 2007;34(10):782–787.
60. Swanson, PE, Fitzpatrick, MM, Ritter, JH, et al. Immunohistologic differential diagnosis of basal cell carcinoma, squamous cell carcinoma, and trichoepithelioma in small cutaneous biopsy specimens. J Cutan Pathol. 1998;25(3):153–159.
61. Thewes, M, Worret, WI, Engst, R, et al. Stromelysin-3: a potent marker for histopathologic differentiation between desmoplastic trichoepithelioma and morphealike basal cell carcinoma. Am J Dermatopathol. 1998;20(2):140–142.
62. Pham, TT, Selim, MA, Burchette, JL, Jr., et al. CD10 expression in trichoepithelioma and basal cell carcinoma. J Cutan Pathol. 2006;33(2):123–128.
63. Katona, TM, Perkins, SM, Billings, SD. Does the panel of cytokeratin 20 and androgen receptor antibodies differentiate desmoplastic trichoepithelioma from morpheaform/infiltrative basal cell carcinoma? J Cutan Pathol. 2008;35(2):174–179.
64. Fernandez-Flores, A. Advanced differentiation in trichoepithelioma and basal cell carcinoma investigated by immunohistochemistry against neurofilaments. Folia Histochem Cytobiol. 2009;47(1):61–64.
65. Takei, Y, Fukushiro, S, Ackerman, AB. Criteria for histologic differentiation of desmoplastic trichoepithelioma (sclerosing epithelial hamartoma) from morphea-like basal-cell carcinoma. Am J Dermatopathol. 1985;7(3):207–221.
66. Julian, CG, Bowers, PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39(2 Pt 1):191–195.
67. Diomedi Camassei, F, Francalanci, P, Boldrini, R, et al. Paratesticular pilomatricoma: a new location. Pediatr Surg Int. 2001;17(8):652–653.
68. Behnke, N, Schulte, K, Ruzicka, T, et al. Pilomatricoma in elderly individuals. Dermatology. 1998;197(4):391–393.
69. Agarwal, RP, Handler, SD, Matthews, MR, et al. Pilomatrixoma of the head and neck in children. Otolaryngol Head Neck Surg. 2001;125(5):510–515.
70. Geh, JL, Moss, AL. Multiple pilomatrixomata and myotonic dystrophy: a familial association. Br J Plast Surg. 1999;52(2):143–145.
71. Leppard, BJ, Bussey, HJ. Gardner’s syndrome with epidermoid cysts showing features of pilomatrixomas. Clin Exp Dermatol. 1976;1(1):75–82.
72. Noguchi, H, Kayashima, K, Nishiyama, S, et al. Two cases of pilomatrixoma in Turner’s syndrome. Dermatology. 1999;199(4):338–340.
73. Viero, RM, Tani, E, Skoog, L. Fine needle aspiration (FNA) cytology of pilomatrixoma: report of 14 cases and review of the literature. Cytopathology. 1999;10(4):263–269.
74. Kaddu, S, Soyer, HP, Hodl, S, et al. Morphological stages of pilomatricoma. Am J Dermatopathol. 1996;18(4):333–338.
75. Rutten, A, Wenzel, P, Goos, M. [Gardner syndrome with pilomatrixoma-like hair follicle cysts]. Hautarzt. 1990;41(6):326–328.
76. Kaddu, S, Beham-Schmid, C, Soyer, HP, et al. Extramedullary hematopoiesis in pilomatricomas. Am J Dermatopathol. 1995;17(2):126–130.
77. Fujioka, M, Gozo, N, Osamu, M, et al. Secondary anetoderma overlying pilomatrixomas. Dermatology. 2003;207(3):316–318.
78. Uchiyama, N, Shindo, Y, Saida, T. Perforating pilomatricoma. J Cutan Pathol. 1986;13(4):312–318.
79. Honda, Y, Oh-i, T, Koga, M, et al. Perforating pilomatricoma: transepithelial elimination or not. J Dermatol. 2002;29(2):100–103.
80. Carlson, JA, Healy, K, Slominski, A, et al. Melanocytic matricoma: a report of two cases of a new entity. Am J Dermatopathol. 1999;21(4):344–349.
81. Marrogi, AJ, Wick, MR, Dehner, LP. Pilomatrical neoplasms in children and young adults. Am J Dermatopathol. 1992;14(2):87–94.
82. Kaddu, S, Soyer, HP, Wolf, IH, et al. Proliferating pilomatricoma. A histopathologic simulator of matrical carcinoma. J Cutan Pathol. 1997;24(4):228–234.
83. Ma, KF, Tsui, MS, Chan, SK. Fine needle aspiration diagnosis of pilomatrixoma. A monomorphic population of basaloid cells with squamous differentiation not to be mistaken for carcinoma. Acta Cytol. 1991;35(5):570–574.
84. Jacobson, M, Ackerman, AB. “Shadow” cells as clues to follicular differentiation. Am J Dermatopathol. 1987;9(1):51–57.
85. LeBoit, PE, Sexton, M. Microcystic adnexal carcinoma of the skin. A reappraisal of the differentiation and differential diagnosis of an underrecognized neoplasm. J Am Acad Dermatol. 1993;29(4):609–618.
86. Noto, G, Pravata, G, Arico, M. “Shadow” cells in proliferating trichilemmal tumors. Am J Dermatopathol. 1990;12(3):319–320.
87. Hitchcock, MG, Ellington, KS, Friedman, AH, et al. Shadow cells in an intracranial dermoid cyst. Arch Pathol Lab Med. 1995;119(4):371–373.
88. Headington, JT. Tumors of the hair follicle. A review. Am J Pathol. 1976;85(2):479–514.
89. Headington, JT. Differentiating neoplasms of hair germ. J Clin Pathol. 1970;23(6):464–471.
90. Sau, P, Lupton, GP, Graham, JH. Trichogerminoma. Report of 14 cases. J Cutan Pathol. 1992;19(5):357–365.
91. Watanabe, S, Torii, H, Matsuyama, T, et al. Trichoblastic fibroma. A case report and an immunohistochemical study of cytokeratin expression. Am J Dermatopathol. 1996;18(3):308–313.
92. Yamamoto, O, Hisaoka, M, Yasuda, H, et al. A rippled-pattern trichoblastoma: an immunohistochemical study. J Cutan Pathol. 2000;27(9):460–465.
93. Aloi, F, Tomasini, C, Pippione, M. Pigmented trichoblastoma. Am J Dermatopathol. 1992;14(4):345–349.
94. Chan, JK, Ng, CS, Tsang, WY. Nodular desmoplastic variant of trichoblastoma. Am J Surg Pathol. 1994;18(5):495–500.
95. Requena, L, Requena, I, Romero, E, et al. Trichogenic trichoblastoma. An unusual neoplasm of hair germ. Am J Dermatopathol. 1990;12(2):175–181.
96. Alsadhan, A, Taher, M, Shokravi, M. Cutaneous lymphadenoma. J Am Acad Dermatol. 2003;49(6):1115–1116.
97. Santa Cruz, DJ, Barr, RJ, Headington, JT. Cutaneous lymphadenoma. Am J Surg Pathol. 1991;15(2):101–110.
98. McNiff, JM, Eisen, RN, Glusac, EJ. Immunohistochemical comparison of cutaneous lymphadenoma, trichoblastoma, and basal cell carcinoma: support for classification of lymphadenoma as a variant of trichoblastoma. J Cutan Pathol. 1999;26(3):119–124.
99. Yamamoto, O, Asahi, M. Cytokeratin expression in trichoblastic fibroma (small nodular type trichoblastoma), trichoepithelioma and basal cell carcinoma. Br J Dermatol. 1999;140(1):8–16.
100. Cordoba, A, Guerrero, D, Larrinaga, B, et al. Bcl-2 and CD10 expression in the differential diagnosis of trichoblastoma, basal cell carcinoma, and basal cell carcinoma with follicular differentiation. Int J Dermatol. 2009;48(7):713–717.
101. Ackerman, ARV, Soyer, HP. Neoplasms with Follicular Differentiation. New York: Ardor Scribendi; 2000.
102. Pinkus, H, Coskey, R, Burgess, GH. Trichodiscoma. A benign tumor related to haarscheibe (hair disk). J Invest Dermatol. 1974;63(2):212–218.
103. Grosshans, E, Dungler, T, Hanau, D. [Pinkus’ trichodiscoma (author’s translation)]. Ann Dermatol Venereol. 1981;108(11):837–846.
104. Coskey, RJ, Pinkus, H. Trichodiscoma. Int J Dermatol. 1976;15(8):600–601.
105. Misago, N, Kimura, T, Narisawa, Y. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol. 2009;36(9):943–951.
106. Scully, K, Bargman, H, Assaad, D. Solitary fibrofolliculoma. J Am Acad Dermatol. 1984;11(2 Pt 2):361–363.
107. Starink, TM, Brownstein, MH. Fibrofolliculoma: solitary and multiple types. J Am Acad Dermatol. 1987;17(3):493–496.
108. Schulz, T, Ebschner, U, Hartschuh, W. Localized Birt-Hogg-Dube syndrome with prominent perivascular fibromas. Am J Dermatopathol. 2001;23(2):149–153.
109. Schaffer, JV, Gohara, MA, McNiff, JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53(2 Suppl 1):S108–S111.
110. Nickerson, ML, Warren, MB, Toro, JR, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002;2(2):157–164.
111. Ayo, DS, Aughenbaugh, GL, Yi, ES, Hand, JL, et al. Cystic lung disease in Birt-Hogg-Dube syndrome. Chest. 2007;132(2):679–684.
112. Schmidt, LS, Warren, MB, Nickerson, ML, et al. Birt-Hogg-Dube syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet. 2001;69(4):876–882.
113. Chung, JY, Ramos-Caro, FA, Beers, B, et al. Multiple lipomas, angiolipomas, and parathyroid adenomas in a patient with Birt-Hogg-Dube syndrome. Int J Dermatol. 1996;35(5):365–367.
114. Walter, P, Kirchhof, B, Korge, B, et al. Flecked chorioretinopathy associated with Birt-Hogg-Dube syndrome. Graefes Arch Clin Exp Ophthalmol. 1997;235(6):359–361.
115. Schulz, T, Hartschuh, W. Birt-Hogg-Dube syndrome and Hornstein-Knickenberg syndrome are the same. Different sectioning technique as the cause of different histology. J Cutan Pathol. 1999;26(1):55–61.
116. Khoo, SK, Bradley, M, Wong, FK, et al. Birt-Hogg-Dube syndrome: mapping of a novel hereditary neoplasia gene to chromosome 17p12-q11.2. Oncogene. 2001;20(37):5239–5242.
117. Schmidt, LS, Nickerson, ML, Warren, MB, et al. Germline BHD-mutation spectrum and phenotype analysis of a large cohort of families with Birt-Hogg-Dube syndrome. Am J Hum Genet. 2005;76(6):1023–1033.
118. De la Torre, C, Ocampo, C, Doval, IG, et al. Acrochordons are not a component of the Birt-Hogg-Dube syndrome: does this syndrome exist? Case reports and review of the literature. Am J Dermatopathol. 1999;21(4):369–374.
119. Vincent, A, Farley, M, Chan, E, et al. Birt-Hogg-Dube syndrome: a review of the literature and the differential diagnosis of firm facial papules. J Am Acad Dermatol. 2003;49(4):698–705.
120. Calonje, E, Guerin, D, McCormick, D, et al. Superficial angiomyxoma: clinicopathologic analysis of a series of distinctive but poorly recognized cutaneous tumors with tendency for recurrence. Am J Surg Pathol. 1999;23(8):910–917.
121. McCalmont, CS, White, WL, Jorizzo, JL. Giant fibromyxoid tumors of the adventitial dermis. Forme fruste of trichodiscoma? Am J Dermatopathol. 1991;13(4):403–409.
122. Nakhleh, RE, Swanson, PE, Wick, MR. Cutaneous adnexal carcinomas with divergent differentiation. Am J Dermatopathol. 1990;12(4):325–334.
123. Rahbari, H, Mehregan, AH. Pilary complex carcinoma: an adnexal carcinoma of the skin with differentiation towards the components of the pilary complex. J Dermatol. 1993;20(10):630–637.
124. Wong, TY, Suster, S. Tricholemmal carcinoma. A clinicopathologic study of 13 cases. Am J Dermatopathol. 1994;16(5):463–473.
125. Swanson, PE, Marrogi, AJ, Williams, DJ, et al. Tricholemmal carcinoma: clinicopathologic study of 10 cases. J Cutan Pathol. 1992;19(2):100–109.
126. Chan, KO, Lim, IJ, Baladas, HG, et al. Multiple tumour presentation of trichilemmal carcinoma. Br J Plast Surg. 1999;52(8):665–667.
127. Wick, M. Neoplastic and pseudoneoplastic lesions of the hair follicles. In: Patterson J, Wick MR, eds. Nonmelanocytic Tumors of the Skin. Washington, DC: American Registry of Pathology; 2006:93.
128. Kurokawa, I, Senba, Y, Nishimura, K, et al. Cytokeratin expression in trichilemmal carcinoma suggests differentiation towards follicular infundibulum. In Vivo. 2006;20(5):583–585.
129. Amaral, AL, Nascimento, AG, Goellner, JR. Proliferating pilar (trichilemmal) cyst. Report of two cases, one with carcinomatous transformation and one with distant metastases. Arch Pathol Lab Med. 1984;108(10):808–810.
130. Saida, T, Oohara, K, Hori, Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatologica. 1983;166(4):203–208.
131. Mori, O, Hachisuka, H, Sasai, Y. Proliferating trichilemmal cyst with spindle cell carcinoma. Am J Dermatopathol. 1990;12(5):479–484.
132. Chen, KT, Taylor, DR, Jr. Pilomatrix carcinoma. J Surg Oncol. 1986;33(2):112–114.
133. Scheinfeld, N. Pilomatrical carcinoma: a case in a patient with HIV and hepatitis C. Dermatol Online J. 2008;14(1):4.
134. De Galvez-Aranda, MV, Herrera-Ceballos, E, Sanchez-Sanchez, P, et al. Pilomatrix carcinoma with lymph node and pulmonary metastasis: report of a case arising on the knee. Am J Dermatopathol. 2002;24(2):139–143.
135. Kondo, T, Tanaka, Y. Malignant pilomatricoma in the parietal area. Pathol Oncol Res. 2006;12(4):251–253.
136. Green, DE, Sanusi, ID, Fowler, MR. Pilomatrix carcinoma. J Am Acad Dermatol. 1987;17(2 Pt 1):264–270.
137. Bremnes, RM, Kvamme, JM, Stalsberg, H, et al. Pilomatrix carcinoma with multiple metastases: report of a case and review of the literature. Eur J Cancer. 1999;35(3):433–437.
138. Sable, D, Snow, SN. Pilomatrix carcinoma of the back treated by Mohs micrographic surgery. Dermatol Surg. 2004;30(8):1174–1176.
139. Gould, E, Kurzon, R, Kowalczyk, AP, et al. Pilomatrix carcinoma with pulmonary metastasis. Report of a case. Cancer. 1984;54(2):370–372.
140. Manivel, C, Wick, MR, Mukai, K. Pilomatrix carcinoma: an immunohistochemical comparison with benign pilomatrixoma and other benign cutaneous lesions of pilar origin. J Cutan Pathol. 1986;13(1):22–29.
141. Lineaweaver, WC, Wang, TN, Leboit, PL. Pilomatrix carcinoma. J Surg Oncol. 1988;37(3):171–174.
142. Hassanein, AM, Glanz, SM. Beta-catenin expression in benign and malignant pilomatrix neoplasms. Br J Dermatol. 2004;150(3):511–516.
143. Lazar, AJ, Calonje, E, Grayson, W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32(2):148–157.
144. Okamura, JM, Barr, RJ. Cutaneous lymphoepithelial neoplasms. Adv Dermatol. 1997;12:277–294. [discussion 295].
145. Mehregan, AH, Rahbari, H. Benigh epithelial tumors of the skin. II: Benign sebaceous tumors. Cutis. 1977;19(3):317–320.
146. Boschnakow, A, May, T, Assaf, C, et al. Ciclosporin A-induced sebaceous gland hyperplasia. Br J Dermatol. 2003;149(1):198–200.
147. Marini, M, Saponaro, A, Remorino, L, et al. Eruptive lesions in a patient with bone marrow transplantation. Int J Dermatol. 2001;40(2):133–135.
148. De Berker, DA, Taylor, AE, Quinn, AG, et al. Sebaceous hyperplasia in organ transplant recipients: shared aspects of hyperplastic and dysplastic processes? J Am Acad Dermatol. 1996;35(5 Pt 1):696–699.
149. Boonchai, W, Leenutaphong, V. Familial presenile sebaceous gland hyperplasia. J Am Acad Dermatol. 1997;36(1):120–122.
150. Weisshaar, E, Schramm, M, Gollnick, H. Familial nevoid sebaceous gland hyperplasia affecting three generations of a family. Eur J Dermatol. 1999;9(8):621–623.
151. Luderschmidt, C, Plewig, G. Circumscribed sebaceous gland hyperplasia: autoradiographic and histoplanimetric studies. J Invest Dermatol. 1978;70(4):207–209.
152. Morioka, S. The natural history of nevus sebaceus. J Cutan Pathol. 1985;12(3-4):200–213.
153. Kucukoduk, S, Ozsan, H, Turanli, AY, et al. A new neurocutaneous syndrome: nevus sebaceus syndrome. Cutis. 1993;51(6):437–441.
154. Carlson, JA, Cribier, B, Nuovo, G, et al. Epidermodysplasia verruciformis-associated and genital-mucosal high-risk human papillomavirus DNA are prevalent in nevus sebaceus of Jadassohn. J Am Acad Dermatol. 2008;59(2):279–294.
155. Stashower, ME, Smith, K, Corbett, D, et al. Basaloid/follicular hyperplasia overlying connective tissue/mesenchymal hamartomas simulating basal cell carcinomas. J Am Acad Dermatol. 2001;45(6):886–891.
156. Alessi, E, Wong, SN, Advani, HH, et al. Nevus sebaceus is associated with unusual neoplasms. An atlas. Am J Dermatopathol. 1988;10(2):116–127.
157. Cribier, B, Scrivener, Y, Grosshans, E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2 Pt 1):263–268.
158. Domingo, J, Helwig, EB. Malignant neoplasms associated with nevus sebaceus of Jadassohn. J Am Acad Dermatol. 1979;1(6):545–556.
159. Xin, H, Matt, D, Qin, JZ, et al. The sebaceous nevus: a nevus with deletions of the PTCH gene. Cancer Res. 1999;59(8):1834–1836.
160. Kimura, T, Miyazawa, H, Aoyagi, T, et al. Folliculosebaceous cystic hamartoma. A distinctive malformation of the skin. Am J Dermatopathol. 1991;13(3):213–220.
161. Badr, A, Lakshmiah, GR. Folliculosebaceous cystic hamartoma of the nipple: a case report. J Cutan Pathol. 2009;36(5):597–600.
162. Brucher, JJ, Franke, I, Ulrich, J, et al. Giant genital variant of folliculosebaceous cystic hamartoma: successful management by CO2 laser and acitretin therapy. Br J Dermatol. 2007;157(4):833–835.
163. Suarez-Penaranda, JM, Vieites, B, Ramirez-Santos, A, et al. Clinicopathological and immnuohistochemical findings in a series of folliculosebaceous cystic hamartoma. J Cutan Pathol. 2009;36(2):251–256.
164. Toyoda, M, Morohashi, M. Folliculosebaceous cystic hamartoma with a neural component: an immunohistochemical study. J Dermatol. 1997;24(7):451–457.
165. Misago, N, Kimura, T, Toda, S, et al. A revaluation of folliculosebaceous cystic hamartoma: the histopathological and immunohistochemical features. Am J Dermatopathol. 2010;32(2):154–161.
166. Raizada, RM, Khan, NU. Aural sebaceous adenomas. J Laryngol Otol. 1986;100(12):1413–1416.
167. Schwartz, RA, Goldberg, DJ, Mahmood, F, et al. The Muir-Torre syndrome: a disease of sebaceous and colonic neoplasms. Dermatologica. 1989;178(1):23–28.
168. Burgdorf, WH, Pitha, J, Fahmy, A. Muir-Torre syndrome. Histologic spectrum of sebaceous proliferations. Am J Dermatopathol. 1986;8(3):202–208.
169. Machin, P, Catasus, L, Pons, C, et al. Microsatellite instability and immunostaining for MSH-2 and MLH-1 in cutaneous and internal tumors from patients with the Muir-Torre syndrome. J Cutan Pathol. 2002;29(7):415–420.
170. Housholder, MS, Zeligman, I. Sebaceous neoplasms associated with visceral carcinomas. Arch Dermatol. 1980;116(1):61–64.
171. Zaim, MT. Sebocrine adenoma. An adnexal adenoma with sebaceous and apocrine poroma-like differentiation. Am J Dermatopathol. 1988;10(4):311–318.
172. Mahalingam, M, Byers, HR. Intra-epidermal and intra-dermal sebocrine adenoma with cystic degeneration and hemorrhage. J Cutan Pathol. 2000;27(9):472–475.
173. Rutten, A, Burgdorf, W, Hugel, H, et al. Cystic sebaceous tumors as marker lesions for the Muir-Torre syndrome: a histopathologic and molecular genetic study. Am J Dermatopathol. 1999;21(5):405–413.
174. Hassanein, AM, Al-Quran, SZ, Kantor, GR, et al. Thomsen-Friedenreich (T) antigen: a possible tool for differentiating sebaceous carcinoma from its simulators. Appl Immunohistochem Mol Morphol. 2001;9(3):250–254.
175. Ansai, S, Mitsuhashi, Y, Kondo, S, et al. Immunohistochemical differentiation of extra-ocular sebaceous carcinoma from other skin cancers. J Dermatol. 2004;31(12):998–1008.
176. Abbas, O, Mahalingam, M. Cutaneous sebaceous neoplasms as markers of Muir-Torre syndrome: a diagnostic algorithm. J Cutan Pathol. 2009;36(6):613–619.
177. Ponti, G, Losi, L, Di Gregorio, C, et al. Identification of Muir-Torre syndrome among patients with sebaceous tumors and keratoacanthomas: role of clinical features, microsatellite instability, and immunohistochemistry. Cancer. 2005;103(5):1018–1025.
178. Chhibber, V, Dresser, K, Mahalingam, M. MSH-6: extending the reliability of immunohistochemistry as a screening tool in Muir-Torre syndrome. Mod Pathol. 2008;21(2):159–164.
179. Kato, N, Ueno, H. Superficial epithelioma with sebaceous differentiation. J Dermatol. 1992;19(3):190–194.
180. Rothko, K, Farmer, ER, Zeligman, I. Superficial epithelioma with sebaceous differentiation. Arch Dermatol. 1980;116(3):329–331.
181. Friedman, KJ, Boudreau, S, Farmer, ER. Superficial epithelioma with sebaceous differentiation. J Cutan Pathol. 1987;14(4):193–197.
182. Akasaka, T, Imamura, Y, Tomichi, N, et al. A case of superficial epithelioma with sebaceous differentiation. J Dermatol. 1994;21(4):264–267.
183. Lee, MJ, Kim, YC, Lew, W. A case of superficial epithelioma with sebaceous differentiation. Yonsei Med J. 2003;44(2):347–350.
184. Troy, JL, Ackerman, AB. Sebaceoma. A distinctive benign neoplasm of adnexal epithelium differentiating toward sebaceous cells. Am J Dermatopathol. 1984;6(1):7–13.
185. Misago, N, Mihara, I, Ansai, S, et al. Sebaceoma and related neoplasms with sebaceous differentiation: a clinicopathologic study of 30 cases. Am J Dermatopathol. 2002;24(4):294–304.
186. Misago, N, Narisawa, Y. Rippled-pattern sebaceoma. Am J Dermatopathol. 2001;23(5):437–443.
187. Nielsen, TA, Maia-Cohen, S, Hessel, AB, et al. Sebaceous neoplasm with reticulated and cribriform features: a rare variant of sebaceoma. J Cutan Pathol. 1998;25(4):233–235.
188. Kazakov, DV, Kutzner, H, Rutten, A, et al. Carcinoid-like pattern in sebaceous neoplasms: another distinctive, previously unrecognized pattern in extraocular sebaceous carcinoma and sebaceoma. Am J Dermatopathol. 2005;27(3):195–203.
189. Wick, MR, Goellner, JR, Wolfe, JT, III., et al. Adnexal carcinomas of the skin. II. Extraocular sebaceous carcinomas. Cancer. 1985;56(5):1163–1172.
190. Moreno, C, Jacyk, WK, Judd, MJ, et al. Highly aggressive extraocular sebaceous carcinoma. Am J Dermatopathol. 2001;23(5):450–455.
191. Antuna, SA, Mendez, JG, Cincunegui, JA, et al. Metastatic lesion of the cervical spine secondary to an extraocular sebaceous carcinoma. Acta Orthop Belg. 1996;62(4):229–232.
192. Howrey, RP, Lipham, WJ, Schultz, WH, et al. Sebaceous gland carcinoma: a subtle second malignancy following radiation therapy in patients with bilateral retinoblastoma. Cancer. 1998;83(4):767–771.
193. Wolfe, JT, III., Yeatts, RP, Wick, MR, et al. Sebaceous carcinoma of the eyelid. Errors in clinical and pathologic diagnosis. Am J Surg Pathol. 1984;8(8):597–606.
194. Southey, MC, Young, MA, Whitty, J, et al. Molecular pathologic analysis enhances the diagnosis and management of Muir-Torre syndrome and gives insight into its underlying molecular pathogenesis. Am J Surg Pathol. 2001;25(7):936–941.
195. Buitrago, W, Joseph, AK. Sebaceous carcinoma: the great masquerader: emgerging concepts in diagnosis and treatment. Dermatol Ther. 2008;21(6):459–466.
196. Rao, NA, Hidayat, AA, McLean, IW, et al. Sebaceous carcinomas of the ocular adnexa: a clinicopathologic study of 104 cases, with five-year follow-up data. Hum Pathol. 1982;13(2):113–122.
197. Chao, AN, Shields, CL, Krema, H, et al. Outcome of patients with periocular sebaceous gland carcinoma with and without conjunctival intraepithelial invasion. Ophthalmology. 2001;108(10):1877–1883.
198. Margo, CE, Grossniklaus, HE. Intraepithelial sebaceous neoplasia without underlying invasive carcinoma. Surv Ophthalmol. 1995;39(4):293–301.
199. Sinard, JH. Immunohistochemical distinction of ocular sebaceous carcinoma from basal cell and squamous cell carcinoma. Arch Ophthalmol. 1999;117(6):776–783.
200. Bayer-Garner, IB, Givens, V, Smoller, B. Immunohistochemical staining for androgen receptors: a sensitive marker of sebaceous differentiation. Am J Dermatopathol. 1999;21(5):426–431.
201. Ostler, DA, Prieto, VG, Reed, JA, et al. Adipophilin expression in sebaceous tumors and other cutaneous lesions with clear cell histology: an immunohistochemical study of 117 cases. Mod Pathol. 2010;23(4):567–573.
202. Mathiak, M, Rutten, A, Mangold, E, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol. 2002;26(3):338–343.
203. Suster, S. Clear cell tumors of the skin. Semin Diagn Pathol. 1996;13(1):40–59.
204. Rangel, J, McCalmont, TH. Intracytoplasmic adipophilin immunopositivity: a pitfall in the distinction of metastatic renal carcinoma from sebaceous carcinoma. J Cutan Pathol. 2010;37(12):1193–1195.
205. Pujol, RM, LeBoit, PE, Su, WP. Microcystic adnexal carcinoma with extensive sebaceous differentiation. Am J Dermatopathol. 1997;19(4):358–362.
206. Requena, L, Sanchez Yus, E, et al. Lymphoepithelioma-like carcinoma of the skin: a light-microscopic and immunohistochemical study. J Cutan Pathol. 1994;21(6):541–548.
207. Goldstein, N. Ephidrosis (local hyperhidrosis). Nevus sudoriferus. Arch Dermatol. 1967;96:67–68.
208. Driban, NE, Cavicchia, JC. Porokeratotic eccrine ostial and dermal duct nevus. J Cutan Pathol. 1987;14(2):118–121.
209. Blanchard, L, Hodge, SJ, Owen, LG. Linear eccrine nevus with comedones. Arch Dermatol. 1981;117(6):357–359.
210. Challa, VR, Jona, J. Eccrine angiomatous hamartoma: a rare skin lesion with diverse histological features. Dermatologica. 1977;155(4):206–209.
211. Imai, S, Nitto, H. Eccrine nevus with epidermal changes. Dermatologica. 1983;166(2):84–88.
212. Moreno, A, Pujol, RM, Salvatella, N. Porokeratotic eccrine ostial and dermal duct nevus. J Cutan Pathol. 1988;15(1):43–48.
213. Neill, JS, Park, HK. Apocrine nevus: light microscopic, immunohistochemical and ultrastructural studies of a case. J Cutan Pathol. 1993;20(1):79–83.
214. Kanitakis, J, Kyamidis, K, Toussinas, A, et al. Pure apocrine nevus: immunohistochemical study of a new case and literature review. Dermatology. 2011;222:97–101.
215. Yasaka, N, Iozumi, K, Nashiro, K, et al. Bilateral periorbital eccrine hidrocystoma. J Dermatol. 1994;21(7):490–493.
216. Simon, RS, Sanches Yus, E. Does eccrine hidrocystoma exist? J Cutan Pathol. 1998;25(3):182–184.
217. Hashimoto, K, Zagula-Mally, ZW, Youngberg, G, et al. Electron microscopic studies of Moll’s gland cyst. J Cutan Pathol. 1987;14(1):23–36.
218. Shields, JA, Eagle, RC, Jr., Shields, CL, de Potter, P, Markowitz, G. Apocrine hidrocystoma of the eyelid. Arch Ophthalmol. 1993;111(6):866–867.
219. Sugiyama, A, Sugiura, M, Piris, A, et al. Apocrine cystadenoma and apocrine hidrocystoma: examination of 21 cases with emphasis on nomenclature according to proliferative features. J Cutan Pathol. 2007;34(12):912–917.
220. Madison, JF, Cooper, PH, Burgdorf, WH. Mucinous syringometaplasia with prominent epithelial hyperplasia and deep dermal involvement. J Cutan Pathol. 1990;17(4):220–224.
221. Bhawan, J, Malhotra, R. Syringosquamous metaplasia. A distinctive eruption in patients receiving chemotherapy. Am J Dermatopathol. 1990;12(1):1–6.
222. El Darouti, MA, Marzouk, SA, El Hadidi, HA, et al. Eccrine syringosquamous metaplasia. Int J Dermatol. 2001;40(12):777–781.
223. Clarke, LE, Ioffreda, M, Abt, AB. Eccrine syringofibroadenoma arising in peristomal skin: a report of two cases. Int J Surg Pathol. 2003;11(1):61–63.
224. Utani, A, Hattori, Y. A reactive acrosyringeal proliferation in a patient with ectodermal dysplasia: eccrine syringofibroadenoma-like lesion. J Dermatol. 1999;26(1):36–43.
225. Henner, MS, Shapiro, PE, Ritter, JH, et al. Solitary syringoma. Report of five cases and clinicopathologic comparison with microcystic adnexal carcinoma of the skin. Am J Dermatopathol. 1995;17(5):465–470.
226. Yung, CW, Soltani, K, Bernstein, JE, et al. Unilateral linear nevoidal syringoma. J Am Acad Dermatol. 1981;4(4):412–416.
227. Nguyen, DB, Patterson, JW, Wilson, BB. Syringoma of the moustache area. J Am Acad Dermatol. 2003;49(2):337–339.
228. Gerdsen, R, Wenzel, J, Uerlich, M, et al. Periodic genital pruritus caused by syringoma of the vulva. Acta Obstet Gynecol Scand. 2002;81(4):369–370.
229. Patrizi, A, Neri, I, Marzaduri, S, et al. Syringoma: a review of twenty-nine cases. Acta Derm Venereol. 1998;78(6):460–462.
230. Singh, A, Mishra, S. Clear cell syringoma—association with diabetes mellitus. Indian J Pathol Microbiol. 2005;48(3):356–357.
231. Furue, M, Hori, Y, Nakabayashi, Y. Clear-cell syringoma. Association with diabetes mellitus. Am J Dermatopathol. 1984;6(2):131–138.
232. Pruzan, DL, Esterly, NB, Prose, NS. Eruptive syringoma. Arch Dermatol. 1989;125(8):1119–1120.
233. Feibelman, CE, Maize, JC. Clear-cell syringoma. A study by conventional and electron microscopy. Am J Dermatopathol. 1984;6(2):139–150.
234. Van der Putte, SC. The pathogenesis of familial multiple cylindromas, trichoepitheliomas, milia, and spiradenomas. Am J Dermatopathol. 1995;17(3):271–280.
235. Yoshida, A, Sato, T, Sugawara, Y, et al. Two cases of multiple eccrine spiradenoma with linear or localized formation. J Dermatol. 2004;31(7):564–568.
236. Ter Poorten, MC, Barrett, K, Cook, J. Familial eccrine spiradenoma: a case report and review of the literature. Dermatol Surg. 2003;29(4):411–414.
237. Ko, JY, Lee, CW, Moon, SH, et al. Giant vascular eccrine spiradenoma: report of a case with immunohistochemical study. J Korean Med Sci. 2006;21(1):172–176.
238. Mambo, NC. Eccrine spiradenoma: clinical and pathologic study of 49 tumors. J Cutan Pathol. 1983;10(5):312–320.
239. Yamakoshi, T, Makino, T, Watanabe, H, et al. A case of giant vascular eccrine spiradenoma with unusual clinical features. Clin Exp Dermatol. 2009;34(7):e250–251.
240. Kazakov, DV, Kutzner, H, Spagnolo, DV, et al. Sebaceous differentiation in poroid neoplasms: report of 11 cases, including a case of metaplastic carcinoma associated with apocrine poroma (sarcomatoid apocrine porocarcinoma). Am J Dermatopathol. 2008;30(1):21–26.
241. Navi, D, Fung, M, Lynch, PJ. Poromatosis: the occurrence of multiple eccrine poromas. Dermatol Online J. 2008;14(1):3.
242. Wilkinson, RD, Schopflocher, P, Rozenfeld, M. Hidrotic ectodermal dysplasia with diffuse eccrine poromatosis. Arch Dermatol. 1977;113(4):472–476.
243. Ichioka, S, Yamada, A. A case of cystic dermal duct tumor corresponding to poroid hidradenoma. J Dermatol. 1993;20(9):554–557.
244. Nakagawa, T, Inai, M, Yamamoto, S, et al. Hidroacanthoma simplex showing variation in the appearance of tumor cells in different nests. J Cutan Pathol. 1988;15(4):238–244.
245. Rahbari, H. Hidroacanthoma simplex—a review of 15 cases. Br J Dermatol. 1983;109(2):219–225.
246. Kakinuma, H, Miyamoto, R, Iwasawa, U, et al. Three subtypes of poroid neoplasia in a single lesion: eccrine poroma, hidroacanthoma simplex, and dermal duct tumor. Histologic, histochemical, and ultrastructural findings. Am J Dermatopathol. 1994;16(1):66–72.
247. Freeman, RG, Knox, JM, Spiller, WF. Eccrine poroma. Am J Clin Pathol. 1961;36:444–450.
248. Hyman, AB, Brownstein, MH. Eccrine poroma. An analysis of forty-five new cases. Dermatologica. 1969;138(1):29–38.
249. Kamiya, H, Oyama, Z, Kitajima, Y. “Apocrine” poroma: review of the literature and case report. J Cutan Pathol. 2001;28(2):101–104.
250. Harvell, JD, Kerschmann, RL, LeBoit, PE. Eccrine or apocrine poroma? Six poromas with divergent adnexal differentiation. Am J Dermatopathol. 1996;18(1):1–9.
251. Aloi, FG, Pippione, M. Dermal duct tumor. Appl Pathol. 1986;4(3):175–178.
252. Hu, CH, Marques, AS, Winkelmann, RK. Dermal duct tumor: a histochemical and electron microscopic study. Arch Dermatol. 1978;114(11):1659–1664.
253. Kuo, HW, Ohara, K. Pigmented eccrine poroma: a report of two cases and study with dermatoscopy. Dermatol Surg. 2003;29(10):1076–1079.
254. Kennedy, C, Bhogal, B, Moss, R, et al. Pigmented intraepidermal eccrine poroma. Br J Dermatol. 1979;101(Suppl 17):76–78.
255. Ishida, M, Hotta, M, Kushima, R, et al. A case of porocarcinoma arising in pigmented hidroacanthoma simplex with multiple lymph node, liver and bone metastases. J Cutan Pathol. 2011;38(2):227–231.
256. Zina, AM, Bundino, S, Pippione, MG. Pigmented hidroacanthoma simplex with porocarcinoma. Light and electron microscopic study of a case. J Cutan Pathol. 1982;9(2):104–112.
257. Perniciaro, C, Muller, SA, Zelickson, BD, et al. Hidroacanthoma simplex: an ultrastructural and immunohistochemical study. J Cutan Pathol. 1994;21(3):274–279.
258. Lee, JY, Lin, MH. Pigmented malignant hidroacanthoma simplex mimicking irritated seborrheic keratosis. J Cutan Pathol. 2006;33(10):705–708.
259. Battistella, M, Langbein, L, Peltre, B, et al. From hidroacanthoma simplex to poroid hidradenoma: clinicopathologic and immunohistochemic study of poroid neoplasms and reappraisal of their histogenesis. Am J Dermatopathol. 2010;32(5):459–468.
260. Pylyser, K, De Wolf-Peeters, C, Marien, K. The histology of eccrine poromas: a study of 14 cases. Dermatologica. 1983;167(5):243–249.
261. Mascaro, J. Considerations sur les tumeurs fibro-epitheliales: le syringofibroadenome eccrine. Ann Dermatol Syphiligr. 1963;90:143–153.
262. Tey, HL. Characterizing the nature of eccrine syringofibroadenoma: illustration with a case showing spontaneous involution. Clin Exp Dermatol. 2009;34(5):e66–68.
263. Castori, M, Ruggieri, S, Giannetti, L, et al. Schopf-Schulz-Passarge syndrome: further delineation of the phenotype and genetic considerations. Acta Derm Venereol. 2008;88(6):607–612.
264. Carlson, JA, Rohwedder, A, Daulat, S, et al. Detection of human papillomavirus type 10 DNA in eccrine syringofibroadenomatosis occurring in Clouston’s syndrome. J Am Acad Dermatol. 1999;40(2 Pt 1):259–262.
265. Cho, E, Lee, JD, Cho, SH. A case of reactive eccrine syringofibroadenoma. Ann Dermatol. 2011;23(1):70–72.
266. Duffy, KL, Bowen, AR, Tristani-Firouzi, P, et al. Eccrine syringofibroadenoma-like change adjacent to a squamous cell carcinoma: potential histologic pitfall in Mohs micrographic surgery. Dermatol Surg. 2009;35(3):519–522.
267. Tey, HL, Chong, WS, Wong, SN. Leprosy-associated eccrine syringofibroadenoma of Mascaro. Clin Exp Dermatol. 2007;32(5):533–535.
268. Kanitakis, J, Zambruno, G, Euvrard, S, et al. Eccrine syringofibroadenoma. Immunohistological study of a new case. Am J Dermatopathol. 1987;9(1):37–40.
269. Blasini, W, Hu, S, Gugic, D, Vincek, V. Papillary eccrine adenoma in association with cutaneous horn. Am J Clin Dermatol. 2007;8(3):179–182.
270. Cooper, PH, Frierson, HF. Papillary eccrine adenoma. Arch Pathol Lab Med. 1984;108(1):55–57.
271. Rulon, DB, Helwig, EB. Papillary eccrine adenoma. Arch Dermatol. 1977;113(5):596–598.
272. Winkelmann, RK, Wolff, K. Solid-cystic hidradenoma of the skin. Clinical and histopathologic study. Arch Dermatol. 1968;97(6):651–661.
273. Johnson, BL, Jr., Helwig, EB. Eccrine acrospiroma. A clinicopathologic study. Cancer. 1969;23(3):641–657.
274. Stanley, RJ, Sanchez, NP, Massa, MC, et al. Epidermoid hidradenoma. A clinicopathologic study. J Cutan Pathol. 1982;9(5):293–302.
275. Cooper, PH. Mitotic figures in sweat gland adenomas. J Cutan Pathol. 1987;14(1):10–14.
276. Patterson, JW, Straka, BF, Wick, MR. Linear syringocystadenoma papilliferum of the thigh. J Am Acad Dermatol. 2001;45(1):139–141.
277. Niizuma, K. Syringocystadenoma papilliferum: light and electron microscopic studies. Acta Derm Venereol. 1976;56(5):327–336.
278. Helwig, EB, Hackney, VC. Syringadenoma papilliferum; lesions with and without naevus sebaceous and basal cell carcinoma. AMA Arch Derm. 1955;71(3):361–372.
279. Ishiko, A, Shimizu, H, Inamoto, N, et al. Is tubular apocrine adenoma a distinct clinical entity? Am J Dermatopathol. 1993;15(5):482–487.
280. Ansai, S, Watanabe, S, Aso, K. A case of tubular apocrine adenoma with syringocystadenoma papilliferum. J Cutan Pathol. 1989;16(4):230–236.
281. Landry, M, Winkelmann, RK. An unusual tubular apocrine adenoma. Arch Dermatol. 1972;105(6):869–879.
282. Umbert, P, Winkelmann, RK. Tubular apocrine adenoma. J Cutan Pathol. 1976;3(2):75–87.
283. Toribio, J, Zulaica, A, Peteiro, C. Tubular apocrine adenoma. J Cutan Pathol. 1987;14(2):114–117.
284. Ahn, BK, Park, YK, Kim, YC. A case of tubular apocrine adenoma with syringocystadenoma papilliferum arising in nevus sebaceus. J Dermatol. 2004;31(6):508–510.
285. Loane, J, Kealy, WF, Mulcahy, G. Perianal hidradenoma papilliferum occurring in a male: a case report. Ir J Med Sci. 1998;167(1):26–27.
286. Tanaka, M, Shimizu, S. Hidradenoma papilliferum occurring on the chest of a man. J Am Acad Dermatol. 2003;48(2 Suppl):S20–S21.
287. Hashimoto, K. Hidradenoma papilliferum. An electron microscopic study. Acta Derm Venereol. 1973;53(1):22–30.
288. Vazmitel, M, Spagnolo, DV, Nemcova, J, et al. Hidradenoma papilliferum with a ductal carcinoma in situ component: case report and review of the literature. Am J Dermatopathol. 2008;30(4):392–394.
289. Hirsch, P, Helwig, EB. Chondroid syringoma. Mixed tumor of skin, salivary gland type. Arch Dermatol. 1961;84:835–847.
290. Mills, SE. Mixed tumor of the skin: a model of divergent differentiation. J Cutan Pathol. 1984;11(5):382–386.
291. Hassab-el-Naby, HM, Tam, S, White, WL, et al. Mixed tumors of the skin. A histological and immunohistochemical study. Am J Dermatopathol. 1989;11(5):413–428.
292. Mentzel, T, Requena, L, Kaddu, S, et al. Cutaneous myoepithelial neoplasms: clinicopathologic and immunohistochemical study of 20 cases suggesting a continuous spectrum ranging from benign mixed tumor of the skin to cutaneous myoepithelioma and myoepithelial carcinoma. J Cutan Pathol. 2003;30(5):294–302.
293. Adachi, T, Oda, Y, Sakamoto, A, et al. Mixed tumor of deep soft tissue. Pathol Int. 2003;53(1):35–39.
294. Valverde, K, Senger, C, Ngan, BY, et al. Eccrine porocarcinoma in a child that evolved rapidly from an eccrine poroma. Med Pediatr Oncol. 2001;37(4):412–414.
295. Bottles, K, Sagebiel, RW, McNutt, NS, et al. Malignant eccrine poroma. Case report and review of the literature. Cancer. 1984;53(7):1579–1585.
296. Mehregan, AH, Hashimoto, K, Rahbari, H. Eccrine adenocarcinoma. A clinicopathologic study of 35 cases. Arch Dermatol. 1983;119(2):104–114.
297. Moreno, A, Salvatella, N, Guix, M, et al. Malignant hidroacanthoma simplex. A light microscopic, ultrastructural, and immunohistochemical study of 2 cases. Dermatologica. 1984;169(4):161–166.
298. Swanson, PE, Cherwitz, DL, Neumann, MP, et al. Eccrine sweat gland carcinoma: an histologic and immunohistochemical study of 32 cases. J Cutan Pathol. 1987;14(2):65–86.
299. Manteaux, A, Cohen, PR, Rapini, RP. Zosteriform and epidermotropic metastasis. Report of two cases. J Dermatol Surg Oncol. 1992;18(2):97–100.
300. Allan, SJ, McLaren, K, Aldridge, RD. Paget’s disease of the scrotum: a case exhibiting positive prostate-specific antigen staining and associated prostatic adenocarcinoma. Br J Dermatol. 1998;138(4):689–691.
301. Hammer, A, Hager, H, Steiniche, T. Prostate-specific antigen-positive extramammary Paget’s disease—association with prostate cancer. Apmis. 2008;116(1):81–88.
302. Pilgrim, JP, Kloss, SG, Wolfish, PS, et al. Primary mucinous carcinoma of the skin with metastases to the lymph nodes. Am J Dermatopathol. 1985;7(5):461–469.
303. Santa-Cruz, DJ, Meyers, JH, Gnepp, DR, et al. Primary mucinous carcinoma of the skin. Br J Dermatol. 1978;98(6):645–653.
304. Mendoza, S, Helwig, EB. Mucinous (adenocystic) carcinoma of the skin. Arch Dermatol. 1971;103(1):68–78.
305. Rosen, T. Cutaneous metastases. Med Clin North Am. 1980;64(5):885–900.
306. Ivan, D, Nash, JW, Prieto, VG, et al. Use of p63 expression in distinguishing primary and metastatic cutaneous adnexal neoplasms from metastatic adenocarcinoma to skin. J Cutan Pathol. 2007;34(6):474–480.
307. Van der Kwast, TH, Vuzevski, VD, Ramaekers, F, et al. Primary cutaneous adenoid cystic carcinoma: case report, immunohistochemistry, and review of the literature. Br J Dermatol. 1988;118(4):567–577.
308. Cooper, PH, Adelson, GL, Holthaus, WH. Primary cutaneous adenoid cystic carcinoma. Arch Dermatol. 1984;120(6):774–777.
309. Headington, JT, Teears, R, Niederhuber, JE, et al. Primary adenoid cystic carcinoma of skin. Arch Dermatol. 1978;114(3):421–424.
310. Seab, JA, Graham, JH. Primary cutaneous adenoid cystic carcinoma. J Am Acad Dermatol. 1987;17(1):113–118.
311. Eckert, F, Pfau, A, Landthaler, M. [Adenoid cystic sweat gland carcinoma. A clinicopathologic and immunohistochemical study]. Hautarzt. 1994;45(5):318–323.
312. Bergman, R, Lichtig, C, Moscona, RA, et al. A comparative immunohistochemical study of adenoid cystic carcinoma of the skin and salivary glands. Am J Dermatopathol. 1991;13(2):162–168.
313. Pavlakis, K, Zoubouli, C, Liakakos, T, et al. Myoepithelial cell cocktail (p63+SMA) for the evaluation of sclerosing breast lesions. Breast. 2006;15(6):705–712.
314. Missall, TA, Burkemper, NM, Jensen, SL, et al. Immunohistochemical differentiation of four benign eccrine tumors. J Cutan Pathol. 2009;36(2):190–196.
315. Holst, VA, Marshall, CE, Moskaluk, CA, et al. KIT protein expression and analysis of c-kit gene mutation in adenoid cystic carcinoma. Mod Pathol. 1999;12(10):956–960.
316. Kao, GF, Helwig, EB, Graham, JH. Aggressive digital papillary adenoma and adenocarcinoma. A clinicopathological study of 57 patients, with histochemical, immunopathological, and ultrastructural observations. J Cutan Pathol. 1987;14(3):129–146.
317. Duke, WH, Sherrod, TT, Lupton, GP. Aggressive digital papillary adenocarcinoma (aggressive digital papillary adenoma and adenocarcinoma revisited). Am J Surg Pathol. 2000;24(6):775–784.
318. Bueno, RA, Jr., Neumeister, MW, Wilhelmi, BJ. Aggressive digital papillary adenocarcinoma presenting as finger infection. Ann Plast Surg. 2002;49(3):326–327.
319. Keramidas, EG, Miller, G, Revelos, K, et al. Aggressive digital papillary adenoma-adenocarcinoma. Scand J Plast Reconstr Surg Hand Surg. 2006;40(3):189–192.
320. Smith, KJ, Skelton, HG, Holland, TT. Recent advances and controversies concerning adnexal neoplasms. Dermatol Clin. 1992;10(1):117–160.
321. Mori, O, Nakama, T, Hashimoto, T. Aggressive digital papillary adenocarcinoma arising on the right great toe. Eur J Dermatol. 2002;12(5):491–494.
322. Ohta, M, Hiramoto, M, Ohtsuka, H. Metastatic microcystic adnexal carcinoma: an autopsy case. Dermatol Surg. 2004;30(6):957–960.
323. Ban, M, Sugie, S, Kamiya, H, et al. Microcystic adnexal carcinoma with lymph node metastasis. Dermatology. 2003;207(4):395–397.
324. Rotter, N, Wagner, H, Fuchshuber, S, et al. Cervical metastases of microcystic adnexal carcinoma in an otherwise healthy woman. Eur Arch Otorhinolaryngol. 2003;260(5):254–257.
325. Carroll, P, Goldstein, GD, Brown, CW, Jr. Metastatic microcystic adnexal carcinoma in an immunocompromised patient. Dermatol Surg. 2000;26(6):531–534.
326. Cooper, PH, Mills, SE. Microcystic adnexal carcinoma. J Am Acad Dermatol. 1984;10(5 Pt 2):908–914.
327. Hoang, MP, Dresser, KA, Kapur, P, et al. Microcystic adnexal carcinoma: an immunohistochemical reappraisal. Mod Pathol. 2008;21(2):178–185.
328. Wick, MR, Cooper, PH, Swanson, PE, et al. Microcystic adnexal carcinoma. An immunohistochemical comparison with other cutaneous appendage tumors. Arch Dermatol. 1990;126(2):189–194.
329. Friedman, KJ. Low-grade primary cutaneous adenosquamous (mucoepidermoid) carcinoma. Report of a case and review of the literature. Am J Dermatopathol. 1989;11(1):43–50.
330. Landman, G, Farmer, ER. Primary cutaneous mucoepidermoid carcinoma: report of a case. J Cutan Pathol. 1991;18(1):56–59.
331. Cooper, PH, Frierson, HF, Jr., Morrison, AG. Malignant transformation of eccrine spiradenoma. Arch Dermatol. 1985;121(11):1445–1448.
332. Rockerbie, N, Solomon, AR, Woo, TY, et al. Malignant dermal cylindroma in a patient with multiple dermal cylindromas, trichoepitheliomas, and bilateral dermal analogue tumors of the parotid gland. Am J Dermatopathol. 1989;11(4):353–359.
333. Warkel, RL, Helwig, EB. Apocrine gland adenoma and adenocarcinoma of the axilla. Arch Dermatol. 1978;114(2):198–203.
334. Wick, MR, Goellner, JR, Wolfe, JT, III., et al. Adnexal carcinomas of the skin. I. Eccrine carcinomas. Cancer. 1985;56(5):1147–1162.
335. Wong, TY, Suster, S, Nogita, T, et al. Clear cell eccrine carcinomas of the skin. A clinicopathologic study of nine patients. Cancer. 1994;73(6):1631–1643.
336. Jakobiec, FA, Austin, P, Iwamoto, T, et al. Primary infiltrating signet ring carcinoma of the eyelids. Ophthalmology. 1983;90(3):291–299.
337. Png, JC, Tung, KH, Wong, YE, et al. Extramammary Paget’s disease: a report of three cases and review of the literature. Ann Acad Med Singapore. 1995;24(4):636–639.
338. Lai, YL, Yang, WG, Tsay, PK, et al. Penoscrotal extramammary Paget’s disease: a review of 33 cases in a 20-year experience. Plast Reconstr Surg. 2003;112(4):1017–1023.
339. Lee, SC, Roth, LM, Ehrlich, C, et al. Extramammary Paget’s disease of the vulva. A clinicopathologic study of 13 cases. Cancer. 1977;39(6):2540–2549.
340. Nadji, M, Morales, AR, Girtanner, RE, et al. Paget’s disease of the skin. A unifying concept of histogenesis. Cancer. 1982;50(10):2203–2206.
341. Rayne, SC, Santa Cruz, DJ. Anaplastic Paget’s disease. Am J Surg Pathol. 1992;16(11):1085–1091.
342. Takeshita, K, Izumoi, S, Ebuchi, M, et al. A case of rectal carcinoma concomitant with Pagetoid lesion in the perianal region—histopathological and electron microscopic observations. Gastroenterol Jpn. 1978;13(2):85–95.
343. Mayes, DC, Patterson, JW, Ramnani, DM, et al. Alpha-methylacyl coenzyme A racemase is immunoreactive in extramammary Paget disease. Am J Clin Pathol. 2007;127(4):567–571.
344. Shah, KD, Tabibzadeh, SS, Gerber, MA. Immunohistochemical distinction of Paget’s disease from Bowen’s disease and superficial spreading melanoma with the use of monoclonal cytokeratin antibodies. Am J Clin Pathol. 1987;88(6):689–695.
345. Bayer-Garner, IB, Reed, JA. Immunolabeling pattern of syndecan-1 expression may distinguish pagetoid Bowen’s disease, extramammary Paget’s disease, and pagetoid malignant melanoma in situ. J Cutan Pathol. 2004;31(2):169–173.
346. Bains, R, Gleason, BC, Thomas, AB, et al. Cystic fibrosis transmembrane conductance regulator is helpful in the distinction of extra-mammary Paget’s disease from squamous cell carcinoma in situ (Bowen’s disease). J Cutan Pathol. 2011;38(7):581–584.
347. Ogawa, E, Okuyama, R, Egawa, T, et al. Ectopic expression of the p53 homologue p63 is linked to squamous metaplasia in extramammary Paget’s disease with invasive adenocarcinoma. Histopathology. 2009;54(3):378–381.
348. Yanai, H, Takahashi, N, Omori, M, et al. Immunohistochemistry of p63 in primary and secondary vulvar Paget’s disease. Pathol Int. 2008;58(10):648–651.