
Top texture: © Laguna Design / Science Source;
Chapter 7: Chromosomes
Chapter Opener: © Power and Syred/Science Photo Library/Getty Images.
7.1 Introduction
A general principle is evident in the organization of all cellular genetic material. It exists as a compact mass that is confined to a limited volume, and its various activities, such as replication and transcription, must be accomplished within this space. The organization of this material must accommodate local transitions between inactive and active states.
The condensed state of nucleic acid results from its binding to basic proteins. The positive charges of these proteins neutralize the negative charges of the nucleic acid. The structure of the nucleoprotein complex is determined by the interactions of the proteins with the DNA (or RNA).
A common problem is presented by the packaging of DNA into phages, viruses, bacterial cells, and eukaryotic nuclei. The length of the DNA as an extended molecule would vastly exceed the dimensions of the compartment that contains it. The DNA (or in the case of some viruses, the RNA) must be compressed exceedingly tightly to fit into the space available. Thus, in contrast with the customary picture of DNA as an extended double helix, structural deformation of DNA to bend or fold it into a more compact form is the rule rather than the exception.
The magnitude of the discrepancy between the length of the nucleic acid and the size of its compartment is evident in the examples summarized in TABLE 7.1. For bacteriophages and eukaryotic viruses, the nucleic acid genome, whether single-stranded or double-stranded DNA or RNA, effectively fills the container (i.e., the viral capsid, which can be rodlike or spherical).
TABLE 7.1 The length of nucleic acid is much greater than the dimensions of the surrounding compartment.
| Compartment | Shape | Dimensions | Type of Nucleic Acid | Length |
|---|---|---|---|---|
| TMV | Filament | 0.008 × 0.3 μm | One single-stranded RNA | 2 μm = 6.4 kb |
| Phage fd | Filament | 0.0006 × 0.85 μm | One single-stranded DNA | 2 μm = 6.0 kb |
| Adenovirus | Icosahedron | 0.07 μm diameter | One double-stranded DNA | 11 μm = 35.0 kb |
| CrypticPhage T4 | Icosahedron | 0.065 × 0.0 μm | One double-stranded DNA | 55 μm = 170.0 kb |
| E. coli | Cylinder | 1.7 × 0.65 μm | One double-stranded DNA | 1.3 mm = 4.2 × 103 kb |
| Mitochondrion (human) | Oblate spheroid | 3.0 × 0.5 μm | ~10 identical double-stranded DNAs | 50 μm = 16.0 kb |
| Nucleus (human) | Spheroid | 6 μm diameter | 46 chromosomes of double-stranded DNA | .8 m = 6 × 109 kb |
For bacteria or eukaryotic cell compartments, the discrepancy is hard to calculate exactly, because the DNA is contained in a compact area that occupies only part of the compartment. The genetic material is seen in the form of the nucleoid in bacteria, and as the mass of chromatin in eukaryotic nuclei at interphase (between divisions), or as maximally condensed chromosomes during mitosis.
The density of DNA in these compartments is high. In a bacterium it is approximately 10 mg/mL, in a eukaryotic nucleus it is approximately 100 mg/mL, and in the phage T4 head it is more than 500 mg/mL. Such a concentration in solution would be equivalent to a gel of great viscosity. We do not entirely understand the physiological implications of such high concentrations of DNA, such as the effect this has upon the ability of proteins to find their binding sites on DNA.
The packaging of chromatin is flexible; it changes during the eukaryotic cell cycle. At the time of division (mitosis or meiosis), the genetic material becomes even more tightly packaged, and individual chromosomes become recognizable.
The overall compression of the DNA can be described by the packing ratio, which is the length of the DNA divided by the length of the unit that contains it. For example, the smallest human chromosome contains approximately 4.6 × 107 base pairs (bp) of DNA (about 10 times the genome size of the bacterium Escherichia coli). This is equivalent to 14,000 μm (= 1.4 cm) of extended DNA. At the point of maximal condensation during mitosis, the chromosome is approximately 2 μm long. Thus, the packing ratio of DNA in the chromosome can be as great as 7,000.
Researchers cannot establish packing ratios with such certainty for the more amorphous overall structures of the bacterial nucleoid or eukaryotic chromatin. The usual reckoning, however, is that mitotic chromosomes are likely to be 5 to 10 times more tightly packaged than interphase chromatin, which indicates a typical packing ratio of 1,000 to 2,000.
Major unanswered questions concern the specificity of higher order DNA packaging. How is DNA folding regulated to produce particular patterns, and how do these patterns relate to core genetic functions such as replication, chromosome segregation, or transcription?
7.2 Viral Genomes Are Packaged into Their Coats
From the perspective of packaging the individual sequence, there is an important difference between a cellular genome and a virus. The cellular genome is essentially indefinite in size; the number and location of individual sequences can be changed by duplication, deletion, and rearrangement. Thus, it requires a generalized method for packaging its DNA—one that is insensitive to the total content or distribution of sequences. By contrast, two restrictions define the needs of a virus. The amount of nucleic acid to be packaged is predetermined by the size of the genome, and it must all fit within a coat assembled from a protein or proteins coded by the viral genes.
A virus particle is deceptively simple in its superficial appearance. The nucleic acid genome is contained within a capsid, which is a symmetrical or quasisymmetrical structure assembled from one or only a few proteins. Attached to the capsid (or incorporated into it) are other structures; these structures are assembled from distinct proteins and are necessary for infection of the host cell.
The virus particle is tightly constructed. The internal volume of the capsid is rarely much greater than the volume of the nucleic acid it must hold. The difference is usually less than twofold, and often the internal volume is barely larger than the nucleic acid.
In its most extreme form, the restriction that the capsid must be assembled from proteins encoded by the virus means that the entire shell is constructed from a single type of subunit. The rules for assembly of identical subunits into closed structures restrict the capsid to one of two types. For the first type, the protein subunits stack sequentially in a helical array to form a filamentous or rodlike shape. For the second type, they form a pseudospherical shell—a type of structure that conforms to a polyhedron with icosahedral symmetry. Some viral capsids are assembled from more than a single type of protein subunit. Although this extends the exact types of structures that can be formed, most viral capsids conform to the general classes of quasicrystalline filaments or icosahedrons.
There are two general solutions to the problem of how to construct a capsid that contains nucleic acid:
The protein shell can be assembled around the nucleic acid, thereby condensing the DNA or RNA by protein–nucleic acid interactions during the process of assembly.
The capsid can be constructed from its component(s) in the form of an empty shell, into which the nucleic acid must be inserted, being condensed as it enters.
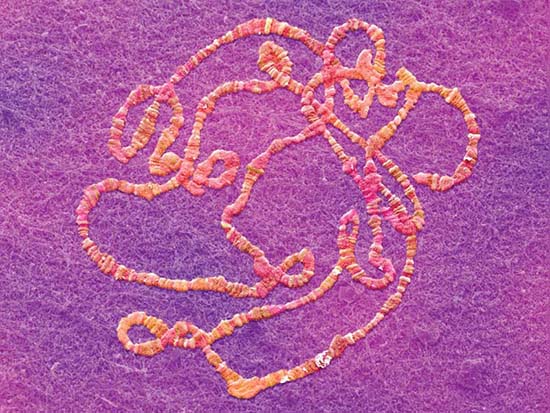
The capsid is assembled around the genome for single-stranded RNA viruses. The principle of assembly is that the position of the RNA within the capsid is determined directly by its binding to the proteins of the shell. The best characterized example is tobacco mosaic virus (TMV). Assembly begins at a duplex hairpin that lies within the RNA sequence. From this nucleation center, assembly proceeds bidirectionally along the RNA until it reaches the ends. The unit of the capsid is a two-layer disk, with each layer containing 17 identical protein subunits. The disk is a circular structure, which forms a helix as it interacts with the RNA. At the nucleation center, the RNA hairpin inserts into the central hole in the disk, and the disk changes conformation into a helical structure that surrounds the RNA. Additional disks are added, with each new disk pulling a new stretch of RNA into its central hole. The RNA becomes coiled in a helical array on the inside of the protein shell, as illustrated in FIGURE 7.1.
FIGURE 7.1 A helical path for TMV RNA is created by the stacking of protein subunits in the virion (the entire virus particle).
The spherical capsids of DNA viruses are assembled in a different way, as best characterized for the phages lambda and T4. In each case, an empty head shell is assembled from a small set of proteins. The duplex genome then is inserted into the head, accompanied by a structural change in the capsid.
FIGURE 7.2 summarizes the assembly of lambda. It begins with a small head shell that contains a protein “core.” This is converted to an empty head shell of more distinct shape. At this point the DNA packaging begins, the head shell expands in size (though it remains the same shape), and finally the full head is sealed by the addition of the tail.

FIGURE 7.2 Maturation of phage lambda passes through several stages. The empty head changes shape and expands when it becomes filled with DNA, diagrammed on the left. The electron micrographs on the right show the particles at the beginning (top) and the end (bottom) of the maturation pathway.
Top photo reproduced from: Cue, D., and Feiss M. 1993. Proc Natl Acad Sci USA 90: 9240–9294. Copyright © 2004 National Academy of Sciences, U.S.A. Bottom photo courtesy of Robert Duda, University of Pittsburgh.
A double-stranded DNA that spans short distances is a fairly rigid rod, yet it must be compressed into a compact structure to fit within the capsid. This packaging can be achieved by a smooth coiling of the DNA into the head or it might require introduction of abrupt bends.
Inserting DNA into a phage head involves two types of reaction: translocation and condensation. Both are energetically unfavorable.
Translocation is an active process in which the DNA is driven into the head by an ATP-dependent mechanism. A common mechanism for translocation is used for many viruses that replicate by a rolling circle mechanism to generate long tails that contain multimers of the viral genome. The best characterized example is phage lambda. The genome is packaged into the empty capsid by the terminase enzyme. FIGURE 7.3 summarizes the process.
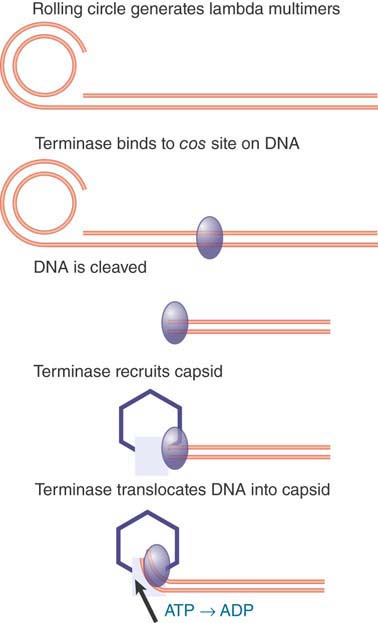
FIGURE 7.3 Terminase protein binds to specific sites on a multimer of virus genomes generated by rolling circle replication. It cuts the DNA and binds to an empty virus capsid, and then uses energy from hydrolysis of ATP to insert the DNA into the capsid.
The terminase was first recognized (and named) for its role in generating the ends of the linear phage DNA by cleaving at cos sites. (The name cos reflects the fact that it generates cohesive ends that have complementary single-stranded tails.) The phage genome encodes two subunits that make up the terminase. One subunit binds to a cos site; at this point it is joined by the other subunit, which cuts the DNA. The terminase assembles into a heterooligomer in a complex that also includes integration host factor (IHF; a dimer that is encoded by the bacterial genome). It then binds to an empty capsid and uses ATP hydrolysis to power translocation along the DNA. The translocation drives the DNA into the empty capsid.
Another method of packaging uses a structural component of the phage. In the Bacillus subtilis phage ϕ29, the motor that inserts the DNA into the phage head is an integral structure that connects the head to the tail. It functions as a rotary motor, where the motor action effects the linear translocation of the DNA into the phage head. The same motor is used to eject the DNA from the phage head when it infects a bacterium.
Less is known about the mechanism(s) of condensation into an empty capsid, except that capsids typically contain “internal proteins” as well as DNA. Such internal proteins might provide some sort of scaffolding onto which the DNA condenses. This would be similar to the use of the proteins of the shell in the plant RNA viruses (e.g., TMV, described earlier in this section).
How specific is the packaging? It cannot depend simply on particular sequences, because deletions, insertions, and substitutions all fail to interfere with the assembly process. The relationship between DNA and the head shell has been investigated directly by determining which regions of the DNA can be chemically crosslinked to the proteins of the capsid. The surprising answer is that all regions of the DNA are more or less equally susceptible. This probably means that when DNA is inserted into the head it follows a general rule for condensing, but the pattern is not determined by particular sequences.
These varying mechanisms of virus assembly all accomplish the same end: packaging a single DNA or RNA molecule into the capsid. Some viruses, however, have genomes that consist of multiple nucleic acid molecules. Reovirus contains 10 double-stranded RNA segments, all of which must be packaged into the capsid. Specific sorting sequences in the segments might be required to ensure that the assembly process selects one copy of each different molecule in order to collect a complete set of genetic information. In the simpler case of phage ϕ6, which packages three different segments of double-stranded RNA into one capsid, the RNA segments must bind in a specific order; as each is incorporated into the capsid, it triggers a change in the conformation of the capsid that creates binding sites for the next segment.
Some plant viruses are multipartite: Their genomes consist of segments, each of which is packaged into a different capsid. An example is alfalfa mosaic virus (AMV), which has four different single-stranded RNAs, each of which is packaged independently into a coat comprising the same protein subunit. A successful infection depends on the entry of one of each type into the cell. The four components of AMV exist as particles of different sizes. This means that the same capsid protein can package each RNA into its own characteristic particle. This is a departure from the packaging of a unique length of nucleic acid into a capsid of fixed shape.
The assembly pathway of viruses whose capsids have only one authentic form might be diverted by mutations that cause the formation of aberrant monster particles in which the head is longer than usual. These mutations show that a capsid protein(s) has an intrinsic ability to assemble into a particular type of structure, but the exact size and shape can vary.
Some of the mutations occur in genes that code for assembly factors, which are needed for head formation, but are not themselves part of the head shell. Such ancillary proteins limit the options of the capsid protein, reducing variation in the assembly pathway. Comparable proteins are employed in the assembly of cellular chromatin (see the chapter titled Chromatin).
7.3 The Bacterial Genome Is a Nucleoid with Dynamic Structural Properties
Although bacteria do not display structures with the distinct morphological features of eukaryotic chromosomes, their genomes nonetheless are organized into definite substructures within the cell. We can see the genetic material as a fairly compact clump (or series of clumps) that occupies about a third of the volume of the cell. FIGURE 7.4 displays a thin section through a bacterium in which this nucleoid is evident.

(a)

(b)
FIGURE 7.4 (a) A thin section shows the bacterial nucleoid as a compact mass in the center of the cell. (b) The nucleoid spills out of a lysed E. coli cell in the form of loops of a fiber.
(a) Photo courtesy of the Molecular and Cell Biology Instructional Laboratory Program, University of California, Berkeley.
(b) © Dr. Gopal Murti/Science Source.
When E. coli cells are lysed, fibers are released in the form of loops attached to the broken envelope of the cell, as shown in Figure 7.4b. The DNA of these loops is not found in the extended form of a free duplex, but instead is compacted by association with proteins.
Increasing numbers of nucleoid-associated proteins (NAPs) that resemble eukaryotic chromosomal proteins have been isolated in archaea and bacteria. Exactly what constitutes a NAP is vague, because some of them might contribute to multiple genetic functions. As a group, NAPs are emerging as antagonistic regulators of gene activity and nucleoid structure. In the gram-negative bacteria, researchers have characterized as many as 12 different NAPs, some of them depicted in FIGURE 7.5.
Most NAPs have DNA-binding activities that can affect the spatial arrangement of DNA through bending, wrapping, or bridging.

(a)
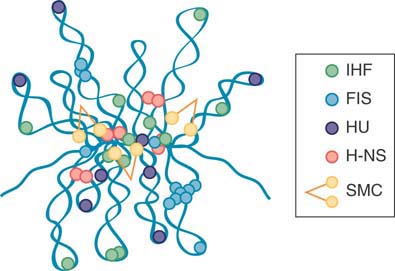
(b)
FIGURE 7.5 Topological organization of the bacterial chromosome. (a) Schematic representation of the bottlebrush model of the nucleoid. This diagram depicts the interwound supercoiled loops emanating from a dense core. The topologically isolated domains are on average 10 kb and therefore are likely to encompass several branched plectonemic loops. (b) Schematic representation of the small nucleoid-associated proteins and the structural maintenance of chromosome (SMC) complexes. These proteins introduce DNA bends and also function in bridging chromosomal loci.
FIGURE 7.6 summarizes how NAPs vary in their function and expression patterns as cells progress through growth phases. The dynamics of individual NAPs and their interactions with one another are becoming increasingly more clear despite the complexity of their multifaceted effects on nucleoid structure and function.
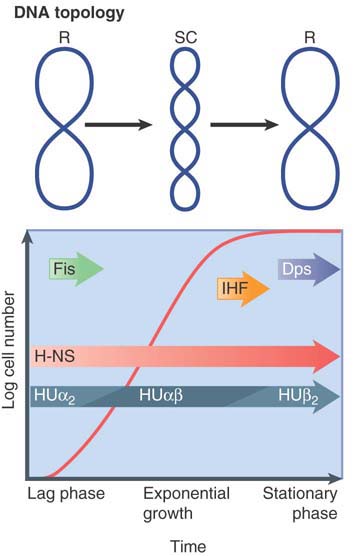
FIGURE 7.6 Growth phase and elements that affect nucleoid structure. A typical growth curve for E. coli growing in batch culture begins with a lag phase followed by the log phase of exponential growth and, finally, stationary phase (when the cells stop growing). Important nucleoid-associated proteins are expressed at different times during the growth curve, as indicated. In addition, there are significant changes in DNA topology: DNA is negatively supercoiled (SC) in log phase cells, whereas it is more relaxed (R) in lag phase and stationary phase cells.
Protein H-NS (histone-like, nucleoid-structuring protein) has a preference for AT-rich DNA and can form DNA-H-NS-DNA bridges, allowing this NAP to simultaneously influence gene promoter activity and nucleoid structure. H-NS is expressed throughout all growth phases. Its interactions with other expression-modulating proteins likely contribute to the ability of H-NS to silence hundreds of genes and form boundaries of microdomains. Recent advances in chromosome conformation capture (C3; also see the chapter titled Chromatin) and high-resolution fluorescence imaging suggest that H-NS might mediate the colocalization of many H-NS-binding sites into two foci. These have been proposed to represent each of two replichores, the left and right arms of the circular genome that are replicated by the bidirectional movement forks from the origin.
Protein HU has two subunits: homodimers or heterodimers of HUα and HUβ. They bend or wrap DNA and play a role in DNA flexibility. These histone-like proteins bind nonspecifically to multiple sites with some preference for distorted DNA regions such as bends, forks, four-way junctions, nicks, or overhangs. Consequently, they are implicated as architectural factors affecting various functions in DNA metabolism.
Other NAPs, such as IHF, Dps, and bacterial condensins, also appear to have multiple or overlapping roles in nucleoid architecture and core genetic processes. One of these is the integration host factor (IHF), first identified as a bacteriophage lambda cofactor for site-specific integration. IHF has since been found to bend DNA and induce U-turns and influence global transcription, not unlike a general transcription factor. The ability of IHF to alter local DNA structure through U-turn formation appears to be a defining feature of its mode of action in replication, phage integration, transposition, and transcription. Another well-characterized and interesting NAP is the DNA protection during starvation protein (Dps). Dps is expressed in the stationary phase and in oxidatively stressed cells, likely functioning to limit DNA damage. MukB and its homologs are chromosome structural maintenance proteins that are now recognized as components of bacterial condensin complexes. Similar to eukaryotic condensins in structure and function, they regulate chromosome condensation and are required for proper segregation of chromosomes during cell division. Genetic evidence establishes a role for these complexes (MukBEF or SMC-ScpAB) in DNA topology and domain delineation.
As a group, the NAP proteins and their expression patterns point to an integrating principle whereby nucleoid structure and gene expression are comodulated during cell growth and reproduction in an environmentally responsive manner. How these packaging functions are coupled to gene positioning and promoter functions to affect bacterial fitness and to what extent such an integrated system imposes evolutionary constraints for bacterial fitness are among the key questions in bacterial functional genomics.
7.4 The Bacterial Genome Is Supercoiled and Has Four Macrodomains
The DNA of the bacterial nucleoid isolated in vitro behaves as a closed duplex structure, as judged by its response to ethidium bromide. This small molecule intercalates between base pairs to generate positive superhelical turns in “closed” circular DNA molecules; that is, molecules in which both strands have covalent integrity. (In “open” circular molecules, which contain a nick in one strand, or with linear molecules, the DNA can rotate freely in response to the intercalation, thus relieving the tension.)
In a natural closed DNA that is negatively supercoiled, the intercalation of ethidium bromide first removes the negative supercoils and then introduces positive supercoils. The amount of ethidium bromide needed to achieve zero supercoiling is a measure of the original density of negative supercoils.
Some nicks occur in the compact nucleoid during its isolation; they can also be generated by limited treatment with DNase. This does not, however, abolish the ability of ethidium bromide to introduce positive supercoils. This capacity of the genome to retain its response to ethidium bromide in the face of nicking reflects the existence of many independent chromosomal domains, and that the supercoiling in each domain is not affected by events in the other domains.
Early data suggested that each domain consists of around 40 kilobases (kb) of DNA, but more recent analysis suggests that the domains can be smaller, about 10 kb each. This would correspond to approximately 400 domains in the E. coli genome. It is likely that there is in fact a range of domain sizes. The ends of the domains appear to be randomly distributed instead of located at predetermined sites on the chromosome.
The existence of separate domains could permit different degrees of supercoiling to be maintained in different regions of the genome. This could be relevant in considering the different susceptibilities of particular bacterial promoters to supercoiling (see the chapter titled Prokaryotic Transcription).
Supercoiling in the genome can in principle take either of two forms:
If a supercoiled DNA is free, its path is unconstrained, and negative supercoils generate a state of torsional tension that is transmitted freely along the DNA within a domain. Torsional tension resulting from negative supercoils can be relieved by unwinding the double helix, as described in the chapter titled Genes Are DNA and Encode RNAs and Polypeptides. The DNA is in a dynamic equilibrium between the states of tension and unwinding.
Supercoiling can be constrained if proteins are bound to the DNA to hold it in a particular three-dimensional configuration. In this case, the supercoils are represented by the path the DNA follows in its fixed association with the proteins. The energy of interaction between the proteins and the supercoiled DNA stabilizes the nucleic acid so that no tension is transmitted along the molecule.
Measurements of supercoiling in vitro encounter the difficulty that constraining proteins might have been lost during isolation. However, various approaches suggest that DNA is under torsional stress in vivo. One approach is to measure the effect of nicking the DNA.
Unconstrained supercoils are released by nicking, whereas constrained supercoils are unaffected. Nicking releases about 50% of the overall supercoiling. This suggests that about half of the supercoiling is transmitted as tension along DNA, with the other half being absorbed by protein binding. Another approach uses the crosslinking reagent psoralen, which binds more readily to DNA when it is under torsional tension. The reaction of psoralen with E. coli DNA in vivo corresponds to an average density of 1 negative superhelical turn/200 bp (σ = −0.05).
We also can examine the ability of cells to form alternative DNA structures; for example, to generate cruciforms (intrastrand base pairing) at palindromic sequences. From the change in linking number that is required to drive such reactions, it is possible to calculate the original supercoiling density. This approach suggests an average density of σ = −0.025, or 1 negative superhelical turn/100 bp.
Thus supercoils do appear to create torsional tension in vivo. There might be variation about an average level, and the precise range of densities is difficult to measure. It is, however, clear that the level is sufficient to exert significant effects on DNA structure—for example, in assisting melting in particular regions such as origins or promoters.
Operating at a larger scale, nucleoid structural features, including macrodomains, have recently been observed using genetic and live imaging techniques.
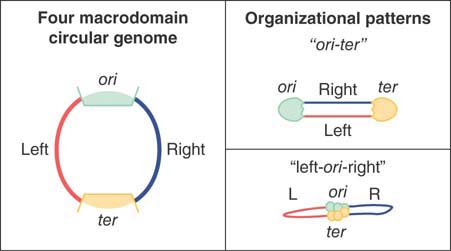
FIGURE 7.7 Large-scale organizational patterns of the macrodomains in bacteria. The domains are delimited by the origin (ori) and termination (ter) regions, creating two different replichores termed left and right.
FIGURE 7.7 shows two large-scale organizational patterns that have been observed in bacteria. The domains are delimited by the origin (ori) and termination (ter) regions, creating two different replichores termed left and right. The two patterns, referred to as ori-ter and left-ori-right, have been observed to be prevalent in different species of bacteria. Interestingly, they have both been shown to occur in Bacillus subtilis, but at different times during cell cycle progression. In this regard, bacterial and eukaryotic genomes display a similar phenomenon in which genome structure and dynamics is linked to progression through the cell cycle and DNA synthesis phases.
7.5 Eukaryotic DNA Has Loops and Domains Attached to a Scaffold
Interphase chromatin is a tangled-appearing mass occupying a large part of the nuclear volume. This is in contrast with the highly organized and reproducible ultrastructure of mitotic chromosomes. What controls the distribution of interphase chromatin within the nucleus?
Some indirect evidence about its nature is provided by the isolation of the genome as a single, compact body. Using the same technique that was developed for isolating the bacterial nucleoid (see the previous section, The Bacterial Genome Is Supercoiled), researchers can lyse nuclei on top of a sucrose gradient. This releases the genome in a form that can be collected by centrifugation. As isolated from Drosophila melanogaster, it can be visualized as a compactly folded fiber (10 nm in diameter) consisting of DNA bound to proteins.
Supercoiling measured by the response to ethidium bromide corresponds to about 1 negative supercoil/200 bp. These supercoils can be removed by nicking with DNase, although the DNA remains in the form of the 10-nm fiber. This suggests that the supercoiling is caused by the arrangement of the fiber in space, and that it represents the existing torsion.
Full relaxation of the supercoils requires 1 nick/85 kb or so, thus identifying the average length of torsionally “closed” DNA. This region could comprise a loop or domain similar in nature to those identified in the bacterial genome. Loops can be seen directly when the majority of proteins are extracted from mitotic chromosomes. The resulting complex consists of the DNA associated with about 8% of the original protein content. As shown in FIGURE 7.8, the protein-depleted chromosomes reveal an underlying structure of a metaphase scaffold that still resembles the general form of a mitotic chromosome, surrounded by a halo of DNA.

FIGURE 7.8 Histone-depleted chromosomes consist of a protein scaffold to which loops of DNA are anchored.
Reprinted from: Paulson, J. R., and Laemmli, U. K. 1977. “The structure of histone-depleted metaphase chromosomes.” Cell 12:817–828., with permission from Elsevier (http://www.sciencedirect.com/science/article/pii/009286747790280X). Photo courtesy of Ulrich K. Laemmli, University of Geneva, Switzerland.
The metaphase scaffold consists of a dense network of fibers. Threads of DNA emanate from the scaffold, apparently as loops of average length 10 to 30 μm (30 to 90 kb). The DNA can be digested without affecting the integrity of the primarily proteinaceous scaffold. In interphase nuclei, this underlying proteinaceous structure is less well defined, but a more broadly dispersed arrangement in the nucleoplasm has been referred to as the nuclear matrix rather than the scaffold.
7.6 Specific Sequences Attach DNA to an Interphase Matrix
Is DNA attached to a matrix via specific sequences? Researchers can empirically define DNA sites attached to proteinaceous structures in interphase nuclei. They are called matrix attachment regions (MARs) or scaffold attachment regions (SARs). The precise functionality of the nuclear matrix and MARs has been a topic of considerable debate. Some observations are clear: The same sequences appear to attach to the protein substructure in both metaphase and interphase cells. Chromatin appears to be attached to an underlying structure in vivo, and there have been many suggestions that this attachment affects aspects of transcription, repair, or replication.
Are particular DNA regions associated with this matrix? FIGURE 7.9 summarizes two approaches to detect specific MARs. Both begin by isolating the matrix as a crude nuclear preparation containing chromatin and nuclear proteins. Researchers can then use different treatments to characterize DNA in the matrix or to identify DNA able to attach to it. The same general approaches can be applied to metaphase scaffold preparations.

FIGURE 7.9 MARs can be identified by characterizing the DNA retained by the matrix isolated in vivo (left) or by identifying the fragments that can bind to the matrix from which all DNA has been removed (right).
To analyze existing MARs that are bound to the matrix in vivo, chromosomal loops can be decondensed by extracting the chromatin proteins. Removal of the DNA loops by treatment with restriction nucleases leaves only the (presumptive) in vivo MAR sequences attached to the matrix.
The complementary approach is to remove all of the DNA from the matrix by treatment with DNase, at which point isolated fragments of DNA can be tested for their ability to bind to the matrix in vitro.
The same sequences should be associated with the matrix in vivo or in vitro. After researchers identify a potential MAR, they can determine the size of the minimal region needed for association in vitro by deletions, aiding in the identification of MAR-sequence-binding proteins.
A surprising feature is the lack of conservation of sequence in MAR fragments. Other than A-T richness, they lack any other obvious consensus sequences. Other interesting sequences, however, often are in the DNA stretch containing the MAR. cis-acting sites that regulate transcription are common, as are 5′ introns and recognition sites for topoisomerase II. It is therefore possible that a MAR serves more than one function by providing a site for attachment to the matrix and containing other sites at which topological changes in DNA are effected.
What is the relationship between the chromosome scaffold of dividing cells and the matrix of interphase cells? Are the same DNA sequences attached to both structures? In several cases, the same DNA fragments that are found within the nuclear matrix in vivo can be retrieved from the metaphase scaffold. Fragments that contain MAR sequences can bind to a metaphase scaffold, so it therefore seems likely that DNA contains a single type of attachment site. In interphase cells the attachment site is connected to the nuclear matrix, whereas in mitotic cells it is connected to the chromosome scaffold. Interestingly, it is also clear that although some MARs are constitutive (continuously bound to the matrix or scaffold), others appear to be facultative and change their interactions with the matrix depending on cell type or other conditions.
The nuclear matrix and chromosome scaffold consist of different proteins, although there are some common components. Topoisomerase II is a prominent component of the chromosome scaffold, and is a constituent of the nuclear matrix, reflecting the importance of topology in both cases.
7.7 Chromatin Is Divided into Euchromatin and Heterochromatin
Each chromosome contains a single, very long duplex of DNA, folded into a fiber that runs continuously throughout the chromosome. Thus, in accounting for interphase chromatin and mitotic chromosome structure, we have to explain the packaging of a single, exceedingly long molecule of DNA into a form in which it can be transcribed and replicated, and can become cyclically more and less compressed.
Individual eukaryotic chromosomes become visible as single compact units during mitosis. FIGURE 7.10 is an electron micrograph of a replicated chromosome isolated and photographed at metaphase. The sister chromatids are evident at this stage, and will give rise to the daughter chromosomes upon their separation starting at anaphase. Each chromatid consists of a large thick fiber with a nubbly appearance. The DNA is 5 to 10 times more condensed in mitotic chromosomes than in interphase chromatin.

FIGURE 7.10 The sister chromatids of a mitotic pair each consist of a fiber (~30 nm in diameter) compactly folded into the chromosome.
© Biophoto Associates/Science Source.
During most of the life cycle of the eukaryotic cell, however, its genetic material occupies an area of the nucleus in which individual chromosomes cannot be distinguished by conventional microscopy. The global structure of the interphase chromatin does not appear to change visibly between divisions or even during the period of replication, when the amount of chromatin doubles. Chromatin is fibrillar, although the overall spatial configuration of the fiber has long been difficult to discern. However, recent advances in high-resolution microscopy, fluorescence in situ hybridization (FISH) staining, and live imaging have finally begun to reveal additional aspects of chromatin structure and nuclear architecture not evident in the last century.
As the nuclear section of FIGURE 7.11 illustrates, we can divide chromatin into two types of material:
In most regions, the chromatin is less densely packed than in the mitotic chromosome. This material, called euchromatin, is relatively dispersed and occupies most of the nucleoplasm.
Some regions of chromatin are very densely packed, displaying a condition comparable to that of the chromosome at mitosis. This material, called heterochromatin, is typically found at centromeres, but occurs at other locations as well, including telomeres and highly repetitive sequences. It passes through the cell cycle with relatively little change in its degree of condensation. It forms a series of discrete clumps, visible in Figure 7.11, with a tendency to be found at the nuclear periphery and at the nucleolus. In some cases, the various heterochromatic regions, especially those associated with centromeres, aggregate into a densely staining chromocenter. The common form of heterochromatin that always remains heterochromatic is called constitutive heterochromatin. In contrast, there is another category of heterochromatin, called facultative heterochromatin, in which regions of euchromatin are converted to a heterochromatic state.

FIGURE 7.11 A thin section through a nucleus stained with Feulgen shows heterochromatin as compact regions clustered near the nucleolus and nuclear membrane.
Photo courtesy of Edmund Puvion, Centre National de la Recherche Scientifique.
The same fibers run continuously between euchromatin and heterochromatin, as these states simply represent different degrees of condensation of the genetic material. In the same way, euchromatic regions exist in different states of condensation during interphase and mitosis. Thus, the genetic material is organized in a manner that permits alternative states to be maintained side by side in chromatin, and allows cyclical changes to occur in the packaging of euchromatin between interphase and division. We discuss the molecular basis for these states in the chapters titled Chromatin and Epigenetics I and II.
The structural condition of the genetic material is correlated with its activity. The common features of constitutive heterochromatin are as follows:
It is permanently or nearly always condensed.
It replicates late in S phase and has a reduced frequency of genetic recombination relative to euchromatic gene-rich areas of the genome.
It often consists of multiple repeats of a few sequences of DNA that are not transcribed or are transcribed at very low levels. (Genes that reside in heterochromatic regions are generally less transcriptionally active than their euchromatic counterparts, but there are exceptions to this general rule.)
The density of genes in this region is very much reduced compared with euchromatin, and genes that are translocated into or near it are often inactivated. The one dramatic exception to this is the ribosomal DNA in the nucleolus, which has the general compacted appearance and behavior of heterochromatin (such as late replication), yet is engaged in very active transcription.
There are numerous molecular markers for changes in the properties of the DNA and protein components (see the chapters titled Epigenetics I and II). They include reduced acetylation of histone proteins, increased methylation at particular sites on histones, and methylation of cytosine bases in DNA. These molecular changes result in the condensation of the chromatin and the recruitment of heterochromatin-specific proteins, which are responsible for maintaining or spreading its inactivity. Although active genes are contained within euchromatin, only a minority of the sequences in euchromatin are transcribed at any time. Thus, location in euchromatin is necessary for most gene expression, but is not sufficient for it.
In addition to the general distributions observed for heterochromatin and euchromatin, studies have addressed whether there is an overall chromosome organization within the nucleus. The answer in many cases is yes; chromosomes appear to occupy distinct three-dimensional spaces known as chromosome territories, as diagrammed in FIGURE 7.12, showing a probabilistic model of the spatial arrangement of human chromosome territories. The chromosomes occupying these territories are not entangled with one another, but do share areas of interaction and some common functional organization. For example, heterochromatic and other silent regions are found primarily at the nuclear periphery, whereas gene-dense regions are internally located. Active genes are often found at the borders of territories, sometimes clustered together in interchromosomal spaces that are enriched in transcriptional machinery, known as transcription factories.
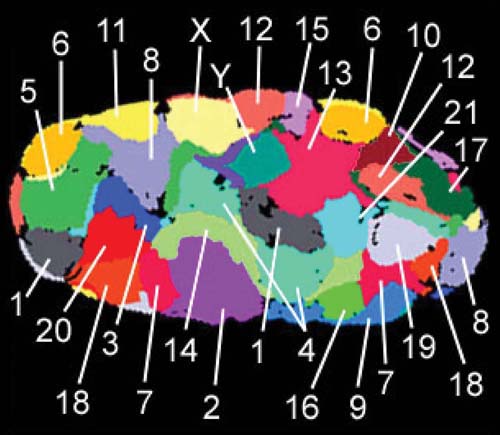
FIGURE 7.12 Chromosomes occupy chromosome territories in the nucleus and are not entangled with one another. This is a false-colored representation of chromosome territories obtained by individually staining chromosomes 1–22, X and Y in a human fibroblast nucleus. Heterochromatic regions, silenced genes, and gene-sparse regions of chromosomes are typically localized to the nuclear periphery. Active genes are often found at the borders of chromosome territories, and active genes from several chromosomes can cluster in interchromosomal territories that are enriched in transcription machinery.
Data from Bolzer, A., et al. 2005. PLoS Biol 3(5): e157.
How chromosome territories are established, and how they vary by cell cycle and cell type, are not yet understood, but advances in super-resolution microscopy, genomics, and mathematical modeling are beginning to reveal the presence of subchromosomal compartments and domains that occur in the historically refractory structural scale between a 30-nm chromatin fiber and whole chromosomes. For instance, researchers can define large chromosomal domains by the time at which they replicate in S phase. Comparing replication-timing profiles of several mammalian cell types reveals that the changes occur in defined units of 400–800 kb called replication domains (RDs). As summarized in FIGURE 7.13, these RDs correspond to structural domains called topologically associated domains (TADs), as revealed by chromatin interaction maps described in the Chromatin chapter. Evidence for this relationship comes from the concomitant switching between RDs and TAD compartments as cells differentiate. In this regard, RDs and TADs might represent chromosomal subdomains or nuclear compartments that act as epigenetic modules preserved across cell types.
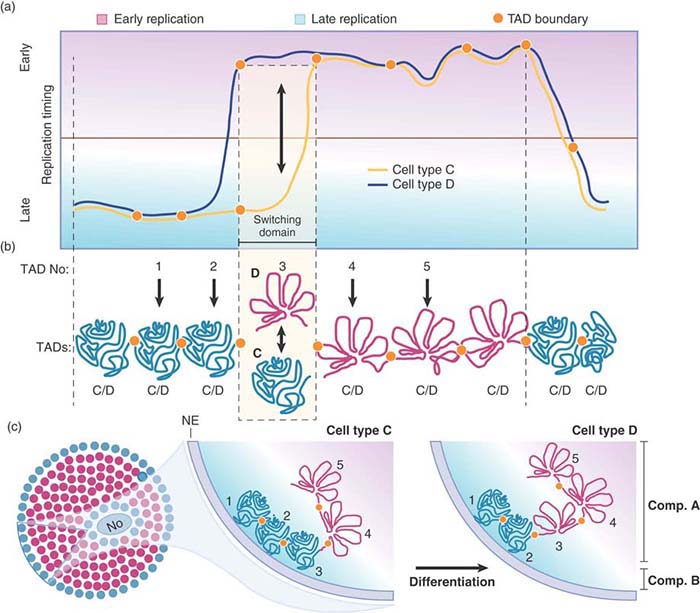
FIGURE 7.13 Chromatin is regulated at the level of defined units during differentiation. (a) Changes in temporal order of replication timing identify units of chromosome structure. Comparing replication timing profiles of two hypothetical cell types (C and D) identifies a replication domain that change replication timing during differentiation (switching domain). (b) Replication domains correspond to TADs. TADs can be early replicating and open (red) or late replicating and closed (green) depending on the cell type. Exemplary TADs are numbered 1 to 5. TADs 1 and 2 are late replicating, and TADs 4 and 5 are early replicating in both cell types. TAD 3 is late replicating in cell type C and early replicating in cell type D. (c) In general, early replicating TADs (red circles) are more open and located in the nuclear interior, and late replicating TADs (green circles) are more compact and located toward the nuclear periphery. During differentiation, TADs that switch replication timing move toward or away from nuclear lamina and undergo a change in compaction depending on the direction of the replicating timing switch.
7.8 Chromosomes Have Banding Patterns
As a result of the diffuse state of chromatin, it is difficult to directly determine the specificity of its organization. Three-dimensional sequence-level mapping techniques are beginning to give us insights into the organization of interphase chromatin. At the level of the chromosome, each member of the complement has a different and reproducible ultrastructure. When mitotic chromosomes are subjected to proteolytic enzyme (trypsin) treatment followed by staining with the chemical dye Giemsa, they generate distinct chromosome-specific patterns called G-bands. FIGURE 7.14 presents an example of the human set.

FIGURE 7.14 G-banding generates a characteristic lateral series of bands in each member of the chromosome set.
Photo courtesy of Lisa Shaffer, Washington State University, Spokane.
Until the development of this technique, researchers could distinguish human chromosomes only by their overall size and the relative location of the centromere. G-banding allows each chromosome to be identified by its characteristic banding pattern. This pattern allows translocations from one chromosome to another to be identified by comparison with the original diploid set. FIGURE 7.15 shows a diagram of the bands of the human X chromosome. The bands are large structures, each approximately 107 bp of DNA, and each of which can include many hundreds of genes.

FIGURE 7.15 The human X chromosome can be divided into distinct regions by its banding pattern. The short arm is p and the long arm is q; each arm is divided into larger regions that are further subdivided. This map shows a low-resolution structure; at higher resolution, some bands are further subdivided into smaller bands and interbands, e.g., p21 is divided into p21.1, p21.2, and p21.3.
The banding technique is of enormous practical use, but the mechanism of banding remains a mystery. All that is certain is that the dye stains untreated chromosomes more or less uniformly. Thus, the generation of bands depends on a variety of treatments, such as proteolytic digestion, that change the response of the chromosome (presumably by extracting the component that binds the stain from the nonbanded regions). Researchers can generate similar bands by using an assortment of other treatments.
Researchers often can distinguish G-bands from interbands by their lower G-C content. If there are 10 bands on a large chromosome with a total content of 100 megabases (Mb), this means that the chromosome is divided into regions averaging 5 Mb in length that alternate between low G-C (band) and high G-C (interband) content. There is a tendency for genes to be enriched in the interband regions. All of this argues for some long-range, sequence-dependent organization.
The human genome sequence confirms this basic observation. FIGURE 7.16 shows that there are distinct fluctuations in G-C content when the genome is divided into small bins (DNA segments or lengths). The average of 41% G-C is common to mammalian genomes. There are regions as low as 30% or as high as 65%. The average length of regions with greater than 43% G-C is 200 to 250 kb. This makes it clear that the band/interband structure does not correspond directly with the more numerous homogeneous segments that alternate in G-C content, although the bands do tend to contain a higher content of low G-C segments. Genes are concentrated in regions of higher G-C content.
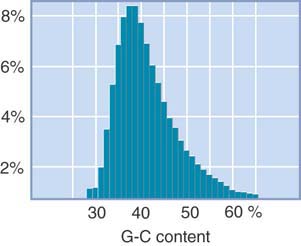
FIGURE 7.16 There are large fluctuations in G-C content over short distances. Each bar shows the percentage of 20-kb fragments with the given G-C content.
7.9 Lampbrush Chromosomes Are Extended
It would be extremely useful to observe gene expression in its natural state in order to see what structural changes are associated with transcription. The compression of DNA in chromatin, coupled with the difficulty of identifying particular genes within intact chromatin, makes it impossible to visualize the transcription of individual active genes, although advances in live imaging and microscopic resolution are beginning to overcome that limitation.
Scientists can observe gene expression directly in certain unusual situations in which the chromosomes are found in a highly extended form that allows individual loci (or groups of loci) to be distinguished. Lateral differentiation of structure is evident in many chromosomes when they first appear for meiosis. At this stage, the chromosomes resemble a series of beads on a string. The beads are densely staining granules, properly known as chromomeres. Chromomeres are larger and distinct from individual nucleosomes, which are also sometimes referred to as beads on a string (see the chapter titled Chromatin). In general, though, there is little gene expression at meiosis, and it is not practical to use this material to identify the activities of individual genes. An exceptional situation that allows the material to be examined is presented by lampbrush chromosomes, which have been best characterized in certain amphibians and birds.
Lampbrush chromosomes are formed during an unusually extended meiosis, which can last up to several months. During this period, the chromosomes exist in a stretched-out form that we can visualize by using a light microscope. At a later point during meiosis, the chromosomes revert to their usual compact size. The extended state provides a unique opportunity to see the structure of the chromosome.
The lampbrush chromosomes are meiotic bivalents, each consisting of paired homologous chromosomes that have been replicated. The sister chromatids remain connected along their lengths and each homolog appears, therefore, as a single fiber. FIGURE 7.17 shows an example in which the homologs have desynapsed and are held together only by chiasmata that indicate points of chromosome crossover. Each sister chromatid pair forms a series of ellipsoidal chromomeres, 1 to 2 μm in diameter, which are connected by a very fine thread. This thread contains the two sister duplexes of DNA and runs continuously along the chromosome, through the chromomeres.

FIGURE 7.17 A lampbrush chromosome is a meiotic bivalent in which the two pairs of sister chromatids are held together at chiasmata (indicated by arrows).
Photo courtesy of Joseph G. Gall, Carnegie Institution.
The lengths of the individual lampbrush chromosomes in the newt Notophthalmus viridescens range from 400 to 800 μm, compared with the range of 15 to 20 μm seen later in meiosis. Thus, the lampbrush chromosomes are about 30 times less compacted along their axes than their somatic counterparts. The total length of the entire lampbrush chromosome set is 5 to 6 μm and is organized into about 5,000 chromomeres.
The lampbrush chromosomes take their name from the lateral loops that extrude from the chromomeres at certain positions. The arrangement of fibers around the chromosome axis resembles the cleaning fibers of a lampbrush (a common tool back when lampbrush chromosomes were first observed in 1882). The loops extend in pairs, one from each sister chromatid. The loops are continuous with the axial thread, representing chromosomal material extruded from its more compact organization in the chromomere. The loops are surrounded by a matrix of ribonucleoproteins that contain nascent RNA chains. Often, a transcription unit can be defined by the increase in the length of the RNP moving around the loop. The loop is an extruded segment of DNA that is being actively transcribed. In some cases, researchers have identified loops corresponding to particular genes. For these cases, the structure of the transcribed gene—and the nature of the product—can allow for a rare situation wherein gene expression can be directly visualized and studied in situ.
7.10 Polytene Chromosomes Form Bands
The interphase nuclei of some tissues of the larvae of dipteran flies contain chromosomes that are greatly enlarged relative to their usual condition. They possess both increased diameter and greater length. FIGURE 7.18 shows an example of a chromosome set from the salivary gland of D. melanogaster. The members of this set are called polytene chromosomes.
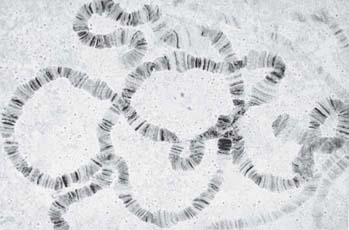
FIGURE 7.18 The polytene chromosomes of D. melanogaster form an alternating series of bands and interbands.
Photo courtesy of José Bonner, Indiana University.
Each member of the polytene set consists of a visible series of bands (more properly, but rarely, described as chromomeres). The bands range in size from the largest, with a breadth of approximately 0.5 μm, to the smallest, at nearly 0.05 μm. (The smallest can be distinguished only under an electron microscope.) The bands contain most of the mass of DNA and stain intensely with appropriate reagents. The regions between them stain more lightly and are called interbands. There are about 5,000 bands in the D. melanogaster set.
The centromeres of all four chromosomes of D. melanogaster aggregate to form a chromocenter that consists largely of heterochromatin. (In the male it includes the entire Y chromosome.) The remaining 75% of the genome is organized into alternating bands and interbands in the polytene chromosomes. The length of the chromosome set is about 2,000 μm. The DNA in extended form would stretch for approximately 40,000 μm, so the packing ratio is 20. This demonstrates vividly the extension of the genetic material relative to the usual states of interphase chromatin or mitotic chromosomes.
What are the chromosomal structural features revealed by these giant chromosomes? Each is produced by the successive replications of a synapsed diploid pair of chromosomes. The replicas do not separate, but instead remain aligned with each other in their extended state. This repeated replication without sister chromatid separation is a process known as endoreduplication. At the beginning of the process, each synapsed pair has a DNA content of 2C (where C represents the DNA content of the individual chromosome). This amount then doubles up to nine times, at its maximum giving a content of 1,024C. The number of doublings is different in the various tissues of the D. melanogaster larva.
We can visualize each chromosome as a large number of parallel fibers running longitudinally that are tightly condensed in the bands and less so in the interbands. It is likely that each fiber represents a single (C) haploid chromosome. This gives rise to the name polytene (“many threads”). The degree of polyteny is the number of haploid chromosomes contained in the giant chromosome.
The banding pattern is characteristic for each strain of Drosophila. The constant number and linear arrangement of the bands were first noted in the 1930s, when it was realized that they form a cytological map of the chromosomes. Rearrangements—such as deletions, inversions, or duplications—result in alterations of the order of bands.
The linear array of bands can be equated with the linear array of genes. Thus, genetic rearrangements, as seen in a linkage map, can be correlated with structural rearrangements of the cytological map. Ultimately, a particular mutation can be located in a particular band. The total number of genic loci in D. melanogaster exceeds the number of bands, so there are probably multiple genes in most or all bands.
The positions of particular genes on the cytological map can be determined directly by the technique of in situ hybridization. The modern version of this protocol using fluorescent probes is described in the chapter titled Methods in Molecular Biology and Genetic Engineering. Although fluorescent probes are currently preferred, when the method was originally developed a radioactive probe representing the gene of interest was used; FIGURE 7.19 summarizes this protocol. A probe representing a gene is hybridized with the denatured DNA of the polytene chromosomes in situ, and the excess unbound probe is washed away. Autoradiography identifies the position or positions of the corresponding genes by the superimposition of grains at a particular band or bands. (The principle is the same when fluorescent probes are used; the only fundamental difference is the detection of the label by fluorescence microscopy.) FIGURE 7.20 shows an example. Using in situ hybridization, it is possible to determine directly the band within which a particular sequence lies.

FIGURE 7.19 Individual bands containing particular genes can be identified by in situ hybridization.
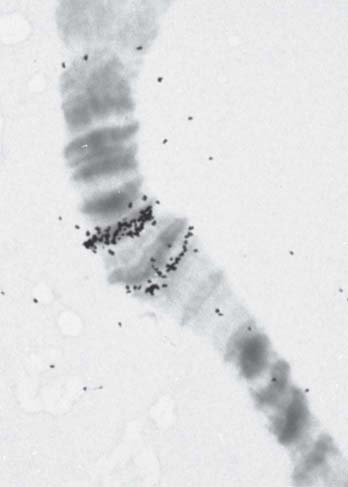
FIGURE 7.20 A magnified view of bands 87A and 87C shows their hybridization in situ with labeled RNA extracted from heat-shocked cells.
Photo courtesy of José Bonner, Indiana University.
7.11 Polytene Chromosomes Expand at Sites of Gene Expression
One of the intriguing features of polytene chromosomes is that researchers can visualize transcriptionally active sites. Some of the bands pass transiently through an expanded state in which they appear like a puff on the chromosome, when chromosomal material is extruded from the axis. FIGURE 7.21 presents examples of some very large puffs (called Balbiani rings).

FIGURE 7.21 Chromosome IV of the insect C. tentans has three Balbiani rings in the salivary gland.
Reprinted from: Daneholt, B. 1975. “Transcription in polytene chromosomes.” Cell 4:1–9, with permission from Elsevier http://www.sciencedirect.com/science/journal/00928674. Photo courtesy of Bertil Daneholt, Karolinska Institutet.
What is the nature of the puff? It consists of a region in which the chromosome fibers unwind from their usual state of packing in the band. The fibers remain continuous with those in the chromosome axis. Puffs usually emanate from single bands, although when they are very large, as typified by the Balbiani rings, the swelling can be so extensive as to obscure the underlying array of bands.
The pattern of puffs is related to gene expression. During larval development, puffs appear and regress in temporal and tissue-specific patterns. A characteristic pattern of puffs is found in each tissue at any given time. Many puffs are induced by the hormone ecdysone that controls Drosophila development. Some puffs are induced directly by the hormone; others are induced indirectly by the products of earlier puffs.
The puffs are sites where RNA is being synthesized. The accepted view of puffing has been that expansion of the band is a consequence of the need to relax its structure in order to synthesize RNA. Puffing has therefore been viewed as a consequence of transcription. A puff can be generated by a single active gene. The sites of puffing differ from ordinary bands in that they accumulate additional proteins, including RNA polymerase II and other proteins associated with transcription. The bands 87A and 87C indicated in Figure 7.20 encode heat-shock proteins and form puffs upon heat shock. We can observe the accumulation of RNA polymerase II at these puffs by immunofluorescence, as shown in FIGURE 7.22.

FIGURE 7.22 Heat-shock-induced puffing at major heat shock loci 87A and C. Displayed is a small segment of chromosome 3 before (left) and after (right) heat shock. Chromosomes are stained for DNA (blue) and for RNA polymerase II (yellow).
Photo courtesy of Victor G. Corces, Emory University.
The features displayed by lampbrush and polytene chromosomes suggest a general conclusion. To be transcribed, the genetic material is dispersed from its usual, more tightly packed state. The question to keep in mind is whether this dispersion at the gross level of the chromosome mimics the events that occur at the molecular level within the mass of ordinary interphase euchromatin.
Do the bands of a polytene chromosome have a functional significance? That is, does each band correspond to some type of genetic unit? You might think that the answer would be immediately evident from the sequence of the fly genome, because by mapping interbands to the sequence it should be possible to determine whether a band has any fixed type of identity. Thus far, however, patterns that identify a functional significance for the bands are unknown.
7.12 The Eukaryotic Chromosome Is a Segregation Device
During mitosis, the sister chromatids move to opposite poles of the cell. Their movement depends on the attachment of the chromosome to microtubules, which are connected at their other end to the poles. The microtubules comprise a cellular filamentous system, which is reorganized at mitosis so that they connect the chromosomes to the poles of the cell. The sites in the two regions where microtubule ends are organized—in the vicinity of the centrioles at the poles and at the chromosomes—are called microtubule organizing centers (MTOCs).
FIGURE 7.23 illustrates the separation of sister chromatids as mitosis proceeds from metaphase to telophase. The region of the chromosome that is responsible for its segregation at mitosis and meiosis is called the centromere. The centromeric region on each sister chromatid is moved along microtubules to the opposite pole. Opposing this motive force, “glue” proteins called cohesins hold the sister chromatids together. Initially the sister chromatids separate at their centromeres, then they are released completely from one another during anaphase when the cohesins are degraded. The centromere is moved toward the pole during mitosis, and the attached chromosome appears to be “dragged along” behind it. The chromosome therefore provides a device for attaching a large number of genes to the apparatus for division. The centromere essentially acts as the luggage handle for the entire chromosome and its location typically appears as a constricted region connecting all four chromosome arms, as can be seen in the photo in Figure 7.11, which shows the sister chromatids at the metaphase stage of mitosis.

FIGURE 7.23 Chromosomes are pulled to the poles via microtubules that attach at the centromeres. The sister chromatids are held together until anaphase by glue proteins (cohesins). The centromere is shown here in the middle of the chromosome (metacentric), but can be located anywhere along its length, including close to the end (acrocentric) and at the end (telocentric).
The centromere is essential for segregation, as shown by the behavior of chromosomes that have been broken. A single break generates one piece that retains the centromere, and another, an acentric fragment, that lacks it. The acentric fragment does not become attached to the mitotic spindle, and as a result it fails to be included in either of the daughter nuclei. When chromosome movement relies on discrete centromeres, there can be only one centromere per chromosome. When translocations generate chromosomes with more than one centromere, aberrant structures form at mitosis. This is because the two centromeres on the same sister chromatid can be pulled toward different poles, thus breaking the chromosome. In some species, though (such as the nematode Caenorhabditis elegans), the centromeres are holocentric, being diffuse and spread along the entire length of the chromosome. Species with holocentric chromosomes still make spindle fiber attachments for mitotic chromosome separation, but do not require one and only one regional or point centromere per chromosome. Most of the molecular analysis of centromeres has been done on canonical point (budding yeast) or regional (fly, mammalian, rice) centromeres.
The regions flanking the centromere often are rich in satellite DNA sequences and display a considerable amount of heterochromatin. The entire chromosome is condensed, though, so centromeric heterochromatin is not immediately evident in mitotic chromosomes. Researchers can, however, visualize it by a technique that generates “C-bands.” For example, in FIGURE 7.24 all the centromeres show as darkly staining regions. Although it is common, heterochromatin cannot be identified around every known centromere, which suggests that it is unlikely to be essential for the division mechanism.

FIGURE 7.24 C-banding generates intense staining at the centromeres of all chromosomes.
Photo courtesy of Lisa Shaffer, Washington State University, Spokane.
The centromeric chromatin comprises DNA sequences, specialized centromeric histone variants, and a group of specific proteins that are responsible for establishing the structure that attaches the chromosome to the microtubules. This structure is called the kinetochore. It is a darkly staining fibrous object of about 400 nm. The kinetochore provides a microtubule attachment point on the chromosome.
7.13 Regional Centromeres Contain a Centromeric Histone H3 Variant and Repetitive DNA
The region of the chromosome at which the centromere forms was originally thought to be defined by DNA sequences, yet recent studies in plants, animals, and fungi have shown that centromeres are specified epigenetically by chromatin structure. Centromere-specific histone H3 (known as Cse4 in yeast, CENP-A in higher eukaryotes, and more generically as CenH3; see the chapter titled Chromatin) appears to be a primary determinant in establishing functional centromeres and kinetochore assembly sites. This finding explains the old puzzle of why specific DNA sequences could not be identified as “the centromeric DNA” and why there is so much variation in centromere-associated DNA sequences among closely related species. FIGURE 7.25 shows the role of the centromeric histone H3, CENP-A, in organizing the centromere at the point of kinetochore attachment. Several working models of the spatial arrangement of chromatin relative to the kinetochore are shown.

FIGURE 7.25 Organization of CENP-A and H3 Nucleosomes in Centromeres. (a) Centromeres are ~40 kb long in chicken, corresponding to 200 nucleosomes per centromere. Of these, 30 are predicted to contain CENP-A (roughly 1 in 6–8 centromeric nucleosomes). Thus, centromeric chromatin is largely composed of nucleosomes containing histone H3. (b and c) The CENP-A chromatin was originally suggested to form an amphipathic organization, with CENP-A on the exterior facing the kinetochore, and H3 largely on the interior. This chromatin was proposed to form either a helix or loop structure. (d) The boustrophedon model of centromeric CENP-A-containing chromatin was proposed based on super-resolution microscopy.
Data from Fukagawa, T., et al. (2014). Dev Cell 30: 496–508doi: (10.1016/j.devcel.2014.08.016.
Centromeres are highly specialized chromatin structures that occupy the same site for many generations, despite the fact that they can be repositioned without DNA transposition. In eukaryotic chromosomes, the centromere-specific histone H3 variant CenH3 replaces the normal H3 histone at sites where centromeres reside and kinetochores attach chromosomes to spindle fibers. This specialized centromeric chromatin is the foundation for binding of other centromere-associated proteins. In addition, other histones at the centromere (including H2A and canonical H3) are subject to posttranslational modifications that are required for normal binding of centromeric proteins and accurate chromosome segregation, indicating that the epigenetic pattern that defines a centromere is complex. This view represents a paradigm shift in how we understand centromere formation, identity, and function. CenH3 is a nucleosomal protein and not a DNA sequence per se; thus, the centromere is now regarded as being primarily epigenetic in its specification. The role of satellite DNA sequences, which are also characteristic of centromeres, remains difficult to ascertain, despite their prevalence and conservation. Research has now turned to understanding the role of nucleosome assembly factors that are specific to CenH3 installation. New questions address matters of specificity, such as how do cells maintain a uniform level of CenH3 at centromeres following replication?
The length of DNA required for centromeric function is often quite long. The short, discrete elements of Saccharomyces cerevisiae appear to be an exception to the general rule. S. cerevisiae is the only case so far in which centromeric DNA can be identified by its ability to confer stability on plasmids. A related approach has been used with the yeast Schizosaccharomyces pombe. S. pombe has only three chromosomes, and the region containing each centromere has been identified by deleting most of the sequences of each chromosome to create a stable minichromosome. This approach locates the centromeres within regions of 40 to 100 kb that consist largely or entirely of repetitious DNA. Attempts to localize centromeric functions in Drosophila chromosomes suggest that they are dispersed in a large region of 200 to 600 kb. The large size of this type of centromere may reflect multiple specialized functions, including kinetochore assembly and sister chromatid pairing.
The size of the centromere in Arabidopsis is comparable. Each of the five chromosomes has a centromeric region in which recombination is very largely suppressed. This region occupies >500 kb. The primary motif comprising the heterochromatin of primate centromeres is the α-satellite DNA, which consists of tandem arrays of a 171-bp repeating unit (see the chapter titled Clusters and Repeats). There is significant variation between individual repeats, although those at any centromere tend to be better related to one another than to members of the family in other locations.
Current models for regional centromere organization and function invoke alternating chromatin domains, with clusters of CenH3 nucleosomes interspersed among clusters of nucleosomes with H3 and some of the histone variant H2A.Z. Different histones are subject to centromere-specific patterns of modification. The CenH3 nucleosomes form the chromatin foundation for recruitment and assembly of the other proteins that eventually comprise a functional kinetochore. The formation of neocentromeres that contain CenH3 but not α-satellite DNA provide important evidence for the idea of centromeres being epigenetically determined. Key questions remain as to the role of repetitive DNA and alternating chromatin domains in forming the large bipartite kinetochore structure on replicated sister centromeres.
7.14 Point Centromeres in S. cerevisiae Contain Short, Essential DNA Sequences
If a centromeric sequence of DNA is responsible for segregation, any molecule of DNA possessing this sequence should move properly at cell division, whereas any DNA lacking it should fail to segregate. This prediction has been used to isolate centromeric DNA in the yeast S. cerevisiae. Yeast chromosomes do not display visible kinetochores comparable to those of multicellular eukaryotes but otherwise divide at mitosis and segregate at meiosis by the same mechanisms.
Genetic engineering has produced plasmids of yeast that are replicated like chromosomal sequences (see the chapter titled The Replicon: Initiation of Replication). They are unstable at mitosis and meiosis, though, and disappear from a majority of the cells because they segregate erratically. Fragments of chromosomal DNA containing centromeres have been isolated by their ability to confer mitotic stability on these plasmids.
A centromeric DNA region (CEN) fragment is identified as the minimal sequence that can confer stability upon such a plasmid. Another way to characterize the function of such sequences is to modify them in vitro and then reintroduce them into the yeast cell where they replace the corresponding centromere on the chromosome. This allows the sequences required for CEN function to be defined directly in the context of the chromosome.
A CEN fragment derived from one chromosome can replace the centromere of another chromosome with no apparent consequence. This result suggests that centromeres are interchangeable. They are used simply to attach the chromosome to the spindle and play no role in distinguishing one chromosome from another.
The sequences required for centromeric function fall within a stretch of about 120 bp. The centromeric region is packaged into a nuclease resistant structure and binds a single microtubule. We may therefore look to the S. cerevisiae centromeric region to identify proteins that bind centromeric DNA and proteins that connect the chromosome to the spindle.
As summarized in FIGURE 7.26, we can distinguish three types of sequence element in the CEN region:
Cell cycle–dependent element (CDE)-I is a sequence of 9 bp that is conserved with minor variations at the left boundary of all centromeres.
CDE-II is a greater than 90% A-T–rich sequence of 80 to 90 bp found in all centromeres; its function could depend on its length rather than exact sequence. Its constitution is reminiscent of some short, tandemly repeated (satellite) DNA (see the chapter titled Clusters and Repeats). Its base composition might cause some characteristic distortions of the DNA double helical structure.
CDE-III is an 11-bp sequence highly conserved at the right boundary of all centromeres. Sequences on either side of the element are less well conserved and might also be needed for centromeric function. (CDE-III could be longer than 11 bp if it turns out that the flanking sequences are essential.)

FIGURE 7.26 Three conserved regions can be identified by the sequence homologies between yeast CEN elements.
Mutations in CDE-I or CDE-II reduce but do not inactivate centromere function; however, point mutations in the central CCG of CDE-III completely inactivate the centromere.
7.15 The S. cerevisiae Centromere Binds a Protein Complex
Can we identify proteins that are necessary for the function of CEN sequences? There are several genes in which mutations affect chromosome segregation and whose proteins are localized at centromeres. FIGURE 7.27 summarizes the contributions of these proteins to the centromeric structure.

FIGURE 7.27 The DNA at CDE-II is wound around an alternative nucleosome containing Cse4, CDE-III is bound by the CBF3 complex, and CDE-I is bound by a Cbf1 homodimer. These proteins are connected by the group of Ctf19, Mcm21, and Okp1 proteins, and numerous other factors serve to link this complex to a microtubule.
The CEN region recruits three DNA-binding factors: Cbf1, CBF3 (an essential four-protein complex), and Mif2 (CENP-C in multicellular eukaryotes). In addition, a specialized chromatin structure is built by binding the CDE-II region to a protein called Cse4, a histone H3 variant (analogous to CENP-A in multicellular eukaryotes), probably in the context of an otherwise normal nucleosome. A protein called Scm3 is required for proper association of Cse4 with CEN. Inclusion of CenH3 histone variants related to Cse4 is a universal aspect of centromere construction in all species. The basic interaction consists of bending the DNA of the CDE-II region around a protein aggregate; the reaction is probably assisted by the occurrence of intrinsic bending in the CDE-II sequence.
CDE-I is bound by a homodimer of Cbf1; this interaction is not essential for centromere function, but in its absence the fidelity of chromosome segregation is reduced about 10×. The 240-kD heterotetramer, CBF3, binds to CDE-III. This interaction is essential for centromeric function.
The proteins bound at CDE-I, CDE-II, and CDE-III also interact with another group of proteins (Ctf19, Mcm21, and Okp1), which in turn link the centromeric complex to the kinetochore proteins (at least 70 individual kinetochore proteins have been identified in yeast) and to the microtubule.
The overall model suggests that the complex is localized at the centromere by a protein structure that resembles the normal building block of chromatin (the nucleosome). The bending of DNA at this structure allows proteins bound to the flanking elements to become part of a single complex. The DNA-binding components of the complex form a scaffold for assembly of the kinetochore, linking the centromere to the microtubule. The construction of kinetochores follows a similar pattern, and uses related components, in a wide variety of organisms.
7.16 Telomeres Have Simple Repeating Sequences
Another essential feature in all chromosomes is the telomere, which “seals” the chromosome ends. We know that the telomere must be a special structure, because chromosome ends generated by breakage are “sticky” and tend to react with other chromosomes, whereas natural ends are stable.
We can apply two criteria in identifying a telomeric sequence:
It must lie at the end of a chromosome (or, at least at the end of an authentic linear DNA molecule).
It must confer stability on a linear molecule subjected to multiple rounds of replication and immune from end-joining DNA repair machinery.
The problem of finding a system that offers an assay for function again has been brought to the molecular level by using yeast. All of the plasmids that survive in yeast (by virtue of possessing autonomously replicating sequence [ARS] and CEN elements) are circular DNA molecules. Linear plasmids are unstable (because they are degraded). Could an authentic telomeric DNA sequence confer stability on a linear plasmid? Fragments from yeast DNA that prove to be located at chromosome ends can be identified by such an assay, and a region from the end of a known natural linear DNA molecule—the extrachromosomal ribosomal DNA (rDNA) of Tetrahymena—is able to render a yeast plasmid stable in linear form.
Telomeric sequences have been characterized from a wide range of eukaryotes. The same type of sequence is found in plants and humans, so the construction of the telomere seems to follow a nearly universal principle (Drosophila telomeres are an exception, consisting of terminal arrays of retrotransposons). Each telomere consists of a long series of short, tandemly repeated sequences. There can be 100 to 1,000 repeats, depending on the organism.
Telomeric sequences can be written in the general form 5′-(T/A)nGm-3′ where n is 1 to 4 and m is >1. FIGURE 7.28 shows a generic example. One unusual property of the telomeric sequence is the extension of the G-T–rich strand, which for 14 to 16 bases is usually a single strand. The G-tail is probably generated because there is a specific limited degradation of the C-A–rich strand.

FIGURE 7.28 A typical telomere has a simple repeating structure with a G-T–rich strand that extends beyond the C-A–rich strand. The G-tail is generated by a limited degradation of the C-A–rich strand.
Some indications about how a telomere functions are given by some unusual properties of the ends of linear DNA molecules. In a trypanosome population, the ends vary in length. When an individual cell clone is followed, the telomere grows longer by 7 to 10 bp (one to two repeats) per generation. Even more revealing is the fate of ciliate telomeres introduced into yeast. After replication in yeast, yeast telomeric repeats are added onto the ends of the Tetrahymena repeats.
Addition of telomeric repeats to the end of the chromosome in every replication cycle could solve the difficulty of replicating linear DNA molecules (discussed in the chapter Extrachromosomal Replicons). The addition of repeats by de novo synthesis would counteract the loss of repeats resulting from failure to replicate up to the end of the chromosome. Extension and shortening would be in dynamic equilibrium.
If telomeres are continually being lengthened (and shortened), their exact sequence might be irrelevant. All that is required is for the end to be recognized as a suitable substrate for addition. This explains how the ciliate telomere functions in yeast.
7.17 Telomeres Seal the Chromosome Ends and Function in Meiotic Chromosome Pairing
Isolated telomeric fragments do not behave as though they contain single-stranded DNA; instead, they show aberrant electrophoretic mobility and other properties.
Guanine bases have an unusual capacity to associate with one another. The single-stranded G-rich tail of the telomere can form G-quadruplex (also called G4 DNA or G quartets) of G residues. Each quartet contains four guanines that hydrogen bond with one another to form a planar structure. Each guanine comes from the corresponding position in a successive TTAGGG repeating unit. FIGURE 7.29 shows an organization based on a crystal structure. The quartet that is illustrated represents an association between the first guanine in each repeating unit. It is stacked on top of another quartet that has the same organization, but is formed from the second guanine in each repeating unit. A series of quartets could be stacked like this in a helical manner. Although the formation of this structure attests to the unusual properties of the G-rich sequence in vitro, it does not demonstrate whether the quartet forms in vivo, for which there is only limited evidence to date.
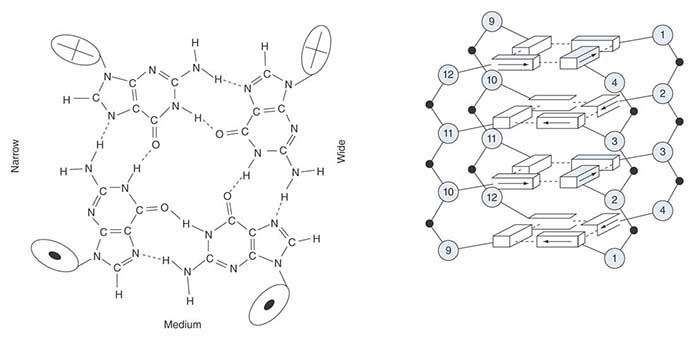
FIGURE 7.29 The crystal structure of a short repeating sequence from the human telomere forms three stacked G quartets. The top quartet contains the first G from each repeating unit. This is stacked above quartets that contain the second G (G3, G9, G15, G21) and the third G (G4, G10, G16, G22).
What feature of the telomere is responsible for the stability of the chromosome end? The schematic in FIGURE 7.30 shows that a loop of DNA forms at the telomere. The absence of any free end might be the crucial feature that stabilizes the end of the chromosome. The average length of the loop in animal cells is 5 to 10 kb. The loop is formed when the 3′ single-stranded end of the telomere (TTAGGG)n displaces the same sequence in an upstream region of the telomere. This converts the duplex region into a structure called a t-loop, where a series of TTAGGG repeats are displaced to form a single-stranded region, and the tail of the telomere is paired with the homologous strand.
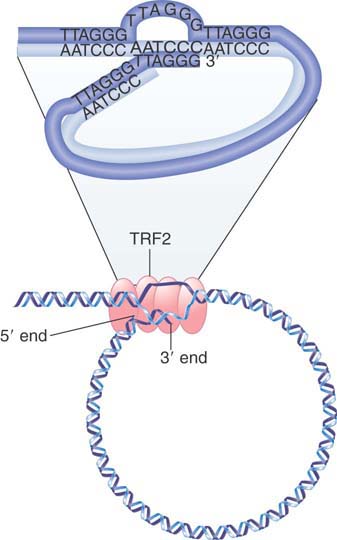
FIGURE 7.30 A loop forms at the end of chromosomal DNA. The 3′ single-stranded end of the telomere (TTAGGG)n displaces the homologous repeats from duplex DNA to form a t-loop. The reaction is catalyzed by TRF2.
© Dr. Gopal Murti/Science Source.
The reaction is catalyzed by the telomere-binding protein TRF2, which together with other proteins forms a complex that stabilizes the chromosome ends. Its importance in protecting the ends is indicated by the fact that the deletion of TRF2 causes chromosome rearrangements to occur.
In mammals, six telomeric proteins (TRF1, TRF2, Rap1, TIN2, TPP1, and POT1) primarily comprise a complex called shelterin, depicted in FIGURE 7.31 Shelterin functions to protect telomeres from DNA damage repair pathways and to regulate telomere length control by telomerase (discussed in the next section). Increasing roles for telomeres in aging, cancer, and cell differentiation reveal that telomeres are more than static caps at the ends of linear chromosomes.

FIGURE 7.31 A schematic of how shelterin might be positioned on telomeric DNA, highlighting the duplex telomeric DNA interactions of TRF1 and TRF2 and the binding of POT1 to the single-stranded TTAGGG repeats. Although one of the shelterin complexes may have the depicted structure, telomeres contain numerous copies of the complex bound along the double-stranded TTAGGG repeat array. It is not known whether all (or even most) shelterins are present in six-protein complexes. Nucleosomes are omitted for simplicity.
Reprinted with permission from the Annual Review of Genetics, Volume 42 © 2008 by Annual Reviews www.annualreviews.org. Courtesy of Titia de Lange, The Rockefeller University.
Besides their role in capping the ends of linear chromosomes, telomeres also have an ancient and conserved function in meiosis, whereby they cluster on the nuclear envelope just prior to homologous chromosome synapsis. This clustering defines the “bouquet” stage of meiosis, as shown in FIGURE 7.32, and represents a once-in-a-life-cycle configuration. The telomere clustering involves motility forces that act across the nuclear envelope via microtubules, actin, or other filamentous systems. Genetic disruption of meiotic telomere clustering results in chromosome recombination and segregation defects, including the production of aneuploid daughter cells or sterility. Interestingly, fruit flies, which lack canonical telomerase-based telomeres, do not exhibit meiotic telomere clustering, but have evolved other mechanisms to ensure homologous chromosome pairing.

FIGURE 7.32 The meiotic telomere cluster is visualized by telomere FISH. Microscopic image of a maize nucleus fixed at meiotic prophase (zygotene stage), subjected to telomere (green) and centromere (white) FISH, and counter-stained for total DNA with DAPI (red). This pseudocolored image is a two-dimensional projection of a three-dimensional, multi-color image dataset.
Photo courtesy of S. P. Murphy and H. W. Bass, Florida State University.
7.18 Telomeres Are Synthesized by a Ribonucleoprotein Enzyme
The telomere has three widely conserved functions:
The first is to protect the chromosome end. Any other DNA end—for example, the end generated by a double-strand break—becomes a target for repair systems. The cell must be able to distinguish the telomere.
The second is to allow the telomere to be extended. If it is not extended, it becomes shorter with each replication cycle (because replication cannot initiate at the very end).
The third is to facilitate meiotic chromosome reorganization for efficient pairing and recombination of homologous chromosomes.
Proteins that bind to the telomeres contribute to the solution of all of these. In yeast, different sets of proteins solve the first two problems, but both are bound to the telomere via the same protein, Cdc13:
The Stn1 protein protects against degradation (specifically, against any extension of the degradation of the C-A strand that generates the G-tail).
A telomerase enzyme extends the C-A–rich strand. Its activity is influenced by two proteins that have ancillary roles such as controlling the length of the extension.
The telomerase uses the 3′–OH of the G+T telomeric strand as a primer for synthesis of tandem TTGGGG repeats. Only dGTP and dTTP are needed for the activity. The telomerase is a large ribonucleoprotein that consists of a templating RNA (encoded by TLC1 in yeast, hTERC in humans) and a protein with catalytic activity (encoded by EST2 in yeast, hTERT in humans). The RNA component is typically short (159 bases long in Tetrahymena, and 451 bases long in humans, though 1.3 kb in yeast) and includes a sequence of 15 to 22 bases that is identical to two repeats of the C-rich repeating sequence. This RNA provides the template for synthesizing the G-rich repeating sequence. The protein component of the telomerase is a catalytic subunit that can act only upon the RNA template provided by the nucleic acid component.
FIGURE 7.33 shows the action of telomerase. The enzyme progresses discontinuously: The template RNA is positioned on the DNA primer, several nucleotides are added to the primer, and then the enzyme translocates to begin again. The telomerase is a specialized example of a reverse transcriptase, an enzyme that synthesizes a DNA sequence using an RNA template (see the chapter titled Transposable Elements and Retroviruses). We do not know how the complementary (C-A–rich) strand of the telomere is assembled, but we can speculate that it could be synthesized by using the 3′–OH of a terminal G-T hairpin as a primer for DNA synthesis.

FIGURE 7.33 Telomerase positions itself by base pairing between the RNA template and the protruding single-stranded DNA primer. It adds G and T bases, one at a time to the primer, as directed by the template. The cycle starts again when one repeating unit has been added.
Telomerase synthesizes the individual repeats that are added to the chromosome ends, but does not itself control the number of repeats. Other proteins are involved in determining the length of the telomere. Some have been identified by the est1 and est3 mutants in yeast, which have altered telomere lengths. These proteins bind telomerase and can influence the length of the telomere by controlling the access of telomerase to its substrate. Researchers have identified proteins that bind telomeres in mammalian cells, including homologs of EST1, but less is known about their functions.
Each organism has a characteristic range of telomere lengths. They are long in mammals (typically 5 to 15 kb in humans) and short in yeast (typically around 300 bp in S. cerevisiae). The basic control mechanism is that the probability that a telomere will be a substrate for telomerase increases as the length of the telomere shortens; we do not know if this is a continuous effect or if it depends on the length falling below some critical value. When telomerase acts on a telomere, it can add several repeating units. The enzyme’s intrinsic mode of action is to dissociate after adding one repeat; addition of several repeating units depends on other proteins that cause telomerase to undertake more than one round of extension. The number of repeats that is added is not influenced by the length of the telomere itself, but instead is controlled by ancillary proteins that associate with telomerase.
The minimum features required for existence as a chromosome are as follows:
Telomeres to ensure survival
A centromere to support segregation
An origin to initiate replication
All of these elements have been put together to construct a yeast artificial chromosome (YAC; see the chapter titled Methods in Molecular Biology and Genetic Engineering). This is a useful method for perpetuating large sequences. It turns out that the synthetic chromosome is stable only if it is longer than 20 to 50 kb. We do not know the basis for this effect, but the ability to construct a synthetic chromosome allows us to investigate the nature of the segregation device in a controlled environment.
7.19 Telomeres Are Essential for Survival
Telomerase activity is found in most dividing cells (such as embryonic cells, stem cells, and in unicellular eukaryotes) and is generally turned off in terminally differentiated cells that do not divide. FIGURE 7.34 shows that if telomerase is mutated in a dividing cell, the telomeres become gradually shorter with each cell division.
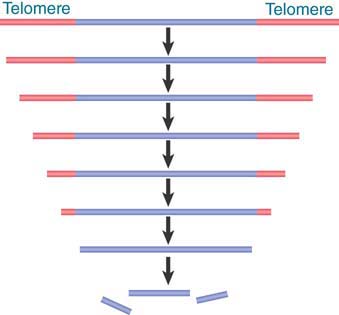
FIGURE 7.34 Mutation in telomerase causes telomeres to shorten in each cell division. Eventual loss of the telomere causes chromosome breaks and rearrangements.
Loss of telomeres has dire effects. When the telomere length reaches zero, it becomes difficult for the cells to divide successfully. Attempts to divide typically generate chromosome breaks and translocations. This causes an increased rate of mutation. In yeast, this is associated with a loss of viability, and the culture becomes predominantly occupied by senescent cells (which are elongated and nondividing, and eventually die).
Some cells grow out of the senescing yeast culture. They have acquired the ability to extend their telomeres by an alternative to telomerase activity. The survivors fall into two groups. The members of one group have circularized their chromosomes: They now have no telomeres, and as a result they have become independent of telomerase. The other group uses unequal crossing-over to extend their telomeres (see FIGURE 7.35). The telomere is a repeating structure, so it is possible for two telomeres to misalign when chromosomes pair. Recombination between the mispaired regions generates an unequal crossing-over (as discussed in the chapter Clusters and Repeats): When the length of one recombinant chromosome increases, the length of the other decreases.

FIGURE 7.35 Crossing-over in telomeric regions is usually suppressed by mismatch-repair systems, but can occur when they are mutated. An unequal crossing-over event extends the telomere of one of the products, allowing the chromosome to survive in the absence of telomerase.
Cells usually suppress unequal crossing-over because of its potentially deleterious consequences. Two systems are responsible for suppressing crossing-over between telomeres. One is provided by telomere-binding proteins. In yeast, the frequency of recombination between telomeres is increased by deletion of the gene TAZ1, which codes for a protein that regulates telomerase activity. The second is a general system that is responsible for mismatch repair. In addition to correcting mismatched base pairs that can arise in DNA, this system suppresses recombination between mispaired regions. Figure 7.35 shows that this includes telomeres. When it is mutated, a greater proportion of telomerase-deficient yeast survives the loss of telomeres because recombination between telomeres generates some chromosomes with longer telomeres.
When eukaryotic cells from multicellular eukaryotes are placed in culture, they usually divide for a fixed number of generations and then enter senescence. The reason appears to be a decline in telomere length because of the absence of telomerase expression. Cells enter a crisis from which some emerge, but typically the cells that emerge have chromosome rearrangements that have resulted from lack of protection of chromosome ends. These rearrangements can cause mutations that contribute to the tumorigenic state. The absence of telomerase expression in this situation is due to failure to express the gene (a normal condition of differentiated cells), and reactivation of telomerase is one of the mechanisms by which these cells then survive continued culture. The vast majority of cancer cells reactivate telomerase, though a small percentage also utilizes unequal recombination to maintain telomeres during prolonged proliferation.
It has long been suggested that within a species, greater telomere length could lead to greater cellular lifespans in tissues and thus to increased lifespan of the organism. Although data to support this has been generally lacking, recent work in zebra finches has shown that telomere length measured very early in life can in fact predict lifespan. It is not yet clear whether these results will apply to other species, including humans, but this work is the first clear evidence that telomere length can in fact correlate with natural aging and lifespan.
Summary
The genetic material of all organisms and viruses takes the form of tightly packaged nucleoprotein. Some virus genomes are inserted into preformed virions, whereas others assemble a protein coat around the nucleic acid. The bacterial genome forms a dense nucleoid, with about 20% protein by mass, but details of the interaction of the proteins with DNA are not known. The DNA is organized into up to 100 domains that maintain independent supercoiling, with a density of unrestrained supercoils corresponding to 1/100 to 200 bp. In eukaryotes, interphase chromatin and metaphase chromosomes both appear to be organized into large loops. Each loop can be an independently supercoiled domain. The bases of the loops are connected to a metaphase scaffold or to the nuclear matrix by specific DNA sites.
Most transcriptionally active sequences reside within the euchromatin that comprises the majority of interphase chromatin. The regions of heterochromatin are packaged about 5 to 10 times more compactly and are mostly transcriptionally inert. All chromatin becomes densely packaged during cell division, when we can distinguish the individual chromosomes. The existence of a reproducible ultrastructure in mammalian chromosomes is indicated by the production of G-bands through treatment with Giemsa stain. The bands are very large regions (about 107 bp) that we can use to map chromosomal translocations or other large changes in structure.
Lampbrush chromosomes of amphibians and polytene chromosomes of insects have unusually extended structures, with packing ratios less than 100. Polytene chromosomes of D. melanogaster are divided into about 5,000 bands. These bands vary in size by an order of magnitude, with an average of around 25 kb. Transcriptionally active regions can be visualized in even more unfolded (“puffed”) structures, in which material is extruded from the axis of the chromosome. This can resemble the changes that occur on a smaller scale when a sequence in euchromatin is transcribed.
The centromeric region contains the kinetochore, which is responsible for attaching a chromosome to the mitotic spindle. The centromere often is surrounded by heterochromatin. Centromeric sequences have been identified only in the yeast S. cerevisiae, where they consist of short, conserved elements. These elements, CDE-I and CDE-III, bind Cbf1 and the CBF3 complex, respectively, and a long A-T–rich region called CDE-II binds the histone H3 variant Cse4 to form a specialized nucleosome. Another group of proteins that binds to this assembly provides the connection to microtubules.
Telomeres make the ends of chromosomes stable. Almost all known telomeres consist of multiple repeats in which one strand has the general sequence Cn (A/T)m, where n > 1 and m = 1 to 4. The other strand, Gn (T/A)m, has a single protruding end that provides a template for addition of individual bases in defined order. The enzyme telomerase is a ribonucleoprotein whose RNA component provides the template for synthesizing the G-rich strand. This overcomes the problem of the inability to replicate at the very end of a duplex. The telomere stabilizes the chromosome end because the overhanging single strand Gn (T/A)m displaces its homolog in earlier repeating units in the telomere to form a loop, so there are no free ends that resemble double-strand breaks.
References
7.2 Viral Genomes Are Packaged into Their Coats
Reviews
Black, L. W. (1989). DNA packaging in dsDNA bacteriophages. Annu. Rev. Immunol. 43, 267–292.
Butler, P. J. (1999). Self-assembly of tobacco mosaic virus: the role of an intermediate aggregate in generating both specificity and speed. Philos. Trans. R. Soc. Lond. B. Bio. Sci. 354, 537–550.
Klug, A. (1999). The tobacco mosaic virus particle: structure and assembly. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 354, 531–535.
Mindich, L. (2000). Precise packaging of the three genomic segments of the double-stranded-RNA bacteriophage phi6. Microbiol. Mol. Biol. Rev. 63, 149–160.
Research
Caspar, D. L. D., and Klug, A. (1962). Physical principles in the construction of regular viruses. Cold Spring Harbor Symp. Quant. Biol. 27, 1–24.
de Beer, T., et al. (2002). Insights into specific DNA recognition during the assembly of a viral genome packaging machine. Mol. Cell 9, 981–991.
Dube, P., et al. (1993). The portal protein of bacteriophage SPP1: a DNA pump with 13-fold symmetry. EMBO. J. 12, 1303–1309.
Fraenkel-Conrat, H., and Williams, R. C. (1955). Reconstitution of active tobacco mosaic virus from its inactive protein and nucleic acid components. Proc. Natl. Acad. Sci. USA 41, 690–698.
Jiang, Y. J., et al. (2000). Notch signalling and the synchronization of the somite segmentation clock. Nature 408, 475–479.
Zimmern, D. (1977). The nucleotide sequence at the origin for assembly on tobacco mosaic virus RNA. Cell 11, 463–482.
Zimmern, D., and Butler, P. J. (1977). The isolation of tobacco mosaic virus RNA fragments containing the origin for viral assembly. Cell 11, 455–462.
7.3 The Bacterial Genome Is a Nucleoid with Dynamic Structural Properties
Reviews
Brock, T. D. (1988). The bacterial nucleus: a history. Microbiol. Rev. 52, 397–411.
Drlica, K., and Rouviere-Yaniv, J. (1987). Histonelike proteins of bacteria. Microbio. Rev. 51, 301–319.
Dillon, S. C., and Dorman, C. J. (2010). Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 8, 185–195.
Dorman, C. J. (2013). Genome architecture and global gene regulation in bacteria: making progress towards a unified model? Nat. Rev. Microbiol. 11, 349–355.
Scolari, V. F., et al. (2015). The nucleoid as a smart polymer. Front.Microbiol. 6, Article 424.
Research
Fisher, J. K., et al. (2013). Four-dimensional imaging of E. coli nucleoid organization and dynamics in living cells. Cell 153, 882–895.
Wang, X., et al. (2014). Bacillus subtilis chromosome dynamics in the bacterial cell cycle. Proc. Natl. Acad. Sci. USA 111, 12877–12882.
7.4 The Bacterial Genome Is Supercoiled and Has Four Macrodomains
Review
Hatfield, G. W., and Benham, C. J. (2002). DNA topology-mediated control of global gene expression in Escherichia coli. Annu. Rev. Genet. 36, 175–203.
Research
Pettijohn, D. E., and Pfenninger, O. (1980). Supercoils in prokaryotic DNA restrained in vitro. Proc. Natl. Acad. Sci. USA 77, 1331–1335.
Postow, L., et al. (2004). Topological domain structure of the Escherichia coli chromosome. Genes Dev. 18, 1766–1779.
7.5 Eukaryotic DNA Has Loops and Domains Attached to a Scaffold
Reviews
Cremer, T., and Cremer, M. (2010). Chromosome territories. Cold Spring Harb. Perspec. Biol. 2010;2, a003889.
Dileep, V., et al. (2015). Large-scale chromatin structure–function relationships during the cell cycle and development: insights from replication timing. Cold Spring Harb. Symp. Quant. Biol. vol LXXX.
Research
Bolzer, A., et al. (2005). Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS. Biol. 3(5), e156.
Liang, Z., et al. (2015). Chromosomes progress to metaphase in multiple discrete steps via global compaction/expansion cycles. Cell 161, 1124–1137.
International Human Genome Sequencing Consortium. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921.
Saccone, S., et al. (1993). Correlations between isochores and chromosomal bands in the human genome. Proc. Natl. Acad. Sci. USA 90, 11929–11933.
Venter, J. C., et al. (2001). The sequence of the human genome. Science 291, 1304–1350.
7.6 Specific Sequences Attach DNA to an Interphase Matrix
Reviews
Chattopadhyay, S., and Pavithra, L. (2007). MARs and MARBPs: key modulators of gene regulation and disease manifestation. Subcell. Bio. 41, 213–230.
Galande, S., et al. (2007). The third dimension of gene regulation: organization of dynamic chromatin loopscape by SATB1. Curr. Opin. Genet. Dev. 17, 408–414.
7.7 Chromatin Is Divided into Euchromatin and Heterochromatin
Review
Geyer, P. K., et al. (2011). Nuclear organization: taking a position on gene expression. Curr. Opin. Cell. Biol. 23, 354–359.
7.12 The Eukaryotic Chromosome Is a Segregation Device
Review
Hyman, A. A., and Sorger, P. K. (1995). Structure and function of kinetochores in budding yeast. Annu. Rev. Cell. Dev. Biol. 11, 471–495.
7.13 Regional Centromeres Contain a Centromeric Histone H3 Variant and Repetitive DNA
Reviews
Fukagawa, T., and Earnshaw, W. C. (2014). The centromere: chromatin foundation for the kinetochore machinery. Dev. Cell 30, 496–508.
Research
Black, B. E., et al. (2004). Structural determinants for generating centromeric chromatin. Nature 430, 578–582.
Depinet, T. W., et al. (1997). Characterization of neo-centromeres in marker chromosomes lacking detectable alpha-satellite DNA. Hum. Mol. Genet. 6, 1195–1204.
Foltz, D. R., et al. (2006). The human CENP-A centromeric nucleosome-associated complex. Nat. Cell Biol. 8, 458–469.
Sun, X., et al. (1997). Molecular structure of a functional Drosophila centromere. Cell 91, 1007–1019.
Yamagishi, Y., et al. (2010). Two histone marks establish the inner centromere and chromosome biorientation. Science 330, 239–243.
7.14 Point Centromeres in S. cerevisiae Contain Short, Essential DNA Sequences
Reviews
Blackburn, E. H., and Szostak, J. W. (1984). The molecular structure of centromeres and telomeres. Annu. Rev. Biochem. 53, 163–194.
Clarke, L., and Carbon, J. (1985). The structure and function of yeast centromeres. Annu. Rev. Genet. 19, 29–56.
Research
Fitzgerald-Hayes, M., et al. (1982). Nucleotide sequence comparisons and functional analysis of yeast centromere DNAs. Cell 29, 235–244.
7.15 The S. cerevisiae Centromere Binds a Protein Complex
Reviews
Bloom, K. (2007). Centromere dynamics. Curr. Opin. Genet. Dev. 17, 151–156.
Kitagawa, K., and Hieter, P. (2001). Evolutionary conservation between budding yeast and human kinetochores. Nat. Rev. Mol. Cell Biol. 2, 678–687.
Research
Lechner, J., and Carbon, J. (1991). A 240 kd multisubunit protein complex, CBF3, is a major component of the budding yeast centromere. Cell 64, 717–725.
Meluh, P. B., and Koshland, D. (1997). Budding yeast centromere composition and assembly as revealed by in vitro cross-linking. Genes Dev 11, 3401–3412.
Meluh, P. B., et al. (1998). Cse4p is a component of the core centromere of S. cerevisiae. Cell 94, 607–613.
Ortiz, J., et al. (1999). A putative protein complex consisting of Ctf19, Mcm21, and Okp1 represents a missing link in the budding yeast kinetochore. Genes Dev 13, 1140–1155.
7.16 Telomeres Have Simple Repeating Sequences
Reviews
Blackburn, E. H., and Szostak, J. W. (1984). The molecular structure of centromeres and telomeres. Annu. Rev. Biochem. 53, 163–194.
Zakian, V. A. (1989). Structure and function of telomeres. Annu. Rev. Genet. 23, 579–604.
Research
Dejardin, J., and Kingston, R. E. (2009). Purification of proteins associated with specific genomic loci. Cell 136, 175–186.
Wellinger, R. J., et al. (1996). Evidence for a new step in telomere maintenance. Cell 85, 423–433.
7.17 Telomeres Seal the Chromosome Ends and Function in Meiotic Chromosome Pairing
Reviews
Palm, W., and de Lange, T. (2008). How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334.
Scherthan, H. (2007). Telomere attachment and clustering during meiosis. Cell. Mol. Life Sci. 64, 117–124.
Research
Bass, H. W., et al. (1997). Telomeres cluster de novo before the initiation of synapsis: a three-dimensional spatial analysis of telomere positions before and during meiotic prophase. J. Cell Biol. 137, 5–18.
Chikashige, Y., et al. (2006). Meiotic proteins bqt1 and bqt2 tether telomeres to form the bouquet arrangement of chromosomes. Cell 125, 59–69.
Griffith, J. D., et al. (1999). Mammalian telomeres end in a large duplex loop. Cell 97, 503–514.
Henderson, E., et al. (1987). Telomeric oligonucleotides form novel intramolecular structures containing guanine-guanine base pairs. Cell 51, 899–908.
Karlseder, J., et al. (1999). p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science 283, 1321–1325.
Parkinson, G. N., et al. (2002). Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 417, 876–880.
van Steensel, B., et al. (1998). TRF2 protects human telomeres from end-to-end fusions. Cell 92, 401–413.
Williamson, J. R., et al.(1989). Monovalent cation-induced structure of telomeric DNA: the G-quartet model. Cell 59, 871–880.
7.18 Telomeres Are Synthesized by a Ribonucleoprotein Enzyme
Reviews
Blackburn, E. H. (1991). Structure and function of telomeres. Nature 350, 569–573.
Blackburn, E. H. (1992). Telomerases. Annu. Rev. Biochem. 61, 113–129.
Blackburn, E. H., et al. (2006). Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med 12, 1133–1138.
Collins, K. (1999). Ciliate telomerase biochemistry. Annu. Rev. Biochem. 68, 187–218.
Smogorzewska, A., and de Lange, T. (2004). Regulation of telomerase by telomeric proteins. Annu. Rev. Biochem. 73, 177–208.
Zakian, V. A. (1995). Telomeres: beginning to understand the end. Science 270, 1601–1607.
Zakian, V. A. (1996). Structure, function, and replication of S. cerevisiae telomeres. Annu. Rev. Genet. 30, 141–172.
Research
Greider, C., and Blackburn, E. H. (1987). The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 51, 887–898.
Murray, A., and Szostak, J. W. (1983). Construction of artificial chromosomes in yeast. Nature 305, 189–193.
Pennock, E., et al. (2001). Cdc13 delivers separate complexes to the telomere for end protection and replication. Cell 104, 387–396.
Shippen-Lentz, D., and Blackburn, E. H. (1990). Functional evidence for an RNA template in telomerase. Science 247, 546–552.
Teixeira, M. T., et al. (2004). Telomere length homeostasis is achieved via a switch between telomerase-extendible and nonextendible states. Cell 117, 323–335.
7.19 Telomeres Are Essential for Survival
Review
Bailey, S. M., and Murname, J. P. (2006). Telomeres, chromosome instability and cancer. Nucleic. Acids. Res. 34, 2408–2417.
Research
Hackett, J. A., et al. (2001). Telomere dysfunction increases mutation rate and genomic instability. Cell 106, 275–286.
Heidinger, B. J., et al. (2012). Telomere length in early life predicts lifespan. Proc. Natl. Acad. Sci. 109, 1743–1748.
Nakamura, T. M., et al. (1997). Telomerase catalytic subunit homologs from fission yeast and human. Science 277, 955–959.
Nakamura, T. M., et al. (1998). Two modes of survival of fission yeast without telomerase. Science 282, 493–496.
Rizki, A., and Lundblad, V. (2001). Defects in mismatch repair promote telomerase-independent proliferation. Nature 411, 713–716.