
Top texture: © Laguna Design / Science Source;
Chapter 19: RNA Splicing and Processing

Chapter Opener: © Laguna Design/Getty Images.
19.1 Introduction
RNA is a central player in gene expression. It was first characterized as an intermediate in protein synthesis, but since then many other RNAs that play structural or functional roles at various stages of gene expression have been discovered. The involvement of RNA in many functions involved with gene expression supports the general view that life may have evolved from an “RNA world” in which RNA was originally the active component in maintaining and expressing genetic information. Many of these functions were subsequently assisted or taken over by proteins, with a consequent increase in versatility and probably efficiency.
All RNAs studied thus far are transcribed from their respective genes and (particularly in eukaryotes) require further processing to become mature and functional. Interrupted genes are found in all groups of eukaryotic organisms. They represent a small proportion of the genes of unicellular eukaryotes, but the majority of genes in multicellular eukaryotic genomes. Genes vary widely according to the numbers and lengths of introns, but a typical mammalian gene has seven to eight exons spread out over about 16 kb. The exons are relatively short (about 100 to 200 bp), and the introns are relatively long (almost 1 kb) (see the chapter titled The Interrupted Gene).
The discrepancy between the interrupted organization of the gene and the uninterrupted organization of its mRNA requires processing of the primary transcription product. The primary transcript has the same organization as the gene and is called the pre-mRNA. Removal of the introns from pre-mRNA leaves an RNA molecule with an average length of about 2.2 kb. Removal of introns is a major part of the processing of RNAs in all eukaryotes. The process by which the introns are removed is called RNA splicing. Although interrupted genes are relatively rare in most unicellular/oligocellular eukaryotes (such as the yeast Saccharomyces cerevisiae), the overall proportion underestimates the importance of introns because most of the genes that are interrupted encode relatively abundant proteins. Splicing is therefore involved in the production of a greater proportion of total mRNA than would be apparent from analysis of the genome, perhaps as much as 50%.
One of the first clues about the nature of the discrepancy in size between nuclear genes and their products in multicellular eukaryotes was provided by the properties of nuclear RNA. Its average size is much larger than mRNA, it is very unstable, and it has a much greater sequence complexity. Taking its name from its broad size distribution, it is called heterogeneous nuclear RNA (hnRNA).
The physical form of hnRNA is a ribonucleoprotein particle, hnRNP, in which the hnRNA is bound by a set of abundant RNA-binding proteins. Some of the proteins may have a structural role in packaging the hnRNA; several are known to affect RNA processing or facilitate RNA export out of the nucleus.
Splicing occurs in the nucleus, together with the other modifications that are made to newly synthesized RNAs. The process of expressing an interrupted gene is reviewed in FIGURE 19.1. The transcript is capped at the 5′ end, has the introns removed, and is polyadenylated at the 3′ end. The RNA is then transported through nuclear pores to the cytoplasm, where it is available to be translated.

FIGURE 19.1 RNA is modified in the nucleus by additions to the 5′ and 3′ ends and by splicing to remove the introns. The splicing event requires breakage of the exon–intron junctions and joining of the ends of the exons. Mature mRNA is transported through nuclear pores to the cytoplasm, where it is translated.
With regard to the various processing reactions that occur in the nucleus, we should like to know at what point splicing occurs vis-à-vis the other modifications of RNA. Does splicing occur at a particular location in the nucleus, and is it connected with other events—for example, transcription and/or nucleocytoplasmic transport? Does the lack of splicing make an important difference in the expression of uninterrupted genes?
With regard to the splicing reaction itself, one of the main questions is how its specificity is controlled. What ensures that the ends of each intron are recognized in pairs so that the correct sequence is removed from the RNA? Are introns excised from a precursor in a particular order? Is the maturation of RNA used to regulate gene expression by discriminating among the available precursors or by changing the pattern of splicing?
Besides RNA splicing to remove introns, many noncoding RNAs also require processing to mature, and they play roles in diverse aspects of gene expression.
19.2 The 5′ End of Eukaryotic mRNA Is Capped
Transcription starts with a nucleoside triphosphate (usually a purine, A or G). The first nucleotide retains its 5′-triphosphate group and makes the usual phosphodiester bond from its 3′ position to the 5′ position of the next nucleotide. The initial sequence of the transcript can be represented as:
5′pppA/GpNpNpNp …
However, when the mature mRNA is treated in vitro with enzymes that should degrade it into individual nucleotides, the 5′ end does not give rise to the expected nucleoside triphosphate. Instead it contains two nucleotides that are connected by a 5′–5′ triphosphate linkage and also bear a methyl group. The terminal base is always a guanine that is added to the original RNA molecule after transcription.
Addition of the 5′ terminal G is catalyzed by a nuclear enzyme, guanylyl-transferase (GT). In mammals, GT has two enzymatic activities, one functioning as the triphosphatase to remove the two phosphates in GTP and the other as the guanylyl-transferase to fuse the guanine to the original 5′-triphosphate terminus of the RNA. In yeast, these two activities are carried out by two separate enzymes. The new G residue added to the end of the RNA is in the reverse orientation from all the other nucleotides:
5′Gppp + 5′pppApNpNp … → Gppp5′–5′ApNpNp … + pp + p
This structure is called a cap. It is a substrate for several methylation events. FIGURE 19.2 shows the full structure of a cap after all possible methyl groups have been added. The most important event is the addition of a single methyl group at the 7 position of the terminal guanine, which is carried out by guanine-7-methyltransferase (MT).

FIGURE 19.2 The cap blocks the 5′ end of mRNA and can be methylated at several positions.
Although the capping process can be accomplished in vitro using purified enzymes, the reaction normally takes place during transcription. Shortly after transcription initiation, Pol II is paused about 30 nucleotides downstream from the initiation site, waiting for the recruitment of the capping enzymes to add the cap to the 5′ end of nascent RNA. Without this protection, nascent RNA may be vulnerable to attack by 5′–3′ exonucleases, and such trimming may induce the Pol II complex to fall off of the DNA template. Thus, the process of capping is important for Pol II to enter the productive mode of elongation to transcribe the rest of the gene. In this regard, the pausing mechanism for 5′ capping represents a checkpoint for transcription reinitiation from the initial pausing site.
In a population of eukaryotic mRNAs, every molecule contains only one methyl group in the terminal guanine, generally referred to as a monomethylated cap. In contrast, some other small noncoding RNAs, such as those involved in RNA splicing in the spliceosome (see the section later in this chapter titled snRNAs Are Required for Splicing), are further methylated to contain three methyl groups in the terminal guanine. This structure is called a trimethylated cap. The enzymes for these additional methyl transfers are present in the cytoplasm. This may ensure that only some specialized RNAs are further modified at their caps.
One of the major functions for the formation of a cap is to protect the mRNA from degradation. In fact, enzymatic decapping represents one of the major mechanisms to regulate mRNA turnover in eukaryotic cells (see the section later in this chapter titled Splicing Is Temporally and Functionally Coupled with Multiple Steps in Gene Expression). In the nucleus, the cap is recognized and bound by the cap binding CBP20/80 heterodimer. This binding event stimulates splicing of the first intron and, via a direct interaction with the mRNA export machinery (TREX complex), facilitates mRNA export out of the nucleus. Once reaching the cytoplasm, a different set of proteins (eIF4F) binds the cap to initiate translation of the mRNA in the cytoplasm.
19.3 Nuclear Splice Sites Are Short Sequences
To focus on the molecular events involved in nuclear intron splicing, we must consider the nature of the splice sites, the two exon–intron boundaries that include the sites of breakage and reunion. By comparing the nucleotide sequence of a mature mRNA with that of the original gene, the junctions between exons and introns can be determined.
No extensive homology or complementarity exists between the two ends of an intron. However, the splice sites do have well-conserved, though rather short, consensus sequences. It is possible to assign a specific end to every intron by relying on the conservation of exon–intron junctions. They can all be aligned to conform to the consensus sequence shown in the upper portion of FIGURE 19.3.

FIGURE 19.3 The ends of nuclear introns are defined by the GU-AG rule (shown here as GT-AG in the DNA sequence of the gene). Minor introns are defined by different consensus sequences at the 5′ splice site, branch site, and 3′ splice site.
The height of each letter indicates the percent occurrence of the specified base at each consensus position. High conservation is found only immediately within the intron at the presumed junctions. This identifies the sequence of a generic intron as:
GU … … AG
Because the intron defined in this way starts with the dinucleotide GU and ends with the dinucleotide AG, the junctions are often described as conforming to the GU-AG rule. (Of course, the coding strand sequence of DNA has GT-AG.)
Note that the two sites have different sequences, and so they define the ends of the intron directionally. They are named proceeding from left to right along the intron as the 5′ splice site (sometimes called the left, or donor, site) and the 3′ splice site (also called the right, or acceptor, site). The consensus sequences are implicated as the sites recognized in splicing by point mutations that prevent splicing in vivo and in vitro.
In addition to the majority of introns that follow the GU-AG rule, a small fraction of introns are exceptions with a different set of consensus sequences at the exon–intron boundaries, as shown in the lower portion of Figure 19.3. These introns were initially described as minor introns that follow the AU-AC role because of the conserved AU-AC dinucleotides at both ends of each intron, as shown in the middle panel of Figure 19.3. However, the major and minor introns are better described as U2-type and U12-type introns, respectively, based on the distinct splicing machineries that process them (see the section later in this chapter titled An Alternative Spliceosome Uses Different snRNPs to Process the Minor Class of Introns). As a result, some introns that appear to follow the GU-AG rule are actually processed as U12-type introns, as indicated in the lower panel of Figure 19.3.
19.4 Splice Sites Are Read in Pairs
A typical mammalian gene has many introns. The basic problem of pre-mRNA splicing results from the simplicity of the splice sites and is illustrated in FIGURE 19.4. What ensures that the correct pairs of sites are recognized and spliced together in the presence of numerous sequences that match the consensus of bona fide splice sites in the intron? The corresponding GU-AG pairs must be connected across great distances (some introns are more than 100 kb long). We can imagine two types of mechanism that might be responsible for pairing the appropriate 5′ and 3′ splice sites:
It could be an intrinsic property of the RNA to connect the sites at the ends of a particular intron. This would require matching of specific sequences or structures, which has been seen in certain insect genes, but this does not seem to be the case for most eukaryotic genes.
It could be that all 5′ sites may be functionally equivalent and all 3′ sites may be similarly indistinguishable, but splicing could follow rules that ensure a 5′ site is always connected to the 3′ site that comes next in the RNA.
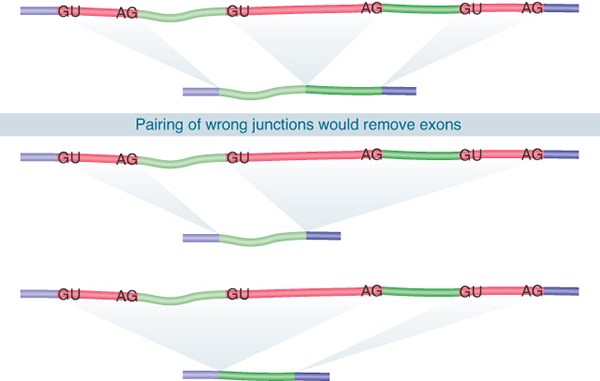
FIGURE 19.4 Splicing junctions are recognized only in the correct pairwise combinations.
Neither the splice sites nor the surrounding regions have any sequence complementarity, which excludes models for complementary base pairing between intron ends. Experiments using hybrid RNA precursors show that any 5′ splice site can in principle be connected to any 3′ splice site. For example, when the first exon of the early SV40 transcription unit is linked to the third exon of mouse β-globin, the hybrid intron can be excised to generate a perfect connection between the SV40 exon and the β-globin exon. Indeed, this interchangeability is the basis for the exon-trapping technique described previously in the chapter titled The Content of the Genome. Such experiments have two general interpretations:
Splice sites are generic. They do not have specificity for individual RNA precursors and individual precursors do not convey specific information (e.g., secondary structure) that is needed for splicing. However, in some cases specific RNA-binding proteins (e.g., hnRNP A1) have been shown to promote splice-site pairing by binding to adjacent prospective splice sites.
The apparatus for splicing is not tissue specific. An RNA can usually be properly spliced by any cell, whether or not it is usually synthesized in that cell. (Exceptions in which there are tissue-specific alternative splicing patterns are presented in the section later in this chapter titled Alternative Splicing Is a Rule, Rather Than an Exception, in Multicellular Eukaryotes.)
If all 5′ splice sites and all 3′ splice sites are similarly recognized by the splicing apparatus, what rules ensure that recognition of splice sites is restricted so that only the 5′ and 3′ sites of the same intron are spliced? Are introns removed in a specific order from a particular RNA?
Splicing is temporally coupled with transcription (e.g., many splicing events are already completed before the RNA polymerase reaches the end of the gene); as a result it is reasonable to assume that transcription provides a rough order of splicing in the 5′ to 3′ direction (something like a first-come, first-served mechanism). Second, a functional splice site is often surrounded by a series of sequence elements that can enhance or suppress the site (see the section later in this chapter titled Splicing Can Be Regulated by Exonic and Intronic Splicing Enhancers and Silencers). Thus, sequences in both exons and introns can also function as regulatory elements for splice-site selection.
We can imagine that, in order to be efficiently recognized by the splicing machinery, a functional splice site has to have the right sequence context, including specific consensus sequences and surrounding splicing-enhancing elements that are dominant over splicing-suppressing elements. These mechanisms together may ensure that splice signals are read in pairs in a relatively linear order.
19.5 Pre-mRNA Splicing Proceeds Through a Lariat
The mechanism of splicing has been characterized in vitro using cell-free systems in which introns can be removed from RNA precursors. Nuclear extracts can splice purified RNA precursors; this shows that the action of splicing does not have to be linked to the process of transcription. Splicing can occur in RNAs that are neither capped nor polyadenylated even though these events normally occur in the cell in a coordinated manner, and the efficiency of splicing may be influenced by transcription and other processing events (see the section later in this chapter titled Splicing Is Temporally and Functionally Coupled with Multiple Steps in Gene Expression).
The stages of splicing in vitro are illustrated in the pathway of FIGURE 19.5. The reaction is discussed in terms of the individual RNA types that can be identified, but remember that in vivo the types containing exons are not released as free molecules but remain held together by the splicing apparatus.

FIGURE 19.5 Splicing occurs in two stages. First the 5′ exon is cleaved off, and then it is joined to the 3′ exon.
FIGURE 19.6 shows that the first step of the splicing reaction is a nucleophilic attack by the 2′–OH on the 5′ splice site. The left exon takes the form of a linear molecule. The right intron–exon molecule forms a branched structure called the lariat, in which the 5′ terminus generated at the end of the intron simultaneously transesterificates to become linked by a 2′–5′ bond to a base within the intron. The target base is an A in a sequence called the branch site.

FIGURE 19.6 Nuclear splicing occurs by two transesterification reactions, in which an –OH group attacks a phosphodiester bond.
In the second step, the free 3′–OH of the exon that was released by the first reaction now attacks the bond at the 3′ splice site. Note that the number of phosphodiester bonds is conserved. There were originally two 5′–3′ bonds at the exon–intron splice sites; one has been replaced by the 5′–3′ bond between the exons and the other has been replaced by the 2′–5′ bond that forms the lariat. The lariat is then “debranched” to give a linear excised intron that is rapidly degraded.
The sequences needed for splicing are the short consensus sequences at the 5′ and 3′ splice sites and at the branch site. Together with the knowledge that most of the sequence of an intron can be deleted without impeding splicing, this indicates that there is no demand for specific conformation in the intron (or exon).
The branch site plays an important role in identifying the 3′ splice site. The branch site in yeast is highly conserved and has the consensus sequence UACUAAC. The branch site in multicellular eukaryotes is not well conserved but has a preference for purines or pyrimidines at each position and retains the target A nucleotide.
The branch site is located 18 to 40 nucleotides upstream of the 3′ splice site. Mutations or deletions of the branch site in yeast prevent splicing. In multicellular eukaryotes, the relaxed constraints in its sequence result in the ability to use related sequences (called cryptic sites) when the authentic branch is deleted or mutated. Proximity to the 3′ splice site appears to be important because the cryptic site is always close to the authentic site. A cryptic site is used only when the branch site has been inactivated. When a cryptic branch sequence is used in this manner, splicing otherwise appears to be normal, and the exons give the same products as the use of the authentic branch site does. The role of the branch site is therefore to identify the nearest 3′ splice site as the target for connection to the 5′ splice site. This can be explained by the fact that an interaction occurs between protein complexes that bind to these two sites.
19.6 snRNAs Are Required for Splicing
The 5′ and 3′ splice sites and the branch sequence are recognized by components of the splicing apparatus that assemble to form a large complex. This complex brings the 5′ and 3′ splice sites together before any reaction occurs, which explains why a deficiency in any one of the sites may prevent the reaction from initiating. The complex assembles sequentially on the pre-mRNA and passes through several “presplicing complexes” before forming the final, active complex, which is called the spliceosome. Splicing occurs only after all the components have assembled.
The splicing apparatus contains both proteins and RNAs (in addition to the pre-mRNA). The RNAs take the form of small molecules that exist as ribonucleoprotein particles. Both the nucleus and cytoplasm of eukaryotic cells contain many discrete small RNA types. They range in size from 100 to 300 bases in multicellular eukaryotes and extend in length to about 1,000 bases in yeast. They vary considerably in abundance, from 105 to 106 molecules per cell to concentrations too low to be detected directly.
Those restricted to the nucleus are called small nuclear RNAs (snRNAs); those found in the cytoplasm are called small cytoplasmic RNAs (scRNAs). In their natural state, they exist as ribonucleoprotein particles (snRNPs and scRNPs). Colloquially, they are sometimes known as snurps and scyrps, respectively. Another class of small RNAs found in the nucleolus, called small nucleolar RNAs (snoRNAs), are involved in processing ribosomal RNA (see the section later in this chapter titled Production of rRNA Requires Cleavage Events and Involves Small RNAs).
The snRNPs involved in splicing, together with many additional proteins, form the spliceosome. Isolated from the in vitro splicing systems, it comprises a 50S to 60S ribonucleoprotein particle. The spliceosome may be formed in stages as the snRNPs join, proceeding through several presplicing complexes. The spliceosome is a large body, greater in mass than the ribosome.
FIGURE 19.7 summarizes the components of the spliceosome. The five snRNAs account for more than a quarter of its mass; together with their 41 associated proteins, they account for almost half of its mass. Some 70 other proteins found in the spliceosome are described as splicing factors. They include proteins required for assembly of the spliceosome, proteins required for it to bind to the RNA substrate, and proteins involved in constructing an RNA-based center for transesterification reactions. In addition to these proteins, another approximately 30 proteins associated with the spliceosome are believed to be acting at other stages of gene expression, which suggests splicing may be connected to other steps in gene expression (see the section later in this chapter titled Splicing Is Temporally and Functionally Coupled with Multiple Steps in Gene Expression).

FIGURE 19.7 The spliceosome is approximately 12 megadaltons (MDa). Five snRNPs account for almost half of the mass. The remaining proteins include known splicing factors, as well as proteins that are involved in other stages of gene expression.
The spliceosome forms on the intact precursor RNA and passes through an intermediate state in which it contains the individual 5′ exon linear molecule and the right-lariat intron–exon. Little spliced product is found in the complex, which suggests that it is usually released immediately following the cleavage of the 3′ site and ligation of the exons.
We may think of the snRNP particles as being involved in building the structure of the spliceosome. Like the ribosome, the spliceosome depends on RNA–RNA interactions as well as protein–RNA and protein–protein interactions. Some of the reactions involving the snRNPs require their RNAs to base pair directly with sequences in the RNA being spliced; other reactions require recognition between snRNPs or between their proteins and other components of the spliceosome.
The importance of snRNA molecules can be tested directly in yeast by inducing mutations in their genes or in in vitro splicing reactions by targeted degradation of individual snRNAs in the nuclear extract. Inactivation of five snRNAs, individually or in combination, prevents splicing. All of the snRNAs involved in splicing can be recognized in conserved forms in all eukaryotes, including plants. The corresponding RNAs in yeast are often rather larger, but conserved regions include features that are similar to the snRNAs of multicellular eukaryotes.
The snRNPs involved in splicing are U1, U2, U5, U4, and U6. They are named according to the snRNAs that are present. Each snRNP contains a single snRNA and several (fewer than 20) proteins. The U4 and U6 snRNPs are usually found together as a di-snRNP (U4/U6) particle. A common structural core for each snRNP consists of a group of eight proteins, all of which are recognized by an autoimmune antiserum called anti-Sm; conserved sequences in the proteins form the target for the antibodies. The other proteins in each snRNP are unique to it. The Sm proteins bind to the conserved sequence A/GAU3–6Gpu, which is present in all snRNAs except U6. The U6 snRNP instead contains a set of Sm-like (Lsm) proteins.
Some of the proteins in the snRNPs may be involved directly in splicing; others may be required in structural roles or just for assembly or interactions between the snRNP particles. About one-third of the proteins involved in splicing are components of the snRNPs. Increasing evidence for a direct role of RNA in the splicing reaction suggests that relatively few of the splicing factors play a direct role in catalysis; most splicing factors may therefore provide structural or assembly roles in the spliceosome.
19.7 Commitment of Pre-mRNA to the Splicing Pathway
Recognition of the consensus splicing signals involves both RNAs and proteins. Certain snRNAs have sequences that are complementary to the mRNA consensus sequences or to one another, and base pairing between snRNA and pre-mRNA, or between snRNAs, plays an important role in splicing.
Binding of U1 snRNP to the 5′ splice site is the first step in splicing. The human U1 snRNP contains the core Sm proteins, three U1-specific proteins (U1-70k, U1A, and U1C), and U1 snRNA. The secondary structure of the U1 snRNA is shown in FIGURE 19.8. It contains several domains. The Sm-binding site is required for interaction with the common snRNP proteins. Domains identified by the individual stem-loop structures provide binding sites for proteins that are unique to U1 snRNP. U1 snRNA interacts with the 5′ splice site by base pairing between its single-stranded 5′ terminus and a stretch of four to six bases of the 5′ splice site.

FIGURE 19.8 U1 snRNA has a base-paired structure that creates several domains. The 5′ end remains single stranded and can base pair with the 5′ splice site.
Mutations in the 5′ splice site and U1 snRNA can be used to test directly whether pairing between them is necessary. The results of such an experiment are illustrated in FIGURE 19.9. The wild-type sequence of the splice site of the 12S adenovirus pre-mRNA pairs at five out of six positions with U1 snRNA. A mutant in the 12S RNA that cannot be spliced has two sequence changes; the GG residues at positions 5 to 6 in the intron are changed to AU. When a mutation is introduced into U1 snRNA that restores pairing at position 5, normal splicing is regained. Other cases, in which corresponding mutations are made in U1 snRNA to see whether they can suppress the mutation in the splice site, suggest this general rule: Complementarity between U1 snRNA and the 5′ splice site is necessary for splicing, but the efficiency of splicing is not determined solely by the number of base pairs that can form.

FIGURE 19.9 Mutations that abolish function of the 5′ splice site can be suppressed by compensating mutations in U1 snRNA that restore base pairing.
The U1 snRNA pairing reaction with the 5′ splicing is stabilized by protein factors. Two such factors play a particular role: The branch point binding protein (BBP, also known as SF1) interacts with the branch point sequence, and U2AF (a heterodimer consisting of U2AF65 and U2AF35 in multicellular eukaryotic cells or Mud2 in the yeast S. cerevisiae) binds to the polypyrimidine tract between the branch point sequence and the invariant AG dinucleotide at the end of each intron. Each of these binding events is not very strong, but together they bind in a cooperative fashion, resulting in the formation of a relatively stable complex called the commitment complex.
The commitment complex is also known as the E complex (E for “early”) in mammalian cells, the formation of which does not require ATP (compared to all late ATP-dependent steps in the assembly of the spliceosome; see the section later in this chapter titled The Spliceosome Assembly Pathway). Unlike in yeast, however, the consensus sequences at the splice sites in mammalian genes are only loosely conserved, and consequently additional protein factors are needed for the formation of the E complex.
The factor or factors that play a central role in this and other spliceosome assembly processes are SR proteins, which constitute a family of splicing factors that contain one or two RNA-recognition motifs at the N-terminus and a signature domain rich with multiple Arg/Ser dipeptide repeats (called the RS domain) at their C-terminus. Their RNA-recognition motifs are responsible for sequence-specific binding to RNA, and the RS domain can bind to both RNA and other splicing factors via protein–protein interactions, thereby providing additional “glue” for various parts of the E complex.
As illustrated in FIGURE 19.10, SR proteins can bind to the 70-kD component of U1 snRNP (the U1 70-kD protein also contains an RS domain, but it is not considered a typical SR protein) to enhance or stabilize its base pairing with the 5′ splice site. SR proteins can also bind to 3′ splice site–bound U2AF (an RS domain is also present in both U2AF65 and U2AF35). These protein–protein interaction networks are thought to be critical for the formation of the E complex. SR proteins copurify with the Pol II complex and are able to kinetically commit RNA to the splicing pathway; thus they likely function as the splicing initiators in multicellular eukaryotic cells.

FIGURE 19.10 The commitment (E) complex forms by the successive addition of U1 snRNP to the 5′ splice site, U2AF to the pyrimidine tract/3′ splice site, and the bridging protein SF1/BBP.
Typical SR proteins are neither encoded in the genome of S. cerevisiae nor needed for splicing by the organism where the splicing signals are nearly invariant, but they are absolutely essential for splicing in all multicellular eukaryotes where the splicing signals are highly divergent. The evolution of SR proteins in multicellular eukaryotes likely contributes to high-efficacy and high-fidelity splicing on loosely conserved splice sites. The recognition of functional splice sites during the formation of the E complex can take two routes, as illustrated in FIGURE 19.11. In S. cerevisiae, where nearly all intron-containing genes are interrupted by a single small intron (between 100 and 300 nucleotides in length), the 5′ and 3′ splice sites are simultaneously recognized by U1 snRNP, BBP, and Mud2, as discussed earlier. This process is referred to as intron definition and is illustrated on the left of Figure 19.11. (Note that the intron definition mechanism applies to small introns in multicellular eukaryotic cells, and thus the figure is drawn with the nomenclature for mammalian splicing factors involved in the process.)

FIGURE 19.11 The two routes for initial recognition of 5′ and 3′ splice sites are intron definition and exon definition.
In comparison, introns are long and highly variable in length in multicellular eukaryotic genomes, and there are many sequences that resemble real splice sites in them. This makes the paired recognition of the 5′ and 3′ splice sites inefficient, if not impossible. The solution to this problem is the process of exon definition, which takes advantage of normally small exons (between 100 and 300 nucleotides in length) in multicellular eukaryotic cells.
As shown on the right side of Figure 19.11, during exon definition the U2AF heterodimer binds to the 3′ splice site and U1 snRNP base pairs with the 5′ splice site downstream from the exon sequence. This process may be aided by SR proteins that bind to specific exon sequences between the 3′ and downstream 5′ splice sites. By an as yet unknown mechanism, the complexes formed across the exon are then switched to the complexes that link the 3′ splice site to the upstream 5′ splice site and the downstream 5′ splice site to the next downstream 3′ splice sites across introns. This establishes the “permissive” configuration that allows later spliceosome assembly steps to occur.
Blockage of this transition is actually a means to regulate the selection of certain exons during regulated splicing (see the section later in this chapter titled Splicing Can Be Regulated by Exonic and Intronic Splicing Enhancers and Silencers). Finally, the exon definition mechanism mediated by SR proteins also provides a mechanism to only allow adjacent 5′ and 3′ splice sites to be paired and linked by splicing.
19.8 The Spliceosome Assembly Pathway
Following formation of the E complex, the other snRNPs and factors involved in splicing associate with the complex in a defined order. FIGURE 19.12 shows the components of the complexes that can be identified as the reaction proceeds.

FIGURE 19.12 The splicing reaction proceeds through discrete stages in which spliceosome formation involves the interaction of components that recognize the consensus sequences.
In the first ATP-dependent step, U2 snRNP joins U1 snRNP on the pre-mRNA by binding to the branch point sequence, which also involves base pairing between the sequence in U2 snRNA and the branch point sequence. This results in the conversion of the E complex to the prespliceosome commonly known as the A complex, and this step requires ATP hydrolysis.
The B1 complex is formed when a trimer containing the U5 and U4/U6 snRNPs binds to the A complex. This complex is regarded as a spliceosome because it contains the components needed for the splicing reaction. It is converted to the B2 complex after U1 is released. The dissociation of U1 is necessary to allow other components to come into juxtaposition with the 5′ splice site, most notably U6 snRNA.
The catalytic reaction is triggered by the release of U4, which also takes place during the transition from the B1 to B2 complex. The role of U4 snRNA may be to sequester U6 snRNA until it is needed. FIGURE 19.13 shows the changes that occur in the base-pairing interactions between snRNAs during splicing. In the U6/U4 snRNP, a continuous length of 26 bases of U6 is paired with two separated regions of U4. When U4 dissociates, the region in U6 that is released becomes free to take up another structure. The first part of it pairs with U2; the second part forms an intramolecular hairpin. The interaction between U4 and U6 is mutually incompatible with the interaction between U2 and U6, so the release of U4 controls the ability of the spliceosome to proceed to the activated state.

FIGURE 19.13 U6/U4 pairing is incompatible with U6/U2 pairing. When U6 joins the spliceosome it is paired with U4. Release of U4 allows a conformational change in U6; one part of the released sequence forms a hairpin and the other part pairs with U2. An adjacent region of U2 is already paired with the branch site, which brings U6 into juxtaposition with the branch. Note that the substrate RNA is reversed from the usual orientation and is shown 3′ to 5′.
For clarity, Figure 19.13 shows the RNA substrate in extended form, but the 5′ splice site is actually close to the U6 sequence immediately on the 5′ side of the stretch bound to U2. This sequence in U6 snRNA pairs with sequences in the intron just downstream of the conserved GU at the 5′ splice site (mutations that enhance such pairing improve the efficiency of splicing).
Thus, several pairing reactions between snRNAs and the substrate RNA occur in the course of splicing. They are summarized in FIGURE 19.14. The snRNPs have sequences that pair with the pre-mRNA substrate and with one another. They also have single-stranded regions in loops that are in close proximity to sequences in the substrate and that play an important role, as judged by the ability of mutations in the loops to block splicing.

FIGURE 19.14 Splicing utilizes a series of base-pairing reactions between snRNAs and splice sites.
The base pairings between U2 and the branch point and between U2 and U6 create a structure that resembles the active center of group II self-splicing introns (see Figure 19.15 in the section titled Pre-mRNA Splicing Likely Shares the Mechanism with Group II Autocatalytic Introns). This suggests the possibility that the catalytic component could comprise an RNA structure generated by the U2–U6 interaction. U6 is paired with the 5′ splice site, and cross-linking experiments show that a loop in U5 snRNA is immediately adjacent to the first base positions in both exons. Although the available evidence points to an RNA-based catalysis mechanism within the spliceosome, contribution(s) by proteins cannot be ruled out. One candidate protein is Prp8, a large scaffold protein that directly contacts both the 5′ and 3′ splice sites within the spliceosome.
Both transesterification reactions take place in the activated spliceosome (the C complex) after a series of RNA arrangements is completed. The formation of the lariat at the branch site is responsible for determining the use of the 3′ splice site, because the 3′ consensus sequence nearest to the 3′ side of the branch becomes the target for the second transesterification.
The important conclusion suggested by these results is that the snRNA components of the splicing apparatus interact both among themselves and with the substrate pre-mRNA by means of base-pairing interactions, and these interactions allow for changes in structure that may bring reacting groups into apposition and may even create catalytic centers.
Although (like ribosomes) the spliceosome is likely a large RNA machine, many protein factors are essential for the machine to run. Extensive mutational analyses undertaken in yeast identified both the RNA and protein components (known as PRP mutants for pre-mRNA processing). Several of the products of these genes have motifs that identify them as a family of ATP-dependent RNA helicases, which are crucial for a series of ATP-dependent RNA rearrangements in the spliceosome.
Prp5 is critical for U2 binding to the branch point during the transition from the E to the A complex; Brr2 facilitates U1 and U4 release during the transition from the B1 to B2 complex; Prp2 is responsible for the activation of the spliceosome during the conversion of the B2 complex to the C complex; and Prp22 helps the release of the mature mRNA from the spliceosome. In addition, a number of RNA helicases play roles in recycling of snRNPs for the next round of spliceosome assembly.
These findings explain why ATP hydrolysis is required from various steps of the splicing reaction, although the actual transesterification reactions do not require ATP. Despite the fact that a sequential series of RNA arrangements takes place in the spliceosome, it is remarkable that the process seems to be reversible after both the first and second transesterification reactions.
19.9 An Alternative Spliceosome Uses Different snRNPs to Process the Minor Class of Introns
GU-AG introns comprise the majority (more than 98%) of splice sites in the human genome. Exceptions to this case are noncanonical splice AU-AC sites and other variations. Initially, this minor class of introns was referred to as AU-AC introns compared to the major class of introns that follow the GU-AG rule during splicing. With the elucidation of the machinery for processing of both major and minor introns, it becomes clear that this nomenclature for the minor class of introns is not entirely accurate.
Guided by years of research on the major spliceosome, the machinery for processing the minor class of introns was quickly elucidated; it consists of U11 and U12 (related to U1 and U2, respectively), a common U5 shared with the major spliceosome, and the U4atac and U6atac snRNAs. The splicing reaction is essentially similar to that of the major class of introns, and the snRNAs play analogous roles: U11 base pairs with the 5′ splice sites; U12 base pairs with the branch point sequence near the 3′ splice site; and U4atac and U6atac provide analogous functions during the spliceosome assembly and activation of the spliceosome.
It turns out that the dependence on the type of spliceosome is also influenced by the sequences in other places in the intron, so that there are some GU-AG introns spliced by the U12-type spliceosome. A strong consensus sequence at the left end defines the U12-dependent type of intron: 5′GAUAUCCUUU … PyAGC3′. In fact, most U12-dependent introns have the GU … AG termini. They have a highly conserved branch point (UCCUUPuAPy), though, which pairs with U12. This difference in branch point sequences is the primary distinction between the major and minor classes of introns. For this reason, the major class of introns is termed U2-dependent introns and the minor class is called U12-dependent introns, instead of AU-AC introns.
The two types of intron coexist in a variety of genomes, and in most cases are found in the same gene. U12-dependent introns tend to be flanked by U2-dependent introns. The phylogeny of these introns suggests that AU-AC U12-dependent introns may once have been more common, but tend to be converted to GU-AG termini, and to U2 dependence, in the course of evolution. The common evolution of the systems is emphasized by the fact that they use analogous sets of base pairing between the snRNAs and with the substrate pre-mRNA. In addition, all essential splicing factors (i.e., SR proteins) studied thus far are required for processing both U2-type and U12-type introns.
One noticeable difference between U2 and U12 types of intron is that U1 and U2 appear to independently recognize the 5′ and 3′ splice sites in the major class of introns during the formation of the E and A complexes, whereas U11 and U12 form a complex in the first place, which together contact the 5′ and 3′ splice sites to initiate the processing of the minor class of introns. This ensures that the splice sites in the minor class of introns are recognized simultaneously by the intron definition mechanism. It also avoids “confusing” the splicing machineries during the transition from exon definition to intron definition for processing the major and minor classes of introns that are present in the same gene.
19.10 Pre-mRNA Splicing Likely Shares the Mechanism with Group II Autocatalytic Introns
Introns in all genes (except nuclear tRNA–encoding genes) can be divided into three general classes. Nuclear pre-mRNA introns are identified only by the presence of the GU … AG dinucleotides at the 5′ and 3′ ends and the branch site/pyrimidine tract near the 3′ end. They do not show any common features of secondary structure. In contrast, group I and group II introns found in organelles and in bacteria (group I introns are also found in the nucleus in unicellular/oligocellular eukaryotes) are classified according to their internal organization. Each can be folded into a typical type of secondary structure.
The group I and group II introns have the remarkable ability to excise themselves from an RNA. This is called autosplicing, or self-splicing. Group I introns are more common than group II introns. There is little relationship between the two classes, but in each case the RNA can perform the splicing reaction in vitro by itself, without requiring enzymatic activities provided by proteins; however, proteins are almost certainly required in vivo to assist with folding (see the Catalytic RNA chapter).
FIGURE 19.15 shows that three classes of introns are excised by two successive transesterifications (shown previously for nuclear introns). In the first reaction, the 5′ exon–intron junction is attacked by a free hydroxyl group (provided by an internal 2′–OH position in nuclear and group II introns or by a free guanine nucleotide in group I introns). In the second reaction, the free 3′–OH at the end of the released exon in turn attacks the 3′ intron–exon junction.

FIGURE 19.15 Three classes of splicing reactions proceed by two transesterifications. First, a free –OH group attacks the exon 1–intron junction. Second, the –OH created at the end of exon 1 attacks the intron–exon 2 junction.
Parallels exist between group II introns and pre-mRNA splicing. Group II mitochondrial introns are excised by the same mechanism as nuclear pre-mRNAs via a lariat that is held together by a 2′–5′ bond. When an isolated group II RNA is incubated in vitro in the absence of additional components, it is able to perform the splicing reaction. This means that the two transesterification reactions shown in Figure 19.15 can be performed by the group II intron RNA sequence itself. The number of phosphodiester bonds is conserved in the reaction, and as a result an external supply of energy is not required; this could have been an important feature in the evolution of splicing.
A group II intron forms a secondary structure that contains several domains formed by base-paired stems and single-stranded loops. Domain 5 is separated by two bases from domain 6, which contains an A residue that donates the 2′–OH group for the first transesterification. This constitutes a catalytic domain in the RNA. FIGURE 19.16 compares this secondary structure with the structure formed by the combination of U6 with U2 and of U2 with the branch site. The similarity suggests that U6 may have a catalytic role in pre-mRNA splicing.

FIGURE 19.16 Nuclear splicing and group II splicing involve the formation of similar secondary structures. The sequences are more specific in nuclear splicing; group II splicing uses positions that may be occupied by either purine (R) or pyrimidine (Y).
The features of group II splicing suggest that splicing evolved from an autocatalytic reaction undertaken by an individual RNA molecule, in which it accomplished a controlled deletion of an internal sequence. It is likely that such a reaction would require the RNA to fold into a specific conformation, or series of conformations, and would occur exclusively in cis-conformation.
The ability of group II introns to remove themselves by an autocatalytic splicing event stands in great contrast to the requirement of nuclear introns for a complex splicing apparatus. The snRNAs of the spliceosome can be regarded as compensating for the lack of sequence information in the intron, and as providing the information required to form particular structures in RNA. The functions of the snRNAs may have evolved from the original autocatalytic system. These snRNAs act in trans upon the substrate pre-mRNA. Perhaps the ability of U1 to pair with the 5′ splice site, or of U2 to pair with the branch sequence, replaced a similar reaction that required the relevant sequence to be carried by the intron. Thus, the snRNAs may undergo reactions with the pre-mRNA substrate—and with one another—that have substituted for the series of conformational changes that occur in RNAs that splice by group II mechanisms. In effect, these changes have relieved the substrate pre-mRNA of the obligation to carry the sequences needed to sponsor the reaction. As the splicing apparatus has become more complex (and as the number of potential substrates has increased), proteins have played a more important role.
19.11 Splicing Is Temporally and Functionally Coupled with Multiple Steps in Gene Expression
Pre-mRNA splicing has long been recognized to take place cotranscriptionally, though the two reactions can take place separately in vitro and have been studied as separate processes in gene expression. Major experimental evidence supporting cotranscriptional splicing came from the observations that many splicing events are completed before the completion of transcription. In general, introns near the 5′ end of the gene are removed during transcription, but introns near the end of the gene can be processed either during or after transcription.
Besides temporal coupling between transcription and splicing, there are probably other reasons for these two key processes to be linked in a functional way. Indeed, the machineries for 5′ capping, intron removal, and even polyadenylation at the 3′ end (see the section later in this chapter titled 3′ mRNA End Processing Is Critical for Termination of Transcription) show physical interactions with the core machinery for transcription. A common mechanism is to use the large C-terminal domain of the largest subunit of Pol II (known as CTD) as a loading pad for various RNA-processing factors, although in most cases it is yet to be defined whether the tethering is direct or mediated by some common protein or even RNA factors (see the Eukaryotic Transcription chapter).
Such physical integration would ensure efficient recognition of emerging splicing signals to pair adjacent functional splice sites during transcription, thus maintaining a rough order of splicing from the 5′ to 3′ direction. The recognition of the emerging splicing signals by the RNA-processing factors and enzymes associated with the elongation Pol II complex would also allow these factors to compete effectively with other nonspecific RNA-binding proteins, such as hnRNP proteins, that are abundantly present in the nucleus for RNA packaging.
If RNA splicing benefits from transcription, why not the other way around? In fact, increasing evidence has suggested so; as illustrated in FIGURE 19.17, the 5′ capping enzymes seem to help overcome initial transcriptional pausing near the promoter; splicing factors appear to play some roles in facilitating transcriptional elongation; and the 3′ end formation of mRNA is clearly instrumental to transcriptional termination (see the section later in this chapter titled 3′ mRNA End Processing Is Critical for Termination of Transcription). Thus, transcription and RNA processing are highly coordinated in multicellular eukaryotic cells.

FIGURE 19.17 Coupling transcription with the 5′ capping reaction. Pol II transcription is initially paused near the transcription start point. Both guanylyl-transferase (GT) and 7-methyltransferase (MT) are recruited to the Pol II complex to catalyze 5′ capping, and the cap is bound by the cap-binding protein complex at the 5′ end of the nascent transcript. These reactions allow the paused Pol II to enter the mode of productive elongation.
RNA processing is functionally linked not only to the upstream transcriptional events but also to downstream steps, such as mRNA export and stability control. It has been known for a long time that intermediately processed RNA that still contains some introns cannot be exported efficiently, which may be due to the retention effect of the spliceosome in the nucleus. Splicing-facilitated mRNA export can be demonstrated by nuclear injection of intronless RNA derived from cDNA or pre-mRNA that will give rise to identical RNA upon splicing. The RNA that has gone through the splicing process is exported more efficiently than the RNA derived from the cDNA, indicating that the splicing process helps mRNA export.
As illustrated in FIGURE 19.18, a specific complex, called the exon junction complex (EJC), is deposited onto the exon–exon junction. This complex appears to directly recruit a number of RNA-binding proteins implicated in mRNA export. Apparently, these mechanisms may act in synergy to promote the export of mRNA coming out of transcription and the cotranscriptional RNA-splicing apparatus. This process may start early in transcription. The cap binding CBP20/80 complex appears to directly bind to the mRNA export machinery (the TREX complex) in a manner that depends on splicing to remove the first intron near the 5′ end to facilitate mRNA export. A key factor in mediating mRNA export is REE (also named Aly, Yra1 in yeast), which is part of the EJC and can directly interact with the mRNA transporter TAP (Mex67 in yeast), as shown in FIGURE 19.19.

FIGURE 19.18 The exon junction complex (EJC) is deposited near the splice junction as a consequence of the splicing reaction.

FIGURE 19.19 An REF protein (shown in green) binds to a splicing factor and remains with the spliced RNA product. REF binds to a transport protein (shown in purple) that binds to the nuclear pore.
The EJC complex has an additional role in escorting mRNA out of the nucleus, which has a profound effect on mRNA stability in the cytoplasm. This is because an EJC that has retained some aberrant mRNAs can recruit other factors that promote decapping enzymes to remove the protective cap at the 5′ end of the mRNA. As illustrated in FIGURE 19.20, the EJC is normally removed by the scanning ribosome during the first round of translation in the cytoplasm. If, however, for some reason a premature stop codon is introduced into a processed mRNA as a result of point mutation or alternative splicing (see the next section, titled Alternative Splicing Is a Rule, Rather Than an Exception, in Multicellular Eukaryotes), the ribosome will fall off before reaching the natural stop codon, which is typically located in the last exon. The inability of the ribosome to strip off the EJC complex deposited after the premature stop codon will allow the recruitment of decapping enzymes to induce rapid degradation of the mRNA. This process is called nonsense-mediated mRNA decay (NMD), which represents an mRNA surveillance mechanism that prevents translation of truncated proteins from the mRNA that carries a premature stop codon (NMD is discussed further in the mRNA Stability and Localization chapter).

FIGURE 19.20 The EJC complex couples splicing with NMD. The EJC can also recruit Upr proteins if it remains on the exported mRNA. After nuclear export, EJC should be tripped off by the scanning ribosome in the first round of translation. If an EJC remains on the mRNA because of a premature stop codon in the front, which releases the ribosome, the EJC will recruit additional proteins, such as Upf, which will then recruit the decapping enzyme (DCP). This will induce decapping at the 5′ end and mRNA degradation from the 5′ to 3′ direction in the cytoplasm.
19.12 Alternative Splicing Is a Rule, Rather Than an Exception, in Multicellular Eukaryotes
When an interrupted gene is transcribed into an RNA that gives rise to a single type of spliced mRNA, the assignment of exons and introns is unambiguous. However, the RNAs of most mammalian genes follow patterns of alternative splicing, which occurs when a single gene gives rise to more than one mRNA sequence. By large-scale cDNA cloning and sequencing, it has become apparent that more than 90% of the genes expressed in mammals are alternatively spliced. Thus, alternative splicing is not just the result of mistakes made by the splicing machinery; it is part of the gene expression program that results in multiple gene products from a single gene locus.
Various modes of alternative splicing have been identified, including intron retention, alternative 5′ splice-site selection, alternative 3′ splice-site selection, exon inclusion or skipping, and mutually exclusive selection of the alternative exons, as summarized in FIGURE 19.21. A single primary transcript may undergo more than one mode of alternative splicing. The mutually exclusive exons are normally regulated in a tissue-specific manner. Adding to this complexity, in some cases the ultimate pattern of expression is also dictated by the use of different transcription start points or the generation of alternative 3′ ends.

FIGURE 19.21 Different modes of alternative splicing.
Alternative splicing can affect gene expression in the cell in at least two ways. One way is to create structural diversity of gene products by including or omitting some coding sequences or by creating alternative reading frames for a portion of the gene. This can often modify the functional property of encoded proteins. For example, the CaMKIIδ gene contains three alternatively spliced exons, as shown in FIGURE 19.22. The gene is expressed in almost all cell types and tissues in mammals. When all three alternative exons are skipped, the mRNA encodes a cytoplasmic kinase that phosphorylates a large number of protein substrates. When exon 14 is included, the kinase is transported to the nucleus because exon 14 contains a nuclear localization signal. This allows the kinase to regulate transcription in the nucleus. When both exons 15 and 16 are included, which is normally detected in neurons, the kinase is targeted to the cell membrane, where it can influence specific ion channel activities.

FIGURE 19.22 Alternative splicing of the CaMKIIδ gene: different alternative exons target the kinase to different cellular compartments.
In other cases, the alternatively spliced products exhibit opposite functions. This applies to essentially all genes involved in the regulation of apoptosis; each gene expresses at least two isoforms, one functioning to promote apoptosis and the other protecting cells against apoptosis. It is thought that the isoform ratios of these apoptosis regulators may dictate whether the cell lives or dies.
Alternative splicing may also affect various properties of the mRNA by including or omitting certain regulatory RNA elements, which may significantly alter the half-life of the mRNA. In many cases, the main purpose of alternative splicing may be to cause a certain percentage of primary transcripts to carry a premature stop codon(s) so that those transcripts can be rapidly degraded. This may represent an alternative strategy to transcriptional regulation to control the abundance of specific mRNAs in the cell. This mechanism is used to achieve homeostatic expression for many splicing regulators in specific cell types or tissues. In such regulation, a specific positive splicing regulator may affect its own alternative splicing, resulting in the inclusion of an exon containing a premature stop codon. This siphons a fraction of its mRNA to degradation, thereby reducing the protein concentration. Thus, when the concentration of such positive splicing regulator fluctuates in the cell, its mRNA concentration will be shifted in the opposite direction.
Although many alternative splicing events have been characterized and the biological roles of the alternatively spliced products determined, the best understood example is still the pathway of sex determination in D. melanogaster, which involves interactions between a series of genes in which alternative splicing events distinguish males and females. The pathway takes the form illustrated in FIGURE 19.23, in which the ratio of X chromosomes to autosomes determines the expression of sex lethal (sxl), and changes in expression are passed sequentially through the other genes to doublesex (dsx), the last in the pathway.

FIGURE 19.23 Sex determination in D. melanogaster involves a pathway in which different splicing events occur in females. Blockages at any stage of the pathway result in male development. Illustrated are tra pre-mRNA splicing controlled by the Sxl protein, which blocks the use of the alternative 3′ splice site, and dsx pre-mRNA splicing regulated by both Tra and Tra2 proteins in conjunction with other SR proteins, which positively influence the inclusion of the alternative exon.
The pathway starts with sex-specific splicing of sxl. Exon 3 of the sxl gene contains a termination codon that prevents synthesis of functional protein. This exon is included in the mRNA produced in males but is skipped in females. As a result, only females produce Sxl protein. The protein has a concentration of basic amino acids that resembles other RNA-binding proteins. The presence of Sxl protein changes the splicing of the transformer (tra) gene. Figure 19.23 shows that this involves splicing a constant 5′ site to alternative 3′ sites (note that this mode applies to both sxl and tra splicing, as illustrated). One splicing pattern occurs in both males and females and results in an RNA that has an early termination codon. The presence of Sxl protein inhibits usage of the upstream 3′ splice site by binding to the polypyrimidine tract at its branch site. When this site is skipped, the next 3′ site is used. This generates a female-specific mRNA that encodes a protein.
Thus, Sxl autoregulates the splicing of its own mRNA to ensure its expression in females, and tra produces a protein only in females; like Sxl, Tra protein is a splicing regulator. tra2 has a similar function in females (but is also expressed in the males). The Tra and Tra2 proteins are SR splicing factors that act directly upon the target transcripts. Tra and Tra2 cooperate (in females) to affect the splicing of dsx. In the dsx gene, females splice the 5′ site of intron 3 to the 3′ site of that intron; as a result, translation terminates at the end of exon 4. Males splice the 5′ site of intron 3 directly to the 3′ site of intron 4, thus omitting exon 4 from the mRNA and allowing translation to continue through exon 6. The result of the alternative splicing is that different Dsx proteins are produced in each sex: The male product blocks female sexual differentiation, whereas the female product represses expression of male-specific genes.
19.13 Splicing Can Be Regulated by Exonic and Intronic Splicing Enhancers and Silencers
Alternative splicing is generally associated with weak splice sites, meaning that the splicing signals located at both ends of introns diverge from the consensus splicing signals. This allows these weak splicing signals to be modulated by various trans-acting factors generally known as alternative splicing regulators. However, contrary to common assumptions, these weak splice sites are generally more conserved across mammalian genomes than are constitutive splice sites. This observation is evidence against the notion that alternative splicing might result from splicing mistakes by the splicing machinery and favors the possibility that many alternative splicing events might be evolutionarily conserved to preserve the regulation of gene expression at the level of RNA processing.
The regulation of alternative splicing is a complex process, involving a large number of RNA-binding trans-acting splicing regulators. As illustrated in FIGURE 19.24, these RNA-binding proteins may recognize RNA elements in exons and introns near the alternative splice site and exert positive and negative influence on the selection of the alternative splice site. Those that bind to exons to enhance the selection are positive splicing regulators and the corresponding cis-acting elements are referred to as exonic splicing enhancers (ESEs). SR proteins are among the best characterized ESE-binding regulators. In contrast, some RNA-binding proteins, such as hnRNP A and B, bind to exonic sequences to suppress splice site selection; the corresponding cis-acting elements are thus known as exonic splicing silencers (ESSs). Similarly, many RNA-binding proteins affect splice-site selection through intronic sequences. The corresponding positive and negative cis-acting elements in introns thus are called intronic splicing enhancers (ISEs) or intronic splicing silencers (ISSs).

FIGURE 19.24 Exonic and intronic sequences can modulate splice-site selection by functioning as splicing enhancers or silencers. In general, SR proteins bind to exonic splicing enhancers and the hnRNP proteins (e.g., the A and B families of RNA-binding proteins [RBPs]) bind to exonic silencers. Other RBPs can function as splicing regulators by binding to intronic splicing enhancers or silencers.
Adding to this complexity are the positional effects of many splicing regulators. The best-known examples are the Nova and Fox families of RNA-binding splicing regulators, which can enhance or suppress splice-site selection, depending on where they bind relative to the alternative exon. For example, as illustrated in FIGURE 19.25, binding of both Nova and Fox to intronic sequences upstream of the alternative exon generally results in the suppression of the exon, whereas their binding to intronic sequences downstream of the alternative splicing exon frequently enhances the selection of the exon. Both Nova and Fox are differentially expressed in different tissues, particularly in the brain. Thus, tissue-specific regulation of alternative splicing can be achieved by tissue-specific expression of trans-acting splicing regulators.
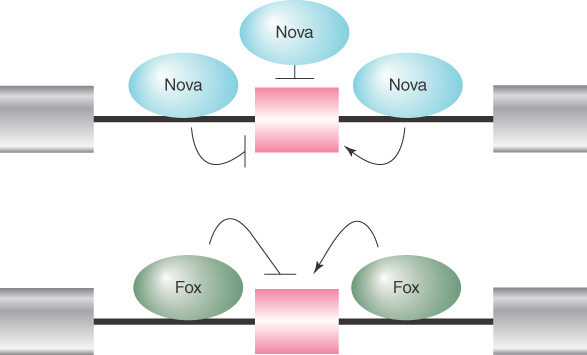
FIGURE 19.25 The Nova and Fox families of RNA-binding proteins can promote or suppress splice site selection in a context-dependent fashion. Binding of Nova to exons and flanking upstream introns inhibits the inclusion of the alternative exon, whereas Nova binding to the downstream flanking intronic sequences promotes the inclusion of the alternative exon. Fox binding to the upstream intronic sequence inhibits the inclusion of the alternative exon, whereas binding of Fox to the downstream intronic sequence promotes the inclusion of the alternative exon.
How a specific alternative splicing event is regulated by various positive and negative splicing regulators is not completely understood. In principle, these splicing regulators function to enhance or suppress the recognition of specific splicing signals by some of the core components of the splicing machinery. The best-understood cases are SR proteins and hnRNA A/B proteins for their positive and negative roles in enhancing or suppressing splice-site recognition, respectively. Binding of SR proteins to ESEs promotes or stabilizes U1 binding to the 5′ splice site and U2AF binding to the 3′ splice site. Thus, spliceosome assembly becomes more efficient in the presence of SR proteins. This role of SR proteins applies to both constitutive and alternative splicing, making SR proteins both essential splicing factors and alternative splicing regulators. In contrast, hnRNP A/B proteins seem to bind to RNA and compete with the binding by SR proteins and other core spliceosome components in the recognition of functional splicing signals.
SR proteins are able to commit a pre-mRNA to the splicing pathway, whereas hnRNP proteins antagonize this process. Given that hnRNP proteins are highly abundant in the nucleus, how do SR proteins effectively compete with hnRNPs to facilitate splicing? Apparently, this is accomplished by the cotranscriptional splicing mechanism inside the nucleus of the cell (see the section earlier in this chapter titled Commitment of Pre-mRNA to the Splicing Pathway). It is thus conceivable that the transcription process can affect alternative splicing. In fact, this has been shown to be the case. Alternative splicing appears to be affected by specific promoters used to drive gene expression, as well as by the rate of transcription during the elongation phase.
Different promoters may attract different sets of transcription factors, which may, in turn, affect transcriptional elongation. Thus, the same mechanism may underlie the influence of promoter usage and transcriptional elongation rate on alternative splicing. The current evidence suggests a kinetic model where a slow transcriptional elongation rate would afford a weak splice site emerging from the elongating Pol II complex sufficient time to pair with the upstream splice site before the appearance of the downstream competing splice site. This model stresses a functional consequence of the coupling between transcription and RNA splicing in the nucleus.
19.14 trans-Splicing Reactions Use Small RNAs
In mechanistic and evolutionary terms, splicing has been viewed as an intramolecular reaction, essentially amounting to a controlled deletion of the intron sequences at the level of RNA. In genetic terms, splicing is expected to occur only in cis. This means that only sequences on the same molecule of RNA should be spliced together.
The upper part of FIGURE 19.26 shows the usual situation. The introns can be removed from each RNA molecule, allowing the exons of that RNA molecule to be spliced together, but there is no intermolecular splicing of exons between different RNA molecules. Although we know that trans-splicing between pre-mRNA transcripts of the same gene does occur, it must be exceedingly rare, because if it were prevalent the exons of a gene would be able to complement one another genetically instead of belonging to a single complementation group.

FIGURE 19.26 Splicing usually occurs only in cis between exons carried on the same physical RNA molecule, but trans-splicing can occur when special constructs that support base pairing between introns are made.
Some manipulations can generate trans-splicing. In the example illustrated in the lower part of Figure 19.26, complementary sequences were introduced into the introns of two RNAs. Base pairing between the complements should create an H-shaped molecule. This molecule could be spliced in cis, to connect exons that are covalently connected by an intron, or it could be spliced in trans, to connect exons of the juxtaposed RNA molecules. Both reactions occur in vitro.
Another situation in which trans-splicing is possible in vitro occurs when substrate RNAs are provided in the form of one containing a 5′ splice site and the other containing a 3′ splice site together with appropriate downstream sequences (which may be either the next 5′ splice site or a splicing enhancer). In effect, this mimics splicing by exon definition and shows that in vitro it is not necessary for the left and right splice sites to be on the same RNA molecule.
These results show that there is no mechanistic impediment to trans-splicing. They exclude models for splicing that require processive movement of a spliceosome along the RNA. It must be possible for a spliceosome to recognize the 5′ and 3′ splice sites of different RNAs when they are in close proximity.
Although trans-splicing is rare in multicellular eukaryotes, it occurs as the primary mechanism to process precursor RNA into mature, translatable mRNAs in some organisms, such as trypanosomes and nematodes. In trypanosomes, all genes are expressed as polycistronic transcripts, like those in bacteria. However, the transcribed RNA cannot be translated without a 37-nucleotide leader brought in by trans-splicing to convert a polycistronic RNA into individual monocistronic mRNAs for translation. The leader sequence is not encoded upstream of the individual transcription units, though. Instead, it is transcribed into an independent RNA, carrying additional sequences at its 3′ end, from a repetitive unit located elsewhere in the genome. FIGURE 19.27 shows that this RNA carries the leader sequence followed by a 5′ splice-site sequence. The sequences encoding the mRNAs carry a 3′ splice site just preceding the sequence found in the mature mRNA.

FIGURE 19.27 The SL RNA provides an exon that is connected to the first exon of an mRNA by trans-splicing. The reaction involves the same interactions as nuclear cis-splicing but generates a Y-shaped RNA instead of a lariat.
When the leader and the mRNA are connected by a trans-splicing reaction, the 3′ region of the leader RNA and the 5′ region of the mRNA in effect comprise the 5′ and 3′ halves of an intron. When splicing occurs, a 2′–5′ link forms by the usual reaction between the GU of the 5′ intron and the branch sequence near the AG of the 3′ intron. The two parts of the intron are covalently linked, but generate a Y-shaped molecule instead of a lariat.
The RNA that donates the 5′ exon for trans-splicing is called the spliced leader RNA (SL RNA). The SL RNAs, which are 100 nucleotides in length, can fold into a common secondary structure that has three stem-loops and a single-stranded region that resembles the Sm-binding site. The SL RNAs therefore exist as snRNPs that count as members of the Sm snRNP class. During the trans-splicing reaction, SL RNA becomes part of the spliced product replacing the original cap and leader (called an outron), as illustrated in the upper panel of FIGURE 19.28. Like other snRNPs involved in splicing (except U6), SL RNA carries a trimethylated cap, which is recognized by the variant cap-binding factor eIF4E to facilitate translation.

FIGURE 19.28 The SL RNA adds a leader to facilitate translation. Coupled with the cleavage and polyadenylation reactions, the addition of the SL RNA is also used to convert polycistronic transcripts to monocistronic units.
In Caenorhabditis elegans, about 70% of genes are processed by the trans-splicing mechanism, which can be further divided into two classes of genes. One class produces monocistronic transcripts that are processed by both cis- and trans-splicing. In these cases, cis-splicing is used to remove internal intronic sequences, and then trans-splicing is employed to provide the 22-nucleotide leader sequence derived from the SL RNA for translation. The other class is polycistronic. In these cases, trans-splicing is used to convert the polycistronic transcripts into monocistronic transcripts in addition to providing the SL leader sequence for their translation, as illustrated in the bottom panel of Figure 19.28.
C. elegans has two types of SL RNA. SL1 RNA (the first to be discovered) is only used to remove the 5′ ends of pre-mRNAs transcribed from monocistronic genes. How does the SL RNA find the 3′ splice site to initiate trans-splicing, and in doing so, how does trans-splicing avoid competition or interference with cis-splicing? The ability to target a functional 3′ splice site is provided by the proteins as part of the SL snRNP. For example, purified SL snRNP from Ascaris, a parasitic nematode, contains two specific proteins, one of which (SL-30kD) can directly interact with the BPB protein at the 3′ splice site. The SL1 RNA is only trans-spliced to the first 5′ untranslated region, and does not interfere with downstream cis-splicing events. This is because only the 5′ untranslated region contains a functional 3′ splice site, but it does not have the upstream 5′ splice site to pair with the downstream 3′ splice site.
The SL2 RNA is used in most cases to process polycistronic transcripts that are separated by a 100-nucleotide spacer sequence between the two adjacent gene units. In a small fraction of genes where the two adjacent gene units are linked without any spacer sequences, the SL1 RNA is used to break them up.
During processing of these polycistronic transcripts by either of the SL snRNAs, the trans-splicing reaction is tightly coupled with the cleavage and polyadenylation reactions at the end of each gene unit. Such coupling appears to be facilitated by direct protein–protein interactions between the SL2 snRNP and the cleavage stimulatory factor CstF that binds to the U-rich sequence downstream of the AAUAAA signal (see the next section, The 3′ Ends of mRNAs Are Generated by Cleavage and Polyadenylation). These mechanisms allow related genes to be coregulated at the level of transcription (because they are transcribed as polycistronic transcripts) and individually regulated after transcription (because individual gene units are separated as a result of RNA processing).
19.15 The 3′ Ends of mRNAs Are Generated by Cleavage and Polyadenylation
It is not clear whether RNA polymerase II actually engages in a termination event at a specific site. It is possible that its termination is only loosely specified. In some transcription units, termination occurs more than 1,000 bp downstream of the site, corresponding to the mature 3′ end of the mRNA (which is generated by cleavage at a specific sequence). Instead of using specific terminator sequences, the enzyme ceases RNA synthesis within multiple sites located in rather long “terminator regions.” The nature of the individual termination sites is largely unknown.
The mature 3′ ends of Pol II transcribed mRNAs are generated by cleavage followed by polyadenylation. Addition of poly(A) to nuclear RNA can be prevented by the analog 3′–deoxyadenosine, which is also known as cordycepin. Although cordycepin does not stop the transcription of nuclear RNA, its addition prevents the appearance of mRNA in the cytoplasm. This shows that polyadenylation is necessary for the maturation of mRNA from nuclear RNA. The poly(A) tail is known to protect the mRNA from degradation by 3′–5′ exonucleases. In yeast, it is suggested that the poly(A) tail also plays a role in facilitating nuclear export of matured mRNA and in cap stability.
Generation of the 3′ end is illustrated in FIGURE 19.29. The RNA polymerase transcribes past the site corresponding to the 3′ end, and sequences in the RNA are recognized as targets for an endonucleolytic cut followed by polyadenylation. RNA polymerase continues transcription after the cleavage, but the 5′ end that is generated by the cleavage is unprotected, which signals transcriptional termination (see the next section, 3′ mRNA End Processing Is Critical for Termination of Transcription).

FIGURE 19.29 The sequence AAUAAA is necessary for cleavage to generate a 3′ end for polyadenylation.
The site of cleavage/polyadenylation in most pre-mRNAs is flanked by two cis-acting signals: an upstream AAUAAA motif, which is usually located 11 to 30 nucleotides from the site, and a downstream U-rich or GU-rich element. The AAUAAA is needed for cleavage and polyadenylation because deletion or mutation of the AAUAAA hexamer prevents generation of the polyadenylated 3′ end (though in plants and fungi there can be considerable variation from the AAUAAA motif).
The development of a system in which polyadenylation occurs in vitro opened the route to analyzing the reactions. The formation and functions of the complex that undertakes 3′ processing are illustrated in FIGURE 19.30. Generation of the proper 3′ terminal structure depends on the cleavage and polyadenylation specific factor (CPSF), which contains multiple subunits. One of the subunits binds directly to the AAUAAA motif and to the cleavage stimulatory factor (CstF), which is also a multicomponent complex. One of these components binds directly to a downstream GU-rich sequence. CPSF and CstF can enhance each other in recognizing the polyadenylation signals. The specific enzymes involved are an endonuclease (the 73-kD subunit of CPSF) to cleave the RNA and a poly(A) polymerase (PAP) to synthesize the poly(A) tail.
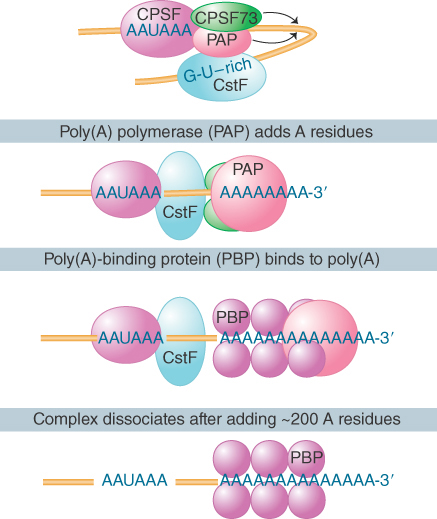
FIGURE 19.30 The 3′ processing complex consists of several activities. CPSF and CstF each consist of several subunits; the other components are monomeric. The total mass is more than 900 kD.
PAP has nonspecific catalytic activity. When it is combined with the other components, the synthetic reaction becomes specific for RNA containing the sequence AAUAAA. The polyadenylation reaction passes through two stages. First, a rather short oligo(A) sequence (about 10 residues) is added to the 3′ end. This reaction is absolutely dependent on the AAUAAA sequence, and poly(A) polymerase performs it under the direction of the specificity factor. In the second phase, the nuclear poly(A) binding protein (PABP II) binds the oligo(A) tail to allow extension of the poly(A) tail to the full length of about 200 residues. The poly(A) polymerase by itself adds A residues individually to the 3′ position. Its intrinsic mode of action is distributive; it dissociates after each nucleotide has been added. However, in the presence of CPSF and PABP II it functions processively to extend an individual poly(A) chain. After the polyadenylation reaction, PABP II binds stoichiometrically to the poly(A) stretch, which by some unknown mechanism limits the action of poly(A) polymerase to about 200 additions of A residues.
Upon export of mature mRNAs out of the nucleus, the poly(A) tail is bound by the cytoplasmic poly(A) binding protein (PABP I). PABP I not only protects the mRNA from degradation by the 3′ to 5′ exonucleases but also binds to the translation initiation factor eIF4G to facilitate translation of the mRNA. Thus, the mRNA in the cytoplasm forms a closed loop in which a protein complex contains both the 5′ and 3′ ends of the mRNA (see the Translation chapter). Polyadenylation therefore affects both stability and initiation of translation in the cytoplasm.
During embryonic development of Xenopus, polyadenylation is carried out in the cytoplasm to provide a maternal control in early embryogenesis. Some stored maternal mRNAs may either be polyadenylated by the poly(A) polymerase in the cytoplasm to stimulate translation or deadenylated to terminate translation. A specific AU-rich cis-acting element (CPE) in the 3′ tail directs the meiotic maturation-specific polyadenylation in the cytoplasm to activate translation of some specific maternal mRNAs. To regulate mRNA degradation, at least two types of cis-acting sequences in the 3′ tail can trigger mRNA deadenylation: embryonic deadenylation element (EDEN), a 17-nucleotide sequence, and ARE elements, which are AU rich, usually containing tandem repeats of AUUUA. A poly(A)-specific RNAase (PARN) is involved in mRNA degradation in the cytoplasm. Of course, mRNA deadenylation is always in competition with mRNA stabilization, which together determine the half-life of individual mRNAs in the cell (see the chapter titled mRNA Stability and Localization).
19.16 3′ mRNA End Processing Is Critical for Termination of Transcription
Information about the termination reaction for eukaryotic RNA polymerases is less detailed than our knowledge of initiation. The 3′ ends of RNAs can be generated in two ways. Some RNA polymerases terminate transcription at a defined terminator sequence in DNA, as shown in FIGURE 19.31. RNA polymerase III appears to use this strategy by having a discrete oligo(dT) sequence to signal the release of Pol III for transcription termination.
FIGURE 19.31 Transcription by Pol III and Pol I uses specific terminators to end transcription.
For RNA polymerase I, the sole product of transcription is a large precursor that contains the sequences of the major rRNA. Termination occurs at two discrete sites (T1 and T2) downstream of the mature 3′ end. These terminators are recognized by a specific DNA-binding Reb1 in yeast or TTF1 in mice. Pol I termination is also associated with a cleavage event mediated by the endonuclease Rnt1p, which cleaves the nascent RNA about 15 to 50 bases downstream from the 3′ end of processed 28S rRNA (see the section later in this chapter titled Production of rRNA Requires Cleavage Events and Involves Small RNAs). In this regard, Pol I termination is mechanistically related to Pol II termination in that both processes may involve an RNA cleavage event.
In contrast to Pol I and Pol III termination, RNA polymerase II usually does not show discrete termination, but continues to transcribe about 1.5 kb past the site corresponding to the 3′ end. The cleavage event at the polyadenylation site provides a trigger for termination by RNA polymerase II, as shown in FIGURE 19.32.

FIGURE 19.32 3′ end formation of Pol II transcripts facilitates transcriptional termination.
Two models have been proposed for Pol II termination. The allosteric model suggests that RNA cleavage at the polyadenylation site may trigger some conformational changes in both the Pol II complex and local chromatin structure. This may be induced by factor exchanges during the polyadenylation reaction, resulting in Pol II pausing and then release from template DNA.
An alternative model known as the torpedo model proposes that a specific exonuclease binds to the 5′ end of the RNA that is continuing to be transcribed after cleavage. It degrades the RNA faster than it is synthesized, so that it catches up with RNA polymerase. It then interacts with ancillary proteins that are bound to the carboxy-terminal domain of the polymerase; this interaction triggers the release of RNA polymerase from DNA, causing transcription to terminate. This model explains why the termination sites for RNA polymerase II are not well defined, but may occur at varying locations within a long region downstream of the site corresponding to the 3′ end of the RNA. The major experimental evidence for the torpedo model is the role of the nuclear 5′–3′ exonuclease Rat1 in yeast or Xrn2 in mammals. Deletion of the gene frequently causes readthrough transcription to the next gene. However, in some experimental systems, mutation of the AAUAAA signal to impair cleavage at the natural polyadenylation site does not necessarily trigger the release of the transcribing Pol II and cause transcriptional readthrough. This evidence, coupled with some local changes in chromatin structure, thus favors the allosteric model.
It has become apparent that the allosteric and torpedo models are not necessarily mutually exclusive; both may reflect some critical aspects associated with Pol II transcriptional termination. By either or both mechanisms, it is clear that transcriptional termination by Pol II is tightly coupled with the 3′ end formation for most mRNAs in eukaryotic cells.
19.17 The 3′ End Formation of Histone mRNA Requires U7 snRNA
Biogenesis of the canonical histones is primarily controlled by the regulation of histone mRNA abundance during the cell cycle. At this G1/S transition, the abundance of histone mRNAs is increased more than 30-fold due to elevated transcription; this process is regulated by the cyclin E/Cdk2 complex (see the chapter titled Replication Is Connected to the Cell Cycle). The rise in histone mRNAs is followed by a rapid decay of histone mRNAs at the end of S phase.
Canonical histone mRNAs are not polyadenylated (except in S. cerevisiae). (Note that some of the histone variants, such as H3.3, are not cell-cycle regulated and are polyadenylated; see the Chromatin chapter.) The formation of their 3′ ends is therefore different from that of the coordinated cleavage/polyadenylation reaction; it depends upon a highly conserved stem-loop structure located 14 to 50 bases downstream from the termination codon and a histone downstream element (HDE) located about 15 nucleotides downstream of the stem-loop. Cleavage occurs between the stem-loop and HDE, leaving five bases downstream of the stem-loop. Mutations that prevent formation of the duplex stem of the stem-loop prevent formation of the end of the RNA. Secondary mutations that restore duplex structure (though not necessarily the original sequence) restore 3′ end formation. This indicates that formation of the secondary structure is more important than the exact sequence.
The reaction forming the histone 3′ end is shown in FIGURE 19.33. Two factors are required to specify the cleavage reaction: The stem-loop binding protein (SLBP) recognizes the stem-loop structure, and the 5′ end of U7 snRNA base pairs with a purine-rich sequence within HDE. U7 snRNP is a minor snRNP consisting of the 63-nucleotide U7 snRNA and a set of several proteins related to snRNPs involved in mRNA splicing (see the section earlier in this chapter titled snRNAs Are Required for Splicing). Unique to U7 snRNP are two Sm-like proteins, LSM10 and LSM11, which replace Sm D1 and D2 in the splicing snRNPs. Prevention of base pairing between U7 snRNA and HDE impairs 3′ processing of the histone mRNAs, and compensatory mutations in U7 snRNA that restore complementarity restore 3′ processing. This indicates that U7 snRNA functions by base pairing with the histone mRNAs.

FIGURE 19.33 Generation of the 3′ end of histone h3 mRNA depends on a conserved hairpin and a sequence that base pairs with U7 snRNA.
Cleavage to generate a 3′ terminus occurs at a fixed distance from the site recognized by U7 snRNA, which suggests that the snRNA is involved in defining the cleavage site. The factor responsible for cleavage is a specific cleavage and polyadenylation specificity factor (CPSF73). Thus, this member of the metallo-β-lactamase family plays a key role in 3′ end formation for both polyadenylated mRNAs and nonpolyadenylated histone mRNAs. Several other proteins have been identified as important for histone 3′ end formation, including CPSF100 and Symplekin, but their specific roles remain to be defined. These additional proteins may provide scaffold functions to stabilize the 3′-end–processing complex.
Interestingly, disruption of U7 base pairing with the target sequences in histone genes or siRNA-mediated depletion of other components involved in the formation of the histone 3′ end all result in transcriptional readthrough and polyadenylation by using a poly(A) signal downstream from the DHE. Thus, similar to the role of mRNA cleavage/polyadenylation in Pol II transcriptional termination on most protein-coding genes, U7-mediated RNA cleavage during 3′ end formation appears to be critical for transcriptional termination on histone genes.
19.18 tRNA Splicing Involves Cutting and Rejoining in Separate Reactions
Most splicing reactions depend on short consensus sequences and occur by transesterification reactions in which breaking and forming bonds are coordinated. The splicing of tRNA genes is achieved by a different mechanism that relies upon separate cleavage and ligation reactions.
Some 59 of the 272 nuclear tRNA genes in the yeast S. cerevisiae are interrupted. Each has a single intron that is located just one nucleotide beyond the 3′ side of the anticodon. The introns vary in length from 14 to 60 bases. Those in related tRNA genes are related in sequence, but the introns in tRNA genes representing different amino acids are unrelated. No consensus sequence exists that could be recognized by the splicing enzymes. This is also true of interrupted nuclear tRNA genes of plants, amphibians, and mammals.
All the introns include a sequence that is complementary to the anticodon of the tRNA. This creates an alternative conformation for the anticodon arm in which the anticodon is base paired to form an extension of the usual arm. An example is shown in FIGURE 19.34. Only the anticodon arm is affected—the rest of the molecule retains its usual structure.

FIGURE 19.34 The intron in yeast tRNAPhe base pairs with the anticodon to change the structure of the anticodon arm. Pairing between an excluded base in the stem and the intron loop in the precursor may be required for splicing.
The exact sequence and size of the intron are not important. Most mutations in the intron do not prevent splicing. Splicing of tRNA depends principally on recognition of a common secondary structure in tRNA rather than a common sequence of the intron. Regions in various parts of the molecule are important, including the stretch between the acceptor arm and D arm, in the TψC arm, and especially in the anticodon arm. This is reminiscent of the structural demands placed on tRNA for translation (see the Translation chapter).
The intron is not entirely irrelevant, however. Pairing between a base in the intron loop and an unpaired base in the stem is required for splicing. Mutations at other positions that influence this pairing (e.g., to generate alternative patterns for pairing) influence splicing. The rules that govern availability of tRNA precursors for splicing resemble the rules that govern recognition by aminoacyl-tRNA synthetases (see the chapter titled Using the Genetic Code).
In a temperature-sensitive mutant of yeast that fails to remove the introns, the interrupted precursor RNAs accumulate in the nucleus. The precursors can be used as substrates for a cell-free system extracted from wild-type cells. The splicing of the precursor can be followed by virtue of the resulting size reduction of the RNA product. This is seen by the change in position of the band on gel electrophoresis, as illustrated in FIGURE 19.35. The reduction in size can be accounted for by the appearance of a band representing the intron.

FIGURE 19.35 Splicing of yeast tRNA in vitro can be followed by assaying the RNA precursor and products by gel electrophoresis.
The cell-free extract can be fractionated by assaying the ability to splice the tRNA. The in vitro reaction requires ATP. Characterizing the reactions that occur with and without ATP shows that the two separate stages of the reaction are catalyzed by different enzymes:
The first step does not require ATP. It involves phosphodiester bond cleavage by an atypical nuclease reaction. It is catalyzed by an endonuclease.
The second step requires ATP and involves bond formation; it is a ligation reaction, and the responsible enzyme activity is described as an RNA ligase.
Splicing of pre-tRNA to remove introns is essential in all organisms, but different organisms use different mechanisms to accomplish pre-tRNA splicing. In bacteria, introns in pre-tRNAs are self-spliced as group I or group II autocatalytic introns. In archaea and eukaryotes, pre-tRNA splicing involves the action of three enzymes: (1) an endonuclease that recognizes and cleaves the precursor at both ends of the intron, (2) a ligase that joins the tRNA exons, (3) and a 2′-phosphotransferase that removes the 2′-phosphate on spliced tRNA.
The yeast endonuclease is a heterotetrameric protein consisting of two catalytic subunits, Sen34 and Sen2, and two structural subunits, Sen54 and Sen15. Its activities are illustrated in FIGURE 19.36. The related subunits, Sen34 and Sen2, cleave the 3′ and 5′ splice sites, respectively. Subunit Sen54 may determine the sites of cleavage by “measuring” distance from a point in the tRNA structure. This point is in the elbow of the (mature) L-shaped structure. The role of subunit Sen15 is not known, but its gene is essential in yeast. The base pair that forms between the first base in the anticodon loop and the base preceding the 3′ splice site is required for 3′ splice-site cleavage.

FIGURE 19.36 The 3′ and 5′ cleavages in S. cerevisiae pre-tRNA are catalyzed by different subunits of the endonuclease. Another subunit may determine location of the cleavage sites by measuring distance from the mature structure. The AI base pair is also important.
An interesting insight into the evolution of tRNA splicing is provided by the endonucleases of archaea. These are homodimers or homotetramers, in which each subunit has an active site (although only two of the sites function in the tetramer) that cleaves one of the splice sites. The subunit has sequences related to the sequences of the active sites in the Sen34 and Sen2 subunits of the yeast enzyme. The archaeal enzymes recognize their substrates in a different way, though. Instead of measuring distance from particular sequences, they recognize a structural feature called the bulge-helix-bulge. FIGURE 19.37 shows that cleavage occurs in the two bulges. Thus, the origin of splicing of tRNA precedes the separation of the archaea and the eukaryotes. If it originated by insertion of the intron into tRNAs, this must have been a very ancient event.

FIGURE 19.37 Archaeal tRNA-splicing endonuclease cleaves each strand at a bulge in a bulge-helix-bulge motif.
The overall tRNA splicing reaction is summarized in FIGURE 19.38. The products of cleavage are a linear intron and two half-tRNA molecules. These intermediates have unique ends. Each 5′ terminus ends in a hydroxyl group; each 3′ terminus ends in a 2′,3′-cyclic phosphate group.
The two half-tRNAs base pair to form a tRNA-like structure. When ATP is added, the second reaction occurs, which is catalyzed by a single enzyme with multiple enzymatic activities:
Cyclic phosphodiesterase activity. Both of the unusual ends generated by the endonuclease must be altered prior to the ligation reaction. The cyclic phosphate group is first opened to generate a 2′-phosphate terminus.
Kinase activity. The product has a 2′-phosphate group and a 3′–OH group. The 5′–OH group generated by the endonuclease must be phosphorylated to give a 5′-phosphate. This generates a site in which the 3′–OH is next to the 5′-phosphate.
Ligase activity. Covalent integrity of the polynucleotide chain is then restored by ligase activity. The spliced molecule is now uninterrupted, with a 5′–3′ phosphate linkage at the site of splicing, but it also has a 2′-phosphate group marking the event on the spliced tRNA. In the last step, this surplus group is removed by a phosphatase, which transfers the 2′-phosphate to NDP to form ADP ribose 1′,2′-cyclic phosphate.

FIGURE 19.38 Splicing of tRNA requires separate nuclease and ligase activities. The exon–intron boundaries are cleaved by the nuclease to generate 2′,3′-cyclic phosphate and 5′–OH termini. The cyclic phosphate is opened to generate 3′–OH and 2′-phosphate groups. The 5′–OH is phosphorylated. After releasing the intron, the tRNA half molecules fold into a tRNA-like structure that now has a 3′–OH, 5′–P break. This is sealed by a ligase.
The tRNA splicing pathway described here is slightly different from that of vertebrates. Before the action of the RNA ligases, a cyclase generates a 2′,3′ cyclic terminus from the initial 3′-phosphomonoester terminus via a 3′ adenylated intermediate. The RNA ligase is also different from that in yeast because it can join a 2′,3′-cyclic phosphodiester and a 5′–OH to form a conventional 3′,5′-phosphodiester bond, but these reactions leave no extra 2′-phosphate.
19.19 The Unfolded Protein Response Is Related to tRNA Splicing
An unusual splicing system that is related to tRNA splicing is the unfolded protein response (UPR) pathway conserved in eukaryotes. As summarized in FIGURE 19.39, the accumulation of unfolded proteins in the lumen of the endoplasmic reticulum (ER) triggers the UPR pathway. This leads to increased transcription of genes encoding chaperones that assist protein folding in the ER. A signal must therefore be transmitted from the lumen of the ER to the nucleus.

FIGURE 19.39 The unfolded protein response occurs by activating special splicing of HAC1 mRNA to produce a transcription factor that recognizes the UPRE.
The sensor that activates the pathway is the inositol-requiring protein Ire1, which is localized in the ER and/or inner nuclear membrane. The N-terminal domain of Ire1 lies in the lumen of the ER where it detects the presence of unfolded proteins, presumably by binding to exposed motifs. The C-terminal half of Ire1 is located in either the cytoplasm or nucleus (because of the continuous membrane of the ER and the nucleus) and exhibits both Ser/Thr kinase activity and a specific endonuclease activity. Binding of unfolded proteins causes aggregation of Ire1 monomers on the ER membrane, leading to the activation of the C-terminal domain on the other side of the membrane by autophosphorylation.
The activated C-terminal endonuclease has, at present, only one (though important) substrate, which is the mRNA encoding the UPR-specific transcription factor Hac1 in yeast (Xbp1 in vertebrates). Under normal conditions, when the UPR pathway is not activated, HAC1 mRNA contains a 252-nucleotide intron (Xbp1 contains a 26-nucleotide intron). The intron in HAC1 prevents the mRNA from being translated into a functional protein in yeast, whereas in mammalian cells the intron in Xbp1 allows translation, but the protein is rapidly degraded by the proteosome. Unusual splicing components are involved in processing this intron. The activated Ire1 endonuclease acts directly on HAC1 mRNA (Xbp1 mRNA in vertebrates) to cleave the two splicing junctions, leaving 2′,3′-cyclic phosphate at the 3′ end of the 5′ exon and 5′–OH at the 5′ end of the 3′ exon. The two junctions are then ligated by the tRNA ligase that acts in the tRNA-splicing pathway. Thus, the entire pathway for processing HAC1 (Xbp1) pre-mRNA resembles the pre-tRNA pathway.
Important differences exist between the two pathways, however. Ire1 and tRNA endonuclease share no sequence homology or subunit composition. The endonuclease activity of IreI is highly regulated in the ER and has only one substrate (HAC1 pre-mRNA). In contrast, tRNA endonuclease has many substrates, all with common tRNA folding, with little preference for sequences surrounding the splice sites.
By using such a tRNA-like pathway to remove the intron in the HAC1 (Xbp1) mRNA, the mature mRNA can be translated to produce a potent basic-leucine zipper (bZIP) transcription factor to bind to a common motif (UPRE) in the promoter of many downstream genes. The gene products protect the cell by increasing the expression of proteins to assist protein folding.
If the UPR system is overwhelmed by unfolded proteins, the activated kinase domain of Ire1 binds to the TRAF2 adaptor molecule in the cytoplasm to activate the apoptosis pathway and kill the cell. Thus, the cell uses an unusual tRNA-processing strategy to respond to unfolded proteins. However, there is no apparent relationship between the Ire1 endonuclease and the tRNA-splicing endonuclease, so it is not obvious how this specialized system would have evolved.
19.20 Production of rRNA Requires Cleavage Events and Involves Small RNAs
The major rRNAs are synthesized as part of a single primary transcript that is processed by cleavage and trimming events to generate the mature products. The precursor contains the sequences of the 18S, 5.8S, and 28S rRNAs. (The nomenclature of different ribosomal RNAs is based on early sedimentation studies conducted on sucrose gradients in the 1970s.) In multicellular eukaryotes, the precursor is named for its sedimentation rate as 45S RNA. In unicellular/oligocellular eukaryotes it is smaller (35S in yeast).
The mature rRNAs are released from the precursor by a combination of cleavage events and trimming reactions to remove external transcribed spacers (ETSs) and internal transcribed spacers (ITSs). FIGURE 19.40 shows the general pathway in yeast. The order of events can vary, but basically similar reactions are involved in all eukaryotes. Most of the 5′ ends are generated directly by a cleavage event. Most of the 3′ ends are generated by cleavage followed by a 3′–5′ trimming reaction. These processes are specified by many cis-acting RNA motifs in ETSs and ITSs and are acted upon by more than 150 processing factors.

FIGURE 19.40 Mature eukaryotic rRNAs are generated by cleavage and trimming events from a primary transcript.
Many ribonucleases have been implicated in processing rRNA, including some specific components of the exosome, which is an assembly of several exonucleases that also participates in mRNA degradation (see the mRNA Stability and Localization chapter). Mutations in individual enzymes usually do not prevent processing, which suggests that their activities are redundant and that different combinations of cleavages can be used to generate the mature molecules.
Multiple copies of the transcription unit for the rRNAs are always available. The copies are organized as tandem repeats (see the Clusters and Repeats chapter). The genes encoding rRNAs are transcribed by RNA polymerase I in the nucleolus. In contrast, 5S RNA is transcribed from separate genes by RNA polymerase III. In general, the 5S genes are clustered, but are separated from the genes for the major rRNAs.
In bacteria, the organization of the precursor differs. The sequence corresponding to 5.8S rRNA forms the 5′ end of the large (23S) rRNA; that is, no processing occurs between these sequences. FIGURE 19.41 shows that the precursor also contains the 5S rRNA and one or two tRNAs. In Escherichia coli, the seven rrn operons are dispersed around the genome; four rrn loci contain one tRNA gene between the 16S and 23S rRNA sequences, and the other rrn loci contain two tRNA genes in this region. Additional tRNA genes may or may not be present between the 5S sequence and the 3′ end. Thus, the processing reactions required to release the products depend on the content of the particular rrn locus.

FIGURE 19.41 The rrn operons in E. coli contain genes for both rRNA and tRNA. The exact lengths of the transcripts depend on which promoters (P) and terminators (t) are used. Each RNA product must be released from the transcript by cuts on either side.
In prokaryotic and eukaryotic rRNA processing, both processing factors and ribosomal proteins (and possibly other proteins) bind to the precursor so that the substrate for processing is not the free RNA but rather a ribonucleoprotein complex. Like pre-mRNA processing, rRNA processing takes place cotranscriptionally. As a result, the processing factors are intertwined with ribosomal proteins in building the ribosomes, instead of first processing and then stepwise assembly on processed rRNAs.
Processing and modification of rRNA requires a class of small RNAs called small nucleolar RNAs (snoRNAs). The S. cerevisiae and vertebrate genomes have hundreds of snoRNAs. Some of these snoRNAs are encoded by individual genes; others are expressed from polycistrons; and many are derived from introns of their host genes. These snoRNAs themselves undergo complex processing and maturation steps. Some snoRNAs are required for cleavage of the precursor to rRNA; one example is U3 snoRNA, which is required for the first cleavage event. The U3-containing complex corresponds to the “terminal knobs” at the 5′ end of nascent rRNA transcripts, which are visible under an electron microscope. We do not know what role the snoRNA plays in cleavage. It could be required to pair with specific rRNA sequences to form a secondary structure that is recognized by an endonuclease.
Two groups of snoRNAs are required for the modifications that are made to bases in the rRNA. The members of each group are identified by very short conserved sequences and common features of secondary structure.
The C/D group of snoRNAs is required for adding a methyl group to the 2′ position of ribose. There are more than 100 2′-O-methyl groups at conserved locations in vertebrate rRNAs. This group takes its name from two short, conserved sequence motifs called boxes C and D. Each snoRNA contains a sequence near the D box that is complementary to a region of the 18S or 28S rRNA that is methylated. Loss of a particular snoRNA prevents methylation in the rRNA region to which it is complementary.
FIGURE 19.42 shows that the snoRNA base pairs with the rRNA to create the duplex region that is recognized as a substrate for methylation. Methylation occurs within the region of complementarity at a position that is fixed five bases on the 5′ side of the D box. It is likely that each methylation event is specified by a different snoRNA; about 40 snoRNAs have been implicated in this modification. Each C+D box snoRNA is associated with three proteins: Nop1 (fibrillarin in vertebrates), Nop56, and Nop58. The methylase(s) have not been fully characterized, although the major snoRNP protein Nop1/fibrillarin is structurally similar to methyltransferases.

FIGURE 19.42 A snoRNA base pairs with a region of rRNA that is to be methylated.
Another group of snoRNAs is involved in base modification by converting uridine to pseudouridine. About 50 residues in yeast rRNAs and about 100 in vertebrate rRNAs are modified by pseudouridination. The pseudouridination reaction is shown in FIGURE 19.43, in which the N1 bond from uridylic acid to ribose is broken, the base is rotated, and C5 is rejoined to the sugar.
FIGURE 19.43 Uridine is converted to pseudouridine by replacing the N1-sugar bond with a C5-sugar bond and rotating the base relative to the sugar.
Pseudouridine formation in rRNA requires the H/ACA group of about 20 snoRNAs. They are named for the presence of an ACA triplet three nucleotides from the 3′ end and a partially conserved sequence (the H box) that lies between two stem-loop hairpin structures. Each of these snoRNAs has a sequence complementary to rRNA within the stem of each hairpin. FIGURE 19.44 shows the structure that would be produced by pairing with the rRNA. Each pairing region has two unpaired bases, one of which is a uridine that is converted to pseudouridine.

FIGURE 19.44 H/ACA snoRNAs have two short, conserved sequences and two hairpin structures, each of which has regions in the stem that are complementary to rRNA. Pseudouridine is formed by converting an unpaired uridine within the complementary region of the rRNA.
The H/ACA snoRNAs are associated with four specific nucleolar proteins: Cbf5 (dyskerin in vertebrates), Nhp2, Nop10, and Gar1. Importantly, Cbf5/dyskerin is structurally similar to known pseudouridine synthases, and thus it likely provides the enzymatic activity in the snoRNA-guided pseudouridination reaction. Many snoRNAs are also used to guide base modifications in tRNAs as well as in snRNAs involved in pre-mRNA splicing, which are critical for their functions in prospective reactions. However, a large number of snoRNAs do not have apparent targets. These snoRNAs are called orphan RNAs. The existence of these orphan RNAs indicates that many biological processes may use RNA-guided mechanisms to functionally modify other expressed RNAs in a more diverse fashion than we currently understand.
Summary
Splicing accomplishes the removal of introns and the joining of exons into the mature sequence of RNA. Four types of reactions have been identified, as distinguished by their requirements in vitro and the intermediates that they generate. The systems include eukaryotic nuclear introns, group I and group II introns, and tRNA introns. Each reaction involves a change of organization within an individual RNA molecule, and is therefore a cis-acting event.
Pre-mRNA splicing follows preferred but not obligatory pathways. Only very short consensus sequences are necessary; the rest of the intron appears largely irrelevant. However, both exonic and intronic sequences can exert positive or negative influence on the selection of the nearby splice site. All 5′ splice sites are probably equivalent, as are all 3′ splice sites. The required sequences are given by the GU-AG rule, which describes the ends of the intron. The UACUAAC branch site of yeast, or a less well conserved consensus in mammalian introns, is also required. The reaction with the 5′ splice site involves formation of a lariat that joins the GU end of the intron via a 2′–5′ linkage to the A at position 6 of the branch site. The 3′–OH end of the exon then attacks the 3′ splice site, so that the exons are ligated and the intron is released as a lariat. Lariat formation is responsible for choice of the 3′ splice site. Both reactions are transesterifications in which phosphodiester bonds are conserved. Several stages of the reaction require hydrolysis of ATP, probably to drive conformational changes in the RNA and/or protein components. Alternative splicing patterns are caused by protein factors that either facilitate use of a new site or that block use of the default site.
Pre-mRNA splicing requires formation of a spliceosome—a large particle that assembles the consensus sequences into a reactive conformation. The spliceosome forms by the process of intron definition, involving recognition of the 5′ splice site, branch site, and 3′ splice site. This applies to small introns, like those in yeast. If, however, introns are large, like those in vertebrates, recognition of the splice sites first follows the process of exon definition, involving the interactions across the exon between the 3′ splice site and the downstream 5′ splice site. This is then switched to paired interactions across the intron for later steps of spliceosome assembly. By either intron definition or exon definition, the initial process of splice site recognition commits the pre-mRNA substrate to the splicing pathway. The pre-mRNA complex contains U1 snRNP and a number of key protein-splicing factors, including U2AF and the branch site binding factor. In multicellular eukaryotic cells, the formation of the commitment (E) complex requires the participation of SR proteins.
The spliceosome contains the U1, U2, U4/U6, and U5 snRNPs, as well as some additional splicing factors. The U1, U2, and U5 snRNPs each contain a single snRNA and several proteins; the U4/U6 snRNP contains two snRNAs and several proteins. Some proteins are common to all snRNP particles. U1 snRNA base pairs with the 5′ splice site, U2 snRNA base pairs with the branch sequence, and U5 snRNP holds the 5′ and 3′ splice sites together via a looped sequence within the spliceosome. When U4 releases U6, the U6 snRNA base pairs with the 5′ splice site and U2, which remains base paired with the branch sequence; this may create the catalytic center for splicing. An alternative set of snRNPs provides analogous functions for splicing the U12-dependent subclass of introns. The catalytic core resembles that of group II autocatalytic introns; as a result, it is likely that the spliceosome is a giant RNA machine (like the ribosome) in which key RNA elements are at the center of the reaction.
Splicing is usually intramolecular, but trans-splicing (intermolecular splicing) occurs in trypanosomes and nematodes. It involves a reaction between a small SL RNA and the pre-mRNA. Nematode worms have two types of SL RNA: One is used for splicing to the 5′ end of an mRNA, and the other is used for splicing to an internal site to break up the polycistronic precursor RNA. The introduction of the SL RNA to the processed mRNAs provides necessary signals for translation.
The termination capacity of RNA polymerase II is tightly linked to 3′ end formation of the mRNA. The sequence AAUAAA, located 11 to 30 bases upstream of the cleavage site, provides the signal for both cleavage by an endonuclease and polyadenylation by the poly(A) polymerase. This is enhanced by the complex bound on the GU-rich element downstream from the cleavage site. Transcription is terminated when an exonuclease, which binds to the 5′ end of the nascent RNA chain created by the cleavage, catches up to RNA polymerase.
All Pol II transcripts are polyadenylated with the exception of histone mRNAs, which neither contain an intron nor receive a poly(A) tail. The 3′ end formation of histone mRNA depends on a stem-loop structure and base pairing of a downstream element with U7 snRNA to result in a cleavage. The stem-loop structure may protect the end, as in bacteria.
tRNA splicing involves separate endonuclease and ligase reactions. The endonuclease recognizes the secondary (or tertiary) structure of the precursor and cleaves both ends of the intron. The two half-tRNAs released by loss of the intron can be ligated by the tRNA ligase in the presence of ATP. This tRNA maturation pathway is exploited by the unfolded protein response pathway in the ER.
rRNA processing takes place in the nucleolus where U3 snRNA initiates a series of actions of endonucleases and exonucleases to cut and trim extra sequences in the precursor rRNA to produce individual ribosomal RNAs. Hundreds to thousands of noncoding RNAs are expressed in eukaryotic cells. In the nucleolus, two groups of such noncoding RNAs, termed snoRNAs, are responsible for pairing with rRNAs at sites that are modified. Group C/D snoRNAs identify target sites for methylation, and group H/ACA snoRNAs specify sites where uridine is converted to pseudouridine.
References
19.1 Introduction
Review
Lewin, B. (1975). Units of transcription and translation: sequence components of hnRNA and mRNA. Cell 4, 77–93.
19.2 The 5′ End of Eukaryotic mRNA Is Capped
Review
Bannerjee, A. K. (1980). 5′ terminal cap structure in eukaryotic mRNAs. Microbiol. Rev. 44, 175–205.
Research
Mandal, S. S., Chu, C., Wada, T., Handa, H., Shatkin, A. J., and Reinberg, D. (2004). Functional interactions of RNA-capping enzyme with factors that positively and negatively regulated promoter escape by RNA polymerase II. Proc. Natl. Acad. Sci. USA 101, 7572–7577.
McCracken, S., Fong, N., Rosonina, E., Yankulov, K., Brothers, G., Siderovski, D., Hessel, A., Foster, S., Shuman, S., and Bentley, D. L. (1997). 5′-capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev. 11, 3306–3318.
19.3 Nuclear Splice Sites Are Short Sequences
Reviews
Padgett, R. A. (1986). Splicing of messenger RNA precursors. Annu. Rev. Biochem. 55, 1119–1150.
Sharp, P. A. (1987). Splicing of mRNA precursors. Science 235, 766–771.
Sharp, P. A., and Burge, C. B. (1997). Classification of introns: U2-type or U12-type. Cell 91, 875–879.
Research
Graveley, B. R. (2005). Mutually exclusive splicing of the insect Dscam pre-mRNA directed by competing intronic RNA secondary structures. Cell 123, 65–73.
Krainer, A. R., Maniatis, T., Ruskin, B., and Green, M. R. (1984). Normal and mutant human b-globin pre-mRNAs are accurately and efficiently spliced in vitro. Cell 36, 993–1005.
19.5 Pre-mRNA Splicing Proceeds Through a Lariat
Review
Sharp, P. A. (1994). Split genes and RNA splicing. Cell 77, 805–815.
Research
Reed, R., and Maniatis, T. (1985). Intron sequences involved in lariat formation during pre-mRNA splicing. Cell 41, 95–105.
Ruskin, B., Krainer, A. R., Maniatis, T., and Green, M. R. (1984). Excision of an intact intron as a novel lariat structure during pre-mRNA splicing in vitro. Cell 38, 317–331.
19.6 snRNAs Are Required for Splicing
Reviews
Guthrie, C. (1991). Messenger RNA splicing in yeast: clues to why the spliceosome is a ribonucleoprotein. Science 253, 157–163.
Guthrie, C., and Patterson, B. (1988). Spliceosomal snRNAs. Annu. Rev. Genet. 22, 387–419.
Maniatis, T., and Reed, R. (1987). The role of small nuclear ribonucleoprotein particles in pre-mRNA splicing. Nature 325, 673–678.
Research
Black, D. L., Chabot, B., Steitz, J. A. (1985). U2 as well as U1 small nuclear ribonucleoproteins are involved in premessenger RNA splicing. Cell 42, 737–750.
Black, D. L., and Steitz, J. A. (1986). Pre-mRNA splicing in vitro requires intact U4/U6 small nuclear ribonucleoprotein. Cell 46, 697–704.
Grabowski, P. J., Seiler, S. R., and Sharp, P. A. (1985). A multicomponent complex is involved in the splicing of messenger RNA precursors. Cell 42, 345–353.
Krainer, A. R., and Maniatis, T. (1985). Multiple components including the small nuclear ribonucleoproteins U1 and U2 are required for pre-mRNA splicing in vitro. Cell 42, 725–736.
19.7 Commitment of Pre-mRNA to the Splicing Pathway
Reviews
Berget, S. M. (1995). Exon recognition in vertebrate splicing. J. Biol. Chem. 270, 2411–2414.
Fu, X.-D. (1995). The superfamily of arginine/serine-rich splicing factors. RNA 1, 663–680.
Reed, R. (1996). Initial splice-site recognition and pairing during pre-mRNA splicing. Curr. Opin. Genet. Dev. 6, 215–220.
Research
Abovich, N., and Rosbash, M. (1997). Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell 89, 403–412.
Berglund, J. A., Chua, K., Abovich, N., Reed, R., and Rosbash, M. (1997). The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell 89, 781–787.
Fu, X.-D. (1993). Specific commitment of different pre-mRNA to splicing single SR proteins. Nature 365, 82–85.
Hoffman, B. E., and Grabowski, P. J. (1992). U1 snRNP targets an essential splicing factor, U2AF65, to the 3′ splice site by a network of interactions spanning the exon. Genes Dev. 6, 2554–2568.
Ibrahim, E. C., Schaal, T. D., Hertel, K. J., Reed, R., Maniatis, T. (2005). Serine/arginine-rich protein-dependent suppression of exon skipping by exonic splicing enhancers. Proc. Natl. Acad. Sci. USA 102, 5002–5007.
Kohtz, J. D., Jamison, S. F., Will, C. L., Zuo, P., Lührmann, R., Garcia-Blanco, M. A., and Manley, J. L. (1994). Protein-protein interactions and 5′ splice-site recognition in mammalian mRNA precursors. Nature 368, 119–124.
Robberson, B. L., and Berget, S. M. (1990). Exon definition may facilitate splice site selection in RNAs with multiple exons. Mol. Cell Biol. 10, 84–94.
Wu, J. Y., and Maniatis, T. (1993). Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell 75, 1061–1070.
19.8 The Spliceosome Assembly Pathway
Review
Burge, C. B., Tushl, T. H., and Sharp, P. A. (1999). Splicing of precursors to mRNAs by the spliceosome. In Gesteland, R. F., and Atkins, J. F., eds. The RNA World, 2nd ed., Cold Spring Harbor Laboratory Press, Plainview, NY, pp. 525–560.
Research
Cheng, S. C., and Abelson, J. (1987). Spliceosome assembly in yeast. Genes Dev. 1, 1014–1027.
Konarska, M. M., and Sharp, P. A. (1987). Interactions between small nuclear ribonucleoprotein particles in formation of spliceosomes. Cell 49, 736–774.
Newman, A., and Norman, C. (1991). Mutations in yeast U5 snRNA alter the specificity of 5′ splice site cleavage. Cell 65, 115–123.
Tseng, C. K., and Cheng, S. C. (2008). Both catalytic steps of nuclear pre-mRNA splicing are reversible. Science 320, 1782–1784.
Yan, C., Hang, J., Wan, R., Huang, M., Wong, C., and Shi, Y. (2015). Structure of a yeast spliceosome at 3.6-angstrom resolution. Science 349, 1182–1191.
Zhuang, Y., and Weiner, A. M. (1986). A compensatory base change in U1 snRNA suppresses a 5′ splice site mutation. Cell 46, 827–835.
19.9 An Alternative Spliceosome Uses Different snRNPs to Process the Minor Class of Introns
Research
Burge, C. B., Padgett, R. A., and Sharp, P. A. (1998). Evolutionary fates and origins of U12-type introns. Mol. Cell 2, 773–785.
Dietrich, R. C., Incorvaia, R., and Padgett, R. A. (1997). Terminal intron dinucleotide sequences do not distinguish between U2- and U12-dependent introns. Mol. Cell 1, 151–160.
Hall, S. L., and Padgett, R. A. (1994). Conserved sequences in a class of rare eukaryotic introns with non-consensus splice sites. J. Mol. Biol. 239, 357–365.
Tarn, W.-Y., and Steitz, J. A. (1996). A novel spliceosome containing U11, U12, and U5 snRNPs excises a minor class AT-AC intron in vitro. Cell 84, 801–811.
Tarn, W.-Y., and Steitz, J. A. (1996). Highly diverged U4 and U6 small nuclear RNAs required for splicing rare AT-AC introns. Science 273, 1824–1832.
19.10 Pre-mRNA Splicing Likely Shares the Mechanism with Group II Autocatalytic Introns
Reviews
Madhani, H. D., and Guthrie, C. (1994). Dynamic RNA-RNA interactions in the spliceosome. Annu. Rev. Genet. 28, 1–26.
Michel, F., and Ferat, J.-L. (1995). Structure and activities of group II introns. Annu. Rev. Biochem. 64, 435–461.
Research
Madhani, H. D., and Guthrie, C. (1992). A novel base-pairing interaction between U2 and U6 snRNAs suggests a mechanism for the catalytic activation of the spliceosome. Cell 71, 803–817.
19.11 Splicing Is Temporally and Functionally Coupled with Multiple Steps in Gene Expression
Reviews
Maniatis, T., and Reed, R. (2002). An extensive network of coupling among gene expression machines. Nature 416, 499–506.
Maquat, L. E. (2004). Nonsense-mediated mRNA decay: splicing, translation and mRNA dynamics. Nature Rev. Mol. Cell Biol. 5, 89–99.
Pandit, S., Wang, D., and Fu, X.-D. (2008). Functional integration of transcriptional and RNA processing machineries. Curr. Opin. Cell Biol. 20, 260–265.
Proudfoot, N. J., Furger, A., and Dye, M. J. (2002). Integrating mRNA processing with transcription. Cell 108, 501–512.
Research
Cheng, H., Dufu, K., Lee, C. S., Hsu, J. L., Dias, A., and Reed, R. (2006). Human mRNA export machinery recruited to the 5′ end of mRNA. Cell 127, 1389–1400.
Das, R., Yu, J., Zhang, Z., Gygi, M. P., Krainer, A. R., Gygi, S. P., and Reed R. (2007). SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol. Cell 26, 867–881.
Le Hir, H., Izaurralde, E., Maquat, L. E., and Moore, M. J. (2000). The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon-exon junctions. EMBO J. 19, 6860–6869.
Lin, S., Coutinho-Mansfield, G., Wang, D., Pandit, S., and Fu, X. D. (2008). The splicing factor SC35 has an active role in transcriptional elongation. Nature Struc. Mol. Biol. 15, 819–826.
Luo, M. L., Zhou, Z., Magni, K., Christoforides, C., Rappsilber, J., Mann, M., and Reed, R. (2001). Pre-mRNA splicing and mRNA export linked by direct interactions between UAP56 and Aly. Nature 413, 644–647.
Zhou, Z., Luo, M. J., Straesser, K., Katahira, J., Hurt, E., and Reed, R. (2000). The protein Aly links premessenger-RNA splicing to nuclear export in metazoans. Nature 407, 401–405.
19.12 Alternative Splicing Is a Rule, Rather Than an Exception, in Multicellular Eukaryotes
Reviews
Black, D. (2003). Mechanisms of alternative premessenger RNA splicing. Annu. Rev. Biochem. 72, 291–336.
Luco, R. F., Allo, M., Schor, I. E., Kornblihtt, A. R., and Misteli, T. (2011). Epigenetics in alternative pre-mRNA splicing. Cell 144, 16–26.
Research
Ge, H., and Manley, J. L. (1990). A protein, ASF, controls cell-specific alternative splicing of SV40 early pre-mRNA in vitro. Cell 62, 25–34.
Krainer, A. R., Conway, G. C., and Kozak, D. (1990). The essential pre-mRNA splicing factor SF2 influences 5′ splice site selection by activating proximal sites. Cell 62, 35–42.
Lynch, K. W., and Maniatis, T. (1996). Assembly of specific SR protein complexes on distinct regulatory elements of the Drosophila doublesex splicing enhancer. Genes Dev. 10, 2089–2101.
Tian, M., and Maniatis, T. (1993). A splicing enhancer complex controls alternative splicing of doublesex pre–mRNA. Cell 74, 105–114.
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., Kingsmore, S. F., Schroth, G. P., and Burge, C. B. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476.
Xu, X.-D., Yang, D., Ding, J. H., Wang, W., Chu, P. H., Dalton, N. D., Wang, H. Y., Bermingham, J. R., Jr., Ye, Z., Liu, F., Rosenfeld, M. G., Manley, J. L., Ross, J., Jr., Chen, J., Xiao, R. P., Cheng, H., and Fu, X. D. (2005). ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 120, 59–72.
19.13 Splicing Can Be Regulated by Exonic and Intronic Splicing Enhancers and Silencers
Review
Blencowe, B. J. (2006). Alternative splicing: new insights from global analysis. Cell 126, 37–47.
Research
Cramer, P., Cáceres, J. F., Cazalla, D., Kadener, S., Muro, A. F., Baralle, F. E., and Kornblihtt, A. R. (1999). Coupling of transcription with alternative splicing: RNA Pol II promoters modulate SF2/ASF and 9G8 effects on an exonic splicing enhancer. Mol. Cell 4, 251–258.
de la Mata, M., Alonso, C. R., Kadener, S., Fededa, J. P., Blaustein, M., Pelisch, F., Cramer, P., Bentley, D., and Kornblihtt, A. R. (2003). A slow RNA polymerase II affects alternative splicing in vivo. Mol. Cell 12, 525–532.
Fairbrother, W. G., Yeh, R. F., Sharp, P. A., and Burge, C. B. (2002). Predictive identification of exonic splicing enhancers in human genes. Science 297, 1007–1113.
Locatalosi, D. D., Mele, A., Fak, J. J., Ule, J., Kayikci, M., Chi, S. W., Clark, T. A., Schweitzer, A. C., Blume, J. E., Wang, X., Darnell, J. C., and Darnell, R. B. (2008). HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456, 464–470.
Sharma, S., Falick, A. M., and Black, D. L. (2005). Polypyrimidine tract binding protein blocks the 5′ splice site-dependent assembly of U2AF and the prespliceosome E complex. Mol. Cell 19, 485–496.
Wang, Z., Rolish, M. E., Yeo, G., Tung, V., Mawson, M., and Burge, C. B. (2004). Systematic identification and analysis of exonic splicing silencers. Cell 119, 831–845.
Yeo, G., Coufal, N. G., Liang, T. Y., Peng, G. E., Fu, X. D., and Gage, F. H. (2008). An RNA code for the Fox2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nature Struc. Mol. Biol. 16, 130–137.
Zhang, X. H., and Chasin, L. A. (2004). Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev. 18, 1241–1250.
Zhu, J., Mayeda, A., and Krainer, A. R. (2001). Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol. Cell 8, 1351–1361.
19.14 trans-Splicing Reactions Use Small RNAs
Review
Nilsen, T. (1993). Trans-splicing of nematode pre-mRNA. Annu. Rev. Immunol. 47, 413–440.
Research
Blumenthal, T., Evans, D., Link, C. D., Guffanti, A., Lawson, D., Thierry-Mieg, J., Thierry-Mieg, D., Chiu, W. L., Duke, K., Kiraly, M., and Kim, S. K. (2002). A global analysis of C. elegans operons. Nature 417, 851–854.
Denker, J. A., Zuckerman, D. M., Maroney, P. A., and Nilsen, T. W. (2002). New components of the spliced leader RNP required for nematode trans-splicing. Nature 417, 667–670.
Fischer, S. E. J., Butler, M. D., Pan, Q., and Ruvkun, G. (2008). trans-splicing in C. elegans generates the negative RNAi regulator ERI-6/7. Nature 455, 491–496.
Hannon, G. J., Maroney, P. A., Denker, J. A., and Nilsen, T. W. (1990). trans-splicing of nematode pre-mRNA in vitro. Cell 61, 1247–1255.
Huang, X. Y., and Hirsh, D. (1989). A second trans-spliced RNA leader sequence in the nematode C. elegans. Proc. Natl. Acad. Sci. USA 86, 8640–8644.
Krause, M., and Hirsh, D. (1987). A trans-spliced leader sequence on actin mRNA in C. elegans. Cell 49, 753–761.
Murphy, W. J., Watkins, K. P., and Agabian, N. (1986). Identification of a novel Y branch structure as an intermediate in trypanosome mRNA processing: evidence for trans-splicing. Cell 47, 517–525.
Sutton, R., and Boothroyd, J. C. (1986). Evidence for trans-splicing in trypanosomes. Cell 47, 527–535.
19.15 The 3′ Ends of mRNAs Are Generated by Cleavage and Polyadenylation
Reviews
Colgan, D. F., and Manley, J. L. (1997). Mechanism and regulation of mRNA polyadenylation. Genes Dev. 11, 2755–2766.
Shatkin, A. J., and Manley, J. L. (2000). The ends of the affair: capping and polyadenylation. Nature Struct. Biol. 7, 838–842.
Wahle, E., and Keller, W. (1992). The biochemistry of 3′-end cleavage and polyadenylation of messenger RNA precursors. Annu. Rev. Biochem. 61, 419–440.
Research
Conway, L., and Wickens, M. (1985). A sequence downstream of AAUAAA is required for formation of SV40 late mRNA 3′ termini in frog oocytes. Proc. Natl. Acad. Sci. USA 82, 3949–3953.
Fox, C. A., Sheets, M. D., and Wickens, M. P. (1989). Poly(A) addition during maturation of frog oocytes: distinct nuclear and cytoplasmic activities and regulation by the sequence UUUUUAU. Genes Dev. 3, 2151–2162.
Gil, A., and Proudfoot, N. (1987). Position-dependent sequence elements downstream of AAUAAA are required for efficient rabbit b-globin mRNA 3′ end formation. Cell 49, 399–406.
Karner, C. G., Wormington, M., Muckenthaler, M., Schneider, S., Dehlin, E., and Wahle, E. (1998). The deadenylating nuclease (DAN) is involved in poly(A) tail removal during the meiotic maturation of Xenopus oocytes. EMBO J. 17, 5427–5437.
McGrew, L. L., Dworkin-Rastl, E., Dworkin, M. B., and Richter, J. D. (1989). Poly(A) elongation during Xenopus oocyte maturation is required for translational recruitment and is mediated by a short sequence element. Genes Dev. 3, 803–815.
Takagaki, Y., Ryner, L. C., and Manley, J. L. (1988). Separation and characterization of a poly(A) polymerase and a cleavage/specificity factor required for pre-mRNA polyadenylation. Cell 52, 731–742.
19.16 3′ mRNA End Processing Is Critical for Termination of Transcription
Review
Buratowski, S. (2005). Connection between mRNA 3′ end processing and transcription termination. Curr. Opin. Cell Biol. 17, 257–261.
Research
Dye, M. J., and Proudfoot, N. J. (1999). Terminal exon definition occurs cotranscriptionally and promotes termination of RNA polymerase II. Mol. Cell 3, 371–378.
Kim, M., Krogan, N. J., Vasiljeva, L., Rando, O. J., Nedea, E., Greenblatt, J. F., and Buratowski, S. (2004). The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432, 517–522.
Luo, W., Johnson, A. W., and Bentley, D. L. (2006). The role of Rat1 in coupling mRNA 3′ end processing to transcription termination: implications for a unified allosteric-torpedo model. Genes Dev. 20, 954–965.
19.17 The 3′ End Formation of Histone mRNA Requires U7 snRNA
Review
Marzluff, W. F., Wagner, E. J., and Duronio, R. J. (2008). Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nature Rev. Genet. 9, 843–854.
Research
Dominski, Z., Yang, X. C., and Marzluff, W. F. (2005). The polyadenylation factor CPSF73 is involved in histone pre-mRNA processing. Cell 123, 37–48.
Kolev, N. G., and Steitz, J. A. (2005). Symplekin and multiple other polyadenylation factors participate in 3′ end maturation of histone +mRNAs. Genes Dev. 19, 2583–2592.
Mowry, K. L., and Steitz, J. A. (1987). Identification of the human U7 snRNP as one of several factors involved in the 3′ end maturation of histone premessenger RNAs. Science 238, 1682–1687.
Pillar, R. S., Grimmler, M., Meister, G., Will, C. L., Lührmann, R., Fischer, U., and Schümperli, D. (2003). Unique Sm core structure of U7 snRNPs: assembly by a specialized SMN complex and the role of a new component, Lsm 11, in histone RNA processing. Genes Dev. 17, 2321–2333.
Wang, Z. F., Whitfield, M. L., Ingledue, T. C., 3rd, Dominski, Z., and Marzluff, W. F. (1996). The protein that binds the 3′ end of histone mRNA: a novel RNA-binding protein required for histone pre-mRNA processing. Genes Dev. 10, 3028–3040.
19.18 tRNA Splicing Involves Cutting and Rejoining in Separate Reactions
Research
Diener, J. L., and Moore, P. B. (1998). Solution structure of a substrate for the archaeal pre-tRNA splicing endonucleases: the bulge-helix-bulge motif. Mol. Cell 1, 883–894.
Di Nicola Negri, E., Fabbri, S., Bufardeci, E., Baldi, M. I., Gandini Attardi, D., Mattoccia, E., and Tocchini-Valentini, G. P. (1997). The eucaryal tRNA splicing endonuclease recognizes a tripartite set of RNA elements. Cell 89, 859–866.
Reyes, V. M., and Abelson, J. (1988). Substrate recognition and splice site determination in yeast tRNA splicing. Cell 55, 719–730.
Trotta, C. R., Miao, F., Arn, E. A., Stevens, S. W., Ho, C. K., Rauhut, R., and Abelson, J. N. (1997). The yeast tRNA splicing endonuclease: a tetrameric enzyme with two active site subunits homologous to the archaeal tRNA endonucleases. Cell 89, 849–858.
19.19 The Unfolded Protein Response Is Related to tRNA Splicing
Review
Lin, J. H., Walter, P., and Benedict Yen, T. S. (2008). Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. Mech. Dis. 3, 399–425.
Research
Gonzalez, T. N., Sidrauski, C., Dörfler, S., and Walter, P. (1999). Mechanism of non-spliceosomal mRNA splicing in the unfolded protein response pathway. EMBO J. 18, 3119–3132.
Sidrauski, C., Cox, J. S., and Walter, P. (1996). tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 87, 405–413.
Sidrauski, C., and Walter, P. (1997). The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90, 1031–1039.
19.20 Production of rRNA Requires Cleavage Events and Involves Small RNAs
Reviews
Alessandro, F., and Tollervey, D. (2002). Making ribosomes. Curr. Opin. Cell. Biol. 14, 313–318.
Filipowicz, W., and Pogacic, V. (2002). Biogenesis of small nucleolar ribonucleoproteins. Curr. Opin. Cell. Biol. 14, 319–327.
Granneman, S., and Baserga, S. L. (2005). Crosstalk in gene expression: coupling and co-regulation of rDNA transcription, preribosome assembly and pre-rRNA processing. Curr. Opin. Cell Biol. 17, 281–286.
Henras, A. K, Plisson-Chastang, C., O’Donohue, M.-F., Chakraborty, A., and Gleizes, P.-E. (2015). An overview of pre-ribosomal processing in eukaryotes. Wiley Interdiscip. Rev. RNA 6, 225–242. doi:10.1002/wrna.1269
Matera, A. G., Terns, R. M., and Terns, M. P. (2007). Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nature Rev. Mol. Cell Biol. 8, 209–220.
Research
Balakin, A. G., Smith, L., and Fournier, M. J. (1996). The RNA world of the nucleolus: two major families of small RNAs defined by different box elements with related functions. Cell 86, 823–834.
Bousquet-Antonelli, C., Henry, Y., G’elugne, J. P., Caizergues-Ferrer, M., and Kiss, T. (1997). A small nucleolar RNP protein is required for pseudouridylation of eukaryotic ribosomal RNAs. EMBO J. 16, 4770–4776.
Ganot, P., Bortolin, M. L., and Kiss, T. (1997). Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell 89, 799–809.
Ganot, P., Caizergues-Ferrer, M., and Kiss, T. (1997). The family of box ACA small nucleolar RNAs is defined by an evolutionarily conserved secondary structure and ubiquitous sequence elements essential for RNA accumulation. Genes Dev. 11, 941–956.
Kass, S., Tyc, K., Steitz, J. A., and Sollner-Webb, B. (1990). The U3 small nucleolar ribonucleoprotein functions in the first step of preribosomal RNA processing. Cell 60, 897–908.
Kiss-Laszlo, Z., Henry, Y., Bachellerie, J. P., Caizergues-Ferrer, M., and Kiss, T. (1996). Site-specific ribose methylation of preribosomal RNA: a novel function for small nucleolar RNAs. Cell 85, 1077–1068.
Kiss-Laszlo, Z., Henry, Y., and Kiss, T. (1998). Sequence and structural elements of methylation guide snoRNAs essential for site-specific ribose methylation of pre-rRNA. EMBO J. 17, 797–807.
Ni, J., Tien, A. L., and Fournier, M. J. (1997). Small nucleolar RNAs direct site-specific synthesis of pseudouridine in rRNA. Cell 89, 565–573.