SET POINT
In spite of great fluctuation in the quantity and frequency of eating, we all maintain remarkable precision between energy expenditure and energy intake. Social factors, emotions, and time of day, as well as taste, satiety, and personal habits, influence our eating patterns, yet we maintain a reasonably stable body weight month after month. This is referred to as energy homeostasis or simply a metabolic “set point.”
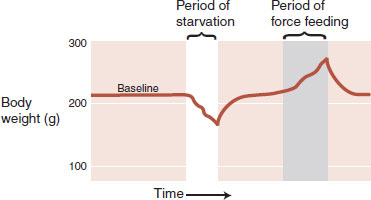
Evidence of a set point comes from a variety of sources. If a rat is deprived of food, then, when offered a normal diet, it will overeat for a short period and return its body weight to its preexisting level. Likewise, after being force-fed to increase body weight, it will limit its food intake to return to normal weight (Figure 13.1). The brain and body work in harmony to keep the body weight at a specific point.
The human corollary to this is the disturbingly low success rates that people have with most diets. In a review of long-term efficacy of dietary treatment interventions for obesity, Ayyad and Anderson found that only 15% of the patients fulfilled at least one of the criteria for success 5 years after the study. Unfortunately, most people who diet will slowly return to their preexisting weight within 1 year.
Several lines of research suggest that the set point is genetically controlled. Adoption studies provide a unique way to separate the effects of genetics from environment on body weight by comparing children with their biologic parents and with the parents from the house in which they were raised. In an analysis of Danish adoptees, Stunkard et al. found a strong relation between weight class of the adoptees and the body mass of the biologic parents. However, there was no correlation between the weight class of the adoptees and their adopted parents, suggesting that genetic makeup, not environment, is a major determining factor in one’s body weight.
Yet, obesity was a rare condition 50 years ago. Clearly, our genes have not evolved in half a century. There must be other explanations for the significant change in body weight that is occurring in first-world nations. The Pima Indians of Mexico and Arizona can provide some insight on this issue.
The Pima Indians separated into two tribes approximately 700 to 1,000 years ago—one tribe remained in Mexico and the other settled in what is now known as Arizona. In spite of their similar genetic makeup, they have remarkably different average body weights. The Arizona Indians have a high prevalence of obesity and non-insulin-dependent diabetes mellitus, whereas their Mexican relatives are not overweight and have little diabetes. The difference can be best explained by understanding the divergent lifestyles these two populations developed.
The Mexican Pima Indians have remained in the mountains, continuing a traditional rural lifestyle. They exert considerable physical energy working the farms and eat a diet high in starch and fiber. The Arizona Indians, on the other hand, were forced to move onto reservations and abandon their former way of life. There they lead more leisurely lives and eat a diet high in fat and sugar.
The obesity problem that the Pima Indians suffer from has been attributed to a “thrifty gene”—a gene that promotes saving and storing calories. Two thousand years ago, such a genetic predisposition would enhance the survival for those struggling with the fluctuations of prosperity and famine. However, with plentiful high caloric, highly palatable food, the “thrifty gene” promotes accumulation and storage beyond what is healthy. So the weight problem is genetic, but influenced by what is available in the environment.
FIGURE 13.1  A rat will return its body weight to its preintervention weight when allowed to eat a regular diet. (Adapted from Keesey RE, Boyle PC. Effects of quinine adulteration upon body weight of LH-lesioned and intact male rats. J Comp Physiol Psychol. 1973;84(1):38-46.)
A rat will return its body weight to its preintervention weight when allowed to eat a regular diet. (Adapted from Keesey RE, Boyle PC. Effects of quinine adulteration upon body weight of LH-lesioned and intact male rats. J Comp Physiol Psychol. 1973;84(1):38-46.)
The concept of a set point helps us understand the difficulties encountered by anyone trying to diet. One of the mechanisms the brain uses to return the body weight to its baseline set point became apparent in the Minnesota Starvation Experiment. This was an experiment conducted with conscientious objectors in Minnesota toward the end of the Second World War to help the military understand the condition of the starving civilians in Europe.
Participants were subjected to a semistarvation diet for 6 months with the goal of losing approximately 25% of their weight. From our perspective, one of the more interesting symptoms these men experienced during the experiment was obsessive thought about food. Not unlike the cravings discussed in the previous chapter, food became the principal topic of their thoughts, conversation, and daydreams. Reading cookbooks and collecting recipes became an intensely interesting pastime for many men. Eating, either mentioned in a book or shown in a movie, was a cue that put the men at heightened risk for breaking their diet. The urge to eat was so powerful that the researchers established a buddy system so that participants would not be tempted to cheat when away from the dorm. Clearly, their brains were focusing on what the body needed.
The brain can also adjust the energy expenditure as another way to maintain a stable body weight. Figure 13.2 shows a thermodynamic perspective of energy expenditure. Energy enters an organism as food and exits as heat or work. Energy is stored as fat or glycogen and mobilized when needed. Total energy expenditure can be subdivided into three components: obligatory energy expenditure, physical activity, and adaptive thermogenesis.
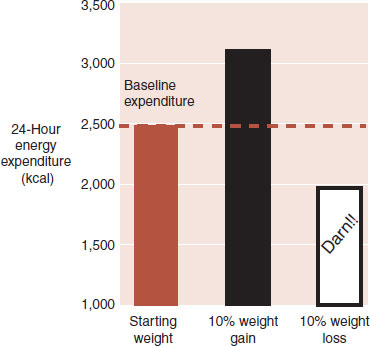
Adaptive thermogenesis is the other major obstacle the brain uses to thwart weight loss. Changes in sympathetic tone alter energy expenditure. For example, activation of the sympathetic nervous system results in catabolic breakdown of adipose tissue and produces energy. Figure 13.3 shows the changes in energy expenditure in healthy subjects after a 10% alteration in body weight. Not only does the brain increase adaptive thermogenesis when the body gains weight but also turns it down in response to weight loss—a frustration many dieters have experienced.
HOMEOSTATIC MECHANISMS
The key point is that despite large day-to-day fluctuations in food intake and energy expenditure, our weight remains within a relatively narrow range. This is accomplished through feedback loops between the brain and the body. Brain lesion and stimulation studies in the 1940s identified the hypothalamus as a major center controlling food intake. Although the initial studies turned out to be overly simplified, the hypothalamus remains an important command center for weight maintenance.
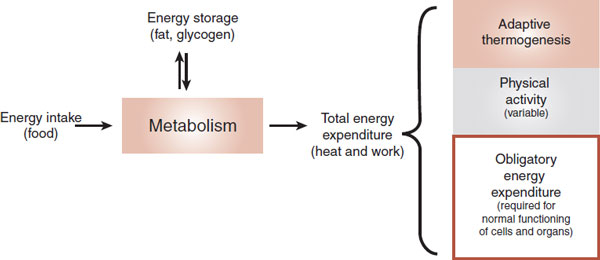
FIGURE 13.2 Energy accumulation and expenditure must be in balance for an organism to maintain a stable body weight. Adaptive thermogenesis is regulated by the brain and can vary in response to changes in diet or temperature. (Adapted from Lowell BB, Spiegelman BM. Towards a molecular understanding of adaptive thermogenesis. Nature. 2000;404(6778):652-660.)
FIGURE 13.3 Total energy expenditure is increased in response to weight gain and decreased when weight is lost. (Adapted from Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med. 1995;332(10):621-628.)
But what are the signals the brain receives and sends? Interrupting or enhancing these signals may provide opportunities for interventions to stem the obesity epidemic. The best way to understand the current conceptualization of the central nervous system (CNS) homeostatic mechanisms is to separate the short-term signals from the long-term ones.
Short-Term Signals
The short-term signals tend to affect meal size rather than the overall energy storage. The signals comprise nutrients and gut hormones in the circulation as well as afferent signals sent up the vagus nerve. These signals to the brain result in the sensation of satiety but do not produce sustained alteration in body adiposity.
Nutrients
Glucose is the primary nutrient that mediates satiety. Hypoglycemia increases hunger sensations and stimulates eating. Glucose infusions will decrease the food intake. Other nutrients in the systemic circulation, such as fats and amino acids, play a real but limited role in signaling to the brain the effects of a recent meal. High levels of these nutrients tell the brain to stop eating, but the brakes are insufficient when the individual has been starving.
Mechanoreceptors
The physical presence of food in the stomach and upper small intestine activates mechanoreceptors. The stomach wall is innervated with stretch receptors that increase in activity in proportion to the volume in the stomach. We usually think of the vagus as a conduit from the brain to the gut, but as much as 80% of the neural traffic is flowing in the opposite direction (see Figure 2.9). The vagus nerve transmits signals about gastric distension to the hindbrain. This may be the reason some patients with vagus nerve stimulators report weight loss.
Gut Hormones
Numerous gut hormones are involved in food intake regulation. The most widely studied hormone is cholecystokinin (CCK). CCK is released from endocrine cells in the mucosal layer of the small intestine in response to fats and proteins. CCK inhibits further food intake through several mechanisms, such as stimulating the vagal nerve and inhibiting gastric emptying. Additionally, there are CCK receptors in the brain. The injection of CCK directly into the ventricles will inhibit eating. CCK appears to have central and peripheral mechanisms to put the brake on a meal.
Although CCK will limit food intake, its long-term administration does not induce significant weight loss. In studies with rats, the repeated administration of CCK resulted in smaller but more frequent meals. Thus, the overall energy balance was not altered. Figure 13.4 summarizes the short-term signals regulating food intake.
There are many other gut hormones that also inhibit eating, for example, glucagon-like peptide-1 (GLP-1) and peptide tyrosine–tyrosine. Ghrelin is of most interest as it is the only gut hormone that stimulates hunger. Produced in the stomach, fasting increases the levels of ghrelin, which then fall after a meal. Peripheral and central administration of ghrelin increases the food intake. In contrast to CCK, there is some evidence that ghrelin has long-term effects on weight and may be a potential culprit in obesity. Some studies suggest that reduced ghrelin production is one of the reasons gastric bypass surgery is so effective (see Figure 13.10).
The Joy of Eating
Eating is more than just sustenance; it is one of the great pleasures of life. The perception of pleasure that we get from some foods is most likely an adaptation that enhanced the survival of our ancestors during lean times. However, with the abundance of inexpensive highly palatable refined foods, genes that favored sweet foods cause us to overeat.
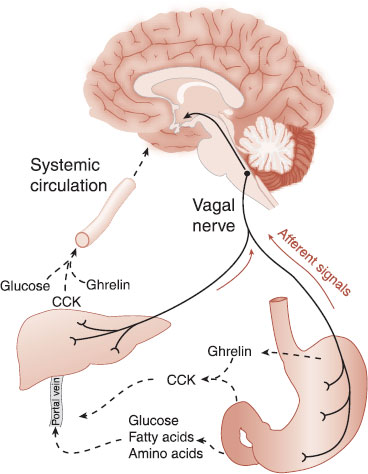
FIGURE 13.4 Short-term signals from the intestines (hormones in the circulation and stimulation of the vagus nerve) signal to the brain that the body is full. CCK, cholecystokinin. (Adapted from Havel PJ. Peripheral signals conveying metabolic information to the brain: short-term and long-term regulation of food intake and energy homeostasis. Exp Biol Med. 2001;226(11):963-977.)
Certain foods increase dopamine at the nucleus accumbens (see Figure 12.6). Additionally, the endogenous opioids appear to be more active during a good meal. Other evidence suggests that eating is a highly valued pleasure that can resemble an addiction. For example, obese individuals have reduced D2 receptors in the striatum and activation of the orbitofrontal cortex when craving food. Clearly, dopamine and the endogenous opioids are signals that influence energy consumption.
Long-Term Signals
Long-term signals tell the brain about the overall energy storage, not just the caloric content of the recent meal. In the 1950s, it was suggested that adipose tissue releases a hormone that signals the hypothalamus about the current state of energy storage.
DISORDERS
YOUR GRANDFATHER’S DIET
A remarkable study from an isolated community in Sweden has shown that a man’s risk of death from cardiovascular disease and diabetes is affected by his grandfather’s diet during his grandfather’s slow growth period—ages 9 to 12 for boys. Using records of harvest success or failure during the 19th century, the researchers found increased risk of heart disease and diabetes if the grandfather lived through bountiful harvests during his slow growth period. Alternatively, grandfathers who grew up during times of famine sired grandchildren who lived longer. The mechanism, derived in part from rodent studies, is believed to be transgenerational epigenetic inheritance (see Figure 6.12).
Termed an adiposity signal, the elusive hormone must have the following three traits:
– Circulate in the blood in proportion to the amount of stored fat
– Cross the blood–brain barrier and stimulate specific receptors in the brain
– Produce changes in caloric intake and energy expenditure when levels of the hormone fluctuate
Leptin
It was not until the discovery of leptin in 1994 that the first adiposity signal was identified. The mutant ob/ob mice (ob = obese) have an alternation in one gene, which results in hyperphagia and weight gain of three to five times the normal. Identifying the locus of the genetic defect allowed the cloning of the protein that the ob/ob mice were missing: leptin.
Leptin is primarily produced in white fat cells and circulates in direct proportion to the total fat load. The largest concentration of leptin receptors is found in the arcuate nucleus of the hypothalamus. Mice that lack leptin are obese. When given exogenous leptin, they reduce the food intake and lose weight (Figure 13.5).
Other hormones have been proposed as adiposity signals, the most prominent of which is insulin. The role of insulin is complicated because its primary function is to enhance the glucose intake into the muscle and adipose tissues. Yet, insulin secretion is influenced by total body fat, and in turn it can elicit a reduction in body weight. Insulin appears to work in parallel with leptin. Figure 13.6 shows a schematic representation of these adiposity signals.
FIGURE 13.5 Two ob/ob mice, both of which are missing the gene to produce leptin. The mouse on the right has received daily leptin injections for 4.5 weeks and weighs about half as much as the mouse on the left. (From Amgen Inc., Thousand Oaks, California.) Photo by John Sholtis.
Arcuate Nucleus
The hypothalamus is at the heart of the regulation of the body’s energy metabolism. So it is no wonder that the largest concentration of leptin receptors is found in the arcuate nucleus of the hypothalamus. The arcuate nucleus is located at the base of the hypothalamus next to the third ventricle (Figure 13.7). Two groups of neurons have been identified within the arcuate nucleus that mediate the leptin signal: proopiomelanocortin (POMC) and neuropeptide Y (NPY).
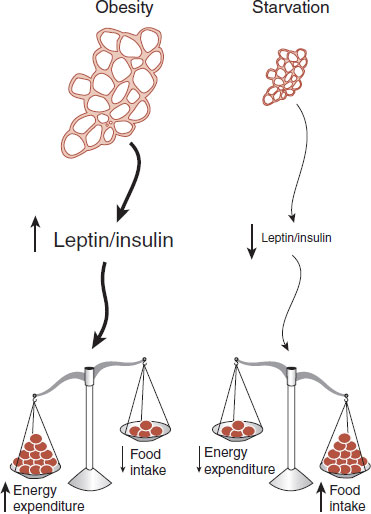
FIGURE 13.6 Adipose tissue secretes hormones in proportion to the total fat stored. The hormones in turn affect the energy expenditure and food intake in relation to their levels in the circulation.
Proopiomelanocortin
The POMC neurons put the brakes on eating. The POMC neuropeptide is cleaved to produce α-melanocyte–stimulating hormone (α-MSH), which is a potent suppressor of food intake. The effects of α-MSH are mediated through the melanocortin (MC) receptors, particularly MC3R and MC4R, which are strongly expressed in the hypothalamus.
High levels of leptin as well as other circulating hormones will stimulate the POMC neurons. Conversely, low levels of leptin inhibit the POMC neurons. Thus, POMC neurons and α-MSH are directly responsive to circulating hormones that signal an excess of adipose tissue.
Neuropeptide Y
The NPY neurons work to increase the food intake and decrease the energy expenditure. The NPY neurons express a peptide called agouti-related peptide (AGRP), which is an antagonist at the MC4 receptor. Consequently, activation of the NPY neurons blocks the effects of α-MSH and the POMC neurons. The role of NPY neurons and AGRP in the accumulation of calories has been demonstrated in many experimental conditions (see Figure 6.11):
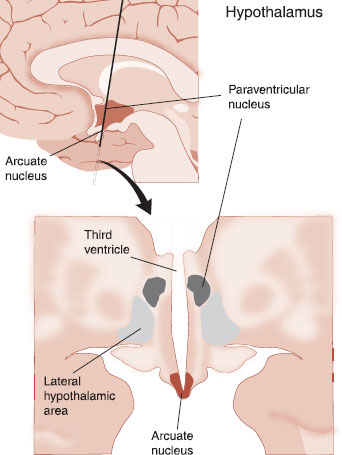
FIGURE 13.7 Two sections of the brain showing the location of the arcuate nucleus within the hypothalamus along with two other important nuclei for the control of energy balance.
1. Stimulation of the NPY neurons increases food intake.
2. Increased expression of AGRP results in increased food intake.
3. During starvation, there is increased activation of the NPY neurons and increased expression of AGRP.
4. Leptin inhibits the NPY neurons and AGRP expression.
Thus, there is an accelerator and brake relationship between the NPY neurons and the POMC neurons that responds to signals from the body about the long-term status of energy storage (Figure 13.8). Of particular relevance to the current epidemic of obesity is the unequal relationship between each set of neurons. While they provide equal stimulation of the downstream effort neurons, only the NPY neurons directly inhibit the POMC neurons. The POMC neurons do not inhibit the NPY neurons. Consequently, there appears to be a slightly greater emphasis on the accumulation of calories. In other words, the accelerator is stronger than the brake, which, from an evolutionary perspective, would seem to enhance the survival in times of low food supply or famine.
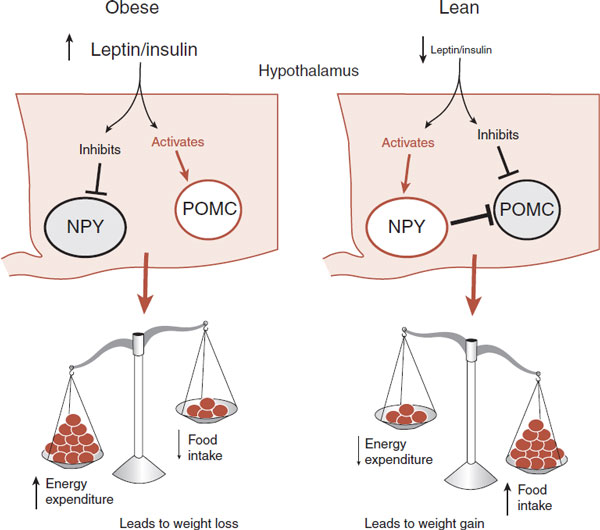
FIGURE 13.8 The presence or absence of adiposity signals, such as leptin and insulin, has opposite effects on the NPY and POMC neurons. In turn, the activation of the NPY and POMC neurons has different effects on energy balance and body weight. NPY, neuropeptide Y; POMC, proopiomelanocortin.
STRESS
Many patients will report that food is a source of comfort when they are “stressed out.” Several lines of evidence suggest there may be a correlation between the hypothalamic–pituitary–adrenal axis and the ability to resist the pleasures of food:
1. Childhood stress is associated with increased weight problems in adolescence and adulthood.
2. Corticotropin-releasing hormone and cortisol stimulate the release of dopamine at the nucleus accumbens, which makes it harder to resist temptations.
3. Glucocorticoids increase fat deposits.
4. Stressed rats given access to sweet water have lower glucocorticoid levels.
Downstream Targets
The downstream effects of the arcuate neurons are numerous and largely remain mysterious. Two important sites are the paraventricular nucleus (PVN) and the lateral hypothalamic (LH) area, also shown in Figure 13.7. The POMC and NPY neurons project in parallel to these sites with corresponding activation and deactivation, depending on the short-term and long-term signals from the periphery.
The effects of the hunger and satiety signals are carried out by three systems: the cerebral cortex (behavior), the endocrine system, and the ANS. The PVN affects the output of the endocrine system and the ANS—both of which affect the energy expenditure. The LH communicates with the cerebral cortex, which in turn modulates the food-seeking behavior. This simplified analysis of the downstream effects of signals from the body is shown in Figure 13.9.
POINT OF INTEREST
Smoking has one legitimate virtue: appetite suppression. Smokers are generally thinner and typically gain about 10 lb when they cease smoking. Recent research has identified the anorexic mechanism. Nicotine binds with a subunit of the nicotine acetylcholine receptors on the POMC neurons. This activates the POMC neurons, which leads to reduced food intake and weight loss. This receptor subunit may be accessible to pharmacologic manipulation turning on one benefit of smoking without the toxic effects.
Endocannabinoids
It has been known for a long time that marijuana stimulates the appetite. The naughty boys at our colleges called it the munchies. Stimulation of the cannabinoid (CB1) receptor by the main active component of marijuana, Δ9-tetrahydrocannabinol, is believed to induce this behavior. Clinicians have successfully utilized this effect when treating anorexic conditions such as AIDS (acquired immunodeficiency syndrome)-related wasting syndrome. With animals, it has been found that the endocannabinoid system is activated with short-term fasting or the presentation of palatable food, thereby inducing appetite.
The CB1 receptor is involved with the regulation of food intake at several levels. First, it enhances the motivation to seek and consume palatable food, possibly by interactions with the mesolimbic pathways. Studies with rodents show that endocannabinoids enhance the release of dopamine at the nucleus accumbens and may synergize the effects of the opioids.
The endocannabinoid system also stimulates food consumption in the hypothalamus. Studies have shown that endocannabinoids are highest in the hypothalamus with fasting and lowest during food consumption. Additionally, the endocannabinoids appear to work in concert with the neurohormones such as leptin and ghrelin to control the appetite. The ultimate effect may be through the CB1 receptors at the PVN and LH. One of the most exciting but disappointing developments in the treatment of obesity was the CB1 receptor blocker rimonabant (the anti-munchie pill). Rimonabant induced about a 10-lb weight loss, but it also led to the emergence of anxiety, depression, and suicidal thinking. The Food and Drug Administration (FDA) rejected the medication and it was withdrawn in Europe.
EATING DISORDERS
Obesity
Fifty years ago, obesity was a rare disease. Readily available palatable food accompanied by a sedentary lifestyle has now resulted in an epidemic of obesity. Most disturbing is the rapid rise in childhood obesity. Clearly, diet and exercise are first-line interventions for this condition, but these efforts are a constant uphill battle against a genetic set point that favors rich, sweet food that historically was in short supply.
After the discovery of leptin, there was great excitement about the prospect of a treatment for obesity. Leptin was shown to reduce obesity in genetically leptin-deficient humans and rodents (Figure 13.5). Unfortunately, the magnitude of weight loss in most obese humans who received exogenous leptin in studies has been modest. In actuality, most obese people have high levels of circulating leptin, but for some reason fail to respond to the signal. This has generated speculation that obesity may be associated with, or even caused by, a resistance to leptin, as is seen with insulin resistance in type 2 diabetes.
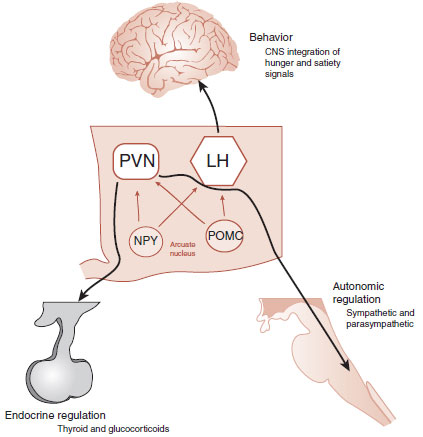
FIGURE 13.9 The PVN and lateral hypothalamus (along with others) influence the three major effort pathways so that the body can maintain a stable energy balance. CNS, central nervous system; PVN, paraventricular nucleus; LH, lateral hypothalamic; NPY, neuropeptide Y; POMC, proopiomelanocortin.
Why cannot we find a pill that tells the brain, “I’m full”? It is not for lack of trying. The fen–phen debacle (fenfluramine/phentermine)—with its modest weight loss and potential for pulmonary hypertension or valvular disease—has spooked the field. Likewise, a recent follow-up of patients who took benzedrine or dexedrine for weight loss in the sixties showed a 60% greater chance of developing Parkinson’s disease. There are a number of stimulant medications such as phentermine and benzphetamine that suppress appetite, but they are used only for short-term weight loss—say 8 to 12 weeks.
The dilemma is to find a medication that is effective and safe for long-term use. We know of four medications that were rejected (or not accepted) by the FDA and one that was voluntarily withdrawn from the market in this short period of time. Recently, the FDA reconsidered and accepted Belviq (lorcaserin), a serotonin 2C receptor agonist that is believed to activate POMC. The benefits are mild—roughly 13-lb weight loss compared with 5 to 6 lb for patients on the placebo. Unfortunately, when it comes to medications that control weight, it is difficult to deceive the brain without unintended damages—or at least that is the case so far.
Gastric Bypass Surgery
One success during the past decade has been gastric bypass surgery. Also called bariatric surgery, it is clearly not only the most effective treatment for morbid obesity but also the most invasive—and not without significant risks. Many patients not only lose substantial weight but also can be cured of secondary problems such as diabetes and sleep apnea. There are two primary methods: gastric banding and Roux-en-Y gastric bypass (Figure 13.10). Both procedures mechanically limit the amount of food in the stomach, which reduces the caloric intake.
Recent research suggests that one of the reasons the Roux-en-Y procedure may be more effective has more to do with changes in gut hormones than mechanical effects. In a small non-randomized study comparing the two procedures, the patients who had the Roux-en-Y showed greater weight loss along with greater changes in gut hormones (Figure 13.10). Not shown in the figure are the other hormones such as GLP-1 and peptide YY, which also showed significant differences at 1 year between the two procedures.
A separate study by a different group investigated brain activation before and after a Roux-en-Y bypass. Subjects were cued with auditory and visual representations of food, while a functional magnetic resonance imaging (MRI) followed their brain response. As expected, the food cues activated the reward pathways discussed in the previous chapter (see Figure 12.6). Remarkably, after the surgery, the reward pathway activation was significantly reduced in response to food cues. In other words, the subjects did not have as much food cravings after the bypass surgery.
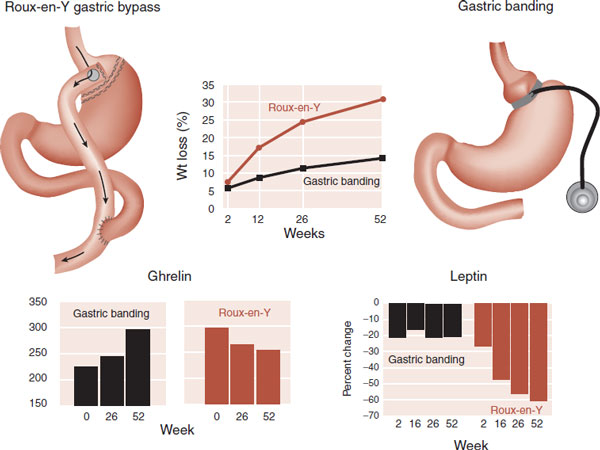
FIGURE 13.10 The superiority of Roux-en-Y gastric bypass surgery to produce substantial weight loss which may be due to changes in hormones that regulate appetite. (Adapted from Korner j, Inabnet W, Febres G, et al. Prospective study of gut hormone and metabolic changes after adjustable gastric banding and Roux-en-Y gastric bypass. Int J Obesity. 2009;33:786-795.)
Taken together, these studies suggest that Rouxen-Y procedure dampens numerous hormones and neurotransmitters that mediate the neural response to food. Maybe one reason it is so hard to find a pill that reduces appetite is because one molecule cannot replicate the panoply of hormones released by the gastrointestinal tract that regulate the body’s energy balance.
Purging
It is tempting to speculate that patients with anorexia and bulimia have set points that favor lean body mass, in other words, some pathology in their hypothalamus. However, most of the studies point to the involvement of other areas of the brain. Specifically, the symptoms of anxiety, perfectionism, and obsessions about body image are more consistent with disorders in the prefrontal cortex and amygdala. Likewise, eating disorder patients who have lost significant weight experience food cravings similar to what the men in the Minnesota Starvation Experiment experienced. Finally, the neuropeptide and neuroendocrine alterations present when patients are restricting intake return to normal levels when the patients recover. Eating disorder patients struggle with a drive to be thin, but this is not likely to be caused by pathological appetite signals.
PSYCHIATRIC MEDICATIONS
Weight gain is one of the most difficult side effects associated with psychiatric treatment and is often a reason patients stop effective treatments. Surprisingly, little is known about the mechanism of this problem. The new antipsychotic agents have been of particular concern. The FDA has required all manufacturers to include black box warnings documenting concerns about weight gain as well as the development of diabetes and hypercholesterolemia.
The mechanism of the weight gain remains a mystery. One compelling study examined the affinity for various receptors (serotoninergic, adrenergic, dopaminergic, histaminergic, and muscarinic) compared with reports of short-term weight gain. They determined that the strongest correlation for gaining weight is with H1 histamine receptor activity (r = −0.72). Table 13.1 shows the relative risk of weight gain from the second-generation antipsychotic agents, as determined by a consensus group that included the American Diabetes Association and the American Psychiatric Association (as well as others), compared with the H1 affinity.
The histamine neurons are known to be involved with energy homeostasis—an effect enhanced by modafinil. Additionally, centrally administered histamine increases the activity of leptin in rodents. Inversely, the blocking effect of the antihistamines may result in weight gain through decreased energy metabolism as well as increased appetite. However, diphenhydramine (Benadryl) is considered weight neutral, so there must be more than just H1 affinity causing the weight problems associated with antipsychotic medications.
TREATMENT
LONGEVITY
Caloric restriction (CR) is the most reproducible intervention to extend life. Rhesus monkeys whose caloric intake is restricted by 30% live longer. The figures (A) show the survival curves for age-related mortality and overall mortality for the monkeys with CR compared to controls. The CR monkeys have less diabetes, cancer, and cardiovascular disease. Furthermore, MRIs show that CR monkeys have more brain volume and less CNS fluid (B). CR appears to prevent cell loss and brain atrophy associated with aging. While beneficial, it is unlikely that humans can muster the self-control to use CR to prevent age-related CNS decline.
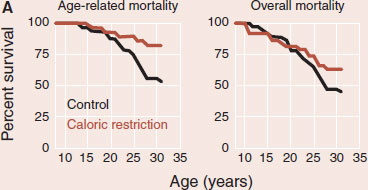
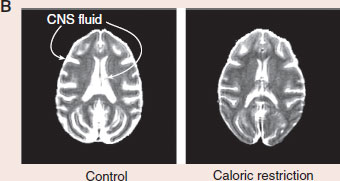
Adapted from Colman RJ, Anderson RM, Johnson SC, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201-204. Brain scans courtesy of Ricki Colman and the University of Wisconsin-Madison.
Patients on antidepressants or mood stabilizers are also often caught between maintaining an ideal body size and the negative consequences of effective treatment. Long-term studies find weight gain and increased risk of diabetes mellitus DM in patients treated with these medications. What could be causing this? One theory is that patients are regaining the weight they lost prior to starting treatment. More likely explanations are that psychiatric mediations increase the food craving or reduce the resting metabolic rate. Unfortunately, there is scant evidence to identify a specific mechanism.
Few psychiatric medications induce weight loss. Bupropion and topiramate have demonstrated beneficial effects on weight, but results are modest and few studies exist. Amphetamines were the original “diet pills” and were freely prescribed in the fifties and sixties. In the early seventies, due to problems with addiction and occasional psychosis, the government deemed this class of medications Schedule II and required new studies to establish effectiveness. These steps, for all intents and purposes, stopped the use of stimulants as diet pills. More recently, the quick removal of dexfenfluramine (Redux) in the 1990s and the massive lawsuits that followed portend further research with stimulants for weight control.
The mechanism by which stimulants induce weight loss is not as straightforward as one would expect. These medications—affectionately called “speed” and “uppers” on the street—actually reduce motor activity: the paradoxical effect. Figure 13.11 documents this effect in a group of boys with no behavioral or learning difficulties and an average IQ of 130. Motor activity was measured by a little sensor the children wore around their ankles during a 2-hour test period. The graph shows that, with few exceptions, the boys reduced the motor activity while on amphetamine. (Some clinicians incorrectly interpret this normal response to stimulants as an indication that the subject has ADHD.)
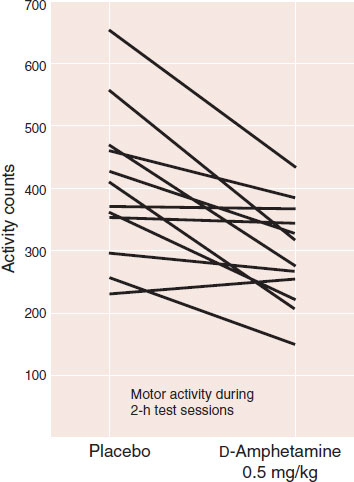
FIGURE 13.11 Physical activity in normal prepubertal boys decreased when they were on amphetamine compared with placebo. (Adapted from Rapoport JL, Buchsbaum MS, Zahn TP, et al. Dextroamphetamine: cognitive and behavioral effects in normal prepubertal boys. Science. 1978;199(4328):560-563.
NUTRITION AND MENTAL HEALTH
There is a growing body of evidence correlating nutrition and mental health. Separate studies in the UK, Spain, and Australia have shown that increased consumption of a Western diet (processed food, refined grains, and sugary products) is associated with more depressive symptoms. Although unproven as a therapeutic intervention, maybe a healthy diet rich in fruits and vegetables should be a prescription we give to every patient.
A more recent study analyzed resting energy expenditure (see Figure 13.3) in 14 normal men and women given methylphenidate. Subjects received 0.5 mg/kg of methylphenidate or placebo in a blinded, crossover study. While on methylphenidate the resting energy of the subjects increased by 7%. Other studies with humans have consistently shown decreased caloric intake while on stimulants. Subjects will consume a normal quantity when eating, but eat less frequently.
The stimulants appear to increase energy expenditure and reduce energy intake—which on the surface is exactly what we want. Unfortunately, the stimulants also have a host of adverse CNS effects. Of interest, some of the recently rejected appetite suppressants also had psychiatric adverse effects such as depression, anxiety, and suicidal ideation—a prime reason the FDA turned down these medications. There is an interesting, and as yet poorly understood, association between medications that reduce appetite and mental health—further evidence that obesity is more than just energy consumption.
QUESTIONS
1. All of the following support the concept of a metabolic “set point,” except
a. Most diets fail.
b. Forced feeding leads to increased energy expenditure.
c. The “thrifty gene” promotes weight gain when high caloric food is readily available.
d. Adaptive thermogenesis is unchanged by caloric intake.
2. Patients with anorexia nervosa and participants in the Minnesota Starvation Experiment share which of the following symptoms?
a. Perfectionistic personality traits.
b. Obsessive thoughts about food.
c. Altered metabolic “set point.”
d. Increased energy expenditure.
3. Short-term signals about hunger and satiety include all of the following, except
a. Leptin.
b. CCK.
c. Glucose.
d. Ghrelin.
4. Adiposity signals must be able to do all of the following, except
a. Cross the blood–brain barrier.
b. Change in relation to the amount of stored fat.
c. Alter afferent signals from the vagus nerve.
d. Inhibit or enhance caloric intake.
5. The largest concentration of leptin receptors are in the
a. Amygdala.
b. Arcuate nucleus.
c. Adrenal cortex.
d. Anterior corticospinal tract.
6. The AGRP does which of the following?
a. Antagonist at the MC4 receptor and stimulates eating.
b. Antagonist at the MC4 receptor and inhibits intake.
c. Agonist at the MC4 receptor and stimulates eating.
d. Agonist at the MC4 receptor and inhibits intake.
7. The Melanocortin system
a. Has been implicated as a cause of AN.
b. Stimulates foraging for food.
c. Potentiates the endocannabinoid receptor.
d. Suppresses the food intake.
8. The effects of the brain’s assessment of caloric needs are carried out by all of the following, except
a. Endocrine hormones.
b. Changes in behavior.
c. Modulation of the “set point.”
d. Alterations in the ANS.
See Answers section at the end of the book.