Canine distemper (CD) is one of the most important viral diseases in ferrets and, as also happens with ferret systemic coronavirus disease, the mortality rate approaches 100%. The availability of highly effective, safe vaccines and better awareness of ferret owners toward the disease have significantly decreased the prevalence of CD in the ferret population, although shelter population and breeding facilities where large numbers of ferrets congregate are still at higher risk for severe disease outbreaks. Besides canine distemper virus (CDV) causing severe natural disease, ferrets have also been used to experimentally reproduce other morbilliviral diseases, including measles and its associated conditions such as immunosuppression and subacute sclerosing panencephalitis [1–4] (see Chapter 26 for a more detailed description; see also Reference 4).
Etiology
Canine distemper virus (CDV) is an enveloped, nonsegmented, negative sense single-stranded RNA virus of the genus Morbillivirus in the family Paramyxoviridae [5]. Other viruses belonging to this genus include measles virus, rinderpest virus, peste des petits ruminants virus, phocine distemper virus (PDV), cetacean morbillivirus, and porpoise morbillivirus (http://www.ictvonline.org). CDV has two envelope glycoproteins, hemagglutinin and fusion protein, mediating receptor binding and membrane fusion, respectively.
The virus causes CD, an acute to subacute contagious systemic disease affecting mainly terrestrial carnivores in the families Canidae, Mustelidae, and Procyonidae [6–8]. Other families within the order Carnivora that can be affected by CDV include Felidae, Viverridae, Herpestidae, Ailuridae, Ursidae, and Hyaenidae [5,6,9–11]. In addition, clinical disease has been reported in pangolins (order Pholidota, family Manidae), peccaries (order Artiodactyla, family Tayassuidae), Rhesus macaques (order Primates, family Cercopithecidae), and several species of seals and sea lions (order Pinnipedia, families Phocidae and Otariidae [5,12–15]). The virus has one serotype and several strains, and severity of the disease depends on the viral strain and host factors such as species and immunocompetency [5,7,16,17]. CDV isolates are serologically homogeneous, but different strains differ in their pathogenicity [5].
Transmission and Epidemiology
Transmission and epidemiology of CDV in ferrets are similar to those in dogs. Transmission of CDV occurs primarily by aerosol or contact with oral, respiratory, or ocular fluids and exudates containing the virus [18]. Close contact between affected and susceptible animals is necessary due to the relative fragility of CDV in the environment [18]. Other routes of transmission, such as skin contact, feces or urine, are less important. However, susceptible ferrets have been experimentally infected by exposure to aerosolized urine from CDV-infected ferrets [19]. Transplacental transmission has been documented in dogs [20], but not in ferrets [7]. The virus can be detected in blood, nasal mucosa, lung, spleen, and cervical lymph nodes as early as 2 days after experimental infection [7]. Viremia persists until the virus is neutralized by antibodies or the ferret dies [7]. In dogs, viral shedding occurs even if animals are subclinically infected, and virus may be shed for up to 90 days after infection, starting about 7 days post infection [6]. However, shorter periods of shedding are more common in field cases [5].
Ferrets that have not been appropriately vaccinated are at the highest risk to contract disease. Contact among recently infected, subclinical or clinically affected animals maintains the virus in a population, and constant introduction of naïve animals provides a susceptible population for infection [5]. Introduction of subclinically infected ferrets into a multianimal household or shelter may lead to severe disease outbreaks [21]. Outbreaks are often reported in geographical areas where most animals come from one or just a few shelters or pet shops [21]. In enzootic areas where animal populations are high, clinical disease is mostly seen in young animals following loss of maternal antibodies at 3–6 months of age. In isolated populations with no protective antibody titers, CDV may cause an epizootic, and outbreaks may be severe, widespread, and affect all ages [5,18].
CDV can cause severe disease in wild carnivore populations and outbreaks have mainly been reported in social species or species with a high density, such as African lions (Panthera leo), seals (Phoca spp.), African wild dogs (Lycaon pictus), black-footed ferrets (Mustela nigripes), raccoons (Procyon lotor), and cetaceans [18,22–25]. In solitary animals such as stone martens, outbreaks of distemper have been reported to be coincident with the breeding season, where these animals have increased contact with conspecifics [18].
Clinical Signs
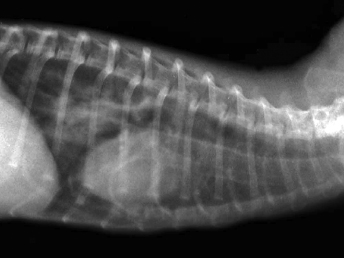
Clinical signs include lethargy, loss of body condition, semiclosed eyes, oculonasal discharge, dyspnea, neurologic signs, and dermatologic problems [1,7,21,26,27]. Lung auscultation can reveal increased lung sounds, and tachypnea can reach 80 breaths/minute in advance cases (reference range, 33–36 breaths/minute [21]). Radiographs may demonstrate a characteristic interstitial pattern (Fig. 20.1). Abdominal palpation may reveal splenomegaly, an unspecific indicator of disease in ferrets [21,26,28,29]. Body temperature may be as high as 41°C (reference range, 37.7–39.1°C) in the early stages of the disease [7]. This initial raise in body temperature may not be consistent [21,29]. Temperature returns to normal at the mid stages of the disease, and animals may become hypothermic just prior to death [21,30]. Experimental infections with ferrets have demonstrated that a temperature increase after infection is sustained, but not very severe, sometimes just up to 39.5°C [30–32]. This may account for the irregular detection of hyperthermia in clinical cases. Transient fever is also seen in ferrets that become infected but are able to clear the disease [30]. In addition, fever is also influenced by the strain of CDV, and some strains may not cause fever [30] or produce variable fever [32].
Fig. 20.1. Thoracic radiograph, ferret with CD. Lung opacities indicative of pneumonia. (Reprinted from Perpiñán et al. Vet Rec 163: 246–250, 2008, with permission from BMJ Group.)
The mortality of ferrets with clinical signs of CD is very high, approaching 100% [7,33], although some animals may recover with treatment [21]. Infections with less pathogenic strains produce a significantly lower mortality rate [34]. Disease outcome is likely affected by multiple factors including age and immune status of the ferret, strain of virus, infective dose, and route of infection.
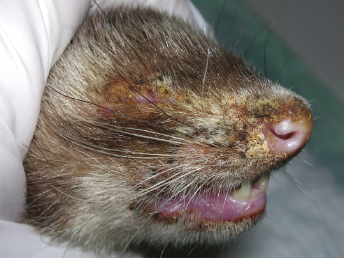
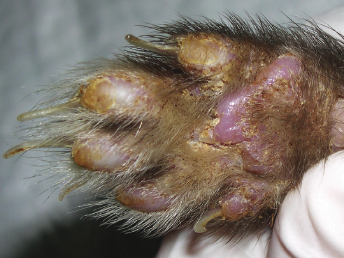
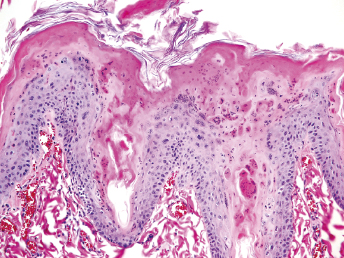
There have been numerous studies of experimental infection of ferrets with CDV. Experimentally inoculated animals have incubation periods of 4–10 days, and mortality at 5–28 days after the onset of the clinical signs is very high [1,27,30,31,34,35]. The course of the disease has been reported to be 12–16 days for ferret-adapted CDV and 21–35 days for infection with wild canine strains [7,21]. Affected animals generally develop systemic disease with respiratory, dermatologic, and neurologic signs. The first clinical signs are anorexia, pyrexia, photophobia, and serous nasal discharge [1,7,17,26–28,30,31,33,34,36–40]. An erythematous, pruritic rash appears on the chin and eventually spreads to the inguinal area (Fig. 20.2). Generalized pruritus, particularly in the dorsal cervical and interscapular areas, and generalized desquamation has only been rarely reported [28,29,41]. A case of CD in a ferret with just dermatologic signs has been reported [29]. The oculonasal exudate becomes mucopurulent and develops into brown, encrusted material surrounding the lips, nose, chin, and eye; the eyelids usually stick shut. Hyperkeratosis of the footpads occurs inconsistently (Fig. 20.3). Secondary bacterial infections, such as pneumonia, can be seen, and the ferret may die of it or live long enough to develop neurologic signs [1,7,17,21,26–28,30,31,33,34,36–40].
Fig. 20.2. Ferret with CD. Severe crusting dermatitis in the face. (Reprinted from Perpiñán et al. Vet Rec 163: 246–250, 2008, with permission from BMJ Group.)Fig. 20.3. Ferret with CD. Hyperkeratosis and crusting of the foot pads is a characteristic lesion of CD in ferrets. (Reprinted from Perpiñán et al. Vet Rec 163: 246–250, 2008, with permission from BMJ Group.)
Neurologic signs occur commonly in the advanced clinical stages of CD in ferrets [7,26,31,41] and are usually the cause of death or euthanasia. However, different CDV strains vary in neurotropism [1,6]. For some strains, neurologic signs are determined by the duration of clinical signs; the longer the course of disease, the more likely ferrets will develop neurologic signs [42]. Clinical signs of nervous system disease include myoclonus, paresis, muscular tremors, convulsions, and coma prior to death. Pneumonia is less common or nonexistent with these neurovirulent strains [26,31]. In contrast, strains with low neurovirulence seldom produce neurologic signs [42]. Such attenuated CDV strains produce mild illness with some deaths caused by pneumonia, but do not develop neurologic signs [34]. Ferrets are at high risk to develop clinical disease following vaccination with attenuated live vaccines [26]. The clinical signs and lesions in ferrets infected with attenuated live vaccines are similar to those produced by wild type CDV.
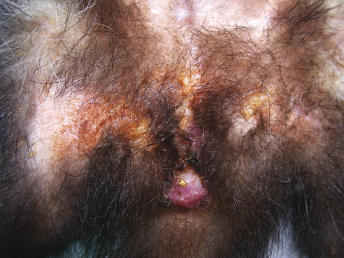
Descriptions of naturally occurring CD in ferrets have rarely been reported in the recent past, except in some shelter and rescue facilities, probably because the disease has a low prevalence as a result of vaccination [7,41]. During natural outbreaks, incubation periods are difficult to determine since the exact time of exposure is unknown [25]. Minimum incubation periods ranged from 11 to 56 days, and the course of the disease ranged from 14 to 34 days [21]. Long courses of disease, up to 7 weeks, have been reported in some cases of natural infections [29,41]. In natural infections, lethargy, often described by owners as increased sleeping times, hyporexia, and inability to gain weight as well as coughing, sneezing, serous oculonasal discharge, semiclosed eyes, and dyspnea have also been reported as early clinical signs. During the course of disease the oculonasal exudate becomes mucopurulent and develops into brown, encrusted material surrounding the lips, nose, chin, and eye (Fig. 20.2). The eyelids usually stick shut. Soiled perineal area and pasty or green feces have occasionally been reported (Fig. 20.4). Generalized and severe pruritus appears in the mid-late course of the disease and is followed by desquamation, particularly in the interscapular and dorsal neck areas, and later by foci of hyperkeratosis (small scabs). Loss of body condition and poor quality of the hair are also common and progressive. In the final stages of the disease, the affected ferrets developed extensive crusting of the head, marked pruritus, and severe respiratory distress. Secondary bacterial infections, such as bacterial pneumonia, may occur and often cause death of the affected ferret [1,17,21,27,28,30,33,34,36–40].
Fig. 20.4. Ferret with CD. Dermatitis of the perineal area. (Reprinted from Perpiñán et al. Vet Rec 163: 246–250, 2008, with permission from BMJ Group.)
Similar differences between natural and experimental CDV infections have been reported in other species such as the black-footed ferret. When naturally infected with CDV, black-footed ferrets had long incubation periods of up to 52 days and a long course of disease of up to 48 days. In such cases, pruritus was intense, and neurologic signs were absent [25]. However, when CD was induced experimentally, pruritus was not observed, animals died with neurologic signs, and the incubation period and the course of disease were shorter and similar to those reported for experimental CD in ferrets [43]. According to these results in ferrets and black-footed ferrets naturally infected with CDV, it can be hypothesized that strains commonly circulating in natural populations are not as pathogenic as the strains used to develop vaccines or to produce experimental infections. This is compatible with those natural strains being dog-adapted.
Pathology
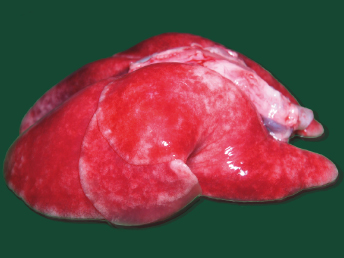
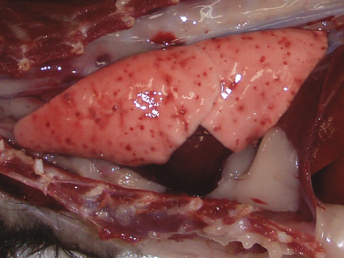
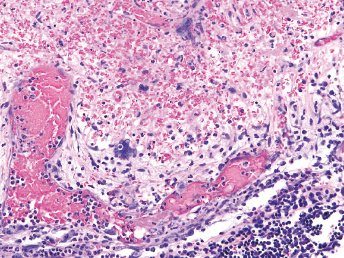
CD is a systemic disease that affects particularly the cutaneous, ocular, respiratory system, and central nervous system (CNS) [5]. Lesions at necropsy are similar to those seen in the dog and are often limited to cutaneous and ocular alterations. Gross lesions include oculonasal discharge, hyperkeratosis of the planum nasale and footpads (Fig. 20.3), and a papular rash beginning on the chin and progressing to a generalized form. Ocular discharge is often the first sign of infection and results in the development of crusts and adherence of the eyelids. Other lesions include thymic atrophy in young animals and catarrhal to mucopurulent rhinitis, catarrhal and/or hemorrhagic tracheitis and bronchitis and severe interstitial pneumonia (Fig. 20.5) that becomes purulent in late stages of the disease [5]. Splenomegaly is a nonspecific finding in ferrets that can also be observed with CD [21].
Fig. 20.5. Lung, ferret with CD. Interstitial pneumonia characterized by wet, heavy, noncollapsed lungs. (Reprinted from Perpiñán et al. Vet Rec 163: 246–250, 2008, with permission from BMJ Group.)
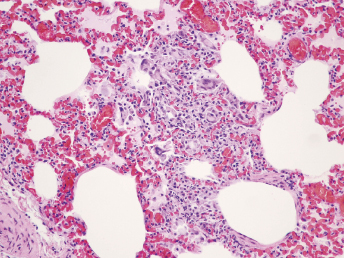
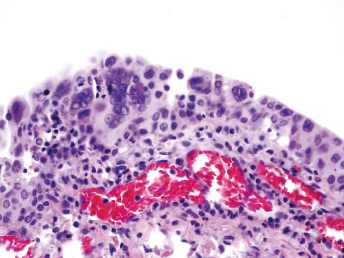
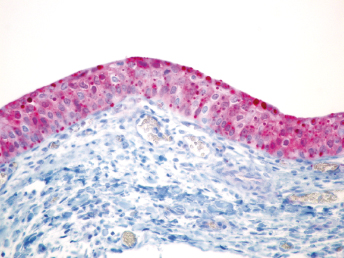
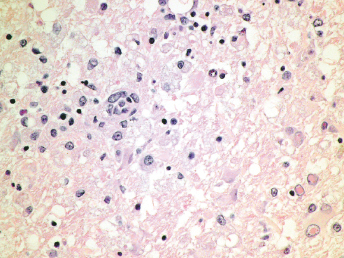
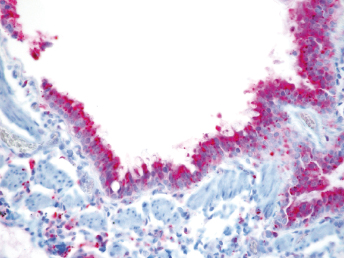
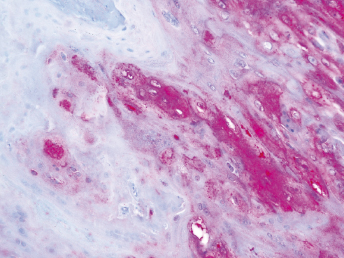
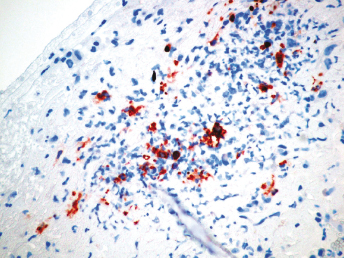
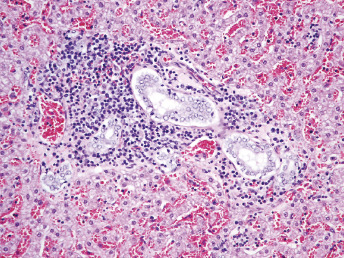
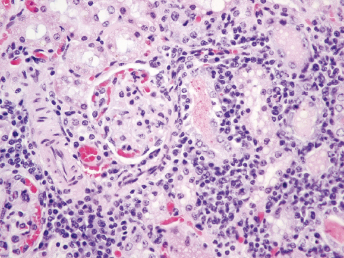
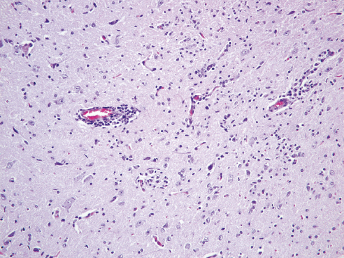
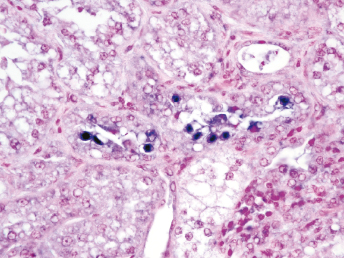
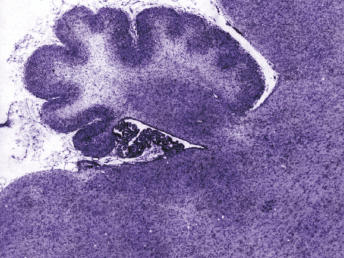
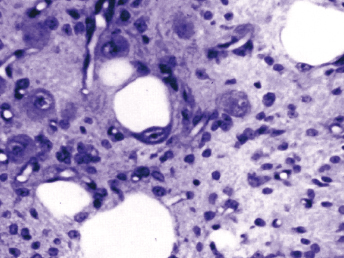
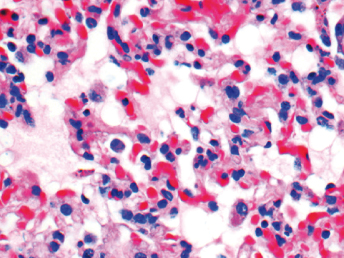
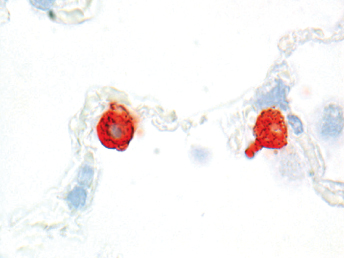
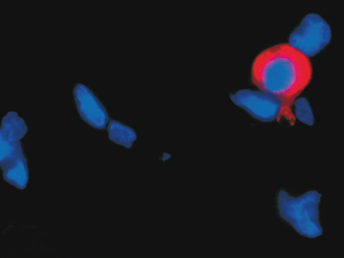
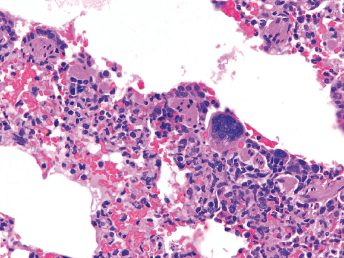
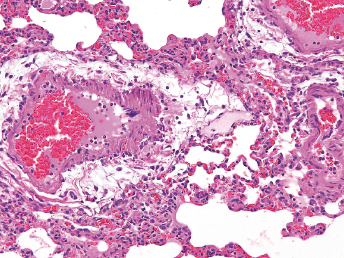
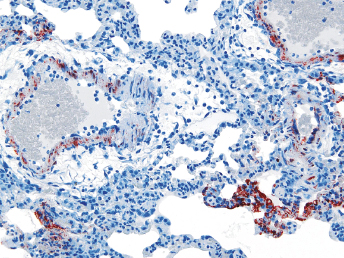
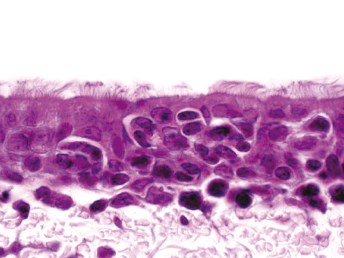
Microscopically, the primary lesion in the lungs is a severe interstitial pneumonia with necrotizing bronchiolitis that is commonly associated with bronchiolar syncytial cells (Fig. 20.6) and eosinophilic intracytoplasmic inclusions (Fig. 20.7) of bronchial epithelial cells [1,21,26]. Suppurative bronchopneumonia due to secondary bacterial infections is commonly observed and may be the primary lesion in young ferrets [5,18]. Brightly eosinophilic, 2–5 μm intracytoplasmic and, occasionally, intranuclear inclusions may be seen in a wide variety of epithelial cells (transitional epithelium of the urinary tract) (Fig. 20.7 and Fig. 20.8), epithelial cells of the gastric mucosa, conjunctiva, gallbladder, liver, pancreas, epididymis, salivary, and adrenal glands, and occasionally in white blood cells and megakaryocytes [1,5,7,26,29]. Inclusions are also found in astroglia and neurons of the CNS (Fig. 20.9). Inclusions occur at much lesser numbers in the epidermis and epithelium of hair follicles, small intestines, and cornea. The primary ocular lesion is a mucopurulent conjunctivitis. Other ocular lesions include corneal ulceration, keratoconjunctivitis sicca, and blepharitis. Lymphoid necrosis may be widespread in the lymph nodes, spleen, and lymph nodules of the intestines. Additionally, multinucleate cells may be found in any of these sites. Nonsuppurative encephalitis with demyelination may be seen in animals with neurologic disease. Microscopic lesions in the skin consist of hyperkeratotic and parakeratotic dermatitis with syncytial cell formation (Fig. 20.10) and intracytoplasmic inclusions (Fig. 20.7) affecting especially not only the footpads, but also hair follicles and sebaceous glands in the areas with the grossly observed cutaneous rash. Myocardial necrosis is rarely observed.
Fig. 20.6. Lung, ferret with CD. Interstitial pneumonia with alveolar walls expanded by monocytes and necrotizing bronchiolitis with syncytial cells. H&E.Fig. 20.7. Bladder, ferret with CD. Lymphoplasmacytic cystitis with syncytial cells and intracytoplasmic eosinophilic viral inclusion bodies. H&E.Fig. 20.8. Urinary bladder, ferret with CD. Distemper viral antigen (red) is detected in the cytoplasm of urothelial cells. Note the strongly labeled intracytoplasmic inclusion bodies. Vector red chromogen, hematoxylin counterstain.Fig. 20.9. Cerebrum, ferret with CD. Lymphoplasmacytic encephalitis with glial nodules and intranuclear eosinophilic viral inclusion bodies. H&E.Fig. 20.10. Foot pad, ferret with CD. Hyperkeratotic dermatitis with syncytial cells. H&E.
Changes in hematologic and biochemical parameters are nonspecific, although lymphopenia is commonly reported in dogs and other animals [5,18]. Ferrets experimentally infected with CDV usually develop leucocytopenia [30–32]. In ferrets with natural distemper, mild nonregenerative anemia is commonly observed, with neutrophilia or lymphopenia being less consistently seen [21]. Plasma biochemistries are usually unremarkable, although protein electrophoresis can reveal increases in α- and β-globulins [21].
Bone marrow cytology may reveal erythroid hypoplasia with eosinophilic intracytoplasmic bodies in erythroid precursors, and spleen aspirates may show extramedullary hematopoiesis [21]. Radiographs may show splenomegaly and changes in the lung compatible with pneumonia in those cases with respiratory signs [21]. Bronchoalveolar lavages can be performed in cases with respiratory involvement to obtain samples for culture and sensitivity [21]. Skin scrapings and culture for dermatophytes may help to differentiate the skin lesions from scabies and dermatophytes, respectively [29].
Pathogenesis
CDV is a pantropic virus that infects a wide variety of epithelial and hematopoietic cells as well as neurons and glial cells. Airborne infections occur most commonly, and after entrance into the upper respiratory tract, the virus replicates in respiratory epithelium and regional lymph nodes. The respiratory tract epithelium and the regional lymphoid tissue are the primary sites of viral replication. The cervical lymph nodes have been shown to be the primary site of experimental infection by some [44]. Following detection of CDV in few dendritic cells on the second day after inoculation, the number of CDV-positive dendritic and lymphoid cells increased greatly as the infection progressed. CDV displays strong lymphotropism, which correlates with the presence of its principal receptor, the signaling lymphocytic activation molecule (CD150), on a variety of immune cells [45,46]. Local manifestation is followed by virus spread via lymphatic vessels to the mediastinal and the mesenteric lymph nodes. Viremia develops within 4–5 days as evidenced by detection of viral particles in white blood cells, but virus has been detected as early as 2 days after infection [36,47]. Viremia persists until viral neutralization or death of the infected ferret [36,47]. Over the next week, hematogenous spread occurs. The virus spreads to essentially all tissues through dissemination by white blood cells, and replication can readily be detected in the spleen, in Kupffer cells in the liver and, after 1 week of infection, in epithelial cells in the kidneys, the gastrointestinal tract, and the urinary bladder. Viral antigen is persistently present, and infected animals become moribund and succumb. As the infection progresses, viral particles aggregate and form larger oval bodies that correspond in size and shape to eosinophilic inclusion bodies that are highly antigenic [48,49]. Neurovirulent strains of CDV that do not kill infected ferrets within 2 weeks and cause neuroinvasion result in neurologic disease. CDV infection of the brain occurs in ferrets through vascular endothelial cell infection as the initial event of CNS invasion as well as rapid and early-onset CDV infection of choroid plexus epithelia and ependymal cells at 4 days post infection [50]. CDV-positive platelet microthrombi have been detected in many capillary beds, including the CNS, suggesting that CDV-positive platelets are an important agent for the transmission of infectious virus to endothelium directly by fusion of platelet membranes to endothelial cells or indirectly by altering microvascular integrity and permeability leading to microthrombosis [50]. Furthermore, an anterograde pathway has been described via the olfactory nerve [51,52]. CNS invasion and infection of glial cells and neurons only occurs after massive infection of the lymphatic system and spread of CDV to epithelial cells throughout the body. In animals that mount an effective cellular and humoral immune response, perivascular lymphocyte cuffing and infiltration of the infected areas occur at the same time as the cutaneous rash and other clinical signs recede. CDV-neutralizing antibodies can be detected in the serum and cerebrospinal fluid (CSF) [51,53,54]. The immune response results in clearance of CDV from the CNS, but can be accompanied by continued demyelination, and the development of neurologic signs in a subset of animals several weeks after recovery from the acute infection [55,56]. This mechanism has been mainly reported in dogs, while most ferrets fail to mount such an immune response and CDV continues to spread through the brain, causing widespread infection of different CNS cell types [53,57]. The distribution of this spread depends on the port of entry, either from the outer layers into the parenchyma or anterogradely from the olfactory nerve through the mitral cells further along the olfactory signaling route [51].
Essentially 100% fatal, virulent CDV results in an accelerated syndrome that closely mimics disease seen in dogs. The virus is shed in the nasal exudates of infected animals and is readily spread by aerosolization. Virus can be detected in nasal secretion from 5 to 13 days post infection. Within a colony of ferrets, once infection is established, it is likely the entire stock will become infected until isolation of clinically healthy stock is undertaken. The disease is profoundly immunosuppressive, with animals that survive the acute catarrhal phase of the disease succumbing to neurologic dysfunction within several weeks.
Ferrets have also been shown to be an excellent model for infection with other morbilliviruses [1,2]. Ferrets experimentally infected with PDV developed characteristic clinical signs of distemper, including fever and rash at 10 days post inoculation, and histologic lesions consistent with infection with morbilliviruses were observed [2]. The most severe lesions were observed in the lungs and were characterized by multifocal to coalescing interstitial pneumonia with severe type II pneumocyte hyperplasia and necrotizing bronchiolitis. Lymphoid depletion was observed in the lymph nodes, thymus, and spleen, and there was a mild ulcerative conjunctivitis and multifocal neuronal degeneration cerebral gliosis [2] Characteristic eosinophilic intracytoplasmic inclusion bodies were detected in bronchial, biliary, and urothelial cells [2]. Similarly, ferrets inoculated with CDV isolated from lions developed fatal disease with clinical signs and morphologic lesions closely resembling CD [1]. Briefly, inoculated ferrets became anorectic, exhibited oculonasal discharge, and became moribund at 5–6, 9–12, and 12–22 days post inoculation, respectively [1]. Ferrets developed severe bilateral conjunctivitis, and inclusion bodies were detected microscopically in epithelial cells of the skin, conjunctiva, gallbladder, liver, pancreas, stomach, trachea, lung, urinary bladder, and kidney [1].
Ferrets have been used as a model for measles virus (MV) infections, including vaccine development, measles-induced immunosuppression, and measles-associated neurologic complications, such as subacute sclerosing panencephalitis [3]. Human infants under 1 year of age often have a poor response to MV vaccination due to the presence of maternal antibodies, which is similar to the situation in ferrets that are also born with maternal antibodies that diminish as the animal ages [58,59]. In ferret kits, born from CDV-vaccinated mothers, the half-life of virus-specific maternal antibodies of approximately 9 days is shorter than in human infants, but overall, the antibody response in ferret kits recapitulates the overall aspects of maternally acquired anti-MV antibodies in human infants [59]. Therefore, ferrets have been extensively used to characterize the interactions of live-attenuated morbillivirus vaccines with maternal antibodies, both as a model and to protect ferrets against CDV [3]. For more details on vaccination of ferrets with CDV, see section on “Treatment and Prevention.”
In ferrets infected with CDV, immunosuppression is sustained throughout the acute phase of the disease and persists for weeks after virus clearance [30,60]. A genetically modified CDV has been used to characterize virus–host interactions in time course studies that demonstrated limitation of the early viral infection to lymphatic tissues with T and B cells as the primary targets [61]. As previously discussed, CDV only spreads to epithelial cells after massive infection of the immune system that is followed by gradual neuroinvasion [51]. The importance of immune cell infection for disease progression was further demonstrated by the inability of a CDV that did not recognize the main CDV receptor CD150, also known as SLAM (signaling lymphocyte activation molecule), which is expressed in lymphocytes and other immune cells, to spread to other cell types [62]. A V gene-defective CDV multiplied with reduced efficiency in lymphocytes and did not inhibit the interferon and cytokine responses [62]. This is similar to MV infection, where a V gene encoded protein mediated a block in cytokine induction in mouse models [63]. The observed early strong interleukin 10 response and the sustained induction of interleukin 2, 4, 6, 12p40, and interferon gamma is similar to response of human patients with naturally acquired MV infection [3,63,64].
MV causes three distinct forms of either early or late central nervous disease: immune-mediated acute disseminated encephalomyelitis (ADEM) that occurs within 2–4 weeks after infection; the rare inclusion body encephalitis (MIBE) that occurs in immunocompromised patients within weeks after recovery of the initial disease; and subacute sclerosing panencephalitis (SSPE) that occurs months to years after the initial infection [3]. For more than 40 years, ferrets have been used as a model to study SSPE [65]. Ferrets inoculated intracerebrally with brain cell cultures from SSPE patients developed acute encephalitis [65]. Second passage of the agent produced more severe disease in ferrets and the authors concluded that their inability to isolate MV would suggest a defective form of the virus [65]. In subsequent studies, the SSPE strains were found to be highly cell-associated and, in contrast to wild-type MV or SSPE strains inoculated within cell-free media, would consistently cause encephalitis in intracerebrally inoculated ferrets [66]. In ferret cell culture, SSPE strains produced multinucleated syncytia identical to those of MV in human cell cultures, and MV antigen was detected in both cytoplasm and in intranuclear inclusion bodies [67]. Further studies established the importance of the cell culture used for virus propagation and the cell-associated SSPE strain for the course of disease [68]. Unfortunately, in contrast to human SSPE cases, microscopic and ultrastructural changes were less severe and sclerosis was not observed, despite severe clinical signs and high numbers of virus in the brain. [69–71]. To reproduce SSPE more accurately, a nonproductive SSPE strain was inoculated into ferrets that had been previously immunized with a live-attenuated MV vaccine [72]. Ferrets developed subacute encephalitis with widespread inflammation in both the white and gray matter in the brain and sometimes in the spinal cord associated with the presence of cell-associated nonproductive MV [72]. Ferrets that developed subacute encephalitis also had significant measles-specific IgG concentrations in the CSF and lacked antibodies against the viral matrix protein despite high titers against other structural proteins, all features more consistent with SSPE [68,72,73]. When cell-associated nonproductive SSPE virus was inoculated intracardially, the virus spread to the brain causing encephalitis within 5–7 days but was not detected in the blood [4]. In the most recent studies, the role of the matrix (M) protein that mediates the interaction between the envelope and internal viral proteins during particle assembly and egress in the pathogenicity of MV was investigated [74]. M mutations have been found in SSPE strains, and can affect the strength of interaction with the envelope glycoproteins, assembly efficiency, and viral spread [74]. Inoculation of ferrets with a recombinant wild-type CDV carrying a vaccine strain M protein resulted in attenuated disease and caused only a mild and transient leukopenia. These data indicate that the differences in particle infectivity and envelope protein sorting mediated by the vaccine M protein contribute to vaccine strain attenuation [74]. By reproducing key features of the various aspects of MV infections, inoculation of ferrets with human isolates provides unique insights into MV pathogenesis [3].
Diagnosis
The diagnosis of CD is usually based on the history, the clinical signs, and fluorescent antibody labeling or real-time polymerase chain reaction (RT-PCR) of conjunctival smears or respiratory secretions [5,18]. Clinical signs are very characteristic, especially when respiratory, dermatologic, and neurologic signs occur simultaneously. The diagnosis may be more challenging during the initial stages of the disease or when only a particular body system is affected. The main differential diagnosis for ferrets presenting only with respiratory signs is influenza, but CD progresses faster and causes more severe clinical signs, including marked anorexia and lethargy [7,33]. Lesions produced by mange (sarcoptic and demodectic) may resemble dermatologic lesions similar to those of CD, but scabies is a rare disease in ferrets, and the distribution of cutaneous lesions in ferrets with CD is quite characteristic [75–78]. Conditions other than CD that cause neurologic signs in ferrets include Aleutian disease, rabies, ferret systemic coronavirus infection, botulism, brain tumors, toxicoses, fungal infections of the brain (cryptococcosis, blastomycosis), and toxoplasmosis [79,80].
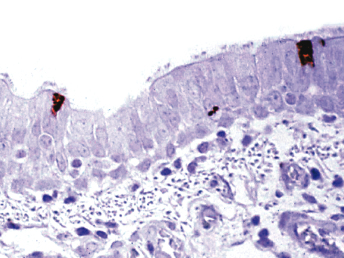
Immunofluorescent antibody labeling is usually performed on cytologic smears prepared from conjunctival, tonsillar, and respiratory epithelium or secretions [5] using monoclonal antibodies directed against the nucleocapsid protein of CDV [81], the major structural protein. However, the technique can also be performed on bone marrow, urine sediment, cells in the CSF, and blood (buffy coat) [5]. Fluorescence in blood mononuclear cells is detected earlier than in conjunctival smears in the course of infection, but false-negative results may be encountered if leukopenia is present [28]. Other diagnostic techniques include immunohistochemistry (Fig. 20.8, Fig. 20.11, and Fig. 20.12), RT-PCR, in situ hybridization, serum antibody testing, viral isolation, and histopathology [5,18,82]. Immunohistochemistry can be performed on frozen or routinely formalin-fixed, paraffin-embedded biopsy, or necropsy samples [5] using monoclonal antibodies directed against the nucleocapsid protein of CDV [81]. In situ hybridization has rarely been used for diagnostic purposes, and only probes that are complementary to virulent CDV are able to detect viral RNA in all tissues, including the brain [83]. RT-PCR can be used to detect CDV RNA in fresh and formalin-fixed tissues, CSF, blood, or any other secretory material or exudates [5]. Animals that die from distemper usually have abundant quantities of virus in spleen, tonsils, lymph nodes, stomach, lung, duodenum, bladder, and brain [5]. Fluorescent antibody and RT-PCR testing on samples from conjunctival smears, mucous membrane scrapings, or blood smears may result in a false positive in the first few weeks post vaccination with a modified live vaccine (MLV) [33]. In cases of suspected morbillivirus infections, a broadly reactive, pan-paramyxovirus RT-PCR that use consensus-degenerate hybrid oligonucleotide primers, combined with sequence analysis, may be useful to identify CDV amplicons [84]. Specific RT-PCRs based on the phosphoprotein gene of CDV have been used to differentiate wild-type from vaccine strains of CDV [84].
Fig. 20.11. Lung, ferret with CD. Distemper viral antigen (red) is detected in the cytoplasm of bronchial epithelial cells. Vector red chromogen, hematoxylin counterstain.Fig. 20.12. Food pad, ferret with CD. Distemper viral antigen (red) is detected in the cytoplasm of syncytial cells and keratinocytes. Vector red chromogen, hematoxylin counterstain.
Viral isolation is possible [5], and may be very useful to distinguish vaccine virus from field strains. In dogs with distemper, CDV has classically been isolated by cocultivation of lymphocytes from suspect animals with mitogen-stimulated dog lymphocytes [85]. Field isolates of CDV also replicate in dog and ferret macrophages [86,87]. In contrast, Vero cells do not allow the propagation of field isolates, whereas cell culture-adapted CDV strains such as the Onderstepoort vaccine strain are able to replicate in many cell lines [6,88]. The marmoset B-cell line B95a has also been shown to be a good host for CDV isolation from clinical cases [89]. Virulence for the natural host may be lost when CDV is adapted to cell culture [90]. As previously discussed, SLAM is the principal cellular receptor for morbilliviruses including CDV replicating in vivo, as evidenced by their common tropism and pathology. This would suggest that SLAM expressing cells are ideally suited for isolation of field strains of CDV [46,91]. Indeed, a stable transfected Vero cell line that expresses canine “signaling lymphocyte activation molecule” (SLAM; CD150) receptors (Vero.DogSLAMtag) has been shown to be highly suitable for isolating and characterizing morbilliviruses [2,91]. CDV replicates faster and produces higher titers when propagated in Vero.DogSLAMtag cells [2]. In contrast, vaccine strains of CDV, which have been passaged on SLAM-negative cells, were found to use an alternative receptor besides SLAM, most likely because of in vitro adaptation [46].
Serology is commonly used in experimental studies [1,31,92,93]; however, it is not particularly useful in clinical cases. Antibodies against CDV may persist for long periods of time after vaccination [5], and therefore, titers in previously vaccinated ferrets provide little information about clinical disease. In addition, clinically affected animals are usually immune suppressed and are unable to develop a proper antibody response [5,7,30]. Infection with CDV can actually lower anti-CDV antibodies in ferrets [27] due to increased antigen–antibody complex formation and clearance, virus-induced B cell immunosuppression, or decreased antibody induction attributable to decreased virus replication in vaccinated ferrets [27]. The neutralization test is considered the gold standard, but enzyme-linked immunosorbent assay (ELISA) to detect IgM can also be used to detect acute infection. IgG titers are ambiguous and can indicate either past or present infection or previous vaccination [5]. In dogs, detection of anti-CDV antibodies in CSF offers definitive evidence of distemper encephalitis. In ferrets, anti-CDV antibodies are not detected in the CSF of vaccinated ferrets or those with systemic CD without CNS disease [5]. It should also be remembered that infection does not equal disease, and 25–75% of dogs infected with CDV develop subclinical disease, but no clinical signs [5]. The same might be true for ferrets.
Treatment
Treatment of ferrets should be started as soon as a presumptive diagnosis of CD is made, although it is unlikely that animals with severe clinical signs or those with neurologic signs will respond [1,21]. Therapy should always include broad spectrum antibiotics, as secondary infections are common (e.g., brochopneumonia) [7,21,33]. If possible, the antibiotic should be selected based on culture and sensitivity of samples obtained through tracheal wash. Administration of vitamin A reduced mortality in ferrets with experimental CD; and should be administered at doses of 50,000 IU (15 mg) of renitol palmitate IM SID for two treatments [94]. Administration of two doses of vitamin A has also been useful to reduce mortality in humans with measles (another Morbillivirus) [95]. High doses of vitamin C were suggested decades ago as a very effective treatment for distemper in dogs [96]. In that study, all dogs affected with CD were treated with vitamin C (at 1 g IV SID for three treatments), and all of them recovered from disease [96]. Although the results of that article are now difficult to believe, treatment with vitamin C (reducing the dose for ferrets) should be considered as toxicity due to treatment seems unlikely. Hyperimmune serum against CD was useful in resolving one initial case of distemper in a ferret [21]; if not commercially available, 1 mL IV of hyperimmune serum from a healthy and appropriately vaccinated ferret can be used, although this has not been proven scientifically [97]. Nutritional support (with easily digestible diets, amino acids, and electrolytes) is important in clinical cases, as affected ferrets lose appetite and develop a poor nutritional status, which reduces the immune function required to fight the disease. Keeping affected ferrets hydrated is also important [28]. Some ferrets present in the final stages of the disease with intense pruritus, and treatment with antihistamines is not always effective; corticosteroids can be administered in these ferrets prior to euthanasia to alleviate pruritus [21]. Other symptomatic treatments with anti-inflammatory drugs or bronchodilators may be indicated in some cases. Isolation of affected cases is mandatory [7].
Vaccination can also be used as a treatment in the face of an outbreak. Vaccination at the same time or a few hours after infection can be effective for disease prevention, but larger doses of vaccinal virus are necessary [98]. The time interval required for development of resistance is inversely related to the dose of modified virus; the more time elapsed between infection and vaccination, the higher the dose of vaccinal virus required and the lower the survival rate [98]. Vaccination is not generally effective once more than 48 hours have elapsed between infection and vaccination, or when the amount of virus is too high or the amount of vaccinial virus too low [98]; however, vaccination in these situations can delay onset of clinical signs and can increase duration of disease [21,98].
Treatment should always be discussed with the owners, and a very poor prognosis should be given in order for them to take an appropriate decision. Euthanasia may be suggested based on poor prognosis and obvious suffering of the ferret in advanced cases (severe respiratory distress, intense pruritus, lethargy, inappetence, etc.). Only those owners fully aware of the prognosis and totally dedicated to the ferret should continue treatment once the disease has progressed.
Prevention
Types of Vaccines
Common vaccination of ferrets against CDV has resulted in a rare occurrence of this disease. There are several types of vaccines and protocols that have been used to vaccinate ferrets, and they will be discussed in this chapter. Inactivated vaccines are inferior in protection to MLVs [92,93], and therefore, they are not commonly used. MLVs of dog or mink origin are able to produce disease [26,28], and they are not used either. It has been reported that CD produced by MLV of canine origin may have particular characteristics different from more typical CD in ferrets. Davidson [28] reported cases where vaccine-induced CD was nonfatal, and Gill et al. [26] reported an outbreak on a ferret farm where animals with postvaccinal distemper were not able to transmit the disease to adjacent nonvaccinated animals.
MLVs of avian origin (i.e., propagated in chick embryo tissues) are considered to be safe in domestic ferrets and other species [6,9,93], but have produced mortality in black-footed ferrets [43]. One of these vaccines was approved for use in ferrets in the United States for several years (Fervac-D, United Vaccines Inc., Madison, WI), but its production was discontinued, probably because of the number of adverse reactions it caused. MLVs of avian origin are available and approved for dogs in many countries: these vaccines are generally safe and effective in ferrets. Many of these vaccines use the strain Onderstepoort. Multivalent dog vaccines (with modified live CDV of avian origin) have been used safely in ferrets, either using the full dose or using a fifth of the full dose [7,97]. Approved ferret distemper vaccines are also available in other countries outside the United States [7].
There is an approved vaccine (in the United States) for CDV in ferrets (PUREVAX ferret distemper, Merial, Duluth, GA). This is a recombinant vaccine that contains only a small portion of the genetic material of the virus and, therefore, cannot produce disease. This vaccine uses an attenuated canary poxvirus as vector; that is, small portions of genetic material of CDV are included in this poxvirus. Safety and efficacy has been demonstrated with this type of vaccines in ferrets and other exotic carnivores [99]. This vaccine may be the best option for vaccinating ferrets and other carnivores against CDV. Unfortunately, this vaccine is not yet available in many countries.
Vaccination Protocols
Immunity given to the kit via the mother can interfere with the vaccination [35]. This interference may be less pronounced with recombinant vaccines [35]. Duration of this passive immunity can be between 6 and 14 weeks [97]. Using this information, a prime vaccination protocol at 6, 10, and 14 weeks has been suggested (assuming the dam is vaccinated for CDV) [7]. However, considering that the disease is usually rare and that many ferrets live indoors for most of their time, a series of 2 vaccinations, separated by 4 weeks, between 6 and 14 weeks of age may be adequate [28]. It has also been demonstrated that when vaccination starts when the ferret is older than 3–4 months old (once passively acquired, neutralizing antibodies are no longer present), a series of 2 vaccinations separated by 4 weeks is enough to provide protection [31,32,35,93]. Other studies reported that just one vaccination after the ferret is older than 3 months of age is enough to provide protection [98].
Immunity against CDV is long lasting after recovery from natural infection or booster vaccination [5]. One study demonstrated that a single vaccination with 1000 EID50 (egg infective dose; one EID50 is the amount of virus that will infect 50% of inoculated eggs) of chicken embryo-modified virus given after 18 weeks of age conferred solid immunity in ferrets for at least 6 years [98]. Available CDV vaccines for dogs or ferrets commonly have >3000 EID50. However, cases of CD have been reported in animals vaccinated with MLVs of avian origin 18 and 24 months before disease [29,97]. Immunity to virulent CDV is prolonged or life-long, but not as absolute after vaccination [5]. The protection obtained after clearing the infection is stronger than after vaccination, but it may still be compromised if the animal is exposed to a highly virulent or large quantity of virus or if it becomes immunocompromised or stressed [5].
Intranasal, oral, and intramuscular routes can also be effective to immunize ferrets [32,35,99], but have little advantage over the subcutaneous route. Leukopenia of several weeks duration can be observed after vaccination [31,35,93]. The veterinarian should design a vaccination protocol based on the general information provided in this chapter and the specific factors affecting his/her veterinary practice (types of vaccines available, laws, outdoor or indoor keeping of ferrets, prevalence of distemper in dogs or wild mammals, practice policy, etc.). A vaccination schedule is found in Chapter 26 of this book.
Adverse Reactions to Vaccination
Adverse reactions to distemper vaccination have been reported in the United States [100,101] but are also common in other countries. Adverse effects happen 5–25 minutes after vaccination and consist of an anaphylactic reaction with generalized hyperemia, hypersalivation, and vomiting [100,101]. Dyspnea, cyanosis, collapse, diarrhea, and hematochezia are also noted in some ferrets [100,101]. Incidence of adverse reactions in the United States has been reported to be 1–5% [100,101], but has reached 60% in other countries, for example, Brazil (José Manuel Mouriño, personal communication). The possibility of an adverse reaction increases with cumulative vaccinations; in one study, the risk increased by 80% with each additional distemper vaccination that ferrets had previously received [101]. Another study detected more adverse reactions in older animals [100]. Treatment of an anaphylactic reaction has included the administration of diphenhydramine (1 mg/kg SC, IM or IV, or 0.5 mg/kg PO), epinephrine (0.1 mL of a 1 : 1000 (1 mg/mL) solution SC or IM), dexamethasone (2 mg/kg SC), fluids, and/or supplemental oxygen [97,100,101]. Other treatment protocols or doses could also be effective. Ferrets generally survive with treatment [100,101].
While adverse reactions are common in some countries or with some vaccines, they are quite rare in other countries or with the use of other types of vaccines. Different vaccine components (e.g., virus strain, adjuvants, manufacturing process) could be the cause for this. As an example, Nobivac D and Nobivac DH have been used extensively in Europe without problems [7], but Nobivac DP causes a high incidence of adverse reactions in Brazil (José Manuel Mouriño, personal communication). All these vaccines are manufactured by Intervet and have the same distemper fraction (Onderstepoort strain, >4000 EID50), which suggests that specific adjuvants may cause vaccine reactions, although the parvovirus fraction in the Novibac DP cannot be ruled out as a cause of adverse reactions in that specific case. It is unknown if genetics has any influence in the incidence of adverse reactions. The second author has never seen a case of adverse reaction in ferrets when vaccinating with recombinant vaccines in the United States and with MLV of avian origin in Spain. In the United States, Fervac-D (United Vaccines Inc.) produced anaphylaxis in many ferrets [7,102], while PUREVAX ferret distemper (Merial) is reported to be much safer [100].
Ferrets that have developed an adverse reaction following vaccination against CDV should probably never be vaccinated again, as immunity after a single vaccination can persist for the full life of a ferret. Brand name vaccines that produce significant numbers of adverse reactions should not be used again and should be exchanged for other products. In those areas/countries with a high prevalence of adverse reactions, veterinarians should consider obtaining signed consent forms prior to vaccinating pet ferrets [102]. Although premedication with antihistamines prior to CDV vaccine is commonly used by some veterinarians and can decrease the incidence and severity of adverse effects, better ways to deal with the problem of adverse reactions is to consider the real exposure of the ferret to CDV, the long-lasting immunity after a single vaccination, the availability of different types and brands of vaccines, and the possibility of evaluating anti-CDV antibodies to assess the need for additional vaccination.
A serologic titler >1 : 100 is considered to be effective by some authors [1,31], although clinical distemper (without mortality) has occurred in hybrid ferrets vaccinated with an inactivated vaccine and having a titer of 1 : 256 [93]. Antibody titers can increase with time without any booster [92,93]. It should be noted that there may be other mechanisms other than serologic titers that protect against clinical distemper (e.g., cytotoxic T lymphocytes), and that the titer does not always correlate with protection to the disease [35]. Vaccines can produce protection without eliciting a strong serologic response [99], and therefore caution should be exercised in assessing immunity to infection solely on the basis of serologic titers [99]. As an example, antibodies are detected by 14 days after vaccination [93], but protection can be achieved only 2 days after vaccination with modified live virus vaccine of avian origin [98]. However, serum titers generally correlate well with the level of protection [5].
Disinfection
CDV is susceptible to ultraviolet light, heat, and drying [5], but it can survive in infected tissues for days at 25°C, weeks at 2–4°C, and years at −70°C [5,7]. It can also survive within a pH range of 4.5–9.0 [7]. Commonly used chemical agents, such as quaternary ammonium compounds (e.g., Roccal®-D or Roccal®-D Plus, Pfizer, New York, NY; directions for use should be followed), 0.75% phenol, 2–5% sodium hydroxide and 0.1% formalin, can also inactivate the virus [7]. Virkon-S (a disinfectant made with peroxides) is also effective against CDV according to the manufacturer's label. Fomites (such as handling gloves) are also able to transmit the disease and should also be disinfected [7].
Ferret Coronaviruses
Over the last decade, coronaviral diseases have emerged as some of the most important viral diseases in ferrets. Following the early recognition in March of 1993 of a novel enteric disease in domestic ferrets along the East Coast of the United States, a coronavirus was quickly implicated as the cause [103]. This disease is now recognized as epizootic catarrhal enteritis (ECE). A novel coronavirus, designated as ferret enteric coronavirus (FRECV), was identified in feces of domestic ferrets clinically diagnosed with ECE [104]. A phylogenetic analysis based on the predicted partial amino acid sequences of the polymerase, spike and membrane proteins, and full sequence of the nucleocapsid protein demonstrated that FRECV is a novel alphacoronavirus that is genetically most closely related to feline coronavirus (FCoV), porcine transmissible gastroenteritis virus, and canine coronavirus [104]. Since 2004, a systemic disease, characterized by feline infectious peritonitis (FIP)-like clinical signs and lesions, has been observed in ferrets across the United States and Europe [105–107]. This disease was also found to be associated with an alphacoronavirus that was most closely related to FRECV, and was designated as ferret systemic coronavirus (FRSCV, [105]). Based on sequence data from a limited number of enteric and systemic strains, FRSCV and FRECV are closely related but are genetically distinct [104,108]. In fact, current data indicate that FRSCV differs significantly more from FRECV than FIPV does from FCoV [108].
Etiology
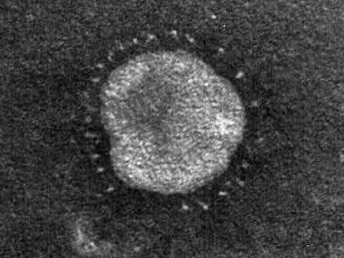
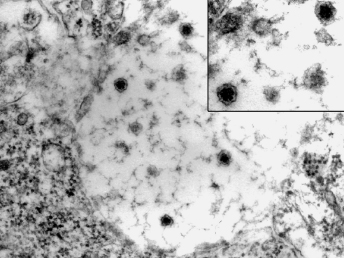
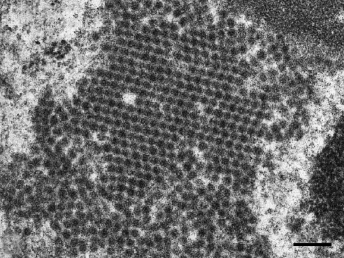
Coronaviruses are large, enveloped, positive-stranded RNA viruses that have been classified in the genus Coronavirus within the family Coronaviridae, order Nidovirales [109]. Coronaviruses have been subdivided based on their sequence data and the subfamily Coronavirinae consists of four genera ([110], http://www.ictvonline.org). Numerous important enteric diseases in domestic animals are caused by group 1 coronaviruses, now classified as alphacoronaviruses, including transmissible gastroenteritis virus (TGE) of swine, FCoV, and canine coronavirus. Both FRECV and FRSCV have also been identified as alphacoronaviruses [104,108]. Whereas FRECV causes ECE, a disease that causes lesions limited to the gastrointestinal tract of ferrets [103,104], FRSCV causes ferret systemic coronaviral disease (FSCD), a disease in ferrets that closely resembles FIP in cats [105,106]. The name coronavirus refers to the characteristic electron microscopic appearance of virions with the viral spike peplomers forming large, bulbous surface projections (Fig. 20.13) that create a crown-like image [109]. These viral spike peplomers populate the surface of coronaviruses and determine their host tropism. The general structure of all coronaviruses is determined by the spike (S), envelope (E), membrane (M), and nucleocapsid (N) proteins [109,110].
Fig. 20.13. High speed concentrate of clarified fecal extract, ferret with ECE. FRECV virion, approximately 120 nm in diameter with regularly spaced array of 20 nm long pin-shaped peplomers. Lead citrate and uranyl acetate stain. (Reprinted from Williams et al., J Am Vet Med Assoc 217: 526–530, 2001, with permission from the American Veterinary Medical Association.)
Transmission and Epidemiology
ECE is a highly contagious diarrheal disease of ferrets with outbreaks usually involving 100% of animals in a household, breeding facility, or shelter. The morbidity of ECE commonly reaches 100%, but overall mortality is low with fewer than 5% of animals dying [103]. Virus is shed in feces and saliva in large quantities over prolonged periods of time. Infection of ferrets occurs primarily through the oral route and ferrets that are losing maternal antibodies are at high risk of becoming infected [104]. Shedding can be intermittent and reinfection may play a significant role in maintaining infection in large ferret populations. Ferrets that are clinically normal but experience a stressful situation may develop mild clinical signs of ECE, and FRECV can be detected in feces by RT-PCR.
During the initial outbreaks before 2000, young ferrets often presented with milder, subclinical disease, whereas older ferrets exhibited more severe clinical signs and had higher mortality rates. The disease quickly spread throughout the United States and to several other countries [103]. Today, the prevalence of FRECV is very high in some ferret populations in the United States and Europe [104,111]. However, severe clinical ECE has rarely been reported in the more recent past [97,112]. In addition, the severity of this disease appears to have been greatly reduced over the past 5 years, compared with the situation in the 1990s [97,112]. Based on our current knowledge, it appears that FRECV has become enzootic, rarely resulting in severe clinical signs. This is supported by serologic data and detection of viral nucleic acid in fecal samples by RT-PCR of clinically normal ferrets. It is therefore important to recognize that positive serology or RT-PCR does not equate with disease [112].
A novel systemic disease, FSCD, infecting ferrets and causing pyogranulomatous perivasculitis and peritonitis, and thereby closely resembling FIP in cats, was first recognized in Spain in 2004 [106]. Subsequently, the disease was diagnosed in ferrets in the United States [105,107]. This systemic coronavirus disease more commonly affects young ferrets, generally less than a year old [105,113]. The disease may initially present in a geographical area as an outbreak, followed by years with low incidence. There have been reports of high incidence of ferrets with FSCD in dense ferret populations, but isolated cases are more commonly reported. The mechanism of transmission is unknown, but ingestion of viral particles is the most likely route.
Clinical Signs
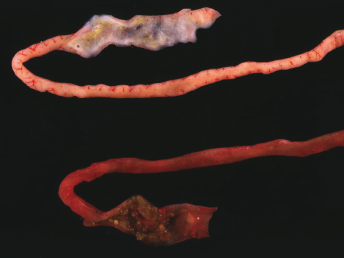
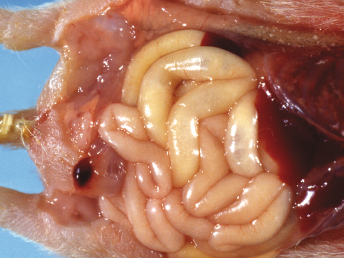
Initial clinical signs of ECE include lethargy, hyporexia or anorexia, and vomiting. Physical examination may reveal lethargy, mesenteric lymphadenopathy, dehydration, and diarrhea [103]. In the original outbreaks, these signs were quickly followed by a profuse, foul smelling, bright green watery diarrhea with a high mucus content, and dehydration (Fig. 20.14). During the more chronic stages of the disease, feces of affected ferrets commonly contained grainy material that resembled birdseeds (Fig. 20.15). This characteristic clinical presentation resulted in the disease being commonly referred to as “green slime disease.” After a detailed review of the gross and microscopic lesions and the discovery of an enteric coronavirus within the affected intestines, the name epizootic catarrhal enteritis (ECE) was introduced [103]. In all ferrets that develop diarrhea, during the acute stages, the feces are typically bright green with a high mucus content [103]. However, similar diarrhea may be observed with other gastrointestinal diseases. As discussed earlier, in the chronic stages of the disease, poor digestion leads to grainy feces resembling bird seeds [103]. Especially in stressed ferrets that develop additional gastric ulcers, brown diarrhea or melena may also be observed [97].
Fig. 20.14. Feces, ferret with ECE. Characteristic bright-green feces with high mucus content from a ferret with ECE. (Courtesy of Dr. Bruce Williams.)Fig. 20.15. Small intestine, ferret with ECE. Sections of jejunum from two ferrets with acute (top) and more chronic ECE (bottom). Acute enteritis is characterized by bright-green diarrhea with high mucus content. The more chronically affected intestine has a thin wall and contains green, “birdseed-like” material.
During the original outbreaks, the clinical history often revealed the introduction of a young ferret that functioned as an asymptomatic carrier into a group with naïve, commonly adult ferrets. Following rapid spread of the disease and high morbidity in such groups of ferrets, the older animals commonly developed more severe disease [103,114]. In the current, more enzootically infected ferret population, severe stress of especially younger ferrets may result in less severe and often intermittent disease.
Clinicopathologic abnormalities have not been reported in cases of ECE, but it is likely to observe changes associated with dehydration. Other changes have also been suggested, such as mild lymphocytosis (due to lymphoplasmacytic inflammation), neutrophilia (due to secondary bacterial infections), increase in lipase and globulins (intestinal inflammation), increases in ALT and GGT (hepatic lipidosis, lymphocytic hepatitis), increases in AST and CK (wasting disease), and hypoalbuminemia (wasting disease) [97]. These are theoretical changes that need to be confirmed in clinical cases.
FSCD has been reported in mostly young ferrets, with the majority being less than 18 months of age [105,113]. Clinical signs in ferrets have been vague and nonspecific, just like the granulomatous or dry form of FIP in cats [106,115,116]. Common clinical signs include diarrhea, weight loss, lethargy, hyporexia or anorexia, and vomiting [105,113]. Gastroenteric disease may cause loss of body condition and moderate to severe emaciation. Occasionally, signs of CNS disease, including hind limb paresis or paraparesis, ataxia, tremors, and seizures have been reported. Some ferrets that present with primary neurologic disease may exhibit head tilt and seizure activity. On abdominal palpation, large abdominal masses, splenomegaly, and renomegaly are commonly detected (Fig. 20.16). Abdominal masses (other than the spleen) may represent enlarged and/or irregular mesenteric lymph nodes, enlarged and/or irregular kidneys, and thickened and/or irregular intestines [105,113]. Peripheral lymphadenopathy has been reported in a few cases, and some ferrets also had fevers ranging from 39.4°C to 40.8°C/102.9°F to 105.4°F [105,113]. Other findings may depend on the organ affected: sneezing, coughing, labored breathing, nasal discharge, dehydration, bruxism, systolic heart murmur, jaundice, focal areas of erythema of the skin, green colored urine, reddened rectal mucosa, and rectal prolapse [105,113]. Ocular signs have not yet been reported.
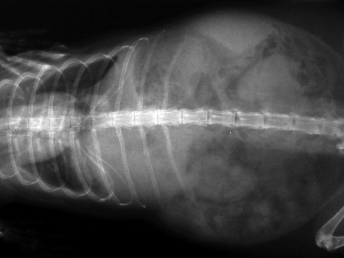
Fig. 20.16. Abdominal radiograph, ferret with FSCD. Fibrinous and granulomatous peritonitis characterized by multifocal radiodense nodules widely distributed throughout the mesentery. (Courtesy of Dr. Katrina Ramsell.)
Hematologic changes are variable and nonspecific, including nonregenerative anemia, neutrophilic leukocytosis, thrombocytopenia, or lymphopenia [105,113]. Polyclonal hypergammaglobulinemia is characteristic of this disease, although concurrent hyperproteinemia does not always occur [113]. When it occurs, hyperproteinemia can be as high as 130 g/L [113]. Globulin levels are usually higher than 42 g/L and represent 75–90% of all the plasma proteins, and gammablobulins are usually greater than 18 g/L and represent 35–60% of plasma proteins [113]. The albumin : globulin ratio is decreased in most cases (0.10 to 0.30) [113]. A moderate increase in α- and β-globulins can also be seen [113].
Serum chemistry values are generally normal, and could change depending on the organ affected: elevations in lipase, blood urea nitrogen, ALT, GGT, and AP have been reported [113]. Radiography (plain and with contrast) and ultrasonography may help characterize abdominal masses [113]. Urine abnormalities can also be noted [105].
Pathology
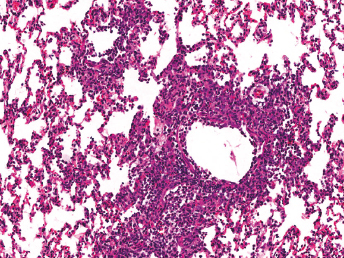
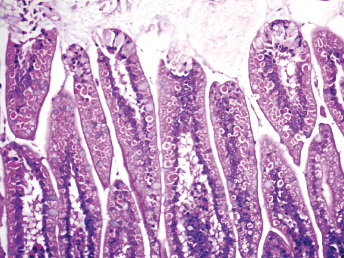
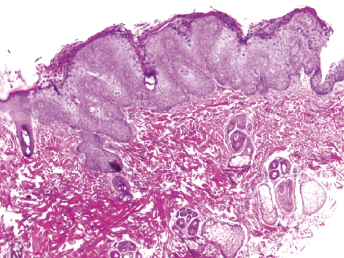
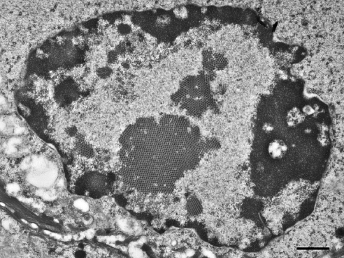
On gross examination of ferrets with ECE, the initial lesions include mucosal hyperemia of affected intestinal loops, which progresses with time to thinning of the intestinal wall. The intestinal contents can range from bright green, mucinous feces in acute stages to grainy fecal material resembling bird seeds in the chronic stages [103]. The most common microscopic lesion is diffuse lymphocytic enteritis with villus atrophy (Fig. 20.17). Vacuolar degeneration and necrosis of the apical epithelium is observed in acutely infected ferrets (Fig. 20.18), whereas fusion and blunting of intestinal villi can be seen at chronic stages [103]. Since the disease progresses segmentally, a combination of all these lesions may be observed throughout the small intestine [103]. Immunohistochemistry using a monoclonal antibody against alphacoronavirus antigen (Fig. 20.19 can be used to detect coronavirus-infected epithelial cells [103,104] Transmission electron microscopy has identified coronavirus-like particles, approximately 120 nm in diameter, in cytoplasmic vacuoles of apical enterocytes (Fig. 20.20) and at the cell surface [103] Similar viral particles have been observed electronmicroscopically in fecal samples from multiple ferrets [103].
Fig. 20.17. Jejunum, ferret with chronic ECE. Characteristic fusion and blunting of villi and prominent lymphocytic enteritis. H&E.Fig. 20.18. Jejunum, ferret with acute ECE. Characteristic vacuolar degeneration of villar enterocytes with intraepithelial lymphocytes. H&E.Fig. 20.19. Jejunum, ferret with ECE. Detection of alphacoronaviral antigen by immunohistochemistry (red) and FRECV nucleic acid by in-situ hybridization (blue), AEC chromogen with hematoxylin counterstain and NBT chromogen with methylen green counterstain. (Reprinted from Wise et al., Virology 349: 164–174, 2006, with permission from Elsevier.)Fig. 20.20. Jejunal enterocytes, ferret with ECE. Highly pleomorphic FRECV virions, approximately 120 nm in diameter, in cytoplasmic vacuoles of apical enterocytes. Lead citrate and uranyl acetate stain. (Reprinted from Williams et al., J Am Vet Med Assoc 217: 526–530, 2001, with permission from the American Veterinary Medical Association.)
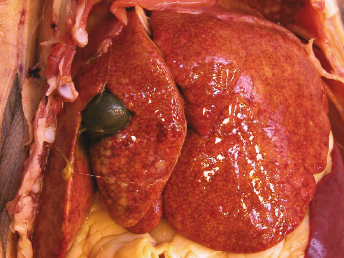
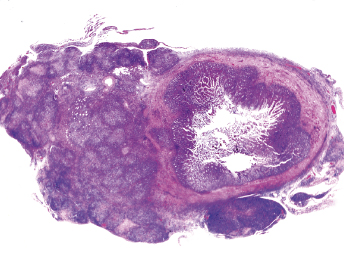
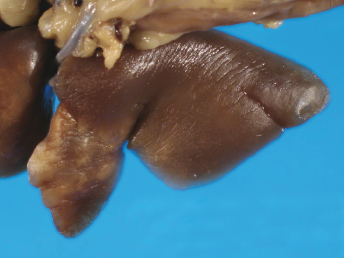
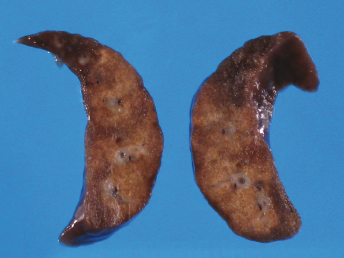
Gross lesions observed in ferrets with FRSCV infection closely resemble those described in cats with the granulomatous or dry form of FIP [105,115,116]. The most commonly observed gross lesion consists of multifocal to coalescing white to tan irregular nodules or plaques (Fig. 20.21) ranging from 0.5 to 2.0 cm in diameter dispersed over serosal surfaces [112]. Nodules are usually oriented along vascular pathways. The peritoneum, particularly the intestinal serosa and the mesentery, are most commonly affected, with the mesentery being multifocally and irregularly thickened (Fig. 20.21) by pale white firm nodules and plaques [105,106,113]. Similar nodules can be commonly found on the surface or extending into the parenchyma of numerous other organs, with the liver, kidneys, spleen, and lung most commonly affected. The mesenteric lymph nodes are affected in most cases [105,113]. They can be enlarged up to eight times their normal size, with a highly irregular capsular surface characterized by dozens of slightly raised white nodules (Fig. 20.22). On cut surface, the normal parenchyma is often replaced by granulomatous inflammation [105]. Other, less-specific gross lesions include the commonly observed splenomegaly, and occasional renomegaly and hepatomegaly [112,113]. Based on current knowledge, ferrets with FRSCV infrequently present with serous effusions into the body cavities that are characteristic of the effusive or wet form of FIP; however, fibrinous exudate is rarely encountered [105,106,115,116]. In animals with neurologic signs, gross lesions within the nervous system were rather limited. Moderate meningeal opacity around the medulla and choroid plexus of the fourth ventricle may be observed. On transverse sections, the choroid plexus can be slightly thickened and viscous exudates may be visible [112].
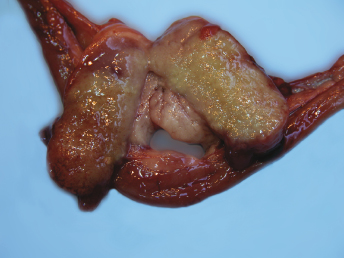
Fig. 20.21. Abdomen, ferret with FSCD. Granulomatous peritonitis and lymphadenitis characterized by multifocal to coalescing, white firm nodules of varying size widely distributed throughout the mesentery and within the enlarged mesenteric lymph node. (Reprinted from Murray et al., Vet Clin N Am: Exot Anim Pract 13: 543–560, 2010, with permission from Elsevier.)Fig. 20.22. Mesenteric lymph node, ferret with FSCD. The parenchyma of the severely enlarged node is effaced by severe, diffuse granulomatous inflammation. (Reprinted from Murray et al., Vet Clin N Am: Exot Anim Pract 13: 543–560, 2010, with permission from Elsevier.)
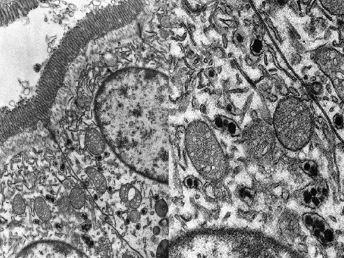
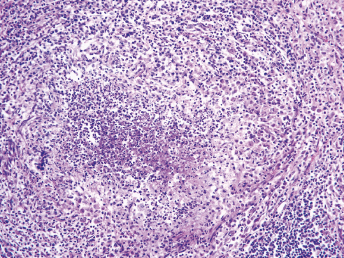
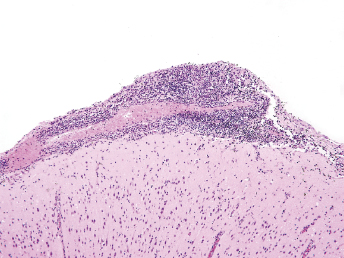
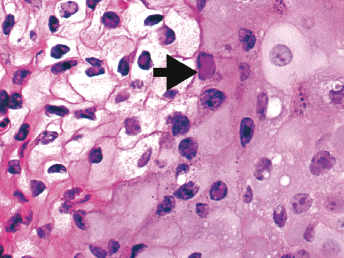
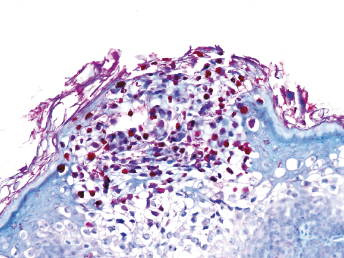
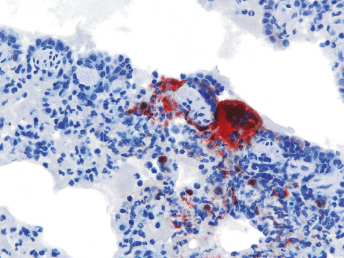
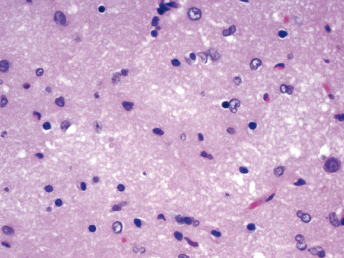
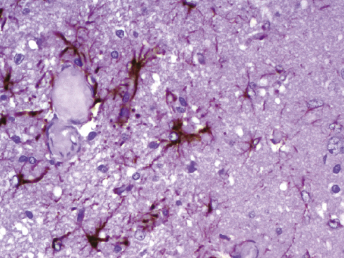
Histologic lesions of FRSCV infection that are characterized by severe pyogranulomatous inflammation (Fig. 20.23) are most commonly observed in the mesentery and along the peritoneal surface [105,106,113]. Pyogranulomatous inflammation commonly encompasses the small intestine (Fig. 20.24) and focally expands or destroys the muscularis and serosa. Pyogranulomas are characterized by central areas of necrosis composed of cellular debris and degenerative neutrophils surrounded by epithelioid macrophages (Fig. 20.25) with additional layers of lymphocytes and plasma cells [105,112]. Rare multinucleated giant cells have been described. Necrosis is an inconsistent feature, but microgranulomas may be composed predominantly of epithelioid macrophages. Variable degrees of fibrosis surround some granulomas. Granulomatous inflammation is often localized around vessels and frequently involves the adventitia, with inflammatory cells migrating into the medial tunics of small veins and venules. Similar areas of multifocal pyogranulomatous inflammation commonly expand and obliterate the normal architecture of the lymph nodes and other infected organs, resulting in nephritis, pancreatitis, adrenalitis, meningitis, myocarditis, and pneumonia. In animals with neurologic signs, the primary lesions may be localized entirely within the brain and consist of a severe pyogranulomatous leptomeningitis (Fig. 20.26), choroiditis, ependymitis, and encephalomyelitis. The inflammatory process is centered on vessels, particularly venules, along the inner and outer surfaces of the brain, with only focal extension into the underlying parenchyma. The most severe parenchymal extension of the inflammatory reaction is usually observed periventricularly. Immunohistochemistry using a monoclonal antibody against alphacoronavirus nucleocapsid antigen shows strong positive intracytoplasmic labeling of macrophages within the center of pyogranulomas. Transmission electron microscopy of areas of pyogranulomatous inflammation revealed macrophages with spherical, enveloped viral particles, 70–140 nm in diameter, in membrane-bound cytoplasmic vacuoles and free within the cytoplasm. Occasionally, circumferential spikes are observed along the outer wall of the virions [112].
Fig. 20.23. Liver, ferret with FSCD. The hepatic capsule is covered by thick strands of fibrin and multifocal, white firm nodules are randomly distributed over the hepatic serosa and commonly extend into the parenchyma consistent with granulomatous inflammation. (Reprinted from Murray et al., Vet Clin N Am: Exot Anim Pract 13: 543–560, 2010, with permission from Elsevier.)Fig. 20.24. Jejunum, ferret with FSCD. Granulomatous peritonitis encompasses the jejunum circumferentially and extends into the mesentery. H&E. (Reprinted from Murray et al., Vet Clin N Am: Exot Anim Pract 13: 543–560, 2010, with permission from Elsevier.)Fig. 20.25. Jejunum, ferret with FSCD. Pyogranulomatous peritonitis with necrotic center composed of cellular debris and degenerative neutrophils surrounded by epithelioid macrophages followed by lymphocytes and plasma cells and a rim of fibroblasts. H&E. (Reprinted from Murray et al., Vet Clin N Am: Exot Anim Pract 13: 543–560, 2010, with permission from Elsevier.)Fig. 20.26. Cerebrum, ferret with FSCD. Pyogranulomatous perivasculitis in meninges. H&E.
Pathogenesis
Our current knowledge of the pathogenesis of ferret coronavirus-associated diseases is rather limited. No experimental reproduction of either of the two diseases has been reported, so most of the current hypotheses are drawn from clinical observations, pathologic examinations, and genetic analysis of the ferret coronaviruses.
In general, coronaviruses attach to specific cellular receptors via the spike protein, and viral membranes fuse with cell membranes [117]. Following entry into the host cell, coronaviruses begin to replicate in the cytoplasm in a membrane-protected microenvironment. The coronavirus genome has a 5′ methylated cap and a 3′polyadenylated-A tail that allow the viral RNA to attach to cellular ribosomes for translation [118]. The coronaviral enzyme replicase facilitates transcription of the viral RNA genome into new copies using the host cells machinery [117]. The viral genome which is composed of all viral proteins is replicated, and a long polyprotein is formed to which all viral proteins are attached. The nonstructural coronaviral enzyme protease separates the proteins in the chain, thereby allowing the virus to encode large numbers of genes with a small number of nucleotides [109,118]. Coronavirus transcription involves a template switch during the extension of a negative copy of the subgenomic mRNAs [109,118]. Coronavirus N protein is required for coronavirus RNA synthesis. Cell macromolecular synthesis may be controlled after coronavirus infection by locating some virus proteins in the host cell nucleus [109,118].
FRECV primarily infects epithelial cells at the tip of the intestinal villi in the jejunum and ileum causing degeneration and necrosis [103,104]. The infection continues to progress segmentally throughout the small intestinal tract, and infection also extends from the villar tips down to the crypts in more severe cases. The primary site of infection is unknown, but large amounts of viral nucleic acid have also been detected in salivary glands, and viral shedding has been confirmed in both feces and saliva [104]. Whether a viremia occurs following enteric infection with FRECV is unknown, but at best, it will be short-lived and at low levels since testing by RT-PCR has not identified viral nucleic acid in blood of ferrets that shed the virus in their feces [104].
Clinically, systemic coronavirus disease in ferrets, with which FRSCV has been associated, closely resembles FIP. Likewise, the gross and histopathologic lesions associated with systemic coronavirus disease in ferrets are nearly identical to those seen in the tissues of cats affected with the granulomatous form of FIP [105,106]. FIP is a fatal, multisystemic, immune-mediated disease of cats caused by virulent mutants of FCoV. The FIP viruses are believed to arise spontaneously from persisting low pathogenic to nonpathogenic feline enteric coronavirus strains [119]. This concept was, however, challenged in a recent publication [120]. Similar to FIP, systemic coronavirus disease in ferrets is characterized by positive immunohistochemical labeling of the cytoplasm of intralesional macrophages (Fig. 20.27) for coronaviral antigen [105,121]. Systemic disease has been consistently associated with FRSCV, but not FRECV [108]. However, partial sequence analysis showed FRSCV to be more similar to FRECV than to other alphacoronaviruses, including FCoV [104,108]. Based on the limited number of FRSCV and FRECV strains analyzed thus far, FRSCV strains differ significantly from FRECV strains in the gene encoding for the spike protein [108]. Additional FRECV and FRSCV strains must be analyzed to either substantiate or modify current data.
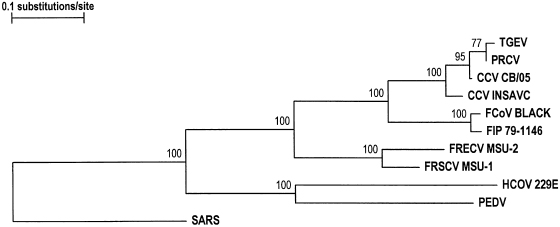
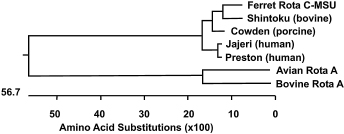
Fig. 20.27. Phylogenetic trees based on deduced partial amino acid sequences of the envelope proteins of FRSCV MSU-1, FRECV MSU-2 and representative strains of alpha coronaviruses. SARS virus was included as the outgroup sequence. Bootstrap values are indicated at the nodes. (Reprinted from Wise et al., Virus Res 149: 42–50, 2010, with permission from Elsevier.)
The similarities in clinical disease and microscopic lesions between FRSCV and FIP virus suggested a similar pathogenesis for FRSCV-associated disease and FIP, but experimental proof is needed. Several coronavirus genes have been implicated in viral virulence. Macrophage tropism of FRSCV is most likely responsible for the differences in virulence and associated disease compared with FRECV as has also been suggested for the increased virulence of FIP virus versus FCoV in cats [109,119,122]. Candidate genes speculated to play a role in the virulence shift include the S and group-specific genes 3abc, 7b, and 7a [123–125]. Based on sequence data of a limited number of FRSCV and FRECV strains, there is a significant difference between the S genes of enteric and systemic strains [108]. Whether this difference is the primary cause of the altered virulence of FRSCV and the associated macrophage tropism is unclear. Mutations in ORF 3c have also been suggested as the basis for change in virulence of FIPV in cats [116]. One study found that the 3c gene of feline coronaviruses was intact in 100% of analyzed FCoV strains, but mutated in approximately 70% of FIPV strains [126]. Ferret coronaviruses carry a single ORF 3 gene and the ORF 3 sequence of a number of strains has been examined [108]. FRECV strains associated with enteric disease all showed an intact ORF 3, while truncated proteins were observed in two out of three FRSCV strains examined [108]. This lead to the conclusion that mutation in 3c cannot be the sole cause of macrophages tropism in either FIPV or FRSCV and the role of ORF 3c mutations in the altered virulence of FRSCV is unknown.
Diagnosis
The clinical signs are suggestive of an infection with ECE, but other diseases, in particular intestinal coccidiosis, need to be excluded [127]. In contrast, hematologic and biochemical findings in ferrets with ECE are nonspecific. For ferrets with suspected systemic coronavirus disease, the typical clinical signs, blood work results, and a polyclonal gammopathy on serum protein electrophoresis are suggestive, but not definitive evidence, of FRSCV-associated disease. Typical hematologic signs in ferrets with systemic coronavirus disease include nonregenerative anemia, hyperglobulinemia, hypoalbuminemia, and thrombocytopenia. Serum protein electrophoretograms show a polyclonal hypergammaglobulinemia [112,113]. Differential diagnoses for hypergammaglobulinemia in domestic ferrets include Aleutian disease, malignant lymphoma, multiple myeloma, and chronic infections or inflammation, for example, Helicobacter, inflammatory bowel disease [112,113]. Counterimmunoelectrophoresis testing for anti-Aleutian disease parvovirus antibodies should be performed to exclude Aleutian disease as a differential [112]. Biochemical changes in ferrets with systemic coronavirus disease are variable and consistent with the observed organ damage, that is, kidneys, liver, pancreas, and the gastrointestinal tract. Serum chemistry abnormalities include elevations in serum lipase, blood urea nitrogen, serum alanine transferase, elevated alkaline phosphatase, and serum gamma glutamyl transferase [105,112,113]. Urinalysis results have only been reported for a few cases of FRSCV-associated disease, but abnormal findings include green urine, proteinuria, blood, and rare bilirubin crystals [105].
Radiographs may show abdominal masses (Fig. 20.16), splenomegaly, and nephromegaly [128]. Patchy densities in the lungs have been reported in at least one ferret [112].
Because no pathognomonic clinical signs exist for FRECV- or FRSCV-associated diseases, diagnostic testing is required to confirm the diagnosis. Commonly used tests to detect ferret coronaviruses include immunohistochemistry, electron microscopy, and RT-PCR.
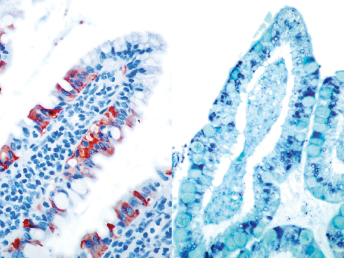
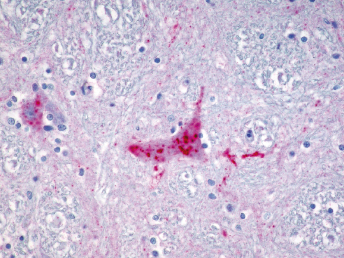
Immunohistochemistry using a monoclonal antibody against alphacoronavirus antigen can be used successfully to detect either FRECV or FRSCV (Fig. 20.19 and Fig. 20.28), but will not differentiate the two viruses [103]. Immunohistochemistry using a monoclonal antibody (FCV370) against feline coronavirus has worked well to demonstrate viral antigen in lesions of formalin fixed tissues [121]. However, currently available RT-PCRs for FCoV will not detect ferret coronaviruses [105].
Fig. 20.28. Jejunum, ferret with FSCD. Detection of alphacoronaviral antigen by immunohistochemistry (red) in epithelioid macrophages within center of pyogranulomatous peritonitis, AEC chromogen with hematoxylin counterstain.
Electron microscopy has proven to be a good way of detecting the 120 nm coronaviral particles in the feces of animals with ECE [103].
RT-PCR is available in some laboratories, and coronavirus consensus primers have been utilized to detect any ferret coronavirus [104]. Unfortunately, coronavirus consensus RT-PCR has only been successful on fresh or frozen material, including feces, but not formalin-fixed tissues [104]. Based on partial sequence analysis of a limited number of coronaviruses associated with either ECE or FSCD, FRSCV and FRECV share at least 96% nucleotide sequence identities in the M, N, and nonstructural protein genes, ORFs 3 and 7b [108]. However, their S proteins showed only 79.6% amino acid sequence identity. This allowed the development of two ferret coronavirus genotype-specific real-time (r)RT-PCR assays and these assays are the current gold standard for differentiating FRSCV from FRECV [108]. Using RT-PCR, FRECV can be detected in samples of small intestine as well as feces and saliva, but has not been detected in serum [103,104,112] In contrast, FRSCV can be detected in a wide range of organs consistent with the distribution of gross and microscopic lesions [105,112]. To determine whether ferrets are shedding FRECV, fecal swabs or samples are preferred. A combination of unfixed and fixed tissues containing granulomatous lesions is used to detect FRSCV in ferrets affected with the systemic form.
In situ hybridization using oligoprobes based on FRECV-specific sequences has also been used (Fig. 20.19) to confirm infection of villar epithelial cells with FRECV in ferrets with ECE [104].
A serologic test to detect antibodies to ferret coronaviruses is also available through some laboratories [112]. The test can be used to determine previous exposure of ferrets to a ferret coronavirus, but until now has only been used to detect evidence of previous FRECV infection. It is important to recognize that overinterpretation of titers can lead to erroneous conclusions as is the case for feline coronaviruses. Therefore, interpretation of titers requires a good understanding of the limitations of this test. Serum antibody tests are frequently used to help diagnose FIP in cats [116,129]. These tests include immuno-fluorescence assay (IFA), virus neutralization (VN), and ELISA, but IFA is most commonly used [129]. High antibody titers are suggestive of FIP, and low or negative antibody titers make FIP less likely [129]. Unfortunately, these tests are not specific for FIP, and many healthy animals will have positive antibody titer from viral exposure, but most of them will not develop disease [116,129]. In cats, an RT-PCR to detect messenger RNA of the M gene was developed for use on blood monocytes [130]. The M gene is only expressed during viral replication. Therefore, the detection of replicating coronavirus in blood monocytes would be highly suggestive of FIP in cats [130]. Unfortunately, the M gene of the ferret coronaviruses is very different from that of the feline coronavirus, and this test will not work for ferrets (Bernhard Kaltenboeck, DVM, PhD, personal communication, 2008).
Virus isolation has not provided good results [103,104].
The definitive diagnosis of either coronavirus infection requires histology to demonstrate the characteristic microscopic lesions associated with intralesional viral antigen or nucleic acid. Since FRECV is enzootic in the ferret population, detection of virus in feces does not equal ECE. Still, clinical signs of the characteristic green diarrhea and detection of FRECV in feces of such animals provides strong circumstantial evidence for a diagnosis of ECE. Intestinal biopsies are a good means of obtaining a definitive diagnosis and thereby prognosis and response to treatment for ferrets with suspected ECE. Microscopic lesions and detection of antigen are more common in the jejunum and the ileum, and FRECV has not been detected in the large intestine or other organs [103,104]. For FRSCV, biopsy samples of enlarged lymph nodes from ferrets with suspected systemic coronavirus disease can provide an antermortem diagnosis; but the nodes should not be completely removed [112,113]. Typical microscopic lesions are very suggestive of a diagnosis of FRSCV-associated disease; however, definitive diagnosis requires positive immunohistochemistry staining of the coronavirus antigen in macrophages within areas of granulomatous inflammation. Immunohistochemistry against alphacoronavirus using the monoclonal antibody FIPV3-70 is effective to demonstrate coronavirus in those tissues [105,106,112]. As previously discussed, this antibody cross-reacts with all alphacoronaviruses, and it will also detect FRECV. Further differentiation of FRSCV from FRECV remains a major challenge and currently only the genotype specific RT-PCRs are available to differentiate FRECV from FRSCV. Immunohistochemical staining is more common in lymph nodes, but can also be seen in kidney, spleen, lung, liver, pancreas, intestine, heart, and adrenal glands correlating with the distribution of gross and microscopic lesions [106].
Ultimately, clinical, pathologic, and molecular diagnostic data must be combined to determine the final diagnosis for either disease.
Treatment
Empiric treatment has included broad-spectrum antibiotics, aggressive fluid therapy, and supportive care [103,114]. After recovering from ECE, some adult ferrets develop persistent, intermittent malabsorption with diarrhea, which can be temporal or permanent [114]. Treatment with a short course of steroids (prednisone 1 mg/kg BID × 14 d) and changing the diet to an easily absorbed food may speed recovery [103,114]. Metronidazole has anti-inflammatory and antibiotic effects and can improve stool consistency (20 mg/kg PO BID) [97]. Metronidazole is not very palatable and can compromise owner's compliance with treatment. The combination of enrofloxacin (5 mg/kg PO BID) and amoxicillin/clavulanic acid (10–20 mg/kg PO BID) can be another good option [97].
Although there are many similarities between feline FIP and ferret systemic coronavirus, no study has demonstrated that the pathogenesis is the same, and therefore, treatments that may work in one disease may not work in the other. Due to its recent description, we still do not know many characteristics of this disease. Actually, most ferrets with systemic coronavirus have treatment initiated but die or are euthanized 2–3 months after diagnosis [113]. Exceptionally, ferrets may survive the disease for more than 3 years [112].
Currently, no cure exists for ferrets with systemic coronavirus [112]. Assuming the disease behaves in a way similar to FIP in cats, treatment should be directed at suppressing the immune response, suppressing inflammatory response and to reduce or eliminate the virus by modulating the immune response [112]. Similar to FIP, intermittent signs of improvement in ferrets with systemic coronavirus are common with any treatment or even with no treatment [113]. Possible therapeutic protocols have been reviewed by Murray et al. [112] and are described in this chapter. However, it should be noted that these protocols are based on cats with FIP, have not been properly evaluated in ferrets, and are anecdotal:
Prednisolone (1–2 mg/kg PO BID, with a gradual reduction of the dose). It suppresses immune response, reduces inflammation, increases appetite, and improves patient demeanor.
Other immunosuppressive drugs, such as cyclophosphamide, chlorambucil, or azathioprine, are not commonly recommended due to common side effects. However, if treatment with corticosteroids does not lower the gamma globulin level, prednisolone and azathioprine may be combined for additional immunosuppression and to reduce the final dose of both drugs.
Polyprenyl (3 mg/kg PO 2–3 times per week) is a new immunostimulant that has showed some success in treatment of dry FIP in cats.
Interferon is an immune modulator, and both feline interferon omega and human interferon alpha have been shown to inhibit feline coronavirus replication in vitro. However, only feline interferon omega has had some effect at increasing survival time in cats with FIP, but studies are not definitive about the benefits of this drug.
Pentoxifylline has anti-inflammatory, immunomodulatory, and antivasculitis effects. Vasculitis is a common lesion in ferrets with systemic coronavirus. Suggested doses in cats are 20–25 mg/kg BID.
Melatonin has anti-inflammatory, immunomodulatory, and antiviral properties. In addition, melatonin stimulates the appetite and has antioxidant effects reducing free radicals.
Doxycycline has antibiotic, anti-inflammatory, and immunomodulatory effects. The dose in ferrets is 10 mg/kg BID.
The combination of prednisone, pentoxifylline, and doxycycline may have synergistic effects on decreasing inflammation and vasculitis.
Supportive care is essential for ferrets with FSCD. Sick ferrets usually do not eat enough to meet their nutritional needs, and therefore need nutritional support by means of a soft, highly energetic, and easily digestible food. Supplementing vitamins may also be beneficial and may improve appetite; cobalamine (vitamin B12) at 250 μg SC weekly or vitamin B complex (1–2 mg/kg SC). Anemia is a frequent clinical finding in ferrets with systemic coronavirus, and supplemental iron and/or erythropoietin may be beneficial. Gastroprotectants can reduce nausea and inappetence, and prevent gastric ulcers caused by the administration of prednisolone; sucralfate can be administered at 75–100 mg/kg TID, 1 hour before feeding or 2 hours after feeding, always before the administration of prednisolone. Cimetidine (10 mg/kg PO TID) is an antacid with beneficial effects on the immune system, but omeprazol and famotidine can also be used and have the advantage of once-a-day administration. The use of antiemetics, fluid therapy, antidiarrheals, other antibiotics, and so on, should depend on the symptoms and circumstances of each clinical case.
Prevention
Epizootic catarrhal enteritis is a highly contagious and easily transmissible disease [97,103], and prevention is aimed at avoiding exposure to ferret enteric coronavirus. Affected ferrets should be isolated from asymptomatic or unexposed ferrets [114]. Infection can occur through contact with infected ferrets, their feces, or fomites [97]. Fecal contamination of the environment can be reduced by thorough cleaning of litter boxes, cages, and bowls [112]. It should be noted that ferrets carry the virus long after clinical signs have resolved, and remain contagious to other ferrets for up to 6 months, perhaps longer [97].
Prevention of FRSCV infection is aimed at avoiding contact with infected ferrets and isolating or euthanizing affected animals in a group.
Disinfection
Coronaviruses persist in the environment for extended periods of time under appropriate conditions [97]. Coronaviruses can be inactivated quite easily with many commonly used disinfectants [131].
Aleutian Disease Virus
Aleutian disease (AD) is a chronic and progressive condition caused by the Aleutian disease virus (ADV) [7,132]. The disease was initially described in farmed American mink (Neovison vison) and later in domestic ferrets [7]. ADV infection has also been detected in wild mustelid species [133–136]. The name originates from mink that were homozygous for the Aleutian gene, resulting in a blue color and also high susceptibility to ADV infection and severe disease [137]. Overt disease has only been primarily described in mink and ferrets. The relationship between infection with ADV and the development of AD is a complex process that depends on many factors, including species, genetic background, immune status of the animal, route of infection, infective dose, and strain of the virus [7,136,138–143]. Furthermore, difficulties in accurate diagnosis of AD complicate the picture.
Etiology
ADV is a parvovirus and the only member of the genus Amdovirus [140]. ADV is distantly related and antigenically different from other parvoviruses, including canine parvovirus, feline parvovirus, and mink enteritis virus [7,144,145]. Parvoviruses are small, nonenveloped, single-stranded DNA viruses [146]. The pathogenicity of different strains of ADV depends on the infected species [139,140]. Ferret-derived strains of ADV are different from mink-derived strains of ADV, and at least three strains different from mink ADV have been identified in ferrets [144,147]. Ferret-derived ADV shares a DNA segment similar to a hypervariable capsid region of mink-derived ADV [148]. However, ferret ADV causes less severe disease in mink than mink ADV, and highly pathogenic mink ADV only produces low levels of an antibody response with clearance of the virus after 12 days in ferrets. Ferrets infected with mink ADV do not develop hypergammaglobulinemia and only develop mild lesions that are not specific for AD [147]. Although ferret-derived ADV strains were originally thought to be mutants of the more virulent mink-derived ADV strains [7], this hypothesis has not been proven, and a feral origin of ADV in wild carnivore species should not be excluded [149].
Transmission and Epidemiology
AD is under most circumstances not a highly contagious disease [150,151]. Transmission may occur through aerosolization or contact with fomites as well as ingestion of feces, urine, saliva, or blood containing ADV [137]. Uninfected mink are often housed for months near or even together with infected animals, and remain uninfected [150–152]. The slow propagation of AD suggests that a threshold dosage is required to transmit virus to susceptible animals [150]. Interestingly, in some cases ADV can spread rapidly under ranch conditions by aerosol and mechanical transmission through fomites [150,153]. Such airborne infection is unlikely to occur commonly in natural circumstances as a high dose of virus is needed to produce infection [150]. Although the virus is present in urine, saliva and feces, large doses of virus or a prolonged exposure to smaller doses seem necessary to infect animals through the oral route [150]. Blood-sucking insects have not shown to be important in the transmission of AD [150].
All mustelids, except for the European badger (Meles meles) and some otter species, are solitary species. For most of the time, an individual mustelid has little contact with conspecifics, except during the breeding season and when a female carries her offspring [154]. Under field conditions, ADV is most commonly transmitted in those circumstances. In captive minks, males can transmit the virus to females by biting during copulation, but positive females rarely transmit ADV to negative males, thus limiting the spread within a population [150]. Vertical transmission has been shown to be of importance in both captive and wild mink ([152], [150]). Vertical transmission is mainly responsible for maintaining the virus in a population and can also produce fetal death and infertility [150]. However, mink with advanced AD do not become pregnant or if they do, abort or resorb the fetuses [150]. Regardless, since wild American mink lead a solitary life, the virus is not easily transmitted in nature by direct and indirect contact, but vertical transmission insures maintenance of the virus from one generation of mink to the next [150].
The seroprevalence of ADV in domestic ferrets ranges from 6% to 60% depending on the study [7,147,155,156] and, according to these studies, the prevalence of ADV is significantly higher in the United States when compared with the United Kingdom. It is probably also higher in situations where large numbers of ferrets are housed together, such as farms, shelters, or research facilities [7,145]. Despite this high seroprevalence in some areas, cases of clinical AD in ferrets are not commonly reported in the literature, nor are they commonly diagnosed in clinical practice. However, cases of AD may be more frequent in ferrets housed in groups [7,145].
Infection (detection of antibodies and viral nucleic acid) has been documented in mink farmers, and disease in humans has been suspected in some cases [157]. Therefore, AD may be considered a zoonosis under situations of high viral load.
Clinical Signs
Clinical findings in ferrets with AD are variable and depend on the organs affected. Dyspnea, posterior paresis, neurologic dysfunction, chronic wasting, melena, renal dysfunction, and sudden death have been reported [7,145,158] and are most commonly observed in adult ferrets 2–4 years of age [137]. Other findings observed in ferrets with AD include cardiac problems, uveitis, splenomegaly, and lymphadenomegaly [145]. The clinical history may not provide any particular useful information, as both individually housed animals and ferrets held in groups may develop clinical signs of AD. Outbreaks of AD have been reported [145]. Ferrets may be infected for years prior to showing clinical signs and immunosuppression due to stress or any other causes may lead to clinical disease manifestation [137].
Hypergammaglobulinemia (gamma globulins >20% of the total protein concentration) is common in clinical cases [7,145], but does not always occur [158]. Hypergammaglobulinemia has been reported to be monoclonal [159] or more monoclonal in ferrets than in minks [148]. However, distinction between monoclonal and polyclonal may not always be possible, particularly in the early stages of the gamma globulin rise. In addition, it should be remembered that other diseases of ferrets can produce hypergammaglobulinemia, such as systemic coronavirus infections, although polyclonal gammopathy is observed with this disease [113].
Results from other tests (hematology, biochemistry, radiography, ultrasonography) are not specific of AD and depend on the organs affected: anemia, chronic inflammation, azotemia, or increase in hepatic enzymes can be seen [160].
Pathology
Gross lesions of AD are seen only late in the course of disease and can include serosanguinous fluid within the intestinal lumen, hepatomegaly, splenomegaly, thymic enlargement, and emaciation [7,161]. Splenomegaly and lymphadenopathy are the most common gross lesions, and splenic infarction as a result of marked splenomegaly may complicate the clinical and pathologic picture (Fig. 20.29). Enlarged, brown-tan kidneys may be present, but kidneys may be small and shrunken depending on the chronicity of the disease. Enlarged livers are pale and may have scattered white nodules. The small intestine may have multifocal mucosal ecchymoses and intraluminal melena. In animals in which the respiratory system is affected, lesions include diffuse pulmonary congestion, serosanguineous pleural effusion, pulmonary edema, pulmonary ecchymoses, and lobular consolidation and collapse. In terminal cases, clotting abnormalities resulting from vasculitis and the marked hypergammaglobulinemia may result in petechiae, ecchymoses, and hematuria. Gross lesions usually are not observed in subclinical infections with ADV.
Fig. 20.29. Abdomen, ferret with AD. Severe splenomegaly with infarction, renomegaly with multifocal petechiae, gastric ecchymoses and hematuria. (Courtesy of the Archives of the Armed Forces Institute of Pathology, original photograph by John Gorham, DVM.)
Histologically, AD is characterized by hyperreactivity of the lymphoid system, with lymphoplasmacytic infiltrates in multiple organs [158,161]. Common microscopic lesions include portal lymphoplasmacytic cholangiohepatitis with or without bile duct hyperplasia and periportal fibrosis (Fig. 20.30), interstitial lymphoplasmacytic nephritis, membranoproliferative glomerulonephrosis, lymphoplasmacytic gastritis, and nonsuppurative encephalitis and myelitis [145]. Especially prominent plasmacytic infiltrates are seen in numerous organs, most prominently in the renal interstitium, hepatic portal areas, and in the splenic red pulp that can be expanded by an almost pure population of plasma cells. Additionally, there may be marked plasmacytosis of numerous lymph nodes and the bone marrow. Plasmacytic infiltrates may also be observed in the thymus and in the lungs. The marked membranous glomerulonephrosis is commonly associated with numerous ectatic protein-filled tubules (Fig. 20.31). Tubular necrosis may be observed in acute cases (Fig. 20.32). In contrast, glomerulosclerosis is commonly seen in ferrets with chronic interstitial nephritis, but there is little evidence of tubular protein casts or plasmacytic infiltrate in such cases. Lymphoplasmacytic perivascular cuffing in the brain and spinal cord may be observed along with lymphoplasmacytic meningitis (Fig. 20.33). Vasculitis may be seen in almost any organ.
Fig. 20.30. Liver, ferret with AD. Periportal lymphoplasmacytic cholangiohepatitis with bile duct hyperplasia. H&E.Fig. 20.31. Kidney, ferret with AD. Interstitial lymphoplasmacytic nephritis with membranoproliferative glomerulonephrosis and intratubular protein casts. H&E.Fig. 20.32. Kidney, ferret with AD. Severe interstitial lymphoplasmacytic nephritis with acute tubular necrosis. H&E.Fig. 20.33. Cerebrum, ferret with AD. Lymphoplasmacytic encephalitis with perivascular cuffing and glial nodules. H&E.
Some specific lesions observed with AD in minks are different in ferrets [7,158] and development of lesions also depends on the strain of virus [147], and therefore can be different in experimental versus natural infections. Lesions such as proliferation of bile ducts that are common in mink with AD, have not been experimentally reproduced in ferrets inoculated with any strain of ADV [147], but have been observed in naturally occurring cases of ADV [158]. It remains uncertain whether this lesion is directly caused by ADV or represents a nonspecific finding secondary to hepatic injury [147]. Germinal center formation in lymphoid cell infiltrates has been observed in ferrets infected with ADV, but not in uninoculated animals [147]. However, the occurrence of mixed lymphocytic and plasmacytic infiltrates in the liver, kidney, or gastrointestinal tract needs to be interpreted with caution, as both experimentally infected ferrets as well as uninoculated control ferrets may have similar lymphoplasmacytic infiltrates [148]. Lesions compatible with AD in noninfected control ferrets have been reported in one study [147]. Whereas 28% of ADV infected ferrets had severe and 66% had mild to moderate lymphoid hyperplasia, 31% of control ferrets also developed mild to moderate lymphoid hyperplasia [147]. In addition, it is common to find lesions in ferrets experimentally infected with ADV that do not produce mortality or clinical signs [147]. Ultimately, detection of ADV within such lesions by PCR or in situ hybridization is required for an accurate diagnosis.
Pathogenesis
Depending on the virulence of ADV strain and host immunity, ADV can cause three types of disease progression: (1) progressive infection, (2) persistent nonprogressive infection, and (3) nonpersistent nonprogressive infection with clearance of the virus [162]. ADV replicates and becomes sequestered in macrophages and dendritic cells and large amounts of viral antigen are usually found in phagocytic cells of the liver and lymphoid organs [163]. Once the animal becomes infected with ADV, a strong antibody response against both structural and nonstructural proteins and an increase in gamma globulins result from the antiviral immune response [164,165]. Infected animals that are unable to clear the virus will have long-term infections that cause immune complexes and lesions due to the deposition of immune complexes in various organs, including the kidneys, liver, arteries, and brain.
ADV infections in ferrets cause similar disease patterns as observed in mink; however, it is important to recognize that ferret strains of ADV are different from mink strains [144] and commonly cause less progressive disease. In susceptible mink strains, ADV infection results commonly in a rapidly life-threatening immune-mediated glomerulonephritis, vasculitis, and hypergammaglobulinemia. In ferrets, there are notable similarities, including an increase in gamma globulins, and in late stages of the disease, an immune complex glomerulonephritis. However, in most ferrets, the disease is usually much more insidious, with a progression of as long as 2 years. Ferrets in the late stages of disease will be hyperproteinemic (8–9 mg/dL, with >20% being composed of gamma globulins).
Ferrets infected with highly pathogenic mink ADV usually cleared the virus 12 days after infection. Infected ferrets developed a low-level antibody response, but no hypergammaglobulinemia, and only mild lesions which were not specific for AD [147]. Even though the gamma globulin levels were significantly higher in ADV-infected ferrets than in control ferrets, the gamma globulin levels in ADV-infected ferrets were generally lower than 1.5 g/dL [147]. In this study, lymphoid hyperplasia was severe in 28% of ADV infected ferrets and mild to moderate in 66%; however, 31% of control ferrets also had mild to moderate lymphoid hyperplasia [147]. The significance of lymphocytic infiltrates as an indicator for an infection with ADV is uncertain, since control ferrets in one study also developed lymphocytic infiltrates in the liver [148]. In contrast, ferrets inoculated with ferret ADV developed a stronger antibody response, more severe rise in gamma globulins, and more severe lesions than when inoculated with mink ADV [147]. However, even with ferret ADV, gamma globulins are generally not high enough to be considered hypergammaglobulinemia, and only a few cases had gamma globulins between 2 and 3 g/dL at day 120 post infection; after that, the level of gamma globulins decreases, although it still remains higher than in control ferrets [147]. Lesions in ferrets inoculated with ferret ADV were more severe than in control ferrets or ferrets inoculated with mink ADV; but did not cause mortality or clinical signs [147]. Antibody levels were positively correlated in some cases to the level of hypergammaglobulinemia and the severity of lesions [147]. Germinal center formation in the lymphoid cell infiltrates was observed in ferrets with ADV infection, but not in control ferrets, and proliferation of bile ducts, a common lesion in mink with AD, was not observed in ferrets inoculated with any strain of ADV [147]. These findings are consistent with the observation that ferrets with persistent antibodies against ADV or intermitted viremia and viral shedding in feces and urine, as confirmed by PCR or lesions of AD present at necropsy, had no history of clinical disease or mortality over several years and eventually died from unrelated conditions [166,167].
Diagnosis
The progression from infection with ADV to the development of AD is a complex process that depends on numerous factors, including species, genetic background, immune status of the animal, route of infection, infective dose, and strain of the virus [7,136,138–143]. In addition, our current understanding of AD is mainly based on observations from mink, and AD in ferrets does not seem to follow exactly the same pattern. Our lack of understanding AD in ferrets complicates the diagnosis and only a combination of several diagnostic tests can lead to a definitive antemortem and postmortem diagnosis.
Traditionally, antemortem diagnosis of AD in ferrets has been based on a combination of compatible clinical signs, positive serology, and hypergammaglobulinemia [7]. However, clinical cases can be misdiagnosed by using this methodology, as hypergammaglobulinemia and clinical signs (and even a biopsy with lymphoplasmacytic infiltrates) are nonspecific toAD, and many ferrets with a positive serology or positive PCR do not actually have AD. Furthermore, the amount of information on AD in ferrets is considerably smaller than that obtained for captive mink, and extrapolation should be done carefully. Therefore, the diagnosis of AD is complicated and some cases reported or diagnosed as AD may not actually be this disease (or at least the diagnosis is not definitive). As an example, a ferret with chronic wasting, lymphadenomegaly, splenomegaly, hypergammaglobulinemia and positive ADV PCR on feces may be suffering disease from systemic coronavirus. This section will describe the different diagnostic tests that can provide useful information for the diagnosis of AD in ferrets, and the more diagnostic tests one combines, the more likely a definitive diagnosis of AD will be.
Following infection with ADV, antiviral antibodies (IgG) bind to ADV and form immune complexes [161,168]. These protein aggregates are deposited in tissues, leading to a number of immune-related phenomena and producing some of the described lesions, clinical signs, and hypergammaglobulinemia [7,161,168]. Antibodies are produced starting on day 15 post infection, and hypergammaglobulinemia is noted at 2–6 months post infection [147], but this may vary with the strain of virus [145]. Ferrets produce lower numbers of antibodies than minks, and it has been suggested that if a ferret reacts positively to an antibody test designed for minks, such result should be interpreted as strongly positive [97]. There is no scientific proof for this statement. The gold standard serologic test is counterimmune electrophoresis [145], but an ELISA has also been developed [139]. It is important to recognize that high titers to ADV do not indicate development of AD and that many seropositive animals will not develop clinical disease.
PCR can detect ADV nucleic acid in blood, saliva, feces, and urine [7,145,153,167]. Some authors believe that the amount of virus in saliva is less than what can be detected in blood [97]. PCR is better and more sensitive in detecting ADV than in situ hybridization [151]; however, in situ hybridization allows for detection of viral nucleic acid within microscopic lesions (Fig. 20.34), thereby confirming the diagnosis of AD. In infected minks, viral DNA is commonly detected earlier by PCR than are antiviral antibodies [151]. Some mink initially have high amounts of viral DNA, then partial clearance, followed by reappearance [151]. Partial viral clearance or sequestration occurs early in infection, associated with the immune response to the virus [151]. It is unknown if the same situation occurs in ferrets. DNA can still be recovered from mild or no lesions [151]. There can be mink with no detectable virus in blood but detectable virus in tissues [151]. Similarly to serology, ferrets with a positive PCR result for ADV may not develop AD. There have been cases where persistently antibody-positive ferrets were followed up for several years and eventually died from unrelated conditions [166]. Another work also reported an asymptomatic ferret positive on ADV serology and PCR (urine, feces, and blood) for at least 2 years, before dying from unrelated disease [167]. In this case, lesions of AD were present at necropsy, but these lesions were not found in the cases reported by Chitty [166].
Fig. 20.34. Kidney, ferret with AD. Renal tubular epithelial cells with intranuclear labeling (blue) for ADV nucleic acid by in-situ hybridization. NBT chromogen, hematoxylin counterstain.
A definitive diagnosis should be based on the demonstration of ADV antigen or antibodies (e.g., using PCR or ISH) within lymphoplasmacytic infiltrates. Hypergammaglobulinemia and high antibody titers are also good diagnostic aids. Antibody titers, amount of viral DNA, hypergammaglobulinemia and severity of lesions are positively correlated in some cases [147,151]. However, it has been shown in minks that correlation between viral DNA and renal lesions, CEP and renal lesions, and renal and hepatic lesions may be poor in some cases [151]. Biopsy of affected organs (liver, kidney, stomach) may provide a definitive antemortem diagnosis.
When ferrets from one study were tested for ADV antibodies, positive animals had gamma globulins significantly higher than negative ones, but even in positive animals, the level of gamma globulins was generally lower than 1.5 g/dL [147]. Ferrets inoculated with ferret ADV strain developed a stronger antibody response, more severe rise in gamma globulins, and more severe lesions than when inoculated with mink ADV [147]. However, even in infections with ferret ADV strain, gamma globulins are generally not high enough to be considered hypergammaglobulinemia, and only a few cases had gamma globulins between 2 and 3 g/dL at day 120 post infection; after that, the level of gamma globulins decreases, although it still remained higher than in uninoculated ferrets [147]. Lesions in ferrets infected with a ferret ADV strain are more severe than uninoculated ferrets or ferrets inoculated with mink ADV; however, these lesions are not able to produce mortality or clinical signs [147]. In that study, the level of antibodies was positively correlated in some cases to the level of hypergammaglobulinemia and the severity of lesions [147]. Despite these experimental studies, clinical cases of AD resulting in mortality have been described in ferrets [155,158,161].
Treatment
Several therapeutic protocols have had some success in treating mink with AD, mainly using immunosuppressive drugs such as cyclophosphamide [169]. A protocol of 10 mg/kg intraperitoneally of cyclophosphamide, 3 times per week for 13 weeks has been effective in suppressing antibody response and suppressing deposition of immune complexes in kidneys of minks [145]. However, this drug can produce significant side effects such as depression, anorexia, cyanosis, and leukopenia [145]. Lower doses were not as effective in treating disease. In ferrets, the use of immunosuppressive doses of oral prednisone could be considered to reduce immune complexes. Melatonin implants have had some protective action against AD, probably due to melatonin's ability to scavenge free radicals, but it could also be due to the induction of antioxidant enzymes or to the modulation of immunity [170].
Meanwhile, it is important to provide supportive treatment (fluids, nutrition) and antibiotics, as sick animals have a reduced ability of establishing a humoral immune response [145]. Overall, most affected ferrets are euthanized once clinical signs are no longer controllable or when clinical signs appear suddenly and are severe (CNS involvement, hemorrhagic enteritis) or when there are other concurrent diseases (insulinoma or adrenal disease).
Prevention
Vaccines have had little success in preventing infection and disease [164,171]. There are no commercially available vaccines, and it does not seem likely they will appear in the near future, although vaccines with antibodies have shown some usefulness in mink [145]. Immunity after infection has not been documented experimentally once the animal no longer has antibodies [153].
Prevention of an infection that is so prevalent, but so rarely produces disease, is very difficult, as euthanasia of carrier ferrets cannot be justified. However, if maintenance of an ADV-free ferret colony is desired, quarantine and available diagnostic tests should be used to avoid introducing ADV-positive ferrets into the group.
Disinfection
Parvoviruses are extremely stable and resistant to most common detergents and disinfectants [146], and persist in the environment for long periods of time [151]. Parvoviruses can survive for months under cool, moist conditions protected from sunlight, and they are very stable when frozen [172]. Parvoviruses have persisted in feces held for 6 months at room temperature, and may remain viable in the natural environment for 9–12 months [172]. Inanimate fomites, insects, birds, and rats have been implicated as mechanical carriers of parvoviruses, and prepared feed may become contaminated with virus [172].
ADV can survive in a formalin concentration of 0.3% at 5°C for 2 weeks [150]. The virus is remarkably stable in tissue suspensions [150] and can be transmitted through fomites such as gloves (by animals biting gloves) or pens [150]. The virus is inactivated using 2% sodium hydroxide (lower doses are ineffective) at 90°C for 30 minutes [150]. Diluted bleach (1 : 30) for a 10-minute minimum exposure time, steam cleaning, boiling, formaldehyde, glutaraldehyde, and chlorine solutions are also useful for eliminating parvoviruses [146]. Hot-water washes will likely be ineffective because parvoviruses may survive for over 7 hours at 80°C and several days at 56°C [172].
Influenza Viruses
Ferrets are highly susceptible to infections with various types of influenza A and B viruses, and it has been long recognized that infected ferrets easily transmit virus to healthy ferrets and develop severe, naturally occurring disease [173–175]. However, reports of naturally occurring influenza in ferrets are rarely published [173,175]. Since ferrets and humans have similar lung physiology and exhibit similar patterns of influenza virus spread throughout the respiratory tract due to a similar distribution of sialic acid influenza receptors, ferrets have been used widely as an experimental model for both human and avian influenza virus infection [176–181]. Currently, ferrets represent the only model that allows studying both pathogenesis and transmission of influenza virus infection, which also makes them an excellent model to test efficacy of vaccines and therapeutic drugs ([182]; see Chapter 25 of this book).
Etiology
Influenza viruses are negative-sense, single-stranded, segmented RNA viruses in the Orthomyxoviridae family, with influenza A viruses causing disease in mammals and birds [183]. Influenza A viruses are classified on the basis of surface glycoproteins into 16 hemagglutinin subtypes (H1–H16) and 9 neuraminidase subtypes (N1–N9). All of them have been reported in avian species, but fewer subtypes have been reported in mammals [176,184].
Most avian influenza viruses cause asymptomatic infection or mild disease in poultry (low pathogenicity avian influenza), and may represent any combination of the 16 HA and 9 NA subtypes [176]. Some of these combinations (H5 and H7) can be highly pathogenic and produce mass mortality in intravenously inoculated chickens [184]. Endemic/enzootic influenzas in mammalian species have been described in humans and pigs (H1N1, H3N2), horses and dogs (H3N8), and rare occurrences of avian influenza subtypes that spill over from birds to mammals [176,184].
Transmission and Epidemiology
Influenza is an anthropozoonotic (and zoonotic) disease, and the clinical history may show that a person within the family has suffered influenza within the previous days before the illness in the ferret, although transmission from ferret to ferret and from pig to ferret is also possible [7,185]. Transmission usually occurs by aerosol droplets, but ingestion of infected material may occur [176,186]. The risk of viral transmission is greatest at the height of pyrexia when infected ferrets can transmit the disease to other ferrets and to humans [177]. The transmissibility of H1N1 virus between ferrets depends on the presence of specific gene segments: HA for direct contact transmission, PB2 for respiratory droplet transmission, and the NP and 3 polymerase complex genes for high-replication efficiency in the respiratory tract [187,188]. Influenza epizootics in ferrets have also been observed during local outbreaks in humans [189].
Clinical Signs
Clinical signs of influenza infection depend on the age of the infected ferret, the virus strain, environmental conditions, the degree of secondary bacterial infections, and other variables [176,177]. The morbidity is usually high and mortality may occur in young or immunocompromised ferrets. Human influenza viruses tend to cause more severe disease than avian viruses, and mortality may be high following infection with highly pathogenic human influenza viruses [179]. Signs of illness are acute and usually last 3–5 days as long as there are no complications [177]. Physical examination of affected ferrets may show temporal fever and signs of mild infection of the upper respiratory tract, such as rapid onset of sneezing and serous oculonasal secretion [7,33,97,190–192]. Severe cases may evolve to bronchitis and pneumonia, and clinical signs may also include lethargy, dyspnea, conjunctivitis, photophobia, otitis, dermatitis around the eyes and nose, and neurologic signs [7,173,193–195]. Influenza virus infections may cause limited enteritis or renal and hepatic dysfunction [196,197]. Neurologic symptoms, such as ataxia, hind limb paresis, and torticollis, have been reported in ferrets experimentally inoculated with highly pathogenic avian influenza A (H5N1) viruses [198].
Consistent hematologic and biochemical changes have not been described in ferrets naturally infected with influenza viruses. However, transient lymphopenia has been observed in ferrets experimentally inoculated with human-origin influenza viruses [195,199]. Radiography may show signs of pneumonia in advanced cases.
Pathology
As with clinical signs, microscopic lesions and distribution of influenza virus within affected tissues of infected ferrets strongly depend on virus strain [176]. A complete summary of clinical signs associated with different strains of influenza viruses has been published elsewhere [182]. Influenza A virus infections most commonly cause rhinitis that progresses to tracheobronchitis and pneumonia (Fig. 20.35). Gross lesions are usually limited to the respiratory tract and are characterized by multifocal or coalescing pulmonary consolidation (Fig. 20.36) involving 20–70% of the lung surface [176]. Microscopic lesions are mainly limited to the respiratory tract. In the upper airways, there is desquamation of nasal epithelium and infiltration of the nasal submucosa with inflammatory cells [176,180]. In the lungs, there is mild bronchitis, marked necrotizing bronchiolitis (Fig. 20.37), and diffuse alveolitis with acute intra-alveolar edema and hemorrhage admixed with intra-alveolar macrophages and fewer neutrophils, erythrocytes, fibrin, and cellular debris [176,180,195,200]. Other lesions consisted of alveolar epithelial cell hyperplasia, peribronchiolar accumulation of mixed inflammatory cells, and epithelial necrosis in submucosal serous glands [179]. Highly pathogenic avian influenza virus H5N1 causes a nonsuppurative necrotizing encephalitis within the cerebrum, cerebellum, brain stem, olfactory bulb, choroid plexus, and meninges that are characterized by neuronal degeneration, neuronophagia, glial nodules, and perivascular accumulation of mononuclear cells and sometimes neutrophils [201,202]. Additional lesions reported include multifocal hepatocellular necrosis associated with hemorrhage, biliary necrosis and hyperplasia, and periportal accumulation of mononuclear or mixed inflammatory cells [203]. In naturally occurring cases of influenza viral disease, lung lesions were characterized by bronchointerstitial pneumonia with necrosuppurative bronchiolitis characterized by patchy areas of alveolar septal thickening, cellular debris in alveoli admixed with neutrophils and macrophages, and attenuation of lining airway epithelium and bronchiogenic abscessation [173].
Fig. 20.35. Lung, ferret infected with influenza virus H1N1. Severe interstitial pneumonia primarily affecting the caudodorsal portion of the lungs. (Courtesy of Sally Davis and Jefferey Taubenberger.)Fig. 20.36. Lung, cross section, ferret infected with influenza virus H1N1. Affected lungs are noncollapsed and have a prominent bronchointerstitial pattern. (Courtesy of Sally Davis and Jefferey Taubenberger.)Fig. 20.37. Lung, ferret infected with influenza virus H1N1. Severe interstitial pneumonia and necrotizing bronchiolitis. H&E. (Courtesy of Sally Davis and Jefferey Taubenberger.)
Following infection with human influenza viruses H1N1 and H3N2, large amounts of virus were detected in the upper respiratory tract and trachea and progressively less in bronchi and bronchioles [178]. In the upper respiratory tract, trachea, bronchi, and bronchioles, virus was predominantly detected in ciliated epithelial cells and in epithelium of submucosal serous glands, whereas in alveoli, virus was detected in type 1 (Fig. 20.38) rather than type 2 pneumocytes and alveolar macrophages [178,181,204]. In ferrets infected with highly pathogenic avian influenza virus H5N1, viral antigen was detected predominantly in type 2 pneumocytes in alveoli and less commonly in type 1 pneumocytes, rarely in the upper respiratory tract and in bronchiolar epithelial cells, but not in tracheal or bronchial epithelial cells [181,205]. Virus antigen expression was also observed in neurons and hepatocytes [200,202].
Fig. 20.38. Lung, ferret infected with influenza virus H1N1. Immunohistochemical detection of viral antigen (brown) in respiratory epithelial cells. DAB chromogen, hematoxylin counterstain. (Courtesy of Sally Davis and Jefferey Taubenberger.)
Pathogenesis
After intranasal inoculation, influenza virus localizes and replicates in great numbers within the nasal mucosa [206]. The influenza virus attaches to sialic acid on the surface of respiratory epithelial cells via an α-2,6- or an α-2,3-glycosidic linkage for human and avian influenza viruses, respectively [207,208]. The ability of certain strains to infect cells of the lower respiratory tract or other organ systems relates primarily to properties of the hemagglutinin glycoprotein [209]. Cleavage of a hemagglutinin precursor molecule is required to activate virus infectivity, and the distribution of activating proteases in the host is one of the determinants of tissue tropism [209]. In contrast to low or nonpathogenic strains where cleavage of hemagglutinins occurs extracellular, hemagglutinins of highly pathogenic strains are cleaved intracellularly by ubiquitous proteases enabling them to infect a wide variety of cells [209]. Even though host immunity plays a crucial role in preventing the spread of the virus outside the respiratory tract, highly pathogenic influenza viruses are potent inducers of proinflammatory cytokines in macrophages, in particular tumor necrosis factor alpha and interferon beta, that contribute to the severity of disease [210–212].
Due to their susceptibility to natural, egg-grown, or mammalian cell-grown influenza A and B viruses, ferrets represent the only model that allows studying both pathogenesis and transmission of influenza virus infection, which also makes them an excellent model to test efficacy of vaccines and therapeutic drugs [182,213,214]. As an experimental model, ferrets have been used to study both uncomplicated upper respiratory tract infection following intranasal inoculation and viral pneumonia that may lead to systemic disease after either intranasal or intratracheal inoculation [176,178–180,204]. To model influenza virus transmission, including transmission of drug-resistant influenza strains, ferrets are usually inoculated intranasally and placed in special cages with controlled air flow that are connected to cages with uninfected ferrets [178,213,215]. The development of an influenza virus aerosol delivery and analytic system that can measure the number of viable influenza viruses in droplets and droplet nuclei exhaled from infected ferrets allows a more accurate reaction of natural influenza infection for studying virus transmission [216]. Ferrets have also been widely used as a model to investigate both vaccines and new antivirals, as a surrogate of influenza virus virulence, as a preliminary for human studies with challenge viruses in quarantine units, and for ferrets as an efficacy model for influenza vaccines [213,217]. Other model systems include primed ferrets, whereby ferrets are infected with a low dose of one virus subtype and allowed to recover for 3–4 weeks before being immunized with a vaccine formulated with a different subtype, resulting in more rapid vaccine-induced immune responses and higher homologous hemagglutination inhibition titers [203,213,218–220]. Ferrets have also been used as models for particular manifestations of influenza infections such as Reye syndrome and inner ear infections [221,222]. There are literally hundreds of studies detailing inoculation of ferrets with various strains of influenza viruses in different experimental settings as indicated earlier, and any attempt to detail these studies would go beyond the scope of this chapter. The various aspects of the ferret model of influenza, including clinical presentation, immunity, pathogenesis, comparative pathology, therapy, transmission, and vaccine testing have been reviewed elsewhere ([176,177,182,213,214,217,223–225]).
Diagnosis
Presumptive diagnosis of naturally occurring influenza is generally based on clinical signs, compatible clinical history, and recovery from illness within 4–5 days [7]. The main differential diagnosis is CD, which is generally a more severe disease [7].
Serological tests, such as ELISA, hemagglutination inhibition assay, and microneutralization assay, are available and antibody production starts at 6–7 dpi [226,227], by which time clinical signs could have disappeared. Serology is rarely used in clinical cases, but commonly used in research. Virus isolation and real-time RT-PCR can be performed using fresh or frozen tissues, nasal swabs, or bronchoalveolar washes [7]. Many laboratories can further classify the identified influenza A virus into the hemagglutinin and neuraminidase subtypes. Histopathology and immunohistochemistry (Fig. 20.38) are also tools available for the postmortem diagnosis of influenza virus infections in formalin-fixed tissues from affected ferrets [173,228]. With the rise of the worldwide concern for influenza, numerous laboratories offer tests to diagnose influenza in animals; veterinarians should be aware of the influenza tests offered by their diagnostic laboratories.
Treatment
Generally, the disease is mild, and treatment consists of supportive care and the administration of antibiotics, cough suppressants, and so on. However, a number of experimental studies have shown that antiviral treatment in ferrets can prevent disease, reduce clinical signs, and reduce internal lesions produced by the virus [201]. Antiviral drugs that have been used in a safe and effective way in ferrets include the neuraminidase inhibitor oseltamivir (Tamiflu® [oseltamivir phosphate], Genentech, South San Francisco, CA; 2.5–5 mg/kg PO BID for 10 days) [201], although higher doses (12.5 mg/kg PO BID) may be needed to treat clinical cases with more pathogenic strains [229,230]. Zanamivir (Relenza®, GlaxoSmithKline, Philadelphia, PA), another neuraminidase inhibitor, has also been used in ferrets with influenza; this drug should be administered by inhalation at doses of 0.3–1 mg/kg BID [231]. The use of monoclonal antibodies against human influenza has also been used successfully in ferrets, both for prevention and treatment of influenza, using doses of 10–30 mg/kg IV once [190]. Interferon α (for humans) reduces severity of clinical signs in ferrets, using doses of 107 units SID for several days [232]; it can be administered intranasally or by injection, but the intranasal route does not seem to be so effective for highly pathogenic strains, probably because it cannot reach the lung [232]. Higher doses of interferon α might be more efficacious [232]. It has also been demonstrated that intranasal gels with low pH (approximately 3.5) can reduce clinical signs of influenza in ferrets, but more studies are needed to recommend this kind of treatment [191]. Cianovirin N (0.25 mg/kg BID intranasally for 5 days) has also been used experimentally with some success in ferrets with influenza [233]. Other antiviral drugs, such as amantadine or rimantadine (inhibitors of the ion cannel M2), produce a higher percentage of resistance, are not as effective against influenza B viruses, and can have adverse effects in the CNS [190,231].
Most of these drugs can be used both for prevention and for treatment of influenza, and they lose efficacy once the ferret has already started showing clinical signs [233]. In addition, effective doses to prevent disease are lower than effective doses to treat clinical signs [229]. New drugs for the treatment of human influenza are being developed constantly, and most of them have been tested in experimental ferrets; therefore, new drugs such as peramivir will be available in the near future to safely and effectively treat ferrets [194,234].
Prevention
Influenza is a zoonotic and anthropozoonotic disease, and it is transmitted from infected ferrets to humans, as well as from infected humans to ferrets [7]. Ferrets are extensively used as experimental models for human influenza, as human strains are pathogenic in this animal, and the ferret's biological response to infection is similar to that produced in humans [7,177]. During the first 3–5 days of infection, at the height of pyrexia, infected ferrets can transmit the disease to other ferrets and to humans [177]. Aerosol droplets are a ready mode of transmission [207]. Therefore, it seems logical to assume that any influenza virus able to produce disease in humans may also produce disease in ferrets, and this may also be applicable to new strains that may arise in the future. In addition, strains that produce severe disease in humans also tend to produce severe disease in ferrets, and vice versa [235].
As with any other viral disease, a thorough cleaning and removal of all organic material from the environment should precede disinfection so that the pathogens come in direct contact with the disinfectant [236]. Various physical forces such as ultraviolet light, heat, and drying can help reduce the load of influenza virus in the environment [236]. Ultraviolet radiation from sunlight is lethal to most microbes, but it does not penetrate effectively beyond the outer contamination layer [236]. Artificial ultraviolet light is used in some laboratories that handle samples of influenza, but its use to disinfect premises with ferrets does not seem feasible. A hot, dry sunny day will cause rapid natural inactivation of influenza virus [236]. Dry heat can be used to inactivate influenza virus, and effective combinations are 32°C for 3 hours and 38°C for 30 minutes [236]. Wet heat (steam) is not as effective as dry heat. Washing with soap and hot water is also effective at removing organic matter and inactivating influenza virus [236]. Many chemical disinfectants are able to inactivate avian influenza virus, including iodophors, hydrogen peroxide, ethanol (70%), bleach (2–3% sodium hypochlorite for a 10- to 30-minute contact time at a temperature not higher than 15°C), phenols, cresols, glutaraldehyde (best at temperatures >10°C), formaldehyde and formalin, acids (hydrochloric and citric), and sodium hydroxide (caustic soda) [236,237]. Phenols, cresols, iodophors, and glutaraldehyde are effective in the presence of organic matter. Quaternary ammonium compounds, sodium hypochlorite, and ethanol are not as effective in the presence of organic matter [236]. Whenever possible, chemicals that are safe for humans and easy to obtain should be used (e.g., bleach, iodophors, quaternary ammonium compounds), leaving other disinfectants that are more toxic for humans (e.g., formaldehyde, glutaraldehyde, sodium hydroxide) for situations where other disinfectants cannot be used [236].
Vaccination of ferrets is not an effort-effective way to prevent disease: the disease is usually mild, and there is a great antigenic variation between different strains [7]. However, vaccines used for humans are considered safe and effective, as many of them have been previously tested in ferrets [238–240]. Animals that recover from infection acquire immunity for at least 5 weeks. Protection is antibody mediated and passively transferred in the colostrum [226].
Ferret Rotaviruses
Ferret rotaviruses are one of the few causes of viral enteritis in ferrets. Besides anecdotal reports of a group A rotavirus and a single report of an atypical rotavirus both causing diarrhea in juvenile ferrets [241], only group C rotavirus has been established as a cause of diarrhea in juvenile ferrets based on genetic analysis [242].
Etiology
Rotaviruses are 55–70 nm nonenveloped, double-stranded RNA viruses in the family Reoviridae and are composed of six structural and five nonstructural proteins [243–245]. Rotaviruses have been divided into seven major serogroups designated A to G based upon antigenic properties and RNA migration patterns in polyacrylamide gels [244–248]. All groups are serologically unrelated and contain their own distinct antigen [249]. Furthermore, a species-specific antigen is present on the outer virion capsid layer [247]. Group A rotaviruses are “typical” rotaviruses that share a common antigen within the inner virion capsid layer [247,250,251], whereas rotaviruses in all other groups have been designated as “atypical” and do not contain the common antigen present in group A viruses [249,251]. Recently, a nucleotide sequence-based, complete genome classification system was developed for group A rotaviruses [252].
In ferrets, there are only reports of a species-specific group C rotavirus identified as Ferret Rota C-MSU (Fig. 20.39, [242]), and a single report of an atypical rotavirus [241], both causing diarrhea in juvenile ferrets. The atypical rotavirus most likely also represents a group C rotavirus since it lacked the common antigen of type A rotaviruses and had an electrophoretic RNA migration pattern characteristic of atypical rotaviruses [241]. The VP6 sequence of the Ferret Rota, C-MSU has been compared with corresponding sequences of known group C and A rotaviruses and was found to be most closely related to the Shintoku (bovine strain) and the Cowden (porcine strain) group C rotaviruses [242,253,254]. Anecdotal reports of group A rotavirus infections causing diarrhea in juvenile ferrets exist, but hard evidence is lacking.
Fig. 20.39. Phylogenetic tree based upon deduced amino acid sequences of the VP6 proteins of Ferret Rota C-MSU; rotavirus group C strains, bovine Shintoku (M88768), porcine Cowden (M94157), human Jajeri (AF325805), and human Preston (M94156); and rotavirus group A strains, avian rotavirus A (D16329) and bovine rotavirus A (K02254). GenBank accession numbers are in parentheses. (Reprinted from Wise et al., Vet Pathol 46: 985–991, 2009, with permission from Sage Publications Inc.)
Transmission and Epidemiology
Rotaviruses cause diarrhea in young ferrets, but milder infections of older animals can occur [242,247,255,256]. Rotaviruses are transmitted by the fecal-oral route, via contact with contaminated fur or skin, surfaces and objects, and possibly by the respiratory route [247,256,257]. Feces shed by an infected animal contain large numbers of infectious viral particles, and actually fewer than 100 of these particles are required to transmit infection [251,257,258]. In ferrets, transmission of rotaviruses to juvenile ferrets occurs primarily via contact with their infected mothers or a contaminated environment. Outbreaks of diarrhea may therefore occur throughout the year in breeding facilities, but are more common in colder months [137]. Infected jills can harbor the virus for long periods of time in their fur, and reinfection is a common occurrence in a breeding facility. Viral particles are quite stable within feces and common disinfectants are ineffective, thereby making it difficult to eradicate the virus once ferrets have become infected [247].
Clinical Signs
Rotaviral disease usually occurs in juvenile ferrets younger than 2 months of age and commonly at 1–3 weeks of age (Fig. 20.40). Yellowish, soft to watery diarrhea leads to quick dehydration and death of some animals. The perianal region and sometimes the fur of the entire body can be matted or stained by feces. Erythema of the anus and perineum has been described for some cases [241]. The morbidity is highest among litters of primiparous jills and can reach 90% but tends to decrease by 10–20% with each gestation [137]. The abdomen is commonly distended by gas- and fluid-filled intestines [242]. Older animals may exhibit mild, transient diarrhea.
Fig. 20.40. Ferret kits. The bodies are thin and dehydrated with distended abdomens. (Reprinted from Wise et al., Vet Pathol 46: 985–991, 2009, with permission from Sage Publications Inc.)
Pathology
Gross lesions of ferrets with group C rotaviral disease are characterized by an enlarged abdomen (Fig. 20.40), dehydration, and thin-walled small intestines that are often distended with gas and fluids (Fig. 20.41, [242]). Microscopically, the small intestine of ferrets with acute disease is characterized by superficial atrophic enteritis [242]. The epithelial cells at the villous tips undergo degeneration and necrosis, resulting in loss or sloughing of affected epithelial cells into the intestinal lumen (Fig. 20.42). In more chronic cases, there is mild blunting and fusion as well as bridging of affected villi and a moderate to severe lymphoplasmacytic infiltrate into the lamina propria [242]. Calcified casts within renal tubules are commonly observed secondary to severe dehydration. All other organs are microscopically unremarkable. Transmission electron microscopy of sections of small intestine demonstrates rotaviral particles within apical vacuoles of affected epithelial cells (Fig. 20.43, [242]). Similar lesions have been observed in other atypical rotavirus infections causing diarrhea in juvenile ferrets [241].
Fig. 20.41. Small intestines, ferret. The small intestines are thin walled and distended with gas and fluid. (Reprinted from Wise et al., Vet Pathol 46: 985–991, 2009, with permission from Sage Publications Inc.)Fig. 20.42. Small intestine, ferret. Microscopically, there is degeneration and necrosis of epithelial cells at the villous tips with loss or sloughing of affected epithelial cells into the lumen. H&E. (Reprinted from Wise et al., Vet Pathol 46: 985–991, 2009, with permission from Sage Publications, Inc.)Fig. 20.43. Small intestinal epithelial cell, ferret. Viral particles are within apical vacuoles, have a viral capsid, and are between 55 and 70 nm in diameter. Lead citrate and uranyl acetate stain. (Reprinted from Wise et al., Vet Pathol 46: 985–991, 2009, with permission from Sage Publications, Inc.)
Pathogenesis
Transmission of rotaviral disease occurs most commonly through the oral route. Inoculation of 2- to 3-week-old ferrets with bacterial free, filtered homogenates from ferrets with rotavirus-associated diarrhea resulted in similar clinical signs [241].
The triple protein coats make ingested rotaviruses highly resistant to low gastric pH and digestive enzymes in the small intestine [257]. After reaching the small intestine, viral particles adhere to epithelial cells along the tips of the intestinal villi [251,257,258]. Cellular invasion follows by receptor mediated endocytosis and rotaviruses form apical endosomes, vesicles lined by a trilaminar membrane in the cytoplasm of apical villar epithelial cells [242,257]. Replication of the rotaviruses in the infected cells causes structural and functional changes of the affected epithelium [251,257,258]. Proteins in the third layer (VP7 and the VP4 spike) disrupt the membrane of the endosome, causing a difference in calcium concentration [257,258]. This leads to a breakdown of VP7 into single protein subunits and formation of a double-layered protein shell composed of VP2 and VP6 that protects the viral RNA and allows the virus to evade host immune responses [257,258]. Viral mRNA transcripts are created by the RNA-dependent RNA polymerase for both protein biosynthesis and gene replication. Most of the rotavirus proteins accumulate in viroplasm, which is localized around the cell nucleus and consists of viral factories formed by two viral nonstructural proteins: NSP5 and NSP2 [257,258]. Newly formed virions migrate to the endoplasmic reticulum where they obtain their third outer layer formed by VP7 and VP4, and infectious virus is released from the cell by lysis [251,257,258].
Diagnosis
RT-PCR on fecal samples or sections of small intestine from infected ferrets is most commonly used to diagnose rotavirus infections. Transmission electron microscopy is utilized in some laboratories to detect characteristic viral particles in feces. The commercially available ELISA kits for group A rotaviruses will not detect atypical ferret rotaviruses [259]. Ferret rotaviruses have not been isolated in cell culture.
Different rotavirus serogroups can be distinguished based on their RNA migration patterns in polyacrylamide gel electrophoresis (PAGE), RNA terminal fingerprint analysis, and serologic assays [244,249,260,261]. With PAGE, the genome segments can be divided into size regions I through IV [262]. Group A rotaviruses have a characteristic 7-8-9 triplet in region III that cannot be found in atypical rotaviruses [246,262]. Group C rotaviruses also have a slower migrating segment 7 than group A rotaviruses [246,262]. These differences result in a characteristic RNA migration pattern for group A rotaviruses of 4-2-3-2 and a characteristic RNA migration pattern for group C rotaviruses of 4-3-2-2 [246,262]. Following identification of rotaviruses by PAGE, serologic comparisons are needed to confirm this grouping [246,262]. RT-PCR to detect group A rotavirus on fecal material is available for diagnostic purposes, and a group C rotavirus-specific RT-PCR assay has been recently developed [242].
Treatment
Treatment is directed toward compensation for the loss of large amounts of fluid and prevention of secondary bacterial infections. Antibiotics may be beneficial in accelerating the recovery and decreasing mortality rates [137]. The young age of affected animals and rapid progression of dehydration makes immediate supportive care, in particular fluid therapy and force feeding, vital for keeping the animals alive.
Prevention
Rotavirus-infected ferrets shed large amounts of virus in their feces. Viral particles are stable in the environment and may be carried in the fur of a shedding animal for extended periods of time [256]. There are some reports indicating group C rotaviruses being shed in lower amounts than group A rotaviruses and being less stable in feces [263]; however, this is of little practical value when dealing with preventing an infection. Common sanitary measures that adequately eliminate bacteria and parasites are ineffective in controlling transmission of rotaviruses, and chlorine solutions, like household bleach, or sodium hydroxide are most effective for disinfection [258].
There is currently no commercial vaccine available. Several anecdotal reports describe success in controlling diarrhea in breeding facilities by force-feeding stool from affected juveniles to pregnant jills. Colostral antibodies can protect neonates from rotaviral diarrhea, but not from infection [247]. Ferret kits require all of their IgA from the mother's milk, which is central in protecting ferrets from disease [264].
Rabies Virus
Rabies is an almost invariably fatal disease of humans and many mammalian species. It remains an important global disease causing more than 55,000 human fatalities annually worldwide and requiring millions of patients to receive postexposure treatment [265]. Whereas rabies is enzootic in different mammalian wildlife species in the United States, it is uncommon in domestic animals [266]. Through constant vaccination programs of dogs and cats against rabies since the 1940s, the dog-specific rabies strains have disappeared in the United States, and rabies cases are recorded from infection with wildlife strains, the vast majority caused by bats [267]. Naturally occurring rabies is an exceedingly rare disease in ferrets, and less than 30 cases have been reported in the United States in the last 50 years [137,268,269]. No case of human rabies caused by a ferret bite has been reported [7].
Etiology
Rabies virus is an enveloped, negative sense, single-stranded RNA virus in the genus Lyssavirus, in the family Rhabdoviridae, order Mononegavirales [270]. The viral genome encodes five genes: nucleoprotein, phosphoprotein, matrix protein, glycoprotein, and RNA polymerase. Lyssaviruses are classified into 12 species based on their genetic similarity to rabies virus and besides rabies virus include the Lagos bat virus, the Kujand virus, the Aravan virus, the Irkut virus, the Mokola virus, the Duvenhage virus, the Shimoni bat virus, the West Caucasian bat virus, the European bat lyssavirus type 1 and type 2, and the Australian bat Lyssavirus [271,272]. The European bat virus isolates have been distinguished from the Duvenhage-virus and were identified as two distinct genotypes [273,274]: genotype 5 (European Bat Lyssavirus type 1, EBLV-1) and genotype 6 (European Bat Lyssavirus type 2, EBLV-2). A phylogenetic analysis of the complete genome of lyssaviruses revealed a strong geographical localization of each genotype, with the greatest genetic diversity in Africa, and an independent origin for the two known genotypes that infect European bats [271]. All lyssavirus genotypes except the Mokola virus have bat reservoirs [275]. Four new lyssavirus genotypes that infect bats in central and southeast Asia have been proposed: Aravan virus, Khujand virus, Irkut virus, and West Caucasian Bat virus [271].
Distinctive rabies virus variants that occur within geographically discrete areas and are maintained through intraspecies transmission of the virus can be identified through monoclonal antibody testing or partial genomic analysis of the nucleoprotein and glycoprotein sequences [276–278]. In an analysis of 192 rabies virus isolates from 55 countries, the authors identified six clades of rabies viruses in nonflying mammals that had distinct geographical distributions, which most likely reflected major physical barriers and little viral movement among geographical regions [279]. Interspecies transmission of rabies virus usually results in dead-end host infections, however, novel reservoirs and closely related rabies virus variants may emerge in long-standing epizootics [275,278,280].
Transmission and Epidemiology
Any mammal can be infected with rabies virus and develop clinical signs, and antibodies against rabies have also been detected in wild birds [281]. Bats, raccoons, skunks, and foxes may carry rabies virus in the United States, and bat rabies is present in all states except Hawaii [282]. Most human cases in the United States are caused by bat rabies viruses, and transmission may occur without awareness of having been exposed to or bitten by a bat [267]. It is therefore important to recognize that ferrets exposed to bats may have become infected, and dead bats found on the premises accessed by the ferret should be submitted for viral testing. Bat species that are most commonly associated with human deaths due to transmission of rabies virus in the United States include silver-haired bats (Lasionycteris noctivagans) and eastern pipistrelle bats (Pipistrellus subflavus), and less commonly, Brazilian free-tailed bats (Tadaida braziliensis; [267]). Raccoon rabies is mainly found in the eastern states of the United States, but human death due to raccoon rabies virus strains is rare [283]. In Europe, serotine bats are considered the principal reservoir species for EBLV-1, and pond and Daubenton's bats the reservoirs for EBLV-2 [284].
Rabies virus is usually present in the saliva of animals with clinical signs of rabies, and the almost exclusive route of transmission is through saliva transmitted by a bite [267]. Rare cases of aerosol transmission in laboratory accidents or in caves containing millions of bats as well as through organ transplants have been reported in humans [285,286].
After a bite exposure, saliva containing infectious rabies virus is deposited in muscle and subcutaneous tissues and the virus travels in peripheral nerves toward the CNS where it causes severe encephalitis and clinical signs [267]. While migrating toward the CNS, the virus cannot be easily detected within the host, and vaccination will still induce cell-mediated immunity to prevent clinical disease [270]. After an affected animal or human starts to show clinical signs, treatment is almost never effective and mortality is over 99% [267].
Since the incidence of rabies infections in ferrets as well as the period of rabies virus shedding in the saliva of infected animals prior to displaying clinical signs were unknown, historically, any ferret bite was considered as a potential exposure to rabies virus. This was exaggerated by reports of ferret-induced injuries, including unprovoked bites of infants and small children, which stirred a large controversy of the suitability of ferrets as pets and their potential risk as rabies transmitters [287–289]. These concerns were largely unfounded and, to the authors' knowledge, there are no reported cases of human rabies secondary to a ferret bite.
Clinical Signs
Rabies is infectious to all mammals, and clinical signs have been best characterized in dogs. There are three recognized clinical stages of rabies viral disease in dogs that are variably present in other mammalian species:
The prodromal stage: a 1- to 3-day period of behavioral changes.
The excitative stage: a 3- to 4-day period that is also known as the furious rabies stage since affected animals tend to be hypersensitive to external stimuli and easily attack and bite at anything. Other clinical signs of furious rabies include hypersalivation, cardiac arrhythmias, priapism, and hydrophobia caused by spasms of diaphragmatic muscles on attempts to drink or at the sight of water.
The paralytic stage: also known as dumb rabies, is characterized by incoordination, hind limb paralysis, drooling, and difficulty swallowing due to damage of motor neurons causing muscle paralysis. Sphincter muscle involvement is common.
Depending on the host and rabies strain, infected animals may present only with clinical signs of either the furious (encephalitic) rabies or the dumb (paralytic) rabies. Rabies is ultimately almost always fatal, and death is usually caused by respiratory arrest. Animals that present with clinical signs of paralytic rabies usually survive longer than those who have encephalitic rabies.
Ferrets tend to present with paralytic rabies, and clinical signs consist of hyperactivity with intermittent periods of lethargy, hypothermia, bladder atony, constipation, inappetence and anorexia, abnormal or frequent vocalization, sneezing, ptyalism, excessive grooming (paresthesia), photophobia, ataxia, tremors, seizures, hind limb weakness, paralysis or spasms in the mandible, and posterior paralysis or ascending paralysis [7,266,274]. In experimental infections, only 10% of affected ferrets developed aggressive behavior [266]. Differential diagnoses include any disease able to produce CNS signs (see section on CD). Since transmission has to involve contact with wildlife or rabid domestic animals, considering the history and determining potential exposure are crucial when clinically differentiating rabies from other neurologic diseases. Unvaccinated ferrets with access to the outdoors are clearly at the highest risk to contract disease [79]. No hematologic or serum biochemical changes are characteristic or specific for rabies in animals [290].
Pathology
There are no gross lesions of rabies. Histopathological changes do not reflect the severity of the clinical disease. Histologically, lesions are limited to the brain and spinal cord and are typical of a viral nonsuppurative polioencephalomyelitis, including perivascular cuffing, vascular congestion, neuronophagia, neuronal degeneration, and focal to diffuse gliosis. The distribution of lesions varies with the rabies virus strain. Lesions are most commonly observed in the cerebrum, brainstem, and spinal cord and may be most severe in the brain stem. Areas of hemorrhage and necrosis and neuronal loss associated with focal microglial nodules and perivascular lymphocytic cuffing are not uncommon in the spinal cord. The dorsal root ganglia commonly have moderate inflammation characterized by lymphocytes and neuronal degeneration. Spinal and cranial nerve ganglia, particularly the trigeminal nerve ganglia, may show necrosis and severe inflammation with infiltration by many lymphocytes. Spongiform lesions may be found in the gray matter, in the neuropil, and in neuronal cell bodies of the thalamus and cerebral cortex. The radial and sciatic nerves may have mild inflammatory changes and focal areas of myelin degeneration.
Intracytoplasmic neuronal inclusions (Negri bodies) are considered pathognomonic for rabies (Fig. 20.44), but these are only seen in about 50–75% of cases [291,292]. They are most commonly found in ganglionic cells of the hippocampus and in Purkinje cells of the cerebellum. The density and distribution of Negri bodies are different in short-time survival versus long-time survival cases. In animals that survive for a longer time period, Negri bodies are not only frequently observed in the hippocampal and hypothalamic regions, they are also common in large numbers in other parts of the brain as well, including ganglion cells of cervical dorsal root ganglia and in the lower cervical sympathetic ganglion. In dogs with rabies, mild inflammation may be seen in the salivary glands. Other organs, such as heart, pancreas, adrenal, and liver may also have foci of inflammatory cells, consisting mainly of lymphocytes.
Fig. 20.44. Cerebellum, ferret with rabies. Nonsuppurative meningoencephalitis with characteristic intracytoplasmic inclusion bodies, Negri bodies, in Purkinje cells. H&E.
Ultrastructurally, viral inclusion bodies are nonmembrane bound, generally located in the perinuclear cytoplasm, and have been characterized into three morphologically distinct forms [291,292]. They can present as round to oval bodies with a compact electron-dense granular matrix and radially arranged, circular electron lucent areas that have electron dense cores or as round to oval bodies that consist of tubular structures embedded in a granular electron-dense matrix [292]. These tubular structures can be observed in longitudinal and transverse sections and have blunt ends, dense walls, and lucent central areas. Besides these two types, inclusions may also present with a simple electron-dense granular matrix with neither the radially arranged circular areas nor the tubular structures [292].
Pathogenesis
Rabies virus is almost exclusively transmitted through bites of a rabid animal [266]. Aerosol exposure and transmission through organ transplantation are rare and of no consequence in ferrets [267]. The virus is transmitted with the saliva of the infected animal into the subcutaneous tissue and muscle where it remains for the majority of the incubation period that may last from 20 to 90 days or longer in some cases [266,267]. Within a muscle or nerve cell, the trimeric spikes on the external side of the rabies viral membrane bind to nicotinic acetylcholine receptors on the postsynaptic membrane of the neuromuscular junction in muscle cells [293]. The number of receptors varies among species and may be responsible for increased susceptibility in some species that have a high quantity of nicotinic acetylcholine receptors, such as foxes [293]. Through pinocytosis, the virus enters into the cell within an endosome where it will start to replicate. Following receptor binding, the virus crosses the neuromuscular junction and travels retrograde in the axons of peripheral nerves to the CNS [294]. After reaching the CNS, the virus is widely disseminated. The impairment of serotonin neurotransmission following infection of raphe nuclei in the brainstem is considered to be the cause of behavioral changes [267,295]. In rabies vector species, the virus spreads from the brain to multiple organs along autonomic and sensory neurons and ultimately reaches the salivary gland and is secreted in the saliva [267].
Ferrets have been experimentally inoculated with skunk, racoon, and European red fox variants of the rabies virus [268,269,296]. In all studies, the main incubation period was around 4–5 weeks, aggressive behavior was rarely reported, and the severity of clinical signs as well as the mortality rate were dose dependent in ferrets inoculated with the skunk and European fox strains [268,269,296]. Ferrets inoculated with a raccoon strain of rabies virus were only moderately susceptible regardless of dose [296]. Animals that survived experimental infection remained clinically normal except for one ferret that had been inoculated with the skunk rabies variant and developed severe paralysis [268,269,296]. A single case of an experimentally inoculated ferret (skunk variant rabies) that developed clinical signs of hind limb paralysis at 81 days post inoculation, but survived with paraplegia and completely cleared viral antigen, has been described [297]. As previously discussed, the route of exposure, intramuscular versus mucosal, the dose of the inoculated virus, and the virus variant may determine fatal versus nonfatal infections [298]. In most cases of documented recovery, as in the ferret reported here, high titers of neutralizing antibodies have been detected [297].
The serologic response of ferrets to rabies virus infection varied depending on the rabies virus variant that was used for inoculation [268,269,296]. Almost half the ferrets inoculated with the skunk rabies variant developed neutralizing antibodies compared with only 10% of ferrets that had been inoculated with the raccoon rabies variant [269,296]. No virus was detected within the saliva of ferrets inoculated with the skunk and European fox strains [268,269]. In contrast, viral shedding in the saliva was reported in almost 50% of ferrets that had been inoculated with the raccoon rabies variant [296]. Viral excretion ranged from 2 days prior to the onset of clinical signs to 6 days after the onset [296]. Viral shedding has also been reported in 1 of 23 ferrets that had been inoculated with a rodent strain of rabies virus [299]. Oral inoculation of ferrets failed to cause infection [298].
Only rabies virus has been reported to cause natural lyssavirus infections in ferrets, but ferrets have been experimentally infected with EBVL-1 and EBVL-2 [274]. Following a short incubation period of less than 2 weeks, ferrets inoculated with EBLV-1 developed clinical signs of rabies, and the mortality rate was dose dependent [274]. Clinical signs included ataxia, paralysis and spasm of the lower jaw, spasmodic mastication and seizures, fatigue, photophobia, inquisitiveness, aggressiveness, cachexia, gasping, and screaming. EBLV-1 was not detected in saliva, and viral RNA could only be detected in the salivary glands of a few animals [274]. Ferrets inoculated with EBLV-2 did not develop clinical signs, and no virus was detected in tissues from these ferrets [274]. All ferrets, inoculated with either EBLV-1 or EBLV-2, seroconverted, and ferrets that did not die from EBLV-1 infection had continuously increasing titers [274]. In ferrets inoculated with EBLV-2, the highest titers were observed 1 month post inoculation, after which a rapid decrease of the titers was detected [274]. The study documents that ferrets are fully susceptible to EBLV-1, but are dead-end hosts and therefore do not actively transmit EBLV-1 to a new host [274].
Diagnosis
Clinical signs including sudden paralysis or an acute change in personality are good indications for rabies in ferrets, especially in animals that have not been vaccinated and had recent contact to wildlife, for example, skunks, raccoons, foxes, or bats, in areas where rabies is enzootic [137,266]. CD is the primary differential for a ferret developing neurologic signs. Unfortunately, many animals may not show typical signs of rabies, and contact of unvaccinated animals with wildlife species in areas where rabies is enzootic may represent an exposure to the virus. This includes contact with bats in areas where bat rabies is prevalent. Unvaccinated ferrets suspected of having rabies should be euthanized and submitted for laboratory testing. Removal of the brain or the head should preferably be done by the laboratory personnel to avoid potential exposure of the collector. Rapid and accurate laboratory diagnosis for animal rabies is important for confirmation and protection of people that had previous contact with the suspected animal.
The diagnosis of rabies can only be performed after necropsy [7]. No antemortem diagnostic tests are sensitive enough to be consistently reliable [290]. In the United States, the Center for Disease Control (CDC) advises on the most current methodology of rabies testing, and more information can be found at http://www.cdc.gov/rabies. Rabies testing is regulated by the departments of community health of each state, and testing is performed according to the CDC guidelines (see Chapter 9). The most commonly used test is the direct fluorescent antibody test on sections of fresh/frozen brain tissue [290]. The test must include tissues from at least two locations in the brain, preferably the brain stem and cerebellum and requires specific laboratory biological safety equipment. Other diagnostic tests include histology, immunohistochemistry (Fig. 20.45), electron microscopy, virus isolation, serology, and RT-PCR [266,290]. Negri bodies, viral cytoplasmic inclusions most commonly observed in Purkinje cells, can be observed on histologic examination, but they are not consistently found in all rabies cases [266]. Immunohistochemistry has similar sensitivity and specificity as the direct fluorescent antibody test, but since it is performed on formalin-fixed sections, it also requires additional time to receive test results and involves higher labor costs [266]. To overcome these issues, the CDC developed a direct rapid immunohistochemistry test that performs similarly to the fluorescent antibody test [300]. Virus isolation is performed through mouse inoculation or cell culture; both have similar sensitivity, but cell culture is faster [266]. Neither method is used for routine laboratory diagnosis. RT-PCR is useful when the sample size is small, such as when collecting saliva or spinal fluid. RT-PCR for rabies diagnosis is as rapid as the direct fluorescent antibody test and as sensitive as the mouse inoculation test. RT-PCR is also used in epidemiologic investigation and outbreak studies, and, when combined with sequencing, allows the differentiation of rabies virus variants [266]. Serologic tests are used to determine vaccine immunogenicity, as some countries require a positive antibody titer for importation [290].
Fig. 20.45. Jejunum, ferret with FSCD. Dection of rabiesvirus antigen by immunohistochemistry (red) in neurons. Vector red chromogen with hematoxylin counterstain.
Treatment
There is no treatment for rabies in animals [7,266]. If a ferret is exposed to a rabid animal, state laws regulate postexposure treatment. Such treatment may consist of euthanasia if the ferret is not vaccinated, or if the ferret is vaccinated with an approved vaccine and vaccine status is current, immediate revaccination and placement in strict isolation for 45 days [290]. According to the CDC, animals with a low probability of rabies such as healthy ferrets with no history of exposure to rabid animals and current vaccine histories do not need to be euthanized after biting a person [301]. An observation period of 10 days may be appropriate to rule out the risk of potential human rabies exposure [290]. This observation period is particularly important if the attacked is unprovoked [290]. However, in areas where rabies is a problem, when people are bitten by stray ferrets or ferrets with CNS signs, the animal should be euthanized and the head submitted for examination. No cases of human rabies caused by ferret bites have been reported in the United States [7].
Prevention
Vaccination of ferrets is effective in 90% of cases [7]. Vaccination is indicated in those areas where rabies is enzootic and ferrets have access to the outdoors. Rabies vaccination is mandatory for ferrets in many states in the United States. Inactivated (killed) vaccines are commonly used, and efficacy of such vaccines has been demonstrated in experimental studies. Of all ferrets that had been subcutaneously vaccinated once with an inactivated rabies virus, only 11% died following challenge with a virulent fox origin rabies virus 1 year later, in contrast to 94% mortality in unvaccinated ferrets [302]. Ferrets that had been vaccinated with an inactivated rabies vaccine showed rapid seroconversion, and virus neutralizing antibodies could be detected for at least 7 months [303].
The recommended vaccination protocol for inactivated rabies vaccines consists of the first vaccination at 3 months of age and then annual boosters [7]. The primary vaccination has to occur at least 28 days prior to exposure to render protection [303]. Ferrets are also considered to be protected immediately after booster vaccination.
A canary pox-vectored rabies vaccine has been tested in ferrets, and the cellular response to the vaccine was much milder than to the adjuvanted rabies vaccines discussed previously [304]. This is of importance since a severe inflammatory reaction, in particular the persistence of lymphocytes and macrophages, has been suggested to play a role in the pathogenesis of vaccine-associated sarcomas [305,306]. Considering the concern for such local vaccine reactions and the potential for the development of vaccine-site sarcomas, the canary pox–vectored vaccine would be less likely to evoke such response. However, canary pox–vectored rabies vaccines are currently not approved for use in ferrets in North America.
Modified live rabies vaccines have not been licensed for ferrets, and there is evidence that such vaccines can induce disease [7].
Adverse Reactions to Vaccination
Rabies vaccination, either administered alone or in combination with distemper vaccine, has been associated with anaphylactic reactions [100]. Characteristics and treatment of these anaphylactic reactions are described under the section on CD in this chapter. The specific component producing anaphylaxis is unknown, and it could be different among manufacturers, countries, and so on. Fibrosarcomas and sarcomas associated with the site of rabies vaccination have been described in ferrets [305,306]. Rabies vaccination is therefore questionable in areas where vaccination is not mandatory or in animals with a very low risk to contract disease.
Disinfection
Rabies virus can remain viable in a carcass for several days at 20°C, although it may survive much longer when the body of the victim is refrigerated [290]. Storage at ultra-low temperatures (−30°C to −80°C) prolongs viral activity for years in untreated fresh-frozen tissue [290]. As an enveloped virus, rabies is inactivated by ultraviolet light, heat, and various concentrations of formalin, phenol, alcohol, halogens, mercurials, mineral acids, and other disinfectants [290].
Pseudorabies
Natural infections of pseudorabies virus (PRV) in ferrets have not been reported [226], which can be most likely attributed to a lack of access to infectious virus, regardless of whether the animals are kept as household pets or for laboratory purposes (see Transmission and Epidemiology). Most reports of experimental infections with pseudorabies in ferrets are more than 40 years old, and the last outbreak of pseudorabies in mustelids was reported in mink in 1986 [307,308]. However, ferrets have been used in experimental studies investigating the neuronal circuitry that controls the activity of phrenic and abdominal motoneurons [309–311].
Etiology
Pseudorabies, also known as Aujeszky's disease, is caused by PRV, which has been classified as suid herpesvirus 1, a member of the Alphaherpesvirinae subfamily. PRV is most closely related to bovine herpesvirus 1, equine herpesvirus 1, and varicella-zoster virus [312]. Alphaherpesviruses are characterized by their rapid lytic growth in cell culture, their neurotropism, their latency in neurons, and their broad host range. PRV is a double-stranded linear DNA virus with a nucleoprotein core that contains the viral genome, an icosahedral capsid, a proteinaceous tegument, and a lipid bilayer envelope which contains virally encoded glycoproteins.
Transmission and Epidemiology
Pigs are the natural host for PRV infections, but the virus can infect a wide range of animal species such as hedgehogs, cattle, sheep, goats, opossums, chickens, pigeons, geese, ducks, buzzards, sparrow hawks, rabbits, guinea pigs, rats, mice, some nonhuman primates such as rhesus macaques, and many carnivorous species including dogs, cats, foxes, polecats, jackals, and ferrets [313,314]. There have been no reports of PRV infecting humans. Historically, PRV was distributed worldwide, but has now been eradicated from numerous countries, including the United Kingdom, Canada, Austria, Sweden, Denmark, and New Zealand [313]. PRV has also been eradicated from domestic pigs in the United States but continues to circulate in feral swine [315]. The prevalence of PRV infections in carnivorous species is currently unknown in the United States. However, there have been recent reports in the United States of PRV infections in dogs that were used to hunt feral swine [316].
In pigs, PRV may be shed in nasal and oral secretions as well as semen and vaginal secretions. The virus is most commonly transmitted by nose-to-nose contact, but coitus, artificial insemination, fomites, or transplacental infection have been reported. Infectious levels of virus remain high in dead pigs [313]. Other species may become infected through direct nose-to-nose contact with pigs shedding the virus, or more commonly by consuming uncooked offal of infected pigs [317]. With no direct contact to PRV-infected feral swine or wild boar or feeding of uncooked offal from either source, the natural transmission of PRV is not likely in most countries.
Clinical Signs
Pigs are the natural host for PRV infections, and clinical signs depend on the route of infection, the age at exposure, the immune status of the infected pig, and the viral strain. Neonatal pigs initially show respiratory signs, but rapidly develop neurologic disease that ultimately results in death. Older pigs primarily present with respiratory disease; neurologic disease is only associated with infections by high doses of highly virulent strains.
In most other species susceptible to infection, PRV typically causes fatal neurologic disease that is often associated with localized pruritus [317]. In addition to neurologic signs such as trembling, ataxia, weakness, inactivity, aggression, or hyperexcitability, clinical signs in carnivorous species typically include facial pruritus and ptyalism as the most common clinical manifestations [316,317]. Ferrets experimentally inoculated with PRV have also been reported to develop dyspnea, restlessness, vomiting, diarrhea, vocalization, muscle stiffness, and death [307,318,319]. Death occurs usually within 4 days or less after the onset of clinical signs [307,318,319].
Pathology
Gross lesions of PRV in most carnivorous species are limited since PRV typically represents as a neurotropic disease [316,317]. In contrast, generalized lymphadenopathy, moderate splenomegaly, and pulmonary edema have been described in experimentally inoculated ferrets [307,318]. The thoracic cavity and the pericardial sac may contain serosanguineous fluid. The mediastinum and thymus are often edematous. Petechiae and ecchymoses may be widespread in the lungs, cardiac muscle, epicardial fat, mediastinum, thymus, diaphragm, gastric wall, and pancreas [307,308,318]. Hemorrhages may also occur throughout the intestines, meninges, and urinary bladder.
Histologically, lesions in carnivorous species are determined by the neurotropic dissemination of the virus [316,317]. The trigeminal ganglia are most commonly affected by severe inflammation characterized by lymphocytes admixed with neutrophils and macrophages. Ganglion cells undergo degeneration and neuronophagia. Nuclei often contain large, eosinophilic inclusion bodies resulting in peripheral marginalization of the chromatin [316,317]. Similar changes may be observed in the facial, glossopharyngeal, and vagal nerves. The metencephalon and myelencephalon, especially the medulla oblongata, are commonly affected by a nonsuppurative encephalitis with typical perivascular cuffing by lymphocytes and glial nodules [316]. Inflammation may be neutrophilic especially when associated with severe neuronophagia. Neuronal degeneration, necrosis, and intranuclear inclusion bodies in neurons and astrocytes are common features. In addition to ganglioneuritis and encephalitis, PRV may also cause myocardial degeneration with ganglionitis of the cardiac autonomic plexi and degeneration of intestinal myenteric ganglia [317]. The facial skin often exhibits an ulcerative dermatitis [316].
In experimentally inoculated ferrets, microscopic lesions were not only limited to the nervous system, but affected the vasculature and multiple large parenchymas indicating hematogenous and lymphatic viral spread [307,318]. Similar lesions have been described in wild mustelids infected by PRV as well as experimentally infected mink [308,320]. Histologic lesions in nonneuronal tissues included dendritic cell proliferation as well as lymphoid cell necrosis in spleen, lymph nodes, tonsils and Peyer's patches, multifocal coagulative necrosis and Kupffer cell proliferation in the liver, cholangitis, and ovarian necrosis [307,318]. Multifocal areas of severe necrotizing and ulcerative dermatitis were commonly associated with phlebitis that corresponded to injection sites [307]. A severe necrotizing arteritis was observed in several tissues, but was most pronounced in the lungs [307]. Principal histologic changes in blood vessels were similar to those observed in with PRV-inoculated mink and consisted of degeneration and necrosis of the walls, especially fibrinoid necrosis of the media, sometimes accompanied by thrombosis and perivascular hemorrhages [307,308]. Cardiac muscle necrosis, hemoglobinuric nephrosis, and jejunal crypt necrosis as observed in experimentally infected mink are not common features of PRV infection in ferrets [307,308].
Lesions in the CNS were characterized by a nonsuppurative meningoencephalitis with lymphocytes infiltrating the meniges and forming perivascular cuffs. Lesions were most consistently found in the parietal and occipital lobes, in the nucleus intercalatus in the dorsal motor nucleus of the vagus nerve just beneath the 4th ventricle in the medulla and less commonly in the hypoglossal nucleus and the spinal vestibular nucleus in the medulla, in the pons, and in the midbrain as well as in the cerebellum [307]. Focal areas of malacia were present in the gray and white matter, and neurons in affected areas underwent chromatolysis and pyknosis. There was mild to marked gliosis with formation of glial nodules. Intranuclear eosinophilic inclusion bodies were occasionally found in neurons and glial cells [307]. Lesions in the spinal cord were most pronounced in the lumbar part and similar to those in the brain, with neuronal degeneration, necrosis, occasional neuronophagia, lymphoid cells causing perivascular cuffing and meningeal infiltration, and marked gliosis with glial nodule formation. Epithelial cells lining the spinal canal were commonly degenerated and desquamated [307]. Axonal swelling and demyelination was occasionally observed in the spinal funiculi. Lesions in the peripheral nervous system were primarily detected in the sciatic nerve and mesenteric ganglia [307]. Intranuclear inclusion bodies were observed in the spinal cord and outside the CNS in the mesenteric ganglia, ganglion cells in sections of adrenals, and in sections of submaxillary salivary gland and pancreas [307].
Lesions were more severe in those ferrets with a longer clinical course. The incubation period seemed related to the viral dose and to the distance between the inoculation site and the CNS [307,321]. Virus was usually recovered from the spinal cord and, less frequently, from the respiratory tract [321].
Pathogenesis
Viral infection starts with cell entry that involves attachment of the viral particle to the cell surface. In PRV, this initial binding step is an interaction between glycoprotein C in the virion envelope and heparan sulfate on the surface of the cell [314]. Following glycoprotein-mediated fusion of the virion envelope with the cell membrane, the capsid and tegument proteins are released into the cell. The latter takes over the host cell protein synthesis, and the capsid and inner tegument proteins are transported to the cell nucleus [314]. Viral DNA replication occurs by a rolling-circle mechanism, and the onset of DNA synthesis signals the start of the late stage of the PRV replication cycle and synthesis of true late proteins. The capsid proteins are transported to the nucleus, where they assemble into the capsid. During primary envelopment, the fully assembled nucleocapsid buds out of the nucleus, temporarily entering the perinuclear space [314]. The nucleocapsid loses its primary envelope and gains its final envelope by associating with tegument and envelope proteins and budding into the trans-Golgi apparatus. The mature virus is brought to the cell surface within a sorting compartment/vesicle derived from the envelopment compartment [314].
The distribution of inflammation in PRV infections is consistent with the mechanism of viral spread. Following primary replication, the virus is taken up by sensory nerve endings and spreads by both anterograde and retrograde axonal transport first to the sensory nerve ganglia and subsequently to the CNS [312]. In addition, cell-to-cell spread and, considering the widely distributed lesions in the vasculature and multiple large parenchymas, hematogenous and lymphatic viral spread via infected monocytes also occurs in ferrets [307,314,318]. In ferrets, the virus has been experimentally administered by oral, intranasal, subcutaneous, intramuscular, or intracardiac routes, and microscopic lesions varied in distribution and severity accordingly [307,321].
It is unknown whether ferrets can become latently infected with PRV. Latency is defined as a status when viral DNA persists, but infectious virus is not produced [312]. The trigeminal ganglion, olfactory bulb, and tonsils are major sites of latency. The molecular mechanisms of latency are still unclear, but glycoprotein E seems to play a central role in the ability of PRV to invade neurons [312].
Because PRV has a broad host range and causes lethal infections in various animal species, but poses no threat to humans, it has been utilized as a model organism to study alphaherpesvirus biology, including the basics of the viral life cycle and the more complex interactions with the host [314,322]. Besides its broad host range, PRV's abilities for transsynaptic spread and self-amplification have allowed its use in an extensive number of neuroanatomical studies seeking to define the architecture of multisynaptic pathways [314]. Ferrets have been utilized in such model systems studying the transneuronal tracing of the motorneurons innervating the diaphragm and abdominal muscles [310]. During PRV injection, studies demonstrated that both muscles receive inputs from neurons in circumscribed regions of the spinal cord and brainstem (Fig. 20.46 and Fig. 20.47), some of which have an overlapping distribution in the magnocellular part of the medullary reticular formation [309,311]. Experimental data suggest that the coactivation of inspiratory and expiratory muscles during emesis or some postural adjustments in ferrets can be elicited through collateralized projections from a single group of brainstem neurons located in the medullary reticular formation [310]. The data suggest that in ferrets, a population of neurons in the brainstem, located mainly in the magnocellular part of the medullary reticular formation, provides inputs to both inspiratory and expiratory motoneurons in the spinal cord Fig. 20.48 [310]. The organization of this circuitry is species-specific. Later studies demonstrated that some medullary reticular formation neurons simultaneously influence the activity of upper airway and respiratory pump muscles, whereas other neurons in this brain stem region independently contribute to regulation of diaphragm and genioglossal muscle contraction [323].
Fig. 20.46. Brainstem, ferret with transneuronal tracing using pseudorabies virus. Distribution of pseudorabies infected neurons in the brainstem, represented by open squares, 4.5 days after virus injection into the diaphragm. Courtesy of Bill Yates. (Reprinted from Billig et al., J Neurosci 20: 7446–7454, 2000 with permission from the Society of Neuroscience.)Fig. 20.47. Brainstem, ferret with transneuronal tracing using pseudorabies virus. Immunohistochemical detection (brown) of pseudorabies virus antigen in premotor neurons in regions of the brainstem following injection of pseudorabies virus into rectus abdominis on the contralateral side. DAB chromogen with modified Kluver-Barrera counterstain. Courtesy of Bill Yates. (Reprinted from Billig et al., J Neurosci 20: 7446–7454, 2000, with permission from the Society of Neuroscience.)Fig. 20.48. Brainstem, ferret with transneuronal tracing using pseudorabies virus. Immunofluorescent detection of pseudorabies virus antigen in premotor neurons in regions of the brainstem. Red cells were only infected by retrograde transynaptic passage of one recombinant viral strain from the diaphragm, green cells were only infected by retrograde transsynaptic passage of a different recombinant virus strain from rectus abdominis, and yellow cells contain both viruses. Courtesy of Bill Yates. (Reprinted from Billig et al., J Neurosci 20: 7446–7454, 2000, with permission from the Society of Neuroscience.)
Diagnosis
Differential diagnoses for ferrets exhibiting clinical neurologic signs combined with pruritus and/or other of the previously described symptoms of PRV infections include rabies, canine distemper, and various toxicoses such as organophosphates, heavy metals, ethylene glycol, strychnine, inorganic arsenic, organomercurial compounds, and chlorinated hydrocarbons [316].
The most commonly performed diagnostic tests for PRV in veterinary diagnostic laboratories include PCR and virus isolation on infected tissue samples and nasal swabs, as well as serologic assays. Tissues reported to be useful for virus isolation or PCR include mesencephalon, medulla oblongata, and cerebellum, haired skin from pruritic areas, salivary gland, pharyngeal mucosa, lung, liver, and adrenal gland. Immunofluorescence and immunohistochemistry are also available to detect virus in tissue sections (Fig. 20.49). Other reported tests include a rabbit bioassay, in situ hybridization, and electron microscopy. The serologic assays consist of latex agglutination tests, serum neutralization assays, and enzyme-linked immunosorbent assays testing for a humoral response to glycoprotein gE [314]. Enzyme-linked immunosorbent assays specifically identifying the presence of gE in an animal's blood are particularly useful because these distinguish an immune response (gE+) to a field strain infection from an immune response (gE–) to a vaccine strain [314]. PCR on the trigeminal ganglion is required to determine latency of the virus.
Fig. 20.49. Brainstem, ferret with transneuronal tracing using pseudorabies virus. Immunohistochemical detection (brown) of pseudorabies virus antigen in premotor neurons in nucleus retroambiguus infected after injecting pseudorabies virus into the diaphragm on the contralateral side. DAB chromogen with modified Kluver-Barrera counterstain. Courtesy of Bill Yates. (Reprinted from Billig et al., J Neurosci 20: 7446–7454, 2000, with permission from the Society of Neuroscience.)
Treatment and Prevention
Alphaherpesviruses are susceptible to most commercially available disinfectants, detergents, and antiseptics. They can be inactivated by temperatures above 56°C within 5 minutes and by temperatures of 37°C within 3 hours [324].
There are no licensed commercial vaccines for PRV in ferrets, and since exposure of either pet or laboratory ferrets to PRV is highly unlikely, no vaccination is necessary. Prevention of PRV infection should be easily achieved by avoiding the feeding of uncooked offal from swine or wild boar to ferrets. Ferrets that develop neurologic signs caused by PRV and left untreated will commonly die. There are no data regarding the mortality of PRV-infected ferrets when treated. Supportive therapy to restore fluid, electrolyte, acid–base and nutritional balances, as well as antibiotic therapy against secondary infections are recommended. In animals with nasal discharge, mucolytic drugs may be considered. A number of antiviral drugs have been used to treat herpesviral infections in other animals, such as ocular disease in cats [324]. These drugs may be considered for the treatment of PRV infected ferrets; however, their efficacy is not supported by any published data.
Bovine Herpesvirus 1
Bovine herpesvirus 1 (BoHV-1) has been isolated from the spleen of a clinically normal ferret, and experimental inoculation of ferrets produced acute and chronic respiratory tract disease.
Etiology
BoHV-1 is a herpesvirus in the family Herpesviridae and the subfamily Alphaherpesviridae. BoHV-1 causes several diseases in cattle worldwide, including rhinotracheitis, vaginitis, balanoposthitis, abortion, conjunctivitis, and enteritis and is a contributing factor to shipping fever. BoHV-1 is most closely related to PRV, equine herpesvirus 1, and varicella-zoster virus [312]. Additional information about alphaherpesviruses has been described under PRV.
Transmission and Epidemiology
Bovine herpesvirus 1 (causative agent of Infectious Bovine Rhinotracheitis [IBR]) has been isolated from the spleen of a clinically normal ferret. The diet of this animal contained 5% of uncooked beef tripe as well as occasional supplementation with other raw beef by-products [325].
Cattle are the natural host of BoHV-1. In cattle, BoHV-1 is spread horizontally through sexual contact, artificial insemination, and aerosol transmission, and vertically across the placenta. The main mode of transmission is direct nose-to-nose contact by mucus-containing virus. The ability of BoHV-1 to evade the hosts' immune system makes it difficult to clear viral infection, and lifelong infections are possible. As with PRV in pigs, latent infection is quite often found in the trigeminal ganglia of the cow. Under conditions of stress or other infectious diseases, latent infections can be reactivated with or without clinical symptoms. Infected animals will be continuous shedders throughout their lifetime when the virus reactivates, and they will continue to shed virus in high titers even when not showing clinical signs. Goats, buffalo, red deer, sheep, swine, and reindeer can act as reservoirs for BoHV-1.
In the only described naturally occuring case of BoHV-1 infection in a clinically normal ferret, the virus was isolated from the liver, the spleen, and the lungs [325]. The diet of this animal consisted of 5% raw beef by-products and the authors speculated that virus-laden raw beef was the source of infection [325].
Clinical Signs
In cattle, BoHV-1 can cause both clinical and subclinical infections, depending on the virulence of the strain. Disease is rarely non-life-threatening, but economically important.
There has been only a single case report of a naturally occuring BoHV-1 infection in a clinically normal ferret [325]. Experimental infection resulted in respiratory signs characterized by sneezing, coughing, and anorexia from days 3 to 7 post inoculation [326]. The virus was readily recovered from the upper and lower respiratory tract, retropharyngeal lymph nodes, and spleen on day 4 post inoculation, but only from pharynx on days 8 and 12 post inoculation. Infected ferrets showed serologic responses comparable to those expected in experimentally exposed cattle.
Pathology
In experimentally inoculated ferrets, a mucopurulent exudate was observed grossly in the nasopharynx and in some animals in the trachea at 4 and 8 days post inoculation [326]. There were petechiae and ecchymoses in the pharyngeal and tracheal mucosa. Histologically, infected ferrets had an acute suppurative pharyngitis characterized by infiltration of neutrophils into the mucosa and submucosa. There was an acute esophagitis affecting the proximal portion of the esophagus characterized by severe ballooning degeneration of the stratified squamous epithelium, intranuclear inclusion bodies in epithelial cells and lymphoplasmacytic inflammation of the submucosa [326]. The administration of 4 mg of dexamethasone intraperitoneally caused recrudescence of viral shedding [326].
Pathogenesis
Alphaherpesviral infections and replication have been reviewed in detail [314], and additional information is provided in the section on PRV. BoHV-1 enters the animal as an aerosol through the mucous membranes of the respiratory tract or venerally in the genital tract [327]. In cattle, indirect transmission may occur through contaminated food or water, infected semen used in articial insemination, or contaminated milking machines [327]. Shedding begins from the nasal mucosa as soon as infection occurs and the virus replicates in the upper respiratory tract. During replication in the respiratory tract, infected epithelial cells will undergo degeneration and necrosis. The epithelial cell necrosis provides entry sites for secondary bacterial infections. Further outcome of the infection depends on the virulence of the infecting strain, dosage, and host immunity, and viral replication may be limited to the entry site or virus may be spread systemically through viremia or anterograde and retrograde within the nervous system [314,327].
In ferrets, the mechanism by which BoHV-1 disseminated to the liver, spleen, and lung tissue has not been elucidated. Experimental infection of ferrets with BoHV-1 by intranasal and intraperitoneal inoculations induced acute and chronic respiratory disease, suggesting a similar viral tropism in ferrets as in cattle [326].
Diagnosis
Differential diagnoses for ferrets exhibiting respiratory signs include canine distemper, influenza virus, mycoplasmosis, and various bacterial infections.
The most commonly performed diagnostic tests for BoHV-1 in veterinary diagnostic laboratories are similar to those employed for PRV and include PCR and virus isolation on infected tissue samples and nasal swabs, as well as serologic assays. Tissues reported to be useful for virus isolation or PCR include nasal mucosa and lung, as well as liver, spleen, brain, and adrenal glands in animals with systemic disease. Immunofluorescence and immunohistochemistry are also available to detect virus in tissue sections. The most commonly utilized serologic assays are the serum neutralization test and indirect and competition ELISA tests [328]. The IgM ELISA is useful for diagnosis of recently infected calves. Antibodies against BoHV-1 in cattle have been shown to persist for a long time at high levels, making it easy to detect infected animals [329]. A commercial anti-BoHV-1 blocking ELISA kit is available to differentiate between vaccinated and naturally infected cattle, but the sensitivity is only 74% [328]. PCR on the trigeminal ganglion is required to determine latency of the virus.
Treatment and Prevention
For cattle, there is a vaccine available that reduces the severity and incidence of disease. In particular, the development of marker vaccines to differentiate between vaccine strains and wild-type virus allows cost-efficient usage of vaccines for synchronous reduction of economic losses and eradication of disease [329]. Some countries in Europe have successfully eradicated the disease by applying strict culling policies.
Considering that BoHV-1 can cause pathology in ferrets, they should not be fed raw meat or meat products [137]. This statements applies to other herpesvirus infections transmitted from other mammalian animal species too, for example, PRV, and eliminating raw meat from a ferret's diet will, if not eliminate, make the likelyhood of transmission of herpesviral diseases to ferrets very low.
BoHV-1 is relatively resistant to environmental influences and remains stable at 4°C for 1 month, but is inactivated at 56°C within 21 minutes, at 37°C within 10 days, and at 22°C within 50 days [328]. BoHV-1 can survive for more than 30 days in feeds. As an enveloped virus, it is sensitive to organic solvents such as chloroform, ether and acetone, and many disinfectants. It is readily inactivated by 0.5% NaOH, 0.01% HgCl2, 1% chlorinated lime, 1% phenolic derivatives, 1% quaternary ammonium bases, 10% Lugol's iodine, and 5% formalin [328].
In conclusion, although several herpesviruses are able to infect and produce clinical signs of disease in ferrets, the lack of reports on natural infections make specific preventative measurements unnecessary especially as long as ferrets are not fed raw beef and pork meat products or have no contact with livestock or other potential carrier species.
Feline Parvovirus
Feline panleukopenia virus (FPV) as well as raccoon parvovirus (RPV) can replicate in experimentally inoculated adult ferrets, but do not cause disease [330]. In contrast, congenital and neonatal infections of ferrets with FPV can cause cerebellar hypoplasia [331,332].
Etiology
FPV is a parvovirus that belongs to the genus Parvovirus within the family Parvoviridae and includes several closely related species-specific viruses that infect carnivorous animals, including canine parvovirus (CPV) with its antigenic types CPV-2a, CPV-2b, and CPV-2c, RPV and mink enteritis virus [333]. FPV is a nonenveloped single-stranded DNA virus that infects organs with rapidly dividing cells, such as the intestine, bone marrow, and lymphoid tissue [334].
Transmission and Epidemiology
The fecal-oral route is the main mode of virus transmission of FPV. In postpartum infected cats, FPV replicates in lymph nodes, thymus, spleen, and intestine, and large quantities are shed in feces [334]. FPV can cross the placenta and infects replicating cells in the developing fetus. Virus acquired in utero may persist as a latent infection in renal and other tissues for months [332]. Since ferret kits can become infected with FPV transplacentally, they may shed the virus after birth even though they would acquire maternal antibodies [335].
Serologic studies of adult ferrets in Finland failed to detect antibodies against FPV, CPV, and mink enteritis virus [336]. Ferrets inoculated orally with CPV isolated from dogs failed to shed virus, to develop disease, or to seroconvert [337]. In contrast, when ferrets were intraperitoneally inoculated with FPV, CPV, RPV, and mink enteritis virus, FPV and RPV replicated in ferrets but did not cause clinical disease or microscopic lesions, and isolated virus could be passaged repeatedly [330].
Clinical Signs
In one study, ferret kits inoculated intraperitoneally at 1 and 2 days after birth with a live FPV vaccine developed severe ataxia at 6–8 weeks post inoculation [331]. Affected animals had general motor neuron impairment characterized by tremors, loss of righting and climbing, but were able to eat and drink [331]. Kits inoculated at 3 days of age as well as kits from dams inoculated at various stages of gestation with the live FPV vaccine showed no clinical signs [331]. This is in contrast to another study where transplacental infection with FPV was achieved in a single fetus of an intraperitoneally inoculated dam and the infected fetus developed cerebellar hypoplasia and mild renal lesions [332]. In other studies, direct inoculation of fetuses at different stages of gestation and intracerebral inoculation of newborn kits also resulted in cerebellar hypoplasia and occasional extraneuronal lesions [332,338]. In utero infections can also result in abortion and mummified fetuses.
Pathology
Gross lesions in affected ferret kits were limited to the cerebellum. Uniform, symmetrical hypoplasia was observed in all animals [331,332]. The vermis was completely collapsed and the hemispheres flattened dorsoventrally.
Microscopically, cerebellar alterations in cats with severe clinical signs were characteristic of hypoplastic end-stage lesions with complete absence of the granular layer and no intranuclear inclusions [331,332]. Commonly, individual folia were disorganized, while others showed no signs of viral attack [331]. In another study, intrauterine inoculation with FPV resulted in lesions of the infected ferret that at 1 day after birth, and 18 days post inoculation of the dam were characterized by depletion of the germinal tissue and absence of the granular cell layer [332]. Similar lesions (Fig. 20.50, Fig. 20.51, and Fig. 20.52) were observed in intracerebrally inoculated newborn kits [338]. There were scattered intranuclear inclusion bodies in germinal cells and pyknotic germinal cell nuclei predominantly in the deep nuclei [332,338]. Immature Purkinje cells formed the dominant element in the cortex. The subependymal cell plate, in particular the periventricular zone, was also severely affected, and germinal cells at all depth had large numbers of intranuclear inclusions [332]. At such sites, ependymal cells had undergone fragmentation, and the ependymal surface was focally effaced. Depolarization of germinal cells had also occurred, leading to surface overgrowth and rosette formation [332].
Fig. 20.50. Cerebellum, ferret, 13 days old, negative control. Unaffected cerebellum with complex pattern of folia, prominence of the external germinal layer, and early state of development of the definitive granular layer. Phosphotungstic acid hematoxylin stain. (Reprinted from Kilham and Margolis, Am J Pathol 48: 991–1011, 1966, with permission from Elsevier.)Fig. 20.51. Cerebellum, ferret, 13 days old, inoculated with feline panleukopenia virus 1 day after birth. Cerebellum with destruction of the external germinal layer, absence of the definitive granular layer, and reduction of total cerebellar mass. Only a small area of cortex at the posterior pole is spared. Phosphotungstic acid hematoxylin stain. (Reprinted from Kilham and Margolis, Am J Pathol 48: 991–1011, 1966, with permission from Elsevier.)Fig. 20.52. Cerebellum, ferret, 52 days old, inoculated with feline panleukopenia virus 1 day after birth. Severe granuloprival hypoplasia with intracytoplasmic vacuolar degeneration of Purkinje cells. Phosphotungstic acid hematoxylin stain. (Reprinted from Kilham and Margolis, Am J Pathol 48: 991–1011, 1966, with permission from Elsevier.)
Pathogenesis
The unique type of ataxia due to cerebellar hypoplasia observed in ferret kits is caused by a direct effect of FPV on the germinal primordium, ultimately causing absence of the granular layer of the cortex that develops in the 3rd trimester of gestation [335]. The ability of the virus to cross the placenta and its affinity for replicating cells, thereby attacking the developing cerebellum, are responsible for the development of this particular clinical disease. FPL causes no disease or lesions in infected dams, but crosses the placenta and destroys the external germinal cell layer of the developing cerebellum which will result in manifestation of ataxia shortly after birth [335]. The damage to the fetal external granular layer has to occur prior to its migration to form the internal granular layer of the cerebellum at a stage of cell replication [339]. Viral infection results in primary loss of granular cells rather than in alterations of synaptic organization [340]. The postmitotic granular cell layer is resistant in older kits to an infection with FPV, and there is no evidence that other replicating cells, for example, hematopoietic cells or crypt epithelial cells, are susceptible to FPV infection in ferret kits.
The capsid of the nonenveloped FPV not only serves as a durable protection against the environment, but it also undergoes sequential conformational changes that allow translocation of the viral genome from its initial host cell nucleus into the nucleus of the subsequent host [341]. FPV binds to neuraminidase-sensitive N-glycolyl neuraminic acid side chains that function as attachment receptors of host cells, but infectious entry is mediated by binding to host species-specific protein domains on cell surface transferrin receptor molecules [342]. The cell entry is accompanied by conformational shifts of the virus that cause exposure of a capsid-tethered phospholipase A2 enzymatic core which will act as an endosomolytic agent to mediate virus translocation across the lipid bilayer into the cell cytoplasm [341]. After entry into the nucleus, FPV can only initiate transcription after the host cell enters the S-phase and will then take over the cell's synthetic pathways.
Diagnosis
FPV antigen can be detected in feces from infected kits with commercial test kits that detect viral antigens such as latex agglutination or immunochromatographic tests. Diagnostic veterinary laboratories perform PCR testing and sequencing on whole blood or feces as well as on tissues following postmortem examination. Whereas serological tests, for example, ELISA or indirect immunofluorescence, are not useful for vaccinated cats since they do not distinguish between vaccination and infection, they are highly useful in ferrets since most animals that have not been in contact with FPV-infected cats will be seronegative [343]. Immunohistochemistry using an antibody against FPV on brain tissues of ferret kits that were euthanized or died due to severe ataxia is highly recommended. FPV can also be diagnosed by virus isolation from blood or feces or by hemagglutination of porcine erythrocytes [343].
Treatment and Prevention
Since no FPV-associated disease has been reported in ferrets that have been inoculated at 3 days of age and older, there is limited concern for ferret owners and laboratory ferret colonies, except those that breed ferrets. FPV has to be viewed as a teratogenic virus in ferrets and contact of ferrets with cats that may shed the virus should be avoided [331,332].
Diseased cats, in particular animals with severe diarrhea, shed large amounts of FPV, quickly contaminating the environment [343]. FPV is a nonenveloped DNA virus that is highly resistant to physical factors and chemical substances [343]. It can remain infectious for months in a contaminated environment.
Both modified-live and inactivated FPV vaccines are used in cats and provide solid immunity against disease [343]. In healthy cats, protection by modified-live vaccines is more rapid, and they are the more popular vaccines [344]. However, modified-live virus vaccines should not be used in pregnant queens and kittens under 4 weeks of age because of the risk of damage to the developing cerebellum [331,332,343]. There are no indications to vaccinate ferrets, but in environments where jills or their newborn kits may get into contact with cats, cats should be vaccinated with an inactivated FPV vaccine.
Ferret Papillomavirus
A single case of a multicentric Bowenoid carcinoma in situ associated with a papillomavirus of unknown origin has been described in a pet ferret [345].
Etiology
Papillomaviruses are small, circular, double-stranded DNA viruses with mucosal and skin tropism associated with neoplastic transformation of infected cells [346]. Papillomaviruses are classified within their own family, the Papillomaviridae, and are further divided into genera based on variations in their highly conserved L1 gene [347]. Alpha papillomaviruses include oncogenic papillomaviruses that cause potentially malignant mucosal lesions and viruses that cause benign mucosal or cutaneous lesions, whereas beta papillomaviruses usually require immunosuppression to cause visible lesions [347]. Most of the human papillomaviruses are classified within the alpha or beta papillomaviruses. Delta papillomaviruses include many mammalian papillomaviruses that not only cause benign fibropapillomas in ungulates, but can also infect multiple species and have been shown to cause equine sarcoids [347]. Some papillomaviruses infecting cats and dogs have been designated as lambda papillomaviruses, however, most recently identified feline and canine papillomaviruses associated with cutaneous neoplasms have not been assigned to a particular genus [346].
The papillomavirus within a Bowenoid carcinoma in situ in a ferret was identified based on light microscopic, immunohistochemical, and electron microscopic features [345]. No molecular analysis was possible since no fresh material had been collected, and it remains unclear to what genus the papillomavirus belongs and whether it represents a novel ferret-specific papillomavirus.
Transmission and Epidemiology
Most papillomaviruses are highly host- and site-specific and lesions are often restricted to limited locations on the body and have been detected in most species that have been extensively studied [346]. Infected species include humans, cattle, dogs, cats, horses, bats, rabbits, dolphins, and deer among others. Immunosuppressed individuals are especially predisposed to the development of cutaneous neoplastic lesions associated with papillomaviruses [346].
The incidence of papillomavirus infections in ferrets and the route of transmission of cutaneous infections are unknown, but transmission through skin abrasions has been proposed as the primary mechanism of transmission of cutaneous papillomaviruses in humans [348]. The successful transmission of bovine papillomavirus type 2 from peripheral blood raises the possibility that papillomaviruses may in some circumstances be transmitted through the hematogenous route [349].
Clinical Signs
The ferret presented with multiple black and tan proliferative skin lesions that resembled keratin plaques (Fig. 20.53), and a secondary bacterial and mycotic infection was diagnosed at the time of surgical biopsy [345]. The animal had an unconfirmed history of insulinoma and adrenal disease, and the clinical significance of the skin lesions is unknown [345].
Fig. 20.53. Skin; ferret. Epidermal plaque characterized by moderate irregular hyperplasia with sharply demarcated margins and occasional involvement of superficial follicular infundibula. H&E. (Reprinted from Rodrigues et al., Vet Pathol 47: 964–968, 2010, with permission from Sage Publications Inc.)
Pathology
Grossly, the ferret had multiple black and tan proliferative skin lesions that resembled keratin plaques.
Histologically, the multifocal thickened cutaneous plaques had sharply demarcated margins and were characterized by marked parakeratotic hyperkeratosis (Fig. 20.53) and moderate irregular epidermal hyperplasia that occasionally involved the superficial follicular infundibula [345]. Cutaneous plaques commonly exhibited full thickness epidermal dysplasia with loss of normal epithelial stratification and loss of nuclear polarity (Fig. 20.54). Several keratinocytes had pleomorphic round to oval nuclei, finely stippled chromatin with one to two prominent nucleoli, and moderate to abundant amphophilic to basophilic homogeneous cytoplasm [345]. A few cells in the stratum corneum had abundant, clear, basophilic, homogeneous to vesicular cytoplasm and vesicular to clumped nuclear chromatin; rare keratinocytes in the superficial stratum granulosum or deep stratum corneum contained 5- to 7-mm amphophilic to eosinophilic intranuclear inclusion bodies (Fig. 20.55 and Fig. 20.56, [345]). Anisocytosis and anisokaryosis were moderate, but mitotic activity was high. Numerous scattered apoptotic cells and occasional multinucleated keratinocytes were observed [345].
Fig. 20.54. Skin; ferret. Full-thickness epidermal dysplasia with loss of normal epithelial stratification and nuclear polarity. H&E. (Reprinted from Rodrigues et al., Vet Pathol 47: 964–968, 2010, with permission from Sage Publications Inc.)Fig. 20.55. Skin; ferret. Rare keratinocytes in the superficial stratum granulosum contain 5- to 7-mm amphophilic to eosinophilic intranuclear inclusion bodies (arrow). H&E. (Reprinted from Rodrigues et al., Vet Pathol 47: 964–968, 2010, with permission from Sage Publications Inc.)Fig. 20.56. Skin; ferret. Immunohistochemical labeling for papillomavirus antigen (red) in nuclei of dysplastic keratinocytes. Vector red chromogen, hematoxylin counterstaining. (Reprinted from Rodrigues et al., Vet Pathol 47: 964–968, 2010, with permission from Sage Publications Inc.)
Electron microscopy revealed large numbers of electron-dense, approximately 50 nm in diameter, hexagonal viral particles (Fig. 20.57 and Fig. 20.58) compatible with papillomavirus forming viral inclusions within nuclei of dysplastic superficial keratinocytes [345].
Fig. 20.57. Skin; ferret. Ultrastructure of dysplastic keratinocyte with large paracrystalline arrays composed of electron-dense viral particles within the nucleus. Lead citrate and uranyl acetate stain. Bar 1/4 1 mm. (Reprinted from Rodrigues et al., Vet Pathol 47: 964–968, 2010, with permission from Sage Publications Inc.)Fig. 20.58. Skin; ferret. Viral particles are hexagonal and approximately 50 nm in size. Lead citrate and uranyl acetate stain. Bar 1/4 200 nm. (Reprinted from Rodrigues et al., Vet Pathol 47: 964–968, 2010, with permission from Sage Publications Inc.)
Pathogenesis
In cats, Bowenoid in situ carcinomas appear as multifocal, crusting, hyperpigmented, roughly circular plaques that develop on haired, pigmented skin and are histologically characterized by irregular epidermal hyperplasia and dysplasia, which can progress to invasive squamous cell carcinomas and are associated with papillomavirus infections [346,350]. Exposure to UV light is not a significant cause of lesion development. It is important to recognize that asymptomatic epidermal infections by papillomaviruses that cause cutaneous viral plaques in humans, cats, and dogs are also common [346].
Infection of basal cells in the epidermis by papillomaviruses can result in a persistent asymptomatic infection [351]. Because viral replication only occurs when infected basal cells become terminally differentiated, papillomaviruses attempt to increase both proliferation of basal cells and terminal keratinocyte differentiation, potentially resulting in formation of a viral papilloma [351,352]. The host immune response will resolve most viral papillomas, but papillomavirus can alter cell growth and differentiation with the potential of malignant transformation of infected cells [346]. It remains uncertain whether cutaneous papillomaviruses cause squamous cell carcinomas in ferrets; however, in cats, Bowenoid carcinoma in situ has been suggested to represent a neoplastic transformation of papillomaviral plaques, and papillomaviruses may play a central role in the development of invasive squamous cell carcinoma [346]. The common presence of papillomaviruses in normal skin makes it difficult to prove that they influence neoplastic transformation and are not innocent bystanders [346].
Diagnosis
There are currently no specific diagnostic tools available for the diagnosis of papillomavirus infections in ferrets. Proliferative cutaneous lesions resembling viral plaques, papillomas, Bowenoid carcinoma in situ, or squamous cell carcinoma should be screened for papillomavirus infections. Immunhistochemistry and in situ hybridization using generic antibodies and probes are available and have been shown to be successful [345] Generic PCR tests combined with sequencing should be utilized to help further characterize papillomaviruses in ferrets.
Treatment and Prevention
Only a single case of a cutaneous papillomavirus-associated neoplastic lesion has been identified in a ferret, and the incidence of commensal infections is unknown. Currently, screening of proliferative skin lesions in ferrets for their potential association with papillomavirus infections and identification of the papillomavirus are important to determine whether future preventive measurements will be necessary and if the development of specific vaccines may be desirable.
SARS
Severe acute respiratory syndrome (SARS) is a newly recognized human respiratory disease that started as a pandemic in the beginning of this century [353]. The causative agent is an enveloped positive-sense single-stranded RNA virus that was identified as a human coronavirus, SARS-CoV [353]. SARS-CoV represents a coronavirus that is distinct from the three existing groups of coronaviruses, but is closest to betacoronaviruses because 19 out of 20 cysteines found in the S1 domain of the spike protein are spatially conserved when compared with the betacoronavirus consensus sequence ([354], http://www.ictvonline.org). Since the natural source of SARS-CoV is unknown, a range of species, including ferrets, were investigated for their susceptibility to SARS-CoV infection to help identify the natural reservoir and to develop an animal model for testing efficacy of vaccines ([355]; see also Chapter 25 of this book).
Intratracheal infection of ferrets with SARS-CoV caused lethargy 2–4 days post inoculation and death of one ferret on day 4 in one study and increased temperature and nasal discharge in two other studies [356–358], but no clinical signs in two other studies [359,360]. SARS-CoV was detected in nasal washes, rectal swabs, and pharyngeal swabs on days 2 to 8 post inoculation and in the trachea, lungs, and tracheobronchial lymph nodes at necropsy between days 2 and 5 post inoculation [356–359]. Lesions were observed affecting 5–10% of the lung parenchyma and were characterized histologically by mild alveolar damage with loss of epithelium from alveolar and bronchiolar walls, thickening of alveolar walls, hyaline membranes in some alveoli and peribronchial and perivascular lymphocyte infiltration, but no syncytial cells [356–359]. SARS-CoV was detected by immunohistochemistry in mononuclear cells within inflamed areas of the lungs [357]. Control animals that were cohoused with experimentally infected ferrets also became infected with SARS-CoV, became lethargic, and developed conjunctivitis. Their viral titers gradually increased from day 2 post inoculation [358].
Angiotensin-converting enzyme 2 (ACE2) has been identified as a receptor for the attachment to and uptake of SARS-CoV in host cells [361]. Ferret ACE2 has been cloned and has been shown to function as efficiently as a SARS-CoV receptor as human ACE2, but more efficiently than mouse ACE2 [362]. In a more recent study, the pathology of SARS-CoV infection was investigated in detail in ferrets with particular focus on the distribution of virus in tissues and lesions in association with the distribution of ACE2 [363]. Inoculated ferrets developed multifocal pulmonary consolidation, and microscopic lesions were characterized by diffuse alveolar damage (Fig. 20.59) associated with SARS-CoV antigen expression (Fig. 20.60 and Fig. 20.61) in type 2 pneumocytes [363]. Alveoli contained variable numbers of alveolar macrophages and neutrophils within a proteinaceous exudate. Alveolar septa were moderately thickened with infiltrating neutrophils and macrophages, and there was multifocal moderate epithelial necrosis, multifocal hyperplasia, and hypertrophy of type 2 pneumocytes and bronchiolar epithelial cells [363]. The bronchus-associated lymphoid tissue was hyperplastic, and there were focal aggregates of lymphoid cells around pulmonary blood vessels. ACE2 expression occurred mainly in type 2 pneumocytes and serous epithelial cells of tracheobronchial submucosal glands [363].
Fig. 20.59. Lung, ferret with SARS. Diffuse alveolar damage with intraluminal protein. H&E. (Reprinted from van den Brand et al., Vet Pathol 45: 551–562, 2008, with permission from Sage Publications Inc.)Fig. 20.60. Lung, ferret with SARS. Immunohistechmical detection of SARS coronavirus antigen (brown) expression in type II pneumocytes. DAB chromogen, hematoxylin counterstain. (Reprinted from van den Brand et al., Vet Pathol 45: 551–562, 2008, with permission from Sage Publications Inc.)Fig. 20.61. Lung, ferret with SARS. Type II pneumocyte with SARS coronavirus antigen expression (red). Immunofluorescence. (Reprinted from van den Brand et al., Vet Pathol 45: 551–562, 2008, with permission from Sage Publications Inc.)
Inflammatory pathways, in particular the role of chemokines in regulating the Th1 response, have been studied in a ferret model of SARS-CoV and demonstrated increased gene expression of CXCL10 and its receptor CXCR3 in lung of infected ferrets similar to the observations in human SARS patients [364]. Since type 1 interferons are essential for the clearance of respiratory viral infections, one study inoculated ferrets either intranasally with SARS-CoV or subcutaneously with interferon alpha 2b to investigate differences in gene signatures [365]. A previously established ferret model of intranasally induced SARS-CoV infection characterized by fever, sneezing, lymphopenia, and gross and microscopic lesions similar to the previously described lesions, was used for comparison [356]. Inoculation of interferon alpha 2b caused STAT1 phosphorylation and upregulation of abundant interferon response genes, chemokine receptors, and other genes that participate in phagocytosis and leukocyte transendothelial migration [365]. In contrast, ferrets inoculated with SARS-CoV showed upregulation of a broader variety of genes involved in cell migration and inflammation. In particular, on day 1, every functional pathway in interferon alpha 2b inoculated ferrets had more down regulated genes than upregulated genes, wheras the SARS-CoV inoculated ferrets had the opposite trend [365].
Prophylactic intraperitoneal inoculation of ferrets with a human IgG1 monoclonal antibody that is reactive with the whole inactivated SARS-CoV has been shown to neutralize it in vitro, reduce viral replication in the lungs of infected ferrets, prevent SARS-CoV-induced gross pathology, and abolish shedding of virus in pharyngeal secretions [359]. In a later study, the investigators extended the breadth of protection by synergistically combining inoculation of the above-described antibody with a noncompeting human monoclonal antibody that recognized a different epitope on the receptor-binding domain of SARS-CoV [366].
The efficacy of various vaccines has been tested in ferret models of SARS-CoV [357,367–369]. Adenovirus vaccines expressing SARS-CoV spike protein have been tested in an intranasal SARS-CoV challenge model in ferrets; vaccination led to a substantial reduction in viral load and prevented severe pneumonia [357]. In another study, ferrets received formalin-inactivated, whole virus vaccine prior to challenge with SARS-CoV [367]. Similar to the previous study, vaccinated ferrets cleared virus earlier than unprotected animals and displayed high levels of neutralizing antibodies 1 week after challenge, with the titers waning at 3 weeks after inoculation, concurrent with clearance of the virus [367]. Vaccination did not significantly reduce microscopic lung lesions. A more recent study compared a whole, killed SARS-CoV vaccine (with and without alum) with an adenovirus-based vector vaccine that encoded the SARS nucleocapsid and spike proteins for their ability to protect ferrets against intranasal SARS-CoV challenge [368]. Both vaccines induced neutralizing antibody responses and reduced viral replication and shedding in the upper respiratory tract, and progression of virus to the lower respiratory tract and the severity of pathologic changes, but did not provide complete protection [368]. Similar results were observed when immunizing ferrets with recombinant, modified vaccinia virus Ankara that expresses the SARS-CoV spike protein; however, vaccination also caused a strong inflammatory response in the liver [369]. These data suggest that a combination of vaccine strategies may be required for effective protection from SARS-CoV.
Henipaviruses
The recently named genus Henipavirus belongs to the Paramyxoviridae family, subfamily Paramyxovirinae, and includes Nipah and Hendra viruses, which are negative-sense, single-stranded RNA viruses [370]. Both viruses can infect various animal species, including humans, and each virus has a wide tissue tropism [371]. Pteropus spp. fruit bats have been established as the natural host for both viruses, and horses and pigs may act as intermediary hosts for human infections [370–372]. Neither virus has been reported to cause naturally occuring disease in ferrets, but ferrets have been used as a model for experimental infection with Nipah virus, and more recently Hendra virus, with the goal of evaluating vaccines or antiviral treatment [373–375].
Ferrets inoculated oronasally with Nipah virus develop fever at 4–7 days post inoculation followed by severe respiratory and neurologic disease resembling the infection in humans [373,374,376]. Clinical signs consisted of severe depression, coughing, serous nasal discharge, orthopnea and expiratory dyspnea, cutaneous ecchymoses and subcutaneous edema of the head, vomiting and hypothermia, urinary incontinence, myoclonus, tremors, and hind limb paresis [373,374]. In one study, ferrets that were inoculated with a low viral dose did not develop clinical signs or gross and microscopic lesions, did not shed detectable virus or viral RNA, and did not seroconvert [373]. At necropsy, there was subcutaneous edema of the head, hemorrhagic lymphadenopathy of submandibular lymph nodes, and petechiae in the lung (Fig. 20.62 and Fig. 20.63) and kidney [373,374]. Histologically, inoculated ferrets developed focal necrotizing alveolitis and pulmonary vasculitis (Fig. 20.64), glomerular necrosis, focal splenic necrosis and severe diffuse subacute inflammation of the organs and connective tissues of the head and neck. Less common lesions included focal cystitis, severe acute necrotizing salpingitis, focal adrenal necrosis, thyroiditis, and nonsuppurative meningitis [373]. Mildly affected lymph nodes had focal mononuclear and neutrophilic inflammation of the capsule, accompanied by a zone of subcapsular lymphocyte depletion, whereas more severely affected lymph nodes had large areas of severe hemorrhagic and coagulative necrosis [373]. Syncytial cells were commonly observed intralesionally and contained abundant antigen (Fig. 20.65) as detected by immunohistochemistry [373]. The primary lesion described in one study consisted of systemic vasculitis with fibrinoid necrosis of the media and prominent endothelial syncytia in the spleen, kidney, lung, lymph node (Fig. 20.66), and meninges [374]. Nipah virus was also demonstrated in all fluids, including blood, and in the adrenal gland, kidney, lung, bronchial lymph node, and spleen [373,374]. Low levels were detected in the bladder, liver, ovary, testes, and brain, with the highest levels being present in the olfactory bulbs [373]. Subsequently, ferrets that had been inoculated oralnasally with a high dose of Nipah virus were treated 10 hours post inoculation with a cross-reactive neutralizing human monoclonal antibody that targets the henipavirus G glycoprotein [373]. All ferrets that received the monoclonal antibody were protected from disease while all controls died, suggesting postexposure passive antibody therapy as a potentially succesful therapeutic approach [373]. This is in contrast to another study, which found chloroquine ineffective against Nipah virus infection in ferrets [374].
Fig. 20.62. Lung, ferret with Nipah virus. Multifocal petechiae throughout the lungs. (Courtesy of Deborah Middleton.)Fig. 20.63. Kidney, ferret with Nipah virus. Multifocal petechiae throughout the renal cortex. (Courtesy of Deborah Middleton.)Fig. 20.64. Lung, ferret with Nipah virus. Bronchiolitis with syncytial bronchiolar epithelial cells. H&E. (Courtesy of Deborah Middleton.)Fig. 20.65. Lung, ferret with Nipah virus. Immunohistochemical detection of Nipah virus (brown) within syncytial bronchiolar epithelial cells. DAB chromogen and hematoxylin counterstain. (Courtesy of Deborah Middleton.)Fig. 20.66. Lymph node, ferret with Nipah virus. Severe necrosis effacing lymphoid tissue with intralesional syncytial cells. H&E. (Courtesy of Deborah of Middleton.)
Experimental Hendravirus infections in ferrets produced results similar to those described with Nipah virus infection [373–375,377]. Ferrets that were inoculated oronasally with a high dose of Hendra virus succumbed 6–9 days post inoculation exposure [375]. Clinical signs were essentially identical to those previously described for Nipah virus-infected ferrets and included severe depression and generalized tremors [375]. Microscopically, experimentally infected ferrets had systemic vasculitis, splenitis, and bronchiolalveolitis (Fig. 20.67, Fig. 20.68, and Fig. 20.69) with syncytial cell formation [375]. Inoculations with low doses of Hendra virus resulted in longer survival times and more pronounced neurologic disease, whereas higher doses resulted in the rapid development of severe respiratory disease [377]. Based on these studies, lowering but not clearing Hendravirus systemically will result in the development of both respiratory and neurologic symptoms, with the neurologic symptoms becoming more pronounced [377]. In total, the experimentally reproduced Nipah virus and Hendra virus disease models in the ferret exhibited both severe respiratory and neurologic disease, and generalized vasculitis, in which the underlying pathology closely resembled Henipavirus-mediated disease seen in humans [378].
Fig. 20.67. Lung, ferret with Hendra virus. Vasculopathy with syncytial endothelial cells. H&E. (Courtesy of Deborah Middleton.)Fig. 20.68. Lung, ferret with Hendra virus. Large amounts of viral antigen (brown) throughout the vascular walls, including endothelial cells and pericytes. DAB chromogen and hematoxylin counterstain. (Courtesy of Deborah Middleton.)Fig. 20.69. Kidney, ferret with Nipah virus. Glomerulopathy with large amounts of intralesional viral antigen (brown). DAB chromogen and hematoxylin counterstain. (Courtesy of Deborah Middleton.)
Parainfluenza Virus
Parainfluenza viruses are negative strand RNA viruses in the family Paramyxoviridae [379]. They are adapted to a wide host range of animals including humans. Human parainfluenza viruses (HPIV) are genetically and antigenically divided into four species within two genera, Respirovirus and Rubulavirus [379]. Respiroviruses include HPIV-1 and HPIV-3, and Rubulaviruses include HPIV-2 and HPIV-4. HPIV-1 to 3 are major causes of lower respiratory tract infections in children and immunocompromised patients [379]. HPIV-2 had been considered closely related to simian virus 5 (SV5) and canine parainfluenza virus (CPIV, [379–381]), but newer sequence data now place SV5 and CPIV in the parainfluenza 5 species (http://www.ictvonline.org). Natural disease of ferrets caused by parainfluenzaviruses has not been reported, but ferrets have been utilized as a model for CPIV, SV5, HPIV-1, HPIV-2, and HPIV-3 infection [382–388].
Intranasal inoculation of neonatal ferrets with HPIV-1 to 3 resulted consistently in deaths of inoculated ferrets in 48–72 hours [386–388]. With serial egg passage of the viruses, the mortality rate decreased, while the degree of infectivity remained the same [387]. The attenuation was accompanied by a reduced capacity of the viruses to induce interferon and decreased growth in human and primate cell cultures [387]. Gross and microscopic lesions were similar and consisted of acute laryngotracheitis (Fig. 20.70 and Fig. 20.71), bronchiolitis, and interstitial pneumonia [386,388]. Lesions in the lung were most severe and characterized by necrotizing bronchiolitis and secondary hyperplasia [388]. Thymic necrosis was described in one animal [388]. Virus was consistently isolated from the lungs. When adult ferrets were inoculated with high and low virulent viruses, no disease resulted, and circulating hemagglutinin-inhibiting antibodies could be detected within 2 weeks of intranasal inoculation [386,388]. Inoculation of pregnant dams with a high cell culture passage virus resulted in protective immunity of their offspring when challenged with virulent strains shortly after birth [388].
Fig. 20.70. Trachea, ferret with canine parinfluenza virus. Focal epithelial hyperplasia with loss of cilia and intraepithelial and submucosal infiltration of mononuclear cells. H&E. (Courtesy of Wolfgang Baumgärtner.)Fig. 20.71. Trachea, ferret with canine parinfluenza virus. Immunohistochemical detection of canine parainfluenza virus antigen (brown) in tracheal epithelial cells. DAB chromogen and hematoxylin counterstain. (Courtesy of Wolfgang Baumgärtner.)
The P/V gene has been suggested to be responsible for those parainfluenza virus mechanisms that counteract cellular host responses, and ferrets have been used as a model to test the effect of P/V gene substitutions on SV5 growth and immune responses [384]. The gene encodes both the phosphoprotein P subunit of the RNA-dependent RNA polymerase and the V protein which counteracts antiviral responses [379,384]. SV5 is a poor inducer of host cell responses in tissue culture of human cells [384]. Products of the P/V gene substitutions convert the noncytopathic SV5 into a highly cytopathic mutant that induces in primary cultures of ferret lung fibroblasts high levels of apoptosis, interferon-beta, and proinflammatory cytokines [384]. Intranasal inoculation of ferrets with wild-type and mutant SV5 resulted in a significantly higher anti-SV5 serum IgG response as well as higher wild-type viral titers in the trachea [384]. The antibody response was dose-dependent for the mutant, but not for the wild-type.
Domestic ferrets can be experimentally infected with canine-derived CPIV isolates, shed virus, and develop mild clinical respiratory disease and lesions similar to those found in dogs [380]. The pathogenesis of canine parainfluenza virus was investigated in a ferret model in a number of studies [382,383,385]. Immunocompetent and immunosuppressed ferrets that were inoculated intranasally with CPIV developed minimal clinical signs that were restricted to the upper respiratory tract and consisted of cough elicited by tracheal compression between 3 and 7 days post inoculation [385]. Lymphoplasmacytic and histiocytic intraepithelial inflammation, cellular degeneration of ciliated epithelial cells, and focal epithelial hyperplasia were observed in the trachea and occasionally the nasal cavity, but no lesions were found in the lungs [385]. Only tracheal epithelial cells were positive for CPIV by immunocytochemistry.
The virulence of a neurotropic strain of CPIV was investigated by intracerebral inoculation of ferret kits [382]. Infection with CPIV resulted in a self-limiting, nonsuppurative ependymitis and choroiditis with associated ependymal cell degeneration accompanied by paraventricular and perivascular cellular infiltrates of lymphocytes and monocytes, predominantly in the fourth ventricle and the cervical spinal cord, and less frequently in the lateral ventricles and Sylvian duct [382]. Multifocal Wallerian degeneration with swollen myelin sheaths, loss of axons, and infrequent intra-axonal gitter cell infiltration were rarely observed in the mesencephalon and vestibular nucleus [382]. Viral antigen was detected by immunohistochemistry only in ependymal and subependymal cells [382]. A second study investigated the virulence of two phenotypically distinct strains of CPIV that showed distinctly different in vitro characteristics [383]. Intracerebral inoculation of ferrets with both strains caused no clinical signs or gross lesions with either strain. Microscopically, moderate lymphocytic and histiocytic choroiditis, meningitis, and ependymitis were observed with the syncytial giant cell and plaque-forming CPIV isolate, but only mild to moderate inflammation was observed with the nonsyncytial giant cell-forming strain [383]. The ependymal cells showed focal degeneration with loss of cilia and cellular detachment, and paraventricular vessels were surrounded by lymphoplasmacytic and histiocytic cell infiltrates in Virchow–Robin spaces [383]. CPIV antigen was detected by immunohistochemistry in ependymal cells lining the ventricles in ferrets infected with both strains isolated, with the nonsyncytial giant cell-forming strain causing less expression [383].
Respiratory Syncytial Virus
Respiratory syncytial virus (RSV) is a nonsegmented negative stranded RNA virus in the subfamily Pneumovirinae of the family Paramyxoviridae [389]. Two major surface proteins, attachment glycoprotein G and fusion glycoprotein F, are responsible for viral attachment and cell membrane fusion, and appear to be the principal targets of host humoral immune response following infection [390]. Based on variations in the G protein, two major antigenic groups, A and B, have been identified, but their importance in severity of disease or reinfection is uncertain [391]. RSV represents the most important cause of infectious pulmonary disease in human infants and is also a major pathogen in immunosuppressed and older individuals [392]. Other related viruses include bovine RSV, ovine RSV, caprine RSV, pneumonia virus of mice, and turkey rhinotracheitis virus [389]. No natural occuring infections of ferrets with RSV have been reported.
Attempts to inoculate adult ferrets with RSV demonstrated replication in the nasal tissues, but not in the lungs, when using high titer inocula [393,394]. In contrast, RSV replicated in the lungs of juvenile ferrets that were inoculated at birth and up to 14 days of age, but not in ferrets that were inoculated at 28 days of age [395]. However, titers within lung tissues diminished faster with increasing age of the inoculated animals. Virus growth in the nose was independent of the age of the animal [395]. Regardless of the age at which ferrets were inoculated with RSV, they did not develop clinical disease, no nasal discharge was seen, and no ferret died from infection [393–395]. Additionally, the humoral antibody response was weak or absent. Inoculated juvenile ferrets developed a mild suppurative and erosive rhinitis with formation of multinucleated syncytial cells and appearance of intracytoplasmic inclusions. RSV antigen was detected in abundance in the respiratory epithelium of the nasal turbinates [393,395]. Lesions were healed within 30 days, but submucosal cellular hyperplasia and irregularities in cartilage remained [393]. In the lungs, focal areas of atelectasis were observed and only small amounts of RSV antigen were found in the cells of the alveolar walls [395]. Organ cultures of ferret tracheas inoculated with RSV demonstrated viral growth in fibroblasts of the lamina propria and serosa, but not in ciliated epithelial cells [396].
The age-dependent decrease of pulmonary permissiveness to infection with RSV was further confirmed by organ culture experiments. When lung cultures from ferrets at 3, 28, and 42 days of age as well as adult ferrets were inoculated with RSV, only organ cultures obtained from 3-day-old ferrets replicated RSV in high titers [397]. The authors concluded that the lung culture results indicated that a cellular response or tissue maturation rather than immunologic factors were responsible for the age dependence of RSV growth in ferret lungs [397]. Others demonstrated that juvenile ferrets can be protected from RSV challenge at 3 days of age by gestational infection of the dams with RSV [398]. The protection levels of juvenile ferrets were proportional to the titers of maternal serum neutralizing antibodies and ferrets whose maternal reciprocal serum neutralizing antibody titers were greater than 640 had essentially no viral replication in nasal tissues [398]. However, passive administration of adult ferret serum with a neutralizing titer of 1 : 1024 or greater, either intraperitoneally or orally, did not confer immunity, indicating a non-antibody-mediated protective mechanism [398]. The lack of bronchiolar involvement and the rapidly decreasing permissiveness of the lung with increasing age severely limits the utility of ferrets as a model to study RSV pathology, but provides a model for age dependence of RSV infection in humans [389,399].
With the attempt to test recombinant adenoviruses to produce live, viral-vectored RSV vaccines, ferret models have become more attractive because of the highly restricted host range of human adenoviruses and the limited permissiveness of other animal models to RSV infection [400]. In one study that tested the immunogenicity and protective efficacy of adenoviral vectored RSV vaccines that expressed either the RSV fusion glycoprotein F, the RSV attachment glycoprotein G, or both, ferrets were found to be a more rigorous challenge model than dogs, and intranasal inoculation of ferrets with the RSV vaccines induced significant adenovirus-type specific immune responses [400]. Vaccination with five different vaccination regimens protected ferrets from RSV infection in a dose-dependent manner and booster immunization conferred complete immunity [400].
Ferret models of RSV infection have also been utilized to study increased airway responsiveness to insults. One proposed mechanism of altered airway function is the disruption of neural pathways that normally mediate contraction or relaxation of airways [401]. The study focused on chronic effects of RSV infection on airway control in young ferrets during a period of rapid somatic growth. Significantly reduced airway nonadrenergic noncholinergic inhibitory responses of infected ferrets persisted to at least 24 weeks of age after the acute infection, demonstrating that RSV produces dysfunction of this neural pathway [401].
Vesicular Stomatitis Virus
There are no reports of naturally occuring infections or recent reports of experimental infections of ferrets with vesicular stomatitis virus. In one study, intranasal, intramuscular, intracardiac, or intraperitoneal infection of ferret dams on day 41 of the gestation period with vesicular stomatitis virus serotype Indiana resulted in transplacental infection of fetuses and caused death of newborns 36 hours after birth [402]. Virus could be isolated and demonstrated by immunohistochemistry in placenta and liver, and to lesser degress from kidney, intestine, and spleen in all newborns regardless of the route of infection [402]. No clinically significant illness was observed in the dams, despite a lymphoplasmacytic endometritis, periportal hepatitis, and splenic hyperplasia [402]. Serum-neutralizing antibody titers of greater than 1 : 40,960 were detected in each of four adult ferrets tested 30 days after inoculation [402]. The lack of clinical signs in inoculated older ferrets is in contrast to the results of other studies [402–404].
In another study investigating the host range of vesicular stomatitis virus, 6- to 8-week-old ferrets were inoculated intranasally, intradermally, intramucosally, intracardially, and by corneal scarification [403,404]. Inoculated ferrets developed anorexia, hypersalivation, nasal discharge, restlessness, pyrexia, and weight loss, and developed pronounced vesicles similar to lesions observed in cattle or horses [403,404]. Lesions consisted of well-demarcated vesicles that became bleeding erosions in the tongue, dental pads, lips, and foot pads [403]. Lesions appeared after 4–5 days and started to reepithelialize after 10 days. Healing proceeded without complications and was quite rapid. Lesions were similar regardless of the route of inoculation, but varied in severity [403]. Only ferrets inoculated by corneal scarification developed ulcerative keratitis and conjunctivitis without vesicular lesions. One of the intracerebrally inoculated ferrets died after developing generalized paralysis. Virus was not isolated from feces and urine, but could be isolated from blood and saliva 24 hours after intranasal inoculation [404].
Recombinant vesicular stomatitis virus-based vectors have been shown to be effective vaccine vectors as demonstrated in a number of animal models. Ferrets have been used in comprehensive preclinical safety studies to assess in vivo replication of such live attenuated recombinant vesicular stomatitis virus vaccine vector candidates that encode human immunodeficiency virus (HIV) gag protein [405]. In one study, 7- to 8-week old ferrets were inoculated intracranially or intranasally with either a wild-type vesicular stomatitis virus or an attenuated recombinant vesicular stomatitis virus vector engineered to express HIV gag protein [405]. All ferrets inoculated intracranially with wild-type vesicular stomatitis virus serotype Indiana exhibited severe clinical signs, including ataxia, tremors, and seizures [405]. Signs in ferrets inoculated intranasally with wild-type virus were similar, solely the onset was slightly delayed [405]. Ferrets inoculated intracranially or intranasally with the attenuated recombinant virus showed no clinical signs or mortality [405]. Only brain and nasal turbinates were collected for further testing 4 hours as well as on days 1, 2, 4, and 8 post inoculation. High levels of genomic RNA and mRNA were detected in tissues from ferrets inoculated with the wild-type virus, but not with the attenuated recombinant virus vector [405].
Retrovirus
Malignant non-Hodgkin's lymphoma and lymphoid leukemias are common in ferrets and various forms have been described [406,407]. In many species, including humans, cattle, chickens, and cats, lymphomas and leukemias are caused by retroviruses [408]. The occurrence of clusters of lymphoma in ferret colonies and the young age of some ferrets developing lymphoma suggest a potential viral cause, but testing for feline leukemia virus and ADV have been negative [409,410]. Whereas gastric MALT-lymphoma has been associated with infection with Helicobacter mustelae, no retrovirus has been identified in other types of lymphoma [411,412]. In one study, lymphoma was experimentally reproduced by intraperitoneal inoculation of ferrets with cells or cell-free inocula from a ferret with spontaneous malignant lymphoma [411]. All ferrets in the study developed mild sustained lymphocytosis within 6 weeks post inoculation and a chronic indolent syndrome featuring profound splenomegaly, lymphocytosis with atypia, and histologically polymorphous lymphoma [411]. Reverse transcriptase activity and retrovirus-like particles were identified in the inocula supporting the hypothesis of retroviral disease causing horizontal transmission of malignant lymphoma in ferrets [411].
Hepatitis E Virus
Hepatitis E virus (HEV) is a single-strand positive-sense RNA virus in the family Hepeviridae [413]. HEV is transmitted by the fecal-oral route and causes liver disease in juveniles and immunecompromised patients [413]. The mortality rates are highest in pregnant women in the third trimester and reach up to 20%. In addition to humans, HEV has been reported in pigs, chicken, mongoose, deer, rabbit, bats, fish, and only recently, in fecal samples of ferrets in the Netherlands [413,414]. No clinical disease was observed in any of the infected ferrets [414]. While increasing numbers of sporadic cases of hepatitis E have been reported in humans around the world and seropositivity for HEV has also been associated with having a pet at home, the risk of zoonotic disease through transmission of HEV from ferrets is completely unknown.
Transmissible Spongiform Encephalopathies
Transmissible spongiform encephalopathies (TSE) are chronic, slowly progressive, but always fatal and currently nontreatable neurodegenerative diseases caused by pathogenic isoforms of prion proteins. TSEs in humans include Creutzfeldt–Jakob disease, Gerstmann–Sträussler–Scheinker syndrome and kuru, and, in animals, scrapie in sheep and goats, chronic wasting disease (CWD) in cervid species, bovine spongiform encephalopathy (BSE) in cattle, and transmissible mink encephalopathy (TME) in mink [415,416]. A normal cellular isoform of these proteins, which is called PrPc, is found on the cellular membrane of neurons. According to the prion hypothesis, a disease-associated self-propagating, misfolded form of PrP, usually identified as PrPSc (for scrapie as the classical TSE) or PRPres (for protease resistant prion protein), is derived from the normal PrPc [417]. There are several ways that TSEs can develop. Sporadic TSEs due to the spontaneous change of normal PrPc into PrPres and inherited TSEs due to inheritance of a defective gene coding for PrPc have only been reported in humans [416]. Infectious forms of TSEs in which introduction of abnormal prions into the body causes conversion of normal PrPc to enzyme-resistant PrPres have been reported in both humans and animals [415,416].
There is no evidence of spontaneously occurring TSEs in ferrets or naturally occurring transmission of prion diseases from other species to ferrets. Microscopic lesions, for example, neuronal vacuolation, suggestive of a prion disease in the brain of ferrets with neurologic disease, were negative for protease-resistant prion protein [418]. However, ferrets have been investigated as an experimental model for a number of TSEs, including transmissible mink encephalopathy (TME) and CWD, and recent data demonstrate that ferrets may be a surrogate small animal model of CWD that can provide insights into the potential for CWD to cross species barriers [419–423]. Mink have been successfully inoculated with BSE, but to our knowledge, similar experiments have not been performed in ferrets [424].
CWD in Ferrets
Clinical Signs
Ferrets inoculated intracranially with primary passage CWD-infected mule deer brain homogenate developed clinical TSE after 15–20 months of inoculation [420,423,425]. The incubation period decreased to only 5 months following serial passage of CWD-infected ferret brain tissue. Clinically, affected ferrets started to isolate from other animals and demonstrated polyphagia and somnolence [420,423]. Intention tremors, hyperreflexia, ataxia, piloerection, lordosis, torticollis, and lethargy were observed as the clinical disease progressed [423]. Orally inoculated ferrets remained clinically unremarkable for up to 2.5 years post inoculation [423]. Inoculation of ferrets with a different CWD homogenate resulted in early signs of decreased arousal, alertness, and exploratory behavior followed by reduced food consumption and grooming [422]. Pruritus, aggressiveness, and hyperphagia were inconsistently observed. Later on, ferrets developed motor dysfunction of the hindquarters or lower spinal ataxia with a wide-based stance [422]. Neurologic signs progressed to generalized ataxia, including crossing of front legs, swaying of the neck, head bobbing, and lowered head carriage [422].
Pathology
Ferrets inoculated intracranially with primary passage CWD-infected mule deer brain homogenate developed no gross lesions, but microscopic lesions of mild spongiform encephalopathy were observed in the terminal stages of disease [420,423]. Spongioform lesions were most prominent in the neuropil and the white matter tracts of the basal ganglia, thalamus, optic chiasm, midbrain, and pons [423]. Perikaryonic vacuoles were rarely observed. Ferrets inoculated after serial passage of CWD-infected ferret brain tissue developed similar, but more intense and destructive TSE lesions, with spongiosis and gliosis increasingly targeting the cerebral cortex (Fig. 20.72 and Fig. 20.73) and cerebellum [423]. Immunohistochemistry detected small, granular deposits of abnormal prion protein affecting neurons and the neuropil in the basal ganglia, thalamus, and cerebellum of ferrets inoculated with the primary passage homogenate [423]. In ferrets with terminal disease, coarse aggregates of PrPres were detected in neurons in the pontine nuclei [423]. Ferrets inoculated with species-adapted CWD brain homogenate had diffuse, fine prion aggregates in the cerebral cortex (Fig. 20.74). In subsequent studies, ferret-adapted CWD was efficiently transmitted to ferrets by intraperitoneal and oral inoculation [422]. In these studies, PrPres was also consistently detected by immunohistochemistry and western blot in germinal centers of lymphoid follicles in the spleen, mesenteric, and retropharyngeal lymph nodes of ferrets inoculated with one of the ferret-adapted CWD inocula, but not the other, regardless of the route of inoculation [422].
Fig. 20.72. Cerebral cortex, ferret with CWD. Spongiform vacuolation, gliosis and focal neuronal necrosis, typical for transmissible spongiform encephalopathy. H&E. (Courtesy of Christina Sigurdson.)Fig. 20.73. Cerebral cortex, ferret with CWD. Immunohistochemical labeling (brown) with an antibody against GFAP shows gliosis on neurons. DAB chromogen, hematoxylin counterstain. (Courtesy of Christina Sigurdson.)Fig. 20.74. Cerebral cortex, ferret with CWD. PrP labeling by immunohistochemistry (brown) revealed punctuate, neuron-associated PrPCWD deposits in affected areas. DAB chromogen, hematoxylin counterstain. (Courtesy of Christina Sigurdson.)
Pathogenesis
Intracerebral inoculation of a CWD brain homogenate into ferrets resulted in disease after an incubation period of 15–20 months, which is similar to what has been described under natural conditions with oral transmission for white-tailed deer, and is consistent with a species barrier between deer and ferret [420,423,425,426]. Inoculation with a homogenate after serial passage in ferrets resulted in significant shortening of the incubation period, which suggested adaptation of the inoculum to a new, ferret-modified prion strain. This conclusion was supported by an altered pattern of deposition of prion proteins in the brain of inoculated ferrets [420,423,425]. Subsequent studies confirmed that ferret adapted CWD could infect ferrets both orally as well as following intracranial and intraperitoneal inoculation [422]. Ferret-adapted CWD could also be transmitted to hamsters [420]. In western blots, the PrPres glycoform profile resembled that of CWD in deer, typified by a dominant diglycosylated glycoform [423]. Furthermore, inoculation with one CWD homogenate resulted in infection of lymphoid tissue, whereas inoculation with another CWD inoculum did not [422]. Replication in lymphoid tissue frequently precedes neural invasion and was presumed to be important in the pathogenesis of ferret CWD [422]. There is increasing support for the existence of cervid CWD subtypes and variants and the difference in the described CWD phenotypes in ferrets may reflect these strain variations [422]. These studies established ferrets as an outbred model to study transmission and pathogenesis of CWD and demonstrated the utility of this model to investigate further CWD strain variants in deer [420,422,423].
TME in Ferrets
A number of studies examined the transmissibility of TME into ferrets to determine the host range of TME, to study its pathogenesis, and to investigate the species barrier between the two closely related mustelids, mink and ferret [419,427]. Most of these studies were done more than 30 years ago and provide significantly less detail in regard to clinical signs and pathology or confirmation of successful reproduction of disease than the more recent studies establishing ferrets as a model for CWD [420,422,423].
In the most recent study, intracerebral inoculation of ferrets with the Stetsonville source of TME resulted in a significantly longer incubation period of 28–38 months when compared with an incubation period of 4 months in mink, confirming a species barrier [419]. The second serial passage of ferret-adapted TME had an incubation period of 8–9 months while the third serial passage reduced the incubation period to 5 months [419]. When comparing the ferret with the mink PrP gene, a silent six base change resulting in two amino acid changes, Phe to Lys at codon 179 and Arg to Gln at codon 224, were identified [419]. These changes may indicate the region of PrP that is responsible for the species barrier effect between mink and ferret [419].
In an earlier study, intrauterine inoculation of ferret fetuses with a TME homogenate of mink brain did not result in clinical disease for 2 years post inoculation [427]. Microscopic examination of these ferrets 2 years post inoculation revealed focal areas of vacuolization bilaterally in the middle zone of the cerebral cortex that were most prominent along their lateral aspects [427]. Hypertrophied astrocytes were present in some vacuolated areas, and vacuoles tended to be multilocular and clustered with an occasional proximity to blood vessels. Neuronal degeneration was not prominent. The distribution of the lesions in ferrets was different from that described in mink, but the more limited lesions in the cerebral cortex of ferrets closely resembled those in mink [427–429]. No testing for PrPres was performed.
In a previous study with 10 ferrets, 9 black and 1 albino, animals were inoculated subcutaneously with different dilutions of a second passage TME brain homogenate [421]. Only the albino ferret developed clinical signs 10 months after inoculation that abated after several months. More specifically, the ferret had difficulties chewing and swallowing its food, some degree of incoordination mainly in the hind legs, and emaciation [421]. Following necropsy 15 months after inoculation, microscopic examination found multiple, large vacuoles in the frontal and parietal cortex that were accompanied by only minimal neuronal degeneration. All black ferrets remained clinically and histologically normal. Homogenates of brain and spleen from the albino ferret were inoculated subcutaneously into mink and black and albino ferrets. Mink inoculated with either homogenate developed TME [421]. The black ferrets remained free from disease for 28 months, whereas the albino ferrets inoculated with either homogenate started to lose weight, developed a roughened hair coat 14–15 months post inoculation, and demonstrated intense biting and scratching that produced considerable loss of hair [421]. Varying degrees of cerebral cortical vacuolation, similar to the previously observed lesions, were described in these ferrets [421]. Interestingly, numerous albino ferrets were not euthanized after the development of clinical signs, and these animals recovered completely within 4–6 weeks and remained normal until 28 months post inoculation. There have been no previous studies documenting recovery of animals infected with TME after showing clinical signs [226].
References
1. Evermann JF, Leathers CW, Gorham JR, McKeirnan AJ, Appel MJG (2001) Pathogenesis of two strains of lion (Panthera leo) morbillivirus in ferrets (Mustela putorius furo). Vet Pathol 38: 311–316.
2. Nielsen O, Smith G, Weingartl H, Lair S, Measures L (2008) Use of Slam transfected Vero cell line to isolate and characterize marine mammal morbilliviruses using an experimental ferret model. J Wildl Dis 44: 600–611.
3. Pillet S, Svitek N, von Messling V (2009) Ferrets as a model for morbillivirus pathogenesis, complications, and vaccines. Curr Top Microbiol Immunol 330: 73–87.
4. Thormar H, Brown HR, Goller NL, Barshatzky MR, Wisniewski HM (1988) Transmission of measles virus encephalitis to ferrets by intracardiac inoculation of a cell-associated SSPE virus strain. APMIS 96: 1125–1128.
5. Greene CE, Appel MJ (2006) Canine distemper. In: Greene CE, ed. Infectious diseases of the dog and cat, 3rd ed. St. Louis, MO: Saunders Elsevier, pp. 25–41.
6. Appel MJG (1987) Canine distemper virus. In: Appel MJG, ed. Virus infections of carnivores. Amsterdam, The Netherlands: Elsevier Science Publishers, pp. 133–159.
7. Fox JG, Pearson RC, Gorham JR (1998) Viral diseases. In: Fox JG, ed. Biology and diseases of the ferret, 2nd ed. Philadelphia: Lippincott Williams & Wilkins, pp. 355–374.
8. Roscoe DE (1993) Epizootology of canine distemper in New-Jersey racoons. J Wildl Dis 29: 390–395.
9. Appel MJG, Summers BA (1995) Pathogenicity of morbilliviruses for terrestrial carnivores. Vet Microbiol 44: 187–191.
10. López-Peña M, Vázquez S, Alemán N, López-Beceiro A, Muñoz F, Pereira JL, Nieto JM (2001) Canine distemper in a genet (Gennetta gennetta), associated with endogenous lipid pneumonia. J Comp Pathol 124: 207–211.
12. Appel MJG, Reggiardo C, Summers BA, Pearce-Kelling S, Mare CJ, Noon TH, Reed RE, Shively JN, Orvell C (1991) Canine distemper virus infection and encephalitis in Javelinas (collared peccaries). Arch Virol 119: 147–152.
13. Barrett I, Wohlsein P, Bidewell CA, Rowell SF (2004) Canine distemper virus in a Californian sea lion (Zatophus californianus). Vet Rec 154: 334.
14. Chin S-C, Guo JC, Chen SY, Yu P-H, Chie C-H, Liu CH, Yeh LS 2008. A review of Formosan pangolin (Manis pentadactyla pentadactyla) necropsies at the Taipei Zoo. Proceedings of the American Association of Zoo Veterinarians. Los Angeles, California, United States. p. 236.
15. Sun ZZ, Li AX, Ye HH, Shi YS, Hu ZM, Zeng L (2010) Natural infection with canine distemper virus in hand-feeding Rhesus monkeys in China. Vet Microbiol 141: 374–378.
16. Nikolin VM, Wibbelt G, Michler FU, Wolf P, East ML (2012) Susceptibility of carnivore hosts to strains of canine distemper virus from distinct genetic lineages. Vet Microbiol 156: 45–53.
17. Stanton JB, Givens L, Evermann JF, Brown CC (2003) Immunohistochemical analysis of two strains of lion (Panthera leo)-adapted canine distemper virus in ferrets (Mustela putorius furo). Vet Pathol 40: 464–467.
18. Williams ES (2001) Canine distemper. In: Williams ES, Barker IK, eds. Infectious diseases of wild mammals, 3rd ed. Ames, IA: Iowa State University Press, pp. 50–59.
19. Shen DT, Gorham JR, Pedersen V (1981) Viruria in dogs infected with canine distemper. Vet Med Small Anim Clin 76: 1175–1177.
20. Krakowka S, Koestner A (1977) Comparison of canine distemper virus strains in gnotobiotic dogs: effects on lymphoid tissues. Am J Vet Res 38: 1919–1922.
21. Perpiñán D, Ramis A, Tomás A, Carpintero E, Bargalló F (2008) Outbreak of canine distemper in domestic ferrets (Mustela putorius furo). Vet Rec 163: 246–250.
22. Kuiken T, Kennedy S, Barrett T, Van de Bildt MW, Borgsteede FH, Brew SD, Codd GA, Duck C, Deaville R, Eybatov T, Forsyth MA, Foster G, Jepson PD, Kydyrmanov A, Mitrofanov I, Ward CJ, Wilson S, Osterhaus AD (2006) The 2000 canine distemper epidemic in Caspian seals (Phoca caspica): pathology and analysis of contributory factors. Vet Pathol 43: 321–338.
23. Lipscomb TP, Schulman FY, Moffett D, Kennedy S (1994) Morbilliviral disease in Atlantic bottlenose dolphins (Tursiops truncatus) from the 1987–1988 epizootic. J Wildl Dis 30: 567–571.
24. Lonergan M, Hall A, Thompson H, Thompson PM, Pomeroy P, Harwood J (2010) Comparison of the 1988 and 2002 phocine distemper epizootics in British harbour seal Phoca vitulina populations. Dis Aquat Organ 88: 183–188.
25. Williams ES, Thorne ET, Appel MJG, Belitsky DW (1988) Canine Distemper in black-footed ferrets (Mustela nigripes) from Wyoming. J Wildl Dis 24: 385–398.
26. Gill JM, Hartley WJ, Hodgkinson NL (1988) An outbreak of post-vaccinal suspected distemper-like encephalitis in farmed ferrets (Mustela putorius furo). N Z Vet J 36: 173–176.
27. Wimsatt J, Jay MT, Innes KE, Jessen MJ, Collins JK (2001) Serologic evaluation, efficacy, and safety of a commercial modified-live canine distemper vaccine in domestic ferrets. Am J Vet Res 62: 736–740.
28. Davidson M (1986) Canine distemper virus-infection in the domestic ferret. Compend Contin Educ Pract Vet 8: 448–453.
29. Zehnder AM, Hawkins MG, Koski MA, Luff JA, Benak J, Lowenstine LJ, White SD (2008) An unusual presentation of canine distemper virus infection in a domestic ferret (Mustela putorius furo). Vet Dermatol 19: 232–238.
30. von Messling V, Springfeld C, Devaux P, Cattaneo R (2003) A ferret model of canine distemper virus virulence and immunosuppression. J Virol 77: 12579–12591.
31. Stephensen CB, Welter J, Thaker SR, Taylor J, Tartaglia J, Paoletti E (1997) Canine distemper virus (CDV) infection of ferrets as a model for testing Morbillivirus vaccine strategies: NYVAC- and ALVAC-based CDV recombinants protect against symptomatic infection. J Virol 71: 1506–1513.
32. Welter J, Taylor J, Tartaglia J, Paoletti E, Stephensen CB (1999) Mucosal vaccination with recombinant poxvirus vaccines protects ferrets against symptomatic CDV infection. Vaccine 17: 308–318.
33. Rosenthal KL (2004) Respiratory diseases. In: Quesenberry KE, Carpenter JW, eds. Ferrets, rabbits, and rodents: clinical medicine and surgery, 2nd ed. St. Louis, MO: Saunders, pp. 72–78.
34. Kauffman CA, Bergman AG, O'connor RP (1982) Distemper virus-infection in ferrets—an animal model of measles-induced immunosuppression. Clin Exp Immunol 47: 617–625.
35. Welter J, Taylor J, Tartaglia J, Paoletti E, Stephensen CB (2000) Vaccination against canine distemper virus infection in infant ferrets with and without maternal antibody protection, using recombinant attenuated poxvirus vaccines. J Virol 74: 6358–6367.
36. Crook E, Gorham JR, McNutt SH (1958) Experimental distemper in mink and ferrets. 1. Pathogenesis. Am J Vet Res 19: 955–957.
37. Gorham JR (1999) Some experiments and field observations of distemper in mink and ferrets. Adv Vet Med 41: 557–570.
38. Jones L, Tenorio E, Gorham J, Yilma T (1997) Protective vaccination of ferrets against canine distemper with recombinant pox virus vaccines expressing the H or F genes of rinderpest virus. Am J Vet Res 58: 590–593.
39. Kilham L, Habermann RT, Herman CM (1956) Jaundice and bilirubinemia as manifestations of canine distemper in raccoons and ferrets. Am J Vet Res 17: 144–148.
40. Shen DT, Gorham JR (1978) Contact transmission of distemper virus in ferrets. Res Vet Sci 24: 118–119.
41. Blair EM, Chambers MA, King HA (1998) Treating distemper in a young ferret. Vet Med 93: 655–658.
42. Bonami F, Rudd PA, von Messling V (2007) Disease duration determines canine distemper virus neurovirulence. J Virol 81: 12066–12070.
43. Carpenter JW, Appel MJG, Erickson RC, Novilla MN (1976) Fatal vaccine-induced canine distemper virus infection in black-footed ferrets. J Am Vet Med Assoc 169: 961–964.
44. Liu C, Coffin DL (1957) Studies on canine distemper infection by means of fluorescein-labeled antibody's: I. Pathogenesis, pathology and diagnosis of distemper in experimentally infected ferrets. Virology 3: 115–131.
45. Sidorenko SP, Clark EA (2003) The dual-function CD150 receptor subfamily: the viral attraction. Nat Immunol 4: 19–24.
46. Tatsuo H, Ono N, Yanagi Y (2001) Morbilliviruses use signaling lymphocyte activation molecules (CD150) as cellular receptors. J Virol 75: 5842–5850.
47. Crook E, Gorham JR, McNutt SH (1959) Experimental distemper in mink and ferrets: 2. Appearance and significance of histopathologic changes. Am J Vet Res 20: 378–383.
48. Coffin DL, Liu C (1957) Studies on canine distemper infection by means of fluorescein-labeled antibody: II. The pathology and diagnosis of the naturally occurring disease in dogs and the antigenic nature of the inclusion body. Virology 3: 132–145.
49. Moulton JE, Brown CH (1954) Antigenicity of canine distemper inclusion bodies as demonstrated by fluorescent antibody technic. Proc Soc Exp Biol Med 86: 99–102.
50. Krakowka S, Axthelum MK, Gorham JR (1987) Effects of induced thrombocytopenia on viral invasion of the CNS in canine distemper virus infection. J Comp Pathol 97: 441–450.
51. Rudd PA, Cattaneo R, von Messling V (2006) Canine distemper virus uses both the anterograde and the hematogenous pathway for neuroinvasion. J Virol 80: 9361–9370.
52. Rudd PA, Bastien-Hamel LE, von Messling V (2010) Acute canine distemper encephalitis is associated with rapid neuronal loss and local immune activation. J Gen Virol 91: 980–989.
53. Summers BA, Greisen HA, Appel MJ (1979) Early events in canine distemper demyelinating encephalomyelitis. Acta Neuropathol (Berl) 46: 1–10.
54. Tipold A, Moore P, Zurbriggen A, Burgener I, Barben G, Vandevelde M (1999) Early T cell response in the central nervous system in canine distemper virus infection. Acta Neuropathol (Berl) 97: 45–56.
55. Summers BA, Greisen HA, Appel MJ (1984) Canine distemper encephalomyelitis: variation with virus strain. J Comp Pathol 94: 65–75.
56. Vandevelde M, Zurbriggen A (2005) Demyelination in canine distemper virus infection: a review. Acta Neuropathol (Berl) 109: 56–68.
57. Vandevelde M, Zurbriggen A, Higgins RJ, Palmer D (1985) Spread and distribution of viral antigen in nervous canine distemper. Acta Neuropathol (Berl) 67: 211–218.
58. Appel MJ, Harris WV (1988) Antibody titers in domestic ferret jills and their kits to canine distemper virus vaccine. J Am Vet Med Assoc 193: 332–333.
59. Gans HA, Arvin AM, Galinus J, Logan L, DeHovitz R, Maldonado Y (1998) Deficiency of the humoral immune response to measles vaccine in infants immunized at age 6 months. J Am Med Assoc 280: 527–532.
60. Schobesberger M, Summerfield A, Doherr MG, Zurbriggen A, Griot C (2005) Canine distemper virus-induced depletion of uninfected lymphocytes is associated with apoptosis. Vet Immunol Immunopathol 104: 33–44.
61. von Messling V, Milosevic D, Cattaneo R (2004) Tropism illuminated: lymphocyte-based pathways blazed by lethal morbillivirus through the host immune system. Proc Natl Acad Sci U S A 101: 14216–14221.
62. von Messling V, Svitek N, Cattaneo R (2006) Receptor (SLAM [CD150]) recognition and the V protein sustain swift lymphocyte-based invasion of mucosal tissue and lymphatic organs by a morbillivirus. J Virol 80: 6084–6092.
63. Fontana JM, Bankamp B, Bellini WJ, Rota PA (2008) Regulation of interferon signaling by the C and V proteins from attenuated and wild-type strains of measles virus. Virology 374: 71–81.
64. Svitek N, von Messling V (2007) Early cytokine mRNA expression profiles predict morbillivirus disease outcome in ferrets. Virology 362: 404–410.
65. Katz M, Rorke LB, Masland WS, Koprowski H, Tucker SH (1968) Transmission of an encephalitogenic agent from brains of patients with subacute sclerosing panencephalitis to ferrets. Preliminary report. N Engl J Med 279: 793–798.
66. Katz M, Rorke LB, Masland WS, Brodano GB, Koprowski H (1970) Subacute sclerosing panencephalitis: isolation of a virus encephalitogenic for ferrets. J Infect Dis 121: 188–195.
67. Thormar H, Jervis GA, Karl SC, Brown HR (1973) Passage in ferrets of encephalitogenic cell-associated measles virus isolated from brain of a patient with subacute sclerosing panencephalitis. J Infect Dis 127: 678–685.
68. Thormar H, Mehta PD, Barshatzky MR, Brown HR (1985) Measles virus encephalitis in ferrets as a model for subacute sclerosing panencephalitis. Lab Anim Sci 35: 229–232.
69. Brown HR, Jervis GA, Thormar H (1977) Ultrastructural and histological studies of brains of ferrets inoculated with subacute sclerosing panencephalitis: similarities to human disease. J Neuropathol Exp Neurol 36: 653–665.
70. Brown HR, Thormar H, Barshatzky M, Wisniewski HM (1985) Localization of measles virus antigens in subacute sclerosing panencephalitis in ferrets. Lab Anim Sci 35: 233–237.
71. Müller D, Katz M, Rorke LB, ter Meulen V, Koprowski H (1973) Immunohistological, microscopical and neurochemical studies on encephalitides: VI. Subacute sclerosing panencephalitis. Histopathological and histochemical analysis of experimental encephalitis induced in ferrets by SSPE viruses. Acta Neuropathol (Berl) 24: 12–29.
72. Mehta PD, Thormar H (1979) Immunological studies of subacute measles encephalitis in ferrets: similarities to human subacute sclerosing panencephalitis. J Clin Microbiol 9: 601–604.
73. Thormar H, Mehta PD, Lin FH, Brown HR, Wisniewski HM (1983) Presence of oligoclonal immunoglobulin G bands and lack of matrix protein antibodies in cerebrospinal fluids and sera of ferrets with measles virus encephalitis. Infect Immun 41: 1205–1211.
74. Dietzel E, Anderson DE, Castan A, von Messling V, Maisner A (2011) Canine distemper virus matrix protein influences particle infectivity, particle composition, and envelope distribution in polarized epithelial cells and modulates virulence. J Virol 85: 7162–7168.
75. Beaufrere H, Neta M, Smith DA, Taylor WM (2009) Demodectic mange associated with lymphoma in a ferret. J Exot Pet Med 18: 57–61.
76. Fox JG (1998) Parasitic diseases. In: Fox JG, ed. Biology and diseases of the ferret, 2nd ed. Baltimore, MD: Lippincott, Williams & Wilkins, pp. 375–391.
77. Noli C, vanderHorst HHA, Willemse T (1996) Demodicosis in ferrets (Mustela putorius furo). Vet Q 18: 28–31.
78. Phillips PH, Ocallaghan MG, Moore E, Baird RM (1987) Pedal Sarcoptes scabiei infestation in ferrets (Mustela putorius furo). Aust Vet J 64: 289–290.
79. Antinoff N (2004) Musculoskeletal and neurologic diseases. In: Quesenberry KE, Carpenter JW, eds. Ferrets, rabbits, and rodents: clinical medicine and surgery, 2nd ed. St. Louis, MO: W.B. Saunders, pp. 115–120.
80. Diaz-Figueroa O, Smith MO (2007) Clinical neurology of ferrets. Vet Clin North Am Exot Anim Pract 10: 759–773.
81. Masuda M, Sato H, Kamata H, Katsuo T, Takenaka A, Miura R, Yoneda M, Tsukiyama-Kohara K, Mizumoto K, Kai C (2006) Characterization of monoclonal antibodies directed against the canine distemper virus nucleocapsid protein. Comp Immunol Microbiol Infect Dis 29: 157–165.
82. Rzezutka A, Mizak B (2002) Application of N-PCR for diagnosis of distemper in dogs and fur animals. Vet Microbiol 88: 95–103.
83. Zurbriggen A, Müller C, Vandevelde M (1993) In situ hybridization of virulent canine distemper virus in brain tissue, using digoxigenin-labeled probes. Am J Vet Res 54: 1457–1461.
84. Schatzberg SJ, Li Q, Porter BF, Barber RM, Claiborne MK, Levine JM, Levine GJ, Israel SK, Young BD, Kiupel M, Greene C, Ruone S, Anderson L, Tong S (2009) Broadly reactive pan-paramyxovirus reverse transcription polymerase chain reaction and sequence analysis for the detection of canine distemper virus in a case of canine meningoencephalitis of unknown etiology. J Vet Diagn Invest 21: 844–849.
85. Appel MJG, Pearce-Kelling S, Summers BA (1992) Dog lymphocyte cultures facilitate the isolation and growth of virulent canine distemper virus. J Vet Diagn Invest 4: 258–263.
86. Appel MJG, Jones OR (1967) Use of alveolar macrophages for cultivation of canine distemper virus. Proc Soc Exp Biol Med 126: 571–574.
87. Poste G (1971) The growth and cytopathogenicity of virulent and attenuated strains of canine distemper virus in dog and ferret macrophages. J Comp Pathol 81: 49–54.
88. Rockborn G (1958) Canine distemper virus in tissue culture. Arch Gesamte Virusforsch 8: 485–492.
89. Kai C, Ochikubo F, Okita M, Iinuma T, Mikami T, Kobune F, Yamanouchi K (1993) Use of B95a cells for isolation of canine distemper virus from clinical cases. J Vet Med Sci 55: 1067–1070.
91. Seki F, Ono N, Yamaguchi R, Yanagi Y (2003) Efficient isolation of wild strains of canine distemper viruses in Vero cell expressing canine SLAM (CD150) and their adaptability to marmoset B95a cells. J Virol 77: 9943–9950.
92. Pavlacik L, Celer V, Kajerova V, Jekl V, Knotek Z, Literak I (2007) Monitoring of antibodies titre against canine distemper virus in ferrets vaccinated with a live modified vaccine. Acta Vet Brno 76: 423–429.
93. Williams ES, Anderson SL, Cavender J, Lynn C, List K, Hearn C, Appel MJG (1996) Vaccination of black-footed ferret (Mustela nigripes) x Siberian polecat (M. eversmanni) hybrids and domestic ferrets (M. putorius furo) against canine distemper. J Wildl Dis 32: 417–423.
94. Rodeheffer C, von Messling V, Milot S, Lepine F, Manges AR, Ward BJ (2007) Disease manifestations of canine distemper virus infection in ferrets are modulated by vitamin A status. J Nutr 137: 1916–1922.
95. Sudfeld CR, Navar AM, Halsey NA (2010) Effectiveness of measles vaccination and vitamin A treatment. Int J Epidemiol 39: 48–55.
96. Belfield WO (1967) Vitamin C in treatment of canine and feline distemper complex. Vet Med Small Anim Clin 62: 345.
97. Lewington JH (2007) Ferret husbandry, medicine and surgery. Philadelphia: Saunders.
98. Burger D, Gorham JR (1964) Response of ferrets and mink to vaccination with chicken embryo-adapted distemper virus. Arch Gesamte Virusforsch 4: 449–461.
99. Wimsatt J, Biggins D, Innes K, Taylor B, Garell D (2003) Evaluation of oral and subcutaneous delivery of an experimental canarypox recombinant canine distemper vaccine in the Siberian polecat (Mustela eversmanni). J Zoo Wildl Med 34: 25–35.
100. Greenacre CB (2003) Incidence of adverse events in ferrets vaccinated with distemper or rabies vaccine: 143 cases (1995–2001). J Am Vet Med Assoc 223: 663–665.
101. Moore GE, Glickman NW, Ward MP, Engler KS, Lewis HB, Glickman LT (2005) Incidence of and risk factors for adverse events associated with distemper and rabies vaccine administration in ferrets. J Am Vet Med Assoc 226: 909–912.
102. Deem SL, Spelman LH, Yates RA, Montali RJ (2000) Canine distemper in terrestrial carnivores: a review. J Zoo Wildl Med 31: 441–451.
103. Williams BH, Kiupel M, West KH, Raymond JT, Grant CK, Glickman LT (2000) Coronavirus-associated epizootic catarrhal enteritis in ferrets. J Am Vet Med Assoc 217: 526–530.
104. Wise AG, Kiupel M, Maes RK (2006) Molecular characterization of a novel coronavirus associated with epizootic catarrhal enteritis (ECE) in ferrets. Virology 349: 164–174.
105. Garner MM, Ramsell K, Morera N, Juan-Salles C, Jimenez J, Ardiaca M, Montesinos A, Teifke JP, Lohr CV, Evermann JF, Baszler TV, Nordhausen RW, Wise AG, Maes RK, Kiupel M (2008) Clinicopathologic features of a systemic coronavirus-associated disease resembling feline infectious peritonitis in the domestic ferret (Mustela putorius). Vet Pathol 45: 236–246.
106. Martinez J, Ramis AJ, Reinacher M, Perpiñán D (2006) Detection of feline infectious peritonitis virus-like antigen in ferrets. Vet Rec 158: 523–523.
107. Juan-Sallés C, Teifke N, Morera N, Jimenez J, Montesinos A, Ardiaca M, Löehr CV, Garner MM (2006) Pathology and immunohistochemistry of a disease resembling feline infectious peritonitis in ferrets (Mustela putorius furo). Proc Amer Col Vet Pathol 84: 845.
108. Wise AG, Kiupel M, Garner MM, Clark AK, Maes RK (2010) Comparative sequence analysis of the distal one-third of the genomes of a systemic and an enteric ferret coronavirus. Virus Res 149: 42–50.
109. Lai MMC, Perlman S, Anderson LJ (2007) Coronaviridae. In: Knipe DM, Howley PM, eds. Fields virology, 5th ed. Philadelphia: Lippincott Williams & Wilkins, pp. 1305–1335.
110. Gonzalez JM, Gomez-Puertas P, Cavanagh D, Gorbalenya AE, Enjuanes L, et al. (2003) A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch Virol 148: 2207–2235.
111. Provacia LB, Smits SL, Martina BE, Raj VS, Doel PV, Amerongen GV, Moorman-Roest H, Osterhaus AD, Haagmans BL (2011) Enteric coronavirus in ferrets, The Netherlands. Emerg Infect Dis 17: 1570–1571.
112. Murray J, Kiupel M, Maes RK (2010) Ferret coronavirus-associated diseases. Vet Clin North Am Exot Anim Pract 13: 543–560.
113. Perpiñán D, López C (2008) Clinical aspects of systemic granulomatous inflammatory syndrome in ferrets (Mustela putorius furo). Vet Rec 162: 180–184.
114. Hoefer HL (2004) General gastrointestinal disorders. In: Quesenberry KE, Carpenter JW, eds. Ferrets, rabbits, and rodents: clinical medicine and surgery. St. Louis, MO: Saunders, pp. 25–33.
115. Hartmann K (2005) Feline infectious peritonitis. Vet Clin North Am Small Anim Pract 35: 39–79.
116. Pedersen NC (2009) A review of feline infectious peritonitis virus infection: 1963–2008. J Feline Med Surg 11: 225–258.
117. Weiss SR, Navas-Martin S (2005) Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Biol Rev 69: 635–664.
118. Perlman S, Netland J (2009) Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7: 439–450.
119. Rottier PJM, Nakamura K, Schellen P, Volders H, Haijema BJ (2005) Acquisition of macrophage tropism during the pathogenesis of feline infectious peritonitis is determined by mutations in the feline coronavirus spike protein. J Virol 79: 14122–14130.
120. Brown MA, Troyer JL, Pecon-Slattery J, Roelke ME, O'Brien SJ (2009) Genetics and pathogenesis of feline infectious peritonitis virus. Emerg Infect Dis 15: 1445–1452.
121. Martinez J, Reinacher M, Perpiñán D, Ramis A (2008) Identification of group I coronavirus antigen in multisystemic granulomatous lesions in ferrets (Mustela putorius furo). J Comp Pathol 138: 54–58.
122. Pedersen NC (1987) Virologic and immunologic aspects of feline infectious peritonitis virus infection. Adv Exp Med Biol 218: 529–550.
123. Haijema BJ, Volders H, Rottier PJM (2004) Live, attenuated coronavirus vaccines through the directed deletion of group-specific genes provide protection against feline infectious peritonitis. J Virol 78: 3863–3871.
124. Vennema H, Poland A, Foley J, Pedersen NC (1998) Feline infectious peritonitis viruses arise by mutation from endemic feline enteric coronaviruses. Virology 243: 150–157.
125. Kennedy M, Boedeker N, Gibbs P, Kania S (2001) Deletions in the 7a ORF of feline coronavirus associated with an epidemic of feline infectious peritonitis. Vet Microbiol 81: 227–234.
126. Chang HW, de Groot RJ, Egberink HF, Rottier PJM (2009) Feline infectious peritonitis: insights into feline coronavirus pathobiogenesis and epidemiology based on genetic analysis of the viral 3c gene. J Gen Virol 91: 415–420.
127. Sledge DG, Bolin SR, Lim A, Kaloustian LL, Heller RL, Carmona FM, Kiupel M (2011) Outbreaks of severe enteric disease associated with Eimeria furonis infection in ferrets (Mustela putorius furo) of 3 densely populated groups. J Am Vet Med Assoc 239: 1584–1588.
128. Dominguez E, Novellas R, Moya A, Espada Y, Martorell J (2011) Abdominal radiographic and ultrasonographic findings in ferrets (Mustela putorius furo) with systemic coronavirus infection. Vet Rec 169: 231.
129. Addie D, Belak S, Boucraut-Baralon C, Egberink H, Frymus T, Gruffydd-Jones T, Hartmann K, Hosie MJ, Lloret A, Lutz H, Marsilio F, Pennisi MG, Radford AD, Thiry E, Truyen U, Horzinek MC (2009) Feline infectious peritonitis. ABCD guidelines on prevention and management. J Feline Med Surg 11: 594–604.
130. Simons FA, Vennema H, Rofina JE, Pol JM, Horzinek MC, Rottier PJ, Egberink HF (2005) A mRNA PCR for the diagnosis of feline infectious peritonitis. J Virol Methods 124: 111–116.
131. Rabenau HF, Kampf G, Cinatl J, Doerr HW (2005) Efficacy of various disinfectants against SARS coronavirus. J Hosp Infect 61: 107–111.
132. Alexandersen S, et al. (1994) Acute interstitial pneumonia in mink kits inoculated with defined isolates of Aleutian mink disease parvovirus. Vet Pathol 31: 216–228.
133. Fournier-Chambrillon C, Aasted B, Perrot A, Pontier D, Sauvage F, Artois M, Cassiède JM, Chauby X, Dal Molin A, Simon C, Fournier P (2004) Antibodies to Aleutian mink disease parvovirus in free-ranging European mink (Mustela lutreola) and other small carnivores from southwestern France. J Wildl Dis 40: 394–402.
134. Sánchez-Migallón Guzmán D, Carvajal A, García-Marín JF, Ferreras MC, Pérez V, Mitchell M, Urra F, Ceña JC (2008) Aleutian disease serology, protein electrophoresis, and pathology of the European mink (Mustela lutreola) from Navarra, Spain. J Zoo Wildl Med 39: 305–313.
135. Mañas S, Ceña JC, Ruiz-Olmo J, Palazón S, Domingo M, Wolfinbarger JB, Bloom ME (2001) Aleutian mink disease parvovirus in wild riparian carnivores in Spain. J Wildl Dis 37: 138–144.
136. Oie KL, Durrant G, Wolfinbarger JB, Martin D, Costello F, Perryman S, Hogan D, Hadlow WJ, Bloom ME (1996) Relationship between capsid protein (VP2) sequence and pathogenicity of Aleutian mink disease parvovirus (ADV): a possible role for raccoons in the transmission of ADV infections. J Virol 70: 852–861.
137. Langlois I (2005) Viral diseases of ferrets. Vet Clin North Am Exot Anim Pract 8: 139–160.
138. Alexandersen S, Larsen S, Cohn A, Uttenthal A, Race RE, Aasted B, Hansen M, Bloom ME (1989) Passive transfer of antiviral antibodies restricts replication of Aleutian mink disease parvovirus in vivo. J Virol 63: 9–17.
139. Allender MC, Schumacher J, Thomas KV, McCain SL, Ramsay EC, James EW, Wise AG, Maes RK, Reel D (2008) Infection with Aleutian disease virus-like virus in a captive striped skunk. J Am Vet Med Assoc 232: 742–746.
140. Cheng F, Chen AY, Best SM, Bloom ME, Pintel D, Qiu JM (2010) The capsid proteins of Aleutian mink disease virus activate caspases and are specifically cleaved during infection. J Virol 84: 2687–2696.
141. Hadlow WJ, Race RE, Kennedy RC (1984) Royal pastel mink respond variously to inoculation with Aleutian disease virus of low virulence. J Virol 50: 38–41.
142. Porter DD, Porter HG, Larsen AE (1990) Aleutian disease parvovirus infection of mink and ferrets elicits an antibody response to A2ND nonstructural viral protein. J Virol 64: 1859–1860.
143. Porter HG, Porter DD, Larsen AE (1981) Aleutian disease in the ferret. Fed Proc 40: 1120–1120.
144. Murakami M, Matsuba C, Une Y, Nomura Y, Fujitani H (2001) Nucleotide sequence and polymerase chain reaction/restriction fragment length polymorphism analyses of Aleutian disease virus in ferrets in Japan. J Vet Diagn Invest 13: 337–340.
145. Stevenson M, Gates L, Murray J, Bloom ME (2001) Aleutian mink disease parvovirus: implications for companion ferrets. Compend Contin Educ Pract Vet 23: 178–185.
146. McCaw DL, Hoskins JD (2006) Canine viral enteritis. In: Greene CE, ed. Infectious diseases of the dog and cat, 3rd ed. St. Louis, MO: Saunders Elsevier, pp. 63–73.
148. Kenyon AJ, Howard E, Buko L (1967) Hypergammaglobulinemia in ferrets with lymphoproliferative lesions (Aleutian disease). Am J Vet Res 28: 1167–1172.
149. Knuuttila A, Uzcátegui N, Kankkonen J, Vapalahti O, Kinnunen P (2009) Molecular epidemiology of Aleutian mink disease virus in Finland. Vet Microbiol 133: 229–238.
150. Gorham JR, Henson JB, Crawford TB, Padgett GA (1976) The epizootiology of Aleutian disease. In: Kimberlin RH, ed. Slow virus diseases of animals and man. Amsterdam: North-Holland Publishing Company, pp. 135–158.
151. Jackson MK, Ellis LC, Morrey JD, Li ZZ, Barnard DL (1996a) Progression of Aleutian disease in natural and experimentally induced infections of mink. Am J Vet Res 57: 1753–1758.
152. Cho HJ, Greenfield J (1978) Eradication of Aleutian disease of mink by eliminating positive counterimmunoelectrophoresis test reactors. J Clin Microbiol 7: 18–22.
153. Jackson MK, Winslow SG, Dockery LD, Jones JK, Sisson DV (1996b) Investigation of an outbreak of Aleutian disease on a commercial mink ranch. Am J Vet Res 57: 1706–1710.
154. Larivière S, Jennings AP (2009) Family Mustelidae. In: Wilson DE, Mittermeier, RA, eds. Handbook of mammals of the world, Vol. 1: Carnivores. Barcelona, Spain: Lynx Edicions.
155. Oxenham M (1990) Aleutian disease in the ferret. Vet Rec 126: 585.
156. Welchman DD, Oxenham M, Done SH (1993) Aleutian disease in domestic ferrets—diagnostic findings and survery results. Vet Rec 132: 479–484.
157. Jepsen JR, d'Amore F, Baandrup U, Clausen MR, Gottschalck E, Aasted B (2009) Aleutian mink disease virus and humans. Emerg Infect Dis 15: 2040–2042.
158. Une Y, Wakimoto Y, Nakano Y, Konishi M, Nomura Y (2000) Spontaneous Aleutian disease in a ferret. J Vet Med Sci 62: 553–555.
159. Palley LS, Corning BF, Fox JG, Murphy JC, Gould DH (1992) Parvovirus-associated syndrome (Aleutian disease) in 2 ferrets. J Am Vet Med Assoc 201: 100–106.
160. Petrie JP, Morrisey JK (2004) Cardiovascular and other diseases. In: Quesenberry KE, Carpenter JW, eds. Ferrets, rabbits, and rodens: clinical medicine and surgery, 2nd ed. St. Louis, MO: W.B. Saunders, pp. 58–71.
161. Daoust PY, Hunter DB (1978) Spontaneous Aleutian disease in ferrets. Can Vet J 19: 133–135.
163. Porter DD, Larsen AE, Porter HG (1969) The pathogenesis of Aleutian disease of mink: in vivo viral replication and the host antibody response to viral antigen. J Exp Med 130: 575–589.
164. Aasted B, Alexandersen S, Christensen J (1998) Vaccination with Aleutian mink disease parvovirus (AMDV) capsid proteins enhances disease, while vaccination with the major non-structural AMDV protein causes partial protection from disease. Vaccine 16: 1158–1165.
165. Bloom ME, Race RE, Wolfinbarger JB (1982) Identification of a non-virion protein of Aleutian disease virus: mink with Aleutian disease have antibody to both virion and nonvirion proteins. J Virol 43: 608–616.
166. Chitty J 2009. Aleutian disease in ferrets: diagnostics and controversies. Proceedings of the Association of Avian Veterinarians (AAV) pre-conference program and Association of Exotic Mammal Veterinarians (AEMV) scientific program. Milwaukee, Wisconsin, United States. pp. 75–78.
167. Pennick KE, Stevenson MAM, Latimer KS, Ritchie BW, Gregory CR (2005) Persistent viral shedding during asymptomatic Aleutian mink disease parvoviral infection in a ferret. J Vet Diagn Invest 17: 594–597.
168. Best SM, Bloom ME (2005) Pathogenesis of Aleutian mink disease parvovirus and similarities to B19 infection. J Vet Med B Infect Dis Vet Public Health 52: 331–334.
169. Cheema A, Gorham JR, Henson JB (1972) Aleutian disease of mink—prevention of lesions by immunosuppression. Am J Pathol 66: 543–552.
170. Ellis LC (1996) Melatonin reduces mortality from Aleutian disease in mink (Mustela vison). J Pineal Res 21: 214–217.
171. Castelruiz Y, Blixenkrone-Moller M, Aasted B (2005) DNA vaccination with the Aleutian mink disease virus NS 1 gene confers partial protection against disease. Vaccine 23: 1225–1231.
172. Barker IK, Parrish CR (2001) Parvovirus infections. In: Williams ES, Barker IK, eds. Infectious diseases of wild mammals. Ames, IA: Iowa State University Press, pp. 131–146.
173. Patterson AR, Cooper VL, Yoon KJ, Janke BH, Gauger PC (2009) Naturally occurring influenza infection in a ferret (Mustela putorius furo) colony. J Vet Diagn Invest 21: 527–530.
174. Smith W, Andrewes CH, Laidlaw PP (1933) A virus obtained from influenza patients. Lancet 222: 66–68.
175. Swenson SL, Koster LG, Jenkins-Moore M, Killian ML, DeBess EE, Baker RJ, Mulrooney D, Weiss R, Galeota J, Bredthauer A (2010) Natural cases of 2009 pandemic H1N1 Influenza A virus in pet ferrets. J Vet Diagn Invest 22: 784–788.
176. Kuiken T, van den Brand J, van Riel D, Pantin-Jackwood M, Swayne DE (2010) Comparative pathology of select agent influenza A virus infections. Vet Pathol 47: 893–914.
177. Maher JA, DeStefano J (2004) The ferret: an animal model to study influenza virus. Lab Anim 33: 50–53.
178. Munster VJ, de Wit E, van den Brand JM, Herfst S, Schrauwen EJ, Bestebroer TM, van de Vijver D, Boucher CA, Koopmans M, Rimmelzwaan GF, Kuiken T, Osterhaus AD, Fouchier RA (2009) Pathogenesis and transmission of swine-origin 2009 A (H1N1) influenza virus in ferrets. Science 325: 481–483.
179. Tumpey TM, Maines TR, Van Hoeven N, Glaser L, Solorzano A, Pappas C, Cox NJ, Swayne DE, Palese P, Katz JM, Garcia-Sastre A (2007) A two-amino acid change in the hemagglutinin of the 1918 influenza virus abolishes transmission. Science 315: 655–659.
180. van den Brand JMA, Stittelaar KJ, van Amerongen G, Rimmelzwaan GF, Simon J, de Wit E, Munster V, Bestebroer T, Fouchier RA, Kuiken T, Osterhaus AD (2010) Severity of pneumonia due to new H1N1 influenza virus in ferrets is intermediate between that due to seasonal H1N1 virus and highly pathogenic avian influenza H5N1 virus. J Infect Dis 201: 993–999.
181. van Riel D, Munster VJ, de Wit E, Rimmelzwaan GF, Fouchier RA, Osterhaus AD, Kuiken T (2007) Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am J Pathol 171: 1215–1223.
182. Belser JA, Katz JM, Tumpey TM (2011b) The ferret as a model organism to study influenza A virus infection. Dis Model Mech 4: 575–579.
183. Cox NJ, Subbarao K (2000) Global epidemiology of influenza: past and present. Annu Rev Med 51: 407–421.
184. Swayne DE, Halvorson DA (2008) Influenza. In: Saif YM, Glisson JR, Fadly AM, McDougald LR, Nolan L, eds. Diseases of poultry, 12th ed. Ames, IA: Blackwell, pp. 153–184.
185. De Vleeschauwer A, Van Poucke S, Braeckmans D, Van Doorsselaere J, Van Reeth K (2009) Efficient transmission of swine-adapted but not wholly avian influenza viruses among pigs and from pigs to ferrets. J Infect Dis 200: 1884–1892.
186. Lipatov AS, Kwon YK, Pantin-Jackwood MJ, Swayne DE (2009) Pathogenesis of H5N1 influenza virus infections in mice and ferret models differs according to respiratory tract or digestive system exposure. J Infect Dis 199: 717–725.
187. van Hoeven N, Belser JA, Szretter KJ, Zeng H, Staeheli P, Swayne DE, Katz JM, Tumpey TM (2009) Pathogenesis of 1918 pandemic and H5N1 influenza virus infections in a guinea pig model: antiviral potential of exogenous alpha interferon to reduce virus shedding. J Virol 83: 2851–2861.
188. Watanabe T, Watanabe S, Shinya K, Kim JH, Hatta M, Kawaoka Y (2009) Viral RNA polymerase complex promotes optimal growth of 1918 virus in the lower respiratory tract of ferrets. Proc Natl Acad Sci U S A 106: 588–592.
189. Fisher JW, Scott P (1944) An epizootic of influenza A in a ferret colony. Can J Public Health 35: 364–366.
190. Friesen RHE, Koudstaal W, Koldijk MH, Weverling GJ, Brakenhoff JPJ, Lenting PJ, Stittelaar KJ, Osterhaus A, Kompier R, Goudsmit J (2010) New class of monoclonal antibodies against severe influenza: prophylactic and therapeutic efficacy in ferrets. PLoS ONE 5: e96.
191. Rennie P, Bowtell P, Hull D, Charbonneau D, Lambkin-Williams R, Oxford J (2007) Low pH gel intranasal sprays inactivate influenza viruses in vitro and protect ferrets against influenza infection. Respir Res 8: e38.
192. Svitek N, Rudd PA, Obojes K, Pillet S, von Messling V (2008) Severe seasonal influenza in ferrets correlates with reduced interferon and increased IL-6 induction. Virology 376: 53–59.
193. Peltola VT, Boyd KL, McAuley JL, Rehg JE, McCullers JA (2006) Bacterial sinusitis and otitis media following influenza virus infection in ferrets. Infect Immun 74: 2562–2567.
194. Yun NE, Linde NS, Zacks MA, Barr IG, Hurt AC, Smith JN, Dziuba N, Holbrook MR, Zhang L, Kilpatrick JM, Arnold CS, Paessler S (2008) Injectable peramivir mitigates disease and promotes survival in ferrets and mice infected with the highly virulent influenza virus, A/Vietnam/1203/04 (H5N1). Virology 374: 198–209.
195. Zitzow LA, Rowe T, Morken T, Shieh WJ, Zaki S, Katz JM (2002) Pathogenesis of avian influenza A (H5N1) viruses in ferrets. J Virol 76: 4420–4429.
196. Glathe H, Lebhardt A, Hilgenfeld M (1984) Intestinal influenza infection in ferrets. Arch Exp Veterinarmed 38: 771–777.
197. Reperant LA, Rimmelzwaan GF, Kuiken T (2009) Avian influenza viruses in mammals. Rev Sci Tech 28: 137–159.
198. Rowe T, Cho DS, Bright RA, Zitzow LA, Katz JM (2003) Neurological manifestations of avian influenza viruses in mammals. Avian Dis 47: 1122–1126.
199. Belser JA, Gustin KM, Maines TR, Blau DM, Zaki SR, Katz JM, Tumpey TM (2011a) Pathogenesis and transmission of triple-reassortant swine H1N1 influenza viruses isolated before the 2009 H1N1 pandemic. J Virol 85: 1563–1572.
200. Yen HL, Lipatov AS, Ilyushina NA, Govorkova EA, Franks J, Yilmaz N, Douglas A, Hay A, Krauss S, Rehg JE, Hoffmann E, Webster RG (2007) Inefficient transmission of H5N1 influenza viruses in a ferret contact model. J Virol 81: 6890–6898.
201. Boltz DA, Rehg JE, McClaren J, Webster RG, Govorkova EA (2008) Oseltamivir prophylactic regimens prevent H5N1 influenza morbidity and mortality in a ferret model. J Infect Dis 197: 1315–1323.
202. Maines TR, Lu XH, Erb SM, Edwards L, Guarner J, Greer PW, Nguyen DC, Szretter KJ, Chen LM, Thawatsupha P, Chittaganpitch M, Waicharoen S, Nguyen DT, Nguyen T, Nguyen HH, Kim JH, Hoang LT, Kang C, Phuong LS, Lim W, Zaki S, Donis RO, Cox NJ, Katz JM, Tumpey TM (2005) Avian influenza (H5N1) viruses isolated from humans in Asia in 2004 exhibit increased virulence in mammals. J Virol 79: 11788–11800.
203. Govorkova EA, Rehg JE, Krauss S, Yen HL, Guan Y, Peiris M, Nguyen TD, Hanh TH, Puthavathana P, Long HT, Buranathai C, Lim W, Webster RG, Hoffmann E (2005) Lethality to ferrets of H5N1 influenza viruses isolated from humans and poultry in 2004. J Virol 79: 2191–2198.
204. Itoh Y, Shinya K, Kiso M, Watanabe T, Sakoda Y, Hatta M, Muramoto Y, Tamura D, Sakai-Tagawa Y, Noda T, Sakabe S, Imai M, Hatta Y, Watanabe S, Li C, Yamada S, Fujii K, Murakami S, Imai H, Kakugawa S, Ito M, Takano R, Iwatsuki-Horimoto K, Shimojima M, Horimoto T, Goto H, Takahashi K, Makino A, Ishigaki H, Nakayama M, Okamatsu M, Takahashi K, Warshauer D, Shult PA, Saito R, Suzuki H, Furuta Y, Yamashita M, Mitamura K, Nakano K, Nakamura M, Brockman-Schneider R, Mitamura H, Yamazaki M, Sugaya N, Suresh M, Ozawa M, Neumann G, Gern J, Kida H, Ogasawara K, Kawaoka Y (2009) In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 460: 1021–1025.
205. Chutinimitkul S, van Riel D, Munster VJ, van den Brand JM, Rimmelzwaan GF, Kuiken T, Osterhaus AD, Fouchier RA, de Wit E (2010) In vitro assessment of attachment pattern and replication efficiency of H5N1 influenza A viruses with altered receptor specificity. J Virol 84: 6825–6833.
206. Basarab O, Smith H (1969) Quantitative studies on the tissue localization of influenza virus in ferrets after intranasal and intravenous or intracardial inoculation. Br J Exp Pathol 50: 612–618.
207. Herlocher ML, Elias S, Truscon R, Harrison S, Mindell D, Simon C, Monto AS (2001) Ferrets as a transmission model for influenza: sequence changes in HA1 of type A (H3N2) virus. J Infect Dis 184: 542–546.
208. Leigh MW, Connor RJ, Kelm S, Baum LG, Paulson JC (1995) Receptor specificity of influenza virus influences severity of illness in ferrets. Vaccine 13: 1468–1473.
209. Steinhauer DA (1999) Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 258: 1–20.
210. Cheung CY, Poon LL, Lau AS, Luk W, Lau YL, Shortridge KF, Gordon S, Guan Y, Peiris JS (2002) Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet 360: 1831–1837.
211. Kreijtz JH, Fouchier RA, Rimmelzwaan GF (2011) Immune responses to influenza virus infection. Virus Res 162: 19–30.
212. Zambon MC (2001) The pathogenesis of influenza in humans. Rev Med Virol 11: 227–241.
213. Boyers A, Herberts C, Lambkin-Williams R, Mann AJ, Oxford J, van der Laan JW (2008) Animal models in influenza vaccine testing. Expert Rev Vaccines 7: e783.
214. Smith H, Sweet C (1988) Lessons for human influenza from pathogenicity studies with ferrets. Rev Infect Dis 10: 56–75.
215. Hers JF, Mulder J (1961) Broad aspects of the pathology and pathogenesis of human influenza. Am Rev Respir Dis 83: 54.
216. Gustin KM, Belser JA, Wadford DA, Pearce MB, Katz JM, Tumpey TM, Maines TR (2011) Influenza virus aerosol exposure and analytical system for ferrets. Proc Natl Acad Sci U S A 108: 8432–8437.
217. Reuman PD, Keely S, Schiff GM (1989) Assessment of signs of influenza illness in the ferret model. J Virol Methods 24: 27–34.
218. Fenton RJ, Clark A, Potter CW (1981) Comparative immunity following infection and immunity with live or inactivated vaccine. Br J Exp Pathol 62: 297–307.
219. Mann A, Marriott AC, Balasingam S, Lambkin R, Oxford JS, Dimmock NJ (2006) Interfering vaccine (defective interfering influenza A virus) protects ferrets from influenza and allows them to develop solid immunity. Vaccine 24: 4290–4296.
220. Govorkova EA, Webby RJ, Humberd J, Seiler JP, Webster RG (2006) Immunization with reverse-genetics-produced H5N1 influenza vaccine protects ferrets against homologous and heterologous challenge. J Infect Dis 194:159–167. Epub 2006 Jun 9.
221. Kilpatrick-Smith L, Hale DE, Douglas SD (1989) Progress in Reye syndrome: epidemiology, biochemical mechanisms and animal models. Dig Dis 7: 135–146.
222. Rarey KE (1985) The ferret as a model for inner ear research. Lab Anim Sci 35: 238–241.
223. Barnard DL (2009) Animal models for the study of influenza pathogenesis and therapy. Antiviral Res 82: A110–A122.
224. Bodewes R, Rimmelzwaan GF, Osterhaus AD (2010) Animal models for the preclinical evaluation of candidate influenza vaccines. Expert Rev Vaccines 9: 59–72.
225. van der Laan JW, Herberts C, Lambkin-Williams R, Boyers A, Mann AJ, Oxford J (2008) Animal models in influenza vaccine testing. Expert Rev Vaccines 7: 783–793.
226. Pearson RC, Gorham JR (1998) Viral disease models. In: Fox JG, ed. Biology and diseases of the ferret, 2nd ed. Baltimore, MD: Lippincott, Williams & Wilkins, pp. 487–497.
227. Rowe T, Leon AJ, Crevar CJ, Carter DM, Xu LL, Ran LS, Fang Y, Cameron CM, Cameron MJ, Banner D, Ng DCK, Ran R, Weirback HK, Wiley CA, Kelvin DJ, Ross TM (2010) Modeling host responses in ferrets during A/California/07/2009 influenza infection. Virology 401: 257–265.
228. Vincent LL, Janke BH, Paul PS, Halbur PG (1997) A monoclonal-antibody-based immunohistochemical method for the detection of swine influenza virus in formalin-fixed, paraffin-embedded tissues. J Vet Diagn Invest 9: 191–195.
229. Govorkova EA, Ilyushina NA, Boltz DA, Douglas A, Yilmaz N, Webster RG (2007) Efficacy of oseltamivir therapy in ferrets inoculated with different clades of H5N1 influenza virus. Antimicrob Agents Chemother 51: 1414–1424.
230. Govorkova EA, Marathe BM, Prevost A, Rehg JE, Webster RG (2011) Assessment of the efficacy of the neuraminidase inhibitor oseltamivir against 2009 pandemic H1N1 influenza virus in ferrets. Antiviral Res 91: 81–88.
231. Herlocher ML, Truscon R, Fenton R, Klimov A, Elias S, Ohmit SE, Monto AS (2003) Assessment of development of resistance to antivirals in the ferret model of influenza virus infection. J Infect Dis 188: 1355–1361.
232. Kugel D, Kochs G, Obojes K, Roth J, Kobinger GP, Kobasa D, Haller O, Staeheli P, von Messling V (2009) Intranasal administration of alpha interferon reduces seasonal influenza A virus morbidity in ferrets. J Virol 83: 3843–3851.
233. Smee DF, Bailey KW, Wong MH, O'Keefe BR, Gustafson KR, Mishin VP, Gubareva LV (2008) Treatment of influenza A (H1N1) virus infections in mice and ferrets with cyanovirin-N. Antiviral Res 80: 266–271.
235. Kang YM, Song BM, Lee JS, Kim HS, Seo SH (2011) Pandemic H1N1 influenza virus causes a stronger inflammatory response than seasonal H1N1 influenza virus in ferrets. Arch Virol 156: 759–767.
236. Birnbaum NG, O'Brien B (2008) Methods for inactivation of avian influenza virus in the environment. In: Swayne DE, ed. Avian influenza. Ames, IA: Blackwell Publishing, pp. 391–405.
237. Rice EW, Adcock NJ, Sivaganesan M, Brown JD, Stallknecht DE, Swayne DE (2007) Chlorine inactivation of highly pathogenic avian influenza virus (H5N1). Emerg Infect Dis 13: 1568–1570.
238. Baras B, Stittelaar KJ, Simon JH, Thoolen R, Mossman SP, Pistoor FHM, van Amerongen G, Wettendorff MA, Hanon E, Osterhaus A (2008) Cross-protection against lethal H5N1 challenge in ferrets with an adjuvanted pandemic influenza vaccine. PLoS ONE 3: e1401.
239. Chen ZY, Santos C, Aspelund A, Gillim-Ross L, Jin H, Kemble G, Subbarao K (2009) Evaluation of live attenuated influenza a virus H6 vaccines in mice and ferrets. J Virol 83: 65–72.
240. Mahmood K, Bright RA, Mytle N, Carter DM, Crevar C, Achenbach JE, Heaton PM, Tumpey TM, Ross TM (2008) H5N1VLP vaccine induced protection in ferrets against lethal challenge with highly pathogenic H5N1 influenza viruses. Vaccine 26: 5393–5399.
241. Torres-Medina A (1987) Isolation of an atypical rotavirus causing diarrhea in neonatal ferrets. Lab Anim Sci 37: 167–171.
242. Wise AG, Smedley RC, Kiupel M, Maes RK (2009) Detection of group C rotavirus in juvenile ferrets (Mustela putorius furo) with diarrhea by reverse transcription polymerase chain reaction: sequencing and analysis of the complete coding region of the VP6 gene. Vet Pathol 46: 985–991.
243. Chang KO, Kim YJ, Saif LJ (1999a) Comparisons of nucleotide and deduced amino acid sequences of NSP4 genes of virulent and attenuated pairs of group A and C rotaviruses. Virus Genes 18: 229–233.
244. Estes MK, Cohen J (1989) Rotavirus gene structure and function. Microbiol Rev 53: 410–449.
245. Estes MK, Kapikian AZ (2007) Rotaviruses. In: Knipe DM, Howley PM, eds. Fields virology, 5th ed. Philadelphia: Lippincott Williams & Wilkins, pp. 1917–1974.
246. Otto P, Schulze P, Herbst W (1999) Demonstration of group C rotaviruses in fecal samples of diarrheic dogs in Germany. Arch Virol 144: 2467–2473.
247. McNulty MS (1978) Rotaviruses. J Gen Virol 40: 1–18.
248. Theil KW, McCloskey CM, Saif LJ, Redman DR, Bohl EH, Hancock DD, Kohler EM, Moorhead PD (1981) Rapid, simple method of preparing rotaviral double-stranded ribonucleic acid for analysis by polyacrylamide gel electrophoresis. J Clin Microbiol 14: 273–280.
249. Pedley S, Bridger JC, Brown JF, McCrae MA (1983) Molecular characterization of rotaviruses with distinct group anitgens. J Gen Virol 64: 2093–2101.
250. Woode GN, Bridger JC, Jones JM, Flewett TH, Bryden AS, Davies HA, White GBB (1976) Morphological and antigenic relationships between viruses (rotaviruses) from acute gastroenteritis of children, calves, piglets, mice and foals. Infect Immun 14: 804–810.
251. Saif LJ (1990) Nongroup A rotaviruses. In: Saif LJ, Theil KW, eds. Viral diarrheas in man and animals. Boca Raton, FL: CRC Press, pp. 73–95.
252. Matthijnssens J, Ciarlet M, McDonald SM, Attoui H, Bányai K, Brister JR, Buesa J, Esona MD, Estes MK, Gentsch JR, Iturriza-Gómara M, Johne R, Kirkwood CD, Martella V, Mertens PP, Nakagomi O, Parreño V, Rahman M, Ruggeri FM, Saif LJ, Santos N, Steyer A, Taniguchi K, Patton JT, Desselberger U, Van Ranst M (2011) Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch Virol 156: 1397–1413.
253. Cooke SJ, Lambden PR, Caul EO, Clarke IN (1991) Molecular cloning, sequence analysis and coding assignment of the major inner capsid protein gene of human group C rotavirus. Virology 184: 781–785.
254. Jiang B, Tsunemitsu H, Gentsch JR, Glass RI, Green KY, Qian Y, Saif LJ (1992) Nucleotide sequence of gene 5 encoding the inner capsid protein (VP6) of bovine group C rotavirus: comparison with corresponding genes of group C, A and B rotaviruses. Virology 190: 542–547.
255. Kim Y, Chang K, Straw B, Saif LJ (1999) Characterization of group C rotaviruses associated with diarrhea outbreaks in feeder pigs. J Clin Microbiol 37: 1484–1488.
256. Woode GN, Crouch CF (1978) Naturally occurring and experimentally induced rotaviral infections of domestic and laboratory animals. J Am Vet Med Assoc 178: 522–526.
257. Greenberg HB, Clark HF, Offit PA (1994) Rotavirus pathology and pathophysiology. Curr Top Microbiol Immunol 185: 255–283.
258. Greenberg HB, Estes MK (2009) Rotaviruses: from pathogenesis to vaccination. Gastroenterology 136: 1939–1951.
259. Maes RK, Grooms DL, Wise AG, Han C, Ciesicki V, Hanson L, Vickers ML, Kanitz C, Holland R (2003) Evaluation of a human group A rotavirus assay of on-site detection of bovine rotavirus. J Clin Microbiol 41: 290–294.
260. Snodgrass DR, Herring AJ, Campbell I, Inglis JM, Hargreaves FD (1984) Comparison of atypical rotaviruses from calves, piglets, lambs and man. J Gen Virol 65: 909–914.
261. Janke BH, Nelson JK, Benfield DA, Nelson EA (1990) Relative prevalence of typical and atypical strains among rotaviruses from diarrheic pigs in conventional swine herds. J Vet Diagn Invest 2: 308–311.
262. Saif LJ, Rosen BI, Parwani AV (1994). Animal rotaviruses. In: Kapikian AZ. Viral infections of the gastrointestinal tract, 2nd ed. New York: Marcel Dekker, pp. 369–407.
263. Chang KO, Nielsen PR, Ward LA, Saif LJ (1999b) Dual infection of gnotobiotic calves with bovine strains of group A and porcine-like group C rotaviruses influences pathogenesis of the group C rotavirus. J Virol 73: 9284–9293.
264. Suffin SC, Prince GA, Mark KB (1979a) Ontogeny of the ferret humoral response. J Immunol 123: 6–9.
265. Meslin FX, Fishbein DB, Matter HC (1994) Rationale and prospects for rabies elimination in developing countries. Curr Top Microbiol Immunol 187: 1–26.
266. Lackay SN, Kuang Y, Fu ZF (2008) Rabies in small animals. Vet Clin North Am Small Anim Pract 38: 851.
267. Jackson AC (2008) Rabies. Neurol Clin 26: 717–726.
268. Blancou J, Aubert JFA, Artois M (1982) Rage experimentale du furet [Mustela (putorius) furo]. Rev Med Vet (Toulouse) 133: 553.
269. Niezgoda M, Briggs DJ, Shaddock J (1997) Pathogenesis of experimentally induced rabies rabies in domestic ferrets. Am J Vet Res 58: 1327–1331.
270. Drew WL (2004) Rabies. In: Ryan KJ, Ray CG, eds. Sherris medical microbiology, 4th ed. New York: McGraw Hill, pp. 597–600.
271. Delmas O, Holmes EC, Talbi C, Larrous F, Dacheux L, Bouchier C, Bourhy H (2008) Genomic diversity and evolution of the lyssaviruses. PLoS ONE 3: e2057.
272. Kissi B, Tordo N, Bourhy H (1995) Genetic polymorphism in the rabies virus nucleoprotein gene. Virology 209: 526–537.
273. Bourhy H, Kissi B, Lafon M, Sacramento D, Tordo N (1992) Antigenic and molecular characterization of bat rabies virus in Europe. J Clin Microbiol 30: 2419–2426.
274. Vos A, Muller T, Cox J, Neubert L, Fooks AR (2004) Susceptibility of ferrets (Mustela putorius furo) to experimentally induced rabies with European Bat Lyssaviruses (EBLV). J Vet Med B Infect Dis Vet Public Health 51: 55–60.
275. Badrane H, Tordo N (2001) Host switching in Lyssavirus history from the Chiroptera to the Carnivora orders. J Virol 75: 8096–8104.
276. Nadin-Davis SA, Casey GA, Wandeler AI (1993) Identification of regional variants of the rabies virus within the Canadian province of Ontario. J Gen Virol 74: 829–837.
277. Nadin-Davis SA, Sampath MI, Casey GA, Tinline RR, Wandeler AI (1999) Phylogeographic patterns exhibited by Ontario rabies virus variants. Epidemiol Infect 123: 325–336.
278. Rupprecht CE, Glickman LT, Spencer P, Wiktor TJ (1987) Epidemiology of rabies virus variants: differentiation using monoclonal antibodies and discriminant analysis. Am J Epidemiol 126: 298–309.
279. Bourhy H, Reynes JM, Dunham EJ, Dacheux L, Larrous F, Que Huong VT, Xu G, Yan J, Miranda MEG, Holmes EC (2008) The origin and phylogeography of dog rabies virus. J Gen Virol 89: 2673–2681.
280. Leslie MJ, Messenger S, Rohde RE, Smith J, Cheshier R, Hanlon C (2006) Bat-associated rabies virus in skunks. Emerg Infect Dis 12: 1274–1277.
281. Gough PM, Jorgenson RD (1976) Rabies antibodies in sera of wild birds. J Wildl Dis 12: 392–395.
282. Blanton JD, Hanlon CA, Rupprecht CE (2007) Rabies surveillance in the United States during 2006. J Am Vet Med Assoc 231: 540–556.
283. Silverstein MA, Salgado CD, Bassin S (2003) First human death associated with raccoon rabies—Virginia, 2003. MMWR Morb Mortal Wkly Rep 52: 1102–1103.
284. Amengual B, Whitby JE, King A, Cobo JS, Bourhy H (1997) Evolution of european bat lyssaviruses. J Gen Virol 78: 2319–2328.
285. Conomy JP, Leibovitz A, McCombs W (1977) Airborne rabies encephalitis: demonstration of rabies virus in the human central nervous system. Neurology 27: 67–69.
286. Gibbons RV (2002) Cryptogenic rabies, bats, and the question of aerosol transmission. Ann Emerg Med 39: 528–536.
287. Applegate JA, Walhout MF (1998) Childhood risks from the ferret. J Emerg Med 16: 425–427.
288. Diesch SL (1982) Reported human injuries or health threats attributed to wild and exotic animals kept as pets (1971–1981). J Am Vet Med Assoc 18: 382.
289. Paisley JW, Lauer BA (1988) Severe facial injuries to infants due to unprovoked attacks by pet ferrets. J Am Med Assoc 259: 2005.
290. Greene CE, Rupprecht CE (2006) Rabies and other Lyssavirus infections. In: Greene CE, ed. Infectious diseases of the dog and cat, 3rd ed. St. Louis, MO: Saunders Elsevier, pp. 167–183.
291. Jensen A, Rabin ER, Wende RD, Melnick JL (1967) A comparative light and electron microscopic study of rabies and hart park virus encephalitis. Exp Mol Pathol 7: 7–10.
292. Sandhyamani S, Roy S, Gode GR, Kalla GN (1981) Pathology of rabies: a light- and electron-microscopical study with particular reference to the changes in cases with prolonged survival. Acta Neuropathol (Berl) 54: 247–251.
293. Baer GM, Shaddock JH, Quirion R (1990) Rabies susceptibility and acetylcholine receptor. Lancet 335: 664–665.
294. Tsiang H (1979) Evidence for an intraaxonal transport of fixed and street rabies virus. J Neuropathol Exp Neurol 38: 286–296.
295. Smart NL, Charlton KM (1992) The distribution of challenge virus standard rabies virus versus skunk street rabies virus in the brains of experimentally infected rabid skunks. Acta Neuropathol (Berl) 84: 501–508.
296. Niezgoda M, Briggs DJ, Shaddock J (1998) Viral excretion in domestic ferrets (Mustela putorius furo) inoculated with a raccoon rabies isolate. Am J Vet Res 58: 1629–1632.
297. Hamir AN, Niezgoda M, Rupprecht CE (2011) Recovery from and clearance of rabies virus in a domestic ferret. J Am Assoc Lab Anim Sci 50: 248–251.
298. Bell JF, Moore GJ (1971) Susceptibility of carnivore to rabies virus administered orally. Am J Epidemiol 93: 176.
299. Foerster U (1979) The adaptability of two rabies virus strains isolated in central Europe to one domesticated and two wild-living species: a contribution to epidemiology of rabies. Part 4: transmission studies on ferret with rodent isolate. Zentralbl Veterinarmed 26: 29–38.
300. Lembo T, Niezgoda M, Velasco-Villa A (2006) Evaluation of a direct, rapid immunohistochemical test for rabies diagnosis. Emerg Infect Dis 12: 310–313.
301. CDC (1999) Human rabies prevention—United States, 1999. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep 48(RR-1): 1–21.
302. Rupprecht CE, Gilbert J, Pitts R (1990) Evaluation of an inactivated rabies virus vaccine in domestic ferrets. J Am Vet Med Assoc 196: 1614.
303. Hoover JP, Baldwin CA, Rupprecht CE (1989) Serologic response of domestic ferrets to canine distemper and rabies virus vaccines. J Am Vet Med Assoc 194: 234–238.
304. Carroll EE, Dubielzig RR, Schultz RD (2002) Cats differ from mink and ferrets in their response to commercial vaccines: a histologic comparison of early vaccine reactions. Vet Pathol 39: 216–227.
305. Munday JS, Stedman NL, Richey LJ (2003) Histology and immunohistochemistry of seven ferret vaccination-site fibrosarcomas. Vet Pathol 40: 288–293.
306. Murray J (1998) Vaccine injection-site sarcoma in a ferret (letter). J Am Vet Med Assoc 214: 955.
307. Ohshima K, Gorham JR, Henson JB (1976) Pathologic changes in ferrets exposed to pseudorabies virus. Am J Vet Res 37: 591–596.
308. Kimman TG, van Oirschot JT (1986) Pathology of Aujeszky's disease in mink. Vet Pathol 23: 303–309.
309. Billig I, Foris JM, Card JP, Yates BJ (1999) Transneuronal tracing of neural pathways controlling an abdominal muscle, rectus abdominis, in the ferret. Brain Res 820: 31–44.
310. Billig I, Foris JM, Enquist LW, Card JP, Yates BJ (2000) Definition of neuronal circuitry controlling the activity of phrenic and abdominal motoneurons in the ferret using recombinant strains of pseudorabies virus. J Neurosci 20: 7446–7454.
311. Yates BJ, Smail JA, Stocker SD, Card JP (1999) Transneuronal tracing of neural pathways controlling activity of diaphragm motoneurons in the ferret. Neuroscience 90: 1501–1513.
312. Mettenleiter TC (2000) Aujeszky's disease (pseudorabies) virus: the virus and molecular pathogenesis—State of the art, June 1999. Vet Res 31: 99–115.
313. Pejsak ZK, Truszczynski MJ (2006) Aujeszky's disease (pseudorabies). In: Straw BE, Zimmerman JJ, D'Allaire S, Taylor DJ, eds. Diseases of swine, 9th ed. Ames, IA: Blackwell, pp. 419–433.
314. Pomeranz LE, Reynolds AE, Hengartner CJ (2005) Molecular biology of pseudorabies virus: impact on neurovirology and veterinary medicine. Microbiol Mol Biol Rev 69: 462–500.
315. Hahn EC, Fadl-Alla B, Lichtensteiger CA (2010) Variation of Aujeszky's disease viruses in wild swine in USA. Vet Microbiol 143: 45–51.
316. Cramer SD, Campbell GA, Njaa BL, Morgan SE, Smith SK, II, McLin WR, IV, Brodersen BW, Wise AG, Scherba G, Langohr IM, Maes RK (2011) Pseudorabies virus infection in Oklahoma hunting dogs. J Vet Diagn Invest 23: 915–923.
317. Boucher JD, Beran G (1977) Pseudorabies in the dog and cat. Iowa State Univ Vet 39: 22–25.
318. Goto H, Gorham JR, Hagen KW (1967) Clinical observation of experimental pseudorabies in mink and ferrets. Jpn J Vet Sci 30: 257–263.
319. Monroe WE (1989) Clinical signs associated with pseudorabies in dogs. J Am Vet Med Assoc 195: 599–602.
320. Trainer DO, Karstadt L (1963) Experimental rabies in some wild North American mammals. Zoonoses Res 2: 135–150.
321. Goto H, Burger D, Gorham J, Jr. (1971) Quantitative studies of pseudorabies virus in mink, ferrets, rabbits and mice. Nihon Juigaku Zasshi 33: 145–153.
322. Enquist LW (1999) Life beyond eradication: veterinary viruses in basic science. Arch Virol Suppl 15: 87–109.
323. Shintani T, Anker AR, Billig I, Card JP, Yates BJ (2003) Transneuronal tracing of neural pathways influencing both diaphragm and genioglossal muscle activity in the ferret. J Appl Physiol 95: 1453–1459.
324. Thiry E, Addie D, Belák S, Boucraut-Baralon C, Egberink H, Frymus T, Gruffydd-Jones T, Hartmann K, Hosie MJ, Lloret A, Lutz H, Marsilio F, Pennisi MG, Radford AD, Truyen U, Horzinek MC (2009) Feline herpesvirus infection. ABCD guidelines on prevention and management. J Feline Med Surg 11: 547–555.
325. Porter DD, Larsen AE, Cox NA (1975) Isolation of infectious bovine rhinotracheitis virus from mustelidae. J Clin Microbiol 1: 112–113.
326. Smith PC (1978) Experimental infectious bovine rhinotracheitis virus infection of English ferrets. Am J Vet Res 39: 1369–1372.
327. Engels M, Ackermann M (1996) Pathogenesis of ruminant herpesvirus infections. Vet Microbiol 53: 3–15.
328. Nandi S, Kumar M, Manohar M, Chauhan RS (2009) Bovine herpes virus infections in cattle. Anim Health Res Rev 10: 85–98.
329. Mettenleiter TC (1996) Conclusions from the symposium. Vet Microbiol 53: 207–211.
330. Parrish CR, Leathers CW, Pearson R, Gorham JR (1987) Comparisons of feline panleukopenia virus, canine parvovirus, raccoon parvovirus, and mink enteritis virus and their pathogenicity for mink and ferrets. Am J Vet Res 48: 1429–1435.
331. Duenwald JC, Holland JM, Gorham JR, Ott RL (1971) Feline panleukopenia: experimental cerebellar hypoplasia produced in neonatal ferrets with live virus vaccine. Res Vet Sci 12: 394–396.
332. Kilham L, Margolis G, Colby ED (1967) Congenital infections of cats and ferrets by feline panleukopenia virus manifested by cerebellar hypoplasia. Lab Invest 17: 465–480.
333. Murphy FA, Fauquet CM, Bishop DHL, Ghabrial SA, Jarvis AW, Martelli GR, Mayo MA, Summers MD, eds. (1995) Virus taxonomy. Sixth report of the International Commitee on Taxonomy of Viruses. New York: Plenum Press and Wien, Austria: Springer Verlag, pp. 169–178.
334. Lamm CG, Rezabek GB (2008) Parvovirus infection in domestic companion animals. Vet Clin North Am Small Anim Pract 38: 837–850.
335. Kilham L, Margolis G, Colby ED (1971) Cerebellar ataxia and its congenital transmission in cats by feline panleukopenia virus. J Am Vet Med Assoc 158: 888–901.
336. Veijalainen P (1986) A serological survey of enteric parvovirus infections in Finnish fur-bearing animals. Acta Vet Scand 27: 159–171.
337. Neuvonen E, Veijalainen P, Kangas J (1982) Canine parvovirus infection in housed raccoon dogs and foxes in Finland. Vet Rec 110: 448–449.
338. Kilham L, Margolis G (1966) Viral etiology of spontaneous ataxia of cats. Am J Pathol 48: 991–1011.
339. Herndon RM, Margolis G, Kilham L (1971b) The synaptic organization of the malformed cerebellum induced by perinatal infection with the feline panleukopenia virus (PLV): II. The Purkinje cell and its afferents. J Neuropathol Exp Neurol 30: 557–570.
340. Herndon RM, Margolis G, Kilham L (1971a) The synaptic organization of the malformed cerebellum induced by perinatal infection with the feline panleukopenia virus (PLV): I. Elements forming the cerebellar glomeruli. J Neuropathol Exp Neurol 30: 196–205.
341. Cotmore SF, Tattersall P (2007) Parvoviral host range and cell entry mechanisms. Adv Virus Res 70: 183–232.
342. Parker JSL, Murphy WJ, Wang D, O'Brien SJ, Parrish CR (2001) Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J Virol 75: 3896–3902.
343. Truyen U, Addie D, Belák S, Boucraut-Baralon C, Egberink H, Frymus T, Gruffydd-Jones T, Hartmann K, Hosie MJ, Lloret A, Lutz H, Marsilio F, Pennisi MG, Radford AD, Thiry E, Horzinek MC (2009) Feline panleukopenia. ABCD guidelines on prevention and management. J Feline Med Surg 11: 538–546.
344. Levy JK, Patterson EV, Reese MJ, Tucker SJ (2006) Impact of vaccination on parvovirus testing in kittens. J Vet Intern Med 20: 711.
345. Rodrigues A, Gates L, Payne HR, Kiupel M, Mansell J (2010) Multicentric squamous cell carcinoma in situ associated with papillomavirus in a ferret. Vet Pathol 47: 964–968.
346. Munday JS, Kiupel M (2010) Papillomavirus-associated cutaneous neoplasia in mammals. Vet Pathol 47: 254–264.
347. de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H (2004) Classification of papillomaviruses. Virology 324: 17–27.
348. Gottschling M, Göker M, Köhler A, Lehmann MD, Stockfleth E, Nindl I (2009) Cutaneotropic human beta-/gamma-papillomaviruses are rarely shared between family members. J Invest Dermatol 129: 2427–2434.
349. Stocco dos Santos RC, Lindsey CJ, Ferraz OP, Pinto JR, Mirandola RS, Benesi FJ, Birgel EH, Pereira CA, Becak W (1998) Bovine papillomavirus transmission and chromosomal aberrations: an experimental model. J Gen Virol 79: 2127–2135.
350. Munday JS, Kiupel M, French AF, Howe L, Squires RA (2007) Detection of papillomaviral sequences in feline Bowenoid in situ carcinoma using consensus primers. Vet Dermatol 18: 241–245.
351. Orth G (2006) Genetics of epidermodysplasia verruciformis: insights into host defense against papillomaviruses. Semin Immunol 18: 362–374.
352. Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K (2004) Mechanisms of human papillomavirus induced oncogenesis. J Virol 78: 11451–11460.
353. Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ; SARS Working Group (2003) A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348: 1953–1966.
354. Eickmann M, Becker S, Klenk HD, Doerr HW, Stadler K, Censini S, Guidotti S, Masignani V, Scarselli M, Mora M, Donati C, Han JH, Song HC, Abrignani S, Covacci A, Rappuoli R (2003) Phylogeny of the SARS coronavirus. Science 302: 1504–1505.
355. Roberts A, Subbarao K (2006) Animal models for SARS. Adv Exp Med Biol 581: 463–471.
356. Chu YK, Ali GD, Jia F, Li Q, Kelvin D, Couch RC, Harrod KS, Hutt JA, Cameron C, Weiss SR, Jonsson CB (2008) The SARS-CoV ferret model in an infection-challenge study. Virology 374: 151–163.
357. Kobinger GP, Figueredo JM, Rowe T, Zhi Y, Gao G, Sanmiguel JC, Bell P, Wivel NA, Zitzow LA, Flieder DB, Hogan RJ, Wilson JM (2007) Adenovirus-based vaccine prevents pneumonia in ferrets challenged with the SARS coronavirus and stimulates robust immune responses in macaques. Vaccine 25: 5220–5231.
358. Martina BE, Haagmans BL, Kuiken T, Fouchier RA, Rimmelzwaan GF, Van Amerongen G, Peiris JS, Lim W, Osterhaus AD (2003) Virology: SARS virus infection of cats and ferrets. Nature 425: 915.
359. ter Meulen J, Bakker AB, van den Brink EN, Weverling GJ, Martina BE, Haagmans BL, Kuiken T, de Kruif J, Preiser W, Spaan W, Gelderblom HR, Goudsmit J, Osterhaus AD (2004) Human monoclonal antibody as prophylaxis for SARS coronavirus infection in ferrets. Lancet 363: 2139–2141.
360. Weingartl HM, Copps J, Drebot MA, Marszal P, Smith G, Gren J, Andova M, Pasick J, Kitching P, Czub M (2004b) Susceptibility of pigs and chickens to SARS coronavirus. Emerg Infect Dis 10: 179–184.
361. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426: 450–454.
362. Zamoto A, Taguchi F, Fukushi S, Morikawa S, Yamada YK (2006) Identification of ferret ACE2 and its receptor function for SARS-coronavirus. Adv Exp Med Biol 581: 519–522.
363. van den Brand JM, Haagmans BL, Leijten L, van Riel D, Martina BE, Osterhaus AD, Kuiken T (2008) Pathology of experimental SARS coronavirus infection in cats and ferrets. Vet Pathol 45: 551–562.
364. Danesh A, Seneviratne C, Cameron CM, Banner D, Devries ME, Kelvin AA, Xu L, Ran L, Bosinger SE, Rowe T, Czub M, Jonsson CB, Cameron MJ, Kelvin DJ (2008) Cloning, expression and characterization of ferret CXCL10. Mol Immunol 45: 1288–1297.
365. Danesh A, Cameron CM, León AJ, Ran L, Xu L, Fang Y, Kelvin AA, Rowe T, Chen H, Guan Y, Jonsson CB, Cameron MJ, Kelvin DJ (2011) Early gene expression events in ferrets in response to SARS coronavirus infection versus direct interferon-alpha2b stimulation. Virology 409: 102–112.
366. ter Meulen J, van den Brink EN, Poon LL, Marissen WE, Leung CS, Cox F, Cheung CY, Bakker AQ, Bogaards JA, van Deventer E, Preiser W, Doerr HW, Chow VT, de Kruif J, Peiris JS, Goudsmit J (2006) Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med 3: e237.
367. Darnell ME, Plant EP, Watanabe H, Byrum R, St Claire M, Ward JM, Taylor DR (2007) Severe acute respiratory syndrome coronavirus infection in vaccinated ferrets. J Infect Dis 196: 1329–1338.
368. See RH, Petric M, Lawrence DJ, Mok CP, Rowe T, Zitzow LA, Karunakaran KP, Voss TG, Brunham RC, Gauldie J, Finlay BB, Roper RL (2008) Severe acute respiratory syndrome vaccine efficacy in ferrets: whole killed virus and adenovirus-vectored vaccines. J Gen Virol 89: 2136–2146.
369. Weingartl H, Czub M, Czub S, Neufeld J, Marszal P, Gren J, Smith G, Jones S, Proulx R, Deschambault Y, Grudeski E, Andonov A, He R, Li Y, Copps J, Grolla A, Dick D, Berry J, Ganske S, Manning L, Cao J (2004a) Immunization with modified vaccinia virus Ankara-based recombinant vaccine against severe acute respiratory syndrome is associated with enhanced hepatitis in ferrets. J Virol 78: 12672–12676.
370. Hsu VP (2007) Nipah and Hendra Viruses. In: Tabor E, ed. Emerging viruses in human populations. Oxford, UK: Elsevier, pp. 179–199.
371. Ksiazek TG, Rota PA, Rollin PE (2011) A review of Nipah and Hendra viruses with an historical aside. Virus Res 162: 173–183.
372. Williamson MM, Torres-Velez FJ (2011) Henipavirus: a review of laboratory animal pathology. Vet Pathol 47: 871–880.
373. Bossart KN, Zhu Z, Middleton D, Klippel J, Crameri G, Bingham J, McEachern JA, Green D, Hancock TJ, Chan YP, Hickey AC, Dimitrov DS, Wang LF, Broder CC (2009) A neutralizing human monoclonal antibody protects against lethal disease in a new ferret model of acute nipah virus infection. PLoS Pathog 5: e1000642.
374. Pallister J, Middleton D, Crameri G (2009) Chloroquine administration does not prevent nipah virus infection and disease in ferrets. J Virol 83: 11979–11982.
375. Pallister J, Middleton D, Wang LF, Klein R, Haining J, Robinson R, Yamada M, White J, Payne J, Feng YR, Chan YP, Broder CC (2011) A recombinant Hendra virus G glycoprotein-based subunit vaccine protects ferrets from lethal Hendra virus challenge. Vaccine 29: 5623–5630.
376. Bossart KN, McEachern JA, Hickey AC, Choudhry V, Dimitrov DS, Eaton BT, Wang LF (2007) Neutralization assays for differentiation of henipavirus serology using Bio-Plex Protein Array system. J Virol Methods 142: 29–40.
377. Rockx B, Bossart KN, Feldmann F, Geisbert JB, Hickey AC, Brining D, Callison J, Safronetz D, Marzi A, Kercher L, Long D, Broder CC, Feldmann H, Geisbert TW (2010) A novel model of lethal Hendravirus infection in African green monkeys and the effectiveness of ribavirin treatment. Virology 84: 9831–9839.
378. Geisbert TW, Feldmann H, Broder CC (2012) Animal challenge models of henipavirus infection and pathogenesis. Curr Top Microbiol Immunol 359: 153–177.
382. Baumgärtner W, Krakowka S, Gorham JR (1989) Canine parainfluenza virus-induced encephalitis in ferrets. J Comp Pathol 100: 67–76.
383. Baumgärtner W, Krakowka S, Durchfeld B (1991) In vitro cytopathogenicity and in vivo virulence of two strains of canine parainfluenza virus. Vet Pathol 28: 324–331.
384. Capraro GA, Johnson JB, Kock ND, Parks GD (2008) Virus growth and antibody responses following respiratory tract infection of ferrets and mice with WT and P/V mutants of the paramyxovirus Simian Virus 5. Virol 376: 416–428.
385. Durchfeld B, Baumgärtner W, Krakowka S (1991) Intranasal infection of ferrets (Mustela putorius furo) with canine parainfluenza virus. Zentralbl Veterinarmed B 38: 505–512.
386. Mascoli CC, Metzgar DP, Larson EJ, Fuscaldo AA, Gower TA (1975) An animal model for studying infection and immunity to and attenuation of human parainfluenza viruses. Dev Biol Stand 28: 414–421.
387. Mascoli CC, Gower TA, Capilupo FA, Metzgar DP (1996) Further studies on the neonatal ferret model of infection and immunity to and attenuation of human parainfluenza viruses. Dev Biol Stand 33: 384–390.
388. Metzgar DP, Gower TA, Larson EF, Fuscaldo AA, Mascoli CC (1974) The effect of parinfluenza virus type 3 on newborn ferrets. J Biol Stand 2: 273–282.
389. Byrd LG, Prince GA (1997) Animal models of respiratory syncytial virus infection. Clin Infect Dis 25: 1363–1368.
390. Krilov LR (2011) Respiratory syncytial virus disease: update on treatment and prevention. Expert Rev Anti Infect Ther 9: 27–32.
391. Hall CB, Walsh EE, Schnabel KC, Long CE, McConnochie KM, Hildreth SW, Anderson LJ (1990) Occurrence of groups A and B of respiratory syncytial virus over 15 years: associated epidemiologic and clinical characteristics in hospitalized and ambulatory children. J Infect Dis 162: 1283–1290.
393. Coates HV, Chanock RM (1962) Experimental infection with respiratory syncytial virus in several species of animals. Am J Hyg 76: 302–312.
394. Pinto CA, Haff RF, Stewart RC (1969) Pathogenesis of and recovery from respiratory svncytial and influenza infections in ferrets. Arch Gesamte Virusforsch 26: 223–237.
395. Prince GA, Porter DD (1976) The pathogenesis of respiratory syncytial virus infection in infant ferrets. Am J Pathol 82: 339–352.
396. Henderson FW, Hu SC, Collier AM (1978) Pathogenesis of respiratory syncytial virus infection in ferret and fetal human tracheas in organ culture. Am Rev Respir Dis 118: 29–37.
397. Porter DD, Muck KB, Prince GA (1980) The age dependence of respiratory syncytial virus growth in ferret lung can be shown in organ and monolayer cultures. Clin Immunol Immunopathol 15: 415–423.
398. Suffin SC, Prince GA, Muck KB, Porter DD (1979b) Immunoprophylaxis of respiratory syncytial virus infection in the infant ferret. J Immunol 123: 10–14.
399. Clyde WA (1980) Experimental models for study of common respiratory viruses. Environ Health Perspect 35: 107–112.
400. Hsu KH, Lubeck MD, Bhat BM, Bhat RA, Kostek B, Selling BH, Mizutani S, Davis AR, Hung PP (1994) Efficacy of adenovirus-vectored respiratory syncytial virus vaccines in a new ferret model. Vaccine 12: 607–612.
401. Colasurdo GN, Hemming VG, Prince GA, Gelfand AS, Loader JE, Larsen GL (1998) Human respiratory syncytial virus produces prolonged alterations of neural control in airways of developing ferrets. Am J Respir Crit Care Med 157: 1506–1511.
402. Suffin SC, Muck KB, Porter DD (1977) Vesicular stomatitis virus causes abortion and neonatal death in ferrets. J Clin Microbiol 6: 437–438.
403. Kowalczykt T, Brandly CA (1954) Experimental infection of dogs, ferrets, chinchillas and hamsters with vesicular stomatitis virus. Am J Vet Res 15: 98–101.
404. Kowalczykt T, Hanson RP, Brandly CA (1955) Infectivity and pathogenicity of vesicular stomatitis virus for ferrets. Am J Vet Res 16: 180–182.
405. Coleman JW, Ogin-Wilson E, Johnson JE, Nasar F, Zamb TP, Clarke DK, Hendry RM, Udem SA (2007) Quantitative multiplex assay for simultaneous detection of the Indiana serotype of vesicular stomatitis virus and HIV gag. J Virol Methods 143: 55–64.
406. Ammersbach M, Delay J, Caswell JL, Smith DA, Taylor WM, Bienzle D (2008) Laboratory findings, histopathology, and immunophenotype of lymphoma in domestic ferrets. Vet Pathol 45: 663–673.
407. Erdman SE, Brown SA, Kawasaki TA, Moore FM, Li X, Fox JG (1996a) Clinical and pathologic findings in ferrets with lymphoma: 60 cases (1982–1994). J Am Vet Med Assoc 208: 1285–1289.
408. Maeda N, Fan H, Yoshikai Y (2008) Oncogenesis by retroviruses: old and new paradigms. Rev Med Virol 18: 387–405.
409. Batchelder MA, Erdman SE, Li X, Fox JG (1996) A cluster of cases of juvenile mediastinal lymphoma in a ferret colony. Lab Anim Sci 46: 271–274.
410. Erdman SE, Kanki PJ, Moore FM, Brown SA, Kawasaki TA, Mikule KW, Travers KU, Badylak SF, Fox JG (1996b) Clusters of lymphoma in ferrets. Cancer Invest 14: 225–230.
411. Erdman SE, Reimann KA, Moore FM, Kanki PJ, Yu QC, Fox JG (1995) Transmission of a chronic lymphoproliferative syndrome in ferrets. Lab Invest 72: 539–546.
412. Erdman SE, Correa P, Coleman LA, Schrenzel MD, Li X, Fox JG (1997) Helicobacter mustelae-associated gastric MALT lymphoma in ferrets. Am J Pathol 151: 273–280.
413. Cossaboom CM, Córdoba L, Cao D, Ni YY, Meng XJ (2012) Complete genome sequence of hepatitis E virus from rabbits in the United States. J Virol 86: 13124–13125.
414. Raj VS, Smits SL, Pas SD, Provacia LB, Moorman-Roest H, Osterhaus AD, Haagmans BL (2012) Novel hepatitis E virus in ferrets, the Netherlands. Emerg Infect Dis 18: 1369–1370.
415. Imran M, Mahmood S (2011) An overview of animal prion diseases. Virol J 8: 493.
416. Watts JC, Balachandran A, Westaway D (2006) The expanding universe of prion diseases. PLoS Pathog 2: e26.
418. Hamir AN, Miller JM, Yaeger MJ (2007) Neuronal vacuolation in an adult ferret. Can Vet J 48: 389–391.
419. Bartz JC, McKenzie DI, Bessen RA, Marsh RF, Aiken JM (1994) Transmissible mink encephalopathy species barrier effect between ferret and mink: PrP gene and protein analysis. J Gen Virol 75: 2947–2953.
420. Bartz JC, Marsh RF, McKenzie DI, Aiken JM (1998) The host range of chronic wasting disease is altered on passage in ferrets. Virology 251: 297–301.
421. Marsh RF, Burger D, Eckroade R, Zu Rhein GM, Hanson RP (1969) A preliminary report on the experimental host range of the transmissible mink encephalopathy agent. J Infect Dis 120: 713–719.
422. Perrott MR, Sigurdson CJ, Mason GL, Hoover EA (2012) Evidence for distinct chronic wasting disease (CWD) strains in experimental CWD in ferrets. J Gen Virol 93: 212–221.
423. Sigurdson CJ, Mathiason CK, Perrott MR, Eliason GA, Spraker TR, Glatzel M, Manco G, Bartz JC, Miller MW, Hoover EA (2008) Experimental chronic wasting disease (CWD) in the ferret. J Comp Pathol 138: 189–196.
424. Robinson MM, Hadlow WJ, Huff TP, Wells GA, Dawson M, Marsh RF, Gorham JR (1994) Experimental infection of mink with bovine spongiform encephalopathy. J Gen Virol 75: 2151–2155.
425. Harrington RD, Baszler TV, O'Rourke KI, Schneider DA, Spraker TR, Liggitt HD, Knowles DP (2008) A species barrier limits transmission of chronic wasting disease to mink (Mustela vison). J Gen Virol 89: 1086–1096.
426. Williams ES, Young S (1992) Spongiform encephalopathies in Cervidae. Rev Sci Tech 11: 551–567.
427. Eckroade RJ, ZuRhein GM, Hanson RP (1973) Transmissible mink encephalopathy in carnivores: clinical, light and electron microscopic studies in raccons, skunks and ferrets. J Wildl Dis 9: 229–240.
428. Burger D, Hartsough GR (1965) Encephalopathy of mink: II. Experimental and natural transmission. J Infect Dis 115: 393–399.
429. Hartsough GR, Burger D (1965) Encephalopathy of mink: I. Epizootiologic and clinical observations. J Infect Dis 115: 387–392.