CHAPTER 12
MUSCLE FATIGUE IN METABOLIC MYOPATHIES
Ronald G.Haller and John Vissing
OBJECTIVES
The aim of this chapter is to provide an overview on:
• the biochemical basis and key clinical features of two classes of human metabolic myopathy;
• studies that have illuminated the mechanism of muscle fatigue when the key pathways of muscle energy production are limited;
• the insights provided by these energy defects for the role of these metabolic pathways in normal muscle fatigue.
INTRODUCTION
In this chapter we will review the implications of two classes of inborn errors of muscle metabolism for muscle fatigue during exercise: muscle mitochondrial defects and disorders of muscle glycogenolysis or glycolysis, utilising as an example of a complete block in muscle glycogenolysis, muscle glycogen phosphorylase deficiency (GSD V, McArdle disease).
MITOCHONDRIAL MYOPATHIES
Mitochondrial myopathies are typically attributable to genetic defects that impair the function of the respiratory chain in skeletal muscle (DiMauro and Schon, 2003). A major clinical consequence of significant respiratory chain defects is a reduced capacity for aerobic exercise, i.e. exercise that is fuelled by the combustion of fats and carbohydrates to carbon dioxide and water, with ATP produced by oxidative phosphorylation. A characteristic of exercise that is fully supported by oxidative phosphorylation is its ability to be sustained for prolonged periods of time, in contrast to exercise that depends upon anaerobic metabolism (phosphocreatine hydrolysis and anaerobic glycogenolysis) to fuel muscle contractions, which rapidly results in muscle fatigue (Allen et al., 2008). Characteristic of anaerobic metabolism is its ability to support rates of ATP turnover that are more than two-fold that of oxidative phosphorylation, so anaerobic metabolism is required for muscle contractions at maximal effort, and when the rate of ATP demand exceeds maximal rates of oxidative phosphorylation (Sahlin, 1986). Accordingly, the limits of an individual’s aerobic capacity is marked by the activation of anaerobic metabolism and is associated with a steep increase in lactate production and in the lactate/pyruvate ratio in working muscle and in blood. In healthy humans, an important variable in determining aerobic capacity is the level of physical conditioning. Trained individuals have a greater aerobic capacity by virtue of having developed the circulatory and muscle mitochondrial adaptations that increase the capacity for oxygen delivery and utilisation (Gollnick and Saltin, 1983). In habitually sedentary individuals, the opposite is true. Hence the range of “normal” aerobic capacity in healthy humans is substantial.
Aerobic capacity is commonly measured in metabolic equivalents termed METS, representing multiples of resting metabolic rate, which by convention is 3.5ml of oxygen per minute per kilogram of body weight (Åstrand and Rodahl, 1986). In our experience, patients with mitochondrial myopathies have an average peak aerobic capacity of approximately 4.5METS, with a large percentage having a peak capacity of less than 4METS (Taivassalo et al., 2003). As a result, affected patients have a greatly restricted range of physical activities that can be supported by oxidative phosphorylation (Figure 12.1). Accordingly, activities considered minor by healthy humans are sufficient to accelerate anaerobic metabolism and cause tachycardia and muscle fatigue in association with marked increases in blood lactate (Figure 12.2A and 12.2B).
Interestingly, the character of muscle fatigue in patients with mitochondrial myopathy is similar to that of healthy subjects (Wiles et al., 1981). Patients do not develop distinctive symptoms of exercise intolerance, but simply have overall low tolerance of aerobic exercise that commonly includes symptoms of exertional shortness of breath and tachycardia. The

Figure 12.1 Correlation between oxygen utilisation expressed in ml per kg per minute (right axis), METS (left axis) and levels of physical activity (X axis)
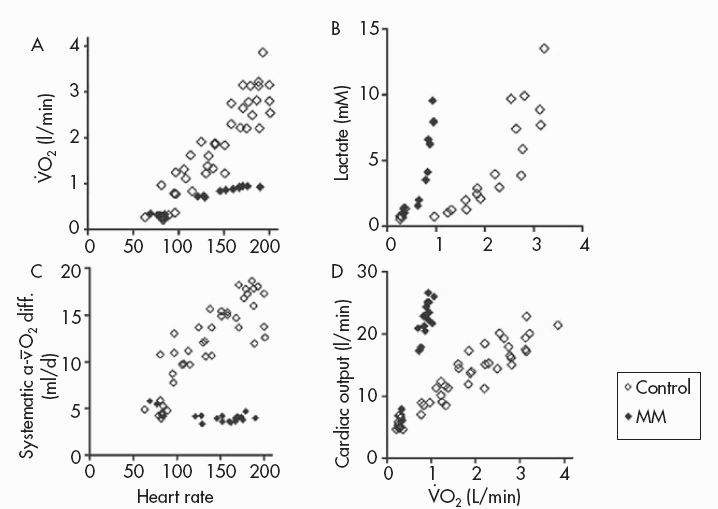
Figure 12.2 Metabolic and physiological effects of a severe defect in muscle mitochondrial metabolism associated with deficiency of multiple iron-sulphur cluster-containing proteins (including succinate dehydrogenase and aconitase and subunits of complex I and III of the respiratory chain (Haller et al., 1991)—a disorder recently shown to be due to a mutation in the iron-sulphur cluster scaffold, ISCU, gene (Mochel et al., 2008)). Results during cycle exercise for an affected patient (filled diamonds) and for control subjects (open diamonds) are shown for: A: 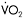 versus heart rate; B: lactate relative to
versus heart rate; B: lactate relative to 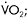 C:. systemic arterio-venous O2 difference relative to heart rate; and D: cardiac output relative to
C:. systemic arterio-venous O2 difference relative to heart rate; and D: cardiac output relative to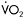
non-specific nature of muscle fatigue that occurs with physical activities that would be expected to be easily tolerated often leads to the mistaken belief that patient symptoms derive from being in poor physical condition.
The metabolic correlates of skeletal muscle fatigue in mitochondrial myopathy include changes in metabolites that are associated with muscle fatigue in healthy subjects. These include depletion of phosphocreatine, increases in muscle and blood lactate and lactate/pyruvate, a decline in muscle pH and increases in muscle levels of ADP and diprotonated phosphate (Matthews et al., 1991). In healthy humans, interventions that increase the oxygen-carrying capacity of blood by increasing red cell mass (blood doping, erythropoietin administration) increase exercise capacity consistent with the notion that oxygen availability rather than levels of functional mitochondria limits oxidative phosphorylation during exercise involving large muscle groups. In contrast, in mitochondrial myopathies, the restriction in oxidative metabolism resides in a defective respiratory chain that limits the ability of mitochondria to utilise available oxygen.
The distinctive pathophysiology of exercise in muscle mitochondrial defects is a mismatch between muscle oxygen utilisation and the circulatory and ventilatory responses to exercise that are normally closely geared to muscle oxygen utilisation (Taivassalo et al., 2003). This mismatch is illustrated by the effects of muscle mitochondrial dysfunction upon the components of oxygen utilisation contained in the Fick equation:
oxygen utilisation= |
|
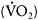 systemic oxygen delivery (cardiac output)×systemic arterio-venous
systemic oxygen delivery (cardiac output)×systemic arterio-venous  O2 difference
O2 differenceAssuming normal O2-carrying capacity (normal levels and function of haemoglobin), arterialised blood contains about 20ml O2, and mixed venous blood 15ml O2 per 100ml of blood, so resting systemic 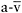 O2 difference is about 5ml. From rest to peak leg exercise, systemic
O2 difference is about 5ml. From rest to peak leg exercise, systemic 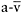 O2 difference in healthy individuals increases three-fold from 5ml to about 15ml·dl−1, and O2 saturation of femoral venous blood falls from about 75% to 25%. This increase in O2 uptake is attributable to the avid mitochondrial utilisation of oxygen, and to the low Km of cytochrome c oxidase for O2 (Richardson et al., 2006). From rest up to moderate levels of exercise, the activation of mitochondrial oxidative phosphorylation lowers intracellular O2 tensions from a pO2 of approximately 30mmHg to a level of 3–5mmHg, establishing a steep O2 gradient from capillaries (pO2 ~35mmHg) to respiring mitochondria (Richardson et al., 2006).
O2 difference in healthy individuals increases three-fold from 5ml to about 15ml·dl−1, and O2 saturation of femoral venous blood falls from about 75% to 25%. This increase in O2 uptake is attributable to the avid mitochondrial utilisation of oxygen, and to the low Km of cytochrome c oxidase for O2 (Richardson et al., 2006). From rest up to moderate levels of exercise, the activation of mitochondrial oxidative phosphorylation lowers intracellular O2 tensions from a pO2 of approximately 30mmHg to a level of 3–5mmHg, establishing a steep O2 gradient from capillaries (pO2 ~35mmHg) to respiring mitochondria (Richardson et al., 2006).
When mitochondrial metabolism limits oxidative phosphorylation, working muscle is unable to normally extract available oxygen from blood, so muscle oxygen levels remain abnormally high and systemic arteriovenous O2 levels remain low during peak exercise (Haller et al., 1991; Bank and Chance, 1994; Taivassalo et al., 2002; Taivassalo et al., 2003). In the most severe mitochondrial defects, systemic  O2 difference remains at or below resting levels (Figure 12.2C) (Taivassalo et al., 2003). Correspondingly, the circulatory delivery of oxygen (cardiac output and blood flow) is greatly exaggerated relative to oxygen utilisation. Normally, the delivery of O2 by the circulation is closely matched to oxygen utilisation by working muscle; but when impaired mitochondrial metabolism limits oxidative metabolism, the increase in cardiac output relative to oxygen utilisation may be 3–5 times normal or even greater (Figure 12.2D) (Haller and Vissing, 1999; Taivassalo et al., 2003). This “hyperkinetic” circulatory response to exercise in mitochondrial myopathy indicates that intact mitochondrial metabolism is required for the normal matching of O2 utilisation and delivery during exercise and suggests that blocked muscle oxidative phosphorylation in working muscle activates circulatory reflexes to drive the circulation during exercise (Haller and Vissing, 1999). Defects in muscle oxidative metabolism also result in exaggerated ventilatory responses to exercise in which ventilation is abnormally high in relation to oxygen utilisation and carbon dioxide production (Flaherty et al., 2001; Taivassalo et al., 2003). Correspondingly, affected patients often experience dyspnea during exercise. In fact, patients with severe mitochondrial myopathies may experience dyspnea as the major symptom limiting physical exertion, despite the fact that the primary metabolic defect is impaired muscle oxidative phosphorylation. The mechanism underlying the prominence of exertional dyspnea in patients with severe oxidative defects that are restricted to skeletal muscle is unknown.
O2 difference remains at or below resting levels (Figure 12.2C) (Taivassalo et al., 2003). Correspondingly, the circulatory delivery of oxygen (cardiac output and blood flow) is greatly exaggerated relative to oxygen utilisation. Normally, the delivery of O2 by the circulation is closely matched to oxygen utilisation by working muscle; but when impaired mitochondrial metabolism limits oxidative metabolism, the increase in cardiac output relative to oxygen utilisation may be 3–5 times normal or even greater (Figure 12.2D) (Haller and Vissing, 1999; Taivassalo et al., 2003). This “hyperkinetic” circulatory response to exercise in mitochondrial myopathy indicates that intact mitochondrial metabolism is required for the normal matching of O2 utilisation and delivery during exercise and suggests that blocked muscle oxidative phosphorylation in working muscle activates circulatory reflexes to drive the circulation during exercise (Haller and Vissing, 1999). Defects in muscle oxidative metabolism also result in exaggerated ventilatory responses to exercise in which ventilation is abnormally high in relation to oxygen utilisation and carbon dioxide production (Flaherty et al., 2001; Taivassalo et al., 2003). Correspondingly, affected patients often experience dyspnea during exercise. In fact, patients with severe mitochondrial myopathies may experience dyspnea as the major symptom limiting physical exertion, despite the fact that the primary metabolic defect is impaired muscle oxidative phosphorylation. The mechanism underlying the prominence of exertional dyspnea in patients with severe oxidative defects that are restricted to skeletal muscle is unknown.
MUSCLE GLYCOLYTIC DEFECTS: MCARDLE DISEASE AND RELATED DISORDERS
Myophosphorylase deficiency (glycogen storage disease type V, McArdle disease) is an autosomal-recessive disorder attributable to mutations in the gene for the muscle form of glycogen phosphorylase on chromosome 11q13 (Aquaron et al., 2007; Deschauer et al., 2007). The effect of all described pathogenic mutations to date is a complete absence of biochemical activity of myophosphorylase, and thus a complete block in muscle glycogen breakdown. Accordingly, McArdle disease provides unique insight into the role of glycogen in muscle energy metabolism.
The classical clinical features of McArdle disease are premature muscle fatigue, electrically silent muscle contractures and rhabdomyolysis (lysis of muscle fibres) produced by exercise that normally would be powered by anaerobic glycogenolysis. Muscle contractures should not be confused with ordinary muscle cramps. They are electrically silent, that is, they are not maintained by muscle or nerve excitation, but rather mimic muscle rigor. With a contracture, muscle is effectively locked in contraction and attempted stretching of the muscle causes severe pain. Recovery commonly is delayed for half an hour or more. Available evidence suggests that muscle contractures are mediated by energy limitations that result in sustained abnormal levels of intracellular calcium (Ruff, 1996). Exertional muscle contractures are routinely associated with muscle fibre necrosis and a breach in sarcolemmal integrity that results in a liberation of muscle cytoplasmic contents into the bloodstream. One of the most dramatic consequences is the release of the muscle haem protein myoglobin from necrotic muscle fibres, resulting in myoglobinuria (causing dark, commonly cola-coloured urine attributable to myoglobin excreted in the urine). Myoglobinuria may result in renal injury, acute tubular necrosis and acute renal failure. These dramatic consequences of exercise in patients with McArdle disease are mimicked by other inborn errors of muscle carbohydrate metabolism, including muscle phosphofructokinase deficiency, muscle phosphoglycerate mutase deficiency, phosphoglycerate kinase deficiency and muscle lactate dehydrogenase deficiency. In each of these disorders, muscle contractions at maximal effort result in catastrophic muscle failure, in which muscle not only fatigues, but develops contractures and muscle necrosis, which, in severe cases, leads to myoglobinuria. These clinical observations indicate that muscle glycogenolysis/glycolysis is not only crucial for supplying energy for muscle contractions during peak exercise, but is also necessary for normal muscle fatigue. That is to say, anaerobic glycogenolysis not only is able to briefly support the most powerful of muscle contractions, but it also is necessary for producing the metabolic milieu in working muscle that safely sets limits to maximal muscle power output so that, after a period of rest and recovery, fatigued skeletal muscle is able to resume normal muscle contractions. Thus, muscle glycogenolysis may be viewed as both the normal engine and the normal breaking system of machina carnis.
In the classical clinical description of McArdle disease, ischemic forearm exercise was employed to demonstrate the characteristic block in muscle lactate production. Ischemic exercise also produces a characteristic pattern of abnormal muscle fatigability (Figure 12.3). The ischemic forearm test is performed by inflating a blood pressure cuff above systolic blood pressure on the arm, and then performing maximal handgrip contractions, commonly with contractions every other second for 60s. In healthy subjects, a gradual decline in maximal voluntary muscle contractions (MVC) occurs so that maximal contractile force declines to about 70% of MVC at the end of the test (Figure 12.3). A similar fatigue pattern is elicited by electrical stimulation of muscle, indicating that contractile force is not limited by voluntary effort. Perhaps not surprisingly, the pattern of fatigue during ischemic exercise in patients with mitochondrial myopathy is similar to healthy subjects (Figure 12.3). In contrast, in patients with muscle phosphorylase deficiency, the pattern of fatigue is highly abnormal. During such testing, contractile force is similar to healthy subjects for the first 5–7 muscle contractions, suggesting that phosphocreatine hydrolysis is able to support muscle energy requirements during this period. Thereafter, a dramatic drop off of contractile force occurs, so that by 15 maximal contractions, force has declined to about 30% of initial MVC, and patients rarely can continue beyond 40s of testing (20 muscle contractions) because of the development of a muscle contracture. Electrically stimulated muscle contractions in affected patients produces a similar pattern of fatigue (Wiles et al., 1981).
The metabolic mechanisms of muscle fatigue, contractures and muscle necrosis when glycogenolysis or glycolysis are impaired have not been established with certainty, but a variety of metabolic abnormalities accompany blocked muscle glycogenolysis (Figure 12.4). These include the characteristic failure of lactate production (Figure 12.4A). Whereas healthy subjects increase blood lactate 4–5-fold during ischemic exercise, venous lactate levels actually fall during forearm exercise in patients with McArdle disease. As a result of the block in lactate production, the normal fall in muscle pH (increase in [H+]) does not occur (Figure 12.4B) (Argov et al., 1987). In healthy subjects, maximal exercise is associated with a steep fall in muscle pH from resting levels of approximately 7.0 to about 6.4. In contrast, such exercise in McArdle disease results in an increase in muscle pH, denoting a decrease in [H+] (Figure 12.4B). The abnormal pH of working phosphorylase-deficient muscle may have a number of deleterious effects. Among the most important of these is its effect on the creatine kinase reaction. Creatine kinase catalyses the reversible reaction by which
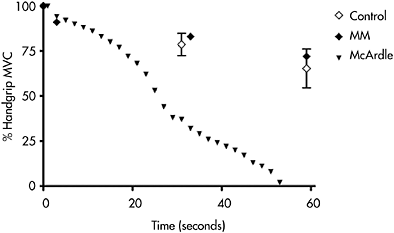
Figure 12.3 Development of fatigue during ischemic handgrip exercise recorded as percentage of original MVC in which subjects are requested to perform a handgrip exercise consisting of 1s MVC followed by 1s of relaxation for 60s. The mean±SD for 12 healthy control subjects (open diamonds) are compared with a patient with a mitochondrial myopathy (closed diamond) and a patient with McArdle disease (closed triangle). The fatigue pattern of patients with other defects of muscle glycolysis is comparable to McArdle disease (not shown)
phosphocreatine (PCr) hydrolysis is coupled to the phosphorylation of ADP. This is a proton-consuming reaction that is normally stimulated by [H+] production via glycolysis. Low levels of [H+] caused by blocked glycogenolysis causes relative inhibition of PCr hydrolysis and results in dramatically elevated levels of ADP during exercise (Radda, 1986). As a consequence, the level of AMP production via myokinase is increased, and the level of AMP deamination via myoadenylate deaminase is accelerated, resulting in high levels of production of IMP and ammonia (Haller and Vissing, 2004a). Accordingly, impaired muscle lactate production in inborn errors of muscle glycogenolysis or glycolysis is associated with an exaggerated increase in ammonia production (Figure 12.4C) that both mirrors the abnormal accumulation of muscle ADP and AMP in affected patients, and is helpful clinically to confirm that blunted lactate production during diagnostic exercise testing is due to a metabolic block rather than poor effort (when the increase in both lactate and ammonia are blunted) (Haller and Vissing, 2004a).
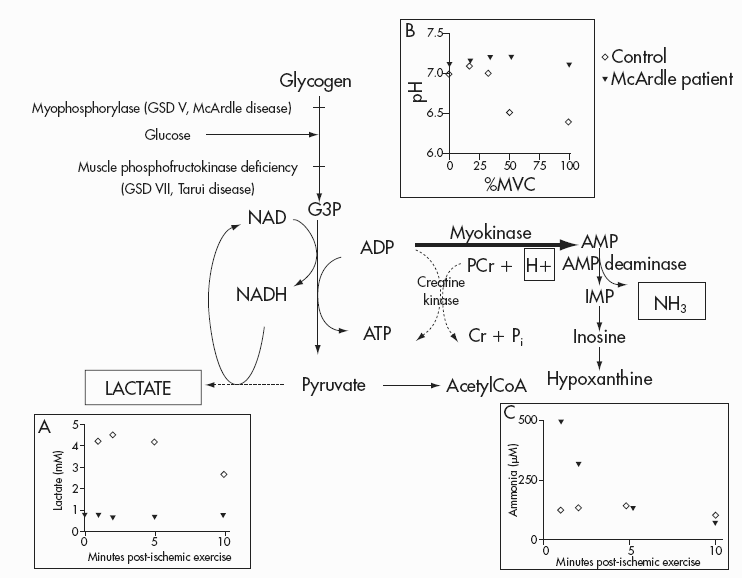
Figure 12.4 Outline of muscle glycogen and glucose metabolism, and the site of metabolic block in McArdle disease and in muscle phosphofructokinase deficiency. Inserts A-C reveal the results of ischemic exercise for control subjects (open diamonds) and a patient with McArdle disease (closed triangle) with respect to lactate production (A) and ammonia production (C). Insert B compares the change in muscle pH during aerobic exercise (measured using 31P magnetic resonance spectroscopy) at different percentages of MVC. Results for such testing in patients with muscle phosphofructokinase (not shown) duplicate those shown for the McArdle patient
An additional important consequence of blocked glycogenolysis is deficient substrate-level phosphorylation of ADP, i.e. blocked ATP production at the level of phosphoglycerate kinase and pyruvate kinase. Substrate level ATP production has been postulated to preferentially supply ATP for the membrane sodium-potassium ATPase, so impaired glycolytic flux might be expected to impair membrane pump function and contribute to the abnormal fatigue and sarcolemmal failure that occurs with intense exercise in McArdle disease and related disorders. The regenerative action potential that underlies sarcolemmal excitability results in an increase in extracellular (and decrease in intracellular) potassium, as well as an increase in intracellular sodium. Maintaining membrane excitability during repeated muscle contraction depends upon the ability of sodium-potassium ATPase, the sodium-potassium pump, to maintain ion gradients and prevent the excessive accumulation of extracellular potassium (Clausen, 1986). Physiological studies have established that abnormal fatigability in McArdle disease is associated with impaired excitation-contraction coupling/sarcolemmal inexcitability, and that the rise in blood and presumably extracellular potassium is exaggerated in relation to the level of muscle contractions in these patients (Cooper et al., 1989). These results implicate sarcolemmal inexcitability and impaired sodium-potassium pump function in the pathophysiology of abnormal muscle fatigue in McArdle disease. Possible mechanisms include: the block in substrate-level phosphorylation that is the preferred source of energy for the sodium-potassium pump (Han et al., 1992; James et al., 1996); absence of acidosis, which may be important for maintaining muscle membrane excitability by decreasing chloride permeability (Pedersen et al., 2004); loss of the osmotic effect from lactate accumulation, which may account for absence of the normal increase in water content of exercised muscle (as monitored by magnetic resonance imaging), and may promote higher than normal concentrations of extracellular potassium in exercising muscle (Fleckenstein et al., 1991); and the exaggerated accumulation of ADP during exercise that may inhibit ATPases, including sodium-potassium ATPase (Ruff and Weissman, 1995). In addition, we have found that sodium-potassium pump levels in muscle samples from McArdle patients, measured by 3H-ouabain binding were only about 70% of control levels (Haller et al., 1998). Sodium-potassium pump levels increase with aerobic training and decrease with deconditioning, so the relative decrease in pump levels could be attributable to relative deconditioning in patients. However, we have found that the increase in pump numbers with aerobic conditioning may be blunted in McArdle patients undergoing exercise training (Haller, Vissing and Clausen, unpublished observation). This suggests that additional factors may be involved, such as disruption of the normal close functional relationship between glycolytic, substrate-level phosphorylation and the activity of the sodium-potassium pump (James et al., 1996).
The fact that fatiguing exercise in McArdle disease and related glycolytic disorders results in muscle contractures and muscle fibre necrosis in addition to impaired excitation-contraction coupling suggests that additional pathogenic factors are involved. A valuable model for studying these issues has been iodoacetate-poisoned muscle that was shown by Lundsgaard to produce muscle contraction and block lactate production in frog skeletal muscle. Iodoacetate blocks glycolysis by inhibiting the enzyme, glyceraldehyde-3-phosphate dehydrogenase. Using iodoacetate-poisoned rat skeletal muscle, Ruff et al. found that muscle contractures in this model are associated with a marked increase in intracellular calcium and with an increase in myofibrillar calcium sensitivity in association with a marked increase in cellular ADP (Ruff and Weissman, 1991; Ruff and Weissman, 1995; Ruff, 1996). Muscle ATP was not substantially reduced, suggesting that ATP depletion per se was not responsible for these results. Interestingly, in this model, the increase in inorganic phosphate that normally parallels the decrease in muscle PCr is blunted compared to normal muscle, owing to the accumulation of phosphorus-containing glycolytic intermediates behind the metabolic block. A similar blunted increase in inorganic phosphate due to a build-up of phosphomonoesters in glycolytic intermediates behind the metabolic block occurs in muscle phosphofructokinase deficiency and in distal glycolytic defects. The accumulation of diprotonated inorganic phosphate (owing to the increase in [H+] when glycolysis is intact) has been implicated in mediating muscle fatigue in healthy humans (Miller et al., 1988), but cannot have a role in the abnormal exertional fatigue, contractures and necrosis in McArdle disease and glycolytic defects, since diprotonated phosphate does not accumulate in the absence of acidosis. Also, inorganic phosphate seems effectively eliminated as a cause of the exertional muscle fatigue in McArdle disease and glycolytic defects, since the pathophysiology of exertional muscle failure in both conditions seems identical, despite marked differences in inorganic phosphate levels (Duboc et al., 1987). A remaining common denominator in the mediation of exertional muscle failure in these conditions is an exaggerated increase in muscle ADP levels, which could operate by inhibiting multiple ATPases.
The foregoing has focused upon the importance of anaerobic glycogenolysis and glycolysis in supplying energy for maximal effort muscle contractures, and the catastrophic consequences for muscle function and integrity when muscle contractions at maximal effort are undertaken when muscle glycogenolysis is blocked. This is in keeping with the common view that blocked glycogenolysis in McArdle disease impairs only anaerobic metabolism and that those symptoms of exercise intolerance in affected patients relate solely to the consequences of this energy limitation. However, the metabolism of glycogen is also necessary to support normal muscle oxidative metabolism, and patients with complete blocks in muscle glycogenolysis (i.e. McArdle disease) or glycolysis (muscle phosphofructokinase (PFK) deficiency, GSD VII, Tarui disease) also have distinctive symptoms attributable to impaired muscle oxidative metabolism (Haller et
al., 1985; Haller and Lewis, 1991). Blocked muscle glycogenolysis results in oxidative phosphorylation being limited by substrate availability, underscoring the critical need for glycogen to support normal peak rates of oxidative metabolism. Available evidence is consistent with the view that blocked glycogenolysis restricts the rate of production of acetyl-CoA and/ or the level of anaplerosis necessary to increase 4-carbon tricarboxylic acid (TCA) cycle intermediates (e.g. fumarate, malate and, ultimately, oxaloac-etate) to support normal peak flux of the TCA cycle, thus restraining the rate of generation of reducing equivalents, NADH and FADH, necessary for normal function of the respiratory chain and normal rates of oxidative phosphorylation (Sahlin et al., 1990; Sahlin et al., 1995). As a result, peak levels of oxidative metabolism in patients with McArdle disease and muscle PFK deficiency are about half or less that of normal (Haller and Lewis, 1991; Haller and Vissing, 2004b). The physiological hallmark of this limitation in muscle oxidative phosphorylation is a restricted capacity of working muscle to extract available oxygen from blood as indicated by a low peak systemic 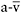 O2 difference, which is associated with an exaggerated increase in the transport of oxygen by the circulation such that the increase in cardiac output relative to O2 utilisation is 2–3 times normal (Haller et al., 1985; Haller and Lewis, 1991).
O2 difference, which is associated with an exaggerated increase in the transport of oxygen by the circulation such that the increase in cardiac output relative to O2 utilisation is 2–3 times normal (Haller et al., 1985; Haller and Lewis, 1991).
A central feature of substrate-limited oxidative metabolism is the variation in oxidative capacity that accompanies changing availability of extra-muscular fuels in response to exercise and diet. This is exemplified by the “second wind” phenomenon that is characteristic of McArdle disease (Braakhekke et al., 1986; Haller and Vissing, 2002; Vissing and Haller, 2003a). In the first 5–8 min of exercise, McArdle patients have a markedly restricted exercise capacity, so relatively minor exertion such as walking at a moderate pace or pedalling a bicycle at a low workload causes fatigue and tachycardia (Haller and Vissing, 2002). However, when exercise is sustained or resumed after a brief rest, patients experience a dramatic improvement in exercise capacity and perceived exertion in association with a marked reduction in exercise heart rate, so that exercise that previously caused fatigue is easily tolerated. Our studies indicate that the markedly low oxidative capacity early in exercise is due to the fact that glycogen is the fuel that normally enables oxidative phosphorylation to increase to peak levels within the first 3–5 min of the transition from rest to exercise. Compounding the oxidative limitation is a relatively low availability of extramuscular fuels—primarily glucose and free fatty acids—during the first minutes of exercise (Haller and Vissing, 2002). Thus, peak oxygen utilisation within the first 5–7 min of exercise in patients with McArdle disease is similar to that of patients with mitochondrial myopathies (Figure 12.5). Like patients with mitochondrial myopathies, McArdle patients have a restricted capacity to extract available oxygen from blood with a peak systemic 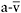 O2 difference that is about half that of healthy subjects tested under the same conditions (Figure 12.5) (Haller et al., 1985; Haller and Vissing, 2002). The dramatically improved exercise capacity accompanied by a steep drop in exercise heart rate that occurs at 7–10min of exercise is attributable to a sudden, large increase in oxidative capacity, marked by an average increase in peak
O2 difference that is about half that of healthy subjects tested under the same conditions (Figure 12.5) (Haller et al., 1985; Haller and Vissing, 2002). The dramatically improved exercise capacity accompanied by a steep drop in exercise heart rate that occurs at 7–10min of exercise is attributable to a sudden, large increase in oxidative capacity, marked by an average increase in peak 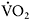 of approximately 25%, which is associated with a similar percentage increase in peak
of approximately 25%, which is associated with a similar percentage increase in peak 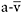 O2 difference (Haller and Vissing, 2002). The drop in heart rate mirrors the fall in exertion perceived by the patient, as heart rate and exercise intensity, expressed as a percentage of an individuals peak
O2 difference (Haller and Vissing, 2002). The drop in heart rate mirrors the fall in exertion perceived by the patient, as heart rate and exercise intensity, expressed as a percentage of an individuals peak 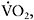 are proportional. For a 35-year-old patient, a heart rate of 175bpm at 5min of exercise indicates the patient was exercising at about 95% of their maximal oxidative capacity. The fall in heart rate to 125 bpm at the same workload after the onset of the second wind indicates exercise at that point is only about 70% of maximal oxidative capacity. Accordingly, after the second wind, the patient is able to exercise at a substantially higher workload because of a higher peak
are proportional. For a 35-year-old patient, a heart rate of 175bpm at 5min of exercise indicates the patient was exercising at about 95% of their maximal oxidative capacity. The fall in heart rate to 125 bpm at the same workload after the onset of the second wind indicates exercise at that point is only about 70% of maximal oxidative capacity. Accordingly, after the second wind, the patient is able to exercise at a substantially higher workload because of a higher peak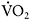 and
and  O2 difference (Haller and Vissing, 2002). Our studies indicate that increased hepatic glucose production and muscle uptake of blood glucose are crucial for the development of a second wind (Vissing et al., 1992;
O2 difference (Haller and Vissing, 2002). Our studies indicate that increased hepatic glucose production and muscle uptake of blood glucose are crucial for the development of a second wind (Vissing et al., 1992;
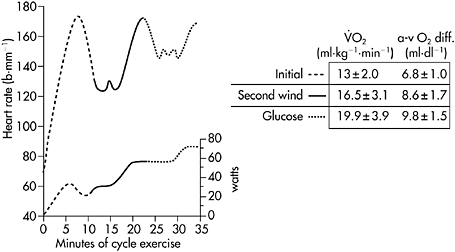
Figure 12.5 Heart rate (upper trace) and corresponding workrate (lower trace) for a patient with McArdle disease during sustained cycle exercise after an overnight fast. In the initial 5–7min of exercise, a 30-watt work level is near maximal and elicits a heart rate approaching 180bpm. Between the seventh and tenth minutes, the patient develops a spontaneous second wind so that exercise that was near maximal before is now easily tolerated at a heart rate that is almost 60bpm lower than before the second wind. Between minutes 15 and 20 the workload is increased, demonstrating that the patient is able to exercise at 55watts at the same heart rate that accompanied 30watts before the second wind. Between minutes 23 and 25, the patient is given an infusion of glucose that doubles blood glucose levels. The glucose infusion is associated with a glucose-induced second wind with improved exercise tolerance and a more than 20bpm drop in heart rate. After glucose, the patient’s maximal workload increases to 70watts. The table at the right shows the corresponding 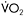 and systemic a-v O2 difference for the patient’s initial, spontaneous second wind, and glucose infusion condition showing a progressive increase in peak
and systemic a-v O2 difference for the patient’s initial, spontaneous second wind, and glucose infusion condition showing a progressive increase in peak 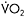 that is parallelled by progressive increases in systemic
that is parallelled by progressive increases in systemic 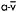 O2 difference consistent with increasing substrate availability for oxidative phosphorylation under second wind and glucose conditions
O2 difference consistent with increasing substrate availability for oxidative phosphorylation under second wind and glucose conditions
Haller and Vissing, 2002). Evidence supporting this conclusion includes the fact that patients with muscle PFK deficiency, who are unable to oxidise muscle glycogen or blood glucose, do not develop a second wind under exercise conditions that consistently produce one in McArdle disease (Haller and Vissing, 2004b).
Although McArdle patients have substantially improved exercise capacity after the onset of the spontaneous second wind, oxidative metabolism remains substrate-limited. This is evident in the fact that increasing blood glucose concentrations by intravenous glucose administration produces another second wind, with a drop in exercise heart rate and an increase in peak 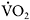 and peak
and peak 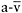 O2 difference (Figure 12.5) (Haller and Vissing, 2002). Although muscle glucose utilisation appears to be necessary for both the spontaneous second wind and glucose-induced, second, second wind, the metabolic mechanism has not been fully elucidated. In addition to direct combustion via the production of acetyl-CoA, glucose may enhance oxidative capacity by augmenting levels of oxaloacetate, thereby promoting the oxidation of fatty acids.
O2 difference (Figure 12.5) (Haller and Vissing, 2002). Although muscle glucose utilisation appears to be necessary for both the spontaneous second wind and glucose-induced, second, second wind, the metabolic mechanism has not been fully elucidated. In addition to direct combustion via the production of acetyl-CoA, glucose may enhance oxidative capacity by augmenting levels of oxaloacetate, thereby promoting the oxidation of fatty acids.
Diet is also an important variable in determining oxidative capacity in McArdle disease. Oral ingestion of sucrose markedly improves exercise capacity, lowers exercise heart rate and abolishes the spontaneous second wind in McArdle disease attributable to increased blood levels of glucose and lactate (Vissing and Haller, 2003b; Andersen et al., 2008; Andersen and Vissing, 2008). In contrast, in muscle PFK deficiency, glucose infusion or a high-carbohydrate meal has the opposite effect, and reduces muscle oxidative capacity (Haller and Lewis, 1991). This relates to the fact that glucose cannot be metabolised by PFK-deficient muscle, and thus has no beneficial effect upon energy production, and to the fact that glucose causes an insulin-mediated reduction in blood levels of free fatty acids, the key oxidative fuel when the metabolism of muscle glycogen and glucose are blocked.
CASE STUDY
Clinical history
A 39-year-old man was referred for evaluation of the cause of symptoms of exertional muscle fatigue, muscle tightness and pigmenturia. The patient’s first recollection of exercise limitations occurred during standardised exercise testing in junior high and high school. Particularly, upper body exercise such as pull-ups, push-ups and rope climbing caused abnormally rapid fatigue, often accompanied by nausea. For example, he could never perform more than 1–2 pull-ups. If such exertion were continued he would develop cramps, in which the affected limb would be locked up, unable to be straightened for minutes to hours, and the attempt to do so would cause severe pain. His legs have less frequently been affected, but maximal effort running, particularly if he had not warmed up could cause similar cramps in his legs. On many occasions, exertional cramping and muscle pain have been followed by dark urine, typically hours after the bout of exertional muscle cramps. He has continued to experience these symptoms over the years with a general downhill course marked by developing symptoms after lesser degrees of exercise.
Despite these problems, the patient has been relatively athletic. He tried out for track, running the mile in high school, though he was never good enough to make the team. His best time for the mile was about 6.5min. In college, he participated in a physical education class, for which he ran three miles per day at a 6.5–7min per mile pace. At age 18, he rode across country on a bicycle tour. He had not cycled significantly before and was one of the slower riders in his group for the first part of the trip, but by the end he was near the leaders. After a hiatus from participating in running or cycling, he notes that he is prone to develop exertional cramps and dark urine until he has trained for a week or two. After high school he spent several years as a “ski bum”. There he noted he fatigued more rapidly than his peers in difficult runs. Particularly steep slopes with moguls that required frequent turns would cause cramping in his thighs.
Diet has no major effect on exercise capacity, though he thinks he may cramp more easily on an empty stomach. No circumstances other than exercise have triggered muscle symptoms. He characterises his muscle as weaker than normal, but illustrates this as tiring out quickly with heavy exertion.
Evaluation: rationale
The history is strongly suggestive of a metabolic muscle disorder, with multiple episodes of pigmenturia suggesting myoglobinuria following maximal effort exercise. Such heavy exercise is described as producing muscle tightness, “cramps” and “locking up” in which the muscle remains contracted from “minutes to hours”, and attempting to stretch the muscle produces pain. These episodes are highly suggestive of muscle contractures, and the maximal effort exercise that triggers them normally is supported by anaerobic metabolism, particularly anaerobic glycogenolysis. However, the history suggests that his aerobic exercise capacity is much higher than in muscle phosphorylase deficiency (McArdle disease), the most common muscle glycolytic disorder. Specifically, the history of being able to run 1–3 miles at a 6.5–7min per mile pace predicts an aerobic capacity far higher than is typical of McArdle disease (Figure 12.1).
In order to confirm the history of a relatively normal aerobic capacity and to determine if the patient has evidence of a spontaneous second wind, a cycle exercise test would be appropriate—ideally with the capacity to measure cardiac output as well as  in order to be able to calculate systemic
in order to be able to calculate systemic 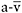 O2 difference to determine if oxygen extraction by working muscle is normal.
O2 difference to determine if oxygen extraction by working muscle is normal.
In addition, it is important to evaluate the patient’s capacity for anaerobic exercise using an ischemic forearm test or a maximal effort aerobic forearm test.
Exercise testing: results
The patient underwent cycle exercise testing which revealed a work capacity of 170watts, a 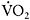 of 2.71 (30ml·kg−1·min−1), peak heart rate of 165bpm, peak cardiac output of 171, and peak
of 2.71 (30ml·kg−1·min−1), peak heart rate of 165bpm, peak cardiac output of 171, and peak 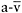 O2 difference of 16ml·dl−1 O2 blood—all values within the normal range (compare with normal values in Figure 12.2) in contrast to typical values found with such testing for patients with McArdle disease (Figure 12.5). In addition, prolonged cycle exercise testing performed on a separate day revealed no evidence of a second wind.
O2 difference of 16ml·dl−1 O2 blood—all values within the normal range (compare with normal values in Figure 12.2) in contrast to typical values found with such testing for patients with McArdle disease (Figure 12.5). In addition, prolonged cycle exercise testing performed on a separate day revealed no evidence of a second wind.
Ischemic forearm testing in which the patient performed a maximal handgrip contraction every 2s revealed the results shown in Figure 12.6. The patient completed the first eight maximal effort contractions normally, but thereafter began to rapidly fatigue and stopped after 17 handgrips (33s) with a contracture affecting the finger flexors (Figure 12.6A). Venous lactate rose from a resting level of approximately 1 mM to 3.2mM at 1 min post-exercise (Figure 12.6B), or about 70% of the increase seen in healthy control subjects (Figure 12.6B). However, the ammonia response was highly abnormal, increasing to a level almost three times that of control subjects (Figure 12.6D). This abnormal ammonia response was dramatically apparent when ammonia relative to lactate values were plotted (Figure 12.6C). The results show that the lactate-ammonia response is linear in healthy subjects, but that the increase in ammonia was dramatically higher relative to the increase in lactate, indicating an abnormally high level of AMP deamination consistent with abnormally high levels of ADP during maximal effort exercise in the patient. These results suggest that the patient has an unusual muscle glycolytic disorder, in which some capacity for glycolysis is preserved. Further evaluation of this patient, employing 31P MRS revealed normal resting spectra. During handgrip exercise, the fall in PCr is normally matched by a proportional increase in inorganic phosphate. In this patient, the increase in inorganic phosphate during exercise was markedly blunted, and there is an anomalous large phosphomonoester (PME) peak indicating the accumulation of phosphorus containing glycolytic intermediates and confirming the fact that the patient has a block in muscle glycolysis (Figure 12.7). Noteworthy as well is the fact that despite the ability to increase lactate during forearm exercise, muscle in the patient does not acidify (Figure 12.7).
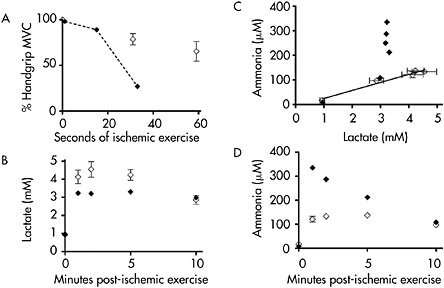
Figure 12.6 Ischemic forearm testing results for control subjects (open diamonds) and the patient described in the case study (closed diamonds) showing: A: rapid muscle fatigue in the patient; B: blunted lactate production in the patient; C: lactate and corresponding ammonia level at rest and during exercise, demonstrating a linear relationship between lactate and ammonia in control subjects and dramatically elevated levels of ammonia relative to lactate in the patient; and D: exaggerated ammonia production in the patient
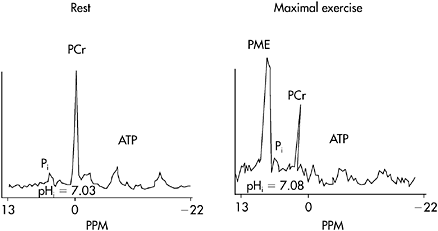
Figure 12.7 31P MRS at rest and during maximal aerobic handgrip in the patient described in the case report. Rest spectra are normal. During heavy exercise, the increase in inorganic phosphate (Pi) is markedly blunted and there appears a large phosphomonoester peak, representing the accumulation of phosphorus containing intermediates of glycolysis behind a metabolic block. Note that muscle pH is abnormally high during heavy exercise (compare with the results for healthy subjects in Figure 12.4)
Interpretation/implications
The diagnosis, in this instance, was achieved by muscle biopsy with muscle enzyme analysis, which indicated that the patient had phosphoglycerate kinase deficiency (5% of normal enzyme activity). The clinical and exercise results in this patient are typical for so-called distal glycolytic defects in which some level of residual enzyme activity is preserved. Remarkably, although a substantial level of increase in lactate occurs with exercise, the level of residual glycolysis is insufficient to cause a substantial fall in muscle pH, and the pattern of fatigue with maximal effort anaerobic exercise in patients with this class of glycolytic defect is virtually the same as in patients with a complete block in glycogenolysis or glycolysis (compare Figure 12.3 and Figure 12.6A). This suggests that the deficit in energy availability relative to energy demand and the metabolic milieu of working muscle during maximal effort anaerobic exercise are similar to those in patients with a complete metabolic block with a characteristic exaggerated increase in ammonia relative to lactate, indicating high levels of ADP, AMP and AMP deamination.
However, there is a significant difference in muscle oxidative capacity in patients with partial, compared to complete, blocks in muscle glycogen or glycogen and glucose utilisation with minor levels of residual glycolysis supporting essentially normal oxidative capacity in patients with partial glycolytic defects.
CONCLUSION
Human genetic disorders of muscle energy metabolism provide unique perspectives from which to view the function of metabolic pathways in muscle physiology and metabolism. Muscle respiratory chain disorders that limit muscle oxidative phosphorylation illuminate the necessity of oxidative metabolism for powering sustained muscle contractions and reveal a key role for mitochondrial metabolism in regulating the delivery of oxygen to working muscles. Disorders of muscle glycogenolysis and glycolysis reveal the crucial role of these metabolic processes for supplying both anaerobic and aerobic energy for muscle contraction. The pathological fatigue that occurs when glycogenolysis and/or glycolysis is blocked implies an important role for these metabolic pathways in normal muscle fatigue.
FIVE KEY PAPERS THAT SHAPED THE TOPIC AREA
Study 1. Wiles, C.M., Jones, D.A. and Edwards, R.H. (1981). Fatigue in human metabolic myopathy. Ciba Foundation Symposium, 82, 264–282.
This is a classical study of muscle fatigue in mitochondrial myopathy and McArdle disease from the laboratory of Professor Richard Edwards, a pioneer in the physiological investigation of human muscle diseases. These studies employ neural stimulation of muscle to eliminate voluntary effort in the investigation of muscle fatigue in these disorders in order to define specific defects of muscle excitation or excitation-contraction coupling.
Study 2. Ruff, R.L. (1996). Elevated intracellular Ca2+ and myofibrillar Ca2+ sensitivity cause iodoacetate-induced muscle contractures. Journal of Applied Physiology, 81, 1230–1239.
This is one of a series of excellent studies utilising the iodoacetate-inhibition of glyceraldehyde-3-phosphate dehydrogenase to illuminate the metabolic mechanism of muscle fatigue and muscle contractures when muscle glycolysis or glycogenolysis is blocked.
Study 3. Haller, R.G. and Vissing, J. (2002). Spontaneous second wind and glucose-induced second, “second wind” in McArdle disease: oxidative mechanisms. Archives in Neurology, 59, 1395–1402
This study reveals that the unavailability of muscle glycogen causes muscle oxidative phosphorylation to be limited by substrate availability and that this oxidative limitation is most severe in the first 5–7 min of exercise; that the spontaneous second wind that occurs after 7–10min of sustained exercise, associated with a fall in exercise heart rate and perceived exertion, is due to a ~25% increase in oxidative capacity; and that muscle oxidative phosphorylation remains substrate-limited after the spontaneous second wind as indicated by the fact that a glucose infusion produces another second wind due to a ~20% further increase in muscle oxidative capacity.
Study 4. Taivassalo, T., Jensen, T.D., Kennaway, N., DiMauro, S., Vissing, J. and Haller R.G. (2003). The spectrum of exercise tolerance in mitochondrial myopathies: a study of 40 patients. Brain, 126, 413–423.
This study evaluated patients with a broad range of impaired muscle oxidative phosphorylation, showing that the key physiological limitation for oxidative metabolism in muscle respiratory chain defects is a limited capacity to extract oxygen from blood (low peak 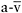 O2 difference) and that
O2 difference) and that 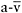 peak O2 difference represents a surrogate marker of the severity of the respiratory chain defect. Furthermore, the study reveals a close, inverse relationship between the severity of impaired muscle oxidative phosphorylation (as indicated by peak
peak O2 difference represents a surrogate marker of the severity of the respiratory chain defect. Furthermore, the study reveals a close, inverse relationship between the severity of impaired muscle oxidative phosphorylation (as indicated by peak 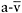 O2 difference in cycle exercise) and the level of exaggerated circulatory and ventilatory responses to exercise in these patients.
O2 difference in cycle exercise) and the level of exaggerated circulatory and ventilatory responses to exercise in these patients.
Study 5. Allen, D.G., Lamb, G.D. and Westerblad, H. (2008). Skeletal muscle fatigue: cellular mechanisms. Physiological Review 88, 287–332.
This is a comprehensive and up-to-date review of studies of cellular mechanisms of muscle fatigue. While fatigue in metabolic myopathies is not a focus of this review, it provides an excellent assessment of the role of muscle metabolism in normal muscle fatigue that is highly relevant to the consideration of the pathophysiology of metabolic muscle disorders.
GLOSSARY OF TERMS
| arterio-venous oxygen difference |
ADP | adenosine diphosphate |
AMP | adenosine monophosphate |
ATP | adenosine triphosphate |
GSD | glycogen storage disease |
GSDV | muscle phosphorylase deficiency |
GSD VII | muscle phosphofructokinase deficiency |
IMP | inosine monophosphate |
MET | metabolic equivalent |
MVC | maximal voluntary contraction |
PCr | phosphocreatine |
PFK | phosphofructokinase |
PME | phosphomonoester |
Pi | inorganic phosphate |
pO2 | partial pressure of oxygen, commonly expressed in millimeters of mercury |
TCA | tricarboxylic acid cycle |
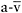 difference
differenceREFERENCES
Allen, D.G., Lamb, G.D. and Westerblad, H. (2008). Skeletal muscle fatigue: cellular mechanisms. Physiological Reviews, 88, 287–332.
Andersen, S.T. and Vissing, J. (2008). Carbohydrate- and protein-rich diets in McArdle disease: effects on exercise capacity. Journal of Neurology and Neurosurgery and Psychiatry, 79, 1359–1363.
Andersen, S.T., Haller, R.G. and Vissing J. (2008). Effect of oral sucrose shortly before exercise on work capacity in McArdle disease. Archives in Neurology, 65, 786–789.
Aquaron, R., Berge-Lefranc, J.L., Pellissier, J.F., Montfort, M.F., Mayan, M., Figarella-Branger, D., Coquet, M., Serratrice, G. and Pouget, J. (2007). Molecular characterization of myophosphorylase deficiency (McArdle disease) in 34 patients from Southern France: identification of 10 new mutations: absence of genotype-phenotype correlation. Neuromuscular Disorders, 17, 235–241.
Argov, Z., Bank, W.J., Maris, J. and Chance, B. (1987). Muscle energy metabolism in McArdle’s syndrome by in vivo phophorus magnetic resonance spectroscopy. Neurology, 37, 1720–1724.
Åstrand, P.O. and Rodahl, K. (1986). Textbook of Work Physiology: Physiological Basis of Exercise. New York: McGraw-Hill.
Bank, W. and Chance, B. (1994). An oxidative defect in metabolic myopathies: diagnosis by non-invasive tissue oxymetry. Annals in Neurology, 36, 830–837.
Braakhekke, J.P., deBruin, M.I., Stegeman, D.F., Wevers, R.A., Binkhorst, R.A. and Joosten, E.M.G. (1986). The second wind phenomenon in McArdle’s disease. Brain, 109, 1087–1101.
Clausen, T. (1986). Regulation of active Na+-K+ transport in skeletal muscle. Physiological Reviews, 66, 542–580.
Cooper, R.G., Stokes, M.J. and Edwards, R.H. (1989). Myofibrillar activation failure in McArdle’s disease. Journal of Neurological Sciences, 93, 1–10.
Deschauer, M., Morgenroth, A., Joshi, P.R., Glaser, D., Chinnery, P.F., Aasly, J., Schreiber, H., Knape, M., Zierz, S. and Vorgerd, M. (2007). Analysis of spectrum and frequencies of mutations in McArdle disease: identification of 13 novel mutations. Journal of Neurology, 254, 797–802.
DiMauro, S. and Schon, E.A. (2003). Mitochondrial respiratory-chain diseases. The New England Journal of Medicine, 348, 2656–2668.
Duboc, D., Jehenson, P., Dinh, S., Marsac, C., Syrota, A. and Fardeau, M. (1987). Phosphorus NMR spectroscopy study of muscular enzyme deficiencies involving glycogenolysis and glycolysis. Neurology, 37, 663–674.
Flaherty, K.R., Wald, J., Weisman, I.M., Zeballos, R.J., Schork, M.A., Blaivas, M., Rubenfire, M. and Martinez, F.J. (2001). Unexplained exertional limitation: characterization of patients with a mitochondrial myopathy. American Journal of Respiratory and Critical Care Medicine, 164, 425–432.
Fleckenstein, J.L., Haller, R.G., Lewis, S.F., Bertocci, L., Payne, J., Barker, B., Payne, J., Parkey, R.W. and Peshock, R.M. (1991). Myophosphorylase deficiency impairs exercise-enhancement on MRI of skeletal muscle. Journal of Applied Physiology, 71, 961–969.
Gollnick, P.D. and Saltin, B. (1983). Skeletal muscle adaptability: significance for metabolism and performance. In L.D.Peachey (ed.) Handbook of Physiology. Bethesda: American Physiological Society, pp. 555–631.
Haller, R.G. and Lewis, S.F. (1991). Glucose-induced exertional fatigue in muscle phosphofructokinase deficiency. The New England Journal of Medicine, 324, 364–369.
Haller, R.G. and Vissing, J. (1999). Circulatory regulation in muscle disease. In B. Saltin, R.Boushel, N.Secker and J.H.Mitchell (eds). Exercise and Circulation in Health and Disease. Champaign: Human Kinetics, pp. 263–273.
Haller, R.G. and Vissing, J. (2002). Spontaneous second wind and glucose-induced second, “second wind” in McArdle disease: oxidative mechanisms. Archives in Neurology, 59, 1395–1402.
Haller, R.G. and Vissing, J. (2004a). Functional evaluation of metabolic myopathy. In A.G.Engel and C.Franzini-Armstrong (eds). Myology, edition III, Vol. 1. New York: McGraw-Hill, pp. 665–679.
Haller, R.G. and Vissing, J. (2004b). Lack of a spontaneous second wind in muscle phosphofructokinase deficiency. Neurology, 62, 82–87.
Haller, R.G., Clausen, T. and Vissing, J. (1998). Reduced levels of skeletal muscle Na+K+-ATPase in McArdle disease. Neurology, 50, 37–40.
Haller, R.G., Henriksson, K.G., Jorfeldt, L., Hultman, E., Wibom, R., Sahlin, K., et al. (1991). Deficiency of skeletal muscle succinate dehydrogenase and aconitase: pathophysiology of exercise in a novel human muscle oxidative defect. Journal of Clinical Investigations, 88, 1197–1206.
Haller, R.G., Lewis, S.F., Cook, J.D. and Blomqvist, C.G. (1985). Myophosphorylase deficiency impairs muscle oxidative metabolism. Annals of Neurology, 17, 196–199.
Han, J.W., Thieleczek, R., Varsanyi, M. and Heilmeyer Jr, L.M. (1992). Compartmentalized ATP synthesis in skeletal muscle triads. Biochemistry, 31, 377–384.
James, J.H., Fang, C.H., Schrantz, J., Hasselgren, P.-O., Paul, R.J. and Fischer, J.E. (1996). Linkage of aerobic glycolysis to sodium-potassium transport in rat skeletal muscle. Journal of Clinical Investigations, 98, 2388–2397.
Matthews, P.M, Allaire, C., Shoubridge, E.A., Karpati, G., Carpenter, S. and Arnold, D.L. (1991). In vivo muscle magnetic resonance spectroscopy in the clinical investigation of mitochondrial disease. Neurology, 41, 114–120.
Miller, R.G., Boska, M.D., Moussavi, R.S., Carson, P.J. and Weiner, M.W. (1988). 31P nuclear magnetic resonance studies of high-energy phosphates and pH in human muscle fatigue. Journal of Clinical Investigations, 31, 1190–1196.
Mochel, F., Knight, M.A., Tong, W.H., Hernandez, D., Ayyad, K., Taivassalo, T., Andersen, P.M., Singleton, A., Rouault, T.A., Fischbeck, K.H., Haller, R.G. (2008). Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. American Journal of Human Genetics, 82, 652–660.
Pedersen, T.H., Nielsen, O.B., Lamb, G.D. and Stephenson, D.G. (2004). Intracellular acidosis enhances the excitability of working muscle. Science, 305, 1144–1147.
Radda, G.K. (1986). The use of NMR spectroscopy for the understanding of disease. Science, 233, 640–645.
Richardson, R.S., Duteil, S., Wary, C., Wray, D.W., Hoff, J. and Carlier, P.G. (2006). Human skeletal muscle intracellular oxygenation: the impact of ambient oxygen availability. Journal of Physiology, 571, 415–424.
Ruff, R.L. (1996). Elevated intracellular Ca2+ and myofibrillar Ca2+ sensitivity cause iodoacetate-induced muscle contractures. Journal of Applied Physiology, 81, 1230–1239.
Ruff, R.L and Weissman, J. (1991). Iodoacetate-induced contracture in rat skeletal muscle: possible role of ADP. American Journal of Physiology, 261, C828–836.
Ruff, R.L. and Weissman, J. (1995). Iodoacetate-induced skeletal muscle contracture: changes in ADP, calcium, phosphate, and pH. American Journal of Physiology, 268, C317–322.
Sahlin, K. (1986). Metabolic changes limiting muscle performance. In B.Saltin (ed.), Biochemistry of Exercise VI. Champaign: Human Kinetics, pp. 323–343.
Sahlin, K., Areskog, N.H., Haller, R.G., Henriksson, K.G., Jorfeldt, L. and Lewis, S.F. (1990). Impaired oxidative metabolism increases adenine nucleotide breakdown in McArdle’s disease. Journal of Applied Physiology, 69, 1231–1235.
Sahlin, K., Jorfeldt, L., Henriksson, K.G., Lewis, S.F. and Haller, R.G. (1995). Tri-carboxylic acid cycle intermediates during incremental exercise in healthy subjects and in patients with McArdle’s disease. Clinical Science (London), 88, 687–693.
Taivassalo, T., Abbott, A., Wyrick, P. and Haller, R.G. (2002). Venous oxygen levels during aerobic forearm exercise: an index of impaired oxidative metabolism in mitochondrial myopathy. Annals of Neurology, 51, 38–44.
Taivassalo, T., Jensen, T.D., Kennaway, N., DiMauro, S., Vissing, J. and Haller, R.G. (2003). The spectrum of exercise tolerance in mitochondrial myopathies: a study of 40 patients. Brain, 126, 413–423.
Vissing, J. and Haller, R.G. (2003a). A diagnostic cycle test in McArdle disease. Annals of Neurology, 54, 539–542.
Vissing, J. and Haller, R.G. (2003b). The effect of oral sucrose on exercise tolerance in McArdle’s disease. The New England Journal of Medicine, 349, 2503–2509.
Vissing, J., Lewis, S.F., Galbo, H. and Haller, R.G. (1992). Effect of deficient muscular glycogenolysis on extramuscular fuel production in exercise. Journal of Applied Physiology, 72, 1773–1779.
Wiles, C.M, Jones, D.A. and Edwards, R.H. (1981). Fatigue in human metabolic myopathy. Ciba Foundation Symposium, 82, 264–282.