Chapter 12
Polymerization Practice
12.1 Introduction
As we have seen in the past few chapters, there are a number of reaction mechanisms used to make polymers. Some reactor designs (semibatch and CSTRs) have been discussed as ways to form uniform or well-characterized polymers and copolymers. This chapter covers some of the important considerations with scaling up polymerizations, starting with bulk polymerization, and takes into account how transport processes (heat and mass transfer) may influence the design of polymerization reactors.
12.2 Bulk Polymerization
The simplest and most direct method of converting monomer to polymer is known as bulk or mass polymerization. A typical feed for a free-radical bulk polymerization might consist of a liquid monomer, a monomer-soluble initiator, and perhaps a chain-transfer agent (no solvent!).
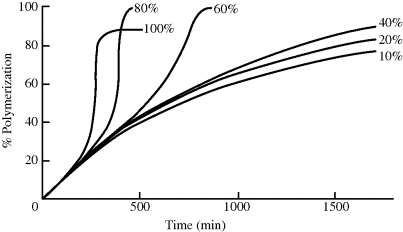
As simple as this seems, some serious difficulties can be encountered, particularly in free-radical bulk polymerizations. One of them is illustrated in Figure 12.1 [1], which indicates the course of polymerization for methyl methacrylate by either bulk polymerization or solution polymerization using various concentrations of benzene, an inert solvent. The reactions were carefully maintained at constant temperature. At low polymer concentrations, the conversion versus. time curves are described by Equation 9.19. As polymer concentrations increase, however, a distinct acceleration of the rate of polymerization is observed which does not conform to the classical kinetic scheme. This phenomenon is known variously as autoacceleration, the gel effect, or the Tromsdorff effect.
The reasons for this behavior lie in the difference between the propagation reaction (Eq. 9.3) and the termination steps (Eqs 9.4a and b), and the extremely high viscosities of the concentrated polymer solutions (104 poise is a ballpark figure–compare with water, which has a viscosity of 0.01 poise). The propagation reaction involves the approach of a small monomer molecule to a growing chain end, whereas termination requires that the ends of two growing chains get together. In bulk polymerizations, at high concentrations of polymer, it becomes exceedingly difficult for the growing chain ends to drag their chains through the entangled mass of dead polymer chains (eventually, the mass may vitrify, meaning that the chains are stuck in place and cannot move around enough to terminate). It is nowhere near as difficult for a monomer molecule to pass through the reaction mass, though. Thus, the rate of the termination reaction is limited not by the nature of the chemical reaction but by the rate at which the reactants can diffuse together, that is, it is diffusion controlled. This lowers the effective termination rate constant kt, and since kt appears in the denominator of Equation 9.13, the net effect is to increase the rate of polymerization. At very high polymer concentrations and below the temperature at which the chains become essentially immobile (or vitrified)—the Tg of the monomer-plasticized polymer—even the propagation step is diffusion limited, hence the bulk polymerization (100% monomer) curve levels off before reaching 100% conversion.
Figure 12.1 Polymerization of methyl methacrylate at 50 °C in the presence of benzoyl peroxide (a thermal initiator) in bulk (100%) or solution polymerization using various concentrations (10–80%) of monomer in benzene.
The difficulties of bulk polymerization are compounded by the inherent nature of the reaction mass. Vinyl monomers have rather large exothermic heats of polymerization, typically between –10 and –21 kcal/mol. Organic systems also have low heat capacities and thermal conductivities, about half those of aqueous solutions. Thus, the temperature can rise very quickly. To top it all off, the tremendous viscosities prevent effective convective (mixing) heat transfer. As a result, the overall heat-transfer coefficients are very low, making it difficult to remove the heat generated by the reaction. This raises the temperature, further increasing the rate of reaction (see Example 9.2) which in turn increases the rate of heat evolution, and can ultimately lead to disaster! To quote Schildknecht [2] on laboratory bulk polymerizations, “If a complete rapid polymerization of a reactive monomer in large bulk is attempted, it may lead to loss of the apparatus, the polymer or even the experimenter.”
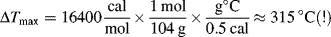
Problems of overheating in bulk polymerizations can be circumvented in several ways:
(Of course, a fifth option would be to use a solvent to aid in absorbing heat, but this is covered in a later section of this chapter.)
Batch bulk polymerization is often used to make objects with a desired shape by polymerizing in a mold. Examples are casting, potting, and encapsulation of electrical components and impregnation of reinforcing agents followed by polymerization. Continuous bulk polymerization is used for the production of thermoplastics by both step-growth and free-radical addition mechanisms. A continuous bulk process is outlined in Figure 12.2 [3]. Conversion is carried to about 40% in a stirred tank. The reaction mass then passes down a tower with the temperature increasing to keep the viscosity at a manageable level and to drive up the conversion. The tower may be a simple gravity-flow device or it may contain slowly rotating spiral blades that scrape the walls, promoting heat transfer and conveying the reaction mass downward. The reaction mass is fed from the tower to a vented extruder at better than 95% conversion. Some additional conversion takes place in the extruder and a vacuum removes unreacted monomer, which is recycled. The extruded strands of molten polymer are then water cooled and chopped to form the roughly 1/8 × 1/8 × 1/8 in (3 × 3 × 3 mm) pellets, which are sold to processors as “molding powder.” Sheets of PMMA are also continuously cast from sirup between polished sheet-metal belts.Most bulk polymerizations are homogeneous. However, if the polymer is insoluble in its monomer and precipitates as the reaction proceeds, the process is sometimes known as heterogeneous bulk or precipitation polymerization. Two examples of such polymers are polyacrylonitrile and poly(vinyl chloride) (PVC). The latter is produced by a heterogeneous bulk process, which allows control of particle size and porosity for optimum plasticizer absorption [4]. Such heterogeneous polymerizations do not follow the kinetic scheme developed in Chapter 9 for homogeneous reactions.
Figure 12.2 Continuous bulk polymerization of styrene. Reprinted by special permission from Reference 3. Copyright © 1962, by McGraw-Hill, Inc., New York, NY 10036.
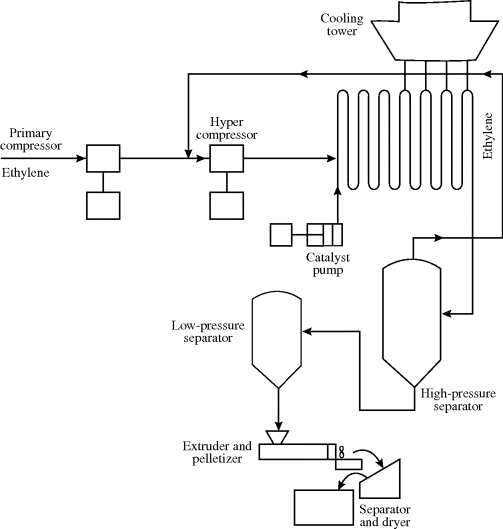
Ethylene and a few other monomers are in the gas phase at atmospheric conditions. Thus, conventional low-density polyethylene (LDPE) is produced at pressures in the range of 15,000–50,000 psi (1000–3400 atm), and temperatures of about 150–300 °C [5]. These pressures are well above the critical pressure of ethylene (731 psi), so the monomer can be considered to be in the “fluid” phase. Depending on the temperature, pressure, and polymer molecular weight, one or two phases may be present as the polymer forms. Peroxide and azo free-radical initiators are used, as are small amounts of oxygen. Oxygen reacts with monomer to form peroxide initiators in situ.
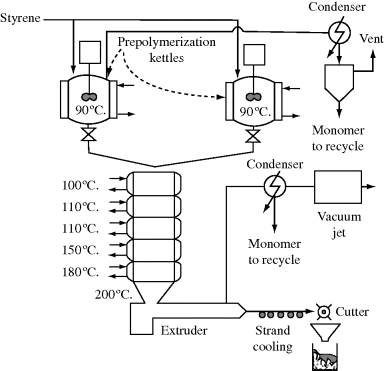
Two types of rectors are used for high-pressure ethylene polymerization: autoclaves and tubular. The former are basically high-pressure CSTRs and the latter are continuous plug-flow reactors. Figure 12.3 illustrates a high-pressure tubular process [6]. The reactors are made of thick-walled pipe, typically from 1 in to 3 in in diameter and from 800 ft to 2500 ft long. They are also heat exchangers, with-heat transfer fluid circulating around the pipe, first to heat the ethylene to reaction temperature and then to remove the large heat of polymerization. Residence times are on the order of a minute, with single-pass conversions of 20% or less. Production rates are in the range of 8–125 million lb/yr per reactor [5].
Separation of polymer from unreacted monomer is basically a matter of reducing the pressure and allowing the monomer to flash off. Given the pressures involved, it is no surprise that the compressors represent a significant fraction of the capital and operating costs of these polymerization processes.
Figure 12.3 Tubular low-density polyethylene process. Reprinted with permission from CHEMTECH 13(4), 223 (1983). Copyright 1983 American Chemical Society.
Bulk polymerization has several advantages:
Among its disadvantages are the following:
12.3 Gas-Phase Olefin Polymerization
Gas-phase polyolefin processes based on Ziegler–Natta catalysts have undergone tremendous growth in the past decade. The popular UNIPOL® fluidized-bed process is shown in Figure 12.4 [6]. It typically operates at pressures in the range of 100–300 psi (7–20 atm) and at temperatures under 100 °C [6]. Monomer, catalyst particles, and hydrogen chain-transfer agent (see Section 10.6) are fed to the fluidized-bed reactor. The upward-circulating monomer suspends the growing polymer particles. The reactors have a characteristic bulge at the top which is a disengaging section. It lowers the velocity of the circulating monomer, keeping the growing polymer particles in the lower reaction zone. The monomer is circulated through a heat exchanger to remove the heat of polymerization. Polymer is discharged from the reactor in granular form, as powdered laundry detergent. It may be used as-is or extruded and pelletized to form the more conventional molding powder.
Figure 12.4 UNIPOL gas-phase polyolefin process. Reprinted with permission from CHEMTECH 13(4), 225 (1983). Copyright 1983 American Chemical Society.
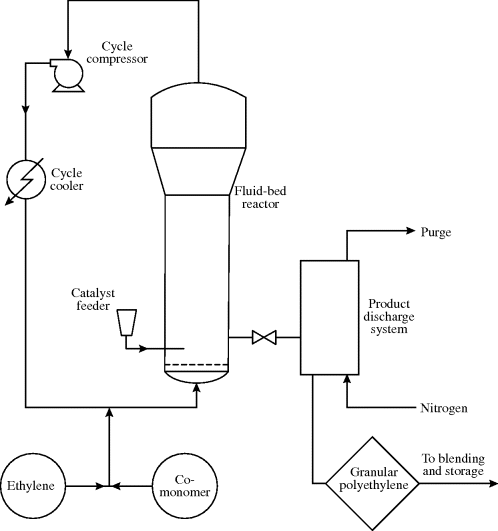
Impact-modified polypropylene is produced by using two reactors in series [7]. The first produces polypropylene particles, which are fed to the second reactor. A mixture of gaseous ethylene and propylene monomers (but no additional catalyst) is fed to the second reactor. The monomers diffuse to the still-active catalyst within the polypropylene particles from the first reactor, where they copolymerize to form a core of ethylene–propylene rubber within a polypropylene shell. This morphology greatly enhances the impact strength over that of polypropylene alone.
Compared to older processes, a number of significant advantages are claimed for the two-stage process above. With modern high-yield catalysts (10,000 g polymer/g catalyst), there is no need to remove the catalyst from the product; thus a difficult and costly step is eliminated. Modern catalysts also produce polypropylene with such a high isotactic content that removal of an atactic fraction, a common step in older processes, is not required. With low pressures, equipment and compression costs are low. No solvent is required. Separation of unreacted monomer is simple. With different catalysts and minor variations in process conditions, virtually the same equipment can be used to manufacture isotactic polypropylene (PP), high-density polyethylene (HDPE), and the so-called linear low-density polyethylene (LLDPE). Overall, major advantages in capital and operating costs, energy requirements, pollution, and superior product properties are cited by the licensors of this process.
12.4 Solution Polymerization
As mentioned earlier, heat transfer problems can be mitigated by using a solvent for polymerization. A solvent can also overcome early polymer vitrification during the reaction. Thus, the addition of an inert solvent to a bulk polymerization mass can minimize many of the difficulties encountered in bulk systems (even though it also decreases the amount of product formed per reactor size). As shown in Figure 12.1, the solvent reduces the tendency toward autoacceleration in free-radical addition. The inert diluent adds its heat capacity without contributing to the evolution of heat and it cuts the viscosity of the reaction mass at any given conversion. In addition, the heat of polymerization may be conveniently and efficiently removed by refluxing the solvent. Thus, the danger of runaway reactions is minimized.
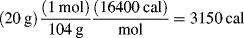
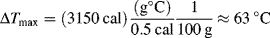
The advantages of solution polymerization are:
Among the disadvantages of solution polymerization are the following:
Solution polymerizations are often used for the production of thermosetting condensation polymers, which are carried to a conversion short of the gel point in the reactor. The crosslinking is later completed in a mold. Such reactions may be carried out in a refluxing organic solvent. The water of condensation is carried overhead along with the solvent vapors. When the vapors are condensed, the water forms a second phase that is decanted before the solvent is returned to the reactor. Not only does this drive the reaction toward higher conversions but also the amount of water evolved provides a convenient measure of conversion (see Example 8.1).
Ionic polymerizations as well as many Ziegler–Natta polymerizations [5] are almost exclusively solution processes. They can be run under conditions such that the polymer product stays in solution, as in the production of stereospecific rubbers. The crystalline polymers polyethylene and isotactic polypropylene are commonly produced at temperatures sufficiently below Tm so that the polymer product is a solid that grows on the catalyst particles as in gas-phase polymerizations. Such processes are known as slurry polymerizations.
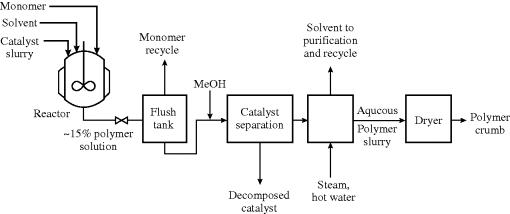
Figure 12.5 sketches a generic process utilizing a Ziegler–Natta catalyst system. Heat removal from the reactor(s) may be accomplished by refluxing the solvent, cooling jackets, external pumparound heat exchangers, or combinations of these. Catalyst deactivation with methanol or acid and separation by filtration or centrifugation are shown, although with modern high-yield catalysts these difficult and expensive steps can be eliminated.
In the Ziegler–Natta solution polymerization process, solvent and unreacted monomer are stripped with hot water and steam and recovered, leaving an aqueous polymer slurry, which is then dried to form a “crumb.” With rubbers, the crumb is compacted and baled; with plastics, it is normally extruded and pelletized. Reactor designs for these processes are interesting and varied [8]. Most processes are continuous operations.
Figure 12.5 Ziegler–Natta solution polymerization process.
12.5 Interfacial Polycondensation
Have you ever seen the example of forming nylon rope in a chemistry demonstration, where the polymer magically appears between two immiscible phases? This is an example of a variation of solution polymerization known as interfacial polycondensation. Besides being used for a “wow” experiment in demonstrations, it has been used in the laboratory for a long time [9], and is also applicable in industrial polymerizations. One monomer of a condensation pair is dissolved in one solvent and the other member of the pair in another solvent (note: this applies to AA and BB monomers, but does not work for AB-type monomers). The two solvents must be insoluble in each other. The polymer is soluble in neither and forms at the interface between them. One of the phases, generally, also contains an agent that reacts with the molecule of condensation to drive the reaction to completion.
An example of such a process is the preparation of nylon 6/10 from hexamethylene diamine and sebacoyl chloride (the acid chloride form of sebacic acid):
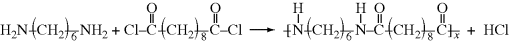
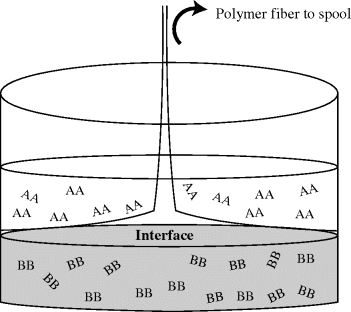
The acid chloride is dissolved in, for example, CCl4, and the diamine in water, along with some NaOH to soak up the HCl that splits out during the reaction (Figure 12.6). In the classic “rope trick” demonstration, the aqueous layer is gently floated on top of the organic layer in a beaker. The reactants diffuse to the interface, where they react rapidly to form a polymer film. With care, the film can be withdrawn from the interface in the form of a continuous, hollow strand that traps considerable liquid. New polymer forms at the interface as the formed polymer is removed and replenished with fresh monomer on both sides. Commercially, it is probably easier to simply stir the phases together.
Figure 12.6 Interfacial polymerization of a nylon. The upper (organic) phase contains sebacoyl chloride (AA monomer) while the lower (aqueous) phase contains hexamethylene diamine (BB monomer). The reaction occurs where the two phases meet.
A major advantage of this technique is that these reactions usually proceed very rapidly at room temperature and atmospheric pressure, in contrast to the long times, high temperature and vacuum pressures usually associated with polycondensations. Another advantage of interfacial polymerizations for step-growth polymerizations is that the stoichiometry of an AA, BB reaction matches at the interface, therefore, we can avoid the problems noted in Chapter 8 with stoichiometric imbalance causing very low molecular weight polymers to form. These must be balanced against the cost of preparing the special monomers, such as the acid chloride above, and the need to separate and recycle solvents and unreacted monomers.
12.6 Suspension Polymerization
Under the discussion of bulk polymerization in Section 2, it was mentioned that one of the ways of facilitating heat removal was to keep one dimension of the reaction mass small. This is carried to its logical extreme in suspension polymerization by suspending the monomer in the form of droplets 0.01–1 mm in diameter in an inert, nonsolvent liquid (almost always water). To achieve these suspended droplets, some physical mixing energy must be added (note that no surfactant is used here, so the droplets do not become an emulsion). However, similar to emulsion polymerization, suspended droplets behave as individual bulk reactors with dimensions small enough such that heat removal is not a problem. The heat can easily be soaked up and removed from the low-viscosity, inert suspension medium.
An important characteristic of these systems is that the suspensions are thermodynamically unstable, and must be maintained with agitation and suspending agents. A typical feed might consist of

Two types of suspending agent are used. A protective colloid is a water-soluble polymer whose function is to increase the viscosity of the continuous water phase. This hydrodynamically hinders coalescence of the monomer droplets, but is inert with regard to the polymerization. A finely divided insoluble inorganic salt such as MgCO3 may also be used. It collects at the droplet–water interface by surface tension and prevents coalescence of the droplets upon collision. Also, a pH buffer is sometimes used to help stability.
The monomer phase is suspended in the water at about a ½ to ¼ monomer/water volume ratio. The reactor is purged with nitrogen and heated to start the reaction. Once underway, temperature control in the reactor is facilitated by the added heat capacity of the water and the low viscosity of the reaction mass, essentially that of the continuous (water) phase, allowing easy heat removal through a jacket heat exchanger.
The size of the product polymer beads depends on the strength of agitation, as well as the nature of the monomer and suspending system. Between 20% and 70% conversion, agitation is critical. Below this range, the organic phase is still fluid enough to redisperse and above it, the particles are rigid enough to prevent agglomeration; but if agitation stops or weakens between these limits, the sticky particles will coalesce or agglomerate in a large mass and finish polymerization that way. Again quoting Schildknecht [10], “After such uncontrollable polymerization is completed in an enormous lump, it may be necessary to resort to a compressed air drill or other mining tool to salvage the polymerization equipment.”
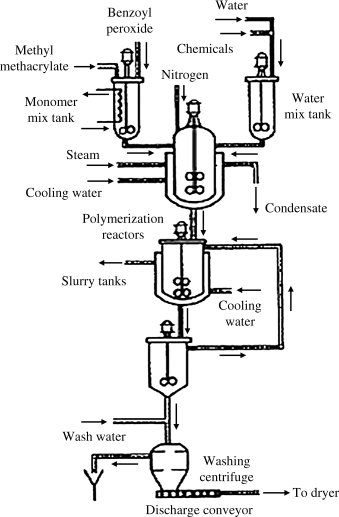
Figure 12.7 [11] shows a typical suspension polymerization process. Since any flow system is bound to have some relatively stagnant corners, it has been impractical to run suspension polymerization continuously on a commercial scale. The stirred tank reactors are usually jacketed, stainless, or glass-lined steel kettles of up to 50,000-gal capacity. The polymer beads are filtered or centrifuged and water-washed to remove the protective colloid and/or rinsed with a dilute acid to decompose the MgCO3. The beads are quite easy to handle when wet, but tend to pick up a static charge when dry, making themselves cling to each other and everything else. These beads can be molded directly, extruded and chopped to form molding powder, or used as-is, for example, as ion-exchange resins or the beads from which polystyrene foam cups and packing peanuts are made.
Ion-exchange resins are basically suspension beads of polystyrene crosslinked by copolymerization with a few percent divinyl benzene (a crosslinking agent), which are then chemically treated to provide the charged groups for functionality in separation and purification. To reduce mass-transfer resistance in the ion-exchange process, an inert solvent may be incorporated in the organic suspension phase. When polymerization is complete, the solvent is removed, leaving a highly porous bead with a large internal surface area (“macroreticular”). The foam beads used in coffee cups are made from linear polystyrene containing an inert liquid blowing agent, usually pentane. Pentane may be added to the monomer prior to polymerization, but more commonly, it is added to the reactor after polymerization and is absorbed by the polystyrene beads. When exposed to steam in a mold, the beads soften and are foamed and expanded by the volatilized blowing agent to form the familiar cups and other foam items.
Figure 12.7 Suspension polymerization of methyl methacrylate. Excerpted by special permission from Reference 11. Copyright © 1966 by McGraw-Hill, Inc., New York, NY 10036.
Commercially, suspension polymerization has been limited to the free-radical addition of water-insoluble liquid monomers. With a volatile monomer such as vinyl chloride, moderate pressures are required to maintain it in the liquid state. It is possible, however, to perform inverse suspension polymerizations with a hydrophilic monomer or an aqueous solution of a water-soluble monomer suspended in a hydrophobic continuous phase.
The major advantages of suspension polymerization then are as follows:
The disadvantages include the following:
12.7 Emulsion Polymerization
Emulsion polymerization was introduced while discussing some advanced polymerization techniques in Chapter 9, but there are some important characteristics of emulsion polymerization that merit discussion for industrial-scale processes. When the supply of natural rubber from the East was cut off by the Japanese in World War II, the United States was left without an essential material (tires for airplanes, especially at that time, needed replacement after every few landings). The success of the Rubber Reserve Program in developing a suitable synthetic substitute and the facilities to produce it in the necessary quantities is one of the all-time outstanding accomplishments of chemists and engineers, comparable to the Manhattan (atomic bomb) Project and the production of penicillin in its significance at the time. The styrene–butadiene copolymer rubber GR-S (government rubber-styrene) or SBR (styrene–butadiene rubber) as it is now called is still the most important synthetic rubber, and much of it is still produced, along with a variety of other polymers, by the emulsion polymerization process developed during the war.
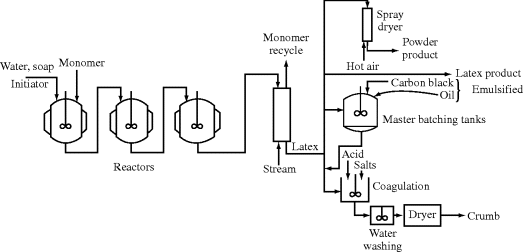
The theory behind the emulsion reaction is discussed in Chapter 9. A generic commercial process is outlined in Figure 12.8. The reactors are usually stainless or glass-lined steel tanks, similar to those used for suspension polymerization. In contrast to suspension polymerization, however, a proper emulsion is thermodynamically stable (due to the surfactant or emulsifying agent), and therefore emulsion polymerizations can be run continuously. Newer processes often have several continuous stirred-tank reactors (CSTRs) in series.
Figure 12.8 Schematic of an emulsion polymerization process.
The product of an emulsion polymerization is known as a latex—polymer particles on the order of 0.05–0.15 μm stabilized by the surfactant. These latexes are often important items of commerce in their own right (think of latex paints or white glue). This process can also be modified slightly for microencapsulation purposes, with the polymer forming a shell surrounding an inner fill material (these capsules are then used in controlled release formulations in a number of fields).
Where the polymer must be mixed with other materials, the process of master batching sometimes allows this to be done conveniently and uniformly. In rubber technology, carbon black and oil are emulsified and mixed with the rubber latex and then the mixture is coagulated together, giving a uniform and intimate dispersion of the additives in the rubber.
The increased recognition of possible adverse physiological effects of even small amounts of residual monomer makes emulsion polymerization attractive in certain applications. The extremely small size of the latex particles provides a very short diffusion path for the removal of small molecules from the polymer, for example, by steam stripping, permitting very low levels of residual monomer to be obtained.
For many applications, the solid polymer must be recovered from the latex. The simplest method is spray drying (a process that produces a fine mist from the solution while evaporating the solvent rapidly with hot air); but since no attempt is made to remove the surfactant, the product is an extremely “dirty” polymer. A latex may be “creamed” by adding a material such as acetone that is at least a partial solvent for the polymer. This makes the particles sticky and causes some agglomeration. The latex is then coagulated by adding an acid, usually sulfuric, which converts the surfactant from its sodium salt to its insoluble hydrogen form, and/or by adding an electrolyte salt, which disrupts the stabilizing double layer on the particles, causing them to agglomerate through electrostatic attraction. The former method leaves much insoluble material adsorbed on the particle surfaces, but in some applications, this may even be beneficial. For example, fatty acids from the surfactant act as lubricants in tire manufacturing. The coagulated polymer “crumb” is then washed, dried, and either baled or processed further.
As with suspension polymerization, commercial emulsion polymerization has pretty much been restricted to the free-radical solution addition of water-insoluble, liquid monomers (with volatile monomers such as butadiene and vinyl chloride, moderate pressures are required to keep them in the liquid phase). Inverse emulsion polymerizations, with a hydrophilic monomer phase dispersed in a continuous hydrophobic base are, however, possible.
The advantages of emulsion polymerization are as follows:
The disadvantages include the following:
12.8 Summary
Much as any industrial chemical plant must consider numerous factors beyond the chemistry of a reaction, industrial polymerization processes require adequate control of temperature (especially to remove large exothermic heats of reaction), provide mixing, and involve appropriate separation techniques. Bulk polymerizations offer pure polymers in a reaction vessel, allowing the greatest possible yield per reactor volume. However, disadvantages such as vitrification, the Tromsdorff effect, and extraction of unreacted monomer have led to other polymerization methods, including solution, suspension, and emulsion polymerization.
Problems
1. Schulz and Harborth, G. Makromol. Chem. 1, 106 (1947).
2. Schildknecht, C.E., Polymer Processes, Interscience, New York, 1956, p. 38.
3. Wohl, M.H., Chem. Eng. August 1, 1962, p. 60.
4. Thomas, J.C., Soc. Plast. Eng. J. 23 (10), 61 (1967).
5. Albright, L.F., Processes for Major Addition-Type Plastics and Their Monomers, McGraw-Hill, New York, 1974.
6. Karol, F.J., CHEMTECH 12 (7), 222 (1983).
7. Haggin, J., Chem. Eng. News March 31, 1986, p. 15; Bisio, A., Chem. Eng. Progr.85 (5), 76 (1989).
8. Gerrens, H., CHEMTECH 12 (7), 434 (1982).
9. Morgan, P.W., Soc. Plast. Eng. J. 15 (6), 485 (1959).
10. Schildknecht, C.E., Polymer Processes, Interscience, New York, 1956, p. 94.
11. Guccione, E., Chem. Eng. June 6, 1966, p. 138.