The Scientific Papers of Biava, et al.
INTRODUCTION
In part one we described the fundamental concept that underlies the promise of a new era in the history of healing and medicine and presented the discovery that is both a confirmation of the validity of this concept and a practical and beneficial breakthrough in its application. Now part two offers a pragmatic follow-up to these considerations. It is intended primarily for medical researchers and practitioners, but it is not without direct interest also to lay persons.
The same as part one, the purpose of the material presented in part two is twofold. On the one hand, it is to disclose the biophysical and biological specifics of the medical breakthrough, enabling practicing physicians to apply it in full knowledge of its scientific basis. On the other, the information communicated here is propaedeutic to opening the full potentials of the breakthrough, so as to usher in a new and beneficial era in the history of medicine and the art of healing. This is much needed, because the challenge to healing and medicine remains critical: despite great advances in the biochemical applications of contemporary medicine, tumoral and other chronic degenerative diseases remain rampant. However, the potentials of the new discovery are enormous. Creative researchers could find new applications of the discovery and find further areas and research domains to which the discovery could be extended.
Practical application and creative development are twin aspects of the challenge of information medicine to medical research and practice. They are priority domains both of contemporary medical science, and of therapeutic practice.
We hope that the scientific papers assembled in this part will prove to be of value to researchers and to practitioners in responding to the challenge and will help to usher in a much needed bright era in the maintenance of health and the healing of disease.
EDITORIALS
The two editorials included in this section offer overviews of new treatment possibilities, following close on the heels of rapid advances at the cutting edge of biochemical research into cell proliferation and differentiation. The new and ever-growing insight into the workings of stem cells led to a greater understanding of the behavior of tumor cells that exhibit similar characteristics and led to the discovery of practical ways of healing, or at least slowing, invasive cancerous growths. These are promising results for the treatment of a variety of other chronic diseases.
Complex Therapeutical Approaches to Complex Diseases

From Current Pharmaceutical Biotechnology, 2015, Volume 16, Number 9
Guest Editor: Pier Mario Biava
This article represents the editorial of a special issue of the journal Current Pharmaceutical Biotechnology, whose title and subject were chosen by Biava as Guest Editor. This special issue consists of various articles, some of which are written by Biava and his collaborators, and other papers by various researchers in different parts of the world. The editorial illustrates the content of this special issue that describes the change of scientific paradigm needed to treat complex diseases such as cancer, chronic inflammatory, and degenerative diseases. The reductionist approach in addressing these complex pathologies reveals all its limitations, as shown by the poor clinical results obtained using this approach. Hence the need to tackle these diseases by adopting a complex and holistic approach.
In the last decades the enormous advances in genetics and biology have made clear that cancer is a very complex disease sustained by many genetic and epigenetic alterations. They activate a great deal of pathological molecular pathways governing relevant physiological processes. In 2000 Hanahan and Weinberg identified six hallmarks of cancer diseases: 1) self-sufficiency in growth signals, 2) insensitivity to antigrowth signals, 3) limitless replicative potential, 4) ability to evade apoptosis, 5) support to angiogenesis, 6) invasion of tissue and ability to give metastasis. In addition, the lost differentiation of cancer cells was proposed recently by Biava P. M. (Current Medicinal Chemistry 2014) to the hallmarks of Hanahan and Weinberg.
Moreover, it was demonstrated that for normal cells to become cancerous, transformation also depends on a complex network of surrounding micro-environmental signals from cell-to-cell “cross-talking” or from soluble extracellular factors. For example, it has been demonstrated that inflammatory cells can sustain, instead of fighting tumor growth. Thus, the whole context is decisive in determining cell fate in line with a complex view of cell biology.
The current special issue describes why chronic diseases, including not only cancer, but also the metabolic syndrome, chronic inflammation, chronic degenerative diseases, etc. make diagnosis, prevention, and targeted therapeutic treatment particularly difficult. Recognition that chronic inflammation may induce genetic, neuro-endocrine, immune, and metabolic changes in a series of diseases is useful for designing new ways for prevention and treatment. A new approach may include reprogramming suppressive immune cells and pro-inflammatory mediators factors, restoring in this way the balance of neuro-endocrine-immune and metabolic network systems disrupted by chronic inflammation.
Differentiation factors, reprogramming therapies and immunotherapy are innovative biological means with more systemic approaches to cancer and chronic degenerative diseases treatment. In particular this special issue records some articles that highlight how growth and differentiation factors taken from a Zebrafish embryo could address the fate of normal and pathological (stem) cells. In fact, these factors taken during the early developmental stage of a Zebrafish embryo may represent a useful tool to enhance stem cell expression of multipotency and activate both telomerase-dependent and telomerase-independent antagonists of cell senescence. On the contrary, these factors taken during the late developmental stages decrease cell viability and direct cells toward senescence. This strategy did not require cumbersome gene manipulation through viral vector mediated gene transfer, or expensive synthetic chemistry. This data shows for the first time that it is possible to direct human mesenchymal stem cells toward different and opposite directions, tuning in specific, physiological ways the regulation of different genes. This is possible only when the specific networks of factors are sufficiently complex because single substances are not able to obtain any significant results.
These data lead us to consider a major shift in scientific paradigm (from reductionism to complexity) for preparing new treatments for chronic degenerative diseases. In fact, these diseases entail unexpected degrees of complexity and disregulation, making the single-moleculeto-specific-target paradigm totally obsolete and inadequate. Rather, only a systemic approach can be envisioned as a successful strategy to deal with such complexity. It is believed that time is ready for a “trans-disciplinary approach” in the treatment of degenerative diseases so that a new culture of collaboration can promote many important innovations and new therapeutic approaches.
Reprogramming of Normal and Cancer Stem Cells
From Current Pharmaceutical Biotechnology, 2011, Volume 12, Number 5
Guest Editor: Pier Mario Biava
This paper represents the editorial of a special issue of the journal Current Pharmaceutical Biotechnology, whose title and subject were chosen by Biava as Guest Editor. This issue consists of fifteen articles, some of which were written by Biava and collaborators, and others by researchers of various scientific institutions. The editorial illustrates the content of this special issue.
Over the last decade there has been an exponential rise in our understanding of the biochemical mechanisms controlling cell proliferation and differentiation. While the four transcription factors Oct4, Sox2, Klf4, and cMyc have shown to be sufficient to induce pluripotency in fibroblasts, there has in addition been much research into the mechanisms and pathways of cell differentiation and the specific properties of stem cells, namely their plasticity and capacity for trans-differentiation. These studies have allowed progress at a very fundamental level, with the prospect of further progress—until recent years quite unimaginable—in the field of reparative, regenerative, and transplant medicine. In fact, from the present time, the genetic engineering production of regulatory factors identified through such research, has allowed the production of new tissues and a new category of cell therapy products, in which the main biological action is carried out by cells or tissues, albeit in the presence of organic or inorganic matrices or coatings. Examples of this type of product are anti-tumor vaccines, in vitro cultivated skin, products made of structural and cellular elements for the reconstruction of bones, cartilages, teeth, etc.
From the best, most analytical and detailed characterization of stem cells, then, it has become clear that some tumor cell behaviors—that have a crucial role in determining their malignity—can be attributed to the presence of cells with characteristics similar to those of stem cells. The field of cancer research is consequently also witnessing a surge in studies designed to identify the metabolic pathways common to tumor and stem cells. This will in turn cast light on which micro-environment factors can direct these pathways toward differentiation and induce cancerous cells to behave less aggressively. From this point of view, over recent years there has been a lively return to studies that were very significant in the 70s and 80s, on the role of the embryonic micro-environment in conditioning tumor cell behavior toward normal phenotypes. This research is now underway, and will in all probability lead to important results over the next few years. Against this background, this special issue on “Reprogramming of Normal and Cancer Stem Cells” focuses on research in terms of conditioning the fate of normal and tumor stem cells with a view to new prospects for the therapies. The issue therefore begins with articles covering the possibility of reprogramming normal stem cells, including through use of biomaterials, and goes on to consider what characteristics of tumor stem cells can allow them to be identified and studied. This is followed by a series of further articles illustrating the role of the micro-environment in conditioning the fate of a tumor cell. A number of metabolic pathways characterizing and common to both stem and tumor cells are examined, in order to gain a better understanding of the possibilities of conditioning the fate of both cell types; in addition, the role played by infectious and inflammatory diseases in the genesis of tumor diseases is also considered.
Today, in fact, we know that inflammatory processes can support rather than hinder tumor growth, and also that pro-inflammatory cytokines can promote tumor proliferation, inhibiting the cell pathways that are able to block the neoplastic growth. The special issue goes on with a series of articles taking a close look at the specific role played by the micro-environment in conditioning the destiny of the tumor stem cells present in some tumors, for example breast and retinoblastoma tumor, and the role played by the use of normal stem cells in treating disorders such as hematological diseases. One article also considers the risks run by some reprogramming techniques: for example, creating embryo cells via parthenogenesis can give rise to tumors. The issue continues with various articles illustrating in close detail the role of the embryonic micro-environment in conditioning the destiny of tumor cells. In this context, one review takes a look at general aspects, while others consider aspects that can help clarify the mechanisms underlying the capacity of factors of this type of microenvironment to reprogram a tumor cell. One mathematical model sets out from a description of the state of cell differentiation, making use of existing data from studies of tumor growth slowing, linked to the use of such factors, with the goal of shedding light on aspects such as fitness, dosage, and administration time for the differentiation factors on improvement in tumor inhibitory response.
Other articles illustrate a number of clinical cases of full regression of hepatocellular carcinomas in intermediate-advanced stages observed following administration of stem cell differentiation factors, and describe the molecular mechanisms that might explain these inhibitory responses on the tumor growth. It should be noted that the randomized and controlled clinical studies launched to date using stem cell differentiation factors are limited to patients with intermediate-advanced stage of hepatocellular carcinomas where other therapies were no longer possible. These factors are at present used only for hepatocellular carcinomas, since it has been demonstrated that substances capable of slowing one tumor’s growth may be inefficacious for another type. Finally, it is important to note that research into the possibility of reprogramming normal and tumor stem cells requires a complex approach to the issue. In fact, the problem requires the study of networks of substances and genes involved in the reprogramming phase, demanding skills in a variety of different areas of research, not simply of medical/biological, but also mathematical/computational and modeling, in view of the complexity and non-linearity of the processes being studied.
A paradigm shift is underway, and the future will witness our engagement in increasing numbers of scientific studies requiring crossdisciplinary skills. The new paradigm and the new ideas were well understood many years ago by Professor John Klavins, who has been a long-time President of the International Academy of Tumor Marker Oncology. Professor Klavins has always sustained my studies on reprogramming cancer cells, though the possibility of controlling tumor growth by using reprogramming factors was not considered realistic at the time I began studying it. I wish to dedicate the following reports to my friend John Klavins.
THE PRINCIPAL REPORTS
Cancer and Cell-Differentiation: A Model to Explain Malignancy
Journal of Tumor Marker Oncology, Fall 2002, Volume 17, Number 3
Pier Mario Biava, Daniele Bonsignorio
The article below is one of several articles in a special issue of the Journal of Tumor Marker Oncology, the scientific official journal of the International Academy of Tumor Marker Oncology. This special issue contains scientific articles written by Biava et al., and the article below is the first article in the issue. It describes a model of cancer, the result of previous research carried out by Biava et al. in the laboratory. In this model tumor cells are described as cancer stem-like cells, which can be reprogrammed using the factors isolated from the embryo micro-environment.
Introduction
The evidence obtained from studying the interactions between tumor cells and embryonic tissues suggests that tumor development in an embryo is reduced or suppressed when the processes of differentiation are in progress.1, 2
In fact, the administration of known carcinogens in the course of cell differentiation in an embryo causes malformations in offspring, but not tumor induction. Once organogenesis is complete, the frequency of tumor induction rises with a concomitant decrease in the rate of malformations.3, 4, 5
These findings could indicate that cancer can be viewed as a developmental deviation susceptible to being controlled by regulators of cells differentiation.
On the basis of this background some experiments on animals were made. These previous experiments have demonstrated that factors present during cell differentiation are able to stop or delay tumor growth in animals. These factors are present in the pregnant uterus of mammals6 and in the embryos of ovipari.7 More recent experiments in vitro showed that pregnant pig and mouse uterus extracts slow down the proliferation rate of several established human tumor cell lines.8 It was clarified that the abnormal growth of cell clones during embryo organogenesis in mammals is prevented by low-molecular weight substances present in pregnant uterus microenvironment. In fact a 5kDa fraction isolated in our laboratory from the pregnant uterus of mammals, named “Life-Protecting Factor,” inhibited the cell proliferation curves of all treated human tumor cell lines as well as the crude pregnant uterus extracts. Therefore, the interactions between mother and embryo seem to be important for the normal development of the embryo and for preventing a pathological cell growth. The embryo itself seems to prevent the abnormal multiplication of tumor cells. In fact it was demonstrated that different tumor cell lines responded with a significant slowing of the proliferation when treated with extracts taken during the stages of cell differentiation, while no slowing effect was observed when they were treated with the extracts taken from a merely multiplicative stage.9
Thus, cell differentiation is a key process in understanding the behavior of both normal and tumor cells. The fact that embryonic development and tumorigenesis are closely correlated is now accepted: they both share several pathways and molecules, which are able to regulate some important genes of the cell cycle. In fact, the main effect of the in vitro treatment of tumor cell lines with the extracts of the oviparous embryos is the activation of p53 expression, as observed by immuno-histochemical and flow cytometry techniques after the treatment of different tumor cell lines with the extracts of fish embryos.7 In addition we record in another article of this issue of the journal the induction of a post-translational regulation of pRb by the Zebrafish embryonic extracts, which is probably responsible for the observed slowing down of the kidney adenocarcinoma proliferation curves in vitro. Embryonic differentiation and tumorigenesis, although they share several metabolic pathways, seem to be opposite processes: the same molecules, which cause cell differentiation in the embryo, seem to be able to oppose the cancer growth. In order to elucidate the mechanisms involved in these two different processes, it is necessary to illustrate an outline and a model of embryonic differentiation and cancerogenesis.
An Outline and a Model of Embryonic Differentiation
Shortly after fertilization, generally in the middle-blastula-gastrula period, the processes of differentiation begin. There are three postulates of cell differentiation:
- Every cell nucleus contains the complete genome established in the fertilized egg. In molecular terms the DNAs of all differentiated cells are identical.
- The unused genes in the differentiated cells are not destroyed or mutated, and they retain the potential for being expressed.
- Only a small percentage of genome is expressed in each differentiated cell and a portion of the RNA synthesized is specific for that cell type.
Briefly, the differentiation, which leads pluripotent embryonic stem cells to specialization, consists in a differential regulation of genes that restricts the expressed genome. The gene configurations of the cells, which rise after each stage of differentiation, differ from the progenitors for some thousands of expressed genes.
Regulators are generally factors that cooperate in a network, and this network promotes and controls the differentiation of each cell type. All cells communicate with each other through this network.
Cell differentiation is a very complex process that takes place at different levels:
A) a differential gene transcription that regulates how the nuclear genes are transcripted into RNA
B) a selective nuclear RNA processing that regulates how the transcript RNAs get into cytoplasm to become messenger RNAs
C) a selective messenger RNA translation that regulates how messenger RNAs in cytoplasm get translated into proteins
D) a differential modification of proteins that regulates how proteins are allowed to function in the cells
Transcription factors are very important in controlling the differential expression of genes, but in eukariotes selective nuclear RNAs’ processes are more important. These selective processes clarify how the same gene can produce two different proteins in different cells or in the same cell at different times. Besides selective degradation, otherwise selective stabilization of messenger RNA is responsible for further specifications of proteins.
Today we have a dynamic vision of the regulation of gene expression. We think that a gene is not an independent and autonomous center of control of the synthesis of proteins; a gene is also controlled directly or indirectly by synthesized proteins.
Certainly, the interactions between nucleus and cytoplasm and between cytoplasm and microenvironment are so wide that they constitute a marvelous example of complexity. Developing embryo is an excellent example of what is called “complex adaptive systems.” In fact an embryo: 1) is a network of many cells acting in parallel, 2) has many levels of organization that are constantly revising and rearranging, 3) has an implicit prediction encoded in its genes and 4) is always in transition and is characterized by perpetual novelty.
Cell differentiation can be better understood by a model described here, which is consistent with the real situation. In this model the number of final gene configurations of cells in the human body (number of types of completely differentiated cells) can be predicted, if we retain that each kind of progenitor cells produces three different daughter cells (different gene configurations) and that there are five stages of differentiation. This corresponds to the real situation: in fact the embryo, after segmentation (morula), differentiates in three layers: ectoderm, endoderm, and mesoderm. Gametes differentiate in a different pathway than somatic cells. After gastrulation, there are four more stages of cell differentiation. For example, on the basis of precise data about some cell lines, like hematopoietic cells, the stages of differentiation are: a) stem cell stage, committed stem cell stage, c) differentiating cell stage, d) differentiated cell stage. If we include the ectodermal, endodermal, and mesodermal cell lines, the stages of differentiation are five. Therefore, the mathematical formula to calculate the number of differentiated cells is:
N = 35
The result is 243, which is the number of the various somatic differentiated cells. To calculate the final number of the differentiated cells we have to add the number of gametes. The sexual cells are 5 in men (spermatogonium, spermatocyte of the first order, spermatocyte of the second order, spermatid, spermatozoon) and 4 in women (ovogonium, ovocyte of the first order, ovocyte of the second order, egg cell). The final result is 252, which is the number of the different kinds of cells in humans.
Cancer as Undifferentiated Mutated Cells: A Model to Explain Malignancy
Tumoral transformation of normal cells is a process relying on a minimal number of stochastic mutational events, comprised between 4 and 7.10 If mutations are introduced into normal cells in a non-stochastic manner, i.e., triggering at precise genes, that number is even reduced.11 Preferential targets of these mutations are genes encoding for key-role effectors of cell cycle regulation and cell signaling, and for growth factors and their receptors; mutations are either gain-of-function, in case of proto-oncogenes, or loss-of-function, in case of tumor suppressor genes.
Anyway, defining the tumoral transformation of a cell simply as an outcome of a sum of gene mutations may be reductive. For normal cells to turn to cancerous, transformation depends also on a complex network of surrounding micro-environmental signals, coming from cell-to-cell “cross-talking” or from soluble extracellular factors. For example, it has been demonstrated that fibroblasts adjacent to prostate epithelium carcinoma cells are able to direct tumor progression,12 that stromal neighbor cells are able to promote malignant transformation of immortalized keratinocytes by releasing proliferative stimuli,13 and that inflammatory cells can sustain instead of fight tumor growth.14 Even pro-inflammatory cytokines were shown to promote cancer cell proliferation by inhibiting tumor suppression pathways.15 Thus, the whole context is decisive in determining cell fate according to a “heterotypic” view of cell biology, as it was called in a recent review.16
According to this view, defining tumorigenesis as a microevolutive process is no more a hazard. A cancer cell acquires, as consequence of this process, some capabilities: 1) self-sufficiency in growth signals, 2) insensitivity to antigrowth signals, 3) capability in evading apoptosis, 4) limitless replicative potential, 5) capability in sustaining angiogenesis, 6) capability in invading tissues and in metastasizing. The acquisition of the enumerated capabilities during the course of tumor progression is usually the consequence of a great variability on the way that cells take to becoming malignant. Nonetheless the hypothesis advanced here is that independent of how the steps in these genetic pathways are arranged, the development of all types of human tumor cells is governed by a final common process. Some authors define “early crisis” and “genetic catastrophe” of a cell as steps that enable an evolving population of premalignant cells to reach malignancy.17 These crises, during which a telomere dysfunction and a DNA damage take place, give rise to different possibilities: 1) cells die, or 2) cells survive after each crisis. The final results are adaptive responses and telomere maintenance in the case of cells survival. Those surviving malignant cells have a) not only increased the level of telomerase, but b) have also activated proto-oncogenes or oncogenes, c) produce growth factors, d) are insensitive to anti-growth signals, e) have several surface antigens, also known as oncofetal antigens, maintained during philogeny, most of which have been identified in the last 30 years.18, 19, 20, 21, 22, 23, 24, 25,26, 27 In other terms the cells that survive a genetic instability period become malignant through the achievement of a new stable genes configuration very similar to those present in an embryo during the periods of multiplication.
In fact, cancer cells and embryonic cells share some molecular pathways and their key-role effectors: for example, the APC/b catenin/TCF/Wnt pathway28, 29 and the Hedgehog/Smoothened/Patched pathway.30 Whereas in the embryonic development these pathways lead cells to successful differentiation, in tumorigenesis their mutated counterparts lead cells to a constant multiplication. This happens because a cancer cell is an undifferentiated cell, in which the mutations present in its genome prevent the cell from completing the whole program of differentiation and development. It is stopped in a step of multiplication, comprised between two stages of differentiation. A cancer cell can be defined as an “undifferentiated mutated cell,” in which the program of differentiation and multiplication are uncoupled. It is like a computer in loop, repeating always the same instructions. Cancer is an example of deterministic chaos. It is a branching process, that conduces a cell, since it does not die, to a rampant genetic instability: the final attractor is a new stable “gene configuration” similar to that present in an embryo during the steps of multiplication, comprised between two stages of differentiation. In this hypothesis, considering the model of cell differentiation previously mentioned, the number of different types of cancer derived from somatic cells can be predicted by the formula:
N = 3 + 32 + 33 + 34 = 120
In order to calculate the final number of different kinds of tumors, it is necessary to add the number of tumors coming from sexual cells and from different embryonic tissues (teratocarcinoma, embryonic carcinoma, corioncarcinoma). Therefore the final amount of all different kinds of tumor is about 130. With regard to malignancy it has to be considered that the most aggressive tumors are represented by cells with “gene configurations” present at early stages of differentiation that carry out the program of multiplication with impressive speed. Besides, it has to be remembered that the current classification of tumors is redundant, because it does not consider that the most malignant types of tumors are constituted by kinds of cells, which have the same “gene configurations.” Finally it has to be remembered that some types of tumors are constituted by different cell clones with “gene configurations” coming from different stages of differentiation.
The Regulation of Cancer Growth: A Model of Complexity
The model of cancer as proposed above is not merely theoretical, but relies on the results of the experiments performed in our lab. Those experiments have shown that molecular factors present during precise stages of cell differentiation are able to inhibit tumor growth. This was demonstrated both in vivo on Lewis cancer and in vitro on several human tumor cell lines. On the contrary, substances present during merely proliferative stages are not effective in delaying the growth curves of several types of tumors. Thus, cell differentiation is a key process for elucidating the behavior of both undifferentiated normal and tumor cells. The mechanisms by which the events of cell differentiation take place rely on a multigenic regulation, so that a more differentiated cell differs from a less differentiated one for the expression of a great number of genes. Furthermore, it has to be marked that, according to the above model, tumor cells have lost an important portion of the program of cell differentiation in a progressive manner.
So said, if ultimately the aim is not the destruction of the tumor cell, but its regulation, it is clear that the goal can be achieved only by providing the cell with all the factors that are able to bring it to differentiation. These factors can be found, but only when life is forming. In fact, during organogenesis the whole repertoire of regulatory molecules is present, which includes 1) DNA transcriptional factors; 2) nuclear RNA selection factors; 3) mRNA translational factors; 4) post-translational protein regulatory factors. As shown, it is possible to use these factors for the genic regulation of tumor cells. A p53-mediated transcriptional regulation and a pRb post-translational regulation were demonstrated, depending on the type of tumor. Thus, it was demonstrated that it is possible to regulate tumor cells, by-passing mutations that give rise to malignancy. This happens only when the network of differentiation is complete enough.
From this point of view it is necessary to focus on the microenvironments and the networks that constitute the biological structures, rather than the single subjects of punctual mechanisms. This does not mean that the research of molecular mechanisms should be left behind, only that there is a need to bring the single partial mechanisms to a synthesis. Indeed, the difficulty of bridging the gap to a new scientific paradigm, that is, shifting our views from reductionism to complexity, has been the main hindrance to a deeper and more complete knowledge of cancer. While studies and researches on stem cells differentiation are proceeding worldwide, the scientific community is ready to accept a change of paradigm. In fact, those studies will be able to show that the mechanisms of differentiation depend on specific differentiating networks. The embryonic microenvironment during precise stages of development is fundamental not only for the differentiation of the normal stem cells but also for the differentiation of tumor cells. The embryo, during organogenesis, is never affected by carcinogenetic processes because, while the life program is under transcription, systems of correction in case of mutations are also active. In fact, it was demonstrated that during cell differentiation the administration of known carcinogens fails to induce the growth of tumors, perhaps because the genome control system is always working.
Recent studies claim that p53 function in the embryo is to prevent malformations, so that some authors have called p53 “guardian of the babies,” as a gene that suppresses the onset of malformations. Anyway, when the stress is too severe and the mutations are too numerous, p53 is no longer able to repair DNA and provokes apoptosis in all cells. These processes also occur in tumor cells when p53 is active. In these regards, tumor cells are similar to mutated embryonic cells.
References
1. Einhorn L. Oncodev Biol Med (1982), 4:219–229.
2. Lakshmi MS & Sherbert GV. Embryonic and Tumor Cells Interactions. Karger. Basel 1974: 380–399.
3. Brent RL. Teratology (1980), 21:281–298.
4. Rice JM. Teratology (1973), 8:113–125.
5. Tomatis L, Mohr V. Transplacental Carcinogenesis. IARCSci. Publ. no.4 Lyon 1973.
6. PM Biava, A Fiorito, C Negro & M Mariani. Cancer Lett (1988), 41:265–270.
7. PM Biava & A Carluccio. J Tumor Marker Oncol (1997), 4:9–15.
8. PM Biava, D Bonsignorio & M Hoxha. J Tumor Marker Oncol (2000), 15:223–233.
9. PM Biava, D Bonsignorio & M Hoxha. J Tumor Marker Oncol (2001), 16:195–201.
10. Renan MJ. Mol Carcinogenesis (1993), 7:139–146.
11. Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW & Weinberg RA. Nature (1999), 400:464–468.
12. Olumi AF, Grossfeld GD, Hayward SW, Carroll, PR, Tlsty TD & Cunha GR. Cancer Res (1999), 59:5002–5011.
13. Skobe M & Fusenig NE. Proc Natl Acad Sci USA (1998), 95:1050–1055.
14. Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Cughey GH & Hanahan D. Genes Dev (1998), 13:1382–1397.
15. Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ & Beach DH. J Exp Med (1999), 190:1375–1382.
16. Hanahan D & Weinberg RA. Cell (2000), 100:57–70.
17. Chin L, Artandi SE, Shen Q, Tam A, Lee S-L, Gottlieb GJ, Greider CW & DePinho RA. Cell (1999), 97:527–538.
18. LeMevel BP & Wells SA Jr. Nature (New Biol), 244 (136): 183–4 (1973).
19. Shah LP, Rees RC & Baldwin RW. Br J Cancer, 33 (6): 577–83 (1976).
20. Steele G Jr & Sjogren HO. Int J Cancer, 14 (4): 435–444 (1974).
21. Ting C-C & Grant JP. J Natl Cancer Inst, 56 (2): 401–4 (1976).
22. Ting C-C, Sanford KK & Price FM. In Vitro, 14 (2): 207–11 (1978).
23. Menard S, Colnaghi MI & Della Porta G. Tumori, 63 (4): 359–66 (1977).
24. Woo J & Cater DB. Biochem J, 128 (5): 1273–84 (1972).
25. Wahlström T, Linder W & Saksela E. Acta Pathol Microbiol Scand, 81 (6): 768–774 (1973).
26. Medawar P & Hunt R. Cancer Res, 36 (9): 3453–4 (1976).
27. Zhang S, Sell S, Livingston PO, Klavins JV. J Tumor Marker Oncol, 12:52 (1997).
28. M Peifer & P Polakis. Science (2000), 287:1606–1609.
29. P Polakis. Genes Dev (2000), 14:1837–1851.
30. PW Ingham. Curr Opin Gen Dev (1998), 8:88–94.
Stem Cell Differentiation Stage Factors from Zebrafish Embryo: A Novel Strategy to Modulate the Fate of Normal and Pathological Human (Stem) Cells
Current Pharmaceutical Biotechnology, 2015, Volume 16, Number 9: 782–92.
Pier Mario Biava, Silvia Canaider, Federica Facchin, Eva Bianconi, Liza Ljungberg, Domenico Rotilio, Fabio Burigana, and Carlo Ventura
This article is important because it describes the different functions of the epigenetic code that have been studied and analyzed in a long research process. In addition to the way in which the cancer cells can be reprogrammed, it also illustrates how it is possible to prevent aging and neurodegeneration and to ameliorate the clinical results in psoriasis patients. The article shows how the current scientific paradigm, based on reductionism, needs to be changed in depth, as it highlights how the results obtained in the prevention of complex pathologies, as these described in the article, are only possible when the information transferred to DNA from the epigenetic code is complete and redundant.
Introduction
Current medical literature acknowledges that embryonic microenvironment is able to suppress tumor development during cell differentiating processes.1, 2 Administration of carcinogenic substances during organogenesis leads in fact to embryonic malformations, but not to offspring tumor growth. However, administration of carcinogenic substances after complete organogenesis causes a rise in offspring tumor development.3, 4, 5 This data indicates that cancer can be considered as a deviation in normal development that can be controlled by factors in embryonic microenvironment during the differentiating stages. Furthermore, it has been demonstrated that teratoma differentiates into normal tissues once implanted in the embryo.6
Recently, it has been shown that implantation of melanoma cells into Zebrafish embryos does not result in tumor development, while in the adult animal, a tumor is formed.7 Moreover, injection of melanoma cells in Zebrafish extra-embryonic membranes originated Zebrafish neuronal cells. This demonstrates that cancer cells can differentiate in normal tissues when implanted in embryos.8 In addition, it was demonstrated that other tumors, including leukemia, liver, and breast tumor cells, can revert into a normal phenotype and/or differentiate into normal tissue when implanted in the embryo.9, 10, 11, 12
The term “reprogramming” was initially introduced to identify the transformation of a normal adult somatic cell into an embryoniclike stem cell, into so-called induced pluripotent stem cells (iPS). The issue of cell reprogramming has now been extended to cancer (stem) cells to define any genetic or epigenetic intervention aimed at inducing differentiation of these cells into a normal phenotype and/or forcing them to become terminally differentiating cells. These interventions focus on the role of the embryonic microenvironment in tumor reprogramming. Intriguingly, it is now evident that the molecular mechanisms underlying normal stem cell differentiation and embryonic development do not stop after birth but are still in part operating and remodeled throughout the adult life to maintain the self-identity and the interplay between tissues and organs. To this end, it has been shown that the transcription factor GATA4 is a critical regulator of both embryonic and postnatal heart development and morphogenic maintenance due to a fine tuning of its structural/regulatory domains.13 Whereas the N-terminal domain of GATA4 is required for inducing cardiogenesis and for promoting postnatal cardiomyocyte survival, distinct residues and domains therein are necessary to mediate these effects.13 Cardiogenic activity of GATA4 requires a 24-amino-acid (aa) region (aa 129 to 152) which is needed for transcriptional synergy and physical interaction with BAF60c. The same region is not essential for induction of endoderm or blood cell markers by GATA4, suggesting that it acts as a cell-type-specific transcriptional activation domain. On the other hand, a serine residue at position 105, which is a known target for mitogen-activated protein kinase (MAPK) phosphorylation, is necessary for GATA4-dependent cardiac myocyte survival and hypertrophy but is entirely dispensable for GATA4-induced cardiogenesis.13
A noteworthy example of morphogenetic flexibility is also provided by the existence of reverse pathways of transformation, from the postnatal stage back to an embryonic-like condition retaining the memory ability to re-differentiate backward to the same original phenotype. A vivid example of such flexibility is shown by the ability of post-natal cardiomyocytes to generate iPS cells with enhanced capacity toward cardiomyogenic re-differentiation.14 Similarly, adult neurogenesis, a process of generating functional neurons from adult neural precursors, has been shown to occur throughout life in restricted brain regions in mammals, including the dentate gyrus of the hippocampus, the subventricular zone of the lateral ventricle, and the rostral migratory stream to the olfactory bulb.15 This discovery is currently boosting emerging principles that have significant implications not only in stem cell biology, developmental neurobiology, and neural plasticity, but, remarkably, in disease mechanisms, including neurodegeneration.
Hence, a kind of memory/projection of the embryonic patterning may be conceived as a relevant background in tissue resident stem cells in the adulthood for the execution of self-healing and “learning” (acquirement of new knowledge) tasks. In this frame, degenerative diseases occurring in any organ (i.e., neurodegenerative diseases) may be viewed as a deviation from the normal potential of tissue resident stem cells to afford self-healing duties and the maintenance of tissue organ identity.
Akin to this perception, here we review several of our experimental findings over the past 20 years on the possibility to reprogram cancer cells in vitro as well as in vivo. In fact, we present results from controlled clinical studies on hepatocellular carcinoma at intermediate-advanced stage based on the treatment with Zebrafish stem cell differentiation stage factors (SCDSFs) taken during precise stages of stem cell differentiating processes.16, 17 We also report on our recent finding that the same SCDSFs obtained at early developmental stages acted as a major controller of stemness and senescence patterning in human adult adipose-derived stem cells.18 Consistent with the concept of considering tissue degeneration a “flexible” deviation from a tissue identity program still entangled with embryogenetic memory, we show our recent findings on the ability of SCDSFs to prevent neurodegeneration in hippocampal cells of CA1 area in mice. Compounding the spectrum of exploitation of SCDSF potential for (stem) cell reprogramming, we recently succeeded in using Zebrafish embryo-extracts to reduce keratosis and ameliorate symptoms in patients affected by psoriasis,19, 20, 21 a T-cell-dependent immune-mediated disease of the skin and joints. Such result is also rewarding due to (i) the recent detection of functional circadian clocks in most, if not all, skin cell types, (ii) the emergence of a close involvement of these circadian clocks in the control of UVB-induced DNA damage and skin cancers, and (iii) the implication for the targeted modulation of stem-cell-mediated immunomodulatory action and control of aging processes.22, 23
Role of SCDSFs in Cancer Cell Lines and in Mice Carcinoma Cells
In vitro effects of SCDSFs on different human tumor cell lines have been investigated in a number of studies.24, 25, 26, 27, 28 Seven different human tumor cell lines (glioblastoma multiforme, melanoma, hepatocarcinoma, kidney adenocarcinoma, colon and breast adenocarcinoma, acute lymphoblastic and leukemia) were treated with factors taken from Zebrafish embryos at different developmental phases, specific of the beginning, intermediate and final embryonic differentiation stages. In general, a reduced growth rate was seen when tumor cells lines were treated with factors drawn during the different developmental stages, ranging from 73 percent reduction for the glioblastoma cells to 26 percent for the melanoma cells. No proliferative effects have been reported, except from a weak tumoral growth with factors extracted at a very early stage of embryonic development in which the differentiation processes did not begin, like morula stage. These data confirm the intuition that in the embryo, during the differentiating stages, there are networks of factors able to readdress tumoral cells toward a normal path. Those networks appear in the very first phases of the gastrulation, while they are absent in merely multiplicative stages.24
Several studies were carried out in order to unravel the molecular mechanisms involved in tumor growth inhibition mediated by Zebrafish embryonic extracts, showing that molecules that have a fundamental role in regulation of the cell cycle, such as p53 and retinoblastoma protein (pRb) were affected. More precisely, a p53 transcriptional regulation took place, highlighted by a considerable increase of the p53 protein expression in some of the tumor cell lines, such as the glioblastoma multiforme and the melanoma.25 In other tumor cell lines, such as kidney adenocarcinoma, the growth reduction was due to changes in phosphorylation of pRb,26 which is known to regulate transcription of E2F-1 and thereby control the cell cycle.
Moreover, apoptotic events as well as cell differentiation events were studied, in order to understand the consequences of cell cycle regulation in tumor cells induced by differentiation factors. The analysis was carried out on colon adenocarcinoma cells, showing activation of an apoptotic pathway dependent on p73, as well as an increase in the cell differentiation marker e-cadherin.27
Finally, in order to ascertain if SCDSFs could synergistically/additively interact with 5-Fluorouracil (5-Fu), whole cell-count, flow-cytometry analysis and apoptotic parameters were recorded in human colon cancer cells (Caco-2) treated with SCDSFs 3 μg/ml in association or not with 5-Fu in the sub-pharmacological therapeutic range (0.01 mg/ml). Cell proliferation was significantly reduced by SCDSFs, meanwhile SCDSF+5-Fu leads to an almost complete growthinhibition. SCDSFs produce a significant apoptotic effect, meanwhile the association with 5-Fu leads to an enhanced additive apoptotic rate at both 24 and 72 hours. SCDSFs alone and in association with 5-Fu trigger both the extrinsic and the intrinsic apoptotic pathways, activating caspase-8, -3 and -7. SCDSFs and 5-Fu alone exerted opposite effects on Bax and Bcl-xL proteins, meanwhile SCDSFs+5-Fu induced an almost complete suppression of Bcl-xL release and a dramatic increase in the Bax/Bcl-xL ratio. These data suggest that Zebrafish embryonic factors could improve chemotherapy efficacy by reducing anti-apoptotic proteins involved in drug-resistance processes.28 Therefore, the molecular mechanisms underlying the tumor growth reduction seen after treatment with SCDSFs can be summarized as follows: the cell cycle stops in G1-S or G2-M phase, according to the tumor type, genetic damage repair and cell re-differentiation, or tumor cells apoptosis if reparation is not possible because of mutation gravity.
The effects of SCDSFs on tumor growth were also observed in vivo after subcutaneous injection of primary Lewis Lung Carcinoma cells into C57BL/6 female syngenic mice weighing 18–20 gr. A single cell suspension of tumor cells was prepared by mechanical dissociation of tumor mass: 50 μL of Dulbecco phosphate buffered saline (DPBS) containing 106 viable tumor cells were mixed with SCDSFs and used in the treated animals, while the control group received 50 mL of DPBS. The growth of the primary tumor was measured with calipers at different days after the injection, and the survival time was recorded. A highly significant difference was noted (p<0.001) between treated and control mice both in terms of primary tumor development and of the survival rate in favor of the treated mice.29
SCDSFs in Clinical Trials on Intermediate-Advanced Hepatocellular Carcinoma (HCC)
From the 1st of January 2001 to the 31st of April 2004 a randomized controlled clinical trial was conducted on 179 patients affected by HCC in an intermediate-advanced stage. Since no further treatments were possible, a product fine-tuned on the basis of the above-mentioned studies was administered. The posology was 30 sublingual drops of the 50 percent epiboly Zebrafish embryo extract three times a day. The sublingual solution was chosen because the composition of the active fraction is composed of low molecular weight proteins (see the data about the protein analysis of SCDSFs on pages 109–111).
Objective tumor response, overall survival, and performance status have been evaluated. Results showed that 19.8 percent of the patients experienced a regression and 16 percent experienced a stabilization with an overall survival of more than 60 percent of the responsive patients after 40 months, compared to 10 percent of the non responsive patients.
A wide improvement of performance status has been registered in a great majority of patients (82.6 percent), also in those who experienced a progression of the disease.16 A more recent study confirms the role of SCDSFs in determining complete response in primitive intermediate advanced liver cancer in 13.1 percent of patients.17
SCDSFs in Human Adipose-Derived Stem Cells (hASCs)
The possibility to address the fate of hASCs, isolated from a microfractured fat tissue obtained with a novel non-enzymatic method and device (Lipogems)30, was explored by exposing them to SCDSFs.18
SCDSFs taken during the late developmental stages (20 somites and pharyngula stages) decreased cell viability and elicited caspase-3 mediated apoptosis. This effect did not involve Bax or Bcl-2 transcription. This phenomenon has long been observed, as shown in the case of Bax-independent, caspase-3-related apoptosis induced by hepatocyte growth factor (HGF) in rat liver epithelial cells and recently confirmed in both malignant and normal cells.31
Unlike SCDSFs taken during the late developmental stages, SCDSFs taken during the early developmental stage (50 percent epiboly stage) did not induce hASC apoptosis, nor did it decrease cell viability. Indeed, SCDSFs of the early developmental stage were able to modulate the stem-cell expression of multipotency, enhancing the stemness genes Oct-4, Sox-2, and c-Myc. In addition to affecting stemness genes that maintain stem-cell identity,32 SCDSFs also elicited transcriptional activation of two major mechanisms capable of counteracting stem-cell senescence, including the gene expression of TERT, the catalytic subunit of telomerase, and the gene transcription of Bmi-1. This is a member of the Polycomb and Trithorax families of repressors which acts as essential factors for self-renewal of adult stem cells, and as a key telomerase independent repressor of cell aging.33
Thus, this study showed that human stem cell exposure to SCDSFs taken during the early developmental stage of Zebrafish embryo may represent a useful tool to enhance stem-cell expression of multipotency and activate both telomerase-dependent and -independent antagonists of cell senescence. On the contrary SCDSFs taken during the late developmental stages decrease cell viability and induce cells toward senescence. This strategy did not require cumbersome gene manipulation through viral vector mediated gene transfer, or expensive synthetic chemistry. This data shows for the first time in the world that it is possible to induce human mesenchymal stem cells toward different and opposite directions, tuning in a specific, physiological way the regulation of different genes.
The Neuroprotective Role of SCDSFs
We present here, for the first time, some recent findings on the ability of SCDSFs to prevent neurodegeneration in hippocampal cells of CA1 area in mice.
In order to evaluate the neuroprotective effect of SCDSFs, murine hippocampal slices of the CA1 area were prepared and cultured as described by Gardoni et al.34 and four Zebrafish embryo solutions were prepared as follows: A (50 percent epiboly plus tail bud stage extracts), B (5 somites stage), C (20 somites plus pharingula stage), and Mix ABC (a mixture of the three solutions A, B and C).24
Organotypic hippocampal slices were treated with N-Methyl-DAspartate (NMDA) 50 μM and 300 μM for 1 hour to induce mortality and a propidium iodide (PI) coloration was performed after 24 hours.35 After fixing, the CA1 area was acquired and mortality was analyzed considering the average PI-fluorescence intensity, using as a term of comparison the maximum cell damage obtained by exposing the organotypic slices to NMDA. We first observed that treatment with NMDA 50 μM and 300 μM induced an increase of mortality of 47 percent and 139 percent respectively compared with the controls (p=0.002 and p=0.0002 respectively).
Then we evaluated the neuroprotective effect of SCDSFs after treatment with three toxic stimuli administered for 1 hour at the 14th day of culture: they were serum deprivation, NMDA 50 μM, and NMDA 300 μM. Analyses were performed 24 hours after treatments.
We noticed that treatment with the Mix ABC (dilution 1:100) sub-ministrated together with each of the three toxic stimuli reduced in a significant manner the neuronal mortality caused by both serum deprivation and NMDA treatments. In fact SCDSFs significantly reduce the neuronal mortality caused by serum deprivation (-31.6 ± 6.2 percent, p=0.005) as shown in figure 1 (see page 106).
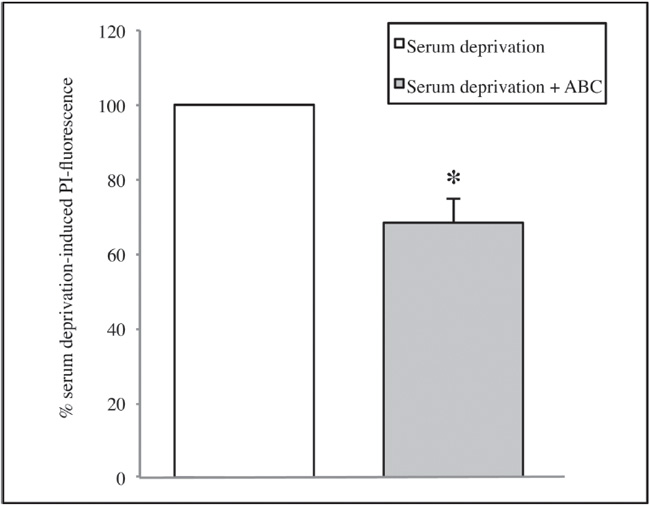
Figure 1. The effect of the Mix ABC on CA1 area cell mortality after 1 hour of serum deprivation (*p=0.005)
Moreover, treatment with NMDA 50 μM significantly increases cell mortality compared with the controls (p=0.002), and SCDSFs significantly reduce the neuronal mortality caused by NMDA 50 μM treatment (p=0.01) as shown in figure 2.
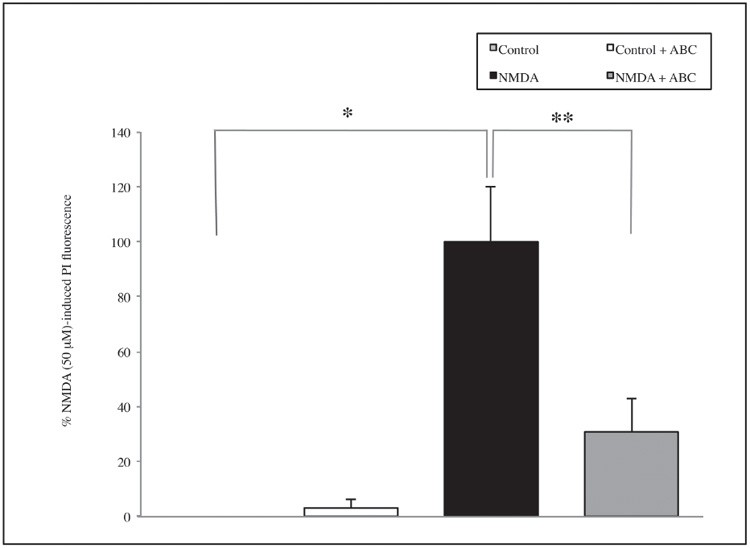
Figure 2. The effect of the Mix ABC on CA1 area cell mortality after 1 hour NMDA 50 μM treatment (*p=0.002; **p=0.01)
Similarly, treatment with NMDA 300 μM significantly increases cell mortality compared with the controls (p=0.0002) and SCDSFs significantly reduce the neuronal mortality caused by NMDA 300 μM treatment (p=0.009) as shown in figure 3.
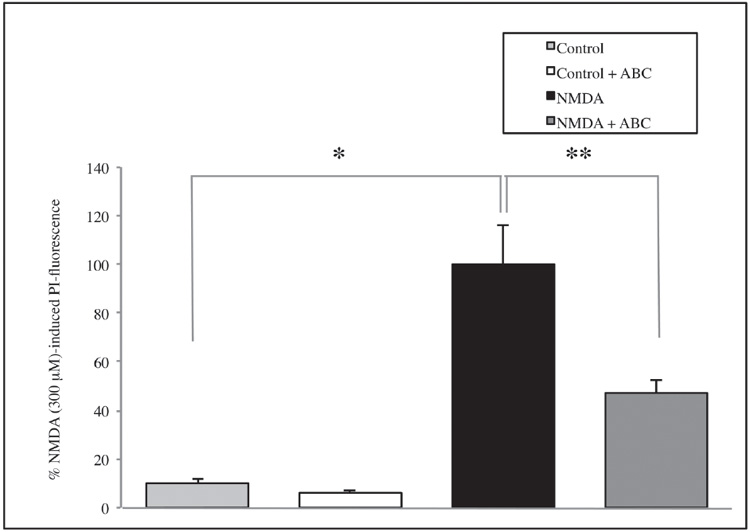
Figure 3. The effect of the Mix ABC on CA1 area cell mortality after 1 hour NMDA 300 μM treatment (*p=0.0002; **p=0.009)
Subsequently, the potential neuroprotective activities of A or B or C were investigated. Also in this case, the experiments showed a reduction in mortality overall for A extract, but results are not enough significant, neither in the serum deprivation group (figure 4) nor in the NMDA group (figure 5) (see page 108). Thus, the whole informational set with a redundance of differentiation stage factors is needed to produce an effective result.
Experimental Research and Clinical Studies on Psoriasis
We also investigated the anti-proliferative effects of SCDSFs by addressing the mitochondrial function (MTT assay) and cell nuclei distribution (Hoechst staining) in epidermal cell cultures stimulated with fetal calf serum (FCS) or epidermal growth factor (EGF). SCDSFs significantly inhibited cell proliferation induced by either approach, although the effect was stronger in cells stimulated with FCS.36 Three clinical trials were conducted to evaluate the efficacy in cases of psoriasis following the administration of a mix of all 5 Zebrafish embryo developmental stage extracts added with Boswelia serrata, 18-beta glycyrrhetic acid, Zanthoxylum alatum, 7-dehydro-cholesterol, and vitamin E. Results show 80 percent clinical objective improvements, with a reduction of keratosis and itch after 20–30 days from the beginning of the treatment.19, 20, 21
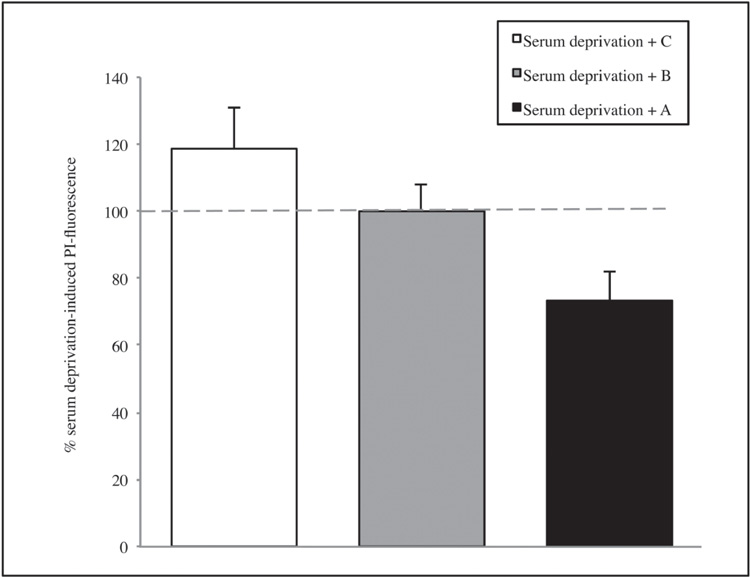
Figure 4. The effect of the single solutions A, B and C on CA1 area of hippocamp after 1 hour of serum deprivation. Values are expressed as percentage of samples treated with serum deprivation without SCDSFs.
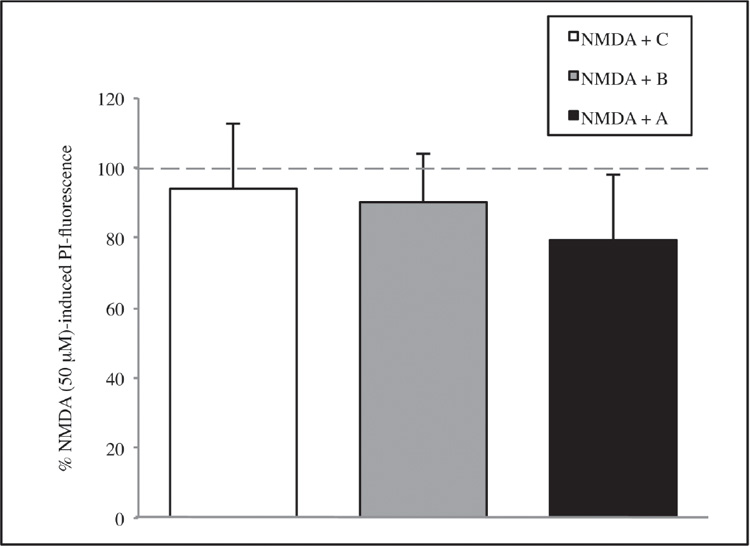
Figure 5. The effect of the single solutions A, B, and C on CA1 area of hippocamp after 1 hour of NMDA 50 μM treatment. Values are expressed as a percentage of samples treated with NMDA 50 μM without SCDSFs.
Protein Analysis of SCDSFs
To better know the content of the SCDSFs that we employed for our researches, we began to perform protein analysis of the extracts, and here we present our first results.
First, protein content of the five Zebrafish embryo extracts resuspended in a glycero-alcoholic solution18, 24 was analyzed on a one-dimensional Sodium Dodecyl Sulphate-PolyAcrylamide Gel Electrophoresis (SDS-PAGE).37 After Coomassie staining,38 the protein amount was evaluated as pixel intensity, and relative abundances were expressed as a percentage of the total intensity. As shown in figure 6, in all five extracts, three main protein clusters are distinguishable according to their molecular weight; i.e., over 45 kDa, around 25–35 kDa, and less than 20 kDa. In any case, the relative protein abundance is different among the five samples.
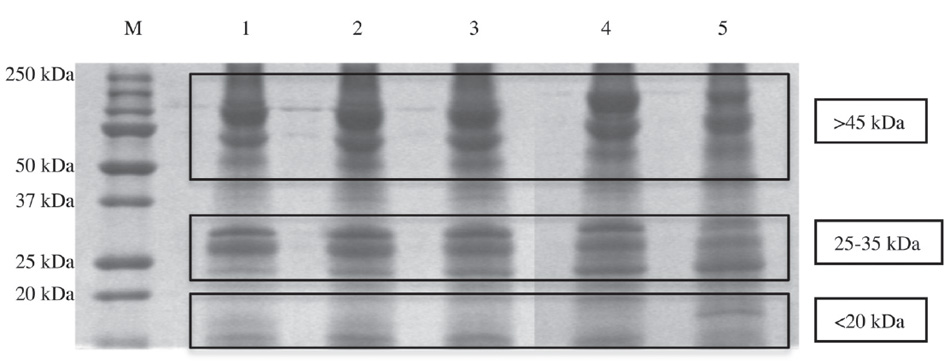
Figure 6. Representative 12 percent SDS-PAGE gel of Zebrafish embryo extracts resuspended in a glycero-alcoholic solution. Lanes: M) Broad-range protein molecular weight markers (in kDa); 1) 50 percent epiboly stage proteins; 2) tail bud stage proteins; 3) 5 somites stage proteins; 4) 20 somites stage proteins; 5) pharingula stage proteins.
At the beginning of the gastrula period (50 percent epiboly stage, Lane 1), the higher molecular weight cluster (> 45 kDa) represents the 45.8 percent of the bands’ intensity; this relative abundance is quite stable at the end of the gastrula period (tail bud stage, Lane 2) with a peak at the beginning of the segmentation, 46.1 percent (5 somites, Lane 3), while at the middle-late segmentation (20 somites, Lane 4 and pharyngula, Lane 5) this percentage composition decreases until the 43.9 percent.
The 25–35 kDa protein cluster abundance is quite stable in the gastrula period (Lanes 1 and 2), around 25.5 percent, while during the segmentation (Lanes 3, 4 and 5) it decreases until the 22.6 percent. At the beginning of the gastrula period (50 percent epiboly stage, Lane 1) the lowest molecular weight cluster (less then 20 kDa) represent the 28.5 percent; the cluster abundance is quite similar among the end gastrulation and early segmentation (Lanes 2 and 3) (29.4 percent) while at the end of the gastrulation stages (20 somites, Lane 4, and pharingula, Lane 5) the percentage increases until 33.5 percent.
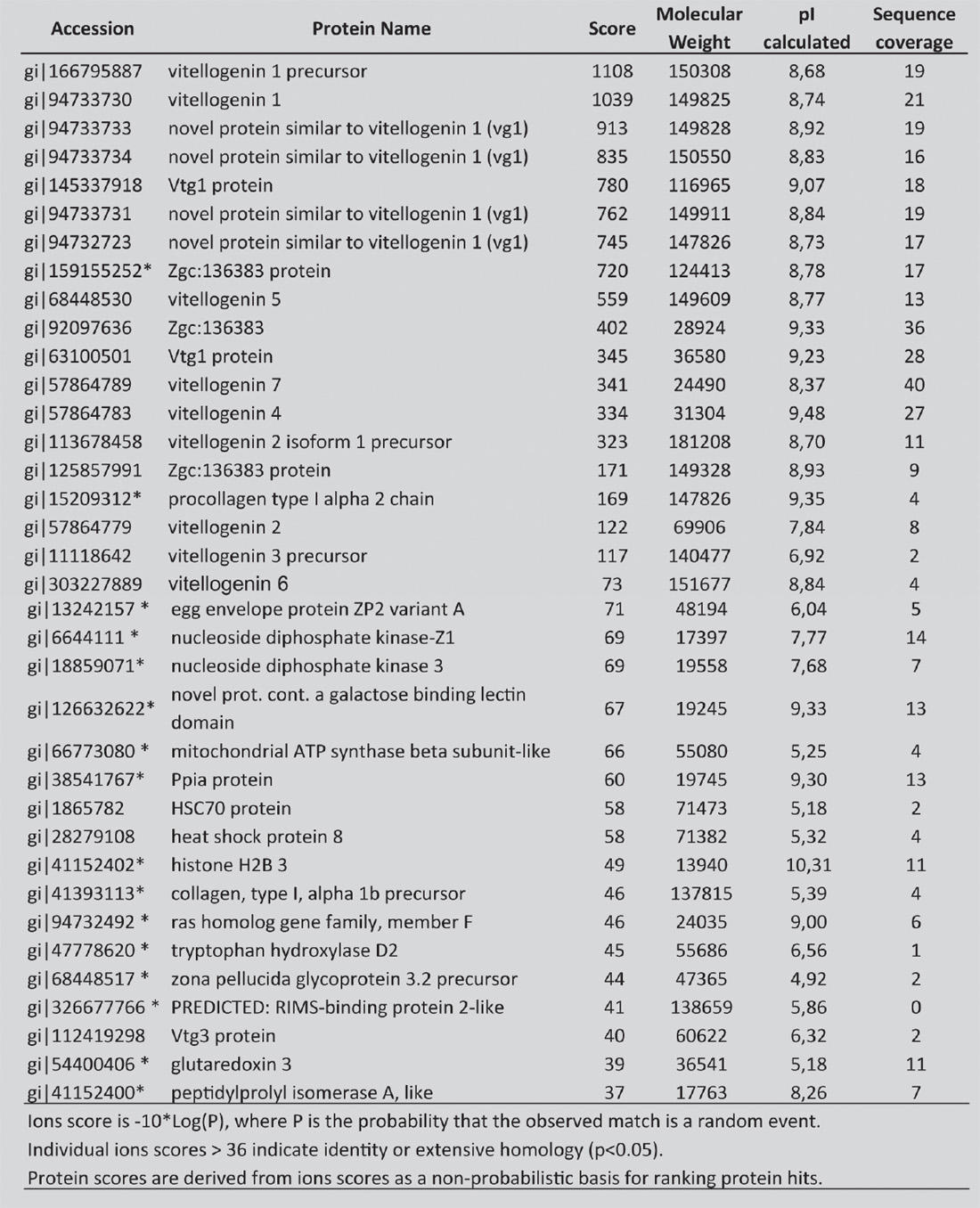
Then, all the proteins extracted from the earliest Zebrafish developmental investigated stage (50 percent epiboly) were identified by using a liquid chromatography–mass spectrometry (LC-MS/MS) analysis, after the in-gel digestion procedure as described by Della Corte and coll.39 We list in table 1 the identified proteins with the correspondent NCBI accession number, the score, their isoelectric point (pI). Individual ions scores >36 indicate identity or extensive homology (p<0.05). Identified proteins include multiple form of yolk protein vitellogenin, heat shock protein (e.g., HSP8 and HSP70) and other proteins that have not been described before (indicated in table 1 with an asterisk).40, 41 These proteins are implicated in many pathways as in signalling, cell cycle regulation, protein trafficking, chaperoning, protein synthesis, and degradation.
TABLE 1. List of Proteins Identified Using the Nano Lc-Esi-Q-Tof in Zebrafish Embryo at Middle-Blastula-Gastrula Stage
With the Specification of Their NCBI Accession Number, Name, Score, Molecular Weight in Daltons, Isoelectric Point (pi) and Percentage Sequence Coverage.
_____________
Proteins highlighted with asterisk (*) had not so far been described in Zebrafish embryo.
Discussion and Conclusions
The use of stem cells differentiation factors in anticancer therapy has enabled one of us to build up a model of cancer corresponding to reality.41 Such a model, conceived in 2002, describes cancer as a consequence of two different processes, i) a process of maturation arrest of stem cells (hierarchical model) and ii) a process of deterministic chaos in which genetic and epigenetic alterations conduce a normal differentiated cell to be malignant (stochastic model). In fact, these two processes are not mutually exclusive, and both have been described.42, 43
Therefore, from this point of view, cancer cells can be defined as “cancer stem-like cells,” that according to their degree of malignancy are considered blocked at a different phase of development. In fact, in tumors with an elevated degree of malignancy, such as acute lymphoblastic and myeloid leukemia, multipotent stem-like cells are present, whereas in tumors with lower malignancy, such as chronic lymphocytic leukemia, cells not yet completely differentiated are present, but toward a final differentiation.
In addition, cancer and stem cells share several characteristics. First, they present oncofetal antigens, maintained during the phylogenesis44 and specific receptor on the cellular membrane on which the stem cells differentiation factors probably act. It has already been mentioned above that such factors could activate pathways of cellular differentiation that lead the cells to differentiate or to die, as usually occurs in the embryo (the apoptotic events in the embryo are many).
Furthermore, cancer and embryonic cells share common metabolic pathways such as APC/beta catenin/TCF/Wnt and the Hedgehog/Smoothened/Patched pathways.
The gene configuration and the metabolism of cancer cells is actually very similar to that of stem cells: they both have active proto-oncogene and produce embryonic growth factors, present oncofetal antigens, and work with an aerobic metabolism.
Nevertheless, cancer cells and stem cells show an important difference. The problem of cancer cells is double: they present genetic mutations that are at the origin of malignancy, and at the same time, they show an imbalance of the epigenetic code. In contrast with normal stem cells, tumor cells are not able to complete their development and to differentiate because they lost information; i.e., they experienced a mutation or epigenetic alterations in their code. The regulation of DNA information using epigenetic regulators such as SCDSFs, taken in the late stages of development of the embryo, transforms the cancer cells into normal cells or causes their apoptosis.
It is now emerging more and more clearly that the transcription factors, the microRNAs, the translational-and post-translational factors, play a fundamental role in the regulation of DNA information and in regulating the cell life. In other words, the epigenetic regulators contained in SCDSFs are able to differentiate and regulate normal stem cells and cancer stem cells, deactivating genes that lead cancer stem cells to proliferate while activating new differentiating pathways.
Our studies have recently been confirmed by other experimental researches performed by some colleagues of the Children’s Hospital of Chicago.12 In particular, they have confirmed that malignant melanoma reverts to a normal phenotype when it is in the environment of Zebrafish embryo. On the other hand there are many studies that highlight the link between tumor malignancy and the presence of cancer stem cells,45 that seem to be resistant to conventional therapy, such as chemoand radiotherapy. In the last 6–7 years scientific works in this field are so numerous that it is almost impossible to name all of them. Here we mention only those researches that demonstrated the presence of tumoral stem cell in breast cancer,46, 47, 48, 49, 50, 51 lung cancer,52, 53, 54, 55 prostate,56, 57, 58 ovary cancer,59, 60, 61, 62, 63 liver cancer,64, 65, 66, 67, 68, 69 stomach cancer,70, 71, 72, 73, 74 colon cancer,75, 76, 77 pancreas cancer,78, 79, 80 glioblastoma multiforme,81, 82, 83 head and neck cancer.84, 85, 86, 87 On the other hand, it is known that malignancy of many haematological tumoral diseases is due to the presence of stem cells.
Regarding the interpretation of the results obtained by using SCDSFs for the prevention of the neurodegenerative and for the treatment of psoriasis, we can assume that: the differentiation factors are epigenetic regulators, that on the one hand prevent the processes and the development of degenerative phenomena and on the other hand regulate the processes of abnormal cellular multiplication, as it comes, for instance, in psoriasis, where the multiplication of cells of the epithelial basal layer is five-to tenfold higher than that considered physiological. In this case we have demonstrated that the differentiation factors reduce the proliferation of the epidermal layers by normalizing it. In addition, other researches demonstrated that it is possible to tune in a fine way the fate of normal stem cells, like human mesenchymal stem cells, using SCDSFs. In fact, if we use in a specific way the different networks of substances present in the different stages of cell differentiation we can induce stem cells toward senescence or apoptosis (late stages of differentiation) or, at the contrary, enhance stem cell expression of multipotency by activating both telomerase-dependent and-independent antagonists of cell senescence (early stage of differentiation). Noteworthy, different modulating effects can be obtained only with a specific network of SCDSFs. From this point of view, the experiments about the prevention of neurodegeneration are enlightening. In fact, to prevent neurodegeneration, first of all we have to enhance stem-cell expression of multipotency, and then we have to induce stem cells toward differentiation in neural cells. For these reasons, all different stage factors expressed during cell differentiation have to be used: only the redundancy of these factors could lead to obtain significant results. These results lead us to consider a major shift in scientific paradigm (from reductionism to complexity) for preparing new treatments for chronic and degenerative diseases. In fact, these diseases entail an unexpected degree of complexity and disregulation, making the single-molecule-to-specific-target paradigm totally obsolete and inadequate. Rather, only a systemic approach can be envisioned as a successful strategy to deal with such complexity. We believe that time is ready for a “metadisciplinary approach” in the treatment of degenerative diseases involving multiple tissues and organs, to help users in a new culture of collaboration from different scientific disciplines join together to combine their knowledge and come up with innovations, new therapeutic approaches, and most of all the development of novel paradigms. This is overdue to provide a reliable effort to help elderly people and everyone who suffers from degenerative diseases or cancer.
List of Abbreviations
Stem Cell Differentiation Stage Factors (SCDSFs), human Adiposederived Stem Cells (hASCs), induced Pluripotent Stem cells (iPS), amino-acid (aa), Mitogen-Activated Protein Kinase (MAPK), Retinoblastoma protein (pRb), 5-Fluorouracil (5-Fu), human Colon cancer cells (Caco-2), N-Methyl-D-Aspartate (NMDA), Propidium Iodide (PI), Dulbecco Phosphate Buffered Saline (DPBS), hepatocellular carcinoma (HCC), Hepatocyte Growth Factor (HGF), Fetal Calf Serum (FCS), Epidermal Growth Factor (EGF), Sodium Dodecyl Sulphate-PolyAcrylamide Gel Electrophoresis (SDS-PAGE), Liquid Chromatography–Mass Spectrometry (LC-MS/MS), Isoelectric point (pI), Heat Shock Protein (HSP).
References
1. Einhorn, L. Are there factors preventing cancer development during embryonic life? Oncodev. Biol. Med., 1983, 4(3), 219–229.
2. Lakshmi, M.S.; Sherbet, G.V. In: Embryonic and Tumor Cell Interactions; Sherbet G.V., Ed.; Karger Basel: New York, 1974; pp. 380–399.
3. Brent, R.L. Radiation Teratogenesis. Teratology, 1980, 21(3), 281–298.
4. Pierce, G.B. The cancer cell and its control by the embryo. Rous-Whipple Award lecture. Am. J. Pathol., 1983, 113(1), 117–124.
5. Yu, C.L.; Tsai, M.H. Fetal fetuin selectively induces apoptosis in cancer cell lines and shows anti-cancer activity in tumor animal models. Cancer Letter, 2001, 166(2), 173–184.
6. Papaioannou, V.E.; McBurney, M.V.; Gardner, R.L.; Evans, M.J. Fate of teratocarcinoma cells injected into early mouse embryos. Nature, 1975, 258(5530), 70–73.
7. Topczewska, J.M.; Postovit, L.M.; Margaryan, N.V.; Sam, A.; Hess, A.R.; Wheaton, W.W.; Nickoloff, B.J.; Topczewski, J.; Hendrix, M.J. Embryonic and tumorigenic pathways converge via Nodal signaling: role in melanoma aggressiveness. Nat. Med., 2006, 12(8), 925–932.
8. Kulesa, P.M.; Kasermeier-Kulesa, J.C.; Teddy, J.M.; Margaryan, N.V.; Seftor, E.A.; Seftor, R.E.; Hendrix, M.J. Reprogramming metastatic melanoma cells to assume a neural crest cell-like phenotype in an embryonic microenvironment. Proc. Natl. Acad. Sci. USA, 2006, 103(10), 3752–3757.
9. Webb, C.G.; Gootwine, E.; Sachs, L. Developmental potential of myeloid leukemia cells injected into rat midgestation embryos. Dev Biol., 1984, 101(1), 221–224.
10. Weaver, V.M.; Petersen, O.W.; Wang, F.; Larabell, C.A.; Briand, P.; Damsky, C.; Bissell, M.J. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol., 1997, 137(1), 231–245.
11. Coleman, W.B.; Wennerberg, A.E.; Smith, G.J.; Grisham, J.W. Regulation of the differentiation of diploid and some aneuploid rat liver epithelial (stemlike) cells by the hepatic microenvironment. Am. J. Pathol., 1993, 142(5), 1373–1382.
12. Postovit, L.M.; Maragaryan, N.V.; Seftor, E.A.; Kirschmann, D.A.; Lipavsky, A.; Wheaton, W.W.; Abbott, D.E.; Seftor, R.E.; Hendrix, M.J. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc. Natl. Acad. Sci. USA, 2008, 105(11), 4329–4334.
13. Gallagher, J.M.; Komati, H.; Roy, E.; Nemer, M.; Latinkic, B.V. Dissociation of cardiogenic and postnatal myocardial activities of GATA4. Mol. Cell Biol., 2012, 32(12), 2214–2223.
14. Rizzi, R.; Di Pasquale, E.; Portararo, P.; Papait, R.; Cattaneo, P.; Latronico, M.V.; Altomare, C.; Sala, L.; Zaza, A.; Hirsch, E.; Naldini, L.; Condorelli, G.; Bearzi, C. Post-natal cardiomyocytes can generate iPS cells with an enhanced capacity toward cardiomyogenic re-differentation. Cell Death Differ., 2012, 19(7), 1162–1174.
15. Ming, G.L.; Song, H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron, 2011, 70(4), 687–702.
16. Livraghi, T.; Meloni, F.; Frosi, A.; Lazzaroni, S.; Bizzarri, T.M.; Frati, L.; Biava, P.M. Treatment with stem cell differentiation stage factors of intermediate-advanced hepatocellular carcinoma: an open randomized clinical trial. Oncol. Res., 2005, 15 (7–8), 399–408.
17. Livraghi, T.; Ceriani, R.; Palmisano, A.; Pedicini, V.; Pich, M.G.; Tommasini, M.A.; Torzilli, G. Complete response in 5 out of 38 patients with advanced hepatocellular carcinoma treated with stem cell differentiation stage factors: case reports from a single centre. Curr. Pharm. Biotechnol., 2011, 12(2), 254–260.
18. Canaider, S.; Maioli, M.; Facchin, F.; Bianconi, E.; Santaniello, S.; Pigliaru, G.; Ljungberg, L.; Burigana, F; Bianchi, F.; Olivi, E.; Tremolada, C.; Biava, P.M.; Ventura, C. Human Stem Cell Exposure to Developmental Stage Zebrafish Extracts: a Novel Strategy for Tuning Stemness and Senescence Patterning. CellR4, 2014, 2(5), e1226.
19. Di Pierro, F.; Negri, M.; Bollero, C. Terapia della psoriasi. Efficacia clinica di un preparato multicomponente. Cosmetic Technology, 2009, 12(2), 13–17.
20. Harak, H.; Frosi, A.; Biava, P.M. Studio clinico sull’efficacia e tollerabilita’ di una crema per uso topico nel trattamento della psoriasi. La Med. Biol., 2012, 3, 27–31.
21. Calzavara-Pinton, P.; Rossi, M. A topical remedy in association with phototherapy. Efficacy evaluation in patients suffering from moderate psoriasis. Hi.tech dermo, 2012, 1, 41–47.
22. Plikus, M.V.; Van Spyk, E.N.; Pham, K.; Geyfman, M.; Kumar, V.; Takahashi, J.S.; Andersen, B. The Circadian Clock in Skin: Implications for Adult Stem Cells, Tissue Regeneration, Cancer, Aging, and Immunity. J Biol. Rhythms., 2015, pii: 0748730414563537.
23. Hou, R.; Liu, R.; Niu, X.; Chang, W.; Yan, X.; Wang, C.; Li, J.; An, P.; Li, X.; Yin, G.; Zhang, K. Biological characteristics and gene expression pattern of bone marrow mesenchymal stem cells in patients with psoriasis. Exp. Dermatol., 2014, 23(7), 521–523.
24. Biava, P.M.; Bonsignorio, D.; Hoxa, M. Cell proliferation curves of different human tumor lines after in vitro treatment with Zebrafish embryonic extracts. J. Tumor Marker Oncol., 2001, 16(3), 195–202.
25. Biava, P.M.; Carluccio, A. Activation of anti-oncogene p53 produced by embryonic extracts in vitro tumor cells. J. Tumor Marker Oncol., 1977, 12(4), 9–15.
26. Biava, P.M.; Bonsignorio, D.; Hoxa, M.; Impagliazzo, M.; Facco, R.; Ielapi, T.; Frati, L.; Bizzarri, M. Post-translational modification of the retinoblastoma protein (pRb) induced by in vitro administration of Zebrafish embryonic extracts on human kidney adenocarcinoma cell line. J. Tumor Marker Oncol., 2002, 17(2), 59–64.
27. Cucina, A.; Biava, P.M.; D’Anselmi, F.; Coluccia, P.; Conti, F.; di Clemente, R.; Miccheli, A.; Frati, L.; Gulino, A.; Bizzarri, M. Zebrafish embryo proteins induce apoptosis in human colon cancer cells (Caco2). Apoptosis, 2006, 11(9), 1617–1628.
28. D’Anselmi, F.; Cucina, A.; Biava, P.M.; Proietti, S.; Coluccia, P.; Frati, L.; Bizzarri, M. Zebrafish stem cell differentiation stage factors suppress Bcl-xL release and enhance 5-Fu-mediated apoptosis in colon cancer cells. Curr. Pharm. Biotechnol., 2011, 12(2), 261–267.
29. Biava, P.M.; Nicolini, A.; Ferrari, P.; Carpi, A.; Sell, S. A systemic approach to cancer treatment: tumor cell reprogramming focused on endocrine-related cancers. Curr. Med. Chem., 2014, 21(9), 1072–1081.
30. Bianchi, F.; Maioli, M.; Leonardi, E.; Olivi, E.; Pasquinelli, G.; Valente, S.; Mendez, A.J.; Ricordi, C.; Raffaini, M.; Tremolada, C.; Ventura, C. A new non enzymatic method and device to obtain a fat tissue derivative highly enriched in pericyte-like elements by mild mechanical forces from human lipoaspirates. Cell Transplant., 2013, 22(11), 2063–2077.
31. Conner, E.A.; Teramoto, T.; Wirth, P.J.; Kiss, A.; Garfield, S.; Thorgeirsson, S.S. HGF-mediated apoptosis via p53/bax-independent pathway activating JNK1. Carcinogenesis, 1999, 20(4), 583–590.
32. Feng, R.; Zhou, S.; Liu, Y.; Song, D.; Luan, Z.; Dai, X.; Li, Y.; Tang, N.; Wen, J.; Li, L. Sox2 protects neural stem cells from apoptosis via up regulating survivin expression. Biochem. J., 2013, 450(3), 459–468.
33. Park, I.K.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 is required for maintenance of adult selfrenewing hematopioetic stem cells. Nature, 2003, 423(6937), 302–305.
34. Gardoni, F.; Bellone, C.; Viviani, B.; Marinovich, M.; Meli, E.; Pellegrini-Giampietro, D.E.; Cattabeni, F.; Di Luca, M. Lack of PSD-95 drives hippocampal neuronal cell death through activation of an alpha CaMKII transduction pathway. Eur. J. Neurosci., 2002, 16(5), 777–786.
35. Pellegrini-Giampietro, D.E.; Cozzi, A.; Peruginelli, F.; Leonardi, P.; Meli, E.; Pellicciari, R.; Moroni, F. 1-Aminoindan-1,5-dicarboxylic acid and (S)-(+)2-(3’-carboxybicyclo [1.1.1] pentyl)-glycine, two mGlu1 receptor-preferring antagonists, reduce neuronal death in in vitro and in vivo models of cerebral ischaemia. Eur. J. Neurosci., 1999, 11(10), 3637–3647.
36. Norata, G.D.; Biava, P.M.; Di Pierro, F. The Zebrafish embryo derivative affects cell viability of epidermal cells: a possible role in the treatment of psoriasis. G. Ital. Dermatol. Venereol., 2013, 148(5), 479–483.
37. Shi, Q.; Jackowski, G. In: Gel Electrophoresis of Proteins: A Practical Approach, 3rd ed.; Hames, Ed.; Oxford University Press Inc: New York, 1998; Chap. 1, 13–29.
38. Kang, D.H.; Gho, Y.S.; Suh, M.K.; Kang, C.H. Highly sensitive and fast protein detection with coomassie brilliant blue in sodium dodecyl sulfatepolyacrylamide gel electrophoresis. Bull. Korean Chem. Soc., 2002, 23, 1511–1512.
39. Della Corte, A.; Tamburrelli, C.; Crescente, M.; Giordano, L.; D’Imperio, M.; Di Michele, M.; Donati, M.B.; De Gaetano, G.; Rotilio, D.; Cerletti, C. Platelet proteome in healthy volunteers who smoke. Platelets, 2012, 23(2), 91–105.
40. Lucitt, M.B.; Price, T.S.; Pizarro, A.; Wu, W.; Yocum, A.K.; Seiler, C.; Pack, M.A.; Blair, I.A.; Fitzgerald, G.A.; Grosser T. Analysis of the Zebrafish proteome during embryonic development. Mol. Cell. Proteomics, 2008, 7(5), 981–994.
41. Biava, P.M.; Bonsignorio, D. Cancer and cell differentiation: a model to explain malignancy. J. Tumor Marker Oncol., 2002, 17(2), 47–54.
42. Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell, 2009, 138(5), 822–829.
43. Visvader, J.E.; Lindeman, G.J. Cancer stem cells: current status and evolving complexities. Cell Stem Cell., 2012, 10(6), 717–728.
44. Biava, P.M; Monguzzi, A; Bonsignorio, D; Frosi, A; Sell, S; Klavins, J.V. Xenopus laevis Embryos share antigens with Zebrafish Embryos and with human malignant neoplasms. J. Tumor Marker Oncol., 2001, 16(3), 203–206.
45. Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer and cancer stem cells. Nature, 2001, 414(6859), 105–111.
46. Chen, J.; Chen, Z.L. Technology update for the sorting and identification of breast cancer stem cells. Chin. J. Cancer, 2010, 29(3), 265–269.
47. Roesler, R.; Cornelio, D.B.; Abujamra, A.L.; Schwartsmann, G. HER2 as a cancer stem-cell target. Lancet Oncol., 2010, 11(3), 225–226.
48. Wu, W. Patents related to cancer stem cell research. Recent Pat. DNA Gene Seq., 2010, 4(1), 40–45.
49. Park, S.Y.; Lee, H.E.; Li, H.; Shipitsin, M.; Gelman, R.; Polyak, K. Heterogeneity for stem cell-related markers according to tumor subtype and histologic stage in breast cancer. Clin. Cancer Res., 2010, 16(3), 876–887.
50. Lawson, J.C.; Blatch, G.L.; Edkins, A.L. Cancer stem cells in breast cancer and metastasis. Breast Cancer Res. Treat., 2009, 118(2), 241–254.
51. Luo, J.; Yin, X.; Ma, T.; Lu, J. Stem cells in normal mammary gland and breast cancer. Am. J. Med. Sci., 2010, 339(4), 366–370.
52. Spiro, S.G.; Tanner, N.T.; Silvestri, G.A.; Janes, S.M.; Lim, E.; Vansteenkiste, J.F.; Pirker, R. Lung cancer: progress in diagnosis staging and therapy. Respirology, 2010, 15(1), 44–50.
53. Gorelik, E.; Lokshin, A.; Levina, V. Lung cancer stem cells as a target for therapy. Anticancer Agents Med. Chem., 2010, 10(2), 164–171.
54. Sullivan, J.P.; Minna, J.D.; Shay, J.W. Evidence for self-renewing lung cancer stem cells and their implications in tumor initiation, progression and targeted therapy. Cancer Metastasis Rev., 2010, 29(1), 61–72.
55. Westhoff, B.; Colaluca, I.N.; D’Ario, G.; Donzelli, M.; Tosoni, D.; Volorio, S.; Pelosi, G.; Spaggiari, L.; Mazzarol, G.; Viale, G.; Pece, S.; Di Fiore, P.P. Alterations of the Notch pathway in lung cancer. Proc. Natl. Acad. Sci. USA, 2009, 106(52), 22293–22298.
56. Lawson, D.A.; Zong, Y.; Memarzadeh, S.; Xin, L.; Huang, J.; Witte, O.N. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc. Natl. Acad. Sci. USA, 2010, 107(6), 2610–2615.
57. Lang, S.H.; Anderson, E.; Fordham, R.; Collins, A.T. Modeling the prostate stem cell niche: an evaluation of stem cell survival and expansion in vitro. Stem. Cells Dev., 2010, 19(4), 537–546.
58. Joung, J. Y.; Cho, K. S.; Kim, J. E.; Seo, H.K.; Chung, J.; Park, W. S.; Choi, M.K.; Lee, K.H. Prostate stem cell antigen mRNA in peripheral blood as a potential predictor of biochemical recurrence of metastatic prostate cancer. J. Surg. Oncol., 2010, 101(2), 145–148.
59. Liu, T.; Cheng, W.; Lai, D.; Huang, Y.; Guo, L. Characterization of primary ovarian cancer cells in different culture systems. Oncol. Rep., 2010, 23(5), 1277–1284.
60. Fong, M.Y.; Kakar, S.S. The role of cancer stem cells and the side population in epithelial ovarian cancer. Histol. Histopathol., 2010, 25(1), 113–120.
61. Murphy, S.K. Targeting ovarian cancer-initiating cells. Anticancer Agents Med. Chem., 2010, 10(2), 157–163.
62. Peng, S.; Maihle, N.J.; Huang, Y. Pluripotency factors Lin 28 and Oct 4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene, 2010, 29(14), 2153–2159.
63. Kusumbe, A. P.; Bapat, S. A. Cancer stem cells and aneuploid populations within developing tumors are the major determinants of tumor dormancy. Cancer Res., 2009, 69(24), 9245–9253.
64. Tomuleasa, C.; Soritau, O.; Rus-Ciuca, D.; Pop, T.; Todea, D.; Mosteanu, O.; Pintea, B.; Foris, V.; Susman, S.; Kacso, G.; Irimie, A. Isolation and characterization of hepatic cells with stem-like properties from hepatocellular carcinoma. J. Gastrointestin. Liver Dis., 2010, 19(1), 61–67.
65. Zou, G.M. Liver cancer stem cells as an important target in liver cancer therapies. Anticancer Agents Med. Chem., 2010, 10(2), 172–175.
66. Lee, T.K.; Castilho, A.; Ma, S.; Ng, I.O. Liver cancer stem cells: implications for new therapeutic target. Liver Int., 2009, 29(7), 955–965.
67. Marquardt, J.U.; Thorgeirsson, S.S. Stem Cells in hepatocarcinogenesis: evidence from genomic data. Semin. Liver Dis., 2010, 30(1), 26–34.
68. Kung, J.W.; Currie, I. S.; Forbes, S. J.; Ross, J. A. Liver development, regeneration, and carcinogenesis. J. Biomed. Biotechnol., 2010, 2010, 984248.
69. Gai, H.; Nguyen, D.M.; Moon, Y.J.; Aguila, J.R.; Fink, L.M.; Ward, D.C.; Ma, Y. Generation of murine hepatic lineage cells from induced pluripotent stem cells. Differentiation, 2010, 79(3), 171–181.
70. Correia, M.; Machado, J.C.; Ristimaki, A. Basic aspects of gastric cancer. Helicobacter, 2009, 14(1), 36–40.
71. Takaishi, S.; Okumura,T.; Tu, S.; Wang, S.S.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the surface marker CD44. Stem Cells, 2009, 27(5), 1006–1020.
72. Nishii, T.; Yashiro, M.; Shinto, O.; Sawada, T.; Ohira, M.; Hirakawa, K. Cancer stem cell-like SP cells have a high adhesion ability to the peritoneum in gastric carcinoma. Cancer Sci., 2009, 100(8), 1397–1402.
73. Chen, Z.; Xu, W. R.; Qian, H.; Zhu, W.; Bu, X.F.; Wang, S.; Yan, Y.M.; Mao, F.; Gu, H. B.; Cao, H. L.; Xu, X. J. Oct4, a novel marker for human gastric cancer. J. Surg. Oncol., 2009, 99(7), 414–419.
74. Kang, D.H.; Han, M.E.; Song, M.H.; Lee, Y.S.; Kim, E.H.; Kim, H.J.; Kim, G.H.; Kim, D.H.; Yoon, S.; Baek, S.Y.; Kim, B.S.; Kim, J.B.; Oh, S.O. The role of hedgehog signaling during gastric regeneration. J. Gastroenterol., 2009, 44(5), 372–379.
75. Yeung, T.M.; Ghandhi, S.C.; Wilding, J.L.; Muschel, R.; Bodmer, W.F. Cancer stem cells from colorectal cancer derived cell lines. Proc. Natl. Acad. Sci. USA, 2010, 107(8), 3722–3727.
76. Gulino, A.; Ferretti, E.; De Smaele, E. Hedgehog signaling in colon cancer and stem cells. EMBO Mol. Med., 2009, 1(6–7), 300–302.
77. Thenappan, A.; Li, Y.; Shetty, K.; Johnson, L.; Reddy, E.P.; Mishra, L. New therapeutic targeting colon cancer stem cells. Curr. Colorectal Cancer Rep., 2009, 5(4), 209.
78. Rasheed, Z.A.; Yang, J.; Wang, Q.; Kowalski, J.; Freed, I.; Murter, C.; Hong, S. M.; Koorstra, J.B.; Rajeshkumar, N. V.; He, X.; Goggins, M.; IacobuzioDonahue, C.; Berman, D. M.; Laheru, D.; Jimeno, A.; Hidalgo, M.; Maitra, A.; Matsui, W. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J. Natl. Cancer Inst., 2010, 102(5), 340–351.
79. Puri, S.; Hebrok, M. Cellular plasticity within pancreas—lessons learned from development. Dev. Cell, 2010, 18(3), 342–356.
80. Quante, M.; Wang, T. C. Stem cells in gastroenterology and hepatology. Nat. Rev. Gastroenterol. Hepatol., 2009, 6(12), 724–737.
81. Sato, A.; Sakurada, K.; Kumabe, T.; Sasajima, T.; Beppu, T.; Asano, K.; Ohkuma, H.; Ogawa, A.; Mizoi, K.; Tominaga, T.; Kitanaka, C.; Kayama, T.; Tohoku Brain Tumor Study Group. Association of stem cell marker CD133 expression with dissemination of glioblastoma. Neurosurg. Rev., 2010, 33(2), 175–183.
82. Di Tomaso, T.; Mazzoleni, S.; Wang, E.; Sovena, G.; Clavenna, D.; Franzin, A.; Mortini, P.; Ferrone, S.; Doglioni, C.; Marincola, F. M.; Galli, R.; Parmiani, G.; Maccalli, C. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin. Cancer Res., 2010, 16(3), 800–813.
83. Ji, J.; Black, K.L.; Yu, J.S. Glioma stem cell research for the development of immunotherapy. Neurosurg. Clin. N. Am., 2010, 21(1), 159–166.
84. Ailles, L.; Prince, M. Cancer stem cells in head and neck squamous cell carcinoma. Methods Mol. Biol., 2009, 568, 175–193.
85. Zhang, P.; Zhang, Y.; Mao, L.; Zhang, Z.; Chen, W. Side population in oral squamous cell carcinoma possesses tumor stem cell phenotype. Cancer Lett., 2009, 277(2), 227–234.
86. Brunner M.; Thurnher, D.; Heiduschka, G.; Grasl, M.Ch.; Brostjan, C.; Erovic, B. M. Elevated levels of circulating endothelial progenitor cells in head and neck cancer patients. J. Surg. Oncol., 2008, 98(7), 545–550.
87. Zhang, Q.; Shi, S.; Yen, Y.; Brown, J.; Ta, J.Q.; Le, A.D. A subpopulation of CD133(+) cancer stem-like cells characterized in human oral squamous cell carcinoma confer resistance to chemotherapy. Cancer Lett., 2010, 289(2), 151–160.
Getting an Insight into the Complexity of Major Chronic Inflammatory and Degenerative Diseases: A Possible New Systemic Approach to Their Treatment
Current Pharmaceutical Biotechnology, 2015, Volume 16, Number 9: 793–803. © Bentham Science Publishers.
P. M. Biava, G. Norbiato
This paper describes why chronic diseases, including not only cancer, but also the metabolic syndrome, chronic inflammation, chronic degenerative diseases, and others, make diagnosis, prevention, and targeted therapeutic treatment particularly difficult. Recognition that chronic inflammation may induce genetic, neuro-endocrine, immune, and metabolic changes in a series of diseases can be used for designing new approaches to treatment. This study suggests a new approach, including reprogramming. The use of stem cell differentiation stage factors as epigenetic reprogramming factors of all cells of the psycho-neuro-endocrine-immune system can restore the balance of a system that has been disrupted by chronic inflammation.
Introduction
Exposure to threats to body homeostasis promotes survival through activation of multiple interacting systems including the behavioral, neuro-endocrine, immune, and metabolic systems that produce an integrated stress response. While the response is initially adaptive, prolonged activation of these systems becomes maladaptive by inducing pronounced changes in body physiology and behavior that have deleterious implications for survival and well-being. The immune system is recognized best for its role in protection against pathogens that cause disease. However, immune activation is not solely relegated to attack against foreign pathogens, but plays an integral role regulating homeostatic conditions related to immune surveillance against tumor genesis and chronic inflammatory diseases. Thus the immune system represents a complex network requiring tight regulatory control and the interaction between neuro-endocrine and immune system is believed to be essential in defining the mechanism regulating infectious and non-infectious disease states.1-2 It is now accepted that the nervous system can receive input from the immune system via the release of cytokines and other immune mediators.3 At the same time, the immune system is also receptive to signals conveyed from neuro-endocrine and neuro-transmitter-mediated signals suggesting a bi-directional interplay between the release of neuro-endocrine hormone factors and cytokines.4-5 The integration of metabolism and immunity can be traced back to an evolutionary need for survival. Accordingly, the primary role of physiological adaptation is to find the energetically most efficient system configuration. In this sense a disease indicates a system-wide deterioration with declining efficiency of energy capture and utilization. The initiation and maintenance of immunity is a metabolically costly process. Sepsis can increase human metabolic rate by 30–60% and maintenance of phagocytes during infection needs approximately the same energy consumption.6 Inflammation, glucocorticoids, and insulin signaling pathways also proved to be involved in metabolic physiological adaptation linked programs.
Glucocorticoids (GCs) are essential endocrine hormones, involved in the regulation of almost all major physiological functions, including energy metabolism, cell proliferation and differentiation, reproduction, immune system, and cardiovascular and brain function. Further GCs, which are with insulin the main metabolic hormones, have an important metabolic function as they provide substrates for oxidative metabolism by increasing hepatic glucose production, adipose tissues lipolysis, and proteolysis. Binding insulin to its receptor triggers the tyrosine phosphorylation of its cellular substrate, such as the insulin receptor substrate family protein7 that is crucial signaling and molecular events for many metabolic effects of insulin.7, 8 Insulin effects are inhibited during stress and inflammation through modification of serine phosphorylation, mediated by intracellular pathways. Modifications that impair action from insulin can be triggered by increased cortisol activity, pro-inflammatory cytokines, and nutrients such as lipids.8 Inhibitory effects of inflammation on peripheral insulin sensitivity have been observed in obesity, in subjects suffering from insulin resistance and type-2 diabetes. GCs play a major role in the body response to stress and inflammation, maintaining a complex tightly balanced system in the brain and periphery that translates the effect of stress on specific tissues. Stress and inflammation are potential factors for a large number of diseases, ranging from peripheral illness such as diabetes, obesity, cardiovascular diseases, cancer,9, 10, 11 to many psychiatric disorders,12 including major depression, schizophrenia, drug addiction, post-traumatic stress disorder, and Alzheimer’s disease.13
The prevalence of these diseases has increased rapidly over the past decades. A great proportion of global disorders involves the failure to resolve inflammation that appears to be chronic from the onset. However, acute and chronic inflammation may coexist over long periods implying continuous re-initiation. Post-inflammatory tissue repair requires the coordinate restitution of different cell type and structures including epithelium, mesenchymal cells, extracellular matrix, and vasculature. So, inappropriate reconstruction of inflammatory tissues may preclude repair, resulting in atrophy, fibrosis, and damages to tissue function, which in turn activates inflammation.
Since inflammatory mechanisms are dominant over homeostatic control, thus inflammatory mediators may alter the homeostatic control leading to alterations of a complex regulatory network in which different adaptive systems, such as the neuro-endocrine-immune-metabolic systems, work continuously in concert establishing new equilibria. This characteristic accounts for the pathological strength of chronic inflammatory responses.9
Energy regulation (EnR) is the most important factor for homeostatic regulation of physiological processes. Neuro-endocrine pathways are involved in EnR. Insulin, insulin-like growth factor, estrogen, androgen, and hosteocalcin are factors that provide energy-rich fuels to stores, while hypothalamic pituitary adrenal axis, the sympathetic system, thyroid hormones, glucagon, and growth hormones provide energy-rich substrate to consumers. It is hypothesized that unresolved inflammation involves the entire body to divert fuels to the activated immune system. As EnR and the neuro-endocrine-immune interplay have not evolved to cope with long-lasting chronic inflammation, the coordination of the interlinked pathways will not function in chronic inflammation as expected for short-lasting diseases. The abnormal control leads to multiple disease-related sequelae, the pathogenesis of which can be explained by alteration of EnR and, subsequently, changes in the neuro-endocrine-immune-metabolic pathways. Physiopathology of energy regulation has been extensively reviewed.14 The biologic complexity of unresolved inflammation and a limited understanding of dysfunctions underlying many chronic diseases, including the metabolic syndrome, brain diseases, and cancer, combine to make the diagnosis, prevention, and targeted therapeutic treatment particularly difficult. Moreover, every anti-inflammatory therapy now in use has one or more serious toxicity potentials, and medical treatments have not focused on pathogenic factors other than inflammation, the regulatory cure of noninfectious infectionassociated diseases.15 Recognition that chronic inflammation may induce epigenetic, genetic, neuro-endocrine, immune, and metabolic changes in a series of diseases is useful for designing new targets for prevention and treatment.16
The current review describes how unresolved inflammation plays an important role in high morbidity diseases including cancer, brain diseases, obesity, and HIV infection. Principal mediators, signaling pathways, biomarkers, and potential therapeutic implications are to be taken into consideration. Owing to the complexity of the networks involved in such diseases, a landscape analysis of inflammatory, immune, endocrine, metabolic, and oncogenic pathophenotypes has been performed, and biological treatment with stem cells differentiation stage factors has been proposed.
Cancer-Related Inflammation
It has long been recognized that the immune system plays a role in the development of tumors. Immune cells can suppress tumor development by killing tumor cells; conversely they can also promote tumor progression. A tumor can be completely eliminated or kept dormant by tumor inhibiting inflammation consisting in production of tumor inhibiting cytokines, in the infiltration of cells of both the innate immune system such as dendritic cells, natural killer cells, and the adaptive system TH1—CD4+ and CD8+.17, 18 In the escape phase a tumor often develops multiple mechanisms to evade anti-tumor cells-mediated destruction19 and induction of immune suppressive microenvironment by tumor and stromal cells that promote cancer cells’ proliferation and migration.20 Approximately 18% of cancer cases worldwide are attributable to infectious diseases caused by bacteria, virus, and parasites, while inflammation can account for approximately 25–50% of human cancer. Key regulators of tumor progression are transcription factors, cytokines, reactive oxygen, nitrogen species (RONs), and gonadal hormones.
As can be seen, the collective immune effects of cytokines, micro RNA and RONs may be either pro-or anti-inflammatory or pro-and anti-tumorigenic, depending on their balance, thus increasing the complexity of the systems involved. The pathophysiology of cancer related inflammation has been extensively reviewed by Mantovani21 and Schetter.22
Reactive Oxygen and Nitrogen Species (RONs)
An inflammatory stimulus leads to the recruitment and activation of various immune cells, including macrophages, neutrofils, and dendritic cells, that release reactive oxygen species and reactive nitrogen species (RONs). These radicals have a role for an efficient immune response that may be either tumorigenic or anti-tumorigenic depending on concentrations.23 Increased RONs may cause genomic instability that contributes to carcinogenesis by mutating proto-oncogenes and tumor suppressor genes. Elevated RONs can post-translationary modify proteins, rendering them auto-antigenic, so increasing angiogenesis and metastatic potential. Parasites, viruses, helicobacter pilory are important risk factors for cholangiocarcinoma, epatocellular carcinoma, cervical and gastric cancer. RONs can be induced by various factors, including inflammatory cytokines and NFkB, hypoxia, and microbial endotoxin.24 Whether nitric oxide (NO) produced by RONs has pro-tumorigenic and anti-tumorigenic effects depends on the status of p53 tumor suppressor gene, which, if activated, leads to growth arrest, DNA repair, apoptosis, and anti-carcinogenic effect. In the absence of functional p53 an inflammatory stimulus can lead to overproduction of (NO) leading to a tumorigenic condition.
Cytokines
Cytokines are signaling molecules that are key mediators of inflammation or immune response. Pro-inflammatory cytokines include IL-1, IL-6, IL-15, IL-17, IL-23, TNFα, and anti-inflammatory cytokines include IL-4, IL-10, IL-13, transforming growth factor, TGFß and interferon (INFα). Depending on the balance of cytokines, their collective effects may be either pro-or anti-inflammatory. Cytokines binding to membrane receptors activate signal transduction pathways that regulate apoptosis, cell proliferation, angiogenesis, and cellular senescence.25 For example: TNFα, by activating the inflammation transcription factor NF-kB can increase tumor growth and the metastatic potential. IL-6 activates the Janus kinase signal transducer and other activators of transcriptor signal pathway that are tumorigenic.26 IL-10 and TGFß are anti-inflammatory cytokines. The main function of IL-10 is to suppress NF-kB which is associated with inhibition of TNFα, IL-6 and IL-12.27
Along with NF-kB, STAT3 and IL-6 are a point of convergence of numerous oncogenic signal pathways.26 The pro-inflammatory cytokines IL-1ß secreted by malignant cells of infiltrating leucocytes increases tumor adhesion and invasion, angiogenesis, and immune suppression.
Micro RNA
Micro RNAs are small, non-coding RNAs that regulate the translation of specific genes in inflammatory and cancer diseases. Stimuli that induce micro RNA expression include direct transcriptional activation or repression from transcriptional enhancers, epigenetic modification of the genome, genomic amplification or dilation, cellular stress, and inflammatory stimuli. Induction or repression of micro RNA expression influences cell fate, cell proliferation, DNA repair, DNA methylation, and apoptosis and provide pro-inflammatory or anti-inflammatory stimuli. Micro RNAs have an essential role in both adaptive and innate immune systems.28
Chronic Inflammation in the Tumor Micro-Environment
Key features of cancer related inflammation (CRI) include the infiltration of a wide number of blood cells, preeminently tumor-associated macrophages (TAMs); the presence of cytokines such as TNFα, IL-1, IL-6; chemokines such as CCL2 and CXCL8; and transcription factors.29-30 Endogenous promoter NF-kB has a key role in regulating innate immunity both in tumors and inflammatory cells. Besides neoplastic cells, a tumor is composed of stroma containing fibroblast, vessels, and leucocytes. Pro-inflammatory cytokines TNF can involve enhanced tumor growth, invasion and inhibition of glucogen synthase kinase 3 Beta, that contribute to tumor development.31-32 The pro-inflammatory cytokine IL-1 Beta, TNalfa, and IL6 are known to stimulate cancer cells to metastasize. IL-1 beta secreted by malignant cells or infiltrating leucocytes increases tumor adhesion and invasion, angiogenesis, and immune suppression. TAMs are principle leucocytes subset driving an amplification of the inflammatory response in the tumor.
Sex Steroid Hormones and Cancer
Breast Cancer
Breast cancer continues to be the most common cancer in women and represents a major issue of public health.33 The majority of estrogen receptors negative (ER-) breast cancers develop resistance to adjuvant hormone therapy and triple negative breast cancer lacks effective targeted treatment. Initially, tumors can be completely eliminated or kept in a dormant state by tumor-inhibiting inflammation characterized by the production of tumor-inhibiting cytokines and infiltration of cells of both the innate immune system such as dendrites cells (DCs) and natural killer (NK) cells and the adaptive immune system such as TH1, CD4+, and CD8+ T-cells.34-35-36 In the escape phase, breast tumors may develop multiple mechanisms to evade immune surveillance that include the creation of autonomous cancer cells to evade anti-tumor cells, cell-mediated destruction, and the induction of an immunosuppressive microenvironment by tumor and/or stroma cells that directly promotes cancer cell proliferation and migration.37 Immune cells can suppress tumor development or inhibit their growth; conversely they can promote tumor progression, establishing an immune suppressive micro-environment.
Biomarkers of breast cancer risks are: insulin resistance, estrogen resistance, hyperinsulinemia, and low grade chronic inflammation, which are major predictors of adiposity, diabetes risks, and breast cancer risks. Both non-tumoral and malignant breast tissue and cells are endowed with key enzymes of steroid metabolism, including 17beta hydroxysteroid dehydrogenase, 5beta-reductase and aromatase. Locally produced or metabolically transformed estrogen may differently affect proliferal activity of breast cancer cells.38 Studies on breast cancer demonstrate that immune network and locally produced estrogens may play a significant role in the development and progression of breast cancer.
Altered insulin receptor binding promotes mitosis and anti-apoptotic effects in breast cancer cells and also tumor cell migration and tumor-associated angiogenesis. Chronically elevated insulin can enhance estrogen bio-activity and promote activities of breast cancer related adipokines.39 Increased systemic reactive oxygen species (ROS) is hypothesized to play a central role in breast carcinogenesis and in carcinogenesis causal pathways linked to obesity.40 Because telomeres (nucleoprotein repeats at the end of chromosomes that protect cells from chromosome instability) suffer disproportionately from oxidative damage, telomere attrition may be considered an important cause of breast cancer. Finally, global DNA hypo-methylation is recognized to be a key epigenetic mechanism associated with increased cancer risks, namely breast cancer risk.41
Prostate Cancer
Prostate cancer (PC) remains one of the most widespread cancers in males. The androgen receptor is the most regulatory transcription factor in cells of prostatic lineage, and this regulatory function is maintained in PC cells. The frontline treatment involves androgen deprivation therapy, which is achieved by blocking androgen production with surgery or androgen antagonist. Suppression of androgens/androgen receptors signaling is effective at improving cancer symptoms and prolonging survival. However, testosterone deficiency leads to the development of the metabolic syndrome suggesting that steps be taken early to manage metabolic complications associated with prostate cancer.42 PC and inflammation are closely linked, so much that cancer patients show both local and systemic changes in inflammatory parameters.
In some cancer types, inflammatory conditions occur before malignant changes, whereas in different types of cancer, an oncogenic alteration generates an inflammatory micro-environment that induces development of tumors.43 Recent studies uncovered the relation among estrogens and androgens, steroid hormones, cytokines IL-1 and IL-2, and prostate cancer. Androgen aromatization to estrogen may play a role in prostate carcinogenesis and tumor progression. Estrogens combined with androgens appear to be required for the malignant transformation of prostate epithelial cells.44 Interestingly, IL-2 produced by macrophages in the tumor micro-environment converts androgen receptor from inhibitor to modulator, thus inducing resistance to hormonal therapies.45
The Brain and the Adaptive Stress System
The perception of a potential threatening situation activates the HPA axis, the paraventricular nucleus of the hypothalamus where corticotrophin-releasing hormone (CRH) and vasopressin stimulate ACTH, which in turn stimulates the adrenal glands to release cortisol in humans. Cortisol is a pleiotropic hormone that is involved in almost every cellular, molecular, and physiological phenotype essential for life. During evolution, several mechanisms appeared to allow a fine-tuned regulation of glucocorticoid signaling that includes glucocorticoid receptors (GRs), mineral corticoid receptors (MRs), and two enzymes, 11Beta hydroxy steroid dehydrogenase type-1 (11beta HSD-1) and type-2 (11Beta HSD-2) that interconvert active cortisol into inactive cortisone. The glucocorticoid receptors have a number of complex interactions with the epigenome. Epigenetic mechanisms are considered essential for the transduction of environmental inputs, like stress, into lasting physiological and behavioral changes. Stress has a number of effects on brain epigenetic mechanisms producing alterations in DNA methylation and histone modifications in most of the stress-sensitive brain regions.46, 47 Many of these changes may be maladaptive and contribute to neuro-degenerative and neuro-psychiatric diseases.
The GR is a ligand-regulated transcription factor member of the nuclear receptor superfamily, which controls gene expression linked to several processes like inflammation, stress response, glucose homeostasis, lipid metabolism, proliferation, and apoptosis development.48 The biological action of glucocorticoids is mediated by intra-cellular GRs that, when bound to homologous ligand, function as DNA-bind protein that enhances or represses basal transcription rate of responsive genes. In most cells GCs promote reduction in GR levels and consequently limit the span of cellular responsiveness to GCs.48 GCs act as the critical negative feedback on all myeloid cells, including those present within the brain parenchyma. An inappropriate feedback of GCs on microglia and high circulating GCs levels in stressed individuals have been associated with deleterious consequences for the brain. Previous studies have proven involvement of G-protein and the signal regulated kinase-CREB pathway in rapid GCs effects.49 This same pathway is also activated by rapid signaling of other steroid receptors, such as oestrogen, androgen, and progesterone receptors and by rapid aldosterone signaling through the MRs in peripheral tissues.50 This suggests that a specific type of G-protein is engaged in multiple hormone actions in the brain and periphery and gives reason of glucocorticoids, mineralocorticoids, and sex hormones involvement in chronic inflammatory diseases and correlated disorders. Gonadal steroids have a similar capacity to produce epigenomic reorganization, suggesting that nuclear hormone receptors are shapers of chromatin structure besides their known role of transcription factors.51
Glucocorticoid Receptors, Neurotransmitter, and Innate Immune Systems in Brain Disease
GCs enter the brain and bind to MRs and GRs. MRs have the function to maintain integrity and stability of limbic circuits and play a potential role in maintaining homeostasis,52 while GRs play a reactive role in stress response, facilitate recovery of brain, distribute energy and maintain circulating levels of cortisol within normality through inhibition of HPA axis.53 The brain has its own innate immune response, which is activated in response to both central and systemic immune challenges.54 Inflammatory signals are known to act on the brain through key behavior-modulating multiple neurotransmitter systems, including serotonin, norepinephrine and dopamine. Activation of brain-neurotransmitters in turn activate pro-inflammatory cytokines, including IL-6 and TNFalfa, which influence the social and affective neural processes that induce depressed mood. GCs play a major role in the response of the brain to inflammatory stress, which depends on an NF-kB signal transduction pathway. Sleep loss induces increases in cellular and genomic markers of inflammation such as IL-6, TNFalpha.55 Parenchymal microglia may produce elevated levels of molecules of the innate system such as IL-6, TNFalfa, and IL-1 beta, that can induce cell death in neuro-degenerative processes.56 It has been hypothesized that the majority of psychiatric diseases are associated with the chronic degeneration of astroglia, which compromise brain homeostatic and defensive capability in diseases like schizophrenia and major depression disorders.57 Research findings suggest a complex tightly balanced system in the brain and periphery that translates the effect of stress on specific tissues. Serious challenge to human capacity to adapt is associated with multiple morbidities, including heart disease, stroke, hypertension, diabetes, depression, obesity, and metabolic syndrome. Inflammation highly contributes to these comorbidities.58
Glucocorticoid Receptor Resistance
GC effect is ultimately determined at the level of the GR. Tissue sensitivity to GC differs among individuals, within tissues of the same individual, and within the same cells.59 A reduced GR expression or binding affinity to its ligand, nuclear translocation, DNA binding or interaction with other transcription factors (NF-kB, AP-1), can lead to a state of GRs resistance.60-61 Epigenetic changes in GR gene resulting in early life behavioral program have been shown in rats and humans.62-63 GR is a severe problem associated with various inflammatory diseases.
Environmental factors that can induce GR resistance include chronic inflammation, exposure to infectious agents, chronic exposure to GCs. A decreased GR sensitivity to GCs has been shown in immune cells of HIV infected individuals.64 Repeated social defeat induces GR resistance in macrophages. Epigenetic regulation such as DNA methylation and micro RNA expression may play a role in inducing glucocorticoid resistance.65 GR resistance may also occur in asthma, rheumatoid arthritis, inflammatory bowel disease, and psoriasis, thyroiditis, lupus-like autoimmune syndrome.66 GR resistance is also found in the induction phase of anti-leukemia therapy, in non-lymphoid malignancy, and in humans small cell lung carcinoma cell lines.67 Insufficient GCs signaling resulting from decreased hormone bioavailability or reduced GR sensitivity may have devastating effects on body function. Such effects might be related in part to the role of glucocorticoids to restraining activation of the immune system and other components of the stress response. Stress-related neuropsychiatric disorders, associated with immune system activation/inflammation, may contribute to stress-related pathology, including alteration in behavior, insulin, cortisol sensitivity, and acquired immune responses.68 Glucocorticoid resistance via impaired GR function reduces inhibitory feedback of HPA axis in inflammatory response, so influencing stress-related illness such as depression, metabolic syndrome, cardiovascular disease and osteoporosis, having inflammation as a common final pathway.
Obesity and the Metabolic Syndrome: The Role of Inflammation
The incidence of obesity is rising steadily throughout the world. Obesity has damaging effects on many organ systems; many of the co-morbid conditions are related to a metabolic syndrome characterized by large waist measure, high triglyceride levels, glucose intolerance, hypertension, and risk factors for the development of type-2 diabetes mellitus, systemic hypertension, coronary heart diseases, and heart failure. The incidence of respiratory disease, gastro-intestinal and musculo-skeletal disorders, thrombo-embolism, stroke, and cancer are increased with obesity.69 Unhealthy eating habits, lack of exercise, and chronic stress as well as chronic inflammation have been considered causes of obesity and its pathological manifestation. Chronic stress has been associated with altered hormone secretion such as hypersecretion of cortisol and catecholamines, pro-inflammatory cytokines, as well as changes in circadian rhythm with evening elevation of cortisol70 at a time during which the sensitivity of tissue to glucocorticoids is increased. This initiates a process that leads to accumulation of total but mostly visceral fat.
Environmental factors, unbalanced maternal nutrition, and an altered feeding behavior during the period of early development have also been suspected to lead to obesity. Stress and the early-life nutritional habit can affect the brain development leading to abnormal feeding behavior and neuro-endocrine alterations long-term.71 Recent evidence has demonstrated alterations in epigenetic status following manipulation of nutrient environment during the developmental period.72 Early life environment, if stressful, can have a life-long influence on these mechanisms, leading to hyperactive HPA axis and high cortisol plasma levels. HPA axis activation was found in adults of both sexes with low birth weight. Interestingly, alterations in DNA methylation of genes involved in cortisol regulation are present in adulthood in association with cardio-metabolic risk.73
All these data suggest that the neuro-endocrine-immune function might be damaged long before the METS onset. Studies have demonstrated that both obesity and metabolic disorders are associated with poorer cognitive performance, cognitive decline, and dementia. Further, there is a clear association between psychiatric medicine and significant weight gain.74 There is growing understanding of a role of hypothalamic pituitary-adrenal axis dysfunction and basal systemic low-grade inflammation in the relationship between psychiatry and obesity. Both hypertrophied adipocytes and adipose tissue-resident lymphocytes and macrophages contribute to increasing circulating levels of pro-inflammatory cytokines, including TNFalfa, and important feeding relating peptides such as leptin and resistin, plasminogen activator inhibitor-1, C-reactive protein, interleukins IL-1 beta and IL-6 in obese individuals.75
Although evidence strongly suggests that GC action has a central role in METS, the state of GC excess is rare and the circulating level of cortisol is normal in the majority of patients with obesity and METS.76 Then it was postulated that an elevated local regeneration of active glucocorticoids by 11ß-HSD1 may be an important factor in the development of complications associated with METS.77 However, subsequent clinical trials with selective inhibitors compounds of 11beta HSD-1 showed only very moderate effect at high drug dose on diabetes and metabolic syndrome, and pre-clinical studies were interrupted.78
Metabolism, like inflammation, has complex control mechanisms. Nuclear receptors are among the most important regulators of metabolism and inflammation.79 Moreover, nuclear receptors can both affect and be affected by reactive oxygen intermediate (ROI) and nitrogen intermediate (RNI) that have dual function in influencing both metabolism and inflammation in obesity. Glucocorticoid receptors and gonadal steroid receptors have similar capacity to produce epigenomic reorganization in adipose cells in addition to their well-established role as transcription factors.79
The increased number of macrophages that infiltrates obese adipose tissue, along with other cells of the stroma, also contribute to production of inflammatory cytokines.80 During early diet induced obesity, there is an adaptive overproduction of vaso-dilators called adipocyte-derived relaxing factors (ADRF) that occurs in perivascular adipose tissue (PVAT). Under physiological conditions PVAT release a number of vasoactive substances such as ADRF, adiponectine, angiotensin, leptin, and nitric oxide that elicit a net beneficial anti-contractile effect on vascular function and are essential for the maintenance of vascular resistance.81 However, in established obesity, PVAT loses its anti-contractile properties by an increase of contractile, oxidative, and inflammatory factors leading to endothelial dysfunction and vascular disease.82 Weight loss by a different approach, including bariatric surgery by gastric bypass, significantly correlates with improvement in blood pressure levels, left ventricular mass, exercise capacity, and glucose tolerance. Thus, chronic inflammation appears to be a clinically important change that occurs in adipose tissue of subjects who become obese. Interestingly, expansion of fat without inflammation does not exert systemic metabolic effects such as systemic insulin resistance.83 Insulin resistance has a key role in the pathogenesis of the metabolic syndrome; it leads to an increased oxidative stress, mitochondrial dysfunction, DNA damage, and cell death. Insulin suppresses endogenous production of glucose while controregulatory hormones such as cortisol and glucagon increase endogenous glucose production. Glucocorticoids increase gluconeogenesis and blunt the suppressive effect of insulin on endogenous glucose production. The mechanisms by which glucocorticoids inhibit insulin activity are still not clear; however, studies on animals suggest an impairment of insulin-signaling cascade, leading to a reduced activation of insulin target protein and genes in liver cells. Modifications that impair the action of insulin can be triggered by cytokines, such as TNFs, indicating that immune mediators can have a crucial regulatory role in the glucose homeostasis.84
Emerging data demonstrate pivotal roles for brain insulin resistance and insulin deficiency as mediators of cognitive impairments and neuro-degeneration, particularly in Alzheimer disease.85 Other inflammatory pathways are induced by cytoplasmatic organelle stress owing to nutrient overload resulting in metabolic stress. In these cases, activation of kinase such as JUN N. terminal kinase (JNK), and IK kinase-beta (IKKbeta) leads to metabolic alterations.8 One such organelle, the endoplasmic reticulum (ER), has been shown to integrate inflammation and stress signal with metabolic status of cells, disruption of which results in disease such as type-2 diabetes.86 Organelle stress in the inflammation contributes to the development of both obesity-associated insulin resistance and chronic metabolic disease. Metabolic and inflammatory pathways may converge at many levels of the organisms, including the levels of cells-surface receptors, nuclear intracellular chaperones, and nuclear receptors. These molecular sites allow for the coordination between nutrient and immune response in order to maintain homeostasis under diverse metabolic, immune conditions. Although low grade inflammation is a known pathological component of obesity, other triggering factors have actually been identified. For example, gut-microbiota have been shown to have a role in the initiation of obesity and insulin resistance.87
Steroid hormones and their receptor signals that potentially modify body fat set-point include estrone, progesterone, aldosterone, retinoids, hydroxycalciferols, and especially their hormone receptors. The role of steroid hormones in obesity has been discussed by Alemany M.88 A high prevalence of METS was shown in men with testosterone deficiency or treated with testosterone receptor antagonist.89
In conclusion, obesity may result from a combination of dysfunction of brain circuits and neuro-endocrine hormones related to pathological over-heating, physical inactivity, and other pathological conditions. It is increasingly appreciated that perinatal events can set an organism on a life-long trajectory for either health or disease, resilience or risk. Extensive research has documented the effects across the life span of over-nutrition, with strong links for an increased risk for obesity, metabolic, endocrine, and immune disorders as well as adverse mental health outcomes.
HIV Infection
It has been noted that during HIV infection, activated immune cells secrete pro-inflammatory cytokines that stimulate the release of systemic glucocorticoids. The triggering of this regulatory loop during inflammatory processes usually provides an important control over the immune system. This did not occur in HIV infected patients that showed an impressive increase of Th-directed cellular immune response in spite of an elevated level of cortisol, suggesting a cortisol resistance syndrome.64, 90 Treatment of these patients with HAART determines a dramatic decline of HIV and immune deficiency–related cause of death, but suppression of HIV replication by HAART is not associated with a reconstitution of the immune function, and this may account for unresolved inflammation in such patients.91 This may lead to a cluster of conditions such as hypertension, high insulinemia, insulin resistance, dyslipidemia, fat redistribution, and increased risk of cardiovascular diseases, stroke, diabetes, and neuro-cognitive and metabolic brain disorders. HIV-associated neuro-cognitive disorders (HAND) are potential consequences of HIV infection, and about half of adults with AIDS suffer from neurological complications related to HIV-1. HAND includes a spectrum of neurological disorders ranging from an asymptomatic form of neuro-cognitive impairments to an intermediate form and the severe form of HIV-associated dementia.
The increased life span of treated patients results in a chronic exposure of the brain to HIV-1 virions and viral proteins leading to inflammation as well as a concomitant chronic inflammation in the peripheral immune system leading to further neurological damage.92 Chronic HIV-viral infection causes life-long antigenic stimulation and the development of a population of differentiated apoptosis-resistant, senescent T-cells, with limited proliferative potential.93 This phenomenon is dysregulated in part by the regressive state of telomeres, the repeated DNA sequences that cap the ends of the chromosomes.94 Reduction in telomere length influences the activity of P53 tumor suppressor pathways, apoptosis, and cell senescence.95 Chronic inflammation may be the most important candidate for telomere length disturbances. In states of inflammation, increases in oxidative stress and cellular division may lead to the accelerated erosion of telomeres, crucial genomic structures that protect chromosomes from decay.96 The final result is an immune system of a limited capacity to recognize novel antigens and hence to prevent disease. HAART treatment is also associated with activation of MR and renin angiotensin aldosterone system, which have hypertensive and pro-inflammatory effects; however, the usefulness of MR antagonists is limited. Efforts are underway to selectively modulate glucocorticoids, mineralocorticoids receptors, and other nuclear receptors. This is facilitated by structural function similarities of these receptors.97
Discussion
As modern society is troubled by complex multifactorial disease, research is called to govern complex realities, including unresolved inflammation, cancer, obesity, and metabolic syndrome and its detrimental cardiovascular complications, as well as depression and other heterogeneous brain disorders.
The brain is the central organ of stress and of adaptation to stressors because it perceives what are threatening or potentially threatening factors and initiates behavioral and physiological response to those challenges. However, it also is a target of stressful experiences and of the hormones and other mediators of the stress response.
In the response to a stressful encounter, the brain activates a complex stress system that engages the organism in an adaptive response to the threatening situation. The adaptive response acts on multiple peripheral tissues and on feedback to the brain. Diseases can occur when the balance between multiple players such as behavioral, neuroendocrine-immune-metabolic systems, and responsive tissues is upset, so that the adaptive systems convert into a maladapted, detrimental chain of events. It is under these conditions that inflammatory control mechanisms are engaged. The problem with inflammation is not how often it starts, but how often it fails to subside. Unresolved inflammation is not the primary cause of major and more frequent chronic diseases, such as cancer, brain disease, obesity, and HIV infection, but it contributes significantly to their pathogenesis.
Epigenetic mechanisms are dominant mechanisms for the transduction of environmental inputs such as stress and inflammation into lasting physiological and behavioral changes, and the development of neuro-psychiatric diseases.
Stress has a number of known effects on epigenetic marks in the brain, producing alterations in DNA methylation and histone modifications in most of the stress-sensitive brain regions, including the hippocampus, amygdala, and pre-frontal cortex.98–99 It has been shown that both acute and chronic stress are altered by methylation of histone. One of the key players in stress is the corticoid hormone receptor family, which affects brain functioning through both delayed genomic and rapid non-genomic mechanisms. Nuclear receptors (NR) superfamily governs diverse biological processes as development, physiology, and disease. Glucocorticoid nuclear receptors play an essential role in the response to environmental stressors. A number of diseases including autoimmune, infectious, and inflammatory disorders as well as certain neuro-psychiatric disorders, such as major depression, have been associated to a decreased responsiveness to glucocorticoids, which is believed to be related to impaired functioning of the glucocorticoid receptors. Glucocorticoid resistance may be a result of impaired GR function, secondary to exposure to inflammatory cytokines as may occur during chronic medical illness.100
Nuclear hormone receptors in general are significant shapers of chromatin structure, in addition to their role as transcription factors. Chronic inflammation associated with immune-mediated disease represents a profound stress factor for the immune system affecting cellular-turnover, replication, and exhaustion. Immune cell longevity is tightly connected to the functional integrity of telomeres, which are regulated by cell multiplication, exposure to oxidative stress, and DNA repair mechanism. The loss of the immune function in aging is associated with conditions that limit life span, such as infectious susceptibility and malignancy.101 Telomeres are the natural end of linear chromosomes, and function to cap chromosomal ends. Short telomeres force the cells to enter into senescence or apoptotic death. Tissues under permanent replicative stress, and brain cells’ ability to build memory, are particularly dependent on cell survival. Notably, chronic infection accelerates erosion of the cell telomeres in immune cells through increased proliferative stress. Reduction of telomeric length of PBMCs seems to be a predictor for progression of atherosclerotic disease, myocardial infarction, and reduction in left ventricular mass. Telomere length has been connected to cancer incidence and cancer mortality.102 Reduction in telomere length influences the activity of P53 tumor suppressor pathways, malignant cell transformation, apoptosis, and cell senescence.95 For reasons that remain merely defined, long-term treated HIV persons have a shortened life span, which is linked to an increased risk of complications, including heart, cancer, liver, kidney, and bone disease and neuro-cognitive decline. This phenomenon is regulated in part by the progressive reduction in telomere length. Notably, patients with type-1 diabetes mellitus (DM) present a reduction in telomeric length in arterial cells. Such reduction correlated strongly with the HbA1C concentration. In addition, telomeric length in arteries and mononuclear cells of patients with uncontrolled DM were significantly shorter than telomeres in mononuclear cells of patients with well-controlled DM.103
The development of strategies that slow down the loss of immune function with progressive cell age has the potential to afford novel therapeutic avenues for a number of degenerative diseases and modifies epigenetic marks contributing to disease developing. Epigenetic silencing of gene transcription by methylation of DNA or modifications of histones is a key event in neoplastic initiation and progression. Alterations in the epigenome have been identified in virtually all types of cancer and involve multiple genes and molecular pathways. It has been proposed that epigenic gene-disregulation may represent a first step in tumor genesis, possibly by affecting the normal differentiation of stem cells and predisposing the cells to additional oncogenic insults. Aberrant DNA methylation and histone acetylation has been linked to a number of age-related disorders, including cancers, obesity, metabolic syndrome, type-2 diabetes, autoimmune disorders, and others. Since epigenetic alterations are reversible, modifying epigenetic marks contributing to disease development may provide an approach to designing new therapies.104 Inhibitors of DNA-methyl transferases or histone diacetilases have been approved for clinical use.105
Therapeutic Interventions
Mesenchymal stem cells (MSCs) represent a promising tool for treatment of chronic inflammatory diseases. MSCs appear to suppress inflammation through secretion of anti-inflammatory cytokines such as IL-10 and GFbeta,106 interferon-gamma,107 soluble human leucocytes antigen-G 92, L-1 receptors agonist,108 and expression of immune regulatory enzymes such as cyclo-oxigenase and indolamine 2,3 deoxygenase.109 MSCs inhibit immune globuline production and arrest B-lymphocytes cell cycle. They also interfere with dendritic cell differentiation, maturation, and function. Based on these properties, MSCs have been used in regenerative medicine, for the treatment of autoimmune diseases,110, 111 and for inhibiting tumor proliferation. MSCs are now used as an easy-to-harvest tool for cell therapy and exhibit robust multipotency and multilineage potential in vitro. Despite these attractive features, MSCs also undergo significant senescence and decline in multipotency expression after multiple passages in culture.112, 113 These findings raise cautionary notes whenever long-passaged MSCs are used in a clinical setting and prompt the need for novel approaches that may oppose senescence and optimize the expression of multipotency in such a promising tool for cell therapy.
Recent studies demonstrated that the exposed MSCs in the presence of Zebrafish growth and differentiation factors harvested at different developmental stages showed that only the late (20 somites and pharingula) developmental stage factors had significantly decreased cell proliferation and viability. These factors determined the activation of a differentiation or proapoptotic program, as shown by the derangement in nuclear morphology and the chromatin condensation, and by the activation of the cascade of caspases. For these reasons the factors taken from these late stages of differentiation of the Zebrafish embryo are used to control tumor growth114, 115, 116, 117, 118, 119, 120, 121 in the clinical treatment of patients, first of all patients affected by hepatocellular carcinoma.122, 123 These factors are also used to slow down the high speed of multiplication of cheratinocites in psoriasis.124 Otherwise, the factors of the first stages of development (middle-blastula-gastrula, tail bud, 5 somites) did not induce MSCs apoptosis, nor did they decrease cell viability. This contrasting behavior may result from a fine equilibrium between first stages-induced transcription of Oct-4, Sox-2, TERT, Bmi-1, and c-Myc, known to inhibit apoptotic pathways and the increase in Bax/Bcl-2 mRNA ratio observed in the first stages-exposed MSCs.125
During Zebrafish embryogenesis, the expression of the pluripotency genes Pou5f1/Oct-4 and Sox-2 is timely regulated by defined factors that are mainly restricted to the early developmental pattern (during the first hpf).126, 127, 128 Our data show that MSCs are able to selectively respond to factors restricted to the very early, but not late, developmental stages of Zebrafish embryo with a transcriptional increase in the same two stemness-related genes necessary for the expression of family members of transcription factors that contribute to the maintenance of human stem cell pluripotency and self-renewal. The over-expression of stemness genes elicited by the first stages of cell differentiation factors was paralleled by an increase in the transcription of both Bmi-1 and TERT. Notably, these findings indicate that both genes exert a major role in counteracting aging processes in vivo and cell senescence in vitro. Bmi-1 is emerging as a major aging repressor and is transcriptionally down-regulated when cells undergo replicative senescence.129 TERT opposes cell senescence by counteracting telomere shortening.130 Studies on brain development in mice have correlated a decrease in TERT expression and activity with decreased neuroblast proliferation and differentiation. Moreover, it has been demonstrated that MSCs or bone marrow stromal stem cells lacking telomerase activity undergo premature cellular senescence, with a progressive decline in the expression of early mesenchymal stem cell markers.
The maintenance of stemness gene expression is also important in the prevention of cell senescence. The singular loss of the Bright/Arid3A transcription factor, the founding member of the ARID family of transcriptions factors, which binds directly to the promoter/enhancer regions of Oct-4, Sox-2, contributing to their repression in both mouse embryonic fibroblasts (MEFs) and mouse embryonic stem cells (ESCs), was found to bypass the cell senescence barrier, leading to MEF reprogramming.131, 132 We show for the first time that human stem cell exposure to early developmental stage of Zebrafish embryo stem cell growth and differentiation factors may represent a useful tool to enhance stem cell expression of multipotency and activate both telomerase-dependent and -independent antagonists of cell senescence. In addition we describe in another article of this special issue that the mix of all stages of cell differentiation taken from Zebrafish embryos are able to prevent in a very significant way the neuro-degeneration of the cell of the hippocampus.
All these effects were obtained without gene manipulation through viral vector mediated gene transfer, or expensive synthetic chemistry. In conclusion, we can say that exposure to early developmental stage of Zebrafish embryo growth and differentiation factors may enhance stem cell expression of multipotency and activate both telomerase-dependent and -independent antagonists of cell senescence. These outcomes may prove rewarding during prolonged expansion in culture, as it occurs in most cell therapy protocols. These factors can also be used as antiaging factors in old patients in clinical use. In the opposite direction, the factors taken from the late stages of cell differentiation of Zebrafish embryo can be used to control cell proliferation of tumor cells and to slow down the high speed of multiplication of cheratinocites as it happens in psoriasis and in many other diseases. Finally, all the network of factors present in all stages of cell differentiation can be used to prevent neuro-degeneration and other degenerative phenomena in chronic diseases and to maintain the homeostasis of the organism under stress and under consequences of stress phenomena (strain).
Conclusion
Complex signal transduction pathways govern regulated development processes of multi-cellular embryos. Aberrant regulation and transduction of embryonal pathways has been implicated in birth defect, alterations in tissue regeneration, stem cell renewal, and cancer growth.133 This might explain vulnerability of most traits of human anatomy and physiology that negatively affect homeostasis. Disruption of homeostatic mechanisms due to the impact of acute life-style and genetic background and early life environment can have a life-long influence on neuro-endocrine mechanisms connecting stress to immune response and metabolism and leads to chronic activation of inflammatory mechanisms. Serious challenge to human capacity to adapt is associated with multiple morbidity, including heart disorders, stroke, hypertension, diabetes, metabolic syndrome, cancer, HIV infection, and neurodegenerative disease.
Epigenetic mechanisms are considered essential for the transduction of environmental stress experiences into chronic inflammatory diseases, leaving traces expressed in the genome. Acute and chronic stress are altered by DNA methylation and histone modifications in stress-sensitive brain regions that may contribute to neuro-degenerative disorders. Aberrant DNA methylation and histone acetylation have been also linked to a number of disorders, including cancer, obesity, metabolic syndrome, type-2 diabetes, and autoimmune disorders. Nuclear receptors are among the most important regulators of metabolism and inflammation: they play an essential role in the response to environmental stressors. Reduced sensitivity of glucocorticoid receptors, induced by epigenetic changes, may lead to cortisol resistance status that has devasting effects on body function involving excessive deterioration of immune and metabolic functions. Glucocorticoid receptors and gonadal steroid receptors have similar capacity to produce epigenomic reorganization in dipose cells in addition to their established role as transcription factors.
Efforts are underway to modulate selectively glucocorticoids, mineralocorticoid receptors, and other nuclear receptors. This is facilitated by the structural function similarities of these receptors.
Chronic infection has been shown to accelerate erosion of telomeres in immune cells. Such alterations have been connected with cancer induction and mortality and changes in P53 tumor suppressor pathways influencing malignant transduction, apoptosis, and cell senescence. Reduction of telomere length has been also observed in long-term treated HIV patients and in type-1 diabetes mellitus, and it is associated with increased risks of severe metabolic, vascular, and brain disturbances.
The final result of non-genomic alterations just described is an immune system of limited capacity to recognize novel antigens and hence to prevent chronic degenerative disorders. Since epigenetic alterations are reversible, modifying epigenetic marks contributing to disease development may provide an approach to design new therapies. We propose here an epigenetic treatment of chronic inflammatory and degenerative diseases using stem cell differentiation stage factors of Zebrafish embryos taken at the final stages of cell differentiation to induce cell differentiation, slow down proliferation of cancer cells, or of cheratinocites in psoriasis. On the contrary we can use stem cell differentiation factors taken at the early stages of cell differentiation to enhance stemness genes and multi-potency, activate both telomerase dependent and independent antagonist of cells senescence, and favor in this way the regeneration of tissues.
Our aim now is to provide novel insights regarding the potential combination of epigenetic reprogramming mechanisms in controlling plasticity and pluripotencies of stem cell populations, and define stem cells differentiation factors capable of repairing different pathological stem cells at the origin of numerous chronic diseases.
References
1. Steiman, L. Elaborate interactions between the immune and nervous systems. Nat. Immunol. 2004, 5:575–581.
2. Dantzer, R.; Bluthé, R. M.; Layé, S.; Bret-Dibat, J. L.; Parnet, P.; Kelley, K. W. Cytokines and sickness behavior. Ann N Y Acad Sci. 1998, 1;840: 586–90.
3. Dantzer, R.; Konsman, J. P.; Bluthé, R. M.; Kelley, K. W. Neural and humoral pathways of communication from the immune system to the brain: parallel or convergent? Auton. Neurosci. 2000, 85(1–3): 60–5.
4. Watkins, L. R.; Maier, SF.; Goehler, L. E. Cytokine-to-brain communication: A review & analysis of alternative mechanisms. Life Sci. 1995, 57(11): 1011–26.
5. Hopkins, S. J.; Rothwell, N. J. Cytokines and the nervous system. Expression and recognition. Trends neurosci. 1995, 18: 83–88.
6. Soltow, Q. A.; Johns, D. P.; Promislow, D. E. A network perspective on metabolism and aging. Integr. Comp.Biol. 2010, 50: 844–54.
7. Taniguchi, C. M.; Emanuelli, B.; Kahn, C. R. Critical nodes in signaling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006, 7(2): 85–96.
8. Hotamisligil, G. S. Inflammation and metabolic disorders. Nature. 2006, 44: 860–867.
9. Hotamisligil, G. S.; Erbay, E. Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol. 2008, 8; 923–934.
10. Libby, P. Inflammation and cardiovascular disease mechanisms. Am. J. Clin Nut. 2006, 83: 456S–460S.
11. Trinchieri, G. Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 2012, 30: 677–706.
12. Wyss-Coray, T.; Mucke, L. Inflammation in neurodegenerative disease—a double-edged sword. Neuron. 2002, 35: 419–32.
13. de Kloet, E. R.; Joëls, M.; Holsboer, F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005, 6(6): 463–75.
14. Straub, R. H.; Del Rey, A.; Basedowsky, H. O. Emerging concepts for the pathogenesis of chronic disabling inflammatory diseases: neuro-endocrineimmune interactions and evolutionary biology. In Ader, R., ed. Psychoneuro immunology. San Diego, CA: Elsevier-Academic Press, 2007, 217–32.
15. Okin, D.; Medzhitov, R. Evolution of inflammatory diseases. Current Biol. 2012, 225 R733–40.
16. Baffy, G.; Loscalzo. J. Complexity and network dynamics in physiological adaptation: an integrated view. Physiology and behavior 2014, 131: 49–56.
17. Schreiber, RD.; Old, LJ.; Smyth, MJ. Cancer Immunoediting: Integrating immunities’ roles in cancer suppression and promotion. Sci. 2011, 331 (6024): 1565–1570.
18. Vasely, MD.; Kershow, MH.; Schreiber, RD. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29(1): 235–271.
19. Shin, MS.; Kim, HS.; Lee, SH. Mutations of tumor necrosis factor-related apoptosis-including ligand receptor-1 and receptor-2 genes in metastatic breast cancer. Cancer res. 2001, 61: 4942–4946.
20. Jiang, X.; Shapiro, DJ. The immune system and the inflammation in breast cancer, Mol.cell.endocrinol 2014, 382(1): 673–682.
21. Mantovani, A.; Romero, P.; Palucka, AK.; Marincola, FM. Tumor immunity: effector response to tumor and role of the microenvironment. Lance 2008, 371(9614): 771–83. doi: 10.1016 /S0140–6736(08)60241–X.
22. Schetter, A. J.; Heegaard, N. H.; Harris, C. C. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis 2010, 31(1):37–49. doi: 10.1093/carcin/bgp272.
23. Pan, J. S.; Hong,M. Z.; Ren, J. L. Reactive oxygen species: a double-edged sword in oncogenesis. World J Gastroenterol. 2009, 14;15(14): 1702–7.
24. Hussain, S. P.; Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int. J Cancer 2007, 1;121(11): 2373–80.
25. Kisley, L. R.; Barrett, B. S.; Bauer, A. K.; Dwyer-Nield, L. D.; Barthel, B.; Meyer, A. M.; Thompson, D. C.; Malkinson, A. M. Genetic ablation of inducible nitric oxide synthase decreases mouse lung tumorigenesis. Cancer Res. 2002, 1;62(23): 6850–6.
26. Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G. Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; Karin, M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 3;15(2):103–13. doi: 10.1016/j.ccr.2009.01.001.
27. Mosser, D. M.; Zhang X. Interleukin-10: new perspectives on an old cytokine. Immunol Rev. 2008, 226:205–18. doi: 10.1111/j.1600-065X.2008.00706.x.
28. Lu, L. F; Liston, A. MicroRNA in the immune system, microRNA as an immune system. Immunology 2009, 127(3):291–8. doi: 10.1111/j.1365-2567.2009.03092.x.
29. Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A. S.; Nizet, V.; Johnson, R. S.; Haddad, G. G.; Karin, M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008, 453(7196):807–11. doi: 10.1038/nature06905. Epub 2008 Apr 23.
30. Balkwill, F. Tumor necrosis factor and cancer. Nat Rev Cancer 2009, 9(5): 361–71. doi: 10.1038/nrc2628. Epub 2009 Apr 3.
31. Popivanova, BK.; Kitamura, K.; Wu, Y.; Kondo,T.; Kagaya,T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J Clin Invest. 2008, 118(2): 560–70. doi: 10.1172/JCI32453.
32. Kulbe, H.; Thompson, R.; Wilson, JL.; Robinson, S.; Hagemann, T.; Fatah, R.; Gould, D.; Ayhan, A.; Balkwill F. The in flammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. 2007, 15;67(2): 585–92.
33. Basu S.; Nachat-Kappes R.; Caldefie-Chézet, F.; Vasson M. P. Eicosanoids and adipokines in breast cancer: from molecular mechanisms to clinical considerations, antioxidants & redox signaling. Antioxid Redox Signal 2013, 18(3): 323–360. doi:10.1089/ars.2011.4408.
34. Schreiber, R. D.; Old, L. J.; Smyth M. J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331(6024): 1565–70. doi: 10.1126/science.1203486.
35. Vesely, M. D.; Kershow, M. H.; Schreiber, R. D.; Smyth M. J. Natural innate and adaptive immunity to cancer. Annu Rev. Immunol. 2011, 29(1): 235–271. doi: 10.1146/annu rev-immunol-031210–101324.
36. Jiang, X.; Shapiro, D. J. The immune system and inflammation in breast cancer. Mol Cell Endocrinol. 2014, 382(1): 673–682. doi: 10.1016/j.mce.2013.06.003.
37. Jiang, X.; Ellison, S. J.; Alarid, Shapiro, D. J. Interplay between the levels of estrogen and estrogen receptors controls of the granzyme inhibitor, proteinase inhibitor-9 and susceptibility to immune surveillance by natural killer cells. Oncogene 2007, 26:4106–4114. doi:10.1038/sj.onc. 1210197.
38. Jiang, X. Harnessing the immune system for the treatment of breast cancer. J. of the Zhejiang University Sci. B (Biomed and Biotecnol) 2014,15(1): 1–15.
39. Rose, D. P.; Vona-Davis, L. The cellular and molecular mechanisms by which insulin influences breast cancer risks and progression. Endocr. relat. cancer 2012, 19(6): R225–41.
40. Crujeiras, A. B.; Diaz-Lagares, A.; Carreira, M. C. Oxidative stress associated to dysfunctional adipose tissue : a potential link between obesity, type-2 diabetes mellitus and breast cancer. Free radic research 2013, 47(4): 243–56.
41. Woo, H. D.; Kim, J. Global DNA hypomethylation in peripheral blood leucocytes as a bio-marker for cancer risk. a meta-analysis. PLoS 1 2012, 7(4): e34–615.
42. Yu, I. C.; Lin, H. I.; Sparks, J. D.; Yen, S.; Chang, C. Androgen receptors role in insulin resistance and obesity in males: the linkage of androgen-deprivation therapy to metabolic-syndrome. Diabetes 2014, 63(10): 3180–8.
43. Nyquist, M. D.; Dehm, SM. Interplay between genomic alterations and androgen receptors signaling during prostate cancer development and progression. Horm Canc. 2013, 4: 61–69. doi:10. 1007/s 12672-013-0131-4.
44. Carruba, G. Estrogen and prostate cancer: an eclipsed truth in an androgen-dominated scenario. J Cell Biochem. 2007, 102(4): 899–911.
45. Zhu, P.; Baek, S. H.; Bourk, E. M.; Ohgi, K. A.; Garcia-Bassets, I.; Sanjo, H.; Akira, S.; Kotol, P. F.; Glass, C. K.; Rosenfeld,M. G.; Rose D. W. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell 2006,124, 615–629.
46. Gross, K. L.; Cydlowsky, J. A. Tissue-specific glucocorticoid action: a family affair. Trends Endocrinol.Metab. 2008, 19: 331–339.
47. Oakley, R. H.; Cydlowsky, J. A. Homologous down regulation of the glucocorticoid receptor: the molecular machinery. Crit Rev Eukaryot Gene Expr. 1993, 3(2): 63–88.
48. Levin, E. R. Rapid signaling by steroid receptors. Regulatory, integrative and comparative physiology. American Journal of Physiology 2008, 295 R 1425R1430 doi: 10.1152/ajpregu.90605.2008.
49. Hammes, S. R. & Levin, E. R. Extranuclear steroid receptors: nature and actions. Endocrine reviews 2007; 28: 726–41.
50. Grossmann, C & Gekle, M. New aspect of rapid aldosterone signaling. Molecular and cellular endocrinology 2009, 308: 53–62.
51. Hunter, R. G.; Gagnidze, K.; McEwen, B.; Pfaff, D. W. Stress and the dynamic genoma: steroids, epigenetics and the transposome. Proc Natl Acad Sci U.S.A. 2014, pii: 201411260.
52. Joëls, M. Functional actions of corticosteroids in the hippocampus. European Journal of Pharmacology 2008, 583: 312–321.
53. De Kloet, E. R.; Reul, G. M. Feedback action and tonic influence of corticosteroids on brain function: a concept arising from the heterogeneity of brain receptor systems. Psycho neuroendocrinology 1987, 12: 83–105.
54. Glezer, I.; Rivest, S. Glucocorticoids: protectors of the brain during innate immune responses. Neuroscientist 2004, 10(6): 538–52.
55. Vgontzas, A. N.; Zoumakis, E.; Bixler, E. O.; Lin, H. M.; Follett, H.; Kales, A.; Chrousos, G. P. Adverse effects of modest sleep restriction on sleepiness, performance, and inflammatory cytokines. J. Clin.Endocr.Metab. 2004, 89: 2119–2126.
56. Scarano, F.; Baltuch, G. Microglia as mediators of inflammatory and degenerative diseases. Annu. Rev. Neurosci. 1999, 22: 219–40.
57. Verkhzhsky, A.; Rodigue, S.; Stardo, L. Astroglia pathology: a central element of neuro psychiatric disease. The Neuro Scientist 2014, 20 (6) 576–578.
58. Nguyen, M. D.; Julien, J. P.; Rivest, S. Innate immunity: the missing link in neuro protection and neuro degeneration. Nat. Rev. neurosci. 2002, (3): 216–27.
59. Lauretsky, H.; Irwin, M. Resiliance and aging. Aging health 2007, 3: 309–323.
60. Chrousos, G. P. Stress and disorders. Nat. Rev. Endocr. 2009, 5: 374–381.
61. van Rossum, E. F.; van den Akker, E. L. Glucocorticoids resistance. Endocr. Dev. 2010, 20: 127–136.
62. Debosscher, K.; van den Berghe W.; Aegheman, G. Crosstalk between nuclear receptors and nuclear factor kB. Oncogene 2006, 25: 6868–6886.
63. Miller, A. H.; Maletic, V.; Raison, C. L. Inflammation and its discontent : the role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009, 65(9): 732–741.
64. Norbiato, G.; Bevilacqua, M.; Vago, T.; Baldi, G.; Chebat, E.; Bertora, P.; Moroni, M.; Galli, M.; Oldenburg, N. Cortisol resistance in acquired immune deficiency syndrome, J.Clin.Endocr.Metab.1992, 74(3): 608–13.
65. Yung, S. H.; Vang Y.; Kim, T.; Tarrav, R.; Reader, B.; Powel, N.; Sheridan, JF. Molecular mechanism of repeated social defeat induced glucocorticoid resistance. Role of the micro RNA. Brain behavior and immunity 2015, 195–206.
66. Yang, N.; Ray, D. W.; Matthews, L. C. Current concepts in glucocorticoid resistance. Steroids 2012, 77(11): 1041–9.
67. Balkwill, F. Tumor necrosis factor and cancer. Nat. Rev. Cancer 2009, 9: 361–371.
68. Raison, C. L.; Miller, A. H. When not enough is too much. Am J. Psychiatry 2003, 160 (9): 1554–65.
69. Haslam, D. W.; James, W. P. Obesity. Lancet 2005, 366(9492): 1197–209.
70. Nicolaides, N. C.; Kyratzi, E.; Lamprokostopoulou, A.; Chrousos, G. P.; Charmandari E. Stress, the stress system and the role of glucocorticoids. Neuroimmunomodulation 2015, 22(1–2): 6–19.
71. Spencer, S. J. Perinatal programming of neuroendocrine mechanisms connecting feeding behavior. Stress frontiers in neurosciences 2013, vol. 7 doi: 10.3389/Fnins.2013.00109.
72. Bruce, K. D.; Hanson, M. A. The developmental origins, mechanisms and implications of metabolic syndrome. The journal of nutrition. 2010, doi: 10.3945/JN.109.111179.
73. Reynolds R. M. Glucocorticoids excess and the developmental origin of disease: two decades of testing the hypothesis—2012 Curd Ricter Harward winner. Psycho neuroendocrinology 2013, 38(1): 1–11.
74. Nguyen, J. C. D.; Killcross, AS.; Jenkins, T. A. Obesity and cognitive decline: role of inflammation and vascular changes. Front Neurosci. 2014, doi: 10.3389/Fnins.2014.00375.
75. Ouchi, N.; Walsh, K.; Lugus, J. J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11(2): 85–97.
76. Anagnostis, P.; Athyros, V. G.; Tziomalos, K.; Karagiannis, A.; Mikhailidis, DP. The pathogenic role of cortisol in the metabolic syndrome: hypothesis. J. Clin. Endocrinol. Metab. 2009, 94: 2692–2701.
77. Stewart, P. M. Tissue specific cushing’s syndrome 11Beta-hydroxy steroid dehydrogenases and the redefinition of corticosteroid hormone action. Eur. J. Endocrinol. 2003, 149: 163–168.
78. Scott, J. S.; Golberg, F. W.; Turnbull, AV. Medical chemistry of inhibitors of 11 Beta-hydroxy-steroid dehydrogenase type-1. J. Med. Chem. 2014, 57: 4466–4486.
79. Nathan, C. Epidemic inflammation: pondering obesity. Mol. Med. 2008, 14(7–8): 485–92.
80. Tilg, H.; Moschen, A. R. Inflammatory mechanisms in the regulation of insulin resistance. Mol. Med. 2008, 14(3–4): 222–231.
81. Gollasch, M.; Dubrovska, G. Parachrine role for periadventitial adipose tissue in the regulation of arterial tone. Trends Pharmacol. Sci. 2004, Vol. 25, no. 12 pp 647–653.
82. Fernandez-Alfonso, MS.; Gill-Ortega, M.; Garcia-Prieto., C. F; Aranguez, I.; Ruiz-Gaio, M.; Somoza, B. Mechanisms of perivascular adipose tissue dysfunction in obesity. International Journal of Endocrinol. 2013, Vol. 2013. Art. I. D. 402053. Page 8. doi: 10.1155/2013/402053.
83. Kim, J. Y.; van de Wal, E.; Laplante, M.; Azara, A. Trujillo, M. E.; Hofmann, S. M. Obesity-associated improvement in metabolic profile through expansion of adipose tissue. J. Clin. Invest. 2007, 117(9): 2621–2637.
84. Peraldi, P.; Hotamisligil, G. S.; Buurman, W. A.; White, M. F.; Spiegelman, B. M. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J Biol Chem. 1996, 271(22): 13018–22.
85. De la Monte, S. M. Insulin resistance and Alzheimer’s disease. BMB reports 2009, 42(8): 475–481.
86. Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A. H.; Iwakoshi, N. N.; Ozdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L. H.; Hotamisligil, G. S. Endoplasmic reticulum stress links obesity, insulin action, and type-2 diabetes. Science 2004, 306: 457–461.
87. Turnbaugh, P. J.; Ley, R. E.; Mahowald, M. A.; Magrini, V.; Mardis, E. R.; Gordon, J. I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444: 1027–1031.
88. Alemany, M. Do the interactions between the glucocorticoids and sex hormones regulate the development of the metabolic syndrome? Front. Endocrinol. 2012, 3: 27.
89. Garcia-Cruz, E.; Leiber Tamajo, A.; Romedo, J.; Piqueras, M.; Luque, P.; Cardenas, A. O.; Alcaraz, A. J. Sex med 2013, 10(10): 2529–38.
90. Norbiato, G.; Bevilacqua, M.; Vago, T.; Taddei, A.; Clerici, M. Glucocorticoids and the immune function in the human immunodeficiency virus infection: a study in hypercortisolemic and cortisol-resistant patients. J Clin Endocrinol Metab. 1997, 82(10):3260–3.
91. Norbiato, G. Endocrine, metabolic, and immunologic components of HIV infection. Ann N Y Acad Sci. 2012, 1262:51–5. doi: 10.1111/j.1749-6632.2012.06620.x.
92. Rao, V. R.; Ruiz, A. P.; Prasad, V. R. Viral and cellular factors underlying neuropathogenesis in HIV associated neurocognitive disorders. AIDS Research and Therapy 2014, 11: 13.
93. Effros, R. B.; Pawelek, G. Replicative senescence of T-cells : does the Hayflick limit lead to immune exhaustion? Immunol. today 1997, 18: 450–454.
94. Blackburn, E. H. Structure and function of telomeres. Nature 1991, 350:569–573.
95. Campisi, J.; d’Adda, D.; Fagagna, F. Cellular senescence : when bad things happen to good cells. Nat. Rev. Mol. Cell. Biol. 2007, 8: 729–40.
96. Wong, J. Y.; De Vivo, I.; Lin, X.; Fang, S. C.; Christiani, D. C. The relationship between inflammatory biomarkers and telomere length in an occupational prospective cohort study. PLoS One 2014;9(1): e87348. doi: 10.1371/journal.pone.0087348. eCollection 2014.
97. Baxter, J. D.; Funder, J. W.; Apriletti, JW.; Webb, P. Toward selective modulating mineralocorticoid receptor function: lessons from other systems. Mol cell endocrinol 2004, 217 (1–2): 151–65.
98. Hunter, R. G.; McEwen, B. S. Stress and anxiety across the life span : structural plasticity and epigenetic regulation. Epigenomics 2013, 5(2): 177–194.
99. Hunter, R. G. Epigenetic effects of stress and corticosteroid in the brain. Front. Cell. Neurosci. 2012, 6: 18.
100. Leader, J. E.; Wang, C.; Fu, M.; Pestell, R. G. Epigenic regulation of nuclear steroid receptors. Biochem. Pharmacol 2006, 72(11): 1589–96.
101. Aubert, G.; Lansdorp, P. M. Telomeres and aging. Physiol.Rev. 2008, 88:557–79.
102. Willeit, P.; Willeit J.; Majer, A.; Weger, S.; Oberhollenzer, F.; Brandstatter, A.; Kronenberg, F; Kieckl, S. Telomere length and risk incidence cancer and cancer mortality. JAMA 2010, 304: 69–75.
103. Uziel, O.; Singer, JA.; Danicek, V.; Sahaar, G.; Berkov, E.; Lucihansky, M.; Fraser, A.; Ram, R.; Lahav, M. Telomeres dynamics in arteries and mononuclear cells in diabetes patients: effect of diabetes and of glycemic control. Exp. Gerontol 2007; 42: 971–8.
104. Lu, Q.; Qiu, X.; Hu, N.; Wen, H.; Su, Y.; Richardson, BC. Epigenetics, disease, and therapeutic interventions. Ageing Res. Rev. 2006, 5(4): 449–67. Epub 2006 Sep 11.
105. Grønbaek, K.; Treppendahl, M.; Asmar, F.; Guldberg, P. Epigenetic changes in cancer as potential targets for prophylaxis and maintenance therapy. Basic Clin Pharmacol. toxicol. 2008, 103(5):389–96. doi: 10.1111/j.1742-7843.2008.00325.x.
106. Nasef A.; Chapel, A.; Mazurier, C.; Bouchet, S.; Lopez, M.; Mathieu, N.; Sensebé, L.; Zhang, Y.; Gorin, N.C.; Thierry, D.; et al. Identification of IL-10 and TGF-beta transcripts involved in the inhibition of T-lymphocyte proliferation during cell contact with human mesenchymal stem cells. Gene Expr. 2007, 3: 217–226.
107. Ryan, J. M.; Barry, F.; Murphy, J. M.; Mahon, B. P. Interferon-gamma does not break, but promotes the immunosuppressive capacity of adult human mesenchymal stem cells. Clin Exp Immunol. 2007, 149: 353–363.
108. Ortiz, L. A.; Dutreil, M.; Fattman, C.; Pandey, A. C.; Torres, G.; Go, K.; Phinney, D. G. Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proc Natl Acad Sci USA 2007, 104: 11002–11007.
109. English, K.; Barry, F. P.; Field-Corbett, C. P.; Mahon, B. P. IFN-gamma and TNF-alpha differentially regulate immunomodulation by murine mesenchymal stem cells. Immunol Lett. 2007, 110: 91–100.
110. Augello, A.; Tasso, R.; Negrini, S. M.; Cancedda, R.; Pennesi G. Cell therapy using allogeneic bone marrow mesenchymal stem cells prevents tissue damage in collagen-induced arthritis. Arthritis Rheum 2007, 56 :1175–1186.
111. Parekkadan, B.; Tilles, A. W.; Yarmush, M. L. Bone marrow-derived mesenchymal stem cells ameliorate autoimmune enteropathy independently of regulatory T cells. Stem Cells 2008, 26: 1913–1919.
112. Hanley E. N., Jr. Human adipose-derived mesenchymal stem cells: serial passaging, doubling time and cell senescence. Biotech Histochem 2012, 87: 303–311.
113. Han, S. M.; Han, S. H.; Coh, Y. R.; Jang, G.; Chan, R. J.; Kang, S. K.; Lee, H. W.; Youn H. Y. Enhanced proliferation and differentiation of Oct4and Sox2-overexpressing human adipose tissue mesenchymal stem cells. Exp Mol Med 2014, 46: e101.
114. Biava, P. M.; Bonsignorio, D.; Hoxa, M. Cell proliferation curves of different human tumor lines after in vitro treatment with Zebrafish embryonic extracts. J. Tumor Marker Oncol., 2001, 16 195–202.
115. Biava, P. M.; Carluccio, A. Activation of anti-oncogene p53 produced by embryonic extracts in vitro tumor cells. J. Tumor Marker Oncol., 1977, 12, 9–15.
116. Biava, P. M.; Bonsignorio, D.; Hoxa, M.; Facco, R.; Ielapi, T.; Frati, L.; Bizzarri, M. Post-traslational modification of the retinoblastoma protein (pRb) induced by in vitro administration of Zebrafish embryonic extracts on human kidney adenocarcinoma cell line. J. Tumor Marker Oncol., 2002, 17(2); 59–64.
117. Biava P. M, Monguzzi A, Bonsignorio D, Frosi A, Sell S, Klavins JV. Xenopus Laevis Embryos share antigens with Zebrafish Embryos and with human malignant neoplasms J. Tumor Marker Oncol. 2001, 16; 203–206.
118. Biava, P. M.; Bonsignorio, D. Cancer and cell differentiation: a model to explain malignancy. J. Tumor Marker Oncol. 2002, 17(3): 47–54.
119. Cucina, A.; Biava, P. M.; D’Anselmi, F.; Coluccia, P.; Conti, F.; Di Clemente, R.; Miccheli, A.; Frati, L.; Gulino, A.; Bizzarri, M. Zebrafish embryo proteins induce apoptosis in human colon cancer cells (Caco2). Apoptosis, 2006, 9, 1617–1628.
120. D’Anselmi F, Cucina A, Biava PM, Proietti S, Coluccia P, Frati L, Bizzarri M. (2011) Zebrafish stem cell differentiation stage factors suppress Bcl-xL release and enhance 5 Fu mediated apoptosis in colon cancer cells. Curr Pharm Biotechnol 2011, 12: 261–267.
121. Biava P.M., Nicolini A., Ferrari P., Sell S. A systemic approach to cancer treatment: tumor cell reprogramming focused on endocrine-related cancers. Curr. Med. Chem. 2014, 9: 1072–1081.
122. Livraghi, T.; Meloni, F.; Frosi, A.; Lazzaroni, S.; Bizzarri, M.; Frati, L.; Biava, P.M. Treatment with stem cell differentiation stage factors of intermediate-advanced hepatocellular carcinoma: an open randomized clinical trial. Oncol Res., 2005, 15 (7,8): 399–408.
123. Livraghi T, Ceriani R, Palmisano A, Pedicini V, Pich MG, Tommasini MA, Torzilli G. Complete response in 5 out of 38 patients with advanced hepatocellular carcinoma treated with stem cell differentiation stage factors: case reports from a single centre. Curr Pharm Biotechnol 2011, 12: 254–260.
124. Norata G,; Biava P.. M.; Di Pierro F. The Zebrafish embryo derivative affects cell viability of epidermal cells: a possible role in the treatment of psoriasis. Giornale Ital. Dermatol. Venereol. 2013, 148 (5):479–483.
125. Canaider, S.; Maioli, M.; Facchin, F.; Bianconi, E.; Santaniello, S.; Pigliaru, G.; Ljungberg, L.; Burigana, F; Bianchi, F.; Olivi, E.; Tremolada, C.; Biava, P. M.; Ventura, C. Human stem cell exposure to developmental stage zebrafish extracts: a novel strategy for tuning stemness and senescence patterning. CellR4, 2014, 2(5), 1226.
126. Lunde K, Belting H. G, Driever W. Zebrafish pou5f1/pou2, homolog of mammalian Oct4, functions in the endoderm specification cascade. Curr Biol 2004; 14: 48–55.
127. Onichtchouk D, Geier F, Polok B, Messerschmidt D. M, Mössner R, Wendik B, Song S, Taylor V, Timmer J, Driever W. Zebrafish Pou5f1-dependent transcriptional networks in temporal control of early development. Mol Syst Biol 2010; 6: 354.
128. Kotkamp K, Mössner R, Allen A, Onichtchouk D, Driever W. A Pou5f1/Oct4 dependent Klf2a, Klf2b, and Klf17 regulatory sub-network contributes to EVL and ectoderm development during zebrafish embryogenesis. Dev Biol 2014; 385: 433–447.
129. Campisi J, Dimri G. P. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol 2003; 23: 389–401.
130. Klapper W, Parwaresch R, Krupp G. Telomere biology in human aging and aging syndromes. Mech Ageing Dev 2001; 122: 695–712.
131. Wilsker D, Patsialou A, Dallas PB, Moran E. ARID proteins: A diverse family of DNA binding proteins implicated in the control of cell growth, differentiation, and development. Cell Growth Differ 2002; 13: 95–106.
132. Popowski M, Templeton TD, Lee BK, Rhee C, Li H, Miner C, Dekker JD, Orlanski S, Bergman Y, Iyer VR, Webb CF, Tucker H. Bright/Arid3A acts as a barrier to somatic cell reprogramming through direct regulation of Oct4, Sox2, and Nanog. Stem Cell Reports 2014; 2: 26–35.
133. Barakat MT, Humke EW, Scott MP. Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol. Med 2010, 16(8): 337–348.
Cancer: A Problem of Developmental Biology; Scientific Evidence for Reprogramming and Differentiation Therapy
Current Drug Targets, 2016, Volume 17, Number 10:1103–10
S. Sell, A. Nicolini, P. Ferrari, P. M. Biava
This article begins with research undertaken in the late seventies that led some researchers to interpret cancer as being due to the presence of embryonic cells blocked in their process of maturation (maturation arrest). It reviews the theory of maturation arrest in the light of recent research on the epigenetic code and its ability to regulate gene expression. Tumor diseases on the basis of more recent research are interpreted as being due to genetic and/or epigenetic alterations that require reprogramming. The article reviews the important reprogramming and differentiation treatments of cancer cells and concludes that tumor diseases are pathologies of stem-cell development and not simply due to maturation arrest. They were caused by many modifications and require treatment by epigenetic reprogramming.
Introduction
In 1978, B. Pierce and colleagues published a book entitled Cancer: A Problem of Developmental Biology.1 Based on their work on embryonal carcinoma, Pierce and colleagues predicted the present concept of tissue determined stem cells2 in which tissue renewal is accomplished by asymmetric division of a stem cell to produce one daughter cell that remains a stem cell and a second daughter cell that becomes a proliferation of transit amplifying cells. The progeny of the transit amplifying cells eventually terminally differentiate into mature cells. According to this model, the main difference between normal tissue renewal and proliferation of cancer cells is that the transit amplifying cells of the cancer do not terminally differentiate as do normal transit amplifying cells, but continue to proliferate. However, Pierce and Wallace3 found that even the proliferating progeny of “stem” cells of a squamous cell carcinoma could give rise to daughter cells that differentiate into mature keratinized cells. In normal tissue renewal, there is equilibrium between the rate of proliferation and the rate of cell death so that the number of cells at any given time is relatively constant. In contrast, in cancer tissue the equilibrium is shifted in favor of proliferation over cell loss so that in cancers the number of cells continues to increase. The key concept is that cancers are maintained by cancer stem cells that give rise to daughter transit amplifying cells that do not mature at a normal rate and continue to increase in number. This is known as maturation arrest.4
An even earlier model of cancer based on an origin from stem cells was the embryonal rest theory of cancer.5 The first theory of the origin of cancer was a “field theory.” Field theories are based on the idea that the tissues surrounding the cells at risk (niche) provide a signal or environment that acts to stimulate the cells to proliferate as cancer cells.5-6 The first idea that cancers might arise from stem cells appeared in the early 19th century.7-8 Although the concept of stem cells was still far in the future, Durante9 and Conheim10 introduced the idea that cancers arise from embryonal tissue that survived in adult organs; i.e., embryonal rests. They proposed that disequilibrium between the embryonal cells in the “rest” and the surrounding tissue allowed these remnants of embryonic tissue to reassume proliferation and produce masses of cells that resembled fetal tissues. This mechanism of cancer development is consistent with a field theory; i.e., a change in the tissue stroma allows cancer to appear but also identifies the cells of origin as stem cells. However, by the early 1900s the embryonic rest theory lost support,6 and in general, interest in cancer research waned as the primary focus for research and clinical studies was infectious diseases. It would take 50 more years before studies on teratocarcinoma would lead to a reassertion of the embryonic rest theory of cancer in the form of the stem-cell theory of cancer.
Teratocarcinoma
Teratocarcinomas consist of mature, differentiated tissues, as well as fetal components: yolk sac and placental elements. The production of alphafetoprotein (AFP) by the yolk sac component and of human chorionic gonadotropin (hCG) by the placental elements suggests that the embryonal cells of a teratocarcinoma are totipotent; i.e., they can differentiate into both adult and embryonic cells. The growth of these tumors may be followed by measuring the serum levels of AFP or hCG. Most of the cells of a teratocarcinoma are mature and nonmalignant; the malignant cells are located in the embryo’s body, a tissue structure that contains undifferentiated embryo tissue. The cancer stem-cell nature of these cells is documented by their ability to form cancers upon transplantation into histocompatible recipients. The normal stem-cell properties of these cells are demonstrated by the fact that transplantation into the inner cell mass of a developing blastocyst results in incorporation of teratocarcinoma, the cells into normal developing tissues.11, 12, 13, 14, 15, 16 Convincing documentation of the tissue stemcell origin of cancer was obtained in the 1960s, when Leroy Stevens was able to produce growth of malignant teratocarcinomas after transplanting normal germinal stem cells from the genital ridge of day 12 SIJ/129 male mice into the testicles of normal 129 adult male mice. In the testicular transplant niche the germinal stem cells grew abnormally and formed tumors, thus supporting both the stem-cell origin of the cancer and the field theory of cancer.17-18 Then about a decade later it was demonstrated that teratocarcinoma cells did not grow when transplanted into a mouse blastocyst. The cells from these transplantable teratocarcinomas injected into normal blastocysts became incorporated into the developing embryos. The resulting adult mice had organs that were made up of a mixture of mature tissues from the normal blastocyst and from the cancer (Chimera). The inner cell mass of the blastocyst is able to reprogram both mature tissue stem cells as well as cancer stem cells.19, 20, 21, 22, 23, 24, 25 Thus, a combined stem-cell field theory of cancer is supported by the observation that teratocarcinomas arise from tissue germinal stem cells if the cells are placed in an environment that allows expression of the malignant phenotype.
Barry Pierce and his co-workers extensively examined the cellular make up of teratocarcinomas.1, 25 From their studies Pierce hypothesized a hierarchical model of cancer, with cancer stem cells giving rise to cancer transit amplifying cells that would exhibit various stages of differentiation culminating in terminally differentiated cells, and this hierarchical model was extended to provide a general thesis for the cells that make up any cancer.1 They postulated that the differentiation state of a cancer depends upon the stage of maturation at which the majority of cells of the cancer become arrested. If maturation arrest occurs at an early stage the tumor will be poorly differentiated and most of the tumor transit amplifying cells will be able to divide; if at a later stage, the tumor will be well differentiated and few of the tumor transit amplifying cells will divide. In either case, the tumor will be maintained by cancer stem cells that provide the self renewing cells of the tumor.
Control of Differentiation by the Embryo’s Microenvironment
The tissues of the embryo are able to induce differentiation of normal and cancer stem cells,26-27 including putative carcinogen induced cancer cells. Exposure of developing embryos to chemical carcinogens may lead to malformations, but not to cancer. Thus, the embryonic microenvironment is able to correct for the mutations induced by carcinogens and prevent cancer formation.28-29 As stated above, the blastocyst environment also controls growth of transplanted malignant cells, whereas malignant cells placed in other sites of the developing embryo, such as the perivitelline space, are not controlled.30, 31, 32 On the other hand, more differentiated tissues of the embryo may also regulate growth of cancer cells that are normally found in that tissue. There is also evidence for diffusible factors produced by blastocyst cells that may induce differentiation of cancer stem cells. The regulatory environment of the blastocyst is not limited to teratocarcinoma. Other cancers shown to be converted to normal developing tissue when placed in an appropriate embryonic microenvironment include leukemia, melanoma, hepatocellular cancer and breast cancer.33, 34, 35, 36, 37 Malignant melanoma cells placed into the extraembryonal membrane of the Zebrafish differentiated into normal neural crest like cells.38 Thus, different embryonal microenvironments may have different differentiation potentials related to normal cellular differentiation. For example, the placenta may also regulate transplanted leukemia cells.39 Other authors obtained similar results with different experiments40-41 and concluded that embryo-mother cross-talk is very important in determining the arrest of tumor growth, because both maternal (decidua) and embryonic tissues contain substances with anti-cancer properties, which cooperate, inhibiting or delaying tumor growth.40 These promising results open new perspectives not only in cancer biology comprehension, but also in leading to a different therapeutic strategy. In fact in the last years many studies have clarified that embryonic cells share fundamental features with tumor cells,42-43 such as proliferation and expression of embryonic proteins (AFP, or ABC transporters), common molecular signals and pathways (i.e., beta-catenin/TCF/WNT Notch, BMP and Hedgehog signals),44, 45, 46 anaerobic metabolism,47 etc. In addition, epithelial-to-mesenchymal transition (EMT) observed in cancer tissue48-49 may also be viewed as a “reactivation” of an embryonic program.50-51
These findings illustrate that cancer is a disease of developmental biology. To understand how the substances taken from developing embryo or other biological substances are able to induce differentiation of cancer cells, we are here reporting important results on reprogramming and differentiation treatment.
Reprogramming and Differentiation Treatment of Cancer Cells
The term “cancer cell reprogramming” is used to define any kind of intervention aimed at transforming cancer cells into terminally differentiated cells. Differentiation treatments have the same goal; that is, to induce terminal differentiation of cancer cells or to force cancer stem cells to become transit amplifying cells that can then be treated by additional treatment such as chemotherapy or radiation. This review reports on advances of these technologies, including our personal contributions. An example of one of the first applications of differentiation therapy is the removal of the block to differentiation in human myeloid leukemia.52-53 The genetic lesions of leukemia result in a block of differentiation (maturation arrest) that allows myeloid leukemic cells to continue to proliferate and/or prevents the terminal differentiation and apoptosis seen in normal white blood cells. In chronic myeloid leukemia, the bcr-abl (t9/22) translocation produces a fusion product that is an activated tyrosine kinase resulting in constitutive activation cells at the myelocyte level. This activation may be inhibited by imatinib mesylate (Gleevec, STI-571), which blocks the binding of ATP to the activated tyrosine kinase, prevents phosphorylation, and allows the leukemic cells to differentiate and undergo apoptosis. In acute promyelocytic leukemia, fusion of the retinoic acid receptor-alpha with the gene coding for promyelocytic protein, the PML-RAR alpha (t15:17) translocation, produces a fusion product that blocks the activity of the promyelocytic protein, which is required for the formation of the granules of promyelocytes and prevents further differentiation. Retinoic acids (RA) bind to the retinoic acid receptor (RAR alpha) component of the fusion product, resulting in degradation of the fusion protein by ubiquitinization. This allows normal PML to participate in granule formation and differentiation of the promyelocytes.
Unfortunately, there are cases of RA resistance. This is a serious problem for patients with acute promyelocytic leukemia who are receiving all-trans retinoic acids. The biologic effects of RA are mediated by two distinct families of transcriptional factors: RA receptors (RARs) and retinoid X receptors (RXRs). RXRs heterodimerize with 1, 25-dihydroxyvitamin D3 [1,25(OH)2D3] receptor (VDR), enabling their efficient transcriptional activation. The cyclin-dependent kinase (cdk) inhibitor p21(WAF1/CIP1) has a vitamin D3–responsive element (VDRE) in its promoter, and 1,25(OH)2D3 enhances the expression of p21(WAF1/CIP1) and induces differentiation of leukemic cell lines. In this case 1,25(OH)2D3 induces increased expression of cdk inhibitors, which mediates a G1 arrest, and this may be associated with differentiation of RA-resistant cells toward mature granulocytes.54 In addiction it is here recorded that the vitamin D anti-tumor effects are inhibited by CYP24A1 gene, which encodes 24-Hydrolase, the key enzyme for degrading many forms of vitamin D, including the most active form 1,25D(3). The inhibition of CYP24A1 by 4,5,6,7 tetrabromobenzimidazole (TBBZ) in human prostate cancer enhances 1,25D(3) mediated anti-tumor effect.55 It is here also recorded that curcumin, a bioactive polyphenol found in curry and several of its analogs, elicits transcriptional activation in retinoic acid and retinoid X receptor (RXR) responsive systems and stimulates RXR homodimerization and vitamin D receptor (VDR)–coactivator interaction.56 Finally, retinoid and their receptors play an important role in controlling the progression of many other tumors.57
The Use of Stem Cell Differentiation Stage Factors (SCDFs)
The application of stem cell differentiation stage factors (SCDFs) to revert cancer cells into a benign phenotype was started many years ago. SCDFs in the form of factors taken from Zebrafish embryos at different stages of development demonstrated a significant inhibition of proliferation of different human tumor lines in vitro (glioblastoma multiforme, melanoma, hepatocellular carcinoma, breast carcinoma, kidney adenocarcinoma, colon adenocarcinoma, acute lymphoblastic leukemia).58 The molecules that play a key role in the regulation process of the cell cycle, such as p5359 and pRb,60 are involved through transcriptional and post-translational processes. In addition, treatment of colon cancer with SCDSFs induces caspase 3 activation, mainly by increasing the release of E2F-1, leading to c-Myc overexpression and the activation of a p73 apoptotic-dependent pathway.
There is also a concurrent significant normalization of the ratio of e-cadherin to beta catenin, with an increase in e-cadherin levels.61 Finally, SCDSFs induce an almost complete growth inhibition of cell proliferation of CaCo2 colon cancer cell line after the concurrent treatment with 5 Fluorouracil,62 suggesting that Zebrafish embryo factors improve chemotherapy efficacy with 5 Fluorouracil. Subcutaneous injection of SCDFs with primary Lewis Lung Carcinoma cells into C57BL/6 female syngeneic mice produces a highly significant difference (P<0.001) between treated and control mice, both in terms of primary tumor growth and survival compared to untreated controls.63 A randomized clinical trial that was conducted between January 1, 2001, and April 30, 2004, on 179 patients with intermediate-advanced hepatocellular carcinoma unresponsive to conventional treatment (transplantation, resection, ablation therapy, or chemoembolization). The patients were treated with a product developed following the aforementioned in vitro and in vivo studies. It contained SCDSFs taken from Zebrafish embryos at a very low concentration (micrograms). This was administered sublingually at a dose of 30 drops to the patients three times a day. Regression occurred in 19.8% of the patients (2.4% of complete regression and 17.4% of partial regression) and 16% disease stabilization, with over 60% survival rate after 40 months in the patients who responded to treatment, compared to 10% in the remaining patients. The performance status improved in 82.6% of the treated patients, including those in advanced stages of the disease.64
Another clinical trial was conducted on 50 patients with “advanced” stage HCC from 2005 to 2010. Complete response was regarded as sustained disappearance of the neoplastic lesions, accompanied by normalization of AFP levels. In 13.1% of the patients with “advanced” stage there was a sustained complete response. No side effects occurred.65 This favorable result using SCDSFs therapy of patients with “advanced” HCC encouraged further testing of other reprogramming treatment.
Other Reprogramming Treatments
OOCYTE EXTRACTS
Extracts taken from prophase amphibian oocytes of axolotl (AOE) and xenopus (XOE) induce re-expression of some tumor suppressor genes. RARB, CST6, CCND2, GAS2 and CDKN2A are silenced or expressed at very low levels in MCF-7 and HCC 1954 cell lines representing luminal and basal breast cancer phenotypes.66 Extracts of AOE and XOE prophase oocytes induce re-expression of these genes in both breast cancer cell lines.
NAIVE HUMAN UMBILICAL CORD MATRIX DERIVED STEM CELLS
Human umbilical cord matrix stem cells (hUCMSCs) are unique stem cells derived from Warton’s jelly (termed Warton’s jelly stromal cells).67 Metastatic growth of MDA-231 human breast carcinoma cells is attenuated in culture and in a mouse xenograft experimental model by un-engineered (naive) human umbilical cord matrix stem cells (hUCMSC).68 In vitro, culture of hUCMSC with MDA-231 carcinoma cells inhibits DNA synthesis, increases the G2 cell population and inhibits colony growth of the carcinoma cells. The inhibited growth of cancer cells appears to be due to blocking of ERK-1/2 and PI3K/Akt signaling with activation of intrinsic apoptosis signals.
REPROGRAMMING TELOMERASE AND THE MAT-8 GENE
A subpopulation of cancer stem-like cells in prostate cancer cell lines and primary prostate cancer tissues that are highly tumorigenic expresses some essential stem-cell-associated transcription factors (Oct3/4, Sox2, Nanog, Klf4, and c-myc).69 Due to their importance in prostate cancer growth, these cancer stem cells seem a likely target for novel strategies for prostate cancer therapy. One of these novel strategies is related to the enzyme telomerase. Malignant cells from many cancers have significant telomerase expression and activity levels that correlate directly with malignant metastatic potential. New gene constructs to reprogram telomerase have been engineered and validated: 1) small interfering RNA against wild type mouse telomerase RNA (alpha MTer-siRNA); 2) mutant-template mouse telomerase RNA (MT-mTer), which encodes incorrect mouse telomeric repeats.
Lentiviral delivery of alpha mTer-siRNA to mouse prostate cancer cells caused growth inhibition and rapid apoptosis in cancer progenitor cells isolated from human prostatectomy specimens.70-71 Transfection of human PC-3 and LNCaP prostate carcinoma cells with small interfering double-stranded RNA (siRNA) oligonucleotides against the MAT-8 gene produced a specific down-regulation of MAT-8 expression and a significant decrease in cellular proliferation of both cancer cell lines.72
Artificial Transcription Factors (ATFs) in Breast Cancer Cells
The epigenetic reprogramming of breast cancer cells may be accomplished by targeted DNA methylation.73 Site specific DNA methylation and prolonged stable repression of the tumor suppressor Maspin and the oncogene Sox2 can be obtained in breast cancer cells by Zincfinger ATFs targeting DNA methyltransferase-3a to the promoters of these genes.
Stimulation of Immunity to Cancer By Reprogramming Tumor-Associated Dendritic Cells
The endocytic activity and immune activation of ovarian cancer-associated dendritic cells (DC) may be selectively increased by treatment with the immunostimulatory miRNA miR-155. Nanoparticles carrying oligonucleotide duplexes mimicking the bulged structure of endogenous pre-miRNA strongly enhanced miR-155 activity without saturating the RNA-induced silencing complex that produced genome-wide transcriptional changes that in turn silenced multiple immunosuppressive mediators. Tumor infiltrating DCs were transformed from immunosuppressive to highly immunostimulatory cells, activating strong antitumor responses that impeded the progression of established ovarian cancer.74
Also the transfer of chromosome 3p fragments in a novel epithelial ovarian cancer cell line model (OV-90) induces tumor suppression. Tumor suppression is associated with a modified transcriptome by microarray analysis. Reprogramming of the transcriptome appears to be a consequence of the chromosome 3 transfer and tumor suppression affected molecular networks sustaining ovarian carcinogenesis.75
Discussion
The use of SCDSFs in anti-tumor treatment has led to a cancer model76 that is consistent with the scientific and clinical evidence. This model integrates that of Pierce and collaborators and views cancer as a consequence of two different processes: 1) maturation arrest and 2) a stochastic process in which genetic and epigenetic alterations conduce a normal differentiated cell to be malignant. These two process are not mutually exclusive, and both have been invoked.77, 78, 79 The process of maturation arrest has already been described in this review. In the stochastic process the initial mechanism could be a mutation or an epigenetic alteration. If the process begins with a mutation, the minimal number of stochastic mutational events which can transform a normal cell in a cancer cell is calculated between four and seven.80 If mutations are introduced into normal cells in a non-stochastic manner, i.e., triggering at precise genes, the number is reduced.81 The preferred targets of these mutations are genes encoding for key-role effectors of cell cycle regulation and cell signaling, and for growth factors and their receptors. Mutations are either gain-of-function, in the case of proto-oncogenes, or loss-of-function, in the case of tumor-suppressor genes.
Nevertheless, defining the transformation of a normal cell into a cancer cell as the result of a sum of mutations may be reductionistic. For normal cells to become cancerous, transformation also depends on a complex network of surrounding microenvironmental signals from cell-to-cell “cross-talking” or from soluble extracellular factors. For example, it is well known that inflammatory cells sustain rather than fight tumor growth82 and that pro-inflammatory cytokines promote cancer cell proliferation by inhibiting tumor-suppression pathways.83 The whole context is thus critical in terms of determining cell fate in line with a complex view of cell biology.84 According to this view, cancer could be defined as a microevolutive process that is usually the consequence of a high variability of the mechanisms used by cells to become malignant. Nonetheless the model of cancer described by one of us76 assumes that regardless of how the steps in these genetic pathways are arranged, development of all types of tumor is governed by a final common process. Some authors define “early crisis” and “genetic catastrophe” of cells as steps enabling the evolving population of pre-malignant cells to reach malignancy. As a result of these crises a cell has two possibilities after each crisis: either die or survive.
In other words, surviving cells begin a chaotic process with a series of multiple bifurcations, at the end of which cells become cancerous. The research we have carried out has shown that the chaotic process is stabilized by what chaos theory refers to as an “attractor,” which leads the tumor cell genome to a new configuration. In other words, the process which causes cancer is a process of deterministic chaos.
Cancer Stem-like Cells
Depending on the degree of malignancy, the cancer cell configuration is similar to that present in stem cells at various stages of development and differentiation. Our experiments have shown that tumor cells are undifferentiated cells, blocked in a multiplication phase between two cell differentiation stages. Tumor cells can therefore be considered as cells in which both genetic and epigenetic changes are usually present, these last changes being linked to the new gene configurations, very similar to those present in stem cells. The above process describes the mechanisms that can transform a normal differentiated cell into a cancer cell, but the mechanisms of carcinogenesis can from the outset concern the normal stem cells present in a specific tissue. In this latter case, the process is simpler but the result is the same: the transformation of a normal cell into a cancer stem-like cell. In this case, the model of Pierce and collaborators (maturation arrest) is sufficient to explain the origin of cancer, while the transformation of a complete differentiated cell in cancer cell requires a stochastic model of deterministic chaos to explain the process. The model cited in this article,76 conceived in 2002, describes both the processes that give rise to all kinds of cancer. In this model the most aggressive tumors are those with genetic configurations present in the early stages of embryonic differentiation, while the well-differentiated tumor cells are those present in the final stages of cell differentiation. The current classifications of tumors are redundant because they do not consider that from an ontogenetic viewpoint, as a cancer that originates in a specific organ becomes increasingly aggressive and f lows into cell types that share the same genetic configuration with cancers of other organs.
Finally, some tumor types are made up of differing cellular clones with varying degrees of malignancy and thus of cells with genetic configurations that derive from various stages of differentiation. So this model of cancer is consistent with the real situation and has been supported by our experiments. They demonstrate that the factors differentiating stem cells are capable of differentiating even tumor cells or causing their apoptosis, by-passing the mutations or correcting the epigenetic alterations that are at the origin of malignancy. In line with this approach, we recall the characteristics shared by both tumor and normal stem cells: tumor cells have oncofetal antigens, which are maintained during phylogenesis,85 and specific receptors on the cell membrane on which stem cell differentiation factors likely act. In addition, tumor and stem cells have common signals and pathways such as APC/beta-catenin/TCF/Wnt pathways, and Hedgehog/Smoothened/Patched pathways, as already described.
The problem with tumor cells is twofold: not only do they harbor genetic mutations, which usually underlie the malignancy, but they also have significant modifications of the epigenetic code. The genetic configuration and metabolism of tumor cells are very similar to those of stem cells in that both have active proto-oncogenes; both produce embryonic growth factors; both, as already mentioned, have oncofetal antigens; and both function with an anaerobic metabolism. The difference between stem and tumor cells lies in the fact that the latter can no longer complete development and differentiate, in view of the mutations. The use of SCDSFs results in tumor cells falling within the scope of normal physiology, blocking the cell cycle and activating metabolic pathways of differentiation or apoptosis. In this way, the mutations—and hence the malignancy—can be by-passed.
In recent years, some studies have linked a tumor’s malignancy and chemoresistance to the presence of cancer stem cells,85 which are responsible for the repopulation of a tumor after chemotherapy.86 Cancer stem-like cells have been identified in various solid tumors, such as glioblastoma multiforme,87, 88, 89 breast cancer,90, 91, 92, 93 lung cancer,94, 95, 96, 97 prostate cancer,98, 99, 100 ovarian cancer,101, 102, 103, 104 liver cancer,105, 106, 107, 108, 109 gastric cancer,110, 111, 112, 113, 114, 115 colon cancer,116, 117, 118 pancreas cancer,119, 120, 121 and squamous carcinoma of the head and neck,122, 123, 124, 125 etc. Moreover, the presence of cancer stem cells has long been known to be characteristic of many hematological malignant diseases.
Conclusions
Ongoing studies are providing important clarification of the different mechanisms of cell differentiation and the various metabolic pathways common to cancer and stem cells. One example is the research carried out at the Children’s Memorial Hospital in Chicago. As mentioned, these studies have confirmed that a malignant melanoma reverts to a normal phenotype when exposed to the microenvironment of the Zebrafish embryo. This occurs because a central nervous system morphogen (called Nodal and a member of the TGF-beta superfamily)—which is re-expressed in the cells of the malignant melanoma and responsible for the melanoma’s aggressiveness—is downregulated by a factor present in an embryonic environment; for example, Lefty.126 This approach falls within the differentiation and reprogramming therapy concept that researchers have for some time hoped to pursue and that wholly supports our studies. The proposed model of cancer76 takes account of the necessary complexity of reprogramming treatments. In fact, cell differentiation mechanisms consist of a multigenic regulation, so that a more differentiated cell differs from a less differentiated one because of the change in expression of a large number of genes.
If the ultimate goal is the differentiation rather than destruction of the cancer cell, this can clearly be achieved only by providing the cell with all the factors that are necessary for reprogramming. These can all be found when life is forming. In fact, during organogenesis the entire repertoire of regulatory molecules able to differentiate stem cells is present (transcriptional, post-transcriptional, translational, posttranslational regulatory factors). Each kind of cancer stem cell can revert to a normal phenotype only when the regulation network of differentiation is specific and complete enough for that kind of cancer cell. As a result, focus should be on microenvironment and networks of the biological structures, rather than on individual, specific mechanisms. This does not mean that research into molecular mechanisms should be disregarded. In fact, any new elucidated molecular pathway illuminates one more piece of the puzzle. Indeed, the difficulty in bridging the gap to a new scientific paradigm, i.e., shifting our view from reductionism to complexity, has been the main barrier to acquiring a more complete knowledge of cancer.
Research on stem-cell differentiation and other work based on a more extended approach to cancer treatment together yield a more complete understanding of the biological processes that sustain tumor growth. These studies are in progress worldwide and the scientific community is now ready to accept the new paradigm, as a number of authors confirm.127 In this vision it was proposed in a recent review128 a new hallmark of cancer: the loss of differentiation of the cancer cell. This confirms that cancer is a problem of developmental biology.

References
1. Pierce GB, Shikes R, Fink LM. Cancer: a problem of developmental biology. Englewood Cliffs, New Jersey; Prentice Hall Inc, 197.
2. Pierce GB, Spears WC. Tumors as caricatures of the process of tissue renewal: prospects for therapy by directing differentiation. Cancer Res 1988; 48: 1196–204.
3. Pierce BB, Wallace C. Differentiation of malignant to benign cells. Cancer Res 1971; 31: 127–34.
4. Sell S, Pierce GB. Biology of disease: Maturation arrest of stem cell differentiation is a common pathway for the cellular origin of teratocarcinomas and epithelial cancers. Lab. Invest 1994;70: 6–21.
5. Shimkin M. Contrary to Nature. NIH, USDOH, 1977.
6. Bainbridge WS. The Cancer Problem, The Macmillan Co. New York, 1914.
7. Recamier JCA. Recherches sur le traitement du cancer: par la compression methodique simple ou combinee, et sur l’histoire general de la meme maladie. Paris Gabon 1829.
8. Remak R. Ein beitrag zur entwickelungsgeschichte der krebshaften geschwulste. Deut Klin 1854;6: 170–74.
9. Durante F. Nesso fisio-pathologico tra la struttura dei nei materni e la genesi di alcuni tumori maligni. Arch Memorie ed Osservazioni di Chirugia Pratica 1874; 11: 217–26.
10. Cohnheim J. Congenitales, quergestreiftes muskelsarkon der nireren. Virchows Arch 1875; 65: 64.
11. Osler W, McCrea T. Modern Medicine: It’s Theory and Practice. Lea and Ferbiger, Philadelphia and New York, 1913.
12. Dixon FJ, Moore RA. Testicular tumors: a clinicopathological study. Cancer 1953; 6: 417–43.
13. Damjanov I. Pathobiology of human germ cell tumors. Recent Results Cancer Research 1991; 123: 1–34.
14. Solter D, Damjanov I. Teratocarcinoma and the expression of ondo-developmental genes. Methods Cancer Res 1979; 18: 277–98.
15. Chan D, Sell S. Tumor Markers. In: Teitz Textbook of Clinical Chemistry; Burtis CA, Ashwood ER Eds. Philadelphia, PA: Saunders 1999, pp 722–49.
16. Peyron A. Sur la presence des cellules genitales primordiales dans les boutons embryononnaires des embryomes parthenogenetiques chez lhomme. Cr R Acad Sci (Paris) 1938: 206: 1680–83.
17. Stevens LC. Experimental production of testicular teratomas in mice. Proc Natl Acad Sci USA 1964;52: 654–61.
18. Stevens LC. Origin of testicular teratomas from primordial germ cells in mice. J Natl Cancer Inst 1967;38: 549–52.
19. Brinster RL. The effect of cells transferred into the mouse blastocyst on subsequent development. J Exp Med 1974:140: 1049–56.
20. Mintz B, Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc Natl Acad Sci USA 1975; 72: 3583–9.
21. Ilmmenesee K. Reversion of malignancy and normalized differentiation of teratocarcinoma cells in mammals. In: Generic mosaics and chimeras in mammals; Russel LC Editor, New York: Plenum 1978; 3–25.
22. Papaionnou VE, McBurney MW, Gardner RL., Evans RL. Fate of teratocarcinoma cells injected into early mouse embryos. Nature 1975; 258: 70–3.
23. Papaioannou VE. Ontogen, pathology, oncology. Int J Dev Biol 1993; 37:33–7.
24. Pierce GB, Dixon FJ. The demonstration of teratogenesis by metamorphosis of multipotential cells. Cancer 1959;12: 573–83.
25. Pierce GM, Dixon FJ, Verney E. Teratocarcinogenic and tissue forming potentials of the cell types comprising neoplastic embryoid bodies. Lab Invest.1960;9: 583–602.
26. Brent RL. Radiation Teratogenesis. Teratology 1980; 21: 281–98.
27. Einhorm L. Are there factors preventing cancer development during embryonic life? Oncodev Biol Med 1982; 4: 219–29.
28. Pierce GB. The cancer cell and its control by the embryo. Am J Pathol 1983;113: 117–24.
29. Pierce GB, Lewis SH, Miller GJ, Morits E, Miller P. Tumorigenicity of embryonal carcinoma as an assay to study control of malignancy by the murine blastocyst. Proc Natl Acad Sci USA 1979: 76; 6649–51.
30. Pierce GB, Podesta A, Wells RS. The role of the blastocyst trophoderm in control of colony formation. In: Teratocarcinoma Stem Cells; Silver S, Strickland S and Martin G, Eds. New York: Cold Spring Harbor Symposium 1983; pp 15–22.
31. Pierce GB, Pantazis CG, Caldwell JE, Wells RS. Specificity of the control of tumor formation by blastocysts. Cancer Res 1982; 1082–7.
32. Gershenson M, Graves K, Carson D, Wells RS, Pierce GB. Regulation of melanoma by the embryonic skin. Proc Natl Acad Sci USA 1986:83: 7307–10.
33. Webb CW, Gootwine E, Sachs L. Developmental potential of myeloid leukemia cells injected into rat midgestation embryos. Dev Biol 1984; 101: 221–4.
34. Coleman WB, Wennerberg AE, Smith GJ, Grisham JW. Regulation of differentiation of diploid and some aneuploid rat liver epithelial (stemlike) cells by the hepatic microenviromment. Am J Pathol 1993;142: 1371–82.
35. Weaver V, Petersen O, Wang F, et al. Reversion of the malignant phenotype of human breast cancer in three-dimensional culture and in vivo by integrin blocking bodies. J Cell Biol 1997; 137: 231–45.
36. Postovit LM, Margaryan NV, Seftor EA, et al. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc Natl Acad Sci USA 2008; 105: 4329–34.
37. Hendrix MJ, Seftor EA, Seftor REB, et al. Reprogramming metastatic tumor cells with the embryonic microenvironment. Nat Rev Cancer 2007; 7: 246–55.
38. Kulesa PM, Kasemeier-Kulesa JC, Teddy JM, et al. Reprogramming metastatic tumor cells to assume a neural crest-like phenotype in an embryonic microenvironment. Proc Natl Acad Sci USA 2006; 103: 3752–7.
39. Gootwine E, Webb, CG, Sachs L. Participation of myeloid leukemia cells injected into embryos in haematopoietic differentiation in adult mice. Nature 1982; 299: 63–5.
40. Biava PM, Fiorito A, Negro C, Mariani M. Effect of treatment with embryonic and uterine tissue homogenates on Lewis lung carcinoma development. Cancer Lett 1988; 41: 265–70.
41. Biava PM, Bonsignorio D, Hoxa M. Life-Protecting Factor (LPF): an anticancer low molecular weight fraction isolated from pregnant uterine mucosa during embryo organogenesis. J Tumor Marker Oncol 2000; 15: 223–33.
42. Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med 2007; 58: 267–84.
43. Lee JT, Herlyn M. Embryogenesis meet tumorigenesis. Nat Med 2006; 12: 882–3.
44. Katoh M, Katoh M. Transcriptional regulation of WNT2B based on the balance of Hedgehog, Notch, BMP and WNT signals. Int J Oncol 2009; 34: 1411–5.
45. Kathoh M, Katoh M. Transcriptional mechanisms of WNT5A based on NF-kappaB, Hedgehog, TGFbeta, and Notch signaling cascades. Int J Mol Med 2009; 23:763–9.
46. Peifer M, Polakis P. WNT signaling in oncogenesis and embryogenesis: a look outside the nucleus. Science 2000, 287: 1606–9.
47. Chritofk HF, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumor growth. Nature 2008; 452: 230–3.
48. Mani SA, Guo W, Liao M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133: 704–15.
49. Carro MS, Lim WK, Javier Alvares M, et al. The transcriptional network for mesenchymal transformation of brain tumor. Nature 2010; 463: 318–25.
50. Borczuk AC, Gorenstein L, Walter KL, et al. Non small cell lung cancer molecular signature recapitulate lung developmental pathways. Am J Pathol 2003; 163: 1949–60.
51. Sell S. On the stem cell origin of cancer. Am. J. Pathology 2010; 176:2584–94.
52. Sell S. Stem cell origin of cancer and differentiation therapy. Crit Rev Oncol Hematol 2004; 51: 1–28.
53. Sell S. Leukemia: stem cells, maturation arrest and differentiation therapies. Stem Cell Rev 2005; 1: 197–205.
54. Muto A, Kizaki M, Yamato K, et al. 1,25 Dihydroxyvitamin D3 induces differentiation of a retinoic acid-resistent acute promyelocitic leukemia cell line (UF-1) associated with espression of p21 ( WAF1/CIP1) and p 27 ( K1P1). Blood 1999; 93: 2225–33.
55. Luo W, Yu WD, Ma Y, et al. Inhibition of protein kinase CK2 reduces Cyp24a1 expression and enhances 1,25-dihydroxyvitamin D (3) antitumor activity in human prostate cancer cells. Cancer Res 2013; 73: 2289–97.
56. Batie S, Lee JH, Jama RA, et al. Synthesis and biological evaluation of halogenated curcumin analogs as potential nuclear receptor selective agonists. Bioorg Med Chem 2013; 21: 693–702.
57. Sun SY, Lotan R. Retinoids and their receptors in cancer development and chemoprevention. Crit. Rev Oncol Hematol 2002, 41: 41–55.
58. Biava PM, Bonsignorio D, Hoxa M. Cell proliferation curves of different human tumor lines after in vitro treatment with Zebrafish embryonic extracts. J Tumor Marker Oncol 2001; 16: 195–202.
59. Biava PM, Carluccio A. Activation of anti-oncogene p53 produced by embryonic extracts in vitro tumor cells. J Tumor Marker Oncol 1977; 12: 9–15.
60. Biava PM, Bonsignorio D, Hoxa M, et al. Post-translational modification of the retinoblastoma protein (pRb) induced by in vitro administration of Zebrafish embryonic extracts on human kidney adenocarcinoma cell line. J Tumor Marker Oncol 2002; 17: 59–64.
61. Cucina A, Biava PM, D’Anselmi F, et al. Zebrafish embryo proteins induce apoptosis in human colon cancer cells (Caco2). Apoptosis 2006; 9: 1617–28.
62. D’Anselmi F, Cucina A, Biava PM, et al. Zebrafish stem cell differentiation stage factors suppress Bcl-xL release and enhance 5 Fu mediated apoptosis in colon cancer cells. Curr Pharm Biotechnol 2011; 12: 261–7.
63. Biava PM, Nicolini A, Ferrari P, Carpi A, Sell S. A systemic approach to cancer treatment: tumor cell reprogramming focused on endocrine-related cancers. Current Med. Chem. 2014, 21: 1072–81.
64. Livraghi T, Meloni F, Frosi A, Lazzaroni S, et al. Treatment with stem cell differentiation stage factors of intermediate-advanced hepatocellular carcinoma: an open randomized clinical trial. Oncol Res 2005; 15: 399–408.
65. Livraghi T, Ceriani R, Palmisano A, et al. Complete response in 5 out of 38 patients with advanced hepatocellular carcinoma treated with stem cell differentiation stage factors: case reports from a single centre. Curr Pharm Biotechnol 2011; 12: 254–60.
66. Allegrucci C, Rushton MD, Dixon JE, et al. Epigenetic reprogramming of breast cancer cells with oocyte extracts. Mol Cancer 2011; 10: 7.
67. Rachakatia RS, Troyer D. Wharton’s jelly stromal cells as potential delivery vehicles for cancer therapeutics. Future Oncol 2009; 5: 1237–44.
68. Ganta C, Chiyo D, Ayuzawa R, et al. Rat umbical cord stem cells completely abolish rat mammary carcinomas with no evidence of metastasis or recurrence 100 days post-tumor cell inoculation. Cancer Res 2009; 69: 1815–20.
69. Xu T, Xu Y, Liao CP, Lau R, Goldkorn A. Reprogramming murine telomerase rapidly inhibits the growth of mouse cancer cells in vitro and in vivo. Mol Cancer Ther 2010; 9: 438–49.
70. Xu T, He K, Wang L, Goldkorn A. Prostate tumor cells with cancer progenitors properties have high telomerase activity and are rapidly killed by telomerase interference. Prostate 2011; 71: 1390–4000.
71. Xu Y, He K, Goldkorn A. Telomerase targeted therapy in cancer and cancer stem cells. Clin Adv. Hematol Oncol 2011; 9: 442–55.
72. Grzmil M, Voigt S, Thelen P, et al. Up-regulated expression of the MAT-8 gene in prostate cancer and its siRNA-mediated inhibition of expression induces a decrease in proliferation of human prostate carcinoma cells. Int J Oncol 2004; 24: 97–105.
73. Stolzenburg S, Rots MG, Beltran AS, et al. Targeted silencing of the oncogenic transcription factor SOX2 in breast cancer. Nucleic Acids Res 2012; 40: 6725–40.
74. Cubillos-Ruiz JR, Baird JR, Tesone AJ, et al. Reprogramming tumor-associated dendritic cells in vivo using miRNA mimetics triggers protective immunity against ovarian cancer. Cancer Res 2012; 72: 1683–93.
75. Quinn MC, Filali-Mouhim A, Provencher DM, Mes-Masson AM, Tonin PN. Reprogramming of the transcriptome in a novel chromosome 3 transfer tumor suppressor ovarian cancer cell line model affected molecular networks that are characteristic of ovarian cancer. Mol Carcinog 2009; 48: 648–61.
76. Biava PM, Bonsignorio D. Cancer and cell differentiation: a model to explain malignancy. J Tumor Marker Oncol 2002; 17: 47–54.
77. Shackeleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell 2009; 138: 822–9.
78. Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res 2012; 22: 457–72.
79. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell 2012; 10: 717–28.
80. Renan MJ. How many mutations are required for tumorigenesis? Implications from human cancer data. Mol Carcinogenesis 1993; 7: 139–46.
81. Han WC, Counter CM, Lundberg AS, et al. Creation of human tumor cells with defined genetic elements. Nature 1999; 400: 464–8.
82. Olumi AF, Grossfeld GD, Hayward SW, et al. Carcinoma associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res 1999; 59: 5002–11.
83. Hudson JD, Shoabi MA, Maestro R, et al. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med 1999; 190: 1375–82.
84. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57–70.
85. Biava PM, Monguzzi A, Bonsignorio D, et al. Xenopus Laevis embryo share antigens with Zebrafish embryos and with human malignant neoplasms. J Tumor Marker Oncol 2001; 16: 203–6.
86. Tannock IF. Cancer: Resistance through repopulation. Nature 2015; 517: 152–3.
87. Sato A, Sakurada K, Kumabe T, et al. Association of stem cell marker CD133 expression with dissemination of glioblastoma. Neurosurg Rev 2010; 33: 175–83.
88. Di Tommaso T, Mazzoleni S, Wang E, et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Cli. Cancer Res 2010; 16: 800–13.
89. Ji J, Black KL, Yu JS. Glioma stem cell research for the development of immunotherapy. Neurosurg Clin N Am 2010: 21: 159–66.
90. Chen J, Chen ZL. Technology update for the sorting and identification of breast cancer stem cells. Chin J Cancer 2010; 29: 265–9.
91. Park SY, Lee HE, Li H, et al. Heterogeneity for stem cell-related markers according to tumor subtype and histologic stage in breast cancer. Cancer Res 2010; 16: 876–87.
92. Lawson JC, Blatch GL, Edkins AL. Cancer stem cells in breast cancer and metastasis. Cancer Res Treat 2009; 8: 241–54.
93. Luo J, Yin X, Ma T, Lu J. Stem cells in normal mammary gland and breast cancer. Am J Med Sci 2010; 339: 366–70.
94. Spiro SG, Tanner NT, Silvestri GA, et al. Lung cancer: progress in diagnosis staging and therapy. Respirology 2010; 15: 44–50.
95. Gorelik E, Lokshin A, Levina L. Lung cancer stem cells as target for therapy. Anticancer Agents Med Chem 2010; 10: 164–71.
96. Sullivan JP, Minna JD, Shay JW. Evidence for self-renewing lung cancer stem cells and their implications in tumor initiation, progression and targeted therapy. Cancer Metastasis Rev 2010; 29: 61–72.
97. Westhoff B, Colaluca IN, D’Ario G, et al. Alteration of the Notch pathway in lung cancer. Proc Natl Acad Sci USA 2010; 87: 457–66.
98. Lawson DA, Zong Y, Memarzadeh S, et al. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc Natl Acad Sci USA 2010; 107: 2610–15.
99. Lang SH, Anderson E, Fordham R, Collin AT. Modeling the prostate stem cell niche: an evaluation of stem cell survival and expansion in vitro. Stem Cell Rev 2010; 19: 537–46.
100. Joung JY, Cho KS, Kim JE, et al. Prostate stem cell antigen mRNA in peripheral blood as a potential predictor of biochemical recurrence of metastatic prostate cancer. J Surg Oncol 2010; 101: 145–8.
101. Liu T, Cheng W, Lai D, Huang Y, Guo L. Characterization of primary ovarian cancer cells in different culture systems. Oncol Rep 2010; 23: 1277–84.
102. Fong MY, Kakar SS. The role of cancer stem cells and the side population in epithelial ovarian cancer. Histol. Histopatol 2010; 25: 113–20.
103. Murphy SK. Targeting ovarian cancer initiating cells. Anticancer Agents Med Chem 2010; 10: 157–63.
104. Pen S, Maihle NJ, Huang Y. Pluripotency factors Lin 28 and Oct 4 identify a sub-population of stem cell-like cells in ovarian cancer. Oncogene 2010; 29: 2153–59.
105. Zou GM. Liver cancer stem cells as an important target in liver cancer therapies. Anticancer Agents Med Chem 2010; 10: 172–175.
106. Lee TK, Castilho A, Ma S, Ng IO. Liver cancer stem cells: implication for new therapeutic target. Liver Int 2009; 29: 955–65.
107. Marquardt JU, Thorgeirsson SS. Stem Cells in hepatocarcinogenesis: evidence from genomic data. Semin Liver Dis 2010; 30: 26–34.
108. Kung JW, Currie IS, Forbes SJ, Ross JA. Liver development, regeneration, and carcinogenesis. J Biomed Biotechnol 2010; 30: 26–34.
109. Gai H, Nguyen DM, Moon YJ, et al. Generation of murine hepatic lineage cells from induced pluripotent stem cells. Differentiation 2010; 79: 171–81.
110. Nishii T, Yashiro M, Shinto O, et al. Cancer stem cell-like SP cells have a high adhesion ability to the peritoneum in gastric carcinoma. Cancer Sci 2009; 100: 1397–402.
111. Chen Z, Xu WR, Quian H, et al. Oct4 a novel marker for human gastric cancer. J Surg Oncol 2009; 34: 1201–7.
112. Kang DH, Han ME, Song MH, et al. The role of hedgehog signaling during gastric regeneration. J Gastroenterol 2009; 44: 372–9.
113. Correia M, Machado JC, Ristimaki A. Basic aspects of gastric cancer. Helicobacter 2009; 14: 36–40.
114. Takaishi S, Okumura T, Tu S, et al. Identification of gastric cancer stem cells using the surface marker CD44. Stem Cells 2009; 59: 106–20.
115. Nishii T, Yashiro M, Shinto O, et al. Cancer stem cell-like SP cells have a high adhesion ability to the peritoneum in gastric carcinoma. Cancer Sci 2009; 100: 1397–402.
116. Yeung TM, Ghandhi SC, Wilding JL, Muschel R, Bodmer WF. Cancer stem cells from colorectal cancer derived cell lines. Proc Natl Acad Sci USA 2010; 107: 3722–7.
117. Gulino A, Ferretti E, De Smaele E. Hedgehog signaling in colon cancer and stem cells. EMBO Mol Med 2009; 1: 300–2.
118. Thenappen A, Li Y, Shetty K, et al. New therapeutic targeting colon cancer stem cells. Curr Colorectal Cancer Rep 2009; 5: 209.
119. Rasheed ZA, Yang J, Wang Q, et al. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J Natl Cancer Inst 2010; 102: 340–51.
120. Puri S, Hebrok M. Cellular plasticity within pancreas-lessons learned from development. Dev Cell 2010; 18: 342–56.
121. Quante M, Wang TC. Stem cells in gastroenterology and hepatology. Nat Rev Gastroenterol Hepatol 2009; 6: 724–37.
122. Ailles L, Prince M. Cancer stem cells in head and neck squamous cell carcinoma. Methods Mol Biol 2009; 568: 175–93.
123. Zhang P, Zhang Y, Mao L, Zhang Z, Chen W. Side population in oral squamous cell carcinoma possesses tumor stem cell phenotype. Cancer Lett 2009; 277: 227–34.
124. Brunner M, Thurnher D, Heiduschka G, et al. Elevated levels of circulating endothelial progenitor cells in head and neck cancer patients. J Surg Oncol 2008; 98: 545–50.
125. Zhang Q, Shi S, Yen Y, et al. A subpopulation of CD133(+) cancer stem-like cells characterized in human oral squamous cell carcinoma confer resistance to chemotherapy. Cancer Lett 2010; 289: 151–60.
126. Strizzi L, Postovit LM, Margaryan NV, et al. Nodal as biomarker for melanoma progression and a new therapeutic target for clinical intervention. Expert Rev Dermatol 2009; 4: 67–78.
127. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646–74.
128. Biava PM, Nicolini A, Ferrari P, Carpi A, Sell S. Systemic approach to cancer treatment: tumor cell reprogramming focused on endocrine-related cancers. Current Med Chem 2014; 21: 1072–81.
ABSTRACTS OF SELECTED STUDIES
Stem Cell Differentiation Stage Factors and Their Role in Triggering Symmetry Breaking Processes during Cancer Development: A Quantum Field Theory Model for Reprogramming Cancer Cells to Healthy Phenotypes
Current Medicinal Chemistry, 2017 Sept 20
P. M. Biava, F. Burigana, R. Germano, P. Kurian, C. Verzegnassi, G. Vitiello
This study interprets results obtained using stem cell differentiation stage factors for reprogramming tumor cells in light of quantum field theory. It examines the effects of reprogramming tumor cells not only in light of the effects on biochemical mechanisms studied in the laboratory, but also in modulating electrochemical phenomena and electrodynamic behavior regarding the molecules of a network of differentiation factors and interstitial water. The study describes how changes in the genetic and epigenetic code are transmitted and amplified in the microenvironment of tumor cell populations, and how changes in the information field of the microenvironment can be used to reprogram tumor cells to healthy, non-proliferative states. Recent investigations have shown that proteins are capable of finding their cognate partners ten and even a hundred times faster than predicted by the Brownian diffusion rates, suggesting that electromagnetic effects can critically affect the time needed for two biomolecular partners to encounter each other.
A long history of research has pursued the use of embryonic factors isolated during cell differentiation processes for the express purpose of transforming cancer cells back to healthy phenotypes. Recent results have clarified that the substances present at different stages of cell differentiation—which we call stem cell differentiation stage factors (SCDSFs)—are proteins with low molecular weight and nucleic acids that regulate genomic expression. The present review summarizes how these substances, taken at different stages of cellular maturation, are able to retard proliferation of many human tumor cell lines and thereby reprogram cancer cells to healthy phenotypes. The model presented here is a quantum field theory (QFT) model in which SCDSFs are able to trigger symmetry breaking processes during cancer development.
These symmetry breaking processes, which lie at the root of many phenomena in elementary particle and condensed-matter physics, govern the phase transitions of totipotent cells to higher degrees of diversity and order, resulting in cell differentiation. In cancers, which share many genomic and metabolic similarities with embryonic stem cells, stimulated re-differentiation often signifies the phenotypic reversion back to health and non-proliferation. In addition to acting on key components of the cellular cycle, SCDSFs are able to reprogram cancer cells by delicately influencing the cancer microenvironment, modulating the electrochemistry and thus the collective electrodynamic behaviors between dipole networks in biomacromolecules and the interstitial water field. Coherent effects in biological water, which are derived from a dissipative QFT framework, may offer new diagnostic and therapeutic targets at a systemic level, before tumor instantiation occurs in specific tissues or organs. Thus, by including the environment as an essential component of our model, we may push the prevailing paradigm of mutation-driven oncogenesis toward a closer description of reality.
The Role of Neuroendocrine Cells in Prostate Cancer: A Comprehensive Review of Current Literature and Subsequent Rationale to Broaden and Integrate Current Treatment Modalities
Current Medicinal Chemistry, 2014, Volume 21, Number 9: 1082–92.
F. Lugnani, G. Simone, P. M. Biava, R. J. Ablin
Neuroendocrine prostate carcinoma (NE-PCa) is a heterogeneous disease. Due to a high prevalence of NE (neuroendocrine) differentiation in patients who receive prolonged androgen deprivation treatment, the real incidence of NE-PCa remains unknown. Similarly, the biological steps from prostate carcinoma (PCa) toward NE differentiation are far less than definitive and, consequently, there is a lack of evidence to support any of the treatments as the “gold standard.”
Materials and Methods
A systematic literature search was conducted using the PubMed, Scopus, and Embase databases to identify original articles and review articles regarding NE-PCa. Keywords were “prostate cancer” and “neuroendocrine.” Articles published between 1995 and 2013 were reviewed and selected with the consensus of all of the authors.
Results
Fifty-one articles were selected by the authors for the purpose of this review. The principle findings were reported into some subsections: Epidemiology, Biological steps of NE differentiation (with some principle articles on animal and in vitro, since there is very little in the literature on human studies); for the treatment options, we had to expand the search on PubMed to a larger timeframe and selection since very little was specifically found in the first criteria: surgery, radiotherapy, ablative techniques, immunomodulation, and epigenetic therapy were then reviewed. A multidisciplinary approach, advocated by many authors, although promising, has failed to demonstrate increased survival rates. Limitations of this review include the lack of a clear definition of NE-PCa and, consequently, the lack of strong evidence provided by a large series with long-term follow-up.
Conclusions
Supported by this extensive review, we propose it is worthwhile to investigate a new multimodal therapeutic approach to address advanced NE-PCa starting from a debunking (with radical intent) of the disease plus epigenetic therapy with stem cell differentiation stage factors (SCDSFs). In addition, immunotherapy can be used to treat the cancer presenting phenotype in association with chemomodulation plus ablative therapies, in case of advanced or recurrent diseases. SCDSFs may be utilized to regulate cancer stem cells, and possible new phenotypes could also be associated with ablative therapies. Hormonal deprivation, radiotherapy, chemotherapy, ex vivo vaccines and targeted therapies could also be used and reserved in case of failure.
A Systemic Approach to Cancer Treatment: Tumor Cell Reprogramming Focused on Endocrine-Related Cancers
Current Medicinal Chemistry, 2014, Volume 21, Number 9: 1072–81.
P. M. Biava, A. Nicolini, P. Ferrari, A. Carpi, S. Sell
The term “cancer cell reprogramming” is used to define any kind of intervention aimed at transforming cancer cells into terminally differentiated cells. Using this approach, new technologies have been applied with different methods for a more systemic approach to cancer treatment. This review reports on advances of these technologies, including our personal contributions, mainly carried out on endocrine-related cancers. Some of the interventions, aimed at reverting cancer cells into a normal phenotype, are based on the evidence that tumor development is suppressed by the embryonic microenvironment. On the basis of this rationale, experiments have been conducted using stem cell differentiation stage factors (SCDSFs) taken at different stages of development of Zebrafish embryos, oocyte extracts, or naive human umbilical cord matrix derived stem cells (UMDSCs). SCDSFs induce significant growth inhibition on different tumor cell lines in vitro, likely because of increases in cell cycle regulatory molecules, such as p53 and pRb.
Treatment with these factors activates apoptosis and differentiation related to caspase-3. This is achieved via p73 apoptotic-dependent pathway activation with a concurrent normalization of the E-cadherin and beta-catenin ratio. Extracts from prophase amphibian oocytes could reprogram relevant epigenetic alterations in MCF-7 and HCC1954 breast cancer cell lines, while un-engineered (naive) human UMDSCs attenuated growth of MDA-231 human breast carcinoma cells. A product prepared for human treatments, containing SCDSFs at very low doses, yielded favorable results in breast cancer and in intermediate-advanced hepatocellular carcinoma. Other reprogramming interventions used in the models of breast, prostate, and ovarian cancer cell lines are described. Finally, current and future perspectives of this novel technology are discussed, and a new hallmark of cancer is suggested: the loss of differentiation of cancer cells.
The Zebrafish Embryo Derivative Affects Cell Viability of Epidermal Cells: A Possible Role in the Treatment of Psoriasis
Giornale Italiano di Dermatologia e Venereologia, 2013 October, Volume 148, Number 5: 479–83.
G. D. Norata, P. M. Biava, F. Di Pierro
In patients affected by psoriasis, use of a topical formula containing a derivative of Zebrafish embryos was associated with reduced skin inflammation and dermal turnover, as well as a generally better outcome. In an attempt to understand the molecular mechanisms lying beyond these findings, we investigated the anti-proliferative effects of the Zebrafish embryo derivative by addressing the mitochondrial function (MTT assay) and cell nuclei distribution (Hoestch staining). In cell cultures stimulated with fetal calf serum (FCS) or epidermal growth factor (EGF), the Zebrafish derivative significantly inhibited cell proliferation induced by either approach, although the effect was stronger in cells stimulated with FCS. These results suggest that the Zebrafish embryo derivative may dampen increased cell proliferation; this observation may be relevant to cutaneous pathologies related to altered proliferative mechanisms, including psoriasis.
Cancer Cell Reprogramming: Stem Cell Differentiation Stage Factors and an Agent-Based Model to Optimize Cancer Treatment
Current Pharmaceutical Biotechnology, 2011, Volume 12, Number 2: 231–242.
P. M. Biava, M. Basevi, L. Biggiero, A. Borgonovo, E. Borgonovo, F. Burigana
Recent tumor research has led scientists to recognize the central role played by cancer stem cells in sustaining malignancy and chemoresistance. A model of cancer presented by one of us describes the mechanisms that give rise to the different kinds of cancer stem-like cells and the role of these cells in cancer diseases. The model implies a shift in the conceptualization of the disease from reductionism to complexity theory. By exploiting the link between the agent-based simulation technique and the theory of complexity, the medical view is here translated into a corresponding computational model. Two main categories of agents characterize the model: 1) cancer stem-like cells and 2) stem cell differentiation stage factors. Cancer cells agents are then distinguished based on the differentiation stage associated with the malignancy. Differentiation factors interact with cancer cells and then, with varying degrees of fitness, induce differentiation or cause apoptosis. The model inputs are then fitted to experimental data and numerical simulations carried out. By performing virtual experiments on the model’s choice variables, a decision-maker (physician) can obtain insights on the progression of the disease and on the effects of a choice of administration frequency and or dose. The model also paves the way to future research, whose perspectives are discussed.
Zebrafish Stem Cell Differentiation Stage Factors Suppress Bcl-Xl, Release and Enhance 5-Fu-Mediated Apoptosis in Colon Cancer Cells
Current Pharmaceutical Biotechnology, 2011, Volume 12, Number 2: 261–267
F. D’Anselmi, A. Cucina, P. M. Biava, S. Proietti, P. Coluccia, L. Frati, M. Bizzarri
Stem cell differentiation stage factors (SCDSF), taken from Zebrafish embryos during the stage in which totipotent stem cells are differentiating into pluripotent stem cells, have been shown to inhibit proliferation and induce apoptosis in colon tumors. In order to ascertain if these embryonic factors could synergistically/additively interact with 5-Fluorouracil (5-Fu), whole cell-count, f low-cytometry analysis, and apoptotic parameters were recorded in human colon cancer cells (Caco2) treated with Zebrafish stem cell differentiation stage factors (SCDSF 3 µg/ml) in association or not with 5-Fu in the sub-pharmacological therapeutic range (0.01 mg/ml). Cell proliferation was significantly reduced by SCDSF, while SCDSF+5-Fu leads to an almost complete growth-inhibition. SCDSF produces a significant apoptotic effect, while the association with 5-FU leads to an enhanced additive apoptotic rate at both 24 and 72 hrs. SCDSF alone and in association with 5-Fu triggers both the extrinsic and the intrinsic apoptotic pathways, activating caspase-8, -3 and -7. SCDSF and 5-Fu alone exerted opposite effects on Bax and Bcl-xL proteins; meanwhile, SCDSF+5-Fu induced an almost complete suppression of Bcl-xL release and a dramatic increase in the Bax/Bcl-xL ratio. These data suggest that Zebrafish embryo factors could improve chemotherapy efficacy by reducing anti-apoptotic proteins involved in drug-resistance processes.
Embryonic Morphogenetic Fields Induce Phenotypic Reversion in Cancer Cells
Current Pharmaceutical Biotechnology, 2011, Volume 12, Number 2: 243–253.
M. Bizzarri, A. Cucina, P. M. Biava, S. Proietti, et al.
Cancer cells introduced into developing embryos can be committed to a complete reversion of their malignant phenotype. It is unlikely that such effects could be ascribed to only a few molecular components interacting according to a simple linear-dynamics model, and they claim against the somatic mutation theory of cancer. Some 50 years ago, Needham and Waddington speculated that cancer represents an escape from a morphogenetic field like those that guide embryonic development. Indeed, disruption of the morphogenetic field of a tissue can promote the onset as well as the progression of cancer. On the other hand, placing tumor cells into a “normal” morphogenetic field—like that of an embryonic tissue—one can reverse malignant phenotype, “reprogramming” tumor into normal cells. According to the theoretical framework provided by the thermodynamics of dissipative systems, morphogenetic fields could be considered as distinct attractors, to which cell behaviors are converging. Cancer-attractors are likely positioned somewhat close to embryonic-attractors. Indeed, tumors share several morphological and ultra-structural features with embryonic cells. The recovering of an “embryonic-like” cell shape might enable the gene regulatory network to reactivate embryonic programs, and consequently to express antigenic and biochemical embryonic characters. This condition confers to cancer an unusual sensitivity to embryonic regulatory cues. Thus, it is not surprising that cancer cells exposed to specific embryonic morphogenetic fields undergo significant modifications, eventually leading to a complete phenotypic reversion.
Cancer, Cell Death, and Differentiation: The Role of Epigenetic Code in Tumor Growth Control
A Nature Conference: Cancer Therapeutics: The Road Ahead October 8–10, 2007, Palazzo dei Congressi di Capri (Italy)
P. M. Biava of Foundation for Research into the Biological Therapies of Cancer, IRCCS Multimedica (Milano)
A. Frosi of Hepatology-Gastroenterology Unit, Sesto S. G. Hospital (Milano)
M. Bizzarri and L. Frati of Department of Experimental Medicine and Pathology, Università La Sapienza (Roma)
Experiments on different human tumor cell lines (glioblastoma multi-forme, melanoma, breast carcinoma, kidney adenocarcinoma, colon adenocarcinoma, acute lymphoblastic leukemia) treated with stem cell differentiation stage factors taken from Zebrafish embyos during the stage of cell differentiation, in which totipotent stem cells differentiate into pluripotent stem cells, demonstrated a significant slowdown in tumor proliferation rate. The same results were obtained when tumor cells were treated with factors present in other different cell differentiation stages of Zebrafish embryos, like 5 somites and 20 somites stages, whereas no slowing effect was observed when tumor cells were treated with the factors taken from a merely multiplicative stage, like morula. In addition we observed a significant decrease in Lewis Lung Carcinoma injected in C57BL/6 mice treated with differentiation factors. Thus cell differentiation could be viewed as a key process in controlling the behavior of tumor cells. Our studies carried out in order to find out which cell regulation pathways are involved in the embryo in this mechanism of tumor growth demonstrated that key role cell cycle regulator molecules, such as p53 and pRb are modified by transcriptional and post-translational processes. Research on apoptosis and differentiation revealed that treatment with stem cell differentiation stage factors induces caspase 3 activation, mainly by increasing the releases of E2F-1, leading to c-Myc overexpression and activation of a p73 apoptotic-dependent pathway. Moreover, a concurrent significant normalization effect on the ratio of e-cadherin/beta catenin expression with increase in e-cadherin levels was observed (data not yet published). Finally, a product prepared for human therapy containing stem cell differentiation stage factors demonstrated 19.8% regression, 16% stable diseases, and a significant difference in survival between the group of patients who responded to treatment versus the group with progression disease (p<0.001) in an open randomized clinical trial on 179 consecutive patients with intermediate-advanced hepatocellular carcinoma. On the basis of these studies, a new vision of cancer related to a complexity model was proposed, confirming the importance of epigenetic modulation by factors present during precise stages of cell differentiation in controlling tumor growth.
Zebrafish Embryo Proteins Induce Apoptosis in Human Colon Cancer Cells (Caco2)
Apoptosis, 2006 September, Volume 11, Number 9: 1617–28.
A. Cucina, P. M. Biava, F. D’Anselmi, P. Coluccia, F. Conti, R. di Clemente, A. Miccheli, L. Frati, A. Gulino, M. Bizzarri
Previous studies have shown that proteins extracted from Zebrafish embryo share some cytostatic characteristics in cancer cells. Our study was conducted to ascertain the biological properties of this protein network. Cancer cell growth and apoptosis were studied in Caco2 cells treated with embryonic extracts. Cell proliferation was significantly inhibited in a dose-dependent manner. Cell-cycle analysis in treated cells revealed a marked accumulation in the G(2)/M phase preceding induction of apoptosis. Embryo proteins induced a significant reduction in FLIP levels and increased caspase-3 and caspase-8 activity as well as the apoptotic rate. Increased phosphorylated pRb values were obtained in treated Caco2 cells: the modified balance in pRb phosphorylation was associated with an increase in E2F1 values and c-Myc over-expression. Our data support previous reports of an apoptotic enhancing effect displayed by embryo extracts, mainly through the pRb/E2F1 apoptotic pathway, which thus suggests that Zebrafish embryo proteins have complex anti-cancer properties.
Treatment with Stem Cell Differentiation Stage Factors of Intermediate-Advanced Hepatocellular Carcinoma: An Open Randomized Clinical Trial
Oncology Research, 2005, Volume 15, Number 7-8: 399–408
T. Livraghi, F. Meloni, A. Frosi, S. Lazzaroni, M. Bizzarri, L. Frati, P. M. Biava
Corresponding Author: P. M. Biava
There is no standard treatment for patients with advanced hepatocellular carcinoma (HCC). We developed a product containing stem cells differentiation stage factors (SCDSF) that inhibits tumor growth in vivo and in vitro. The aim of this open randomized study was to assess its efficacy in patients with HCC not suitable for resection, transplantation, ablation therapy, or arterial chemoembolization. A total of 179 consecutive patients were enrolled. We randomly assigned the patients to receive either SCDSF or only conservative treatment. Primary end points were tumor response and survival. Secondary end points were performance status and patient tolerance. Randomization was stopped at the second interim analysis (6 months) of the first 32 patients recruited when the inspection detected a significant difference in favor of treatment (p = 0.037). The responses to the therapy obtained in 154 additional patients confirmed previous results. Evaluation of survival showed a significant difference between the group of patients who responded to treatment versus the group with progression of disease (p<0.001). Of the 23 treated patients with a performance status (PS) of 1, 19 changed to 0. The study indicated the efficacy of SCDSF treatment of the patients with intermediate-advanced HCC.