[T]he laws of mechanical affinity may be used for the most complete physical separation of the substances soluble in certain fluids. The green pigment of the leaves, the chlorophyll, is known to be a mixture of pigments, the complexity of which was differently estimated by different investigators. Chromatographic analysis is called upon to settle finally this degree of complexity.… Amongst the adsorption means I can provisionally recommend precipitated CaCO3 which gives the most beautiful chromatograms.
—Mikhail Tswett, 1872–1919, Italian-Russian botanist and inventor of chromatography
From Berichte der Deutschen Botanischen Gesellschaft 24 (1906)
Lodged in the earth’s outermost layer, ephemeral scratch on a mineral skin, life plays cards with a handful of elements—builds molecular extravaganzas of carbon and hydrogen, oxygen, nitrogen, sulfur, or precious phosphorus, and forms the pieces to the parts that, assembled, define it. When the game is over, the cards reshuffled, the parts dismantled—membranes ruptured, shells dissolved, bones ground to dust—a few of those organic molecules remain in the sediments and rocks, bearing witness to the distant moments of their creation.
Imagine the most humble bit of life, a microscopic alga basking in the sun-graced surface of the sea. Think of the tiny animal that grazes on the alga, dismantling its parts, using the pieces and discarding the difficult-to-digest fats and sturdy membrane lipids in tiny pellet-like feces that sink slowly into the dark waters of the deep sea—a thousand meters, two, three, maybe more. Imagine the bacteria that cling to the pellets as they settle onto the seafloor, zealous recyclers of organic molecules, using some and transforming others, leaving them stripped down or broken but still recognizable among the generic mineral bits of shell and clay that accumulate, particle by particle, year by year, layer by layer. Dig down, dig back, through meters and kilometers of sediments, through millennia and epochs, and you’ll find them yet, those molecular relics, testaments to that tiny, light-loving bit of bygone life.
What do those molecules know, what do they have to say? Might they remember their maker’s name and environment, how that tiny alga lived and died? Was it rich or poor, food plentiful or scarce, the water warm or cold? Perhaps there was a current from the south, or cold nutrient-rich waters upwelling from the deep. Maybe there was a drought in Africa and dry winds blew nutrient-laden dust over the Atlantic, the continent’s misfortune a literal windfall for marine algae. Perhaps a meteor fell that year and the light went out of the sky, the temperature dropped suddenly, and the world died in a blink. Or perhaps it was an epoch of tectonic activity and volcanic turmoil, and the atmosphere was laden with CO2 from the planet’s molten depths—the earth’s greenhouse fully insulated, temperatures soaring. Might those molecules tell a cautionary tale, perhaps even have some advice for us?
Nature’s molecules can be fascinating in their own right, for their particular blend of form and function, the way the carbon atoms twist and turn, the rings they form, the tempting reactivity of a double bond or the weak link to an oxygen atom. For some chemists, an organic molecule is a challenging puzzle to be solved, or a beautiful sculpture to be duplicated, or improved upon, or put to use. But the organic chemist who looks at a rock—a dull gray-white outcropping of limestone, for example—and wonders about the molecules made by algae or bacteria 200 million years ago is likely to have another take altogether. These are chemists who hang out with geologists and biologists, oceanographers and climatologists. Microbiologists, and even molecular biologists. Paleontologists, archaeologists, and environmental scientists. They are thinking about time—distant past, recent past, present, and future. The organic chemistry they practice is a far cry from that laid out in the French chemist Marcellin Berthelot’s definitive nineteenth-century treatise, based on man’s ability to create and manipulate molecules: “Chemistry creates its object. This creative quality, resembling that of art itself, distinguishes it essentially from natural and historical sciences. The latter have an object given in advance and independent of the will and action of the scientist.”* On the contrary, the organic chemistry of rocks, mud, and sea is both a natural and a historical science in which molecules are created, and manipulated over time, by Nature. It’s a brand of natural history designed for the twenty-first century, when humankind is the most significant player in global cycles and molecules are our unwitting pawns. Ironically, it was developed in the latter half of the twentieth century, when it went against the prevailing trend toward specialization and divergence of disciplines that Berthelot’s definition heralded.
If molecules can be witnesses, then the possibilities for what they have to tell us are endless. Not only can they tell us what the climate was like during the last ice age, but they can provide clues as to what the early humans were eating at the time; they can tell us about the advent of life on Earth, or the presence of life on Mars, about the source of pollution in a bay, or of the gasoline an arsonist used to ignite a fire. This idea of molecules as informants, as biological markers or “biomarkers” that carry information through time and space, is relatively recent, the product of largely extant generations of scientists, several of which are spanned by the three coauthors of this book. When I—the writer and lapsed scientist among us—was a graduate student in California in the mid-1980s amino acids extracted from ancient sediments were providing clues to the dinosaurs’ demise and a British research group found that fossil lipid molecules could provide a record of sea surface temperatures during the last ice age. When my coauthor Jürgen Rullkötter interviewed for his first job with Germany’s new Institute of Petroleum and Organic Geochemistry in the 1970s, chemists were just beginning to understand that the structures of sterane molecules extracted from mudstone might hold clues to the accumulations of oil hidden deep below the surface of the earth. And when our collaborator, main character, and inspiration, Geoff Eglinton, was a student in Manchester, England, back in the 1940s, chemists were chemists, geologists were geologists, and he was hard put to find anyone interested in small traces of organic molecules in rocks.
Like any good organic chemistry student, Geoff spent his time flirting with explosions in the laboratory and learning how to construct new and ever more exotic molecules from scratch. But as a youth he had read Darwin’s Voyage of the Beagle and spent many an hour poring over natural history books, and he yearned to connect the disembodied chemistry he was learning in the laboratory to the “real” world outside. One day, when he was hiking in Derbyshire with the university mountaineering club, he noticed a stinky, rubbery, brown tar oozing out the side of a cliff. At the time, Geoff says, it seemed really weird to him that this gooey brown stuff was leaking out of the dull gray limestone. The geologists in the club told him the stone was compressed marine sediments, made up of the calcium carbonate shells of tiny animals and microscopic algae—zooplankton and phytoplankton that had lived in the surface water of the ocean several hundred million years ago. Wondering if the brown goo contained traces of chlorophyll or other organic molecules made by these ancient organisms, Geoff scooped a bit into a tin and took it home.
“Didn’t you know about Treibs?” I asked Geoff when he told me this story. A decade earlier, the German chemist Alfred Treibs had asked similar questions about deposits of petroleum and the oils that came out of certain types of sedimentary rocks. But no, Geoff says, he didn’t know about Treibs’s work. Petroleum was outside the realm of inquiry for most chemists at the time, something formed deep within the earth—the domain of geologists. When he showed his brown goo to his chemistry professor and expressed an interest in finding out what was in it, the professor just laughed and told him he’d better concentrate on getting his degree first, because it would be a complicated, if not impossible, task to separate such a complex mixture of compounds, let alone figure out what they were. It wasn’t until Geoff was a lecturer with his own research group at the University of Glasgow, dutifully engaged in the enterprise of constructing and transforming organic molecules as he’d been trained to do, that he would find himself in a position to contemplate the composition of anything even faintly resembling the Derbyshire goo.
The Glasgow chemistry department of the 1950s and early 1960s was known for its synthetic organic chemists, but there were also a few distinguished natural products chemists, people who were searching for interesting molecules made by nature. They isolated compounds from exotic plants and fungi, following leads from biologists and anthropologists in an attempt to find compounds that were useful as pharmaceuticals, food flavorings, perfumes, or cosmetics. It was by no means a systematic attempt to compile a natural history of life’s molecules, but as the catalog of chemical structures and their sources grew, that was what it amounted to—an aspect of the enterprise that Geoff found particularly attractive. Though he counted himself among the synthetic chemists, he was in charge of a new instrument that was particularly helpful in figuring out chemical structures, the infrared spectrometer, and the natural products chemists often brought him their new compounds for analysis. What, he wondered, was the ultimate fate of all these interesting compounds the organisms made? Were they entirely ephemeral, recycled to CO2 as soon as the organisms died? Or did some end up buried and preserved in the sediments?
But the natural products chemists were no more interested in such queries than the synthetic chemists. Guy Ourisson, a French natural products chemist who occasionally collaborated with his counterparts in Glasgow, says that he had a geologist friend who pestered him for almost a decade to analyze rock and sediment samples, before he finally acquiesced. His friend reasoned that even if most sediments contained less than 3% organic matter, the earth had such a large mass of sedimentary rock that it would contain 10,000 times more organic matter than all the living organisms together. I recently came across a 1973 paper where Ourisson—known to me in the 1980s as the founder of one of the world’s leading biomarker laboratories—quotes these same facts and exclaims, with the zeal of the convert, “Organic chemists should therefore mostly study rocks!” But in the 1950s and 1960s, he tells me, he found the concept entirely unappealing. He wanted to know about the molecules of life, and who could imagine that a rock would have anything important to say on the matter? Who would want to study the intractable stuff his geologist friend was offering, when there was a world full of plants and animals that only needed to be tossed into a blender and extracted with solvent to yield exciting molecular discoveries? It was challenge enough to isolate, purify, and determine the chemical structure of a compound in such an extract, let alone in the horrendous, impossible-to-separate mixture of compounds that must have accumulated in the sediments.
Geoff, nonetheless, was still curious. He wrote to his brother and asked him to send another sample of the Derbyshire tar, thinking he might give it a go. But when it arrived and he took another look at it, his reaction was much like Ourisson’s. “It looked so unpromising,” he says. “I didn’t really know what to do with it.” He stored it away, along with all the leftovers from the samples the Glasgow chemists gave him for infrared analysis, just in case. Meanwhile, the instruments that would eventually allow chemists to separate such an impossible mixture of organic compounds had just come on the market, and Geoff had acquired one for his laboratory in Glasgow. It would be another few years before it occurred to him that he might use the new gas chromatograph to analyze something like his stinky old tar, but he was, in fact, already headed in that direction.
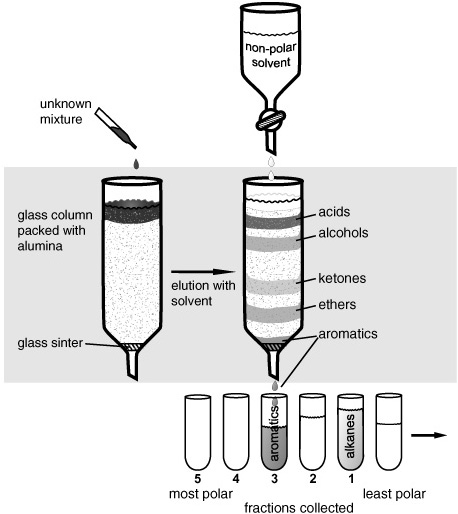
The gas chromatographs that came on the market in the mid-1950s were based on chromatographic principles that organic chemists, in their ongoing quest to obtain pure substances for analysis, had been using and refining for almost half a century. They could grind up a plant or an insect and attempt to dissolve it in various liquids, using the fact that like dissolved like to extract different types of molecules into different solvents. Then they could evaporate off the liquids, reextract the residual solids, and eventually, if they were lucky, obtain a few pure crystals of a single component from the hundreds in the original mix. But chromatography allowed the systematic separation of Nature’s messy mixtures into clans of molecules, and the clans into families, and even, eventually, into individuals. The Russian botanist Mikhail Tswett recognized the basic concept at the turn of the last century when he used column chromatography to separate the pigments in a leaf: he poured an extract of a leaf into the top of a glass tube packed with finely ground chalk, washed it down with solvent, and noted that the pigments moved at different speeds through the column, such that the green extract separated into bands of green, blue-green, yellow, and orange. The premise was prosaic enough: push a bunch of molecules through a column of fine-grained solid with a flood of solvent, and different types of molecules will move at different rates, like runners at a race who start out together but reach the finish line at different times—small and light move faster than large and heavy, graceful faster than awkward, solvent-lovers faster than solvent-phobes—depending on what solvent you use and what sort of stationary phase you pack in the tube. Alumina or silica packing clings to more polar or water-soluble molecules such as alcohols or acids; large, cumbersome, water-insoluble hydrocarbons move fast in solvents like hexane or toluene. Choose well, refine the process, and if you collect what comes out the end of the tube in increments, you have relatively simple mixtures of molecules with similar traits, or even pure compounds.
Column chromatography
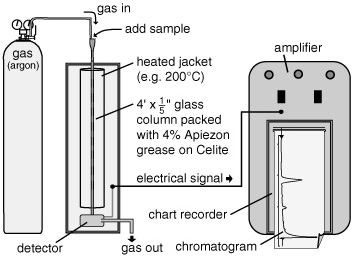
There are myriad variations on the theme. If, instead of a column, one uses a glass plate covered with a thin layer of silica, places a drop of the sample mixture near the bottom edge of the plate, and stands it in a pool of solvent, one has thin layer chromatography, where the solvent crawls upward and the compounds separate and stick to the silica along the way. Their positions can then be compared with those in a standard mix of known compounds, or the silica can be sliced up and the compounds analyzed separately. If one replaces the glass column or plate with a long, thin tube, and the silica or alumina stationary phase with oil or grease, and then vaporizes the sample mixture and pushes it through with a flow of gas instead of solvent, then one has a gas-liquid chromatograph, now colloquially known as a “GC.” Here the mixture separates according to the molecules’ size, volatility, and preference for the gas or liquid phase. Various sorts of detectors can be connected to the GC, so that as the organic compounds exit the tube they generate an electronic signal, which, in turn, makes a pen slide up and down on the moving graph paper of a chart recorder, thus indicating both the amount of compound and length of time it took to make its way through the tube.
The GC Geoff experimented with in the late 1950s had been designed for food and essential oil analyses. It was generally used for mixtures of relatively light, easily vaporized compounds that would move with the gas, molecules with fewer than 14 carbon atoms—but Geoff tried his out on anything he could get his hands on. “We just wanted to see what the thing could do,” he says, sounding even now, with his shock of white hair and beard, like a child with a new toy he’s been caught abusing. One of the perks of his infrared spectroscopy expertise—acquired during an unbearably hot summer he spent as a postdoc at the University of Ohio, where the only cool place to be was in the infrared facility—was that he had a huge cache of strange new compounds to choose from, things the designers of the GC had never even imagined. “We rooted around in there, picking out anything that looked interesting. It’s a wonder we didn’t poison ourselves, since half the time we didn’t know what it was we were analyzing.”
In these early GCs, the oil stationary phase was coated onto a fine powder and packed into a long, narrow metal tube that was housed in an oven or heated jacket. Geoff tried reducing the oil coating to a bare minimum, increased the flow of gas through the column, decreased his sample size, cranked the temperature up as high as he dared, and started pushing through bigger and bigger molecules.
Gas chromatograph, 1950s
He found he could separate mixtures of long-chain hydrocarbons up to 30 carbons long. He even tried running the 27-carbon cholesterol, not really expecting it to get through the column, with its bulky ring structure—and was astounded to see a peak appear on the chromatogram. “It was a rather fat one,” Geoff says, “took a while to come through … but there it was. After that we started running all the big molecules we could find.” The bulky cholesterol molecule is ubiquitous in animals, where it serves as a rigid insert in the otherwise flexible cell membrane. But it was its special role as a culprit in heart disease that made it a hot topic among biomedical researchers in 1959, when Geoff and his small research group published a description of their GC analysis. They received thousands of requests for reprints of the brief paper—and, more importantly, they received samples of all sorts of things to analyze with the new method.
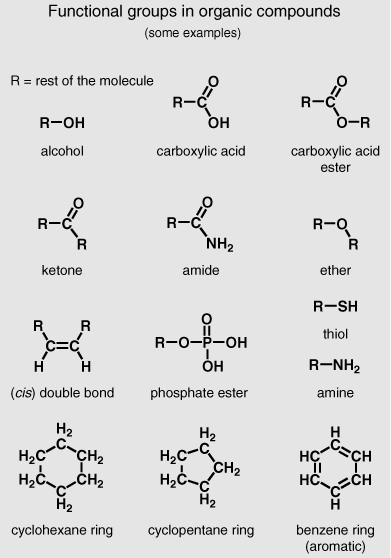
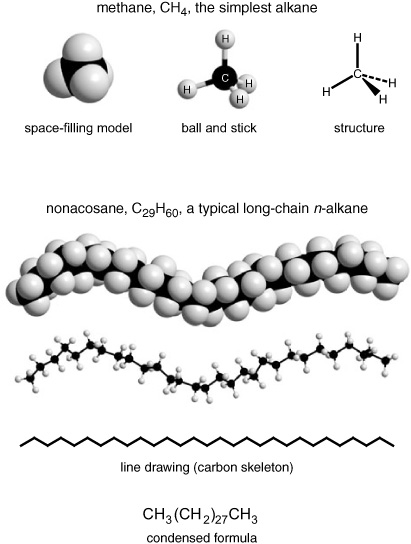
One very famous Cambridge plant biochemist who had spent a good part of his career studying the wax coatings of leaves offered Geoff his legacy: dozens of cigarette tins full of leaf waxes that had been extracted and recrystallized from hundreds of species of plants. Each wax contained a complex mixture of compounds and before the GC could separate them, they had to be passed through a simple alumina chromatography column and divided into families based on polarity. The individual compounds in each family could then be separated on the GC and tentatively identified from a comparison of their behavior—how long they took to get through the column at a given temperature—with that of known compounds in a standard mixture. The easiest family of compounds to analyze, and by far the most prevalent in all of the leaf waxes, was that of the normal alkanes. From most organic chemists’ and biochemists’ point of view, these were the most boring compounds imaginable: straight chains of carbon and hydrogen atoms, distinguished only by their different lengths, with no branches or interesting rings or flat double bonds, no oxygen or nitrogen or sulfur atoms to break the monotony.
Geoff remembers that one chemist who shared his laboratory in Glasgow for a time found it so incomprehensible that he finally paused by Geoff’s lab bench and exclaimed: “Why are you people working on n-alkanes?! They don’t do anything!!” Geoff says he mumbled something in reply, but he can’t remember what. The man was right: the n-alkanes are boring chemically—they don’t do anything. They have no reactive functional groups dangling like charms from the ends of their chains—no water-loving oxygen atoms as in the hydroxyl groups of the alcohols or the carboxyl groups of the acids, no ether links or sweet reactive esters to attract a water molecule or enzyme, not even a double bond or a ketone’s carbonyl group. Even biochemically, once these leaf-wax alkanes are made, they just sit there on the outside of a leaf with their long sturdy chains lined up side by side and layer upon layer in wax crystals. With no reactive double bonds or other weak links for enzymes to attack, they are unattractive to bacteria and fungi, and with no oxygen atoms or functional groups, they are decidedly nonpolar and hydrophobic, completely immiscible with water.
Normal alkanes: different representations
used to understand molecular behavior
About all one could say about the leaf-wax n-alkanes at the time was that their hydrophobia and general inertness made them particularly well suited to their job of protecting plant leaves from dehydration and disease: they kept water inside and pathogens outside. From a purely chemical point of view, they were decidedly uninteresting. But they were so prevalent, so easy to observe, that Geoff couldn’t just ignore them. He was exploring, like the nineteenth-century naturalists who traveled the world describing and naming all the plants and animals they could find. Geoff had embarked with his chromatographic ship to explore the molecular world of those same creatures, and he couldn’t very well ignore all the dull brown finches and sparrows just because the tanagers were more beautiful or the eagles more spectacular. Perhaps, like the Galapagos finches that inspired Darwin’s monumental observation that “one species had been taken and modified for different ends,” these boring n-alkanes would prove more enlightening than the fantastic ring structures Geoff’s colleagues were building and isolating.
Usually, it’s the thrill of conquest or challenge of complicated new structures that attracts organic chemists: like a bird or a beautiful woman, a molecule is more interesting and desirable for being exotic or elusive, or, as one prominent Swedish organic chemist was fond of saying, “a tough nut to crack.” The n-alkanes offered no structural challenges and were easy to identify: their peaks in the chromatograms appeared in order, shortest chains first, longer later, and one only needed to compare their positions with those of a standard mix of n-alkanes that one had synthesized from scratch, a relatively easy task. But as Geoff looked at his chromatograms, which showed the makeup of the entire mixture of n-alkanes in the wax from a given leaf, he began thinking of molecules in a new way. Rather than the beauty of a single character in the series, for they were all rather homely, what stood out in such an analysis were the patterns of peaks in the chromatogram: in the arrays of n-alkanes from the leaf waxes, it was the different distributions of the same set of molecules, the variations within this one homologous series, that caught his attention. Each chromatogram was a graphic representation, a picture or a fingerprint of a particular species, characterized by the absence or presence and relative amounts of the homologues. It was these patterns and the relationships between them that was interesting, and that would eventually lead Geoff and a handful of his contemporaries into a new way of considering the mixtures of organic molecules they obtained from plants and animals, and, ultimately, rocks and sediments.
A chemist friend who worked in the food industry told Geoff that such patterns were being used to classify the mixtures of fatty acids—which are simply long-chain n-alkanes with a carboxylic acid group at the end—in different animal fats and vegetable oils, and it occurred to Geoff that the patterns of peaks in his chromatograms of leaf wax n-alkanes might follow the taxonomy of the plants. They might provide new information about differences and similarities between species, even about the evolutionary pathways that led to their differentiation. To test this idea, he needed waxes from species that would show a clear evolutionary pattern, something like Darwin’s Galapagos finches. He needed a large group of closely related genera of plants, species that had evolved in an isolated environment with a minimal amount of differentiation.… What he needed, of course, was an island.
Gas chromatograms of leaf wax alkanes
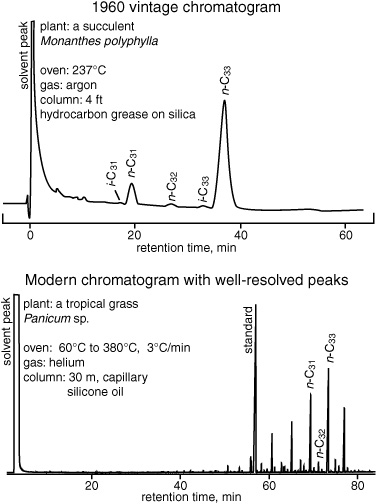
“We had a postdoc in the lab,” Geoff says, “who’d come from this way out place, the island of Gomera, in the Canaries. You can’t think of a place farther from the center of science at the time. He told us they used a whistling language to communicate across the island’s hills and valleys, because there were so few phones.… I think that’s what got me interested in the Canaries, to tell the truth, this fellow’s stories.” Once he started thinking about it, however, the Canary Islands seemed just the thing. They were relatively undeveloped, so there probably wouldn’t be a lot of introduced species; the botany had been well studied; and they were volcanic and geologically young, so the species would be recently differentiated and tightly grouped.
Of course, it wasn’t an easy thing to mount a scientific expedition to such a place, but a chance encounter at a university banquet in Cambridge around this time presented an opportunity: Geoff found himself sitting next to the laboratory manager for the chemistry department, who, it turned out, had been involved in providing instruments to the only prominent organic chemist on the island of Tenerife, at the university where Geoff’s Gomera postdoc had been trained. Over dinner, the Cambridge laboratory manager told Geoff that the chemist had funding, but couldn’t get new instruments past the local customs office. He suggested that Geoff, as a foreigner, could take a new GC into the country with him and then resell it to the chemist, bypassing customs. In return, the Tenerife chemist would pay part of Geoff’s project expenses and provide laboratory facilities. So that’s what Geoff did: he got a grant from the Carnegie Foundation, took a leave from teaching at Glasgow, and set off with his young family and his Ph.D. student, Dick Hamilton, on a scientific-cum-smuggling expedition to the wilds of Tenerife.
Tenerife in 1960 was not extensively developed. Nor was it, to Geoff’s dismay, particularly wild. He had chosen the perfect group of native plants for study, a subfamily of drought-resistant succulents that have particularly thick waxy leaf coatings. In a recent article, botanists had even compared its evolution to that of the Galapagos finches. Unfortunately, most of Tenerife was under cultivation, and the pristine native flora had long been overrun by introduced banana and palm trees. This was a far cry from the isolated Galapagos! But once word got round that the British scientists were looking for native vegetation, the locals guided them into the island’s isolated wild canyons, and the botanist at its magnificent Botanical Gardens offered to identify their finds.
Analysis of the n-alkanes was relatively easy: dip the leaves in chloroform to extract the waxes; pour the extracts into chromatography columns packed with alumina; collect the least polar fraction, composed mostly of n-alkanes and their one-branched brothers, the iso-alkanes; and then run this hydrocarbon fraction on the GC. The amount of each alkane could be determined from the size of its peak in the chromatogram. Geoff and his student prepared histograms that showed the relative amounts of different homologues for each species, and then they compared them, looking for patterns that might link and differentiate the taxonomic groups.
Comparing alkane histograms, 1962
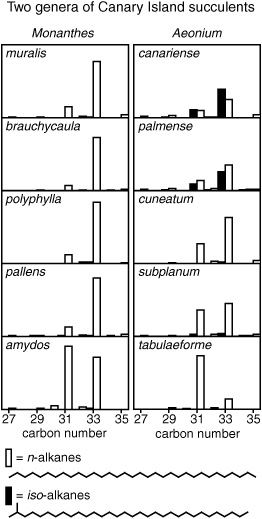
There were patterns, but they were not as distinct as one might have hoped. Each species did indeed have a characteristic distribution of alkanes, an individual fingerprint that might allow one to identify an otherwise hard-to-distinguish species. But the patterns didn’t reveal the evolutionary links or common ancestors of the related species within a genus.
“Were you disappointed?” I asked Geoff the other day, a little disappointed myself, as I looked at histograms from the Canaries expedition. Geoff shrugs off the question, but I wonder if he really remembers or if it’s only now, in hindsight, that he recognizes the importance of the other, seemingly mundane observations in his 1962 paper. The attempt to use leaf wax hydrocarbons as a taxonomic tool was only marginally successful, but the study revealed some of their essential properties as molecular chroniclers—as biomarkers—before the concept even existed: that the n-alkanes range from 25 to 35 carbons long, with the 27-, 29-, and 31-carbon compounds particularly prominent; that those with odd numbers of carbon atoms dominate the mixture by a factor of 10 over even-carbon-number chains; and that no matter how old the plant or where it grew or what time of year one chose to analyze it, the species fingerprints remain true, the n-alkane distributions nearly constant.
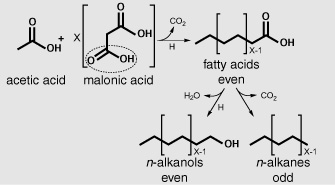
This family of long-chain n-alkanes with odd numbers of carbon atoms would turn out to be specific to land plants, found not only in their leaf waxes, but in their stems, flowers, and pollen. The odd-over-even preference is easily explained by our knowledge of how organisms build unbranched carbon chains, a core biosynthetic process that appears to be common to all forms of life. Unbranched chain structures are assembled two carbon atoms at a time: acetic acid is combined with malonic acid, losing a carbon to form a four-carbon acid, which then latches onto another malonic acid, losing a carbon to form the six-carbon acid, and so on. Accordingly, the fatty acids and the alcohols—biosynthesized via a simple reduction of the acids—have carbon chains with primarily even numbers of carbon atoms. The n-alkanes like those in the leaf waxes, however, are produced by snipping the carboxyl carbon off the end, which results in chains with odd numbers of carbon atoms.
The same physical qualities that make these long, water-phobic hydrocarbon chains such good leaf protectors also make them resistant to breakdown in the environment once the plant dies. Once they hit the dirt, so to speak, they go where it goes, eroded by rain, washed into the sea by rivers, blown out to sea with the dust—indeed, small particles of wax are even blown directly off the surface of leaves and carried long distances on the wind. The presence of long-chain n-alkanes in marine sediments, far from their origin in both time and place, thus records the movements of wind and water from land to sea, and even, if one looks more closely at the carbon atoms in their chains, the ecosystems and climates where the plants that made them formed. Some four decades after Geoff’s Tenerife smuggling exploit, he and Jürgen are once again engaged in a study that has them poring over histograms of leaf wax alkanes. This time, their extracts come not only from the leaves of living plants, but from sediments buried beneath the floor of the South Atlantic Ocean; this time, the patterns tell a story that spans nearly 200,000 years of changing climate and vegetation across an entire continent. But more of that later. First, we need to take a look at the intervening decades of molecular exploration and discovery by an ever-expanding legion of rock-, mud-, and sea-loving organic chemists, among others.
Biosynthesis of unbranched carbon chains:
odd vs. even numbers of carbon atoms
* See Chimie organique fondée sur la synthèse, Mallet-Bachelier (Paris), published in 1860.