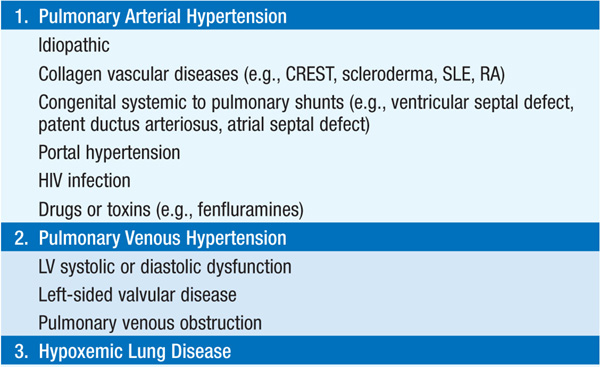
Elevation of pulmonary artery (PA) pressure due to pulmonary vascular or parenchymal disease, increased left heart filling pressures, or a combination. Table 136-1 lists etiologies by underlying categories.
TABLE 136-1 CATEGORIES OF PULMONARY HYPERTENSION

Exertional dyspnea, fatigue, angina (due to RV ischemia), syncope, peripheral edema.
Jugular venous distention, RV lift, increased P2, right-sided S4, tricuspid regurgitation. Peripheral cyanosis and edema are late manifestations.
CXR shows enlarged central PA. ECG may demonstrate RV hypertrophy and RA enlargement. Echocardiogram shows RV and RA enlargement; RV systolic pressure can be estimated from Doppler recording of tricuspid regurgitation (Chap. 120). Pulmonary function tests identify underlying obstructive or restrictive lung disease; impaired CO diffusion capacity is common. Chest CT identifies contributing interstitial lung disease or pulmonary thromboembolic disease. ANA titer is elevated in collagen vascular diseases. HIV testing should be performed in individuals at risk. Cardiac catheterization accurately assesses PA pressures, cardiac output, and pulmonary vascular resistance, and it identifies underlying congenital vascular shunts; during procedure, response to short-acting vasodilators can be assessed.
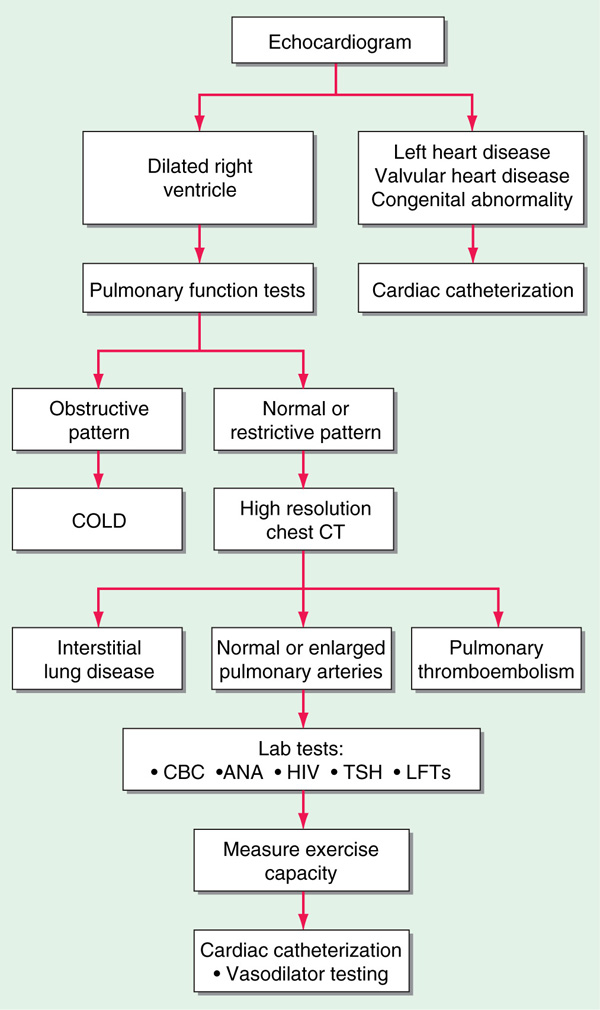
Figure 136-1 summarizes workup of pt with unexplained pulmonary hypertension.
FIGURE 136-1 An algorithm for the workup of a pt with unexplained pulmonary hypertension. All potential etiologies and associated conditions must be investigated in a pt with clinical findings consistent with pulmonary hypertension. ANA, antinuclear antibodies; CBC, complete blood count; COLD, chronic obstructive lung disease; HIV, human immunodeficiency virus; LFTs, liver function tests; TSH, thyroid-stimulating hormone.
Uncommon (2 cases/million), very serious form of pulmonary hypertension. Most pts present in fourth and fifth decades, female > male predominance; up to 20% of cases are familial. Major symptom is dyspnea, often with insidious onset. Mean survival <3 years in absence of therapy.
Prominent a wave in jugular venous pulse, right ventricular heave, narrowly split S2 with accentuated P2. Terminal course is characterized by signs of right-sided heart failure.
CXR: RV and central pulmonary arterial prominence. Pulmonary arteries taper sharply. PFTs: usually normal or mild restrictive defect. ECG: RV enlargement, right axis deviation, and RV hypertrophy. Echocardiogram: RA and RV enlargement and tricuspid regurgitation.
Other disorders of heart, lungs, and pulmonary vasculature must be considered. Lung function studies will identify chronic pulmonary disease causing pulmonary hypertension and cor pulmonale. Interstitial diseases (PFTs, CT scan) and hypoxic pulmonary hypertension (ABGs, Sao2) should be excluded. Perfusion lung scan should be considered to exclude chronic pulmonary embolism (PE). Rarely, pulmonary hypertension is due to parasitic disease (schistosomiasis, filariasis). Cardiovascular disorders to be excluded include pulmonary artery and pulmonic valve stenosis, ventricular and atrial shunts with secondary pulmonary vascular disease (Eisenmenger syndrome), and clinically silent mitral stenosis.
Limit physical activities, use diuretics for peripheral edema, O2 supplementation if PO2 reduced, and chronic warfarin anticoagulation (target INR = 2.0–3.0).
If short-acting vasodilators are beneficial during acute testing in catheterization laboratory, pt may benefit from high-dose calcium channel blocker (e.g., nifedipine, up to 240 mg/d, or amlodipine up to 20 mg/d); must monitor for hypotension or worsening of right heart failure during such therapy.
Additional approved therapies for PAH include:
1. Endothelin receptor antagonists: bosentan (62.5 mg PO bid × 1 month, then 125 mg PO bid) and ambrisentan (5–10 mg daily) significantly improve exercise tolerance. Hepatic transaminases should be monitored. Bosentan is contraindicated in pts taking cyclosporine (which greatly increases bosentan plasma levels) or glyburide (combination is associated with increased hepatic transaminases).
2. Phosphodiesterase-5 inhibitors: sildenafil (20–80 mg PO tid) and tadalafil (40 mg daily) also improve exercise tolerance in PAH. Do not prescribe concurrently with nitrovasodilators; the combination could result in marked hypotension.
3. Prostaglandins (iloprost by inhalation, epoprostenol by continuous IV infusion, and treprostinil by IV, SC, or inhalation routes) improve symptoms, exercise tolerance, and, in the case of epoprostenol, survival. The most common side effect is flushing.
For selected pts with persistent right heart failure, lung transplantation can be considered.

For a more detailed discussion, see Rich S: Pulmonary Hypertension, Chap. 250, p. 2076, in HPIM-18.