Hypercalcemia from any cause can result in fatigue, depression, mental confusion, anorexia, nausea, constipation, renal tubular defects, polyuria, a short QT interval, and arrhythmias. CNS and GI symptoms can occur at levels of serum calcium >2.9 mmol/L (>11.5 mg/dL), and nephrocalcinosis and impairment of renal function occur when serum calcium is >3.2 mmol/L (>13 mg/dL). Severe hypercalcemia, usually defined as >3.7 mmol/L (>15 mg/dL), can be a medical emergency, leading to coma and cardiac arrest.
The regulation of the calcium homeostasis is depicted in Fig. 187-1. The causes of hypercalcemia are listed in Table 187-1. Hyperparathyroidism and malignancy account for 90% of cases.
TABLE 187-1 CLASSIFICATION OF CAUSES OF HYPERCALCEMIA
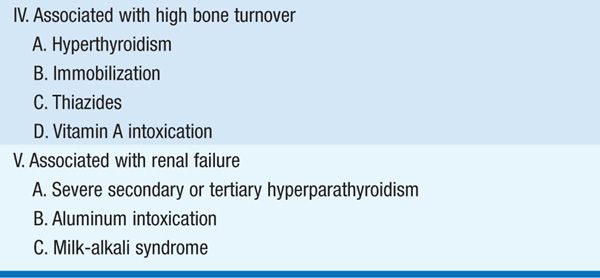
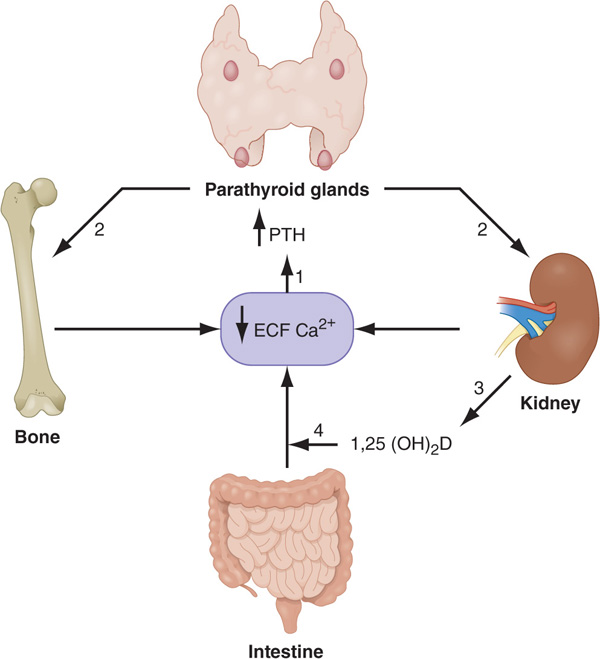
FIGURE 187-1 Feedback mechanisms maintaining extracellular calcium concentrations within a narrow, physiologic range [8.9–10.1 mg/dL (2.2–2.5 mM)]. A decrease in extracellular (ECF) ionized calcium (Ca2+) triggers an increase in parathyroid hormone (PTH) secretion (1) via activation of the calcium sensor receptor on parathyroid cells. PTH, in turn, results in increased tubular reabsorption of calcium by the kidney (2) and resorption of calcium from bone (2) and also stimulates renal 1,25(OH)2 D production (3). 1,25(OH)2 D, in turn, acts principally on the intestine to increase calcium absorption (4). Collectively, these homeostatic mechanisms serve to restore serum calcium levels to normal.
Primary hyperparathyroidism is a generalized disorder of bone metabolism due to increased secretion of parathyroid hormone (PTH) by an adenoma (81%) or rarely a carcinoma in a single gland, or by parathyroid hyperplasia (15%). Familial hyperparathyroidism may be part of multiple endocrine neoplasia type 1 (MEN 1), which also includes pituitary and pancreatic islet tumors, or of MEN 2A, in which hyperparathyroidism occurs with pheochromocytoma and medullary carcinoma of the thyroid.
Hypercalcemia associated with malignancy is often severe and difficult to manage. Mechanisms for this include excess production and release of PTH-related protein (PTHrP) in lung, kidney, and squamous cell carcinoma (humoral hypercalcemia of malignancy); local bone destruction in myeloma and breast carcinoma; activation of lymphocytes leading to release of IL-1 and TNF in myeloma and lymphoma; or an increased synthesis of 1,25(OH)2D in lymphoma.
Several other conditions have been associated with hypercalcemia. These include sarcoidosis and other granulomatous diseases, which lead to increased synthesis of 1,25(OH)2D; vitamin D intoxication from chronic ingestion of large vitamin doses (50–100 × physiologic requirements); lithium therapy, which results in hyperfunctioning of the parathyroid glands; and familial hypocalciuric hypercalcemia (FHH) due autosomal dominant inheritance of an inactivating mutation in the calcium-sensing receptor, which results in inappropriately normal or even high secretion of PTH, despite hypercalcemia and enhanced renal calcium resorption. Severe secondary hyperparathyroidism associated with end-stage renal disease may progress to tertiary hyperthyroidism, in which PTH hypersecretion becomes autonomous, causes hypercalcemia, and is no longer responsive to medical therapy.
Most pts with mild to moderate hyperparathyroidism are asymptomatic, even when the disease involves the kidneys and the skeletal system. Pts frequently have hypercalciuria and polyuria, and calcium can be deposited in the renal parenchyma (nephrocalcinosis) or form calcium oxalate stones. The characteristic skeletal lesion is osteopenia or osteoporosis; rarely, the more severe disorder osteitis fibrosa cystica occurs as a manifestation of long-standing, more severe hyperparathyroidism. Increased bone resorption primarily involves cortical rather than trabecular bone. Hypercalcemia may be intermittent or sustained, and serum phosphate is usually low but may be normal.
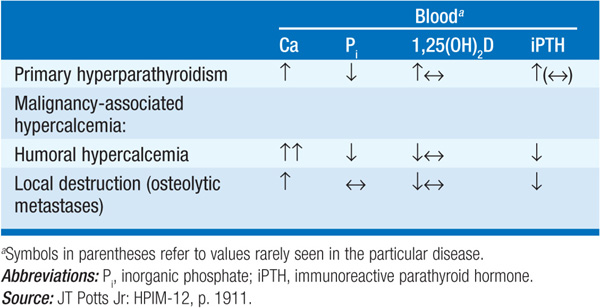
Primary hyperparathyroidism is confirmed by demonstration of an inappropriately high PTH level for the degree of hypercalcemia. Hypercalciuria helps to distinguish this disorder from FHH, in which PTH levels are usually in the normal range and the urine calcium level is low. Differentiation between primary hyperparathyroidism and FHH is important because the latter does not respond to parathyroid surgery. Levels of PTH are low in hypercalcemia of malignancy (Table 187-2).
TABLE 187-2 DIFFERENTIAL DIAGNOSIS OF HYPERCALCEMIA: LABORATORY CRITERIA
Total serum calcium should be corrected when serum albumin is abnormal [addition of 0.2 mM (0.8 mg/dL) to calcium value for every 1.0-g/dL decrement in albumin below 4.1 g/dL, or the converse for an increase in albumin]. Alternatively, ionized calcium can be measured. Third-generation PTH assays should be used for PTH measurement, especially in pts with renal impairment.
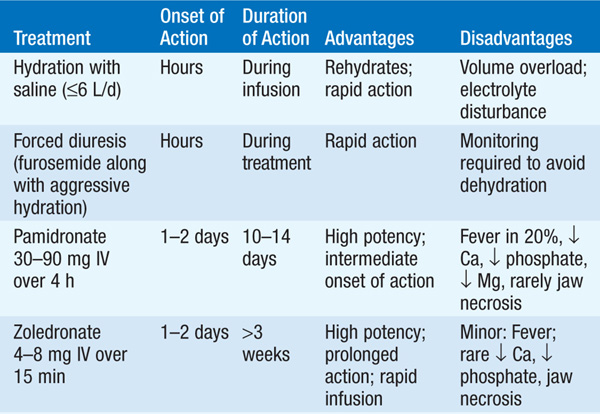
The type of treatment is based on the severity of the hypercalcemia and the nature of the associated symptoms. Table 187-3 shows general recommendations that apply to therapy of severe hypercalcemia [levels of >3.2 mmol/L (>13 mg/dL)] from any cause.
TABLE 187-3 THERAPIES FOR SEVERE HYPERCALCEMIA
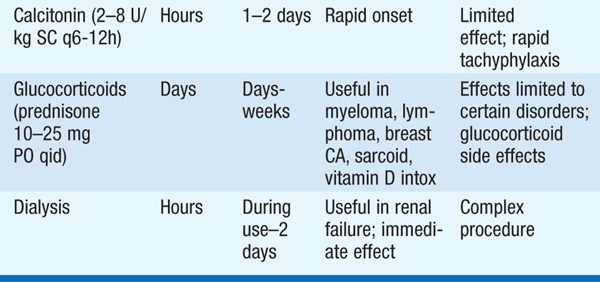
In pts with severe primary hyperparathyroidism, surgical parathyroidectomy should be performed promptly. Asymptomatic disease may not require surgery; usual surgical indications include age <50, nephrolithiasis, creatinine clearance <60 mL/min, reduction in bone mass (T score <–2.5), or serum calcium >0.25 mmol/L (>1 mg/dL) above the normal range. A minimally invasive approach may be used if preoperative localization via sestamibi scans with SPECT or neck ultrasound demonstrates a solitary adenoma and intraoperative PTH assays are available. Otherwise, neck exploration is required. Surgery in a center experienced in parathyroid interventions is recommended. Postoperative management requires close monitoring of calcium and phosphorus, as transient hypocalcemia is common. Calcium supplementation is given for symptomatic hypocalcemia.
Hypercalcemia of malignancy is managed by treating the underlying tumor. Adequate hydration and parenteral bisphosphonates can be used to reduce calcium levels. Long-term control of hypercalcemia is difficult unless the underlying cause can be eliminated.
No therapy is recommended for FHH. Secondary hyperparathyroidism should be treated with phosphate restriction, the use of nonabsorbable antacids or sevelamer, and calcitriol. Tertiary hyperparathyroidism requires parathyroidectomy.
Chronic hypocalcemia is less common than hypercalcemia, but is usually symptomatic and requires treatment. Symptoms include peripheral and perioral paresthesia, muscle spasms, carpopedal spasm, tetany, laryngeal spasm, seizure, and respiratory arrest. Increased intracranial pressure and papilledema may occur with long-standing hypocalcemia, and other manifestations may include irritability, depression, psychosis, intestinal cramps, and chronic malabsorption. Chvostek’s and Trousseau’s signs are frequently positive, and the QT interval is prolonged. Both hypomagnesemia and alkalosis lower the threshold for tetany.
Transient hypocalcemia often occurs in critically ill pts with burns, sepsis, and acute renal failure; following transfusion with citrated blood; or with medications such as protamine and heparin. Hypoalbuminemia can reduce serum calcium below normal, although ionized calcium levels remain normal. The above-mentioned correction (see “Hypercalcemia”) can be used to assess whether the serum calcium concentration is abnormal when serum proteins are low. Alkalosis increases calcium binding to proteins, and in this setting direct measurements of ionized calcium should be used.
The causes of hypocalcemia can be divided into those in which PTH is absent (hereditary or acquired hypoparathyroidism, hypomagnesemia), PTH is ineffective (chronic renal failure, vitamin D deficiency, anticonvulsant therapy, intestinal malabsorption, pseudohypoparathyroidism), or PTH is overwhelmed (severe, acute hyperphosphatemia in tumor lysis, acute renal failure, or rhabdomyolysis; hungry bone syndrome following parathyroidectomy). The most common forms of chronic severe hypocalcemia are autoimmune hypoparathyroidism and postoperative hypoparathyroidism following neck surgery. Chronic renal insufficiency is associated with mild hypocalcemia compensated for by secondary hyperparathyroidism. The cause of hypocalcemia associated with acute pancreatitis is unclear.
Symptomatic hypocalcemia may be treated with IV calcium gluconate (bolus of 1–2 g IV over 10–20 min followed by infusion of 10 ampoules of 10% calcium gluconate diluted in 1 L D5W infused at 30–100 mL/h). Management of chronic hypocalcemia requires a high oral calcium intake, usually with vitamin D supplementation (Chap. 188). Hypoparathyroidism requires administration of calcium (1–3 g/d) and calcitriol (0.25–1 μg/d), adjusted according to serum calcium levels and urinary excretion. Restoration of magnesium stores may be required to reverse hypocalcemia in the setting of severe hypomagnesemia.
Mild hypophosphatemia is not usually associated with clinical symptoms. In severe hypophosphatemia, pts may have muscle weakness, numbness, paresthesia, and confusion. Rhabdomyolysis may develop during rapidly progressive hypophosphatemia. Respiratory insufficiency can result from diaphragm muscle weakness.
The causes of hypophosphatemia include: decreased intestinal absorption (vitamin D deficiency, phosphorus-binding antacids, malabsorption); urinary losses (hyperparathyroidism, hyperglycemic states, X-linked hypophosphatemic rickets, oncogenic osteomalacia, alcoholism, or certain toxins); and shifts of phosphorus from extracellular to intracellular compartments (administration of insulin in diabetic ketoacidosis or by hyperalimentation or refeeding in a malnourished pt). In syndromes of severe primary renal phosphate wasting (X-linked hypophosphatemic rickets, autosomal dominant hypophosphatemic rickets, oncogenic osteomalacia), the phosphatonin hormone FGF23 (fibroblast growth factor 23) plays a key pathogenetic role.
TREATMENT Hypophosphatemia
Mild hypophosphatemia can be replaced orally with milk, carbonated beverages, or Neutra-Phos or K-Phos (up to 2 g/d in divided doses). For severe hypophosphatemia [0.75 mmol/L; (<2.0 mg/dL)], IV phosphate may be administered at initial doses of 0.2–0.8 mmol/kg of elemental phosphorus over 6 h. The total body phosphate depletion cannot be predicted from the serum phosphate level; careful monitoring of therapy is therefore required. Hypocalcemia should be corrected first, and the dose reduced 50% in hypercalcemia. Serum calcium and phosphate levels should be measured every 6–12 h; a serum calcium × phosphate product of >50 must be avoided.
In adults, hyperphosphatemia is defined as a level >1.8 mmol/L (>5.5 mg/dL). The most common causes are acute and chronic renal failure, but it may also be seen in hypoparathyroidism, vitamin D intoxication, acromegaly, acidosis, rhabdomyolysis, and hemolysis. The clinical consequences of severe hyperphosphatemia are hypocalcemia and calcium phosphate deposition in tissues. Depending on the location of tissue calcifications, serious chronic or acute complications may ensue (e.g., nephrocalcinosis, cardiac arrhythmias). Therapy consists of treating the underlying disorder and limiting dietary phosphorus intake and absorption. Oral aluminum phosphate binders or sevelamer may be used, and hemodialysis should be considered in severe cases.
Hypomagnesemia usually indicates significant whole body magnesium depletion. Muscle weakness, prolonged PR and QT intervals, and cardiac arrhythmias are the most common manifestations of hypomagnesemia. Magnesium is important for effective PTH secretion as well as the renal and skeletal responsiveness to PTH. Therefore, hypomagnesemia is often associated with hypocalcemia.
Hypomagnesemia generally results from a derangement in renal or intestinal handling of magnesium and is classified as primary (hereditary) or secondary (acquired). Hereditary causes include both disorders of absorption (rare) and those of renal loss (e.g., Bartter’s and Gitelman syndromes). Secondary causes are much more common, with renal losses being due to volume expansion, hypercalcemia, osmotic diuresis, loop diuretics, alcohol, aminoglycosides, cisplatin, cyclosporine, and amphotericin B, and gastrointestinal losses most commonly resulting from vomiting and diarrhea.
TREATMENT Hypomagnesemia
For mild deficiency, oral replacement in divided doses totaling 20–30 mmol/d (40–60 meq/d) is effective, although diarrhea may result. Parenteral magnesium administration is usually needed for serum levels <0.5 mmol/L (<1.2 mg/dL), with a continuous infusion of magnesium chloride IV to deliver 50 mmol/d over a 24-h period (dose reduced by 50–75% in renal failure). Therapy may be required for several days in order to replete tissue magnesium stores; serum Mg should be monitored every 12–24 h during treatment. Other electrolyte disturbances should be treated simultaneously. Pts with associated seizures or acute arrhythmias can be given 1–2 g of magnesium sulfate IV over 5–10 min.
Hypermagnesemia is rare but can be seen in renal failure when pts are taking magnesium-containing antacids, laxatives, enemas, or infusions, or in acute rhabdomyolysis. The most readily detectable clinical sign of hypermagnesemia is the disappearance of deep tendon reflexes, but hypocalcemia, hypotension, paralysis of respiratory muscles, complete heart block, and cardiac arrest can occur. Treatment includes stopping the preparation, clearing the intestines of residual offending laxatives or antacids with magnesium-free enemas or cathartics, dialysis against a low magnesium bath, or, if associated with life-threatening complications, 100–200 mg of elemental calcium IV over 1–2 h.

For a more detailed discussion, see Bringhurst FR, Demay MB, Krane SM, Kronenberg HM: Bone and Mineral Metabolism in Health and Disease, Chap. 352, p. 3082; Khosla S: Hypercalcemia and Hypocalcemia, Chap. 46, p. 360; and Potts JT Jr, Jüppner H: Disorders of the Parathyroid Gland and Calcium Homeostasis, Chap. 353, p. 3096, in HPIM-18.