CHAPTER 196
Ataxic Disorders
CLINICAL PRESENTATION
Symptoms and signs may include gait impairment, visual blurring due to nystagmus, unclear (“scanning”) speech, hand incoordination, intention tremor (i.e., with movement). Differential diagnosis: Unsteady gait associated with vertigo from vestibular nerve or labyrinthine disease can resemble gait instability of cerebellar disease but produces a sensation of movement, dizziness, or light-headedness. Sensory disturbances also can simulate cerebellar disease; with sensory ataxia, imbalance dramatically worsens when visual input is removed (Romberg sign). Bilateral proximal leg weakness also can rarely mimic cerebellar ataxia.
APPROACH TO THE PATIENT Ataxia
Causes are best grouped by determining whether ataxia is symmetric or focal and by the time course (Table 196-1). Also important to distinguish whether ataxia is present in isolation or is part of a multisystem neurologic disorder. Acute symmetric ataxia is usually due to medications, toxins, viral infection, or a postinfectious syndrome (especially varicella). Subacute or chronic symmetric ataxia can result from hypothyroidism, vitamin deficiencies, infections (Lyme disease, tabes dorsalis, prions), alcohol, other toxins, or an inherited condition (see below). An immune-mediated progressive ataxia is associated with antigliadin antibodies; biopsy of the small intestine may reveal villous atrophy of gluten enteropathy. Elevated serum anti–glutamic acid decarboxylase (GAD) antibodies have been associated with a progressive ataxic syndrome affecting speech and gait. Progressive nonfamilial cerebellar ataxia after age 45 suggests a paraneoplastic syndrome, either subacute cortical cerebellar degeneration (ovarian, breast, lung, Hodgkin’s) or opsoclonus-myoclonus (neuroblastoma, breast, lung).
TABLE 196-1 ETIOLOGY OF CEREBELLAR ATAXIA
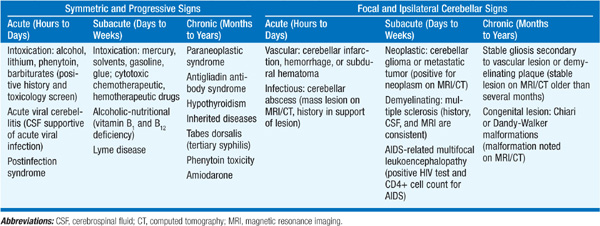
Unilateral ataxia suggests a focal lesion in the ipsilateral cerebellar hemisphere or its connections. An important cause of acute unilateral ataxia is stroke. Mass effect from cerebellar hemorrhage or swelling from cerebellar infarction can compress brainstem structures, producing altered consciousness and ipsilateral pontine signs (small pupils, sixth or seventh nerve palsies); limb ataxia may not be prominent. Other diseases producing asymmetric or unilateral ataxia include tumors, multiple sclerosis, progressive multifocal leukoencephalopathy (immunodeficiency states), and congenital malformations.
INHERITED ATAXIAS
May be autosomal dominant, autosomal recessive, or mitochondrial (maternal inheritance); more than 30 disorders recognized (see Table 373-2, pp. 3337–3340, in HPIM-18). Friedreich’s ataxia is most common; autosomal recessive, onset before age 25; ataxia with areflexia, upgoing toes, vibration and position sense deficits, cardiomyopathy, hammer toes, scoliosis; linked to expanded trinucleotide repeat in the intron of gene encoding frataxin; a second form is associated with genetically determined vitamin E deficiency syndrome. Common dominantly inherited ataxias are spinocerebellar ataxia (SCA)1 (olivopontocerebellar atrophy; “ataxin-1” gene) (Fig. 196-1), SCA2 (ataxin-2; pts from Cuba and India) and SCA3 (Machado-Joseph disease); all may manifest as ataxia with brainstem and/or extrapyramidal signs; SCA3 may also have dystonia and amyotrophy; genes for each disorder contain unstable trinucleotide repeats in coding region.

FIGURE 196-1 Sagittal MRI of the brain of a 60-year-old-man with gait ataxia and dysarthria due to SCA1, illustrating cerebellar atrophy (arrows). MRI, magnetic resonance imaging; SCA1, spinocerebellar ataxia type 1.
EVALUATION
Diagnostic approach is determined by the nature of the ataxia (Table 196-1). For symmetric ataxias, drug and toxicology screens; vitamin B1, B12, and E levels; thyroid function tests; antibody tests for syphilis and Lyme infection; antigliadin and anti-GAD antibodies; paraneoplastic antibodies (Chap. 84); and CSF studies often indicated. Genetic testing is available for many inherited ataxias. For unilateral or asymmetric ataxias, brain MRI or CT scan is the initial test of choice; CT is insensitive for nonhemorrhagic lesions of the cerebellum.
• The most important goal is to identify treatable entities including hypothyroidism, vitamin deficiency, and infectious causes.
• Parainfectious ataxia can be treated with glucocorticoids.
• Ataxia with antigliadin antibodies and gluten enteropathy may improve with a gluten-free diet.
• Paraneoplastic disorders are often refractory to therapy, but some pts improve following removal of the tumor or immunotherapy (Chap. 84).
• Vitamins B1, B12, and E should be administered to pts with deficient levels.
• The deleterious effects of phenytoin and alcohol on the cerebellum are well known, and these exposures should be avoided in pts with ataxia of any cause.
• There is no proven therapy for any of the autosomal dominant ataxias; family and genetic counseling are important.
• There is preliminary evidence that idebenone, a free-radical scavenger, can improve myocardial hypertrophy in Friedreich’s ataxia; there is no evidence that it improves neurologic function.
• Cerebellar hemorrhage and other mass lesions of the posterior fossa may require emergent surgical treatment to prevent fatal brainstem compression.

For a more detailed discussion, see Rosenberg RN: Ataxic Disorders, Chap. 373, p. 3335, in HPIM-18.