Chapter 4
Subcellular Organization of the Nervous System
Organelles and Their Functions
Cells have many features in common, but each cell type also possesses a functional architecture related to its unique physiology. In fact, cells may become so specialized in fulfilling a particular function that virtually all subcellular components may be devoted to that function. For example, the machinery inside mature mammalian erythrocytes is completely dedicated to the delivery of oxygen to the tissues and the removal of carbon dioxide. Toward this end, this cell has evolved a specialized plasma membrane, an underlying cytoskeletal matrix that molds the cell into a biconcave disk, and a cytoplasm rich in hemoglobin. Modification of the cell machinery extends even to the discarding of structures such as the nucleus and the protein synthetic apparatus, which are not needed after the red blood cell matures. In many respects, the terminally differentiated, highly specialized cells of the nervous system exhibit comparable commitment; the extensive development of subcellular components reflects the roles that each plays (Brady & Tai, 2012).
The neuron serves as the cellular correlate of information processing, and, in aggregate, all neurons act together to integrate responses of the entire organism to the external world. It is therefore not surprising that the specializations found in neurons are more diverse and complex than those found in any other cell type. Single neurons commonly interact in specific ways with hundreds of other cells—other neurons, astrocytes, oligodendrocytes, immune cells, muscle, and glandular cells (see Chapter 3). Some of these other cell types in the nervous system, such as the myelinating glia (oligodendrocytes and Schwann cells) or astrocytes are inextricably linked to the neuron both functionally and structurally, representing critical partners in building and maintaining a nervous system, with highly specialized subcellular architecture. Here, we define the major functional domains of the neuron, describe the subcellular elements that compose the building blocks of these domains, and examine the processes that create and maintain neuronal functional architecture.
Axons and Dendrites: Unique Structural Components of Neurons
Neurons and glial cells are remarkable for their size and complexity, but they do share many features with other eukaryotic cells (Peters, Palay, & Webster, 1991). As discussed in Chapter 3, the perikaryon, or cell body, contains a nucleus and its associated protein synthetic machinery. Most neuronal nuclei are large and typically contain a preponderance of euchromatin. This is consistent with the need to create and maintain a large cellular volume. Because protein synthesis must be kept at a high level just to maintain the extensive volume and surface area comprising the perikaryon, dendrites, and axon, transcription levels in neurons are generally high. In turn, the wide variety of different polypeptide constituents associated with cellular domains in a neuron requires that a large number of different genes be transcribed constantly.
When specific mRNAs have been synthesized and processed, they move from the nucleus into a subcellular region that can be termed the translational cytoplasm (Lasek & Brady, 1982) comprising cytoplasmic (“free”) and membrane-associated polysomes, the intermediate compartment of the smooth endoplasmic reticulum, and the Golgi complex. The constituents of translational cytoplasm are thus associated with the synthesis and processing of proteins. Neurons in particular have relatively large amounts of translational cytoplasm to accommodate a high level of protein synthesis. This protein synthetic machinery is arranged in discrete intracellular “granules” termed Nissl substance, after the histologist who first discovered these structures in the nineteenth century. The Nissl substance is actually a combination of stacks of rough endoplasmic reticulum (RER), interposed with rosettes of free polysomes (Fig. 4.1). This arrangement is unique to neurons, and its functional significance is unknown. Most, but by no means all, proteins used throughout the neuron are synthesized in the perikaryon. During or after synthesis and processing, proteins are packaged into membrane-limited organelles in the Golgi complex, incorporated into cytoskeletal elements, or remain as soluble constituents of the cytoplasm. After proteins have been packaged appropriately, they are transported to their sites of function.
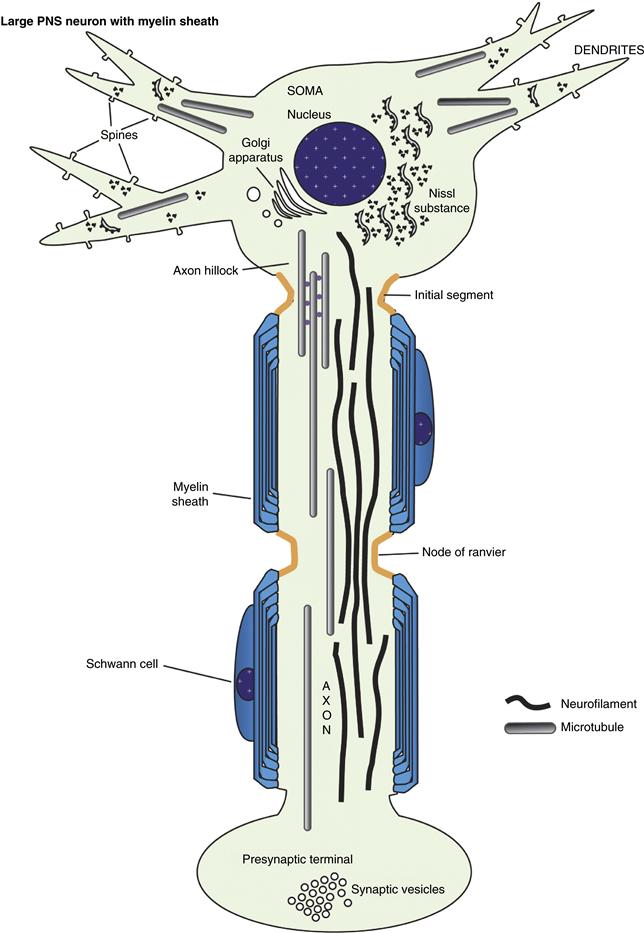
Figure 4.1 Basic elements of neuronal subcellular organization. The neuron consists of a soma, or cell body, in which the nucleus, multiple cytoplasm-filled processes termed dendrites, and the (usually single) axon are placed. The neuron is highly extended in space; a neuron with a cell body of the size shown here could easily maintain an axon several miles in length! The unique shape of each neuron is the result of a cooperative interplay between plasma membrane components (the lipid matrix and associated proteins) and cytoskeletal elements. Most large neurons in vertebrates are myelinated by oligodendrocytes in the CNS and by Schwann cells in the PNS. The compact wraps of myelin encasing the axon distal to the initial segment permit the rapid conduction of the action potential by a process termed “saltatory conduction” (see Chapter 3).
With a few exceptions, vertebrate neurons have two discrete functional domains or compartments, the axonal and the somatodendritic compartments, each of which encompasses a number of distinct microdomains. The axon is perhaps the most familiar functional domain of a neuron and is classically defined as the cellular process by which a neuron makes contact with a target cell to transmit information, providing a conducting structure for transmitting the action potential to a synapse, a specialized subdomain for transmission of a signal from neuron to target cell (neuron, muscle, etc.), most often by release of appropriate neurotransmitters. Consequently, most axons end in presynaptic terminal specializations, although a single axon may have many (hundreds or even thousands in some cases) presynaptic specializations known as en passant synapses along its length. Characteristics of presynaptic terminals are presented in greater detail later.
The axon is the first neuronal process to differentiate during development. A typical neuron has only a single axon that proceeds some distance from the cell body before branching extensively. Usually the longest process of a neuron, axons come in many sizes. In a human adult, axons range in length from a few micrometers for small interneurons to a meter or more for large motor or sensory neurons, and they may be even longer in large animals (such as giraffes, elephants, and whales). In mammals and other vertebrates, the longest axons generally extend approximately half the body length.
Axonal diameters also are quite variable, ranging from 0.1 to 20 µm for large myelinated fibers in vertebrates. Invertebrate axons grow to even larger diameters, with the giant axons of some squid species achieving diameters in the millimeter range. Invertebrate axons reach such large diameters because they lack the myelinating glia that speed conduction of the action potential. As a result, axonal caliber must be large to sustain the high rate of conduction needed for the reflexes that permit escape from predators and capture of prey. Although axonal caliber is closely regulated in both myelinated and nonmyelinated fibers, this parameter is critical for those organisms that are unable to produce myelin.
The region of the neuronal cell body where the axon originates has several specialized features. This domain, called the axon hillock, is distinguished most readily by a deficiency of Nissl substance. Therefore, protein synthesis cannot take place to any appreciable degree in this region. Cytoplasm in the vicinity of the axon hillock may have a few polysomes but is dominated by the cytoskeletal and membranous organelles that are being delivered to the axon. Microtubules and neurofilaments begin to align roughly parallel to each other, helping to organize membrane-limited organelles destined for the axon. The hillock is a region where materials either are committed to the axon (cytoskeletal elements, synaptic vesicle precursors, mitochondria, etc.) or are excluded from the axon (RER and free polysomes, dendritic microtubule-associated proteins). The molecular basis for this sorting is not understood, although recent studies suggest that actin and microtubule-based cytoskeletal elements may play a role in this sorting at the protein level (Arnold, 2009). Cytoplasm in the axon hillock does not appear to contain a physical “sizing” barrier (like a filter) because large organelles such as mitochondria enter the axon readily, whereas only a small number of essentially excluded structures such as polysomes are occasionally seen only in the initial segment of the axon and not in the axon proper. A known exception to this general rule is during development when local protein synthesis does take place at the axon terminus or growth cone. Nevertheless, new methods of detecting gene transcripts, including so-called “deep sequencing,” are beginning to suggest that even in mature axons mRNAs may be more abundant than originally thought, in spite of the fact that classical ribosomes or even polysomes are difficult to detect. In the mature neuron, the physiological significance of this barrier must be considerable because axonal structures are found to accumulate in this region in many neuropathologies, including those due to degenerative diseases (such as amyotrophic lateral sclerosis) and to exposure to neurotoxic compounds (such as acrylamide).
The initial segment of the axon is the region of the axon adjacent to the axon hillock (Palay, Sotelo, Peters & Orkand, 1968; Grubb & Burrone, 2010). Microtubules generally form characteristic fascicles, or bundles, in the initial segment of the axon. These fascicles are not seen elsewhere in the neuron. The initial segment and, to some extent, the axon hillock also have a distinctive specialized plasma membrane. Initially, the plasmalemma was thought to have a thick electrondense coating actually attached to the inner surface of the membrane, but this dense undercoating is in reality separated by 5–10 nm from the plasma membrane inner surface and has a complex ultrastructure. Neither the composition nor the function of this undercoating is known. Curiously, the undercoating is present in the same regions of the initial segment as the distinctive fasciculation of microtubules, although the relationship is not understood. The plasma membrane is specialized in the initial segment and axon hillock in that it contains voltage-sensitive ion channels in large numbers together with several other proteins known to play a part in either clustering the voltage-gated sodium channels (ankyrin G), or stabilizing them (Neurofascin 186). It has generally been thought that most action potentials are initiated in this domain, but it may also have a role in modulating action potentials that arise elsewhere.
Ultimately, axonal structure is geared toward the efficient conduction of action potentials at a rate appropriate to the function of that neuron. This can be seen from both the ultrastructure and the composition of axons. Axons are roughly cylindrical in cross section with little or no taper. As discussed later, this diameter is maintained by regulation of the cytoskeleton. Even at branch points, daughter axons are comparable in diameter to the parent axon. This constant caliber helps ensure a consistent rate of conduction. Similarly, the organization of membrane components is regulated to this end. Voltage-gated ion channels are distributed to maximize conduction. Sodium channels are distributed more or less uniformly in small nonmyelinated axons, but are concentrated at high density in the regularly spaced unmyelinated gaps, known as nodes of Ranvier (Sherman & Brophy, 2005; Susuki & Rasband, 2008). The nodes are flanked by specialized domains known as the paranodal axoglial junctions, which, as their name suggests, are sites where junctional complexes form between the terminal loops of the myelin sheath and the axolemma. These play an important role in both clustering sodium channels at the node and restricting them there in mature nerve fibers. Disruption of the septate junctions between the paranodal loops of myelin and the axonal membrane impair rapid nerve impulse conduction. An axon so organized will conduct an action potential or train of spikes long distances with high fidelity at a defined speed. These characteristics are essential for maintaining the precise timing and coordination seen in neuronal circuits.
Most vertebrate neurons have multiple dendrites arising from their perikarya. Unlike axons, dendrites branch continuously and taper extensively with a reduction in caliber in daughter processes at each branching. In addition, the surface of dendrites is covered with small protrusions, or spines, that are postsynaptic specializations. Although the surface area of a dendritic arbor may be quite extensive, dendrites in general remain in the relative vicinity of the perikaryon. A dendritic arbor may be contacted by the axons of many different and distant neurons or innervated by a single axon making multiple synaptic contacts.
The base of a dendrite is continuous with the cytoplasm of the cell body. In contrast to the axon, Nissl substance extends into dendrites, and certain proteins are synthesized predominantly in dendrites (Martin & Zukin, 2006). There is evidence for the selective placement of some mRNAs in dendrites as well. For example, whereas RER and polysomes extend well into the dendrites, the mRNAs that are transported and translated in dendrites are a subset of the total neuronal mRNA, deficient in some mRNA species (such as neurofilament mRNAs) and enriched in mRNAs with dendritic functions (such as microtubule-associated protein, MAP2, mRNAs). Also, certain proteins appear to be targeted, postsynthesis, to the dendritic compartment as well.
The shapes and complexity of dendritic arborizations may be remarkably plastic. Dendrites appear relatively late in development and initially have only limited numbers of branches and spines. As development and maturation of the nervous system proceed, the size and number of branches increase. The number of spines increases dramatically, and their distribution may change. This remodeling of synaptic connectivity may continue into adulthood, and environmental effects can alter this pattern significantly. Eventually, in the aging brain, there is a reduction in complexity and size of dendritic arbors, with fewer spines and thinner dendritic shafts. These changes correlate with changes in neuronal function during development and aging.
As defined by classical physiology, axons are structural correlates for neuronal output, and dendrites constitute the domain for receiving information. The functional and morphological hallmarks of axons and dendrites are listed in Table 4.1. A neuron without an axon or one without dendrites might therefore seem paradoxical, but such neurons do exist. Certain amacrine and horizontal cells in the vertebrate retina have no identifiable axons, although they do have dendritic processes that are morphologically distinct from axons. Such dendritic processes may have both pre- and postsynaptic specializations or may have gap junctions that act as direct electrical connections between two cells. Similarly, the pseudounipolar sensory neurons of dorsal root ganglia (DRG) have no dendrites. In their mature form, these DRG sensory neurons give rise to a single axon that extends a few hundred micrometers before branching. One long branch extends to the periphery, where it may form a sensory nerve ending in muscle spindles or skin. Large DRG peripheral branches are myelinated and have the morphological characteristics of an axon, but they contain neither pre- nor postsynaptic specializations. The other branch extends into the central nervous system, where it forms synaptic contacts. In DRG neurons, the action potential is generated at distal sensory nerve endings and is then transmitted along the peripheral branch to the central branch and the appropriate central nervous system (CNS) targets, bypassing the cell body.
Table 4.1 Functional and Morphological Hallmarks of Axons and Dendritesa
| Axons | Dendrites |
| With rare exceptions, each neuron has a single axon | Most neurons have multiple dendrites arising from their cell bodies |
| Axons appear first during neuronal differentiation | Dendrites begin to differentiate only after the axon has formed |
| Axon initial segments are distinguished by a specialized plasma membrane containing a high density of ion channels and distinctive cytoskeletal organization | Dendrites are continuous with the perikaryal cytoplasm, and the transition point cannot be distinguished readily |
| Axons typically are cylindrical in form with a round or elliptical cross section | Dendrites usually have a significant taper and small spinous processes that give them an irregular cross section |
| Large axons are myelinated in vertebrates, and the thickness of the myelin sheath is proportional to the axonal caliber | Dendrites are not myelinated, although a few wraps of myelin may occur rarely |
| Axon caliber is a function of neurofilament and microtubule numbers with neurofilaments predominating in large axons | The dendritic cytoskeleton may appear less organized, and microtubules dominate even in large dendrites |
| Microtubules in axons have a uniform polarity with plus ends distal from the cell body | Microtubules in proximal dendrites have mixed polarity, with both plus and minus ends oriented distal to the cell body |
| Axonal microtubules are enriched in tau protein with a characteristic phosphorylation pattern | Dendritic microtubules may contain some tau protein, but MAP2 is not present in axonal compartments and is highly enriched in dendrites |
| Ribosomes are excluded from mature axons, although a few may be detectable in initial segments | Both rough endoplasmic reticulum and cytoplasmic polysomes are present in dendrites, with specific mRNAs being enriched in dendrites |
| Axonal branches tend to be distal from the cell body | Dendrites begin to branch extensively near the perikaryon and form extensive arbors in the vicinity of the perikaryon |
| Axonal branches form obtuse angles and have diameters similar to the parent stem | Dendritic branches form acute angles and are smaller than the parent stem |
| Most axons have presynaptic specializations that may be en passant or at the ends of axonal branches | Dendrites are rich in postsynaptic specializations, particularly on the spinous processes that project from the dendritic shaft |
| Action potentials are usually generated at the axon hillock and conducted away from the cell body | Some dendrites can generate action potentials, but more commonly they modulate the electrical state of the perikaryon and initial segment |
| Traditionally, axons are specialized for conduction and synaptic transmission—that is, neuronal output | Dendritic architecture is most suitable for integrating synaptic responses from a variety of inputs—that is, neuronal input |
aNeurons typically have two classes of cytoplasmic extensions that may be distinguished using electrophysiological, morphological, and biochemical criteria. Although some neuronal processes may lack one or more of these features, enough parameters can generally be defined to allow unambiguous identification.
Summary
Neurons are polarized cells that are specialized for membrane and protein synthesis, as well as for conduction of the nerve impulse. In general, neurons have a cell body, a dendritic arborization that is usually located near the cell body, and an extended axon that may branch considerably before terminating to form synapses with other neurons.
Protein Synthesis in Nervous Tissue
Both neurons and glial cells have strikingly extended morphologies. This cytoarchitecture is ideal for a tissue whose functions depend on multiple intercellular contacts locally and at great distances. Protein and lipid components are synthesized and assembled into the membranes of these cell extensions through pathways of membrane biogenesis that have been elucidated primarily in other cell types, including the yeast Saccharomyces cerevisiae. However, some adaptations of these general mechanisms have been necessary, due to the specific requirements of cells in the nervous system. Neurons, for example, have devised mechanisms for ensuring that the specific components of the axonal and dendritic plasma membranes are selectively delivered (targeted) to each plasma membrane subdomain.
The distribution to specific loci of organelles, receptors, and ion channels is critical to normal neuronal function. In turn, these loci must be “matched” appropriately to the local microenvironment and specific cell–cell interactions. Similarly, in myelinating glial cells during the narrow developmental window when the myelin sheath is being formed, these cells synthesize vast sheets of insulating plasma membrane at an unbelievably high rate. To understand how the plasma membrane of neurons and glia might be modeled to fit individual functional requirements, it is necessary to review the progress that has been made so far in our understanding of how membrane components and organelles are generated in eukaryotic cells.
There are two major categories of membrane proteins: integral and peripheral. Integral membrane proteins, which include receptors for neurotransmitters (e.g., acetylcholine receptor subunits) and polypeptide growth factors (e.g., the dimeric insulin receptor) as well as ion channels, have segments that are either embedded in the lipid bilayer or bound covalently to molecules that insert into the membrane, such as those proteins linked to glycosyl phosphatidylinositol at their C termini (e.g., Thy–1). A protein with a single membrane-embedded segment and an N terminus exposed at the extracellular surface is said to be of type I, whereas type II proteins retain their N termini on the cytoplasmic side of the plasma membrane. Peripheral membrane proteins are localized on the cytoplasmic surface of the membrane and do not cross any membrane during their biogenesis. They interact with the membrane either by means of their associations with membrane lipids or the cytoplasmic tails of integral proteins or by means of their affinity for other peripheral proteins (e.g., platelet-derived growth factor receptor-Grb2-Sos-Ras complex). In some cases, they may bind directly to the polar head groups of the lipid bilayer (e.g., myelin basic protein).
Integral Membrane and Secretory Polypeptides Are Synthesized de Novo in the Rough Endoplasmic Reticulum
Subcellular destinations for integral and peripheral membrane proteins may be determined by their sites of synthesis. In the secretory pathway, integral membrane proteins and secretory proteins are synthesized in the rough endoplasmic reticulum, whereas the mRNAs encoding peripheral proteins are translated on cytoplasmic “free” polysomes, which are not membrane associated but may interact with cytoskeletal structures.
The pathway by which secretory proteins are synthesized and exported was first postulated through the elegant ultrastructural studies on the pancreas by George Palade and colleagues (Palade, 1975). Pancreatic acinar cells were an excellent choice for this work because they are extremely active in secretion, as revealed by the abundance of their RER network, a property they share with neurons. Nissl deduced, in the nineteenth century, that pancreatic cells and neurons would be found to have common secretory properties (Fig. 4.2), because of similarities in the distribution of the Nissl substance.
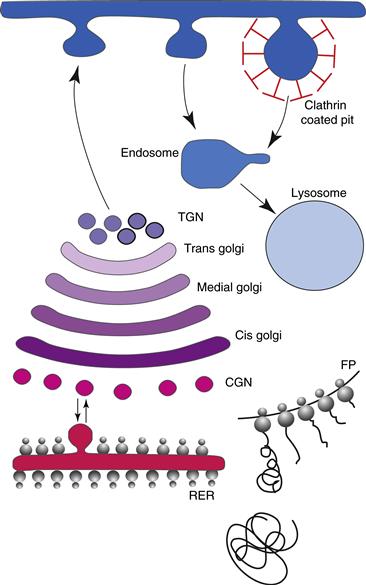
Figure 4.2 The secretory pathway. Transport and sorting of proteins in the secretory pathway occur as they pass through the Golgi complex before reaching the plasma membrane. Sorting occurs in the cis-Golgi network (CGN), also known as the intermediate compartment, and in the trans-Golgi network (TGN). Proteins exit from the Golgi complex at the TGN. The default pathway is the direct route to the plasma membrane. Proteins bound for regulated secretion or for transport to endosomes and from there to lysosomes are diverted from the default path by means of specific signals. In endocytosis, one population of vesicles is surrounded by a clathrin cage and is destined for late endosomes.
Pulse–chase autoradiography revealed that newly synthesized secretory proteins move from the RER to the Golgi apparatus, where the proteins are packaged into secretory granules and transported to the plasma membrane, from which they are released by exocytosis. Pulse–chase studies in neurons reveal a similar sequence of events for proteins transported into the axon (Brady, 1985). Unraveling the detailed molecular mechanisms of the pathway began with the successful reconstitution of secretory protein biosynthesis in vitro and the direct demonstration that, very early during synthesis, secretory proteins are translocated into the lumen of RER vesicles, prepared by cell fractionation, termed microsomes. A key observation here was that the fate of the protein was sealed as a result of encapsulation in the lumen of the RER at the site of synthesis. This cotranslational insertion model provided a logical framework for understanding the synthesis of integral membrane proteins with a transmembrane orientation.
The process by which integral membrane proteins are synthesized closely follows the secretory pathway, except that integral proteins are of course not released from the cell, but instead remain within cellular membranes (Rapoport, 2007). Synthesis of integral proteins begins with synthesis of the nascent chain on a polysome that is not yet bound to the RER membrane (Fig. 4.3). Emergence of the N terminus of the nascent protein from the protein synthesizing machinery allows a ribonucleoprotein, a signal recognition particle (SRP), to bind to an emergent hydrophobic signal sequence and prevent further translation (Walter & Johnson, 1994). Translation arrest is relieved when SRP docks with its cognate receptor in the RER and dissociates from the signal sequence in a process that requires GTP. Synthesis of transmembrane proteins on RER is an extremely energy-efficient process. The passage of a fully formed and folded protein through a membrane is, thermodynamically, formidably expensive; it is infinitely “cheaper” for cells to thread amino acids, in tandem, through a membrane during initial protein synthesis.
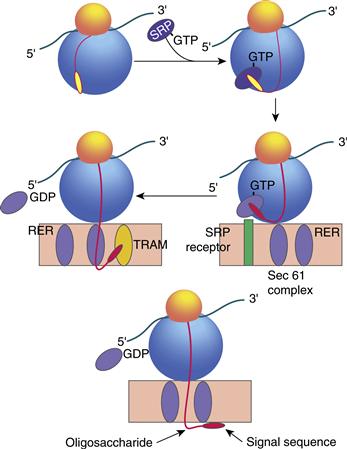
Figure 4.3 Translocation of proteins across the rough endoplasmic reticulum (RER). Integral membrane and secretory protein synthesis begins with partial synthesis on a free polysome not yet bound to the RER. The N terminus of the nascent protein emerges and allows a ribonucleoprotein, signal recognition particle (SRP), to bind to the hydrophobic signal sequence and prevent further translation. Translation arrest is relieved once the SRP docks with its receptor at the RER and dissociates from the signal sequence in a process that requires GTP. Once protein synthesis resumes, translocation occurs through an aqueous pore termed the translocon, which includes translocating chain-associating membrane protein (TRAM) and translocon-associated protein (TRAP). The signal sequence is removed by a signal peptidase located in the lumen of the RER.
Protein synthesis then resumes, and the emerging polypeptide chain is translocated into the RER membrane through a conceptualized “aqueous pore” termed the translocon. Several proteins have been identified in cross-linking experiments as possible components of the translocon, including translocating chain-associating membrane protein (TRAM) and translocon-associated protein a (TRAP), which is a component of the mammalian homologue of the yeast sec 61 complex. Many others are strikingly similar to proteins that were originally discovered in yeast, revealing the common conserved nature of this process in organisms as diverse as yeasts and humans.
A few polypeptides deviate from the common pathway for secretion. For example, certain peptide growth factors, such as basic fibroblast growth factor and ciliary neurotrophic factor, are synthesized without signal peptide sequences but are potent biological modulators of cell survival and differentiation. These growth factors appear to be released under certain conditions, although the mechanisms for such release are still controversial. One possibility is that release of these factors may be associated primarily with cellular injury.
Two cotranslational modifications are commonly associated with the emergence of the polypeptide on the luminal face of the RER (Fig. 4.3). First, an N-terminal hydrophobic signal sequence that is used for insertion into the RER is usually removed by a signal peptidase. Second, oligosaccharides rich in mannose sugars are transferred from a lipid carrier, dolichol phosphate, to the side chains of asparagine residues (Kornfeld & Kornfeld, 1985). The asparagines must be in the sequence N X T (or S), and they are linked to mannose sugars by two molecules of N-acetylglucosamine. Although the prevention of glycosylation of some proteins causes their aggregation and accumulation in the RER and Golgi apparatus, for most glycoproteins, the significance of glycosylation is not apparent. Neither is it a universal feature of integral membrane proteins: some proteins, such as the proteolipid proteins of CNS myelin, neither lose their signal sequence nor become glycosylated, although they do appear to be posttranslationally modified by the addition of the fatty acid, palmitic acid. One clear case of a proved function for a carbohydrate moiety is the targeting of proteins to lysosomes in the trans-Golgi by means of the mannose 6-phosphate receptor (Braulke & Bonifacino, 2009). Intercellular adhesion molecules, such as selectins, which affect the sticking of lymphocytes to blood vessel walls, appear to interact with lectin-like proteins through their oligosaccharide chains. Similarly, the sialic acid side chains of the neural cell adhesion molecule (NCAM) are essential for modulation of cell–cell adhesion mediated by NCAMs. Thus, for the vast majority of polypeptides destined for release from the cell (secretory polypeptides), an N-terminal “signal sequence” first mediates the passage of the protein into the RER and is cleaved immediately from the polypeptide by a signal peptidase residing on the luminal side of the RER. For proteins destined to remain as permanent residents of cellular membranes (and these form a particularly important and diverse category of plasma membrane proteins in neurons and myelinating glial cells), however, many variations on this basic theme have been found. Simply stated:
1. Signal sequences for membrane insertions need not be only N-terminal; those that lie within a polypeptide sequence are not cleaved.
2. A second type of signal, a “halt” or “stop” transfer signal, functions to arrest translocation through the membrane bilayer. The halt transfer signal is also hydrophobic and is usually flanked by positive charges. This arrangement effectively stabilizes a polypeptide segment in the RER membrane bilayer.
3. The sequential display in tandem of insertion and halt transfer signals in a polypeptide as it is being synthesized ultimately determines its disposition with respect to the phospholipid bilayer, and thus its final topology in its target membrane. By synthesizing transmembrane polypeptides in this way, virtually any topology may be generated.
Newly Synthesized Polypeptides Exit from the RER and Are Moved through the Golgi Apparatus
When the newly synthesized protein has established its correct transmembrane orientation in the RER, it is incorporated into vesicles and must pass through the Golgi complex before reaching the plasma membrane (Fig. 4.2). For membrane proteins, the Golgi serves two major functions: it sorts and targets proteins, and it performs further posttranslational modifications, particularly on the oligosaccharide chains that were added in the RER (Glick & Nakano, 2009). Sorting takes place in the cis-Golgi network (CGN), also known as the intermediate compartment, and in the trans-Golgi network (TGN), whereas sculpting of oligosaccharides is primarily the responsibility of the cis-, medial-, and trans-Golgi stacks. The TGN is a tubulovesicular network wherein proteins are targeted to the plasma membrane or to organelles.
In addition to the processing of carbohydrates in the Golgi, posttranslational modifications can take place in other subcellular compartments. Some protein glycosylations are modified further post-Golgi in components of the smooth endoplasmic reticulum or transport vesicles, as described later in this section. Finally, some neuropeptides (vasopressin, enkephalins, etc.) are synthesized as sequence domains in large precursor proteins that must be cleaved in transit by specific proteases to form the biologically active form.
The CGN serves an important sorting function for proteins entering the Golgi from the RER. Because most proteins that move from the RER through the secretory pathway do so by default, any resident endoplasmic reticulum proteins must be restrained from exiting or returned promptly to the RER from the CGN should they escape (Dudek et al., 2009). Although no retention signal has been demonstrated for the endoplasmic reticulum, two retrieval signals have been identified: a Lys-Asp-Glu-Leu or KDEL sequence in type I proteins and the Arg-Arg or RR motif in the first five amino acids of proteins with a type II orientation in the membrane. The KDEL tetrapeptide binds to a receptor called Erd 2 in the CGN, and the receptor–ligand complex is returned to the RER. There may also be a receptor for the N arginine dipeptide; alternatively, this sequence may interact with other components of the retrograde transport machinery, such as microtubules.
Movement of proteins between Golgi stacks proceeds by means of vesicular budding and fusion (Glick & Nakano, 2009). Through the use of a cell-free assay containing Golgi-derived vesicles, the essential mechanisms for budding and fusion have been shown to require coat proteins (COPs) in a manner that is analogous to the role of clathrin in endocytosis. Currently, two main types of COP complex, COPI and COPII, have been distinguished. Although both have been shown to coat vesicles that bud from the endoplasmic reticulum, they may have different roles in membrane trafficking. Coat proteins provide the external frame-work into which a region of a flattened Golgi cisternae can bud and vesiculate. A complex of these COPs forms the coatomer (coat protomer) together with a p200 protein, and a family of GTP-binding proteins called ADP-ribosylation factors (ARFs), originally named for their role in the action of cholera toxin. Immunolocalization of one of the coatamer proteins, β-COP, predominantly to the CGN and cis-Golgi indicates that these proteins may also take part in vesicle transport into the Golgi (Fig. 4.4A). The function of ARF is to drive the assembly of the coatamer and therefore vesicle budding in a GTP-dependent fashion. Dissociation of the coat is triggered when a GTPase-activating protein (GAP) in the Golgi membrane stimulates hydrolysis of the GTP bound to ARF. The cycle of coat assembly and disassembly can continue when the replacement of GDP on ARF by GTP is catalyzed by a guanine nucleotide exchange factor (GEF). The importance of this GDP–GTP exchange to normal vesicular traffic is illustrated dramatically by the effects of brefeldin A, a fungal metabolite that specifically inhibits GTP exchange and disperses the Golgi complex by preventing the return of Golgi components from the intermediate compartment.
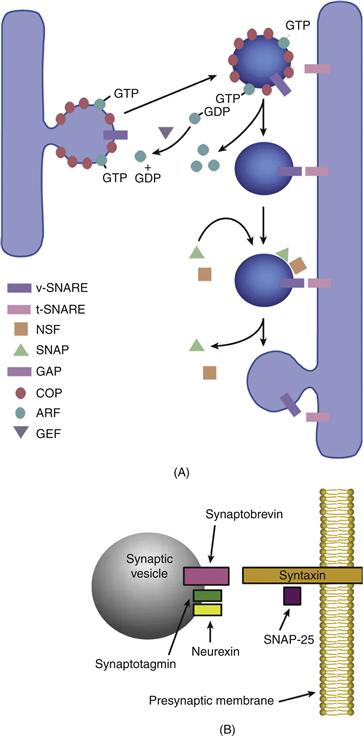
Figure 4.4 A. General mechanisms of vesicle targeting and docking in the ER and Golgi. The assembly of coat proteins (COPs) around budding vesicles is driven by ADP-ribosylation factors (ARFs) in a GTP-dependent fashion. Dissociation of the coat is triggered when hydrolysis of the GTP bound to ARF is stimulated by a GTPase activating protein (GAP) in the Golgi membrane. The cycle of coat assembly and disassembly can continue when the replacement of GDP on ARF by GTP is catalyzed by a guaninenucleotide exchange factor (GEF). Fusion of vesicles with their target membrane in the Golgi is regulated by a series of proteins, N-ethyl-maleimide-sensitive factor (NSF), soluble NSF attachment proteins (SNAPs), and SNAP receptors (SNAREs), which together assist the vesicle in docking with its target membrane. SNAREs on the vesicle (v-SNAREs) are believed to associate with corresponding t-SNAREs on the target membrane. B. Mechanisms of vesicle targeting and docking in the synaptic terminal. The synaptic counterpart of v-SNARE is synap-tobrevin (also known as vesicle-associated membrane protein), and syntaxin corresponds to t-SNARE. SNAP-25 is an accessory protein that binds to syntaxin. Synaptotagmin is believed to be the Ca2+ sensitive regulatory protein in the complex that binds to syntaxin. Neurexins appear to have a role in conferring Ca2+ sensitivity to these interactions.
Fusion of vesicles with their target membrane in the Golgi apparatus is regulated by a series of proteins, N-ethylmaleimide-sensitive factor (NSF), soluble NSF attachment proteins (SNAPs), and SNAP receptors (SNAREs), which together assist the vesicle in docking with its target membrane. In addition, Rabs, a family of membrane-bound GTPases, act in concert with their own GAPs, GEFs, and a cytosolic protein that dissociates Rab–GDP from membranes after fusion called guanine-nucleotide dissociation inhibitor. Rabs are believed to regulate the action of SNAREs, the proteins directly engaged in membrane–membrane contact prior to fusion. The tight control necessary for this process and the importance of ensuring that vesicle fusion takes place only at the appropriate target membrane may explain why eukaryotic cells contain so many Rabs, some of which are known to specifically take part in the internalization of endocytic vesicles at the plasma membrane (Fig. 4.2).
Exocytosis of the neurotransmitter at the synapse must occur in an even more finely regulated manner than endocytosis (Sudhof, 2004). The proteins first identified in vesicular fusion events in the secretory pathway (namely NSF, SNAPs, and SNAREs or closely related homologues) appear to play a part in the fusion of synaptic vesicles with the active zones of the presynaptic neuronal membrane (Fig. 4.4B).
The synaptic counterpart of vSNARE is synaptobrevin [also known as vesicle-associated membrane protein (VAMP)], and syntaxin corresponds to t-SNARE. SNAP-25 is an accessory protein that binds to syntaxin. In the constitutive pathway, such as between the RER and Golgi apparatus, assembly of the complex at the target membrane promotes fusion. However, at the presynaptic membrane, Ca2+ influx is required to stimulate membrane fusion at the presynaptic membrane. Synaptotagmin is believed to be the Ca2+-sensitive regulatory protein in the complex that binds syntaxin. Neurexins appear to have a role in regulation as well, because, in addition to interacting with synaptotagmin, they are the targets of black widow spider venom (α)-latrotoxin, which deregulates the Ca2+-dependent exocytosis of the neurotransmitter.
The comparison between secretion in slow-releasing cells, such as the pancreatic β-cell, and neurotransmitter release at the neuromuscular junction is much like the comparison between a hand-held pocket calculator for balancing a checkbook and a state-of-the-art desktop computer. Two differences stand out. First, the speed of neurotransmitter release is much greater both in release from a single vesicle and in total release in response to a specific signal. Releasing the contents of a single synaptic vesicle at a mouse neuromuscular junction takes from 1 to 2 ms, and the response to an action potential involving the release of many synaptic vesicles is over in approximately 5 ms. In contrast, releasing the insulin in a single secretory granule by a pancreatic β-cell takes from 1 to 5 s, and the full release response may take from 1 to 5 min. A 103- to 105-fold difference in rate is an extraordinary range, making neurotransmitter release one of the fastest biological events routinely encountered, but this speed is critical for a properly functioning nervous system.
A second major difference between slow secretion and fast secretion is seen in the recycling of vesicles. In the pancreas, secretory vesicles carrying insulin are used only once, and so new secretory vesicles must be assembled de novo and released from the TGN to meet future requirements. In the neuron, the problem is that the synapse may be at a distance of 1 m or more from the protein synthetic machinery of the perikaryon, and so newly assembled vesicles even traveling at rapid axonal transport rates (see later) may take more than a day to arrive. Now, the number of synaptic vesicles released in 15 min of constant stimulation at a single frog neuromuscular junction has been calculated to be on the order of 105 vesicles, but a single terminal may have only a few hundred vesicles at any one time. These measurements would make no sense if synaptic vesicles had to be replaced constantly through new synthesis in the perikaryon, as is the case with insulin-carrying vesicles. The reason that these numbers are possible is that synaptic vesicles are taken up locally by endocytosis, refilled with neurotransmitter, and reutilized at a rate fast enough to keep up with normal physiological stimulation levels. This takes place within the presynaptic terminal, and evidence shows that these recycled synaptic vesicles are used preferentially (Sudhof, 2004). Such recycling does not require protein synthesis because the classical neurotransmitters are small molecules, such as acetylcholine, or amino acids, such as glutamate, that can be synthesized or obtained locally. Significantly, neurons have fast and slow secretory pathways operating in parallel in the presynaptic terminal (Sudhof, 2004). Synapses that release classical neurotransmitters (acetylcholine, glutamate, etc.) with these fast kinetics also contain dense core granules containing neuropeptides (calcitonin gene related peptide, substance P, etc.) that are comparable to the secretory granules of the pancreatic β-cell. These are used only once because neuropeptides are produced from large polypeptide precursors that must be made by protein synthesis in the cell body. The release of neuropeptides is relatively slow; as is the case in endocrine release, neuropeptides serve primarily as modulators of synaptic function. The small, clear synaptic vesicles containing the classic neurotransmitters can in fact be depleted pharmacologically from the presynaptic terminal, while the dense core granules remain. These observations indicate that even though fast and slow secretory mechanisms have many similarities and may even have common components, in neurons they can operate independent of one another (Xu & Xu, 2008).
Proteins Exit the Golgi Complex at the trans-Golgi Network
Transport of cargoes in vesicles beyond the TGN is believed to involve coat proteins other than the classical COPs. These are the adaptins, which form Adaptor Protein (AP) complexes. The APs were until recently considered to comprise four families (AP-1-4) where AP-1 and AP-2 segregated their cargoes to clathrin-coated vesicles, whereas AP-3 and AP-4 did not appear to require clathrin. In general it is believed that AP-1 and AP-4 are involved in moving vesicles between the TGN and endosomes, AP-2 is specialized for endocytosis, and AP-3 ferries cargoes from endosomes to lysosomes. Interestingly, mutations in AP-4 cause cerebral palsy, intellectual disability, and spastic paraplegia. Recently a fifth AP complex, AP-5, has been identified that also appears to be involved in endosomal trafficking. Defects in AP-5 have been linked to spastic paraplegia (Hirst et al., 2011).
Most of the N-linked oligosaccharide chains acquired at the RER are remodeled in the Golgi cisternae, and while the proteins are in transit, another type of glycosyl linkage to serine or threonine residues through N-acetylgalactosamine can also be made. Modification of existing sugar chains by a series of glycosidases and the addition of further sugars by glycosyl transferases occur from the cis to the trans stacks. Some of these enzymes have been localized to particular cisternae. For example, the enzymes (β)-1,4-galactosyltransferase and (α)-2,6-sialyltransferase are concentrated in the trans-Golgi. How they are retained there is a matter of some debate. One idea is that these proteins are anchored by oligomerization. Another view is that the progressively rising concentration of cholesterol in membranes more distal to the ER in the secretory pathway increases membrane thickness, which in turn anchors certain proteins and causes an arrest in their flow along the default route.
The default or constitutive pathway seems to be the direct route to the plasma membrane taken by vesicles that bud from the TGN (Fig. 4.2). This is how, in general, integral plasma membrane proteins reach the cell surface. Proteins bound for regulated secretion or for transport to endosomes and from there to lysosomes are diverted from the default path by means of specific signals. It has been assumed that the sorting of proteins for their eventual destination takes place at the TGN itself. However, recent analyses of the three-dimensional structure of the TGN have provoked a revision of this view. These studies have shown that the TGN is tubular, with two major types of vesicles that bud from distinct populations of tubules. The implication is that sorting may already have occurred in the trans-Golgi prior to the proteins’ arrival at the TGN. One population of vesicles consists of those surrounded by the familiar clathrin cage, which are destined for late endosomes. The β-COP protein and related coatomer proteins active in more proximal regions of the secretory pathway are absent from the TGN.
Endocytosis and Membrane Cycling Occurs in the trans-Golgi Network
Two types of membrane invagination occur at the surface of mammalian cells and are clearly distinguishable by electron microscopy (Doherty & McMahon, 2009). The first type is a caveola, which has a thread-like structure on its surface made of the protein caveolin. Caveolae mediate the uptake of small molecules such as vitamin folic acid by a process called potocytosis. They may also have a role in concentrating proteins linked to the plasma membrane by the glycosylphos-phatidylinositol anchor. Demonstration of the targeting of protein tyrosine kinases to caveolae by the tripeptide signal MGC (Met-Gly-Cys) also suggests that caveolae may function in signal transduction cascades.
The other type of endocytic vesicle at the cell surface is that coated with the distinctive meshwork of clathrin triskelions (Doherty & McMahon, 2009). The triskelion comprises three copies of a clathrin heavy chain and three copies of a clathrin light chain. The ease with which these triskelions can assemble into a cage structure demonstrates how they promote the budding of a vesicle from a membrane invagination. Clathrin binds selectively to regions of the cytoplasmic surface of membranes that are selected by adaptins. The AP-2 complex, which is primarily active at the plasma membrane, consists of 100-kDa α and β subunits and two subunits of 50 and 17 kDa each. AP-1 complexes localize to the TGN and have γ and subunits of 100 kDa together with smaller polypeptides of 46 and 19 kDa. Adaptins bind to the cytoplasmic tails of membrane proteins, thus recruiting clathrin for budding at these sites.
A further component of the endocytic complex at the plasma membrane is GTPase dynamin, which seems to be required for the normal budding of coated vesicles during endocytosis. Dynamins are a family of 100-kDa GTPases found in both neuronal and nonneuronal cells and may interact with the AP-2 component of a clathrin-coated pit (Doherty & McMahon, 2009). Dynamin I is found primarily in neurons, whereas dynamin II has a widespread distribution. Oligomers of dynamin form a ring at the neck of a budding clathrin-coated vesicle, and GTP hydrolysis appears to be necessary for the coated vesicle to pinch off from the plasma membrane. The existence of a specific neuronal form of dynamin may be a manifestation of the unusually rapid rate of synaptic vesicle recycling.
The primary function of clathrin-coated vesicles at the plasma membrane is to deliver membrane proteins together with any ligands bound to them to the early endosomal apparatus. The other major site of action of clathrin is in vesicles that bud from the TGN carrying lysosomal enzymes en route to late endosomes. Early endosomes have a tubulovesicular morphology. Receptors that will be recycled back to the plasma membrane partition into the tubules; those endocytosed proteins destined for lysosomes concentrate in the vesicular regions. Recycling seems to be the default pathway, whereas proteins must be actively targeted to lysosomes. However, the precise signals for this are unknown.
Regulation of membrane cycling in the endosomal compartment is likely to include the Rab family of small GTP-binding proteins (Zerial & McBride, 2001). Indeed, each stage of the endocytic pathway may have its own Rab protein to ensure efficient targeting of the vesicle to the appropriate membrane. Rab6 is believed to have a role in transport from the TGN to endosomes, whereas Rab9 may regulate vesicular flow in the reverse direction. In neurons, Rab5a has a role in regulating the fusion of endocytic vesicles and early endosomes and appears to function in endocytosis from both somatodendritic domains and the axon. The association of the protein with synaptic vesicles in nerve terminals, attached presumably by means of its isoprenoid tail, also suggests that early endosomal compartments may have a role in the packaging and recycling of synaptic vesicles.
Lysosome Is the Target Organelle in Several Inherited Diseases that Affect the Nervous System
Lysosomes were first isolated and characterized as a distinct organelle fraction bounded by a single membrane and separable from mitochondria by differential and sucrose gradient centrifugation. Because of their high content of acid hydrolases, the classic view is that lysosomes are organelles of terminal degradation. Indeed, the latency of hydrolase activity before membrane permeabilization by agents such as nonionic detergents has been used biochemically as a measure of the purity and intactness of lysosomal preparations. However, in addition to their well-established function in lipid and protein breakdown, tubulovesicular lysosomes may overlap in sorting functions with early endosomes, particularly during antigen processing in macrophages.
Inherited deficiencies in lipid metabolism in the lysosome often have particularly devastating consequences on the nervous system because of the abundance of the lipid-rich membrane myelin. Metachromatic leukodystrophy is an autosomal recessive disease caused by a deficiency in arylsulfatase A activity, which is also responsible for degrading the myelin lipid cerebroside sulfate (sulfatide). Oligodendrocytes accumulate sulfatide in metachromatic granules, causing severe disruption of myelination. Peripheral nerve myelination is also affected, as are other organs that normally contain much lower amounts of sulfatide, such as the kidney, the liver, and the endocrine system. Krabbe disease, or globoid cell leukodystrophy, is also a dysmyelinating disease in which there is an almost complete lack of oligodendrocytes and therefore myelin, caused by a deficiency in the β-galactosidase responsible for hydrolyzing galactocerebroside to ceramide and galactose. Galactocerebroside is particularly abundant in myelin, constituting about 25% of myelin lipid. Mice that lack galactocerebroside have the ability to assemble multilamellar myelin; however, this myelin does not support adequate nerve conduction, and is not stable. Unlike metachromatic leukodystrophy, Krabbe disease is limited to the CNS and peripheral nervous system (PNS). However, why a buildup of galactocerebroside should prove particularly toxic to oligodendrocytes is not entirely clear. One hypothesis is that a metabolite of galactocerebroside, galactosphingosine (psychosine), is the primary culprit.
How are proteins destined to operate in lysosomes targeted to these organelles? Soluble lysosomal hydrolase enzymes acquire a phosphorylated mannose on their oligosaccharide chains by a two-step process in the Golgi apparatus (Braulke & Bonifacino, 2009). This mannose 6-phosphate label is recognized by specific mannose 6-phosphate receptors, which carry the proteins to late endosomes. In contrast, lysosomal membrane proteins are targeted by means of cytoplasmic tail signals that contain either leucine or tyrosine of type LJ or type YXXJ or NXXY, where J is any hydrophobic amino acid. The LJ signal seems to be essential for efficient delivery directly to endosomes, whereas the second type of signal seems to be more important in the recovery of proteins destined for lysosomes from the plasma membrane. The majority of lysosomal membrane proteins have the YXXJ but do not have the LJ signal. The implication of these observations is that many of the lysosomal membrane proteins make their way to lysosomes from the TGN endosomes through the plasma membrane.
What are the receptors for the type LJ or the type YXXJ or NXXY motifs at the TGN? Because transport from the TGN to the endosomes occurs in clathrin-coated vesicles, the proteins that link such vesicles to membranes, the adaptins, may play a role. The weight of the evidence suggests that AP-1 recognizes the LJ sequence, whereas AP-2 identifies the YXXJ and NXXY motifs. Once the ligands are bound, these adaptins would direct transport of their respective ligands to the endosomes from the TGN or through the plasma membrane, respectively. At present, it is not clear how proteins in the late endosome, such as mannose 6-phosphate receptors, that cycle back to the Golgi are sorted from those whose ultimate destination is a lysosome.
How are Peripheral Membrane Proteins Targeted to Their Appropriate Destinations?
Peripheral membrane proteins are synthesized in the same type of free polysome in which the bulk of the cytosolic proteins are made. However, the cell must ensure that these membrane proteins are sent to the plasma membrane rather than allowed to attach in a haphazard way to other intracellular organelles. The fact that complex machinery has evolved to ensure the correct delivery of integral membrane proteins suggests that some equivalent targeting mechanism must exist for proteins that attach to the cytoplasmic surface of the plasma membrane. Such proteins are translated on “free” polysomes, but these polysomes are associated with cytoskeletal structures and are not distributed uniformly throughout the cell body. In a number of cases, mRNAs that encode soluble cytosolic proteins are concentrated in discrete regions of the cell, resulting in a local accumulation of the translated protein close to the site of action. For some peripheral membrane proteins, this is the plasma membrane.
Evidence that this mechanism might operate in peripheral membrane protein synthesis came from studies showing biochemically and by in situ hybridization that mRNAs encoding the myelin basic proteins are concentrated in the myelinating processes that extend from the cell body of oligodendrocytes (Colman, Kreibich, Frey, & Sabatini, 1982). Myelin basic protein may be a special case because of its very strong positive charge and consequent propensity for binding promiscuously to the negatively charged polar head groups of membrane lipids. Nevertheless, the fact that actin mRNAs are localized to the leading edge of cultured myocytes and mRNA for the microtubule-associated protein MAP2b is concentrated in the dendrites of neurons suggest that targeting by local synthesis is more common than originally thought. This mechanism is probably less important for peripheral membrane proteins that associate with the cytoplasmic surface of the plasma membrane by means of strong specific associations with proteins already located at the membrane because such proteins would act as specific receptors. Because only selected cytoplasmic mRNAs are localized to the periphery, the process is specific. However, no mRNAs are localized exclusively to the periphery, and a significant fraction is typically localized proximal to the nucleus in a region rich with translational and protein-processing machinery of the cell (the Nissl substance or translational cytoplasm).
Special Mechanisms Are Used to Target Proteins to Mitochondria and Peroxisomes
The inner membrane of the mitochondrion is the site of oxidative phosphorylation in which the step-by-step transfer of electrons from oxygen intermediary metabolites to molecular oxidation is coupled to proton transport and ATP synthesis. Thus, this organelle has an essential role in providing the large amount of ATP required for the electrical activity of neurons. The fact that, in a resting adult, about 40% of the total energy consumption is required for ion pumping in the CNS accounts for the exquisite sensitivity of the brain to damage from oxygen deprivation. The sensitivity of neurons to interruptions in the provision of ATP by the mitochondrion is also seen in cases of uremia, where a buildup of ammonium ions depletes the Krebs cycle of α-oxoglutaric acid by converting it into glutamate.
Although the mitochondrion has its own circular DNA that encodes some proteins, most mitochondrial proteins are synthesized in the nucleocytoplasmic system (Endo & Yamano, 2009). This poses the problem of how these proteins once made in the cytoplasm gain entry into the mitochondrion. Furthermore, because the mitochondrion has an inner and an outer membrane, some proteins must cross two membranes to gain access to the inner matrix. This group of proteins includes the enzymes of the Krebs cycle and the fatty acid β-oxidation pathway.
Unlike proteins inserted into the RER, mitochondrial proteins can be imported either posttranslationally or cotranslationally with the use of a cleavable amphipathic helical signal sequence usually at the N terminus. At the RER, the signal sequence of a nascent polypeptide chain can be translocated across the membrane because the polypeptide remains small and unfolded due to the arrest of translation caused by a signal recognition particle. However, mitochondrial proteins are typically synthesized on cytoplasmic or free polysomes and must be folded at least partially to prevent degradation. For posttranslational import, mitochondria rely on a group of molecular chaperones to prevent complete folding of the polypeptides. These hsp70 and hsp60 proteins were originally identified because they are upregulated during heat shock. Their role in binding to proteins and maintaining them in specific conformations helps explain why these proteins have an important function in protecting proteins against the stress of elevated temperatures, as well as facilitating the proper folding of newly synthesized polypeptides. In yeast, a second protein, Ydj1p, whose bacterial homologue DnaJ regulates chaperone function has been identified. Ydj1p possesses an isoprenoid tail linked to its C-terminal amino acid, which may serve to anchor the protein to the outer membrane. The third factor that has been implicated is the mitochondrial stimulation factor, which is a heterodimer possessing an ATP-dependent protein “unfoldase” activity. This factor may be more important in cotranslational import where polysomes are known to be associated with the mitochondrial outer membrane.
Most of our current understanding of protein translocation from the mitochondrial outer membrane inward has come from studies on either the fungus Neurospora crassa or the yeast S. cerevisiae. In both yeast and higher eukaryotes, the partially folded polypeptide targeted for the mitochondrion may be stabilized by a cytoplasmic chaperone that is a member of the hsp70 family, but this interaction is not required for import. However, several proteins in the outer membrane form an essential complex that acts as a receptor and pore for protein translocation. This complex can in turn interact with an inner membrane complex at specialized contact sites that minimize the distance across the two membranes, thereby facilitating the movement of proteins to the inner matrix. Although both pores can function independently, they contact and cooperate when there is a transmembrane potential across the inner membrane. This accounts for early observations showing that importation of subunits of the F1-ATPase, an inner membrane protein, required an active electron transport chain but did not need ATP synthesis. The mitochondrial import sequence extends through the pore into the inner matrix, where a second member of the hsp70 family binds and facilitates movement into the inner matrix. After proteins have crossed into the inner matrix, they must dissociate from hsp70 in order to fold properly, a process that requires another kind of molecular chaperone, hsp60.
Peroxisomes are so named because they contain oxidases that generate H2O2 and the enzyme catalase, which is responsible for detoxifying it. In addition, these organelles contain many other enzymes that take part in lipid, purine, and amino acid metabolism. Peroxisomes are of interest because of the number of inherited diseases associated with defects either in certain enzymes or indeed in the assembly of the organelle itself. Some of these diseases manifest as particularly damaging to the nervous system and include adrenoleukodystrophy (accumulation of very long chain fatty acids due to insufficient lignoceryl-CoA ligase activity caused by inefficient import of the protein) and Refsum disease (buildup of phytanic acid due to defective α-oxidation), both of which cause demyelination.
Like many mitochondrial proteins, peroxisomal proteins are imported posttranslationally (Ma, Agrawal, & Subramani, 2011). Although cytosolic factors are implicated in peroxisomal biogenesis, no peroxisomal chaperones analogous to the hsp70 family have yet been shown to function in protein import. Therefore, unfolding and refolding are assumed not to play a role in the accumulation of proteins inside the peroxisome. Among these cytosolic proteins is presumed to be the receptor for the tripeptide C-terminal import signal SKL (Ser-Lys-Leu) known as peroxisomal targeting signal 1 (PTS1). In addition to C-terminal PTS1, some peroxisomal proteins have a cleavable N-terminal sequence called PTS2, which signals their import. A quite distinct translocation machinery appears to operate for PTS1 and PTS2 proteins. Two possible receptor proteins in the peroxisomal membrane, one of which is the adrenoleukodystrophy protein (ALDP), have been identified. ALDP is a member of a larger family known as ABC ATP-dependent membrane transporters.
A characteristic feature of peroxisomal biogenesis is that it is stimulated by drugs whose detoxification requires peroxisomal activity. It is possible that mature peroxisomes are recruited from a pool of precursor organelles, and there is some evidence for the existence of such a population in rat liver. Although the mature organelle appears to be spherical, electron microscopic evidence suggests a peroxisomal reticulum in which synthesis and protein import may take place. Mature peroxisomes might then arise from this reticulum by a process of budding.
Cytoplasmic Proteins Are also Compartmentalized
Membrane-bound organelles are the most familiar form of compartmentation in cells, but cytoplasmic regions of the cell containing metabolic compartments exist as well. Regions of the neuronal or glial cytoplasm may have highly specialized polypeptide compositions that are important for function. For example, the neuronal phosphoprotein synapsin is highly enriched in presynaptic terminals, where it participates in the localization and targeting of synaptic vesicles. Similarly, calmodulin and the glycolytic enzyme aldolase have been localized in muscle cells to the region of the I-band, where they are thought to facilitate coupling of ATP production to contractility.
As mentioned earlier, cytoplasmic proteins are synthesized on cytoplasmic polysomes, termed “free” polysomes to reflect an absence of underlying ER membrane, even though they may be restricted to specific domains of the cell cytoplasm. This restriction is particularly obvious in the neuronal perikaryon, where both cytoplasmic polysomes and membrane-associated polysomes are concentrated in areas near the nucleus and Golgi complex. In addition, cytoplasmic polysomes containing specific mRNAs may be localized to certain regions of the cell, such as the proximal dendrite (those encoding the microtubule-associated protein MAP-2) and the processes of oligodendrocytes (those encoding myelin basic protein). In contrast, the protein synthetic machinery of the polysome appears to be effectively excluded from the mature axon. Therefore, cytoplasmic polysomes are representative of cytoplasmic compartmentation for proteins and nucleic acids.
In most cases, localized cytoplasmic proteins interact with cytoskeletal structures in the cytoplasm (see next section), but macromolecular complexes that form in order to make a cellular process more efficient or free from error have been described. Evidence exists that glycolytic enzymes of neurons and muscle cells may be organized in a labile complex that facilitates energy metabolism, but the existence of such complexes remains controversial.
Perhaps the best characterized cytoplasmic macromolecular complex is the proteasome, which is a large protein complex (2 × 106 Da, sedimenting as a 20S particle) that contains several distinct enzymatic activities, including catalytic sites for both ubiquitin-dependent and ubiquitin-independent proteolysis (Hochstrasser, 1995). Ubiquitin is a small, highly conserved polypeptide that is added covalently to cytoplasmic proteins targeted for degradation. The catalytic core of the proteasome is a barrel-shaped structure formed by four heptameric stacked rings, but additional proteins (about 16 polypeptides) may interact with the 20S core to form a larger 26S particle. Because proteasomes constitute the primary cytoplasmic pathway for protein degradation (i.e., nonlysosomal pathways), they serve a number of important physiological functions, including regulation of cell proliferation and processing of antigens for presentation. In the nervous system, however, proteasomes are likely to be most important for homeostasis, allowing turnover of cytoplasmic polypeptides at specific sites so that the elaborate cellular extensions of neurons and glia may be maintained.
Cytoplasmic proteins may also be compartmentalized effectively by posttranslational modification. Two types of modification may be particularly important for this kind of compartmentalization. Local activation of kinases can lead to the phosphorylation of proteins in specific domains of the neuron. For example, the reversible phosphorylation of synapsin in the presynaptic terminal appears to be responsible for the targeting of synaptic vesicles to the terminal and for the mobilization of vesicles during prolonged stimulation. An impressive variety of cytoplasmic protein kinases that may be selectively activated to modify serines or threonines presented in distinctive consensus sequences have been described. Distinct from these serine or threonine kinases, a number of other kinases that specifically modify tyrosines can be found in the brain. In some cases, the tyrosine kinase is linked directly to a membrane-spanning receptor and phosphorylates cytoplasmic proteins in the vicinity of the receptor after activation. Completing the cycle of phosphorylation and dephosphorylation are a number of phosphatases with varying specificities. The properties and physiological roles for kinases and phosphatases are discussed in greater detail later.
A second common posttranslational modification of cytoplasmic proteins is the addition of carbohydrate moieties. Whereas modification of membrane-associated proteins in the Golgi complex proceeds by the addition of complex carbohydrates through N linkages on selected asparagines, glycosylated cytoplasmic proteins have simpler carbohydrates added through O linkages to serine or threonine hydroxyls. This modification was first recognized as a feature of many nuclear proteins and components of the nuclear membrane, but subsequent studies showed that a number of cytoplasmic proteins also have O-linked carbohydrates. Unlike phosphorylation, relatively little is known about the functional significance of cytoplasmic glycosylation. Remarkably, however, serines and threonines subject to O-linked glycosylation would also be good sites for phosphorylation by various kinases as well. This congruence raises the possibility that glycosylation and phosphorylation of some cytoplasmic proteins may serve complementary functions.
Summary
Membrane biogenesis and protein synthesis in neurons and glial cells are accomplished by the same mechanisms that have been worked out in great detail in other cell types. Integral membrane proteins are synthesized in the rough endoplasmic reticulum, and peripheral membrane proteins are products of cytoplasmic-free ribosomes that are found in the cell sap. For transmembrane proteins and secretory polypeptides, synthesis in the RER is followed by transport to the Golgi apparatus, where membranes and proteins are sorted and targeted for delivery to precise intracellular locations. It is likely that the neuron and glial cell have evolved additional highly specialized mechanisms for membrane and protein sorting and targeting because these cells are so greatly extended in space, although these additional mechanisms have yet to be fully described. The basic features of the process of secretion, which includes neurotransmitter delivery to presynaptic terminals, are beginning to be understood as well. The key features of this process are apparently common to all cells, including yeast, although the neuron has developed certain specializations and modifications of the secretory pathway that reflect its unique properties as an excitable cell.
Cytoskeletons of Neurons and Glial Cells
The cytoskeleton of eukaryotic cells is an aggregate structure formed by three classes of cytoplasmic structural proteins: microtubules (tubulins), microfilaments (actins), and intermediate filaments. Each of these elements exists concurrently and independently in overlapping cellular domains. Most cell types contain one or more examples of each class of cytoskeletal structure, but there are exceptions. For example, mature mammalian erythrocytes contain no microtubules or intermediate filaments, but they do have highly specialized actin cytoskeletons. Among cells of the nervous system, the oligodendrocyte is unusual in that it contains no cytoplasmic intermediate filaments. Typically, each cell type in the nervous system has a unique complement of cytoskeletal proteins that are important for the differentiated function of that cell type (Pigino, Song, Kirkpatrick, & Brady, 2011).
Although the three classes of cytoskeletal elements interact with each other and with other cellular structures, all three are dynamic structures rather than passive structural elements. Their aggregate properties form the basis of cell morphologies and plasticity in the nervous tissue. In many cases, biochemical specialization in the cytoskeleton is characteristic of a particular cell type, function, and developmental stage. Each type of cytoskeletal element has unique functions that are essential for a working nervous system.
Microtubules Are an Important Determinant of Cell Architecture
Microtubules are nearly ubiquitous components of the cytoskeleton in eukaryotes. They play key roles in intracellular transport, are a primary determinant of cell morphology, are the structural correlate of the mitotic spindle, and form the functional core of cilia and flagella. Microtubules are very abundant in the nervous system, and tubulin subunits of microtubules may constitute more than 10% of total brain protein. As a result, many fundamental properties of microtubules have been defined by using microtubule protein prepared from brain extracts. At the same time, the microtubule cytoskeleton of the neuron has a variety of biochemical specializations that meet the unique demands imposed by the size and shape of the neuron.
Of the various functions defined for microtubules, intracellular transport and the generation of cellular morphology are the most important roles played by microtubules in cells of the nervous system. In part, this comes from their ability to organize cytoplasmic polarity. Microtubules in vitro are dynamic, polar structures with plus and minus ends that correspond to the fast- and slow-growing ends, respectively. In contrast, both stable and labile microtubules can be identified in vivo, where they help define both microscopic and macroscopic aspects of intracellular organization in cells. Microtubule organization, stability, and composition in nervous tissue are all highly regulated in the nervous system.
By electron microscopy, microtubules appear as hollow tubes 25 nm in diameter and in axons can be up to hundreds of micrometers in length. High resolution electron micrographs also reveal that the walls of microtubules typically comprise 13 protofilaments formed by a linear arrangement of globular subunits, although microtubules with 12 to 14 protofilaments exist in some tissues and organisms. Globular subunits in the walls of a microtubule are heterodimers of α- and β-tubulin, whereas a variety of microtubule-associated proteins bind to the surface of microtubules.
Neuronal microtubules are remarkable for their genetic and biochemical diversity. Multiple genes exist for both α-and β-tubulins. These genes are expressed differentially according to cell type and developmental stage. Some of these genetic isotypes are expressed ubiquitously, whereas others are only turned on at specific times in development, in specific cell types, or both. Most tubulin genes are expressed in nervous tissue, and some appear to be enriched or specific to neurons. When specific isotypes are prepared in a pure form, they show variability in assembly kinetics and ability to bind ligands. However, when more than one isotype is expressed in a single cell, such as a neuron, they coassemble into microtubules with mixed composition.
A variety of posttranslational modifications of tubulins have been described, the most common of which are tyrosination–detyrosination, acetylation–deacetylation, and phosphorylation. The first two of these pathways are associated intimately with the assembly of microtubules, but relatively little is known about physiological functions for any of these modified tubulins. Most α-tubulin isotypes are synthesized with a Glu-Tyr dipeptide at the C terminus (Tyr-tubulin), but the tyrosine can be removed by a specific tubulin carboxypeptidase after assembly into a microtubule, leaving a terminal glutamate (Glu-tubulin). When microtubules containing detyrosinated α-tubulins are disassembled, the liberated α-tubulins are retyrosinated rapidly by a specific tubulin tyrosine ligase. The result is that microtubules that have been assembled for an extended period of time will tend to be rich in Glu-tubulin. The tyrosination state of α-tubulin does not affect its assembly–disassembly kinetics in vitro, but evidence suggests that detyrosination may affect the interaction of microtubules with other cellular structures. In parallel with detyrosination, α-tubulins are also substrates for a specific acetylation reaction. Acetylation of tubulin was initially described for flagellar tubulins, but subsequent work demonstrated that this modification was widespread in neurons and many other cell types. Because the acetylase acts preferentially on α-tubulin assembled into microtubules, long-lived or stable microtubules tend to be rich in acetylated α-tubulin. However, the distribution of microtubules rich in acetylated tubulin may not be identical with that of Glu-tubulin. Acetylated α-tubulin is also deacetylated rapidly upon disassembly of microtubules, although acetylation does not alter the stability of microtubules in vitro.
In contrast with these modifications, tubulin phosphorylation involves a β-tubulin and appears to be restricted to an isotype expressed preferentially in neurons and neuron-like cells. A variety of kinases have been shown to phosphorylate tubulin in vitro, but the endogenous kinase has not been identified. The effect of phosphorylation on assembly is unknown, but phosphorylation is upregulated during neurite outgrowth. As with α-tubulin modifications, the physiological role of phosphorylation on neuronal β-tubulin has yet to be determined. A variety of additional posttranslational modifications have been reported, but their significance and distribution in the nervous system are not well documented.
The biochemical diversity of microtubules is increased through the association of different MAPs with different populations of microtubules (Table 4.2). The significance of microtubule diversity is not completely understood, but it may include functional differences as well as variations in assembly and stability. In particular, MAP composition may be used to define specific neuronal domains. For example, one type of MAP, MAP-2, appears to be restricted to dendritic regions of the neuron, whereas another class of MAPs, tau proteins, are modified differentially in axons. A recently identified isoform of MAP-2 is similar to MAP-2c but includes an additional repeat within the microtubule-binding site; hence it is known as 4-repeat MAP-2c, or MAP-2d.57. Oligodendrocyte progenitors transiently express this novel isoform of MAP-2c in their cell bodies but not in their processes, suggesting that MAP-2d might have a role separate from its known capacity to bundle microtubules (Vouyiouklis & Brophy, 1995).
Table 4.2 Major Microtubule Proteins and Microtubule Motors in Mammalian Brain
MAPs in nervous tissue fall into two heterogeneous groups: tau proteins and high molecular weight MAPs. Tau proteins have been the subject of intense interest because posttranslationally modified tau proteins are the primary polypeptide constituents of neurofibrillary tangles from the brains of Alzheimer patients. Tau proteins appear to be neuronal MAPs, although reports of tau immunoreactivity outside neurons have appeared. Tau proteins bind to microtubules during assembly–disassembly cycles with a constant stoichiometry and can promote microtubule assembly and stabilization. However, recent evidence suggests a role for tau protein as a scaffold for signaling molecules and localizing these pathways to the microtubule that may be more important than stabilization in the mature neuron. Tau exists in a number of molecular weight isoforms that vary with region of the nervous system and developmental stage. For example, tau proteins in the adult CNS are typically from 60 to 75 kDa, whereas PNS axons contain a higher molecular mass tau of approximately 100 kDa. The different isoforms of tau protein are generated from a single mRNA by alternative splicing, and additional heterogeneity is produced by phosphorylation.
In contrast with tau MAPs, high molecular weight MAPs are a diverse group of largely unrelated proteins found in a variety of tissues, although some are brain specific. Most have apparent molecular mass of 1300 kDa or more and form side arms protruding from the walls of microtubules. Many of them may participate in microtubule assembly and cytoskeletal organization but may also serve as scaffolds. Traditionally, the high molecular weight MAPs comprise five polypeptides: MAPs 1a, 1b, 1c, 2a, and 2b. MAP-2 proteins are closely related and are located primarily in dendrites. In contrast, the three polypeptides known as MAP-1 are unique polypeptides with little sequence homology. MAP-1c is a cytoplasmic form of dynein (see the section on molecular motors later in this chapter). MAPs 1a and 1b are widespread and appear to be regulated developmentally. MAPs 1a, 1b, and 2 are all thought to play important roles in stabilizing and organizing the microtubule cytoskeleton.
In most cell types, cytoplasmic microtubules appear to be relatively dynamic structures, although stable microtubules or microtubule segments are found in all cells. In nonneuronal cells such as astrocytes and other glial cells, microtubules are typically anchored in centrosomal regions that serve as microtubule organizing centers. As a result, their cytoplasmic microtubules are oriented so that plus ends are distal to the cell center. The biochemistry of microtubule organizing centers is not fully understood, but they contain a novel tubulin subunit, γ-tubulin, which is thought to function as a nucleating site for microtubules. In contrast, dendritic and axonal microtubules of neurons are not continuous with the microtubule organizing center, so alternate mechanisms must exist for stabilization and organization of these microtubules. The situation is complicated further by the fact that dendritic and axonal microtubules differ in both composition and organization. Studies show that both axonal and dendritic microtubules are nucleated at the microtubule-organizing center but are subsequently released for delivery to the appropriate compartment through the action of the microtubule severing protein, katanin (Roll-Mecak & McNally, 2010). Surprisingly, axonal and dendritic compartments are not equivalent. There are two striking differences. First, MAPs of dendritic and axonal microtubules are different in both identity and phosphorylation state. Second, microtubule orientation in axons is similar to that seen in other cell types with the plus end distal, but microtubules in dendrites may exhibit both polarities. Perhaps due to these differences, dendritic microtubules are less likely to be aligned with one another and appear less regular in their spacing.
Stabilization of axonal and dendritic microtubules is essential because of the volume of cytoplasm and distance from sites of protein synthesis for tubulin. Because microtubules play critical roles in both dendritic and axonal function, mechanisms to ensure their proper extent and organization must exist. A common side effect of one class of antineoplastic drugs, the vinca alkaloids, underscores the importance of microtubule stability in axons. Vincristine and other vinca alkaloids act by destabilizing spindle microtubules, but dosage must be monitored carefully to prevent the development of peripheral neuropathies due to loss of axonal microtubules.
Axonal microtubules contain a particularly stable subset of microtubule segments that are resistant to depolymerization by antimitotic drugs, cold, and calcium. The stable microtubule segments are biochemically distinct and may constitute more than half of the axonal tubulin. Stable domains in microtubules may serve to regulate the axonal cytoskeleton by nucleating and organizing microtubules as well as stabilizing them. The biochemical basis of microtubule stability is not completely understood but probably includes posttranslational modification of the tubulin, the presence of stabilizing proteins, or both. There are indications that levels of cold-insoluble tubulin correlate with axonal plasticity. In contrast, relatively little is known about the regulation of dendritic microtubules, but local synthesis of MAP-2 in dendrites may play a role in regulating their stability.
Microfilaments and the Actin-Based Cytoskeleton Are Involved in Intracellular Transport and Cell Movement
The actin cytoskeleton is universally present in eukaryotes, although actin microfilaments are most familiar as the thin filaments of skeletal muscle. Microfilaments (Table 4.3) play a critical role in contractility for both muscle and nonmuscle cells. Actin and its contractile partner myosin are particularly abundant in nervous tissue relative to other nonmuscle tissues (Letourneau, 2009). In fact, one of the earliest descriptions of nonmuscle actin and myosin was in brain (Berl, Puszkin, & Nicklas, 1973). In neurons, actin microfilaments are most abundant in presynaptic terminals, dendritic spines, growth cones, and the subplasmalemmal cortex. Although concentrated in these regions, microfilaments are also present throughout the cytoplasm of both neurons and glia in the form of short filaments from 4 to 6 nm in diameter and from 400 to 800 nm in length.
Table 4.3 Selected Proteins of the Microfilament Cytoskeleton in Brain
Actins
α-actin (smooth and skeletal muscle)
β-actin and γ-actin (neuronal and nonneuronal cells)
Actin monomer-binding proteins
Profilin
Thymosin 4 and 10
Capping proteins
EB1, EB3
Ezrin/radixin/moesin
Schwannomin/merlin
Gelsolin and other microfilament severing proteins
Gelsolin
Villin
Cross-linking and bundling proteins
Spectrin (fodrin)
Dystrophin, utrophin, and related proteins
α-Actinin
Tropomyosin
Myosins I, II, V, VI, VII
As with tubulin, multiple actin genes exist in both vertebrates and invertebrates. Four α-actin human genes have been cloned. Each of these α-actin genes is expressed specifically in a different muscle cell type (skeletal, cardiac, vascular smooth, and enteric smooth muscle). In addition to α-actins, two nonmuscle actin genes (β-and γ-actin) are present in humans. β-Actin and γ-actin genes are expressed ubiquitously, and both are abundant in nervous tissue. The functional significance of these different genetic isotypes is not clear because the actins are highly conserved proteins. Across the range of known actin sequences, the amino acids are identical at approximately two of three positions. Even the positions of introns within different actin genes are highly conserved across many species and genes. Despite this high degree of conservation, differences in the distribution of specific isotypes within a single neuron have been reported. For example, β-actin may be enriched in growth cones. The prominent actin bundles seen in fibroblasts and some other nonneuronal cells in culture are not characteristic of neurons, and most neuronal actin microfilaments are less than 1 μm in length.
Many microfilament-associated proteins have been described in nervous tissue (myosin, tropomyosin, spectrin, α-actinin, etc.), but less is known about their distribution and normal function in neurons and glia. Myosins and myosin-associated proteins are considered later in the section on molecular motors, but several categories of actin-binding proteins can be defined (Table 4.3). Monomer actin-binding proteins such as profilin and thymosin β4 or β10 are abundant in the developing brain and are thought to help regulate the amount of actin assembled into microfilaments by sequestering actin monomers. These monomers can be mobilized rapidly in response to appropriate signals. For example, phosphatidylinositol 4,5-bisphosphate causes the actin–profilin complex to dissociate, freeing the monomer for microfilament assembly. Such regulation may play a key role in growth cone motility, where actin assembly is an important mechanism for filopodial extension.
Several proteins that can cap actin microfilaments, serving to anchor them to other structures or to regulate microfilament length, have been identified. The ezrin–radixin–moesin gene family encodes barbed-end capping proteins that are concentrated at sites where the microfilaments meet the plasma membrane, suggesting a role in anchoring microfilaments or linking them to extracellular components through membrane proteins. A mutation in a member of this family expressed in Schwann cells, merlin or schwannomin, is responsible for the human disease neurofibromatosis type 2. Development of numerous tumors with a Schwann cell lineage in neurofibromatosis type 2 suggests that this microfilament-binding protein acts as a tumor suppressor.
Whereas some membrane proteins can interact directly with actin microfilaments of the membrane cytoskeleton, others interact with the actin cytoskeleton through intermediaries such as spectrin. Proteins such as spectrin (fodrin), α-actinin, and dystrophins cross-link, or bundle, microfilaments, giving rise to higher order complexes. Spectrin is enriched in the cortical membrane cytoskeleton and is thought to have a role in the localization of integral membrane proteins such as ion channels and receptors. Dystrophin is the best-known member of a family of related proteins that all appear to be essential for the clustering of receptors in muscle and nervous tissue. A mutation in dystrophin is responsible for Duchenne muscular dystrophy. Positioning of integral membrane proteins on the cell surface is likely to be an essential function of the actin-rich membrane cytoskeleton, acting in concert with a class of proteins that contain the protein-binding module, the PDZ domain, a motif that typically interacts with the C-terminus of transmembrane proteins.
Members of the gelsolin family have multiple activities. They cannot only cap the barbed end of a microfilament, but also sever microfilaments and nucleate microfilament assembly under some circumstances. These severing-capping proteins may be essential for reorganizing the actin cytoskeleton. Because gelsolin severing activity is Ca2+ activated, it may provide a mechanism for altering the membrane cytoskeleton in response to Ca2+ transients. Other second messengers, such as phosphatidylinositol 4,5-bisphosphate, may also serve as regulators of gelsolin function, suggesting an interplay between different classes of actin-binding proteins such as gelsolin and profilin. Oligodendrocytes are the only nonneuronal cells in the CNS that express significant amounts of the actin-binding and microfilament-severing protein gelsolin (Tanaka & Sobue, 1994).
Proteins with other functions may also interact directly with actin or actin microfilaments. For example, the enzyme DNase I binds actin tightly, inhibiting both DNase activity and actin assembly. The physiological function of this interaction is unclear, but it has proved a useful tool for probing actin structure and function. Some membrane proteins, such as the epidermal growth factor receptor, bind actin microfilaments directly, which may be important in anchoring these membrane components at a particular location on the cell surface. Other cytoskeletal structures may have specific interactions. Both MAP-2 and tau microtubule-associated proteins have been shown to interact with actin microfilament in vitro and have the potential to mediate interactions between microtubules and microfilaments. Finally, the synaptic vesicle-associated phosphoprotein, synapsin I, has a phosphorylation-sensitive interaction with microfilaments that appears to be important for the targeting and storage of synaptic vesicles in the presynaptic terminal. Many of these interactions have been defined by in vitro–binding studies, and their physiological significance is not always clearly established. However, there is little doubt that interactions occur between the actin cytoskeleton and a variety of other cellular structures.
The presence of actin as a major component of both pre- and postsynaptic specializations, as well as in the growth cone, gives the actin cytoskeleton special significance in the nervous system. The enrichment of microfilaments and associated proteins in the membrane cytoskeleton means that they are the cytoskeletal components most subject and most responsive to changes in the local external environment of the neuron.
Microfilaments also play a critical role in positioning the various receptors and ion channels at specific locations on the neuronal surface, such as the initial segment (Grubb & Burrone, 2010). Although many studies have emphasized enrichment of the microfilament cytoskeleton at the plasma membrane, microfilaments are also abundant in the deep cytoplasm. In many respects, the microfilaments may be best regarded as a uniquely plastic component of the neuronal cytoskeleton that plays a critical role in local trafficking of both cytoskeletal and membrane components.
Intermediate Filaments Are Prominent Constituents of Nervous Tissue
Intermediate filaments of the nervous system appear as solid, ropelike fibrils from 8 to 12 nm in diameter that may be many micrometers long (Herrmann, Bar, Kreplak, Strelkov, & Aebi, 2007). Intermediate filament proteins constitute a superfamily of five classes, which have distinctive patterns of expression specific to cell type and developmental stage (Table 4.4).
Table 4.4 Intermediate Filament Proteins of the Nervous System
| Class and Name | Cell Type |
| Types I and II | |
| Acidic and basic keratins | Epithelial and endothelial cells |
| Type III | |
| Glial fibrillary acidic protein | Astrocytes and nonmyelinating Schwann cells |
| Vimentin | Neuroblasts, glioblasts, fibroblasts, etc. |
| Desmin | Smooth muscle |
| Peripherin | Asubset of peripheral and central neurons |
| Type IV | |
| NF triplet (NFH, NFM, NFL) | Most neurons, expressed at highest level in large myelinated fibers |
| α-Internexin | Developing neurons, parallel fibers of cerebellum |
| Nestin | Early neuroectodermal cells. |
| The most divergent member of this class; some have classified it as a sixth type | |
| Type V | |
| Nuclear lamins | Nuclear membranes |
Type I and type II intermediate filament proteins are keratins, which are hallmarks of epithelial cells. Keratins are not associated with nervous tissue and will not be considered further here. In contrast, all nucleated cells contain type V intermediate filament proteins, the nuclear lamins. Lamins are encoded by the most evolutionarily divergent of the intermediate filament genes, with a distinctive pattern of introns and exons, as well as having a different polypeptide domain structure. Intermediate filaments in the nervous system are all produced by either type III or type IV intermediate filament proteins.
Type III intermediate filaments are a diverse family that includes, among others, vimentin (characteristic of fibroblasts and many embryonic tissues such as embryonic neurons) and glial fibrillary acidic protein (GFAP, a marker for astrocytes and Schwann cells). Type III intermediate filament subunits typically have a molecular mass between 45 and 60 kDa and consist of a conserved rod domain and relatively small gene-specific amino- and carboxy-terminal sequences. As a result, intermediate filaments formed from type III subunits form smooth filaments without side arms. Type III polypeptides can form homopolymers but may also coassemble with other type III intermediate filament subunits.
Another type III intermediate filament protein, peripherin, is unique to neurons and may be coexpressed with neurofilament triplet proteins. Peripherin has a characteristic expression during development and regeneration in specific neuronal populations. It has been shown to coassemble with neurofilament triplet proteins both in vitro and in vivo, where presumably it can substitute for the low molecular weight neurofilament (NFL). However, whether coassembly is generally the case is not known. Physiological roles for neuron-specific type III intermediate filament polypeptides are uncertain. Unlike type IV intermediate filaments, intermediate filaments made from type III subunits tend to disassemble more readily under physiological conditions. Thus, the presence of type III intermediate filament subunit proteins may produce more dynamic structures, which could be important during development or regeneration.
Although other type III intermediate filament proteins are found in the nervous system, they are generally restricted to glia or to neurons at early stages of differentiation. Vimentin is abundant in a wide variety of cells during early development, including both glioblasts and neuroblasts. Some Schwann cells and astrocytes contain vimentin. Curiously, mature oligodendrocytes do not appear to have any cytoplasmic intermediate filaments; an exception to the general rule that metazoan cells contain all three classes of cytoskeletal structures. Oligodendrocyte precursors do, however, express vimentin and may express GFAP transiently.
In neurons, intermediate filaments typically have side arms that limit packing density, whereas glial intermediate filaments lack side arms and may be very tightly packed. Neuronal intermediate filaments have an unusual degree of metabolic stability, which makes them well suited to the role of stabilizing and maintaining neuronal morphology. The existence of neurofilaments was established for many years before much was known about their biochemistry or function. Neurofilaments could be seen in early electron micrographs, and many traditional histological procedures visualize neurons as a result of a specific interaction of metals with neurofilaments. Recent work on the biochemistry and molecular genetics of intermediate filaments has illuminated many aspects of their function in the nervous system.
The primary type of intermediate filament in neurons is formed from three subunits, the neurofilament triplet, each encoded by a separate gene. Neurofilament triplet proteins are type IV intermediate filament proteins, which are generally expressed only in neurons and have a characteristic domain structure seen in both primary sequence and gene structure. The polypeptides were initially identified from axonal transport studies. Apparent molecular mass for the neurofilament subunits vary widely across species, but mammalian forms typically range from 180 to 200 kDa for the high molecular weight subunit (NFH), from 130 to 170 kDa for the medium subunit (NFM), and from 60 to 70 kDa for the NFL. Interestingly, Schwann cells in damaged peripheral nerves also transiently express NFM and NFL. Neurofilament subunits are phosphorylated in axons, with NFM and NFH having unusually high levels of phosphorylation. In some species, NFH has 50 or more repeats of a consensus phosphorylation site at its carboxy terminus, and levels of NFH phosphorylation indicate that most of these sites are phosphorylated in vivo. This high level of phosphorylation in neurofilament subunit tail domains is a distinctive characteristic of neurofilaments.
A second motif characteristic of neurofilaments is the presence of a glutamate-rich region in the tail adjacent to the core rod domain. This glutamate region has particular significance for neuroscientists because it appears to be the basis for the reaction of the classic neurofibrillary silver stains for neurons. These stains were first introduced in the late nineteenth century and have been used extensively by neurohistologists and neuroanatomists from Ramon y Cajal’s time to the present day. However, the molecular basis of these neurofibrillary stains was not known until 1968, when F. O. Schmitt showed that neurofibrils were formed by 10-nm-diameter neurofilaments. Remarkably, the ability of isolated neurofilament subunits to react with silver histological stains is retained even after separation in gel electrophoresis for neurofilaments from organisms as diverse as humans, squid, and the marine fanworm, Myxicola. Conservation of the glutamate-rich domain suggests both an important functional role for this motif and the early divergence of neurofilaments from the other intermediate filament families.
Neurofilaments formed from neurofilament triplet proteins play a critical role in determining axonal caliber. As mentioned earlier, neurofilaments have characteristic side arms, unique among intermediate filaments; these side arms are formed by NFM and NFH carboxy-terminal regions. Although all three neurofilament subunits contribute to the neurofilament central core, the side arms are formed only by the NFM and NFH subunits. Phosphorylation of NFH and NFM side arms alters charge density on the neurofilament surface, repelling adjacent neurofilaments with a similar charge. The high density of surface charge due to phosphate groups on neurofilaments makes it difficult to imagine a stable interaction between neurofilaments and other structures of like charge. Although many reports refer to cross bridges between neurofilaments, direct studies of interactions between neurofilaments provide little evidence of stable crosslinks between neurofilaments or between neurofilaments and other cytoskeletal structures. However, dynamic interactions between neurofilaments and cellular structures or proteins may be critical for many aspects of neurofilament function and metabolism.
Alteration in expression levels for neurofilament subunits or mutations in neurofilament genes can lead to specific neuropathologies. Overexpression of genes encoding normal NFH or expression of some mutant NFL genes in transgenic mouse models leads to the accumulation of neurofilaments in the cell body and proximal axon of spinal motor neurons. These accumulations are similar to those seen in amyotrophic lateral sclerosis and related motor neuron diseases, leading to the hypothesis that the disruption of normal neurofilament function is a common intermediate in the pathogenesis of motor neuron disease. Similarly, an early indicator of neuropathies caused by neurotoxins such as acrylamide and hexanedione is the accumulation of neurofilaments in either proximal or distal regions of the axon. Disruption of neurofilament organization is a hallmark of pathology for many degenerative diseases of the nervous system, particularly those affecting large myelinated axons such as those of spinal motor neurons. Although altering neurofilament organization can produce pathology, the question of whether neurofilament defects are a primary event in pathogenesis or a manifestation of an underlying metabolic pathology remains controversial in most cases.
Another member of the type IV intermediate filament family, α-internexin, is also expressed only in neurons. Unlike the triplet proteins, α-internexin is expressed preferentially early in development of the nervous system and then disappears from most neurons during maturation. Intermediate filaments containing α-internexin do persist in portions of the adult nervous system, such as the branched axons of granule cells in the cerebellar cortex. Evidence show that α-internexin can coassemble with members of the neurofilament triplet, but it also forms homopolymeric filaments. The primary sequence of α-internexin has features in common with both NFL and NFM that are thought to form the basis for assembly properties distinct from those of other type IV intermediate filaments.
The final type of intermediate protein present in the nervous system is nestin, which is expressed transiently during early development. Nestin is also expressed in Schwann cells and in the progenitors of oligodendrocytes, which appear late in the development of the embryonic nervous system. Remarkably, nestin appears to be expressed almost exclusively in ectodermal cells after commitment to the neuroglial lineage, but prior to terminal differentiation. At 1250 kDa, nestin is the largest intermediate filament subunit and is the most divergent in sequence with several distinctive features, leading some to classify nestin as a sixth type of intermediate filament protein, whereas others group it with type IV intermediate filaments. Relatively little is known about the assembly properties of nestin in vivo or the physiological function of nestin intermediate filaments in neuroectodermal cells.
How Do the Various Cytoskeletal Systems Interact?
Each class of cytoskeletal structures may be found without the others in some cellular domains, but all three classes—microtubules, microfilaments, and intermediate filaments—coexist in many domains. As a result, they inevitably interact. This is not to say that individual cytoskeletal elements are necessarily cross-linked to one another. As mentioned earlier, microtubules and neurofilaments have highly phosphorylated side arms that project from their surfaces. The high density of negative charge on the surface tends to repel structures with a like charge such as other microtubules and neurofilaments. This does not mean that microtubules and neurofilaments do not interact with each other, but it does suggest that such interactions may be transient.
One location containing longer microfilaments and more elaborate organization is the growth cone, which contains bundles of microfilaments in the filopodia, as well as a more dispersed actin network. Neurofilaments are largely excluded from the growth cone, typically extending no further than the neck of the growth cone. In contrast, microtubules and microfilaments play complementary roles in the growth cone itself. Microfilaments are critical in sprouting but appear less critical for elongation, at least over short distances. Disruption of microtubules in the distal neurite does not affect sprouting but does inhibit neurite elongation. It seems likely that microtubules and microfilaments also act synergistically in the function of the axon initial segment. The tips of microtubules are often associated with proteins that appear to regulate their interaction with sub-plasma membrane structures. At the axon initial segment the proteins EB1 and EB3 appear to link microtubules with ankyrin-G, a key interactor with the spectrin-actin cytoskeleton and that is known to play a crucial role in the assembly of this critical neuronal domain.
Summary
The intracellular framework that gives shape to the neuron and glial cell is the cytoskeleton, a complicated set of filaments and tubules and their associated proteins. These organelles are responsible as well for intracellular movement of materials and, during development, for cell migration and plasma membrane extension within nervous tissue.
Molecular Motors in the Nervous System
Until 1985, our knowledge of molecular motors in vertebrate cells of any type was restricted to myosins and flagellar dyneins. Myosins had been identified in nervous tissue, but their functions were uncertain. Because the preponderance of evidence indicated that fast axonal transport was microtubule based, there was considerable interest in dyneins in cell cytoplasm. Despite a number of studies, no evidence for a functional cytoplasmic dynein emerged. Worse yet, the characteristic properties of fast organelle movements appeared inconsistent with both myosins and dyneins. Over the past decade, however, we have developed a good but still incomplete understanding of how these motors may work inside cells.
Myosins and dyneins can be distinguished pharmacologically by their differential susceptibility to inhibitors of ATPase activity, but the spectrum of inhibitors active against fast axonal transport fails to match the properties of either myosin or dynein. The most striking difference between inhibitor effects on axonal transport and on myosin or dynein motors was seen in the effect of a nonhydrolyzable analog of ATP. Adenylyl-imidodiphosphate (AMP-PNP) is a weak competitive inhibitor of both myosin and dynein, requiring a 10- to 100-fold excess of analog. In contrast, within minutes of AMP-PNP perfusion into isolated axoplasm, both anterograde and retrograde axonal transport stop. Inhibition by AMP-PNP occurs even in the presence of stoichiometric concentrations of ATP. Organelles moving in both directions freeze in place and remain attached to microtubules. AMP-PNP weakens the interaction of myosin with microfilaments and of dynein with microtubules, but stabilizes the binding of membrane-bound organelles to microtubules. Thus, the effects of AMP-PNP indicate that movement of membrane-bound organelles in fast axonal transport must require another type of motor, distinct from myosins and dyneins.
The effects of AMP-PNP both demonstrated the existence of a new type of mechanochemical ATPase and provided a basis for identifying its constituent polypeptides. Binding of ATPase to microtubules should be increased by AMP-PNP and decreased by ATP. Polypeptides meeting this criterion were soon identified. The new ATPase was named kinesin, based initially on an ability to move microtubules across glass coverslips as first described in axoplasmic extracts. Studies soon established that kinesin was a microtubule-activated ATPase with minimal basal activity. This combination of ATPase activity and motility in vitro confirmed that kinesin was a new class of microtubule-based motor (Brady & Sperry, 1995).
Kinesin has now been identified in a variety of organisms and tissues, leading to an extensive characterization of many biochemical, pharmacological, immunochemical, and molecular properties. Electron microscopic and biophysical analyses reveal kinesin as a long, rod-shaped protein, approximately 80 nm in length. Neuronal kinesin is a heterotetramer with two heavy chains (molecular mass 115–130 kDa) and two light chains (62–70 kDa). Localization of antibodies specific for kinesin subunits by high-resolution electron microscopy of bovine brain kinesin indicates that the two heavy chains are arranged in parallel, forming the heads and much of the shaft, whereas light chains are localized to the fan-shaped tail region (Fig. 4.5).
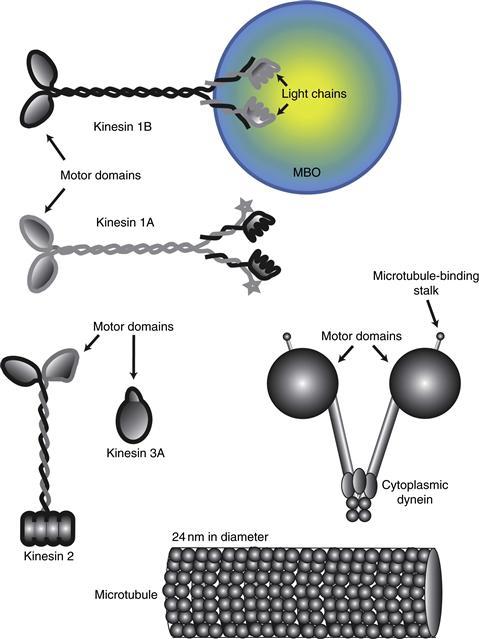
Figure 4.5 Examples of microtubule motor proteins in the mammalian nervous system. The first microtubule motor identified in nervous tissue was a kinesin 1, but showed 3 kinesin 1 genes including a neuron-specific form (kinesin 1A). Motor domains are well conserved by tail domains and appear to be specialized for interaction with various targets, such as different membrane-bound organelles. After the sequence of the kinesin heavy chain was established, the presence of additional genes that contained sequences homologous to the motor domain of kinesin was soon recognized. The molecular organization of these various motor proteins is diverse, including monomers (Kinesin 3A), trimers (Kinesin 2), and tetramers (ubiquitous and neuron-specific kinesins). Cytoplasmic dynein may interact with membrane-bound organelles and cytoskeletal structures. Genetic methods have established that there may be more than 40 kinesin-related genes and 16 dynein heavy chains genes in a single organism.
A variety of approaches have demonstrated that the ATP-binding and microtubule-binding domains of kinesin are in the head regions of the heavy chains, whereas the light chains in the tail region of kinesin appear to bind to membranes. When in vitro motility assays are employed for analysis of brain kinesins, movements are directed toward the plus ends of microtubules. Because axonal microtubules are oriented with their plus ends distal from the cell body, this movement would be appropriate for a motor that moves organelles in the anterograde direction.
Neuronal kinesin appears associated with a variety of membrane-bound organelles, including synaptic vesicles, mitochondria, coated vesicles, and lysosomes. The interaction of kinesin and other molecular motors with membrane surfaces is not well understood. In the case of kinesin, the interaction is thought to involve the light chains of kinesin along with the carboxy termini of the heavy chains.
Kinesins have now been shown to be a family of related proteins with a highly conserved domain that includes ATP- and microtubule-binding domains (Hirokawa, Noda, Tanaka, & Niwa, 2009). Many of these kinesin-related polypeptides appear associated with cell division, and kinesin-related proteins in vertebrate tissues are not well characterized. However, multiple members of the kinesin superfamily are expressed in both adult and developing brains. This proliferation of motor proteins has dramatically altered the questions being asked about motor function in the brain. The discovery that ncd, a kinesin-related protein from Drosophila, can move structures toward the minus end of microtubules increases the number of potential functions that kinesin family members might serve in nervous tissue still further, perhaps including a role in retrograde transport. Further study is needed to establish specific functions for each member of the kinesin superfamily expressed in neurons or glial cells.
As an indirect result of the discovery of kinesin, one of the high molecular weight microtubule-associated proteins of brain, MAP-1c, was found to be the long sought cytoplasmic form of dynein (Allan, 2011). Both MAP-1c dynein and kinesin can be isolated from bovine brain by incubation of microtubules with nucleotide-free soluble extracts. Both are bound to microtubules under these conditions and released by ATP. MAP-1c dynein moved microtubules in vitro with a polarity opposite that seen with kinesin and was identified as a two-headed cytoplasmic dynein using both structural and biochemical criteria. Concurrently, a similar protein was identified in nematodes.
MAP-1c dyneins form a 40-nm-long complex of molecular mass 1.6 × 106 Da, which includes two heavy chains and a number of light chains (Figs. 4.8 and 4.9). Less information is available about the distribution and properties of MAP-1c dyneins than about kinesin. Immunocytochemical studies in nonneuronal cells showed immunoreactivity on mitotic spindles. In addition, a punctate pattern of immunoreactivity also present in interphase cells was thought to be due to dynein bound to membrane-bound organelles. Dyneins are widely thought to be the motor for fast retrograde axonal transport but are also a candidate for a motor in slow axonal transport.
Figure 4.6 Examples of myosin motor proteins found in mammalian brain. Myosin heavy chains contain the motor domain, whereas light chains regulate motor function. Myosin II was the first molecular motor characterized biochemically from skeletal muscle and brain. Genetic approaches have now defined more than 15 classes of myosin, many of which are found in brain. Myosin II is a classic two-headed myosin forming thick filaments in nonmuscle cells. Myosin I motors have single motor domains, but may interact with actin microfilaments or membranes. Myosin V has multiple binding sites for calmodulin that act as light chains. Mutations in other classes of myosin have been linked to deafness. Myosins I, II, and V have been detected in growth cones as well as in mature neurons.
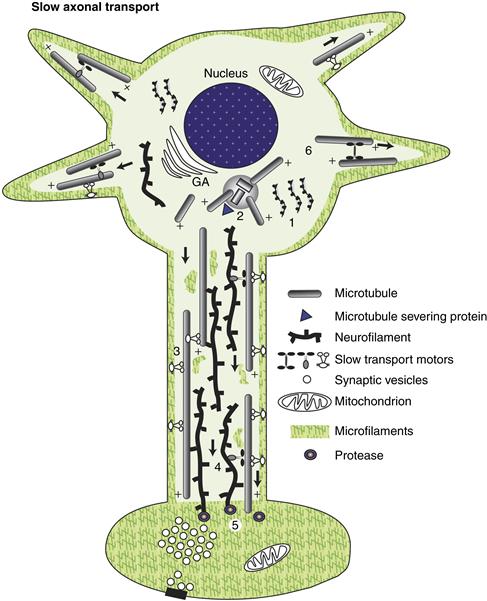
Figure 4.7 Slow axonal transport represents the delivery of cytoskeletal and cytoplasmic constituents to the periphery. Cytoplasmic proteins are synthesized on free polysomes and organized for transport as cytoskeletal elements or macromolecular complexes (1). Microtubules are formed by nucleation at the microtubule-organizing center near the centriolar complex (2) and then released for migration into the axon or dendrites. The molecular mechanisms are not as well understood as those for fast axonal transport, but slow transport appears to be unidirectional with no retrograde component. Studies suggest that motors like cytoplasmic dynein may interact with the axonal membrane cytoskeleton to move the microtubules with their plus ends leading (3). Neurofilaments may not be able to move on their own but may hitchhike on microtubules (4). Other cytoplasmic proteins may do the same or may be moved by other motors. Once cytoplasmic structures reach their destinations, they are degraded by local proteases (5) at a rate that allows either growth (in the case of growth cones) or maintenance of steady-state levels. The different composition and organization of the cytoplasmic elements in dendrites suggest that different pathways may be involved in the delivery of cytoskeletal and cytoplasmic materials to the dendrite (6). In addition, some mRNAs are transported into the dendrites, but not into axons.
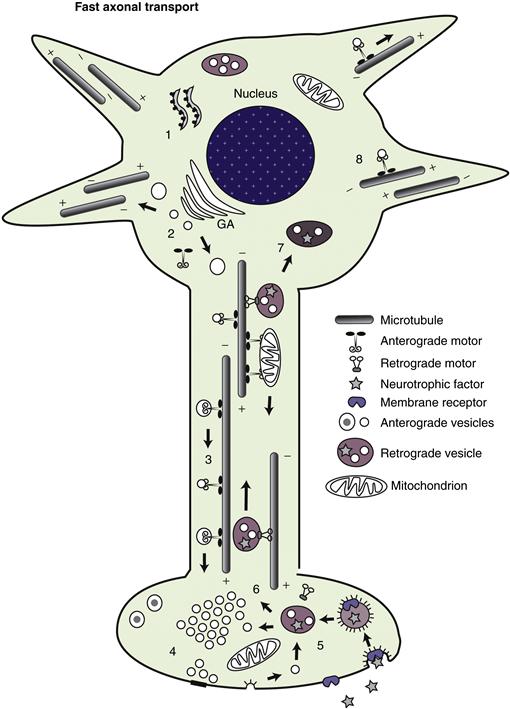
Figure 4.8 Fast axonal transport represents the transport of membrane-associated materials, having both anterograde and retrograde components. For anterograde transport, most polypeptides are synthesized on membrane-bound polysomes, also known as rough endoplasmic reticulum (1), and then transferred to the Golgi apparatus for processing and packaging into specific classes of membrane-bound organelles (2). Proteins following this pathway include both integral membrane proteins and secretory polypeptides in the lumen of vesicles. Cytoplasmic peripheral membrane proteins such as kinesins are synthesized on the cytoplasmic or free polysomes. Once vesicles have been assembled and the appropriate motors associate with them, they are moved down the axon at a rate of 100–400 mm per day (3). Different membrane structures are delivered to different compartments and may be regulated independently. For example, dense core vesicles and synaptic vesicles are both targeted for the presynaptic terminal (4), but the release of vesicle contents involves distinct pathways. After vesicles merge with the plasma membrane, their protein constituents are taken up by coated pits and vesicles via the receptor-mediated endocytic pathway and delivered to a sorting compartment (5). After proper sorting into appropriate compartments, membrane proteins are either committed to retrograde axonal transport or recycled (6). Retrograde moving organelles are morphologically and biochemically distinct from anterograde vesicles. These larger vesicles have an average velocity about half that of anterograde transport. The retrograde pathway is an important mechanism for the delivery of neurotrophic factors to the cell body. Material delivered by retrograde transport typically fuses with cell body compartments to form mature lysosomes (7), where most constituents are recycled. However, neurotrophic factors and neurotrophic viruses can act at the level of the cell body. Although evidence shows that vesicle transport also occurs into dendrites (8), less is known about this pocess. Dendritic vesicle transport is complicated by the fact that dendritic microtubules may have mixed polarity.
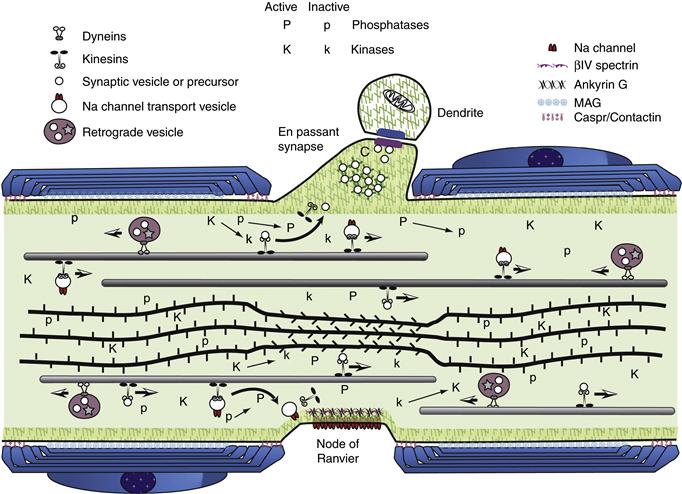
Figure 4.9 Axonal dynamics in a myelinated axon from the peripheral nervous system (PNS). Axons are in a constant flux with many concurrent dynamic processes. This diagram illustrates a few of the many dynamic events occurring at a node of Ranvier in a myelinated axon from the PNS. Axonal transport moves cytoskeletal structures, cytoplasmic proteins, and membrane-bound organelles from the cell body toward the periphery (from right to left). At the same time, other vesicles return to the cell body by retrograde transport (retrograde vesicle). Membrane-bound organelles are moved along microtubules by motor proteins such as the kinesins and cytoplasmic dyneins. Each class of organelles must be directed to the correct functional domain of the neuron. Synaptic vesicles must be delivered to a presynaptic terminal to maintain synaptic transmission. In contrast, organelles containing sodium channels must be targeted specifically to nodes of Ranvier for saltatory conduction to occur. Cytoskeletal transport is illustrated by microtubules (rods in the upper half of the axon) and neurofilaments (bundle of rope-like rods in the lower half of the axon) representing the cytoskeleton. They move in the anterograde direction as discrete elements and are degraded in the distal regions. Microtubules and neurofilaments interact with each other transiently during transport, but their distribution in axonal cross sections suggests that they are not stably cross-linked. In axonal segments without compact myelin, such as the node of Ranvier or following focal demyelination, a net dephosphorylation of neurofilament side arms allows the neurofilaments to pack more densely. Myelination is thought to alter the balance between kinase (K indicates an active kinase; k is an inactive kinase) and phosphatase (P indicates an active phophatase; p is an inactive phosphatase) activity in the axon. Most kinases and phosphatases have multiple substrates, suggesting a mechanism for targeting vesicle proteins to specific axonal domains. Local changes in the phosphoryation of axonal proteins may alter the binding properties of proteins. The action of synapsin I in squid axoplasm suggests that dephosphorylated synapsin cross-links synaptic vesicles to microfilaments. When a synaptic vesicle encounters the dephosphorylated synapsin and actin-rich matrix of a presynaptic terminal, the vesicle is trapped at the terminal by inhibition of further axonal transport, effectively targeting the synaptic vesicle to a presynaptic terminal. Similarly, a sodium channel-binding protein may be present at nodes of Ranvier in a high-affinity state (i.e., dephosphorylated). Transport vesicles for nodal sodium channels (Na channel vesicle) would be captured upon encountering this domain, effectively targeting sodium channels to the nodal membrane. Interactions between cells could in this manner establish the functional architecture of the neuron.
Myosins from muscle were the first molecular motors identified, but in recent years interest in nonmuscle myosins has increased (Foth, Goedecke, & Soldati, 2006). Nonmuscle myosins may be categorized as belonging to one of eight classes, but only a subset has been clearly demonstrated in the nervous system. Nonmuscle myosins in this subset share considerable homology in their motor domains but diverge widely in other domains.
The most familiar of the myosins are the myosin II proteins (Fig. 4.6), which are found in the thick filaments of smooth and skeletal muscle but are also present in nonmuscle cells. Two heavy chains of myosin II form a dimer that may interact with other myosin II dimers to form bipolar filaments. Under tissue culture conditions, many cells contain bundles of actin microfilaments, known as stress fibers, that exhibit a characteristic distribution of myosin II into distinct patterns that may be sarcomeric equivalents, but stress fibers are not apparent in neurons and other cells of the nervous system in situ. However, bipolar thick filaments assembled from myosin II dimers can be isolated from nervous tissues. Many of the cellular contractile events described in nonneuronal cells, such as the contractile ring in mitosis, are thought to include myosin II. Although brain myosin II was one of the first nonmuscle myosins to be described, relatively little is known about the function of myosin II in neurons. Myosin II has been localized in the neurites of neurons in primary culture.
Myosin I proteins have a single, smaller heavy chain that does not form filaments but possesses a homologous actinactivated ATPase domain. One exciting aspect of myosin I is its ability to interact directly with membrane surfaces, which may generate movements of plasma membrane components or intracellular organelles. Myosin I has been purified from neural and neuroendocrine tissues. At least three genes in this family have been found in mammals, and multiple forms are present in brain. An interesting aspect of myosin IB is its expression in the stereocilia of hair cells in the cochlea and vestibular system, where it may play a role in mechanotransduction. Both myosin I and myosin II molecules have been proposed to have a role in the motility of lamelipodia at the leading edge of growth cones, but they are also expressed at substantial levels in adult nervous tissue in which growth cones are rare.
The mouse mutation dilute, which affects coat color, was shown to result from a mutation in a gene that encodes a novel myosin heavy chain distinct from both myosins I and II. Similar myosin molecules have been identified in other cell types and organisms and are classified as myosin V. The change in coat color seen in the dilute mouse is due to an inability of skin dendritic pigment cells to deliver the pigment to developing hairs. There are complex neurological deficits in dilute mutants, including seizures that eventually lead to death in severely altered alleles of the dilute mutation. Myosin V was also discovered independently in extracts from chicken and mammalian brain. Myosin V is thought to play a role in the delivery of membrane proteins from the microtubule-based transport system to the plasma membrane. However, neurons in dilute mice without one allele of myosin V clearly develop axons and make connections. The seizures do not begin until early adulthood.
Representatives from two more classes of myosin have been identified in nervous tissue. Genes for a myosin VI and a myosin VIIA have been identified in brain as well as in other tissues. Both have been implicated in forms of congenital deafness. Myosin VI appears to be the gene responsible for Snell’s Waltzer deafness, and myosin VIIA has been identified as the gene responsible for a human disease involving both deafness and blindness, Usher syndrome type 1B. Both of these myosins are expressed in mechanosensory hair cells of the cochlea and vestibular apparatus, where they exhibit a different localization from each other and from myosin IB.
The diversity of brain myosins and their distinctive localization suggests that the various myosins may have narrowly defined functions. However, relatively little is known about specific neuronal functions for myosins despite intensive study of myosins in the nervous system. The axonal transport of myosin II-like proteins has been described, but little further progress has been made on the functions of myosin II in the mature nervous system. Even less is known about myosin I in the nervous system. However, myosins likely play roles in growth cone motility, synaptic plasticity, and even neurotransmitter release. There are few instances in our knowledge of neuronal function in which we fully understand the role played by specific molecular motors, but members of all three classes are abundant in nervous tissue. This proliferation of different motor molecules and their isoforms suggests that some physiological activities may require multiple classes of motor molecules.
Summary
The concept is now firmly in place that neurons and glial cells, like other cells, contain molecular motors responsible for moving discrete populations of molecules, particles, and organelles through intracellular compartments.
Building and Maintaining Cells of the Nervous System
The functional architecture of neurons comprises many specializations in cytoskeletal and membranous components. Each of these specializations is dynamic, constantly changing, and being renewed at a rate determined by the local environment and cellular metabolism. The processes of axonal transport represent a key to understanding neuronal dynamics and provide a basis for exploring neuronal development, regeneration, and neuropathology. Recent advances are important sources of insight into the molecular mechanisms underlying axonal transport, although many questions remain.
Slow Axonal Transport Moves Soluble Components and Cytoskeletal Structures
Slow axonal transport has two major components, both representing the movement of cytoplasmic constituents (Fig. 4.7). The cytoplasmic and cytoskeletal elements of the axon in axonal transport move at rates at least two orders of magnitude more slowly than fast transport. Slow component a is composed largely of cytoskeletal proteins, neurofilaments, and microtubule protein. Slow component b is a complex and heterogeneous rate component, including hundreds of distinct polypeptides ranging from cytoskeletal proteins such as actin (and tubulin in some nerves) to soluble enzymes of intermediary metabolism (such as glycolytic enzymes). Many characteristics of axonal transport have been described, and these characteristics provide the foundation for our understanding of mechanisms.
Neurofilaments and microtubules move as discrete cytological structures (Baas & Buster, 2004). Studies on the transport of neurofilament protein indicate that little degradation or metabolism occurs until neurofilaments reach nerve terminals, where they are degraded rapidly. Comparable results have been obtained in studies labeling microtubule protein by radioactivity or fluorescence. Under favorable conditions, movement of individual microtubules can be detected in neurites or growth cones. Both radiolabeling studies and direct observations of individual microtubules indicate that all microtubules and neurofilaments move down the axon, but the motor protein involved is uncertain. Differential metabolism appears to be a key to the targeting of cytoplasmic and cytoskeletal proteins. Proteins with slow degradative rates accumulate and reach higher steady-state concentrations. Alteration of degradation rates changes the steady-state concentration of a protein. The concentration of actin in presynaptic terminals is explained by a slower turnover in terminals relative to neurofilament proteins and tubulin, and inhibition of calpain causes neurofilament rings to appear in presynaptic terminals. Differential turnover may be accomplished by specific proteases or posttranslational modifications that affect the susceptibility to degradation.
The coherent movement of neurofilaments and microtubule proteins provides strong evidence for the “structural hypothesis.” For example, pulse-labeling experiments show that radiolabeled neurofilament proteins move as a bell-shaped wave with little or no trailing of neurofilament protein. This fits with the observed stability of neurofilaments under physiological conditions. Thus, any soluble pool of neurofilament subunits is negligible. Similarly, the coherent transport of tubulin and MAPs makes sense only if microtubules are moved because MAPs do not interact with unpolymerized tubulin.
A striking demonstration of microtubule movement can be seen with fluorescent analogs of tubulin (Brown, 2003). When local segments of an axon are photoactivated, short fluorescent microtubule segments can be seen to move down the growing neurite. These microtubules move rapidly (1–2 µm/sec) with frequent pauses that produce a net anterograde transport of microtubules at 1–2 mm/day and similar rates are observed for neurofilaments. When studies of axonal transport using radiolabeled amino acids are combined with direct observations of individual microtubules or neurofilaments by video microscopy, there is little doubt that microtubules and neurofilaments can and do move in the axon as intact, individual cytoskeletal elements.
More recently, similar methods have been used to document the transport of classically soluble, cytoplasmic proteins like glycolytic enzymes and synuclein (Roy, Winton, Black, Trojanowski, & Lee, 2007). As with microtubules and neurofilaments, these movements are intermittent and rapid. The extended pauses lead to a net rate of anterograde transport of approximately 2–4 mm/day, consistent with rates obtained in pulse-chase radiolabel studies.
Fast Axonal Transport Is the Means by Which Membrane Vesicles and their Contents Are Rapidly Moved Long Distances within a Neuron
Early biochemical and morphological studies established that the material moving in fast axonal transport was associated with membrane-bound organelles (Fig. 4.8) (Brady, 1993). A variety of materials could be shown to move in fast transport. In anterograde transport, materials being moved include membrane-associated enzyme activities, neurotransmitters, and neuropeptides. Many of the materials moving down the axon in anterograde transport are returned in retrograde transport, in some cases after modification in the terminal. In addition, a number of exogenous materials taken up in the distal regions of the axon are moved back to the cell body by retrograde transport. Exogenous materials in retrograde transport include neurotrophic factors, such as nerve growth factor, and viral particles invading the nervous system. The uptake of neurotrophic factors may play a critical role in the process of regeneration.
Electron microscopic analysis of materials accumulated at a ligation or crush demonstrated that organelles moving in the anterograde direction were morphologically distinct from those moving in the retrograde direction (Tsukita & Ishikawa, 1980). Consistent with the ultrastructural differences, radiolabel and immunocytochemical studies indicate that there are both quantitative and qualitative differences between anterograde and retrograde moving material. Differences between anterograde and retrograde transport indicate that some processing or repackaging events must occur as part of turnaround in axonal transport. Turnaround processing appears to require a proteolytic event because certain protease inhibitors inhibit turnaround without affecting anterograde and retrograde transport.
Biochemical and morphological approaches resulted in considerable progress toward a description of the materials being transported in fast axonal transport but were not suitable for identifying the molecular motors used in translocation. A different technology that permitted direct observation of organelle movements and precise control of experimental conditions was required. Such experiments became possible with the advent of video microscopic techniques (Brady, 1991).
An early use of video-enhanced contrast (VEC) microscopy was to characterize the bidirectional movement of membrane-bound organelles in giant axons from the squid Loligo pealeii. Years before, studies had shown that axoplasm could be extruded from the giant axon as an intact cylinder. Properties of the isolated axoplasm had been characterized in some detail, making VEC microscopic analysis of axoplasm a natural choice. Remarkably, fast axonal transport continued unabated in isolated axoplasm for hours. Isolated axoplasm from the giant axon has no plasma membrane or other permeability barriers but can be readily maintained in an active state. Combining VEC microscopy with isolated axoplasm permitted rigorous dissection of the mechanisms for fast axonal transport with the use of biochemical and pharmacological approaches. A number of insights into axonal transport mechanisms have resulted from these studies.
How Is Axonal Transport Regulated?
The diversity of polypeptides in each axonal transport rate component and the coherent movement of proteins having many different molecular weights produce a conundrum. How can so many different polypeptides move down the axon as a group? In theory, one could propose that each protein has a motor of its own. In that case, each rate component might represent movements due to a specific class of motors or to variable affinities for a smaller number of motors. However, the relatively small numbers of motor molecules and the logistical difficulties associated with such a model effectively preclude this possibility.
The structural hypothesis mentioned earlier was formulated in response to the observation that rate components of axonal transport move as discrete waves, each with a characteristic rate and a distinctive composition (Figs. 4.7 and 4.8). The hypothesis is deceptively simple: Axonal transport represents the movement of discrete cytological structures (Brady, 1993). Proteins in axonal transport do not move as individual polypeptides. Instead, they move as part of a cytological structure or in association with a cytological structure. The only assumption made is that a limited number of elements can interact directly with transport motors, so transported material must be packaged appropriately to be moved. The different rate components result from packaging of transported material into different cytologically identifiable structures. In other words, membrane-associated proteins move as membrane-bound organelles (vesicles, mitochondria, etc.), whereas tubulin and MAPs move as microtubules, and neurofilaments move as neurofilaments.
The structural hypothesis does not require movement of a cross-linked microtubule–neurofilament complex in the form of a solid axoplasmic column. Evidence for existence of a cross-linked complex of microtubules and neurofilaments is not compelling in any case. Instead, the hypothesis specifically predicts that individual microtubules and neurofilaments move rather than tubulin dimers or neurofilament monomers. No assumptions are made about higher-order interactions between cytoskeletal structures or membranous structures. Indeed, one variant of the structural hypothesis proposes that cytoskeletal proteins move in the form of small oligomers rather than polymers, although there is no evidence for the presence of such oligomers in vivo. For example, to the limits of detection, neurofilaments exist only as the polymer under in vivo conditions. Similarly, tubulin interchanges between dimer and polymer with no known oligomeric intermediate. The movement of microtubules, neurofilaments, and complexes of soluble proteins is in fact what is observed (Baas & Buster, 2004; Roy et al., 2007).
Because synthesis of proteins takes place at some distance from many functional domains of a neuron, transport to distal regions of the neuron is necessary, but not sufficient, for proper function. Specific materials must also be delivered to their proper site of utilization and should not be left in inappropriate locations. For example, because a synaptic vesicle has no known function in axons or the cell body, it must be delivered to a presynaptic terminal along with other components necessary for regulated neurotransmitter release. The traditional picture places the presynaptic terminal at the end of an axonal process. Such images imply that a synaptic vesicle need only move along the axonal microtubules until it reaches microtubule ends in the presynaptic terminal. However, many CNS synapses are not at the end of an axon. Numerous terminals are located sequentially along a single axon, making en passant contacts with multiple target cells along the way. Targeting of synaptic vesicles then becomes a more complex problem and targeting ion channels or neurotransmitter receptors to nodes of Ranvier or other appropriate sites on the neuronal surface is equally challenging.
Although the specific details of this targeting are not well understood, a simple model for the targeting of synaptic vesicles serves to illustrate how such targeting may occur (Fig. 4.9). Synapsin I is a cytoplasmic protein enriched in presynaptic terminals that can bind reversibly to both actin microfilaments and synaptic vesicles. Both binding activities are regulated by the phosphorylation of synapsin, and this phosphorylation is increased during stimulation. If nonphosphorylated synapsin were present at the border between the axon and the presynaptic terminal, then a fraction of the synaptic vesicles (or their precursors) traveling down the axon in fast axonal transport would be bound and cross-linked to the actin cytoskeleton that is enriched in the presynaptic terminal. These bound synaptic vesicles would become part of the reserve pool in the terminal at some distance from active zones. As the terminal was stimulated, local calmodulin-activated kinases would phosphorylate synapsin and allow these reserve synaptic vesicles to be mobilized. Consistent with this model, dephosphorylated synapsin has been shown to inhibit fast axonal transport when introduced directly into the axoplasm at concentrations comparable to those seen in the presynaptic terminal (McGuinness et al., 1989).
The role of kinases in this targeting appears to be a common feature in the regulation of axonal transport (Morfini, Szebenyi, Richards, & Brady, 2001). Local activation of kinases satisfies several criteria that any mechanism for targeting to specific neuronal subdomains must address. Specifically, the mechanism must be spatially restricted because distances to the cell body can be quite large. There must be some means to connect the targeting signal to an external microenvironment, such as an appropriate target cell. Finally, there must be a way of distinguishing subdomains so that synaptic vesicles are not delivered to nodes of Ranvier and voltage-gated sodium channels are not all targeted to the presynaptic terminal. The careful segregation of different organelles and polypeptides to different regions within a neuron suggests that highly efficient targeting mechanisms do exist.
Summary
A well-studied feature of the neuron is the phenomenon of axonal transport, which has been described in both anterograde and retrograde directions. The axonal transport system is responsible for the delivery of materials from the cell body to distant parts of the neuron, for membrane retrieval and circulation, and for uptake of materials from presynaptic terminals and dendrites and their delivery to the cell soma. The precise molecular mechanisms by which anterograde and retrograde transport can be targeted within an individual dendrite and/or axon are not understood at present.
Neurons and glial cells may have unusually large cell volumes enclosed within extensive plasma membrane surfaces. Nature has evolved a number of “universal” mechanisms in other systems and adapted them for the special needs of nervous tissue cells. The synthesis and delivery of components, and in particular proteins, to cytoplasmic organelles and cell surface subdomains engage general and evolutionarily conserved molecular mechanisms and pathways that are employed in single-cell yeasts as well as in cells in complex nervous tissue.
Once synthesized and sorted, most intracellular organelles (vesicles destined for axonal or dendritic domains, mitochondria, cytoskeletal components) must be distributed, and targeted, to precise intracellular locations. Because they are so extended in space, neurons and glial cells have adapted and developed to a high degree common mechanisms that operate to distribute components within all cells. In the neuron, movement of materials within the axon has been the central focus of most studies. The phenomenon of axoplasmic transport is now in some measure understood at the molecular level.
Dedication
We are very sad to report that David Colman died suddenly during the preparation of this edition. As a major contributor to our current understanding of myelination, David was a highly respected scientific colleague and, most of all, a good friend. We dedicate this chapter to his memory.
References
1. Allan VJ. Cytoplasmic dynein. Biochemical Society Transactions. 2011;39:1169–1178.
2. Arnold DB. Actin and microtubule-based cytoskeletal cues direct polarized targeting of proteins in neurons. Science Signaling. 2009;2:49.
3. Baas PW, Buster DW. Slow axonal transport and the genesis of neuronal morphology. Journal of Neurobiology. 2004;58:3–17.
4. Berl S, Puszkin S, Nicklas WJK. Actomyosin-like protein in brain. Science. 1973;179:441–446.
5. Brady S. Axonal transport methods and applications. In: Boulton A, Baker G, eds. Neuromethods, general neurochemical techniques. Clifton, NJ: Humana Press; 1985:419–476.
6. Brady ST. Molecular motors in the nervous system. Neuron. 1991;7:521–533.
7. Brady ST. Axonal dynamics and regeneration. In: Gorio A, ed. Neuroregeneration. New York City, NY: Raven Press; 1993:7–36.
8. Brady ST, Sperry AO. Biochemical and functional diversity of microtubule motors in the nervous system. Current Opinion in Neurobiology. 1995;5:551–558.
9. Brady ST, Tai L. Cell biology of the nervous system. In: Brady ST, Siegel G, Albers RW, Price D, eds. Basic neurochemistry: Principles of molecular, cellular and medical neurobiology. 2011:3–25. Boston, MA: Elseiver.
10. Braulke T, Bonifacino JS. Sorting of lysosomal proteins. Biochimica et Biophysica Acta, 1793 2009;605–614.
11. Brown A. Live-cell imaging of slow axonal transport in cultured neurons. Methods in Cell Biology. 2003;71:305–323.
12. Colman DR, Kreibich G, Frey AB, Sabatini DD. Synthesis and incorporation of myelin polypeptides into CNS myelin. Journal of Cell Biology. 1982;95:598–608.
13. Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annual Review of Biochemistry. 2009;78:857–902.
14. Dudek J, Benedix J, Cappel S, et al. Functions and pathologies of BiP and its interaction partners. Cellular and Molecular Life Sciences. 2009;66:1556–1569.
15. Endo T, Yamano K. Multiple pathways for mitochondrial protein traffic. Biological chemistry. 2009;390:723–730.
16. Foth BJ, Goedecke MC, Soldati D. New insights into myosin evolution and classification. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3681–3686.
17. Glick BS, Nakano A. Membrane traffic within the Golgi apparatus. Annual Review of Cell and Developemental Biology. 2009;25:113–132.
18. Grubb MS, Burrone J. Building and maintaining the axon initial segment. Current Opinion in Neurobiology. 2010;20:481–488.
19. Herrmann H, Bar H, Kreplak L, Strelkov SV, Aebi U. Intermediate filaments: From cell architecture to nanomechanics. Nature Reviews Molecular Cell Biology. 2007;8:562–573.
20. Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nature Reviews Molecular Cell Biology. 2009;10:682–696.
21. Hirst J, Barlow LD, Francisco GC, et al. The fifth adaptor protein complex. PLoS biology. 2011;9:e1001170.
22. Hochstrasser M. Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Current Opinion in Cell Biology. 1995;7:215–223.
23. Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annual Review of Biochemistry. 1985;54:631–664.
24. Lasek RJ, Brady ST. The Axon: A prototype for studying expressional cytoplasm. In: Organization of the cytoplasm. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1982;113–124.
25. Letourneau PC. Actin in axons: Stable scaffolds and dynamic filaments. Results and Problems in Cell Differentiation. 2009;48:65–90.
26. Ma C, Agrawal G, Subramani S. Peroxisome assembly: Matrix and membrane protein biogenesis. The Journal of Cell Biology. 2011;193:7–16.
27. Martin KC, Zukin RS. RNA trafficking and local protein synthesis in dendrites: An overview. Journal of Neuroscience. 2006;26:7131–7134.
28. McGuinness TL, Brady ST, Gruner J, Sugimori M, Llinas R, Greengard P. Phosphorylation-dependent inhibition by synapsin I of organelle movement in squid axoplasm. Journal of Neuroscience. 1989;9:4138–4149.
29. Morfini G, Szebenyi G, Richards B, Brady ST. Regulation of kinesin: Implications for neuronal development. Developmental Neuroscience. 2001;23:364–376.
30. Palade G. Intracellular aspects of the process of protein synthesis. Science. 1975;189:347–358.
31. Palay SL, Sotelo C, Peters A, Orkand PM. The axon hillock and the initial segment. Journal of Cell Biology. 1968;38:193–201.
32. Peters A, Palay SL, Webster HD. The fine structure of the nervous system: Neurons and their supporting cells 3rd ed. New York, NY: Oxford University Press; 1991.
33. Pigino G, Song Y, Kirkpatrick LL, Brady ST. The cytoskeleton of neurons and glia. In: Brady ST, Siegel G, Albers RW, Price D, eds. Basic neurochemistry: Principles of molecular, cellular and medical neurobiology. 2011;101–118. Boston, MA: Elsevier.
34. Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450:663–669.
35. Roll-Mecak A, McNally FJ. Microtubule-severing enzymes. Current Opinion in Cell Biology. 2010;22:96–103.
36. Roy S, Winton MJ, Black MM, Trojanowski JQ, Lee VM. Rapid and intermittent cotransport of slow component-b proteins. Journal of Neuroscience. 2007;27:3131–3138.
37. Sherman DL, Brophy PJ. Mechanisms of axon ensheathment and myelin growth. Nature Reviews Neuroscience. 2005;6:683–690.
38. Sudhof TC. The synaptic vesicle cycle. Annual Review of Neuroscience. 2004;27:509–547.
39. Susuki K, Rasband MN. Molecular mechanisms of node of Ranvier formation. Current Opinion in Cell Biology. 2008;20:616–623.
40. Tanaka J, Sobue K. Localization and characterization of gelsolin in nervous tissues: Gelsolin is specifically enriched in myelin-forming cells. Journal of Neuroscience. 1994;14:1038–1052.
41. Tsukita S, Ishikawa H. The movement of membranous organelles in axons Electron microscopic identification of anterogradely and retrogradely transported organelles. Journal of Cell Biology. 1980;84:513–530.
42. Vouyiouklis DA, Brophy PJ. Microtubule-associated proteins in developing oligodendrocytes: transient expression of a MAP2c isoform in oligodendrocyte precursors. Journal of Neuroscience Research. 1995;42:803–817.
43. Walter P, Johnson AE. Signal sequence recognition and protein targeting to the endoplasmic reticulum membrane. Annual Review of Cell Biology. 1994;10:87–119.
44. Xu T, Xu P. Searching for molecular players differentially involved in neurotransmitter and neuropeptide release. Neurochemical Research. 2008;33:1915–1919.
45. Zerial M, McBride H. Rab proteins as membrane organizers. Nature Reviews Molecular Cell Biology. 2001;2:107–117.