
MUTANT MODELS OF Nrg1 AND ErbB4
Abnormalities of Brain Structures, Functions, and Behaviors Relevant to Schizophrenia
KEY CONCEPTS
Structure and Function of Neuregulin 1
The neuregulin proteins (NRGs) represent a large family of growth factors encoded by four individual genes (NRG1-4). Of these four genes, NRG1 is the best characterized. NRG1 is located on the short arm of chromosome 8, where Stefansson et al. (2002) identified NRG1 as a putative schizophrenia susceptibility gene. In the central nervous system (CNS), NRG1 plays critical roles in neuronal migration, synapse formation, and plasticity; the last of these is based on NRG1 regulation of neurotransmitter receptor expression and function (Falls, 2003; Corfas et al., 2004; Harrison & Law, 2006; Mei & Xiong, 2008). Alterations in regulation of one or more of these key processes by NRG1 are thought to contribute to the pathophysiology of schizophrenia.
The NRG1 proteins are classified into six subtypes (Types I–VI) based on type-specific sequences and the structural domains located in the NH2-terminal region (figure 15.1A) (Falls, 2003; Mei & Xiong, 2008). Within the NH2-terminal region, the immunoglobulin (Ig)-like domain is present in Types I, II, IV, and V, whereas a hydrophobic cysteine-rich domain (CRD) is present only in Type III. Alternative splicing in the juxtamembrane stalk region and the intracellular domain (ICD) generates additional variants of each major subtype. Differential levels and patterns of the expression of the three major types, I–III, occur in various tissues including the brain (Meyer & Birchmeier, 1995). In addition, mice with targeted mutations disrupting certain classes of NRG1 isoforms have distinct defects in neural development (Falls, 2003). Together, these results demonstrate isoform-specific expression and function of NRG1.
Proteolytic Processing of NRG1
A majority of NRG1 isoforms are produced as single-pass transmembrane proproteins. However, Type III NRG1 pro-proteins span the membrane twice because of the presence of the hydrophobic CRD. Proteolytic processing in the extracellular, juxtamembrane region of NRG1 releases the ecotodomain containing the epidermal growth factor (EGF)-like domain, which participates in paracrine signaling by activating ERBB kinases (figure 15.1B) (Falls, 2003; Mei & Xiong, 2008). For Type III NRG1 isoforms, the cleaved ecto-domain containing the EGF-like domain remains tethered to the membrane by the CRD and is believed to signal through ERBB receptor kinases in a contact-dependent, juxtacrine manner (Wolpowitz et al., 2000; Leimeroth et al., 2002; Falls, 2003). Type III NRG1 undergoes a second, intramembranous, γ-secretase-dependent cleavage to release ICD from the membrane (Bao et al., 2003). The released ICD translocates to the nucleus and regulates gene expression (Bao et al., 2003, 2004). Thus, Type III NRG1 can signal by means of the activation of ERBB kinases and by means of the ICD.
NRG1 Signaling Through ERBB Receptor Tyrosine Kinases
NRG1 serves as one of the ligands of ERBB receptor tyrosine kinases. EGFR/ ERBB1, ERBB2, ERBB3, and ERBB4 are members of the ERBB kinase family. Binding of NRG1 to homo- or hetero-dimerized ERBB receptors by way of the EGF-like domain leads to stimulation of the kinase activity of ERBB, activation of intracellular signaling networks, and induction of cellular responses. Among the ERBB family members, EGFR and ERBB2 do not bind NRG1 but do have functional kinase domains. In contrast, ERBB3 binds to NRG1 but has an impaired kinase function. Most importantly, ERBB4 interacts with NRG1 and has a functional kinase domain. Since EGFR and ERBB2 do not bind NRG1, the catalytically active ERBB dimers that are most capable of transducing NRG1 signals are ERBB2-ERBB3, ERBB2-ERBB4, ERBB3-ERBB4, and ERBB4-ERBB4.
Figure 15.1. Illustration of different types of NRG1 and their proteolytic processing events. (A) The NRG1 proteins are classified into six major types based on distinct NH2-terminal type-specific sequences that arise from the unique usage of multiple promoters. Types I, II, IV, and V NRG1 are sometimes referred to as Ig-NRG1 because of the presence of an immunoglobulin (lg)-Iike domain, which is connected to the epidermal growth factor (EGF)-Iike domain by a spacer region (S) in certain isoforms. Type III NRG1 has a cysteine-rich domain (CRD) and is therefore sometimes referred to as CRD-NRG1. Because the CRD is hydrophobic, it acts as the second transmembrane anchor in addition to the first transmembrane domain (TMc) at the COOH-terminus. The NH2-terminal regions of Types III and VI NRG1 are connected directly to the EGF-Iike domain. The EGF-Iike domain present in all six types of NRG1 proteins interacts with the ERBB receptor tyrosine kinases. Alternative splicing of sequences within the stalk region and the intracellular domain at the COOH-terminus yields different isoforms of each type of NRG1. The asterisk marks the stop codon.
(B) A majority of NRG1 isoforms are synthesized as single-pass transmembrane pro-proteins, with the NH2-terminal region containing the EGF-Iike domain located on the extracellular side. However, Type III NRG1 spans the membrane twice and the NH2-terminal and COOH terminal regions are both located on the cytoplasmic side. Proteolytic processing (represented by the light-shaded lightning arrow) of NRG1 pro-proteins by tumor necrosis factor-α converting enzyme (TACE), β-site of amyloid precursor protein cleaving enzyme (BACE), or meltrin β releases the ecto-domain containing the EGF-Iike domain, which binds to and activates ERBB receptor tyrosine kinase. In the case of Type III NRG1, the cleaved ecto-domain is tethered to the membrane by the CRD and activates ERBB kinase in a contact-dependent manner. Moreover, Type III NRG1 undergoes a second intramembranous cleavage that is, at least in part, γ-secretase-dependent (indicated by the dark-shaded lightning arrow), and this results in the release of NRG1 intracellular domain (ICD) from the membrane (Bao et al., 2003). The released ICD translocates to the nucleus, where it regulates gene expression (Bao et al., 2003, 2004). Thus, Type III NRG1 can signal bidirectionally, through the activation of ERBB kinases and through the ICD. Although the processing of Types IV, V, and VI isoforms of NRG1 pro-proteins is less clear, it most likely resembles that of Types I and II. (Source: Authors, based on Falls [2003]; Mei & Xiong [2008].)
NRG1 and Neurotransmission
In view of the diverse set of cognitive functions and behaviors that are thought to be compromised in schizophrenia, it is not surprising that many neurotransmitter systems and neuromodulatory circuits have been implicated in its etiology. As such, it is important to consider how the dysregulation of NRG1/ERBB signaling might disrupt neurotransmitter systems affected in schizophrenia.
The schizophrenia literature has focused on alterations in four major transmitter systems and circuits as key players in the etiology and symptomatology of schizophrenia. The first consists of the meso-limbic and meso-cortical dopamine signaling systems that have been implicated because of the profound response of patients to pharmacotherapeutics that target the dopamine system. The second candidate dysregulated system involves cortical inhibitory interneurons that utilize gamma-aminobutyric acid for neurotransmission (i.e., GABAergic interneurons).The third neurotransmitter system implicated in schizophrenia is based on recent literature focusing on an important role for alterations in cortico-limbic glutamatergic synaptic transmission in schizophrenia pathology, with particular emphasis on N-methyl-D-aspartate (NMDA) receptor pathways. Finally, we discuss alterations in cholinergic signaling, which have long been noted as being associated with the pathophysiology of schizophrenia, and also the strong comorbidity of schizophrenia with nicotine abuse.
NRG1 and Dopaminergic Transmission
The literature on the neurobiology of schizophrenia strongly implicates alterations in dopamine signaling at the core of the disease. The important role of imbalances in dopamine signaling is supported by (1) findings of hyperdopaminergic signaling (increased D2 receptor activation) in subcortical areas and (2) findings consistent with hypodopaminergic signaling in cortical regions. The former is thought to underlie the “positive” symptoms of schizophrenia, whereas the latter may be responsible for the impairments in cognitive functions and behavior (Guillin, Abi-Dargham, & Laruelle, 2007). Attempts to dissect how the imbalance in dopamine signaling could lead to the manifestations of positive and negative symptoms have been avidly pursued in animal models. In fact, the selective manipulation of schizophrenia susceptibility genes reveals an important role for NRG1/ERBB signaling components in dopamine transmission. Several studies implicate NRG1/ ERBB signaling in the development of central dopaminergic neurons (Steiner et al., 1999). Infusion of exogenous NRG1 (beta peptide) induces substantial increases in striatal dopamine release (Yurek et al., 2004). Perhaps most intriguing are recent probes into the effects of NRG1/ERBB signaling in the regulation of long-term potentiation (LTP) in the hippocampus—an established model for dissecting the synaptic underpinnings of memory. Addition of soluble NRG1 regulates LTP by increasing dopamine release, leading to activation of dopamine D4 receptors (Kwon et al., 2008). The shift in dopamine signaling, in turn, regulates glutamatergic transmission, leading to an inhibition or reversal of glutamatergic LTP in the hippocampus (Neddens et al., 2009). Another study manipulated NRG1/ERBB signaling by expressing a dominant negative ERBB4 receptor and found altered dopaminergic tone, as evidenced by increased levels of dopamine receptors (D1R, D2R) and DAT transporter (Roy et al., 2007).
NRG1 and GABAergic Synapses
Postmortem analyses of brains from schizophrenia patients have demonstrated reduction in the synapses between GABAergic, fast-spiking chandelier interneurons and layer 3 pyramidal neurons (Lewis, 2000; Volk & Lewis, 2002; Hashimoto et al., 2003, 2007; Lewis, Volk, & Hashimoto, 2004; Lewis, Hashimoto, & Volk, 2005). Chandelier interneurons are believed to impose synchrony on excitatory output from the cortex, and deficits in chandelier neuronal function are likely to contribute to impairments in higher-level cortical processing or the integration of thalamo-cortical signaling, or both.
Several studies have implicated NRG1 signaling in the development, maintenance, or plasticity of GABAergic cortical interneurons. During early embryonic development, the tangential migration of interneurons into the neocortex requires NRG1/ERBB4 signaling (Flames et al., 2004; Flames & Marin, 2005; Lopez-Bendito et al., 2006). Adult mice that are heterozygous for disruption of Type III Nrg1 have a decreased number of GABAergic interneurons that stain strongly for parvalbumin (PV) within the prelimbic and infralimbic cortex (Johnson, 2007). In vitro, NRG1 signaling regulates the expression of GABA-A receptors (Rieff et al., 1999; Okada & Corfas, 2004). Cortical GABAergic axon terminals contain ERBB4, and activation of presynaptic ERBB4 modulates the probability of GABA release (Mei & Xiong, 2008; Woo et al., 2007). In addition, in PV-ErbB4−/− mice a selective loss of ERBB4 in PV-positive interneurons prevents NRG1 from stimulating GABA release (Wen et al., 2010). Thus, NRG1/ERBB4 signaling modulates the strength of GABAergic transmission. Given that alterations in GABA signaling appear to depend on the type, timing, and method of NRG1 manipulation, animal models present an important opportunity to sort out the differential role of NRG1/ERBB signaling during development and in adulthood.
NRG1 and Glutamatergic Plasticity
Functional imaging, pharmacological, and postmortem studies all point to aberrant cortical glutamatergic transmission in schizophrenia (Harrison et al., 2003; Laruelle et al., 2003; Coyle, 2004; Coyle & Tsai, 2004; Harrison & Weinberger, 2005) NRG1/ERBB signaling plays an important role in the development and plasticity of thalamo-cortical and cortico-cortical glutamatergic circuits. First, NRG1/ERBB signaling appears to be a key player in the radial migration of differentiating excitatory pyramidal neurons (Anton et al., 1997) as well as in the targeting of glutamatergic thalamo-cortical projections (Lopez-Bendito et al., 2006). In the adult, NRG1/ERBB signaling modulates activity-dependent synaptic plasticity by regulating NMDA and AMPA receptor phosphorylation and trafficking (Ozaki et al., 1997; Stefansson et al., 2002; Gu et al., 2005; Kwon et al., 2005; Hahn et al., 2006; Bjarnadottir et al., 2007; Li et al., 2007). Although the results are complex and to some extent contradictory, these studies point to the need for a fine balance of NRG1/ERBB signaling in which both deficient and excessive signaling interfere with synaptic plasticity (Gu et al., 2005; Kwon et al., 2005;Hahn et al., 2006; Bjarnadottir et al., 2007; Li et al., 2007; Role & Talmage, 2007). Such findings also underscore the important role of studies in animal models where different aspects of the signaling can be selectively disrupted or up-regulated and where the precise nature of the change or changes in glutamatergic synapses can be assessed directly.
NRG1 and Cholinergic Modulation of Circuits
The CHRNA7 gene, which encodes the α7 subunit of the nicotinic acetylcholine receptors, a7*nAChRs, has been linked to schizophrenia and, in particular, to endophenotypes associated with sensory gating deficits (Martin et al., 2007). Recent genomewide association studies (GWAS) that identified DNA copy number variations associated with schizophrenia converged on multiple regions of 15 q, with the common domain being the upstream regulatory domain of the CHRNA7 gene (International Schizophrenia Consortium, 2008). Postmortem studies have also demonstrated region-specific reductions in a7*nAChRs in patients with schizophrenia and further showed that these reductions were associated with risk single nucleotide polymorphisms (SNPs) in NRG1 (Leonard et al., 2002; Gault et al., 2003; Mathew et al., 2007; Stephens et al., 2009). Animal studies demonstrate that NRG1 signaling regulates the expression and presynaptic targeting of a7*nAChRs (Yang et al., 1998; Liu et al., 2001; Kawai, Zago, & Berg, 2002; Hancock et al., 2008; Talmage, 2008; Zhong et al., 2008). In view of the strong comorbidity of smoking and schizophrenia, a systematic investigation into the mechanisms by which NRG1 signaling alters cholinergic and cholinoceptive systems is important. In particular, studies linking alterations in NRG1/ERBB signaling components and the functional profile of cholinergic receptor expression in circuits affected in schizophrenia should be pursued.
NRG1 and ERBB4 as Schizophrenia Susceptibility Genes
In 2002, a group from deCode reported an association between schizophrenia and a cluster of sequence polymorphisms near the 5-prime end of NRG1 (Stefansson et al., 2002). This so-called Icelandic risk haplotype (IceHAP) was estimated to confer a population-attributable risk of 16%. NRG1 has been evaluated specifically as a schizophrenia susceptibility gene in more than 30 subsequent studies, about 60% of which showed positive association either with the IceHAP or other polymorphisms spanning essentially the entire locus. In at least two instances, risk-conferring interactions between NRG1 and ERBB4 sequence polymorphism were reported (Norton et al., 2006; Shiota et al., 2008). Altered hippocampal or prefrontal cortical expression of NRG1 isoforms and ERBB4 isoforms, or both, is associated with specific risk alleles in postmortem brains (Law et al., 2006, 2007; Nicodemus et al., 2009). The observed changes are predicted to alter signaling in the NRG1/ERBB4 system. The increase in Type I and IV NRG1 expression would increase paracrine signaling, whereas the decrease in Type III NRG1 expression would decrease juxtacrine NRG1 signaling (figure 15.1). Increased expression of the JMa/CYT-1 isoform of ERBB4 would selectively increase signaling via phosphatidylinositol 3-kinase (PI3K) (figure 15.2).
Further evidence in support of a role for altered NRG1 function in the etiology of schizophrenia comes from a series of studies demonstrating the link between specific NRG1 risk alleles and particular schizophrenia-associated endophenotypes, including enlarged lateral ventricles (Mata et al., 2009), altered hippocampal activity measured with functional magnetic resonance imaging (fMRI) (Kircher et al., 2009b), verbal fluency (Kircher et al., 2009a), P300 event-related potential (ERP) (Bramon et al., 2008), various psychosocial measures (e.g., Keri et al., 2009), and expression of CHRNA7 (Mathew et al., 2007). Whether, and how, changes in the levels and type of NRG1/ERBB4 signaling contribute to these phenotypes is not clear, but these findings raise an important set of questions that can be addressed in mouse genetic models. Progress along these avenues is discussed in detail below.
Mutant Mouse Models
Whereas ERBB2 and ERBB3 require heterodimerization for kinase activation, ERBB4 can undergo homodimerization to transduce NRG1 signals, indicating that neuronal expression of ERBB4, in particular, may be of functional significance. There is evidence that genetic interaction between variants of NRG1 and ERBB4 loci increases susceptibility to schizophrenia (Norton et al., 2006; Shiota et al., 2008). Furthermore, ERBB2 and ERBB3 are not known schizophrenia susceptibility genes (Kanazawa et al., 2007; Watanabe et al., 2007), and mice heterozygous for targeted disruption of ErbB2 or ErbB3 do not show behaviors reminiscent of schizophrenia (Gerlai, Pisacane, & Erickson, 2000). Hence, we will focus on NRG1 and ERBB4 functional alterations associated with schizophrenia by reviewing studies of the currently available mutant mouse models of Nrg1 and ErbB4.
Many mouse lines carrying mutations in the Nrg1 or ErbB4 gene have been generated. Homozygous deletions of Nrg1 or ErbB4 lead to embryonic or perinatal lethality because of heart malformations, thus limiting their usefulness to studies of early brain development. However, heterozygous mutant mice with targeted disruption of Nrg1 or ErbB4 are viable and fertile, and these animals have been used for investigating the effects of reduced NRG1 or ERBB4 expression in adult brains. Additionally, several mouse lines carrying conditional mutations of ErbB4 or a dominant negative allele of ErbB4 are available for functional characterization of NRG1/ERBB signaling. Table 15.1 lists heterozygous Nrg1 mutant mouse models that eliminate all or specific isoforms of Nrg1 as well as a transgenic mouse line over-expressing Type I Nrg1. Table 15.2 lists ErbB4 mutant mice that have been analyzed. A variety of structural and functional studies as well as behavioral assays have been performed on Nrg1 and ErbB4 mutant mice to probe the function of CNS circuits underlying schizophrenia-associated endophenotypes.
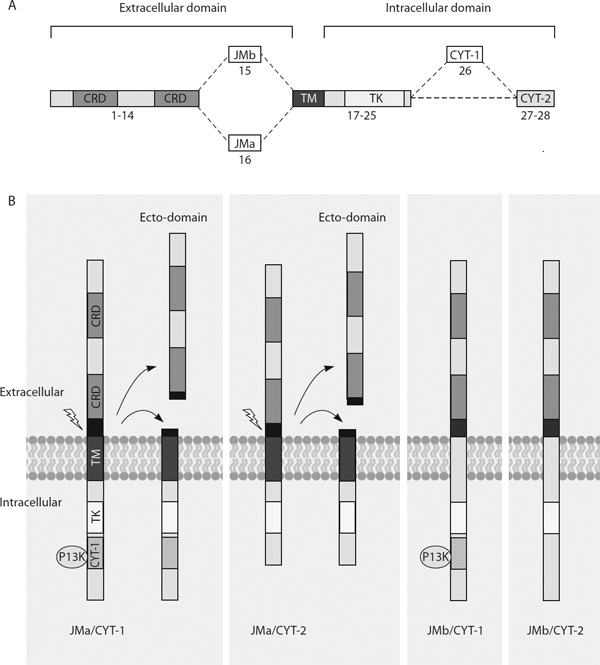
Figure 15.2. Structural organization and processing of four ERBB4 splice variants.
(A) Differential usage of exon 15 and 16 encoding the extracellular juxtamembrane sequences results in the generation of the juxtamembrane-b (JMb) or juxtamembrane-a (JMa) isoforms, respectively. The JMa and JMb isoforms can be further differentiated on the basis of the presence (cytoplasmic-1, CYT-1) or absence (cytoplasmic-2, CYT-2) of exon 26-encoded sequences within the cytoplasmic region. Therefore, four ERBB4 isoforms are JMa/CYT-1, JMa/CYT-2, JMb/CYT-1, and JMb/CYT-2. Corresponding exon numbers are shown at the bottom of the domain structures. CRD = cysteine-rich domain; TK = tyrosine kinase domain; TM = transmembrane domain.
(B) The ERBB4 isoforms interact with NRG1 through their ecto-domains. Following ligand-binding, the JMa isoform but not the JMb isoform is proteolytically processed (represented by the lightning arrow) by TACE to produce two fragments: a soluble ecto-domain that is subsequently shed, and a second fragment that contains the intracellular cytoplasmic domain. Within the intracellular domain, only the CYT-1 isoform contains the interaction motif that recruits the p85 subunit of phosphatidylinositol 3-kniase (PI3K), which subsequently activates Akt kinase. (Source: Authors, based on Falls [2003]; Mei & Xiong [2008].)
Anatomic or Structural Defects and Alterations in Synaptic Functions in Nrg1 and ErbB4 Mutant Mice
Enlargement of Lateral Ventricles
The lateral ventricles represent a brain region that circulates the cerebrospinal fluid and helps protect against physical trauma. A review of structural MRI data of schizophrenia patients has revealed enlargement of the lateral ventricles in approximately 80% of the studies (McCarley et al., 1999). Ventriculomegaly is therefore the most robust MRI finding in schizophrenia. Recently, a variant of NRG1 was associated with increased lateral ventricle volume in patients with first-episode schizophrenia (Mata et al., 2009). It is interesting that Type III-Nrg1+/− mutant mice also exhibit age-dependent, enlarged lateral ventricles (Chen et al., 2008).
Selective Loss of Parvalbumin-Positive GABAergic Interneurons in the Prefrontal Cortex
Abnormal neural synchrony due to deficits in the GABAergic neurotransmission has been implicated in schizophrenia. Consistent with this is a selective reduction of immunoreactivity in a specific subtype of inhibitory GABAergic interneuron—PV-positive interneurons—in the prefrontal cortex of patients with schizoprenia (Beasley & Reynolds, 1997; Hashimoto et al., 2003; Lewis, Hashimoto, & Volk, 2005). Similarly, there is a striking decrease in the PV-positive, but not the calbindin-positive, population of GABAergic interneurons in the infralimbic and prelimbic areas of the prefrontal cortex of adult Type III-Nrg1+/− mutant mice (Johnson, 2007). This cell- and region-specific effect of reduced levels of Type III NRG1 was further demonstrated by a lack of changes in the PV-positive GABAergic interneuron population in the dorso-peduncular prefrontal area (Johnson, 2007). In addition, young (postnatal day 20) ErbB4+/− HER4heart mutant mice (table 15.2) show a decrease in the number of GABAergic interneurons at midcortical and caudal cortical levels and in the hippocampus (Flames et al., 2004). It remains to be seen whether there will be a reduction in the number of PV-positive GABAergic interneurons in the schizophrenia-linked prefrontal cortical region in ErbB4+/− HER4heart mutant animals after the inhibitory circuitry has fully developed in adulthood.
Table 15.1 Summary of Nrg1 Mouse Lines
EGF-Nrg1+/− mice
|
EGF-Nrg1+/− mice
|
Ig-Nrg1+/− mice
|
TM-Nrg1+/− mice
|
Type III (or CRD)-Nrg1+/− mice
|
Nrg1α−/− mice
|
Nrg1Type I-tg mice
|
SOURCE: Authors.
Table 15.2 Summary of ErbB4 Mouse Lines
ErbB4+/− mice
|
ErbB4flox/− Nes-Cre mice
|
ErbB4flox/− hGFAP-Cre mice
|
DN-ErbB4 mice
|
ErbB2/B4-CNSko mice
|
ErbB4−/− HER4heart mice
|
PV-ErbB4−/− mice
|
SOURCE: Authors.
Decreased Spine Density of Pyramidal Neurons in the Subiculum of the Ventral Hippocampus
Dendritic spines are the major sites of postsynaptic contact of excitatory synapses in the cortex. Moreover, spines are believed to be the anatomic structures involved in learning and memory (Bourne & Harris, 2007). Postmortem studies of schizophrenia have revealed decreased density of dendritic spines in the subicular region of the ventral hippocampus as well as in the prefrontal cortex, suggesting impaired synaptic function in these regions (Garey et al., 1998; Glantz & Lewis, 2000; Rosoklija et al., 2000). A similar reduction in the spine density of apical dendrites of pyramidal neurons in the subiculum of the ventral hippocampus has been shown in Type III-Nrg1+/− mutant mice (Chen et al., 2008). In addition, there is a decrease in the spine density of pyramidal neurons in the CA1 and prefrontal cortical regions in ErbB2/B4-CNSko mutant mice (Barros et al., 2009). Clozapine, the atypical antipsychotic drug used to treat schizophrenia patients (Krakowski et al., 2006), increases spine density of hippocampal neurons in both wild-type mice and ErbB2/B4-CNSko mutants, thereby normalizing the spine defects in the mutant mice (Barros et al., 2009). Finally, addition of recombinant NRG1 to cultured hippocampal neurons from wild-type mice leads to an increase in spine density (Barros et al., 2009). Together, these studies demonstrate a crucial role of NRG1/ERBB signaling in spine development, and they implicate altered NRG1/ERBB function in the abnormal neuroconnectivity that may underlie schizophrenia.
Functional Alterations in the Hippocampal and Prefrontal Cortical Regions
The anatomic and structural alterations observed in schizophrenia likely translate into functional and behavioral alterations. fMRI mapping of neuronal activity has been used to study functional changes associated with schizophrenia and revealed alterations in the hippocampal CA1 subfield, orbitofrontal cortex, and dorsolateral prefrontal cortex of schizophrenia patients (Schobel et al., 2009), as well as regional deficits in activation of the hippocampus, anterior cingulate, and precuneus during working memory tasks in individuals carrying NRG1 risk alleles (Kircher et al., 2009a, 2009b). Similarly, abnormalities in the hippocampus and prefrontal cortex have also been reported in fMRI studies of Type III-Nrg1+/− mutant mice (Chen et al., 2008).
NRG1 and Myelination
Postmortem microarray studies by Hakak et al. (2001) revealed decreased expression of the NRG1 receptor, ERBB3, as well as other genes predominantly expressed in mature oligodendrocytes in tissue from patients with schizophrenia. These findings raised the possibility that reductions in the number or function of oligodendrocytes might contribute to the disease. Studies in Nrg1 mutant mice and in the in vitro culture systems have demonstrated that NRG1 signaling is essential for proper myelination of peripheral nerves by Schwann cells (Michailov et al., 2004; Taveggia et al., 2005; Nave & Salzer, 2006). However, a role for NRG1 signaling in CNS myelination by oligodendrocytes is less clear. Minor and track-specific deficits have been seen in Type III-Nrg1+/− mutant mice, and inhibition of ERBB signaling in oligodendrocytes by the expression of a dominant negative ErbB4 transgene reduces the CNS myelination (Roy et al., 2007; Taveggia et al., 2008). In contrast, when Brinkmann et al. (2008) totally eliminated ERBB or NRG1 expression in the CNS using conditional gene deletion, they failed to see any effect on CNS myelination. In sum, these studies make it unlikely that NRG1 is a major regulator of CNS myelination and leave open the question of the role of disrupted oligodendroctye function in the etiology of schizophrenia.
Behavioral Abnormalities of Nrg1 and ErbB4 Mutant Mice
Behavioral Studies of Hypomorphic Nrg1 Mutant Animals
HYPERLOCOMOTOR ACTIVITY. Hyperactivity and stereotypic behaviors are often measured in animal models of schizophrenia-associated phenotypes. Studies on a variety of Nrg1 mutant animals revealed hyperlocomotor activity. TM-Nrg1+/− and EGF-Nrg1+/− mutant mice display increased locomotion in a novel open-field environment (Gerlai, Pisacane, & Erickson, 2000; Stefansson et al., 2002; O’Tuathaigh et al., 2006, 2007; Boucher et al., 2007; Karl et al., 2007; Duffy et al., 2008; van den Buuse et al., 2009). Moreover, as opposed to the results in wild-type animals, hyperlocomotor activity observed in TM-Nrg1+/− mutant mice is not affected by the psychotropic drug amphetamine (van den Buuse et al., 2009). In line with a hyperactive phenotype, TM-Nrg1+− and EGF-Nrg1+− mutant mice show an increased number of arm-compartment entries in various behavioral paradigms (Boucher et al., 2007; O’Tuathaigh et al., 2007; Duffy et al., 2008; Ehrlichman et al., 2009; Moy et al., 2009) and EGF-Nrg1+/− mutant animals demonstrate better motor performance (i.e., balance and coordination) on the accelerating rotarod than their wild-type counterparts in the rotarod paradigm (Gerlai, Pisacane, & Erickson, 2000). Clozapine normalizes hyperlocomotor activity in TM-Nrg1+/− mutant mice (Stefansson et al., 2002), providing indirect evidence that the hyperactivity observed in Nrg1+/− mutant mice is related to altered dopaminergic signaling.
Studies of Ig-Nrg1+/− mutant animals, however, did not reveal increased locomotor activity in the open field test or running-wheel activity assay (Rimer et al., 2005). Type III Nrg1+/− mutant mice also show normal locomotor activity in the open field or home cage environment (Chen et al., 2008; Luca, E., Role, L. W., & Talmage, D. A., unpublished results). In addition, both TM-Nrg1+/− and Type III-Nrg1+/− mutant mice show normal hyperlocomotor responses (i.e., equivalent to wild-type littermate controls) to amphetamine challenge (van den Buuse et al., 2009; Chen, Y. J., Talmage, D. A. & Role, L. W., unpublished results). It is likely that the discrepancy between the locomotor results from mutants heterozygous for TM domain-, EGF domain-, Ig domain-, or Type III-specific Nrg1 mutations reflects disruption of a greater number of Nrg1 isoforms by the TM and EGF domain deletions.
It is interesting that EGF-Nrg1+/− mutant animals, which are hyperactive in an open field environment, show normal locomotor activity in a home cage environment (Gerlai, Pisacane, & Erickson, 2000; Duffy et al., 2008; Ehrlichman et al., 2009). This raises the possibility that the hyperlocomotion phenotype reflects altered responses to stressful situations such as exposure to a novel open field.
ABNORMAL EXPLORATORY BEHAVIOR AND IMPAIRED HABITUATION. Impaired processing of novel environmental stimuli and disruption of habituation are considered relevant to the cognitive dysfunction observed in schizophrenia (Grossberg, 2000). It is interesting that male TM-Nrg1+/− mutant mice exhibit gender-specific abnormalities in certain aspects of their exploratory behaviors, such as higher levels of sifting (i.e., sifting movements of the front paws through cage bedding material) and reduced grooming in an open field-type environment (O’Tuathaigh et al., 2006). During the subsequent habituation phase of exploration, male TM-Nrg1+/− mutants show increased grooming (O’Tuathaigh et al., 2006). Moreover, adult (four to seven months of age) TM-Nrg1+/− mutant animals show an age-specific increase in the frequency of exploration-related behaviors, such as head dipping and vertical activity in an open field activity test, a light-dark emergence test, and a hole board task (Boucher et al., 2007; Karl et al., 2007). In sum, this increase in the frequency of exploratory behaviors is in line with the hyperlocomotor activity phenotype of TM-Nrg1+/− mutant mice.
IMPAIRED SOCIAL INTERACTION. Impaired social interaction is a key feature of the symptoms of schizophrenia (Freedman, 2003). Increased aggression is observed in some schizophrenia patients (Krakowski et al., 2006). Several studies on Nrg1-deficient mouse models have revealed alterations in social approach and increased aggression. TM-Nrg1+/− and EGF-Nrg1+/− mutant mice exhibit a selective disruption in their behavioral response to social novelty by showing no preference to the novel conspecific in a resident-intruder paradigm (O’Tuathaigh et al., 2007, 2008; Ehrlichman et al., 2009; Moy et al., 2009). TM-Nrg1+/− mutant animals also display more nonsocial, aggressive exploratory behaviors toward the conspecific (O’Tuathaigh et al., 2007, 2008).
PREPULSE INHIBITION, LATENT INHIBITION, AND MISMATCH NEGATIVITY DEFICITS. Prepulse inhibition (PPI) and latent inhibition (LI) paradigms are used for assaying information processing deficits related to cognitive dysfunction in schizophrenia. PPI is a behavioral paradigm in which there is a decrease in the magnitude of startle response to an intense stimulus when preceded by a weaker prestimulus. PPI is commonly used as a measure of sensory-motor gating, which is a preattentional mechanism critical to selective processing of external stimuli (Swerdlow, Braff, & Geyer, 2000). Patients with schizophrenia show deficits in PPI (Powell & Geyer, 2002; Weike et al., 2000). Analyses of Type III-Nrg1+/− and TM-Nrg1+/− mutant mice reveal PPI deficits (Stefansson et al., 2002; Chen et al., 2008). Moreover, chronic nicotine administration in Type III-Nrg11+I mutant animals ameliorates PPI deficits (Chen et al., 2008), a result reminiscent of the nicotine effect on the functional impairments of schizophrenia (George et al., 2006; Kumari & Postma, 2005; Postma et al., 2006). Although one report of PPI in TM-Nrg1+/− mutant animals showed a deficit (Stefansson et al., 2002), two other studies demonstrated normal PPI (Boucher et al., 2007; van den Buuse et al., 2009). The observed difference in PPI phenotype could be due to variations in the genetic background of the mouse lines. Studies of EGF-Nrg1+/− mutants also did not reveal a baseline PPI deficit (Duffy et al., 2008; Ehrlichman et al., 2009). However, EGF-Nrg1+/− mutant mice exhibit a PPI deficit following the administration of the NMDA receptor antagonist, MK801 (Duffy et al., 2008). Treatment of TM-Nrg1+/− mutant animals with the 5-HT1A receptor agonist, 8-OH-DPAT, similarly resulted in the disruption of PPI, along with a marked reduction of startle amplitude (van den Buuse et al., 2009). Together, these results imply that NRG1 has a role in sensorimotor gating involving glutamatergic, cholinergic, and serotonergic neurotransmission, which have been shown to contribute to the etiology and pathophysiology of schizophrenia.
LI measures an animal’s ability to ignore irrelevant stimuli. In one form of the LI test, prior exposure to a tone makes the tone less likely to reduce ambulatory activity in a test chamber. The reduction in LI observed in schizophrenia is thought to reflect a selective attention deficit (Lubow, 2005). Ig-Nrg1+/− mutant animals display more greatly impaired LI than littermate controls by showing a greater decrease in ambulatory distance traveled in response to the acoustic conditioned stimulus (Rimer et al., 2005).
Mismatch negativity (MMN) is a behavioral paradigm that assesses the mismatch response in electroencephalography (EEG) to an odd stimulus within a sequence of otherwise regular stimuli. Auditory MMN can occur in response to deviance in sound pitch, intensity, or duration and has been shown to be impaired in patients with schizophrenia (Shelley et al., 1991). Auditory MMN is disrupted in EGF-Nrg1+/− mutant mice, indicating abnormal auditory sensory processing in these mutants (Ehrlichman et al., 2009).
DEFICITS IN LEARNING AND WORKING MEMORY. Patients with schizophrenia show a broad range of cognitive dysfunction, including deficits in learning and working memory (Nuechterlein et al., 2004). To assess short-term or working memory, Type III-Nrg1+/− mutant mice were tested in delayed, alternating choice T-maze tasks. In these tasks, using alternating trials, animals learn to find the arm that is baited. Type III-Nrg1+/− mutant mice had been shown to perform this task with less accuracy than control animals, thereby demonstrating cognitive dysfunction (Chen et al., 2008).
The Barnes maze task is another behavioral paradigm for assessing working memory in which performance is dependent on the memory of the location of an escape hole. During initial learning sessions of this test, male TM-Nrg1+/− mutant mice spend more time finding and entering the escape hole, and then commit a greater number of errors, indicating impaired spatial learning or memory (O’Tuathaigh et al., 2007).
Contextual fear conditioning involves placing an animal in a novel test environment to deliver an aversive stimulus such as a foot shock during the training session and then returning it to the home cage. During the testing period, on returning to the same test environment, the animal shows a freezing response to fear if it had learned to associate the test environment with the aversive stimulus. When tested in the contextual fear paradigm, EGF-Nrg1+/− mutant mice display less freezing than the controls, indicating impaired memory function (Ehrlichman et al., 2009).
In sum, NrgI mutant mice exhibit performance abnormalities on tasks designed to probe phenotypes observed in patients with schizophrenia. Although some behaviors are altered in a majority of Nrg1 mutants studied, others are only found in animals with disruption of certain Nrg1 isoforms. In general, Nrg1 mutant animals display hyperactivity in a novel open field, have abnormal exploratory behavior and impaired habituation, show altered information processing and sensorimotor gating, and have impaired social interactions and disrupted working memory.
Behavioral Studies of Transgenic Mice Over-Expressing Type I NRG1
Results from postmortem studies revealing elevated levels of Type I NRG1 transcripts in the hippocampus and prefrontal cortex of patients with schizophrenia (Hashimoto et al., 2004; Law et al., 2006) have stimulated the generation of transgenic mice over-expressing Type I Nrg1 (i.e., Nrg1Type I-tg). The Nrg1Type I-tg animals show robust over-expression of Type I NRG1 in the brain, particularly in the hippocampus and cortex, but not in the striatum or cerebellum (Deakin et al., 2009). These animals were also studied in behavioral assays similar to paradigms used for Nrg1 hypomorphs. Female Nrg1Type I-tg mice are slightly less active and show mild increases in some anxiety measures (Deakin et al., 2009). Nrg1Type I-tg mice have a movement-related tremor (Michailov et al., 2004; Brinkmann et al., 2008; Deakin et al., 2009) and show poor performance on the accelerating rotarod (Deakin et al., 2009). In addition, these animals exhibit PPI deficits indicative of sensorimotor gating deficits, but with an increased baseline acoustic startle response (Deakin et al., 2009). Thus, behavioral changes observed in Nrg1Type I-tg mice are not entirely reciprocal with those detected in Nrg1 hypomorphs. Nonetheless, these results strongly support the idea that an optimal level of NRG1/ ERBB signaling is essential for normal behavior and synaptic plasticity (Role & Talmage, 2007).
Behavioral Studies of ErbB4 Mutant Animals
In comparison to analyses on Nrg1 mutant animals, there are relatively fewer studies on the schizophrenia-related behavioral phenotypes of ErbB4 mutant mice.
LOCOMOTOR ACTIVITY AND MOTOR PERFORMANCE. Both ErbB4+/− mutant mice and conditional mutants of ErbB4 have been analyzed for the locomotor phenotype. When evaluated in an open field test, ErbB4+/− mutant animals exhibit a hyperlocomotor phenotype that is less robust than that observed in TM-Nrg1+/− mutant mice (Stefansson et al., 2002). Mice with a conditional deletion of ErbB4 in astrocytes (i.e., ErbB4flox/− hGFAP-Cre mutant mice) as well as mice with a selective loss of ErbB4 in PV-positive interneurons (i.e. PV-ErbB4−/− mutant mice) also show enhanced locomotor activity (Moy et al., 2009; Wen et al., 2010). However, double mutant mice with conditional deletions of ErbB2 and ErbB4 alleles in astrocytes (i.e., ErbB2/B4-CNSko mutant mice) display normal locomotor behavior (Barros et al., 2009). Although mice with a nervous-system-specific conditional mutation of ErbB4 (i.e, ErbB4flox/- Nes-Cre mutant mice) show hyperactivity in the open field before weaning (postnatal day 21), these animals become hypoactive at an older age (postnatal day 66) (Golub, Germann, & Lloyd, 2004). DN-ErbB4 mutant mice also display hypoactivity when exploring an open field (Roy et al., 2007). It is not clear why locomotor activity phenotypes are not consistent in mice with disrupted ERBB4 signaling, but the age of animals tested is an important variable. In the open field test, the locomotor activity of three- to four-month-old wild-type male mice is almost twice as high as that of four- to six-month-old wild-type male animals (Karl et al., 2007). In studies demonstrating a normal or hypoactive phenotype, some or all test animals in cohorts were less than four months of age (Golub, Germann, & Lloyd, 2004; Roy et al., 2007; Barros et al., 2009). Therefore, it is likely that the inclusion of younger animals with higher locomotor activity may raise the baseline activity levels used for comparison.
The accelerating rotarod paradigm measures motor performance (i.e., balance and coordination). EGF-Nrg1+/− mutant animals show enhanced performance on the rotarod (Gerlai et al., 2000), whereas Erbb4flox/− hGFAP-Cre and Erbb4flox/− Nes-Cre mutant mice display normal performance in this test (Thuret et al., 2004; Moy et al., 2009) indicating normal sensorimotor coordination.
ABNORMAL SOCIAL INTERACTIONS. A variety of ErbB4 mutant mice have been analyzed in social interaction assays similar to those used for Nrg1+/− mutants. Whereas TM-Nrg1+/− and EGF-Nrg1+/− mice exhibit a selective disruption in their response to social novelty by showing no preference for the novel stranger (O’Tuathaigh et al., 2007, 2008), Erbb4flox/− hGFAP-Cre mice show normal responses in the resident-intruder paradigm (Moy et al., 2009). The ErbB1/B4-CNSko mice, however, display increased aggression—including biting, kicking, and wrestling—toward novel conspecifics similar to the reported behavior of TM-Nrg1+/− mutant animals in a resident-intruder paradigm (O’Tuathaigh et al., 2007, 2008; Barros et al., 2009). The aggressive behavior of ErbB2/B4-CNSko mutant mice is normalized by clozapine treatment (Barros et al., 2009). It is interesting that DN-ErbB4 mutant mice exhibit social dysfunction by showing increased latency in investigating a novel intruder and significantly less dominance-related mounting behavior (Roy et al., 2007). The observed social dysfunctions in DN-ErbB4 mutants are reminiscent of the negative symptoms of social withdrawal seen in patients with chronic schizophrenia (Blanchard & Cohen, 2006).
PREPULSE INHIBITION DEFICITS. PV-ErbB4−/− mutant mice and male ErbB2/B4-CNSko mice show schizophrenia-like PPI deficits that have been previously found in Type III-Nrg1+/− and TM-Nrg1+/− mutant animals (Stefansson et al., 2002; Chen et al., 2008; Barros et al., 2009; Wen et al., 2010). The hyperactive ErbB4+/− mutant animals also display minor PPI deficits (Stefansson et al., 2002). The PPI deficits of male ErbBi/B4-CNSko mice are alleviated by clozapine treatment (Barros et al., 2009). In addition, the disrupted PPI in PV-ErbB4−/− mice is ameliorated by diazepam, a GABA enhancer, further indicating that alterations in ERBB4 regulation of GABAergic neurotransmission may underlie schizophrenia-associated PPI deficits (Wen et al., 2010).
DEFICITS IN LEARNING AND MEMORY. Both TM-Nrg1+/− and Type III-Nrg1+/− mutant mice exhibit impaired short-term or working memory (O’Tuathaigh et al., 2007; Chen et al., 2008). Male Erbb4flox/− mice show similar deficits in learning and memory in the Morris water navigation task. In this paradigm, the animal is released into a pool of water and learns to escape from it by using visual cues placed around the pool to locate a platform that is submerged in the water. The measure of learning is the latency in finding the hidden platform. Male ErbB4flox/− mutant animals display longer latencies; this indicates poor spatial learning and memory performance (Golub, Germann, & Lloyd, 2004). PV-ErbB4−/− mutant mice also were evaluated for deficits in working memory based on their performance in the automated radial eight-arm maze task. During the training phase, food-restricted mice were trained to retrieve food pellets from the end of each arm. Mice were allowed to freely access eight arms baited with food pellets during the test phase and were analyzed for the number of wrong entries (i.e., repeated entries to an already visited arm or omission of an arm) and the number of correct entries within the first entries. Compared to controls, PV-ErbB4−/− mutant mice show increased wrong entries and decreased correct entries, thereby demonstrating impaired working memory (Wen et al., 2010).
Collectively, studies of behavioral phenotypes of ErbB4 mutant mice have revealed altered locomotor activity, abnormal social interactions, sensorimotor gating deficits, and impaired working memory. The abnormal phenotypes present in ErbB4 mutants are shared by various Nrg1 mutants, and these results reinforce the idea that NRG1/ERBB4 interactions play a prominent role in the development or function, or both, of neural circuits underlying behaviors altered in schizophrenia. The conditional ErbB4 mutant mouse lines with genetic disruptions of ErbB4 in neurons, oligodendrocytes, astrocytes, or PV-positive interneurons will undoubtedly prove very useful in studying schizophrenia-associated alterations that may potentially be caused by disrupted functions of specific cell populations.
Conclusion
In summary, the anatomic, structural, and functional alterations found in Nrg1 and ErbB4 mutant mice most likely contribute to schizophrenia-related behavioral abnormalities in these animals. Thus, genetic susceptibility involving NRG1/ ERBB signaling contributes to an array of circuit and behavioral changes that are strikingly reminiscent of schizophrenia phenotypes. Together with results from linkage analyses and association studies, these findings provide strong evidence for altered NRG1 and ERBB function in schizophrenia. Furthermore, transgenic mice that model alterations in schizophrenia at the levels of anatomy, structure, synaptic function, and behavior should aid significantly the understanding of the etiology and pathophysiology of this disabling disease, thereby enhancing the development of novel treatment therapeutics.
KEY AREAS FOR FUTURE RESEARCH
Selected Readings
Bao, J., Wolpowitz, D., Role, L. W., & Talmage, D. A. (2003). Back signaling by the Nrg-1 intracellular domain. Journal of Cell Biology 161(6): 1133–1141.
Chen, Y.-J. J., Johnson, M. A., Lieberman, M. D., Goodchild, R. E., Schobel, S., Lewandowski, N., Rosoklija, G., Liu, R.-C., Gingrich, J. A., Small, S., et al. (2008). Type III neuregulin-1 is required for normal sensorimotor gating, memory-related behaviors, and corticostriatal circuit components. Journal of Neuroscience 28(27): 6872–6883.
Falls, D. L. (2003). Neuregulins: Functions, forms, and signaling strategies. Experimental Cell Research 284(1): 14–30.
Mei, L. & Xiong, W. C. (2008). Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nature Reviews Neuroscience 9(6): 437–452.
Role, L. W. & Talmage, D. A. (2007). Neurobiology: New order for thought disorders. Nature 448(7151) : 263–265.
Stefansson, H., Sigurdsson, E., Steinthorsdottir, V., Bjornsdottir, S., Sigmundsson, T., Ghosh, S., Brynjolfsson, J., Gunnarsdottir, S., Ivarsson, O., Chou, T. T., et al. (2002). Neuregulin 1 and susceptibility to schizophrenia. American Journal of Human Genetics 71(4): 877–892.
References
Anton, E. S., Ghashghaei, H. T., Weber, J. L., McCann, C., Fischer, T. M., Cheung, I. D., Gassmann, M., Messing, A., Klein, R., Schwab, M. H., et al. (2004). Receptor tyrosine kinase ErbB4 modulates neuroblast migration and placement in the adult forebrain. Nature Neuroscience 7(12): 1319–1328.
Anton, E. S., Marchionni, M. A., Lee, K. F., & Rakic, P. (1997). Role of GGF/neuregulin signaling in interactions between migrating neurons and radial glia in the developing cerebral cortex. Development 124(18): 3501–3510.
Bao, J., Lin, H., Ouyang, Y., Lei, D., Osman, A., Kim, T. W., Mei, L., Dai, P., Ohlemiller, K. K., & Ambron, R. T. (2004). Activity-dependent transcription regulation of PSD-95 by neuregulin-1 and Eos. Nature Neuroscience 7(11): 1250–1258.
Bao, J., Wolpowitz, D., Role, L. W., & Talmage, D. A. (2003). Back signaling by the Nrg-1 intracellular domain. Journal of Cell Biology 161(6): 1133–1141.
Barros, C. S., Calabrese, B., Chamero, P., Roberts, A. J., Korzus, E., Lloyd, K., Stowers, L., Mayford, M., Halpain, S., & Muller, U. (2009). Impaired maturation of dendritic spines without disorganization of cortical cell layers in mice lacking NRG1/ErbB signaling in the central nervous system. Proceedings of the National Academy of Sciences U.S.A. 106(11): 4507–4512.
Beasley, C. L. & Reynolds, G. P. (1997). Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics. Schizophrenia Research 14(3): 349–355.
Bjarnadottir, M., Misner, D. L., Haverfield-Gross, S., Bruun, S., Helgason, V. G., Stefansson, H., Sigmundsson, A., Firth, D. R., Nielsen, B., Steffansdottir, R., et al. (2007). Neuregulin1 (NRG1) signaling through Fyn modulates NMDA receptor phosphorylation: Differential synaptic function in NRG1+/-knock-outs compared with wild-type mice. Journal of Neuroscience 17(17): 4519–4529.
Blanchard, J. J. & Cohen, A. S. (2006). The structure of negative symptoms within schizophrenia: Implications for assessment. Schizophrenia Bulletin 31(2): 238–245.
Boucher, A. A., Arnold, J. C., Duffy, L., Schofield, P. R., Micheau, J., & Karl, T. (2007). Heterozygous neuregulin 1 mice are more sensitive to the behavioural effects of Delta(9)-tetrahydrocannabinol. Psychopharmacology (Berl) 192(3): 325–336.
Bourne, J. & Harris, K. M. (2007). Do thin spines learn to be mushroom spines that remember? Current Opinion in Neurobiology 17(3): 381–386.
Bramon, E., Dempster, E., Frangou, S., Shaikh, M., Walshe, M., Filbey, F. M., McDonald, C., Sham, P., Collier, D. A., & Murray, R. (2008). Neuregulin-1 and the P300 waveform—a preliminary association study using a psychosis endophenotype. Schizophrenia Research 103(1–3): 178–185.
Brinkmann, B. G., Agarwal, A., Sereda, M. W., Garratt, A. N., Muller, T., Wende, H., Stassart, R. M., Nawaz, S., Humml, C., Velanac, V., et al. (2008). Neuregulin-1/ErbB signaling serves distinct functions in myelination of the peripheral and central nervous system. Neuron 59(4): 581–595.
Chen, S., Velardez, M. O., Warot, X., Yu, Z. X., Miller, S. J., Cros, D., & Corfas, G. (2006). Neuregulin 1-erbB signaling is necessary for normal myelination and sensory function. Journal of Neuroscience 16(12): 3079–3086.
Chen, Y. J., Johnson, M. A., Lieberman, M. D., Goodchild, R. E., Schobel, S., Lewandowski, N., Rosoklija, G., Liu, R. C., Gingrich, J. A., Small, S., et al. (2008). Type III neuregulin-1 is required for normal sensorimotor gating, memory-related behaviors, and corticostriatal circuit components. Journal of Neuroscience 28(27): 6872–6883.
Corfas, G., Roy, K., & Buxbaum, J. D. (2004). Neuregulin 1-erbB signaling and the molecular/cellular basis of schizophrenia. Nature Neuroscience 7(6): 575–580.
Coyle, J. T. (2004). The GABA-glutamate connection in schizophrenia: Which is the proximate cause? Biochemical Pharmacology 68(8): 1507–1514.
Coyle, J. T. & Tsai, G. (2004). NMDA receptor function, neuroplasticity, and the pathophysiology of schizophrenia. International Review of Neurobiology 59: 491–515.
Deakin, I. H., Law, A. J., Oliver, P. L., Schwab, M. H., Nave, K. A., Harrison, P. J., & Bannerman, D. M. (2009). Behavioural characterization of neuregulin 1 type I overexpressing transgenic mice. Neuroreport 20(17): 1523–1528.
Duffy, L., Cappas, E., Scimone, A., Schofield, P. R., & Karl, T. (2008). Behavioral profile of a heterozygous mutant mouse model for EGF-like domain neuregulin 1. Behavioral Neuroscience 122(4): 748–759.
Ehrlichman, R. S., Luminais, S. N., White, S. L., Rudnick, N. D., Ma, N., Dow, H. C., Kreibich, A. S., Abel, T., Brodkin, E. S., Hahn, C. G., et al. (2009). Neuregulin 1 transgenic mice display reduced mismatch negativity, contextual fear conditioning and social interactions. Brain Research 1294:116–127.
Erickson, S. L., O’Shea, K. S., Ghaboosi, N., Loverro, L., Frantz, G., Bauer, M., Lu, L. H., & Moore, M. W. (1997). ErbB3 is required for normal cerebellar and cardiac development: A comparison with ErbB2-and heregulin-deficient mice. Development 124(24): 4999–5011.
Falls, D. L. (2003). Neuregulins: Functions, forms, and signaling strategies. Experimental Cell Research 284(1): 14–30.
Flames, N., Long, J. E., Garratt, A. N., Fischer, T. M., Gassmann, M., Birchmeier, C., Lai, C., Rubenstein, J. L., & Marin, O. (2004). Short- and long-range attraction of cortical GABAergic interneurons by neuregulin-1. Neuron 44(2): 251–261.
Flames, N. & Marin, O. (2005). Developmental mechanisms underlying the generation of cortical interneuron diversity. Neuron 46(3): 377–381.
Freedman, R. (2003). Schizophrenia. New England Journal of Medicine 349(18): 1738–1749.
Garcia-Rivello, H., Taranda, J., Said, M., Cabeza-Meckert, P., Vila-Petroff, M., Scaglione, J., Ghio, S., Chen, J., Lai, C., Laguens, R. P., et al. (2005). Dilated cardiomyopathy in Erb-b4-deficient ventricular muscle. American Journal of Physiology, Heart and Circulatory Physiology 289(3): H1153–H1160.
Garey, L. J., Ong, W. Y., Patel, T. S., Kanani, M., Davis, A., Mortimer, A. M., Barnes, T. R., & Hirsch, S. R. (1998). Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. Journal of Neurology, Neurosurgery, and Psychiatry 65(4):446–453.
Gassmann, M., Casagranda, F., Orioli, D., Simon, H., Lai, C., Klein, R., & Lemke, G. (1995). Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 378(6555): 390–394.
Gault, J., Hopkins, J., Berger, R., Drebing, C., Logel, J., Walton, C., Short, M., Vianzon, R., Olincy, A., Ross, R. G., et al. (2003). Comparison of polymorphisms in the alpha7 nicotinic receptor gene and its partial duplication in schizophrenic and control subjects. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 123(1): 39–49.
George, T. P., Termine, A., Sacco, K. A., Allen, T. M., Reutenauer, E., Vessicchio, J. C., & Duncan, E. J. (2006). A preliminary study of the effects of cigarette smoking on prepulse inhibition in schizophrenia: Involvement of nicotinic receptor mechanisms. Schizophrenia Research 87(1–3): 307–315.
Gerlai, R., Pisacane, P., & Erickson, S. (2000). Heregulin, but not ErbB2 or ErbB3, heterozygous mutant mice exhibit hyperactivity in multiple behavioral tasks. Behavioural Brain Research 109(2): 219–227.
Glantz, L. A. & Lewis, D. A. (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Archives of General Psychiatry 57(1): 65–73.
Golub, M. S., Germann, S. L., & Lloyd, K. C. (2004). Behavioral characteristics of a nervous system-specific erbB4 knock-out mouse. Behavioural Brain Research 153(1): 159–170.
Grossberg, S. (2000). The imbalanced brain: From normal behavior to schizophrenia. Biological Psychiatry 48(2): 81–98.
Gu, Z., Jiang, Q., Fu, A. K., Ip, N. Y., & Yan, Z. (2005). Regulation of NMDA receptors by neuregulin signaling in prefrontal cortex. Journal of Neuroscience 25(20): 4974–4984.
Guillin, O., Abi-Dargham, A., & Laruelle, M. (2007). Neurobiology of dopamine in schizophrenia. International Review of Neurobiology 78: 1–39.
Hahn, C. G., Wang, H. Y., Cho, D. S., Talbot, K., Gur, R. E., Berrettini, W. H., Bakshi, K., Kamins, J., Borgmann-Winter, K. E., Siegel, S. J., et al. (2006). Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nature Medicine 12(7): 824–828.
Hakak, Y., Walker, J., Li, C., Wong, W., Davis, K., Buxbaum, J., Haroutunian, V., & Fienberg, A. (2001). Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proceedings of the National Academy of Sciences U.S.A. 98(8): 4746–4751.
Hancock, M. L., Canetta, S. E., Role, L. W., & Talmage, D. A. (2008). Presynaptic type III neuregulin1-ErbB signaling targets (alpha)7 nicotinic acetylcholine receptors to axons. Journal of Cell Biology 181(3): 511–521.
Harrison, P. J. & Law, A. J. (2006). Neuregulin 1 and schizophrenia: Genetics, gene expression, and neurobiology. Biological Psychiatry 60(2): 132–140.
Harrison, P. J., Law, A. J., & Eastwood, S. L. (2003). Glutamate receptors and transporters in the hippocampus in schizophrenia. Annals of the New York Academy of Sciences 1003: 94–101.
Harrison, P. J. & Weinberger, D. R. (2005). Schizophrenia genes, gene expression, and neuropathology: On the matter of their convergence. Molecular Psychiatry 10(1): 40–68.
Hashimoto, R., Straub, R. E., Weickert, C. S., Hyde, T. M., Kleinman, J. E., & Weinberger, D. R. (2004). Expression analysis of neuregulin-1 in the dorsolateral prefrontal cortex in schizophrenia. Molecular Psychiatry 9(3): 299–307.
Hashimoto, T., Arion, D., Unger, T., Maldonado-Aviles, J. G., Morris, H. M., Volk, D. W., Mirnics, K., & Lewis, D. A. (2007). Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Molecular Psychiatry 13(2): 147–161.
Hashimoto, T., Volk, D. W., Eggan, S. M., Mirnics, K., Pierri, J. N., Sun, Z., Sampson, A. R., & Lewis, D. A. (2003). Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. Journal of Neuroscience 23(15): 6315–6326.
Hippenmeyer, S., Vrieseling, E., Sigrist, M., Portmann, T., Laengle, C., Ladle, D. R., & Arber, S. (2005). A developmental switch in the response of DRG neurons to ETS transcription factor signaling. PLoS Biology 3(5): e159.
International Schizophrenia Consortium. (2008). Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455(7210): 237–241.
Johnson, M. (2007). The role of Type III Neuregulin 1 in birth and migration of neurons in the embryonic and adult forebrain interneurons. Ph.D. dissertation, Columbia University, New York.
Kanazawa, T., Glatt, S. J., Tsutsumi, A., Kikuyama, H., Koh, J., Yoneda, H., & Tsuang, M. T. (2007). Schizophrenia is not associated with the functional candidate gene ERBB3: Results from a case-control study. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 144B(1) : 113–116.
Karl, T., Duffy, L., Scimone, A., Harvey, R. P., & Schofield, P. R. (2007). Altered motor activity, exploration and anxiety in heterozygous neuregulin 1 mutant mice: Implications for understanding schizophrenia. Genes, Brain and Behavior 6(7): 677–687.
Kawai, H., Zago, W., & Berg, D. K. (2002). Nicotinic alpha 7 receptor clusters on hippocampal GABAergic neurons: Regulation by synaptic activity and neurotrophins. Journal of Neuroscience 22(18): 7903–7912.
Keri, S., Kiss, I., Seres, I., & Kelemen, O. (2009). A polymorphism of the neuregulin 1 gene (SNP8NRG243177/rs6994992) affects reactivity to expressed emotion in schizophrenia. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 150B(3): 418–420.
Kircher, T., Krug, A., Markov, V., Whitney, C., Krach, S., Zerres, K., Eggermann, T., Stocker, T., Shah, N. J., Treutlein, J., et al. (2009a). Genetic variation in the schizophrenia-risk gene neuregulin 1 correlates with brain activation and impaired speech production in a verbal fluency task in healthy individuals. Human Brain Mapping 30(10): 3406–3416.
Kircher, T., Thienel, R., Wagner, M., Reske, M., Habel, U., Kellermann, T., Frommann, I., Schwab, S., Wolwer, W., von Wilmsdorf, M., et al. (2009b). Neuregulin 1 ICE-single nucleotide polymorphism in first episode schizophrenia correlates with cerebral activation in fronto-temporal areas. European Archives of Psychiatry and Clinical Neuroscience 259(2): 72–79.
Krakowski, M. I., Czobor, P., Citrome, L., Bark, N., & Cooper, T. B. (2006). Atypical antipsychotic agents in the treatment of violent patients with schizophrenia and schizoaffective disorder. Archives of General Psychiatry 63(6): 622–629.
Kramer, R., Bucay, N., Kane, D. J., Martin, L. E., Tarpley, J. E., & Theill, L. E. (1996). Neuregulins with an Ig-like domain are essential for mouse myocardial and neuronal development. Proceedings of the National Academy of Sciences U.S.A. 93(10): 4833–4838.
Kumari, V. & Postma, P. (2005). Nicotine use in schizophrenia: The self medication hypotheses. Neuroscience and Biobehavioral Reviews 29(6): 1021–1034.
Kwon, O. B., Longart, M., Vullhorst, D., Hoffman, D. A., & Buonanno, A. (2005). Neuregulin-1 reverses long-term potentiation at CA1 hippocampal synapses. Journal of Neuroscience 25(41): 9378–9383.
Kwon, O. B., Paredes, D., Gonzalez, C. M., Neddens, J., Hernandez, L., Vullhorst, D., & Buonanno, A. (2008). Neuregulin-1 regulates LTP at CA1 hippocampal synapses through activation of dopamine D4 receptors. Proceedings of the National Academy of Sciences U.S.A. 105(40): 15587–15592.
Laruelle, M., Kegeles, L. S., & Abi-Dargham, A. (2003). Glutamate, dopamine, and schizophrenia: From pathophysiology to treatment. Annals of the New York Academy of Sciences 1003: 138–158.
Law, A. J., Kleinman, J. E., Weinberger, D. R., & Weickert, C. S. (2007). Disease-associated intronic variants in the ErbB4 gene are related to altered ErbB4 splice-variant expression in the brain in schizophrenia. Human Molecular Genetics 16(2): 129–141.
Law, A. J., Lipska, B. K., Weickert, C. S., Hyde, T. M., Straub, R. E., Hashimoto, R., Harrison, P. J., Kleinman, J. E., & Weinberger, D. R. (2006). Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5’ SNPs associated with the disease. Proceedings of the National Academy of Sciences U.S.A. 103(17): 6747–6752.
Leimeroth, R., Lobsiger, C., Lussi, A., Taylor, V., Suter, U., & Sommer, L. (2002). Membrane-bound neuregulin1 type III actively promotes Schwann cell differentiation of multipotent Progenitor cells. Developmental Biology 246(2): 245–258.
Leonard, S., Gault, J., Hopkins, J., Logel, J., Vianzon, R., Short, M., Drebing, C., Berger, R., Venn, D., Sirota, P., et al. (2002). Association of promoter variants in the alpha7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Archives of General Psychiatry 59(12): 1085–1096.
Leu, M., Bellmunt, E., Schwander, M., Farinas, I., Brenner, H. R., & Muller, U. (2003). Erbb2 regulates neuromuscular synapse formation and is essential for muscle spindle development. Development 130(11): 2291–2301.
Lewis, D. A. (2000). GABAergic local circuit neurons and prefrontal cortical dysfunction in schizophrenia. Brain Research, Brain Research Reviews 31(2–3): 270–276.
Lewis, D. A., Hashimoto, T., & Volk, D. W. (2005). Cortical inhibitory neurons and schizophrenia. Nature Reviews Neuroscience 6(4): 312–324.
Lewis, D. A., Volk, D. W., & Hashimoto, T. (2004). Selective alterations in prefrontal cortical GABA neurotransmission in schizophrenia: A novel target for the treatment of working memory dysfunction. Psychopharmacology (Berl) 174(1): 143–150.
Li, B., Woo, R. S., Mei, L., & Malinow, R. (2007). The neuregulin-1 receptor erbB4 controls glutamatergic synapse maturation and plasticity. Neuron 54(4): 583–597.
Li, L., Cleary, S., Mandarano, M. A., Long, W., Birchmeier, C., & Jones, F. E. (2002). The breast proto-oncogene, HRGalpha regulates epithelial proliferation and lobuloalveolar development in the mouse mammary gland. Oncogene 21(32): 4900–4907.
Liu, X., Hwang, H., Cao, L., Buckland, M., Cunningham, A., Chen, J., Chien, K. R., Graham, R. M., & Zhou, M. (1998). Domain-specific gene disruption reveals critical regulation of neuregulin signaling by its cytoplasmic tail. Proceedings of the National Academy of Sciences U.S.A. 95(22): 13024–13029.
Liu, Y., Ford, B., Mann, M. A., & Fischbach, G. D. (2001). Neuregulins increase alpha7 nicotinic acetylcholine receptors and enhance excitatory synaptic transmission in GABAergic interneurons of the hippocampus. Journal of Neuroscience 21(15): 5660–5669.
Long, W., Wagner, K. U., Lloyd, K. C., Binart, N., Shillingford, J. M., Hennighausen, L., & Jones, F. E. (2003). Impaired differentiation and lactational failure of Erbb4-deficient mammary glands identify ERBB4 as an obligate mediator of STAT5. Development 130(21) : 5257–5268.
Lopez-Bendito, G., Cautinat, A., Sanchez, J. A., Bielle, F., Flames, N., Garratt, A. N., Talmage, D. A., Role, L. W., Charnay, P., Marin, O., et al. (2006). Tangential neuronal migration controls axon guidance: A role for neuregulin-1 in thalamocortical axon navigation. Cell 125 (1): 127–142.
Lubow, R. E. (2005). Construct validity of the animal latent inhibition model of selective attention deficits in schizophrenia. Schizophrenia Bulletin 31(1): 139–153.
Martin, L. F., Leonard, S., Hall, M. H., Tregellas, J. R., Freedman, R., & Olincy, A. (2007). Sensory gating and alpha-7 nicotinic receptor gene allelic variants in schizoaffective disorder, bipolar type. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 144B(5): 611–614.
Mata, I., Perez-Iglesias, R., Roiz-Santianez, R., Tordesillas-Gutierrez, D., Gonzalez-Mandly, A., Vazquez-Barquero, J. L., & Crespo-Facorro, B. (2009). A neuregulin 1 variant is associated with increased lateral ventricle volume in patients with first-episode schizophrenia. Biological Psychiatry 65(6): 535–540.
Mathew, S. V., Law, A. J., Lipska, B. K., Davila-Garcia, M. I., Zamora, E. D., Mitkus, S. N., Vakkalanka, R., Straub, R. E., Weinberger, D. R., Kleinman, J. E., et al. (2007). Alpha7 nicotinic acetylcholine receptor mRNA expression and binding in postmortem human brain are associated with genetic variation in neuregulin 1. Human Molecular Genetics 16(23): 2921-2932.
McCarley, R. W., Wible, C. G., Frumin, M., Hirayasu, Y., Levitt, J. J., Fischer, I. A., & Shenton, M. E. (1999). MRI anatomy of schizophrenia. Biological Psychiatry 45(9): 1099–1119.
Mei, L. & Xiong, W. C. (2008). Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nature Reviews Neuroscience, 9(6): 437–452.
Meyer, D. & Birchmeier, C. (1995). Multiple essential functions of neuregulin in development. Nature 378(6555): 386–390.
Michailov, G. V., Sereda, M. W., Brinkmann, B. G., Fischer, T. M., Haug, B., Birchmeier, C., Role, L., Lai, C., Schwab, M. H., & Nave, K. A. (2004). Axonal neuregulin-1 regulates myelin sheath thickness. Science 304(5671): 700–703.
Moy, S. S., Ghashghaei, H. T., Nonneman, R. J., Weimer, J. M., Yokota, Y., Lee, D., Lai, C., Threadgill, D., & Anton, E. S. (2009). Deficient NRG1-ERBB signaling alters social approach: Relevance to genetic models of schizophrenia. Journal of Neurodevelopmental Disorders 1(4): 302–312.
Nave, K. A. & Salzer, J. L. (2006). Axonal regulation of myelination by neuregulin 1. Current Opinion in Neurobiology 16(5): 492–500.
Neddens, J., Vullhorst, D., Paredes, D., & Buonanno, A. (2009). Neuregulin links dopaminergic and glutamatergic neurotransmission to control hippocampal synaptic plasticity. Communicative and Integrative Biology 2(3): 261–264.
Nicodemus, K. K., Law, A. J., Luna, A., Vakkalanka, R., Straub, R. E., Kleinman, J. E., & Weinberger, D. R. (2009). A 5’ promoter region SNP in NRG1 is associated with schizophrenia risk and type III isoform expression. Molecular Psychiatry 14(8): 741–743.
Norton, N., Moskvina, V., Morris, D. W., Bray, N. J., Zammit, S., Williams, N. M., Williams, H. J., Preece, A. C., Dwyer, S., & Wilkinson, J. C. (2006). Evidence that interaction between neuregulin 1 and its receptor erbB4 increases susceptibility to schizophrenia. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 141B(1): 96–101.
Nuechterlein, K. H., Barch, D. M., Gold, J. M., Goldberg, T. E., Green, M. F., & Heaton, R. K. (2004). Identification of separable cognitive factors in schizophrenia. Schizophrenia Research 72(1): 29–39.
Okada, M. & Corfas, G. (2004). Neuregulin1 downregulates postsynaptic GABAA receptors at the hippocampal inhibitory synapse. Hippocampus 14(3): 337–344.
O’Tuathaigh, C. M., Babovic, D., O’Meara, G., Clifford, J. J., Croke, D. T., & Waddington, J. L. (2007). Susceptibility genes for schizophrenia: Characterisation of mutant mouse models at the level of phenotypic behaviour. Neuroscience and Biobehavioral Reviews 31(1): 60–78.
O’Tuathaigh, C. M., Babovic, D., O’Sullivan, G. J., Clifford, J. J., Tighe, O., Croke, D. T., Harvey, R., & Waddington, J. L. (2007). Phenotypic characterization of spatial cognition and social behavior in mice with “knockout” of the schizophrenia risk gene neuregulin 1. Neuroscience 147(1): 18–27.
O’Tuathaigh, C. M., O’Connor, A. M., O’Sullivan, G. J., Lai, D., Harvey, R., Croke, D. T., & Waddington, J. L. (2008). Disruption to social dyadic interactions but not emotional/ anxiety-related behaviour in mice with heterozygous “knockout” of the schizophrenia risk gene neuregulin-1. Progress in Neuropsychopharmacology & Biological Psychiatry 32(2): 462–466.
O’Tuathaigh, C. M., O’Sullivan, G. J., Kinsella, A., Harvey, R. P., Tighe, O., Croke, D. T., & Waddington, J. L. (2006). Sexually dimorphic changes in the exploratory and habituation profiles of heterozygous neuregulin-1 knockout mice. Neuroreport 17(1): 79–83.
Ozaki, M., Sasner, M., Yano, R., Lu, H. S., & Buonanno, A. (1997). Neuregulin-beta induces expression of an NMDA-receptor subunit. Nature 390(6661): 691–694.
Postma, P., Gray, J. A., Sharma, T., Geyer, M., Mehrotra, R., Das, M., Zachariah, E., Hines, M., Williams, S. C., and Kumari, V. (2006). A behavioural and functional neuroimaging investigation into the effects of nicotine on sensorimotor gating in healthy subjects and persons with schizophrenia. Psychopharmacology (Berl) 184(3–4): 589–599.
Powell, S. B. & Geyer, M. A. (2002). Developmental markers of psychiatric disorders as identified by sensorimotor gating. Neurotoxicity Research 4(5–6): 489–502.
Rieff, H. I., Raetzman, L. T., Sapp, D. W., Yeh, H. H., Siegel, R. E., & Corfas, G. (1999). Neuregulin induces GABA(A) receptor subunit expression and neurite outgrowth in cerebellar granule cells. Journal of Neuroscience 19(24): 10757–10766.
Rimer, M., Barrett, D. W., Maldonado, M. A., Vock, V. M., & Gonzalez-Lima, F. (2005). Neuregulin-1 immunoglobulin-like domain mutant mice: Clozapine sensitivity and impaired latent inhibition. Neuroreport 16(3): 271–275.
Role, L. W. & Talmage, D. A. (2007). Neurobiology: New order for thought disorders. Nature 448(7151): 263–265.
Rosoklija, G., Toomayan, G., Ellis, S. P., Keilp, J., Mann, J. J., Latov, N., Hays, A. P., & Dwork, A. J. (2000). Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders: Preliminary findings. Archives of General Psychiatry 57(4): 349–356.
Roy, K., Murtie, J. C., El-Khodor, B. F., Edgar, N., Sardi, S. P., Hooks, B. M., Benoit-Marand, M., Chen, C., Moore, H., O’Donnell, P., et al. (2007). Loss of erbB signaling in oligodendrocytes alters myelin and dopaminergic function, a potential mechanism for neuropsychiatric disorders. Proceedings of the National Academy of Sciences U.S.A. 104(19): 8131–8136.
Schobel, S. A., Lewandowski, N. M., Corcoran, C. M., Moore, H., Brown, T., Malaspina, D., & Small, S. A. (2009). Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Archives of General Psychiatry 66(9): 938–946.
Shelley, A. M., Ward, P. B., Catts, S. V., Michie, P. T., Andrews, S., & McConaghy, N. (1991). Mismatch negativity: An index of a preattentive processing deficit in schizophrenia. Biological Psychiatry 30(10): 1059–1062.
Shiota, S., Tochigi, M., Shimada, H., Ohashi, J., Kasai, K., Kato, N., Tokunaga, K., & Sasaki, T. (2008). Association and interaction analyses of NRG1 and ERBB4 genes with schizophrenia in a Japanese population. Journal of Human Genetics 53(10): 929–935.
Stefansson, H., Sigurdsson, E., Steinthorsdottir, V., Bjornsdottir, S., Sigmundsson, T., Ghosh, S., Brynjolfsson, J., Gunnarsdottir, S., Ivarsson, O., Chou, T. T., et al. (2002). Neuregulin 1 and susceptibility to schizophrenia. American Journal of Human Genetics 71(4): 877–892.
Steiner, H., Blum, M., Kitai, S. T., & Fedi, P. (1999). Differential expression of ErbB3 and ErbB4 neuregulin receptors in dopamine neurons and forebrain areas of the adult rat. Experimental Neurology 159(2): 494–503.
Stephens, S. H., Logel, J., Barton, A., Franks, A., Schultz, J., Short, M., Dickenson, J., James, B., Fingerlin, T. E., Wagner, B., et al. (2009). Association of the 5’-upstream regulatory region of the alpha7 nicotinic acetylcholine receptor subunit gene (CHRNA7) with schizophrenia. Schizophrenia Research 109(1–3): 102–112.
Swerdlow, N. R., Braff, D. L., & Geyer, M. A. (2000). Animal models of deficient sensorimotor gating: What we know, what we think we know, and what we hope to know soon. Behavioural Pharmacology 11(3–4): 185–204.
Talmage, D. A. (2008). Mechanisms of neuregulin action. Novartis Foundation Symposium 189: 74–84; discussion 84–93.
Taveggia, C., Thaker, P., Petrylak, A., Caporaso, G. L., Toews, A., Falls, D. L., Einheber, S., & Salzer, J. L. (2008). Type III neuregulin-1 promotes oligodendrocyte myelination. Glia 56(3): 284–293.
Taveggia, C., Zanazzi, G., Petrylak, A., Yano, H., Rosenbluth, J., Einheber, S., Xu, X., Esper, R. M., Loeb, J. A., Shrager, P., et al. (2005). Neuregulin-1 type III determines the ensheathment fate of axons. Neuron 47(5): 681–694.
Thuret, S., Alavian, K. N., Gassmann, M., Lloyd, C. K., Smits, S. M., Smidt, M. P., Klein, R., Dyck, R. H., & Simon, H. H. (2004). The neuregulin receptor, ErbB4, is not required for normal development and adult maintenance of the substantia nigra pars compacta. Journal of Neurochemistry 91(6): 1302–1311.
Tidcombe, H., Jackson-Fisher, A., Mathers, K., Stern, D. F., Gassmann, M., & Golding, J. P. (2003). Neural and mammary gland defects in ErbB4 knockout mice genetically rescued from embryonic lethality. Proceedings of the National Academy of Sciences U.S.A. 100(14): 8281–8286.
Tronche, F., Kellendonk, C., Kretz, O., Gass, P., Anlag, K., Orban, P. C., Bock, R., Klein, R., & Schütz, G. (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nature Genetics 23(1): 99–103.
van den Buuse, M., Wischhof, L., Lee, R. X., Martin, S., & Karl, T. (2009). Neuregulin 1 hypomorphic mutant mice: Enhanced baseline locomotor activity but normal psychotropic drug-induced hyperlocomotion and prepulse inhibition regulation. International Journal of Neuropsychopharmacology 12(10): 1383–1393.
Volk, D. W. & Lewis, D. A. (2002). Impaired prefrontal inhibition in schizophrenia: Relevance for cognitive dysfunction. Physiology and Behavior 77(4–5): 501–505.
Watanabe, Y., Fukui, N., Nunokawa, A., Muratake, T., Kaneko, N., Kitamura, H., & Someya, T. (2007). No association between the ERBB3 gene and schizophrenia in a Japanese population. Neuroscience Research 57(4): 574–578.
Weike, A. I., Bauer, U., & Hamm, A. O. (2000). Effective neuroleptic medication removes prepulse inhibition deficits in schizophrenia patients. Biological Psychiatry 47(1): 61–70.
Wen, L., Lu, Y. S., Zhu, X. H., Li, X. M., Woo, R. S., Chen, Y. J., Yin, D. M., Lai, C., Terry, A. V., Jr., Vazdarjanova, A., et al. (2010). Neuregulin 1 regulates pyramidal neuron activity via ErbB4 in parvalbumin-positive interneurons. Proceedings of the National Academy of Sciences U.S.A. 107(3): 1211–1216.
Wolpowitz, D., Mason, T. B., Dietrich, P., Mendelsohn, M., Talmage, D. A., & Role, L. W. (2000). Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron 25(1): 79–91.
Woo, R. S., Li, X. M., Tao, Y., Carpenter-Hyland, E., Huang, Y. Z., Weber, J., Neiswender, H., Dong, X. P., Wu, J., Gassmann, M., et al. (2007). Neuregulin-1 enhances depolarization-induced GABA release. Neuron 54(4): 599–610.
Yang, X., Kuo, Y., Devay, P., Yu, C., & Role, L. (1998). A cysteine-rich isoform of neuregulin controls the level of expression of neuronal nicotinic receptor channels during synaptogenesis. Neuron 20(2): 255–270.
Yurek, D. M., Zhang, L., Fletcher-Turner, A., & Seroogy, K. B. (2004). Supranigral injection of neuregulin1-beta induces striatal dopamine overflow. Brain Research 1028(1): 116–119.
Zhong, C., Du, C., Hancock, M., Mertz, M., Talmage, D. A., & Role, L. W. (2008). Presynaptic type III neuregulin 1 is required for sustained enhancement of hippocampal transmission by nicotine and for axonal targeting of alpha7 nicotinic acetylcholine receptors. Journal of Neuroscience 28(37): 9111–9116.
Zhuo, L., Theis, M., Alvarez-Maya, I., Brenner, M., Willecke, K., & Messing, A. (2001). hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 31(2): 85–94.