KEY CONCEPTS
Schizophrenia is a devastating neuropsychiatric disorder with a lifetime prevalence of about 1% in most studied populations. It is characterized by positive psychotic symptoms such as hallucinations, delusions, and disorganized behavior; negative symptoms such as social withdrawal and apathy; and impaired cognition. Early onset, poor response to medication, frequent relapse, and chronic course impose a considerable burden on sufferers, their families, and society. Genetic epidemiology studies in schizophrenia have revealed a heritability of about 80% (Sullivan, Kendler, & Neale, 2003). However, identification of genetic risk factors for schizophrenia has been elusive despite decades of research in the etiology of the disease.
Genetic Architecture of Psychiatric Disorders
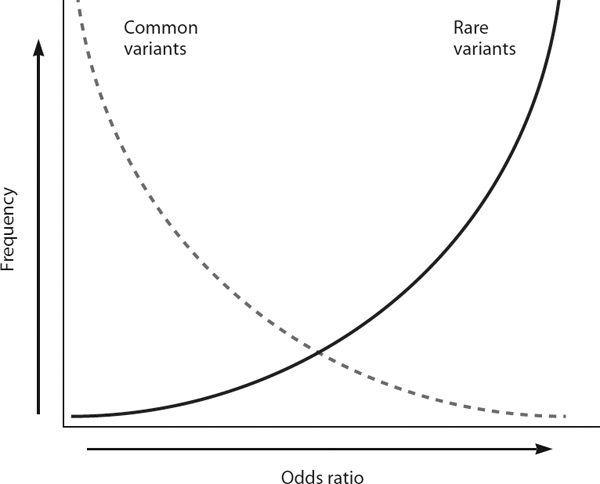
Psychiatric disorders, like other common diseases, are multifactorial in nature and have complex genetic etiologies. The genetic architecture underlying disease susceptibility is characterized by both the frequency and the penetrance of risk alleles (figure 8.1). The common disease-common allele (CDCA) hypothesis emphasizes the importance of relatively common alleles, each of small effect, acting together to increase disease risk. The common disease-rare allele (CDRA) hypothesis conversely emphasizes the impact of individually rare yet highly penetrant alleles. It is likely that both common and rare alleles contribute to the risk of psychiatric disorders, although the relative impact of each remains unknown.
Modern association studies based on the CDCA hypothesis exploit ancestral genetic variants that are now common (> 5% minor allele frequency) in the population. These studies can focus on candidate genes based on a priori functional evidence or identify candidates in an unbiased manner using genomewide association studies (GWAS) (Altshuler, Daly, & Lander, 2008). Nearly 800 candidate genes have been investigated for association with schizophrenia by more than 1,400 studies (see www.szgene.org). Although many of these are considered strong susceptibility genes, none have unequivocal statistical support (Karayiorgou & Gogos, 2006; Sanders et al., 2008; Goldstein, 2009). In addition, unbiased GWAS of psychiatric disorders have identified candidate loci for schizophrenia, as well as bipolar disorder and autism (Ferreira et al., 2008; O’Donovan et al., 2008; Wang et al., 2009). These loci await confirmation in larger-scale studies. It is interesting to note that these loci do not include any top candidate genes, such as Neuregulin 1 or Dysbindin.
The CDRA hypothesis did not receive much traction until recently, primarily because of the rarity of families afflicted with psychiatric disorders in a strictly Mendelian fashion. Taking into account variable penetrance and de novo occurrences, however, one sees that rare alleles may contribute substantially to both familial and sporadic cases (Bodmer & Bonilla, 2008; ISC, 2008; Xu et al., 2008). It is not surprising that the alleles with the strongest statistical support for association with schizophrenia are rare, highly penetrant structural mutations. Identification of rare alleles in families traditionally relied on linkage studies because that approach was successful in locating rare mutations in Mendelian inherited diseases. Unfortunately, linkage studies have been less conclusive for complex conditions such as psychiatric disorders. Ironically, although the limited success of linkage studies inspired the use of GWAS for identifying common risk alleles, it was the “unexpected” use of GWAS data for copy number variation detection that, as explained in the next section, provided the strongest supporting evidence to date for the “rare variant” hypothesis.
Copy Number Variation in the Human Genome
Recently, the rapid development of new whole-genome scanning technologies has started to fill the resolution gap between the traditional cytogenetic analysis (> 2 Mb) and mutation analysis by DNA sequencing (< 1 kb). As a result, a large amount of previously undetected variation has been revealed in the human genome. One of the most intriguing types of variation, the extent of which was previously unappreciated, is copy number variation, defined as deletions or duplications larger than 1 kb.
In one of the first attempts to evaluate the prevalence of CNVs in the general population, Iafrate and colleagues used bacterial artificial chromosome probe-based array-comparative genomic hybridization (BAC-CGH) with a resolution of 1 Mb on 55 individuals, some of whom had known chromosomal abnormalities that served as an internal control of the array’s sensitivity (Iafrate et al., 2004). The scan discovered 255 CNVs beyond the known abnormalities, of which about 10% had a greater than 10% prevalence. CNV location was significantly associated with segmental duplications (i.e., long segments of DNA with near-identical sequence). Independently, Sebat et al. (2004) developed another CGH system called representational oligonucleotide microarray analysis (ROMA) with an estimated resolution of 35 kb to investigate the extent of variation among normal individuals. In 20 normal individuals, they identified 221 CNVs in total, of which 76 were unique (Sebat et al., 2004). In line with the Iafrate et al. study, CNV locations were significantly associated with segmental duplication, suggesting a possible mechanism underlying these rearrangements. In a subsequent study of 270 controls from the HapMap sample, Redon et al. determined that CNVs cover approximately 12% of the genome in the general population (Redon et al., 2006) and encompass more nucleotide content per genome than single nucleotide polymorphisms (SNPs), highlighting their importance in contributing to total genetic variation. Overall, these and other studies unambiguously demonstrated that variation in copy number is a common source of genetic heterogeneity in the general population and that array-based CNV detection methods can be successful in discovering this type of genetic variation in the human genome.
Copy Number Variation and Schizophrenia
The 22q11.2 Microdeletion and Susceptibility to Schizophrenia
A microdeletion of 22q11.2 was the first CNV described in schizophrenia (Karayiorgou et al., 1995). Since its discovery 15 years ago, a strong and specific relationship has been established between the presence of the 22q11.2 microdeletion and psychosis in schizophrenia or schizoaffective disorder (Karayiorgou et al., 1995; Xu et al., 2008). During late adolescence and early adulthood, up to one-third of all individuals carrying this deletion develop schizophrenia or schizoaffective disorder (Pulver et al., 1994; Murphy, Jones, & Owen, 1999; Gothelf et al., 2007). A number of studies have indicated that 22q11.2 deletion syndrome (22q11DS) accounts for up to 1–2% of schizophrenia cases (Karayiorgou et al., 1995; Xu et al., 2008; ISC, 2008; Stefansson et al., 2008) and represents to date the only confirmed recurrent structural mutation responsible for introducing sporadic cases of schizophrenia into the population. This strong bidirectional association between schizophrenia and the 22q11.2 microdeletion makes this deletion one of the risk factors of greatest effect on schizophrenia, with a greater risk conferred only by being the child of two parents with schizophrenia or the monozygotic co-twin of an affected individual. Significantly, there are no major clinical differences in the core schizophrenia phenotype between individuals with schizophrenia that are 22q11.2 microdeletion carriers and those who are not (Bassett et al., 1998; Bassett et al., 2003). Moreover, many of these individuals have no obvious congenital abnormalities, and most do not have serious intellectual disabilities, making them indistinguishable from other schizophrenia patients recruited into research samples. The structural and neurocognitive anomalies that distinguish the 22q11.2 carriers who develop schizophrenia from those who do not remain unknown.
Preliminary results from a number of underpowered cross-sectional studies suggest that distinguishing features are consistent with those most consistently reported for general schizophrenia. These include larger lateral and third ventricles; generalized decreases in gray and white matter volumes, especially in frontal and temporal lobes (Chow et al., 2002; van Amelsvoort et al., 2004a); and a number of cognitive impairments that may reflect differences in the development and function of frontal brain regions (van Amelsvoort et al., 2004b; Chow et al., 2006).
Recent Copy Number Variant Discoveries in Schizophrenia
The original finding at the 22q11.2 locus raised an interesting and important question of whether the role of CNVs in the genetic makeup of complex psychiatric disorders is more widespread. Owing to rapid developments in high-density microarray technologies designed to screen for structural variants across the whole human genome and advances in statistical analysis methods, several groups in rapid succession provided evidence that rare CNVs contribute to the genetic etiology of schizophrenia and autism.
A 30 kb resolution SNP array of 359 schizophrenia and control trios (a total of 1,077 individuals) determined that de novo CNVs are significantly more common in sporadic cases than controls (10% as opposed to 1.3%) (Xu et al., 2008). Inherited rare CNVs were 1.5 times more common in sporadic cases than in controls, representing a smaller but significant increase (from 20% to 30%). Comparison of the genes affected by de novo CNVs demonstrated enrichment of neural development and RNA processing pathways. Rare inherited CNVs were also shown to be associated with familial schizophrenia. Xu et al. (2009) showed that rare inherited CNVs are almost twice as common in familial cases of schizophrenia as in sporadic cases or controls. This discovery resulted from comparing the results of a high-resolution SNP array scan on 48 schizophrenia probands plus their parents and affected relatives to those of a scan on more than 300 controls and sporadic cases, followed by validation using independent approaches. Nine of 12 families with inherited CNVs showed evidence of CNV cosegregation. The familial cases were 2.7 times more likely than controls to have genic CNVs (CNVs overlapping at least one gene, either partly or in its entirety), but there was no enrichment of de novo CNVs. These findings provided strong empirical evidence supporting the notion that multiple genetic variants, including individually rare ones that affect many different genes, contribute to the genetic risk of schizophrenia. They also highlighted some important differences in the genetic architecture of familial and nonfamilial forms of the disease: although the overall frequency of carriers of all rare structural variants is the same between the familial and sporadic cases (approximately 40%), the type of variants is markedly different. Sporadic schizophrenia is characterized by a marked enrichment of rare new mutations and only a modest increase in the rate of rare inherited CNVs, which do not appear to affect genes preferentially. By contrast, familial schizophrenia is characterized by enrichment in rare inherited genic CNVs (predicted to have higher penetrance), whereas new mutations are less prominent.
Two case-control studies that did not distinguish between familial and sporadic cases also provided evidence for a collective enrichment of rare CNVs in schizophrenia. Walsh and colleagues used ROMA on 150 individuals with schizophrenia plus controls followed by SNP-based validation and refinement (Walsh et al., 2008). They reported a significant threefold increase in rare, genic CNVs among case subjects (15% as opposed to 5%) and a fourfold increase among early-onset case subjects as opposed to controls (20% to 5%). In an independent trio sample of 83 early-onset cases, three assays for CNVs determined that rare, genic CNVs were present in 28% of cases, whereas they were present in 13% of nontransmitted parental chromosomes—a significant enrichment. The affected genes in case subjects were significantly enriched for pathways in brain development and neural function, including genes such as ERBB4 and Neurexin1 (NRXN1). Another study by the International Schizophrenia Consortium analyzed 3,391 individuals with schizophrenia and controls using SNP genotype arrays to identify CNVs greater than 100 kb (ISC, 2008). When rare (less than 1% frequency) CNVs were considered, individuals with schizophrenia had 1.15 times as many controls and 1.41 times as many genes affected by CNVs. This study confirmed that deletions in 22q11.2 were significantly associated with schizophrenia, and it identified two additional CNVs at 15q13.3 and 1q21.1 that were significantly enriched among case subjects.
Stefansson et al. (2008) provided additional evidence for a role of CNVs in schizophrenia, taking a two-step strategy. First, they identified 66 de novo CNVs in over 7,500 control European and Chinese trios and pairs (Stefansson et al., 2008). Next, they checked for associations between these 66 mutations and schizophrenia in 1,433 SGENE (Schizophrenia GENE) consortium cases and more than 33,000 controls. Three deletions at 1q21.1, 15q11.2, and 15q13.3 were nominally enriched in individuals with schizophrenia or psychosis, two of which were also reported in the ISC scan (see the previous discussion earlier in this section; ISC, 2008). On association analysis in 3,285 additional cases and 7,951 controls, all three CNVs (but not SNPs within the regions) were significantly associated with psychosis with high odds ratios. However, only 1q21.1 was significantly associated with a strict definition of schizophrenia. Several other lower-scale studies tried to assess the relevance of rare CNVs to the etiology of schizophrenia. A BAC-CGH scan of 93 Bulgarian schizophrenia trios followed by SNP array verification identified 13 rare CNVs in case subjects; these were not found in controls or the CNV database (Kirov et al., 2008). The most interesting findings were a deletion at 2p16.3 involving Neurexin1, carried by affected siblings and their unaffected mother; a de novo duplication at 15q13.1; and a deletion at 16p12.2 inherited from a parent who suffers from an affective disorder. Another analysis of 54 Dutch cases with a SNP array demonstrated CNVs affecting four candidate genes: MYT1L, CTNND2, ASTN2, and NRXN1 (Vrijenhoek et al., 2008). On screening 752 additional cases and 706 controls for CNVs affecting these four genes, CNVs in MYT1L, ASTN2, and NRXN1 were found only in case subjects. A study with 471 affected individuals from the United Kingdom using a SNP array and CGH validation determined that CNVs with frequency less than 1% and length greater than 1 Mb were 2.26 times more frequent in case subjects than in controls (Kirov et al., 2009). Four regions demonstrated significant association with broad schizophrenia diagnosis upon a preliminary meta-analysis of the Stefansson et al. and ISC studies: 17p12, 1q21.1, 15q11.2, and 15q13.3. Ingason et al. (2009) examined 4,345 schizophrenia patients and 35,079 controls from eight European populations for duplications and deletions at the 16p13.1 locus, using microarray data. They found a threefold excess of duplications and deletions in schizophrenia cases as opposed to controls, with duplications present in 0.30% of cases versus 0.09% of controls and deletions in 0.12% of cases and 0.04% of controls. The implicated region could be divided into three intervals defined by flanking repeats. Duplications spanning the first and second intervals showed the most significant association with schizophrenia. Notably, in a single Icelandic family, a duplication spanning these intervals was present in two cases of schizophrenia as well as in individual cases of alcoholism, attention deficit hyperactivity disorder, and dyslexia. Finally, recurrent microdeletions and microduplications of a 600 kb genomic region at an adjacent chromosomal locus (16p11.2) have been implicated in childhood-onset developmental disorders. McCarthy et al. (2009) reported an association between 16p11.2 microduplications and schizophrenia in two large cohorts. The microduplication was detected in 0.63% of cases and 0.03% of controls from the initial cohort, and in 0.34% of cases and 0.04% of controls from the replication cohort, resulting in a 14.5-fold enrichment in the combined sample.
Overall, these studies suggest that rare structural rearrangements collectively contribute to schizophrenia risk. Thus, rare CNVs represent an important source of genetic heterogeneity in the etiology of complex psychiatric diseases such as schizophrenia.
Determining the Pathogenic Nature of CNVs and Affected Genes
The identification of CNVs as an important source of variation in the human genome and the connection between rare CNVs and psychiatric disorders raises the question of how to distinguish abnormal structural mutations from neutral polymorphisms and establish a causal relationship between pathogenic CNVs and their corresponding phenotypes. Establishing causality may be easier for recurrent CNVs if a sufficient number of patients is available. In such cases, establishing a bidirectional association between CNVs and disease along the lines described here for the 22q11.2DS is important to strengthen causality and facilitate development of diagnostic assays. Specifically, in addition to enrichment among cases, unambiguous association between a CNV and schizophrenia requires demonstration of relatively high penetrance among CNV carriers (higher than the baseline rate of psychosis in mental retardation), as well as meeting full diagnostic criteria indistinguishable from those of individuals with schizophrenia that are not CNV carriers.
Establishing causality is harder for private CNVs found in only a single individual, which represent the majority of CNVs described to date in patient cohorts. In such cases, determining a possible causal connection between a specific CNV locus and a disease phenotype depends on a number of factors. For example, a rare de novo CNV is more likely to be pathogenic than a CNV inherited by an unaffected parent. For inherited CNVs, cosegregation with disease is a strong indicator of causal connection between disease and the CNV. In particular, the observation that all affected members of a family carry a rare CNV is a strong indication of pathogenicity. (It is not detrimental if unaffected members also carry it if there is incomplete penetrance.) The issue of whether a potentially pathogenic CNV segregates to all affected members within a family has not received much attention in the current literature, which is heavily based on case-control studies. This is a source of concern, as it may lead to false findings. Finally, CNVs that affect the coding region or the splicing pattern of a given gene are more likely to be pathogenic than, for example, intronic or intergenic CNVs. Overall, support for a pathogenic role can be offered by the location of a CNV in a given gene as well as by observation of recurrent incidence of independent CNVs in more than one exon of the same gene or members of the same gene family.
Help toward establishing causality may be provided by reference CNV maps across the whole genome and by online databases that are being established. The Database of Genomic Variants (DGV) (see http://projects.tcag.ca/variation) is such an accumulation of data, which as of this writing holds reports on over 8,400 CNV loci. It also provides public access to the Genome Structural Variation Consortium results from high-resolution CNV scans on 40 individuals with European or African ancestry. By offering a compendium of current findings, this database is especially valuable to determine whether newly discovered CNVs are unique to one’s case population or found at relatively high frequency among controls in prior reports. However, because the CNV annotations in this database are generated by different labs, which often use different platforms and analysis algorithms, the false positive detection rate, CNV frequency, CNV size, and CNV breakpoints might vary, and all these could affect the final outcomes when used to establish a causal relation between CNVs and disease phenotypes. Another database, the Wellcome Trust Case-Control Consortium (WTCCC) (see www.wtccc.org.uk), is a collaborative effort among 50 groups in the United Kingdom, originally used to accumulate SNP genotype information across a variety of diseases for over 17,000 individuals. As this information can also be used to identify CNVs, it represents an important collection for genomic rearrangements. More recently, Conrad et al. provided a population-based CNV map by using a tiling oligonucleotide microarray of 42 million probes on 800 individuals from different ethnic groups (Conrad et al., 2010). Another recently launched international research effort, the 1000 Genomes Project, aims to establish a complete and detailed catalogue of human genetic variations, which in turn can be used for association studies relating genetic variation to disease. Once accomplished, the project will provide a map with more than 95% of the variants (e.g., SNPs, CNVs, insertions or deletions), as well as minor allele frequencies as low as 1% across the genome and 0.1–0.5% in gene regions and a high-resolution reference map for future CNV-based association studies.
Besides these reference CNV maps, several databases have also been established to collect pathogenic CNVs observed in various diseases: the Database of Chromosomal Imbalance and Phenotype in Human using Ensemble Resources (DECIPHER), the Chromosome Abnormality Database (CAD), the Mendelian Cytogenetics Network Online Database, and the European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations (ECARUCA). The accumulation of recurrent CNVs in certain phenotypes will provide information on the potential connections between related diseases and might help dissect some aspects of psychiatric diseases by establishing the causal connection between intermediate phenotypes and underlying molecular mechanisms.
In addition to the challenges in establishing causality between CNVs and psychiatric diseases, accumulating evidence points toward the difficulty in pinpointing causal relationships between a specific gene or genes affected by CNVs and disease phenotypes. Even though the methods for CNV discovery are relatively well developed, a remaining problem is how to determine the exact boundaries of CNVs identified in both control populations and patient genomes. Without precise information on CNV boundaries, in many cases it might be very difficult to refine the candidate regions and determine the affected genes. High-density arrays and deep sequencing technology now provide improved resolution to fully determine the CNV boundaries in a high-throughput fashion. This technical issue notwithstanding, there are at least two ways in which the removal or duplication of genes from a region deleted or duplicated by a CNV could be acting: (1) a single dosage-sensitive gene may exert a major effect, or (2) the CNV-associated neurobehavioral phenotypes may stem from the cumulative effect of a number of gene products, acting either additively or synergistically. According to the latter model, although one or a few loci may have a greater phenotypic impact, it is the cumulative effect of the imbalance of several genes within the CNV that determines the overall phenotype. This scenario is likely to complicate gene identification efforts using traditional human genetic approaches.
Candidate Risk Genes Within Copy Number Variants
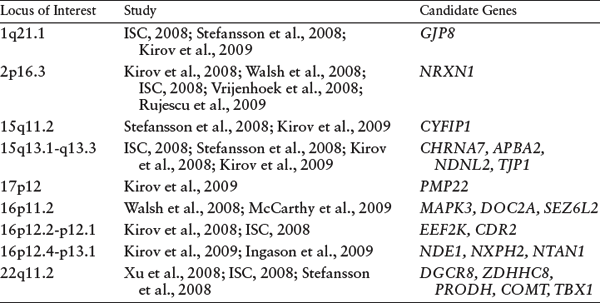
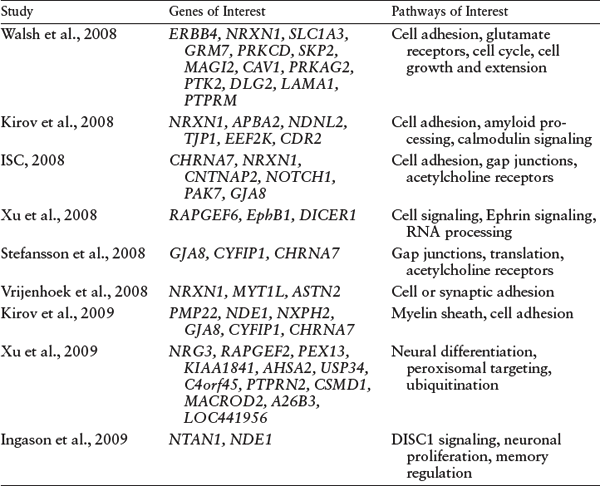
Although no large-scale systematic meta-analysis exists of the several genomewide screens to identify schizophrenia-associated CNVs, there are recurrent CNV reports as well as results from preliminary meta-analyses of a subset of the studies that have been conducted to date (Itsara et al., 2009; Kirov et al., 2009), which are summarized in table 8.1. These recurrent CNVs as well as other, rarer CNVs cover many potential candidate genes involved in neural structure and function (table 8.2).
The 22q11.2 deletion locus contains approximately 30 genes. Some potential candidates including COMT, PRODH, ZDHH8, DGCR8, and TBX1 are already under investigation, and several behavioral and neuronal deficits have been associated with these genes in their respective animal models (Paterlini et al., 2005; Paylor et al., 2006; Mukai et al., 2008; Stark et al., 2008).
Another interesting candidate gene is Neurexin1 (NRXN1), which was identified as disrupted by CNVs at 2p16.3 in several recent genomewide array scans of schizophrenia cohorts. A targeted screen of all three Neurexin genes in 2,977 schizophrenia patients and 33,746 controls from seven European populations did not identify CNVs affecting NRXN2 or NRXN3 but described a significant association with schizophrenia and CNVs that disrupt exons of NRXN1 (Rujescu et al., 2009). Because NRXN1 is a cell-surface receptor involved in the formation of synaptic contacts in the central nervous system, it could have widespread effects on brain structure and connectivity (Reissner et al., 2008). It is interesting that Contactin-associated protein 2 (CNTNAP2 or CASPR2) at 7q35, another member of the Neurexin family, was recently reported to be deleted in cases of schizophrenia (Friedman et al., 2008). CNTNAP2 has also been reported as a genetic susceptibility factor in autism in CNV, linkage, and association studies (Alarcon et al., 2008; Arking et al., 2008; Bakkaloglu et al., 2008; Rossi et al., 2008). Deficits in CNTNAP2 have also been associated with other psychiatric conditions, including Tourette’s syndrome, obsessive-compulsive disorder, mental retardation (Verkerk et al., 2003), epilepsy (Strauss et al., 2006), and typical specific language impairment in children (Vernes et al., 2008). In the peripheral nervous system, CNTNAP2 regulates potassium channel clustering near nodes of Ranvier (Poliak & Peles, 2003). More recently, Abrahams et al. (2007) investigated the potential function of CNTNAP2 in the superior temporal gyrus and cerebral cortex in midgestation human fetal brains by using gene profiling and followed by selective in situ hybridization. Their results suggested that human CNTNAP2 expression was enriched in circuits involved in higher cortical functions, including language. The functional implication of CNTNAP2 in the central nervous system and recurrence of the deficits in schizophrenia, autism, and other psychiatric conditions make it a very interesting candidate.
Table 8.1 Loci Overlapping in CNV Reports
SOURCE: Authors.
Table 8.2 Candidate Genes from CNV Reports
SOURCE: Authors.
Several genes involved in neural function were affected by CNVs in the Xu et al. studies (Xu et al., 2008; Xu et al., 2009). Notably, two members of the RAPGEF family, RAPGEF2 and RAPGEF 6, were affected by independent CNVs (Xu et al., 2008; Xu et al., 2009). This gene family is implicated in neuronal migration and arborization. Additional CNVs affected Neuregulin-3 (NRG3) (Xu et al., 2009) and EphB1 (Xu et al., 2008) a receptor for ephrin, which participates in axon guidance.
Another interesting set of candidates is related to microRNA biogenesis and microRNA-mediated translation control. Besides DGCR8 within 22q11DS, one of the top candidate genes within the 15q11.2 CNV region is CYFIP1, which binds the fragile X mental retardation protein FMRP1 and translation initiation factor eIF4E (Napoli et al., 2008). Both FMRP1 and eIF4E have been implicated in microRNA-mediated translational control machinery (Jin et al., 2004; Pillai et al., 2005). More interesting, DICER1, another microRNA processing enzyme, has also been shown to be affected by a de novo CNV at 14q32 (Xu et al., 2008).
Three studies reported schizophrenia-associated CNVs in 16p11.2-p13.1, which is a locus previously associated with autism, bipolar disorder, and mental retardation. Possible candidate genes in this region include NDE1, which binds DISC1, a well-known schizophrenia susceptibility gene; NXPH2, which binds Neurexins; EEF2K, a key kinase downstream of calmodulin; and CDR2, which is a target of autoantibodies in cerebellar diseases. A 17p12 deletion was found in four of five affected family members in the Kirov et al. study, as well as two other schizophrenia cases without neurologic symptoms (Mizuguchi et al., 2008; Kunugi et al., 2008; Kirov et al., 2009). This locus contains peripheral myelin protein 22 (PMP22), which causes Charcot-Marie-Tooth Syndrome, a hereditary neuropathy. Finally, 1q21.1 is a region previously reported to be deleted in cases of mental retardation and autism; one interesting candidate gene within this region is gap junction protein alpha 8/connexin50, which is associated with cataracts and corneal abnormalities.
In addition to the genes discussed here, there are several excellent candidate genes located within rarer CNVs (table 8.2) implicating a number of biological pathways, including glutamate, dopamine, and acetylcholine signaling pathways, as well as aspects of cell-cell and cell-matrix adhesion and signaling. Additionally, CNV disrupted loci may contain regulatory elements, transcription factors, or microRNAs that affect genes at a distance; thus, the known genes may not represent the full extent of the genetic information affected by these genomic rearrangements.
KEY AREAS FOR FUTURE RESEARCH
Selected Readings
Cirulli, E. T. & Goldstein, D. B. (2010). Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nature Reviews Genetics 11(6): 415–425.
Goldstein, D. B. (2009). Common genetic variation and human traits. New England Journal of Medicine 360(17): 1696–1698.
ISC. (2008). Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455(7210): 237–241.
Karayiorgou, M., Simon, T. J., & Gogos, J. A. (2010). 22q11.2 microdeletions: Linking DNA structural variation to brain dysfunction and schizophrenia. Nature Reviews Neuroscience 11(6): 402–416.
Redon, R., Ishikawa, S., Fitch, K. R., Feuk, L., Perry, G. H., Andrews, T. D., Fiegler, H., Shapero, M. H., Carson, A. R., Chen, W., et al. (2006). Global variation in copy number in the human genome. Nature 444(7118): 444–454.
Sebat, J., Lakshmi, B., Troge, J., Alexander, J., Young, J., Lundin, P., Maner, S., Massa, H., Walker, M., Chi, M., et al. (2004). Large-scale copy number polymorphism in the human genome. Science 305(5683): 525–528.
Xu, B., Roos, J. L., Levy, S., van Rensburg, E. J., Gogos, J. A., & Karayiorgou, M. (2008). Strong association of de novo copy number mutations with sporadic schizophrenia. Nature Genetics 40(7): 880–885.
References
Abrahams, B. S., Tentler, D., Perederiy, J. V., Oldham, M. C., Coppola, G., & Geschwind, D. H. (2007). Genome-wide analyses of human perisylvian cerebral cortical patterning. Proceedings of the National Academy of Sciences U.S.A. 104(45): 17849–17854.
Alarcon, M., Abrahams, B. S., Stone, J. L., Duvall, J. A., Perederiy, J. V., Bomar, J. M., Sebat, J., Wigler, M., Martin, C. L., Ledbetter, D. H., et al. (2008). Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. American Journal of Human Genetics 82(1): 150–159.
Altshuler, D., Daly, M. J., & Lander, E. S. (2008). Genetic mapping in human disease. Science 322(5903): 881–888.
Arking, D. E., Cutler, D. J., Brune, C. W., Teslovich, T. M., West, K., Ikeda, M., Rea, A., Guy, M., Lin, S., Cook, E. H., et al. (2008). A common genetic variant in the Neurexin superfamily member CNTNAP2 increases familial risk of autism. American Journal of Human Genetics 82(1): 160–164.
Bakkaloglu, B., O’Roak, B. J., Louvi, A., Gupta, A. R., Abelson, J. F., Morgan, T. M., Chawarska, K., Klin, A., Ercan-Sencicek, A. G., Stillman, A. A., et al. (2008). Molecular cytogenetic analysis and resequencing of Contactin associated protein-like 2 in autism spectrum disorders. American Journal of Human Genetics 82(1): 165–173.
Bassett, A. S., Chow, E. W., AbdelMalik, P., Gheorghiu, M., Husted, J., & Weksberg, R. (2003). The schizophrenia phenotype in 22q11 deletion syndrome. American Journal of Psychiatry 160(9): 1580–1586.
Bassett, A. S., Hodgkinson, K., Chow, E. W., Correia, S., Scutt, L. E., & Weksberg, R. (1998). 22q11 deletion syndrome in adults with schizophrenia. American Journal of Medical Genetics 81(4): 328–337.
Bodmer, W. & Bonilla, C. (2008). Common and rare variants in multifactorial susceptibility to common diseases. Nature Genetics 40(6): 695–701.
Chow, E. W., Watson, M., Young, D. A., & Bassett, A. S. (2006). Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophrenia Research 87(1–3): 270–278.
Chow, E. W., Zipursky, R. B., Mikulis, D. J., & Bassett, A. S. (2002). Structural brain abnormalities in patients with schizophrenia and 22q11 deletion syndrome. Biological Psychiatry 51(3): 208–215.
Conrad, D. F., Pinto, D., Redon, R., Feuk, L., Gokcumen, O., Zhang, Y., Aerts, J., Andrews, T. D., Barnes, C., Campbell, P., et al. (2010). Origins and functional impact of copy number variation in the human genome. Nature 464(7289): 704–712.
Ferreira, M. A., O’Donovan, M. C., Meng, Y. A., Jones, I. R., Ruderfer, D. M., Jones, L., Fan, J., Kirov, G., Perlis, R. H., Green, E. K., et al. (2008). Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nature Genetics 40(9): 1056–1058.
Friedman, J. I., Vrijenhoek, T., Markx, S., Janssen, I. M., van der Vliet, W. A., Faas, B. H., Knoers, N. V., Cahn, W., Kahn, R. S., Edelmann, L., et al. (2008). CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Molecular Psychiatry 13(3): 261–266.
Goldstein, D. B. (2009). Common genetic variation and human traits. New England Journal of Medicine 360(17): 1696–1698.
Gothelf, D., Feinstein, C., Thompson, T., Gu, E., Penniman, L., Van Stone, E., Kwon, H., Eliez, S., & Reiss, A. L. (2007). Risk factors for the emergence of psychotic disorders in adolescents with 22q11.2 deletion syndrome. American Journal of Psychiatry 164(4): 663–669.
Iafrate, A. J., Feuk, L., Rivera, M. N., Listewnik, M. L., Donahoe, P. K., Qi, Y., Scherer, S. W., & Lee, C. (2004). Detection of large-scale variation in the human genome. Nature Genetics 36(9): 949–951.
Ingason, A., Rujescu, D., Cichon, S., Sigurdsson, E., Sigmundsson, T., Pietilainen, O. P., Buizer-Voskamp, J. E., Strengman, E., Francks, C., Muglia, P., et al. (2009). Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Molecular Psychiatry 16(1): 17–25.
ISC. (2008). Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455(7210): 237–241.
Itsara, A., Cooper, G. M., Baker, C., Girirajan, S., Li, J., Absher, D., Krauss, R. M., Myers, R. M., Ridker, P. M., Chasman, D. I., et al. (2009). Population analysis of large copy number variants and hotspots of human genetic disease. American Journal of Human Genetics 84(2): 148–161.
Jin, P., Zarnescu, D. C., Ceman, S., Nakamoto, M., Mowrey, J., Jongens, T. A., Nelson, D. L., Moses, K., & Warren, S. T. (2004). Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nature Neuroscience 7(2): 113–117.
Karayiorgou, M. & Gogos, J.A. (2006). Schizophrenia genetics: Uncovering positional candidate genes. European Journal of Human Genetics 14(5): 512–519.
Karayiorgou, M., Morris, M. A., Morrow, B., Shprintzen, R. J., Goldberg, R., Borrow, J., Gos, A., Nestadt, G., Wolyniec, P. S., & Lasseter, V. K. (1995). Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proceedings of the National Academy of Sciences U.S.A. 92(17): 7612–7616.
Kirov, G., Grozeva, D., Norton, N., Ivanov, D., Mantripragada, K. K., Holmans, P., Craddock, N., Owen, M. J., & O’Donovan, M. C. (2009). Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Human Molecular Genetics 18(8): 1497–1503.
Kirov, G., Gumus, D., Chen, W., Norton, N., Georgieva, L., Sari, M., O’Donovan, M. C., Erdogan, F., Owen, M. J., Ropers, H. H., et al. (2008). Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Human Molecular Genetics 17(3): 458–465.
Kunugi, H., Ozeki, Y., Mizuguchi, T., Hirabayashi, N., Ogawa, M., Ohmura, N., Moriuchie, M., Haradae, N., Matsumotob, N., & Kunugi, H. (2008). A case of schizophrenia with chromosomal microdeletion of 17p11.2 containing a myelin-related gene PMP22. Open Psychiatry Journal 2: 1–4.
McCarthy, S. E., Makarov, V., Kirov, G., Addington, A. M., McClellan, J., Yoon, S., Perkins, D. O., Dickel, D. E., Kusenda, M., Krastoshevsky, O., et al. (2009). Microduplications of 16p11.2 are associated with schizophrenia. Nature Genetics 41(11): 1223–1227.
Mizuguchi, T., Hashimoto, R., Itokawa, M., Sano, A., Shimokawa, O., Yoshimura, Y., Harada, N., Miyake, N., Nishimura, A., Saitsu, H., et al. (2008). Microarray comparative genomic hybridization analysis of 59 patients with schizophrenia. Journal of Human Genetics 53(10): 914–919.
Mukai, J., Dhilla, A., Drew, L. J., Stark, K. L., Cao, L., MacDermott, A. B., Karayiorgou, M., & Gogos, J. A. (2008). Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nature Neuroscience 11(11): 1302–1310.
Murphy, K. C., Jones, L. A., & Owen, M. J. (1999). High rates of schizophrenia in adults with velo-cardio-facial syndrome. Archives of General Psychiatry 56(10): 940–945.
Napoli, I., Mercaldo, V., Boyl, P. P., Eleuteri, B., Zalfa, F., De Rubeis, S., Di Marino, D., Mohr, E., Massimi, M., Falconi, M., et al. (2008). The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell 134(6): 1042–1054.
O’Donovan, M. C., Craddock, N., Norton, N., Williams, H., Peirce, T., Moskvina, V., Nikolov, I., Hamshere, M., Carroll, L., Georgieva, L., et al. (2008). Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nature Genetics 40(9): 1053–1055.
Paterlini, M., Zakharenko, S. S., Lai, W. S., Qin, J., Zhang, H., Mukai, J., Westphal, K. G., Olivier, B., Sulzer, D., Pavlidis, P., et al. (2005). Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nature Neuroscience 8(11): 1586–1594.
Paylor, R., Glaser, B., Mupo, A., Ataliotis, P., Spencer, C., Sobotka, A., Sparks, C., Choi, C. H., Oghalai, J., Curran, S., et al. (2006). Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: Implications for 22q11 deletion syndrome. Proceedings of the National Academy of Sciences U.S.A. 103 (20): 7729–7734.
Pillai, R. S., Bhattacharyya, S. N., Artus, C. G., Zoller, T., Cougot, N., Basyuk, E., Bertrand, E., & Filipowicz, W. (2005). Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science 309(5740): 1573–1576.
Poliak, S. & Peles, E. (2003). The local differentiation of myelinated axons at nodes of Ranvier. Nature Reviews Neuroscience 4(12): 968–980.
Pulver, A. E., Nestadt, G., Goldberg, R., Shprintzen, R. J., Lamacz, M., Wolyniec, P. S., Morrow, B., Karayiorgou, M., Antonarakis, S. E., Housman, D., et al. (1994). Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. Journal of Nervous Mental Disorders 182(8): 476–478.
Redon, R., Ishikawa, S., Fitch, K. R., Feuk, L., Perry, G. H., Andrews, T. D., Fiegler, H., Shapero, M. H., Carson, A. R., Chen, W., et al. (2006). Global variation in copy number in the human genome. Nature 444(7118): 444–454.
Reissner, C., Klose, M., Fairless, R., & Missler, M. (2008). Mutational analysis of the Neurexin/neuroligin complex reveals essential and regulatory components. Proceedings of the National Academy of Sciences U.S.A. 105(39): 15124–15129.
Rossi, E., Verri, A. P., Patricelli, M. G., Destefani, V., Ricca, I., Vetro, A. Ciccone, R., Giorda, R., Toniolo, D., Maraschio, P., et al. (2008). A 12Mb deletion at 7q33–q35 associated with autism spectrum disorders and primary amenorrhea. European Journal of Medical Genetics 51(6): 631–638.
Rujescu, D., Ingason, A., Cichon, S., Pietilainen, O. P., Barnes, M. R., Toulopoulou, T., Picchioni, M., Vassos, E., Ettinger, U., Bramon, E., et al. (2009). Disruption of the Neurexin 1 gene is associated with schizophrenia. Human Molecular Genetics 18(5): 988–996.
Sanders, A. R., Duan, J., Levinson, D. F., Shi, J., He, D., Hou, C., Burrell, G. J., Rice, J. P., Nertney, D. A., Olincy, A., et al. (2008). No significant association of 14 candidate genes with schizophrenia in a large European ancestry sample: Implications for psychiatric genetics. American Journal of Psychiatry 165(4): 497–506.
Sebat, J., Lakshmi, B., Troge, J., Alexander, J., Young, J., Lundin, P., Maner, S., Massa, H., Walker, M., Chi, M., et al. (2004). Large-scale copy number polymorphism in the human genome. Science 305(5683): 525–528.
Stark, K. L., Xu, B., Bagchi, A., Lai, W. S., Liu, H., Hsu, R., Wan, X., Pavlidis, P., Mills, A. A., Karayiorgou, M., et al. (2008). Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nature Genetics 40(6): 751–760.
Stefansson, H., Rujescu, D., Cichon, S., Pietilainen, O. P., Ingason, A., Steinberg, S., Fossdal, R., Sigurdsson, E., Sigmundsson, T., Buizer-Voskamp, J. E., et al. (2008). Large recurrent microdeletions associated with schizophrenia. Nature 455(7210): 232–236.
Strauss, K. A., Puffenberger, E. G., Huentelman, M. J., Gottlieb, S., Dobrin, S. E., Parod, J. M., Stephan, D. A., & Morton, D. H. (2006). Recessive symptomatic focal epilepsy and mutant Contactin-associated protein-like 2. New England Journal of Medicine 354(13): 1370–1377.
Sullivan, P. F., Kendler, K. S., & Neale, M. C. (2003). Schizophrenia as a complex trait: Evidence from a meta-analysis of twin studies. Archives of General Psychiatry 60(12): 1187–1192.
van Amelsvoort, T., Daly, E., Henry, J., Robertson, D., Ng, V., Owen, M., Murphy, K. C., & Murphy, D. G. (2004a). Brain anatomy in adults with velocardiofacial syndrome with and without schizophrenia: Preliminary results of a structural magnetic resonance imaging study. Archives of General Psychiatry 61(11); 1085–1096.
van Amelsvoort, T., Henry, J., Morris, R., Owen, M., Linszen, D., Murphy, K., & Murphy, D. (2004b). Cognitive deficits associated with schizophrenia in velo-cardio-facial syndrome. Schizophrenia Research 70(2–3): 223–232.
Verkerk, A. J., Mathews, C. A., Joosse, M., Eussen, B. H., Heutink, P., & Oostra, B. A. (2003). CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics 82(1): 1–9.
Vernes, S. C., Newbury, D. F., Abrahams, B. S., Winchester, L., Nicod, J., Groszer, M., Alarcón, M., Oliver, P. L., Davies, K. E., Geschwind, D. H., et al. (2008). A functional genetic link between distinct developmental language disorders. New England Journal of Medicine 359(22): 2337–2345.
Vrijenhoek, T., Buizer-Voskamp, J. E., van der Stelt, I., Strengman, E., GROUP Consortium, Sabatti, C., Geurts van Kessel, A., Brunner, H. G., Ophoff, R. A., & Veltman, J. A. (2008). Recurrent CNVs disrupt three candidate genes in schizophrenia patients. American Journal of Human Genetics 83(4): 504–510.
Walsh, T., McClellan, J., McCarthy, S., Addington, A., Pierce, S., Cooper, G., Nord, A., Kusenda, M., Malhotra, D., Bhandari, A., et al. (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320(5875): 539-543.
Wang, K., Zhang, H., Ma, D., Bucan, M., Glessner, J. T., Abrahams, B. S., Salyakina, D., Imielinski, M., Bradfield, J. P., Sleiman, P. M. A., et al. (2009). Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459(7246): 528–533.
Xu, B., Roos, J. L., Levy, S., van Rensburg, E. J., Gogos, J. A., & Karayiorgou, M. (2008). Strong association of de novo copy number mutations with sporadic schizophrenia. Nature Genetics 40(7): 880–885.
Xu, B., Woodroffe, A., Rodriguez-Murillo, L., Roos, J. L., van Rensburg, E. J., Abecasis, G. R., Gogo, J., & Karayiorgou, M. (2009). Elucidating the genetic architecture of familial schizophrenia using rare copy number variant and linkage scans. Proceedings of the National Academy of Sciences U.S.A. 106(39): 16746–16751.