6 | PRINCIPLES OF MOLECULAR BIOLOGY
STEVEN E. HYMAN AND ERIC J. NESTLER
Advances in molecular biology and in understanding the genetics of complex human phenotypes have opened a new world of possibilities for research on mental disorders and their treatment. However, because the syndromes with which psychiatry is ultimately charged are expressed at the level of behavior, molecular and genetic work must be complemented by ongoing research in the more integrative aspects of neuroscience and behavioral science described elsewhere in this textbook. Thus, for example, the pathophysiology of mental disorders depends on the complex interaction of genetic (bottom-up) factors and environmental (top-down) factors affecting the development and subsequent function of the brain. Many scientific disciplines must contribute to that understanding.
Despite substantial evidence that genes and environment are inseparable partners in brain development and plasticity—and therefore in the formation of personalities, talents, and all other aspects of behavior, including vulnerability to mental illness and very likely responsiveness to treatments for mental illness—simplistic versions of the nature–nurture debate never seem to disappear. It is true that from the time a one-cell embryo is formed from the fusion of sperm and egg, the genetic endowment of that individual is fixed. An important focus of current research, however, is to find out precisely what that means with respect to behavioral traits. It is now becoming increasingly clear that behavioral phenotypes, including vulnerability to serious mental disorders such as schizophrenia, bipolar disorder, depression, and autism, result from the interaction of multiple (perhaps hundreds of) genes, very likely acting at different times during brain development in interaction with the environment. An important goal of psychiatric research is therefore to understand how genetic information is read out during development and how the brain changes as a result of stochastic or random processes during development, as well as in response to experience of the sensory world and other environmental inputs, such as drugs, infections, and injuries.
The goal of this chapter is to describe the fundamental molecular processes by which information is encoded in the genome and how this information is expressed within an environmental context. We describe what genes are, how they function, and how their expression is regulated by signals from outside the cell. This chapter shows that the control of gene expression by extracellular signals is a critical arena for gene–environment interactions relevant to psychiatry (Hyman and Nestler, 1993). Chapter 7 builds on this foundation by describing epigenetic regulation in psychiatry, namely, how stable changes in the structure of chromatin (the combination of DNA and proteins that comprise the nucleus) at particular genes—and hence in the behaviors those genes regulate—can be produced without changes in the genetic code itself.
NUCLEIC ACIDS
Deoxyribonucleic acid (DNA) contains the genetic blueprints of cells, that is, the information to produce ribonucleic acid (RNA) and proteins, which in turn create the fundamental structural and functional properties of our cells. The latest estimate is that the human genome contains approximately 25,000 genes, which use about 1.5% of ~3,000,000,000 base pairs of DNA. Deoxyribonucleic acid exists as a double helix, each strand of which is an unbranched chain built out of small building blocks called nucleotide bases. DNA and RNA are synthesized from four types of nucleotide bases. The four nucleotides that make up DNA are the purines, adenine (A) and guanine (G), and the pyrimidines, cytosine (C) and thymine (T), each containing a deoxyribose sugar group. In RNA the pyrimidine uracil (U) takes the place of thymine, and the sugar group is ribose instead of deoxyribose. Individual nucleotides are joined into strands of DNA or RNA via the phosphate groups that form a phosphodiester linkage.
The alternating deoxyribose sugar and phosphate groups that connect the bases of each DNA strand form a “sugar-phosphate backbone” on the outside of the double helix with the bases arrayed on the inside of the double helix (Fig. 6.1). In DNA, the nucleotide base A is paired with (or is complementary to) T on the opposite strand, and G is paired with C. In RNA, U, which is structurally similar to T, is also complementary to A. The rules of base pairing observed in DNA result from the fact that only complementary pairs of nucleotides form a maximum number of stabilizing hydrogen bonds. Any other arrangement of bases destabilizes the structure of the DNA. A critical property of a linear polymer such as DNA (or RNA) is that it can serve as a template for the synthesis of other macromolecules. The principle of complementary base pairing provides the mechanism by which information can be transferred. An enzyme, a DNA polymerase in the case of DNA replication, or RNA polymerase in the case of transcription of DNA into RNA, can proceed down a template strand of DNA, adding a nucleotide base complementary to the base on the template strand as it forms a new strand of nucleic acid (Fig. 6.1).
Although the actual enzymatic steps involved in the replication of DNA are quite complex, the overall principles are simple. Replication begins with separation of the two complementary DNA strands in a local region. Each strand then serves as a template for a new DNA molecule by the sequential polymerization of nucleotides. Eventually the replication process generates two complete DNA double helices, each identical in sequence to the original. Replication of DNA is said to be semiconservative because each daughter DNA molecule contains one of the original parental strands plus one newly synthesized strand. Transcription of DNA into RNA is conceptually similar except that only one DNA strand serves as a template and, when synthesis of the RNA is complete, it is released and the DNA strands can reanneal into their stable double-helical structure.
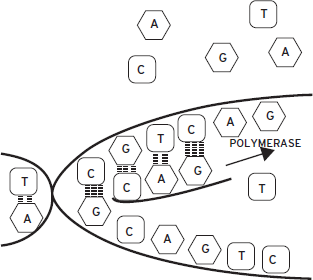
Figure 6.1 Schematic of complementary base pairing of deoxyribonucleic acid (DNA). Two complementary strands of DNA hybridize with one another to form a double helix. The sugar-phosphate backbones of the two strands are found on the outside of the double helix; the bases are found on the inside. Formation of a DNA double helix is stabilized when hydrogen bonds form between complementary bases of the two strands. Two hydrogen bonds form when A is across from T; three hydrogen bonds form when G is across from C. Other appositions of bases are destabilizing and do not occur. A strand of DNA can serve as a template for a second strand of nucleic acid (DNA or ribonucleic acid [RNA]). A polymerase enzyme assembles individual nucleotides into a new strand using the first strand as a template. The sequence of the new strand is therefore determined by the template, permitting the transfer of information across generations. Complementary base pairing is also the fundamental principle in the synthesis of RNA from DNA and in many experimental situations in which it is desirable to detect the presence of a specific DNA or RNA strand.
INFORMATION FLOWS FROM DNA TO RNA TO PROTEIN
DNA carries information in its linear sequence of nucleotides. Although the linear polynucleotide structure of DNA is well suited for the stable storage of information and for self-replication, its chemical simplicity and its relatively rigid helical structure limit its biological functions. Thus, the information contained within DNA must be read out to yield RNA and proteins. Like DNA, RNA is chemically quite simple (i.e., composed of four nucleotides), but because it is a flexible single strand, free to fold into numerous conformations, it is functionally more versatile than DNA.
Messenger RNA (mRNA) functions as an intermediate between the sequence of DNA that comprises the transcribed regions of genes and the sequence of proteins. Not all RNA serve as mRNA, however. Other RNAs function in varied roles in cells. Ribosomes, the organelles on which proteins are synthesized, are constructed out of complexes of several types of ribosomal RNA (rRNAs) with proteins. Transfer RNAs (tRNAs) transport specific amino acids to the ribosomes for incorporation into proteins during the process in which mRNA is translated into protein. More recently, scientists have characterized several other types of non-coding RNAs, categorized as long or short based on their length. These non-coding RNAs are considered a form of epigenetic regulation and are discussed in greater detail in Chapter 7. Briefly, long non-coding RNAs, generally characterized as >200 nucleotides in length, regulate chromatin structure and gene expression (Taft et al., 2010). The most prevalent short non-coding RNAs, termed microRNAs (miRNAs), bind to specific mRNAs and inhibit their translation and/or promote their degradation (Bartel, 2009; Vo et al., 2010). The discovery of miRNAs has been translated into a powerful tool called RNA interference or RNAi, which uses small hairpin RNAs (shRNAs), which are cleaved into small inhibitory RNAs, to experimentally suppress expression of a targeted mRNA and protein.
Like DNA, mRNA carries information encoded in its linear sequence of nucleotides. DNA and mRNA specify amino acid building blocks for proteins in linear stretches of three nucleotides. Proteins consist of unbranched chains of amino acid building blocks. An amino acid is a small molecule that contains an amino group (NH2) and a carboxylic acid or carboxy group (COOH) plus a variable side chain. The side chains used by the 20 common amino acids differ markedly in size, shape, hydrophobicity, and charge. Amino acids are linked to each other by peptide bonds that join the amino group of one amino acid to the carboxy group of another amino acid.
Within the portion of an mRNA that is translatable into a protein, each successive group of three nucleotide bases (called a codon) specifies either one amino acid or termination of the protein chain. The rules specifying the correspondence between a codon and an amino acid are called the genetic code. Because RNA is a linear polymer of 4 nucleotides, there are 43 or 64 possible codons but only 20 amino acids. As a result, although each codon specifies only a single amino acid, most amino acids are specified by more than one codon. The genetic code is therefore said to be degenerate. With only a few minor exceptions, the code has been conserved across evolution. The codons in an mRNA molecule do not interact directly with the amino acids they specify; the translation of mRNA into protein depends on the presence of tRNAs that serve as adapter molecules that recognize a specific codon and the corresponding amino acid (Fig. 6.2).
The ribosome is a structure composed of proteins and structural rRNAs; these organelles provide a structure on which tRNAs can interact (via their anticodons) with the codons of an mRNA in sequential order. The ribosome finds a specific start site on the mRNA that sets the reading frame and then moves along the mRNA molecule progressively, translating the nucleotide sequence one codon at a time, using tRNAs to add amino acids to the growing end of the polypeptide chain (Fig. 6.3).

Figure 6.2 Transfer RNA (tRNA): Transfer RNA (tRNA) is a single strand of RNA that folds on itself through the apposition of complementary base pairs and the subsequent formation of hydrogen bonds, indicated by the dashed lines in the figure. One of the loops formed contains the anticodon, the sequence of three nucleotides on the tRNA that binds to the complementary codon on a messenger RNA molecule. For the anticodon AGA shown in the figure, the corresponding codon on the mRNA would be UCU. The free end of the tRNA binds to a specific amino acid. Each tRNA, with a given anticodon, binds only one type of amino acid determined by the genetic code. In the case shown in the figure, the amino acid bound would be serine. From Hyman and Nestler (1993) reprinted with permission from The Molecular Foundations of Psychiatry, (Copyright ©1993). American Psychiatric Publishing.
Over the past decade, it has become clear that a small number of mRNAs, along with associated protein translation machinery, are transported to dendrites of neurons, where they mediate rapid changes in protein synthesis in response to synaptic activity (Wang et al., 2010). This transport is mediated by the presence of specific sequences in the 3’-untranslated region of certain mRNAs, which are targeted by RNA-binding proteins to form RNA granules for transport. Fragile X mental retardation protein (FMRP) is an example of one of these RNA-binding proteins, which plays an important role in controlling dendritic protein synthesis. Loss of FMRP expression causes Fragile X mental retardation syndrome (see Chapter 7).
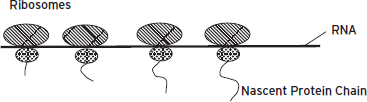
Figure 6.3 Protein translation. Ribosomal subunits bind together on mature messenger ribonucleic acids (mRNAs) to form actively translating ribosomes. The ribosome begins adding amino acids when it reaches a “start codon” on the mRNA and processes down the mRNA, one codon at a time, adding the appropriate amino acid as it is delivered by a transfer RNA (tRNA). When a “stop codon” is reached, the ribosome releases the polypeptide chain and dissociates from the mRNA. Each mRNA that is being actively translated has multiple ribosomes moving sequentially down its length, forming a polyribosome complex. From Hyman and Nestler (1993) reprinted with permission from The Molecular Foundations of Psychiatry, (Copyright ©1993). American Psychiatric Publishing.
In contrast with nucleic acids, which are constructed of four bases that are chemically similar, proteins are constructed out of 20 very different amino acids. By incorporating so many different types of amino acids, each with its chemically diverse side chains, proteins have extraordinary functional versatility, unlike DNA or RNA. The specific properties of proteins depend not only on the linear sequence of their amino acid building blocks (primary structure), but also on the tendency of certain combinations of amino acids to form intrinsic structural motifs (secondary structures, for example, α helices or β sheets) and by their folded three-dimensional characteristics (tertiary structure). In addition, proteins form stable interactions (complexes) with other proteins (quaternary structure). In such cases, the individual polypeptide chains are called subunits. The folding of proteins, and the interactions of proteins with each other and with other molecules such as nucleic acids, is highly regulated by chemical modifications of the protein, most often of particular side chains. For example, one ubiquitous mechanism of regulation of protein function is by the covalent addition of a phosphate group (by specific enzymes called protein kinases) to the hydroxyl groups found in serine, threonine, or tyrosine side chains (Chapters 4 and 5). Cells may contain tens of thousands of distinct proteins, each with unique structural and functional properties, including neurotransmitter receptors, neuropeptides, ion channels, enzymes, and a very large number of other types of proteins.
GENES AND CHROMOSOMES
As a first approximation, genes were initially defined as stretches of DNA that encode a single protein or a single functional RNA, such as an rRNA or a tRNA. This rule breaks down quickly, however, because there are mechanisms, such as alternative splicing of the primary RNA transcript into different mRNAs, that may intervene between a given gene and a finished protein. Moreover, individual protein precursors may be cleaved into numerous functionally active peptides. As a result of these and other processes, many individual genes actually encode multiple proteins, although we still do not know what fraction of all genes function in this manner.
Genes are arrayed on extremely long chains of DNA called chromosomes. Eukaryotic chromosomes contain not only genes, but also large amounts of intergenic DNA. Indeed, within the human genome, the ~25,000 genes take up perhaps only 3% of chromosomal DNA. Moreover, genes are not distributed equally along the chromosomes but are often found in clusters. Some chromosomes are “gene rich” and others are relatively “gene poor.” Within intergenic regions there is a great deal of DNA with unique sequences of unknown function, as well as long stretches of tandemly repeated sequences known as satellite DNA. Included within these regions are retrotransposons, such as LINE-1 (long interspersed nuclear element-1) repeats, which are transcribed by RNA polymerase and encode several enzymes including reverse transcriptase (an enzyme that synthesizes a strand of DNA from RNA). The resulting DNA strands can then incorporate into the genome, where they can disrupt normal gene expression. An interesting line of current research is to understand the role of LINE-1 and related retrotransposons as a novel mechanism controlling neural gene expression (see Chapter 7).
The chromosomes of eukaryotic cells are so long (~2 meters if stretched out linearly) that they would not fit in the nucleus in their extended form. Thus, stretches of the DNA that are not being actively transcribed are tightly packed into a conformation described as a coiled coil, which permits the chromosomes to fit within the nucleus. To create packed conformations, lengths of DNA are coiled around structural and regulatory proteins, of which the most important are the histones. These structures are termed nucleosomes. Regions of DNA that are being actively transcribed into RNA may be greater than 1000-fold more extended than regions that are transcriptionally quiescent. The unwinding of tightly packed nucleosomes, which enables their expression, is mediated by highly complex modifications of histones and many other nuclear proteins, which are discussed in detail in Chapter 7 (Borrelli et al., 2008).
GENE EXPRESSION
As described in the preceding paragraphs, proteins are not synthesized directly from the DNA that encodes them, but in two sequential processes, transcription of DNA into mRNA, which occurs in the nucleus, and translation of the mRNA into protein according to the rules of the genetic code, which occurs in the cytoplasm. Transcription of protein-coding genes can be divided into three major steps. First, the enzyme primarily involved in RNA synthesis, RNA polymerase, must interact with the gene at an appropriate transcription start site and begin transcribing (initiation). Second, the RNA polymerase must successfully transcribe an appropriate length of RNA (elongation). Third, transcription of the RNA must terminate appropriately. The resulting RNA is then posttranscriptionally processed. It receives a stretch of adenines, a poly-(A) tail, which makes it more stable in the cell. This process of polyadenylation is dynamically regulated and can exert a powerful influence on expression of the resulting mRNAs (Proudfoot, 2011). It is also spliced to remove internal sequences (introns) that are not appropriate for translation into protein in that particular cell. The spliced “exportable” sequences (exons) exit the nucleus to be translated into protein in the cytoplasm (Fig. 6.4). Alternative splicing vastly increases the number of protein products produced by the cell, with certain genes, for example, the neurexins, generating hundreds of splice variants (Padgett, 2012).
TRANSCRIPTIONAL CONTROL
We have now reviewed the flow of information from gene to protein. Every step along the way is regulated, but the major control point in reading out the information contained within the genome is transcription initiation. Indeed, this is the step at which environmental signals often exert powerful regulatory control on gene expression, during development and in mature cells. How does this come about?
In addition to encoding information that ultimately directs the synthesis of proteins, genes contain regulatory information. Every cell in our bodies contains all of our genes in its nucleus, but not every gene is active in every cell. Our cells differ—indeed, their identity is defined—by the fact that each type of cell in the body expresses only a subset of the entire complement of genes. In any given cell, some genes are “on,” being read out to make mRNA and hence proteins, and the rest are silent. Thus, for example, in the red blood cell precursors of the bone marrow, the genes that encode globins, proteins that are the critical building blocks of hemoglobin, are actively making globin-encoding mRNA. In our midbrains, a subset of cells are making a protein called tyrosine hydroxylase, the rate-limiting enzyme for the synthesis of catecholamines, like dopamine and norepinephrine. It would not be adaptive for these cells to synthesize large amounts of hemoglobin, just as red blood cell precursors need not use energy to synthesize tyrosine hydroxylase. The processes by which certain genes are “silenced” in a tissue-specific manner during development are now increasingly understood at the chromatin or epigenetic level (Chapter 7).

Figure 6.4 RNA splicing. Horizontal black lines represent deoxyribonucleic acid (DNA) regulatory regions and introns. Black, gray, and white rectangles represent exons. The region to the left of the first exon is the 5′ regulatory region of the gene, but cis-regulatory elements are also found in introns and sometimes downstream of the last exon. The primary transcript, also known as heterogeneous ribonucleic acid (hnRNA), contains exons and introns and gives rise, in the case shown, to two alternatively spliced messenger ribonucleic acids (mRNAs): one containing exons 1, 2, and 4, and one containing exons 1, 3, and 4. These splice variants, after export from the nucleus and translation in the cytoplasm, give rise to distinct proteins.
Regulatory sequences within genes work by virtue of their ability to bind specific proteins. Certain regulatory sequences specify the beginnings and ends of segments of DNA that can be transcribed into RNA. Other regulatory sequences determine in what cell types and under what circumstances their gene can be read out. DNA sequences that subserve such control functions are often called cis-regulatory elements (Fig. 6.5). The term cis refers to the fact that the relevant regulatory sequences are physically linked, on the DNA, to the region being controlled, which is usually a segment of DNA that can be transcribed into RNA. The proteins that bind to cis elements have been described as trans-acting factors because they may be encoded anywhere in the genome rather than on the same stretch of DNA that they regulate. Those proteins that are involved in specifying whether, and under what circumstances, a gene will be transcribed are more generally known as transcription factors. Many transcription factors bind DNA directly; others interact only indirectly via protein–protein interactions with factors that bind DNA.
Those cis-regulatory elements that specify the site within a gene at which transcription starts, and on which the complex of proteins that forms the basal transcription apparatus is assembled, are called the basal or core promoter. The core promoters for many protein-encoding genes consist of a sequence motif rich in the nucleotides A and T, called the TATA box. The TATA box is generally located ~30 nucleotides upstream of the actual site at which transcription of DNA into RNA is initiated. In the nervous system, however, numerous genes do not contain identifiable TATA boxes, which means that their transcription start sites are defined differently. In eukaryotes, transcription of protein-coding genes is carried out by the enzyme RNA polymerase II, which does not directly contact DNA but interacts with the complex of proteins that assembles at the TATA box or distinct transcription start site (Fig. 6.5).
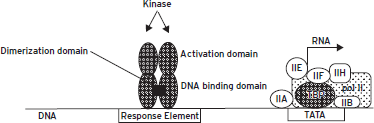
Figure 6.5 Cis- and trans-regulation. The figure shows two cis-regulatory elements (open rectangles) along the stretch of deoxyribonucleic acid (DNA) (thin line). The element to the left represents a response element that serves as a binding site for a hypothetical transcription factor that binds as a homodimer. The other is the thymine adenine thymine adenine (TATA) element, shown binding the TATA binding protein (TBP). Multiple general transcription factors and ribonucleic acid (RNA) polymerase II (pol II) associate with TBP. This basal transcription apparatus recruits RNA polymerase II into the complex and also forms the substrate for interactions with activator proteins, such as those binding to the activator elements shown. The transcription factor shown binding to the response element is a substrate for a protein kinase that phosphorylates its activation domain (see text).
SEQUENCE-SPECIFIC TRANSCRIPTION FACTORS INTERACT WITH THE BASAL TRANSCRIPTION APPARATUS TO PRODUCE SIGNIFICANT LEVELS OF TRANSCRIPTION
The basal transcription complex assembled at the TATA box or other transcription start site is only sufficient to initiate low levels of transcription of DNA into RNA. To achieve significant levels of gene expression, the basal transcription complex requires help from additional transcription factors that recognize and bind to cis-regulatory elements found elsewhere within the gene. Cis-elements that exert control near the core promoter itself have been called promoter elements, and those that act at a distance—often several hundred to more than 1,000 nucleotide base pairs away—have been called enhancer elements, but the commonly made distinction between promoter and enhancer elements is artificial. Both are composed of small “modular” sequences of DNA (generally 7–12 base pairs in length), each of which is a specific binding site for one or more transcription factors. Multiple cis-regulatory elements arrayed throughout the control regions of a gene, and the proteins they bind, act in combinatorial fashion to give each gene its distinct patterns of expression and regulation.
Transcription factors that are tethered to DNA by binding cis-elements are often described as sequence-specific transcription factors. Most such proteins have a modular structure composed of physically separate domains: a domain that recognizes and binds a specific DNA sequence, an activation domain that interacts with the basal transcription complex to activate or inhibit it, and a multimerization domain that permits the formation of homo- and heteromultimers with other transcription factors (Fig. 6.5). Many transcription factors are active only as dimers or higher order complexes. Within transcription factor dimers, whether homodimers or heterodimers, it is common for both partners to contribute jointly to the DNA binding domain and the activation domain. Sequence-specific transcription factors may contact the basal transcription complex directly; in many cases, they interact through the mediation of adapter proteins. In either case, transcription factors that bind cis-regulatory elements at a distance from the core promoter can interact with the basal transcription apparatus because the DNA forms loops that bring distant regions in contact with each other (Fig. 6.6A and B). Once bound to DNA, transcription factors serve as scaffolds for the recruitment of many types of activator or repressor proteins that serve to epigenetically activate or repress gene expression, respectively (see Chapter 7).
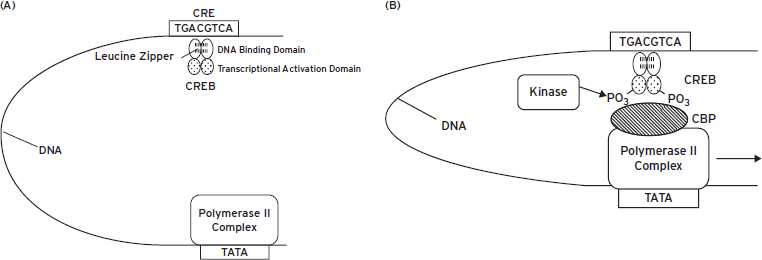
Figure 6.6 Looping of deoxyribonucleic acid (DNA) permits activator (or repressor) proteins binding at a distance to interact with the basal transcription apparatus. In the figure, the basal transcription apparatus is shown as a single box bound at the thymine adenine thymine adenine (TATA) element. The activator protein, cyclic adenosine monophosphate (cAMP)-response element binding protein (CREB), is shown bound as a homodimer to its cognate cis-regulatory element, the cAMP-response element (CRE), at a distance from the TATA element (panels A and B). Upon phosphorylation, many activators such as CREB are able to recruit adapter proteins that mediate between the activator and the basal transcription apparatus. An adapter protein that binds phosphorylated CREB is called CREB binding protein (CBP), a form of histone acetyltransferase (HAT) that acetylates nearby histones to facilitate gene transcription (see Chapter 7). With the recruitment of the adapter, a mature transcription complex forms that permits the synthesis of ribonucleic acid (RNA) by RNA polymerase II (panel B).
REGULATION OF GENE EXPRESSION BY EXTRACELLULAR SIGNALS
All regulation of gene expression by extracellular signals requires a mechanism that carries a signal from the cell membrane to the nucleus. In many cases, intracellular signaling molecules, such as protein kinases, serve this function. This is illustrated by CREB (cAMP response element binding protein), which mediates many of the effects of the cyclic adenosine monophosphate (cAMP) system, and other intracellular pathways, on gene expression (Altarejos and Montminy, 2011). CREB is bound under basal cellular conditions to its cognate cis-regulatory element (the cyclic AMP-response element, or CRE). Stimuli that increase levels of cAMP activate cAMP-dependent protein kinase (PKA), which results in liberation of the kinase’s catalytic subunits (Chapter 4). A portion of the free catalytic subunit then enters the nucleus, where it phosphorylates CREB, permitting it to activate transcription. Similarly, stimuli that activate other intracellular signaling cascades (e.g., Ca2+ or extracellular signal-regulated kinase [ERK] pathways) lead to the activation of other protein kinases, which also phosphorylate and activate CREB.
In other cases, a transcription factor, itself activated at the cell membrane or in the cytoplasm, then translocates to the nucleus. This is illustrated by the transcription factor NF-κB (nuclear factor κB), which is involved, among other things, in the activation of genes involved in inflammatory responses but also implicated in diverse neural phenomena (Hayden and Ghosh, 2012). Under basal conditions, NF-κB is retained in the cytoplasm by its inhibitory binding protein (IκB); this interaction masks a protein sequence within the NF-κB molecule that serves as a nuclear localization signal. Phosphorylation of IκB by IκB-kinase (IκK) leads to dissociation of NF-κB, which permits it to enter the nucleus, where it binds to its response elements; IκB is then digested within the cytoplasm.
The preceding discussion highlights that the critical nuclear translocation step can involve the transcription factor itself or another signaling molecule. Either mechanism can activate a third scenario: Some transcription factors are expressed only at very low levels when cells are in their unstimulated state. Their genes contain response elements that lead to their synthesis when cells are activated. Thus, gene expression may occur in cascades, with the stimulation of preexisting transcription factors leading to the expression of genes encoding (among many other genes) additional transcription factors, which can then stimulate yet other genes. Activation of genes by preexisting transcription factors occurs more rapidly than activation of genes by factors that must be synthesized de novo. Thus, not surprisingly, different neurotransmitters, drugs, and other stimuli may activate gene expression with widely varying time courses ranging from minutes to many hours.
Many transcription factors are members of families, presumably related by evolution, with related structures and functions. In the following section we illustrate the function of representative families of transcription factors, which serve as instructive examples of the diverse mechanisms governing the regulation of transcription in the nervous system.
THE CREB FAMILY OF TRANSCRIPTION FACTORS
CREB was the first-discovered and best-characterized member of a family of related proteins that bind to a particular DNA sequence termed the CRE, as mentioned (Altarejos and Montminy, 2011). The family is composed of CREB, the ATFs (activating transcription factors), and the CREMs (cAMP-response element modulators). CREB itself plays a major role in mediating the effects of cAMP and Ca2+ and of those neurotransmitters that act through cAMP or Ca2+, on gene expression. A large number of genes contain CREs, including the gene encoding the transcription factor c-Fos and the genes encoding tyrosine hydroxylase, BDNF (brain-derived neurotrophic factor), and several neuropeptides, such as proenkephalin, prodynorphin, somatostatin, and VIP (vasoactive intestinal polypeptide). Members of the CREB/ATF family bind to CREs as dimers. The dimerization domain used by the CREB/ATF proteins and several other families of transcription factors is called a leucine zipper, an alpha helical motif in which every seventh residue is a leucine. The proteins form a coil in which electrostatic interactions stabilize dimer formation.
The methods used to characterize CREs, and indeed all cis-regulatory elements, involve deleting them, or mutating them more subtly, and then reintroducing them into eukaryotic cells in culture by transfection. When a critical base in a DNA regulatory element is mutated, it weakens or destroys the binding site for the relevant transcription factor. If the protein can no longer bind, the gene can no longer be activated by the physiological stimulus under investigation, such as cAMP. By comparing response element sequences that have been investigated by mutagenesis within many genes, an idealized consensus sequence can be derived. The consensus nucleotide sequence of the CRE is TGACGTCA, with the nucleotides CGTCA absolutely required.
The consensus CRE sequence illustrates an important principle, the palindromic nature of many transcription factor binding sites. In the sequence TGACGTCA, it can be readily observed that the sequence on the two complementary strands, which run in opposite directions, is identical. Many cis-regulatory elements are perfect or approximate palindromes permitting binding of transcription factors in the form of dimers, where each member of the dimer recognizes one of the half-sites.
The primary mechanism by which CREB is regulated is through its phosphorylation on a single serine residue (Ser133) by any of several protein kinases, including PKA, Ca2+/calmodulin-dependent protein kinase IV (CaMKIV), and several kinases in the ERK cascade (see Chapter 4). CREB is constitutively synthesized so that it exists in neurons under basal conditions, although its expression can be regulated under certain circumstances, such as in response to psychotropic drug treatments. Nonphosphorylated CREB is localized to the nucleus, where it is bound to its response elements without considerable transcriptional activity (Fig. 6.6A). Phosphorylation of CREB activates its transcriptional activity by permitting CREB to interact with CBP (Fig. 6.6B). As noted in the preceding, CBP is called an adapter protein because it intervenes between a sequence-specific transcription factor CREB and the basal transcription apparatus, thus activating transcription. Despite its name, CBP provides this adapter function for several regulated transcription factors in addition to CREB family proteins.
The mechanism by which neurotransmitters that increase cAMP levels regulate gene expression via CREB is straightforward. Neurotransmitter-receptor stimulation increases levels of cAMP and of activated PKA. Activated PKA (that is, free catalytic subunits of the enzyme) is then translocated into the nucleus, where it phosphorylates and activates CREB. Phosphorylation of CREB, in turn, serves to activate the expression of genes that contain CREB bound to their promoter regions. Similar mechanisms operate for neurotransmitters (or nerve impulses) that activate CREB via stimulation of cellular Ca2+ signals and for neurotrophic factors that activate CREB via stimulation of ERK cascades.
The convergence of multiple signaling pathways on CREB may be very significant for the function of the nervous system. For example, associative memory appears to result from the integration of multiple signals that converge on target neurons, integration that could be achieved in part via CREB phosphorylation. Indeed, experiments performed by numerous laboratories on multiple species from Drosophila to rats, in which CREB was inactivated by different experimental methods, yield organisms with deficits in long-term memory, while increases in CREB function produce animals with enhanced memory (e.g., Josselyn, 2010; Barco and Marie, 2011). CREB has also been implicated in many additional psychiatric phenomena including drug addiction and depression (Blendy, 2006; Cao et al., 2010; Carlezon et al., 2005).
CREB illustrates yet another important principle of transcriptional regulation. As described the preceding section, CREB is a member of a larger family of related proteins. The ATFs and CREMs (distinct CREM products are generated from a single CREM gene via alternative splicing) bind CREs as dimers; many can dimerize with CREB itself. Activating transcription factor-1 (ATF1) appears to be very similar to CREB; it can be activated by the cAMP and Ca2+ pathways. Many of the other ATF proteins and CREM isoforms appear to activate transcription; however, certain CREMs, such as one called ICER (inducible cAMP repressor), act to repress it. Inhibitory CREM isoforms lack a glutamine-rich transcriptional activation domain found in CREB. Thus, certain CREB–CREM heterodimers might bind CREs but fail to activate transcription. Some inhibitory CREMs (e.g., ICER) are induced by CREB itself but with a delayed time course. In this way, these proteins may help terminate genes that had been activated by a CREB signal. Work in recent years has begun to demonstrate the involvement of ATFs and CREMs in an individual’s adaptations to numerous environmental stimuli, such as stress and drugs of abuse (e.g., Green et al., 2008).
THE AP-1 FAMILY OF TRANSCRIPTION FACTORS
Another group of transcription factors that plays a central role in the regulation of neural gene expression by extracellular signals is the AP-1 proteins (Morgan and Curran, 1995). The name AP-1 was originally applied to a transcriptional activity, then called Activator Protein-1, that was subsequently found to be composed of multiple proteins that bind as heterodimers (and a few as homodimers) to the DNA sequence TGACTCA, the AP-1 sequence. AP-1 proteins are divided into two groups, the Fos and Jun families. Like the CREB/ATF family, AP-1 proteins dimerize via a leucine zipper. The consensus AP-1 sequence is a heptamer, which forms a palindrome flanking a central C or G. The AP-1 sequence differs from the CRE sequence by only a single base. Yet this one base difference strongly biases protein binding away from CREB (which requires an intact CGTCA motif) to the AP-1 family of proteins and means that, under most circumstances, this sequence will not confer cAMP responsiveness on a gene. Instead, AP-1 sequences tend to confer responsiveness to growth factor–stimulated signaling pathways such as the ERK pathways and to the protein kinase C pathway. Indeed, the AP-1 sequence was historically described as a TPA-response element (TRE) because the phorbol ester 12-O-tetradecanoyl-phorbol-13-acetate (TPA), which activates protein kinase C, strongly induces gene expression via AP-1 proteins. It is a staggering illustration of the specificity of cellular regulation that a single base change (from a CRE to an AP-1 site) in a gene thousands of bases long can result in such a profound change in gene regulation.
AP-1 proteins generally bind DNA as heterodimers composed of one member each of the Fos and Jun families. The Fos family includes c-Fos, Fra-1 (Fos-related antigen-1), Fra-2, and FosB (which gives rise to full-length FosB plus a truncated splice variant termed ∆FosB). The Jun family includes c-Jun, JunB, and JunD. Heterodimers form between members of the Fos and Jun families via the leucine zipper. The potential complexity of transcriptional regulation is greater still because some AP-1 proteins can heterodimerize via the leucine zipper with members of the CREB–ATF family (e.g., ATF2 with c-Jun).
Jun proteins, in particular JunD, are expressed constitutively at appreciable levels in neural cells. In contrast, all Fos proteins are expressed at low or even undetectable levels under basal conditions, but with stimulation, can be induced to high levels of expression. Thus, unlike genes that are regulated by constitutively expressed transcription factors such as CREB, genes that are regulated by c-Fos/c-Jun heterodimers require new transcription and translation of these AP-1 proteins.
GENES ENCODING FOS AND JUN FAMILY PROTEINS ARE OFTEN TERMED IMMEDIATE EARLY GENES
Genes, such as the c-Fos gene itself, that are activated rapidly (within minutes), transiently, and without requiring new protein synthesis are frequently referred to as cellular immediate early genes (IEGs) (Morgan and Curran, 1995) (see Chapter 4). Genes that are induced or repressed more slowly (over hours), and are dependent on new protein synthesis, may be described as late response genes. The term IEG was initially applied to describe viral genes that are activated immediately upon infection of eukaryotic cells by commandeering host cell transcription factors for their expression. Viral IEGs generally encode transcription factors needed to activate viral late gene expression. This terminology has been extended to cellular (i.e., nonviral) genes with varying success. The terminology is problematic because there are many cellular genes induced independently of protein synthesis, but with a time course intermediately between those of classical IEGs and late response genes. In fact, some genes may be regulated with different time courses or requirements for protein synthesis in response to different extracellular signals. Moreover, it must be recalled that many cellular genes regulated as IEGs encode proteins that are not transcription factors (e.g., prodynorphin or BDNF). Despite these caveats, the concept of IEG-encoded transcription factors in the nervous system has been useful heuristically. Because of their rapid induction from low basal levels in response to neuronal depolarization (the critical signal being Ca2+ entry) and to second messenger and growth factor pathways, several IEGs have been used as cellular markers of neural activation, permitting novel approaches to functional neuroanatomy. This includes not only the Fos and Jun families of transcription factors, but also Zif268 (also called Egr1), which belongs to a distinct family of transcription factors that binds to its own response elements. Examples of the use of c-Fos as a marker of neuronal activation include induction of c-Fos in dorsal horn of the spinal cord by nociceptive stimuli, in motor and sensory thalamus by stimulation of sensory cortex, in supraoptic/paraventricular nuclei by water deprivation, and in numerous brain regions by acute and chronic opiate and cocaine administration, as well as in response to a number of other psychotropic drug treatments.
THE COMPOSITION OF AP-1 COMPLEXES CHANGES OVER TIME
Following acute stimulation of cells, different members of the Fos family are induced with varying time courses of expression, which leads to a progression of distinct AP-1 protein complexes over time (Nestler, 2008). It is believed that these changes produce varying patterns of expression of AP-1-regulated genes, thereby permitting neurons to adapt to the pattern of stimulation to which they are being subjected.
Under resting conditions, c-Fos mRNA and protein are barely detectable in most neurons; however, c-Fos gene expression can be induced dramatically in response to a variety of stimuli. As just one example, experimental induction of a grand mal seizure causes marked increases in levels of c-Fos mRNA in the rat brain within 30 minutes and induces substantial levels of c-Fos protein within 2 hours. c-Fos is highly unstable and is degraded back to low, basal levels within 4–6 hours. Administration of cocaine or amphetamine causes a similar pattern of c-Fos expression in the striatum. In either of these stimulus paradigms, other Fos-like proteins are also induced, but with a longer temporal latency than c-Fos; their peak levels of expression lag behind c-Fos by approximately 1 hour. Moreover, expression of these proteins persists a bit longer than that of c-Fos but still returns to basal levels within 8–12 hours.
With repeated stimulation, however, the c-Fos gene, and to a lesser extent the genes for other Fos-like proteins, become refractory to further activation (i.e., their expression becomes desensitized). However, in most systems, isoforms of ∆FosB continue to be expressed (Nestler, 2008). These isoforms exhibit very long half-lives in brain (1–2 weeks). As a result, ∆FosB accumulates in specific neurons in response to repeated perturbations and persists long after cessation of these perturbations (Fig. 6.7). Such stable accumulation of ∆FosB, which is responsible for desensitizing expression of c-Fos, has been shown to play an important role in mediating the long-term effects of drugs of abuse and several other treatments on the nervous system.
THE C-FOS GENE IS ACTIVATED BY MULTIPLE SIGNALING PATHWAYS
The precise intracellular mechanisms by which extracellular stimuli induce c-Fos expression are now well understood (Fig. 6.8). Stimuli that depolarize neurons (e.g., seizure activity, glutamate) induce c-Fos through a Ca2+-dependent mechanism that involves the phosphorylation, by CaMKIV, of a CREB protein that is already present in the cell and bound to CREs in the c-Fos gene. Neurotransmitters that activate the cAMP pathway in target neurons phosphorylate CREB on the same amino acid residue via PKA. Phosphorylation of CREB, as outlined in the previous section, activates its transcriptional activity and leads to increased c-Fos expression.
The c-Fos gene can also be induced by the ERK pathway, which is activated by many types of growth factors, via at least two distinct mechanisms, which may operate to different extents in different cell types. One mechanism, discussed in the preceding description of CREB, involves the phosphorylation of CREB by ribosomal S6 kinase (RSK) directly downstream of ERK. A second mechanism involves the phosphorylation and activation, also by ERK, of a different transcription factor termed Elk-1 (E twenty-six [ETS]-like transcription factor 1). Elk-1 binds, along with still another transcription factor, serum response factor (SRF), to the serum response element (SRE) within the c-Fos gene. A third mechanism mediates c-Fos induction by cytokines via activation of the JAK (Janus kinase)-STAT (signal transducers and activators of transcription) pathway. The c-Fos gene contains a response element called the SIE (SIF [sis-inducible factor]-inducible element), which binds and is activated by STAT transcription factors. Cytokine regulation of JAK-STAT signaling is described in greater detail in what follows.
The activation of IEG products such as c-Fos in response to a large number of stimuli raises the question of how specificity of response is achieved. First, specificity is partly achieved by the particular neural circuitry involved; that is, c-Fos and the other proteins are induced only along those particular neural pathways activated in response to the stimulus. Second, specificity is achieved by specialization within neuronal cell types. For example, in particular cell types not every gene that contains an appropriate binding site (e.g., an AP-1 site) to which c-Fos can bind is in a chromatin configuration that permits access to c-Fos-containing complexes (Chapter 7). Third, individual transcription factors generally cannot act alone to induce or repress the expression of a given gene. Multiple types of transcription factors, binding to distinct regulatory elements within a gene’s promoter region, must often act in concert to produce significant effects on gene expression.
Fourth, as alluded to the above-described point, protein products of many IEGs including c-Fos can bind DNA with high affinity only after binding to other transcription factors to form heterodimers. Such interactions are well exemplified by c-Fos and c-Jun. By itself, c-Fos is unable to bind DNA with high affinity. c-Jun homodimers can bind DNA but do so with relatively low affinity. However, c-Fos/c-Jun heterodimers bind to the AP-1 site with high affinity to regulate transcription. In contrast, heterodimers of c-Fos and JunB appear to be relatively inactive. Due to the existence of numerous members of the Fos and Jun families, complex regulatory schemes can readily be imagined by which a great deal of specificity in regulating cellular genes can be attained.
Although the primary mechanism by which Fos family members are regulated appears to be at the level of their transcription, the proteins are also regulated by phosphorylation. This is best illustrated by c-Fos itself, which is heavily phosphorylated on several closely spaced serine residues in the C-terminal region of the protein by PKA and CaMKs and by protein kinase C. c-Fos phosphorylation appears to be a critical regulatory mechanism for the protein: The difference between normal c-Fos (cellular Fos) and its viral counterpart v-Fos (which is oncogenic) is a frame-shift mutation that deletes the serine residues from the viral protein. It has been suggested that phosphorylation of c-Fos triggers the protein’s ability to suppress its own transcription, thereby providing key regulatory feedback control of the expression of this transcription factor. Another example is the phosphorylation of Jun proteins by Jun-kinases (JNKs). Jun-kinases are activated by cellular forms of stress and, like ERK, are considered to be part of the MAP (mitogen-activated protein)-kinase family (see Chapter 4). The resulting phosphorylation of Jun transcription factors is thought to be an important part of a cell’s adaptation to stress.
REGULATION OF GENE EXPRESSION BY CYTOKINES AND NONRECEPTOR PROTEIN TYROSINE KINASES
Cytokines subserve a wide array of functions within and outside of the nervous system (Chapters 1 and 2). Such cytokines include leukemia inhibitory factor (LIF), ciliary neurotrophic factor (CNTF), and interleukin-6 (IL-6), to name just a few. Recall from Chapter 4 that each of these cytokines produces its biological effects through activation of the gp130 class of plasma membrane receptor and the subsequent activation of the JAK-STAT pathway, noted earlier (Horvath, 2000; Ihle, 2001). The crucial feeding peptide, leptin, also acts via a JAK-STAT linked receptor.
The gp130-linked receptors for these cytokines and related signals do not contain protein kinase domains. Rather, the cytoplasmic portions of the receptors interact with nonreceptor protein tyrosine kinases of the JAK family, which include JAK1, JAK2, and Tyk2. Cytokine receptors show some specificity for the types of JAKs activated in a given cell type. Signal transduction to the nucleus involves the tyrosine phosphorylation, by the JAKs, of one or more of a family of transcription factor proteins called STATs. Upon phosphorylation, STAT proteins translocate to the nucleus, where they bind appropriate cytokine response elements present in target genes. The transcriptional activity of STAT proteins can be illustrated by regulation of c-Fos, as depicted in Fig. 6.8. Analogous mechanisms are involved in mediating the ability of cytokines to regulate many neural genes, including those for VIP and several other neuropeptides.
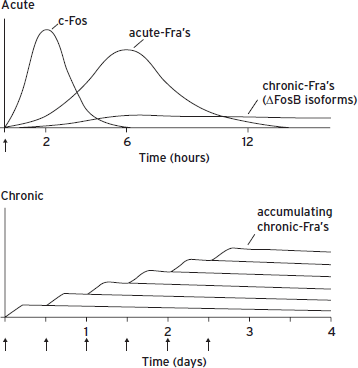
Figure 6.7 Scheme for the gradual accumulation of ∆FosB versus the rapid and transient induction of c-Fos and several other Fos family proteins in the brain. (Top) Several waves of Fos family proteins are induced by many acute stimuli in neurons. c-Fos is induced rapidly and degraded within several hours of the acute stimulus, whereas others (FosB, ∆FosB, FRA-1 [Fos-related antigen-1], FRA-2) are induced somewhat later and persist somewhat longer than c-Fos. In contrast, ∆FosB is induced (although at low levels) following a single acute stimulus but persists in brain with a half-life of 1–2 weeks due to its unique stability. In a complex with Jun family proteins, these waves of Fos family proteins form AP-1 binding complexes with shifting composition over time. (Bottom) With repeated (e.g., twice daily) stimulation, each acute stimulus induces a low level of ∆FosB. This is indicated by the lower set of overlapping lines, which indicate ∆FosB induced by each acute stimulus. The result is a gradual increase in the total levels of ∆FosB with repeated stimuli during a course of chronic treatment. This is indicated by the increasing stepped line in the graph. The increasing levels of ∆FosB with repeated stimulation result in the gradual induction of a long-lasting AP-1 complex, which underlies persisting forms of neural plasticity in the brain. Adapted from Nestler (2008).
WNT SIGNALING CASCADES AND THE REGULATION OF GENE EXPRESSION
Wnt (Wingless) is a secreted protein that activates a seven-transmembrane receptor, termed Frizzled (Fzd) (Inestrosa and Arenas, 2010). (Many of the names given to proteins in this pathway are based on morphological phenotypes seen upon deletion of homologous genes in Drosophila.) Wnt activation of Fzd leads to the activation of Disheveled (Dvl) by promoting its polymerization. The primary downstream signaling mechanism of activated Dvl is termed the “canonical” pathway (Fig. 6.9), where Dvl polymerization triggers its binding to Axin, which in turn triggers the AKT (a serine/threonine kinase)–mediated phosphorylation and inhibition of GSK3β (glycogen synthase kinase-3β). GSK3β phosphorylates several downstream targets, including β-catenin, which functions as a transcription factor (Inestrosa and Arenas, 2010). GSK3β phosphorylation of β-catenin promotes β-catenin’s degradation; hence, Wnt-Dvl, by inhibiting GSK3β, activates β-catenin–mediated transcription. Dvl polymerization also triggers the activation of Rac1, a small GTPase, which promotes the translocation of β-catenin to the nucleus. β-Catenin is a required cofactor for the transcription factors TCF (T cell transcription factor) and LEF (lymphoid enhancer factor). TCF/LEF binds to TCF/LEF response elements (also known as WREs or Wnt response elements) present in numerous genes to regulate gene expression. The Wnt-Dvl-GSK3β-β-catenin signaling cascade has been implicated in numerous neuropsychiatric syndromes and is the target for several types of psychotropic drugs, in particular, mood stabilizers such as lithium (e.g., Li and Jope, 2010; Wilkinson et al., 2011). There is thus keen interest in identifying the target genes of this cascade in limbic brain regions that mediate these clinically important behavioral effects.
STEROID HORMONE RECEPTORS ARE LIGAND-ACTIVATED TRANSCRIPTION FACTORS
The differentiation of many cell types in the brain is established by exposure to steroids. For example, exposure to estrogen or testosterone during critical developmental periods results in sexually dimorphic development of certain nuclei. The steroid hormone receptor superfamily (also called the nuclear receptor superfamily) includes the receptors for lipid-soluble molecules that diffuse readily across cell membranes, such as glucocorticoids, gonadal steroids, mineralocorticoids, retinoids, thyroid hormone, vitamin D, and cholesterol-related steroids (Chawla et al., 2001). They act on their receptors within the cell cytoplasm, in marked distinction to the other types of intercellular signals described that act on plasma membrane receptors (see Chapter 4). Another unique feature of the nuclear receptor superfamily is that their receptors are actually transcription factors. Like other transcription factors described, the steroid hormone receptors are modular in nature. Each has a transcriptional activation domain at its amino terminus, a DNA binding domain, and a hormone binding domain at its carboxy terminus. The DNA binding domains recognize different types of steroid hormone response elements within the regulatory regions of genes. The DNA binding domains of the steroid hormone receptors are described as zinc finger domains, a cysteine-rich motif that contains a zinc ion. This motif is used by many other transcription factors (so-called zinc finger proteins), but by few factors that are regulated by extracellular signals.
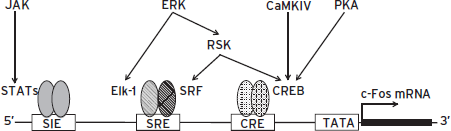
Figure 6.8 Regulation of c-Fos gene transcription. A cAMP response element (CRE) binds cAMP response element binding protein CREB, a serum response element (SRE) binds serum response factor (SRF) plus another transcription factor termed Elk-1 (E twenty-six [ETS]-like transcription factor 1) or ternary complex factor (TCF), and a SIF (sis-inducible factor)-inducible element (SIE) binds signal transducers and activators of transcription (STAT proteins). These three elements represent a small number of all known transcription factor–binding sites in the c-Fos gene. Proteins that bind at these sites are constitutively present in cells and are activated by phosphorylation. CREB can be activated by protein kinase A, Ca2+/calmodulin-dependent protein kinases (CaMKs), or ribosomal S6 kinases (RSKs); Elk-1 can be activated by extracellular signal-regulated kinases (ERKs); and STATs can be activated by Janus kinases (JAKs). Because the activation of c-Fos by any of multiple signaling pathways requires only signal-induced phosphorylation rather than new protein synthesis, it can be triggered rapidly by a wide array of stimuli.
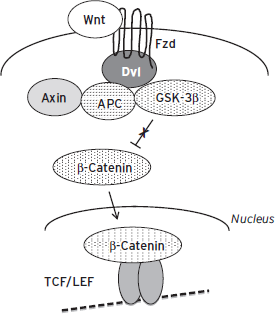
Figure 6.9 Regulation of gene expression by Wnt (Wingless) signaling cascades. Wnt activates the Frizzled (Fzd) receptor, which through Disheveled (Dvl) binds a complex composed of adenomatous polyposis coli (APC), Axin, glycogen synthase kinase-3β (GSK-3β), and other proteins. This leads to the phosphorylation and inhibition of GSK-3β and to the stabilization and accumulation of β-catenin, which then translocates to the nucleus (a process facilitated by Dvl), where it binds to TCF (T cell transcription factor) and LEF (lymphoid enhancer factor) transcription factors to regulate expression of specific genes. A growing number of neural genes have been recognized in recent years as targets for Wnt signaling.
Once bound by hormone, activated steroid hormone receptors translocate into the nucleus, where they bind to their cognate response elements. Such binding then increases or decreases the rate at which these target genes are transcribed, depending on the precise nature and DNA sequence context of the element. Steroid hormone receptors can therefore be considered ligand-activated transcription factors. In recent years, steroid hormone receptors have been shown to regulate the transcription of genes that do not contain steroid response elements by forming protein-protein interactions with other transcription factors, for example, AP-1 and CREB proteins. This discovery reveals highly complex regulatory mechanisms by which steroid hormones control gene expression (Biddie et al., 2011).
Although the primary mechanism by which the transcriptional activity of steroid hormone receptors is regulated is through their ligand binding and consequent nuclear translocation, the receptors are also regulated in vivo at transcriptional and posttranslational levels. The total amount of the receptors expressed in specific target tissues can be altered by numerous types of environment stimuli. For instance, levels of expression of the glucocorticoid receptor are under highly dynamic regulation within several limbic brain regions in response to chronic stress, and can mediate life-long changes in stress vulnerability (Zhang and Meaney, 2010).
DISCLOSURES
Dr. Nestler serves on the scientific advisory boards of PsychoGenics, Inc., Merck Research Laboratories, and Berg Pharma. He also is an ad hoc consultant for Johnson & Johnson.
Dr. Hyman serves on the Novartis Science Board and has advised Astra-Zeneca within the last year. Both advisory roles focus on early stage drug discovery. His research is funded by the Stanley Foundation.
REFERENCES
Altarejos, J.Y., and Montminy, M. (2011). CREB and CRTC co-activators: sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 12:141–151.
Barco, A., and Marie, H. (2011). Genetic approaches to investigate the role of CREB in neuronal plasticity and memory. Mol. Neurobiol. 44:330–349.
Bartel, D.P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136:215–233.
Biddie, S.C., John, S., et al. (2011). Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol. Cell 43:145–155.
Blendy, J.A. (2006). The role of CREB in depression and antidepressant treatment. Biol. Psychiatry 59:1144–1150.
Borrelli, E., Nestler, E.J., et al. (2008). Decoding the epigenetic language of neuronal plasticity. Neuron 60:961–974.
Carlezon, W.A., Jr., Duman, R.S., et al. (2005). The many faces of CREB. Trends Neurosci. 28:436–445.
Cao, J.-L., Vialou, V.F., et al. (2010). Essential role of the cAMP-CREB pathway in opiate-induced homeostatic adaptations of locus coeruleus neurons. Proc. Natl. Acad. Sci. USA 107:17011–17016.
Chawla, A., Repa, J.J., et al. (2001). Nuclear receptors and lipid physiology: opening the X-files. Science 294:1866–1870.
Green, T.A., Alibhai, I.N., et al. (2008). Induction of activating transcription factors ATF2, ATF3, and ATF4 in the nucleus accumbens and their regulation of emotional behavior. J. Neurosci. 28:2025–2032.
Hayden, M.S., and Ghosh, S. (2012). NF-κB, the first quarter century: remarkable progress and outstanding questions. Genes Dev. 26:203–234.
Horvath, C.M. (2000). STAT proteins and transcriptional responses to extracellular signals. Trends Biochem. Sci. 25:496–502.
Hyman, S.E., and Nestler, E. (1993). The Molecular Foundations of Psychiatry. Washington, DC: American Psychiatric Association.
Ihle, J.N. (2001). The Stat family in cytokine signaling. Curr. Opin. Cell Biol. 13:211–217.
Inestrosa, N.C., and Arenas, E. (2010). Emerging roles of Wnts in the adult nervous system. Nat. Rev. Neurosci. 11:77–86.
Josselyn, S.A. (2010). Continuing the search for the engram: examining the mechanism of fear memories. J. Psychiatry Neurosci. 35:221–228.
Li, X., and Jope, R.S. (2010). Is glycogen synthase kinase-3 a central modulator in mood regulation? Neuropsychopharmacology 35:2143–2154.
Morgan, J.I., and Curran, T. (1995). Immediate-early genes: ten years on. Trends Neurosci. 18:66–77.
Nestler, E.J. (2008). Transcriptional mechanisms of addiction: role of delta-FosB. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363:3245–3255.
Padgett, R.A. (2012). New connections between splicing and human disease. Trends Genet. 28:147–154.
Proudfoot, N.J. (2011). Ending the message: poly(A) signals then and now. Genes Dev. 25:1770–1782.
Taft, R.J., Pang, K.C., et al. (2010). Non-coding RNAs: regulators of disease. J. Pathol. 220:126–139.
Vo, N.K., Cambronne, X.A., et al. (2010). MicroRNA pathways in neural development and plasticity. Curr. Opin. Neurobiol. 20:457–465.
Wang, D.O., Martin, K.C., et al. (2010). Spatially restricting gene expression by local translation at synapses. Trends Neurosci. 33:173–182.
Wilkinson, M.B., Dias, C., et al. (2011). A novel role of the WNT-Dishevelled-GSK3β signaling cascade in the mouse nucleus accumbens in a social defeat model of depression. J. Neurosci. 31:9084–9092.
Zhang, T.Y., and Meaney, M.J. (2010). Epigenetics and the environmental regulation of the genome and its function. Annu. Rev. Psychol. 61:439–466.