13 | THE BRAIN AND ITS EPIGENOME
AMANDA C. MITCHELL, YAN JIANG, CYRIL J. PETER, KI A. GOOSENS, AND SCHAHRAM AKBARIAN
INTRODUCTION
Psychiatric disorders, including autism, mood and anxiety, or psychosis spectrum disorders, substance abuse, and addiction each lack a unifying molecular or cellular pathology, and most cases are believed to be of multifactorial etiology with numerous environmental and genetic components involved. This, taken together with the fact that laboratory animal models, including rats and mice, do not reflect the full complexities surrounding disorders of higher cognition and emotion (Nestler and Hyman, 2010), poses a formidable challenge to the quests of understanding the pathophysiology of disease and developing efficient therapies for the majority of patients. Consider that conventional psychopharmacology, including drugs targeting monoamine signaling, for example, dopaminergic, serotonergic, and noradrenergic pathways, elicits an insufficient therapeutic response in one half or less of patients diagnosed with schizophrenia and related illnesses (Lehman et al., 2004), or depression and anxiety (Krishnan and Nestler, 2010). Thus, it will be necessary to further explore the neurobiology and molecular pathology of mental disorders, in order to develop novel treatment strategies of higher efficacy.
One promising avenue of research that is moving center stage in basic and clinical neurosciences alike is epi- (Greek for “over,” “above”) genetics (Labrie et al., 2012). Not long ago epigenetics was mainly viewed through the prism of a single mark, DNA cytosine methylation, in the context of cell division related to, for example, embryonic development or cancer. This work might suggest that epigenetics bears little relevance for the postnatal and mature brain with its large proportion of postmitotic and differentiated cells. At least four independent lines of evidence fuel the current interest in neuroepigenetics, making it repeatedly a “hot topic” at recent annual conventions of neuroscientists and psychiatrists. First, based on human and animal brain studies, it is becoming increasingly clear that epigenetic markings, including DNA methylation and many types of histone modifications, remain “plastic” throughout all periods of development and aging, with ongoing dynamic regulation even in neurons and other differentiated cells. Changes in neuronal activity, learning, and memory, including the establishment of reward- and addiction-related behaviors, and numerous other paradigms all have been shown to be associated with DNA methylation and histone modification changes at specific genomic sequences in brain chromatin (Day and Sweatt, 2011; Robison and Nestler, 2011). These principal insights immediately propelled epigenetics into the forefront of brain research, as it provides a molecular system operating at the genome–environment intersect, and obviously is a topic of interest deeply rooted in the concepts of modern psychology and the behavioral sciences. Second, recent work has revealed that each of the causative mutations in a subset of monogenetic neurological disorders (including but not limited to Rubinstein-Taybi, Kleefstra, Rett, and other syndromes) disrupts the function of a protein involved in the regulation of chromatin structure and function (Haggarty and Tsai, 2011). These findings from clinical genetics indicated that the developing brain, indeed, is sensitive to dysregulation of the epigenetic machinery, and that some neurological conditions could arise from more widespread chromatin defects affecting the immature brain. Moreover, similar types of mutations were subsequently found in some cases with adult-onset psychosis or dementia, which implies that “chromatin disorders” encompass a much wider range of neuropsychiatric disease, contrasting the traditional view that they reflect “static” lesions confined to the developing nervous system (Jakocevski and Akbarian, 2012). Third, a subset of chromatin-modifying drugs—compounds with inhibitory activity directed against histone deacetylases are a well-known example—demonstrated a promising therapeutic potential in animal models for cognitive and emotional disease (Machado-Vieira et al., 2011), and even for neurodegenerative conditions (Fischer et al., 2010). Finally, even some of the most optimistic estimates on the role of protein-coding sequences as genetic risk factors for major psychiatric disease predict that only 25%–50% of sporadic cases of autism and schizophrenia carry disease-associated mutations altering protein sequence and function (O’Roak et al., 2011; Xu et al., 2011). Therefore, a subset of the remaining disease-associated variants are thought to involve regulatory and probably noncoding DNA, and by exploring the pattern and distribution of various epigenetic markings (which, in concert, define the functional architecture of chromatin, including open or silenced euchromatin, constitutive heterochromatin, etc.), one could expect to obtain important clues about the molecular pathology associated with the disease-related DNA variants (Houston et al., 2012). In the following, we will, after a concise introduction to the various markings and molecules that define a cell’s epigenome, touch upon each of four points raised in the preceding, and then finish this chapter with a brief discussion about how epigenetic technologies and discoveries could have a lasting impact on our understanding of the neurobiology and heritability of mental disorders.
THE EPIGENOME—GENERAL PRINCIPLES
The elementary unit of chromatin is the nucleosome, or 146 bp of genomic DNA wrapped around an octamer of core histones, connected by linker DNA and linker histones. The collective set of covalent DNA and histone modifications and variant histones provides the major building blocks for the “epigenome,” or the epigenetic landscapes that define the functional architecture of the genome, including its organization into many tens of thousands of transcriptional units, clusters of condensed chromatin, and other features that are differentially regulated in different cell types and developmental stages of the organism (Li and Reinberg, 2011; Rodriguez-Paredes and Esteller, 2011). An in-depth description of all epigenetic markings would be far beyond this review chapter, but multiple recent excellent reviews on this topic provide an excellent starting point for the reader interested to learn more about these topics (Ederveen et al., 2011; Kinney et al., 2011; Zhou et al., 2011). Here, we confine the discussion to a subset of the epigenetic markings repeatedly explored in the human and animal brain.
Common terminology used in chromatin studies includes (1) nucleosomes, comprised of a protein octamer of four small proteins, the nucleosome core histones, around which 146 bp of DNA is wrapped around. Transcription start sites are often defined by a nucleosome-free interval, probably for increased access of the transcriptional initiation complex and other regulators of gene expression. Arrays of nucleosomes, connected by linker DNA and linker histones, comprise the 10-nm “beads-on-a-string” chromatin fiber; (2) euchromatin defines loose chromatin typically at sites of actively transcribed genes and units poised for transcription; (3) heterochromatin defines tightly packed nucleosomal arrays. Constitutive heterochromatin remains highly condensed in most interphase nuclei. Examples include pericentric and telomeric repeat DNA, the inactivated X chromosome (“Barr body”) of female somatic cells, and other chromosomal structures often found in close proximity to the nuclear envelope and also around the nucleolus (see Fig. 13.1). Facultative heterochromatin includes silenced genes that upon differentiation or other stimuli could switch to a state of active transcription.
DNA (HYDROXY)-METHYLATION
Two related but functionally very different types of DNA modifications, methylation (m) and hydroxymethylation (hm) of cytosines in CpG dinucleotides, provide the bulk of the epigenetic modifications in vertebrate DNA (Kriaucionis and Heintz, 2009). There are additional types of DNA modifications, which are mostly chemical intermediates in the context of mC5 and hmC5 (cytosines methylated at the carbon 5 position) synthesis and breakdown (He et al., 2011; Ito et al., 2011). While the majority of DNA (hydroxy)-methylation is found at sites of CpG dinucleotides and, more generally, in the CpG-enriched sequences of the genome, a recent study in rat cerebral cortex reported that the amount of mC5 found at nonCpG sites is far higher than previously assumed (Xie et al., 2012). The mC5 and hmC5 markings show a differential (but not mutually exclusive) pattern of genomic occupancy. The hmC5 mark is concentrated toward the 5′ end of genes and the proximal-most portion of transcriptional units, and broadly correlates with local gene expression levels (Jin et al., 2011; Song et al., 2011). On the other hand, less than 3% of mC5 markings are positioned around the 5′ end of genes (Maunakea et al., 2010). The classical concept on the transcriptional regulatory role of DNA methylation, which also has guided many brain-related studies, is that promoter-bound repressive chromatin remodeling complexes negatively regulate transcription (Sharma et al., 2005). There are many studies that report changes in promoter DNA methylation (mostly in conjunction with decreased gene expression) in preclinical models of psychosis, depression, and addiction, as well as in brain tissue in subjects diagnosed with one of these conditions. Interestingly, however, while the largest amount, or 97%, of mC5s are found in intra- and intergenic sequences and within DNA repeats (Maunakea et al., 2010), only few of these studies have explored brain DNA methylation changes at repeat DNA and other sequences outside of promoters.

Figure 13.1 The epigenome, from nucleus to nucleosome. Schematic illustration of a gene poised for transcription by polymerase II (Pol II) initiation complex, with nucleosome-free interval at transcription start site (TSS). Distal enhancer sequence which in looplike structure moves in close proximity to active gene. Marks a small subset of heterochromatic portions of the genome, including silenced gene and heterochromatic structures bordering the nuclear envelope and pore complex, and also the nucleolar periphery. A small subset of representative histone variants and histone H3 site-specific lysine (K) residues at N-terminal tail (K4, K9, K27, K36, K79) and H4K20 residue are shown as indicated, together with panel of mono- and trimethyl, or acetyl modifications that differentiate between active promoters, transcribed gene bodies, and repressive chromatin, as indicated. DNA cytosines that are hydroxymethylated at the C5 position are mostly found at active promoters, while methylated cytosines are positioned within body of actively transcribed genes and around repressed promoters and in constitutive heterochromatin.
The role of hmC5, however, in transcriptional regulation still remains controversial. Yi Zhang’s group (Wu et al., 2011) performed genome-wide profile of 5hmC in both wild-type and Tet1-depleted mouse embryonic stem (ES) cells, and their data suggested that 5hmC is enriched at both gene bodies of actively transcribed genes and extended promoter regions of Polycomb-repressed developmental regulators. There is also evidence that besides TSSs, hmC5 is also enriched in enhancer region marked by H3K4me1 and H3K27ac in human embryonic stem cells (Stroud et al., 2011; Szulwach et al., 2011a). From a Nature paper published (Williams et al., 2011), it was reported that hmC5 was enriched by both TSSs and gene bodies, and its catalytic enzyme TET1 interacts with Sin3A corepressor complex and is involved in transcriptional repression of a significant portion of polycomb group target genes.
HISTONE MODIFICATIONS
The epigenetic regulation of chromatin by virtue of chemical histone modifications is even more complex than DNA methylation discussed previously, and it is now thought that there are far more than 100 amino acid residue-specific posttranslational modifications (PTMs) in a typical vertebrate cell (Tan et al., 2011), including mono (me1), di (me2)-, and tri (me3) methylation, acetylation, and crotonylation; polyADP-ribosylation and small protein (ubiquitin, SUMO) modification of specific lysine residues; and as arginine (R) methylation and “citrullination,” serine (S) phosphorylation, tyrosine (T) hydroxylation, and several others (Kouzarides, 2007; Tan et al., 2011; Taverna et al., 2007). These site- and residue-specific PTMs are typically explored in the context of chromatin structure and function, with an epigenetic histone code (a combinatorial set of histone PTMs that differentiates between promoters, gene bodies, enhancer, and other regulatory sequences, condensed heterochromatin, and so on [Zhou et al., 2011]). For an overview on the principle (but by far not an exhaustive illustration of all molecular markings defining the epigenome), see Figure 13.1. It is important to emphasize that histone PTMs rarely occur in isolation, and instead multiple histone PTMs appear to be coregulated and, as a group, define the aforementioned chromatin states (Berger, 2007). Many active promoters, for example, are defined by high levels of histone H3 lysine 4 methylation and various histone lysine acetylation markings (Zhou et al., 2011). Furthermore, there is also evidence for a coordinated and sequential regulation; phosphorylation of histone H3 at the serine (S)10 position often serves as a trigger for subsequent acetylation of neighboring lysine residues H3K9 and H3K14 in the context of transcriptional activation, while at the same time blocking repression-associated methylation of H3K9 (Nowak and Corces, 2004).
HISTONE VARIANTS
In addition to the core histones H2A/H2B/H3/H4, histone variants such as H3.3, H2A.Z, and H2A.X exist (Fig. 13.1). The role of these variant histones, which differ from the canonical histone only at very few amino acid positions, is often discussed in the context of replication-independent expression and assembly (Woodcock, 2006), and several histone variants robustly affect nucleosome stability and compaction (Jin and Felsenfeld, 2007). One popular model postulates that during the process of gene expression, RNA polymerase and the transcriptional activation and elongator complexes destabilize nucleosomes, which in turn promotes nucleosome remodeling and variant histone incorporation, which then further potentiate or stabilize gene expression (Bintu et al., 2011; Sutcliffe et al., 2009).
THE EPIGENOME IS PACKAGED INTO HIGHER ORDER CHROMATIN STRUCTURES
Epigenetic decoration of nucleosomes, including the DNA and histone modifications, and histone variants described previously, in itself, would fall short to adequately describe the epigenome, or even the localized chromatin architecture at any given (genomic) locus. This is because nucleosomal organization leads to only a 7-fold increase in packaging density of the genetic material, as compared with naked DNA; however, the actual level of compaction in the vertebrate nucleus in interphase (which defines the nucleus during the time period a cell is not dividing, including postmitotic cells such as neurons) is about three orders of magnitude higher (Belmont, 2006). The chromosomal arrangements in the interphase nucleus are not random, however. Specifically, loci at sites of active gene expression are more likely to be clustered together and positioned toward a central position within the nucleus, while heterochromatin and silenced loci move more toward the nuclear periphery (Cremer and Cremer, 2001; Duan et al., 2010). Chromatin loopings, in particular, are among the most highly regulated “supranucleosomal” structures and are associated with transcriptional regulation, by, for example, positioning distal regulatory enhancer or silencer elements that—in the linear genome—are positioned potentially many hundred kilobases apart from a gene, to interact directly with that specific promoter (Gaszner and Felsenfeld, 2006; Wood et al., 2010). Despite the growing realization of the importance of these and other higher order chromatin structures for transcriptional regulation, this is an area where very little is known about regulation in the nervous system, let alone potential alterations in psychiatric disease. Until recently, there were only three studies in the literature that explored loop formations in brain tissue (Dhar et al., 2009; Horike et al., 2005; Jiang et al., 2010), with a few additional papers using the brain as negative control for their studies on the sensory epithelium of the nose (Lomvardas et al., 2006) or the hematopoietic system (Simonis et al., 2006). However, to date, nothing is known about chromatin loopings in human brain.
Three-dimensional chromatin architectures are commonly mapped using derivatives of chromosome conformation capture (3C). This technique was originally developed for simple eukaryote systems such as yeast (Dekker et al., 2002) but has been further advanced to include 4C, 5C, HiC, and ChIA-PET (Simonis et al., 2007), allowing the mapping of chromosomal architectures across many megabases, or in the case of HiC and ChIA-PET, even genome-wide. At its core, the technique explores physical interactions between DNA fragments separated by interspersed sequence (chromosome architecture in cis) or between sequences positioned in different chromosomes (interactions in trans). Cross-linked chromatin is digested with a specific restriction enzyme, religated and amplified using primer pairs for which forward and reverse primers match to different portions of the genomic locus of interest. It will be important to clarify in the nearby future whether the 3C technique and its derivatives are applicable to human postmortem brain tissue. If so, then the exploration of chromatin loopings in normal and diseased brain could potentially provide valuable insights about the chromosomal architectures surrounding any given locus, including potential aberrations in the context of disease-associated polymorphisms or DNA structural variants. Presently, the exploration of many regulatory sequences is simply not possible because they are, to put it simply, not “visible” in the linear genome sequence, because of their spatial separation from transcription start sites or annotated genes by many hundreds of kilobases, potentially. To highlight the potential use of chromosome conformation capture in the context of psychiatric disease, consider the example of the major histocompatibility complex (MHC) locus, which long has been implicated in psychiatric disease (Smeraldi et al., 1976), conferring significant genetic risk to schizophrenia and related disease as most recently shown in three large genome-wide association studies (GWASs) published jointly in 2009 (Purcell et al., 2009; Shi et al., 2009; Stefansson et al., 2009). These studies identified up to 45 disease-associated SNPs (single-nucleotide polymorphisms) in the 26–33 megabase region of the MHC loci on chromosome 6. Strikingly, 50% of these SNPs are not located near genes. In fact, the strongest SNP at rs13194053 (p = 9.54 × 10−9) is more than 29 kb away from its nearest gene, HIST1H2AH. A functional role for many intergenic regions would not be surprising since many intergenic regions alter expression of upstream and downstream genes (Kleinjan and van Heyningen, 2005), and the mechanistic workup of these SNPs will certainly require application of chromosome conformation capture and related technologies.
CHROMATIN MARKINGS REMAIN PLASTIC THROUGHOUT THE LIFE SPAN OF THE HUMAN BRAIN, WITH IMPLICATIONS FOR THE NEUROBIOLOGY OF PSYCHIATRIC DISEASE
DEVELOPMENTAL PLASTICITY OF BRAIN EPIGENOMES
Most or perhaps all epigenetic markings studied to date, including DNA methylation, are now thought to be reversible and subject to bidirectional regulation in somatic tissues including brain, and there is no a priori reason for the unidirectional accumulation of a specific epigenetic mark while the brain is maturing and aging (Cheung et al., 2010; Guo et al., 2011; Klose and Zhang, 2007; Loenarz and Schofield, 2011; Miller and Sweatt, 2007; Ooi and Bestor, 2008). Nonetheless, multiple lines of evidence suggest that there is substantial reorganization of chromatin structures during the course of postnatal development and aging. Human cerebral cortex, for example, shows complex and gene-specific changes in the amount of methyl-cytosine (mC5; cytosines are methylated at the carbon 5 position). There is a fast rise in mC5 at many promoters during the transition from peri- to postnatal ages that continues at a slower pace into old age in conjunction with subtle changes (mostly a decline) in expression of transcripts originating from these promoters (Hernandez et al., 2011; Numata et al., 2012; Siegmund et al., 2007). Such age-related epigenetic drifts could impact vulnerability to neurodegenerative disease. A fascinating example has been recently reported for the cerebellum of the mouse, where levels of the mC5 derivative, hydroxymethyl-cytosine 5hmC, increase by 10-fold from postnatal Week 1 to adulthood (Szulwach et al., 2011b). Notably, among the genes that are affected by increasing 5hmC amounts at their promoters during cerebellar maturation, pathways for aging-related neurodegenerative diseases and angiogenesis were overrepresented and included at least 15 genes linked to hereditary forms of spinocerebellar ataxia, a neurological syndrome defined by severe motor dysfunction with the degeneration of cerebellar Purkinje neurons and other systems (Szulwach et al., 2011b). Also, of relevance, ten-eleven translocation (TET) proteins are responsible for converting mC5 to hmC5, and the active domains of these proteins belong to the same dioxygenase superfamily as hypoxia-inducible factor (HIF), an oxygen sensor that has been ascribed with a key role in angiogenesis and oxidative stress responses (Szulwach et al., 2011b). It will be extremely interesting to explore whether oxidation and other stress factors in the cellular environment, via TET-mediated regulation of DNA methylation levels, could leave a lasting imprint on chromatin structures in neurons or glia.
Like the aforementioned dynamic changes in DNA methylation during the course of development and aging, the epigenetic landscapes of histone modifications also undergo substantial reorganization across the life span of the human brain. For example, histone methylation markings that differentiate between open and repressive chromatin surrounding NMDA receptor gene promoters show highly dynamic changes in cerebellar cortex during the transition from perinatal stages and infancy to adulthood (Stadler et al., 2005) that reflect, in part, development changes in levels of the corresponding gene transcripts (Akbarian et al., 1996). Furthermore, hundreds of loci undergo histone methylation changes in cortical neurons during the first few years of life (Cheung et al., 2010). The brains from mice that are prone to accelerated senescence (the SAMP8 line) and have learning and memory deficits show age-related drifts in histone PTMs: these epigenetic drifts are defined by a loss of the markings associated with active gene expression, such as histone H4 lysine 20 monomethyl (H4-K20me1) and H3-K36me3 (Fig. 13.1), in conjunction with a robust rise in the repressive mark, H3-K27me3 (Wang et al., 2010). The hippocampus of aged, 16-month-old wild-type mice shows deficits in acetylated histone H3-lysine 12 (H4K12) (Peleg et al., 2010), a histone PTM that is broadly correlated with the transcriptional elongation process (Hargreaves et al., 2009). In addition, drugs with histone deacetylase inhibitor (HDACi) activity induce upregulation of H4-K12ac dramatically and thereby could improve hippocampal-dependent learning and memory in aged mice (Peleg et al., 2010). It is possible that age-related drifts in brain epigenomes negatively affect neuronal (Fischer et al., 2010; Lu et al., 2004) and oligodendroglial (Copray et al., 2009) transcriptomes, thereby contributing to a decline in the signaling capacity of nerve cells, defects in axon myelination, and other molecular defects that have been linked to cognitive disorders of the adult brain, including those that like Alzheimer’s disease are associated with neurodegeneration (Yankner et al., 2008) and others such as schizophrenia that are not accompanied by ongoing loss of nerve cells (Tang et al., 2009). Taken together, these findings leave little doubt that brain epigenomes are indeed subject to dynamic changes throughout all periods of maturation and aging, which may have important implications for the neurobiology of disease.
Posttraumatic stress disorder (PTSD) is probably a good example to illustrate how current concepts in epigenetics influence thoughts about pathophysiology of psychiatric disease. Obviously, from a heuristic perspective, it is very attractive to design working hypotheses that attribute a key role for epigenetic markings inside the nucleus of neurons and other brain cells subserving a memory function that, in response to an intense “environmental” influence (e.g., trauma), convey lasting alterations in a subject’s emotional and physical health and resilience (Zovkic and Sweatt, 2012). Indeed, some recent studies in human subjects point to the promising potential of such types of working models. For example, a recent study on peripheral cells collected pre- and postdeployment from U.S. military service members identified global DNA methylation levels in repetitive DNA sequences, including LINE-1 and Alu repeat elements, as biomarkers that were significantly associated with resilience or, conversely, vulnerability to PTSD (Rusiecki et al., 2012). Similarly, studies in civilian/urban populations discovered that changes in blood DNA methylation signatures in PTSD subjects selectively affected cytokine and steroid signaling, immune defense, and inflammation-related genes, consistent with various lines of evidence implicating some degree of peripheral immune dysregulation in this disorder (Smith et al., 2011; Uddin et al., 2010). Based on the aforementioned studies in blood, one would predict that brain chromatin, too, is involved in the neurobiology of PTSD, and human postmortem brain shows distinct DNA and histone methylation changes in amydgala and hippocampus, ventral striatum, and other anatomical structures with a critical role for emotion, affect, and memory (Zovkic and Sweatt, 2012). Indeed, there is excessive methylation of the glucocorticoid receptor gene promoter NR3C1 and ribosomal DNA repeats in the hippocampus of adult suicide victims who also suffered childhood abuse (McGowan et al., 2008, 2009). Furthermore, the prefrontal cortex of suicide completers exhibits a shift from open to repressive chromatin-associated histone methylation for the TRKB neurotrophin high-affinity receptor, and for various genes regulating polyamine metabolism (Ernst et al., 2009; Fiori and Turecki, 2010). Furthermore, downregulated histone deacetylase 2 (HDAC2) expression in ventral striatum of subjects diagnosed with depression is thought to lead to an overall increase in histone H3 acetylation in this mesolimbic structure (Covington et al., 2009). In the aforementioned studies, many of these epigenetic changes are associated with decreased expression of the corresponding gene transcripts, which reaffirms the importance of epigenetic mechanisms and transcriptional regulation for the pathophysiology of psychiatric disease.
While postmortem brains of psychiatric disease cases are notoriously hampered by the fact that most subjects received psychoactive medication prior to death, some evidence from animal studies would suggest that at least a subset of the chromatin changes in diseased brain, as mentioned previously, are not mere epiphenomena due to medication or postmortem confounds such as tissue autolysis but, instead, closely associated with the disease process. To mention just two examples, the hippocampal glucocorticoid receptor gene Nr3c1 shows excessive methylation not only in patients (McGowan et al., 2009), but also in adults rats brought up with suboptimal maternal care (“low licking” versus “high licking” mothers) (Weaver et al., 2004), and chronic social defeat stress in mice and rats elicits histone acetylation changes in ventral striatum and hippocampus very similar to those encountered in depressed human subjects (Covington et al., 2009; Hollis et al., 2010).
MONOGENETIC ETIOLOGIES OF NEUROPYCHIATRIC DISEASE INCLUDES MUTATIONS IN PROTEINS INVOLVED IN READING, WRITING, OR ERASURE OF EPIGENETIC MARKINGS
There are many hundreds of genes that encode proteins that either write, erase, or read the molecular markings of the epigenome (Filippakopoulos et al., 2010; Janzen et al., 2010); however, we want to make the reader aware that some experts feel this type of terminology can be misleading, especially because the regulation of many epigenetic markings could turn out to be only a “cog” in the chromatin-remodeling machinery but not a key driver (Henikoff and Shilatifard, 2011). The genome encodes three DNA methyltransferases, DNMT1, DNMT3a, and DNMT3b, that establish and maintain DNA methylation markings, and in addition, there are complex DNA demethylation pathways involving mC5 hydroxylation and oxidation via ten-eleven translocation dioxygenases, or activation-induced deaminase (AID)/APOBEC-mediated deamination of mC5 or hmC5, followed by base excision repair-mediated replacement with (unmethylated) cytosine (Bhutani et al., 2011; Guo et al., 2011). Furthermore, the collective set of histone methyltransferases (KMTs) and demethylases (KDMs) together could easily account for >100 genes in a mammalian genome, suggestiong that the molecular framework to establish and erase histone methylation marks is likely to be extremely complex and probably differentially regulated across various cell types, or developmental stages, of the organism (Copeland et al., 2009; Rotili and Mai, 2011). Reader proteins, which bind to a specific epigenetic mark, are defined by their characteristic “reader module”; well-studied examples include the methyl-CpG binding domain (MBD) for mC5-DNA, the bromodomain for lysine acetylation, and “chromo,” “Tudor,” malignant brain tumor (“MBT),” “WD40repeat,” plant homeodomain (PHD) finger domains targeting methylated lysines or arginines in a residue-specific manner (Taverna et al., 2007). For the “open-chromatin” mark histone H3-trimethyl-lysine 4 (H3K4me3), these include many components of the RNA polymerase II–associated transcriptional initiation complex, while other marks such as H3K9me3 are primarily targeted by transcriptional repressors and regulators of chromatin condensation (Vermeulen et al., 2010).
The collective set of reader, writer, and eraser proteins includes at least 15 genes that are associated with monogenetic forms of neurodevelopmental or adult-onset neuropsychiatric disease (Jakocevski and Akbarian, 2012). While an exhaustive discussion of these various neurodevelopmental syndromes would be beyond the scope of this book chapter, it is important to point out that while chromatin defects in the brain were until very recently considered static lesions of early development that occurred in the context of rare genetic syndromes, it is now clear that mutations and maladaptations of the epigenetic machinery cover a much wider continuum, including adult-onset neurodegenerative disease. For example, while hypomorphic (partial loss-of-function) mutations in the DNA methyltransferase DNMT3B were already known to cause a multiorgan syndrome—Immunodeficiency, Centromere Instability, Facial anomalies (ICF 1)—that includes mental retardation and defective brain development (Hansen et al., 1999; Okano et al., 1999), it was only recently discovered that select mutations in DNA methyltransferase–coding regions, including DNMT1, are responsible for some cases of hereditary sensory and autonomic neuropathy, type 1 (HSAN1) (Klein et al., 2011), a rare neurodegenerative condition characterized by various neuropathies and early-onset dementia in the third or fourth decade of life. In other pedigrees, DNMT1 mutations were linked to narcolepsy and late-onset deafness and cerebellar ataxia (Winkelmann et al., 2012). Likewise, structural variants in the X-linked gene MECP2, encoding the methyl-CpG-binding protein 2, not only cause Rett syndrome (RTT), a disorder of early childhood with an incidence of 1 in 10,000 that is associated with cognitive deficits and a broad range of neurological symptoms (Amir et al., 1999; Chouery et al., 2011), but are also thought to be responsible for some cases of autism and schizophrenia with onset in childhood or adolescence (Piton et al., 2011). Furthermore, consider the DNA variants and mutations encompassing the KMT1D gene (9q34.3), encoding a histone H3-lysine 9 specific methyltransferase initially recognized as the causative gene responsible for a distinct neurodevelopmental and multiorgan syndome, the Kleefstra mental retardation syndrome (Kleefstra et al., 2009). Meanwhile, however, KMT1D mutations are also responsible for some cases with schizophrenia (Kirov et al., 2012), and various nonspecific psychiatric phenotypes and even neurodegenerative disease in the postadolescence period (Verhoeven et al., 2011).
Taken together, these findings leave little doubt that mutations that fall within the coding sequence or otherwise affect levels of expression of a select set of regulatory proteins involved in DNA or histone methylation could cause neuropsychiatric disease even after brain development has largely been completed, including some cases diagnosed with psychosis or early-onset dementia. One could speculate that DNMT1, MECP2, KMT1D, and other monogenetic causes of neuropsychiatric disease (Jakocevski and Akbarian, 2012) could then also play a wider role in the pathophysiology of autism, schizophrenia, and other illnesses, outside of what has been discussed—and among the overall population of psychiatric patients very rarely occurring—cases with mutations and deletions of these chromatin regulatory proteins. For example, a recent postmortem study on 16 cases on the autism spectrum, exploring genome-wide occupancies of histone H3-tri-methyl-lysine 4 (H3K4me3), a mark sharply upregulated at transcription start sites (see Fig. 13.1) in prefrontal neurons, reported an abnormally broad histone methylation profile (“spreading”) at the 5′ end of many hundreds or thousands of genes for four of their cases, while the remaining twelve cases showed much more limited alterations at a few loci only (Shulha et al., 2012). While purely speculative at this point, it remains possible that these subsets of autism cases with an apparently more generalized abnormality in prefrontal histone methylation profiles are affected by defects in the pathways governing the writing, reading, or erasure of H3K4me3 and related markings. These may include pathways that are critical for the orderly activity of DM5C/SMCX/JARID1C, the X-linked H3K4-specific histone demethylase that is also responsible for some cases of mental retardation, autism, and other neurodevelopmental disease, or the H3K4 methyltransferase MLL1 previously implicated in the neurobiology of schizophrenia (Adegbola et al., 2008; Akbarian and Huang, 2009; Huang et al., 2007; Iwase et al., 2007).
“EPIGENETIC DRUG” DEVELOPMENT IN PSYCHIATRY—READY FOR PRIME TIME?
While most major psychiatric disorders, including the broader range of autism, mood, or psychosis spectrum disorders, each lack a unifying neuropathology, the pathophysiology almost certainly involves dysregulated gene expression in cerebral cortex and other brain regions. Starting with the initial reports on gene expression changes in prefrontal cortex and hippocampus of subjects diagnosed with schizophrenia, a large number of postmortem brain studies have been published, collectively suggesting that distinct sets of gene transcripts are frequently, albeit never consistently, expressed at altered levels in at least a subset of psychiatric disease cases, when compared with control brain cohorts. Well-known examples, as they pertain to schizophrenia or mood disorder, involve transcripts for GABAergic inhibitory signaling, or myelination and other oligodendrocyte-specific function, and in some studies more generalized transcriptome changes compromising metabolic activities, as well as many markers of pre- and postsynaptic neurotransmission (Akbarian and Huang, 2006; Aston et al., 2004; Benes, 2010; Charych et al., 2009; Dracheva et al., 2004; Duncan et al., 2010; Guidotti et al., 2005; Hakak et al., 2001; Hashimoto et al., 2008; Katsel et al., 2005; Martins-de-Souza et al., 2009; Regenold et al., 2007; Sibille et al., 2009; Tkachev et al., 2003; Woo et al., 2008).
Therefore, drugs that interfere with chromatin-bound proteins involved in transcriptional regulation could be of interest both to preclinical researchers interested in modeling the aforementioned gene expression deficits in the animal, as well as to groups in academia or industry that are interested in exploring novel psychopharmacologic treatments. In this context, it is worth mentioning that sodium valproate, one of the most frequently prescribed drugs in neurology and psychiatry largely due to its anticonvulsive and mood-stabilizing properties, is a weak but broadly acting inhibitor of histone deacetylase enzymes (HDAC) (Guidotti et al., 2011). Histone acetylation is viewed as a facilitative signal for transcription, while HDAC cleaves off the acetyl groups from the histone lysine residues and is commonly associated with repressive chromatin remodeling (Sharma et al., 2006). Thus, HDAC inhibitors (HDACi) are thought to upregulate gene expression at some loci, by shifting the balance toward acetylation (of promoter-bound histones). For example, it has been suggested that valproate-induced histone hyperacetylation may exert some of its therapeutic effects via transcriptional upregulation at “GABAergic” and other neuronal genes (Guidotti et al., 2011). In animal experiments, histone deacetylase inhibitors improve learning and memory function in a variety of paradigms, including at advanced age and also in mice with mutation in Creb-binding protein, CBP (which is mutated in subjects with Rubinstein-Taybi syndrome). Other preclinical work strongly suggests that HDACi may exert therapeutic effects in depression and related psychiatric illnesses (Covington et al., 2009; Morris et al., 2010; Schroeder et al., 2007). Whether HDACi’s would emerge in the future, indeed, as novel psychopharmacologic treatment options for the treatment of psychiatric disorders is not yet clear. However, it is the broad therapeutic potential of HDACi in the animal model, which goes far beyond the aforementioned psychiatric conditions and includes acute and chronic neurodegeneratived disease including acute brain injury and stroke, as well as Parkinson’s, Alzheimer’s, and Lou Gehrig’s (motor neuron) disease, and various triplet repeat disorders including Huntington’s chorea and spinocerebellar ataxia (Baltan et al., 2011; Chuang et al., 2009; Fischer et al., 2010; Tsou et al., 2009).
Similar to the HDACi previously mentioned, there is some evidence for the therapeutic potential of drugs affecting histone methylation, but it remains unclear whether these findings would in the future bear fruit and lead to novel psychiatric treatment options. Of interest are small molecules such as BIX-01294, which inhibit a select set of histone methyltransferases (HMTs), including histone H3K9-specific HMT G9a/Glp (Kubicek et al., 2007). The H3K9 methylation mark, particularly the di- and trimethylated forms, are associated with repression and negative regulation of transcription, and consequently, expression of some genes in brains is increased after exposure to BIX-01294 (Kubicek et al., 2007). Behavioral changes after BIX-01294 have been reported as well, including increased reward and addiction behavior in the context of cocaine and other stimulant exposure (Maze et al., 2010). The drug’s mechanism of action could, at least in part, involve the inhibition of G9a/Glp-mediated repressive chromatin remodeling at the promoters of Bdnf, Cdk5, Arc, and other genes, which then in turn could lead to increased spine density and synaptic connectivity (Maze et al., 2010). Like the histone-modifying drugs previously discussed, several structurally unrelated DNA methylation inhibitors, including cytidine analogues 5-azacytidine or compounds such as N-phthalyl-L-tryptophan/RG108, when administered directly into brain tissue of mice and rats, alter synaptic plasticity and hippocampal learning and memory and are thereby associated with powerful modulation of reward- and addiction-related behavior (Han et al., 2010; LaPlant et al., 2010; Levenson et al., 2006; Lubin et al., 2008; Miller et al., 2010; Miller and Sweatt, 2007). Whatever the underlying mechanism of action, we predict that, as in cancer treatment and other areas of medicine where presently worldwide hundreds of clinical trials involve epigenetic drug targets, in psychiatry, too, the therapeutic potential of chromatin-modifying drugs will soon be tested on a broader basis, given the plethora of promising findings that are currently emerging from preclinical and translational research.
EPIGENETICS AS A TOOL TO EXPLORE DISEASE-ASSOCIATED DNA STRUCTURAL VARIANTS AND POLYMORPHISMS OUTSIDE OF PROTEIN-CODING SEQUENCES
As already discussed, a significant portion of (psychiatric) disease-associated mutations and polymorphisms are thought to cause functional changes other than alterations in protein-coding sequence. This poses a potential challenge, as the functional role of the “normal” DNA sequence and the (disease-related) structural variants may be hard to discern by sequence analyses alone. To provide the reader with an illustrative example of the challenging tasks that lie ahead for psychiatric (epi)genetics, consider the example of the major histocompatibility complex locus, which long has been implicated in mental illness, conferring significant genetic risk for schizophrenia and related diseases as most recently shown in three large genome-wide association studies published jointly in 2009 (Purcell et al., 2009; Shi et al., 2009; Stefansson et al., 2009). Intergenic disease–associated SNPs identified in the MHC region of chromosome 6 may interact via proteins with functional regions of the genome. Indeed, when open chromatin-associated histone methylation markings, including the H3K4 tri- and mono-methyl mark, and the genomic occupancy pattern of the transcription factor (and potential barrier protein preventing the uncontrolled spread of heterochromatin), CTCF (CCCTC-binding factor), was profiled in neuronal chromatin from human prefrontal cortex and in peripheral cell lines, it became apparent that several schizophrenia-associated SNPs are highly enriched with one or several of these markings (Fig. 13.2). Because the H3K4 tri- and mono-methyl markings are often associated with transcriptional regulation, including promoter- and enhancer-like functions, one could speculate that the risk alleles are probably interfering with local transcription. While this is purely speculative at this point in time, the preceding example illustrates how the study of epigenetic markings bears the potential to provide important clues on the role of otherwise poorly characterized DNA sequences outside of coding regions.
Obviously, the study of chromatin structures could also provide important information for DNA sequences for which a functional role is already known, such as an annotated promoter. One of the best-known examples involves the fragile X mental retardation (FMR1) gene in fragile X mental retardation syndrome, where the abnormal expansion of a CGG codon from (normally) 5–40 repeats from 50 to over 200 triggers excessive promoter DNA methylation, effectively shutting down gene expression by silencing the surrounding chromatin (Oberle et al., 1991). This is true even for genes with a much more subtle contribution to disease risk, such as GAD1 encoding glutamic acid decarboxylase (67kDa) GABA synthesis enzyme, for which some halplotypes and polymorphisms, positioned within a few Kb from the GAD1 transcription start site, confer genetic risk for accelerated loss of frontal lobe gray matter (Addington et al., 2005; Straub et al., 2007) and, via epistatic interaction with catechol-o-methyltransferase (COMT) alleles, regulate synaptic dopamine and modulate overall GABA tissue levels in the prefrontal cortex (Marenco et al., 2010). The same genetic variants surrounding the GAD1 promoter recently emerged as a major driver for the disease-related decline in GAD67 transcript and the epigenetic decoration of the proximal GAD1 promoter in subjects with schizoprenia, including the balance between “open” and “repressive” histone methylation markings histone H3 trimethyl-lysines, K4me3 and K27me3 (Huang et al., 2007). These findings, taken together, clearly illustrate the potential of epigenetic approaches to shed light on the functional impact of structural variants involving regulatory, noncoding DNA, both for rare mutations with high disease risk/penetrance (e.g., FMR1 in fragile X) or for common variants that make only much smaller contribution to the overall disease risk (e.g., GAD1 in schizophrenia).
SYNOPSIS AND OUTLOOK
“Neuroepigenetics” is a new discipline (Day and Sweatt, 2010) that presently takes center stage in the field of mental health research, mainly because: (1) Recent findings suggest that the epigenetic landscapes of the human brain remain “plastic” throughout all periods of brain development and aging, with ongoing dynamic regulation occurring even in neurons and other postmitotic constituents (Cheung et al., 2010; Hernandez et al., 2011; Numata et al., 2012; Siegmund et al., 2007). (2) The range of neurological conditions due to a primary chromatin defect extends far beyond the early developmental period and may even include a subset of cases with adult-onset psychosis, or dementia and other neurodegenerative disease (Klein et al., 2011; Winkelmann et al., 2012). (3) Chromatin-modifying drugs could lead to novel treatments for neurological and psychiatric disease (BalTan et al., 2011; Chuang et al., 2009; Fischer et al., 2010; Peter and Akbarian, 2011; Tsou et al., 2009). (4) Exploration of chromatin structures could be expected to uncover, in a substantial portion of cases, the functional impact of disease-relevant mutations in regulatory and other sequences that are otherwise difficult to “capture” by DNA sequence analyses alone. Indeed, the important role of epigenetically regulated noncoding DNA was ascertained by recent bioinformatical studies showing that many noncoding DNA sequences are generally deficient of SNP and undergo a purifying selection (Tolstorukov et al., 2011).
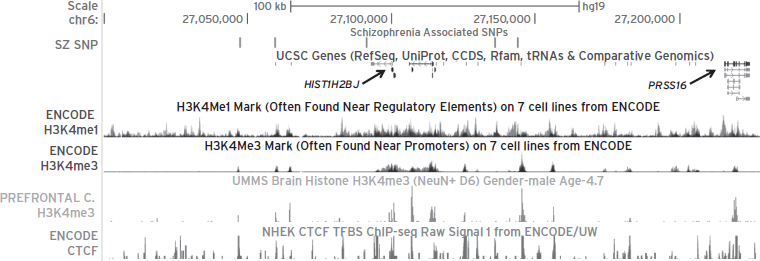
Figure 13.2 Epigenetic profiles encompassing the MHC region on chromosome 6, which harbors strong linkage disequilibrium (r2 = 0.52–0.77) of five schizophrenia single-nucleotide polymorphisms (SNPs) in the major histocompatibility complex (MHC) region (27,000,000–27,300,000) (Purcell et al., 2009; Stefansson et al., 2009). The most significant SNP rs6913660 is located greater than 50 kb from the nearest gene (HIST1H2BJ). The chr 6:27,000,000–27,300,000 region on the UCSC genome browser is shown with six tracks represented: (i) five schizophrenia-associated SNPs in linkage disequilibrium in the following order: rs6904071, rs926300, rs6913660, rs13219181, and rs1319453; (ii) UCSC genes; (iii) histone 3 lysine 4 monomethylation (H3K4me1) marks on seven cells from ENCODE; (iv) histone 3 lysine 4 trimethylation (H3K4me3) marks on seven cell lines from ENCODE; (v) H3K4me3 marks from prefrontal cortex neuron in a 4.7-year-old boy; and (vi) CTCF ChIP-seq data from the NHEK cell line. Notice that several schizophrenia-associated SNPs are found within H3K4me1, H3K4me3, and CTCF peak sequences, indicating possible physical interactions between these various chromatin fragments.
Finally, it is worth mentioning that, based on next-generation sequencing of epigenetic markings in sperm, perhaps as much as 4% of the human genome could maintain nucleosomal organization and many types of epigenetic decoration when transmitted through the germline. This includes many loci considered of critical importance for early pre- and postimplantation development, imprinted gene clusters, microRNA clusters, homeobox (HOX) gene clusters, and the promoters of many stand-alone developmental transcription and signaling factors (Hammoud et al., 2010). These and related findings will most certainly further stimulate research aimed at uncovering evidence for epigenetic heritability of psychiatric disease, including depression, schizophrenia, and addiction, to name a few, which all have in common that for a majority of subjects no straightforward genetic risk architecture has been identified.
Without doubt, psychiatric epigenetics will remain a most productive area of research for many years to come.
DISCLOSURE
The authors declare no conflicts of interests to disclose.
Work conducted in the authors’ laboratories is sponsored by the National Institutes of Health to S. A., the Brain Behavior Research Foundation to S. A., the NIMH (R01 MH084966 to K. A. G.), and the U.S. Army Research Laboratory and the U.S. Army Research Office (grant 58076-LS-DRP) to K. A. G.
REFERENCES
Addington, A.M., Gornick, M., et al. (2005). GAD1 (2q31.1), which encodes glutamic acid decarboxylase (GAD67), is associated with childhood-onset schizophrenia and cortical gray matter volume loss. Mol. Psychiatry 10:581–588.
Adegbola, A., Gao, H., et al. (2008). A novel mutation in JARID1C/SMCX in a patient with autism spectrum disorder (ASD). Am. J. Med. Genet. A 146A, 505–511.
Akbarian, S., and Huang, H.S. (2006). Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain. Res. Rev. 52:293–304.
Akbarian, S., and Huang, H.S. (2009). Epigenetic regulation in human brain-focus on histone lysine methylation. Biol. Psychiatry 65:198–203.
Akbarian, S., Sucher, N.J., et al. (1996). Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J. Neurosci. 16:19–30.
Amir, R.E., Van den Veyver, I.B., et al. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23:185–188.
Aston, C., Jiang, L., et al. (2004). Microarray analysis of postmortem temporal cortex from patients with schizophrenia. J. Neurosci. Res. 77:858–866.
Baltan, S., Murphy, S.P., et al. (2011). Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J. Neurosci. 31:3990–3999.
Belmont, A.S. (2006). Mitotic chromosome structure and condensation. Curr. Opin. Cell. Biol. 18:632–638.
Benes, F.M. (2010). Amygdalocortical circuitry in schizophrenia: from circuits to molecules. Neuropsychopharmacol. 35:239–257.
Berger, S.L. (2007). The complex language of chromatin regulation during transcription. Nature 447:407–412.
Bhutani, N., Burns, D.M., et al. (2011). DNA demethylation dynamics. Cell 146:866–872.
Bintu, L., Kopaczynska, M., et al. (2011). The elongation rate of RNA polymerase determines the fate of transcribed nucleosomes. Nat. Struct. Mol. Biol. 18:1394–1399.
Charych, E.I., Liu, F., et al. (2009). GABA(A) receptors and their associated proteins: implications in the etiology and treatment of schizophrenia and related disorders. Neuropharmacology 57:481–495.
Cheung, I., Shulha, H.P., et al. (2010). Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Natl. Acad. Sci. USA 107:8824–8829.
Chouery, E., Ghoch, J.A., et al. (2011). A novel deletion in ZBTB24 in a Lebanese family with Immunodeficiency, Centromeric Instability, and Facial Anomalies Syndrome Type 2. Clin. Genet. 82:480–493.
Chuang, D.M., Leng, Y., et al. (2009). Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 32:591–601.
Copeland, R.A., Solomon, M.E., et al. (2009). Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 8:724–732.
Copray, S., Huynh, J.L., et al. (2009). Epigenetic mechanisms facilitating oligodendrocyte development, maturation, and aging. Glia 57:1579–1587.
Covington, H.E., III, Maze, I., et al. (2009). Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 29:11451–11460.
Cremer, T., and Cremer, C. (2001). Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2:292–301.
Day, J.J., and Sweatt, J.D. (2010). DNA methylation and memory formation. Nat. Neurosci. 13:1319–1323.
Day, J.J., and Sweatt, J.D. (2011). Epigenetic mechanisms in cognition. Neuron 70:813–829.
Dekker, J., Rippe, K., et al. (2002). Capturing chromosome conformation. Science 295:1306–1311.
Dhar, S.S., Ongwijitwat, S., et al. (2009). Chromosome conformation capture of all 13 genomic loci in the transcriptional regulation of the multisubunit bigenomic cytochrome C oxidase in neurons. J. Biol. Chem. 284:18644–18650.
Dracheva, S., Elhakem, S.L., et al. (2004). GAD67 and GAD65 mRNA and protein expression in cerebrocortical regions of elderly patients with schizophrenia. J. Neurosci. Res. 76:581–592.
Duan, Z., Andronescu, M., et al. (2010). A three-dimensional model of the yeast genome. Nature 465:363–367.
Duncan, C.E., Webster, M.J., et al. (2010). Prefrontal GABA(A) receptor alpha-subunit expression in normal postnatal human development and schizophrenia. J. Psychiatr. Res. 44:673–681.
Ederveen, T.H., Mandemaker, I.K., et al. (2011). The human histone H3 complement anno 2011. Biochim. Biophys. Acta 1809:577–586.
Ernst, C., Chen, E.S., et al. (2009). Histone methylation and decreased expression of TrkB.T1 in orbital frontal cortex of suicide completers. Mol. Psychiatry 14:830–832.
Filippakopoulos, P., Qi, J., et al. (2010). Selective inhibition of BET bromodomains. Nature 468:1067–1073.
Fiori, L.M., and Turecki, G. (2010). Genetic and epigenetic influences on expression of spermine synthase and spermine oxidase in suicide completers. Int. J. Neuropsychopharmacol. 13:725–736.
Fischer, A., Sananbenesi, F., et al. (2010). Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol. Sci. 31:605–617.
Gaszner, M., and Felsenfeld, G. (2006). Insulators: exploiting transcriptional and epigenetic mechanisms. Nat. Rev. Genet. 7:703–713.
Guidotti, A., Auta, J., et al. (2005). GABAergic dysfunction in schizophrenia: new treatment strategies on the horizon. Psychopharmacol. (Berl) 180:191–205.
Guidotti, A., Auta, J., et al. (2011). Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology 60:1007–1016.
Guo, J.U., Su, Y., et al. (2011). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145:423–434.
Haggarty, S.J., and Tsai, L.H. (2011). Probing the role of HDACs and mechanisms of chromatin-mediated neuroplasticity. Neurobiol. Learn. Mem. 96:41–52.
Hakak, Y., Walker, J.R., et al. (2001). Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. USA 98:4746–4751.
Hammoud, S.S., Purwar, J., et al. (2010). Alterations in sperm DNA methylation patterns at imprinted loci in two classes of infertility. Fertil. Steril. 94:1728–1733.
Han, J., Li, Y., et al. (2010). Effect of 5-aza-2-deoxycytidine microinjecting into hippocampus and prelimbic cortex on acquisition and retrieval of cocaine-induced place preference in C57BL/6 mice. Eur. J. Pharmacol. 642:93–98.
Hansen, R.S., Wijmenga, C., et al. (1999). The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA 96:14412–14417.
Hargreaves, D.C., Horng, T., et al. (2009). Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138:129–145.
Hashimoto, T., Bazmi, H.H., et al. (2008). Conserved regional patterns of GABA-related transcript expression in the neocortex of subjects with schizophrenia. Am. J. Psychiatry 165:479–489.
He, Y.F., Li, B. et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333:1303–1307.
Henikoff, S., and Shilatifard, A. (2011). Histone modification: cause or cog? Trends Genet. 27:389–396.
Hernandez, D.G., Nalls, M.A., et al. (2011). Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum. Mol. Genet. 20:1164–1172.
Hollis, F., Wang, H., et al. (2010). The effects of repeated social defeat on long-term depressive-like behavior and short-term histone modifications in the hippocampus in male Sprague-Dawley rats. Psychopharmacol. (Berl) 211:69–77.
Horike, S., Cai, S., et al. (2005). Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 37:31–40.
Houston, I., Peter, C.J., et al. (2012). Epigenetics in the human brain. Neuropsychopharmacol. 38:183–197.
Huang, H.S., Matevossian, A., et al. (2007). Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J. Neurosci. 27:11254–11262.
Ito, S., Shen, L., et al. (2011). Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333:1300–1303.
Iwase, S., Lan, F., et al. (2007). The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 128:1077–1088.
Jakocevski, M., and Akbarian, S. (2012). Epigenetic mechanisms in neurodevelopmental and neurodegenerative disease. Nat. Med. 18:1194–1204.
Janzen, W.P., Wigle, T.J., et al. (2010). Epigenetics: tools and technologies. Drug Discov. Today Technol. 7:e59–e65.
Jiang, Y., Jakovcevski, M., et al. (2010). Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J. Neurosci. 30:7152–7167.
Jin, C., and Felsenfeld, G. (2007). Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 21:1519–1529.
Jin, S.G., Wu, X., Li, A.X., et al. (2011). Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 39:5015–5024.
Katsel, P., Davis, K.L., et al. (2005). Variations in myelin and oligodendrocyte-related gene expression across multiple brain regions in schizophrenia: a gene ontology study. Schizophr. Res. 79:157–173.
Kinney, S.M., Chin, H.G., et al. (2011). Tissue-specific distribution and dynamic changes of 5-hydroxymethylcytosine in mammalian genomes. J. Biol. Chem. 286:24685–24693.
Kirov, G., Pocklington, A.J., et al. (2012). De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 17:142–153.
Kleefstra, T., van Zelst-Stams, W.A., et al. (2009). Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J. Med. Genet. 46:598–606.
Klein, C.J., Botuyan, M.V., et al. (2011). Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 43:595–600.
Kleinjan, D.A., and van Heyningen, V. (2005). Long-range control of gene expression: emerging mechanisms and disruption in disease. Am. J. Hum. Genet. 76:8–32.
Klose, R.J., and Zhang, Y. (2007). Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 8:307–318.
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128:693–705.
Kriaucionis, S., and Heintz, N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324:929–930.
Krishnan, V., and Nestler, E.J. (2010). Linking molecules to mood: new insight into the biology of depression. Am. J. Psychiatry 167:1305–1320.
Kubicek, S., O’Sullivan, R.J., et al. (2007). Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 25:473–481.
Labrie, V., Pai, S., et al. (2012). Epigenetics of major psychosis: progress, problems and perspectives. Trends Genet. 28:427–435.
LaPlant, Q., Vialou, V., et al. (2010). Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat. Neurosci. 13:1137–1143.
Lehman, A.F., Lieberman, J.A., et al. (2004). Practice guideline for the treatment of patients with schizophrenia, second edition. Am. J. Psychiatry 161:1–56.
Levenson, J.M., Roth, T.L., et al. (2006). Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 281:15763–15773.
Li, G., and Reinberg, D. (2011). Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev. 21:175–186.
Loenarz, C., and Schofield, C.J. (2011). Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem. Sci. 36:7–18.
Lomvardas, S., Barnea, G., et al. (2006). Interchromosomal interactions and olfactory receptor choice. Cell 126:403–413.
Lu, T., Pan, Y., et al. (2004). Gene regulation and DNA damage in the ageing human brain. Nature 429:883–891.
Lubin, F.D., Roth, T.L., et al. (2008). Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 28:10576–10586.
Machado-Vieira, R., Ibrahim, L., et al. (2011). Histone deacetylases and mood disorders: epigenetic programming in gene-environment interactions. CNS Neurosci. Ther. 17:699–704.
Marenco, S., Savostyanova, A.A., et al. (2010). Genetic modulation of GABA levels in the anterior cingulate cortex by GAD1 and COMT. Neuropsychopharmacol. 35:1708–1717.
Martins-de-Souza, D., Gattaz, W.F., et al. (2009). Alterations in oligodendrocyte proteins, calcium homeostasis and new potential markers in schizophrenia anterior temporal lobe are revealed by shotgun proteome analysis. J. Neural. Transm. 116:275–289.
Maunakea, A.K., Nagarajan, R.P., et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466:253–257.
Maze, I., Covington, H.E., III, et al. (2010). Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327:213–216.
McGowan, P.O., Sasaki, A., et al. (2008). Promoter-wide hypermethylation of the ribosomal RNA gene promoter in the suicide brain. PLoS One 3, e2085.
McGowan, P.O., Sasaki, A., et al. (2009). Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 12:342–348.
Miller, C.A., Gavin, C.F., et al. (2010). Cortical DNA methylation maintains remote memory. Nat. Neurosci. 13:664–666.
Miller, C.A., and Sweatt, J.D. (2007). Covalent modification of DNA regulates memory formation. Neuron 53:857–869.
Morris, M.J., Karra, A.S., et al. (2010). Histone deacetylases govern cellular mechanisms underlying behavioral and synaptic plasticity in the developing and adult brain. Behav. Pharmacol. 21:409–419.
Nestler, E.J., and Hyman, S.E. (2010). Animal models of neuropsychiatric disorders. Nat. Neurosci. 13:1161–1169.
Nowak, S.J., and Corces, V.G. (2004). Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet. 20:214–220.
Numata, S., Ye, T., et al. (2012). DNA methylation signatures in development and aging of the human prefrontal cortex. Am. J. Hum. Genet. 90:260–272.
Oberle, I., Rousseau, F., et al. (1991). Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252:1097–1102.
Okano, M., Bell, D.W., et al. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99:247–257.
Ooi, S.K., and Bestor, T.H. (2008). The colorful history of active DNA demethylation. Cell 133:1145–1148.
O’Roak, B.J., Deriziotis, P., et al. (2011). Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 43:585–589.
Peleg, S., Sananbenesi, F., et al. (2010). Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 328:753–756.
Peter, C.J., and Akbarian, S. (2011). Balancing histone methylation activities in psychiatric disorders. Trends. Mol. Med. 17:372–379.
Piton, A., Gauthier, J., et al. (2011). Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol. Psychiatry 16:867–880.
Purcell, S.M., Wray, N.R., et al. (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460:748–752.
Regenold, W.T., Phatak, P., et al. (2007). Myelin staining of deep white matter in the dorsolateral prefrontal cortex in schizophrenia, bipolar disorder, and unipolar major depression. Psychiatry Res. 151:179–188.
Robison, A.J., and Nestler, E.J. (2011). Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci. 12:623–637.
Rodriguez-Paredes, M., and Esteller, M. (2011). Cancer epigenetics reaches mainstream oncology. Nat. Med. 17:330–339.
Rotili, D., and Mai, A. (2011). Targeting histone demethylases: a new avenue for the fight against cancer. Genes and Cancer 2:663–679.
Rusiecki, J.A., Chen, L., et al. (2012). DNA methylation in repetitive elements and post-traumatic stress disorder: a case-control study of US military service members. Epigenomic. 4:29–40.
Schroeder, F.A., Lin, C.L., et al. (2007). Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol. Psychiatry 62:55–64.
Sharma, R.P., Grayson, D.R., et al. (2005). Chromatin, DNA methylation and neuron gene regulation—the purpose of the package. J. Psychiatry Neurosci. 30:257–263.
Sharma, R.P., Rosen, C., et al. (2006). Valproic acid and chromatin remodeling in schizophrenia and bipolar disorder: preliminary results from a clinical population. Schizophr. Res. 88:227–231.
Shi, J., Levinson, D.F., et al. (2009). Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460:753–757.
Shulha, H.P., Cheung, I., et al. (2012). Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Arch. Gen. Psychiatry 69:314–324.
Sibille, E., Wang, Y., et al. (2009). A molecular signature of depression in the amygdala. Am. J. Psychiatry 166:1011–1024.
Siegmund, K.D., Connor, C.M., et al. (2007). DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One 2, e895.
Simonis, M., Klous, P., et al. (2006). Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat. Genet. 38:1348–1354.
Simonis, M., Kooren, J., et al. (2007). An evaluation of 3C-based methods to capture DNA interactions. Nat. Methods 4:895–901.
Smeraldi, E., Bellodi, L., et al. (1976). Further studies on the major histocompatibility complex as a genetic marker for schizophrenia. Biol. Psychiatry 11:655–661.
Smith, A.K., Conneely, K.N., et al. (2011). Differential immune system DNA methylation and cytokine regulation in post-traumatic stress disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156B:700–708.
Song, C.X., Szulwach, K.E., et al. (2011). Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 29:68–72.
Stadler, F., Kolb, G., et al. (2005). Histone methylation at gene promoters is associated with developmental regulation and region-specific expression of ionotropic and metabotropic glutamate receptors in human brain. J. Neurochem. 94:324–336.
Stefansson, H., Ophoff, R.A., et al. (2009). Common variants conferring risk of schizophrenia. Nature 460:744–747.
Straub, R.E., Lipska, B.K., et al. (2007). Allelic variation in GAD1 (GAD67) is associated with schizophrenia and influences cortical function and gene expression. Mol. Psychiatry 12:854–869.
Stroud, H., Feng, S., et al. (2011). 5-hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 12:R54.
Sutcliffe, E.L., Parish, I.A., et al. (2009). Dynamic histone variant exchange accompanies gene induction in T cells. Mol. Cell Biol. 29:1972–1986.
Szulwach, K.E., Li, X., et al. (2011a). Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet. 7:e1002154.
Szulwach, K.E., Li, X., et al. (2011b). 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14:1607–1616.
Tan, M., Luo, H., et al. (2011). Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146:1016–1028.
Tang, B., Chang, W.L., et al. (2009). Normal human aging and early-stage schizophrenia share common molecular profiles. Aging Cell 8:339–342.
Taverna, S.D., Li, H., et al. (2007). How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14:1025–1040.
Tkachev, D., Mimmack, M.L., et al. (2003). Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet 362:798–805.
Tolstorukov, M.Y., Volfovsky, N., et al. (2011). Impact of chromatin structure on sequence variability in the human genome. Nat. Struct. Mol. Biol. 18:510–515.
Tsou, A.Y., Friedman, L.S., et al. (2009). Pharmacotherapy for Friedreich ataxia. CNS Drugs 23:213–223.
Uddin, M., Aiello, A.E., et al. (2010). Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc. Natl. Acad. Sci. USA 107:9470–9475.
Verhoeven, W.M., Egger, J.I., et al. (2011). Kleefstra syndrome in three adult patients: further delineation of the behavioral and neurological phenotype shows aspects of a neurodegenerative course. Am. J. Med. Genet. A 155A:2409–2415.
Vermeulen, M., Eberl, H.C., et al. (2010). Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142:967–980.
Wang, C.M., Tsai, S.N., et al. (2010). Identification of histone methylation multiplicities patterns in the brain of senescence-accelerated prone mouse 8. Biogerontology 11:87–102.
Weaver, I.C., et al. (2004). Epigenetic programming by maternal behavior. Nat. Neurosci. 7:847–854.
Williams, K., Christensen, J., et al. (2011). TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 473:343–348.
Winkelmann, J., Lin, L., et al. (2012). Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum. Mol. Genet. 21:2205–2210.
Woo, T.U., Kim, A.M., et al. (2008). Disease-specific alterations in glutamatergic neurotransmission on inhibitory interneurons in the prefrontal cortex in schizophrenia. Brain Res. 1218:267–277.
Wood, A.J., Severson, A.F., et al. (2010). Condensin and cohesin complexity: the expanding repertoire of functions. Nat. Rev. Genet. 11:391–404.
Woodcock, C.L. (2006). Chromatin architecture. Curr. Opin. Struct. Biol. 16:213–220.
Wu, H., D’Alessio, A.C., et al. (2011). Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 25:679–684.
Xie, W., Barr, C.L., et al. (2012). Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 148:816–831.
Xu, B., Roos, J.L., et al. (2011). Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat. Genet. 43:864–868.
Yankner, B.A., Lu, T., et al. (2008). The aging brain. Annu. Rev. Pathol. 3:41–66.
Zhou, V.W., Goren, A., et al. (2011). Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 12:7–18.
Zovkic, I.B., and Sweatt, J.D. (2012). Epigenetic mechanisms in learned fear: implications for PTSD. Neuropsychopharmacology 38:77–93.