23 | CORTICAL GABA NEURONS IN SCHIZOPHRENIA
ALLISON A. CURLEY AND DAVID A. LEWIS
Cognitive impairments are a core feature of schizophrenia and the best predictor of functional outcome; however, current pharmacotherapies offer only limited cognitive improvement. Therefore, knowledge of the abnormalities in brain circuitry that give rise to the cognitive abnormalities is critical for developing new treatments. Higher order cognitive processes, such as working memory, the ability to transiently retain and manipulate a limited amount of information in order to guide thought or behavior, are strongly associated with neural network oscillations, the synchronized firing of large assemblies of cells, that are dependent on signaling via the inhibitory neurotransmitter γ-aminobutyric acid (GABA) (Bartos and Elgueta, 2012). The cells that synthesize and release GABA, appropriately termed GABA neurons (or interneurons), exhibit a variety of abnormalities in schizophrenia. Thus, alterations in GABA neurons are thought to, at least in part, underlie the cognitive deficits of schizophrenia. Consequently, in this chapter we provide: (1) an introduction to interneurons and their role in cognition, (2) a description of the GABAergic alterations found in schizophrenia, (3) a review of potential mechanisms underlying these alterations, and (4) a discussion of the consequences of impaired GABA signaling. The prefrontal cortex (PFC) is a brain area especially critical for cognition, and where interneurons have been most extensively studied in schizophrenia, and thus we concentrate on this region here.
INTRODUCTION TO INTERNEURONS
SUBTYPES OF GABA NEURONS
Excitatory pyramidal cells comprise 75–80% of the neurons in the primate neocortex, and the remaining 20–25% are GABA neurons. In contrast to pyramidal cells that mainly send their axons over long distances, interneurons primarily project locally. GABA neurons in the cerebral cortex can be divided into different subclasses based on their electrophysiological, molecular, and anatomical properties (Ascoli et al., 2008) (Fig. 23.1). Electrophysiologically, interneurons are classified as fast-spiking (FS) or non-fast-spiking (non-FS), based on their firing patterns in response to depolarization above spiking threshold. FS cells exhibit a nearly constant interval between spikes. In contrast, non-FS cells typically show an adapting firing pattern, such that the interspike interval increases with stimulus duration. Interneuron subpopulations can also be distinguished by the molecular markers they express. With a few exceptions, the calcium binding proteins parvalbumin (PV), calbindin (CB), and calretinin (CR) are found in distinct cell types. FS cells contain PV (hereafter referred to as PV neurons), do not contain any neuropeptides, and give rise to axons that target the perisomatic region, the cell body and proximal dendrites, of pyramidal cells. PV neurons comprise ~25% GABA neurons in the primate PFC (but a much larger proportion of GABA neurons in rodent cortex), and PV-positive cell bodies and axon terminals are present in the highest density in layers deep 3–4. PV cells are divided into two main classes: (1) basket (PVb) cells, whose axons target the soma, proximal dendrites, and spines of pyramidal cells, and (2) chandelier (PVch) cells, whose axon terminals form distinctive vertical arrays termed cartridges that exclusively target the axon initial segment (AIS) of pyramidal cells.
Non-FS cells are more heterogeneous and include a number of distinct subpopulations (Fig. 23.1). One subpopulation contains the neuropeptide cholecystokinin (CCK). A subset of these cells also contains CR, and a separate subset expresses the cannabinoid 1 receptor (CB1R). The latter are known as CCK basket (CCKb) cells. All CCKb cells target the perisomatic region of pyramidal cells, and in the primate neocortex, CCK-immunoreactive cell bodies are principally localized to layers 2-superficial 3, and their axon terminals are present mainly in layers 2, 4, and 6. A second subtype of non-FS cells expresses CB and the neuropeptide somatostatin (SST), and furnishes axons that synapse onto the distal dendrites of pyramidal cells. SST cells are present in all layers of the cortex, as well as the underlying white matter. A third subtype contains CR, is found primarily in layers 1 and 2, and provides axon terminals that mainly innervate pyramidal neuron dendrites and other interneurons.
GABA SYNTHESIS
GABA is synthesized by the 67 kiloDalton and 65 kiloDalton isoforms of glutamic acid decarboxylase (GAD67 and GAD65, respectively) which decarboxylate glutamate to produce GABA (Fig. 23.2). GAD65 and GAD67 are encoded by separate genes (GAD1 and GAD2, respectively) on different chromosomes, and undergo different posttranslational modifications (Akbarian and Huang, 2006). Many, but not all, GABA neurons appear to contain both isoforms. GAD67 is distributed throughout the neuron, whereas GAD65 is primarily located in axon terminals. The activity of both enzymes is regulated by their cofactor pyridoxal 5’-phosphate, which activates or inactivates the enzyme when it is bound and released, respectively. GAD67 is saturated with cofactor and therefore largely exists in the active form, while GAD65 is mostly inactive (with low levels of cofactor binding). Thus, the activity of GAD67 is principally regulated by expression and appears to be responsible for the majority of cortical GABA synthesis, producing up to 90% of the GABA in mouse brain. In contrast, GAD65 seems to be mainly active during conditions of high synaptic demand (Patel et al., 2006).

Figure 23.1 Diversity of cortical GABA neurons. GABAergic interneurons can be classified based on morphological (A), molecular (B), and electrophysiological (C) properties. Some interneurons express the calcium binding proteins parvalbumin (PV) or calretinin (CR), whereas others contain the neuropeptides somatostatin (SST) or cholecystokinin (CCK). (A) PV and CCK neurons target the perisomatic region of pyramidal cells, while SST and CR neurons target pyramidal neuron dendrites. PV neurons can be divided into chandelier (PVch) and basket (PVb) cells based on their morphology. The axon terminals of PVch cells exclusively target the pyramidal cell axon initial segment, while the terminals of PVb cells synapse onto the soma and proximal dendrites. (B) The different interneuron subtypes are distributed distinctively across the layers of the cortex, as evidenced by the different expression patterns of their mRNAs. (C) PV cells exhibit a fast spiking (FS) firing pattern, characterized by a high firing frequency and constant interval between action potentials, while the remaining subclasses are classified as non-FS cells that fire at a lower frequency and exhibit progressively increasing intervals between action potentials. (Image adapted from Gonzalez-Burgos et al. Am J Psychiatry (2007) 164 (1):12; and Hashimoto et al. Mol Psychiatry (2008) 13:147–161.)
GABA RECEPTORS
Released GABA exerts its effects through binding to the two major types of GABA receptors—GABAA and GABAB receptors (Fig. 23.2). The ionotropic GABAA receptor is composed of 5 subunits that form a central ion pore, with the most common composition being two α, two β, and one γ subunit. The subunit composition of GABAA receptors determines important functional and pharmacological properties, such as the kinetics of the postsynaptic signal decay (Gonzalez-Burgos et al., 2011). Activation of these receptors results in an increased chloride conductance, with the direction of ion flow being determined by the intracellular chloride concentration resulting from the activity of the chloride transporters NKCC1 and KCC2, that bring chloride into and out of the cell, respectively. In the majority of cases, chloride flows inward through GABAA receptors and hyperpolarizes the membrane, producing the classic inhibitory postsynaptic response (Fig. 23.2).
GABAB receptors form heterodimers, each composed of a GABAB1 and GABAB2 subunit, and both subunits are required to produce a functional receptor. Activation of metabotropic GABAB receptors stimulates the opening of potassium channels and closing of calcium channels via G proteins, resulting in hyperpolarization. GABAB receptors are located both pre- and postsynaptically.
ROLE OF GABA NEURONS IN NETWORK OSCILLATIONS AND COGNITION
A single interneuron not only makes multiple contacts onto a pyramidal cell, but also contacts many different pyramidal cells (Gonzalez-Burgos and Lewis, 2008). This places interneurons in a position to exert strong inhibitory control over large numbers of pyramidal cells, so that the firing of a single interneuron transiently silences many pyramidal cells. When the pyramidal cells are then released from inhibition, they fire in concert. This inhibitory mechanism of controlling and timing the activity of large numbers of pyramidal cells by interneurons is crucial for proper cortical function. It maintains neural activity within a functional range, preventing the runaway cortical activity of seizures, and exerts an important influence on the timing of neural activity. In addition, the synchronized firing of networks of interconnected interneurons and pyramidal cells produces rhythmic oscillations of different frequencies.
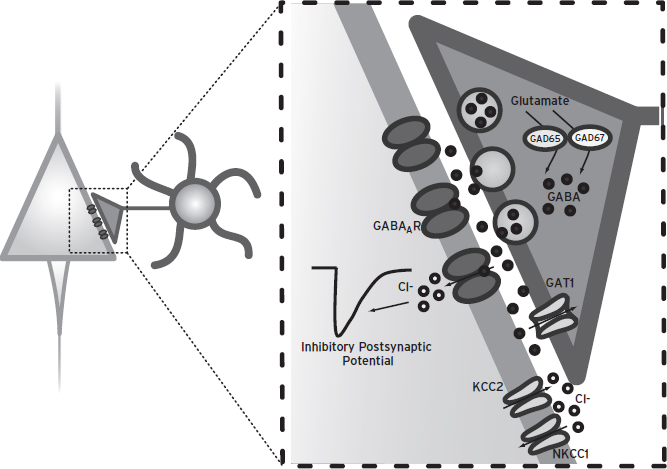
Figure 23.2 Basics of ionotropic GABA neurotransmission. GABA is synthesized from glutamate by the enzyme GAD67 or GAD65. After release from synaptic vesicles into the synaptic cleft, GABA binds to postsynaptic GABAA receptors. GABAA receptor activation stimulates the opening of chloride channels. The direction and magnitude of the chloride current produced by GABAA receptor activation is regulated by the transporters NKCC1 and KCC2, which uptake and extrude chloride, respectively. In the majority of cases, chloride flows inward and hyperpolarizes the membrane, producing the classic inhibitory postsynaptic potential. GABA membrane transporter 1 (GAT1) removes GABA from the synaptic cleft, thereby regulating the concentration of GABA that reaches postsynaptic receptors.
Different subtypes of GABA neurons appear to play distinct roles in the generation of oscillations of different frequencies (Klausberger and Somogyi, 2008). PVb neurons are known to be crucial in the generation of γ oscillations, the synchronized activity of networks of pyramidal neurons at 30–80 Hz (Gonzalez-Burgos et al., 2010). For example, although the activity of multiple interneuron subtypes is associated with γ oscillations, they are most tightly coupled to the firing of PVb neurons. Moreover, recent evidence using optogenetic techniques has demonstrated that suppression and activation of PV neurons suppresses and generates, respectively, γ activity in vivo, indicating that PV neurons are both necessary and sufficient for the generation of γ oscillations (Sohal et al., 2009). The GABAA α1-containing receptors that predominate at PVb cell synapses exhibit a fast decay of the inhibitory synaptic potential (IPSP) that is consistent with the firing rate of γ oscillations (Gonzalez-Burgos and Lewis, 2008). In contrast, CCKb neurons are the interneuron subtype that is most strongly coupled to the slower θ oscillations (4–7 Hz) (Klausberger and Somogyi, 2008). In addition, the GABAA α2-containing receptors that are prominent at CCKb synapses onto pyramidal cells in rodents exhibit a slower IPSP decay, consistent with θ oscillations.
PFC θ and γ oscillations are associated with higher order cognitive processes such as working memory (Curley and Lewis, 2012). For example, θ and γ band activity is induced during the delay period of working memory tasks, and the power of θ and γ synchrony increases in proportion to working memory load. In addition, injection of a GABAA receptor antagonist into the PFC disrupts working memory performance in monkeys (Lewis et al., 2005). Thus, CCKb-and PVb-mediated θ and γ oscillations, respectively, are crucial to normal cognitive function.
INTERNEURON ALTERATIONS IN SCHIZOPHRENIA
CORTICAL GABA SYNTHESIS AND UPTAKE ARE ALTERED
The first evidence of altered cortical GABA signaling in schizophrenia came from early findings of decreased activity of GAD and reduced GABA release and uptake in postmortem tissue samples (Blum and Mann, 2002). However, the most consistent evidence supporting altered GABA neurotransmission is reports of lower levels of the mRNA encoding GAD67 in the PFC of subjects with schizophrenia. Lower GAD67 mRNA in the PFC was first demonstrated in 1995 by Akbarian and colleagues using in situ hybridization, and has since been widely replicated using DNA microarray, quantitative PCR (qPCR), and in situ hybridization (Blum and Mann, 2002; Gonzalez-Burgos et al., 2010). Thus far, 10 studies have found deficits in tissue-level GAD67 mRNA in the PFC in schizophrenia, with the average deficit ranging in magnitude from 12% to 68%.
On a cellular level, the density of GAD67 mRNA-positive neurons is approximately 25–35% lower across PFC layers 1–5 of subjects with schizophrenia (Lewis et al., 2005). In the remaining GAD67-positive neurons, the expression level per neuron is not different from that of comparison subjects. Since total neuron number is unaltered in the PFC, these data suggest that GABA neurons are not missing in schizophrenia, but that the majority express normal levels of GAD67 mRNA and a subset express levels so low as to not be detectable.
Although a GAD67 mRNA deficit in the PFC has been widely and consistently reported, knowledge of protein levels is of particular importance given that mRNA and protein levels are not necessarily correlated, since a number of factors regulate transcription and translation (Blum and Mann, 2002). For example, pharmacological manipulation of GABA levels is associated with changes in GAD67 protein but not mRNA. However, similar to the mRNA deficit, levels of GAD67 protein are also lower in the PFC in schizophrenia, and in the largest study to date, the magnitudes of the tissue-level reductions in mRNA and protein (15% and 10%, respectively) were similar (Curley et al., 2011).
In contrast to the reductions in GAD67, the mRNA and protein levels of GAD65 have been reported to be unchanged or only slightly reduced in the PFC in schizophrenia (Lewis et al., 2012). Moreover, the density of GAD65-immunoreactive (-IR) terminals is also unaltered. Thus, schizophrenia is characterized by a preferential deficit of GAD67, but not of GAD65, in the PFC. Given that GAD67 mediates the majority of GABA synthesis in the cortex, lower GAD67 in schizophrenia is thought to result in a reduction of cortical GABA levels that significantly impairs synaptic transmission and inhibition of postsynaptic targets.
Although it is possible that compensatory increases in GAD65 may normalize GABA levels in the illness, this scenario is unlikely for a number of reasons. First, as described earlier, most reports of GAD65 levels in schizophrenia have found no alteration. Second, GAD67 knockout mice exhibit normal levels of GAD65 mRNA and protein, and markedly lower levels of GABA (Asada et al., 1997). Third, recent data indicate that some interneurons primarily use GAD67 while others rely mainly on GAD65 (Lewis et al., 2012), suggesting that interneurons do not use GAD65 and GAD67 interchangeably.
Other aspects of GABA neurotransmission in the PFC are also altered in schizophrenia. For example, expression of the mRNA encoding the GABA membrane transporter 1 (GAT1), which removes GABA from the extracellular space, is decreased in the PFC of schizophrenia subjects, and the density of GAT1 mRNA-positive neurons is lower (Lewis et al., 2005). In concert with the findings of unchanged neuron number in the PFC, this latter finding suggests lower GAT1 expression per neuron, such that in some neurons the level falls below detectability, rather than a reduction in the number of GAT1-expressing neurons in schizophrenia. The deficits in GAD67 and GAT1 may be occurring in the same cells, since examination of the same matched pairs of control and schizophrenia subjects found that the within-pair differences in GAD67 mRNA-positive and GAT1 mRNA-positive neuron density were similar in laminar pattern and significantly correlated in the same cohort of subjects. Thus, schizophrenia is characterized by alterations in both the synthesis and reuptake of cortical GABA (Lewis et al., 2005).
PARVALBUMIN NEURONS ARE PARTICULARLY AFFECTED IN SCHIZOPHRENIA
Given that different subtypes of interneurons possess distinct electrical, molecular, and anatomical properties, the functional outcome of impaired GABA neurotransmission in schizophrenia depends on the subpopulation(s) of neurons affected. PV cells are one subtype of GABA neuron that is known to contain lower GAD67 mRNA in schizophrenia (Lewis et al., 2012). Dual label in situ hybridization has shown that approximately 50% of PV mRNA-positive neurons lack detectable levels of GAD67. In addition, PV mRNA levels are significantly decreased, and laminar analyses have revealed that this reduction occurs principally in layers 3 and 4, layers where lower GAD67 mRNA is also present.
However, in contrast to GAD67 mRNA, although the expression of PV mRNA per neuron is decreased, neither the density of neurons with detectable levels of PV mRNA nor the density of PV-immunoreactive neurons is altered in schizophrenia (Lewis et al., 2012). Although some studies have reported a lower density of PV-immunoreactive neurons in schizophrenia, these results may have been confounded by a reduced sensitivity for PV immunoreactivity resulting from the tissue processing method utilized. Moreover, the lower density of PV-immunoreactive neurons was reported in layers 3–5, where the largest decrement in PV mRNA is found. Thus, levels of PV protein are likely to have fallen below detectability in conditions of reduced sensitivity, and thus the reported findings likely do not reflect lower PV neuron number, but rather an inability to visualize all of the neurons that are present (Stan and Lewis, 2012). In addition, in matched pairs of schizophrenia and control subjects, the within-subject pair difference in PV mRNA expression per neuron was positively correlated with the difference in density of GAD67 mRNA-positive neurons, suggesting that GAD67 is dramatically lower in a subpopulation of neurons that contain lower, but still detectable, levels of PV mRNA (Lewis et al., 2005).
CONVERGENT EVIDENCE POINTS TO WEAKER INHIBITION FROM PV BASKET CELLS
A recent study demonstrated that the axon terminals of PVb cells exhibit an approximately 50% reduction in GAD67 protein (Lewis et al., 2012). In addition to lower GABA synthesis, several lines of evidence suggest that PVb cells exhibit other presynaptic alterations that result in weaker inhibition onto pyramidal cells in the PFC of subjects with schizophrenia (Lewis et al., 2012) (Fig. 23.3). First, mRNA and protein levels of µ opioid receptors are increased in the PFC of subjects with schizophrenia. Perisomatic µ opioid receptors, which are localized to PVb cells, hyperpolarize the cell body through the activation of inwardly rectifying potassium channels, making the cell less likely to fire, and µ opioid receptors localized to axon terminals lead to the suppression of vesicular GABA release. Therefore, the greater complement of µ opioid receptors in schizophrenia could contribute to increased suppression of GABA release and result in deficient PVb cell output (Lewis et al., 2012).
On the postsynaptic side of the PVb-pyramidal cell synapse, additional alterations are present that may further lower the PVb-mediated inhibition of pyramidal neurons (Fig. 23.3). First, PVb-pyramidal neuron synapses are populated by α1-containing GABAA receptors, and lower α1 mRNA has been demonstrated in schizophrenia PFC by several studies (Lewis et al., 2012). In addition, mRNA levels of the β2 and γ2 subunits, which coassemble with the α1 subunit, are also lower in schizophrenia. The lower levels of α1 and β2 are most prominent in layers 3–4, exhibiting the same laminar pattern as the PV mRNA deficit. Furthermore, in PFC layer 3, α1 mRNA levels are significantly reduced by 40% in pyramidal cells, but unchanged in interneurons. These data suggest that the number of GABAA receptors postsynaptic to PVb cell inputs is selectively lower in pyramidal cells, contributing to weaker PVb inhibition of pyramidal cells in schizophrenia (Lewis et al., 2012).

Figure 23.3 Schematic summary of alterations in neuronal circuitry in the PFC of subjects with schizophrenia. (A) The perisomatic inhibition of pyramidal (P) neurons by parvalbumin basket (PVb) cells is lower due to (1) lower GAD67 mRNA and protein, and therefore less GABA synthesis; (2) higher levels of µ opioid receptor expression in PVb cells that reduces their activity and suppresses GABA release; (3) reduced expression of cholecystokinin (CCK) mRNA, which stimulates the activity of, and GABA release from, PVb cells; and (4) less mRNA for, and presumably fewer, postsynaptic GABAA α1 receptors in pyramidal neurons. (B) The perisomatic inhibition of pyramidal neurons by cholecystokinin-expressing basket (CCKb) cells is enhanced due to lower levels of CCK and cannabinoid 1 receptor (CB1R) mRNAs that reduce depolarization-induced suppression of inhibition (DSI). Levels of GAD67 in CCKb cells are unknown, but are thought to be very low, relative to PV cells, in the healthy state. (C) PV-expressing chandelier (PVch) cells have decreased GABA membrane transporter 1 (GAT1) protein in their axon terminals and increased postsynaptic GABAA α2-containing receptors at pyramidal neuron axon initial segments. The levels of GAD67 protein in PVch cells in schizophrenia are not known. (D) Somatostatin (SST)-containing cells contain lower mRNA levels of SST, and expression of its receptor, SSTR2, is also lower. Levels of GAD67 in SST cells have not been measured. (E) Calretinin (CR)-containing cells are thought to be unaffected, since levels of CR mRNA and protein are unchanged. GAD67 levels in CR cells are unknown.
Finally, inhibitory inputs onto pyramidal cells may be less hyperpolarizing in schizophrenia. The chloride transporters NKCC1 and KCC2 partially control the driving force of chloride entry into pyramidal neurons, thereby determining the strength of the postsynaptic response to GABA. The mRNA levels of two phosphatases, OSXR1 and WNK3, which phosphorylate NKCC1 and KCC2, increasing and decreasing their function, respectively, are markedly elevated in the PFC of subjects with schizophrenia. Thus, increased activity of NKCC1 and decreased activity of KCC2 in schizophrenia would result in a higher than normal intracellular chloride concentration, leading to a reduced chloride driving force, and subsequently less hyperpolarization of pyramidal neurons when GABA is released from PVb cells (Lewis et al., 2012).
PV CHANDELIER CELLS ARE ALSO ALTERED IN SCHIZOPHRENIA
PVch cells also exhibit alterations in schizophrenia (Fig. 23.3) (Lewis et al., 2012). The density of PVch cartridges that are immunoreactive for GAT1 is 40% lower in schizophrenia, although the density of other GAT1-immunoreactive structures is unchanged. In addition, the density of GABAA α2-containing axon initial segments is increased in the PFC of subjects with schizophrenia, and this increase is significantly correlated with the reduced density of GAT1 cartridges. These reciprocal alterations have been interpreted as coordinated compensations attempting to counteract a deficit in GAD67 in PVch cells, since lower GABA reuptake and an increased probability of GABAA receptor binding would both augment GABA signaling. However, to date, GAD67 levels in PVch cells have not been measured.
CCK BASKET CELLS ARE ALSO ALTERED IN SCHIZOPHRENIA
Abnormalities in PV neurons alone may not account for the lower GAD67 mRNA and protein levels observed in schizophrenia. In addition to the reductions in layers 3–4 in the PFC, lower levels of GAD67 mRNA have also been observed in layers 1, 2, and 5, where relatively few PV-expressing GABA neurons are located and where PV mRNA expression is unaltered in schizophrenia (Lewis et al., 2005). These findings suggest that other subsets of interneurons residing outside layers 3–4 may also exhibit reductions in GAD67. One candidate population is CCKb cells, whose cell bodies are principally localized to layers 2-superficial 3 (Fig. 23.3). Lower levels of CCK and CB1R mRNAs, and lower CB1R protein levels, are present in the PFC of schizophrenia subjects. Furthermore, in matched pairs of schizophrenia and control subjects, the within-subject pair difference in GAD67 mRNA levels was positively correlated with differences in both CCK and CB1 mRNAs, suggesting that the GAD67 mRNA deficit in layers 2–3 is present in CCKb cells. However, GAD67 levels have not yet been directly assessed in CCKb cells in schizophrenia. A recent finding that CB1R-expressing axon terminals in monkey PFC tissue have very low levels of GAD67 protein suggests that CCKb cells may rely mainly on GAD65 for GABA synthesis. Consistent with this hypothesis, nearly all CCK-expressing, but only a few PV-expressing, cells in mouse neocortex express GAD65. Thus, CCKb cells may not contain lower GAD67 in schizophrenia (Curley and Lewis, 2012).
CCKb cells are known to participate in a process termed depolarization-induced suppression of inhibition (DSI). In this phenomenon, elevated calcium levels produced from depolarization stimulate the retrograde release of endocannabinoids from pyramidal cells. The resulting binding of endocannabinoids to presynaptic CB1Rs located on CCKb cell terminals activates the receptors, inhibits presynaptic calcium channels, and suppresses GABA release from CCKb terminals, producing a reduced inhibition of the original depolarized pyramidal cell and other nearby cells (Wilson and Nicoll, 2002). DSI is present throughout development and into adulthood in monkey PFC. Thus, the lower levels of CB1R present in schizophrenia may result in less DSI and consequently greater inhibition of pyramidal cells by CCKb cells (Curley and Lewis, 2012). In fact, markers of endocannabinoid synthesis and degradation are unaltered in schizophrenia, consistent with the idea that lower CB1R levels in the illness are not downregulated in response to increased endocannabinoid levels. Furthermore, endocannabinoid-mediated suppression of GABA release from CCKb cells can also be accomplished through application of CCK, and therefore lower CCK levels in schizophrenia may also contribute to increased CCKb cell output (Curley and Lewis, 2012). In addition, since CCKb cell synapses onto pyramidal cells predominantly feature GABAA α2-containing receptors, the increased expression of α2 mRNA that is present in the PFC of subjects with schizophrenia is consistent with an increased inhibitory effect of CCKb cells. However, since α2-containing receptors are also present at PVch cell synapses, whether the higher levels of GABAA α2-containing receptors are also present at CCKb cell synapses remains to be determined (Curley and Lewis, 2012).
Alternatively, if GAD67 is lower in CCKb cells in schizophrenia, reduced CCK and CB1R levels may represent a compensation designed to increase GABA neurotransmission. Consistent with this idea, germ-line reductions of GAD67 in mouse PFC are associated with lower CB1R levels, and the alterations in GAD67 and CB1R are positively correlated, similar to schizophrenia. However, since GAD67 protein levels are normally very low in CCKb axon terminals from monkey PFC, germ-line reductions in GAD67 may lower CB1R levels by acting at the circuit level rather than within CCKb cells (Curley and Lewis, 2012).
SST NEURONS ARE ALSO AFFECTED IN SCHIZOPHRENIA
A fourth population of interneurons that exhibit alterations in schizophrenia is cells expressing SST (Fig. 23.3). Lower SST mRNA is present in the PFC of subjects with schizophrenia (Fung et al., 2010), and the reductions are significantly correlated with the deficit observed in GAD67 mRNA in the same subject cohort. Lower levels of somatostatin receptor 2 mRNA are also present in the PFC of subjects with schizophrenia (Beneyto et al., 2012). However, GAD67 levels in SST cells have not been directly examined in schizophrenia, and thus the nature of their involvement in the disease pathology is currently unclear.
CALRETININ CELLS DO NOT SEEM TO BE ALTERED IN SCHIZOPHRENIA
The ~40–50% of interneurons that express CR in the primate PFC do not appear to be affected in schizophrenia (Fig. 23.3). CR mRNA, CR-immunoreactive neurons, and CR-immunoreactive axon terminals are all unaltered in the illness (Lewis et al., 2005). In contrast to PVb, PVch, CCKb, and SST cells, CR cells primarily target other GABA neurons, and thus the fact that these cells seem to be spared in schizophrenia is suggestive of a selective deficit in the inhibition of pyramidal cells, but not of interneurons, in the illness.
ALTERATIONS IN GABA NEURONS ARE FOUND IN OTHER BRAIN REGIONS
An important question that arises from the findings of GABA neuron abnormalities in the PFC is whether these changes are specific to this brain region or are also present in other cortical areas. In fact, reductions in GAD67 mRNA have been reported (although not yet replicated in every location) in the primary motor, primary visual, anterior cingulate, and orbitofrontal cortices, as well as the superior temporal gyrus and hippocampus (Gonzalez-Burgos et al., 2010; Thompson Ray et al., 2011). In addition, other markers of GABA neurotransmission, including PV, GAT1, SST, and GABAA α1, are lower across multiple cortical regions in schizophrenia, with similar magnitudes of reduction observed across brain regions for each transcript (Lewis et al., 2012). Lower GABAB receptor immunoreactivity has been reported in the hippocampus, entorhinal cortex, and inferior temporal cortex of subjects with schizophrenia (Mizukami et al., 2002). Importantly, similar to the PFC, CR and GAD65 mRNA are unaltered across multiple cortical regions (Lewis et al., 2012; although axon terminal GAD65 protein levels are lower in primary auditory cortex, see Moyer et al., 2012). Thus, the changes observed in the PFC do not appear to be specific to this brain region, and similar GABA neuron alterations are likely to be present in a variety of cortical regions.
IN VIVO MEASUREMENT OF GABA LEVELS HAS YIELDED INCONSISTENT RESULTS
Despite an abundance of postmortem data suggesting reduced GABA synthesis and alterations in other aspects of GABA neurotransmission in the illness, in vivo measurements of cortical GABA using proton magnetic resonance spectroscopy have yielded conflicting results, with decreased, increased, and unchanged levels reported in different cortical regions (Kegeles et al., 2012; Maddock and Buonocore, 2012). Thus, it is currently unknown whether impaired GABA synthesis is accompanied by lower GABA levels in schizophrenia. Further complicating the issue, magnetic resonance spectroscopy studies assess total tissue levels of GABA, not those associated with synapses, and therefore the relationship between these studies and GABAergic neurotransmission in schizophrenia remains unclear.
ALTERATIONS IN GABA NEURONS ARE DUE TO THE DISEASE PROCESS OF SCHIZOPHRENIA
An important consideration is whether the alterations in markers of GABA neurotransmission described earlier are specific to the disease process of schizophrenia, or represent a consequence of illness chronicity, treatment, or comorbid diagnoses (Lewis et al., 2005). In general, studies in animals exposed to medications used in the treatment of schizophrenia, and the comparison of subjects with schizophrenia on or off such medications at the time of death, suggest that altered GABA-related gene expression in schizophrenia is not attributable to medication effects. Similarly, alcohol use does not seem to affect GAD67, PV, or GAT1 mRNA levels (Lewis et al., 2005), and nicotine and cannabis use also do not account for the lower GAD67 mRNA levels in schizophrenia (Curley et al., 2011).
Furthermore, since GAD67 expression is activity- dependent (Akbarian and Huang, 2006), lower expression might just index the less stimulating social, occupational, and intellectual environment associated with illness chronicity. However, a recent study found no association between GAD67 mRNA expression and age-corrected length of illness (Curley et al., 2011). In addition, the same study found no association between predictors of illness severity or measures of functional outcome and GAD67 mRNA levels.
Another important consideration is whether the observations in schizophrenia are specific to the illness, or are shared among other psychiatric diseases such as bipolar disorder (BPD) and major depressive disorder (MDD). Current evidence suggests that all three disorders do share some common GABAergic alterations, although they are also characterized by differences in their pattern of alterations, and by substantial heterogeneity in findings across studies. For example, lower GAD67 has also been found in BPD and MDD (Guidotti et al., 2000; Thompson Ray et al., 2011), and lower PV has been reported in BPD (Torrey et al., 2005) but not MDD (Beasley et al., 2002; Rajkowska et al., 2007). In contrast, another recent study in the PFC found lower SST levels in MDD and reduced PV in BPD, but no change in GAD67, GAD65, or CR mRNA expression in either illness (Sibille et al., 2011). In addition, microarray studies that examined the expression patterns of large groups of genes have found alterations in a number of GABA-related genes in MDD and BPD (Kato et al., 2007; Klempan et al., 2009). Thus, while these major psychiatric illnesses do seem to be characterized by alterations in GABA neurons, the exact degree of overlap between illnesses remains to be determined.
MECHANISMS UNDERLYING ALTERATIONS IN GABA NEUROTRANSMISSION IN SCHIZOPHRENIA
A thorough interpretation of the GABA neurotransmission deficit in schizophrenia requires knowledge of the underlying mechanism(s). Since lower GAD67 is the most robust finding in postmortem tissue, here we consider some of the candidate mechanisms for this reduction.
ALLELIC VARIATION IN GAD1
GAD1, the gene that encodes GAD67, is located on chromosome 2q31 (Akbarian and Huang, 2006). Single nucleotide polymorphisms (SNPs) in GAD1 have been implicated in the risk for adult- and childhood-onset schizophrenia (Marenco et al., 2010), and have also been associated with GAD67 mRNA levels (Straub et al., 2007), indicating that alterations in the gene may underlie lower GAD67 mRNA levels in schizophrenia. Existing data suggest several possible mechanisms by which these allelic variants may alter GAD67 mRNA levels. For example, although no risk SNPs are located within the coding region of GAD1 (Akbarian and Huang, 2006), one located in the promoter region is associated with altered transcription factor binding, which may lead to impaired promoter function (Zhao et al., 2007). In addition, epigenetic mechanisms, which control gene activity without altering DNA sequence, may also contribute. Methylation of certain lysine residues on nuclear histone proteins, around which DNA is wound, result in shifts between active and repressed transcription (Tsankova et al., 2007). In female schizophrenia subjects, GAD1 risk SNPs are associated with lower PFC GAD67 mRNA and a shift from active to repressed histone methylation (Huang et al., 2007), providing a potential mechanism through which allelic variation may lower GAD67 mRNA levels. However, this mechanism may be specific to females, and thus the exact role of epigenetic mechanisms in GAD1 transcription requires further study.
Despite this evidence, low penetrance, small effect sizes, and a lack of concordance between studies regarding the specific SNPs involved suggest that allelic variation in the GAD1 gene likely plays only a minor role as a causative factor underlying lower GAD67 in schizophrenia (Insel, 2010). Further confusing the issue, schizophrenia GAD1 risk alleles have recently been associated with increased GABA levels in healthy subjects (Marenco et al., 2010), and therefore the exact role of GAD1 risk alleles in the pathogenesis of GAD67 deficits in schizophrenia remains to be determined.
REDUCED SIGNALING THROUGH TRKB
In contrast to a primary genetic effect, lower GAD67 expression may be a consequence of an upstream mechanism. One such mechanism that could lead to a reduction in GAD67, as well as in PV and GAT1, in schizophrenia is reduced signaling through the TrkB receptor and its ligand brain-derived neurotrophic factor (BDNF) (Lewis et al., 2005). TrkB/BDNF signaling is crucial for interneuron development, especially in PV cells, which comprise the majority of TrkB-expressing interneurons. Treatment with BDNF in vitro upregulates GAD and promotes GABA release, and BDNF-overexpressing mice exhibit accelerated PV interneuron maturation (Balu and Coyle, 2011).
Both TrkB and BDNF mRNA and protein are lower in the PFC of subjects with schizophrenia (Balu and Coyle, 2011). In fact, in matched pairs of control and schizophrenia subjects, the within-pair differences in TrkB mRNA are positively correlated with those of both GAD67 and PV mRNAs. In addition, mice with a genetic knockdown of TrkB, but not BDNF, exhibit lower levels of GAD67 and PV mRNAs, suggesting that reduced signaling through the TrkB receptor may underlie lower levels of both mRNAs in schizophrenia. Furthermore, knockdown of TrkB produces a reduced density of GAD67 mRNA-positive cells, without a change in the mRNA levels per cell, similar to the pattern observed in schizophrenia. Although GAT1 expression has not been examined in these animals, it also appears to be modulated by the TrkB/BDNF signaling pathway. In hippocampal cells, inhibition of tyrosine kinases lowers GAT1 phosphorylation and GABA reuptake, while application of BDNF increases GAT1 function (Law et al., 2000).
NMDA HYPOFUNCTION IN PV NEURONS
Glutamate-mediated excitatory neurotransmission occurs through a variety of fast ionotropic receptors (NMDA, AMPA, and kainate), as well as the slower G protein-coupled metabotropic receptors. The NMDA receptor hypofunction hypothesis of schizophrenia originated from the observation that NMDA receptor antagonists recapitulate many clinical features of schizophrenia. Although attempts to document alterations in the level of NMDA receptor subunits in schizophrenia have been mixed, the hypothesis has been strengthened by findings that several schizophrenia risk genes, such as neuregulin 1 and ErbB4, affect NMDA receptor signaling, and by preclinical and clinical studies demonstrating that NMDA receptor enhancement ameliorates disease symptoms (Gonzalez-Burgos and Lewis, 2012).
Interneurons are known to depend on NMDA-mediated excitation, and thus lower glutamatergic drive onto GABA neurons may result in subsequent reductions in markers of inhibitory neurotransmission (Lewis and Moghaddam, 2006). The vast majority (80–90%) of PV neurons express NR1 receptors (Huntley et al., 1994; González-Albo et al., 2001), and so PV neurons are thought to be particularly affected by lower NMDA receptor signaling. In fact, NMDA receptor antagonists lower GAD67, PV, and GAT1 mRNAs (Behrens and Sejnowski, 2009; Bullock et al., 2009). In addition, reduced levels of GAD67 and PV protein are observed in mice with a selective knockout of the NR1 receptor on ~50% of cortical and hippocampal interneurons, presumably mostly PV neurons, during early postnatal development (Gonzalez-Burgos and Lewis, 2012).
CONSEQUENCE OF REDUCED EXCITATION
GAD67 is activity-dependent (Akbarian and Huang, 2006), and reductions in excitatory signaling have been shown to produce many of the same alterations in inhibitory function observed in schizophrenia. For example, monocular deprivation of the lateral geniculate nucleus of the thalamus results in lower GAD67 mRNA in the primary visual cortex of primates. Thus, one hypothesis is that the reduced excitatory activity in schizophrenia is an “upstream” pathology that produces a compensatory downregulation of inhibitory activity, in an attempt to maintain the balance of excitatory and inhibitory activity (E/I balance) needed for proper cortical functioning. E/I balance is important to prevent both runaway excitatory activity (too much excitation) and a dying out of cortical activity (too much inhibition). Thus, reduced excitatory activity of pyramidal cells may result in a compensatory downregulation of inhibitory activity onto these neurons, as evidenced by the multiple different types of reductions in effectors of GABA neurotransmission (Lewis et al., 2012).
In fact, a number of alterations are present in excitatory pyramidal cells in schizophrenia (Glausier and Lewis, 2012). First, there is a significant reduction in the density of dendritic spines, a finding that is most pronounced in layer 3. In addition, although the total number of pyramidal neurons is unchanged in the PFC, somal size is significantly lower, and neuropil and dendritic tree size are reduced. In conjunction with these morphological alterations, certain molecules that regulate spine formation and maintenance are also altered in schizophrenia. For example, mRNA levels of the Rho GTPase cell division cycle 42 (Cdc42), which promotes spine formation, are lower in the PFC of subjects with schizophrenia, and the decrease in Cdc42 mRNA is significantly correlated with the reduction in layer 3 spine density. The inhibition of Cdc42 effector protein 3 (Cdc42EP3) by Cdc42 is thought to enable synaptic potentiation, and mRNA levels of Cdc42EP3 are increased in schizophrenia. Thus, the combination of lower Cdc42 and higher Cdc42EP3 levels may contribute to the spine loss observed in schizophrenia (Glausier and Lewis, 2012).
CONSEQUENCES OF ALTERATIONS IN GABA NEURONS IN SCHIZOPHRENIA: NETWORK OSCILLATIONS AND WORKING MEMORY
As previously reviewed, schizophrenia is associated with alterations in a variety of interneuron subtypes. The abnormalities in two of these cell types, PVb and CCKb cells, are particularly interesting in light of their role in neural network oscillations associated with cognition, and here we consider the consequences of alterations in these cells in schizophrenia. Lower levels of CCK and CB1R in CCKb cells are hypothesized to strengthen the inhibition of postsynaptic pyramidal cells. On the other hand, lower levels of GAD67 and increased levels of μ opioid receptors in PVb cells are thought to weaken the inhibition of postsynaptic pyramidal cells. In addition, PVb cells receive input from CCKb cells, and application of CCK can activate PV neurons (Krook-Magnuson et al., 2012); thus, lower CCK in schizophrenia may also contribute to weaker PVb cell activity. Lower levels of GABAA α1-containing receptors and a reduced chloride driving force in pyramidal neurons may also decrease the strength of hyperpolarizing inputs from PVb cells and exacerbate this reduction in inhibition. Thus, a convergence of evidence suggests that the relative strengths of pyramidal cell inhibition from CCKb and PVb cells are increased and decreased, respectively, in schizophrenia (Curley and Lewis, 2012).
An important determinant of the functional consequences of these alterations is whether the disturbances in each cell type affect the same pyramidal neurons (Curley and Lewis, 2012). Although CCK mRNA levels in schizophrenia have not been examined in a laminar fashion, CB1R immunoreactivity is significantly lower in layers deep 3–4 and 6. The density of PV-immunoreactive axon terminals is also lower in layers deep 3–4, suggesting that the alterations in both PVb and CCKb terminals are present in the middle cortical layers, despite the locations of their cell bodies in different layers. On the other hand, recent data suggest that all pyramidal neurons receive PVb cell inputs, whereas only some receive CCKb inputs and show DSI (Krook-Magnuson et al., 2012), which may indicate that, while all pyramidal cells are affected by impaired inhibition from PVb cells, only some are affected by alterations in CCKb cell inputs. However, whether the affected PVb and CCKb cells target the same pyramidal neurons in schizophrenia remains to be determined (Curley and Lewis, 2012).
CCKb and PVb cells play an important role in the θ and γ oscillations, respectively (Klausberger and Somogyi, 2008), that underlie working memory function. Although in healthy subjects the amplitude of θ and γ oscillations increases in proportion to working memory load, subjects with schizophrenia exhibit impairments in θ and γ band power and working memory performance. Thus, changes in the inhibitory output from CCKb and PVb cells, and the resulting shift in their relative control of pyramidal cells, could disrupt normal network functioning and θ and γ band power. Moreover, since θ and γ oscillations are coupled during memory tasks, a disturbance in one frequency may result in corresponding deficits in the other frequency. Thus, alterations in the perisomatic inhibition of PFC pyramidal neurons by CCKb and PVb neurons are a plausible mechanism underlying θ and γ oscillation deficits and cognitive impairments in schizophrenia (Curley and Lewis, 2012).
CONCLUSIONS
Schizophrenia is characterized by a number of cortical interneuron alterations. Abnormalities are present in PVb, PVch, CCKb, and SST, but not CR cells, and occur in a number of cortical regions in addition to the PFC. These alterations appear to reflect the disease process of schizophrenia, as they are not a consequence of illness chronicity, treatment, or comorbid diagnoses. A number of plausible mechanisms could underlie the most robust finding, a reduction in GAD67 mRNA and protein, including allelic variation in GAD1, reduced signaling through the TrkB receptor, NMDA receptor hypofunction in PV neurons, or reduced excitation. The alterations in two cell types in particular, PVb and CCKb cells, are particularly important given their roles in θ and γ oscillations, respectively, and provide a possible mechanism underlying the oscillation deficits and cognitive impairments of schizophrenia.
DISCLOSURES
Dr. Lewis currently receives grant/research support from Bristol-Myers Squibb and Pfizer. He is a consultant for Bristol-Myers Squibb and Concert.
Dr. Curley has no conflicts of interest to disclose.
REFERENCES
Akbarian, S., and Huang, H.S. (2006). Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res. Rev. 52:293–304.
Asada, H., Kawamura, Y., et al. (1997). Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc. Natl. Acad. Sci. USA. 94:6496–6499.
Ascoli, G.A., Alonso-Nanclares, L., et al. (2008). Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat. Rev. Neurosci. 9:557–568.
Balu, D.T., and Coyle, J.T. (2011). Neuroplasticity signaling pathways linked to the pathophysiology of schizophrenia. Neurosci. Biobehav. Rev. 35:848–870.
Bartos, M., and Elgueta, C. (2012). Functional characteristics of parvalbumin- and cholecystokinin-expressing basket cells. J. Physiol. 590:669–681.
Beasley, C.L., Zhang, Z.J., et al. (2002). Selective deficits in prefrontal cortical GABAergic neurons in schizophrenia defined by the presence of calcium-binding proteins. Biol. Psychiatry 52:708–715.
Behrens, M.M., and Sejnowski, T.J. (2009). Does schizophrenia arise from oxidative dysregulation of parvalbumin-interneurons in the developing cortex? Neuropharmacol. 57:193–200.
Beneyto, M., Morris, H.M., et al. (2012). Lamina- and cell-specific alterations in cortical somatostatin receptor 2 mRNA expression in schizophrenia. Neuropharmacol. 62:1598–1605.
Blum, B.P., and Mann, J.J. (2002). The GABAergic system in schizophrenia. Int. J. Neuropsychopharmacol. 5:159–179.
Bullock, W.M., Bolognani, F., et al. (2009). Schizophrenia-like GABAergic gene expression deficits in cerebellar Golgi cells from rats chronically exposed to low-dose phencyclidine. Neurochem. Int. 55:775–782.
Curley, A.A., Arion, D., et al. (2011). Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: clinical, protein, and cell type-specific features. Am. J. Psychiatry 168:921–929.
Curley A.A., and Lewis D.A. (2012). Cortical basket cell dysfunction in schizophrenia. J. Physiol. 590:715–724.
Fung, S.J., Webster M.J., et al. (2010). Expression of interneuron markers in the dorsolateral prefrontal cortex of the developing human and in schizophrenia. Am. J. Psychiatry 167:1479–1488.
Glausier, J.R., and Lewis D.A. (2012). Dendritic spine pathology in schizophrenia. Neuroscience.
González-Albo, M.C., Elston, G.N., et al. (2001). The human temporal cortex: characterization of neurons expressing nitric oxide synthase, neuropeptides and calcium-binding proteins, and their glutamate receptor subunit profiles. Cereb. Cortex. 11:1170–1181.
Gonzalez-Burgos, G., Fish, K.N., et al. (2011). GABA neuron alterations, cortical circuit dysfunction and cognitive deficits in schizophrenia. Neural Plast. 2011:723184.
Gonzalez-Burgos, G., Hashimoto, T., et al. (2010). Alterations of cortical GABA neurons and network oscillations in schizophrenia. Curr. Psychiatry Rep. 12:335–344.
Gonzalez-Burgos, G., and Lewis, D.A. (2008). GABA neurons and the mechanisms of network oscillations: implications for understanding cortical dysfunction in schizophrenia. Schizophr. Bull. 34:944–961.
Gonzalez-Burgos, G., and Lewis, D.A. (2012). NMDA receptor hypofunction, parvalbumin-positive neurons and cortical gamma oscillations in schizophrenia. Schizophr. Bull. 38:950–957.
Guidotti, A., Auta J., et al. (2000). Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch. Gen. Psychiatry 57:1061–1069.
Huang, H.S., Matevossian, A., et al. (2007). Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J. Neurosci. 27:11254–11262.
Huntley, G.W., Vickers, J.C., et al. (1994). Distribution and synaptic localization of immunocytochemically identified NMDA receptor subunit proteins in sensory-motor and visual cortices of monkey and human. J. Neurosci. 14:3603–3619.
Insel, T.R. (2010). Rethinking schizophrenia. Nature 468:187–193.
Kato, T., Kakiuchi, C., et al. (2007). Comprehensive gene expression analysis in bipolar disorder. Can. J. Psychiatry 52:763–771.
Kegeles, L.S., Mao X., et al. (2012). Elevated prefrontal cortex γ-aminobutyric acid and glutamate-glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry 69:449–459.
Klausberger, T., and Somogyi, P. (2008). Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321:53–57.
Klempan, T.A., Sequeira, A., et al. (2009). Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression. Mol. Psychiatry 14:175–189.
Krook-Magnuson, E., Varga, C., et al. (2012). New dimensions of interneuronal specialization unmasked by principal cell heterogeneity. Trends. Neurosci. 35:175–184.
Law, R.M., Stafford, A., et al. (2000). Functional regulation of gamma-aminobutyric acid transporters by direct tyrosine phosphorylation. J. Biol. Chem. 275:23986–23991.
Lewis, D.A., Curley, A.A., et al. (2012). Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 35:57–67.
Lewis, D.A., Hashimoto, T., et al. (2005). Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 6:312–324.
Lewis, D.A., and Moghaddam, B. (2006). Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch. Neurol. 63:1372–1376.
Maddock, R.J., and Buonocore, M.H. (2012). MR spectroscopic studies of the brain in psychiatric disorders. Curr. Top. Behav. Neurosci. 11:199–251.
Marenco, S., Savostyanova, A.A., et al. (2010). Genetic modulation of GABA levels in the anterior cingulate cortex by GAD1 and COMT. Neuropsychopharmacol. 35:1708–1717.
Mizukami, K., Ishikawa, M., et al. (2002). Immunohistochemical localization of GABAB receptor in the entorhinal cortex and inferior temporal cortex of schizophrenic brain. Prog. Neuropsychopharmacol. Biol. Psychiatry 26:393–396.
Moyer, C.E., Delevich, K.M., et al. (2012). Reduced glutamate decarboxylase 65 protein within primary auditory cortex inhibitory boutons in schizophrenia. Biol. Psychiatry 72:734–743.
Patel, A.B., de Graaf, R.A., et al. (2006). Evidence that GAD65 mediates increased GABA synthesis during intense neuronal activity in vivo. J. Neurochem. 97:385–396.
Rajkowska, G., O’Dwyer, G., et al. (2007). GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology 32:471–482.
Sibille, E., Morris, H.M., et al. (2011). GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. Int. J. Neuropsychopharmacol. 14:721–734.
Sohal, V.S., Zhang, F., et al. (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459:698–702.
Stan, A., and Lewis, D.A. (2012). Altered cortical GABA neurotransmission in schizophrenia: insights into novel therapeutic strategies. Curr. Pharm. Biotechnol. 13:1557–1562.
Straub, R.E., Lipska, B.K., et al. (2007). Allelic variation in GAD1 (GAD67) is associated with schizophrenia and influences cortical function and gene expression. Mol. Psychiatry 12:854–869.
Thompson Ray, M., Weickert C.S., et al. (2011). Decreased BDNF, trkB-TK+ and GAD(67) mRNA expression in the hippocampus of individuals with schizophrenia and mood disorders. J. Psychiatry. Neurosci. 36:195–203.
Torrey, E.F., Barci, B.M., et al. (2005). Neurochemical markers for schizophrenia, bipolar disorder, and major depression in postmortem brains. Biol. Psychiatry 57:252–260.
Tsankova, N., Renthal, W., et al. (2007). Epigenetic regulation in psychiatric disorders. Nat. Rev. Neurosci. 8:355–367.
Wilson, R.I., and Nicoll, R.A. (2002). Endocannabinoid signaling in the brain. Science 296:678–682.
Zhao, X., Qin, S., et al. (2007). Systematic study of association of four GABAergic genes: glutamic acid decarboxylase 1 gene, glutamic acid decarboxylase 2 gene, GABA(B) receptor 1 gene and GABA(A) receptor subunit beta2 gene, with schizophrenia using a universal DNA microarray. Schizophr. Res. 93:374–384.