25 | NEURODEVELOPMENT AND SCHIZOPHRENIA
ESTER J. KWON, * TAKAHIRO SODA, * AND LI-HUEI TSAI
INTRODUCTION
Schizophrenia affects approximately 1% of the world’s population and is a major contributor to both years lost to disability and health care costs. Despite the significant impetus to understand the etiology of schizophrenia and to develop therapeutics to ease the burden of disease, progress has been limited. This may be due to the complexity of the underlying genetics as well as heterogeneity within and between psychiatric diagnoses. What is clear is that there is a genetic component to schizophrenia risk as well as environmental factors that are important for the manifestation of disease. At this point, the genetic causes of schizophrenia remain largely elusive except for highly penetrant but rare variants, which account only for a small fraction of schizophrenia cases. The most notable example is disrupted in schizophrenia-1 (DISC1), which was identified in a Scottish family with a near Mendelian inheritance of psychiatric diseases. Although rare variants are not the genetic cause of schizophrenia for the vast majority of cases, they may be instructive for the identification of underlying pathways that are dysregulated in schizophrenia.
A NEURODEVELOPMENTAL MODEL OF SCHIZOPHRENIA
A neurodevelopmental model of schizophrenia has been hypothesized by numerous researchers. This model postulates that some of the key aspects of brain development that normally occur both pre- and postnatally are not occurring correctly, either in time or space. Complex neural circuitry needs to form and be modulated by experience. Classically, proliferation, migration, arborization and myelination occur prenatally. Elaboration and refining of dendritic trees and synapses as well as myelination of the nervous system continues through the first two-decades of life. There are opportunities for genetic and environmental abnormalities and variation to profoundly influence the trajectories of all of these critical functional processes. Temporal correlation is noted between the prodromal aspects of schizophrenia, in particular abnormalities in cognition and in the neuroanatomic development of regions of the cerebral cortex. As reviewed in earlier chapters, there is some evidence suggesting that reduced interneuron activity may be involved. Some of the key evidence that developmental processes are awry and their potential causes is reviewed in the following and in the subsequent two chapters.
ENVIRONMENTAL EFFECTS DURING DEVELOPMENT
The 50% concordance of schizophrenia between monozygotic twins demonstrates that there is a clear genetic component to disease onset, and the incomplete concordance highlights that there are also other factors in play. Many epidemiological observations associated with increased risk of schizophrenia suggest exposures that might influence brain development. For a comprehensive review see Brown (2011).
Significant effort has been put into correlating pre- and perinatal disturbances to the development of schizophrenia. Numerous studies have linked phenomena such as maternal nutrition and infection, season of birth, and obstetrical complications to the manifestation of schizophrenia later in life (Lewis and Levitt, 2002). Studies of children born from mothers who were in early pregnancy during the Dutch Hunger winter have shown association with schizophrenia, as well as with depression and mood disorders, increasing the risk of schizophrenia approximately twofold. The association of famine with schizophrenia was also observed in the 1959–1961 Chinese famines in two separate regions, Liuzhou and Wuhu (Rapoport et al., 2012). Improvements in imaging technology and longitudinal studies with collection of tissue and blood samples will aid the understanding of how these factors contribute to schizophrenia onset in the future.
An example of association with an environmental factor that may be present during adolescence, and that has now been replicated in several carefully controlled studies is being in a minority group position. Studies examining association of schizophrenia with being in a minority group position show that development of psychosis is dependent on the proportion of their own ethnic group present in the area. Furthermore, the association is seen in first- and second-generation migrants and across several ethnic groups, indicating that it is not the ethnicity or migration itself that is causing the association. Other examples include growing up in an urban environment and experiencing developmental trauma (van Os et al., 2010). These three factors clearly have components of stress, although it is still not understood what types of stress and during which developmental times stresses occur that are related to the onset of psychosis. In addition to these environmental factors, there are also examples of more acute triggers that track with schizophrenia. A meta-analysis of people who have antibodies to toxoplasma gondii has found associations to schizophrenia that replicated previous findings, showing that toxoplasma gondii infection conferred an odds ratio of ~2.7 (Torrey et al., 2012). The presence of antibodies indicated an infection up to the point of analysis, which was after onset of disease; work remains to be done pinpointing whether there is a time window of infection that is relevant for the development of psychosis. Another factor thought to be associated with the onset of psychosis is the use of cannabis. Although reverse causation is a concern when studying cannabis use, a meta-analysis of studies where baseline psychosis was a criterion for exclusion found that there was an increased risk of psychosis with cannabis use associated with an odds ratio of 1.4 (Moore et al., 2007).
Each of these associations implicate periods of stress, for example, nutritional, immunological, or social, in the onset of schizophrenia. These associated factors serve as proxies for what stimulus is really affecting the molecular basis of schizophrenia progression, which remains to be uncovered in future studies. Epigenetics (e.g., DNA methylation, histone acetylation, and noncoding RNA) has been shown to be able to have a strong influence in the case of maternal nutrition and the development of obesity and diabetes and may be important in the role of neurodevelopmental influences in schizophrenia (Bale et al., 2010).
NEUROPATHOLOGY ASSOCIATED WITH SCHIZOPHRENIA
The macroscopic phenotypes that have most predictably tracked with schizophrenia are ventricular enlargement and slight reductions in brain volume and weight as measured in whole brain imaging studies or in postmortem samples (Lawrie and Abukmeil, 1998). Other endophenotypes of schizophrenia include structural changes in various brain regions, changes in gray matter volume, cortical thickness, and neuronal integrity (Harrison and Weinberger, 2005).
Brain imaging studies of rare childhood onset schizophrenia cases (1/500 cases of schizophrenia), which are often similar to more severe outcomes of schizophrenia, provide an opportunity to understand how brain development may be perturbed in schizophrenia, and they are less likely to be influenced by environmental factors such as substance abuse, which is known to cause structural changes to the brain. Longitudinal studies were conducted starting in 1991 at the NIMH, where more than 100 child-onset schizophrenia patients and their siblings underwent brain imaging four–five times every two years. The average age of onset was 10 years, and there was an equal distribution between males and females (Gogtay et al., 2011). These studies show that phenotypes are preserved between child and adult onset schizophrenia, such as increased lateral ventricular sizes, and decreased overall gray matter, hippocampus, and amygdala volumes (Gogtay and Rapoport, 2008). In the course of normal development, gray matter volumes follow an inverted U-shape curve, where they increase throughout childhood and peak at puberty, whereas white matter volumes steadily increase into adulthood. The NIH longitudinal study showed that in child onset schizophrenia gray matter loss occurs throughout development and that white matter development is retarded when compared with control cases. These data support the concept that schizophrenia is a progressive disease with perturbations throughout neurodevelopment that lead up to its onset in early adulthood.
Another longitudinal study was conducted on ultra- high-risk schizophrenia candidates, as determined by their mental health state and family history of mental illness, in Melbourne, Australia (Gogtay et al., 2011). The candidates were on average 19 years of age and 40% developed schizophrenia within 12 months of the initiation of the study, allowing for the imaging of brains and discovery of perturbations during the period of onset between those candidates who developed psychosis and those who did not. They found that there was gray matter loss in the left medial and inferior temporal regions, the anterior cingulated cortex, and in the left orbitofrontal cortex in patients who developed psychosis, which was not seen in those who did not develop psychosis (Gogtay et al., 2011).
Although there is a decrease in brain volume, a significant change in number of neurons has not been observed. One hypothesis is that the decrease in volume is due to the loss of neuronal processes, such as axons, dendrites, and dendritic spines, referred to collectively as the neuropil. Analyses of postmortem brains have shown a marked decrease in dendritic spines in schizophrenic patients when compared with controls, especially in cortical pyramidal neurons (Garey, 2010). A diffusion-weighted magnetic resonance imaging technique called diffusion tensor imaging (DTI) is used to image fiber pathways and connection integrity of white matter in living subjects. In white matter, the flow of water is largely restricted to the tracts and therefore has directionality that can be used as a proxy for the integrity of the fibers.
Before the onset of the majority of schizophrenia cases in late adolescence/early adulthood, there are cognitive deficits that can be observed before diagnosis is made (Gold and Weinberger, 1995). Furthermore, these cognitive deficits are found in undiagnosed family members who have not been exposed to treatment, indicating that there is a genetic disposition for cognitive dysfunction that may be used as a hallmark of severity of disease. The most consistent finding resulting from functional magnetic resonance imaging (fMRI) studies in schizophrenic patients is changes in activity in the dorsolateral prefrontal cortex (DLPFC) during working memory tasks. Schizophrenic patients have decreased performance on working memory tasks, which is correlated with decreased activity in the DLPFC. One nuance is that for tasks in which schizophrenic patients have similar performance as control subjects, there is an increased activity in the DLPFC (Lewis et al., 2005). The impairment in working memory has led investigators to posit several hypotheses based on neurotransmitters and connectivity.
OLIGODENDROCYCTES, MYELIN, DEVELOPMENT, AND SCHIZOPHRENIA
White matter integrity is measured in DTI by fractional anisotrophy, which is representative of myelination and tract organization. Over the many DTI studies performed on schizophrenia cohorts, there are decreases in fractional anisotrophy measurements in white matter across many brain regions, most consistently in the cingulate, corpus callosum, and frontal lobes (Thomason and Thompson, 2011). Moreover, in a study of prodromal ultra-high-risk psychosis candidates, there were decreases in fractional anisotrophy that preceded disease onset (Karlsgodt et al., 2009). In addition, there is a well-characterized reduction in expression of oligodendrocytes and myelin related genes in postmortem brain samples. Structural analyses in postmortem brain have also shown both a decreased density of oligodendrocytes and damaged myelin sheaths in schizophrenia cases compared to controls. Furthermore, a wide variety of candidate genes involved in oligodendrocyte development have found modest associations with increased risk of schizophrenia (see Takahashi et al., 2011, for review).
The temporal process of myelination is correlated with the developmental trajectory of schizophrenia. Myelination is incomplete at birth and develops gradually, with some cortical areas not being completed until adulthood. For example, regions expected to be of particular relevance to schizophrenia such as cortical and hippocampal pathways are not fully myelinated until adulthood. Diseases whose primary pathology is in white matter, such as the leukodystrophies, are often accompanied by psychosis, which suggests a shared altered basic role for myelination and oligodendrocytes. Furthermore, an appealing hypothesis is that oligodendrocyte and myelin dysfunction exerts its role in schizophrenia by altering key features of synaptic activity such as synaptic plasticity or the production of synchronous oscillating activities implicated in establishing aspects of cognition (Walterfang et al., 2011).
NEUROTRANSMITTERS AND ANTIPSYCHOTIC DRUGS
There are several theories associating dysfunction of neurotransmitter pathways and schizophrenia based on neuropharmacological studies, which include the GABA, NMDA, and dopamine theories. It is important to note that none of these are theories are exclusive, neither among themselves nor to the other theories presented, which will be discussed in a subsequent chapter.
Antipsychotic drugs used to treat schizophrenia are effective in mitigating the positive symptoms, such as hallucinations and delusions. The first widely used antipsychotic, chlorpromazine, blocks the dopamine D2 receptor. Derivative first- and second-generation antipsychotics by and large also target the dopamine D2 receptor. Though the D2 receptor has been generally established as the site of action for antipsychotics, the mechanism of action is less clear. There are several pathways downstream of the dopamine D2 receptor, and it is becoming apparent that the localization of dopamine D2 receptor interaction proteins, in addition to the location of the D2 receptor itself, may impact which downstream pathways become affected.
The D2 receptor acts as a classical G protein–coupled receptor, and has been associated with the Gαi/o Gβγ heterotrimeric G protein complex. Activation of this pathway is associated with the inhibition of downstream cAMP signaling. Coupling of these receptors to presynaptic K+ channels has been shown to lead to the inhibition of neurotransmitter release. In addition, it has been reported that these receptors and D1/D2 heterooligomeric receptors activate downstream Ca2+ signaling via coupling to GqGβγ and the activation of phospholipase C.
The D2 receptor has also been shown to activate another signaling pathway, the phosphatidylinositol (3,4,5)-triphosphate kinase (PI3K)/AKT/mammalian Target of Rapamycin (mTOR) pathway, a pathway that regulates the Wnt signaling cascade. The P13K/AKT/mTor pathway is also activated by another class of membrane-bound receptors, the receptor tyrosine kinases. In the baseline state, AKT is bound to phosphatidylinositol(3,4)-bisphosphate (PIP2). Upon activation of a receptor tyrosine kinase or a G protein–coupled receptor, PI3K phosphorylates PIP2 to form PIP3. AKT bound to PIP3 is then phosphorylated by mTOR complex 2 (mTORC2), then phosphorylated by phosphinositide dependent kinase 1 (PDPK1). The phosphorylation by both kinases activates AKT to then phosphorylate its many downstream targets, including mTOR (implicated in autism), GSK3β (involved in the Wnt signaling cascade), BAD (a pro-apoptotic mitochondrial protein), and IκB kinase (IKK; regulator of NFκB mediated transcription).
The intersection of the AKT pathway with the pharmacology of psychiatric disorder treatment is notable. Dopamine D2 receptors have been shown to modulate AKT signaling by recruiting a signaling complex that results in the inactivation of AKT. Upon D2 receptor activation and phosphorylation by G protein–coupled receptor kinase (GRK), β-arrestin and protein phosphatase 2A (PP2A) are recruited to the cell membrane, where they interact with and dephosphorylate AKT. Antipsychotics, which are antagonists to the D2 receptor, block this signaling cascade and allow AKT to remain active (Freyberg et al., 2010).
Lithium, a treatment for bipolar disorder that can also be prescribed in conjunction with antipsychotics for the treatment of schizophrenia, is shown to lead to inhibition of glycogen synthase kinase β (GSK3β). It has been shown that lithium activates AKT to phosphorylate GSK3β, leading to GSK3β inhibition. Lithium has also been proposed to solubilize AKT from the β-arrestin/PP2A complex, which mediates portions of downstream D2 receptor signaling functions (Beaulieu, 2012). Thus, both antipsychotic and lithium treatments secondarily affect the Wnt pathway via affecting AKT activity.
RARE AND COMMON GENETIC VARIATION IN SCHIZOPHRENIA
Recent genetic evidence has found that de novo duplications and deletions of DNA, termed copy number variations (CNVs), increase the risk of schizophrenia. As noted in an earlier chapter, many of these CNVs have been associated with a wide variety of developmental phenotypes. One early CNV study found novel CNVs greater than 100 kilobases in schizophrenic patients and their families and found an increased number of CNVs in patients, with a disproportionate number in genes related to neurodevelopmental pathways (Walsh et al., 2008).
International collaborative efforts have resulted in findings from genome-wide association studies (GWASs) with cohorts on the scale of tens of thousands of control and schizophrenia cases (Ripke et al., 2011; Stefansson et al., 2009). While further GWAS findings are anticipated, even the early studies suggest that there may be some convergence of gene candidates involved in neurodevelopmental phenomena and synaptic processes. The haploinsufficiency of one identified gene identified as genome-wide significant by GWAS, TCF4, leads to Pitt-Hopkins mental retardation in humans (Brockschmidt et al., 2007) and is also involved in the specification of specific subsets of neural progenitors in the mouse (Flora et al., 2007). Another genome-wide significant GWAS identified gene, CACNA1C, encodes a subunit of an L-type calcium channel leading to the intriguing theory that there may be aberrant synaptic transmission underlying the brain activation changes observed in functional neuroimaging studies (Bigos et al., 2010). There is a major ongoing effort to understand the biology of schizophrenia GWAS candidates and whether they are dysregulated and/or dysfunctional in the context of human disease.
Collectively, gene candidates of schizophrenia identified by studying both CNVs and common risk SNPs are converging on pathways that regulate neurodevelopmental and synaptic phenomena. It is also important to note that there is emerging evidence of a shared set of risk factors between psychiatric disorders including schizophrenia and bipolar, as well as autism. There are also several recurrent CNVs that are common between schizophrenic and autistic patients, such as 1q21.1, 15q11.2, 15q13.3, 17q12 (Malhotra and Sebat, 2012; Mefford et al., 2008; Stefansson et al., 2008).
PATHWAYS
Based on the neuropharmacology and neuropathological findings, there have been several theories suggested to understand the etiology of schizophrenia including the GABA, dopamine, and glutamate hypotheses. Although each of these pathways is likely to contribute to and/or be resultant from schizophrenia pathology, none alone is sufficient to explain the onset of disease and the presence of the spectrum of symptoms displayed by schizophrenic patients. An alternative way of understanding the etiology of schizophrenia and disease-associated genetic loci is to examine their involvement in dysregulated biochemical pathways. One such study compared significant and non-significant SNPs from the International Schizophrenia Consortium to pathways described in the Kyoto Encyclopaedia of Genes and Genomes (O’Dushlaine et al., 2011). The only significantly associated pathway found was the cell-adhesion molecule pathway, which is important for synaptic formation and cell signaling. One caveat to this approach is that the functions of many genes identified in CNV and GWAS studies are yet unknown, and thus insights gained from these types of analyses are just emerging. It may therefore be more fruitful at this early stage of understanding schizophrenia genetics to specify genes that are unmistakably linked to disease and study their function as a starting point to understand the context of common risk alleles that have lower effect sizes and occur in complex constellations.
As noted earlier, perturbation of neurodevelopmental processes in schizophrenia is a theory that is accumulating supporting evidence. There are pre- and perinatal environmental factors described in the preceding that are thought to associate with risk of schizophrenia as well as the changes in neuroanatomical structures and cognition observed before the manifestation of disease. There are also many gene candidates of neuropsychiatric disease that perturb biochemical pathways associated with neurogenesis and neuronal migration as well as spine morphogenesis, which are discussed in the following and are summarized in Table 25.1. The confluence of genetic and environmental factors on neurodevelopmental pathways is depicted in Figure 25.1.
EARLY DEVELOPMENT PATHWAYS
DISC1
DISC1 was identified in a Scottish pedigree in which it was disrupted by a balanced translocation, t(1:11)(q42.1;q14.3). The translocation segregated with psychiatric disorders such as schizophrenia, major depression, and bipolar disease with a combined OR 7.1. Mouse models expressing a transgene of human DISC1 recapitulating the Scottish translocation exhibit increased ventricle size, decreased gray matter volume, and changes in dendritic arborization in cortical and hippocampal neurons, mimicking the endophenotypes found in humans. Mouse models of DISC1 and their neuroanatomical, behavioral, and cellular phenotypes are summarized in the review by Jaaro-Peled (2009). The mice also exhibit behavioral abnormalities such as hyperactivity, increased immobility in the forced swim test, decreased sociability, and impaired working memory. Mouse models have also been demonstrated to have reduced embryonic and adult hippocampal neurogenesis. DISC1 is also implicated in proper integration of newborn neurons in the adult dentate gyrus.
DISC1 is a pleiotropic protein with numerous interaction partners affecting multiple biochemical signaling pathways that impact neuronal development and function. Some validated interacting partners for DISC1 include NDE1, NDEL1, LIS1, Kalirin-7, ATF4/5, GSK3β, and the PDE4 family of phosphodiesterases. The study of DISC1 is instructive since it is implicated in psychiatric disease and has a major role in early developmental phenomena and synaptic pathways.
WNT PATHWAY
The Wnt family consists of 19 secreted lipoproteins that act as morphogens and play important roles in the development and patterning of various limbs and organs. Wnts bind to their cell surface receptor, the Frizzled (Fz) family of seven transmembrane G protein–coupled receptors, of which 10 have been identified to date. The various Wnt and Fz proteins have distinct and overlapping patterns throughout the body, which is thought to account for the variability of phenotypes that result upon loss of function, and multiple downstream pathways have been shown to be activated downstream of Wnt binding.
TABLE 25.1. Genetic loci, effect size, affected genes, and their known role in neuronal development. A select table highlighting the known risk loci with putative effects on genes affecting neuronal development

CANONICAL WNT SIGNALING
The canonical Wnt signaling pathway is defined by the involvement of the transcriptional coactivator β-catenin and the downstream signaling cascades that are associated with it. β-catenin is normally confined to the cell membrane as part of the adherens junctions complex in neuronal progenitors in the central nervous system. Upon Wnt ligand binding, Fz proteins bind to disheveled proteins (Dvl). In the absence of Wnt binding, Dvl is part of a destruction complex in conjunction with axis inhibitor (Axin), adenomatous polyposis coli (APC), casein kinase 1 alpha (CK1α), and GSK3β. This destruction complex binds and phosphorylates β-catenin at multiple sites, designating it for proteosomal degradation and preventing the accumulation of β-catenin in the absence of Wnt. In the presence of Wnt stimulation, Fz-associated Dvl polymerizes and binds Axin through DIX domains. Axin binding recruits CK1α and GSK3β to the LRP5/6 complex, resulting in their phosphorylation and increased affinity for Axin. The interaction with LRP5/6 directly inhibits the phosphorylation of β-catenin by GSK3β, allowing for the cytosolic accumulation of β-catenin and its subsequent nuclear localization.
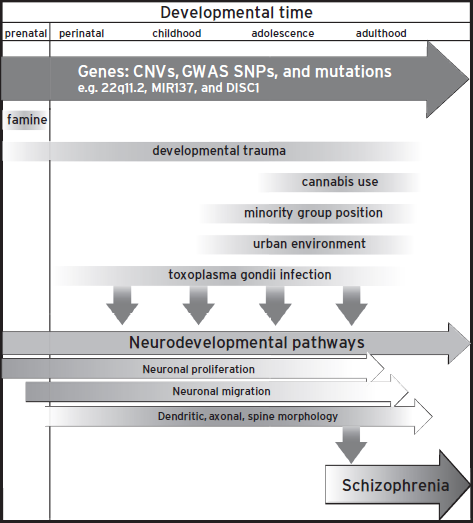
Figure 25.1 Factors that influence schizophrenia. Genes and environmental components affect neurodevelopmental pathways throughout life spans.
In the nucleus, β-catenin acts as a transcriptional co-activator of the T-cell factor and lymphoid enhancer-binding protein (TCF/LEF) transcription factors, which control the dynamics of the cell cycle partly via the regulation of cyclinD1. TCF/LEF has three identified domains: a high mobility group (HMG) box domain that is crucial for DNA interaction, a caspase activated deoxyribonuclease (CAD) domain necessary for binding to Groucho/TLE, and an N-terminal β-catenin binding domain. In the absence of nuclear β-catenin, the TCF/LEF occupying TCF/LEF DNA binding sites is bound to tetrameric Groucho/TLE, which recruits transcriptional repressors, such as histone deacetylases (HDACs), keeping the DNA in a condensed, repressed state. β-catenin directly competes with, and displaces Groucho/TLE (Daniels and Weis, 2005), and recruits general transcriptional activators such as the histone acetyltransferase CBP, the SWI/SNF complex protein Brg-1, and TATA- binding protein, as well as a more specific core complex consisting of Pygopus and Legless/BCL9 (Logan and Nusse, 2004). CBP acetylates histone residues in the promoter region, and the recruitment of Pygopus and Legless/BCL9 leads to the methylation of H3K9 residues by SET-1 methyltransferases, altering the chromatin structure from a condensed, transcriptionally repressed state to a more relaxed and open state, allowing for transcription (Fiedler et al., 2008). A comprehensive review of the Wnt signaling pathway in development can be found here (van Amerongen and Nusse, 2009).
ROLE OF THE CANONICAL WNT SIGNALING PATHWAY IN NEURONAL DEVELOPMENT
Numerous proteins within the canonical Wnt signaling pathway have been linked with neuronal development. GSK3 inactivation, achieved by a conditional knockout of both GSK3α and β resulted in a larger brain with an expansion of the progenitor pool with a concomitant reduction in the thickness of the cortex, indicating that GSK3 promotes progenitor proliferation while suppressing neuronal differentiation (Kim et al., 2009). Overexpression of stabilized β-catenin also results in mice with an increase in brain size, and an increased fraction of cells reentering the cell cycle, essentially phenocopy the GSK3 knockout phenotype, while mice with β-catenin mutations which selectively abolish its transactivation function have defects in forebrain development, accompanied by a decrease in the neuronal progenitor pool (Valenta et al., 2011), reiterating the crucial role of the proteins in the canonical Wnt pathway in the maintenance of the neuronal progenitor pool.
In summary, many factors crucial to the canonical Wnt pathway, as well as numerous interacting proteins with the pathway, have been shown to play crucial roles in neurogenesis, neuronal migration, axon/dendrite growth and morphology, and synaptic development. For a more extensive review of Wnt signaling and development, see Kim and Snider (2011).
DISC1 AND THE WNT SIGNALING PATHWAY
DISC1 has been shown to bind to and inhibit the activity of GSK3β, one of two forms of an enzyme that is known to lead to the phosphorylation of many downstream proteins that are crucial for the pathophysiology of many nervous system disorders. One GSK3β target is TAU, which forms aggregates in many nervous system disorders including Alzheimer’s disease (TAU forms the neurofibrillary tangles that are used in the Braak and Braak postmortem staging of disease progress), Parkinson’s disease, and tuberous sclerosis. Another GSK3β target is β-catenin, the crucial component of the canonical Wnt signaling pathway. DISC1 promotes the maintenance of the neuronal progenitor pool by inhibiting GSK3β, allowing cytoplasmic β-catenin to accumulate, and transduce canonical Wnt signaling. The loss of DISC1 leads to decreased neuronal progenitor proliferation by promoting early cell cycle exit in neurons (Mao et al., 2009; Ming and Song, 2009). DISC1 provides further evidence that the Wnt signaling pathway may be involved in both neurodevelopment and psychiatric disorders.
WNT PATHWAY AND PSYCHIATRIC DISEASE GENE CANDIDATES
Several players in the Wnt signaling pathways have been associated with psychiatric disease, in addition to DISC1 (Fig. 25.2). Glycosylation is a form of regulation of Wnt ligands and their receptors, and perturbations of glycosylation are known to affect their function, localization, and signaling. SNPs in genes that are involved in glycan synthesis (Kegg pathway: hsa01030) associate with schizophrenia (O’Dushlaine et al., 2011), which raises a possibility that a part of the phenotypes caused by perturbations in this molecular pathway act via disruption of the Wnt pathway. Furthermore, several candidate genes, such as MMP16 and Tbx1, are transcriptionally regulated via Wnt activation of TCF/LEF transcription (Huh and Ornitz, 2010; Lowy et al., 2006). Mice hemizygous for Tbx1, located in the 22q11.2 microdeletion, display behavioral phenotypes that are associated with psychiatric disease (Hiramoto et al., 2011; Paylor et al., 2006).
Chromodomain helicase DNA binding protein 8 (CHD8), associated with autism in several exome sequencing studies (Neale et al., 2012; O’Roak et al., 2012), is also recruited to TCF/LEF sites along with β-catenin. CHD8 has been shown to counter the activating activity of β-catenin, by binding to β-catenin and also Histone1, and thus limiting the relaxation of chromatin. BCL9, mentioned earlier as a core component of the transcriptional complex mediating Wnt signaling, is one of the genes in the 1q21.1 recurrent CNV, in which microdeletions have been associated with schizophrenia and microcephaly, and microduplications associated with autism and macrocephaly. Furthermore, ADA2A, in the 17q12 microdeletion associated with autism, is known to be an acetyltransferase required for transcriptional activity and proliferative effects mediated by β-catenin (Yang et al., 2008).
Microdeletions of 3q29 have been associated with microencephaly, dysmorphology, and autistic features, and more recently schizophrenia (Mulle et al., 2010). Several genes in this locus have roles in the Wnt pathway. Discs, large homolog 1 (DLG1 or Sap97) is one of the binding partners of Fz and is known to signal downstream of Wnt binding by binding to APC, affecting cell proliferation. p21 protein (Cdc42/Rac)-activated kinase 2 (PAK2), also in the 3q29 CNV, also serves to phosphorylate and promote the nuclear localization of β-catenin (Zhou et al., 2011).
Neuregulin 1 and its receptor ErbB4, are other well-studied candidates found to have SNPs associated with schizophrenia and play roles in neuronal migration and synapse formation (Mei and Xiong, 2008). ErbB4 is a receptor tyrosine kinase, and its family members have been shown to activate cascades that phosphorylate and lead to the intracellular localization of β-catenin: overexpression of ErbB4 Cyt2 increased nuclear β-catenin and TCF-LEF transcriptional activity (Muraoka-Cook et al., 2009).
WNT SIGNALING AND CELL MORPHOLOGIES
Binding of Wnt to Fz leads to activation of pathways involved in planar cell polarity, acting through Dvl, activating RhoA and Ras-related C3 botulinum toxin substrate 1 Rac1 (Gao et al., 2011; Shafer et al., 2011). Rac1 activation then activates c-Jun N-terminal kinase (JNK) downstream of Wnt7b (Rosso et al., 2005). JNK activation has been shown to be crucial for the proper development of axons and dendrites (de Anda et al., 2012; Rosso et al., 2005), and to enhance or inhibit canonical Wnt activation, depending on the Wnt ligand (Billiard et al., 2005). Tao Kinase 2, in the 16p11.2 region, which has CNVs that associate with both autism and schizophrenia (deletion and duplication, respectively), also plays a role in the activation of JNK and affects basal dendrite formation (de Anda et al., 2012; McCarthy et al., 2009).
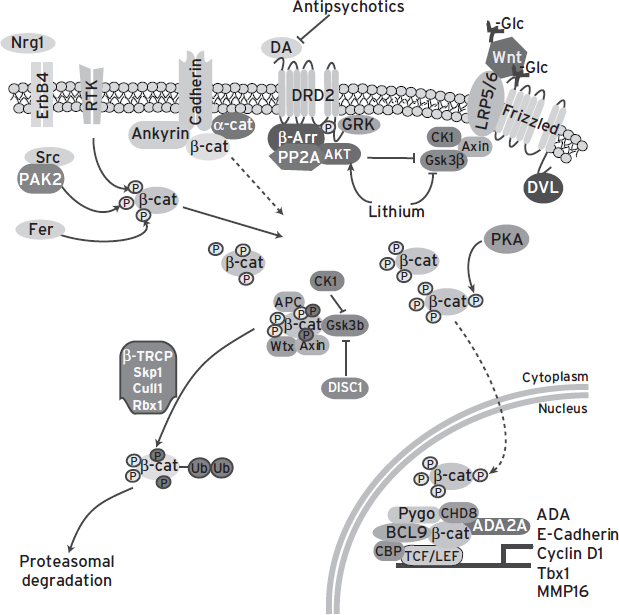
Figure 25.2 Many genes associated with psychiatric disorders regulate or are regulated by the Wnt signaling pathway.
The proteins in the canonical Wnt pathway also have an effect on postmitotic neurons independent of their effect on β-catenin. One of the alternative β-catenin independent pathways that is downstream of LRP5/6- Frizzled signaling depends on the inhibition of GSK3β. GSK3β is known to destabilize microtubules by phosphorylating microtubule proteins such as TAU, MAP1B, and MAP2. Inhibition of GSK3β stabilizes microtubules, promoting axon growth and growth cone remodeling. The phosphorylation of collapsin response mediator protein 2 (CRMP2) by GSK3 is crucial for neuronal polarity, and CRMP2 has been shown to be involved in determining axon/dendrite fate (Yoshimura et al., 2005). Knockdown of Axin results in the abolishment of axon formation in postmitotic neurons. APC, part of the destruction complex in Wnt signaling, also plays a role in neurite extension and neuronal migration. In this manner, the Wnt pathway is able to exert effects on neuronal morphology.
Binding of Wnt to Ryk has been shown to directly activate the Src pathway, similar to the activation of many other receptor tyrosine kinases. The activation of Src, in turn, has been shown to be crucial for axon guidance. Ryk has also been shown to interact directly to Fz8, as well as Dvl, and to potentiate the canonical Wnt signaling pathway, suggesting that the presence of Ryk in conjunction with Fz may either stabilize the Dvl-Fz interaction, or enhance Axin interaction with Fz (Lu et al., 2004). The convergence of neuropharmacological and genetic evidence makes the Wnt pathway(s), if not a promising target, at least a convenient framework within which the function of psychiatric risk gene polymorphisms and rare variants can be tested.
DISC1, ITS BINDING PARTNERS, AND NEURONAL MIGRATION
Lissencephaly is characterized by a thickened cortex and loss of the organized layers of the brain, which results in a smoothened brain. Because synchronized neuronal migration during embryogenesis is responsible for the laminar structure of the brain, it is thought that this process is disrupted in lissencephaly. The majority of autosomal dominant forms of lissencephaly were found to be due to deletions in chromosome 17, where gene mapping identified LIS1 as the critical gene. Mouse models show Lis1-dosage dependent defects in neuronal migration and cortical lamination (Hirotsune et al., 1998). The centrosome has been shown to play a central role in neuronal migration, which involves the molecular motor dynein. Lis1 regulates microtubule function and dynein activity, both of which are coordinated during neuronal migration. A yeast two-hybrid screen identified Ndel1 as a binding partner of Lis1. It has been shown that Ndel1 allows for the interaction of Lis1 and dynein and is required for dynein function. Ndel1 was also identified in a yeast two-hybrid screen to interact with Disc1. Ndel1 has been shown to be involved in neuronal migration, neuronal localization, and neurite and axon development (Kamiya et al., 2006). Disrupting DISC1/Ndel1 interaction disrupts neurite outgrowth in PC12 cells (Pletnikov et al., 2007), and also prevents the colocalization of DISC1 and Lis1, which localizes with the dynein heavy chain, indicating that DISC1 plays an important role in the function of Ndel1/Lis1 complex by regulating its localization. In addition, Nde1 has also been associated with DISC1/Ndel1/Lis1 to regulate neuronal proliferation, migration, and neurite extension (Bradshaw et al., 2011) and is found in the 16p13.1 CNV (Ingason et al., 2011).
It has been shown that DISC1 also interacts with this complex via its interaction with DIX domain containing-1 (Dixdc1), the third mammalian gene discovered to contain a Disheveled-Axin (DIX) domain (Dixdc1). DISC1, Dixdc1, and Ndel1 form a tripartite interaction, and the presence of either DISC1 or DIXDC1 seems to be able to properly localize Ndel1, indicating that these two proteins may have redundant effects on neuronal migration (Singh et al., 2010). This also indicates that a partial loss of function of either of these two genes can be offset by the function of the other, potentially explaining why differences in the ability of DISC1 variants to bind to Ndel does not result in an appreciable difference in human disease burden.
A molecular switch between the Wnt signaling and neuronal migration function of DISC1 has been recently uncovered (Ishizuka et al., 2011). DISC1 was found to be a target of PKA-mediated phosphorylation, downstream of cAMP activation. The study identified two putative PKA mediated phosphorylation sites on DISC1, and discovered that one of the sites, at serine 710, is responsible for the differential localization and binding properties observed in neuronal progenitors versus migrating neurons. DISC1 in neuronal progenitors is unphosphorylated at this site, and binds to and inhibits GSK3β activity: upon phosphorylation at this site, DISC1 increases its affinity for BBS1 and localizes at the centrosome. Crucially, the authors demonstrated that the phospho-dead form of DISC1 (S710A) rescued the proliferative but not the migrational phenotype observed in DISC1 knockdown conditions, while the phospho-mimetic form of DISC1 (S710E) rescued the migrational phenotypes of DISC1 function (Ishizuka et al., 2011). It is becoming clear that DISC1 coordinates multiple signaling cascades.
SYNAPTIC PATHWAYS
Spines are the postsynaptic entity of most excitatory synapses, and their morphological characteristics are important for imparting their functional properties and plasticity. There is recent evidence that has indicated that dendrites and spines are dysregulated in neurological diseases that are accompanied by cognitive impairment, such as autism spectrum disorders and schizophrenia (Penzes et al., 2011). Dysregulation of pathways that regulate the growth, pruning, and maintenance of dendritic spines is a particularly intriguing hypothesis for explaining components of schizophrenia etiology, since the onset is in early adulthood, when there is active synaptic pruning. In addition, analysis of postmortem brain samples show that there is decreased spine number on cortical pyramidal neurons in schizophrenia patients compared to controls (Garey et al., 1998).
Discussed earlier was the role of DISC1 in neurogenesis and migration in the developing brain. DISC1 is observed to be present at 40% of synapses in postmortem tissue and primary cultures, and enriched in the postsynaptic density after subcellular fractionation (Brandon and Sawa, 2011). In primary neuron culture, it has been shown that DISC1 knockdown leads to increases in spine size and decreases in spine density, which is also seen in various DISC1 mouse models. In addition to this, there are increases in mini excitatory postsynaptic current frequencies, and knockdown of GluN1 leads to decreases in DISC1 levels. Yeast two-hybrid screens with DISC1 found that its binding partners were enriched for synaptic proteins and proteins associated with NMDA-dependent signaling. It has been subsequently found that binding partners of DISC1 are important for the regulation of spine morphologies, and DISC1 is involved in their regulation.
KALIRIN
A binding partner of DISC1 identified was Kalirin, which is a Rac GTP nucleotide exchange factor. The KALRN gene encodes several isoforms, of which Kalirin-7 is the most abundant form in the rat. It contains a PDZ binding motif and interacts with several other proteins via their PDZ domain. Kalirin-7 is localized to postsynaptic densities of excitatory synapses, and Kalirin-7 clusters are generally positively stained with PSD-95, AMPA, and NMDA receptors in the rat brain (Ma et al., 2008b). Kalirin-7 is known to be crucial for spine morphogenesis and function. In vitro overexpression in hippocampal cultures causes increases in spine density, while shRNA-mediated knockdown causes the converse (Ma et al., 2003). KALRN-/- knockout mice have decreases in spine density and dendritic complexity and show impairments in long-term potentiation in the Schaeffer collateral, decreases in spontaneous excitatory postsynaptic currents, and deficits in several animal behaviors associated with schizophrenia (working memory, social interaction, and prepulse inhibition) (Ma et al., 2008a). It has been determined that in the basal state, DISC1 coordinates interaction between Kalirin-7 and PSD-95, which prevents Kalirin-7 activation of Rac1. Upon NMDA receptor activation, a fraction of Kalirin-7 dissociates from DISC1 is able to activate Rac1, whose constitutive activation is known to lead to decreases in spine size. In humans, there are decreased Kalirin mRNA levels in the DLPFC in schizophrenic postmortem brain tissue. In addition, Kalirin has been associated with schizophrenia in a Japanese GWAS and sequencing of its exons has identified rare mutations which also associate with schizophrenia (Brandon and Sawa, 2011; Kushima et al., 2012).
CHANNEL PROPERTIES AND SYNAPTIC TRANSMISSION
In a cross-analysis GWAS between schizophrenia and bipolar disease, ANK3 and CACNA1C were found to be reach genome-wide significance (Ripke et al., 2011). ANK3 encodes for ankyrin 3, a protein found in the axon initial segment which is important for clustering Ca++ and Na+ channels into electrically active areas of the axon (Pan et al., 2006), while CACNA1C encodes a subunit for an L-type voltage-gated calcium channel. Interestingly, ankyrin 3 is also known to be a component of the cadherin complex, and disruption of the cadherin complex is known to lead to mislocation of β-catenin (Orsulic et al., 1999), suggesting it may have also have a role in regulating Wnt signaling. Neurogranin, implicated in bipolar disorder and schizophrenia (Stefansson et al., 2009), is a key regulator of the Ca2+ binding messenger protein calmodulin, limiting its activity by sequestration. Based on the association of these genes in both schizophrenia and bipolar disorder, channel properties and synaptic transmission may be a shared feature of these two diseases.
There are also intersections of calcium signaling and the Wnt pathway. Wnt activates the G proteins associated with Fz proteins to modulate downstream calcium signaling. The Fz proteins are coupled to the Gαi/o Gβγ heterotrimeric G protein complex (Katanaev et al., 2005), and Wnt binding to Fz liberates the Gβγ subunits to activate a calcium-dependent pathway that involves the activation of phospholipase C cascade. The perturbation of these signaling pathways can have consequences beyond this pathway, and can impact the canonical Wnt pathway as well.
MICRORNA PATHWAYS
microRNAs (miRNAs) have been identified as a new regulatory layer that guides synaptic morphologies and functions; they have activity dependent regulation and can have differential subcellular localization (Siegel et al., 2011). Some 30% of humans who have deletion of 22q11.2 develop schizophrenia. Children with this deletion have cognitive dysfunction and similar macroscopic changes to brain structure such as decreased total brain, gray matter, and white matter volumes. Mouse models of this deletion show increased prepulse inhibition and decreased hippocampal dependent learning (Karayiorgou et al., 2010). One intriguing candidate present in 22q11.2 is Dcgr8, a protein important in the miRNA processing machinery. Dcgr8 knockout mice have decreased primary dendrites and spines as well as a decrease in functional glutamatergic synapses.
The strongest association signal in a meta-analysis of a schizophrenia GWAS was found in the putative primary transcript of the microRNA-137 (miR-137) (Ripke et al., 2011). Moreover, four predicted targets of miR-137 reached genome-wide significance and were demonstrated to have sequence-specific downregulation by miR-137 (Kwon et al., 2011), indicating that GWASs may have identified a pathway dysregulated in schizophrenia. miR-137 has been demonstrated to play a role in the dendritic arborization and spine density of adult newborn neurons and on embryonic and adult neurogenesis, although with opposite effects between the two periods (Smrt et al., 2010; Sun et al., 2011; Szulwach et al., 2010). The differential action of miR-137 between embryonic and adult neurogenesis may hold clues to how schizophrenia onset may switch in early adulthood.
CONCLUSION
There is growing evidence that schizophrenia is a neurodevelopmental disorder. The disease progression of schizophrenia is well underway before manifestation; there is cognitive impairment, decreased connectivity and organization of white matter tracts in DTI studies, and gross anatomical changes that can be detected before onset. Significant progress has been made in understanding the genetics of schizophrenia in the past five years, and there are many emerging lines of evidence networking existing theories of schizophrenia etiology with genes that contribute risk to schizophrenia. Among the various emerging gene candidates from GWAS and CNV studies, several are associated with neurodevelopmental pathways. It is also becoming evident that there are many intersections between neuropsychiatric disorders, such as schizophrenia, bipolar disorder, autism spectrum disorder, and major depressive disorder. Treating the genetics as an aggregate of causative genes with effect sizes, just as the diagnosis of many psychiatric disorders is an aggregate of symptoms with magnitudes, may be useful in parsing their complexity moving forward. At this stage of unraveling the genetics of psychiatric disorders, studying unambiguously associated genes to elucidate pathways that underlie disease may set the framework for appreciating the complexity of gene candidates identified by GWASs. It is also worth noting that these molecular pathways that are important during neurodevelopment are also active throughout adulthood, and that a single protein can have pleiotropic effects; persistent dysregulation/dysfunction of a protein can have lasting effects and affect different pathways depending on developmental time. As geneticists continue genome-wide sequencing studies, such as CNV studies, GWASs, exome sequencing, and whole genome sequencing, and basic biologists uncover functions of identified genes and the effect of the risk alleles on these functions, there is no doubt new discoveries about the etiology of psychiatric disorders will become apparent, and novel therapeutic targets will emerge. In the future, personalized medicine tailored to the unique genetic burden of individual patients may be used to prevent developmental abnormalities that later lead to increased susceptibility to schizophrenia.
DISCLOSURES
Dr. Kwon has no conflicts of interest to disclose. She is funded by a Simons postdoctoral fellowship.
Dr. Soda has no conflicts of interest to disclose. He was supported by award Number T32GM07753 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Dr. Tsai serves on the Scientific Advisory Board of Sirtris Pharmaceuticals, Inc. and is a consultant for Lilly UK. She receives grant support from the NIH, Howard Hughes Medical Institute, and the Simons Foundation.
REFERENCES
Bale, T.L., Baram, T.Z., et al. (2010). Early life programming and neurodevelopmental disorders. Biol. Psychiatry 68:314–319.
Beaulieu, J.M. (2012). A role for Akt and glycogen synthase kinase-3 as integrators of dopamine and serotonin neurotransmission in mental health. J. Psychiatry Neurosci. 37:7–16.
Bigos, K.L., Mattay, V.S., et al. (2010). Genetic variation in CACNA1C affects brain circuitries related to mental illness. Arch. Gen. Psychiatry 67:939–945.
Billiard, J., Way, D.S., et al. (2005). The orphan receptor tyrosine kinase Ror2 modulates canonical Wnt signaling in osteoblastic cells. Mol. Endocrinol. 19:90–101.
Bradshaw, N.J., Soares, D.C., et al. (2011). PKA phosphorylation of NDE1 is DISC1/PDE4 dependent and modulates its interaction with LIS1 and NDEL1. J. Neurosci. 31:9043–9054.
Brandon, N.J., and Sawa, A. (2011). Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat. Rev. Neurosci. 12:707–722.
Brockschmidt, A., Todt, U., et al. (2007). Severe mental retardation with breathing abnormalities (Pitt-Hopkins syndrome) is caused by haploinsufficiency of the neuronal bHLH transcription factor TCF4. Hum. Mol. Genet. 16:1488–1494.
Brown, A.S. (2011). The environment and susceptibility to schizophrenia. Prog. Neurobiol. 93:23–58.
Daniels, D.L., and Weis, W.I. (2005). Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat. Struct. Mol. Biol. 12:364–371.
de Anda, F.C., Rosario, A.L., et al. (2012). Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat. Neurosci. 15:1022–1031.
Fiedler, M., Sanchez-Barrena, M.J., et al. (2008). Decoding of methylated histone H3 tail by the Pygo-BCL9 Wnt signaling complex. Mol. Cell. 30:507–518.
Flora, A., Garcia, J.J., et al. (2007). The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc. Natl. Acad. Sci. USA 104:15382–15387.
Freyberg, Z., Ferrando, S.J., et al. (2010). Roles of the Akt/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am. J. Psychiatry 167:388–396.
Gao, B., Song, H., et al. (2011). Wnt signaling gradients establish planar cell polarity by inducing Vangl2 phosphorylation through Ror2. Dev. Cell. 20:163–176.
Garey, L. (2010). When cortical development goes wrong: schizophrenia as a neurodevelopmental disease of microcircuits. J. Anat. 217:324–333.
Garey, L.J., Ong, W.Y., et al. (1998). Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J. Neurol. Neurosurg. Psychiatry 65:446–453.
Gogtay, N., and Rapoport, J.L. (2008). Childhood-onset schizophrenia: insights from neuroimaging studies. J. Am. Acad. Child Adolesc. Psychiatry 47:1120–1124.
Gogtay, N., Vyas, N.S., et al. (2011). Age of onset of schizophrenia: perspectives from structural neuroimaging studies. Schizophr. Bull. 37:504–513.
Gold, J.M., and Weinberger, D.R. (1995). Cognitive deficits and the neurobiology of schizophrenia. Curr. Opin. Neurobiol. 5:225–230.
Harrison, P.J., and Weinberger, D.R. (2005). Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol. Psychiatry 10:40–68; image 45.
Hiramoto, T., Kang, G., et al. (2011). Tbx1: identification of a 22q11.2 gene as a risk factor for autism spectrum disorder in a mouse model. Hum. Mol. Genet. 20:4775–4785.
Hirotsune, S., Fleck, M.W., et al. (1998). Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat. Genet. 19:333–339.
Huh, S.H., and Ornitz, D.M. (2010). Beta-catenin deficiency causes DiGeorge syndrome-like phenotypes through regulation of Tbx1. Development 137:1137–1147.
Ingason, A., Rujescu, D., et al. (2011). Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol. Psychiatry 16:17–25.
Ishizuka, K., Kamiya, A., et al. (2011). DISC1-dependent switch from progenitor proliferation to migration in the developing cortex. Nature 473:92–96.
Jaaro-Peled, H. (2009). Gene models of schizophrenia: DISC1 mouse models. Prog. Brain. Res. 179:75–86.
Rapoport, J.L., Giedd, J.N., and Gogtay N. (2012). Neurodevelopmental model of schizophrenia: update 2012. Mol. Psych. 17(12):1228–1238.
Kamiya, A., Tomoda, T., et al. (2006). DISC1-NDEL1/NUDEL protein interaction, an essential component for neurite outgrowth, is modulated by genetic variations of DISC1. Hum. Mol. Genet. 15:3313–3323.
Karayiorgou, M., Simon, T.J., et al. (2010). 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nat. Rev. Neurosci. 11:402–416.
Karlsgodt, K.H., Niendam, T.A., et al. (2009). White matter integrity and prediction of social and role functioning in subjects at ultra-high risk for psychosis. Biol. Psychiatry 66:562–569.
Katanaev, V.L., Ponzielli, R., et al. (2005). Trimeric G protein-dependent frizzled signaling in Drosophila. Cell 120:111–122.
Kim, W.Y., and Snider, W.D. (2011). Functions of GSK-3 signaling in development of the nervous system. Front. Mol. Neurosci. 4:44.
Kim, W.Y., Wang, X., et al. (2009). GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 12:1390–1397.
Kushima, I., Nakamura, Y., et al. (2012). Resequencing and association analysis of the KALRN and EPHB1 genes and their contribution to schizophrenia susceptibility. Schizophr. Bull. 38:552–560.
Kwon, E., Wang, W., et al. (2011). Validation of schizophrenia-associated genes CSMD1, C10orf26, CACNA1C and TCF4 as miR-137 targets. Mol. Psychiatry.
Lawrie, S.M., and Abukmeil, S.S. (1998). Brain abnormality in schizophrenia. A systematic and quantitative review of volumetric magnetic resonance imaging studies. Br. J. Psychiatry 172:110–120.
Lewis, D.A., Hashimoto, T., et al. (2005). Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 6:312–324.
Lewis, D.A., and Levitt, P. (2002). Schizophrenia as a disorder of neurodevelopment. Annu. Rev. Neurosci. 25:409–432.
Logan, C.Y., and Nusse, R. (2004). The Wnt signaling pathway in development and disease. Annu. Rev. Cell. Dev. Biol. 20:781–810.
Lowy, A.M., Clements, W.M., et al. (2006). Beta-catenin/Wnt signaling regulates expression of the membrane type 3 matrix metalloproteinase in gastric cancer. Cancer. Res. 66:4734–4741.
Lu, W., Yamamoto, V., et al. (2004). Mammalian Ryk is a Wnt coreceptor required for stimulation of neurite outgrowth. Cell 119:97–108.
Ma, X.M., Huang, J., et al. (2003). Kalirin, a multifunctional Rho guanine nucleotide exchange factor, is necessary for maintenance of hippocampal pyramidal neuron dendrites and dendritic spines. J. Neurosci. 23:10593–10603.
Ma, X.M., Kiraly, D.D., et al. (2008a). Kalirin-7 is required for synaptic structure and function. J. Neurosci. 28:12368–12382.
Ma, X.M., Wang, Y., et al. (2008b). Kalirin-7 is an essential component of both shaft and spine excitatory synapses in hippocampal interneurons. J. Neurosci. 28:711–724.
Malhotra, D., and Sebat, J. (2012). CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 148:1223–1241.
Mao, Y., Ge, X., et al. (2009). Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136:1017–1031.
McCarthy, S.E., Makarov, V., et al. (2009). Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 41:1223–1227.
Mefford, H.C., Sharp, A.J., et al. (2008). Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 359:1685–1699.
Mei, L., and Xiong, W.C. (2008). Neuregulin 1 in neural development, synaptic plasticity and schizophrenia. Nat. Rev. Neurosci. 9:437–452.
Ming, G.L., and Song, H. (2009). DISC1 partners with GSK3beta in neurogenesis. Cell 136:990–992.
Moore, T.H., Zammit, S., et al. (2007). Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 370:319–328.
Mulle, J.G., Dodd, A.F., et al. (2010). Microdeletions of 3q29 confer high risk for schizophrenia. Am. J. Hum. Genet. 87:229–236.
Muraoka-Cook, R.S., Sandahl, M.A., et al. (2009). ErbB4 splice variants Cyt1 and Cyt2 differ by 16 amino acids and exert opposing effects on the mammary epithelium in vivo. Mol. Cell. Biol. 29:4935–4948.
Neale, B.M., Kou, Y., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485:242–245.
O’Dushlaine, C., Kenny, E., et al. (2011). Molecular pathways involved in neuronal cell adhesion and membrane scaffolding contribute to schizophrenia and bipolar disorder susceptibility. Mol. Psychiatry 16:286–292.
O’Roak, B.J., Vives, L., et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485:246–250.
Orsulic, S., Huber, O., et al. (1999). E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J. Cell. Sci. 112 ( Pt 8), 1237–1245.
Pan, Z., Kao, T., et al. (2006). A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 26:2599–2613.
Paylor, R., Glaser, B., et al. (2006). Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc. Natl. Acad. Sci. USA 103:7729–7734.
Penzes, P., Cahill, M.E., et al. (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14:285–293.
Pletnikov, M.V., Xu, Y., et al. (2007). PC12 cell model of inducible expression of mutant DISC1: new evidence for a dominant-negative mechanism of abnormal neuronal differentiation. Neurosci. Res. 58:234–244.
Rapoport, J.L., Giedd, J.N., and Gogtay, N. (2012). Neurodevelopmental model of schizophrenia: update 2012. Mol. Psych. 17(12):1228–1238.
Ripke, S., Sanders, A.R., et al. (2011). Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 43:969–976.
Rosso, S.B., Sussman, D., et al. (2005). Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat. Neurosci. 8:34–42.
Shafer, B., Onishi, K., et al. (2011). Vangl2 promotes Wnt/planar cell polarity-like signaling by antagonizing Dvl1-mediated feedback inhibition in growth cone guidance. Dev. Cell. 20:177–191.
Siegel, G., Saba, R., et al. (2011). microRNAs in neurons: manifold regulatory roles at the synapse. Curr. Opin. Genet. Dev. 21:491–497.
Singh, K.K., Ge, X., et al. (2010). Dixdc1 is a critical regulator of DISC1 and embryonic cortical development. Neuron 67:33–48.
Smrt, R.D., Szulwach, K.E., et al. (2010). MicroRNA miR-137 regulates neuronal maturation by targeting ubiquitin ligase mind bomb-1. Stem Cells 28:1060–1070.
Stefansson, H., Ophoff, R.A., et al. (2009). Common variants conferring risk of schizophrenia. Nature 460:744–747.
Stefansson, H., Rujescu, D., et al. (2008). Large recurrent microdeletions associated with schizophrenia. Nature 455:232–236.
Sun, G., Ye, P., et al. (2011). miR-137 forms a regulatory loop with nuclear receptor TLX and LSD1 in neural stem cells. Nat. Commun. 2:529.
Szulwach, K.E., Li, X., et al. (2010). Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J. Cell. Biol. 189: 127–141.
Takahashi, N., Sakurai, T., et al. (2011). Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Prog. Neurobiol. 93:13–24.
Thomason, M.E., and Thompson, P.M. (2011). Diffusion imaging, white matter, and psychopathology. Annu. Rev. Clin. Psychol. 7:63–85.
Torrey, E.F., Bartko, J.J., et al. (2012). Toxoplasma gondii and other risk factors for schizophrenia: an update. Schizophr. Bull. 38:642–647.
Valenta, T., Gay, M., et al. (2011). Probing transcription-specific outputs of beta-catenin in vivo. Genes. Dev. 25:2631–2643.
van Amerongen, R., and Nusse, R. (2009). Towards an integrated view of Wnt signaling in development. Development 136:3205–3214.
van Os, J., Kenis, G., et al. (2010). The environment and schizophrenia. Nature 468:203–212.
Walterfang, M., Velakoulis, D., et al. (2011) Understanding aberrant white matter development in schizophrenia: an avenue for therapy? Expert. Rev. Neurother. 11:971–987
Walsh, T., McClellan, J.M., et al. (2008). Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320:539–543.
Yang, M., Waterman, M.L., et al. (2008). hADA2a and hADA3 are required for acetylation, transcriptional activity and proliferative effects of beta-catenin. Cancer. Biol. Ther. 7:120–128.
Yoshimura, T., Kawano, Y., et al. (2005). GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120:137–149.
Zhou, L., Ercolano, E., et al. (2011). Merlin-deficient human tumors show loss of contact inhibition and activation of Wnt/beta-catenin signaling linked to the PDGFR/Src and Rac/PAK pathways. Neoplasia 13:1101–1112.