33 | PATHOGENESIS OF DEPRESSION: CLINICAL STUDIES
MAURA A. FUREY, DANIEL C. MATHEWS, AND CARLOS A. ZARATE Jr.
INTRODUCTION
Since the first antidepressant was introduced, a vast number of agents that regulate serotonin or norepinephrine have been developed, primarily as refinements over previous versions. With the exception of improvements in safety and side effect profiles, no significant progress in the efficacy of one class of antidepressant over another has occurred. Considerable effort has been made in industry, academia, and government to identify alternative drugs targets. Indeed, a number of compounds that produce promising results in preclinical studies failed to translate to an effective antidepressant at the clinical level; examples include NK1 and CRF antagonists (Madaan and Wilson, 2009). There are many reasons for the lack of success in developing novel treatments, most of which are beyond the scope of this chapter. One reason related to the chapter topic is the lack of disease biomarkers that predict clinical efficacy (Leuchter et al., 2010). A biomarker has been defined as “a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention” (Frank and Hargreaves, 2003). Recent biomarker development has focused on existing antidepressants, and thus is limited by the inherent limitations of conventional treatments.
A potentially more successful approach would be to study treatments that are radically different from existing antidepressants, such as ketamine and scopolamine, where rapid antidepressant effects are observed in contrast to the slow response time associated with conventional treatments. The rapid clinical response observed with ketamine and scopolamine offers a unique opportunity to evaluate biomarkers quickly with the goal of gaining a better understanding of the pathogenesis of depression and identifying neural signatures of treatment response. Biomarkers can be of great significance as they can prioritize resources and allow for earlier testing in proof-of-concept studies with novel therapeutic agents, especially if a target is well identified. Biomarker studies vary greatly by approach and can target predictors of treatment response based on clinical laboratory markers (e.g., genetic and epigenetic markers, neurotransmitters, hormones, cytokines, neuropeptides, and enzymes), electrophysiological measures (e.g., electroencephalography [EEG] measures, sleep EEG, evoked potentials, magnetic encephalography [MEG], and skin conductance) functional neuroimaging indices (e.g., magnetic resonance imaging [MRI], functional MRI, magnetic resonance spectroscopy [MRS], positron emission tomography [PET], and single photon emission computed tomography [SPECT]), as well as details in clinical history (Wiedemann, 2011).
An improved understanding of the pathogenesis of depression has the potential to contribute to the development of improved therapies. Moreover, insights gained with this strategy would in turn help basic scientists focus on the presumptive molecular and cellular mechanisms that underlie these radically improved treatments. In fact, clinical studies with ketamine have stimulated preclinical research to understand the molecular and cellular underpinnings that might be involved in ketamine’s rapid antidepressant effects (Aan Het Rot et al., 2012).
Consequently, this chapter will not review the serotonergic or noradrenergic systems, but instead will selectively summarize recent developments from well-designed clinical trials on other targets, such as the glutamatergic and cholinergic systems. The chapter will also characterize potential biological markers that may predict treatment response to these agents. In this chapter, when available, we review the randomized, placebo-controlled pharmacological trials in MDD and BD that have been conducted exploring these novel targets/systems. We briefly review uncontrolled studies only when the results lead to a randomized controlled clinical trial, and thus the uncontrolled study provides relevant historical context to the testing of the target being explored. We do not review trials with non-pharmacological approaches (e.g., deep brain stimulation and transcranial magnetic stimulation).
HISTORY OF THE GLUTAMATERGIC SYSTEM
Understanding the role of glutamatergic neurotransmission has developed gradually over time, particularly when compared to the current levels of interest for research related to the glutamate system and focus on potential applications. High concentrations of glutamate in the brain were first recognized in the 1930s, which led to several trials in the 1940s utilizing dietary glutamate and glutamine for the potential treatment of epilepsy and learning disorders. In the early 1950s, a pivotal animal study demonstrated that an injection of glutamate into the brain of a rat produced convulsions, which led to speculation that glutamate might be a primary excitatory neurotransmitter in the mammalian brain. Subsequent studies demonstrated that glutamate mediated excitatory action via multiple receptors, which were initially classified as N-methyl-d-aspartate (NMDA) and non-NMDA receptors. The latter was subdivided into what are now α-amino-3-hydroxy- 5-methyl-4-isoxazolepropionic acid (AMPA) and kainate receptors after learning that agonists and antagonists preferentially interacted with these receptors. By 1977, a study validated NMDA receptors as synaptic receptors, and in the following 20 years key preclinical studies confirmed that NMDA antagonists inhibited long-term potentiation in the hippocampus (important in learning and memory) and mitigated behavioral deficits seen in inescapable stress paradigms comparable to that of clinically effective antidepressants. While much of the early impetus in psychiatry was led by these preclinical studies, recent remarkable clinical observations—particularly the antidepressant effects observed with ketamine—have resulted in increased interest in the identification of potential targets within the complex and dynamic framework of the glutamatergic system (Murrough, 2012; Watkins and Jane, 2006).
GLUTAMATERGIC RECEPTORS AND BASIC PHYSIOLOGY
Glutamate is found in exponentially higher concentrations than monoamines and is considered the most abundant neurotransmitter in the brain. The excitatory effects of glutamate are primarily balanced by another neurotransmitter, γ-aminobutyric acid (GABA), which mediates the greater part of fast inhibitory neurotransmission. This tight regulation of glutamate is of significance as undue glutamate excitotoxicity is implicated in several neuropsychiatric disorders. Most neuronal glutamate is generated either de novo via the transamination of α-oxogluturate through the Krebs cycle or by recycling from the glutamate/glutamine cycle. In a simplified model, glutamate acts in three different cell compartments—the presynaptic neuron, postsynaptic neuron, and surrounding glial cells. This model also involves other targets involved in the regulation of synaptic and extrasynaptic glutamate levels. Broadly, glutamate primarily activates diverse ionotropic (NMDA, α-amino-3-hydroxy- 5-methyl-4-isoxazolepropionic (AMPA), and kainate (KA) receptors) and eight types of G protein–coupled metabotropic (mGluR) receptors. Other targets include excitatory amino acid transporters (EAATs), which provide glutamate clearance from extracellular space, soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE) complexes, which are thought to play a role in the structural aspects of synaptic vesicle exocytosis, vesicular glutamate transporters (VGLUTs), which are responsible for the uptake of glutamate into the synaptic vesicle, and cytoplasmic postsynaptic density proteins.
Of note, while glial cells have several functions and far outnumber neurons, they are especially crucial in the clearance and recycling of neurotransmitters, such as glutamate via the glutamate/glutamine cycle. The coupling between glutamatergic neurons and surrounding glial cells is fundamental, as impaired glial cell activity may lead to neuronal toxicity or increased glutamatergic activation. Mounting evidence suggests that frontal cortical areas of subjects with major depression or bipolar disorder have lower numbers of glial cells than non-psychiatric controls. Glial loss in the prefrontal cortex induces depressive-like behaviors similar to chronic stress in preclinical models. Other pathophysiological findings associated with glutamatergic neurotransmission have been drawn from postmortem, gene expression, and neuroimaging studies, such as magnetic resonance spectroscopy (MRS). (For a more complete review of the anatomy of the glutamatergic system and its role in depression and moods disorders, see Machado-Vieira et al., 2012; Machado-Vieira, Manji, 2009.) The complex physiological regulation of glutamate may provide an array of targets for future pharmacologic development and drug discovery.
CLINICAL TRIALS WITH GLUTAMATERGIC AGENTS
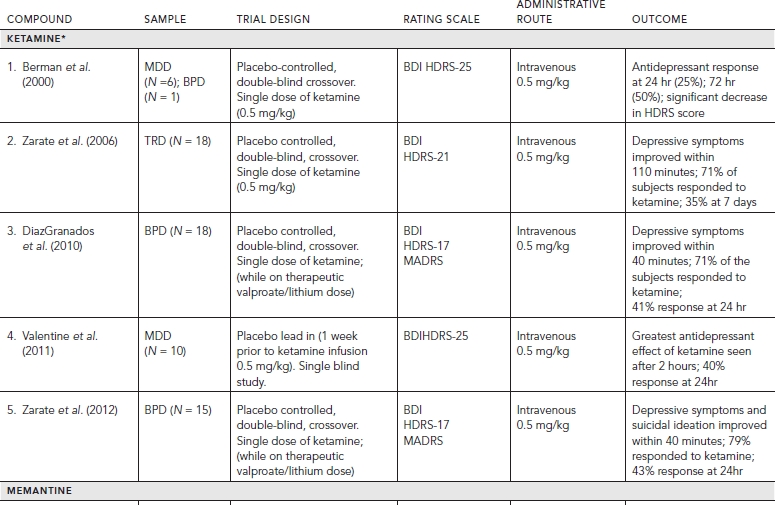
Placebo-controlled clinical trials (and select uncontrolled trials) with glutamatergic targets are summarized in Table 33.1.
NMDA ANTAGONISTS—KETAMINE
Of the three major subtypes of ionotropic receptors, NMDA and AMPA receptors have shown compelling evidence of their role in antidepressant action. A significant “proof of concept” translation of NMDA antagonists with MDD patients in clinical trials has been notable particularly with ketamine. Ketamine is a noncompetitive, high affinity NMDA antagonist that was first developed by Parke-Davis in 1963 as a safer anesthetic compared to phencyclidine (PCP). Ketamine is metabolized by the liver into two primary metabolites, norketamine (major metabolite) and dehydronorketamine (DHNK—minor, inactive metabolite) with a half-life of two to two and a half hours. A series of other metabolites of ketamine have been identified recently, some lasting up to three days (Zarate et al., 2012a). Several preclinical studies demonstrated that ketamine produces antidepressant- or anxiolytic-like effects in various behavior models of depression (e.g., forced swim test, tail suspension test, learned helplessness, etc.) (Tokita et al., 2012).
The first reported placebo-controlled, double-blinded, randomized trial (n = 7) utilizing a single intravenous (IV) dose of ketamine (0.5 mg/kg infusion over 40 minutes) in patients with MDD demonstrated a significant reduction in depressive symptoms 72 hours postinfusion. Profound but transient cognitive deficits and euphoria also were induced by the ketamine infusion. A larger double-blind, placebo-controlled, crossover study at the National Institutes of Mental Health (NIMH) replicated these findings by showing that a single ketamine infusion (0.5 mg/kg over 40 minutes) had rapid and relatively sustained antidepressant effects (lasting one to two weeks) in patients with treatment-resistant MDD. Subjects in this study were medication free at least two weeks prior to infusion and on average failed six prior antidepressants. Effect size for the drug difference was large (d = 1.46) after 24 hours and moderate to large (d = 0.68) after one week. Thirty-five percent of subjects maintained response for at least one week. Adverse effects included perceptual disturbances, confusion, dizziness, euphoria, derealization, and transient elevation in blood pressure with most symptoms peaking at 40 minutes and ceasing 80 minutes postinfusion.
TABLE 33.1. Controlled trials targeting the glutamatergic system
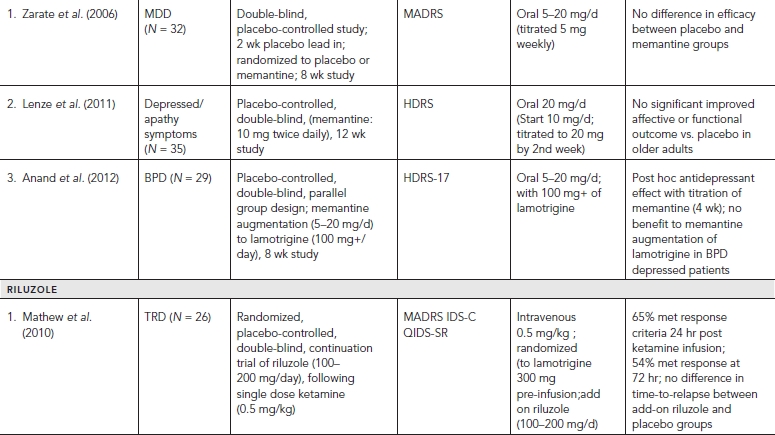
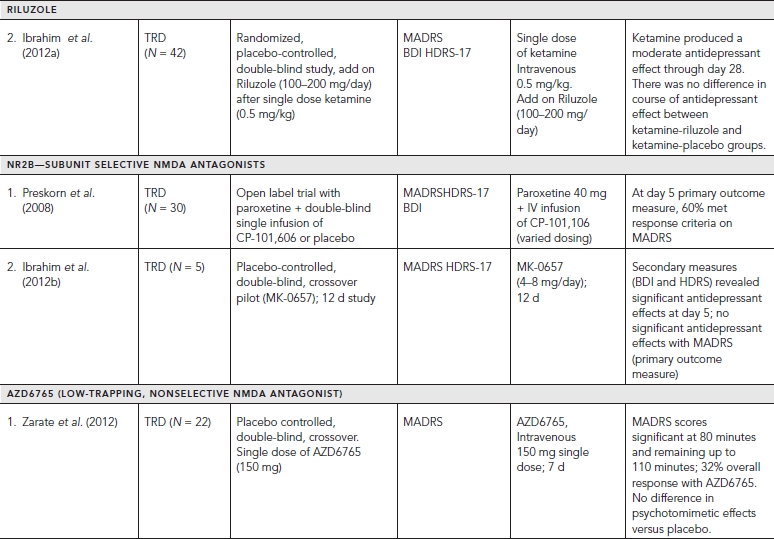
In a similarly designed study, ketamine was added to therapeutic levels of lithium or valproate and also produced rapid antidepressant effects in patients with treatment-resistant bipolar depression. A recent study replicated these latter findings in bipolar depression and also demonstrated that ketamine can rapidly improve suicidal ideation in patients for up to three days following a single intravenous dose (d = .89). Similar, albeit uncontrolled, open label and naturalistic studies (e.g., emergency room settings) utilizing ketamine have shown significant and rapid antisuicidal effects in depressed patients. Interestingly, one of these studies had preliminary results showing ketamine to have rapid beneficial effects on suicidal cognition as measured by the Implicit Association Test—a reliable behavioral measure assessing implicit suicidal associations (Price et al., 2009). Taken together, ketamine’s use in antisuicidal effects likely will be an area of future interest and research.
Despite ketamine’s positive safety profile as a widely used anesthetic agent for children, an important limitation of ketamine’s clinical application in larger populations is its acute neuropsychiatric side effects (Green et al., 1998). Equally important are the questions of tolerability and sustained efficacy with repeat ketamine treatments. To this regard, only small, open-label studies have shown ketamine to be fairly well tolerated with repeated/sustained antidepressant effects (Aan Het Rot et al., 2010; Zanicotti et al., 2012). Frequency of repeat infusions were similar to outpatient ECT scheduling (Monday-Wednesday-Friday) with relapse rates varying significantly after the last dose from 6 days to greater than 45 days (Murrough et al., 2011).
Strategies considered to attenuate ketamine’s side effect profile mainly include augmentation with other drugs (pre or post ketamine). In an early study, healthy volunteers were administered haloperidol (5 mg), a typical antipsychotic, two hours prior to a ketamine infusion, but the results were unremarkable (Krystal et al., 1999). More recently, a small (n = 14), randomized, double-blind study evaluated the potential of riluzole—an inhibitor of glutamate release (reviewed in Zarate and Manji, 2008)—to prevent postketamine relapse in patients with treatment resistant depression (TRD). The study also assessed whether pretreatment with the anticonvulsant and mood-relapse preventive agent lamotrigine would attenuate the psychotomimetic effects and enhance antidepressant activity of ketamine based on an earlier study showing that lamotrigine attenuated the psychotomimetic side effects in healthy volunteers (Anand et al., 2000). In this study, lamotrigine failed to reduce the transient psychotomimetic side effects associated with ketamine in TRD, or enhance its antidepressant effects. Likewise, riluzole did not differ from placebo in preventing postketamine relapse (Mathew et al., 2010).
A larger, four-week, double-blind, randomized, placebo- controlled study (Ibrahim et al., 2012) also was conducted to evaluate the effect of riluzole use following a single ketamine infusion. Four to six hours after a single infusion of ketamine (0.5 mg/kg), 42 subjects with treatment-resistant MDD were randomized to double-blind treatment with either riluzole (100–200 mg/day; n = 21) or placebo (n = 21) for four weeks. Although the effect size of improvement with ketamine was initially large and remained moderate throughout the 28-day trial, the difference between the riluzole and placebo treatment groups was not significant for postketamine relapse. These two studies continue to the highlight the complexity of augmentation strategies for both ketamine’s tolerability and sustained efficacy.
Despite the attention and interest from ketamine’s early trials, the administration of ketamine for use in the clinic remains under study. Larger, controlled trials will be required to evaluate safety with repeated use of ketamine, and continued research will be required to discover successful relapse prevention or strategies to sustain antidepressant effects. Similarly, more studies will be needed to assess whether alternative delivery routes (e.g. intranasal, intramuscular) might improve pharmacokinetics, with continued assessment of optimal dosing regiments, and improved knowledge and understanding of ketamine’s underlying mechanism of actions. (All of the previously noted controlled studies, plus those conducted to date in depression, and additional discussion regarding the maintenance of ketamine’s antidepressant response, and its effects on suicidal ideation are reviewed in Aan Het Rot et al., 2012; Mathew et al., 2012.)
PUTATIVE MOLECULAR MECHANISMS UNDERLYING KETAMINE’S ANTIDEPRESSANT RESPONSE
A growing body of preclinical evidence investigating ketamine’s rapid antidepressant effects has led to novel findings and an increasing body of knowledge toward the neuroplastic effects of this treatment. In addition to NMDA receptor antagonism, ketamine involves enhanced throughput of AMPA receptors, and some inhibitory effects at muscarinic acetylcholine receptors. Prominent preclinical studies have demonstrated ketamine to rapidly phosphorylate/activate the mammalian target of rapamycin (mTOR) pathway with resulting increased levels of synaptic signaling proteins and synaptic plasticity (e.g., new spine synapse formation) in the prefrontal cortex (PFC) of rodents. Included in this cascade of postsynaptic events, literature discusses mixed results for an increased activation of brain-derived neurotrophic factor (BDNF) with ketamine’s antidepressant response. Reduced levels of BDNF have been associated with depression, and BDNF’s essential role in cell differentiation, nerve growth, and neuronal survival research makes it a likely area of investigation. Preclinical work by others has also implicated the potential role of glycogen synthase kinase-3 beta (GSK-3β) inhibition in the hippocampus and PFC after ketamine infusions. Patients with MDD are thought to have increased GSK-3β activity, and inhibition of GSK-3β is also thought to play a key role in the therapeutic mechanism of antidepressants and mood stabilizers. Overall, further research will still be required to elucidate the neurobiology underlying ketamine’s rapid antidepressant response (Aan Het Rot et al., 2012; Mathew et al., 2012; Murrough, 2012).
BIOMARKERS OF ANTIDEPRESSANT RESPONSE TO KETAMINE
Given the heterogeneity of depression and often “trial and error” approach to current treatments, positive predictors of antidepressant response to ketamine would be of great significance and practical value to both patients and clinicians. Several recent studies are investigating ketamine responders via a multimodal systems level approach, and utilize a range of potential markers from genes to the reversal of the complex behavioral phenotype (i.e., genes, gene expression, synaptic plasticity indices, cellular components, neural circuits, behavioral features of depression) using various experimental methods (neuroimaging, genetics, electrophysiological measures, clinical features, etc.). The goal is to both inform regarding the likelihood of response and provide insights into the mechanisms of action of ketamine. This section will briefly highlight some the more prominent studies.
One neurophysiologic study utilized magnetoencephalography (MEG) to measure rostral anterior cingulate cortex (rACC) activity as drug free patients with MDD were presented with a fearful face paradigm. Previous research has shown that higher pretreatment ACC metabolism predicts antidepressant response in sleep deprivation (also known to induce rapid, but brief antidepressant effects) and several other antidepressant treatments (e.g., SSRIs). In the MEG study, higher pretreatment levels of rACC activity correlated positively with the magnitude of subsequent antidepressant response to a ketamine, while healthy individuals showed reduced activity in this region (Salvadore et al., 2009). Another MEG study incorporating a working memory task demonstrated that patients who showed the least amount of engagement in the pregenual ACC as task difficulty increased showed the largest subsequent response to treatment, within four hours of ketamine administration. Pretreatment functional connectivity between the pregenual ACC and the left amygdala also negatively correlated with the antidepressant following ketamine. These findings concur with functional studies in healthy volunteers, where pgACC regional blood flow increases with emotional tasks, but decreases during cognitive tasks that demand attention. The results suggest that conservation of this pattern may predict better treatment outcome (Salvadore et al., 2010). Both MEG studies overall contribute to the extant literature supporting the role of ACC activity as a promising predictor of response to antidepressant treatment (Pizzagalli, 2011).
Two electrophysiologic studies utilizing sleep measures have also shown interesting findings. One study (n = 30, TRD patients) investigated the acute effects of ketamine on depressive symptoms evaluating EEG slow wave activity (SWA), individual slow wave parameters, and plasma BDNF (230 min postinfusion). Earlier research had demonstrated a relationship between SWA and cortical synaptic activity of cortical neurons suggesting its potential role as a surrogate marker of central synaptic plasticity. Decreased production of sleep slow waves is also a core feature observed in depression. Results of the study showed early sleep SWA (during the first non-REM episode) and BDNF levels increased compared to baseline with ketamine responders having changes to BDNF proportional to changes in EEG parameters. Consistent with an earlier finding (Machado-Vieira, Yuan et al., 2009) from the same lab was no difference in BDNF levels from responders and non-responders. The earlier study did note their lack of placebo control and other cautions, such as interpreting peripheral BDNF as a marker of central BDNF. The study overall suggests that sleep SWA parameters and BDNF may serve as future non-invasive measures for testing novel antidepressants and that ketamine increases synaptic strengthening/efficacy (Duncan et al., 2012). Building on the latter study, baseline delta sleep ratio (DSR) was examined to see if it could predict rapid response to a ketamine infusion in individuals with TRD. DSR is the ratio of slow wave activity between the first two non-REM sleep episodes, and this measure has also been shown to be lower in depressed patients than healthy controls. Findings from the study (n = 30) showed a significant positive correlation between baseline DSR and reduced MADRS scores from baseline to day 1 (low baseline scores predicted better mood response). These results are notable, as some traditional antidepressants have been found to normalize slow wave sleep and DSR. Sample size was a known limitation of the findings, and larger replications of the study were recommended by the authors. Nevertheless, this preliminary data could lead to DSR being a useful biomarker of response to ketamine in the future (Duncan et al., 2012).
In regard to genetic studies, a recent study (n = 62) demonstrated that MDD patients with the Val/Val BDNF allele were more likely to have an increased antidepressant response to ketamine (from baseline HAM-D score to 210/230 min postinfusion) than Met carriers. These results also support previous clinical data, where Val/Val mice exhibited increased antidepressant effect to ketamine (and prefrontal cortex synaptogenesis) on the basis of this single polymorphism. The Val66Met SNP is found in 20–30% of humans and has been linked to psychiatric disorders and impaired trafficking/regulation of BDNF (Laje et al., 2012).
Another innovative approach to study both the pathophysiology of MDD and ketamine response has been proton MRS. This research tool allows for the quantification of amino acid neurotransmitters in the brain, and previous MRS research has shown that GABA and Glx (composite peak formed by glutamate and glutamine) are decreased in the medial and dorsal anterolateral PFC of patients with MDD. Subsequently, 14 drug free patients with MDD who were scanned before receiving a single infusion of ketamine showed an association (230 min postinfusion) between lower Glx/ glutamate ratio (a surrogate marker of glutamine) and a greater improvement in response to ketamine treatment. Pretreatment GABA or glutamate did not correlate with improved depressive symptoms, however pretreatment Glx/glutamate in the dorsomedial/dorsal anterolateral PFC was negatively correlated with improvement in depressive symptoms. As glutamine is primarily localized in glia, the authors hypothesized that the decreased Glx/glutamate ratio may reflect a reduced number of glial cells, further signifying that this “neuropathological construct” (Salvadore et al., 2011) may be associated with antidepressant responsiveness to ketamine.
A potential clinical predictor of response to ketamine is family history of alcohol dependence. It is important to acknowledge that family history could represent a biomarker (i.e., genetic, epigenetic influences) or be the result of environment. A recent study found that patients with MDD who had a family history of alcohol dependence had a better short-term response to ketamine infusion than subjects with no family history of alcohol dependence (Phelps et al., 2009). The positive family history group (FHP) had a significantly higher response rate (67%) at 230 minutes (postinfusion) than the group without alcohol dependence family history (FHN) (18%; p = .02). In addition, patients with FHP had fewer dysphoric symptoms postinfusion than those with FHN. Previous clinical studies also found compared to healthy controls, subjects with alcohol dependence experienced fewer perceptual differences and decreased dysphoric mood during ketamine infusion (Krystal et al., 2003). Surprisingly, the healthy controls with a positive family history of alcohol also showed fewer ketamine-induced perceptual alterations (Petrakis et al., 2004). Of note, self-reported history of alcohol use disorders or family history of major depression did not predict response. Possible mechanisms explaining these effects include familial, epigenetic, or genetic variations in the NMDA subunit NR2A, which may have the greatest relevance for human alcohol dependence (Schumann et al., 2008).
Lastly, ketamine infusions in healthy volunteers immediately induced changes similar to those gradually induced by monoaminergic-based antidepressant agents as assessed using quantitative electroencephalography (QEEG). The observed reduction in prefrontal theta cordance (a measure that correlates with cerebral perfusion) may represent a potential marker/predictor for the antidepressant effects of ketamine, a hypothesis that could be tested readily in future studies with depressed populations (Horacek et al., 2010; Hunter et al., 2007).
NMDA—SUBUNIT-SELECTIVE NR2B ANTAGONISTS
As noted earlier, non-competitive NMDA receptor antagonists like ketamine and PCP can produce psychotomimetic effects when used acutely. These observations promoted investigation into subunit-selective NMDA receptor agents to potentially mitigate the adverse effects, while promoting a therapeutic effect. Structurally, NMDA receptors are comprised of four tetrameric proteins: 2 NR1 subunits and 2 NR2 subunits. NR2 subunits are further divided by four subtypes: NR2A-D. Prior preclinical and clinical literature demonstrated the NR2B-selective NMDA antagonist. CP-101,606 to be well tolerated without prominent psychotropic side effects in a study evaluating traumatic brain injury (Merchant et al., 1999).
Building on this knowledge, Preskorn and colleagues undertook a “proof of concept” study with a recent randomized, placebo-controlled, double-blind trial, evaluating the antidepressant efficacy of the NR2B subunit-selective NMDA receptor antagonist CP-101,606 in 30 treatment-refractory MDD subjects. The study showed that a single infusion of CP-101,606 administered adjunctively to paroxetine, had greater antidepressant effects as compared with placebo (saline) infusion, with 78% of treated responders maintaining their response (50% reduction in HDRS score) for at least one week after infusion (mean difference, 8.6; 80% confidence interval, -12.3 to -4.5) (P < 0.10). The authors did note that the initial dose of 0.75 mg/kg was reduced to 0.5 mg/kg during the study given several initial research subjects experienced moderate to severe dissociative symptoms (Preskorn et al., 2008). All of the initial dissociative reactions resolved within six hours of the discontinuation of the infusion. No clinically significant change in labs was noted, however blood pressure changes similar to ketamine trials were observed (i.e., changes ≥20 mm Hg for diastolic or 30 mm Hg for systolic from baseline). Although the lower dose of CP-101,606 produced a rapid and significant antidepressant response without producing a dissociative reaction, the study highlights the challenges of optimal dosing for similar compounds. More studies will be necessary to replicate these findings and the overall efficacy and safety of compounds selective for the NR2B receptor (Preskorn et al., 2008).
Subsequently, an oral formulation of the selective NMDA NR2B receptor antagonist (MK-0657) was administered using a randomized, double-blind, placebo-controlled, crossover pilot study to evaluate the potential antidepressant efficacy and tolerability of this agent in treatment-resistant MDD patients. Following a one-week drug-free period, MDD patients were randomized to receive either MK-0657 monotherapy (4–8 mg/day) or placebo for 12 days. Due to discontinuation of the compound’s development by the manufacturer and recruitment challenges, only five patients completed both the MK-0657 and placebo arms of the study. Secondary efficacy scales as assessed by the Hamilton Depression Rating Scale (HAM-D) and Beck Depression Inventory (BDI) demonstrated significant antidepressant effects as early as day 5 in patients receiving MK-0657 compared to placebo; however, no improvement was noted when symptoms were assessed with the primary efficacy measure (MADRS) (Ibrahim et al., 2012). Of note, MK-0657 increased plasma BDNF levels compared to placebo 9 days after treatment was initiated, indicative of a biological effect typically seen with several antidepressants. Although a small study, the role of BDNF may be involved as a potential biomarker of a NR2B antagonist’s treatment effect. No dissociative or serious adverse effects were observed. While larger studies will be needed for confirmation, preliminary data suggests that an oral formulation of an NR2B antagonist may have antidepressant properties (Ibrahim et al., 2012).
NMDA (NONSELECTIVE LOW TRAPPING CHANNEL BLOCKER)
More recently, an IV formulation of a low-trapping, nonselective NMDA channel blocker (AZD6765) produced rapid antidepressant effects in a placebo-controlled study in TRD patients. Following a two-week drug-free period, MDD patients received IV infusions of either AZD6765 (150 mg) or saline solution one week apart with a randomized, double-blind, crossover design. Antidepressant effects were seen within 80 minutes with MADRS scores remaining significant for 110 minutes. Results overall demonstrated that 32% of subjects responded to AZD6765, and 15% to placebo at some point in the trial. Furthermore, 18% of patients reached remission on AZD6765 versus 10% on placebo at some point in the study. Interestingly, no difference was observed between the groups with regard to psychotomimetic or dissociative effects (Zarate et al., 2012b). Onset of antidepressant effects were comparable to that of ketamine, but the duration was more short-lived. Subunit selectivity and trapping blockade may explain some AZD6765’s observed differences compared to ketamine. Similarly, metabolites of ketamine may contribute to its sustained antidepressant effects (Zarate et al., 2012a). Results from larger placebo-controlled, multicenter trials with AZD6765 are pending (NCT01482221).
OTHER GLUTAMATERGIC MODULATORS: MEMANTINE AND RILUZOLE
Memantine
Memantine, a derivative of amantadine, is already FDA approved for the treatment of moderate to severe Alzheimer’s disease and is considered a low-affinity, non-competitive, open-channel NMDA receptor antagonist. Memantine has essentially no psychotomimetic effects at therapeutic doses (5–20 mg/day), a key distinction from ketamine. In addition, memantine has been utilized clinically for over 15 years showing good tolerability in large patient populations. A review of associated preclinical data within depression and anxiety models (e.g., decreased immobility time) is discussed in greater detail by Parsons et al. (1999). Of note, despite amantadine’s remarkable antidepressant effects observed in patients with the Borna disease virus, it will not be reviewed in this chapter given a lack of data in any randomized controlled trials and due to its likely involvement with multiple pharmacologic mechanisms (Huber et al., 1999).
Unfortunately, most controlled clinical trials have not demonstrated memantine to have robust antidepressant effects. In an eight-week double blind placebo-controlled trial (n = 32), memantine (5–20 mg/day) failed to improve symptoms of depression in patients with MDD (Zarate et al., 2006). More recently, a proof of concept study (n = 29) also failed to show any benefit of memantine augmentation (5–20 mg/day) of lamotrigine (stable dose of at least 100 mg/daily for four weeks prior to randomization) for patients with bipolar depression over an eight-week trial (primary outcome time point). However, memantine was observed to have a significant antidepressant effect early on in the treatment (up to four weeks) during its titration (Anand et al., 2012). Lastly, a 12-week, double-blind placebo controlled pilot study (n = 35) evaluated memantine (10 mg twice daily) for treatment in late-life depression and apathy after a disabling medical event, but no affective or functional improvement was observed when compared with placebo (Lenze et al., 2011).
With regard to positive outcome trials, a larger study (n = 80) alcohol-dependent patients with MDD were randomized to memantine 20 mg/day or escitalopram 20 mg/day. Memantine reduces alcohol cravings in preclinical studies, and alcohol dependence commonly is comorbid with major depression. The study concluded that both treatments significantly reduced depression and anxiety (primary outcome measures) (Muhonen et al., 2008). However, a major limitation of this study was the lack of a placebo control. Alcohol dependence may modulate the antidepressant response that is mediated via NMDA receptors, as long-term alcohol use increases the number (Nagy, 2004) and alters the function of glutamate NMDA receptors (Petrakis et al., 2004). Interestingly, a seven-day, placebo-controlled randomized single-blinded psychopharmacology trial study (n = 127) that utilized three different antiglutamatergic strategies (lamotrigine 25 mg 4x/day, memantine 10 mg 3x/day, or topiramate 25 mg 4x/day) for ethanol detoxification demonstrated significant improvements for alcohol withdrawal symptoms and dysphoric mood (Krupitsky et al., 2007).
In an effort to better understand memantine’s potential antidepressant role, a recent healthy volunteer model of emotional processing attempted to assess the neuropsychological profile of action associated with memantine (10 mg dose; typical dose in clinical studies is at least 20 mg). Overall, the results suggest that memantine produces an early anxiogenic response in the emotion-potentiated startle similar to that seen in studies considering a single dose of the SSRI citalopram. No other significant difference was observed with emotional or non-emotional information processing. Earlier studies have also shown that SSRI treatments initially increase the emotion-potentiated startle effect with acute administration, but that this effect is reversed after seven daily treatments (Browning et al., 2007; Grillon et al., 2007). Given the study only tested one dose; the authors did note that a future study (to 7 days) may help confirm the current profile of effects and provide more clinical utility. Nevertheless, the authors also suggest that the limited neuropsychological profile associated with memantine may be consistent with the clinical data previously reported, where memantine is no more effective than placebo (Pringle et al., 2012). As already noted, similar but longer studies would help clarify some of these interpretations.
Riluzole
Riluzole currently is approved by the US Food and Drug Administration for treating amyotrophic lateral sclerosis. This agent crosses the blood-brain barrier and modulates the glutamatergic system by inhibiting glutamate release (via inhibition of voltage-dependent sodium channels) and enhancing both glutamate reuptake and AMPA trafficking and glutamate transporters. Riluzole is thought to have neuroprotective and plasticity enhancing properties given its ability to stimulate neurotrophic factors, such as nerve growth factor (NGF), BDNF, and glial cell line–derived neurotrophic factor (GDNF) in cultured astrocytes. A number of publications also discuss its use in neurodegenerative disorders. Combined with its glutamatergic modulating properties, Riluzole has had increased investigation in off label uses in both psychiatric and neurologic disorders (Zarate and Manji, 2008).
In clinical trials, riluzole monotherapy for depression has been evaluated only in open-label trials. One trial (n = 19) with TRD and a one-week medication-free period demonstrated significant improvement in depression during weeks three through six of treatment (50–200 mg/day, mean dose = 168.8 mg/day). Improvements were also seen in scales measuring anxiety. The most common side effects were similar (e.g., headache, gastrointestinal distress, and constipation) to those seen in trials of patients with amyotrophic lateral sclerosis. Similar, positive antidepressant and anxiolytic effects were reported with riluzole (50–200 mg/day) as an augmentation antidepressant agent, but only in smaller open-label trials evaluating effects in bipolar depression, generalized anxiety disorder, and obsessive-compulsive disorder (OCD).
Given some of antidepressant findings of riluzole mentioned, two double-blind, randomized, placebo-controlled studies proceeded to apply riluzole as a potential add-on strategy to maintain ketamine’s rapid antidepressant effects (see ketamine section for details). Both studies administered riluzole (100–200 mg/day) orally, and both trials failed to show any difference in time to relapse from placebo after a single ketamine infusion. Nevertheless, both trials did replicate earlier ketamine findings showing it to be well tolerated and having rapid antidepressant effects. Overall, double-blind, placebo-controlled clinical trials will still be necessary to confirm some of the promising findings observed in these open-label studies. For a more systematic review of literature regarding riluzole, see Zarate and Manji (2008).
THE CHOLINERGIC SYSTEM
HISTORY OF THE CHOLINERGIC SYSTEM
The cholinergic system was the first of the neurotransmitters to be identified, and as a result has received a tremendous amount of investigation. Early research characterized the role of the cholinergic system at the neuromuscular junction, in the pathophysiology of Alzheimer’s disease, and in cognitive functions including memory and attention. The cholinergic system also is the target of nerve gas, a fact that was accidentally uncovered in Germany in 1936 as researchers were attempting to improve on available insecticides, or organophosphates. These agents target acetylcholinesterase, an enzyme responsible for terminating effects of acetylcholine in the synapse, resulting in continued neural transmission and continued contractions at the neuromuscular junction. After a drop of the potent agent tabun was dropped in the laboratory, the investigator and his assistant began to experience dizziness and severe shortness of breath. These observations led to the development of nerve gas for military purposes during World War II, although it was never used during the war.
The cholinergic neurotransmitter system was implicated in mood disorders in the 1970s, when Janowsky and colleagues demonstrated that the acute administration of the anticholinesterase physostigmine to currently manic bipolar patients results in the rapid development of depressive symptoms (reviewed in Janowsky et al., 1994). Similarly, when physostigmine was administered to currently depressed patients with major depressive disorder, depressive symptoms worsened acutely. Physostigmine produces an increase in available acetylcholine, and thus is non-selective relative to cholinergic receptor type. As a result of this general implication of cholinergic function, interest has been given to both nicotinic and muscarinic cholinergic agents as potential antidepressants. Nonetheless, while the cholinergic system was identified in the early 1970s as a potential target for antidepressant agents, little attention had been given to this system until recently.
CHOLINERGIC RECEPTORS AND BASIC PHYSIOLOGY
The cholinergic neurotransmitter system is supported by two general types of receptors, the nicotinic and the muscarinic receptors (Albuquerque et al., 2009; Pringle et al., 2011). Muscarinic receptors, which are sensitive to muscarine as well as to acetylcholine, are G protein–coupled receptors and thus act through second messenger systems. Five muscarinic subtypes have been described to date, simply called M1 through M5. Muscarinic receptors are located throughout the central nervous system and in the postganglionic neurons of the parasympathetic division of the autonomic nervous system. Nicotinic receptors, which are responsive to nicotine as well as acetylcholine, are ionotropic and thus act by opening or closing ion channels in response to endogenous stimulation by acetylcholine. The nicotinic receptors are made up of five subunits, which surround a central pore. These receptors can be separated broadly based on their location in the nervous system into two subtypes, muscle-type and neuronal-type nicotinic receptor. Muscle-type nicotinic receptors, found at the neuromuscular junction, are either of the embryonic form or the adult form, each with slightly different compositions of the five subunits in the receptors. The neuronal-type has various combinations of 12 possible receptor subunits.
CLINICAL TRIALS WITH CHOLINERGIC AGENTS
Placebo-controlled clinical trials (and select uncontrolled trials) with cholinergic targets are summarized in Table 33.2.
MUSCARINIC CHOLINERGIC AGENTS
Interest in cholinergic muscarinic agents developed as these receptors explicitly were implicated in aspects of sleep disturbances observed in patients with mood disorders. Specifically, decreased REM latency and increased REM density are observed in this patient population, and these sleep features are mediated through cholinergic muscarinic receptors (reviewed in Janowsky et al., 1994). The pattern of sleep disturbance is consistent with excessive muscarinic activity, and thus indicated increased muscarinic cholinergic function in patients with affective disorders.
Biperiden
Early studies looked at the antidepressant properties of a selective M1 antagonist, biperiden (Kasper et al., 1981). In an uncontrolled, open label study evaluating oral biperiden in “severely depressed” patients, the authors report significant improvement in depressive symptoms by the end of study. These findings however did not replicate in a controlled study of biperiden, where no significant benefit was observed over the control condition (Gillin et al., 1995). However, the original study is of interest, as the authors reported that baseline results of a dexamethasome suppression test significantly predicted subsequent clinical response to biperiden.
Scopolamine
More recently, in the context of a dose-finding study designed to evaluate the impact of a muscarinic blocker on cognitive features of mood disorders, unipolar and bipolar patients received intravenous infusions of the cholinergic muscarinic antagonist scopolamine (Furey and Drevets, 2006). Critically, all patients participating in these studies were free of psychoactive medications for a minimum of two weeks, and as patients were not taken off of existing medication, most were free of medications for a longer period of time. Over the course of this three-dose, four-infusion, placebo-controlled pilot study, depressive symptoms notably improved. Despite the small sample size (n = 8), the response was sufficiently large to warrant a clinical trial to properly assess the antidepressant potential of scopolamine.
A clinical trial was conducted (Furey and Drevets, 2006) that included a total of seven infusions, and began with a single-blind placebo lead-in infusion. Patients then were randomized into either a P/S or a S/P series, whereby P = a block of three placebo infusions and S = a block of three scopolamine infusions; these infusions were administered under double-blind conditions. Full clinical assessments always preceded infusions, and one follow-up assessment was obtained three to five days after the last infusion for a total of eight assessments. This crossover design allowed for all patients to receive treatment.
TABLE 33.2. Trials targeting the cholinergic system
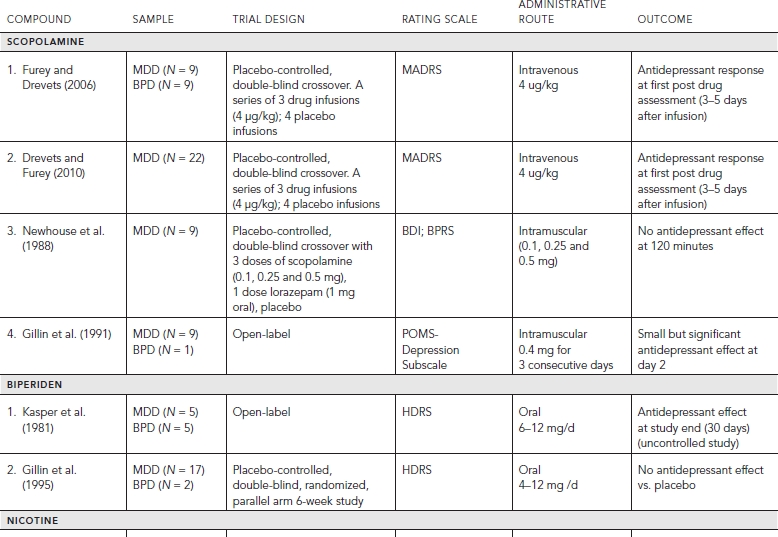
A rapid antidepressant response was observed in both the P/S and the S/P patient groups. The P/S group showed little change in MADRS while receiving placebo in the first study block, and subsequently had a significant reduction in MADRS with the first assessment following the first infusion of scopolamine (F = 94.1, p < 0.0001, Cohen’s d = 3.4), demonstrating a rapid antidepressant response. Importantly the magnitude of reduction in MADRS increased significantly over the series of three infusions (p = 0.04). The S/P group received scopolamine in the first study block, and again showed a significant reduction in MADRS with the first assessment following the first infusion of scopolamine (F = 34.8, p < 0.0001; Cohen’s d = 2.2). This group also showed further improvement over the series of three scopolamine infusions suggesting that (p = 0.002), together with the P/S group, patients will improve further with additional infusions of scopolamine. In the S/P group, the clinical benefit experienced during the scopolamine infusions in block 1 persisted to the end of the study, demonstrating that the antidepressant effects continue in the absence of additional treatment for at least two weeks. Importantly, the groups differed in study block 1 (F = 26.7, p < 0.0001; Cohen’s d = 2.7) demonstrating that drug and placebo separated, and this separation occurred by the first evaluation in study block 1 (t = 2.5, p = 0.02).
These clinical findings have been replicated (Drevets and Furey, 2010) in an independent sample of patients. The experimental design was unchanged, but the patient sample was limited to patients with major depressive disorder. In this study the overall pattern of rapid antidepressant response was identical, thus providing additional support to these clinical findings. These studies together indicate that the cholinergic muscarinic receptors offer a new neurobiological target for antidepressant treatment that has the potential to produce rapid antidepressant responses.
Patterns of response associated with patient subgroups have been considered in a combined/increased patient sample. In patients studied to date, the magnitude of response is similar for unipolar and bipolar patients, as well as for patients with and without comorbid anxiety disorder. The only patient subgroups that identified differences in response outcome were based on patient gender. While men and women showed significant improvement in symptoms following scopolamine administration, women showed significantly larger responses than men (Furey et al., 2010).
Putative Molecular Mechanism Underlying Scopolamine’s Antidepressant Effect
The antidepressant effects observed following scopolamine administration appear to be consistent with the hypothesis that cholinergic hypersensitivity contributes to the pathophysiology of mood disorders. However, the response latency of one to three days together with the persistence of the antidepressant response may suggest that the mechanism of action includes effects beyond the direct influence on muscarinic receptors.
Elevated glutamatergic transmission is associated with the pathophysiology of mood disorders, and like other antidepressant treatments, scopolamine reduces NMDA receptor activity. Muscarinic receptor stimulation enhances NMDA receptor gene expression (Liu et al., 2004), and thus the increased muscarinic receptor sensitivity seen in mood disorders may lead to increased NMDA receptor activity. In rat brain, the administration of scopolamine has been shown to reduce mRNA concentrations for NMDA receptor types 1A and 2A (Liu et al., 2004), and thus may reduce NMDA receptor activity via this mechanism.
A recently described effect of ketamine on synaptic plasticity (described previously) that is thought to underlie the observed antidepressant effects also has been shown following scopolamine administration (Li et al., 2011-Online). Like ketamine, scopolamine administration in rats activates the mTOR pathway, which leads to increases in synaptic signaling protein expression and increases in the number and function of new spine synapses in prefrontal cortical areas. Rapamycin, which blocks mTOR signaling, disrupts both the scopolamine induced increase in synaptogenesis as well as the antidepressant behavioral response observed in rodent models of depression. The timing and the magnitude of these effects following scopolamine administration are similar to those seen following ketamine administration. The authors hypothesize that increases in mTOR signaling depend on increases in extracellular glutamate levels, and both ketamine and scopolamine have been shown to increase glutamate concentration. This work suggests that the rapid antidepressant effects observed following both scopolamine and ketamine administration may involve influences on synaptic plasticity.
Biomarkers of Antidepressant Response to Scopolamine
Biomarkers of treatment response to scopolamine have been identified using functional neuroimaging. Baseline neuroimaging assessments attained prior to drug administration have been utilized to predict subsequent response to scopolamine. As highlighted earlier, the pursuit and identification of biomarkers of treatment response offers the potential to provide insight into the underlying pathophysiology of mood disorder, and subsequently lead to improved clinical treatment.
The role of the cholinergic system in cognitive functions, and the existing literature on cholinergic modulation during cognitive tasks and functional brain response, renders cognitive studies in patients with cholinergic dysfunction potentially very informative. The role of cholinergic activity in executive functions such as working memory (WM) and attention has been hypothesized to act via stimulus processing mechanisms (reviewed in Furey, 2011). Working memory refers to a process by which information (visual or auditory) is encoded, maintained for a short period of time, and is then used during recall or recognition. The information subsequently is not long available (i.e., no long-term storage). The preclinical literature indicates that increasing cholinergic activity through the direct application of acetylcholine has effects on stimulus processing, and alters signal-to-noise (S/N) mechanisms to enhance the representation of target stimuli. Functional imaging studies in humans also have demonstrated that increased cholinergic function increases response to task-relevant stimuli in visual processing areas, much like an increased S/N at the brain region or brain network level, and results in improved performance on WM and attention tasks. Thus, cholinergic function likely influences cognition by modifying neural representations of task-relevant stimuli.
The role of the cholinergic system specifically in WM processes has been well studied, and the results similarly highlight the impact of cholinergic function specifically on the processing of task-relevant stimuli. The best-characterized cognitive feature in mood disorders is described as a negative processing bias, where negative emotional information is preferentially processed over positive information. The excessive cholinergic activity in mood disorders may underlie this negative processing bias, and potentially could be evident in measures obtained with functional neuroimaging methodologies (reviewed in Furey, 2011).
A WM task was utilized to determine if levels of neural activity to specific stimulus information in visual processing brain areas could predict subsequent treatment response to the cholinergic muscarinic agent, scopolamine (Frankel et al., 2011-Online). A face WM task was used that included two task conditions: one which instructed participants to encode and remember the identity of the face and another which instructed participants to encode and remember the emotional expression in the face. In both conditions, the face to encode was presented, followed by a memory delay period, and then by a test image (Frankel et al., 2011-Online). The task was to indicate if the test image matched the encoded image based on the attended (i.e., identity or emotion) stimulus feature.
Functional magnetic resonance imaging (fMRI) was conducted in patients with MDD and healthy individuals during multiple sessions using the infusion schedule described previously. Blood oxygen-level dependent (BOLD) signal was measured, a surrogate measure of neural activity, as participants performed the two conditions in the WM task. Response magnitude was estimated separately for each task component and for each task condition from the data collected during the baseline session, prior to scopolamine infusions. The BOLD estimates of neural activity were correlated with the magnitude of subsequent treatment response to scopolamine (percent change in MADRS from baseline to study end).
Only the two stimulus processing conditions (i.e., stimulus encoding and test components) when patients were processing emotional expressions in the faces produced significant findings. Importantly, these correlations were observed exclusively in visual processing areas. Specifically, significant negative correlations were observed bilaterally in middle occipital cortex (MOC) between BOLD response and the magnitude of treatment response. While the analyses for the emotion encoding and emotion test components were conducted independently, the areas of significant correlation in MOC were largely overlapping, highlighting the selectivity of these particular brain regions in this effect. Importantly, no correlation emerged when patients were processing the same stimuli but encoding face identity, highlighting the selectivity of this effect to emotion processing.
These findings indicate that in MDD, the baseline, pretreatment levels of neural activity in MOC during the processing of emotional information reflects the potential for a clinical response to scopolamine. These results may suggest that the level of underlying cholinergic dysfunction is expressed in levels of neural response to emotional information, and that this underlying level of cholinergic dysfunction represents a biomarker for subsequent response to a cholinergic muscarinic antagonist agent. Healthy participants on average show higher levels of activity in these same MOC regions than MDD patients. Importantly, those patients who do not respond to scopolamine have BOLD estimates that fall well within the range observed in healthy participants, consistent with the interpretation that those patients who do not respond to scopolamine also do not show processing dysfunction in these areas. Similarly, those who do respond to scopolamine show increased levels of dysfunction in these brain regions at baseline.
BOLD estimates also were obtained from the scanning session that followed the scopolamine infusion, and thus reflects the acute response to muscarinic blockade. To determine the extent to which neural response in visual cortex (with a particular interest in MOC) during the same stimulus processing components of the emotion WM task is susceptible to cholinergic modulation in a manner that predicted treatment outcome, the change in BOLD response was calculated (BOLD drug—BOLD baseline), and this delta BOLD measure was correlated with subsequent treatment response. The change in BOLD response following acute scopolamine administration (thus prior to antidepressant effects) during stimulus processing components of the emotion working memory task also correlated with subsequent treatment response. The brain regions of significance were found to substantially overlap with the MOC regions identified in the analyses described previously (Frankel et al., 2011-Online). These findings may suggest that the visual processing brain regions that show predictive value at baseline also respond to cholinergic modulation in a manner that predicts treatment outcome, an interpretation that would support the conclusion that baseline levels of neural dysfunction in MOC are cholinergically mediated.
NICOTINIC CHOLINERGIC AGENTS
In addition to the early literature implicating cholinergic receptors in general, interest in nicotinic cholinergic receptors as possible targets for antidepressant agents originated based on the observation in 2005 that smoking rates were substantially higher in major depressive disorder (between 40% and 60%) than in the general population (22%) (reviewed in Philip et al., 2010). This observation led to the hypothesis that increased levels of smoking reflected an effort to self-medicate, and that nicotine may have antidepressant effects. Related observations included that smokers with a history of major depression have more difficulty quitting smoking, and patients with depression are at greater risk for developing a major depressive episode during smoking cessation (Philip et al., 2010). Conversely, smokers also have reduced levels of monoamine oxidase (MAO), an enzyme involved in the breakdown of monoamines including serotonin, norepinephrine, and dopamine, which is proposed to result in depressed mood (Philip et al., 2010).
Nicotine
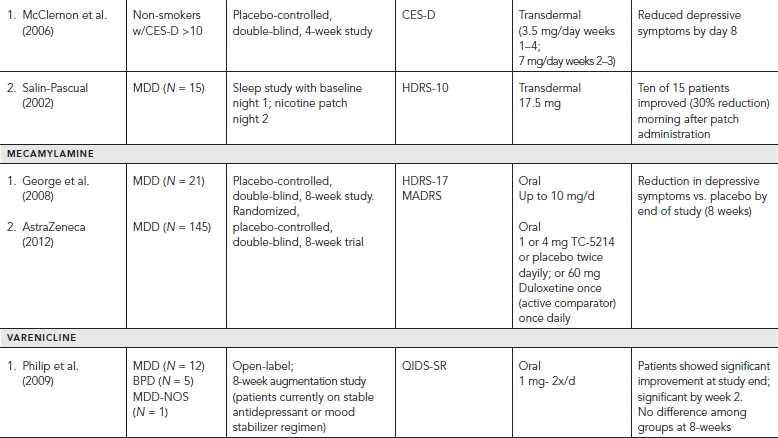
An open label clinical trial using transdermal nicotine in depressed smokers provided some evidence that nicotine may have potential for producing antidepressant effects (Salin-Pascual, 2002). The authors report that 10 of 15 non-smoking patients with major depressive disorder showed a 30% improvement in depressive symptoms. While the group as a whole did not show a significant response, this change was observed the morning after administration of the nicotine patch, suggesting a potential rapid antidepressant response. Several replications in uncontrolled studies have been reported (reviewed in Salin-Pascual, 2002). A placebo-controlled study also was conducted (McClernon et al., 2006), which showed that depressive symptoms improved by day 8 of nicotine administration, but this study was conducted in a mildly depressed cohort (not reaching criteria for MDD) and thus offers limited utility with regard to implications for patients with depression.
As health concerns prevent the clinical use of nicotine as an antidepressant agent, these clinical findings primarily have led to the evaluation of the antidepressant potential of other agents that target nicotinic receptors. A paradox within this literature does emerge. Some antidepressant medications used clinically for smoking cessation, including bupropion, a norepinephrine and dopamine reuptake inhibitor, and nortriptyline, a tricyclic antidepressant that inhibits reuptake of norepinephrine and serotonin, also have nicotinic antagonistic properties. Thus agents with nicotinic agonist effects and agents with nicotinic antagonist effects reportedly produce antidepressant effects. A potential explanation has been offered (reviewed by Philip et al., 2010), which suggests that nicotine initially activates nicotinic receptors but this activation is rapidly followed by desensitization. Continued binding to the nicotinic receptor potentially leads to ongoing desensitization, which is thought to result in chronic antagonism. Thus, antidepressant effects are thought to result from nicotinic antagonistic action.
Mecamylamine
The potential for the non-selective nicotinic cholinergic receptor antagonist mecamylamine to produce antidepressant effects has been evaluated. In a population of adolescents with Tourette’s syndrome and comorbid major depression antidepressant effects were reported, which led to a placebo-controlled clinical trial in patients with MDD (George et al., 2008). MDD patients showing little to no response after receiving a minimum of three months of SSRI treatment were randomized to placebo or mecamylamine (10 mg/d) for an eight-week trial. More patients receiving mecamylamine showed significant improvement (>50% reduction in symptoms) (45%) than those receiving placebo (10%) at end of study, suggesting that MDD patients who are SSRI refractory showed an antidepressant response to mecamylamine.
A double-blind, placebo controlled phase-II study also was conducted with MDD patients who were refractory follow a six-week trial of the SSRI citalopram. Patients were randomized to 5–10 mg/day of mecamylamine or to placebo for a 10-week trial. The group receiving mecamylamine showed more improvement than those receiving placebo (reviewed in Bacher et al., 2009). Nonetheless, a multicenter randomized, double-blind, placebo controlled phase-II study of a non-selective nicotinic receptor antagonist (TC-5214; S-mecamylamine) failed to show antidepressant efficacy in an eight-week trial (AstraZeneca, 2012), raising questions as to the potential utility of these agents.
Varenicline
In a clinical trial designed to evaluate the effect on smoking cessation, varenicline, a nicotinic agonist, was found to alleviate negative symptoms associated with nicotine withdrawal including depression (reviewed in Philip et al., 2010). This observation lead to an eight-week open label augmentation study in currently depressed patients with MDD or BD who were unresponsive to current treatment. Fourteen of 18 patients completed the study, and showed improvement in depressive symptoms as compared to baseline. Eight patients met criteria for full response, six of whom also met criteria for remission. Interestingly, observed improvement in depressive symptoms correlated with patient smoking cessation (reviewed in Philip et al., 2010).
OTHER POTENTIAL TARGETS AND FUTURE CONSIDERATIONS
While this chapter has focused primarily on the glutamatergic and cholinergic systems given recent promise with rapid acting agents in these respective classes, this should not minimize the research being done in other potential areas including the dysregulation of dopamine neurotransmission (Dunlop and Nemeroff, 2007; Nestler and Carlezon, 2006), the pathophysiology of the hypothalamic-pituitary-adrenal axis (HPA) (Pariante and Lightman, 2008; Wasserman et al., 2010), circadian rhythm regulation (Adrien, 2002; Harvey, 2011), stress hormone regulation (Holsboer and Ising, 2010; Nugent et al., 2011), inflammatory/neuroimmune system (Leonard and Maes, 2012; Miller et al., 2009; Muller et al., 2011), involvement of corticotrophin-releasing factors (Holsboer, 2000; Paez-Pereda et al., 2011), tachykinin/neurokinin (NK-1, NK-2) receptors (Herpfer and Lieb, 2005; Mantyh, 2002), and neurotrophic factors (Autry and Monteggia, 2012; Voleti and Duman, 2012).
Despite advances in the previously mentioned areas, there is still limited success with randomized, placebo-controlled trials with non-monoamine based compounds. For example, corticotropin-releasing factor (CRF, a peptide containing 41 amino acids) and its primary receptor (CRF-1) are well known to regulate behavioral responses to stress. CRF is also distributed throughout the CNS with its interneurons widely localized in the neocortex to areas (prefrontal, cingulate, and insular cortices) well known to be involved in depression and affective disorders. Its relation to impaired regulation of the HPA axis further supports rationale to target this system. While an initial open label trial of NBI-30775/R121919, a non-peptidic tricyclic CRF-1 receptor antagonist with high oral bioavailability and blood-brain barrier penetration, showed initial promising results, clinical development later stopped because of a reversible increase of liver enzymes in healthy controls during an unpublished safety study with high dosages (despite absence of CRF-1 receptor expression in the liver). Phase II controlled clinical trials with CP-316,311 (Pfizer) and BMS-562086 (Bristol-Myers Squibb, pexacerfont) also did not provide therapeutic results over placebo (Paez-Pereda et al., 2011). Results are currently pending regarding SSR-125543A another CRF-1 antagonist with recent completion of a Phase II clinical trial of 580 Russian subjects with MDD (Connolly and Thase, 2012). Despite these lackluster results, published results seem to indicate that CRF-1 receptor antagonism is safe and not intrinsically associated with adverse side effects. Interestingly, it should be noted that none of the mentioned studies included a stratification of patients for HPA abnormalities (Paez-Pereda et al., 2011). Similar to CRF-1 antagonists, NK-1 antagonists have also had disappointing results with a pooled analysis (>2,500 patients) of five, eight-week placebo-controlled clinical trials finding no benefit of the NK-1 antagonist aprepitant over placebo (Connolly and Thase, 2012). Lastly, with regard to inflammation, a recent proof-of-concept study (n = 60) demonstrated that tumor necrosis factor (TNF) antagonism does not have generalized efficacy in TRD (Raison et al., 2013). The study incorporated infusions of the TNF antagonist infliximab (5 mg/kg) in a double-blind, placebo-controlled trial and included outpatients that were both medication-free and on a consistent antidepressant regimen. Despite the negative primary findings, an association was observed between high baseline concentrations of the inflammatory biomarker high-sensitivity C-reactive protein (hs-CRP) and subsequent improved response to infliximab. In addition, infliximab-treated patients with a low level of inflammation appeared to do worse than placebo-treated patients. As a result, the study suggests there may be a subgroup of patients with TRD who have increased inflammation and respond to TNF antagonism, but not to placebo. Future development with inflammatory biomarkers may prove promising for personalizing antidepressant treatment. Opium and its derivatives have an extensive history dating several centuries back, both in medicinal and recreational settings. While opioids are utilized largely for the management of pain, endogenous opioid peptides and their receptors have been identified as potential candidates for novel antidepressant treatments given their expression in notable brain areas known to play major roles in affective disorders (e.g., ventral tegmental area, nucleus accumbens [NAc], PFC). Likewise, antidepressants that increase the availability of noradrenaline and serotonin through the inhibition of the reuptake of these monoamines also enhance the opioid pathway (Berrocoso et al., 2009). Of the main receptor subtypes (delta, kappa, mu, and nociceptin), the delta opioid agonists (DOP) are gaining increasing attention, as recent preclinical evidence has provided promising results. DOP agonists have both reduced analgesic properties and less reinforcing/respiratory depression effects than those observed with classic μ receptors (Jutkiewicz, 2006). Currently, two DOP agonist trials (Phase II development) have been completed (AZD-2327 and AZD-7268) for the treatment of MDD with pending efficacy results (Connolly and Thase, 2012). For an excellent and more complete review of novel and emerging drugs/targets for depression, such as sigma receptors, the interested reader should refer to Connolly and Thase (2012).
While barriers to clinical translation for psychiatric drugs are beyond the scope of this chapter, some key issues include the limited predictive value of preclinical models and the ever present challenge of target validation for clinical validity. For example, with CRF-1 antagonism, it is not known if the compounds given in trials are actually blocking CRF-1 receptors in the brain (i.e., target and functional engagement). In this example, we are largely assuming the effects in a preclinical model will extend to functional effects in humans (Potter, 2012). With regard to target validation, a target is often defined as a molecular or cellular structure involved in the pathology of interest that the intervention (e.g., drug) is meant to act on. This is clearly challenging in psychiatry and other CNS areas, as establishing valid targets requires a sound understanding of the underlying pathophysiology. Developing a drug against a specific target can often take several years with escalating costs in the billions of dollars. As a result, “reducing failures early in development is far more important than filling a pipeline with poorly chosen late-stage products likely to fail, and fail expensively” (Smith, 2003). For an optimistic future, collaboration will certainly be required, and an open-source model may provide for more rapid data sharing and better prioritizing of successful targets. Such steps may “de-risk” some of the current barriers for future “bench to bedside” therapeutics. Ideally, continuing advances in other tools such as molecular genetics, systems neuroscience, and translational medicine will lead to innovative tools and biomarkers to ultimately better serve patients suffering with depression and other debilitating disorders. For a more detailed discussion on novel CNS drug development, see Potter (2012).
EXPERIMENTAL MEDICINE
Experimental medicine characterizes a different method designed to understand the pathophysiology underlying mood disorders that merits comment (reviewed in Pringle et al., 2011). This approach proposes a cognitive neuropsychological model to understand antidepressant action, and suggests that pharmacologically mediated changes in emotional processing biases early in treatment contribute to subsequent clinical improvements in mood. Moreover, the delay in treatment response seen following conventional treatment is explained as the time during which the patient learns new associations following social interactions. Much of this work has been conducted in healthy volunteers, demonstrating that conventional antidepressant agents modulate neural responses to emotional information following several days of administration, well before clinical response generally is observed in clinical populations. These findings provide important insights into potential mechanisms of action that require follow-up evaluation in patient groups.
In patient populations, this approach has demonstrated that early in treatment and prior to clinical improvement, antidepressant agents modulate neural responses during emotional processing as measured using fMRI. Similarly, effects on behavioral measures that demonstrate emotional processing biases have been reported at three hours following the first administration of an antidepressant in patients with MDD. As these changes are associated exclusively with drug administration, the question as to whether these early changes predict subsequent clinical response to treatment remains empirical.
MDD is a severe, disabling, and potentially life-threatening medical illness that disrupts the lives of millions of individuals worldwide. Despite a considerable number of antidepressant agents developed over the years, significant limitations exist with regard to the utility and efficacy of available drugs. Critically, no specific agent or class of agents has proven to be more efficacious than any other. In addition, a considerable lag in the onset of therapeutic response is observed, often requiring weeks to months for full therapeutic response, thus leaving patients at risk for ongoing impairment and elevated risk of self-harm. Progress in the development of new and better medications that are particularly geared toward accelerating the onset of improvement of depressive symptoms has stalled despite considerable efforts from industry, academia, and government. The translation from targets in preclinical models to clinic populations has not succeeded as of yet in delivering better treatments. One approach for addressing these shortcomings is to focus on new neurophysiological targets, with the goal of identifying rapid acting interventions, and to simultaneously incorporate biomarkers of treatment response to facilitate the understanding of underlying mechanism.
This chapter primarily reviews two drugs that are radically unique and distinct from current antidepressants due to their rapid antidepressant effects; ketamine and scopolamine. Ketamine produces antidepressant effects within hours, while scopolamine produces effects within a few days. Research to date for both drugs is unique in that controlled studies have been conducted and replicated demonstrating rapid antidepressant effects. The inability to replicate clinical experiments recently has been highlighted as a concern in psychiatry. In addition, the evidence showing rapid efficacy of these compounds is augmented with multimodal biomarkers. Using rapid onset antidepressants permits the simultaneous study of multiple biomarkers and thus allows for the examination of biological signatures of rapid improvement using a systems level approach, extending from genes to behavioral phenotype across the cellular, molecular, neurochemical, and circuit domains. This approach permits the careful study of correlations of multiple biomarkers, precisely timed at the onset of antidepressant effects, thus potentially offering an opportunity to obtain greater insight into the molecular and cellular changes that accompany treatment response. In this way, ketamine and scopolamine are experimental tools being used to uncover clues that can then be characterized more precisely to determine what molecular targets are involved in these rapid antidepressant effects. Indeed, this strategy has already led to significant advances in the last several years reinvigorating and solidifying industry, academia, and government partnerships with moving forward toward the development of drugs with a rapid onset of antidepressant action. Specifically with the antidepressant response to ketamine, the systems level approach has led to the identification of genetics correlates associated with SNP on BDNF, molecular correlates identifying the involvement of mTOR, GSK-3 inhibition, and eEF2 as putative targets, the identification of brain areas and brain function associated with clinical improvement, such as increases in slow wave activity during sleep, and cortical excitability gamma power in sensory regions. Although these findings are promising, much more is needed before we gain a comprehensive understanding of biological underpinning of rapid antidepressant effects. These insights will be critical to the development of the next generation of treatments if they are to provide faster, more effective options than existing treatments offer. Similar approaches are being applied to the cholinergic antagonist scopolamine, and we anticipate that data gleaned from this approach will contribute to that of ketamine and other agents with a rapid onset of action to identify the neural and molecular requirements for fast acting treatments.
DISCLOSURES
The authors gratefully acknowledge the support of the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health (IRP-NIMHNIH; Bethesda, MD, USA), and thank the 7SE Research Unit of the NIMH-NIH for their support. Role of funding source: This review was supported by the IRP-NIMH-NIH. The NIMH had no further role in the writing of this chapter, or in the decision to submit the chapter for publication.
The authors gratefully acknowledge the support of the IRP-NIMH-NIH, and the NARSAD Independent Investigator Award and Brain and Behavior Foundation Bipolar Research Award (Dr. Zarate).
Dr. Mathews has no conflict of interest to disclose, financial or otherwise.
Dr. Zarate is listed as a co-inventor on a patent application for the use of ketamine and its metabolites in major depression. Dr. Zarate has assigned his rights in the patent to the US government but will share a percentage of any royalties that may be received by the government.
Dr. Furey is listed as a co-inventor on a patent application for the use of scopolamine and its metabolites in major depression. Dr. Furey has assigned his rights in the patent to the US Government but will share a percentage of any royalties that may be received by the government.
REFERENCES
Aan Het Rot, M., Collins, K.A., et al. (2010). Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol. Psychiatry 67(2):139–145.
Aan Het Rot, M., Zarate, C.A., Jr., et al. (2012). Ketamine for depression: where do we go from here? Biol. Psychiatry 15:15.
Adrien, J. (2002). Neurobiological bases for the relation between sleep and depression. Sleep Med. Rev. 6(5):341–351.
Albuquerque, E.X., Pereira, E.F., et al. (2009). Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol. Rev. 89(1):73–120.
Anand, A., Charney, D.S., et al. (2000). Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of N-methyl-d-aspartate receptor antagonists. Arch. Gen. Psychiatry 57(3):270–276.
Anand, A., Gunn, A.D., et al. (2012). Early antidepressant effect of memantine during augmentation of lamotrigine inadequate response in bipolar depression: a double-blind, randomized, placebo-controlled trial. Bipolar Disord. 14(1):64–70.
AstraZeneca. A study to assess the safety and effect of TC-5214 in patients with major depressive disorder. ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US) 2012. [cited 2013 Jan 03]. Available from: http://www.clinicaltrials.gov/ct2/show/NCT01288079 NLM Identifier: NCT01288079
Autry, A.E., and Monteggia, L.M. (2012). Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev. 64(2):238–258.
Bacher, I., Wu, B., et al. (2009). Mecamylamine—a nicotinic acetylcholine receptor antagonist with potential for the treatment of neuropsychiatric disorders. Expert. Opin. Pharmacother. 10(16):2709–2721.
Berman, R.M., Cappiello, A., et al. (2000). Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47(4):351–354.
Berrocoso, E., Sanchez-Blazquez, P., et al. (2009). Opiates as antidepressants. Curr Pharm. Des. 15(14):1612–1622.
Browning, M., Reid, C., et al. (2007). A single dose of citalopram increases fear recognition in healthy subjects. J. Psychopharmacol. 21(7):684–690.
Connolly, K.R., and Thase, M.E (2012). Emerging drugs for major depressive disorder. Exp. Opin. Emerg. Drugs. 17(1):105–126.
Diazgranados, N., Ibrahim, L., et al. (2010). A randomized add-on trial of an N-methyl-d-aspartate antagonist in treatment-resistant bipolar depression. Arch. Gen. Psychiatry 67(8):793–802.
Drevets, W.C., and Furey, M.L. (2010). Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol. Psychiatry 67(5):432–438.
Duncan, W.C., Sarasso, S., et al. (2012). Concomitant BDNF and sleep slow wave changes indicate ketamine-induced plasticity in major depressive disorder. Int. J. Neuropsychopharmacol. 7:1–11.
Duncan, W.C., Selter, J., et al. (2012). Baseline delta sleep ratio predicts acute ketamine mood response in major depressive disorder. J. Affect. Disord. [Epub ahead of print.]
Dunlop, B.W., and Nemeroff, C.B. (2007). The role of dopamine in the pathophysiology of depression. Arch. Gen. Psychiatry 64(3):327–337.
Frank, R., and Hargreaves, R. (2003). Clinical biomarkers in drug discovery and development. Nat. Rev. Drug. Discov. 2(7):566–580.
Frankel, E.L., Drevets, W.C., et al. (2011-Online). Neural activity in visual processing areas during WM predicts antidepressant response to scopolamine. Paper presented at the Society of Neurosciences.
Furey, M.L. (2011). The prominent role of stimulus processing: cholinergic function and dysfunction in cognition. Curr. Opin. Neurol. 24(4):364–370.
Furey, M.L., and Drevets, W.C. (2006). Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch. Gen. Psychiatry 63(10):1121–1129.
Furey, M.L., Khanna, A., et al. (2010). Scopolamine produces larger antidepressant and antianxiety effects in women than in men. Neuropsychopharmacology 35(12):2479–2488.
George, T.P., Sacco, K.A., et al. (2008). Nicotinic antagonist augmentation of selective serotonin reuptake inhibitor-refractory major depressive disorder: a preliminary study. J. Clin. Psychopharmacol. 28(3):340–344.
Gillin, J.C., Lauriello, J., et al. (1995). No antidepressant effect of biperiden compared with placebo in depression: a double-blind 6-week clinical trial. Psychiatry Res. 58(2):99–105.
Gillin, J.C., Sutton, L., et al. (1991). The effects of scopolamine on sleep and mood in depressed patients with a history of alcoholism and a normal comparison group. Biol. Psychiatry 30(2):157–169.
Green, S.M., Rothrock, S.G., et al. (1998). Intramuscular ketamine for pediatric sedation in the emergency department: safety profile in 1,022 cases. Ann. Emerg. Med. 31(6):688–697.
Grillon, C., Levenson, J., et al. (2007). A single dose of the selective serotonin reuptake inhibitor citalopram exacerbates anxiety in humans: a fear-potentiated startle study. Neuropsychopharmacology 32(1):225–231.
Harvey, A.G. (2011). Sleep and circadian functioning: critical mechanisms in the mood disorders? Annu. Rev. Clin. Psychol. 7:297–319.
Herpfer, I., and Lieb, K. (2005). Substance P receptor antagonists in psychiatry: rationale for development and therapeutic potential. CNS Drugs 19(4):275–293.
Holsboer, F. (2000). The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23(5):477–501.
Holsboer, F., and Ising, M. (2010). Stress hormone regulation: biological role and translation into therapy. Annu. Rev. Psychol. 61:81–109, C101–C111.
Horacek, J., Brunovsky, M., et al. (2010). Subanesthetic dose of ketamine decreases prefrontal theta cordance in healthy volunteers: implications for antidepressant effect. Psychol. Med. 40(9):1443–1451.
Huber, T.J., Dietrich, D.E., et al. (1999). Possible use of amantadine in depression. Pharmaco. Psychiatry 32(2):47–55.
Hunter, A.M., Cook, I.A., et al. (2007). The promise of the quantitative electroencephalogram as a predictor of antidepressant treatment outcomes in major depressive disorder. Psychiatr. Clin. North. Am. 30(1):105–124.
Ibrahim, L., Diaz Granados, N., et al. (2012a). Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: results from a 4-week, double-blind, placebo-controlled study. Neuropsychopharmacology 37(6):1526–1533.
Ibrahim, L., Diaz Granados, N., et al. (2012b). A randomized, placebo- controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J. Clin. Psychopharmacol. 32(4):551–557.
Janowsky, D.S., Overstreet, D.H., et al. (1994). Is cholinergic sensitivity a genetic marker for the affective disorders? Am. J. Med. Genet. 54(4):335–344.
Jutkiewicz, E.M. (2006). The antidepressant-like effects of delta-opioid receptor agonists. Mol. Interv. 6(3):162–169.
Krishnan, V., and Nestler, E.J. (2008). The molecular neurobiology of depression. Nature 455(7215):894–902.
Krishnan, V., and Nestler, E.J. (2010). Linking molecules to mood: new insight into the biology of depression. Am. J. Psychiatry 167(11):1305–1320.
Krupitsky, E.M., Rudenko, A.A., et al. (2007). Antiglutamatergic strategies for ethanol detoxification: comparison with placebo and diazepam. Alcohol. Clin. Exp. Res. 31(4):604–611.
Krystal, J.H., D’Souza, D.C., et al. (1999). Interactive effects of subanesthetic ketamine and haloperidol in healthy humans. Psychopharmacology. (Berl.) 145(2):193–204.
Krystal, J.H., Petrakis, I.L., et al. (2003). NMDA receptor antagonism and the ethanol intoxication signal: from alcoholism risk to pharmacotherapy. Ann. NY Acad. Sci. 1003:176–184.
Laje, G., et al. (2012). Brain-derived neurotrophic factor Val66Met polymorphism and antidepressant efficacy of ketamine in depressed patients. Biol. Psychiatry 72:e27–28.
Lenze, E.J., Skidmore, E.R., et al. (2011). Memantine for late-life depression and apathy after a disabling medical event: a 12-week, double-blind placebo-controlled pilot study. Int. J. Geriatr. Psychiatry 16(10).
Leonard, B., and Maes, M. (2012). Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci. Biobehav. Rev. 36(2):764–785.
Leuchter, A.F., Cook, I.A., et al. (2010). Biomarkers to predict antidepressant response. Curr. Psychiatry Rep. 12(6):553–562.
Li, N., Voleti, B., et al. (2011-Online). Rapid antidepressant actions of scopolamine require mTOR signalling and synaptogenesis. Paper presented at the Society for Neurosciences.
Liu, H.F., Zhou, W.H., et al. (2004). [Muscarinic receptors modulate the mRNA expression of NMDA receptors in brainstem and the release of glutamate in periaqueductal grey during morphine withdrawal in rats]. Sheng Li Xue Bao 56(1):95–100.
Machado-Vieira, R., Ibrahim, L., et al. (2012). Novel glutamatergic agents for major depressive disorder and bipolar disorder. Pharmacol. Biochem. Behav. 100(4):678–687.
Machado-Vieira, R., Manji, H.K., et al. (2009a). The role of the tripartite glutamatergic synapse in the pathophysiology and therapeutics of mood disorders. Neuroscientist 15(5):525–539.
Machado-Vieira, R., Yuan, P., et al. (2009b). Brain-derived neurotrophic factor and initial antidepressant response to an N-methyl-d-aspartate antagonist. J. Clin. Psychiatry 70(12):1662–1666.
Madaan, V., and Wilson, D.R. (2009). Neuropeptides: relevance in treatment of depression and anxiety disorders. Drug News Perspect. 22(6):319–324.
Mantyh, P.W. (2002). Neurobiology of substance P and the NK1 receptor. J. Clin. Psychiatry 63(Suppl 11):6–10.
Mathew, S.J., Murrough, J.W., et al. (2010). Riluzole for relapse prevention following intravenous ketamine in treatment-resistant depression: a pilot randomized, placebo-controlled continuation trial. Int. J. Neuropsychopharmacol. 13(1):71–82.
Mathew, S.J., Shah, A., et al. (2012). Ketamine for treatment-resistant unipolar depression: current evidence. CNS Drugs 26(3):189–204.
McClernon, F.J., Hiott, F.B., et al. (2006). Transdermal nicotine attenuates depression symptoms in nonsmokers: a double-blind, placebo-controlled trial. Psychopharmacology (Berl.) 189(1):125–133.
Merchant, R.E., Bullock, M.R., et al. (1999). A double-blind, placebo-controlled study of the safety, tolerability and pharmacokinetics of CP-101,606 in patients with a mild or moderate traumatic brain injury. Ann. NY Acad. Sci. 890:42–50.
Miller, A.H., Maletic, V., et al. (2009). Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 65(9):732–741.
Muhonen, L.H., Lonnqvist, J., et al. (2008). Double-blind, randomized comparison of memantine and escitalopram for the treatment of major depressive disorder comorbid with alcohol dependence. J. Clin. Psychiatry 69(3):392–399.
Murrough, J.W. (2012). Ketamine as a novel antidepressant: from synapse to behavior. Clin. Pharmacol. Ther. 91(2):303–309.
Murrough, J.W., Perez, A.M., et al. (2011). A case of sustained remission following an acute course of ketamine in treatment-resistant depression. J. Clin. Psychiatry 72(3):414–415.
Nagy, J. (2004). Renaissance of NMDA receptor antagonists: do they have a role in the pharmacotherapy for alcoholism? IDrugs 7(4):339–350.
Nestler, E.J., and Carlezon, W.A., Jr. (2006). The mesolimbic dopamine reward circuit in depression. Biol. Psychiatry 59(12):1151–1159.
Newhouse, P.A., Sunderland, T., et al. (1988). The effects of acute scopolamine in geriatric depression. Arch. Gen. Psychiatry 45(10):906–912.
Nugent, N.R., Tyrka, A.R., et al. (2011). Gene-environment interactions: early life stress and risk for depressive and anxiety disorders. Psychopharmacology (Berl.) 214(1):175–196.
Paez-Pereda, M., Hausch, F., et al. (2011). Corticotropin releasing factor receptor antagonists for major depressive disorder. Exp. Opin. Investig. Drugs 20(4):519–535.
Pariante, C.M., and Lightman, S.L. (2008). The HPA axis in major depression: classical theories and new developments. Trends. Neurosci. 31(9):464–468.
Parsons, C.G., Danysz, W., et al. (1999). Memantine is a clinically well tolerated N-methyl-d-aspartate (NMDA) receptor antagonist—a review of preclinical data. Neuropharmacology 38(6):735–767.
Petrakis, I.L., Limoncelli, D., et al. (2004). Altered NMDA glutamate receptor antagonist response in individuals with a family vulnerability to alcoholism. Am. J. Psychiatry 161(10):1776–1782.
Phelps, L.E., Brutsche, N., et al. (2009). Family history of alcohol dependence and initial antidepressant response to an N-methyl-d-aspartate antagonist. Biol. Psychiatry 65(2):181–184.
Philip, N.S., Carpenter, L.L., et al. (2010). Nicotinic acetylcholine receptors and depression: a review of the preclinical and clinical literature. Psychopharmacology (Berl.) 212(1):1–12.
Philip, N.S., Carpenter, L.L., et al. (2009). Varenicline augmentation in depressed smokers: an 8-week, open-label study. J. Clin. Psychiatry 70(7):1026–1031.
Pizzagalli, D.A. (2011). Frontocingulate dysfunction in depression: toward biomarkers of treatment response. Neuropsychopharmacology 36(1):183–206.
Potter, W.Z. (2012). New era for novel CNS drug development. Neuropsychopharmacology 37(1):278–280.
Preskorn, S.H., Baker, B., et al. (2008). An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-d-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J. Clin. Psychopharmacol. 28(6):631–637.
Price, R.B., Nock, M.K., et al. (2009). Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol. Psychiatry 66(5):522–526.
Pringle, A., Browning, M., et al. (2011). A cognitive neuropsychological model of antidepressant drug action. Prog. Neuropsychopharmacol. Biol. Psychiatry 35(7):1586–1592.
Pringle, A., Parsons, L., et al. (2012). Using an experimental medicine model to understand the antidepressant potential of the N-Methyl-d-aspartic acid (NMDA) receptor antagonist memantine. J. Psychopharmacol. 16:16.
Raison, C.L., Rutherford, R.E., et al. (2013) A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: The role of baseline inflammatory biomarkers. Arch. Gen. Psychiatry 70(1):31–41.
Salin-Pascual, R.J. (2002). Relationship between mood improvement and sleep changes with acute nicotine administration in non-smoking major depressed patients. Rev. Invest. Clin. 54(1):36–40.
Salvadore, G., Cornwell, B.R., et al. (2009). Increased anterior cingulate cortical activity in response to fearful faces: a neurophysiological biomarker that predicts rapid antidepressant response to ketamine. Biol. Psychiatry 65(4):289–295.
Salvadore, G., Cornwell, B.R., et al. (2010). Anterior cingulate desynchronization and functional connectivity with the amygdala during a working memory task predict rapid antidepressant response to ketamine. Neuropsychopharmacology 35(7):1415–1422.
Salvadore, G., van der Veen, J.W., et al. (2011). An investigation of amino-acid neurotransmitters as potential predictors of clinical improvement to ketamine in depression. Int. J. Neuropsychopharmacol. 1:1–10.
Schumann, G., Johann, M., et al. (2008). Systematic analysis of glutamatergic neurotransmission genes in alcohol dependence and adolescent risky drinking behavior. Arch. Gen. Psychiatry 65(7):826–838.
Smith, C. (2003). Drug target validation: hitting the target. Nature 422(6929):341,343,345 passim.
Tokita, K., Yamaji, T., et al. (2012). Roles of glutamate signaling in preclinical and/or mechanistic models of depression. Pharmacol. Biochem. Behav. 100(4):688–704.
Valentine, G.W., Mason, G.F., et al. (2011). The antidepressant effect of ketamine is not associated with changes in occipital amino acid neurotransmitter content as measured by [(1)H]-MRS. Psychiatry Res. 191(2):122–127.
Voleti, B., and Duman, R.S. (2012). The roles of neurotrophic factor and Wnt signaling in depression. Clin. Pharmacol. Ther. 91(2):333–338.
Wasserman, D., Wasserman, J., et al. (2010). Genetics of HPA-axis, depression and suicidality. Eur. Psychiatry 25(5):278–280.
Watkins, J.C., and Jane, D.E. (2006). The glutamate story. Br. J. Pharmacol. 147(Suppl 1):S100–S108.
Wiedemann, K. (2011). Biomarkers in development of psychotropic drugs. Dialogues. Clin. Neurosci. 13(2):225–234.
Zanicotti, C.G., Perez, D., et al. (2012). Mood and pain responses to repeat dose intramuscular ketamine in a depressed patient with advanced cancer. J. Palliat. Med.
Zarate, C.A., Jr., Brutsche, N., et al. (2012a). Relationship of ketamine’s plasma metabolites with response, diagnosis, and side effects in major depression. Biol. Psychiatry 18:18.
Zarate, C.A., Jr., Mathews, D., et al. (2012b). A randomized trial of a low-trapping nonselective N-methyl-d-aspartate channel blocker in major depression. Biol. Psychiatry.
Zarate, C.A., and Manji, H.K. (2008). Riluzole in psychiatry: a systematic review of the literature. Exp. Opin. Drug. Metab. Toxicol. 4(9):1223–1234.
Zarate, C.A., Jr., Singh, J.B., et al. (2006). A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am. J. Psychiatry 163(1):153–155.