41 | NEUROBIOLOGY OF FEAR AND ANXIETY: CONTRIBUTIONS OF ANIMAL MODELS TO CURRENT UNDERSTANDING
CHRISTOPHER K. CAIN, GREGORY M. SULLIVAN, AND JOSEPH E. LEDOUX
INTRODUCTION
Animal models are experimental preparations developed in one species for the purpose of studying phenomena occurring in another species (McKinney 2001). Since human studies are often impractical or unethical, researchers depend on animal models to study the neurobiological mechanisms of fear and anxiety and to evaluate new treatments, especially drugs. There are approximately 100 animal models of fear and anxiety in the literature. We will focus on the models that have contributed most to our current understanding of the neurobiological mechanisms underlying fear and anxiety, and those with the greatest potential to expand our knowledge of pathological anxiety.
Although we will not attempt to summarize the contribution of animal models to the discovery and development of new anxiolytic drugs, it will become apparent that researchers in industry and academia have largely focused on different animal models. We will argue that this strategy, although useful for a time, is now unnecessary and may be impeding progress for both groups. The path forward, in our opinion, is to focus on animal models amenable to studies of the brain circuits and neural processes that mediate threat processing and defensive responding. This may be essential for advancing our understanding of neurobiological processes relevant to anxiety, identifying the causes of pathological anxiety and predicting how drugs may influence these relevant circuits and dysfunctional processes.
LANGUAGE AND CONCEPTUAL FRAMEWORK
A challenge for research on animal models of human psychiatric disorders is developing an appropriate and useful language to relate preclinical and clinical findings. On the one hand, animal models are only useful to the extent that they resemble a human problem, and similar language can improve communication between clinicians and preclinical researchers. On the other hand, these models are limited by differences in the brains of humans and other animals, and imprecision in nomenclature can complicate interpretation and application of preclinical findings. It is particularly important to be clear about which aspects of human anxiety can be studied using animal models. It will also be helpful to define a general conceptual framework for discussing the relationship between fear, anxiety, and animal models of human disorders.
DEFENSIVE RESPONDING, THREAT PROCESSING, AND FEELINGS
Criteria for diagnosing anxiety disorders often use terms that refer to emotional symptoms, such as afraid, worried, stressed, anxious, distressed, or concerned (DSM-IV-TR 2000). Human anxiety disorders are defined by such emotional symptoms, along with behavioral and physiological symptoms. Because emotional symptoms involve feelings, subjective internal states of consciousness, that are not observable and can only be communicated through language, they are difficult to approach scientifically. Animals with less complex brains are unlikely to experience feelings in the way humans do, and do not possess language to relay feelings. It is therefore extremely important that we resist the temptation to anthropomorphize and view animal models as a tool to reveal mechanisms of feelings; they cannot. Among the perils of such, this can lead to overinterpretation of preclinical results and/or false rejection of animal models that fail to predict human feelings.
A more productive route is to focus on evolutionarily conserved brain systems that detect and respond to threats or actual harm. These systems control the physiological and behavioral defensive responses (DRs) that contribute to anxiety disorder symptoms when dysfunctional. For instance, humans and rats exhibit many of the same behavioral (e.g., freezing, fight, escape), autonomic (e.g., cardiovascular or respiratory), and endocrine (e.g., hormone release) responses when in danger. Core components of the underlying neurocircuitry also show a striking degree of overlap. For example, in both species, the amygdala plays a crucial role in detecting and responding to threats, the hippocampus provides critical contextual information, and the ventromedial prefrontal cortex (vmPFC) can suppress threat responses. The accompanying cellular, hormonal, and molecular processes involved are also remarkably similar. Thus, it is appropriate to model human anxiety-related DRs in other mammals because they are similar, controlled by conserved networks that function unconsciously, and are objectively observable (LeDoux, 2012). Further, human feelings of fear and anxiety depend on DRs. This will be the focus of the chapter. When we have occasion to refer to conscious feelings, these will labeled as fearful feeling, conscious feeling, and so on. In order to make a subtle but important distinction, we will use the term threat when referring to responses to cues that predict harm, whereas the broader term defensive will refer to responses triggered by threats or actual harm.
TOPOGRAPHY OF DEFENSIVE RESPONDING
Fear and anxiety are used in many ways, sometimes interchangeably but sometimes to label distinct reactions to aversive stimulation. Fear is usually used to denote a state elicited by clearly defined environmental threats, either innately aversive or learned. This fear state functions to cope with immediate threats and is more intense than anxiety, but also shorter lived. For example, rodents will often freeze when threatened, but freezing subsides quickly when the threat is removed. Anxiety often describes threat responding elicited by more diffuse cues. Anxiety states can last longer than fear and function to cope with more distant, or poorly defined, threats. For example, rodents hug the walls in an unfamiliar or brightly lit environment (thigmotaxis), a behavior that likely evolved to thwart detection by predators. Since they often operate on very different time scales, fear responses can be more reflexive, whereas anxiety can include more complex cognitive processes exemplified in humans by anticipation and worry.
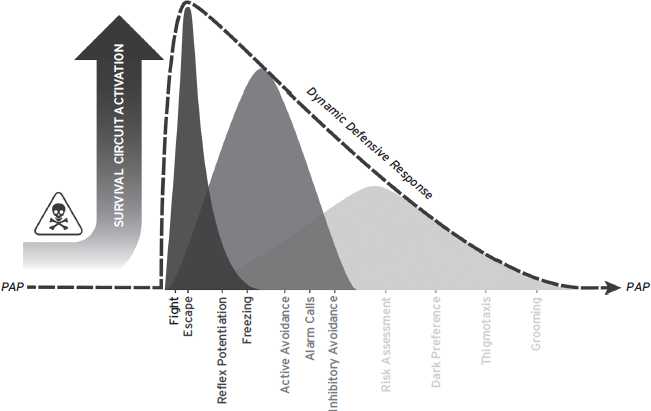
Although fear and anxiety often refer to different aspects of threat responding, sometimes mediated by different brain regions, they are not always clearly dissociable in natural situations or laboratory experiments. The distinction is clouded further when one considers drug classifications or diagnostic criteria for human disorders. Therapeutic agents are routinely categorized as anxiolytic/antianxiety with no category for antifear drugs. Further, although most consider anxiety states to be weaker than fear, many human anxiety disorders are defined by the intensity and form of fear responses. Thus, in addition to anthropomorphic perils, the terms fear and anxiety fail to clearly define alternate mental states with distinct response profiles. Functional behavior system (FBS) approaches may provide a better framework for considering threat-related defensive responding. Rather than attempt to divide fear and anxiety into separate phenomena, FBS approaches assume that both result from activation of defense or survival circuits that evolved to protect organisms from harm, especially predators (Blanchard et al., 1989; LeDoux, 2012). For instance, predatory imminence theory (PIT) suggests that DRs are arranged hierarchically along a continuum, and the particular responses elicited depend on the proximity of the threat (Timberlake and Fanselow, 1994). In PIT, threats activate DRs and divert animals from their preferred activity pattern. DRs at the low end of the spectrum, like thigmotaxis, are elicited when danger is possible but not imminent. DRs at the high end of the spectrum, like fighting or escape, are elicited by imminent threats, like an attacking predator. DRs function to protect the animal from threats or actual harm, and return the animal to its preferred activity pattern.
FBS approaches are useful because they remove ambiguity in other anxiety/fear distinctions but still capture much of the nuance implicit in other definitions. They nicely explain the progression of defensive responding as threats escalate. Threats that are unlikely, poorly defined, or distant serve to weakly activate survival circuits, resulting in relatively minor deviations from the preferred activity pattern. As threats become more likely, better defined, and closer, survival circuit activation increases, and animals deviate further from their preferred activity pattern. Ultimately, the strongest DR, defensive fighting, is elicited when all earlier responses fail to prevent threat escalation. It should be apparent that weaker DRs can occur over a much greater time frame whereas stronger DRs are often reflexive reactions to immediate threats (Fig. 41.1).
Before proceeding, a brief discussion of triggering stimuli and DR forms is warranted. Some triggering stimuli are innate; however, plasticity in survival circuits allows novel stimuli to gain control of DRs through experience. DRs also reflect an interaction between survival circuit activation and environmental options. For example, animals cannot flee in small laboratory testing chambers with no exits. Lastly, although mammalian DRs are remarkably similar in function and form (Blanchard et al., 1989), the exact behaviors are usually species specific (so-called species-specific defense responses or SSDRs; Timberlake and Fanselow, 1994). This simply means that a mouse will exhibit caution, freeze, flee, or fight differently than a monkey or human. Thus, although survival circuits are highly conserved neural systems for detecting and responding to danger, exact responses are usually unlearned, hard-wired reactions determined by evolution within a particular niche.
ANXIETY MODELS AMENABLE TO NEUROCIRCUIT ANALYSIS
Not all models are amenable to circuit analysis. Animal models can only contribute to our understanding of precise neurobiological mechanisms of defensive responding if a neural circuit mediating specific DRs can be identified and exploited. In line with the FBS approach outlined in the previous section, models amenable to neurocircuit analysis take advantage of discrete, well-defined triggering stimuli and innate, stereotyped DRs. This anchors the behavior to a neural system at the entry to, and exit from, the central nervous system. Models in which the stimulus conditions are diffuse or the responses lack stereotyped expression provide less guidance for studies that seek to identify key structures between the sensory and motor systems that underlie DRs.
Circuits are identified using a number of techniques. For instance, various lesions can demonstrate the necessity of specific brain regions. Disconnection lesions are particularly useful for showing that two different brain regions operate in series within a circuit to mediate a specific behavior. Extracellular recording of single neuronal units in awake, behaving animals is also extremely valuable for identifying information flow in neurocircuits. Other techniques, such as local infusion of drugs, brain stimulation, intracellular recording, or imaging of activity-dependent gene expression and/or energy metabolism, can provide converging evidence for the existence of a defined behavioral neurocircuit. Here it becomes evident why discrete triggering stimuli and stereotyped responses are so important. By relating the timing of neural responses to the occurrence of stimuli and DRs, one can track information flow through a circuit beginning with stimuli that activate the circuit and ending with neural activity causing precise behavioral responses (Fig. 41.2). Once a circuit is identified, the role of specific brain areas, cells, synapses, molecules, and genes can be elucidated. This strategy, pioneered in studies of invertebrates (e.g., Bailey et al., 2000), has been successfully applied to studies of mammals (Davis et al., 1982; Medina et al., 2002).
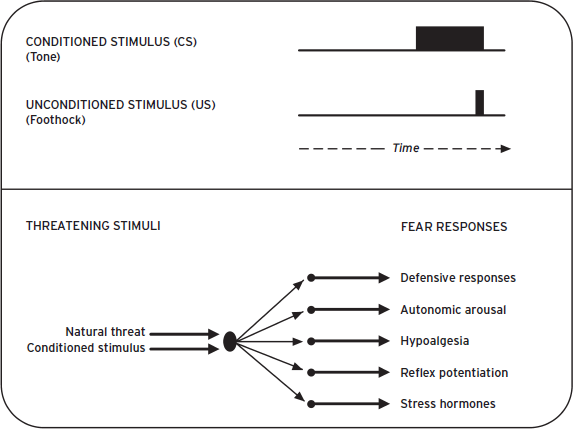
Figure 41.1 Threats trigger survival circuit activation and initiate defensive responding. Defensive responses function to prevent harm, minimize exposure to threats, and return the organism to its preferred activity pattern (PAP). DRs are organized hierarchically, and more imminent threats lead to greater survival circuit activation; more intense, reflexive DRs; and more total time away from the PAP.

Figure 41.2 Examples of animal models amenable to neurocircuit analysis. (A) Acoustic startle is an innate, and extremely fast, defensive reflex (8 ms from sound onset to initiation of muscular response). Once lesion studies identified components of the underlying circuit, neural stimulation studies helped establish the order of these nuclei within the circuit, by measuring the latency of startle responding following stimulation (Davis et al., 1982). VCN: ventral cochlear nucleus; LL: lateral lemniscus; RPC: nucleus reticularis pontis caudalis; MLF: medial longitudinal fasciculus; SC: spinal cord. (B) Pavlovian threat processing involves rapid sensory processing and amygdala activation. Recording of CS-evoked neural activity helped identify this core component of the mammalian survival circuit. Again, latency measures helped establish the order of nuclei within the circuit. aThal: auditory thalamus; aCtx: auditory cortex; LA: lateral amgyala; BA: basal amygdala; CE: central amygdala. (C) A very precise neural circuit has been identified for Pavlovian eyeblink conditioning in the cerebellum, and postconditioning, CS-evoked activity in the interpositus nucleus initiates eyeblink CRs (Medina et al., 2002).
NORMAL VERSUS PATHOLOGICAL FEAR AND ANXIETY
Fear and anxiety are normal, adaptive responses to threatening environmental challenges. When threats activate survival circuits, behavior is restricted to responses compatible with SSDRs, and autonomic and endocrine processes divert bodily resources away from nonessential processes (e.g., digestion) and toward systems needed to execute defense (e.g., musculature and brain). Ideally, the degree of activation is proportional to the threat, and an adaptive, or normal, response will subside soon after the threat is gone. This can be illustrated by the contribution of corticosteroids to defensive responding. Aversive stimuli can trigger an endocrine stress response. This includes the rapid release of corticosteroids into the peripheral circulation, which has a wide variety of effects on bodily processes, including (1) increasing blood glucose and blood pressure, (2) enhancing perception and memory, and (3) suppressing nonessential immune and reproductive functions. Corticosteroids, through a hypothalamic-pituitary-adrenal (HPA) axis feedback loop, also play a crucial role in limiting the duration of the stress response. The system is rapidly activated to deal with danger and then turned off when the threat is gone. Thus, when functioning properly, threat processing circuits promote survival and maximize time in the preferred activity pattern.
Pathological fear and anxiety can refer to either impaired or facilitated threat responding. For instance, patients with amygdala damage have deficits detecting and responding to threats. Some, like patient S. M., who has no amygdala function due to Urbach-Weithe disease, reportedly also fail to experience fearful feelings (Feinstein et al., 2011). Impaired amygdala function has real-life consequences; S. M. has a history of being victimized and abused, likely a result of her inability to recognize dangerous situations and respond appropriately.
Although rare disorders can produce defensive systems that are pathologically hyporesponsive, the vast majority of anxiety disorders involve systems that are hyperresponsive (Rosen and Schulkin, 1998). Thus, pathological fear and anxiety usually refer to threat responding that is exaggerated, inappropriate, or prolonged (Fig. 41.3). Exaggerated refers to DRs that are stronger than the situation requires. This could mean responding with full-blown stress responses in unfamiliar environments when only vigilance is warranted. Or perhaps responding to an unlikely threat as if it were 100% guaranteed to happen. Inappropriate DRs are triggered by stimuli that are not actually dangerous. Prolonged refers to DRs, including HPA axis activation, which persist well after the threat is diminished. Together, these factors describe states of fear and anxiety that are more intense, more frequent, and more often present in daily life.
ANIMAL MODELS OF FEAR AND ANXIETY
Since human anxiety disorders are varied and complex, animal models do not typically attempt to mimic full DSM syndromes (McKinney, 2001). Rather, they are adapted to specify and study key components of clinical syndromes. Researchers have approached this problem in many different ways, with varying degrees of success. While much has been learned about threat processing, defensive behavior, and learning in studies of invertebrates and other mammals (Bailey et al., 2000; Kalin, 2004), research on rodents has arguably contributed the most systematic and detailed information and will be the focus here. Ultimately, animal models are judged based on how well they reveal neurobiological mechanisms contributing to human anxiety and how well they predict the efficacy of treatments.
VALIDITY OF ANIMAL MODELS
Validity refers to the degree that a given model is useful for some purpose (McKinney, 2001; Willner, 1991). Face validity refers to phenomenological similarities between the model and the human psychiatric condition. Animal models have a high degree of face validity when etiological factors, symptoms, underlying physiology, and treatment effects are similar to the human disorder. Construct validity refers to the theoretical rational for linking a process in a model to a process hypothesized to be important in a human disorder. Due to shared aspects of their phylogenetic histories, humans and other animals are often homologous at molecular, cellular, synaptic, and circuitry levels of analysis. Finally, a model is considered to have good predictive validity when it can anticipate similar effects in humans. In the realm of drug development, predictive validity is the ability to identify and rank drugs for therapeutic efficacy in humans.
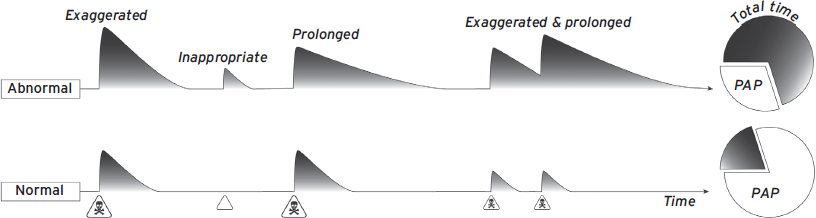
Figure 41.3 Schematic representation of pathological defensive response profiles. Note that abnormalities reduce the amount of time spent in the preferred activity pattern (PAP).
Validity arguments are fundamentally epistemological and should not be used as the sole basis for evaluating the usefulness of an animal model. This is summed up nicely here:
Animal models are tools for our use: They are not developed as part of a beauty contest, with a prize for the most convincing. If a model cannot readily be used, it is of little value, however elegant. Thus, the successful construction of a valid model should not be seen as an end in itself, but as a useful step in the investigation of a scientific problem. As such, validity can be assessed only in relation to the broader objectives of the research program.
WILLNER (1991)
This last point is important. Different models may be useful for different purposes, depending on scientific and practical considerations. For instance, a model for rapid screening of drugs may have great predictive validity but little obvious construct validity. However, this could still be useful for developing new treatments, even if it may not be useful for unraveling neurobiological mechanisms of human anxiety. Thus, validity assessments are judgments made with appropriate context and based on available data, not exact measurements.
STATE VERSUS TRAIT ANXIETY
State anxiety is a temporary condition that is elicited by a stimulus or situation. Trait anxiety is an enduring condition that occurs across multiple situations or tests but is not necessarily pathological. Usually, tasks that assess trait anxiety are more valuable, as human disorders, and the rodent equivalents, are thought to be fairly stable. Unfortunately, many unconditioned animal models assess state anxiety, as is suggested by the relatively poor reliability in individual performance within or between tasks (Andreatini and Bacellar, 2000).
NORMAL VERSUS ABNORMAL RESPONDING
Most animal models evaluate “normal” fear and anxiety processes, where subjects are grouped and effects are reported as variation around a mean. Preexisting variability, genetic or otherwise, is averaged out. Evaluation of normal behavior is important; however, these studies may not adequately model the processes that cause human anxiety disorders. Anxiety disorders affect only a portion of the population, and for specific disorders like PTSD, only a subset of traumatized individuals develop pathological anxiety (Yehuda, 2007). Thus, animal models that examine abnormal responding, or outliers, may be of particular value. Researchers approach this problem using two basic strategies: (1) by performing standard assays but focusing on individuals with abnormal responses that mirror human pathology or (2) by introducing manipulations that skew group averages toward an abnormal response profile. Examples of these strategies will be discussed later.
ANIMAL MODELS OF NORMAL FEAR VERSUS ANXIETY
Animal models can be roughly divided into those that have an explicit learning component, and those that do not. Here we will distinguish between unconditioned and conditioned anxiety models. Note, however, that this terminology is not perfect; some unconditioned assays are referred to as spontaneous, unlearned, or innate anxiety in the literature (Takahashi et al., 2008), and some models involve nonassociative sensitization learning, which is not commonly considered “conditioning.” Where relevant, these distinctions will be pointed out.
UNCONDITIONED MODELS
Unconditioned models examine DRs elicited by a novel test situation, not cues associated with prior aversive experience (Blanchard et al., 1989; Takahashi et al., 2008). These assays utilize ethologically relevant behavioral end points that are thought to be sensitive indices of an animal’s natural anxiety/fear. Although there are many different unconditioned models of fear and anxiety, in the next section we discuss more commonly used rodent assays.
Innate Aversion to Light or Open Spaces
Rodents are nocturnally active small prey animals, and several popular unconditioned assays capitalize on their natural tendency to avoid brightly lit, open spaces. These assays elicit DRs akin to the preencounter behaviors of PIT, which likely evolved to protect animals in uncertain or weakly threatening situations. It has been argued that these weaker DRs reflect a degree of survival circuit activation that, in humans, corresponds to worry or mild anxiety (Craske, 1999; Rau and Fanselow, 2007). Examples are the elevated plus maze (EPM), light/dark, and defensive withdrawal tests. These assays are popular because they are easy to run, are cost-effective, require no learning phase, and reliably predict anxiolytic activity of some drugs (e.g., benzodiazepines) in humans. However, the imprecise nature of triggering stimuli (brightness/openness with no clear onset/termination) and resultant DRs (e.g., percentage of time spent in bright/open area, usually over many minutes) make it very difficult to define the underlying neural circuit. Further, these assays are sensitive to subtle variations and often produce inconsistent findings (Hogg, 1996). Perhaps most problematic, they often fail to detect activity of nonbenzodiazepine anxiolytics (Kehne and Cain, 2010).
Light-enhanced startle is a related assay, but it has an advantage over those to the preceding because it is amenable to circuit analysis (Davis et al., 1982). In brief, this assay examines how an innate aversion to bright light can potentiate defensive reflexes by influencing neural processing in the well-characterized acoustic startle circuit (see Fig. 41.2). Light-enhanced startle has helped implicate specific brain regions (e.g., bed nucleus of the stria terminalis, or BNST), and molecules (e.g., corticotropin-releasing factor type 1, or CRF1, receptor), in the mediation of anxiety (Walker et al., 2009).
Social Behaviors
The unconditioned social interaction test measures the tendency of one animal to investigate and interact with a novel conspecific when the two are placed in a closed, brightly lit arena (File and Seth, 2003). The separation-induced ultrasonic vocalization model in rat pups is another example. In both tests the eliciting stimulus and resultant response are social in nature. Social anxiety assays have been successfully used to rapidly screen and characterize anxiolytic agents with diverse mechanisms of action, showing some superiority over alternative unconditioned anxiety models. The social interaction test has also been reliable enough to implicate some general brain areas in anxiety, such as the amygdala, hippocampus, and brain stem neuromodulatory centers. However, no precise neural circuit has emerged that would allow for detailed analyses of cellular, synaptic, and molecular mechanisms for these effects. Subtle procedural variations can also cause inconsistent results between laboratories.
Antipredator Models
Given that DRs evolved primarily to protect against predation, stimuli associated with predatory animals are also useful for modeling fear and anxiety (Blanchard et al., 1989; Takahashi et al., 2008). In these assays, predator stimuli are presented to rodents, and DRs ranging from defensive fighting down to weaker responses like risk assessment are measured. Pharmacological studies of antipredator responses have been conducted; however, these assays are primarily used to map the neural circuits underlying innate fear/anxiety, often for comparison with the circuitry mediating conditioned threat responses. Gene expression and lesion studies suggest that unconditioned responses to predator cues depend on slightly different brain regions (e.g., medial amygdala, BNST) from conditioned responses. These assays may be particularly useful for modeling ethologically relevant human phobias (Rosen, 2004).
Summary of Unconditioned Models
Unconditioned fear/anxiety assays are popular for evaluating drugs with anxiolytic potential. However, they may not be ideal models of human psychopathology because most assess state, rather than trait, anxiety, and they can be sensitive to very minor procedural variations (e.g., time of day, lighting conditions). Perhaps more problematic, they are not amenable to circuit analysis and neurobiological studies. The light-enhanced startle and antipredator assays are exceptions. These are unique because response triggers and DRs can be discrete and precisely defined, thus allowing for examination of underlying circuits and neural mechanisms. For instance, light-enhanced startle takes advantage of the known acoustic startle circuit and evaluates how anxiogenic manipulations interact to facilitate defensive reflexes. Antipredator studies have been similarly successful in defining differences between the brain regions mediating learned versus unlearned fear/anxiety. Together unconditioned assays have implicated the extended amygdala, septohippocampal system, PFC, and various brainstem neuromodulator centers (e.g., locus coeruleus or raphe nuclei) in anxiety-like responding (Gray and McNaughton, 2000; Karpova et al., 2011; Knapska et al., 2007), though precise neurobiological mechanisms are still largely unknown.
ASSOCIATIVE CONDITIONING MODELS
Brain systems that detect and respond to threats are not simply innately wired stimulus–response mechanisms; they are also plastic, allowing organisms to learn new predictors of danger. Learning can involve nonassociative and/or associative plasticity. Nonassociative learning occurs when repeated exposure to a single stimulus leads to stronger (sensitization) or weaker (habituation) responding. Both sensitization and habituation have been studied in relation to anxiety, however, usually as modulators of associative responses. We therefore emphasize associative learning processes here.
Associative learning is critical for establishing DRs to novel/innocuous stimuli, and for learning to cope with, or suppress, these responses. Two classes of associative learning with particular relevance to fear and anxiety are Pavlovian threat conditioning (PTC) and aversive instrumental conditioning. We refer to conditioned threats instead of the traditional conditioned fears for reasons outlined earlier in the Defensive Responding, Threat Processing, and Feelings section and elsewhere (LeDoux, 2012). PTC is particularly important for many reasons; the conditions for learning are simple, learning is rapid and long-lasting, a basic neurocircuit has been identified, and a great deal is known about the underlying synaptic, cellular, and molecular processes. PTC may also be a prerequisite for other critical forms of anxiety-related learning such as avoidance. Finally, PTC is a powerful tool for investigating how established pathological memories, and/or responses, may be regulated or suppressed through new learning.
Pavlovian Threat Conditioning
Behavioral Aspects of PTC
When neutral conditioned stimuli (CSs) are temporally paired with naturally aversive unconditioned stimuli (USs) PTC occurs (Fig. 41.5). Any sensory stimulus can serve as a CS, including social and complex configural cues like contexts, and USs can be any unpleasant or painful stimulus. Auditory CSs and footshock USs are commonly employed in laboratory experiments partly because researchers can precisely control the delivery of these stimuli. Prior to conditioning, subjects respond weakly to the CS. After conditioning, CS-alone presentations elicit a cassette of DRs including freezing, autonomic reactions, neuroendocrine responses, as well as potentiation of somatic reflexes like startle and eyeblink, collectively referred to as conditioned fear/threat responses (CRs; Fig. 41.4). CRs indicate that associative learning took place, provided that similar responses fail to occur when the CS and US are not paired (unpaired) during conditioning. Although CRs can sometimes resemble unconditioned responses to the US (e.g., startle), CRs nearly always take a different form. CRs can be generated with as little as one CS–US pairing and can last a lifetime. PTC also establishes the CS as secondary incentive that can support instrumental avoidance and higher order Pavlovian conditioning (discussed later).
PTC Neurocircuitry
The amygdala is a core component of the mammalian threat-processing circuitry and is essential for learning, storing, and expressing PTC memories (Cain and LeDoux, 2008) (Fig. 41.5). It is composed of a dozen or so bilateral nuclei that reside in the temporal lobes. Of these, three have been the focus of much of the research on PTC: the lateral (LA), central (CE), and basal (BA) nuclei. LA neurons receive converging auditory (CS) and somatosensory (US) information from thalamic and cortical processing regions as early as 12 ms following stimulus onset. LA neurons in turn communicate with CE both directly and indirectly via projections to BA and intercalated cells. Although CE contributes to learning and memory (Gozzi et al., 2010; Haubensak et al., 2010; Wilensky et al., 2006), CE is best known for its role in controlling expression of Pavlovian CRs (Ciocchi et al., 2010; Johansen et al., 2011; Knobloch et al., 2012). BA appears to have a complex role in the expression of CRs that is only beginning to be understood (Amano et al., 2011; Herry et al., 2010). Because the role of LA is best understood, it will be the focus of the discussion here.
Figure 41.4 Protocol for inducing Pavlovian threat conditioning and common defensive (fear) responses.

Figure 41.5 Core neural circuit mediating Pavlovian threat conditioning and extinction. Pavlovian freezing is used as an example for simplicity. (Left) The amygdala plays a critical role in detecting and responding to conditioned threats. Postconditioning, threats activate LA neurons, which in turn activate CE both directly and indirectly (via projections to BA, or to the intercalated cell masses which causes disinhibition of CE). CE outputs project to downstream effector regions (e.g., ventral PAG) that mediate specific DRs (e.g., freezing). Hippocampal inputs to BA provide contextual information, and prelimbic-PFC neurons help sustain conditioned responding over longer intervals. (Right) Extinction learning counteracts threat responding in at least three ways: (1) by strengthening feedforward inhibition in LA, (2) through infralimbic-PFC activation of intercalated cells, and (3) through a subset of BA cells that project to CE interneurons and inhibit CE output. Hippocampus plays a critical role in gating extinction according to context and via connections to il-PFC and BA extinction neurons.
LA is essential for learning, consolidation, expression, reconsolidation, extinction, and many other aspects of PTC (Cain and LeDoux, 2008; Johansen et al., 2011). Several recent findings provide especially strong support for the central role of LA in PTC. For instance, using optogenetics we recently demonstrated that artificial PTC memories could be created in rats by pairing standard auditory CSs with depolarization of LA neurons (Johansen et al., 2010). CS presentations after training elicited freezing, even though rats never received any footshocks. Although freezing was weaker than observed after real conditioning with footshock USs, that the rats froze at all was a testament to the importance of LA for PTC learning. Further, lesions of LA conducted 16 months after PTC, nearly the entire adult lifetime of the rat, severely disrupt conditioned freezing (Gale et al., 2004). Finally, using a clever combination of molecular tools, researchers recently demonstrated how a subset of LA neurons (~20%) outcompete their neighbors to store PTC plasticity, and were able to identify and erase PTC memories by ablating only those neurons (Josselyn, 2010). Together these findings firmly implicate LA in the learning, storage, and expression of PTC memories.
Extra-amygdala regions also make important contributions to PTC learning and expression. The hippocampus plays an important role in conditioning to more complex contextual cues, most likely through CS-related projections to BA (Maren and Fanselow, 1995). However, hippocampus plays a time-limited role in expression of contextual CRs; systems-level consolidation processes transfer this memory to more cortical brain regions like the anterior cingulate (Teixeria et al., 2006). Prelimbic PFC also appears to help maintain expression of CRs like freezing even after short-latency neural responses in amygdala adapt during CS presentations (Burgos-Robles et al., 2009).
Synaptic Plasticity in LA
Associative pairing of the tone and shock induces synaptic plasticity between CS afferents and LA neurons (for review, see Johansen et al., 2011). PTC also results in synaptic plasticity in structures afferent to the LA (e.g., thalamus, cortex; Quirk et al., 1997; Weinberger, 1995). However, these are unlikely to be essential for threat conditioning at the level of behavior for three reasons: (1) inactivation of the LA prevents learning and memory, indicating that these structures alone cannot support PTC, (2) plasticity in these afferent structures appears to depend on LA function, and (3) conditioning-related plasticity in LA emerges before changes in afferent regions. Together these findings suggest that PTC changes the way a CS is processed in an emotional circuit involving LA, and this plasticity allows the CS to control expression of DRs after the aversive experience (Cain and LeDoux, 2008; Fig. 41.6).
Molecular Mechanisms of PTC in LA
With mounting anatomical, physiological, and behavioral evidence implicating the LA in PTC, many researchers have focused their efforts on deciphering the molecular signaling cascades important for learning/memory in this region. The majority of studies employ genetic and pharmacological manipulations coupled with PTC to determine the function of specific molecules. Manipulations carefully timed with respect to training and testing allow researchers to distinguish between involvement in learning, short-term memory (STM), and long-term memory (LTM) processes. Related studies have also probed the molecular mechanisms of LTP, usually using in vitro brain slice preparations while stimulating sensory afferents and recording in the LA. However, we will omit coverage of in vitro LTP as this topic has been covered in detail elsewhere and the results are generally in agreement with in vivo manipulations. A detailed review of the large body of molecular work related to PTC is beyond the scope of this chapter (for review, see Johansen et al., 2011); however, we will highlight the contributions of a few key molecular players to illustrate how long-lasting plasticity between sensory afferents and LA neurons is achieved.
There appear to be several important molecular stages to PTC-related plasticity in LA. First, receptors and ion channels at the synapse translate presynaptic activity into postsynaptic activation of signaling cascades by elevating intracellular calcium concentrations. NMDA (N-methyl-d-aspartate) receptors, L-type calcium channels, and metabotropic glutamate receptors are crucial for this process. αCaMKII (calcium/calmodulin-dependent protein kinase two) is particularly important for short-term memory/plasticity and may covalently modify existing synaptic proteins, like the alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor, to facilitate glutamatergic transmission. PKA (protein kinase A) and MAPK (mitogen-activated protein kinase) are important for long-term memory/plasticity. When activated, they translocate to the nucleus to stimulate CREB (cAMP-responsive element binding protein)-mediated transcription. Long-term changes in synaptic transmission are ultimately achieved by transcription of the appropriate genes, translation of new proteins, and incorporation of these new proteins at the synapse. For instance, the production and synaptic insertion of new AMPA receptors are critical for PTC (Rumpel et al., 2005). Transcription and translation may also help form new synapses between sensory afferents and LA neurons (Ostroff et al., 2010). Together, molecular work in the PTC pathway demonstrates that synaptic receptors/channels, intracellular kinases, and nuclear machinery respond to CS–US pairings in a coordinated fashion to change CS processing in the LA and allow new stimuli to control defensive responding.
Exciting new work illustrates how molecular alterations in amygdala networks can alter threat processing and possibly contribute to human psychopathology. For instance, the discovery of an acid-sensing ion channel in LA that detects CO2-related changes in pH and elicits DRs has exciting implications for understanding neuropathology in panic disorder, since CO2-enriched air can precipitate panic attacks (Ziemann et al., 2009). And other studies demonstrate that allelic variants in genes affecting catecholamine catabolism (COMT) and serotonin reuptake (5-HTT) strongly predict hyperresponsiveness of defense-related neurons and behavior, including those responsible for threat conditioning, consistent with the notion of stable trait anxiety (Domschke and Dannlowski, 2010).
Variations of the PTC Procedure
he simple PTC procedure described in the preceding is the most common tool for studying fear and anxiety reactions. It is simple because it usually involves discrete, unimodal CSs and USs with few pairings (<10) given with 100% contingency (all CS and US presentations are paired). There are countless variations on the PTC procedure that could help elucidate specific aspects of fear and anxiety. Here we will mention just a few.
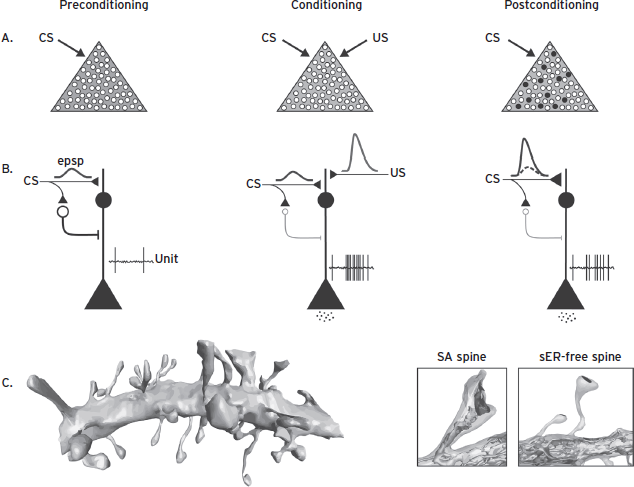
Figure 41.6 The identification of a neurocircuit has greatly facilitated studies of cellular processes important for defensive learning. For instance, approximately 20% of LA neurons outcompete their neighbors during conditioning, via a CREB-dependent mechanism, to learn and store PCT plasticity (A). Prior to conditioning, CS presentations result in weak depolarization of LA neurons and little to no activation of downstream brain areas mediating DRs. However, with CS–US pairings, neurons are strongly depolarized, resulting in initiation of an LTP-like process that strengthens the synapses between CS afferents and LA neurons. Following conditioning, CS presentations strongly depolarize LA neurons, trigger action potentials, and release neurotransmitters to activate downstream brain areas mediating expression of DRs. Recent work suggests the importance neuromodulators like dopamine and norepinephrine to the induction PTC plasticity in LA, both by direct action on excitatory cells and by suppression of feedforward GABAergic inhibition (Ehrlich et al., 2009) (B). Identification of this critical component of the neurocircuit has also allowed for detailed anatomical analyses of LA dendrites (C). For instance, studies using serial electron microscopy to reconstruct segments of LA dendrites after conditioning suggest that larger and more stable spines (SA spines) increase in frequency following conditioning and smaller (smooth ER-free) spines increase postsynaptic density area.
The standard PTC procedure produces robust freezing in rodents, suggesting that CSs can strongly activate survival circuits and produce relatively high-level DRs, similar to those associated with human fear. Recent studies have attempted to model the uncertainty of anxiety more effectively by reducing CS/US contingency, increasing the CS–US delay, or by introducing an interval between the end of the CS and the US. Other variations use complex cues to signal the US, such as contextual stimuli. Much less is known about the neurobiology of these phenomena, but they are promising approaches for unraveling the neural mechanisms of anxiety and structures such as the BNST, hippocampus, and ACC, which have been implicated (Waddell et al., 2006; Walker et al., 2009).
Still another PTC variation assesses how aversive CSs suppress other nondefensive responses such as bar pressing for food. The most common of these assays is called conditioned suppression or the conditioned emotional response (CER) test. These assays are more complicated, require much more training, and have the added layer of food/water deprivation that can muddle interpretation of results. The neural mechanisms of CER and simple PTC are similar, but there are subtle differences. Recently it has been suggested that conditioned suppression is a unique form of Pavlovian-instrument transfer (Balleine and Killcross, 2006) and thus may not be an ideal way to assess PTC directly.
Studies of retrieval-induced learning may also explain the development of some key anxiety disorder symptoms and suggest novel strategies for treatment. As in PTC, cues associated with trauma can reactivate traumatic memories. Such reminders can be severely disruptive, triggering intrusive images and thoughts that accompany a progressive worsening of anxiety symptoms, especially for memory-based disorders like PTSD (Southwick et al., 1999). Preclinical studies indicate that retrieval renders fear memories labile, probably to allow for the incorporation of new information, which is followed by a new consolidation process (reconsolidation). There is ample evidence in the literature that retrieved memories can be reconsolidated in a stronger (fear incubation) or weaker state (Cain et al., 2012). Models that exploit these processes to examine new treatment options will be discussed later.
Summary of Pavlovian Threat Conditioning
PTC has arguably enjoyed the most success as an animal model of fear and anxiety. The PTC model has good face, construct, and predictive validity, at least in terms of normal human fear and anxiety. The ability to identify and dissect a basic neural circuit has undoubtedly contributed to this model’s success. Procedural variations may even allow for studies of weaker, anxiety-like DRs that were previously thought to require unconditioned anxiety models like the EPM. This system, along with variations in the procedure and/or additional learning phases that depend on PTC, may hold the most potential for modeling key aspects of human pathological anxiety as well.
Instrumental Avoidance Conditioning
Unlike PTC, where subjects learn about relationships between environmental stimuli and react accordingly, instrumental paradigms allow the subjects to control exposure to aversive stimuli. There are many variations on the instrumental theme, but here we will focus on two common assay types: passive or inhibitory avoidance (IA) and active avoidance (AA). Avoidance is a hallmark of many human anxiety disorders; however, like fear and anxiety, avoidance mechanisms evolved because they were adaptive and helped protect organisms from threats. But excessive avoidance may interfere with normal functioning and become maladaptive. Here we will discuss normal avoidance and summarize what is known about the underlying neurobiology. Later sections will examine how avoidance can become maladaptive.
Inhibitory Avoidance
IA learning involves withholding a response that was previously punished, typically by shock. Although PTC undoubtedly occurs in these situations, the animal has control over exposure to the shock and shock-paired cues; thus, IA is instrumental in nature. There are many obvious parallels between IA and human psychopathology in the realm of anxiety. For example, agoraphobics inhibit actions that expose them to places in which escape would be difficult or embarrassing. Acrophobics inhibit actions that expose them to heights. Some unconditioned assays discussed earlier (e.g., EPM) may also be considered inhibitory avoidance of innately aversive stimuli. Conflict paradigms, like the Vogel test, also fall into this category. In these assays, rats are punished for appetitive actions (e.g., licking a water spout) with US presentations (e.g., shock). Conflict tests are billed as anxiety models, with the “anxiety” response reflected in the ratio between pre- and postpunishment responding (Gray and McNaughton, 2000). IA paradigms are widely used and influential animal models of anxiety and fear. IA tests are commonly used to screen and compare drugs with potential to combat anxiety in humans, primarily because they are very sensitive to benzodiazepine anxiolytics (especially conflict paradigms).
The greatest successes of IA may be its contributions to our understanding of the role neuromodulatory systems play in consolidation of explicit memories and expression of punishment-based memories. Countless drugs have been administered after IA training that either facilitate or impair subsequent memory. Drugs that facilitate IA are seen as cognitive enhancers; those that impair IA may have anxiolytic potential (or be amnestic). The most common finding is that postconditioning amygdala processes modulate the consolidation of explicit memories stored elsewhere in the brain—probably by stimulating neuromodulatory centers (McGaugh, 2004). This is usually demonstrated by infusing drugs directly into the amygdala after IA training and showing LTM effects. Although the neurocircuitry of IA remains somewhat elusive, it is clear that it differs from that of simple Pavlovian PTC; IA depends on extra-amygdala brain regions that are not required for simple PTC (e.g., septohippocampal system), and posttraining amygdala manipulations that impair IA have no effect on LTM for PTC (Gray and McNaughton, 2000; Wilensky et al., 2006). As IA may be the most common form of avoidance cited as pathological in human anxiety disorders, more work is clearly needed to precisely define the neural circuits, and synaptic, cellular, and molecular processes mediating this form of instrumental learning.
Active Avoidance
For many decades following Pavlov’s work, conditional fear and its neural basis were commonly studied with active avoidance protocols (Cain and LeDoux, 2008). In a typical experiment, rats were presented with tone–shock pairings on one side of a two-compartment chamber. Movement to the opposite side (shuttling) terminated the tone and prevented a shock presentation. These avoidance responses (ARs) served as the dependent measure of fear learning, and animals exhibited responses more frequently and with shorter latencies as training progressed.
Avoidance proved to be a difficult paradigm for the analysis and explanation of learning mechanisms. This was partly because AA learning trials were those that successfully avoided the shock, and it was difficult to explain how absence of the US could reinforce the response. Debates still rage about the mechanisms of AA learning; however, one prominent early theory remains, called two-factor, two-process, or sometimes fear theory (Rescorla and Solomon, 1967). In brief, these theories assume that PTC occurs on early training trials, before an AR is acquired. Then, on subsequent trials, fear is aroused by the CS. When an AR is performed, the CS terminates, fear diminishes, and no US is encountered. These theories suggest that fear reduction associated with CS termination somehow reinforces the response. Although still controversial, there are reports that rodents can learn to escape an aversive CS even when no shock is present during training (escape from fear paradigms; e.g., Cain and LeDoux, 2007). These theories are popular with some partly because they provide an entry into the AA neurocircuit through the known Pavlovian threat conditioning circuitry.
Instrumental AA, like PTC and IA, is a normal, adaptive behavior that gives subjects control over threats in dangerous environments. Indeed, AA mechanisms may even contribute to beneficial active coping strategies that prevent unnecessary fear (discussed later). However, like PTC and IA, AA can become maladaptive and contribute to human pathology.
Early studies found that impairing amygdala function disrupts the acquisition and recall of AA, although null effects or even facilitated AA were also reported. More recent investigations, focusing on subregions of the amygdala and other brain structures, paint a clearer picture (Fig. 41.7). Whereas LA and BA are critical for AA learning in several preparations, CE processes appear to constrain or oppose AA learning, possibly because CE mediates competing threat reactions like freezing (Lazaro-Munoz et al., 2010). Interestingly, several labs have shown that the amygdala dependence of AA is transient; when animals are trained beyond asymptotic performance, amygdala lesions no longer impair AA responding (Gabriel et al., 2003). This suggests that with overtraining AA may become habitual and depend on other, extra-amygdala regions. Dorsal striatum and motor cortex have been suggested as sites with possible significance; however, this remains unconfirmed. Recent unpublished findings from our lab also implicate the vmPFC in AA. This makes sense since Pavlovian reactions like freezing must be suppressed to allow for AA learning, and vmPFC is perfectly situated to suppress CE-mediated responses and allow performance of instrumental actions (Amano et al., 2010).
Two bodies of work, one older and one more recent, provide several key insights into how AA learning may be achieved in relation to the PTC circuitry. Michael Gabriel’s laboratory implicated amygdala processes in AA very early on, in addition to other brain regions such as the ACC, thalamus, and sensory cortex (Gabriel et al., 2003). Interestingly, measuring training-induced neural activity changes (TIA) during AA training in rabbits, this group showed that LA plasticity precedes plasticity in other regions, and TIA effects in other regions depend on LA plasticity. This emphasizes the importance of LA to AA learning, which is consistent with AA evolving from prior PTC. New studies from multiple laboratories suggest that a distinct population of inhibitory cells in the lateral CE may also play a key role in gating fear (and possibly enabling AA). These interneurons express PKCδ and oxytocin receptors, and function to inhibit the output cells of the medial CE and allow for more active DRs (Ciocchi et al., 2010; Haubensak et al., 2010).
Summary of Instrumental Aversive Conditioning
Avoidance has a clear relationship to human anxiety disorders as many are partly defined by excessively avoidant behaviors and thoughts. IA has been widely used to investigate drugs that may have utility in treating human anxiety and has enjoyed reasonable predictive validity. Although IA suffers as a model for studying precise neurobiological mechanisms because it lacks a clearly identified neural circuit, this paradigm has greatly contributed to our understanding of the role that neuromodulators play in consolidation of explicit memories and expression of punishment memories. AA research also has clinical implications, both as a problem (e.g., obsessive-compulsive disorder) and as a potential positive coping mechanism (discussed later). However, until recently AA research has stagnated. A resurgence of AA research, conducted using the successful blueprint of PTC, is already providing a much better understanding of the behavior, brain circuits, and neural mechanisms involved. Clearly more research is needed on both forms of avoidance given their role in human pathological anxiety.
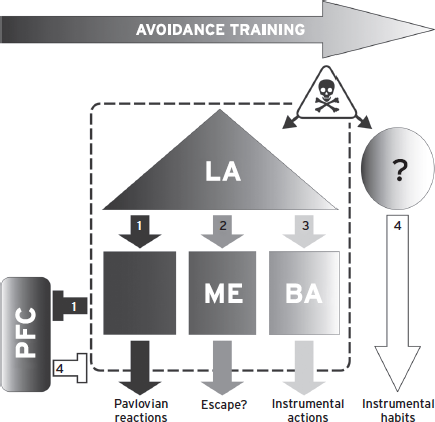
Figure 41.7 Working model of active avoidance. Early in AA training, failure to avoid leads to PCT trials and Pavlovian reactions mediated by LA and CE (1). CE also suppresses PFC activity. As training progresses, animals may first learn to escape via processes that depend on LA and medial amgydala (ME) (2). Eventually, il-PFC activity suppresses CE outputs and allows for AA learning, which depends on LA and BA processes (3–4). Overtraining of AA responses leads to amygdala-independent, and possibly habitual, AA (4). It is not yet known which brain region(s) mediate overtrained AA behavior; however, striatal and motor systems are likely candidates (Cain and LeDoux., 2008).
Inhibition of Anxiety and Fear
Animal models can also focus on forms of learning that reduce or suppress DRs. The preceding sections described situations where survival circuits are activated and defensive responding is elicited. But as in most systems, what goes up must come down, and the mammalian brain has multiple ways to constrain and/or reverse the activation of defense responses. This is true of both behavioral and physiological responding. Here we will discuss a few key examples.
Negative Feedback in the HPA Axis
The hypothalamic-adrenal-pituitary (HPA) axis is a major component of the acute stress response, a highly activated physiological state that prepares the organism by diverting energy and resources from nonessential processes (e.g., digestion) and mobilizing them to prime sensorimotor processes necessary for defensive responding. A properly functioning HPA axis turns itself off once the threat is gone, partly via glucocorticoid-mediated inhibition of hypothalamic and pituitary processes. In some human disorders, dysregulation of the HPA axis is implicated, and stress responding can be prolonged, leading to a number of adverse effects on bodily processes, health, and defensive responding (McEwen, 2004). HPA axis dysregulation can be assessed with tests like the dexamethasone suppression test. These models demonstrate that pathology can result from a failure of inhibition, not just excessive excitation, of DRs (Kehne and Cain, 2010).
Latent Inhibition and Extinction
Minor modifications of the basic Pavlovian PTC procedure can profoundly influence the degree of survival circuit activation and the form of defensive responding. PTC occurs because the CS reliably predicts the US, and subsequent CS presentations allow the animal to anticipate unpleasant outcomes. But what if the CS does not always predict the US? In latent inhibition (LI), subjects are first exposed to a series of stimuli with no bad outcome. Then, standard PTC occurs using the same stimuli. DRs to the CS are nearly always weaker than the control condition, where no preexposure to the stimuli occurs. Extinction is a similar procedure but in reverse. Following PTC, subjects are repeatedly exposed to the CS without the anticipated US. This training contradicts the predictive validity of the CS and creates a new inhibitory CS–NoUS association (Bouton et al., 2006). Then, on subsequent trials conditioned responding is greatly diminished, or absent, depending on the amount of training. It is believed that extinction doesn’t erase the original CS–US association but, rather, that behavior postextinction is the net result of competition between excitatory and inhibitory CS memories. Thus, one could imagine a scenario where one or both of these processes are impaired, leading to abnormally strong or persistent CRs even if basic PTC processes are normal.
Although some neurobiological studies have been conducted on LI in preclinical models, most of these relate to disorders like schizophrenia, where this learning process is disrupted. However, in the past 15 years there has been an explosion of fear extinction research, and we now know a great deal about its neural mechanisms (Milad and Quirk, 2012) (Fig. 41.5). For instance, vmPFC is essential for rapidly expressing extinction memories. Neural activity in vmPFC increases with extinction training, and electrical or growth factor (BDNF [brain-derived neurotrophic factor]) stimulation of vmPFC can induce extinction. vmPFC projects to inhibitory intercalated cells in the amygdala that can suppress CE-mediated threat reactions. LA and BA also play key roles in extinction learning. Several studies indicate that NMDA receptors, BDNF, calcineurin, MAP kinase, cannabinoid receptors, and GABA (gamma-aminobutyric acid) receptors in these regions participate in fear extinction learning (Myers and Davis, 2007). And BA neurons also appear to select between high- and low-fear states postextinction (Herry et al., 2010). Opioid-mediated input from the periacqueductal gray (PAG) may provide the error signal driving extinction plasticity (McNally et al., 2004). Finally, the hippocampus plays a critical role in the context-dependent expression of fear extinction; extinction is strongest in the place where CS-alone presentations occurred, and the hippocampus provides this critical signal (Bouton et al., 2006). Thus, animal models of extinction are seen by many as critical to understanding human pathological anxiety and barriers to its treatment (Cain et al., 2012; Jovanovic and Ressler, 2010). Indeed, a recent study of fear extinction revealed deficits in fear extinction recall and vmPFC activity when PTSD patients were challenged with an extinguished CS, consistent with preclinical findings (Milad and Quirk, 2012).
Conditioned Inhibition (CI)
The CI procedure shares many features with extinction, but unlike extinction, where the CS comes to have ambiguous meaning, CI training produces a stimulus that has purely inhibitory properties. CI training produces a “safety signal” whose presence indicates that no harm will come.
CI learning is complicated and a bit difficult to study, but a few recent publications in rodents suggest that plasticity in the amygdala and striatum play a role. For instance, mice show weakened responses in LA to a conditioned inhibitor, but strengthened responses in the overlying striatum (Rogan et al., 2005). Rats trained similarly show reductions in synapse size in the LA (Ostroff et al., 2010). Thus, like extinction, conditioned inhibitors have the potential to identify safe circumstances and suppress DRs, and dysfunctional CI mechanisms may present as excessive fear/anxiety, even if those processes are operating normally. Impaired CI learning has recently been observed in patients with PTSD (Jovanovic et al., 2012), supporting the notion that dysfunctional inhibitory learning can lead to inappropriate responding (generalization) in anxious people.
ANIMAL MODELS OF PATHOLOGICAL FEAR AND ANXIETY
As discussed, the bulk of animal model research focuses on normal, group-averaged responses in naïve animals. This research is important for gaining a solid understanding of the basic neurobiological processes that operate in human fear and anxiety. However, there is growing appreciation that these models cannot say much about the pathologies causing human disorders, especially if they fail to probe trait anxiety.
Researchers have recognized these problems for years and have taken many different approaches toward improving animal models of human anxiety disorders. Unfortunately, nearly every group uses its own method to coax rats into responding abnormally, which makes comparisons between models extremely difficult. Nevertheless, some important insights have been gained from these efforts, and we will attempt to summarize some major methods for modeling pathological fear and anxiety in animals. The list is not meant to be exhaustive or to suggest that one model is superior to another—it is merely meant to illustrate how researchers have used multimodal or hybrid approaches to examine pathological, rather than normal, defensive responding.
PHYSIOLOGICAL MANIPULATIONS
Physiological models have been used to examine how alterations in survival circuit neurons affect defensive responding. This is usually achieved by delivering chemicals or sensitizing electrical stimulation (i.e., kindling) to induce hyperexcitability in specific brain regions (e.g., amygdala, hypothalamus; Rosen and Schulkin, 1998). For example, models considered relevant to panic disorder (PD) combine treatments with panicogenic drugs with tests of unconditioned or conditioned DRs in rats (Shekhar et al., 2003; Sullivan et al., 2003). In one such model, the respiratory stimulant doxapram is administered prior to threat conditioning. Doxapram induces hyperventilation and panic attacks in most PD patients, by activating carotid body chemoreceptors to produce a (false) visceral signal to the brain of poor air. Doxapram treatment leads to facilitation of PTC, which has been linked to amygdala hyperexcitability and enhanced CRF release in CE. Because the circuitry of PTC is well characterized, it is possible to test the hypothesis that this visceral signal induces panic-like symptoms by altering threat processing in specific amygdala microcircuits.
DEVELOPMENTAL MANIPULATIONS
Developmental models have assessed the effects of early life stress on fearful and anxious behaviors manifested later in life. In rodents, for example, early maternal deprivation permanently alters central CRF systems as well as related behavioral and hormonal responses to stressors throughout life (Sullivan, 2003). Ecologically informed studies of maternal availability have likewise identified similar outcomes in nonhuman primates. Bonnet macaque monkeys raised in variable foraging demand conditions respond to novel situations with increased anxiety compared with age-matched monkeys raised as low-foraging demand controls. As adults, these monkeys exhibit significant differences in cerebrospinal fluid levels of monoamines, somatostatin, and CRF. These findings agree with evidence from humans that early exposure to severe stress is a risk factor for the development of mood and anxiety disorders (Heim and Nemeroff, 2002). Far less researched, but of equal importance, are indications that mild early-life stressors can also foster resilience (stress inoculation), perhaps by facilitating vmPFC-mediated inhibition of amygdala and emotional regulation processes (Lyons et al., 2010).
STRESS-ENHANCED FEAR LEARNING IN ADULTS
Behavioral manipulations in adulthood that affect structure and function of survival circuits often involve exposure to acute or chronic stressors that alter defensive responding, especially PTC. These models are sometimes referred to by the abbreviation: stress-enhanced fear learning (SEFL; e.g., Rau and Fanselow, 2007). Stress is achieved in many ways, including restraint, immobilization, cold exposure, electric shock, predator exposure, and underwater submersion. Exposure to these stressors in adulthood increases aggression, alters context-elicited DRs, affects performance on the EPM, and attenuates spatial memory. Restraint stress also facilitates PTC, results in dendritic atrophy in hippocampus and PFC, suppresses neurogenesis in the dentate gyrus, and induces dendritic hypertrophy and sprouting of new synapses in the amygdala (McEwen, 2004). These stress-induced aspects of neural plasticity suggest a putative link between remodeling of defensive circuits and the development of anxiety as modeled in adult rodents. Chronic stress and underwater submersion have also been used in rodent research to mimic the requisite stressful events of traumatic anxiety disorders such as PTSD. Generally speaking, SEFL effects are interpreted as interactions between nonassociative sensitization processes and associative PTC processes. Thus, stressful manipulations may produce hyperexcitability in neural circuits that lower the threshold for defensive responding, and/or enhance PTC learning/consolidation. Such interpretations model key aspects of psychopathology theories in humans, such as Roger Pitman’s “overconsolidation” theory of PTSD (for discussion, see Cain et al., 2012).
GENETIC MANIPULATION
Genetic manipulations before assessment of behavioral DRs in rodent models have generally been achieved in two ways. One is selective breeding of rat strains with the goal of enhancing or attenuating particular measures of fear and anxiety. For instance, the Wistar-Kyoto (WKY) rat strain has an extreme AA phenotype that may model aspects of human anxiety disorders. WKY rats show abnormally rapid acquisition in a difficult AA task and also fail to show the normal “warm-up” effect characteristic of other strains (Servatius et al., 2008). The other strategy is creation of transgenic animals that have altered expression of particular genes that code for proteins in brain circuits of interest, and evaluating performance on fear and anxiety assays. An example comes from studies of PTC in the serotonin (5-HT) 1A-receptor knockout mouse. When previously context-conditioned 5-HT1A-null mice are tested in the same context but with novel cues added, they do not exhibit the normal CR decrement seen in wild-type mice (Klemenhagen et al., 2006). This difference in sensitivity to novel stimuli has been likened to a memory bias for threatening cues in an otherwise neutral environment, as may occur in human patients with PD or PTSD. Given the well-described role of hippocampus in providing a representation of context to amygdala, focused investigation of hippocampus–amygdala interactions involving the 5-HT1A receptor is suggested.
Naturally occurring genetic polymorphisms related to anxiety have also been studied, and again, the 5-HT system is implicated. In particular, an insertion/deletion event in the 5-HT transporter linked polymorphic region (5-HTTLPR) is known to produce long (l) and short (s) alleles in humans and rhesus monkeys. In vitro, the short allele decreases transcriptional efficiency, which may alter expression of the 5-HT transporter and disrupts control of extracellular concentrations of 5-HT in neural circuits. Humans homozygous for the short allele (s/s) show more anxiety, muscle tension, shyness, and avoidance compared with humans who are homozygous (l/l) or heterozygous (l/s) for the long allele, and greater activation of survival circuits when challenged with threats (Domschke and Dannlowski, 2010).
INDIVIDUAL DIFFERENCES
It has been proposed that consideration be given to individual differences in susceptibility and resilience in animal models of anxiety disorders (Yehuda and LeDoux, 2007). This is particularly important in models for PTSD, as it is well documented that similar exposures to a horrific trauma results in development of PTSD for a relatively small proportion of those exposed. Of note, phenotypic variability in most animal models of fear and anxiety is obscured by the usual practice of reporting the variation around the mean. Recent work shows that “fear reactivity” in conditioned animals is normally distributed in unselected Sprague-Dawley (SD) rats (Yehuda and LeDoux, 2007; Fig. 41.8). When high-fear reactivity rats are extinguished, two additional groups emerge: a fast-extinguishing group and a slow-extinguishing group. The slow-extinguishing group is proposed to have relevance for PTSD, which involves a failure to recover from trauma and is associated with impaired fear extinction recall and reductions in vmPFC activity (Milad and Quirk, 2012).
Related work has recently been conducted with AA in unselected SD rats (Lazaro-Munoz et al., 2010). In many AA studies, a subset of rats show extremely poor AA acquisition and are usually excluded from further analyses. However, these studies demonstrated that poor avoidance was associated with abnormally high expression of competing Pavlovian reactions like freezing. CE lesions that eliminated freezing rescued AA performance without further training. This phenotype may also model a deficit in certain human anxiety disorders, as excessive Pavlovian reactions prevented the poor avoiders from performing a response that could prevent them from harm, even though they had apparently learned that they could avoid harm. Similar to microarray analyses of gene expression in selectively bred rats, these trait-selected rats offer opportunities to correlate gene expression with fearful and anxious phenotypic expression. Thus, models that consider individual phenotypic differences in fearful and anxious responding will likely be among the more effective for modeling anxiety disorders and identifying brain circuits and neuronal processes mediating abnormal anxiety.
MODELS FOCUSED ON TREATMENT AND COPING PROCESSES
Although several of the previous sections allude to implications for treatment of human anxiety disorders, some animal models explicitly attempt to model processes important for learning to suppress, or cope with, fear. The best accepted animal model of treatment learning is extinction. Exciting newer ideas involve disrupting reconsolidation and AA to understand active coping mechanisms for gaining control over threatening circumstances.
Fear Extinction
Extinction processes can be relevant to human treatment in many ways. Generally speaking, extinction training contradicts previously held beliefs or expectations. This creates a new inhibitory memory that competes with a conditioned threat memory for control of behavior. The result is a reduction, or elimination, of conditioned responding. Extinction can be used to reverse expression of both Pavlovian and instrumental CRs, although therapists typically focus on extinction of Pavlovian fear. Consider, for instance, a subject who exhibits pathological avoidance of some fear-eliciting stimulus. The therapist may seek to extinguish fear of this stimulus; however, this can be difficult since avoidance is typically strong, and one cannot extinguish CS fear if the subject avoids the CS altogether. In this case, a therapist may employ response prevention techniques followed by fear extinction. Response prevention means blocking ARs and essentially forcing exposure to the fear-arousing stimulus. Then, exposure, sometimes prolonged exposure (Foa, 2011), is used to extinguish CS fear. If successful, CS fear will diminish along with the motivation to avoid. Extinction is a component of most successful cognitive-behavioral therapies (CBTs) for fear and anxiety disorders, and there is good evidence that extinction-based treatments provide measurable relief to patients with wide-ranging anxiety disorders (Craske, 1999).
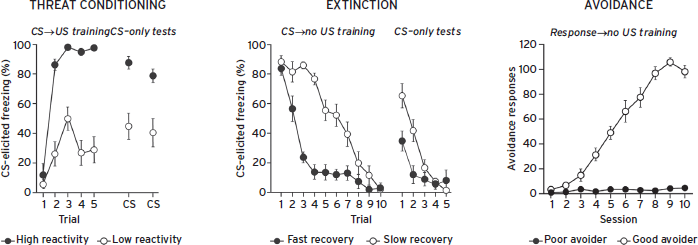
Figure 41.8 Exploiting natural response variation in key defensive learning tasks may facilitate studies modeling pathological fear/anxiety processes. In each task, the top and bottom 20% are shown.
Unfortunately, fear extinction has some notable drawbacks. First, some anxiety disorders are accompanied by impairments in fear extinction (Milad and Quirk, 2012). Second, it has been known since Pavlov that extinction memories are “fragile,” and more recent preclinical studies reliably show that expression of extinction is transient. The phenomena of renewal, reinstatement, and spontaneous recovery all show how extinction treatments are temporary—CRs return when the context changes, new stressors are experienced, or time passes (Bouton et al., 2006). Thus, it is not surprising that preclinical and clinical researchers have been intensely researching ways to facilitate extinction learning and make it more durable.
Although this topic alone could fill a book, we will highlight several important lines of animal model research that could directly affect the efficacy and permanence of extinction-based therapies. First, many laboratories are investigating how the timing of exposures can enhance extinction. It appears that some degree of temporal massing of CS exposures (sometimes called flooding) can enhance initial extinction learning, and once extinction learning begins, sessions spaced out in time facilitate consolidation/LTM (Cain et al., 2003; Li and Westbrook, 2008). Specific variations on this theme have even been reported to produce erasure of PCT memories, presumably by blocking the reconsolidation of reactivated memories with extinction training (Quirk et al., 2010).
Many other studies are evaluating the effects of biological proteins and psychoactive drugs on fear extinction, especially drugs commonly used to treat anxiety (Milad and Quirk, 2012). Several findings are worth highlighting here. First, a number of agents have been found to enhance extinction learning/memory when given before or after CS exposures. These include d-cycloserine, yohimbine, fluoxetine, venlafaxine, valproic acid, cannabidiol, estradiol, corticosterone, and BDNF. Several others have been found to impair fear extinction, most notably benzodiazepines (e.g., valium), blockers of α1 adrenergic (e.g., prazosin) and μ-opioid (e.g., naloxone) receptors (Bernardi and Lattal, 2010; Bouton et al., 2006; McNally et al., 2004). Particularly exciting are findings that extinction therapy can lead to fear memory erasure if combined with appropriate drugs, like SSRIs (Karpova et al., 2011). Such effects suggest that it may be possible to return the defensive circuitry to a developmental state where extinction routinely produces erasure of aversive memories (Quirk et al., 2010). Finally, very recent preclinical studies suggest that deep brain stimulation of specific regions, like vmPFC or ventral striatum, can enhance extinction recall (Rodriguez-Romaguera et al., 2012).
Fear Reconsolidation and Incubation
Interest is gaining in a newer line of preclinical research on the neural mechanisms of reconsolidation and incubation. In brief, memories for conditioned threats become labile when reactivated, perhaps to update the memory trace before it is reconsolidated. This raises the exciting possibility that clinicians can modify or even block the storage of reactivated fear memories. Reconsolidation blockade is typically achieved with drugs that interfere with protein synthesis (Nader et al., 2000); however, these are not suitable for use in humans. Studies are underway to evaluate other ways to blunt reconsolidation. These include drugs that affect protein synthesis and memory such as β-adrenergic receptor blockers (e.g., propranolol; Debiec and LeDoux, 2006), and behavioral interference or extinction procedures (Quirk et al., 2010). In a related phenomenon, fear incubation, recurring activations can enhance reconsolidation processes and produce progressively stronger fear memories—reminiscent of anxiety disorders like PTSD where intrusive memories become progressively more disruptive (Pickens et al., 2009). Although little is known about the neural mechanisms of fear incubation, norepinephrine makes the clearest contribution (Debiec et al., 2011). Reconsolidation can be a double-edged sword when it comes to treatment, however, as cognitive enhancers can cause decreases or increases in threat responding, depending on the amount of CS exposure and the relative recruitment of extinction versus reconsolidation/incubation processes (Lee et al., 2006).
Avoidance, Escape, and Active Coping
A new twist on an old animal model of fear and anxiety suggests that aspects of human active coping can be modeled in laboratory rodents. Although instrumental escape and avoidance are normal and adaptive learning processes, they have a very negative connotation today, partly because they are listed as defining features of many human anxiety disorders. However, escape/avoidance contributions to human pathology reflect the adoption of responses that disrupt life activities, most likely inhibitory avoidance responses that cause one to disengage from the world. Although escape and active avoidance (E-AA) mechanisms can also contribute to pathology, they may represent an ideal way to cope with environmental threats. In E-AA, subjects learn a specific response or action that escapes environmental threats and prevents harm. Thus, E-AA gives subjects control over environmental threats, allowing them to operate in dangerous environments yet stay safe. Lack of control is also a defining feature of many mood and anxiety disorders (DSM-IV 2000). In this sense, adaptive E-AA responses resemble active coping strategies that contribute to resilience and facilitate recovery following trauma (Stewart and Yuen, 2011). Adaptive E-AA responses have some other added benefits, too: they prevent the expression of Pavlovian reactions indicative of human fear and anxiety, and, unlike extinction, this suppression appears to persist, as long as the AA response is available (Cain and LeDoux, 2007; Helmreich et al., 2012; Solomon and Wynne, 1954). AA mechanisms and individual differences were discussed previously, but more research on instrumental E-AA and active coping mechanisms may support another evidence-based treatment method in humans, which may be especially useful for those who respond poorly to other forms of therapy (van der Kolk, 2006).
BEHAVIORAL INHIBITION THEORY OF ANXIETY
In 1982, Jeffrey Gray put forth a theory of anxiety based largely on the notion that classic anxiolytics, like alcohol, barbiturates, and (especially) benzodiazepines, could be useful tools for identifying good animal models of anxiety (Gray and McNaughton, 2000). Since only some DRs were sensitive to these drugs, Gray viewed fear and anxiety as separate response classes, and his definition of anxiety was distinct from that discussed earlier. According to Gray, anxiety reflected a conflict between motivations to approach certain stimuli and avoid others. Thus, anxiety was not simply weaker defensive responding to less certain or imminent threats, but a state triggered when one must choose to move toward threats in order to approach other, usually appetitive, goals. For instance, a rat may suppress foraging when presented with a predator odor but still engage in other active behaviors. Gray viewed this behavioral inhibition (BI) as distinct from fear-elicited DRs like freezing, which suppress all actions.
BI theory was very influential and affected the development and usage of animal models of anxiety. For instance, benzodiazepines suppressed DRs measured with conflict tests (e.g., Vogel test), IA, and several of the now-popular unconditioned assays (e.g., EPM) but often failed to suppress Pavlovian CRs and AA. Thus, conflict tests, IA, and unconditioned assays were deemed superior animal models of anxiety.
Gray also reasoned that “seat of anxiety” could be identified using brain lesions that produced a behavioral profile similar to the classic anxiolytics. This led to the septohippocampal (SH) theory of BI and anxiety, since lesions of this system impaired punishment effects, IA, and other anxiety-like behaviors in the unconditioned assays. This logic also led Gray to initially reject the notion that the amygdala played a central role in anxiety, since amygdala lesions impaired PCT and AA (unlike the anxiolytic drugs) and sometimes failed to affect the BI assays. In later years, Gray reformulated his theory to include a relatively minor role for the amygdala, mainly because fear-potentiated startle was shown to depend on the amygdala and was also sensitive to anxiolytics from diverse mechanistic classes (Davis, 1990).
Although influential, especially in the realm of drug development, the septohippocampal BI theory has steadily lost ground as new findings accumulate (LeDoux, 2002). First, SH injections of classic anxiolytics often failed to affect BI. Second, the classic anxiolytics all modulated GABAergic neurotransmission, and new anxiolytics were discovered that were effective in treating human anxiety but ineffective in prominent BI models (e.g., β-blockers, 5HT1A agonists, SSRIs). Third, an enormous body of research suggested that the core function of the hippocampus related to declarative, spatial/contextual, and working memory, not conflict resolution as proposed by Gray’s BI theory. Thus, SH lesion effects could be explained better by theories hypothesizing that this system provided contextual information to the amygdala threat–response system. Fourth, an equally large body of preclinical and clinical work implicated the amygdala, extended amygdala, and prefrontal cortex in defensive responding and human anxiety. Finally, the septohippocamal physiological mechanism for BI proposed by Gray, theta rhythms, does not appear to play a central role in primate, including human, anxiety. To the contrary, detailed mechanisms of neural processing and plasticity have been discovered in the amygdala, PFC, and hippocampus that can explain the role of neurotransmitter systems, and the action of anxiolytic drugs targeting these systems, in anxiety (see preceding section). These brain regions are now routinely implicated in human anxiety with imaging and neural recordings.
Although BI theory has run into some difficulties, we are not suggesting that the animal models used to support this theory are unrelated to fear and anxiety. Assays that involve punishment, IA, and innately aversive stimuli surely activate survival circuits and DRs relevant to human fear and anxiety. However, recent work strongly suggests that models based on PTC are capable of activating survival circuits to a similar degree, and these models are much better tools for studying the underlying neurobiological mechanisms mediating defensive responding—at both ends of the behavioral spectrum. The notion that approach–avoidance conflicts make a unique contribution to anxiety may still have merit, however, this conflict is probably inherent in all defensive deflections from the preferred activity pattern.
CONCLUSIONS
Current treatments for human pathological anxiety are inadequate; although some individuals respond favorably to psychotherapy and/or pharmacotherapy, both treatment approaches have unacceptably low long-term success rates (Cukor et al., 2010). Meanwhile, the needs of anxiety patients continue to grow, as does the already staggering cost to society (Smith, 2011). Given this unmet need and potential for massive profits, why then are pharmaceutical companies dropping drug development programs for anxiety at an alarming rate? The short answer: it’s too risky. When the drug discovery/development process takes 10–20 years, with an average cost of $1.3 billion, these companies are understandably risk averse (Paul et al., 2010). Although there are many contributing factors, the inability of standard animal models to accurately predict efficacy in human anxious populations plays a major role (Smith, 2011).
Generally speaking, preclinical drug discovery leans heavily on pharmacology-based animal models like those used to support BI theory. These benzodiazepine-sensitive assays primarily test conflict behaviors, IA, and unconditioned responses to innately aversive stimuli. Although these assays probe a wide range of anxiety-related behavior, all represent difficult models for studying underlying neural circuits and precise neurobiological mechanisms. Thus, they select for “pharmacologic isomorphism” but often cannot assess the mode of action for new agents, predict enhanced efficacy over current standards, or explain failures in the clinic that could guide future decisions and refine anxiety models (Geyer and Markou, 2002).
In contrast to the recent troubles in drug discovery, studies of the neurobiology of fear and anxiety have had great success over the past few decades (Johansen et al., 2011). This branch of research relies much more heavily on models amenable to neurocircuit analysis. For instance, Pavlovian threat conditioning uses discrete, controllable stimuli to induce learning, activate survival circuits, and elicit defensive responding. DRs typically studied include reflexive or stereotyped behaviors with clear, measurable onset and termination (Davis, 1990; Medina et al., 2002). These features greatly facilitate circuit analysis by allowing researchers to relate neural responses to triggering stimuli and behavior. Circuit-based models like PTC have vastly improved our understanding of the detailed cellular, synaptic, molecular, and genetic contributions to defensive responding and learning.
Although it was once believed that PTC could not elicit the anxiety-like responses that form the basis of many human disorders, recent work suggests otherwise. For instance, conditioning with reduced contingency, long duration CSs, complex CSs, or conditioned reinforcers can all produce less imminent/certain threats that elicit anxiety-like responses. Additional training phases can also model inhibitory (e.g., CI or extinction) or instrumental (e.g., AA) learning processes that likely make direct contributions to recovery and coping processes. Perhaps most important, circuit-based models like PTC allow one to hypothesize how a new treatment may achieve a positive effect in the context of known neurobiological and psychological processes.
Circuit-based models of anxiety are also consistent with recent efforts to reclassify human mental disorders. There is a growing realization that diagnostic criteria for anxiety disorders based on therapist interviews and personal reports of emotional symptoms may not accurately reflect underlying pathology in core anxiety processes. Thus, efforts such as the NIMH Research Domain Criteria Project (RDoC) aim to study basic dimensions of observable behavior and neurobiological measures related to pathological anxiety. The ultimate goal is to provide a knowledge base about core anxiety processes that will inform new diagnoses and treatment approaches. Increasing efforts to apply the preclinical circuit-based approach to new studies in experimental animals and people is likely to facilitate this process and remove much of the uncertainty associated with subjective emotional measures and models relying too heavily on validation by existing drugs.
DISCLOSURES
Dr. Cain has no conflicts of interest to disclose.
Dr. Sullivan serves as a member of the Scientific Advisory Board of Tonix Pharmaceuticals, Inc. and has served as a consultant to Ono Pharma USA, Inc.
Dr. LeDoux has no conflicts of interest to disclose.
REFERENCES
Amano, T., Duvarci, S., et al., (2011). The fear circuit revisited: contributions of the basal amygdala nuclei to conditioned fear. J. Neurosci. 31:15481–15489.
Amano, T., Unal, C.T., et al., (2010). Synaptic correlates of fear extinction in the amygdala. Nat. Neurosci. 13:489–494.
Andreatini, R., and Bacellar, L.F. (2000). Animal models: trait or state measure? The test-retest reliability of the elevated plus-maze and behavioral despair. Prog. Neuropsychopharmacol. Biol. Psychiatry 24:549–560.
Bailey, C.H., Giustetto, M., et al., (2000). Is heterosynaptic modulation essential for stabilizing Hebbian plasticity and memory? Nat. Rev. Neurosci. 1:11–20.
Balleine, B.W., and Killcross, S. (2006). Parallel incentive processing: an integrated view of amygdala function. Trends Neurosci. 29:272–279.
Bernardi, R.E., and Lattal, K.M. (2010). A role for alpha-adrenergic receptors in extinction of conditioned fear and cocaine conditioned place preference. Behav. Neurosci. 124:204–210.
Blanchard, R.J., Blanchard, D.C., et al., (1989). Ethoexperimental approaches to the study of defensive behavior. In: Blanchard, R.J., Brain, P.F., Blanchard, D.C., and Parmigiani, S., eds. Ethoexperimental Approaches to the Study of Behavior. Dordrecht, the Netherlands: Kluwer Academic, pp. 114–136.
Bouton, M.E., Westbrook, R.F., et al., (2006). Contextual and temporal modulation of extinction: behavioral and biological mechanisms. Biol. Psychiatry. 60:352–360.
Burgos-Robles, A., Vidal-Gonzalez, I., et al., (2009). Sustained conditioned responses in prelimbic prefrontal neurons are correlated with fear expression and extinction failure. J. Neurosci. 29:8474–8482.
Cain, C.K., Blouin, A.M., et al., (2003). Temporally massed CS presentations generate more fear extinction than spaced presentations. J. Exp. Psychol. Anim. Behav. Process. 29:323–333.
Cain, C.K., and LeDoux, J.E. (2007). Escape from fear: a detailed behavioral analysis of two atypical responses reinforced by CS termination. J. Exp. Psychol. Anim. Behav. Process. 33:451–463.
Cain, C.K., and LeDoux, J.E. (2008). Brain mechanisms of Pavlovian and Instrumental Aversive Conditioning. In: Nutt, D.J., Blanchard, R.J., Blanchard, D.C., and Griebel, G., eds. Handbook of Anxiety and Fear. Amsterdam: Elsevier Academic, pp. 103–125.
Cain, C.K., Maynard, G.D., et al., (2012). Targeting memory processes with drugs to prevent or cure PTSD. Exp. Opin. Investig. Drugs 21(9):1323–1350.
Ciocchi, S., Herry, C., et al., (2010). Encoding of conditioned fear in central amygdala inhibitory circuits. Nature 468:277–282.
Craske, M.G. (1999). Anxiety Disorders: Psychological Approaches to Theory and Treatment. Boulder, CO: Westview Press.
Cukor, J., Olden, M., et al., (2010). Evidence-based treatments for PTSD, new directions, and special challenges. Ann. NY Acad. Sci. 1208:82–89.
Davis, M. (1990). Pharmacological and anatomical analysis of fear conditioning. NIDA Res. Monogr. 97:126–162.
Davis, M., Gendelman, D.S., et al., (1982). A primary acoustic startle circuit: lesion and stimulation studies. J. Neurosci. 2:791–805.
Debiec, J., Bush, D.E., et al., (2011). Noradrenergic enhancement of reconsolidation in the amygdala impairs extinction of conditioned fear in rats: a possible mechanism for the persistence of traumatic memories in PTSD. Depress. Anxiety 28:186–193.
Debiec, J., and LeDoux, J.E. (2006). Noradrenergic signaling in the amygdala contributes to the reconsolidation of fear memory: treatment implications for PTSD. Ann. NY Acad. Sci. 1071:521–524.
Domschke, K., and Dannlowski, U. (2010). Imaging genetics of anxiety disorders. NeuroImage 53:822–831.
Ehrlich, I., Humeau, Y., et al., (2009). Amygdala inhibitory circuits and the control of fear memory. Neuron 62:757–771.
Feinstein, J.S., Adolphs, R., et al., (2011). The human amygdala and the induction and experience of fear. Curr. Biol. 21:34–38.
File, S.E., and Seth, P. (2003). A review of 25 years of the social interaction test. Eur. J. Pharmacol. 463:35–53.
Foa, E.B. (2011). Prolonged exposure therapy: past, present, and future. Depress. Anxiety 28:1043–1047.
Gabriel, M., Burhans, L., et al., (2003). Consideration of a unified model of amygdalar associative functions. Ann. NY Acad. Sci. 985:206–217.
Gale, G.D., Anagnostaras, S.G., et al., (2004). Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J. Neurosci. 24:3810–3815.
Geyer, M.A., and Markou, A. (2002). The role of preclinical models in the development of psychotropic drugs. In: Davis, K.L., Charney, D., Coyle, J.T., and Nemeroff, C., eds. Neuropsychopharmacology: The Fifth Generation of Progress. Philadelphia: Lippincott, Williams, & Wilkins, pp. 445–455.
Gozzi, A., Jain, A., et al., (2010). A neural switch for active and passive fear. Neuron 67:656–666.
Gray, J.A., and McNaughton, N. (2000). The Neuropsychology of Anxiety: An Enquiry into the Functions of the Septo-Hippocampal System. New York: Oxford University Press.
Haubensak, W., Kunwar, P.S., et al., (2010). Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature 468:270–276.
Heim, C., and Nemeroff, C.B. (2002). Neurobiology of early life stress: clinical studies. Semin. Clin. Neuropsychiatry 7:147–159.
Helmreich, D.L., Tylee, D., et al., (2012). Active behavioral coping alters the behavioral but not the endocrine response to stress. Psychoneuroendocrinology 37(12):1941–1948.
Herry, C., Ferraguti, F., et al., (2010). Neuronal circuits of fear extinction. Eur. J. Neurosci. 31:599–612.
Hogg, S. (1996). A review of the validity and variability of the elevated plus-maze as an animal model of anxiety. Pharmacol. Biochem. Behav. 54:21–30.
Johansen, J.P., Cain, C.K., et al., (2011). Molecular mechanisms of fear learning and memory. Cell 147:509–524.
Johansen, J.P., Hamanaka, H., et al., (2010). Optical activation of lateral amygdala pyramidal cells instructs associative fear learning. Proc. Natl. Acad. Sci. USA 107:12692–12697.
Josselyn, S.A. (2010). Continuing the search for the engram: examining the mechanism of fear memories. J. Psychiatry Neurosci. 35:221–228.
Jovanovic, T., Kazama, A., et al., (2012). Impaired safety signal learning may be a biomarker of PTSD. Neuropharmacology 62:695–704.
Jovanovic, T., and Ressler, K.J. (2010). How the neurocircuitry and genetics of fear inhibition may inform our understanding of PTSD. Am. J. Psychiatry 167:648–662.
Kalin, N.H. (2004). Studying non-human primates: a gateway to understanding anxiety disorders. Psychopharmacol. Bull. 38(Suppl. 1):8–13.
Karpova, N.N., Pickenhagen, A., et al., (2011). Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science 334:1731–1734.
Kehne, J.H., and Cain, C.K. (2010). Therapeutic utility of non-peptidic CRF1 receptor antagonists in anxiety, depression, and stress-related disorders: evidence from animal models. Pharmacol. Ther. 128:460–487.
Klemenhagen, K.C., Gordon, J.A., et al., (2006). Increased fear response to contextual cues in mice lacking the 5-HT1A receptor. Neuropsychopharmacology 31:101–111.
Knapska, E., Radwanska, K., et al., (2007). Functional internal complexity of amygdala: focus on gene activity mapping after behavioral training and drugs of abuse. Physiol. Rev. 87:1113–1173.
Knobloch, H.S., Charlet, A., et al., (2012). Evoked axonal oxytocin release in the central amygdala attenuates fear response. Neuron 73:553–566.
Lazaro-Munoz, G., LeDoux, J.E., et al., (2010). Sidman instrumental avoidance initially depends on lateral and basal amygdala and is constrained by central amygdala-mediated Pavlovian processes. Biol. Psychiatry 67:1120–1127.
LeDoux, J.E. (2002). Synaptic Self: How Our Brains Become Who We Are. New York: Viking.
LeDoux, J.E. (2012). Rethinking the emotional brain. Neuron 73:653–676.
Lee, J.L., Milton, A.L., et al., (2006). Reconsolidation and extinction of conditioned fear: inhibition and potentiation. J. Neurosci. 26:10051–10056.
Li, S.H., and Westbrook, R.F. (2008). Massed extinction trials produce better short-term but worse long-term loss of context conditioned fear responses than spaced trials. J. Exp. Psychol. Anim. Behav. Process. 34:336–351.
Lyons, D.M., Parker, K.J., et al., (2010). Animal models of early life stress: implications for understanding resilience. Dev. Psychobiol. 52:402–410.
Maren, S., and Fanselow, M.S. (1995). Synaptic plasticity in the basolateral amygdala induced by hippocampal formation stimulation in vivo. J. Neurosci. 15:7548–7564.
McEwen, B.S. (2004). Protection and damage from acute and chronic stress: allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann. NY Acad. Sci. 1032:1–7.
McGaugh, J.L. (2004). The amygdala modulates the consolidation of memories of emotionally arousing experiences. Annu. Rev. Neurosci. 27:1–28.
McKinney, W.T. (2001). Overview of the past contributions of animal models and their changing place in psychiatry. Semin. Clin. Neuropsychiatry 6:68–78.
McNally, G.P., Pigg, M., et al., (2004). Opioid receptors in the midbrain periaqueductal gray regulate extinction of pavlovian fear conditioning. J. Neurosci. 24:6912–6919.
Medina, J.F., Christopher Repa, J., et al., (2002). Parallels between cerebellum- and amygdala-dependent conditioning. Nat. Rev. Neurosci. 3:122–131.
Milad, M.R., and Quirk, G.J. (2012). Fear extinction as a model for translational neuroscience: ten years of progress. Annu. Rev. Psychol. 63:129–151.
Myers, K.M., and Davis, M. (2007). Mechanisms of fear extinction. Mol. Psychiatry 12:120–150.
Nader, K., Schafe, G.E., et al., (2000). Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406:722–726.
Ostroff, L.E., Cain, C.K., et al., (2010). Fear and safety learning differentially affect synapse size and dendritic translation in the lateral amygdala. Proc. Natl. Acad. Sci. USA 107:9418–9423.
Paul, S.M., Mytelka, D.S., et al., (2010). How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 9:203–214.
Pickens, C.L., Golden, S.A., et al., (2009). Long-lasting incubation of conditioned fear in rats. Biol. Psychiatry 65:881–886.
Quirk, G.J., Armony, J.L., et al., (1997). Fear conditioning enhances different temporal components of tone-evoked spike trains in auditory cortex and lateral amygdala. Neuron 19:613–624.
Quirk, G.J., Pare, D., et al., (2010). Erasing fear memories with extinction training. J. Neurosci. 30:14993–14997.
Rau, V., and Fanselow, M.S. (2007). Neurobiological and neuroethological perspectives on fear and anxiety. In: Kirmayer, L.J., Lemelson, R., and Barad, M., eds. Understanding Trauma: Integrating Biological, Clinical, and Cultural Perspectives. New York: Cambridge University Press, pp. 27–40.
Rescorla, R.A., and Solomon, R.L. (1967). Two process learning theory: relationships between Pavlovian conditioning and instrumental learning. Psychol. Rev. 74:151–182.
Rodriguez-Romaguera, J., Do Monte, F.H., et al., (2012). Deep brain stimulation of the ventral striatum enhances extinction of conditioned fear. Proc. Natl. Acad. Sci. USA 109:8764–8769.
Rogan, M.T., Leon, K.S., et al., (2005). Distinct neural signatures for safety and danger in the amygdala and striatum of the mouse. Neuron 46:309–320.
Rosen, J.B. (2004). The neurobiology of conditioned and unconditioned fear: a neurobehavioral system analysis of the amygdala. Behav. Cogn. Neurosci. Rev. 3:23–41.
Rosen, J.B., and Schulkin, J. (1998). From normal fear to pathological anxiety. Psychol. Rev. 105:325–350.
Rumpel, S., LeDoux, J., et al., (2005). Postsynaptic receptor trafficking underlying a form of associative learning. Science 308:83–88.
Servatius, R.J., Jiao, X., et al., (2008). Rapid avoidance acquisition in Wistar-Kyoto rats. Behav. Brain Res. 192:191–197.
Shekhar, A., Sajdyk, T.J., et al., (2003). The amygdala, panic disorder, and cardiovascular responses. Ann. NY Acad. Sci. 985:308–325.
Smith, K. (2011). Trillion-dollar brain drain. Nature 478:15.
Solomon, R.L., and Wynne, L.C. (1954). Traumatic avoidance learning: the principles of anxiety conservation and partial irreversibility. Psychol. Rev. 61:353.
Southwick, S.M., Bremner, J.D., et al., (1999). Role of norepinephrine in the pathophysiology and treatment of posttraumatic stress disorder. Biol. Psychiatry 46:1192–1204.
Stewart, D.E., and Yuen, T. (2011). A systematic review of resilience in the physically ill. Psychosomatics 52:199–209.
Sullivan, G.M., Apergis, J., et al., (2003). Rodent doxapram model of panic: behavioral effects and c-Fos immunoreactivity in the amygdala. Biol. Psychiatry 53:863–870.
Sullivan, R.M. (2003). Developing a sense of safety: the neurobiology of neonatal attachment. Ann. NY Acad. Sci. 1008:122–131.
Takahashi, L.K., Chan, M.M., et al., (2008). Predator odor fear conditioning: current perspectives and new directions. Neurosci. Biobehav. Rev. 32:1218–1227.
Teixeria, C.M., Pomedli, S.R., et al., (2006). Involvement of the anterior cingulate cortex in the expression of remote spatial memory. J. Neurosci. 26:7555–7564.
Timberlake, W., and Fanselow, M.S. (1994). Symposium on behavior systems: learning, neurophysiology, and development. Psychon. B. Rev. 1:403–519.
van der Kolk, B.A. (2006). Clinical implications of neuroscience research in PTSD. Ann. NY Acad. Sci. 1071:277–293.
Waddell, J., Morris, R.W., et al., (2006). Effects of bed nucleus of the stria terminalis lesions on conditioned anxiety: aversive conditioning with long-duration conditional stimuli and reinstatement of extinguished fear. Behav. Neurosci. 120:324–336.
Walker, D.L., Miles, L.A., et al., (2009). Selective participation of the bed nucleus of the stria terminalis and CRF in sustained anxiety-like versus phasic fear-like responses. Prog. Neuropsychopharmacol. Biol. Psychiatry 33:1291–1308.
Weinberger, N.M. (1995). Retuning the brain by fear conditioning. In: Gazzaniga, M.S., ed. The Cognitive Neurosciences. Cambridge, MA: MIT Press, pp. 1071–1090.
Wilensky, A.E., Schafe, G.E., et al., (2006). Rethinking the fear circuit: the central nucleus of the amygdala is required for the acquisition, consolidation, and expression of pavlovian fear conditioning. J. Neurosci. 26:12387–12396.
Willner, P. (1991). Methods for assessing the validity of animal models of psychopathology. In: Boulton, A., Baker, G., and Martin-Iverson, M., eds. Neuromethods: Animal Models in Psychiatry I. New York: Humana Press, pp. 1–23.
Yehuda, R., and LeDoux, J. (2007). Response variation following trauma: a translational neuroscience approach to understanding PTSD. Neuron 56:19–32.
Ziemann, A.E., Allen, J.E., et al., (2009). The amygdala is a chemosensor that detects carbon dioxide and acidosis to elicit fear behavior. Cell 139:1012–1021.