52 | THE GENETIC BASIS OF ADDICTIVE DISORDERS
DAVID GOLDMAN
Addictions share mechanisms subject to both genetic and environmental influences. Conversely, environmental, genetic, and psychophysiological vulnerability factors and individual choice play important, but differing roles, in addicted individuals, including ones addicted to the same agent. The inheritance of addictions ranges from moderate (40%) to high (70%), representing consequences of alleles (sequence variants) shared by descent. At all phases of the multistep process of addiction, gene × environment interactions, including correlations between gene and environment, shape vulnerability. However, neither exposure nor consequent dependence necessarily leads to addiction. Many individuals use addictive agents without becoming addicted. Physical dependence (tolerance) often occurs to drugs administered in the course of medical care, followed by withdrawal and no consequent addiction.
Genetic susceptibility and resilience to addiction are attributable to alleles that are both substance-specific (e.g., variants that alter metabolism) and non-specific. Alleles that moderate reward, stress resiliency and executive cognitive control are among the factors that are non-specific to any particular agent. Cross-inheritance and shared etiology also partly explain the tendency of addictions to co-occur with other psychiatric diseases. From verified examples, alleles that alter propensity and resilience to addiction are diverse in molecular mechanisms, as well as in their abundances and effect sizes. Some are common, but most are probably rarer, and the strongest effects are probably produced by rarer alleles.
The genetic evolutionary context of addiction is poorly understood. However, there is as yet no evidence that vulnerability and protective alleles were directly selected by addiction itself, except in artificially selected model organisms wherein it must be admitted that the effects of selection are often surprisingly rapid. However, certain alleles that alter propensity to addiction appear to have been maintained at high frequency by balanced selection. These include alcohol metabolic enzyme variants in ADH1B and ALDH2, as well as alleles that alter cognition and emotion. Many common alleles that increase liability probably have counterbalancing positive values, if not in modern times then in the past. Certain genes influencing propensity to addiction can be accurately described as genes “for” behavior, although their effects on vulnerability to addictions may be secondary.
Three pathways to identify genes that alter propensity to addictions are candidate gene approaches, candidate gene approaches targeted to relevant psychophysiology and genome-wide approaches. The latter two paradigmatic methods have been partially successful and are to some extent convergent, as genome-wide approaches are applied to psychophysiology and mechanisms of genes identified by GWA (genome-wide association) are defined. The combination of genetic tools with neuroscience techniques such as brain imaging enables exploration of mechanisms by which alleles alter risk. Functional alleles that partially account for interindividual differences in stress resiliency, and thereby addiction, are found at SLC6A4, COMT, NPY, and MAOA, all having been linked to activity of brain regions mediating stress response and emotion. Linkage analysis, including GWA, has enabled the hypothesis-free search for common variants moderating vulnerability. Via GWA, Asp398Asn, a common missense variant of CHRNA5, was discovered to influence risk of smoking and lung cancer consequent to smoking. Confirmation of this discovery validated the potential of GWA in addictions. However, at an early stage when GWA has been applied to relatively small numbers of samples, crudely phenotyped samples, and samples of heterogeneous origins, more than 95% of the genetic variance (heritability) in propensity remains unexplained, and replicated gene findings are primarily from candidate gene analyses.
The predictive value of any particular allele or genotype is low. The primary reasons for mismatch between heritability and strength of effect of individual alleles are twofold. Variation in several psychophysiologic processes including drug metabolism, sensation, reward, anxiety and resilience, and executive cognition contribute to vulnerability, and each is itself a manifestation of the action of many genes in contexts of exposure. Second, addictions are currently defined as end-stage diagnoses, and the effects of genetic variants are diluted or confounded when queried against an amalgam of phenotypes with different etiologies. Recent advances in genomics including massively parallel sequencing at low cost have opened the possibility for systematic searches for rare variants that may have stronger effects. For example, stop codons moderating addiction through the mechanism of impulsivity, also a risk factor in other psychiatric diseases, have been discovered within both MAOA and HTR2B.
The predictive value of genotypes for treatment response will determine their value for development of personalized medicine, one goal of gene identification being individualization of prevention and treatment of addictions. Genotypes may have different predictive value for treatment response than for diagnosis, and the use of genetic predictors will require controlled studies with predefined treatment outcomes. However, and in advance of these prospective clinical trials, genotypes have been identified that appear useful for predicting treatment response for alcoholism and nicotine addiction. For example, an OPRM1 variant is associated with treatment response to naltrexone in alcoholism, and altered brain reward responses.
Addictions, including substance use disorders (SUDs), follow exposure to a wide variety of agents, and addictions are a worldwide phenomenon sparing no culture. Exposure to a diverse array of agents may lead to an addicted state through partially overlapping neurobiological pathways. However, although most or all people are exposed to addictive agents at multiple points in their lives only a minority become addicted. Even tolerance and dependence are not equivalent to addiction. For example, most patients receiving opioid drugs do not become addicted, although temporary tolerance and dependence are elicited. In both vulnerable and resilient (less vulnerable) individuals, repetitive exposures induce long-lasting neuroadaptative changes that alter behavior. In some individuals, tolerance, craving, withdrawal, and motivational shifts lead to persistent and uncontrolled patterns of use that constitute addiction. Addiction accesses different vulnerability mechanisms. Initially, motivation to drug seeking is driven by impulsivity and positive reward. Later, compulsivity and negative affect may more strongly motivate use. As defined in DSM IV, and probably in the next DSM, addictions are “end-stage” diagnoses based on behavior that began to emerge years earlier from preexisting vulnerability and exposure. The trajectory of addiction culminates in long-lasting and potentially irreversible neuroadaptative changes and constellations of behaviors and social impairments. Both the probability of initial use and progression are influenced by the nature of the addictive agent including its mode of administration, distribution, metabolism, and psychoactive properties, by intrinsic factors such as sex, age, age at first use, preexisting addictive disorder or other mental illness, and by extrinsic factors including parenting and childhood adversity, peer influences, social support and drug availability. Genes act within a matrix of factors.
Progress has been made to define clinical categories of addicted individuals who may also be more likely to share mechanisms of vulnerability, and both developmental trajectory and personality features have been incorporated into these schema. Genetic liability to addiction varies both quantitatively and qualitatively across the lifespan. Quantitatively, peer influences and family environment are most important for initial exposure and early patterns of use, and genetic factors and psychopathology are more important in the transition to problematic use. During adolescence the heritability of addiction rises, peaking in young adulthood and declining again in older age (Kendler et al., 2008). Qualitatively, childhood conduct disorder and antisocial behavior are associated with early abuse and dependence. These behaviors were incorporated into the Type I/Type II typology for alcoholism together with early age of onset and personality factors reflecting variation in brain function (Cloninger, 1987). The role of conduct disorder and antisociality on the one hand and anxiety on the other have long been recognized and some addiction-associated behaviors have been broadly described as internalizing (associated with behavioral inhibition), and others as externalizing (associated with disinhibition). However, regardless of whether they are antisocial and impulsive, addicted individuals, for example alcoholics, tend to be anxious (Ducci et al., 2007). Overall, their greater anxiety may be caused by drug-induced allostatic change. Therefore it is important that adoptive studies and twin studies have pointed to the genetic transmission of vulnerability via both the externalizing and internalizing domains of behavior.
INHERITANCE OF ADDICTIONS
Family, adoption, and twin studies convergently establish the existence of genetic variation that determines genetic liability to addictions. Weighted mean heritabilities for addictions computed from large, epidemiologically ascertained cohorts of twins point to moderate to high heritabilities (0.39–0.72) (Goldman et al., 2005). These heritability values are within-population estimates and have not been deflated by including pairs of related individuals who would specifically capture variation in vulnerability that occurs across countries or across time, for example when an addictive agent becomes more readily available and vulnerability shifts as it might for example when use of an addictive agent is legalized. Both “no pathological drug use” and “initiation of use” are heritable, indicating that genetic variation also influences initiation (Kendler et al., 1999). Paradoxically, easing access to addicted agents may increase heritability by decreasing the environmental variance.
MODE OF INHERITANCE
Heritability directly implicates genes in causation; however, any particular heritable trait may be intractable to genetic analysis because of complexity of causation. In contrast, less heritable traits that are more narrowly defined or closer to the action of a gene may be more successfully parsed at the level of specific genetic loci that contribute to them. Therefore, two somewhat interrelated themes of this chapter are the mode of inheritance of addictions and the deconstruction (or redefinition) of addictions using neuroscience phenotypes on which genes may act more directly.
Genetic complexity arises from incomplete penetrance of alleles, phenocopies, variable expressivity, pleiotropy, gene–environment interactions, genetic heterogeneity, polygenicity, and epistasis. An epistatic model in which combinations of genetic variants determine addiction would seem consistent with the complex molecular architecture of the brain. However it is also possible that the molecular complexity of behavior could lead to a high degree of heterogeneity of genetic causation, with additive effects of the vulnerability variants. Epistatic interaction between alleles will tend to produce high MZ:DZ (monozygotic/dizygotic) twin concordance ratios, with identical twins resembling each other on the basis of common sharing of a constellation of variants but dizygotic twins being discordant because of the likelihood that some essential element of the constellation is not transmitted to the co-twin, disrupting the genotypic combination. For example if several dominantly acting alleles at different loci are required, the odds of this genotype being shared in a monozygotic co-twin would be 100% but the odds of sharing in a dizygotic co-twin (or full sibling) would be (1/2)n, where n is the number of alleles. Incompatible with the epistatic model, the risk of addiction in dizygotic twins and first-degree relatives of addicted probands (index cases) has been found to fall off proportionally to degree of relationship, rather than exponentially. For example, MZ:DZ concordance ratios are approximately 2:1 for various addictive disorders (Goldman et al., 2005). Also there is enhanced risk in individuals with first-degree relatives who are addicted, regardless of whether they have been adopted away in infancy. Two factors complicating this interpretation of MZ/DZ ratios are assortative mating, which can increase the likelihood of multilocus allelic combinations in first degree relatives, and multiple allelic combinations that might lead to the same phenotype. However, under the epistatic model there is as yet no good explanation for the linear correspondence between risk and degree of relationship to an affected proband.
A critical test of the epistatic model is the additivity of alleles with proven relationship to addiction. However, as yet few addiction genes have been identified. Perhaps by chance, but perhaps because additively acting loci are in some way more easily discovered, gene × gene interactions in addictions are thus far consistent with the genetic heterogeneity and gene-gene additivity models. Two missense variants in ADH1B (Arg48) and ALDH2 (Lys487) diminish risk of alcoholism via alcohol-induced flushing. Surprisingly, these alleles act additively (Thomasson et al., 1991) despite the fact that they affect consecutive steps in alcohol metabolism. Functional loci within HTR3B and the serotonin transporter (SLC6A4) together alter serotonin function but appear to act additively on risk for alcoholism comorbid with other SUDs (Enoch et al., 2011). Two variants associated with nicotine addiction, one in the CHRNA5-CHRNA3-CHRNB4 nicotinic acetylcholine receptor subunit cluster found by GWA and the other in the TTC12-ANKK1-DRD2 cluster, which includes DRD2, a dopamine receptor important in nicotine reward apparently, also appear to act additively (Ducci et al., 2011). The discovery of epistatic interactions in addiction may await the detection of novel loci as well as measurement of new addiction-related phenotypes on which allele effects may be epistatic; however, the quantitative genetic (transmission) and molecular genetic (gene action) data available thus far do not support reconception of addiction to take into account epistasis.
CHANGES IN GENE EFFECTS ACROSS THE LIFESPAN
The risk, heritability, and specific factors that determine vulnerability to addiction vary across the lifespan and during development. Heritabilities of alcoholism, cannabis addiction, and nicotine addiction are low in early adolescence but gradually increase, peaking in young adulthood and declining with older age (Kendlet et al., 2008). Reciprocally, family environment is most important in childhood and declines as adolescents increasingly shape their social environment. For addictions, and perhaps paradoxically, genotype becomes relatively more important as people are able to make their own choices and shape their environments in line with intrinsic predisposition. Another explanation for the greater importance of inheritance in older children is that some genetic factors are important only after repetitive exposure to addictive agents. An intriguing possibility is that some alleles only alter responses of the adult or adolescent brain. CHRNA5-CHRNA3-CHRNB4 genotypes have been reported to exert a stronger effect on smoking behavior in adulthood than in adolescence and to moderate the risk of a severe nicotine addiction in people who have already initiated use. In contrast, the TTC12-ANKK1-DRD2 and MAOA genes appear to influence personality characteristics, such as novelty seeking and impulsivity, that promote initiation of use (Ducci et al., 2012).
SHARED AND UNSHARED INHERITANCE
Addictive disorders tend to co-occur (Kessler et al., 1997). However, the observation of comorbidity is insufficient to demonstrate causality. For example, comorbidity could be caused by common environmental factors, or the first addiction could increase the risk of the second. Comorbidity sometimes can arise because of the pleitropic action of a single allele—one variant/multiple manifestations. The question of shared and agent-specific pathways to addiction will ultimately be answered at the level of individual genetic loci, some that alter risk for addiction to a specific agent and others that alter a mechanism, or mechanisms, common to multiple addictive agents. Substance-specific genes include metabolic enzymes for alcohol (ALDH2, ADH1B) and nicotine (CYP) as well as genes encoding gatekeeper molecules such as drug receptors (e.g. nicotinic receptors). Other genes influence mechanisms intrinsic to the action of multiple addictive agents or modulate an individual’s likelihood of exposure or ability to quit.
Diverse aspects of addiction neurobiology that have been discovered to be modulated by functional genetic variation include anxiety, resilience, impulsivity, and reward. The genes implicated include rare stop codons and other functional variation in monoamine oxidase A (MAOA) and the serotonin HTR2B receptor, a serotonin transporter (SLC6A4) VNTR (HTTLPR) and a catechol-O-methyl transferase (COMT) missense variant Val158Met. These loci are also implicated in the shared genetic liability between addictions and other psychiatric diseases, and connect the inheritance of addictions to variation in specific aspects of brain function. Nevertheless, few genes influencing addiction vulnerability have been discovered, and therefore most knowledge of the relative importance of shared and unshared factors in inheritance is from quantitative studies of inheritance. Fortunately, these quantitative studies have been highly informative for the overall importance of shared and unshared genetic vulnerability factors.
Adoption, family, and twin studies distinguish between correlation and causation by measuring cross-transmission (Goldman and Bergen, 1998). Several large, methodologically sound twin studies have detected substantial cross-transmission of risk of addictions. For example, the risks of alcoholism and smoking are cross-transmitted. There is apparently more than one shared factor. In the Virginia twin sample two shared factors were found, one being identified as an illicit agent factor relevant to cannabis and cocaine dependence and the second being a licit agent factor mainly explaining vulnerability to alcohol, caffeine, and nicotine (Kendler et al., 2007).
Addictions are also cross-transmitted with other mental illnesses with which they are frequently comorbid. Addictions are cross-transmitted with externalizing disorders, including conduct disorder (CD), antisocial personality disorder, borderline personality disorder, and attention deficit hyperactivity disorder (ADHD) (Kendler et al., 2003; Krueger et al., 2002). Preexisting CD and ADHD are risk factors for addiction. Anxiety and depression may be preexisting or consequent (Kendler et al., 2003). Some longitudinal studies have also found that anxiety disorders and anxiety-related personality traits predict alcohol problems in adolescents and young adults. The connection between addictions and internalizing disorders is interwoven with the role of genes in resilience, stress exposure being a powerful determinant of both addictions and internalizing disorders, as discussed next.
GENE × ENVIRONMENT INTERACTION IN ADDICTION
Two ways gene–environment independence is frequently violated are gene by environment interaction and gene by environment correlation.
GENE × ENVIRONMENT CORRELATION
Genotypes modulate the likelihood of exposures leading to correlation (r) of gene and environment, and creating a “genetics of the environment.” Note that gene × environment correlation is caused by a unidirectional interaction, environment not modulating genotype, although it may lead to widespread epigenetic change modulating gene expression. An example of gene × environment correlation is the ability of CHRNA5 Asn398 to increase risk of lung cancer (Thorgeirsson et al., 2008) via its influence on heavy smoking, thereby leading to increased exposure of the lung to carcinogens. However, and as a possible example of pleiotropy, nicotine could also directly modulate cancer risk at the cellular level.
GENE × ENVIRONMENT INTERACTION
The effect of an environmental exposure can be modified by genotype (Caspi and Moffitt, 2006). Again, gene × environment interaction is caused by a unidirectional interaction, environment not modulating genotype, although it may lead to widespread epigenetic change modulating gene expression. Childhood adversity is an important risk factor for addiction and comorbid diseases, including antisocial personality disorder (ASPD), CD, borderline personality disorder, and anxiety disorders. As discovered by Rutter and others, not all people exposed to severe early trauma develop psychopathology, indicating differences in resiliency. Loci modifying stress resiliency are likely to be numerous and are at this point largely unknown. Several that have been identified are monoamine oxidase A (MAOA) (Ducci et al., 2008), the serotonin transporter (SLC6A4) (Caspi et al., 2003; Hariri et al., 2002), COMT (Zubieta et al., 2003), neuropeptide Y (NPY, an anxiolytic neuropeptide) (Zhou et al., 2008), and FKBP5 (an accessory protein for the glucocorticoid receptor) (Binder et al., 2008). These are functional loci, and most have been demonstrated to alter the relevant molecular and neuroscience-based intermediate phenotypes for stress response and emotion, as will next be discussed.
INTERMEDIATE PHENOTYPES IN ADDICTION
One strategy to discover gene effects for complex diseases that are amalgams of distinctly different pathologies is to split the lumped disease phenotypes or deconstruct them into etiologic components.
If it were readily feasible to parse addictions into subtypes based on clinical features, this would undoubtedly have already been done, because of the burden to society that addictions pose, and commendable efforts have been made in this area. However only a few clinical subtypes (e.g., Cloninger Type I/Type II) have been identified; these subtypes are themselves amalgams. Additional measures are needed. These include measures that do not reflect causal processes but are indicators of severity or response; for example, neuroimaging and neuropsychological indicators can track long-term damage consequent to exposure and that may impede recovery. These can be termed biomarkers. Other measures are intermediate phenotypes that access mediating mechanisms of genetic and environmental influences on the brain. For addictions these would include measures of emotional response and stress resiliency, reward, executive cognitive control and impulsivity, and metabolism and pharmacodynamic response. Furthermore, heritable, disease-associated, intermediate phenotypes are termed endophenotypes (Gottesman and Gould, 2003). Whether a particular biomarker, intermediate phenotype, or endophenotype is useful clinically or in genetic studies depends on multiple factors, including measurement properties, frequency, sensitivity, specificity, independence from other measures, and whether the measurement can be practically obtained from living patients or populations and families seen in the clinic or that might be enrolled in genetic studies.
Alcohol-induced flushing is an intermediate phenotype that is practically useful both clinically and for genetic studies. It is a protective alcohol-related endophenotype influenced by specific alleles mediating variation in ethanol metabolism, as will be discussed. Flushing is a clinically accessible, pharmacokinetic endophenotype that can be more thoroughly characterized and explained via psychophysiological measures, assays of alcohol metabolism, and genotyping of the variants that alter alcohol metabolism. Low pharmacodynamic response to alcohol is a genetically influenced intermediate phenotype that predicts enhanced risk (Heath et al., 1999). Level of response is mainly a result of intrinsic pharmacodynamic variation in neuronal response to alcohol rather than variation in metabolism (Schuckit et al., 1998). Some functional variants have been associated with low response to alcohol, including variants in the serotonin transporter gene (SLC6A4) and the gene encoding the α6 subunit of the gamma-aminobutyric acid receptor A (GABRA6) (Hu et al., 2005). However, the genetic origins of variation in alcohol response are largely, or even entirely, unknown.
Many other intermediate phenotypes relevant to addictions are known, and several have been shown to be more strongly influenced by gene action than addiction itself. Focusing on brain-related measures, electrophysiologic, neuropsychologic, neuroendocrinologic, and neuroimaging measures have been tied to addictions. Electrophysiologic measures including both baseline EEG and evoked responses are both addiction-associated and heritable, baseline EEG power in different spectral bands being highly heritable (van Beijsterveldt and van Baal, 2002). Neuroimaging accesses the structure and activity of circuits underlying emotion, reward, and craving. Studies incorporating neuroimaging have indirectly linked genes to processes and neuronal networks relevant in addiction. Interindividual differences in emotional response are predicted by amygdala activation after exposure to emotional imagery and stressful stimuli, and thus the study of fMRI-detected metabolic activation of amygdala and functionally related regions such as hippocampus has captured effects of genes that influence emotion (Bevilacqua and Goldman, 2011). As will be discussed, SLC6A4 and MAOA are among the genes influencing emotion and more strongly amygdala activation. Activations of frontal cortex by executive cognitive tasks are modulated by genetic variants of both COMT and MAOA, thus providing an important clue, again by indirect means, to impairments in impulse control that are important in predisposition to addiction. Recently it was discovered that a functional polymorphism of the mu-opioid receptor gene (OPRM1 Asn398Asp) modulates activation of the ventral striatum by reward, and this same polymorphism has directionally consistent effects on naltrexone response in alcoholics treated with this medication (Anton et al., 2008; Oslin et al., 2003) and controversial effects on risk of addictions. Depending on the properties of intermediate phenotypes including proximity to gene action, measurement error, and specificity, they offer an important starting point for unraveling the genetics of addiction, and arguably most of the successes in gene identification are directly tied to intermediate phenotypes and functional alleles that alter them in ways that are directionally consistent with physiology.
GENE IDENTIFICATION FOR ADDICTION
In candidate gene studies, genes influencing relevant physiology are rationally selected based on pathways, mechanisms and molecules of addiction or the pharmacokinetics and pharmacodynamics of the drug. A functional locus may be identified and specifically studied, representing a candidate locus, or allele. In genome-wide studies, the genome is interrogated in hypothesis-free fashion via an expanding array of methods including GWA, linkage in families, and genome and exome sequencing. For addictions, genes have primarily been identified via the candidate gene approach. However, GWA and deep sequencing has yielded several “hits,” including a functional locus altering nicotine addiction vulnerability. Both methods are increasingly convergent as detailed information is developed on the locations of functional elements, for example in the ENCODE project, which aims to identify the functional elements genome-wide.
ALCOHOL METABOLIZING GENES: ADH1B AND ALDH2
Polymorphisms of alcohol dehydrogenase IB (ADH1B) and aldehyde dehydrogenase 2 (ALDH2) influence alcohol consumption and risk of alcohol use disorders as well as damage secondary to alcohol exposure. In adults these enzymes catalyze consecutive steps in ethanol metabolism although other enzymes including catalase and cytochrome P450 also play significant roles. Acetaldehyde, the product of ADH, is toxic and potently releases histamine thereby triggering flushing, an aversive reaction that can include headache, nausea, and palpitations. If aldehyde dehydrogenase is blocked by medications including metronidazole and disulfiram (a drug that is actually used to help alcoholics maintain abstinence), flushing follows the ingestion of small quantities of alcohol. Acetaldehyde is a mutagen, leading to increased risk of upper GI cancer in moderate drinkers who carry the dominantly acting ALDH2 Lys487 allele, as some 500 million people do (Brooks et al., 2009).
At ADH1B, the His48 allele increases catalytic efficiency of the enzyme. In East Asian populations both ADH1B His48 and ALDH2 Lys487 are highly abundant, and His48 is present in many individuals of Jewish ancestry. A protective effect of His48 on alcohol dependence has been demonstrated in European and African populations as well as East Asians, in whom this relationship was originally observed (Bierut et al., 2012), and both the ADH1B and ALDH2 flushing alleles are associated with enhanced risk of upper GI cancers.
The ADH1B and ALDH2 His48 and Lys487 alleles are ancient in the human lineage. They occur on characteristic and highly diverged haplotypes at these genes, are found at high frequency in particular populations, and are unlikely to have been selected to high frequency to protect against alcoholism. It has been hypothesized that they have been selected to high frequencies in East Asians because of their ability to alter susceptibility to protozoal infections, including infections of the gut such as amebiasis (Goldman and Enoch, 1990). This hypothesis is unproven; however, metronidazole is used to treat protozoal infections and potently inhibits aldehyde dehydrogenase.
GENES MODERATING MONOAMINE NEUROTRANSMITTERS
Monoamine neurotransmitters modulate emotionality, cognition, and reward. Therefore, it would be predicted that genes that contain functional loci that can alter function of monoamine neurotransmitters should alter liability to addiction. Congruently, catechol-O-methyltransferase (COMT) and the serotonin transporter (SLC6A4), and MAOA (as already mentioned) have been implicated in vulnerability to addiction and other psychiatric diseases that share these etiologic mechanisms.
COMT plays an important role in the regulation of dopamine in the prefrontal cortex and metabolizes dopamine, norepinephrine, and other catechols in various brain regions. The Val158Met polymorphism alters COMT activity via an effect on enzyme stability, the Met158 allele being three- to fourfold less active (Lachman et al., 1996). Because of the higher activity of the Val158 allele, it is predicted that it leads to lower frontal dopamine levels, and it has been associated with dopamine-modulated cognitive differences including inefficient frontal lobe function as seen by neuroimaging (Egan et al., 2001). Amphetamine improved executive cognitive function in val/val homozygotes, but not met/met homozygotes indicating that baseline dopamine levels of val/val homozygotes may be suboptimal for frontal cortical function (Mattay et al., 2003). Reciprocally, and leading to COMT being called a Warrior versus Worrier gene (Goldman, 2012), the Met158, allele has been associated with decreased stress resilience and increased emotionality, including anxiety in women (Enoch et al., 2003). Met 158 was associated with increased pain–stress induced brain activations as well a slower pain threshold and stronger affective response to pain (Zubieta et al., 2003). The evidence for association of COMT to addiction is mixed, including negative studies, studies implicating Val158, and others implicating Met158 (Tammimaki and Mannisto, 2010). These divergent results may be reconciled if the Val158Met polymorphism, which does affect behavior, has countervailing effects on different mechanisms of vulnerability to addiction. The Val158 allele was in excess among methamphetamine, nicotine, and polysubstance addicts (Vandenbergh et al., 1997). On the other hand, the Met158 allele was more common in late-onset alcoholics in Finland (Tiihonen et al., 1999).
Via reuptake, the serotonin transporter (SLC6A4) regulates synaptic serotonin. Reflecting the diverse effects of this neurotransmitter on mood, appetite, and impulse control, serotonin-specific reuptake inhibitors are widely prescribed for a variety of different mental illnesses. HTTLPR, a polymorphism located in the promoter region of this gene and involving variable numbers of an imperfect repeat located in tandem (a VNTR), probably remains the most studied locus in psychiatric genetics. Three relatively common HTTLPR alleles are functional, altering transcriptional efficiency (Hu et al., 2006) and expression of the transporter expression in the brain (Heinz et al., 2000). Reduction of function HTTLPR genotypes has been associated with anxiety, depression, and alcoholism, but inconsistently in the case of each disease. Overall, the predictive effect of HTTLPR on any clinically measured behavior is modest. However, the effects appear stronger in the context of stress exposure moderating impact of stressful life events on depression and suicidal behavior. Carriers of low-transcribing HTTLPR genotypes exhibited more depression and suicidality following stressful life events (Csapi et al., 2003).
Although contradictory meta-analyses have not clarified the GxE relationship of HTTLPR to behavior, neuroscience evidence has supported a role for HTTLPR regulation of emotion and response to stress, and provided clues to mechanism. Low activity HTTLPR genotypes predict stronger metabolic responses of brain regions intrinsically involved in processing of emotional stimuli and predict smaller volumes of some of these structures and diminished connectivity with regions that modulate their activity. The low-activity “s” allele predicts increased amygdala reactivity to emotional stimuli under the passive viewing condition (Hariri et al., 2002) and also predicts reduced amygdala volume and reduced functional coupling between the amygdala and ventromedial prefrontal cortex (Heinz et al., 2005). The effects of reduction of function serotonin transporter alleles on emotion have been validated in two animal models. An orthologous polymorphism designated rs-5HTTLPR is found in the Rhesus macaque (Macaca mulatta). In macaques, early life stress exposure together with rs-HTTLPR again leads to enhanced stress response and emotionality later in life. Carriers of the low-expression genotype that were separated from their mothers at an early age were more stress reactive and drank more alcohol later in life (Barr et al., 2004), a difference that appears to be in part mediated by the hypothalamic–pituitary–adrenal axis.
GENOMIC APPROACHES IN ADDICTIONS
GENOME-WIDE ASSOCIATION
Genome-wide association is a genomic approach to the analysis of genetically influenced traits that queries all genes and unknown functional genetic elements in a hypothesis-free way for their potential effects. The importance of hypothesis-free, global approaches is emphasized by discoveries by the ENCODE consortium and others that there are many previously unrecognized functional elements in the genome, and many of these have poorly understood regulatory functions. Furthermore, GWA, when performed with appropriate attention to confounds such as ethnic stratification and other systematic errors that can upward bias statistical results, and with confirmation and validation, for example by identification of the functional locus, is a direct route to making causal inferences about the roles of genes in behavior. In other words, the implication of the genetic linkage is more than correlational: a locus, or one nearby the linked locus, modulates the behavior. The genome-wide significance threshold is usually approximately 10-8 corresponding to a single locus p value of 0.05. Within this statistical framework, GWA will detect the effects of common alleles (MAF >5%) of at least moderate effect (e.g., odds ratio >1.2), requiring thousands of cases and controls for such small effect sizes. As later discussed, less common alleles have been linked to addictions by resequencing and by linkage studies in families and founder populations in whom particular rare alleles are common. A salient advantage of GWA is that data can be combined across studies using a common pool of SNPs that are either directly genotyped or imputable.
Addictions GWA has yielded a confirmed functional locus for nicotine addiction, located in the CHRNA5- CHRNA3-CHRNB4 gene-cluster on chromosome 15 (Bierut et al., 2007; Thorgeirsson et al., 2008). These genes encode subunits of the nicotinic acetylcholine receptor (nAChR), which is a ligand-gated ion channel activated by nicotine. A functional locus influencing nicotine addiction was identified in this region. CHRNA5 Asp398Asn alters nicotine dependence/heavy smoking, pleasurable response to smoking, smoking quantity, and smoking persistence (Bierut et al., 2007; Thorgeirsson et al., 2008), as well as secondary susceptibilities to lung cancer and vascular disease (Amos et al., 2008) and smoking cessation (Munafo et al., 2011). The locus has a modest effect on addiction itself. Each copy of Asn398 accounts for ~0.5% of the variance in cigarettes smoked/day but more strongly predicts the strength of connectivity between anterior cingulate and ventral striatum (Hong et al., 2010) a circuit that in turn modulates nicotine craving, as shown in Figure 52.1.
Furthermore, (α4β2)2α5 receptors that differed only in containing the CHRNA5 Asn398 allelic form had altered response to agonist. Other studies have shown that Asp398 lowers calcium permeability and increases short-term desensitization, but does not alter receptor sensitivity to activation (Kuryatov et al., 2011). Whether the Asp398Asn polymorphism alters nicotine response or connectivity of reward circuits, or both, remains an open question.
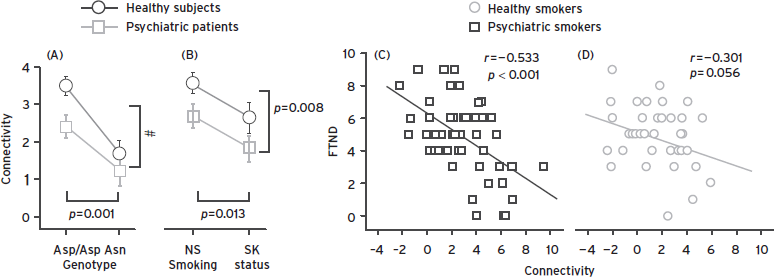
Figure 52.1 The CHRNA5 Asn398 allele, identified as a risk factor in nicotine addiction by GWA, predicts weakness of a dorsal anterior cingulate/ventral striatal circuit whose weakness predicts nicotine craving. Right panel: Strength of the circuit, as measured by resting state functional connectivity (rsFC) predicts nicotine craving. Middle panel: the CHRNA5 Asn398 allele predicts smoking status. Left panel: The Asn398 allele predicts weaker connectivity of the circuit. (From Hong et al. (2010). A genetically modulated, intrinsic cingulate circuit supports human nicotine addiction. Proc Natl Acad Sci U S A. 107(30):13509–13514.)
For alcohol consumption, a meta-analysis of GWA in 12 European populations, totaling 26,316 individuals, identified the autism susceptibility 2 gene (AUTS2). The SNP implicated may moderate AUTS2 expression in prefrontal cortex, and expression of AUTS2 (Schumann et al., 2011). Several genome-wide significant loci were identified for resting EEG traits that are addiction-associated (Hodgkinson et al., 2010), illustrating the power of combining GWAS with intermediate phenotypes.
RARE AND UNCOMMON VARIANTS
The common disease/common variant paradigm has been disturbed by the failure of GWA to account for a substantial portion of inherited variation of complex disease, and by discovery of extensive rare and uncommon genetic variation in humans. These rarer variants of stronger effect may account for a large portion of genetic vulnerability to common diseases. Advances in DNA sequencing technologies facilitate the detection of rare variants; however, the full impact of this approach depends on study of effects of these variants in founder populations and families where they are common, statistical combination of multiple rare allele with the same probable functional impact, and further development and use of other convergent information such as evidence of effect on molecular function or effects of homologous variants in model organisms including the mouse.
Rare variants relevant to addiction are known within the serotonin receptor 2B gene (HTR2B) (Bevilacqua et al., 2011), MAOA (Brunner et al., 1993), and CYP, the CYP2A6 variants even predicting fMRI responses to smoking cues (Tang et al., 2012). Both HTR2B and MAOA influence impulsivity, findings involving these genes having close parallels in animal models, and the CYP alleles have pharmacogenetic effects on response to nicotine that are also lawfully expected due to alteration of nicotine metabolism.
MAOA, located on the X chromosome, encodes monoamine oxidase A, which metabolizes monoamine neurotransmitters including norepinephrine, dopamine, and serotonin. The main effect of a common allele leading to lower MAOA expression is enhanced impulsivity, an important mediating trait in addictions. A stop codon found in eight males in one Dutch family led to impulsivity, carrier females being unaffected (Brunner et al., 1993) A common MAOA variable-number tandem repeat polymorphism can lead to lower MAOA enzyme activity and has been a model for understanding the role of context in gene effects on complex behaviors in which impulsivity plays an important role. Gene × environment interaction was observed in a longitudinally studied cohort of boys, with the lower expression allele and childhood adversity together predicting vulnerability to conduct disorder (Caspi et al., 2002) as confirmed by others. In women, who unlike males can be heterozygous for the low activity MAOA allele, results are mixed, and may depend on severity of adversity. A parallel GxE effect on ASPD was observed in a sample of Native American women with a combined effect of childhood sexual abuse and low activity MAOA genotype on risk of both alcoholism and ASPD (Ducci et al., 2008), and an allele dosage effect in the presence of childhood sexual abuse. Complicating the relationship of MAOA to impulsivity and aggression are other releasers of these behaviors. Testosterone independently predicts aggression, but males with high expression MAOA genotypes did not show the effect, which was limited to males with the low expression MAOA genotype that is apparently permissive for this behavior (Sjoberg et al., 2008). Similarly, MAOA genotype can interact with alcohol consumption, another disinhibitor of behavior (Tikkanen et al., 2009), and as will next be described, alcohol consumption also leads to impulsive aggression in carriers of a stop codon in HTR2B (Bevilacqua et al., 2010).
HTR2B is a serotonin receptor that, in part because of genetic studies implicating it in behavior, has been discovered to be widely expressed in the brain wherein it is found on approximately 40% of dopamine neurons in VTA, where it regulates dopamine release (L. Maroteaux, submitted). A stop codon disabling HTR2B (Q20*) is common in Finland, but rare or absent in other populations surveyed worldwide. The stop codon is linked to severe impulsive aggression, ASPD, and alcoholism, with an effect on impulsive aggression that is strongly modulated, or dependent, on inebriation. This HTR2B stop codon was discovered by sequencing impulsive and aggressive offenders who underwent psychiatric evaluation because of the extreme nature of their crimes. The stop codon is several times as common in these individuals as in Finnish controls, and is cotransmitted with impulsive behavior, including alcoholism, in families. Carriers of the stop codon who committed violent crimes did so while inebriated with alcohol, and were cognitively normal. In the Finnish population, most carriers are unaffected, indicating the *20 is a factor in impulsive behavior and addiction but not sufficient itself. Htr2b -/- mice exhibit higher novelty seeking and impulsivity, and enhanced responses to activating drugs including a D1 dopamine receptor agonist and cocaine.
Variants, many of which are rare, of CYP2A6 (cytochrome P450, Family 2, subfamily A, polypeptide 6) alter risk of nicotine addiction via metabolism, the enzyme accounting for 70% of initial nicotine metabolism.
CONCLUSIONS
Vulnerability to addictive agents is widespread because addictive agents activate natural reward systems and abuse is kindled by variations in executive cognitive control and emotional response that are widespread in populations. Addictive agents of a variety of types are readily accessible, and in fact access is increasing, with legalization of gambling and marijuana, development of designer drugs and new ways to administer drugs, and the nearly universal availability of the internet and electronic devices to which people can become addicted. For these reasons, it seems paradoxical that addictions are moderately to highly heritable. However, there are many sources of resilience against addiction. These include social norms, positive family and peer influences, the diversity of rewarding activities available to most people, and conscious decisions to reduce harm and limit exposures. Because there are many routes to addictions, protective and predisposing genetic variants have diverse actions. Furthermore, no single genetic variant is likely to be highly predictive for an addiction, and indeed no highly predictive locus emerged from GWA studies that provide a global view of the relative contribution of individual loci.
Our state of knowledge of the complex causal landscape of genetic effects on addictions is limited and insufficient for diagnostic prediction. However, the status quo is subject to change. Although it would be unwise to predict a timeline, it is likely that addictions will be redefined via a combination of neuroscience-based measures, improved understanding of the behavioral interface with the environment, and by cataloging the genetic variations that influence risk and understanding their mechanisms.
Addiction is a categorical end-diagnosis, assuming a cutoff between normal and abnormal, although many non-addicted individuals have the same predisposing factors and even subthreshold addictive behaviors. Other complex diagnostic amalgams, syndromes, and chief complaints have been successfully refined and redefined at the etiological level. For example, deafness was deconstructed and reconstructed into a series of better defined diseases through a combination of clinical, neuroanatomic, psychophysiologic, cellular, and genetic approaches. We recognize that the same functional consequence—namely hearing loss—has many distinct causes ranging from exposure to chemicals and loud noises to adverse panoply of genetic variants, with profound implications for prevention and intervention. As compared to the ear, the brain is a more complex organ. However, the brain’s structures, functions, and outputs, both behavioral and molecular, are increasingly amenable to measurement, and as discussed earlier, the genetic study of these measures, many which represent intermediate phenotypes for addiction, offers a different and potentially better path to the identification of genes that alter vulnerability, as illustrated (Fig. 52.2). However, and as also indicated in the figure, because addictions are clinically defined, the genes that exert large effects on intermediate phenotypes such as executive cognition, stress resilience, and reward may not be strong clinical predictors of addictions as presently defined. In similar fashion, none of the deafness genes explains a large portion of deafness as a whole.

Figure 52.2 As presently defined, addictions are heritable but remote to the action of particular genes. This is because of intervening effects of environmental context and different mechanisms of vulnerability. These mechanisms, including differences in brain emotional response, reward, executive cognitive function, and drug metabolism have become partially accessible in the form of molecular and psychophysiologic intermediate phenotypes.
What might be the impact of gene identification in addiction? The CFTR gene does not strongly predict pneumonia, or fever, because it is found in only a fraction of patients presenting with these problems. However, CFTR is the defining factor in cystic fibrosis, a disease that with fuller understanding also involves pancreatic insufficiency, infertility, and non-genetic factors that critically shape clinical course. Although a gene therapy for cystic fibrosis has not emerged, careful attention to non-genetic factors in cystic fibrosis patients has revolutionized the care, and clinical course, of this distinct genetically defined subgroup. The same general approach may also be effective in addictions—the genetically-based diagnosis serving to identify a population to which intensive intervention has to be targeted. For addictions, there are now some positive indications that identification of specific genetic influences is a step towards individualization of treatment. For example, a common functional missense variant of the mu-opioid receptor (OPRM1 Asn40Asp) is associated with altered reward function (Ramchandani et al., 2011) and alcoholic carriers of the Asp40 allele showed greater clinical improvement when treated with the opioid antagonist naltrexone (Anton et al., 2008; Oslin et al., 2003). Similarly, CHRNA5 Asn398Asp (Munafo et al., 2011) and other genes have been reported to influence response to smoking cessation treatment.
In conclusion, addictions are multistage, chronic, and relapsing diseases with heritabilities that range from 0.39 (hallucinogens) to 0.72 (cocaine). Genes influence each stage of the disease. Because addictions are in parts volitional, inborn, and determined by experience, they pose unique medical and moral challenges. The genetic basis of addictions is largely unknown; however the inheritance of addictions and knowledge of a few specific genes altering vulnerability have established that these are illnesses that can be understood via neuroscience-based chains of causality. Neither heritability of addictions nor the existence of addiction genes establishes that addictions are diseases, because many benign traits are also heritable, but inheritance and gene discoveries point to chains of causality that lead some people and not others to addiction. Many putative addiction genes have been discovered by association studies although few appear are well-validated. Encouragingly, among the small group of genes whose effects appear validated (e.g., OPRM1, ADH1B, ALDH2, MAOA, SLC6A4, NPY, COMT, CHRNA5 and CYP2A6) are several that have potential clinical utility, for example to understand alcohol-related flushing and upper GI cancer risk (ADH1B and ALDH2), variation in nicotine metabolism (CYP26), and naltrexone treatment response (OPRM1). These genes act through a variety of mechanisms: by altering drug metabolism, by altering affinity of receptors for addictive drugs (CHRNA5, which may also alter circuitry of reward), and by altering stress response, emotion, and behavioral control, as do genes such as MAOA, NPY, HTR2B, COMT, and HTTLPR. The genes that influence addiction repeat and enhance broader themes in the neuroscience of addiction.
DISCLOSURES
Dr. Goldman has no conflicts of interest to disclose.
REFERENCES
Amos, C.I., Wu, X., et al. (2008). Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat. Genet. 40(5):616–622.
Anton, R.F., Oroszi, G., et al. (2008). An evaluation of mu-opioid receptor (OPRM1) as a predictor of naltrexone response in the treatment of alcohol dependence: results from the Combined Pharmacotherapies and Behavioral Interventions for Alcohol Dependence (COMBINE) study. Arch. Gen. Psychiatry 65(2):135–144.
Barr, C.S., Newman, T.K., et al. (2004). Interaction between serotonin transporter gene variation and rearing condition in alcohol preference and consumption in female primates. Arch. Gen. Psychiatry 61(11):1146–1152.
Bevilacqua, L., and Goldman, D. (2011). Genetics of emotion. Trends Cogn. Sci. 15:401–408.
Bevilacqua, L., Doly, S., et al. (2011). A population-specific HTR2B stop codon predisposes to severe impulsivity. Nature 468(7327):1061–1066.
Bierut, L.J., Goate, A.M., et al. (2012). ADH1B is associated with alcohol dependence and alcohol consumption in populations of European and African ancestry. Mol. Psychiatry 4:445–450.
Bierut, L.J., Madden, P.A., et al. (2007). Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum. Mol. Genet. 16(1):24–35.
Binder, E.B., Bradley, R.G., et al. (2008). Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA 299(11):1291–1305.
Brooks, P.J., Goldman, D., et al. (2009). Alleles of alcohol and acetaldehyde metabolism genes modulate susceptibility to oesophageal cancer from alcohol consumption. Hum. Genomics 3(2):103–105.
Brunner, H.G., Nelen, M., et al. (1993). Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science 262(5133):578–580.
Caspi, A., and Moffitt, T.E. (2006). Gene–environment interactions in psychiatry: joining forces with neuroscience. Nat. Rev. Neurosci. 7(7):583–590.
Caspi, A., McClay, J., et al. (2002). Role of genotype in the cycle of violence in maltreated children. Science 297(5582):851–854.
Caspi, A., Sugden, K., et al. (2003). Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301(5631):386–389.
Cloninger, C.R. (1987). Neurogenetic adaptive mechanisms in alcoholism. Science 236(4800):410–416.
Ducci, F., Enoch, M.A., et al. (2007). Increased anxiety and other similarities in temperament of alcoholics with and without antisocial personality disorder across three diverse populations. Alcohol 41(1):3–12.
Ducci, F., Enoch, M.A., et al. (2008). Interaction between a functional MAOA locus and childhood sexual abuse predicts alcoholism and antisocial personality disorder in adult women. Mol. Psychiatry 13(3):334–347.
Ducci, F., Kaakinen, M., et al. (2011). TTC12-ANKK1-DRD2 and CHRNA5-CHRNA3-CHRNB4 influence different pathways leading to smoking behavior from adolescence to mid-adulthood. Biol. Psychiatry 69:650–660.
Egan, M.F., Goldberg, T.E., et al. (2001). Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc. Natl. Acad. Sci. USA 98(12):6917–6922.
Enoch, M.A., Gorodetsky, E., et al. (2011). Functional genetic variants that increase synaptic serotonin and 5-HT3 receptor sensitivity predict alcohol and drug dependence. Mol. Psychiatry 16(11):1139–1146.
Enoch, M.A., Xu, K., et al. (2003). Genetic origins of anxiety in women: a role for a functional catechol-O-methyltransferase polymorphism. Psychiatr. Genet. 13(1):33–41.
Goldman, D., and Bergen, A. (1998). General and specific inheritance of substance abuse and alcoholism. Arch. Gen. Psychiatry 55(11):964–965.
Goldman, D., and Enoch, M.A. (1990). Genetic epidemiology of ethanol metabolic enzymes: a role for selection. World. Rev. Nutr. Diet. 63:143–160.
Goldman, D., Oroszi, G., et al. (2005). The genetics of addictions: uncovering the genes. Nat. Rev. Genet. 6(7):521–532.
Goldman, D. (2012). Our Genes, Our Choices. Boston: Elsevier.
Gottesman, I.I., and Gould, T.D. (2003). The endophenotype concept in psychiatry: etymology and strategic intentions. Am. J. Psychiatry 160(4):636–645.
Hariri, A.R., Mattay, V.S., et al. (2002). Serotonin transporter genetic variation and the response of the human amygdala. Science 297(5580):400–403.
Heath, A.C., Madden, P.A., et al. (1999). Genetic differences in alcohol sensitivity and the inheritance of alcoholism risk. Psychol. Med. 29(5):1069–1081.
Heinz, A., Braus, D.F., et al. (2005). Amygdala-prefrontal coupling depends on a genetic variation of the serotonin transporter. Nat. Neurosci. 8(1):20–21.
Heinz, A., Jones, D.W., et al. (2000). A relationship between serotonin transporter genotype and in vivo protein expression and alcohol neurotoxicity. Biol. Psychiatry 47(7):643–649.
Hodgkinson, C.A., Enoch, M.A., et al. (2010). Genome-wide association identifies candidate genes that influence the human electroencephalogram. Proc. Natl. Acad. Sci. USA 107(19):8695–8700.
Hong, L.E., Hodgkinson, C.A., et al. (2010). A genetically modulated, intrinsic cingulate circuit supports human nicotine addiction. Proc. Natl. Acad. Sci. USA 107(30):13509–13514.
Hu, X., Oroszi, G., et al. (2005). An expanded evaluation of the relationship of four alleles to the level of response to alcohol and the alcoholism risk. Alcohol Clin. Exp. Res. 29(1):8–16.
Hu, X.Z., Lipsky, R.H., et al. (2006). Serotonin transporter promoter gain-of-function genotypes are linked to obsessive-compulsive disorder. Am. J. Hum. Genet. 78(5):815–826.
Kendler, K.S., Karkowski, L.M., et al. (1999). Genetic and environmental risk factors in the aetiology of illicit drug initiation and subsequent misuse in women. Br. J. Psychiatry 175:351–356.
Kendler, K.S., Myers, J., et al. (2007). Specificity of genetic and environmental risk factors for symptoms of cannabis, cocaine, alcohol, caffeine, and nicotine dependence. Arch. Gen. Psychiatry 64(11):1313–1320.
Kendler, K.S., Prescott, C.A., et al. (2003). The structure of genetic and environmental risk factors for common psychiatric and substance use disorders in men and women. Arch. Gen. Psychiatry 60(9):929–937.
Kendler, K.S., Schmitt, E., et al. (2008). Genetic and environmental influences on alcohol, caffeine, cannabis, and nicotine use from early adolescence to middle adulthood. Arch. Gen. Psychiatry 65(6):674–682.
Kessler, R.C., Crum, R.M., et al. (1997). Lifetime co-occurrence of DSM-III-R alcohol abuse and dependence with other psychiatric disorders in the National Comorbidity Survey. Arch. Gen. Psychiatry 54(4):313–321.
Krueger, R.F., Hicks, B.M., et al. (2002). Etiologic connections among substance dependence, antisocial behavior, and personality: modeling the externalizing spectrum. J. Abnorm. Psychol. 111(3):411–424.
Kuryatov, A., Berrettini, W., et al. (2011). Acetylcholine receptor (AChR) alpha5 subunit variant associated with risk for nicotine dependence and lung cancer reduces (alpha4beta2)alpha5 AChR function. Mol. Pharmacol. 79(1):119–125.
Lachman, H.M., Papolos, D.F., et al. (1996). Human catechol-O-methyltransferase pharmacogenetics: description of a functional polymorphism and its potential application to neuropsychiatric disorders. Pharmacogenetics 6(3):243–250.
Mattay, V.S., Goldberg, T.E., et al. (2003). Catechol O-methyltransferase val158-met genotype and individual variation in the brain response to amphetamine. Proc. Natl. Acad. Sci. USA 100(10):6186–6191.
Munafo, M.R., Johnstone, E.C., et al. (2011). CHRNA3 rs1051730 genotype and short-term smoking cessation. Nicotine Tob. Res. 13: 982–988.
Oslin, D.W., Berrettini, W., et al. (2003). A functional polymorphism of the mu-opioid receptor gene is associated with naltrexone response in alcohol-dependent patients. Neuropsychopharmacology 28(8):1546–1552.
Ramchandani, V.A., Umhau, J., et al. (2011). A genetic determinant of the striatal dopamine response to alcohol in men. Mol. Psychiatry 16(8):809–817.
Schuckit, M.A. (1998). Biological, psychological and environmental predictors of the alcoholism risk: a longitudinal study. J. Stud. Alcohol. 59(5):485–494.
Schumann, G., Coin, L.J., et al. (2011). Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proc. Natl. Acad. Sci. USA 108(17):7119–7124.
Sjoberg, R.L., Ducci, F., et al. (2008). A non-additive interaction of a functional MAO-A VNTR and testosterone predicts antisocial behavior. Neuropsychopharmacology 33(2):425–430.
Tammimaki, A.E., and Mannisto, P.T. (2010). Are genetic variants of COMT associated with addiction? Pharmacogenet. Genomics. 20(12):717–741.
Tang, D.W., Hello, B., et al. (2012). Genetic variation in CYP2A6 predicts neural reactivity to smoking cues as measured using fMRI. Neuroimage. 60(4):2136–2143.
Thomasson, H.R., Edenberg, H.J., et al. (1991). Alcohol and aldehyde dehydrogenase genotypes and alcoholism in Chinese men. Am. J. Hum. Genet. 48(4):677–681.
Thorgeirsson, T.E., Geller, F., et al. (2008). A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 452(7187):638–642.
Tiihonen, J., Hallikainen, T., et al. (1999). Association between the functional variant of the catechol-O-methyltransferase (COMT) gene and type 1 alcoholism. Mol. Psychiatry 4(3):286–289.
Tikkanen, R., Sjoberg, R.L., et al. (2009). Effects of MAOA-genotype, alcohol consumption, and aging on violent behavior. Alcohol Clin. Exp. Res. 33(3):428–434.
van Beijsterveldt, C.E., and van Baal, G.C. (2002). Twin and family studies of the human electroencephalogram: a review and a meta-analysis. Biol Psychol. 61(1–2):111–138.
Vandenbergh, D.J., Rodriguez, L.A., et al. (1997). High-activity catechol-O-methyltransferase allele is more prevalent in polysubstance abusers. Am. J. Med. Genet. 74(4):439–442.
Zhou, Z., Zhu, G., et al. (2008). Genetic variation in human NPY expression affects stress response and emotion. Nature 452(7190):997–1001.
Zubieta, J.K., Heitzeg, M.M., et al. (2003). COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science 299(5610):1240–1243.