62 | STRUCTURAL, FUNCTIONAL, AND MOLECULAR NEUROIMAGING BIOMARKERS FOR ALZHEIMER’S DISEASE
JAMES B. BREWER, JORGE SEPULCRE, AND KEITH A. JOHNSON
Alzheimer disease (AD) is currently a major public health challenge and one that poses an even greater threat for the future as the population ages. The role of neuroimaging in AD diagnosis and in the general assessment of cognitive impairment in the elderly has greatly evolved over the past two decades. In the past, structural neuroimaging was used merely to support the physical exam in the exclusion of focal lesions that could contribute to a patient’s cognitive impairment. In most cases, any such discrete lesion sufficient to impair cognition would also be detectable through a careful examination, so the majority of clinical neuroimaging interpretations were negative and unhelpful. In fact, the added benefit of neuroimaging used in this manner was, and continues to be, controversial, as evidenced by variation in professional society guidelines regarding the use of neuroimaging in this setting (Knopman et al., 2001; Rabins et al., 2007). However, the development of magnetic resonance imaging (MRI) and positron emission tomography (PET) biomarkers that directly assess the degree of regional neurodegeneration or dysfunction and, more recently, neuroimaging biomarkers that assess pathological hallmarks of AD have brought transformative change to the field and shifted interest to earlier pathophysiological events. While much effort remains focused on the study of AD dementia and mild cognitive impairment (MCI), now preclinical stages have become a major focus. The move toward expanding the scope of inquiry into preclinical stages and younger age groups is motivated in part by the recognition that the presymptomatic phase of the illness is of 10–20 years duration and in part by the related need to enable earlier disease modifying intervention. The evolving view of AD pathophysiology has recently been fueled by rapid developments in amyloid-beta (Aβ) imaging and fluid biomarkers; however, simultaneous advances have been realized in other types of AD biomarkers. Neuroimaging biomarkers are now routinely incorporated into AD clinical trials and are increasingly used in clinical practice. This chapter will broadly describe the development and use of these biomarkers as they relate to AD.
ESTIMATES OF NEURODEGENERATION IN ALZHEIMER’S DISEASE
QUANTITATIVE MAGNETIC RESONANCE IMAGING BIOMARKERS
VOLUMETRIC MAGNETIC RESONANCE IMAGING
The slowly progressive neurodegeneration of AD is reflected in brain structural changes that can be appreciated at the macroscopic level. Medial temporal regions, such as the entorhinal cortex and hippocampus, are typically affected earliest, consistent with the hallmark memory problems that usually accompany disease onset. Macroscopic structural change broadly mirrors the pathological spread of the disease, with atrophy subsequently apparent in lateral temporal as well as medial and lateral parietal association cortex followed by frontal regions and, finally, primary sensorimotor cortices (McDonald et al., 2009). Though the changes are slow, over time the accumulation of atrophy is readily apparent to visual inspection (Fox et al., 1996; Scheltens et al., 1992).
Thus, in the absence of a suitable biofluid marker of neural damage and the unacceptable invasiveness of brain biopsy, a great deal of research has focused on the promise of neuroimaging and direct visualization of brain structure to assess the likelihood of neurodegeneration in individual patients. Typically, such approaches have leveraged the improvements in image quality afforded by MRI, though the quantitative nature of computed tomography (CT), including high spatial fidelity, and recent advances in achievable image spatial resolution, tissue contrast, and overall quality might suggest future promise for CT in quantitative assessment of neurodegeneration. The discussion that follows regarding structural neuroimaging will nonetheless focus on approaches using MRI.
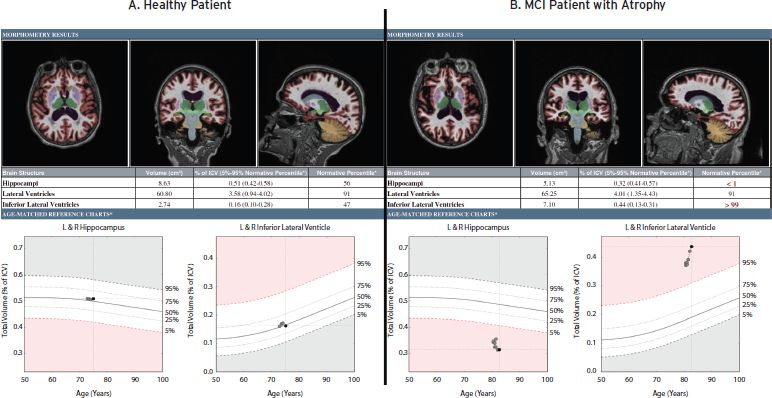
Direct assessment of putative brain atrophy can make use of semiquantitative approaches to rate severity of volume loss or employ tools for quantification of brain structure volumes, volumetric MRI (vMRI), to provide measurements that could be tracked over time or compared to a normative database (for review, see Jack, 2011). In clinical practice, volumetrics may inform the clinical assessment by either supporting or calling into question the impression, previously based solely on clinical history and examination, that neurodegenerative disease is present and possibly causing the complaint (Fig. 62.1).
It is not surprising that the hippocampus has been targeted as the structure most likely to provide a reliable volumetric biomarker of neurodegeneration in AD. Damage to hippocampal tissue is severe early in the disease and, owing to its somewhat cylindrical structure, hippocampal borders are relatively easy to delineate for volume measurement. However, it should be noted that hippocampal atrophy is not specific to AD, nor is all AD associated with severe hippocampal atrophy (for review of focal variants of AD, see Kramer and Miller, 2000). Some degree of hippocampal volume loss is expected even in healthy aging, and the structure’s volume is further correlated with overall intracranial volume. Thus, the effects of age and intracranial volume, and possibly gender and race, must be accounted for. An individual’s prior history of brain trauma, alcoholism, drug abuse, and vascular risk factors such as hypertension and smoking, would also likely influence the measure, so it is unlikely that a distinct “cutoff” in hippocampal volume could be identified that will reliably predict AD risk across patients.
Instead, hippocampal volume is more likely to be useful as a measure that helps assess the likelihood that neurodegeneration is present rather than a diagnostic for AD. As such, the measure should be seen by treating physicians as one additional data point to assist in their clinical impression. Most clinicians are familiar with tests that shape, rather than define, a clinical impression, and such tests are valuable nonetheless. An example might be the measurement of hemoglobin in the setting of a patient complaint of fatigue. A normal hemoglobin directs attention to etiologies other than anemia, and a finding of low hemoglobin supports, but does not assure, that anemia is causing the fatigue. The assessment and management depend heavily on the clinical setting and ancillary factors associated with the measurement. In practical terms, this means that the relevance of a vMRI finding to a particular case will be determined by the treating physician, rather than by the radiologist. However, the radiologist can provide additional qualitative information from the images that lend further value to the quantitative information.
Despite intense focus on translation of hippocampal volumetry to the clinical realm for AD assessment, ancillary measures of atrophy may be critical to improve interpretability of hippocampal volume. Several research groups have examined the value of combining regional volumetric measures across cortical and subcortical structures to more completely describe the spatial pattern of changes associated with AD (Davatzikos et al., 2008; Dickerson et al., 2009; McEvoy et al., 2009; Vemuri et al., 2011). A pattern with broad consistency across techniques emerges, where atrophy is prominent in medial and lateral temporal and parietal regions, moderate in frontal regions, and minimal in primary sensorimotor regions. The combination of regions, naturally, improves classifier sensitivity and specificity beyond that achieved through the use of a single region. However, the ability to translate such advanced approaches to the clinical environment has not yet been demonstrated and may not be practical. In particular, those that rely on support vector machines to obtain data-derived regions with highest classifier performance might be overly reliant on features of the data set used to train the classifier, and so performance may not necessarily generalize to the wider population seen in clinical practice. Further, given anatomical variation in cortical folding, derivation of regional cortical volume or thickness is more challenging and computationally expensive than derivation of volumes for most subcortical structures, such as the hippocampus and ventricular subregions.
Figure 62.1 Example volumetric reports for two subjects enrolled in a longitudinal quantitative imaging study. (A) A 75-year-old healthy subject who remained stable and was without evidence of hippocampal neurodegeneration or temporal horn enlargement during scanning over two years.(B) An 82-year-old, cognitively impaired subject who progressed from MCI to AD at the third year of follow-up. This patient was a non-carrier of APOE4 genotype who, by CSF testing, had elevated phospho-tau and reduced levels of amyloid beta 42 in cerebrospinal fluid. vMRI shows evidence of hippocampal neurodegeneration and ex vacuo dilatation of the temporal horns during scanning over two years.
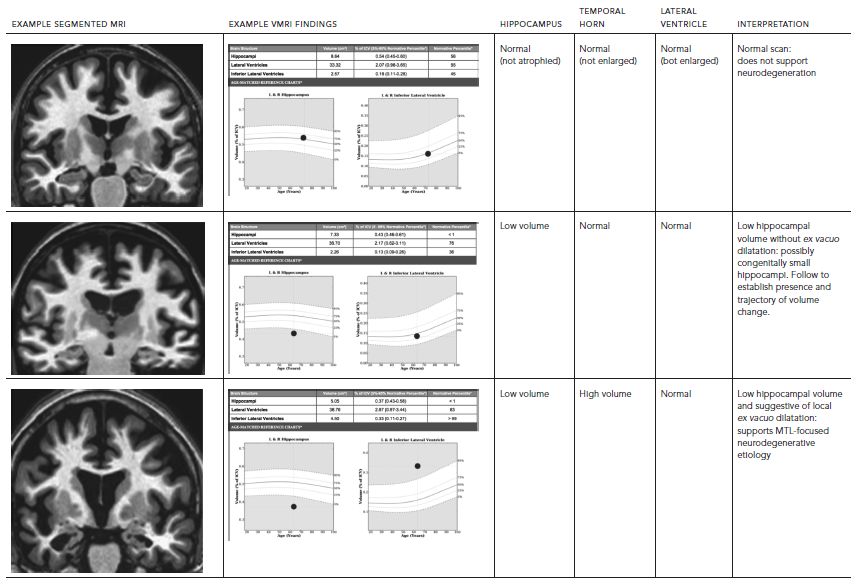
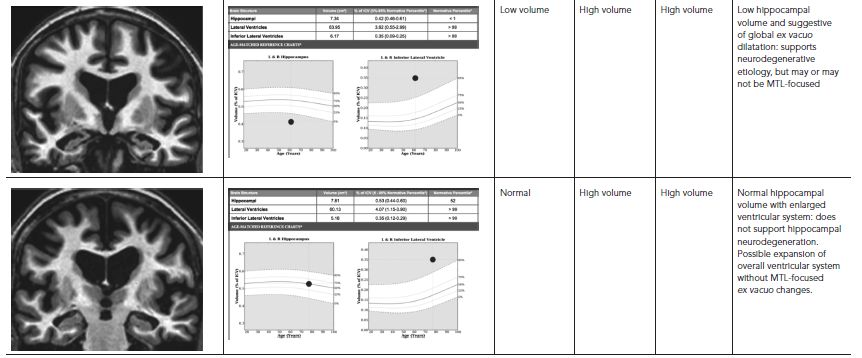
Indeed, some contextual information about a hippocampal volume measurement can be provided by additionally assessing temporal horn volume. Hippocampal and wider medial temporal lobe (MTL) degeneration is often associated with ex vacuo expansion of the temporal horn of the lateral ventricle. Therefore, temporal horn volumetry with comparison to normative values can provide complementary evidence of concurrent ex vacuo dilatation of this ventricular structure. Such a finding could support that the individual’s hippocampus was previously larger and had undergone degeneration, as opposed to having been congenitally small. Further, measurement of the entire ventricular system and its relationship to norms can assist interpretation of the temporal horn volume. The combination of measures would inform assessment of likelihood that the temporal horn enlargement is more specifically due to regional ex vacuo changes (if, along with low hippocampal volume, only the temporal horn is abnormally large), rather than due to general ventricular system expansion (if both temporal horn and the entire ventricular system are abnormally large). Evidence of MTL-focused atrophy might provide an early sign of neurodegenerative disease, including AD, which typically affects these structures first. Table 62.1 provides examples of possible clinical interpretations when hippocampal, temporal horn, and overall lateral ventricle volumes are examined in combination.
Finally, it should be noted that, even when a typical pattern is noted or when markedly out of the normal range, a single measurement in time cannot definitively point to a neurodegenerative etiology, since many of the aforementioned traumatic or congenital factors are associated with low volumes of structures not seen to progressively deteriorate. Therefore, the most powerful evidence for ongoing neurodegeneration might be demonstration of accelerated progressive deterioration in structure volumes through longitudinal imaging. Thus, beyond comparing a patient’s brain to a normative database, a patient’s brain volumes would be quantitatively compared to those measured for that individual at a prior time. Differences in scanner hardware and software adds significant variability to measures of very subtle volume change across time, and, even at a single site, equipment changes are frequent in clinical practice, so this remains a significant challenge to obtaining accurate measurement of change across time. To be most relevant to clinical practice, meaningful information would need to be obtained across a relatively brief period of follow-up, preferably around one year. This would require highly robust methods, including correction for subtle scanner-specific distortions that could assess anatomical changes with high enough precision to reliably distinguish across individual subjects miniscule differences in rate of atrophy (e.g., hippocampal volume loss ranges from 1–5% across healthy elderly, MCI, and AD). Such precision is a significant challenge, particularly if the equipment changes between scans. Nevertheless, equipment manufacturers are becoming more aware of the potential value of quantitative neuroimaging, which lends hope that procedures could be standardized to improve accuracy of measurements despite equipment and software variability.
Though several caveats remain to be considered, working groups gathered through efforts of the Alzheimer’s Association and the National Institute on Aging recently published guidelines for revised diagnostic criteria for AD and MCI that incorporate vMRI and other biomarkers (Albert et al., 2011; McKhann et al., 2011). Tools for vMRI have already been established in large radiological practices to assess the degree to which an individual’s brain structure volumes fit within the normative range adjusted for age, sex, and intracranial volume. Relatively recent Food and Drug Administration approval of an automated vMRI tool has permitted further research of its applicability in unselected clinical cohorts encountered through everyday practice as well as in the highly selected cohorts studied in AD clinical trials.
DIFFUSION-WEIGHTED IMAGING
Magnetic resonance physics allows flexible approaches to examine water molecule behavior within tissue. Structural neuroimaging may focus on achieving high anatomical detail through examining magnetic contrast related to tissue content, itself, or it may focus on measuring more general effects on contrast due to surrounding tissue structure or magnetic properties. In diffusion-weighted imaging, the effect that surrounding tissue structure has on water movement is paramount. MRI contrast can be achieved by examining the degree of water movement within the region; water movement is restricted by surrounding anatomy, and the more restricted, the greater the signal. In directional diffusion weighted imaging, such as diffusion tensor imaging or diffusion spectrum imaging, both the magnitude and direction of water movement is collected, informing whether water movement is more hindered in one direction versus another.
As white matter tracts in the brain are one of the major anatomical features that might direct water to diffuse in one direction (parallel the white matter) relative to another (perpendicular to the white matter), directional diffusion weighted imaging has become a favored research tool with which to examine white matter integrity in neurodegenerative diseases such as AD (for review, see Oishi et al., 2011). Indeed prior cognitive research has described AD as a disease of “disconnection” between disparate cortical brain areas, so the ability to examine fiber tract disruption is a compelling reason to examine the effects of AD on white matter tracts as evidenced by changes in diffusion-weighted signal.
However, it remains to be seen whether degeneration of fiber tracts is best measured through changes in directionality, as opposed to magnitude, of diffusion. When comparing impaired patients to healthy controls, studies have found regional group differences in fractional anisotropy, a measure of directional diffusion, yet some studies have found even more robust differences in mean diffusivity, a directionless measure of diffusion magnitude. Neurodegeneration in AD seems more likely to cause loss of fibers within defined tracts rather than a shift in direction of those tracts. Thus directional measures, though useful for identifying tract location, might be less powerful for detecting tract degeneration than simpler measures of diffusion magnitude. Further, the directional diffusion signal can be influenced by a number of subtle factors, so export of this tool to the clinical realm will face a number of challenges, not least of which will be the development of normative ranges for determining whether a patient’s scan is normal or abnormal when placed in context with the general healthy population. Nevertheless, the approach shows promise as a technique that shows a complementary anatomy not seen in conventional structural imaging.
TABLE 62.1. Influence of temporal horn and lateral ventricle measures on the interpretation of volumetric MRI (vMRI) findings
FUNCTIONAL MAGNETIC RESONANCE IMAGING
Though other approaches exist, most functional MRI is based on the Blood Oxygen Level-Dependent (BOLD) effect, which allows MRI sensitivity to variations in relative levels of oxyhemoglobin and deoxyhemoglobin that vary in association with regional brain activity (for review, see Brown, Perthen, Liu, & Buxton, 2007). Given the probability that changes in neural function precede frank neurodegeneration and changes in structure, functional MRI (fMRI) has been studied extensively as a potential biomarker to identify the earliest brain changes associated with AD. Broadly, methods applied to fMRI can be divided into two categories: task-dependent fMRI and task-free, or “resting-state,” fMRI (rsfMRI).
In task-dependent fMRI, subjects in the scanner might be asked to perform a defined “activation” task that alternates with or occurs separately from a “control” task while BOLD signal is recorded. The control task differs from the activation task in a way that is designed to isolate and identify the brain activity linked to a targeted cognitive function, which is a cognitive function used during the activation task, but not during the control task. This “cognitive subtraction” technique suffers from a number of factors that limit interpretation, particularly when applied to a diseased population. For example, an observed signal difference between AD and controls might be related to uncontrolled variation in task difficulty or attentional resources that must be dedicated to performing the task, despite matched performance across groups. This adds to more general concerns about factors of subject motion and brain atrophy that must be accounted for in all neuroimaging biomarkers of AD. Such factors might be particularly problematic for fMRI biomarkers, because the effect of confounders can be amplified by the analysis approach (Seibert and Brewer, 2011).
In rsfMRI, subjects are instructed to simply lie still in the scanner, perhaps with guidance to keep their eyes open throughout the session, but functional data are nonetheless collected without regard to any specific mental activity. Instead, it is the interregional correlation in BOLD signal fluctuations that provides the marker of interest. The pattern of interregional correlation is remarkably consistent, in that signal from a particular brain region tends to be tightly linked with signal from other regions that show relatively stable correlations across subjects and scanning sessions. These stable interregional correlations have been termed “functional connectivity” and the phrase “functional network” is often used to refer to regions linked by their covariance in BOLD signal. Several distinct functional networks have been described, including the attentional/salience network of bilateral frontal regions and anterior cingulate gyrus, the sensorimotor network of bilateral primary motor and sensory cortex, and the default mode network of medial and lateral parietal, medial and lateral temporal, and medial frontal cortex.
The latter default network is the most studied and, owing to its remarkable overlap with the spatial distribution of amyloid deposition and atrophy in AD (Buckner et al., 2009; Greicius et al., 2004; Raichle et al., 2001; Sepulcre et al., 2010), has garnered the most interest for its potential as an early biomarker of the disease. A consistent finding is that interregional correlations in the default network are reduced in AD. However, concerns remain about the general effects of atrophy and motion on the ability to identify functional correlations themselves. A more compelling finding is the identification of default network reductions in amyloid positive individuals who are asymptomatic, who would not be expected to exhibit large differences in atrophy or motion from those who are amyloid negative (Sperling et al., 2009). Default network correlations have been noted even to be reduced in individuals gene positive for autosomal dominant AD before symptoms have begun (Sperling et al., 2012). Such findings suggest the tremendous potential for fMRI as an early biomarker of AD, though its translation to use in the clinical setting will require a great deal more research.
MAGNETIC RESONANCE SPECTROSCOPY
Magnetic resonance spectroscopy (MRS) has long showed promise for detection of regional chemical changes associated with neurodegeneration, inflammation, or gliosis, and so it has been a technique of interest for detecting such changes in AD. It provides information about relative concentrations of key chemicals, such as N-acetylaspartate, creatinine, and myoinositol, and there is strong evidence that the regional concentration of these chemicals varies with the neurodegeneration seen in AD (for review, see Tran et al., 2009). While specialized research laboratories have consistently demonstrated robust discrimination between clinical groups, suggesting potential value in assessment of individual cases, improved standardization of approach is needed before MRS can be widely applied in multisite clinical trials or generally in the clinical setting.
POSITRON EMISSION TOMOGRAPHY
FLUORODEOXYGLUCOSE IMAGING
CLINICAL USE OF FLUORODEOXYGLUCOSE PET
Fluorodeoxyglucose (FDG) PET is a marker of brain metabolism with limited but established utility in differentiating AD from other brain pathologies that manifest as cognitive impairment. Under normal circumstances, the physiological level of glucose use reflected by FDG PET is due primarily to regional synaptic activity, and FDG uptake in nonhuman primates has been correlated with levels of synaptophysin in histological studies (Rocher et al., 2003). Since glucose consumption is by far the major contributor to the brain’s energy budget, FDG uptake reflects the full range of brain energy requirements, including protein and lipid synthesis, as well as maintenance of electrochemical gradients used in neural activity (Raichle et al., 2001). Alterations in synaptic activity detectable with FDG reflect chronic synaptic dysfunction (or loss) as well as perturbed functional status observed within shorter time intervals, such as during specific cognitive or motor task performance.
Fluorodeoxyglucose metabolism is assessed regionally with PET so that anatomic patterns of relative hypometabolism may be both visually evident and expressed quantitatively; quantification may involve arterial blood sampling or may alternatively use internal tissue reference standards. No single pathognomonic FDG PET finding specifies the presence of the clinical syndrome of AD dementia or the presence of AD pathology. However, the anatomic pattern of relative FDG hypometabolism in AD is characteristic and is considered to be an endophenotype of AD, that is, a consistently affected hypometabolic group of brain structures that has been strongly associated with both the clinical syndrome of AD dementia and the postmortem finding of definite AD (Jagust et al., 2007). The AD endophenotype of FDG hypometabolism comprises the posterior midline cortices of the parietal (precuneus) and posterior cingulate, the inferior parietal lobule, posterolateral portions of the temporal lobe, hippocampus, and medial temporal cortices (Foster et al., 1983; Minoshima et al., 1997). This pattern is present in AD, is linearly related to dementia severity, and is associated with subsequent clinical decline and conversion to AD (Chetelat et al., 2003; Jagust et al., 2007). It is seen less severely or consistently in MCI, and to some extent, in Aβ positive normal elderly (Caselli et al., 2008; Cohen et al., 2009; Langbaum et al., 2009). Because the AD-like pattern of FDG hypometabolism is associated both with the clinical features of established AD dementia, and with AD pathology at postmortem (Jagust et al., 2007), it may be used to differentiate AD from frontotemporal lobar degeneration (FTLD) and to some extent from dementia with Lewy bodies (DLB). Specifically, it is typical to find frontotemporal hypometabolism in FTLD, and occipital—in addition to the AD-like temporoparietal—hypometabolism, in DLB. Other applications have been proposed for FDG PET, particularly to detect individuals who are likely to develop AD dementia in the future (Chetelat et al., 2003) ; however, this has not yet been successfully implemented in clinical practice.
UNDERSTANDING THE ALZHEIMER’S DISEASE BRAIN WITH FLUORODEOXYGLUCOSE
In recent years neuroimaging techniques such as FDG PET and fMRI that are capable of revealing different aspects of the brain function have provided key insights about functional organization and its spatial distribution in AD. For instance, the metabolic-anatomic endophenotype of AD described earlier is sometimes asymmetric and predominantly temporoparietal in the early stages, but later progresses to involve prefrontal and heteromodal areas largely overlapping with the aforementioned default mode network.
As AD progresses from presymptomatic to established AD dementia, regional glucose metabolism gradually worsens (Bateman et al., 2012; Jack, 2011) and the spatial pattern of progression may be conditioned by anatomic interconnectivity, in parallel with the progression of pathological processes (Arnold et al., 1991; Pearson and Powell, 1989). AD neurodegeneration involves specific functional networks of the human brain (Greicius et al., 2004; Seeley et al., 2009), and this feature has led to a new reformulation of the old hebbian principal: “not only neurons that fire together wire together, but also neurons that wire together die together” Buckner, R.L., (Sepulcre et al., 2012). It may be possible to track FDG metabolic changes along specific anatomic pathways that relate to underlying pathologic progression, and also to relate these findings to evidence of regional atrophy (Villain et al., 2010).
Some investigators have reported that FDG hypometabolism is detectable along the AD trajectory prior to the appearance of cognitive symptoms and signs of neurodegeneration in individuals at increased risk for AD (Jack et al., 2010; Jagust et al., 2006). Thus, the AD-like pattern of hypometabolism or hypoperfusion predicts cognitive decline in subjects that eventually convert to AD (Jagust et al., 2006) and in carriers of autosomal dominant AD mutations (Bateman et al., 2012; Johnson et al., 2012). It is also associated with the progression of the clinical dementia rating scale sum-of-boxes in both AD and MCI subjects (Chen et al., 2010). Importantly, several studies have found that glucose alterations are not merely caused by concurrent atrophy, and therefore regional FDG reduction is an independent piece of information in AD (Johnson et al., 2012). Moreover, FDG metabolism continues to decline even in advanced stages of the disease when other pathological factors reach a plateau (Engler et al., 2006; Jack et al., 2010). It is likely that these declines reflect the combined effects of several potentially overlapping processes; these include specific genetic effects, mitochondrial dysfunction, oxidative stress, excitotoxicity, synaptic and neuronal failure triggered by Aβ, neurofibrillary tangle accumulation, and/or other factors, together with other downstream degenerative processes. It is likely that FDG PET is sensitive to several components of the neurodegenerative process, and thus separate biomarker representation may be required to track specific processes.
Finally, several studies have shown that FDG hypometabolism matches, to some extent, the brain distribution of amyloid deposits, particularly in the default mode network (Cohen et al., 2009; Engler et al., 2006; Klunk et al., 2004). However, this relationship is not settled and other groups have not found meaningful or strong associations particularly at later stages of the disease (Furst et al., 2012; Rabinovici et al., 2010). In general, glucose hypometabolism in frontal regions is less evident than in temporoparietal cortex, whereas amyloid uptake is usually somewhat greater in frontal regions Ikonomovic, M.D., (Klunk et al., 2004). Therefore, the association of amyloid deposition with reductions of glucose utilization remains unclear. Many important challenges remain unsolved in AD research and one of them is the integration of structural, functional, and molecular neuroimaging findings.
SUMMARY AND LIMITATIONS OF FLUORODEOXYGLUCOSE PET
FDG PET is a widely available technology that may help diagnostic accuracy in neurodegenerative illnesses, particularly when earlier stages of cognitive impairment are possibly due to AD pathology or to FTLD. However, further work is needed to determine the diagnostic value of FDG when assessing preclinical stages of AD. Like other imaging biomarkers, FDG does not indicate clear spatial patterns that distinguish between normal aging and MCI or AD (Caselli et al., 2008; Chetelat et al., 2003; Langbaum et al., 2009). As with these other modalities, the observed heterogeneity in FDG patterns of metabolism may be the result of individual differences, particularly in cognitive reserve, or comorbid conditions of aging such as vascular disease, as well as to idiosyncratic factors. Neural dysfunction likely precedes neurodegenerative changes, and so FDG PET holds great promise for monitoring cognitive transition from preclinical to established AD (Furst et al., 2012; Jack et al., 2010), Nevertheless, the sensitivity of FDG PET relative to that of vMRI remains to be established despite the existence of head-to-head comparisons performed in the same patients (Karow et al., 2010). Sensitivity of a biomarker depends not only on acquisition modality, but also on analysis techniques, which continue to evolve. Therefore, it is still unknown which variable will best be associated with cognitive symptoms for continued translation into clinical use in the earliest stages of disease. Although the availability of FDG PET has increased dramatically over the past decade and the standardization of methods has improved substantially, costs of PET technology remain high, and cost effectiveness in dementia diagnosis has been difficult to demonstrate.
AMYLOID IMAGING
CLINICAL USE OF AMYLOID IMAGING
Amyloid-β (Aβ) dysmetabolism and subsequent deposition is a defining neuropathological feature of AD (Braak and Braak, 1991; Selkoe, 2006), and the ability to detect brain Aβ deposits during life has revolutionized clinical research in AD Ikonomovic, M.D., (Klunk et al., 2004). The in vivo visualization of brain Aβ deposition with amyloid PET is virtually equivalent to demonstration of the pathology at autopsy, as has been repeatedly demonstrated (Bacskai et al., 2003; Ikonomovic et al., 2008; Klunk et al., 2004; Sojkova et al., 2011) for rare exceptions, see Klunk, 2011. Amyloid PET detects fibrillar amyloid because PET tracers bind to the beta-sheet protein structure that forms when Aβ polymerizes (Ikonomovic et al., 2008). Amyloid PET can thus detect the Aβ that is a major component of the damage occurring in patients with cognitive impairment caused by AD and is always seen in AD dementia patients at autopsy. A negative amyloid PET scan generally indicates very few or no amyloid deposits and greatly reduces the likelihood that any cognitive impairment is caused by AD. A positive scan indicates that moderately to severely elevated numbers of β-amyloid deposits are present. A fundamental concept of amyloid PET is that the images indicate presence of Aβ deposits, but do not reliably distinguish between normal, MCI, and AD dementia, and thus do not relate directly to these traditional clinical diagnostic categories (Johnson et al., 2012). In other words, it is critically important to note that a positive amyloid PET scan does not by itself establish any clinical diagnosis, including that of AD dementia. The test may be positive in normal older individuals as well as in other clinical entities such as DLB.
The highly stereotyped anatomic pattern of amyloid PET ligand binding to areas of high connectivity forms the basis for the amyloid PET endophenotype of AD (Arnold et al., 1991; Braak and Braak, 1991; Buckner et al., 2009). Thus, areas that are highly interconnected, such as precuneus, posterior cingulate, inferior parietal and lateral temporal cortices (i.e., portions of the DMN), are typically affected, but the earliest and most heavily involved is often the middle frontal cortex, which is part of the cognitive control network. The time course of amyloid deposition typically involves these vulnerable regions; however, there is substantial variability in regional deposition at early stages, and at later stages, additional brain regions including primary cortices are also affected.
Most published reports to date involve the C-11-labeled agent N-methyl 11C-2-(4-methylaminophenyl)-6-hydroxybenzothiazole, also known as Pittsburgh Compound-B (PIB; half-life 20 minutes) Ikonomovic, M.D., (Klunk et al., 2004). Other compounds made with the more convenient F-18 radio-label (half-life 110 minutes) are increasingly available for research and even clinical use (Johnson et al., 2012).
The reported correspondence of amyloid positivity with traditional clinical diagnoses is as follows: more than 90% of clinically diagnosed AD patients and approximately 60% of MCI patients are classified as amyloid positive with PET (Johnson et al., 2012). Similar proportions have been reported using CSF biomarkers of Aβ (Fagan et al., 2007).
Longitudinal studies are just now emerging, but when MCI subjects have been followed over one to three years after PET, approximately one-half of those who were amyloid positive at baseline converted to AD dementia and approximately 10% of the amyloid negative subjects converted to AD dementia. These studies are ongoing, and continued clinical and histopathological follow-up will be required because it is possible that the clinical diagnoses would not be confirmed at autopsy.
A substantial fraction of apparently healthy, elderly cognitively normal subjects have Aβ deposits detected by PET (10% to 50%) (Jack et al., 2010; Johnson et al., 2012), and such individuals are termed preclinical AD based on the hypothesis that the progressive accumulation of amyloid places them at higher risk for developing clinical syndrome of AD dementia (Jack et al., 2010). The magnitude and timing of such risk remains the subject of active investigation; however, several AD prevention clinical trials are proceeding on the basis of AD risk defined in this way. The analysis of early changes and preclinical stages of AD is critical to understand the transition to symptomatic forms of the disease. Recent data have shown a continuum of accumulation of in vivo amyloid protein from early stages (i.e., elderly cognitively normal controls) to symptomatic phases of the disease (Jack et al., 2010; Villain et al., 2012; Villemagne et al., 2011) (see Fig. 62.2).
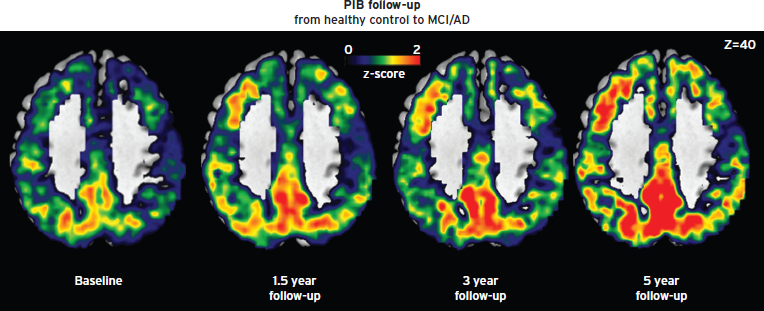
Figure 62.2 Longitudinal PET amyloid imaging of an individual subject who progressed from healthy to dementia.
Amyloid PET may have substantial clinical utility in differential diagnosis of established dementia, particularly in younger individuals in whom the prognosis and clinical management would differ depending on the underlying pathology. For example, the underlying pathology of FTLD does not involve Aβ, and amyloid PET is reported to be negative in clinical FTLD that has been confirmed at autopsy Furst, A.J., (Rabinovici et al., 2011). Other entities in the differential diagnosis of dementia that could potentially be informed by amyloid PET include late life depression or other psychiatric disorder with cognitive impairment, prion disease (Villemagne et al., 2009) and semantic dementia with tauopathy (Drzezga et al., 2008). The situation for parkinsonian dementing syndromes is somewhat more complex, because amyloid deposition is a common but not universal feature of DLB, and may or may not be seen in Parkinson’s disease with dementia (Gomperts et al., 2008). Similarly, posterior cortical atrophy (Migliaccio et al., 2009), progressive aphasia Furst, A.J., (Rabinovici et al., 2008) and corticobasal syndrome Furst, A.J., (Rabinovici et al., 2011) present substantial clinical and histopathological heterogeneity and may not at present be distinguishable with amyloid PET.
SUMMARY AND LIMITATIONS OF AMYLOID IMAGING
Amyloid PET is an emerging technology and the FDA approved an amyloid radiotracer, Florbetapir F18, in April of 2012. At the time of this writing (mid-2012), amyloid PET has not been widely deployed in clinical practice. Whatever clinical value that could be expected is entirely dependent upon the availability of good quality images and accurate interpretation. Although clinical FDG PET scanning is generally available for nonbrain indications, experience with brain imaging is quite variable and depends on local circumstances.
Since essentially no one is free of risk for developing amyloid deposition, the clinical utility of amyloid PET could theoretically extend to nearly any circumstance in which the underlying basis of suspected neurodegenerative disease could be Aβ. However, the high cost of PET and its uncertain impact on clinical management have limited enthusiasm for widespread adoption of (both FDG and) amyloid PET when expert dementia specialist evaluation is available. While utility at this time remains to be precisely defined, the availability of disease modifying AD therapy will almost certainly change the level of demand. At present, appropriate use criteria are still being discussed and refined, but would optimally integrate amyloid PET technology into the existing framework of dementia evaluation, so that amyloid status can be placed in the appropriate context of medical, neurological, neuropsychological, and neuroimaging data.
ADNI AND STANDARDIZATION OF NEUROIMAGING BIOMARKERS
The successful incorporation of neuroimaging biomarkers into multi-site clinical trials has relied upon unprecedented levels of collaboration across the major equipment manufacturers to identify approaches that would yield consistency sufficient to provide meaningful enrichment and outcome measures. Much of the groundwork for these efforts was provided through the Alzheimer’s Disease Neuroimaging Initiative (ADNI). The ADNI included an intensive preparatory phase to develop and assess image preprocessing steps that would enhance longitudinal stability of the measures. Variability across imaging sites, manufacturers, and equipment upgrades remains a source of noise in multi-site imaging studies, but the preparatory phase of ADNI and its cross-institutional collaborative efforts were critical to establishing the potential for the incorporation of neuroimaging biomarkers in large-scale clinical trials, which is already nearly universal. As of April 2012, 298 papers were published or in press using the ADNI study open access data, and the image repository had dispensed more than one million image downloads (http://www.adni-info.org/scientists/Pdfs/09_Green_Data_and_Publications.pdf). Since the initial ADNI study, a number of spinoff and continuation studies have begun within and beyond the field of AD. As further testament to the success and influence of the study, a number of international efforts have begun that have been modeled on the methods of ADNI (Fig. 62.3).
Figure 62.3 Map representation of worldwide studies examining biomarkers of AD. Multisite imaging and biofluid biomarker studies are planned or underway in Europe, China, Taiwan, South Korea, Japan, and Australia. These studies were influenced by the pivotal ADNI study in North America. (Source: World map template from http://presentationmagazine.com.)
ADDED VALUE OF BIOMARKERS IN COMBINATION
A significant advance attributable to ADNI and its readily available dataset is the more complete description of the regional structural and metabolic changes associated with AD, a finding that has been consistent across a number of analysis approaches. Results from ADNI have suggested that structural, metabolic, and amyloid biomarkers may be used in a complementary manner, given a putative temporal progression of biomarker positivity across the development of the disease (Jack et al., 2010). Such hypothesized progression of disease markers in typical, late onset, AD remains, as yet, unsupported through directed experimentation and therefore highly controversial, but several studies that have examined a combination of biomarkers for predicting progression of symptoms toward dementia have found them to provide additive information that may be related to differential sensitivity and dynamic changes that vary with disease stage. Specifically, it appears that amyloid deposition is detectable 10–20 years before the onset of symptoms, whereas tau positivity within the CSF and vMRI changes are relatively concurrent with cognitive changes detectable with specialized neuropsychological testing (see, e.g., Heister et al., 2011; Bateman et al., 2012). Thus, while amyloid positivity suggests that a patient is at higher risk for development of AD, atrophy or hypometabolism suggests that the patient has entered the neurodegenerative stage of disease and is at risk for imminent clinical decline (Fig. 62.4).
The apparent complementary and differential disease-stage sensitivity of amyloid testing and vMRI or FDG PET provides additional leverage for clinical trial design, such that amyloid testing can be incorporated at screening to enrich the trial with subjects that have objective evidence of the targeted disease (or even the targeted protein), and baseline vMRI or FDG PET can provide complementary information about disease stage. Further, the imaging studies provide information that is relatively independent from and orthogonal to the cognitive complaint that led to subject selection, whereas cognitive measures provide information that overlaps with complaint and so could yield positive results even for etiologies that are not neurodegenerative. Consistent with this, studies using ADNI data have shown remarkable power advantages for enrichment based on baseline atrophy that could not have been achieved using any of the available cognitive measures from ADNI (McEvoy et al., 2010). As such, quantitative PET and MRI neuroimaging show promise for enriching clinical trials of prodromal AD with individuals likely to progress to AD and to decline in the period of study (Jack et al., 2008; Kovacevic et al., 2009; McEvoy et al., 2009).
In addition to potential usages in clinical trial enrichment, vMRI and amyloid biomarkers appear to provide complementary secondary outcome measures when acquired across time. Neuroimaging of brain structure through vMRI provides a measure that does not vary based on day-to-day fluctuations in the cognitive abilities of subjects that are caused by wakefulness, medication effects, motivation, and the like, and it has been shown to be less variable across time than cognitive measures (Weiner et al., 2012). Amyloid measures, though less sensitive to disease progression, may support putative effects of the therapy on disease pathology. The combination of regional amyloid measures with regional measures of brain atrophy may be especially powerful, because changes in amyloid burden can be colocalized with structural neuroimaging to examine interactions between amyloid and atrophy as well as a study drug’s effects on each. Neuroimaging biomarkers might therefore support claims that a medication’s effect on cognition is likely due to halted neurodegeneration or reduction of pathological burden rather than due to a brief symptomatic benefit.
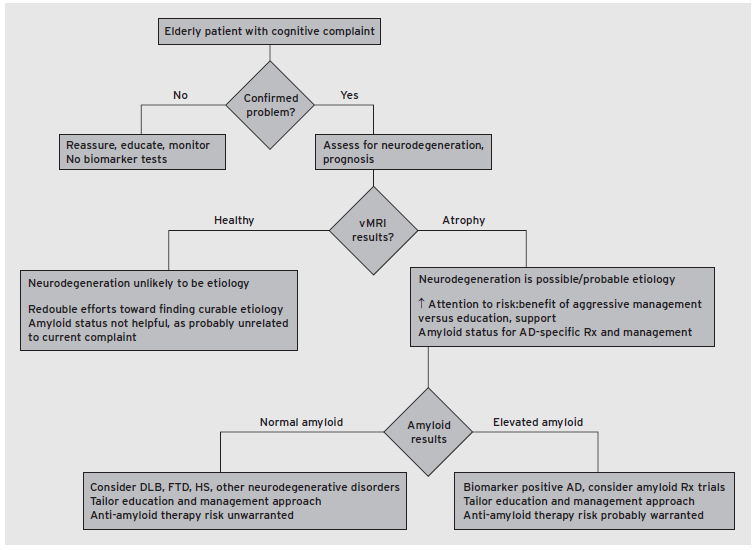
Figure 62.4 Example decision tree presenting one approach to incorporating vMRI and amyloid biomarkers in clinical practice. HS, Hippocampal sclerosis. (McEvoy, L.K., & Brewer, J.B. (2012). Biomarkers for the clinical evaluation of the cognitively impaired elderly: amyloid is not enough. Imag Med, 4(3), 14.)
CONCLUSIONS
Neuroimaging in the assessment of the elderly with cognitive impairment has changed from its previously limited role, in ruling out discrete lesions, to an integral role, in quantitatively assessing the cardinal components of neurodegeneration, neural dysfunction, and pathological burden observable in AD. Both MRI and PET benefit from wide flexibility in application, given the various anatomical, functional, and pathological features that might be assessed with each. The path toward clinical application was guided primarily by large scale, multisite pharmaceutical trials, in which a more direct assessment of the effects of therapies was sought. Such efforts demonstrated the feasibility of obtaining these measures across sites in clinical practice, though a great deal of work remains to determine how best to integrate the information into clinical assessment, and how each can be used to inform predictive prognosis and guide management in individual patients (Brewer, 2009). Though publicly accessible databases of clinical trial enrollees have assisted in these efforts, the recent availability of highly standardized acquisition techniques and FDA-approved quantitative imaging approaches will allow such research to proceed in the unselected populations encountered in clinical practice.
DISCLOSURES
Dr. Brewer is supported by NINDS K02 NS067427, NIA U01 AG10483, NIA P50 AG005131, NIA R01 AG034062. He is an investigator for and receives research funds from Janssen Alzheimer Immunotherapy. He also has received research funds from General Electric Medical Foundation; holds stock options in Cortechs Labs., Inc; and has served on advisory boards for Elan, Avanir, Bristol-Myers-Squibb, and Lilly Biomarker Business Unit.
Dr. Sepulcre has no conflicts of interest to disclose.
Dr. Johnson has served as a site investigator for Avid, Pfizer, Janssen, Bristol-Myers-Squibb, and as a consultant to Bayer, Bristol-Myers-Squibb, Genzyme, and Siemens.
REFERENCES
Albert, M.S., DeKosky, S.T., et al. (2011). The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7(3):270–279.
Arnold, S.E., Hyman, B.T., et al. (1991). The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb. Cortex 1(1):103–116.
Bacskai, B.J., Hickey, G.A., et al. (2003). Four-dimensional multiphoton imaging of brain entry, amyloid binding, and clearance of an amyloid-beta ligand in transgenic mice. Proc. Natl. Acad. Sci. USA 100(21):12462–12467.
Bateman, R.J., Xiong, C., et al. (2012). Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 367(9):795–804.
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82(4):239–259.
Brewer, J.B. (2009). Fully-automated volumetric MRI with normative ranges: translation to clinical practice. Behav. Neurol. 21(1):21–28.
Brown, G.G., Perthen, J.E., et al. (2007). A primer on functional magnetic resonance imaging. Neuropsychol. Rev. 17(2):107–125.
Buckner, R.L., Sepulcre, J., et al. (2009). Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer’s disease. J. Neurosci. 29(6):1860–1873.
Caselli, R.J., Chen, K., et al. (2008). Correlating cerebral hypometabolism with future memory decline in subsequent converters to amnestic pre-mild cognitive impairment. Arch. Neurol. 65(9):1231–1236.
Chen, K., Langbaum, J.B., et al. (2010). Twelve-month metabolic declines in probable Alzheimer’s disease and amnestic mild cognitive impairment assessed using an empirically pre-defined statistical region-of-interest: findings from the Alzheimer’s Disease Neuroimaging Initiative. Neuroimage 51(2):654–664.
Chetelat, G., Desgranges, B., et al. (2003). Mild cognitive impairment: Can FDG-PET predict who is to rapidly convert to Alzheimer’s disease? Neurology 60(8):1374–1377.
Cohen, A.D., Price, J.C., et al. (2009). Basal cerebral metabolism may modulate the cognitive effects of Abeta in mild cognitive impairment: an example of brain reserve. J. Neurosci. 29(47):14770–14778.
Davatzikos, C., Fan, Y., et al. (2008). Detection of prodromal Alzheimer’s disease via pattern classification of magnetic resonance imaging. Neurobiol. Aging 29(4):514–523.
Dickerson, B.C., Bakkour, A., et al. (2009). The cortical signature of Alzheimer’s disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb. Cortex 19(3):497–510.
Drzezga, A., Grimmer, T., et al. (2008). Imaging of amyloid plaques and cerebral glucose metabolism in semantic dementia and Alzheimer’s disease. Neuroimage 39(2):619–633.
Engler, H., Forsberg, A., et al. (2006). Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain 129(Pt 11):2856–2866.
Fagan, A.M., Roe, C.M., et al. (2007). Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch. Neurol. 64(3):343–349.
Foster, N.L., Chase, T.N., et al. (1983). Alzheimer’s disease: focal cortical changes shown by positron emission tomography. Neurology 33(8):961–965.
Fox, N.C., Freeborough, P.A., et al. (1996). Visualisation and quantification of rates of atrophy in Alzheimer’s disease. Lancet 348(9020):94–97.
Furst, A.J., Rabinovici, G.D., et al. (2012). Cognition, glucose metabolism and amyloid burden in Alzheimer’s disease. Neurobiol. Aging 33(2):215–225.
Gomperts, S.N., Rentz, D.M., et al. (2008). Imaging amyloid deposition in Lewy body diseases. Neurology 71(12):903–910.
Greicius, M.D., Srivastava, G., et al. (2004). Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. Proc. Natl. Acad. Sci. USA 101(13):4637–4642.
Heister, D., Brewer, J.B., et al. (2011). Predicting MCI outcome with clinically available MRI and CSF biomarkers. Neurology 77(17):1619–1628.
Ikonomovic, M.D., Klunk, W.E., et al. (2008). Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 131(Pt 6):1630–1645.
Jack, C.R., Jr. (2011). Alliance for aging research AD biomarkers work group: structural MRI. Neurobiol. Aging 32(Suppl 1):S48–S57.
Jack, C.R., Jr., Knopman, D.S., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9(1):119–128.
Jack, C.R., Jr., Lowe, V.J., et al. (2008). 11C PiB and structural MRI provide complementary information in imaging of Alzheimer’s disease and amnestic mild cognitive impairment. Brain 131(Pt 3):665–680.
Jagust, W., Gitcho, A., et al. (2006). Brain imaging evidence of preclinical Alzheimer’s disease in normal aging. Ann. Neurol. 59(4):673–681.
Jagust, W., Reed, B., et al. (2007). What does fluorodeoxyglucose PET imaging add to a clinical diagnosis of dementia? Neurology 69(9):871–877.
Johnson, K.A., Fox, N.C., et al. (2012). Brain imaging in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2(4) :a006213.
Karow, D.S., McEvoy, L.K., et al. (2010). Relative capability of MR imaging and FDG PET to depict changes associated with prodromal and early Alzheimer disease. Radiology 256(3):932–942.
Klunk, W.E. (2011). Amyloid imaging as a biomarker for cerebral beta-amyloidosis and risk prediction for Alzheimer dementia. Neurobiol. Aging 32(Suppl 1):S20–S36.
Klunk, W.E., Engler, H., et al. (2004). Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol. 55(3):306–319.
Knopman, D.S., DeKosky, S.T., et al. (2001). Practice parameter: diagnosis of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 56(9):1143–1153.
Kovacevic, S., Rafii, M.S., et al. (2009). High-throughput, fully automated volumetry for prediction of MMSE and CDR decline in mild cognitive impairment. Alzheimer Dis. Assoc. Disord. 23(2):139–145.
Kramer, J.H., and Miller, B.L. (2000). Alzheimer’s disease and its focal variants. Semin. Neurol. 20(4):447–454.
Langbaum, J.B., Chen, K., et al. (2009). Categorical and correlational analyses of baseline fluorodeoxyglucose positron emission tomography images from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Neuroimage 45(4):1107–1116.
McDonald, C.R., McEvoy, L.K., et al. (2009). Regional rates of neocortical atrophy from normal aging to early Alzheimer disease. Neurology 73(6):457–465.
McEvoy, L.K., and Brewer, J.B. (2012). Biomarkers for the clinical evaluation of the cognitively impaired elderly: amyloid is not enough. Imag. Med. 4(3):14.
McEvoy, L.K., Edland, S.D., et al. (2010). Neuroimaging enrichment strategy for secondary prevention trials in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 24(3):269–277.
McEvoy, L.K., Fennema-Notestine, C., et al. (2009). Alzheimer disease: quantitative structural neuroimaging for detection and prediction of clinical and structural changes in mild cognitive impairment. Radiology 251(1):195–205.
McKhann, G.M., Knopman, D.S., et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7(3):263–269.
Migliaccio, R., Agosta, F., et al. (2009). Clinical syndromes associated with posterior atrophy: early age at onset AD spectrum. Neurology 73(19):1571–1578.
Minoshima, S., Giordani, B., et al. (1997). Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann. Neurol. 42(1):85–94.
Oishi, K., Mielke, M.M., et al. (2011). DTI analyses and clinical applications in Alzheimer’s disease. J. Alzheimers Dis. 26(Suppl 3):287–296.
Pearson, R.C., and Powell, T.P. (1989). The neuroanatomy of Alzheimer’s disease. Rev. Neurosci. 2(2):101–122.
Rabinovici, G.D., Furst, A.J., et al. (2010). Increased metabolic vulnerability in early-onset Alzheimer’s disease is not related to amyloid burden. Brain 133(Pt 2):512–528.
Rabinovici, G.D., Jagust, W.J., et al. (2008). Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann. Neurol. 64(4):388–401.
Rabinovici, G.D., Rosen, H.J., et al. (2011). Amyloid vs FDG-PET in the differential diagnosis of AD and FTLD. Neurology 77(23):2034–2042.
Rabins, P.V., Blacker, D., et al. (2007). American Psychiatric Association practice guideline for the treatment of patients with Alzheimer’s disease and other dementias, second edition. Am. J. Psychiatry 164(12 Suppl):5–56.
Raichle, M.E., MacLeod, A.M., et al. (2001). A default mode of brain function. Proc. Natl. Acad. Sci. USA 98(2):676–682.
Rocher, A.B., Chapon, F., et al. (2003). Resting-state brain glucose utilization as measured by PET is directly related to regional synaptophysin levels: a study in baboons. Neuroimage 20(3):1894–1898.
Scheltens, P., Leys, D., et al. (1992). Atrophy of medial temporal lobes on MRI in “probable” Alzheimer’s disease and normal ageing: diagnostic value and neuropsychological correlates. J. Neurol. Neurosurg. Psychiatry 55(10):967–972.
Seeley, W.W., Crawford, R.K., et al. (2009). Neurodegenerative diseases target large-scale human brain networks. Neuron 62(1):42–52.
Seibert, T.M., and Brewer, J.B. (2011). Default network correlations analyzed on native surfaces. J. Neurosci. Methods 198(2):301–311.
Selkoe, D.J. (2006). The ups and downs of Abeta. Nat. Med. 12(7):758–759; discussion 759.
Sepulcre, J., Liu, H., et al. (2010). The organization of local and distant functional connectivity in the human brain. PLoS Comput. Biol. 6(6):e1000808.
Sepulcre, J., Sabuncu, M.R., et al. (2012). Network assemblies in the functional brain. Curr. Opin. Neurol. 25(4):384–391.
Sojkova, J., Driscoll, I., et al. (2011). In vivo fibrillar beta-amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Arch. Neurol. 68(2):232–240.
Sperling, R.A. (2012). Presentation of Resting fMRI results from DIAN. Alzheimer’s Association International Conference, Vancouver, BC.
Sperling, R.A., Laviolette, P.S., et al. (2009). Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63(2):178–188.
Tran, T., Ross, B., et al. (2009). Magnetic resonance spectroscopy in neurological diagnosis. Neurol. Clin. 27(1):21–60, xiii.
Vemuri, P., Simon, G., et al. (2011). Antemortem differential diagnosis of dementia pathology using structural MRI: Differential-STAND. NeuroImage 55(2):522–531.
Villain, N., Chetelat, G., et al. (2012). Regional dynamics of amyloid-beta deposition in healthy elderly, mild cognitive impairment and Alzheimer’s disease: a voxelwise PiB-PET longitudinal study. Brain 135(Pt7):2126–2139.
Villain, N., Fouquet, M., et al. (2010). Sequential relationships between grey matter and white matter atrophy and brain metabolic abnormalities in early Alzheimer’s disease. Brain 133(11):3301–3314.
Villemagne, V.L., McLean, C.A., et al. (2009). 11C-PiB PET studies in typical sporadic Creutzfeldt-Jakob disease. J. Neurol. Neurosurg. Psychiatry 80(9):998–1001.
Villemagne, V.L., Pike, K.E., et al. (2011). Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann. Neurol. 69(1):181–192.
Weiner, M.W., Veitch, D.P., et al. (2012). The Alzheimer’s Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimers Dement. 8(Suppl 1):S1–S68.