74 | NEUROPATHOLOGY AND SYNAPTIC ALTERATIONS
IN NEURODEVELOPMENTAL DISORDERS
MARA DIERSSEN AND SALVADOR MARTÍNEZ
The term neurodevelopmental disorder has been classically restricted to intellectual disability (ID) (Table 74.1). However, epidemiological studies increasingly demonstrate that most psychiatric disorders begin during childhood or adolescence (Kessler et al., 2007). Moreover, even though the notion of critical periods is still accepted, our concept of development in temporal terms has dramatically changed in the last years, and neurodevelopment now is considered to encompass the period from fetal life into adolescent brain maturation and synaptic pruning. This change in our conceptualization of neurodevelopment has led to an increased interest in understanding the role of neurodevelopmental processes, not only in ID, but also in mental illnesses such as schizophrenia and autism spectrum disorders (Kendler et al., 2005; McGrath et al., 2003). Mental disorders that become symptomatic in childhood or adolescence are affected by early environmental conditions that may interact with genetic risk factors. This increases the potential to identify genes leading to deviations from normal development, at prodromal stages in which intervention might be particularly useful.
Significantly, neurodevelopmental disorders have been found to share a number of similarities in terms of common genetic risks, co-occurrence of neurodevelopmental symptom domains, cognitive processing deficits, early onset, and chronic course. Schizophrenia, autism, and intellectual disabilities, for example, are spectrums of diseases with broad sets of causes that have overlapping phenotypes and genetics, which is suggestive of common deficits. Other disorders of childhood and adolescence, such as attention deficit/hyperactivity disorder (ADHD), eating disorders, or the separation anxiety disorders (Krueger and South, 2009), may not overlap in terms of risks and manifestations.
Structural brain development in healthy children follows complex, regionally heterochronous trajectories (Giedd et al., 1999). In gray matter development, whether measured by cortical volume or thickness, there is a phase of early increase, followed by a late childhood/adolescent phase of adjustment, before the cortex settles into adult dimensions. White matter has a more sustained pattern of expansion, persisting through adolescence. Given the complexities of these trajectories and their exquisite genetic control, it is not surprising that small disturbances can result in disturbances in cognition, affect, and behavior. For example, a disorder may be characterized by a delay in the pattern of typical development, or may be associated with differences in the temporal occurrence of the neural changes. Another possibility is a more profound deviance in the basic shape of the typical developmental trajectory. Obviously, these anomalies are not mutually exclusive. (A trajectory could incorporate elements of delay but also be altered in speed.) Anomalies in developmental trajectories are linked with ADHD or schizophrenia (Arango et al., 2008; Hoftman and Lewis, 2011), but also autism (Courchesne et al., 2007) and neurodegenerative disorders (Grilli et al., 2003).
This chapter gives a general view of the fetal and postnatal development that can be disrupted in an age-sensitive manner. It also discusses some of the common cellular and molecular targets that may explain similarities and differences in the phenotypes and comorbidity.
THE PRENATAL DEVELOPMENT OF THE REGIONAL DIVERSITY OF THE BRAIN
NEURAL TUBE FORMATION
The structural and functional complexity of the brain derives from precise orchestration of molecular and cellular mechanisms regulating the main developmental processes of neural progenitors: proliferation, differentiation, and synaptogenesis, in a temporospatial pattern defined into the matrix of embryonic development and perinatal life. The vertebrate central nervous system originates from the embryonic dorsal ectoderm. Differentiation of the neural plate epithelium from the ectoderm constitutes the first phase of complex processes called gastrulation and neurulation, which culminates in the formation of the neural tube.
Neurulation is a fundamental event of embryogenesis that culminates in the formation of the neural tube, which is the precursor of the brain and spinal cord. The bending of the neural plate involves the elevation of the neural folds establishing a trough-like space called the neural groove (20 days of gestation), which becomes the lumen of the primitive neural tube after closure of the neural groove (between 22 and 28 days of gestation). With the closure of anterior and posterior neuropores, the first phase of neural tube formation is completed, and internal cavities of the neural tube are no longer in connection with amniotic fluid. In addition, the neural folds generate the specialized cells of the neural crest. Then the most anterior portion of the neural tube balloons into three primary vesicles: the forebrain (prosencephalon), midbrain (mesencephalon), and hindbrain (rhombencephalon; Fig. 74.1). By the time the posterior end of the neural tube closes, secondary bulges—the optic vesicles—have extended laterally from each side of the developing forebrain. Then the prosencephalon becomes subdivided into the anterior secondary prosencephalon (telencephalon and hypothalamus) and the more caudal diencephalon.
TABLE 74.1. Disorders that are currently proposed for the diagnostic category of Neurodevelopmental Disorders
in DSM-5
| A 00–01 Intellectual Developmental Disorders |
| A 00 Intellectual Developmental Disorder |
| A 01 Intellectual or Global Developmental Delay Not Elsewhere Classified |
| A 02–04 Communication Disorders |
| A 02 Language Disorder |
| A 03 Speech Disorder |
| A 04 Social Communication Disorder |
| A 05 Autism Spectrum Disorder |
| A 05 Autism Spectrum Disorder |
| A 06–07 Attention Deficit/Hyperactivity Disorder |
| A 06 Attention Deficit/Hyperactivity Disorder |
| A 07 Attention Deficit/Hyperactivity Disorder Not Elsewhere Classified |
| A 08 Specific Learning Disorder |
| A 08 Specific Learning Disorder |
Anomalies of neurulation normally generate severe malformations in the embryos and are classified as dysraphias. Several genetic anomalies have been detected underlying these malformations, including mutations in VANGL1 and VANGL2 (OMIM 610132 and 600533, respectively) as well as SCRIB (OMIM 607733) and SHH (OMIM 600725). When the process of fusion of neural folds does not occur at any level of the neural grove craniorachischisis results. When the closure skipped areas localize at different levels, anencephaly, exencephaly, or meningomyelocele (from rostral to caudal) results.
After the posterior neuropore closes (28 days of gestation), the second phase of caudal neural tube formation begins. This process requires the formation of the caudal cell mass. Neural progenitors differentiate around vesicles in contact with caudal neural tube, and then these vesicles coalesce and fuse with the central canal (30 to 50 days of gestation); and finally there is a regressive process in the tail (50 to 100 days of gestation). Abnormal development of the caudal neural tube formation generates defects of closure and malformation in the lumbo-sacrococcygeal levels of the spinal cord: myelocystocele, diastematomyelia, and myelomeningocele. Myelomeningocele is a frequent malformation commonly associated with multiple human syndromes of genetic origin, including Joubert syndrome, DiGeorge syndrome, Waardenburg syndrome, and orofaciodigital syndrome, some of which have psychiatric manifestations, as well as toxic agents associated with another tube defect, folate sensitivity.
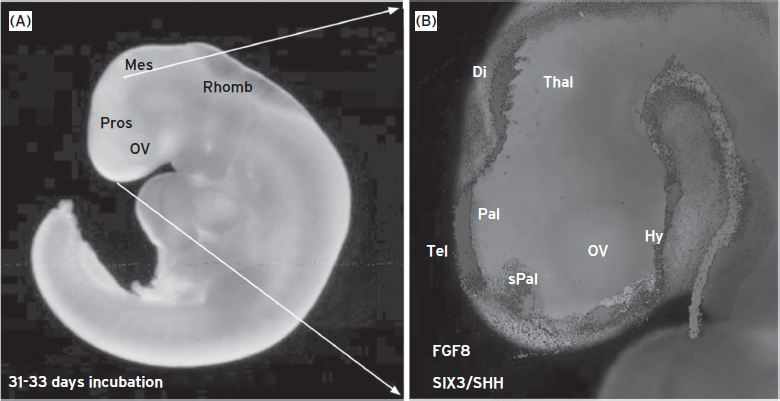
Figure 74.1 Human embryo at postneurulation stage. (A) Lateral view of a human embryo of 31–32 days of incubation. The main brain regions were detected: Prosencephalon (Pros), mesencephalon (Mes), rhombencephalon (Rhomb) and optic vesicle (ov). (B) Magnification of the anterior brain area where different gene expression patterns have been represented by different gray shades. The expression patterns were translated from mouse embryos sections at equivalent stage of neural tube development (E11.5). Di, diencephalon; Hy, hypothalamus; OV, optic vesicle; Pal, pallium; sPal, subpallium; Tel, telencephalon; Thal, thalamus. (Data from Developing Mouse Brain in the Allen Brain Atlas website: http://www.brain-map.org/.)
NEURAL TUBE REGIONALIZATION
The discovery that putative regulatory genes (mainly coding for transcription factors) are expressed in regionally restricted patterns in the developing forebrain has provided new tools for defining histogenetic domains and their boundaries at higher resolution (see Fig. 74.1; Puelles and Rubenstein, 2003). This molecular regionalization has unveiled the morphological significance of numerous gene expression patterns in the neural tube, suggesting the existence of molecular subdivisions of the main AP and DV zones, representing histogenetically specified domains of neural precursors. Genetic alterations associated with brain regionalization anomalies frequently occur with important structural alterations in brain morphogenesis that finally appear as congenital malformations such as holoprosencephaly (OMIM 236100; caused by combinations of mutations in genes as SIX3; Fig. 74.1), ZIC2, and GLI2, which are transcription factors coding for positional information in the prosencephalon at early stages of neural tube development. But smaller genetic alterations underlying copy number variants could produce subtle variations in regionalization that modify regional morphogenesis and synaptogenesis of the affected region, which may represent a predisposition to develop intellectual disabilities and/or psychiatric disorders (revised in Malhotra and Sebat, 2012).

Figure 74.2 Morphogenetic signals and telencephalic regionalization. (A) Schematic representation of neural tube (lateral view) where the main brain regions have been identified and morphogenetic signals regulating telencephalic regionalization have been represented by colors and arrows. Wnt and Bmp are dorsalizing signals, Fgf8 is a rostralizing signal, and Shh is a ventralizing signal, acting upon dorsal (pallial) telencephalon to specify cortical functional areas in the epithelium. (B,C) Schematic representation of a section in anteroposterior (B) and coronal (C) planes of the dorsal telencephalon (pallium and subpallium) that were color coded in neuroepithelial cells. Colored arrows in the ventricle and the dashed area (B) represent morphogenetic gradients that were translated through the neural wall by radial migration of neural cells into the different cortical regions (radial arrows and color gradient domains). (D–F) Sagittal sections showing the gradient expression pattern of two transcription factors: Pax6 regulated by rostral and ventralizing signals (D), and Emx2 regulated by dorsalizing signals (E).
F, A combinatory Photoshop reconstruction of both gradient patterns.
The longitudinal study of this molecular/structural causal association in the neural tube of experimental models has shown how the expression of particular genes is directly related to neural morphogenetic and cytogenetic development. For instance Gbx2 expression in mouse embryos is associated with the generation of thalamic neurons, which develop into the thalamocortical projection (Fig. 74.2) (Miyashita-Lin et al., 1999), whereas Lhx8 and Tbr1 expression are associated with the development of basal forebrain cholinergic neurons (Pombero et al., 2011). Thus, elaborated cellular interactions regulate the establishment of the common complex structural pattern of the developing brain in vertebrates. Distinct neural and glial identities are acquired by neuroepithelial cells according to their relative positions in the neural tube wall, through a progressive restriction of their histogenetic potential, under the influence of local environmental signals. Evidence for controlling morphogenetic processes at specific locations of the developing neural crest and neural tube has suggested the concept of morphogenetic organizers. Such centers regulate by their own signaling the choices of identity and regional polarity of neural precursors in neighboring neuroepithelial regions and may even function as target-derived presynaptic organizers to specify the type of synaptic terminals (Terauchi et al., 2010).
Three regions in the neural plate and tube have been identified as putative secondary organizers (Fig. 74.2A). Two of these organizers control prosencephalic regionalization: the anterior neural ridge (ANR) at the anterior end of the neural plate, and the zona limitans intrathalamicae (ZLI) in the middle of the diencephalon. In addition, the isthmic organizer (IsO) at the mid-hindbrain boundary controls mesencephalic and rostral rhombencephalic (including cerebellar) regionalization. To focus the present chapter on mental function related structures, we describe only the mechanisms of anterior prosencephalic regionalization and concentrate on the function of the anterior neural ridge (ANR).
ANTERIOR NEURAL RIDGE
The most anterior secondary organizer, the ANR, is a morphologically indistinct median sector at the junction between the neural plate and non-neural ectoderm. The ANR controls prosencephalic regionalization and proliferation and it was demonstrated that some genes expressed in this region control others that are necessary for telencephalic and hypothalamic specification (Fig. 74.2A). In particular, the Fgf8 gene is expressed very early in ANR cells, and has been shown to be crucial for specification of the anterior areas of the forebrain and telencephalon. Fgf8 hypomorphic mutations in both mouse and zebrafish result in a small telencephalon and midline anomalies. Prosencephalic regionalization by FGF8 signal operates at least in part through inhibition of Otx2 and Emx2 expression, in cooperation with BMP4, WNT, and SHH signaling molecules. Modifications in this interactive molecular network originate important anomalies in brain and skull development. For instance, decreasing FGF activity caused by mutations in FGF receptors (FGFR1 and FGFR2) produces severe brain alterations and different forms of craniosynostosis (including Apert syndrome; OMIM 101200) (revised in Stocchi et al., 2003; Rice, 2005; Storm et al., 2006). Less severe alterations in this FGF signaling have been described in hypogonadotropic hypogonadism (OMIM 146110), Jackson–Weiss syndrome (OMIM 123150), Kallmann syndrome 2 (OMIM 147950), Pfeiffer syndrome (OMIM 101600), and Trigonocephaly 1 (OMIM 190440). These syndromic forms of FGF signaling anomalies show limb and cephalic patterning alterations (because of FGF expression in ectodermal apical ridge of the limb bud and branchial arches), together different degrees of sensorial—hearing and smelling—deficits (because of the requirement of FGF signaling in cephalic placode development). Moreover, different degrees of developmental retardation and ID (with or without telencephalic structural phenotype) are also present that can be attributed to anomalies in cortical regionalization derived from expansion deficits of the cortical surface (Rash et al., 2011). Morphogenetic influences derived from secondary organizers regulate progenitor proliferation in the developing cortex. For instance, ID can be associated with anomalies in the regulation of centrosomic distribution and mitotic spindle orientation of neuroepithelial precursors, disrupting spatial and temporal rates of neuronal progenitor proliferation and differentiation, and causing a reduction of normal cortical surface and cerebral volume. Alterations of these processes course with primary microcephaly—MCPH1 gene (MIM 251200)—or secondary microcephaly associated to neural migration alterations: LIS1 (OMIM 601545) and NDEL1 (OMIM 607538) (Guerrini and Marini, 2006; Hippenmeyer et al., 2010).
Another gene coding morphogenetic information is sonic hedgehog (SHH), which is expressed in the subpallium slightly later than Fgf8 expression in the ANR. Abundant data suggest that SHH signaling is both necessary and sufficient for the specification of the ventral pattern throughout the nervous system. After abundant experimental demonstrations in animal models, it is widely accepted that normal patterning in the telencephalon depends on the “ventral” repression of GLI3 function by SHH and, and conversely on the “dorsal” repression of SHH signaling by GLI3. Different types of GLI3 mutations in humans produce craniofacial and brain anomalies together with other patterning anomalies. The SHH signal in the subpallium has also been shown to be involved in the regulatory activity of NKX2.1, a homeodomain gene required for the development of the telencephalic subpallium as well as the hypothalamus, the latter being ventral part of the forebrain (see Fig. 74.1). The DLX2 gene codes for a transcription factor expressed in the subpallium, prethalamus, and thalamus, and regulates the expression of ARX (Colasante et al., 2008) in prosencephalic progenitors (Fig. 74.3E–G). It is an essential gene in the proliferation and migration of neural progenitors. Mutations of ARX frequently have been involved in X-linked ID (OMMIM 300419) and epilepsy. The hypothalamus is the main brain center regulating homeostasis by controlling autonomous nervous and hormonal systems, where molecular patterns define neurons with specific functional properties; for instance, SIM1 is a transcription factor expressed in the hypothalamus, in the paraventricular hypothalamic nucleus (Fig. 74.3A–D). SIM1 haploinsufficiency is associated with hyperphagia and obesity. In addition, in close relation to this ventral signal coded by SHH, a longitudinal column of epithelial progenitors is specified along the whole neural tube to produce oligodendroglial progenitors, revealed by the expression of PLP/dm20, PDGFalpha, and OLIG1/2. Although very little is known about the underlying physiopathological mechanisms, extensive knowledge has been accumulated about myelin developmental anomalies in relation to mental disorders and ID (Dong and Greenough, 2004; Takahashi et al., 2011).
All these recent findings on the molecular regionalization of the neuroepithelium support the protomap model for neocortical areal specification, in which neocortical progenitor cells become patterned by extracellular signals into molecular domains that in turn generate area-specific neurons. The protomap is thought to be underpinned by spatial differences in progenitor cell identity that is reflected by regional specific genes at the transcriptional level (Fukuchi-Shimogori and Fukuchi-Shimogori and Grove, 2001; Grove and Fukuchi-Shimogori, 2003). Furthermore, cross-regulation among the rostral (FGF-signal dependent), dorsal (BMP- and WNT-signal dependent), and ventral (SHH-signal dependent) secondary inductive centers plays an essential role in patterning the early telencephalon. Modulation of this cross-regulation has the potential to regulate the relative size of structures whose morphogenesis is controlled by a given patterning center (Hamasaki et al., 2004). For instance, a reduction in FGF8 signaling reduces the ratio of the frontal motor versus sensory regions of the neocortex. Therefore, controlling the relative strength and range of a given patterning signal may provide a fundamental mechanism to modify the relative sizes of brain subdivisions during evolution and in disease states. Nevertheless, very little is known about the underlying processes that generate local reduction or lack of progenitors in specific cortical regions, as is the case of schizencephaly, in which mutations of EMX2, SIX3, and SHH have been reported (Aronica et al., 2012).
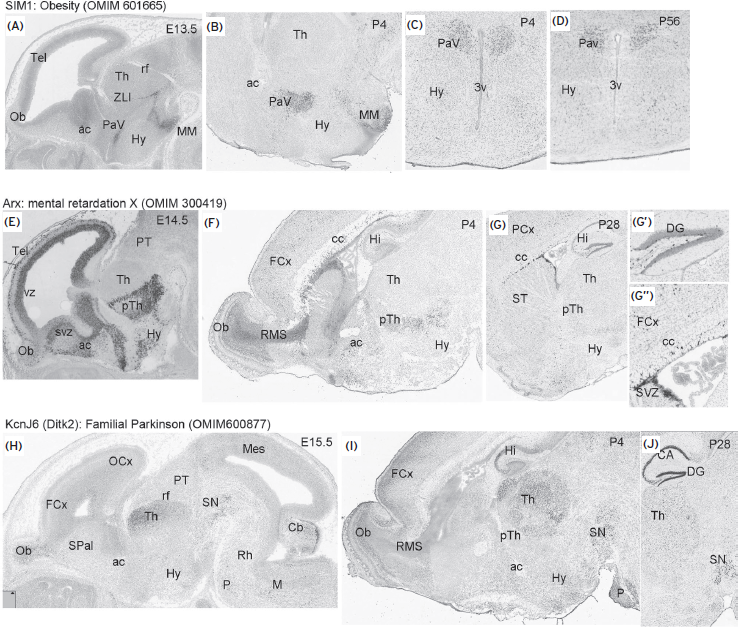
Figure 74.3 Gene expression patterns in mouse brain (from developing mouse brain in the Allen Brain Atlas website: http://www.brain-map.org/.) (A–D) Hypothalamic expression of Sim1 gene at embryonic (E13.3; A, sagittal section) and postnatal P4 (B, sagittal sections; C: coronal section) and P56 (coronal section) in the paraventricular nucleus (PaV). (E–G) Expression pattern of Arx gene at embryonic (E14.5), and postnatal stages (P28) in sagittal sections. G′ and G′′ are high-power pictures of (G) where Arx transcripts accumulated in subgranular cells of the dentate gyrus (DG) in the hippocampus, subependymal region (SVZ), and cortical cells above corpus callosum (cc). (H–J) Expression pattern of KcnJ6 gene in the developing (H) and postnatal (I,J) thalamus (Th), hippocampus (Hi, CA, and DG) and substantia nigra (SN). ac, anterior commissure; cc, corpus callosum; DG, dentate gyrus; FCx, frontal cortex; Hy, hypothalamus; Mes, mesencephalon; MM, mammillare nuclei; OB, olfactory bulb; OCx, occipital cortex; P, pons; PaV, paraventricular nucleus; PT, pretectum; pTh, prethalamus; rf, retroflexus tract; RMS, rostral migratory stream; SN, substantia nigra; SPal, subpallium; SVZ, subventricular zone; ST, atriatum; Th, thalamus; vz, ventricular zone; ZLI, zona limitans; 3V, third ventricle.
Once the developmental processes have operated, both spontaneous and sensory-driven neural activity are essential for instructively guiding the process of synapse development. These effects of neuronal activity are transduced in part through regulation of a set of activity-dependent transcription factors that coordinate a program of gene expression required for the formation and maturation of synapses (West and Greenberg, 2011). A number of human neurodevelopmental disorders have been linked to anomalies in the expression of these activity-regulated genes, such as Fragile X syndrome, Angelman syndrome, and ASD, in which it seems that excitatory and inhibitory synaptic equilibrium in the cortical regions is distorted. Indeed, the transcription of these activity-dependent genes regulates the fine-tuning of the adequate distribution of ion channels in specific neuronal populations. This is the case of GYRK2 (KCNJ6), a gene regulating GABAergic (inhibitory synaptic function) in the substantia nigra, thalamus, and hippocampus. It has been related to familial Parkinson disease and Down syndrome functional alterations (Fig. 74.3H–J).
A mechanistic model can be proposed to understand brain regionalization in which morphogenetic signals from different organizer regions interact in regulative networks to establish initial territories of specified progenitor cells, establishing the molecular domains in which specific functional areas will develop (see Fig. 74.2). Then, the same or different signals, acting at different temporospatial scales, may stabilize the final molecular codes in these regions and regulate the production of specific cell types. Then, young neural cell follows specific migratory patterns of radial and/or tangential movements to generate the cellular diversity of neural structures. This cellular diversity is required for the establishment of neuronal connectivity networks and intercellular trophic support (e.g., neuron–glia interactions). Eventually, ambient influences, acting through posttranscriptional mechanisms, can modify the structural and functional properties of such neural systems, both in positive (adaptive) or negative (toxic) directions (see following sections).
NEURAL MIGRATION IN THE CORTEX
AND SYNAPTOGENESIS DURING EMBRYONIC STAGES
The neurons and glial progenitors generated from the ventricular and subventricular layers of the pallium migrated to populate specific radial strata in the mantle layer (Fig. 74.4F,G). This migration is known as radial migration because it follows the radial axis in the brain wall, and migrating young neurons are guided to the pial (external) surface by radial glia fibers (Fig. 74.4A). In the pallium the first migration from the ventricular progenitors form a cellular layer known as the cortical plate (50 to 54 gestation days in humans), which represents the primordium of the cortex (embryonic days 12.5 to 15.5 in mouse development). Then at three months of gestation in human embryos the cells of layers IV, V, and VI are generated and migrate into the cortical plate (Fig. 74.4D), which correspond to E15.5 to 18.5 in mouse development. Structural maturation beyond this time is complex and varies in the different regions of the pallium, following a general lateromedial gradient; that is, starting from the lateral pallium (allocortex) at 70 to 80 days and progressing dorsally. Although the superficial neurons (layers II and III) are still generating, the neurons in the cortical plate become grouped into the six layers, corresponding to those of the adult cortex between six and seven gestational months (Fig. 74.4E; perinatal development in the mouse cortex). Intercalated with or after radial migration, other directions also can be followed by migratory neural cells, which do not follow radial glia axis and are known as tangential migration (Fig. 74.4F,G; revised in Molnar and Clowry, 2012).
We previously described that cellular migrations are essential processes necessary to develop cellular diversity in brain areas and require precisely coordinated movements in time and space. Cell–cell and cell–substrate interactions underlie the guidance mechanisms of these migrations during brain development, by signal–receptor interaction coded by ephrins and their receptors (revised in Rodger et al., 2012), SLIT proteins, ROBO receptors (see Fig. 74.4A,B), neuregulin1, and ErB4 receptors, as well as FGF8 and FGF receptors (FgfR; Pombero et al., 2011). Finally, positioning of cells in the specific layers of growing cortical areas is dependent on the Reelin (RELN) signaling cascade generated from Cajal-Retzuis cells, which are produced very early in development in layer I (revised in Frotscher, 2010). Actually, the RELN signal is received in migrating cells by apolipoprotein E (Apo-ER2) or very low-density lipoprotein (VLDLR) receptors and transduced by tyrosine phosphorylation of the Disabled-1 (DAB1) protein. Disruption of the RELN gene in humans results in severe brain abnormalities, including lissencephaly and cerebellar hypoplasia, and patients show ataxia, ID, and seizures. It is interesting to note that important tangential migration of neurons coming from the subpallial ganglionic eminences generate early cortical GABAergic interneurons, starting to invade the pallium at six weeks of gestation and generating deep layer interneurons (predominant from six to 15 gestation weeks), whereas during the second half of gestation, the cortical subventricular and subgranular zones (progenitor proliferating layers especially prominent in primates and humans) originate locally the most numerous superficial interneurons, which are completed by interneurons in layer I that originate in the cortical surface by the subpial granular layer (revised in Bystron et al., 2008).
Developmental defects in neuronal positioning and synaptic connectivity are commonly found in neurological and psychiatric diseases, and they are believed to underlie many cognitive and affective disorders (Barkovich et al., 2005; Harrison and Weinberger, 2005; Tabares-Seisdedos et al., 2006). Several mouse mutants are currently available that model at least some aspects of human developmental brain disorders that might be related to similar structural alterations. With the identification of the genes mutated in these animals and the study of the cellular basis of their phenotypes, we have taken significant strides toward an understanding of the mechanisms controlling proper brain development and the consequences of their dysfunction. In particular, mouse mutants deficient in the Reelin and Lis1 expressions have provided valuable insights into the mechanisms of cortical development. Absence of Reelin expression in the spontaneous mutant mouse reeler leads to extensive defects in neuronal position and dendrite development. In humans, loss of RELN (OMIM 600514) results in a type of lissencephaly with severe cortical and cerebellar malformation (Norman-Roberts type lissencephaly; revised by D’Arcangelo, 2006). Genetic and biochemical studies using mouse mutants suggest that the Lis1 protein may participate in the Reelin signaling pathway, controlling cortical development by its interaction with the dynein system and, therefore, neuronal motility.
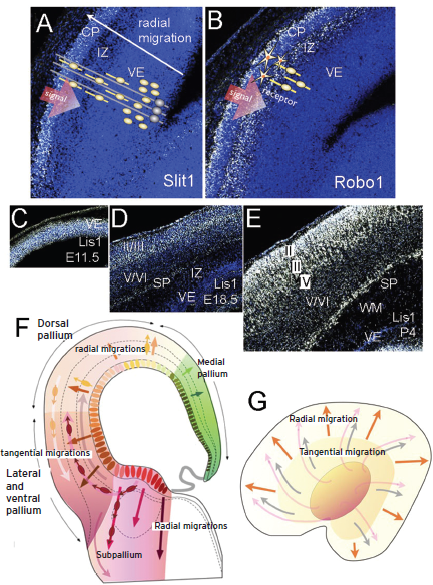
Figure 74.4 Neuronal migration in the pallium: cortical development. (A,B) Signal: Slit1 expression in E15.5 mouse embryo in the cortical plate. Neural progenitors proliferate in the ventricular epithelium (VE) and subventricular zone (yellow ovoid cells), radial glia cells are schematized in gray with long radial processes crossing the neural wall from ventricular to pail surface. Migrating neurons were distributed in between radial glia processes in the intermediate zone (IZ; yellow ovoid soma with processes) toward the cortical plate (CP). In an equivalent section the receptor of Slit1, Robo1 is expressed in migrating cells of the IZ.
(C–E) Expression of Lis1 gene in developing and postnatal mouse cortex. (F) Schematic representation of a coronal section were different lateromedial (ventrodorsal) pallial domains are represented by colored fields. Radial migration is represented by radial arrows and tangential migrations are represented by concentric distributed arrows. Migrating cells were represented by ovoid soma and arrows. (G) Planar representation of the telencephalic vesicle where radial migrations are represented by orange arrows; pallio-pallial tangential migration is represented by grey arrows; subpallio-pallial tangential migration, coming from the subpallium (central orange ovoid).
A recent classification proposed for developmental cortical malformations in humans by Barkovich and co-workers have listed the human genetic mutations identified as malformations owing to abnormal neuronal migration (Barkovich et al., 2005). Most of the genes identified in human’s cortical malformations also have been proved as functionally relevant to neuronal migration in mice; suggesting that in these situations the mouse models are of great value for analyzing the physiopathological mechanisms of these human diseases (Webber et al., 2009). Indeed, it is of special relevance that alterations in the Reelin-dependent genetic cascade activate complex molecular interactions regulating cell migration and microtubule transport. Mutation in some of the molecules involved in this interactive cascade produced important alterations in cell migration and, subsequently, in cortical structure, characterized phenotypically by different degrees of cortical dysplasia, such as the double-cortex syndrome within lissencephaly spectrum (Cardoso et al., 2003), whereas mutations in some other molecules (e.g., DSC1) or moderate genetic alteration of lissencephaly critical region genes produce functional alterations without important structural malformation that may manifest as schizophrenic or bipolar disease symptoms. A recent metaanalysis showed that more than 290 genes have been involved in mental retardation associated with syndromes and metabolic or neurological disorders (Chelly et al., 2006). Although it was estimated that most of the genes remain to be discovered, a table of known monogenic causes of mental retardation shows that neuronal migration and synaptic function–related genes predominated over other functions. Therefore, we can speculate about the importance of neuronal connectivity (synaptogenesis) with or without evident migratory alterations during human development as a substrate of mental retardation and/or predisposition to develop a mental disease. Copy number variants in developmental genes controlling principal neurodevelopmental processes, such are brain regionalization, neural cell migration/differentiation, and synaptogenesis have been postulated underlying the neurobiological roots of mental disorder predisposition (Insel, 2010; Malhotra and Sebat, 2012).
POSTNATAL NEURAL DEVELOPMENT
As discussed, the major developmental processes in the human telencephalon following closure of the neural tube are neuronal proliferation, astrocytic proliferation, neuronal migration, neurite (i.e., axonal and dendritic) growth, neuronal apoptosis, axon myelination, synaptogenesis, and neurite pruning. All of these processes begin during gestation, but the development of the brain circuitry requires the coordination of a complex set of postnatal neurodevelopmental events, such as neurite proliferation, axonal myelination, neurite pruning, and synaptogenesis, which exert their major effects during postgestational development (Giedd and Rapoport, 2010). Thus, the early postnatal period is crucial for brain development. The newborn brain at two to four weeks of age is approximately 36% the size of an adult brain and grows to about 80% of adult size by two years of age (Knickmeyer et al., 2008). Possibly, this dramatic brain growth is not primarily caused by postnatal cortical neurogenesis and neuronal migration (Bhardwaj et al., 2006; Shankle et al., 1999), but by the expansion of glia and myelination. At two to four weeks of age, the primary cortical areas, including motor, somatosensory, visual, and auditory cortices are well defined, but instead, association cortices at this age can be less clearly identified. Thus, the development of gray matter connections, especially in sensorimotor and visual cortices and the onset of myelination, drives the striking development of the brain during the perinatal and early postnatal period.
ENVIRONMENTAL INFLUENCES ON BRAIN DEVELOPMENT
During development, neural networks are shaped by experience-dependent processes that selectively strengthen and prune connections, creating and restructuring synaptic maps, or even changing dendritic architecture locally. The final neuronal circuits, primary mediators of the brain’s diverse functional capacities, and their connectivity, rely on experience-dependent sculpting (Maffei and Turrigiano, 2008; Tau and Peterson, 2010). This environmental sensitivity is especially important during the so-called critical periods; sensitive temporal windows of elevated plasticity allowing the structural consolidation of neuronal circuits and their connectivity. It was long believed that the potential for organization or reorganization existed only during these critical periods in early development, so that over time neural connections become more stable, forming widely distributed, interconnected networks involving balanced excitation and inhibition and structural stabilizers such as myelin. However, the successful treatments for adults with stroke or amblyopia suggest that the potential for circuit reorganization persists well into adulthood; thus, the final neuronal circuits are neither present at birth nor are they invariant through life (Michel, 2012). Besides, it has been proposed that each functional modality (from basic visual processing to language and social skills) has a different postnatal “critical” period.
Thus, the early postnatal and infancy periods are times of opportunity, but also of great vulnerability for the developing brain. Early disruption of proper sensory or social experiences results in mis-wired circuits that will respond suboptimally to normal experiences in the future. These deviations of the typical trajectories of maturation of neural circuits can also be caused by genetic alterations intrinsically impairing the response of the system to the environment, or to the combined effects of adverse experience on genetically abnormal brain maturation, leading to pathogenesis through both genetic and epigenetic mechanisms.
In this scenario, epigenetic mechanisms reveal as a critical determinant in disease predisposition and outcome. The term epigenetics has referred to heritable traits that are not mediated by changes in DNA sequence, and more broadly to any change in gene function not associated with sequence variation and promoted by the environment to influence or “program” gene expression or patterns that may or may not be heritable (Nicol-Benoit et al., 2012). Although often restricted to chromatin modifications specifying the sets of genes to be expressed or repressed, epigenetic mechanisms also include other gene expression controllers, such as noncoding RNAs, including microRNAs. Increasing evidence shows epigenetic processes are widespread in the brain and undergo dynamic regulation in both the developing and postmitotic neurons. Epigenetic gene control is in fact an intrinsic mechanism for normal tissue development. The epigenetic marking of chromatin provides a ubiquitous means for cells to shape and maintain their identity, and to react to environmental stimuli via specific remodeling. In mature, differentiated neurons in the central nervous system, epigenetic codes are critical for basic cellular processes such as synaptic plasticity, and play critical roles in encoding experience and environmental stimuli into stable, behaviorally meaningful changes in gene expression (Day and Sweatt, 2011; Graff and Mansuy, 2008).
In humans, because maturation processes in the brain continue well into adolescence, the influence of the epigenetic programming on disease risk and pathogenesis is critical. For example, early life environmental threats, such as stress (McEwen et al., 2012), influence the long-term outcome of neurodevelopmental disorders. Mutations in epigenetic components are associated with multisystem disease syndromes in human beings, all of which involve the nervous system; thus, the alteration of the epigenetic machinery may be a common process in many neurological diseases. In fact, anomalies in the epigenome have been demonstrated to be critical factors in a number of cognitive and behavioral disorders (Lockett et al., 2010). The epigenetic mechanisms that contribute to these deficits include the deregulation of essential components of the epigenetic machinery, alterations in the expression of genes important for cognition and behavior by epigenetic mechanisms, instability at trinucleotide repeats, and the breakdown of major epigenetic processes such as imprinting and X-chromosome inactivation. Some illustrative examples include Rett syndrome (caused by mutations in the methylated DNA binding protein MecP2), Rubinstein-Taybi syndrome (caused by mutations in the histone acetyltransferase CBP), and Coffin-Lowry syndrome (caused by mutations in histone phosphorylase; see Urdinguio et al., 2009 for a review).
NEURAL PLASTICITY AS A COMMON MECHANISM IN NEURODEVELOPMENTAL DISORDERS
Neural plasticity can be defined as the ability of a neuron or network to functionally or structurally alter in response to changes in input or activity. During the postnatal period, but also in infancy and adolescence, neural plasticity is one important mechanism in circuit development and refinement. Often, an outcome of neuronal plasticity is a structural plasticity manifested as a change of neuronal morphology (Urbanska et al., 2012). The number and branching pattern of dendrites are strictly correlated with the function of a particular neuron and the geometry of the connections it receives. The development of proper dendritic tree morphology depends on the interplay between genetic programming and extracellular signals. Spinogenesis is also an important target for structural plasticity. Spines are tiny actin-rich dendritic protrusions that harbor excitatory synapses. If plasticity mechanisms operate abnormally—either through long-lasting epigenetic modifications or disruption of functional mechanisms—neuronal networks improperly develop in response to activity-dependent experience. In psychiatric disorders and ID syndromes, plasticity malfunction leads to aberrant morphology and/or number of excitatory dendritic spines, a sign that is considered pathognomonic of ID, suggesting important changes in structural plasticity. Of course, the birth, death, and cellular characteristics of neurons, as well as the formation and reformation of their axons, dendrites, and synapses are also possible contributing factors.
In fact, most neurodevelopmental disorders can be considered synaptic plasticity disorders, in which different genomic causes result in abnormal synaptic development. Accumulation of this abnormal synaptic development, over time, leads to a characteristic and consistent behavioral and/or cognitive phenotype (Zoghbi, 2003).
SYNAPTOGENIC PROTEINS IN
MENTAL ILLNESS
As described, neurons are connected by synapses in complex networks that regulate specific physiological and behavioral outcomes in adulthood. For this reason, circuit development is considered a fundamental unit for regulating specific physiological and behavioral outcomes in adulthood. Synapses are the basic units of neural connectivity and communication in the brain in which synaptic transmission relies on the coordinated function of highly specialized structures on both sides of the cleft, involving membranous organelles, cytoskeleton, and vast protein networks. Postsynaptic neurotransmitter receptors, with associated scaffolding and signaling molecules, should be precisely aligned on the dendrite opposite chemically matched presynaptic vesicles with regulated release and recycling machinery in the axon. If we consider that a typical neuron may contain 1,000 to 10,000 synapses, and each synapse contains more than 1,000 protein components (Bayes et al., 2011) and that the complex neuronal networks derived from this connectivity will regulate behavioral outcomes in a dynamic manner, it becomes clear that synaptogenesis is a highly sensitive process.
Multiprotein complexes in the synapsis are organized into molecular networks that detect and respond to patterns of neural activity. These synaptic organizing proteins mediate plasticity and dynamic synaptic changes during postnatal brain maturation and contribute to synaptic specificity, pairing specific presynaptic and postsynaptic cells, and controlling where and when synapses are formed (Siddiqui and Craig, 2011) and destroyed. Many synaptogenic genes are at high risk for predisposing to neurodevelopmental disorders (Valnegri et al., 2012; van Bokhoven, 2011). Synaptic gene alterations include copy number variants, protein-truncating frame shifts, and function-altering missense variants, some de novo, contributing to the disease through pre- and/or postsynaptic alterations. Although in the last two decades a number of synaptic genes have been discovered whose mutations cause ID and a number of neuropsychiatric disorders, we are still far from identifying the impact of these mutations on brain development and neuronal function. One prominent example is the so-called synaptic organizing proteins, which include synaptic adhesion complexes and secreted factors (Betancur et al., 2009). The synaptogenic adhesion complexes are composed of transmembrane presynaptic and postsynaptic partners that bind in trans across the cleft, as in presynaptic neurexin (NRXN) and postsynaptic neuroligin, for example (Bottos et al., 2011). Such cleft-spanning synaptic organizing complexes often have bidirectional activity, inducing presynaptic and postsynaptic differentiation, and mediating cell adhesion and alignment of the pre- and postsynaptic specializations. Since the initial linkage of mutations in neuroligins 3 and 4 to autism, evidence has rapidly accumulated for the contribution of neuroligin and neurexin variants to disorders such as schizophrenia and ID. NRXN bind multiple structurally diverse partners across the cleft. Neuroligin-1 is the major glutamatergic neuroligin and binds only β-neurexins. Neuroligin-2 functions specifically at GABAergic synapses and appears to bind all neurexins. Of particular interest is the replicated finding of SHANKS deficits, which directly implicates glutamatergic synapse dysfunction in both autism and Asperger syndrome (Buxbaum, 2009). This finding is supported by the replicated findings with NRXN1 and NLGNS/4, which can also play a role in excitatory synapse formation, maintenance, and plasticity. NRXN1 deletions have been associated with autistic spectrum disorders, and confer risk for schizophrenia, suggesting an etiological overlap between both disorders.
Recent studies have identified leucine-rich repeat transmembrane neuronal proteins LRRTMs to be trans-synaptic partners for neurexins (Linhoff et al., 2009). LRRTM1 and LRRTM2 are glutamatergic postsynaptic proteins and bind α and β neurexins and compete with neuroligin-1 for an overlapping face of β-neurexin (−S4). Given their broadly overlapping expression patterns, neuroligin-1, LRRTM1, and LRRTM2 are likely to coexist at many glutamatergic postsynaptic sites (Wright and Washbourne, 2011). Other proteins have also been well documented for their role in experience-dependent plasticity such as ephrins and EphRs or the Wnt family and could thus be good candidates for neurodevelopmental disorders (Attwood et al., 2012). Other synaptogenic proteins have not been systematically studied but associations were also found for LRRTM1 and GluRδ1 with schizophrenia and SynCAM1, SHANK3, and CNTNAP2 with autism.
SPINE PATHOLOGY
A close look onto the surfaces of the dendritic processes of neurons allows the observation of small protrusions called dendritic spines, small protrusions (<2 Am) that contain postsynaptic densities (PSD), which are major sites of excitatory synapse. Typically, adult dendritic spines are composed of a bulbous head with a thin neck. In younger neurons, dendritic shafts are covered by thin, long protrusions called dendritic filopodia, which are thought to be precursors of spines. Filopodia typically lack “heads,” whereas mature spines have a distinct head and neck, giving rise to different morphologies named mushroom-like, stubby, thin, and branched. At the cellular level, the primary function of dendritic spines is to compartmentalize local synaptic signaling pathways and restrict the diffusion of postsynaptic molecules, insulating signaling molecules to a specific PSD. Dendritic spines and filopodia show rapid actin-based motility in the time scale of seconds (Yuste, 2011). This confers highly dynamic properties to the spines, which morphology can change very rapidly upon different types of stimulation by remodeling the architecture of their actin cytoskeleton (Penzes and Rafalovich, 2012).
During brain development, the initial dendritic arborization and outgrowth of dendritic spines precede synapse formation, and the number and morphology of the spines change dramatically as synaptic contacts mature. This happens as a response to environmental inputs, activity, and experience that strengthen the immature synaptic connections. On the contrary, inactive synapses become weaker and are eventually eliminated (use it or lose it). Thus, elimination of spines and change of their morphology also seem to be essential to establish neuronal circuits in early life.
Abnormal protrusion morphology in different brain areas seems to be a hallmark of many syndromic or non-syndromic mental disorders (van van Spronsen and Hoogenraad, 2010; von Bohlen Und Halbach, 2010). The first link between neurodevelopmental disorders (e.g., ID) and aberrant spines was reported by Purpura (1974) and showed a significant increase of abnormally long, thin spines on dendrites of cortical neurons in children with ID. Later, many ID syndromes have been reported presenting alterations in spine morphology and synapse number and have been linked to mutations in synaptic proteins involved directly or indirectly in the stabilization of the spine structure, including different signaling pathways, epigenetic regulators, and local mRNA translation at the synapse (Dierssen and Ramakers, 2006). For example, as stated, mutations in synaptic scaffolding protein SHANK3 and adhesion molecules neuroligin-3 and -4 have been linked to autism spectrum disorder but further evidence also suggests that other neurodevelopmental disorders involve defects in the regulation of actin cytoskeleton (Dierssen and Ramakers, 2006; Goellner and Aberle, 2012; von Bohlen Und Halbach, 2010). For example, the schizophrenia risk factor DISC1 regulates dendritic spine morphology via Rac1 and mutations in the cofilin kinase PAK3 (p21-activated kinase) gene lead to X-linked mental retardation. Moreover, decreased levels of PAK3 relate to synaptic dysfunction in
Alzheimer’s disease and possibly FXS, and PAK3 inhibition in mice causes cofilin pathology and memory impairment, consistent with a potential causal role of PAK defects in cognitive deficits in Alzheimer’s disease. The inability to preserve the spine structure causes profound alterations in neural circuit formation and can thus alter information processing, learning, and memory, and produce behavioral impairment (Lott and Dierssen, 2010). However, whether this phenotype is a cause or derives from the mental disturbance and whether we can use this feature as a translational end point to investigate the therapeutic effects of pharmacological interventions are still open questions.
Besides altered spinogenesis, altered spine pruning can also account for mis-wired circuit formation. In 1982, Feinberg (Feinberg, 1982, 1990) proposed that schizophrenia is a disease of aberrant synaptic pruning, based on findings of progressive postnatal “synaptic pruning” and aberrant synaptic elimination in early adolescence. This hypothesis was further expanded by the proposal that hyperpruning of collateral axons in the prefrontal cortex and defective pruning of certain brain structures leads to the aberrant connectivity of schizophrenia. This was further demonstrated by Golgi-impregnation studies revealing a region- and disease-specific decrease in dendritic spine density in dorsolateral prefrontal cortex layer III pyramidal cells in subjects with schizophrenia (Glantz and Lewis, 2000). Aberrant synaptic conductance and/or efficacy in schizophrenics are also observed in the subcortical gray matter, where striatal spines were reduced in size by approximately 30%. Another good example of pruning alterations is Fragile X syndrome (FXS). In a FXS mouse model, the Fmr1 knockout mice, the persistent increase in spine density in dentate gyrus in the absence of a defined pruning period argues for a role of FMRP in processes controlling developmental pruning. However, the disruption of the regulation of forming, stabilizing, or removing dendritic spines is a common phenotype in many neurodevelopmental disorders. Changes in the cytoskeletal structure are important for the protrusion morphology and play a role in many forms of ID that have been related to LIMK1, oligophrenin-1, PAK3, and ARHGEF6. Small GTPases, such as Rho, Rac, and Cdc42, are proteins known for their effects on the actin cytoskeleton and microtubule organization. Different mental retardation–associated genes have been identified that encode modulators of the Rho GTPases. Disturbances in these genes can lead to mental retardation and—on the morphological level—to alterations in dendritic spines (von Bohlen Und Halbach, 2010). A good example is Down syndrome (for a comprehensive review see Dierssen et al., 2009). Postmortem studies show that patients with Down syndrome start their lives with an apparently normal neuronal architecture that progressively degenerates, showing reduced dendrites and degenerative changes in older age. Significantly, abnormalities of synaptic density and length, fewer contact zones, a reduction of dendritic spines, and shorter basilar dendrites are observed in Down syndrome brains. Several candidate genes within the Down syndrome critical region are involved in synaptic plasticity with particular impact on dendritic function and spine motility and plasticity. Among those, DSCR1, DYRK1A, or ITSN1 may be candidates to explain the dendritic spine functional and structural alterations. Interestingly, Intersectin controls local formation and branching of actin filaments and Dyrk1A phosphorylates actin-binding proteins, and may also have a role in shaping the interaction of the spine membrane with the actin cytoskeleton (see Dierssen et al., 2009, 2012).
EXCITATORY/INHIBITORY IMBALANCE
In the mammalian cortex roughly 80% of neurons are excitatory (pyramidal cells) and 20% are inhibitory interneurons (Rubenstein and Merzenich, 2003). Pyramidal cells specialize in transmitting information between different cortical areas and from cortical areas to other regions of the brain, whereas interneurons primarily contribute to local neural assemblies, where they provide inhibitory input and shape synchronized oscillations. Complex brain circuitries comprise hierarchical networks of these excitatory and inhibitory neurons (for a review see Birke and Draguhn, 2010). Establishing and maintaining the appropriate ratio of excitatory versus inhibitory synapses is a critical factor that enables circuit threshold definition and balances output responsiveness. In fact, excitation and inhibition are balanced globally in cortical networks but their ratio may vary dramatically with brain region, development, or aging, and ratios do not necessarily reflect the excitation–inhibition synaptic balance per individual neuron. Balance between feedback or feedforward inhibition and recurrent excitation can give rise to balanced network amplification (Carvalho and Buonomano, 2009; Murphy and Miller, 2009) or stability (Marino et al., 2005). In sensory cortices, such a balance serves to increase temporal precision and reduce the randomness of cortical operation (Cruikshank et al., 2007; Pouille et al., 2009).
Excitatory and inhibitory synapses show divergent features. Excitatory synapses target on mature mushroom-shaped spines containing a prominent postsynaptic density (PSD) after preferential contacting with an exploratory filopodium. In contrast, inhibitory GABAergic synapses are present on dendritic shafts, nerve cell somata, and axon initial segments, lacking postsynaptic thickening, and arise from axon–dendrite contacts with no apparent protrusive activity. At the ultrastructural level, excitatory contacts are asymmetric with an electron-dense postsynaptic density opposing a presynaptic active zone, whereas inhibitory contacts appear relatively symmetrical, with presynaptic vesicles clustered opposite a synaptic cleft without a robust postsynaptic density. During developmental stages neural circuit development is regulated by the balance of excitatory and inhibitory neurotransmission (Zhang et al., 2011), so that disruption of this balance leads to aberrant states, which cause severe dysfunction when chronically unresolved (Marin, 2012).
An increased ratio of excitation/inhibition (E/I) is observed in sensory, mnemonic, social, and emotional systems of autistic patients where it is thought to contribute to the prevalence of poor signal-to-noise ratios, resulting in hyperexcitable, non-tunable cortical circuits (Gatto and Broadie, 2010). These results suggest a shift in the excitation–inhibition ratio favoring an elevated preponderance of glutamatergic connections with inhibitory insufficiency. Excitation-dominant synaptic imbalance may underlie the pathophysiology of other intellectual disabilities. Fragile X syndrome exhibits significant GABAergic deficits leading to hyperexcitability as well as synaptic imbalance neurophysiology (Coghlan et al., 2012). The opposite is observed in Down syndrome (Dierssen et al., 2009; Dierssen, 2012). Various Down syndrome mouse models exhibit a reduced ratio of hippocampal excitation–inhibition, leading to overinhibition with an increased number of inhibitory synapses as well as a reduced density of excitatory synapses. Moreover, large morphological changes are seen in the granular cell dentate gyrus to the CA3 pathway in Ts65Dn mice along with a reduction in synapse density, and reduction of mushroom spines with multivesicular bodies in the middle molecular layer of dentate gyrus. As a consequence, the Ts65Dn dentate gyrus shows enhanced GABAergic synaptic transmission, owing to increased presynaptic release of GABA and increased postsynaptic GABAB receptor–Kir3.2 signaling (Best et al., 2012).
Abnormal GABAergic neurotransmission in the prefrontal cortex of individuals with schizophrenia has also been suggested to cause the cognitive disturbances (Lewis et al., 2012). Indeed, deficits in cortical inhibition have been reported both in vivo and in analyses of postmortem brain tissue from patients with schizophrenia, and cognitive functions such as working memory, which is clearly altered in schizophrenic patients, seems to depend on normal interneuron performance. It was originally suggested that the GABAergic deficits that are observed in schizophrenia might be caused by a deficit in the number of interneurons in the prefrontal and cingulate cortices that was speculated to reduce GABAergic inputs to pyramidal cells. The subsequent finding that such patients had increased GABAA receptor binding activity in pyramidal cells was thought to be consistent with this idea, but other studies failed to find similar interneuron reductions in all individuals with schizophrenia. It is now accepted that GABAergic deficits preferentially occur at the level of specific synapses. Interneurons mediate the precise gating of information through specific signaling pathways by controlling—both spatially and temporally—the amounts of excitatory and inhibitory inputs that individual neurons receive. Thus, the disruption of the balance between excitation and inhibition produced by interneuron reduction has been suggested to lead to gating defects that are related to cognitive impairment (Vogels and Abbott, 2007).
Little is known how the E/I ratio is dynamically adjusted through the postnatal critical periods, when the strength of excitatory and inhibitory synapses is rapidly changing. Some authors have suggested that the tightly regulated E/I ratios in adults’ cortex is a result of drastic changes in relative weight of inhibitory but not excitatory synapses during critical period, and the local inhibitory structural changes are the underpinning of altered E/I ratio across postnatal development (Zhang et al., 2011). Mutation of genes that normally sculpt and maintain the excitatory/inhibitory balance results in severe dysfunction, causing neurodevelopmental disorders including autism, schizophrenia, and ID (van van Spronsen and Hoogenraad, 2010; Yin et al., 2012). For example, altered GAD67 expression is found in postmortem brain tissue from individuals affected of several neurodevelopmental disorders and those with childhood-onset schizophrenia, suggesting interneuron anomalies. Postmortem studies revealed that neurons in autistic patients present reduced level of glutamic acid decarboxylase (GAD), the rate-limiting enzyme in GABA synthesis. GABAA receptors are also reduced at mRNA and protein levels, showing altered α1–5 and β1 subunits in the parietal cortex (Brodmann area [BA]), α1 in the frontal cortex (BA 9), and α1 and β3 in the cerebellum of autistic patients postmortem tissue (Coghlan et al., 2012). Genetic studies of subjects with ASD show abnormalities among gene loci containing synaptic imbalance candidates, including the GABA β3 receptor subunit and genes encoding the neurorexin, neuroligin, and Shank proteins (Kohl and Paulsen, 2010). Such mutations may result in defective synaptic connections and/or functional synaptogenesis, leading to the selective loss of either excitatory or inhibitory synapses, without compensatory changes, or genetic conditions that actively favor the formation or maintenance of one class of synapse relative to the other.
In fact, the E/I imbalance may also affect the shape of the dendritic spines, because the plastic changes in the neuronal cytoskeleton are triggered in mature neurons in response to excitatory neurotransmission (for review see Bosch and Hayashi, 2012). Glutamate is one of the excitatory neurotransmitters that can induce long-term potentiation (LTP) mainly driving the strengthening of the connection between a presynaptic and postsynaptic neuron for a long period. The binding of glutamate to the N-methyl-d-aspartic (NMDA) receptor increases Ca2+ levels and triggers short-lasting activation of proteins, such as CamKII, that can phosphorylate the α-amino-
3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor to modulate insertion of more AMPA receptors at the postsynaptic membrane. The gain or loss of AMPA receptors at the postsynaptic membrane is related to the strength of the synapse and also the morphology of the spines. Long-term depression (LTD) is the opposite process, being the weakening of the synapse, reflected by a reduced number of ion receptors at the postsynaptic membrane. Glutamatergic synapses also undergo dynamic changes during postnatal brain maturation. These activity-dependent rearrangements of the actin network mediate structural changes of dendritic spines and can be mediated by certain proteins. For example, activity-regulated cytoskeleton-associated protein (Arc) (also termed Arg3.1) is an immediate-early gene (IEG) induced in response to sensory experience, learning, LTP, spatial exploration, and novelty, and might be a good candidate for the excessive AMPA receptor internalization. It has been suggested that Arc is an mRNA target of FMRP and that the protein levels of Arc are increased in the absence of FMRP. Arc synthesis is required for mGluR-LTD induction by increasing the AMPA receptor internalization rate. Therefore, Arc may play a major role in the excessive AMPA receptor internalization and altered protrusion morphology in FXS (Niere et al., 2012). Genetic factors involved in these processes might function in synaptic plasticity, learning, enrichment, exercise, and neurogenesis. Also, the DSCR1 gene overexpressed in fetal and adult DS brains inhibits calcineurin, which is involved in synaptic plasticity and has a role in the transition from short- to long-term memory through perturbation in long-term potentiation and long-term depression (Hoeffer et al., 2007). Other interesting examples are drebrin, an actin-binding protein thought to regulate assembly and disassembly of actin filaments, thereby changing the shape of spines, the levels of which are reduced in the early second trimester in the brains of patients with Down syndrome (Weitzdoerfer et al., 2001) and Dyrk1A (Altafaj et al., 2008; Martinez de Lagran et al., 2012), and are associated with altered synaptic plasticity and learning and memory deficits. Changes in levels of expression of these genes may lead to changes in the timing and synaptic interaction between neurons during development, which can lead to suboptimal functioning of neural circuitry and signaling at that time and in later life.
CONCLUSIONS
Brain development is a non-linear dynamic process in which small initial differences may yield large later effects. The basic layout of the brain is established by genetic programs and intrinsic activity and is actively refined by the environment as well as gene–environment interaction. This takes place in an age-sensitive manner with underlying mechanisms including developmental changes in gene expression, epigenetic modifications, synaptic arborization, pruning, and behavioral maturation. A hypothesis that is gaining predominance proposes that a spectrum of diseases such as schizophrenia, autism, and intellectual disabilities may have common deficits that explain their overlapping phenotypes and genetics. In this context, the idea is gaining support that the disruption of general developmental processes, alteration of the establishment of neural circuits in the developing brain and neural plasticity, or the imbalance of excitation and inhibition in specific neural circuits might be responsible for some of the clinical features of these disorders. Recent studies in animal models demonstrate that the molecular basis of such disorders is linked to common defects in development leading to schizophrenia, autism, or intellectual disabilities (Box 74.1).
BOX 74.1 NEURODEVELOPMENTAL DISORDERS REVISITED
The translation of discoveries in basic neuroscience to clinical problems has been frustrating owing in part to the reliance on categorical, symptom-based diagnostic systems, such as the Diagnostic and Statistical Manual of Mental Disorders (DSM; http://www.dsm5.org/) and the International Classification of Diseases (ICD). These systems have been very useful for increasing the reliability of clinical diagnoses, but could complicate the search for genetic, behavioral, and neural elements that define etiology and pathogenetic mechanisms. An alternative conceptualization and classification has been proposed in the Research Domain Criteria initiative (RDoC; http://www.nimh.nih.gov/research-funding/rdoc/index.shtml). The Research Domain Criteria initiative is aimed at establishing new ways of classifying psychopathology based on dimensions of observable behavior and neurobiological measures.
Since 2007, the diagnostic and categorization criteria in psychiatry have been extensively revised based on comprehensive review of scientific advancements, targeted research analyses, and clinical expertise. The release of the final, approved DSM-5 is expected in May 2013. In DSM-5 the category of Neurodevelopmental Disorders contains diagnoses that were listed in DSM-IV under the chapters of Disorders Usually First Diagnosed in Infancy, Childhood, or Adolescence, and Anxiety Disorders. Among the recent revisions is the proposal to now include two superordinate categories of Language Disorder and Speech Disorder and one category for Social Communication Disorder. Speech Disorder now includes specifiers for the specific type of speech difficulty. Learning Disorder has been changed to Specific Learning Disorder, and the previous types of Learning Disorders (Dyslexia, Dyscalculia, and Disorder of Written Expression) are no longer being recommended. The type of Learning Disorder will instead be specified as noted in the diagnosis. Finally, a category of ADHD Not Elsewhere Classified has been added (see Table 74.I). The RDoC scheme, in contrast, is represented as a two-dimensional matrix with rows representing the “dimensions of observable behavior and neurobiological measures,” such as “cognition” that includes attention, perception, working memory, declarative memory, language behavior, and cognitive (effortful) control, and columns representing various “units” (levels) of analysis, including genes, molecules, cells, circuits, behaviors, or paradigms (see Table I in Sarah et al., 2012).
DISCLOSURES
Dr. Dierssen’s laboratory is supported by the Spanish Ministry of Science and Innovation SAF2010-16427, FIS PS09102673-(CureFXS), and PI11/00744, Jerôme Lejeune Foundation, Fundación Areces and Alicia Koplowitz, FRAXA Foundation and Generalitat de Catalunya SGR1313 and CIBERER, an initiative of the Instituto de Salud Carlos III (ISCIII). The funders had no role in preparation of the manuscript.
Dr. Martínez. The funders had no role in preparation of the manuscript.
REFERENCES
Altafaj, X., Ortiz-Abalia, J., et al. (2008). Increased NR2A expression and prolonged decay of NMDA-induced calcium transient in cerebellum of TgDyrk1A mice, a mouse model of Down syndrome. Neurobiol. Dis. 32:377–384.
Arango, C., Moreno, C., et al. (2008). Longitudinal brain changes in early-onset psychosis. Schizophr. Bull. 34:341–353.
Aronica, E., Becker, A.J., et al. (2012). Malformations of cortical development. Brain Pathol. 22:380–401.
Attwood, B.K., Patel, S., et al. (2012). Ephs and ephrins: emerging therapeutic targets in neuropathology. Int. J. Biochem. Cell. Biol. 44:578–581.
Barkovich, A.J., Kuzniecky, R.I., et al. (2005). A developmental and genetic classification for malformations of cortical development. Neurology 65(12):1873–1887.
Bayes, A., van de Lagemaat, L.N., et al. (2011). Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 14:19–21.
Best, T.K., Cramer, N.P., et al. (2012). Dysfunctional hippocampal inhibition in the Ts65Dn mouse model of Down syndrome. Exp. Neurol. 233:749–757.
Betancur, C., Sakurai, T., et al. (2009). The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 32:402–412.
Bhardwaj, R.D., Curtis, M.A., et al. (2006). Neocortical neurogenesis in humans is restricted to development. Proc. Natl. Acad. Sci. USA 103:12564–12568.
Birke, G., and Draguhn, A. (2010). No simple brake: the complex functions of inhibitory synapses. Pharmacopsychiatry 43(Suppl 1):S21–31.
Bosch, M., and Hayashi, Y. (2012). Structural plasticity of dendritic spines. Curr. Opin. Neurobiol. 22:383–388.
Bottos, A., Rissone, A., et al. (2011). Neurexins and neuroligins: synapses look out of the nervous system. Cell. Mol. Life. Sci. 68:2655–2666.
Buxbaum, J.D. (2009). Multiple rare variants in the etiology of autism spectrum disorders. Dia. Clin. Neurosci. 11:35–43.
Bystron, I., Blakemore, C., et al. (2008). Development of the human cerebral cortex: Boulder Committee revisited. Nat. Rev. Neurosci. 9:110–122.
Cardoso, C., Leventer, R.J., et al. (2003). Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am. J. Hum. Genet. 72:918–930.
Carvalho, T.P., and Buonomano, D.V. (2009). Differential effects of excitatory and inhibitory plasticity on synaptically driven neuronal input-output functions. Neuron 61:774–785.
Chelly, J., Khelfaoui, M., et al. (2006). Genetics and pathophysiology of mental retardation. Eur. J. Hum. Genet. 14:701–713.
Chi, C.L., Martinez, S., et al. (2003). The isthmic organizer signal FGF8 is required for cell survival in the prospective midbrain and cerebellum. Development 130:2633–2644.
Coghlan, S., Horder, J., et al. (2012). GABA system dysfunction in autism and related disorders: from synapse to symptoms. Neurosci. Biobehav. Rev. 36:2044–2055.
Colasante, G., Collombat, P., et al. (2008). Arx is a direct target of Dlx2 and thereby contributes to the tangential migration of GABAergic interneurons. J. Neurosci. 28:10674–10686.
Courchesne, E., Pierce, K., et al. (2007). Mapping early brain development in autism. Neuron 56:399–413.
Cruikshank, S.J., Lewis, T.J., et al. (2007). Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat. Neurosci. 10:462–468.
D’Arcangelo, G. (2006). Reelin mouse mutants as models of cortical development disorders. Epilepsy Behav. 8:81–90.
Day, J.J., and Sweatt, J.D. (2011). Epigenetic modifications in neurons are essential for formation and storage of behavioral memory. Neuropsychopharmacol. 36:357–358.
Dierssen M. (2012). Down syndrome: the brain in trisomic mode. Nat. Rev. Neurosci 13(12):844–858.
Dierssen, M., Herault, Y., et al. (2009). Aneuploidy: from a physiological mechanism of variance to Down syndrome. Physiol. Rev. 89:887–920.
Dierssen, M., and Ramakers, G.J. (2006). Dendritic pathology in mental retardation: from molecular genetics to neurobiology. Genes Brain Behav. 5 (Suppl 2):48–60.
Dong, W.K., and Greenough, W.T. (2004). Plasticity of nonneuronal brain tissue: roles in developmental disorders. Ment. Retard. Dev. Disabil. Res. Rev. 10:85–90.
Feinberg, I. (1990). Cortical pruning and the development of schizophrenia. Schizophr. Bull. 16:567–570.
Feinberg, I. (1982). Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J. Psychiatr. Res. 17:319–334.
Frotscher, M. (2010). Role for Reelin in stabilizing cortical architecture. Trends Neurosci. 33:407–414.
Fukuchi-Shimogori, T., and Grove, E.A. (2001). Neocortex patterning by the secreted signaling molecule FGF8. Science 294:1071–1074.
Gatto, C.L., and Broadie, K. (2010). Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front. Syn. Neurosci. 2:4.
Giedd, J.N., Blumenthal, J., et al. (1999). Brain development during childhood and adolescence: a longitudinal MRI study. Nat. Neurosci. 2:861–863.
Giedd, J.N., and Rapoport, J.L. (2010). Structural MRI of pediatric brain development: what have we learned and where are we going? Neuron 67:728–734.
Glantz, L.A., and Lewis, D.A. (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57:65–73.
Goellner, B., and Aberle, H. (2012). The synaptic cytoskeleton in development and disease. Dev. Neurobiol. 72:111–125.
Graff, J., and Mansuy, I.M. (2008). Epigenetic codes in cognition and behaviour. Behav. Brain Res. 192:70–87.
Grilli, M., Ferrari Toninelli, G., et al. (2003). Alzheimer’s disease linking neurodegeneration with neurodevelopment. Funct. Neurol. 18:145–148.
Grove, E.A., and Fukuchi-Shimogori, T. (2003). Generating the cerebral cortical area map. Annu. Rev. Neurosci. 26:355–380.
Guerrini, R., and Marini, C. (2006). Genetic malformations of cortical development. Exp. Brain. Res. 173:322–333.
Hamasaki, T., Leingartner, A., et al. (2004). EMX2 regulates sizes and positioning of the primary sensory and motor areas in neocortex by direct specification of cortical progenitors. Neuron 43:359–372.
Harrison, P.J., and Weinberger, D.R. (2005). Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol. Psychiatry 10:40–68; image 45.
Hippenmeyer, S., Youn, Y.H., et al. (2010). Genetic mosaic dissection of Lis1 and Ndel1 in neuronal migration. Neuron 68:695–709.
Hoeffer, C.A., Dey, A., et al. (2007). The Down syndrome critical region protein RCAN1 regulates long-term potentiation and memory via inhibition of phosphatase signaling. J. Neurosci. 27:13161–13172.
Hoftman, G.D., and Lewis, D.A. (2011). Postnatal developmental trajectories of neural circuits in the primate prefrontal cortex: identifying sensitive periods for vulnerability to schizophrenia. Schizophr. Bull. 37:493–503.
Insel, T.R. (2010). Rethinking schizophrenia. Nature 468:187–193.
Kendler, K.S., Kuhn, J.W., et al. (2005). The interaction of stressful life events and a serotonin transporter polymorphism in the prediction of episodes of major depression: a replication. Arch. Gen. Psychiatry 62:529–535.
Kessler, R.C., Amminger, G.P., et al. (2007). Age of onset of mental disorders: a review of recent literature. Curr. Opin. Psychiatry 20:359–364.
Knickmeyer, R.C., Gouttard, S., et al. (2008). A structural MRI study of human brain development from birth to 2 years. J. Neurosci. 28:12176–12182.
Kohl, M.M., and Paulsen, O. (2010). The roles of GABAB receptors in cortical network activity. Adv. Pharmacol. 58:205–229.
Krueger, R.F., and South, S.C. (2009). Externalizing disorders: cluster 5 of the proposed meta-structure for DSM-V and ICD-11. Psychol. Med. 39:2061–2070.
Lewis, D.A., Curley, A.A., et al. (2012). Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 35:57–67.
Linhoff, M.W., Lauren, J., et al. (2009). An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron 61:734–749.
Lockett, G.A., Wilkes, F., et al. (2010). Brain plasticity, memory and neurological disorders: an epigenetic perspective. Neuroreport 21:909–913.
Lott, I.T., and Dierssen, M. (2010). Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. Lancet Neurol. 9:623–633.
Maffei, A., and Turrigiano, G. (2008). The age of plasticity: developmental regulation of synaptic plasticity in neocortical microcircuits. Prog. Brain Res. 169:211–223.
Malhotra, D., and Sebat, J. (2012). CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148:1223–1241.
Marin, O. (2012). Interneuron dysfunction in psychiatric disorders. Nat. Rev. Neurosci. 13:107–120.
Marino, J., Schummers, J., et al. (2005). Invariant computations in local cortical networks with balanced excitation and inhibition. Nat. Neurosci. 8:194–201.
Martinez de Lagran, M., Benavides-Piccione, R., et al. (2012). Dyrk1A influences neuronal morphogenesis through regulation of cytoskeletal dynamics in mammalian cortical neurons. Cereb. Cortex 22(12):2867–2877.
McEwen, B.S., Eiland, L., et al. (2012). Stress and anxiety: structural plasticity and epigenetic regulation as a consequence of stress. Neuropharmacology 62:3–12.
McGrath, J.J., Feron, F.P., et al. (2003). The neurodevelopmental hypothesis of schizophrenia: a review of recent developments. Ann. Med. 35:86–93.
Michel, G.F. (2012). Using knowledge of development to promote recovery of function after brain damage. Dev. Psychobiol. 54:350–356.
Miyashita-Lin, E.M., Hevner, R., et al. (1999). Early neocortical regionalization in the absence of thalamic innervation. Science 285:906–909.
Molnar, Z., and Clowry, G. (2012). Cerebral cortical development in rodents and primates. Prog. Brain Res. 195:45–70.
Murphy, B.K., and Miller, K.D. (2009). Balanced amplification: a new mechanism of selective amplification of neural activity patterns. Neuron 61:635–648.
Nicol-Benoit, F., Le-Goff, P., et al. (2012). Epigenetic memories: structural marks or active circuits? CMLS 69:2189–2203.
Niere, F., Wilkerson, J.R., et al. (2012). Evidence for a fragile X mental retardation protein-mediated translational switch in metabotropic glutamate receptor-triggered Arc translation and long-term depression. J. Neurosci. 32:5924–5936.
Penzes, P., and Rafalovich, I. (2012). Regulation of the actin cytoskeleton in dendritic spines. Adv. Exp. Med. Biol. 970:81–95.
Pombero, A., Bueno, C., et al. (2011). Pallial origin of basal forebrain cholinergic neurons in the nucleus basalis of Meynert and horizontal limb of the diagonal band nucleus. Development 138:4315–4326.
Pouille, F., Marin-Burgin, A., et al. (2009). Input normalization by global feedforward inhibition expands cortical dynamic range. Nat. Neurosci. 12:1577–1585.
Puelles, L., and Rubenstein, J.L. (2003). Forebrain gene expression domains and the evolving prosomeric model. Trends Neurosci. 26:469–476.
Purpura, D.P. (1974). Dendritic spine “dysgenesis” and mental retardation. Science 186:1126–1128.
Rash, B.G., Lim, H.D., et al. (2011). FGF signaling expands embryonic cortical surface area by regulating Notch-dependent neurogenesis. J. Neurosci. 31:15604–15617.
Rice, D.P. (2005). Craniofacial anomalies: from development to molecular pathogenesis. Curr. Mol. Med. 5:699–722.
Rodger, J., Salvatore, L., et al. (2012). Should I stay or should I go? Ephs and ephrins in neuronal migration. Neurosignals 20:190–201.
Rubenstein, J.L., and Merzenich, M.M. (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2:255–267.
Shankle, W.R., Rafii, M.S., et al. (1999). Approximate doubling of numbers of neurons in postnatal human cerebral cortex and in 35 specific cytoarchitectural areas from birth to 72 months. Pediatr. Dev. Pathol. 2:244–259.
Siddiqui, T.J., and Craig, A.M. (2011). Synaptic organizing complexes. Curr. Opin. Neurobiol. 21:132–143.
Storm, E.E., Garel, S., et al. (2006). Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development 133:1831–1844.
Tabares-Seisdedos, R., Escamez, T., et al. (2006). Variations in genes regulating neuronal migration predict reduced prefrontal cognition in schizophrenia and bipolar subjects from mediterranean Spain: a preliminary study. Neuroscience 139:1289–1300.
Takahashi, N., Sakurai, T., et al. (2011). Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Prog. Neurobiol. 93:13–24.
Tau, G.Z., and Peterson, B.S. (2010). Normal development of brain circuits. Neuropsychopharmacol. 35:147–168.
Terauchi, A., Johnson-Venkatesh, E.M., et al. (2010). Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature 465:783–787.
Urbanska, M., Swiech, L., et al. (2012). Developmental plasticity of the dendritic compartment: focus on the cytoskeleton. Adv. Exp. Med. Biol. 970:265–284.
Urdinguio, R.G., Sanchez-Mut, J.V., et al. (2009). Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol. 8:1056–1072.
Valnegri, P., Sala, C., et al. (2012). Synaptic dysfunction and intellectual disability. Adv. Exp. Med. Biol. 970:433–449.
van Bokhoven, H. (2011). Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 45:81–104.
van Spronsen, M., and Hoogenraad, C.C. (2010). Synapse pathology in psychiatric and neurologic disease. Curr. Neurol. Neurosci. Rept. 10:207–214.
Vogels, T.P., and Abbott, L.F. (2007). Gating deficits in model networks: a path to schizophrenia? Pharmacopsychiatry 40(Suppl 1):S73–77.
von Bohlen Und Halbach, O. (2010). Dendritic spine abnormalities in mental retardation. Cell Tissue Res. 342:317–323.
Webber, C., Hehir-Kwa, J.Y., et al. (2009). Forging links between human mental retardation-associated CNVs and mouse gene knockout models. PLoS Genet 5:e1000531.
Weitz Doerfer, R., Dierssen, M., et al. (2001). Fetal life in Down syndrome starts with normal neuronal density but impaired dendritic spines and synaptosomal structure. J. Neural. Trans. 61:59–70.
West, A.E., and Greenberg, M.E. (2011). Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 3:6.
Wright, G.J., and Washbourne, P. (2011). Neurexins, neuroligins and LRRTMs: synaptic adhesion getting fishy. J. Neurochem. 117:765–778.
Yin, D.M., Chen, Y.J., et al. (2012). Synaptic dysfunction in schizophrenia. Adv. Exp. Med. Biol. 970:493–516.
Yuste, R. (2011). Dendritic spines and distributed circuits. Neuron 71:772–781.
Zhang, Z., Jiao, Y.Y., et al. (2011). Developmental maturation of excitation and inhibition balance in principal neurons across four layers of somatosensory cortex. Neuroscience 174:10–25.
Zoghbi, H.Y. (2003). Postnatal neurodevelopmental disorders: meeting at the synapse? Science 302:826–830.