76 | INTELLECTUAL DISABILITY SYNDROMES
CHARLES E. SCHWARTZ, FIORELLA GURRIERI, AND GIOVANNI NERI
Genetic syndromes that include intellectual disability (ID) as one component manifestation are each individually rare but, taken together, constitute a large group of causally heterogeneous and phenotypically variable conditions. It is difficult, if not impossible, to establish their exact number, given the existence of many “private” conditions, especially those caused by chromosomal imbalances. It is equally hard to provide prevalence estimates, except for some syndromes that are more prevalent and better known, such as Noonan syndrome, Fragile X syndrome, DiGeorge syndrome, Rett syndrome, and Williams syndrome, to mention just a few. Fortunately, significant information can be derived from open access databases (e.g., OMIMFiguce in Man at http://www.ncbi.nlm.nih.gov/omim, Orphanet at http://www.orpha.net, and DECIPHER at http://decipher.sanger.ac.uk) that collect and store relevant data from the medical literature.
From the point of view of the clinical geneticist, who deals with these rare patients and their families, a useful nosological classification of ID syndromes is by genetic etiology. Knowledge of the genetic cause is the sine qua non for accurate counseling and estimate of recurrence risks. However, even with the help of the most advanced technology, such as chromosome microarray (CMA) and whole exome or other forms of next-generation sequencing, the cause remains unknown probably in one-third or more of the cases seen in genetic clinics. The main etiological classes are listed in Table 76.1, together with tentative prevalence estimates among live born.
Understanding the genes underlying ID is also of great interest for neurobiological studies because they can lead to a molecular and cellular understanding of pathobiology, and gene products represent potential therapeutic targets. One of the most well-known examples of these possibilities arise from Fragile X syndrome, in which animal models with disruptions of the causal gene (FMR1) have led to an understanding of synaptic deficits in the disorder, and to novel therapeutics that are beginning to show promise (see, e.g., Berry-Kravis et al., 2012).
The diagnostic approach to the patient who is affected with a syndromic form of ID requires the application of a number of steps. Short of a “gestalt” diagnosis, which may occasionally apply to cases of well-known syndromes, the approach includes the collection of a (at least) three-generation family tree, prenatal and postnatal history, physical examination, relevant imaging data of brain, heart, kidneys, and so on, and functional data, such as EEG. Laboratory tests can be specific for the diagnosis of given syndromes in the presence of a specific clinical suspicion. Otherwise, CMA and analysis of the FMR1 gene are usually considered first-line tests. Next-generation sequencing is becoming more and more applicable to those cases in which all other approaches failed to reach a causal diagnosis. In the end, it should be made clear to the parents and caregivers alike that lack of a causal diagnosis does not preclude the application of symptomatic, as well as rehabilitating treatments, as deemed appropriate on the basis of the phenotypic manifestations and the patient’s needs.
This chapter briefly describes a few selected examples of ID syndromes. The selection was made with the purpose of illustrating various molecular pathways and/or cellular compartments whose disruption as a consequence of genetic mutation cause a disruption of a developmental pathway and, ultimately, an ID syndrome.
This arbitrary selection leaves out numerous syndromic ID conditions even though their causative genes have been identified after incredible research. To get a feel of the genetic burden in ID, some estimates have been attempted: X-linked intellectual disability (XLID) accounts for 5% to 10% of ID in males. More than 150 syndromes, the most common of which is the Fragile X syndrome, have been described. A large number of families with nonsyndromal XLID, 95 of which have been regionally mapped, have been described as well. Mutations in 102 X-linked genes have been associated with 81 of these XLID syndromes and 35 forms of nonsyndromal XLID (Lubs et al., 2012). The distinction between syndromic and non-syndromic ID is not precise, and several genes, initially identified in syndromic conditions, have later been associated with non-syndromic forms (e.g., ARX, CASK, JARID1C, FGD1, and ATRX).
As for autosomal ID, mutations in approximately 450 genes have been reported, of which 50 genes are associated with nonsyndromic ID and 400 genes are associated with syndromic ID (van van Bokhoven, 2011). In spite of the rapid progress in disease gene identification, it is difficult to predict how many additional ID genes remain to be identified. Based on the number of approximately 100 known XLID genes (syndromic as well as non-syndromic), a total of 1,500 to 2,000 genes might be a reasonable estimate (van Bokhoven, 2011). The lack of knowledge is even more expansive when it comes to the biological consequences of these mutations on brain functions. Mutant genes encode proteins with a variety of functions including chromatin remodeling, presynaptic and postsynaptic activity, intracellular trafficking, neurogenesis, and neuronal migration. Interestingly, most of the pathways and about half of the genes identified as being involved in ID have also been found to be altered in patients with autism spectrum disorders (ASD) suggesting that these two neurodevelopmental disorders share a common genetic basis (Betancur, 2011).
TABLE 76.1. Etiological classification of intellectual disability syndromes
| CAUSE | PERCENTAGE |
| Genomic imbalance (detected cytogenetically or by array-CGH) | 20 |
| Mendelian mutations | 7 |
| Environmental | 23 |
| Unknown but likely of genetic origin | 50 |
In the following sections, the selected syndromes are grouped by mode of inheritance: autosomal dominant, autosomal recessive, and X-linked.
AUTOSOMAL DOMINANT
CARDIO-FACIO-CUTANEOUS SYNDROME
Cardio-facio-cutaneous syndrome (CFCS) was first described by Reynolds et al., in 1986. Twenty years later came the demonstration that it is caused by disruption of the RAS-ERK signaling pathway (Narumi et al., 2007; Rodriguez-Viciana et al., 2006), placing it nosologically within the group of RASopathies (Rauen et al., 2010). Although initially it was considered by some authors to be a variant of the Noonan syndrome, the discovery of the causal genes proved it to be a distinct entity, as maintained by Neri et al. (1991).
The incidence of the syndrome is unknown. Since 1986, about 100 cases were reported in the medical literature and more than 100 unpublished cases are known to CFC International, Inc., a family support group operating worldwide (http://www.cfcsyndrome.org). This number probably accounts for a fraction of cases because it excludes most mildly affected individuals. All affected subjects to date are sporadic, most likely because of new dominant mutations, as supported by the observation of a paternal age effect (Roberts et al., 2006).
Cardio-facio-cutaneous syndrome has a distinctive clinical presentation starting at pregnancy, which can be uneventful, but is often marked by polyhydramnios. Hypotonia and serious and often longstanding feeding difficulties are hallmarks of the early postnatal period. Feeding problems are especially troublesome, with poor sucking, aspiration, gastroesophageal reflux, oral aversion, hyperemesis, and gastrointestinal dysmotility, leading to serious and long-lasting failure to thrive. These problems are significant enough to require nasogastric tube feeding, or gastrostomy tube placement in some cases. Assisted feeding may be required into middle childhood or beyond. The physical phenotype is characterized by short stature with relative macrocephaly, distinctive facial appearance with high forehead, bitemporal narrowing, downslanting and wide-spaced palpebral fissures, ptosis, short nose with depressed nasal root, low-set ears with creases on the lobes, sparse curly hair, sparse or absent eyebrows and lashes, ectodermal findings especially follicular keratosis, hyperkeratosis, hyperelastic skin, cutaneous vascular malformations, pigmented nevi, and congenital heart defects, pulmonic stenosis, hypertrophic cardiomyopathy, and atrial septal defects being the most frequent. Intellectual disability, of moderate to severe degree, is almost invariably present. Many children display tactile defensiveness, short attention span, irritability, stubbornness, and obsessive behavior. A minority act aggressively toward others. Nonetheless, parents often comment on a warm and loving personality and an enjoyment of social interactions, particularly music and dance. Epilepsy can be a serious complication in about one-third of cases. It can present as infantile spasms, and absence, generalized, or partial complex seizures. On brain imaging, ventriculomegaly, reduced white matter, and thinning of the corpus callosum are the most common findings. Life expectancy is shortened on average because of the early death of individuals with severe cardiac involvement (Armour and Allanson, 2008).
Noonan syndrome and Costello syndrome, especially the former, are the main differential diagnoses, and there is marked phenotypic overlap among the three conditions. Individuals with Noonan syndrome have normal intelligence or mild psychomotor involvement, less common and less severe failure to thrive, and a history of bleeding disorder. Differences between Costello syndrome and CFCS include skin papillomata, deep palmar and plantar creases, a significant likelihood of chaotic atrial arrhythmia or multifocal atrial tachycardia, and most important, an increased risk of malignant tumors such as rhabdomyosarcoma, transitional cell carcinoma of the bladder, and neuroblastoma.
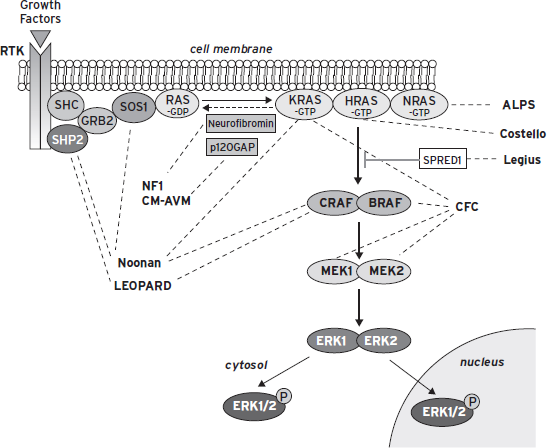
In 2006, Niihori et al. and Rodriguez-Viciana et al. discovered the genes whose mutations cause CFCS. These are BRAF, KRAS, MEK1, and MEK2. Their protein products are components of the RAS-ERK signaling pathway, which is implicated in growth factor-mediated cell proliferation, differentiation, and apoptosis, and plays a crucial role in embryonic development. RAS genes encode GTP-binding proteins that function as on-off switches to activate or inhibit downstream molecules. BRAF mutations account for the majority of mutation-positive cases (65.8%), followed by MEK1 and 2 and, more rarely, KRAS. Dysregulation of the RAS/ERK pathway is the common underlying mechanism of the phenotypic relatedness of CFCS to Noonan and Costello syndrome, whose genes encode proteins that are also part of the RAS/ERK pathway (Fig. 76.1; Table 76.2).
The role of the RAS/ERK pathway in cellular function is the subject of several recent reviews, including Tidyman and Rauen (2009). In terms of the role of this pathway in the dynamic aspects of CNS function, there is good evidence for a critical role of RAS/ERK signaling in synaptic plasticity and, hence, in learning and memory processes (Mazzucchelli and Brambilla, 2000). This impact on synaptic plasticity is very likely directly related to the impairments in intellectual functioning, and the frequent manifestations of autism spectrum disorder in CFCS.
BRAF and KRAS somatic mutations are often reported in human cancers. These are thought to be stronger gain-of-function mutations that would be embryo-lethal if present in the zygote. In CFCS, mutations are milder, compatible with (abnormal) development. The same is thought to be true for Noonan and Costello syndrome mutations. Yet, one would expect that patients affected with these syndromes would be more prone to cancer. This is true for Costello syndrome, but not for Noonan and CFCS, with some exceptions. However, it must be noted that all RASopathies patients may one day benefit from the intensive search of new cancer drugs acting as inhibitors of the RAS/ERK pathway, such as lonafarnib, sorafenib, statins, and so on. If and when an effective pharmacological treatment will become available based on these premises, it will be an example of successful translational medicine.
Figure 76.1 The ras/mitogen-activated protein kinase (MAPK) signaling pathway. This pathway is critical for cell proliferation, differentiation, motility, apoptosis, and senescence. The dashed lines indicate the genes associated with various developmental syndromes. Of interest, some of the syndromes can result from a mutation in more than one of the genes in this pathway. (Adapted from Tidyman, W.E. and Rauen, K.A. (2009). The RASopathies: Developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 19:230–236. Used with permission from Elsevier.)
TABLE 76.2. The RASopathies: syndromes and genes*
| SYNDROMES | OMIM | GENES |
| Noonan | 163950 | PTPN11, KRAS, RAF1, SOS1, NRAS |
| Noonan-multiple lentigines | 151100 | PTPN11, BRAF, RAF1 |
| Costello | 218040* | HRAS |
| Cardio-facio-cutaneous (CFC) | 115150 | BRAF, MEK1, MEK2, KRAS |
| Noonan-like with anagen hair | 607721 | SHOC2 |
| Noonan-like, with or without juvenile myelomonocytic leukemia | 613563 | CBL |
| Neurofibromatosis 1 | 162200 | NF1 |
| Legius (neurofibromatosis type1-like syndrome) | 611431 | SPRED1 |
KABUKI SYNDROME
Kabuki syndrome (KS, OMIM #147920), first described in 1981 with the designation of Kabuki make-up syndrome, is characterized by ID, unusual face, large and protruding ears, and postnatal growth delay (Niikawa et al., 1981). The prevalence was estimated at 1:32,000 in the Japanese population (Niikawa et al., 1988), a figure likely to apply also to other ethnicities. Based on the observation of a large number of patients, Niikawa et al. (1988) established the following diagnostic criteria: (1) typical face with long palpebral fissures and eversion of the lateral third of the lower eyelid; arched eyebrows, sparse or notched in their lateral third; depressed nasal tip; large, protruding ears; (2) skeletal anomalies, including cleft and butterfly vertebrae, narrow intervertebral spaces, scoliosis, brachymesophalangy of hands and fifth finger clinodactyly; (3) persistence of fetal pads on fingertips; (4) mild-to-moderate ID; (5) postnatal growth delay. The neonatal period is characterized by feeding difficulties, which may require tube feeding or gastrostomy, and consequent failure to thrive (Adam, et al., 2011).
A host of different physical anomalies can be present, adding to the complexity of the phenotype (Adam et al., 2011). Nearly half of the individuals with KS have a congenital heart defect, especially coarctation of the aorta (Kawame et al., 1999). Almost three-fourths have a highly arched palate, and one-third have a cleft lip and/or palate. Hypodontia is common, with missing lateral and central incisors. Hearing loss is also common, usually caused by chronic otitis media, but a few patients with true sensorineural hearing loss have been observed. Gastrointestinal anomalies are rare, whereas the genitourinary system is affected in about 25% of individuals with KS. Reported anomalies include fused and/or misplaced kidneys, duplication of collecting system, hypospadias, and cryptorchidism in males (Adam et al., 2011). In females, premature thelarche is fairly common. Brain structural anomalies seem to be rare, with the possible exception of Chiari I malformation, that has been reported in several patients. Immune dysfunction with hypogammaglobulinemia has been reported in a significant proportion of patients (Hoffman et al., 2005).
Most patients with KS have hypotonia, joint laxity, and delay of motoric development. The neuropsychiatric manifestations of KS are interesting. Although mild to moderate ID is reported in more than 90% of patients, the personality of KS children is usually described as pleasant and easy. Verbal and nonverbal reasoning, as well as receptive language seem to represent an area of relative strength, as opposed to expressive language and visuospatial orientation, which show considerable impairment (Mervis et al., 2005). In contrast with many other ID syndromes, autism or autistic spectrum disorders have been reported only rarely. Epilepsy is not a major concern, affecting less than one-third of patients with KS, who tend to respond well to antiepileptic medication.
Kabuki syndrome is an autosomal dominant disorder, with most cases caused by new mutations of the MLL2 gene, a gene of 54 exons, encoding MLL2, a large protein of 5,262 amino acid residues, which belongs to the SET family of proteins. MLL2 is a histone 3 lysine 4 (H3-K4) methyltransferase, a regulator of gene transcription through epigenetic modulation. H3-K4 methylation is an epigenetic mark of euchromatin, favoring gene transcription. H3-K4 demethylation signals a transition from euchromatin to heterochromatin, inhibiting gene transcription. Loss of Mll2 in mice causes cellular apoptosis and growth retardation, resulting in embryo lethality. The causative role of MLL2 in KS was discovered in 2010 by Ng et al., who reported mutations in 10 unrelated patients utilizing whole exome sequencing. Mutations are found in about three-fourths of tested patients. The majority are nonsense or frameshift mutations, predicting a truncated protein and suggesting that haploinsufficiency is the underlying pathogenic mechanism. Mutations are distributed over the entire length of the gene, but are more prevalent in the 3′ exons, leading to loss of the SET domain. Some canonical cases of KS without MLL2 mutation suggest possible genetic heterogeneity of the syndrome.
There are more than 25 known histone methyltransferases in the human genome and several (including NSD1, discussed in the following section) have been implicated as genes involved in developmental delay syndromes. There are a similar number of histone demethylase genes, and some of these have also been implicated in developmental delay syndromes.
SOTOS SYNDROME
Sotos syndrome (SoS OMIM #117550) is characterized by overgrowth of prenatal onset persisting during the first years of life, advanced bone age, and characteristic facial features (Baujat and Cormier-DaBaujat and Cormier-Daire, 2007). Birth weight averages 4,200 g in males and 4,000 g in females, whereas mean birth length is 55.6 cm in males and 57.3 cm in females. The newborn period is affected by poor feeding and congenital hypotonia, the latter persisting into adult life. Occipitofrontal head circumference (OFC) tends to exceed the 97th centile by the age of one year. Final height is usually within normal range because of the advanced bone age, present in 84% of patients with SoS. Advanced bone age is common to many overgrowth syndromes, including Weaver and Beckwith-Wiedemann syndrome. However, SoS can be distinguished for the presence of a peculiar (disharmonic) metacarpophalangeal profile. Hand and foot length is often above the 97th centile; dental eruption can occur early; puberty can be precocious. Facial characteristics consist of marked frontal bossing (in 97.5% of patients), high frontal hairline (97.5%), frontoparietal balding, downslanting palpebral fissures (90%), narrow bitemporal diameter, full cheeks, and high palate (70%). The face gradually lengthens with age and the jaw becomes more prominent, with a pointed chin. Pes planus is frequent, and strabismus is found in nearly 40% of patients. The IQ is below average (mean of 78, ranging from 40 to 129), with delay in expressive language. Autistic behavior or other psychological problems, including abnormal social behavior and anxiety, may also be noted (Baujat and Cormier-Daire, 2007; Sarimski, 2003). Seizures have been reported to occur in 50% of cases. Brain magnetic resonance imaging can show a typical pattern of anomalies, including prominence of the trigonus (90%) and the occipital horns (75%), as well as ventriculomegaly (63%). The supratentorial extracerebral fluid spaces and those of the posterior fossa are increased for age in 70% of patients (Schaefer et al., 1997). Heart defects (i.e., septal defects and patent ductus arteriosus) are observed in 8% of patients with SoS, but seem to be as frequent as 35%–41% in patients of Japanese ethnicity. Gastrointestinal and urogenital anomalies have also been reported. Upper respiratory tract infections, especially otitis media, are frequent and may lead to conductive hearing loss. As in most overgrowth syndromes, increased risk of cancer is a concern. Cohen critically reviewed neoplasms reported to occur with SoS (Cohen, 2003). One-third of patients with neoplasia, all males, developed lympho-hematological malignancies (lymphoma or leukemia), which seem to represent the most frequent neoplasias in SoS. Many cases occurred after the age of five (10/22; 45%) or 10 years (4/22; 18%). Thus, it is not easy to give an overall recommendation for cancer surveillance in SoS.
The most frequent cause of SoS was discovered by Kurotaki and colleagues in 2002, who found rearrangements of the NSD1 gene in SoS patients harboring a chromosomal translocation, encompassing band 5q35. They also identified one nonsense, three frameshift, and 20 submicroscopic deletions of the same gene among 42 sporadic cases (77%). This initial observation was followed by many other reports, confirming that haploinsufficiency of NSD1 is the major cause of SoS (Niikawa, 2004). A recurrent 2.2-Mb microdeletion at 5q35, including NSD1 and adjacent genes, is the primary cause of SoS in Japan, accounting for more than 50% of cases. In Europe and United States intragenic mutations cause 60% to 80% of SoS cases, whereas microdeletions are responsible for no more than 10% of cases. Partial NSD1 deletions, encompassing one or more exons, were detected in SoS cases (6%) without NSD1 mutation or 5q35 microdeletion (Douglas et al., 2005). In general, children carrying an NSD1 deletion have severe ID, with no language, major delay in motor milestones, and autistic features. By contrast, in patients carrying NSD1 point mutations, ID is usually mild to moderate with verbal skills being more affected. There have been suggestions that NSD1 mutations may also cause the Weaver syndrome, a view that is hard to maintain after the discovery that the original cases of Weaver syndrome are caused by mutation of the EZH2 gene (Gibson et al., 2012).
The function of the NSD1 gene (nuclear-receptor-binding SET-domain-containing protein 1) is not entirely clear. Its protein product is likely to act as transcriptional regulator of other genes by various mechanisms, including methylation of histone lysines H3-K36 and H4-K20 (see the preceding), as well as interaction with the transcriptional repressor Nizp1. One may hypothesize that haploinsufficiency of NSD1 could result in silencing of putative growth regulating genes, the final consequence being accelerated growth. A growth-related role of NSD1 is further supported by its expression in fetal tissues (Kurotaki et al., 2001).
AUTOSOMAL RECESSIVE
BARDET–BIEDL SYNDROME
This condition was first described in 1866 by Laurence and Moon as an entity characterized by ID, hypogenitalism, retinopathy, and late onset paraplegia. The same clinical phenotype was independently described by Bardet in 1920 and Biedl in 1922. Because of this, the syndrome has been often named with two different eponyms: Laurence–Moon syndrome and Bardet–Biedl syndrome (BBS). Because of the considerable phenotypic overlap, it has been suggested that the two conditions are allelic. Bardet–Biedl syndrome is now the overall used term. Bardet–Biedl syndrome is a pleiotropic genetic disorder with significant clinical variability.
Inheritance is mostly autosomal recessive, although in some instances BBS may be an oligogenic disorder. The prevalence of BBS varies markedly between populations: from 1:160,000 in northern European populations to 1:13,500 in isolated Arabic populations in which a higher level of consanguinity exists.
Bardet–Biedl syndrome is genetically heterogeneous: 16 BBS genes accounting for approximately 80% of clinically diagnosed BBS have been identified so far. The majority of pathogenic mutations are found in BBS1 and BBS10 (23.2% and 20%, respectively). Genotype–phenotype correlations are poor. Some genes seem to have ethnic specific frequency but no mutated genes are detected exclusively in a single ethnic population (Hjortshoj et al., 2010). Whereas most patients show regular autosomal recessive inheritance, in several instances (about 10%) a triallelic pattern of inheritance (three mutations in BBS genes are required for the phenotype to be manifest, or alternatively a third disease locus acts as a modifier) is observed (Beales et al., 2003; Eichers et al., 2004). This irregular pattern of inheritance may raise problems in counseling the parents of affected BBS children for recurrence risk. It has been suggested to counsel anyway according to the autosomal recessive recurrence risk (Abu-Safieh et al., 2012). All the BBS genes encode proteins that are part of the BBsome, localized at the base of the primary cilium and involved in the organization of ciliogenesis and maintenance of the cilium. Overall, BBS can be considered a disease of immotile cilia. Defects in immotile cilia are characterized clinically by retinitis pigmentosa, polydactyly, situs inversus, learning difficulties, and cystic kidneys, liver, and pancreas (Gerdes et al., 2009) (Table 76.3).
The BBS phenotype evolves with age and therefore the diagnosis is not usually suspected until late childhood. The only typical feature at birth may be postaxial polydactyly, rarely associated with brachydactyly or syndactyly. The most common diagnostic finding, which should prompt investigation for BBS, is the development of rod-cone dystrophy. This usually manifests around five years of age but may be present at two. Clinically, patients with BBS experience a gradual onset of night blindness, followed by photophobia and loss of central and color vision. Obesity is another major clinical finding (72%–86% of patients with BBS) mostly developing after the first year. Type 2 diabetes and other signs of metabolic syndrome are frequently observed. Hypogonadism, delayed puberty, hypogenitalism, and infertility are other issues. Speech problems and developmental delay (that can involve specific areas) are common in BBS. Behavior is an issue, as BBS patients may show obsessive-compulsive behavior, difficulties in socialization, autistic features, or psychosis. Renal abnormalities can be a major cause of morbidity and mortality. The renal abnormalities are variable but classically manifest with cystic tubular disease and anatomical malformations. Conductive hearing loss secondary to chronic otitis media is frequent and may worsen the speech defect. Other manifestations include heart malformations and cardiomyopathies, gastrointestinal issues (Hirschsprung disease), hypodontia and enamel hypoplasia, anosmia, ataxia, and poor coordination (Forsythe and Beales, 2012).
It has been suggested that some phenotypic manifestations also occur in mutation heterozygotes, specifically, obesity, increased risk for renal cancers and malformations, as well as retinal dysfunction (Beales et al., 2000; Kim et al., 2007).
TABLE 76.3. The ciliopathies: syndromes and genes
| SYNDROMES | OMIM | GENES |
| Short rib polydactyly | type II-III-V | 263530, 263520, 263510, 26986 IFT80, DYNC2H1, WDR35, NEK1 |
| Asphyxiating thoracic dystrophy | 611263, 613091, 613819, 614376 | IFT80, DYNC2H1, TTC21B, WDR19 |
| Ellis Van Creveld, Weyers acrofacial dysostosis | 604831, 614376, 193530 | EVC, EVC2 |
| Sensenbrenner | 613610, 614099, 614378 | WDR19, IFT122, WDR35, IFT43 |
| Bardet–Biedl, McKusick–Kauffman | 209900, 236700 | BBS1–12, BBS15, CEP290, TMEM67, MGC1203, MKS1 |
| Joubert syndrome | 213300 | CEP290, RPGRIPL1, NPHP1, TMEM67, ARL13B, TMEM216, OFD1, AHI1 |
| Leber congenital amaurosis | 204000 | CEP290, RPGRIPL1, |
| Senior–Løken syndrome | 266900 | CEP290, NPHP1, INVS, NPHP3, NPHP4, NPHP5 |
| Orofaciodigital syndrome | 311200 | TMEM216, OFD1 |
| Meckel–Gruber syndrome | 249000, 267010 | CEP290, NPHP3, TMEM67, RPGRIPL1, |
| Nephronophthisis | 256100 | CEP290, NPHP1, INVS, NPHP3, NPHP4, NPHP5, TMEM67, RPGRIPL1 |
Because of the many issues in BBS, a multidisciplinary approach is required to manage this pleiotropic condition. Because there is not yet a targeted treatment, the clinical intervention should be focused on symptoms as they arise. In particular, the renal function needs to be monitored and the risk for diabetes insipidus (which is frequent in BBS) should be taken into account. Ophthalmological follow-up is also recommended.
The clinical features of BBS significantly overlap with the manifestations of other ciliopathies. Among those, Alstrom and McKusick–Kauffman syndromes manifest significant overlap with BBS. However, hearing loss and absence of polydactyly differentiate Alstrom syndrome from BBS, and the high prevalence of urogenital anomalies in McKusick–Kauffman syndrome that normally lacks obesity, rod-cone dystrophy, and learning difficulties differentiate this latter condition from BBS.
It is interesting to consider how ciliopathies may contribute to mental illness; several genes in this group have been associated with developmental delay syndromes, including ID and ASD. The means by which cilia are involved in neurodevelopmental processes and by which ciliopathies lead to neurodevelopmental disorders are areas of active research. In addition to the role of motile cilia in mobilizing cerebrospinal fluid, primary cilia, found on most neurons and astrocytes, play roles as modulators of signal transduction during both brain development and homeostasis (Lee and Gleeson, 2011).
COHEN SYNDROME
Cohen syndrome (CS) is a rare autosomal-recessive disorder, first described in 1973, characterized by hypotonia, obesity, ID, characteristic facial appearance, and narrow hands and feet. There is consistent clinical heterogeneity, particularly evident when comparing patients of different ethnic backgrounds, especially when evaluating specific system phenotypes separately, such as the ophthalmic and central nervous systems (Douzgou and Petersen, 2011). The incidence of CS is unknown; however, this condition is overrepresented in Finland owing to the peculiar population structure in genetic isolates. The Finnish CS phenotype is remarkably homogeneous, whereas clinical variability is the rule in non-Finnish patients with CS. Some ethnic group shows peculiarities; for instance, the Amish CS neonatal phenotype is characterized in 50% of cases by respiratory distress syndrome, jaundice, and acidosis, whereas a high-pitched voice and an age-related loss of the left ventricular function have been recurrently reported only in the Finnish CS cohort.
The inheritance is autosomal-recessive and the causative gene, named VPS13B or COH1, encodes a Golgi-associated matrix protein required for Golgi integrity (Kolehmainen et al., 2003; Seifert et al., 2011). The mutational spectrum is wide and mutations can be present in different portions of the gene; missense, non-sense, and frameshift mutations as well as intragenic deletions have been found in CS patients. Non-Finnish patients are almost always compound heterozygotes (Katzaki et al., 2007). Not much is known of the pathogenesis of CS, but because high levels of urinary hyaluronic acid have been reported, it has been suggested that CS is a connective tissue disorder.
Typical craniofacial features include down-slanting palpebral fissures, thick eyebrows, convex profile of nose, and short philtrum with open mouth and prominent lips exposing the upper central incisors. These features become more evident as the child gets older. In addition, patients with CS exhibit short stature, generalized hypotonia, truncal obesity (evident after five years), delayed puberty, and cryptorchidism. Related to hypotonia is the frequent finding of kyphoscoliosis. Microcephaly can be of prenatal onset and ID, moderate to severe, is usually observed. Seizures are rare and a happy affectionate predisposition is typical.
The hands and feet are narrow, with slender fingers and joint hyperextensibility. Ophthalmological involvement is quite constant. Myopia can develop in up to 90% of patients, and retinal pigmentary degeneration can occur in late childhood in about 80% of cases. However, these lesions only rarely lead to blindness. Neutropenia is also a recurrent, although not constant, laboratory finding. Among these clinical features, some can be considered good indicators for the presence of a mutation in the causative gene: postnatal microcephaly, chorioretinal dystrophy, and neutropenia. It has been suggested that, given the large size of the gene, the mutational analysis should not be indicated in the absence of these features. The follow-up of young patients could be a satisfactory alternative unless there are reproductive issues (El El Chehadeh et al., 2010).
The phenotype in CS is quite distinctive, although large-scale mutational analysis has documented the existence of atypical cases. In any case, the diagnosis may be not evident until late childhood when the cardinal features of the condition manifest. A group of ID conditions to be considered as those of syndromes with hypotonia, obesity, and mental retardation, including Prader-Willi syndrome, which clearly has a completely different natural history and clinical appearance. If the phenotypic appearance is not straightforward, quantitative genomic alterations should be ruled out by CMA before considering the diagnosis of non-typical CS.
SMITH–LEMLI–OPITZ SYNDROME
Smith–Lemli–Opitz syndrome (SLOS, #OMIM 270400) was first described in 1964, as a multiple congenital anomalies-mental retardation (MCA-MR) syndrome characterized by microcephaly, peculiar face, developmental delay, syndactyly between the second and third toe, and genital anomalies (Smith et al., 1964). The clinical presentation may be variable in severity, ranging from neonatal death to survival into childhood and adolescence with severe ID and behavioral impairment.
Smith–Lemli–Opitz syndrome is a rare disorder (estimated incidence is between 1:20,000 and 1:60,000 in different populations), but it is not possible to rule out a higher incidence because of mildly affected individuals in whom the condition is difficult to recognize. In any case, it is considered among the most frequent autosomal recessive disorders, with an estimated carrier frequency of 1% to 2% in the general population.
The inheritance is autosomal recessive and the causative gene, named DHCR7, encodes the 3-beta-hydroxysterol-
delta-7-reductase, an enzyme converting 7-dehydrocholesterol into cholesterol. This defects leads to high plasma levels of 7-dehydrocholesterol and low cholesterol (Irons et al., 1993). Therefore, the clinical manifestations of SLOS are caused by a defect in cholesterol biosynthesis, a metabolic pathway that is crucial during embryonic development from early on (Porter and Herman, 2011). Because cholesterol is also required for the proper function of the sonic hedgehog (SHH) pathway, some clinical manifestations commonly related to SHH dysfunction, such as holoprosencephaly, can be observed in SLOS. The mutational spectrum of the SLOS gene is wide and mutations in the transmembrane domain or the C-terminal region have been associated with milder phenotypes.
Prenatally, growth retardation, oligohydramnios, and decreased fetal movements can be observed as well as breech presentation and perinatal asphyxia. Increased nucal translucency can be detected by ultrasound (Quelin et al., 2012). Low birth weight is almost always present and failure to thrive is experienced in a high percentage of patients. Death in the neonatal period may occur because of severe feeding difficulties and hepatic dysfunction. Those who survive into childhood usually have short stature and severe intellectual impairment associated with behavioral issues, including autism, aggressiveness, sleep disturbances, and hyperactivity.
Physical anomalies in SLOS can be present in almost any part of the body (DeBarber et al., 2011; Nowaczyk et al., 2012). The brain is often underdeveloped with microcephaly and a constellation of anomalies, including holoprosencephaly, agenesis of the corpus callosum, ventricular dilatation, and cerebellar vermis hypoplasia. Reduced myelination in the cerebral hemispheres, cranial nerves, and peripheral nerves also occur. The face appears round-shaped with hypertelorism, down-slanting palpebral fissures and ptosis, upturned and wide nose, and micrognathia. The mouth shows broad alveolar ridges, small tongue, and rarely lingual cysts. Strabismus and cataract can also be observed. The extremities are also commonly affected, showing most frequently second to third toe syndactyly, postaxial polydactyly, and shortness of the first metacarpals and metatarsals. Positional foot abnormalities are often present as well as hip dislocation. Abnormalities of the external genitalia, mostly reflecting a failure of masculinization, are frequently observed, including cryptorchidism, hypospadias, micropenis, bifid scrotum, ambiguous genitalia, and even sex reversal. The urinary tract can be obstructed or hypoplastic and renal dysplasia (cysts or hypoplasia) can be present. Cardiovascular malformations occur in about 50% of the patients, mostly PDA, VSD, ASD, or tetralogy of Fallot. Other findings include pancreatic islet cells hyperplasia, unilobate lungs, and photosensitivity.
The clinical diagnosis can be confirmed with a biochemical test measuring plasma concentrations of cholesterol (that are usually very low in SLOS) and 7-dehydrocholesterol (increased in SLOS). High levels of 7-dehydrocholesterol can be detected in the amniotic fluid and chorionic villus specimens as well (Kratz and Kelley, 1999).
Supplementary cholesterol is rationally expected to supply the tissues and also reduce the toxic levels of 7-dehydro-cholesterol. Simvastatin, aimed at further decreased sterol synthesis, has been also used as potential therapy. Although reported in the literature, the beneficial effects on behavior of dietary cholesterol supplementation have not been formally documented through a randomized clinical trial. Larger studies are still needed to clearly demonstrate the utility of treatment of this metabolic disorder (Tierney et al., 2010).
Chromosomal disorders should be ruled out in newborns with features compatible with the SLOS phenotype. Also, some clinical overlapping exists with Meckel syndrome (in which the cystic kidney dysplasia is typical) and Pallister-Hall syndrome (in which the finding of a hypothalamic hamartoblastoma is quite unique).
X-LINKED
ALLAN–HERNDON–DUDLEY SYNDROME
Allan–Herndon–Dudley syndrome (AHDS, OMIM #300523) was first described in 1944 and was the second X-linked ID syndrome to appear in the literature (Allan et al., 1944). The authors described a large North Carolina (USA) family with 24 affected males over multiple generations. The major clinical findings were hypotonia, muscle hypoplasia, general muscle weakness, which resulted in a “limber neck” (noted by family members), dysarthria, and ataxia. Speech was also limited and there was significant developmental retardation.
The incidence of AHDS is unknown. It is considered one of the more prominent X-linked conditions with more than 25 families having been described, most from the United States and Western Europe. A review of most of the families found that in half of them, there was only one affected male (Schwartz and Stevenson 2007). This same review allowed for a good clinical description of AHDS and an idea of the natural history of the syndrome.
Initially, males with AHDS have birth measurements within the normal range. Most maintain normal postnatal growth. However, muscle hypoplasia was evident early in life and persisted into adulthood.
Facial features appear to be consistent across families. A myopathic face or hypotonic facial appearance with an open mouth and tented upper lip is usually present. There is bitemporal narrowing in most males and the face becomes tall and narrow in adulthood. The ears appear to be simple and cupped.
Neurologically, males with ADHS are hypotonic at birth. The postnatal weakness persists and progresses to spasticity with age. Dystonia and athetoid movements are present. Hyperreflexia becomes evident in adulthood.
Cognitive function is delayed from early in life. There is significant delay in acquiring language and all males with AHDS have severe ID.
The gene for AHDS was mapped to the Xq13-q21 region of the X chromosome when Schwartz and colleagues re-studied the original family (Schwartz et al., 1990). In 2004, both Dumitrescu and colleagues (2004) and Friesema and colleagues (2004) reported families and unrelated males who presented with neurological findings similar to those observed in AHDS. Additionally, the affected individuals had decreased serum T4 levels and significantly elevated serum levels of T3. However, TSH levels in serum were in the high normal range. Both groups identified mutations in the monocarboxylate transporter 8 gene (MCT8, SLC16A2) in their patients. Subsequently, Schwartz and colleagues (2005) showed that mutations in MCT8 were also present in their collection of families with AHDS.
MCT8 is critical for the metabolism of thyroid hormone in the brain. The protein functions as a transporter of T3 into neurons. Without T3, many genes within the neuron are not properly regulated. Thus, the presence of severe cognitive impairment in AHDS patients is not unexpected. However, other aspects of the clinical phenotype observed in AHDS patients do not reflect a defect in thyroid function. Additional studies are needed to explain this disconnect.
The presence of elevated serum T3 levels provides a convenient method for testing for AHDS in at-risk individuals such as males with hypotonia and developmental delay. The prevalence of MCT8 is not known, but with a reliable and rapid test, many AHDS families have been identified. Thus, the prevalence of AHDS may be high relative to other X-linked intellectual disability syndromes.
ALPHA-THALASSEMIA INTELLECTUAL DISABILITY SYNDROME
Alpha-thalassemia intellectual disability or ATRX syndrome (OMIM #301040) is an X-linked recessive condition first noted clinically by Weatherall and colleagues in 1981 when they reported an association of hemoglobin H inclusion bodies in patients with ID. The possibility that this represented an X-linked syndrome was evident by 1990 when Wilkie and colleagues (1991) reported additional patients who did not have any abnormality of the alpha-globin genes located at 16p. The patients were all males and presented with a common phenotype of microcephaly, a hypotonic face (hypotelorism, small triangular nose, tented upper lip, open mouth) and severe ID with mild hemoglobin H disease.
Since the initial descriptions by Weatherall et al. (1981) and Wilkie et al. (1991), many additional patients and families with ATRX have been characterized and a fairly comprehensive review was compiled by Gibbons and Higgs (2000). The clinical spectrum of ATRX consists of hypotonia present in infancy, distinctive facial features of an open mouth with a tented upper lip and prominent lower lip, small upturned triangular nose, and hypertelorism. These facial features become “coarser” with age. Some patients have skeletal anomalies and urogenital anomalies such as cryptorchidism and hypospadias. Speech is severely delayed and maybe absent. There is growth delay and microcephaly is present. One feature noted in the original patients, hemoglobin H inclusion bodies, is not consistently present. Thus, although this was thought to be a useful test for ATRX, many proven cases are not positive for this finding and the test is no longer routinely done in males suspected of having ATRX.
Once ATRX was considered to be an XLID syndrome, Gibbons and colleagues mapped the gene to Xq12-q21.3 by linkage analysis in 1992 (Gibbons et al., 1992). They identified the candidate gene, XNP (X-linked nuclear protein) in 1995 (Gibbons et al., 1995). The availability of a gene test allowed screening of XLID syndromes linked to the Xq13-q21 region as well as some syndromes whose clinical phenotype overlapped with ATRX. As a result, other XLID syndromes were shown to also result from mutations in XNP: Carpenter–Waziri, Holmes–Gang, and Chudley–Lowry. Two families with the diagnoses of Juberg–Marsidi and Smith–Fineman–Myers syndromes were found to have mutations in XNP (Villard et al., 1996, 2000). However, the families first described with these syndromes (Juberg and Marsidi, 1980; Smith et al., 1980) have not been tested for XNP mutations. In fact, the original Juberg-Marsidi family was recently found to have a mutation in the HUWE1 gene, whereas XNP testing was negative (Abidi and colleagues, unpublished data).
The prevalence of ATRX is unknown in the general population. However, close to 200 males with ATRX syndrome have been reported; thus, it is reasonable to expect the prevalence to be relatively high, about 1% to 2% in males with ID.
One clinical feature that is now considered a hallmark of ATRX is that female carriers of an XNP mutation have highly skewed X-inactivation, which probably explains the lack of a clinical phenotype in these females. This distinction, skewed X-inactivation and lack of clinical findings in carriers, allows one to clinically differentiated ATRX from another XLID syndrome, Coffin–Lowry (OMIM #303600). This is critical since both can be confused in early childhood.
Some attempt has been made to construct a genotype/phenotype correlation because of the clinical spectrum of the syndrome and the number of patients with mutations. Unfortunately, this correlation has proved to be less than successful. There does appear to be some association between the loss of the C-terminal end of the protein and the presence of severe urogenital abnormalities (Gibbons and Higgs, 2000). Also, Badens and colleagues (2006) did observe that mutations in the PHD-like domains tended to result in more severe psychomotor delay and more severe urogenital abnormalities. However, overall, no clear genotype/phenotype association has been observed.
One recent observation has been made that may be significant. A fairly common R37X mutation has been found in families who present with a milder form of ATRX (Abidi et al., 2005; Guerrini et al., 2000). This may result from the presence of a small amount of full-length protein as detected by Western blotting and the utilization of an initiation site downstream from the mutation (Abidi et al., 2005; Howard et al., 2004). The XNP/ATRX gene was initially cloned and partially characterized by Stayton and colleagues in 1994 (Stayton et al., 1994). They determined it was homologous to members of the helicase II superfamily. Picketts and colleagues (1996) characterized the XNP/ATRX protein further establishing it was a member of the SNF2-like subgroup of proteins, contained helicase domains, a nuclear localization signal, and a C-terminal glutamine-rich region characteristic of a transcription factor. Villard and colleagues (1997) further determined the protein has three zinc finger motifs in the 5′ end of the gene. Last, Gibbons and colleagues (1997) identified a cystine-rich motif, similar to a putative PHD zinc finger domain, in the N-terminal region. With the presence of these various domains, it is not surprising that the XNP/ATRX protein has chromatin remodeling and DNA translocase activities (Gibbons et al., 2003) and with its widespread expression in during embryogenesis (Stayton et al., 1994) mutations in XNP give rise to a multisystem syndrome. This places ATRX into a class of ID and ASD genes that affect regulation of gene expression.
OPITZ–KAVEGGIA SYNDROME (FG SYNDROME)
Opitz–Kaveggia syndrome, better known as FG syndrome (FGS, OMIM #305450) is an X-linked recessive syndrome first reported in 1974 (Opitz and Kaveggia, 1974). The report was based on a single family with macrocephaly, ID, distinctive facial features, hypotonia, broad thumbs, and imperforate anus. The facial features were comprised of hypertelorism, small ears, frontal upsweep of the hair, broad forehead, and down-slanted palpebral fissures. Additionally, partial agenesis of the corpus callosum was found in one of the affected males, but this analysis was not conducted in all affected individuals.
Since the initial FGS description, many other families have been reported to have clinical features of FG syndrome. These publications had expanded the FGS phenotype and led to the identification of multiple FG syndromic loci on the X-chromosome. Genetic heterogeneity as well as clinical variability made the clinical diagnosis difficult and at the same time rather common among males with ID, macrocephaly, and constipation. This was the situation until 2007 when Risheg and colleagues identified the same mutation (c.2881C>T, p.R961W) in the MED12 gene in six families with an FGS diagnosis (Risheg et al., 2007). The six families included a male from the original family of Opitz and Kaveggia.
After the identification of the MED12 mutation, many males with a diagnosis of FGS were screened for mutations in MED12. This did lead to the identification of other males with the same R961W mutation, but it became evident that it was difficult to confirm the FGS diagnosis in males with a rather non-specific phenotype (Lyons et al., 2009). Based on these results and the fact that the original family had the R961W MED12 mutation, it was proposed that only males with this alteration be designated as having Opitz–Kaveggia syndrome. To simplify the diagnosis, Clark and colleagues (2009) developed a clinical algorithm for diagnosis based on a clinical evaluation of 23 patients with the R961W mutation as compared with 48 patients who had a clinical diagnosis of FGS but were mutation negative.
The clinical features of Opitz-Kaveggia syndrome are defined as a male with intellectual disability, developmental disability, small ears, a characteristic face (long narrow face with a tall forehead, upsweep of the frontal hairline, open mouth), congenital anomalies of the corpus callosum, anus, heart or skeleton, macrocephaly, early hypotonia, constipation, and an affable, eager-to-please personality (Clark et al., 2009; Graham et al., 2008; Lyons et al., 2009).
The utilization of the clinical diagnosis algorithm developed by Clark and colleagues (2009) has already proved to be useful. Rump and colleagues (2011) screened MED12 in a Dutch family with three affected male cousins based on the males fitting the clinical criteria for Opitz-Kaveggia syndrome. They found a novel MED12 mutation, C.2873G>A (p.G958E) in the family. This mutation is three amino acids away from the R961W mutation and is in the same stretch of the highly conserved Leu-Ser rich domain of the MED12 protein.
The prevalence of FGS is unknown. Before the identification of MED12 as a causative gene, the FGS diagnosis was not uncommon in males with ID, macrocephaly, and constipation; thus, it was thought to be a significant contributor to X-linked ID. Now, it is likely to be a relatively rare XLID entity, probably not exceeding 0.5% of males with ID. Gene testing over the next few years will better address this point. However, it needs to be noted that another syndrome, Lujan syndrome (OMIM #309520) also has a mutation (p.N1007S) in MED12 (Schwartz et al. 2007) and at least two other MED12 mutations have been identified in FGS patients (Schwartz et al., unpublished data). Thus, although the FGS associated MED12 mutation may be rare, other mutations in the gene may be responsible for ID in the male population.
The MED12 protein is a member of the large mediator complex that plays an important regulatory role in the activity of RNA polymerase II (Knuesel, Meyer, Donner, et al., 2009). Mediator also acts as to regulate transcription initiation (Knuesel, Meyer, Bernecky, et al., 2009). Recently, it was shown that Mediator is part of a protein interaction network comprised of G9a histone methyltransferase and REST (RE1 silencing transcription factor) (Ding et al., 2008). This network suppresses neuronal gene expression in non-neuronal cells. Of interest, Ding et al., (2008) have shown the R961W MED12 mutation disrupts the REST corepressor function of Mediator, suggesting a pathologic basis for the MED12 mutation.
CONCLUSIONS
There seems to be a common denominator to the pathophysiology of the ID syndromes described in this chapter, expandable to many other conditions, all being disorders of development. All causative genes are in essence “master” genes affecting the expression of other genes either by disrupting signaling pathways, affecting chromatin structure, or altering structures (e.g., the cilium) that are essential for the normal functioning of the cell. The knowledge gained points investigators to other gene defects to explore in patients who do not harbor mutations in the presumptive candidate gene based on the phenotype. In spite of the complexity of the mechanisms involved, the knowledge of the molecular details and protein interactions promises to open the way to pharmacological interventions aimed at specifically correcting or at least ameliorating the basic defect or its metabolic consequences in ID syndromes.
ACKNOWLEDGMENTS
We thank Debra Marler for assisting with the preparation of the manuscript. Dedicated to the memory of Ethan Francis Schwartz, 1996–1998.
DISCLOSURES
Dr. Schwartz: To my knowledge, all of my possible conflicts of interest and those of my coauthors, financial or otherwise, including direct or indirect financial or personal relationships, interests, and affiliations, whether or not directly related to the subject of the chapter, are as follows. I am funded by NICHD and SCDDSN (South Carolina Department of Disabilities and Special Needs). This work was supported in part by a grant from the National Institutes of Health (NICHD) [HD26202 to C.E.S.].
Dr. Gurrieri: No conflicts of interest to disclose.
Dr. Neri: No conflicts of interest to disclose.
REFERENCES
Abidi, F.E., Cardoso, C., et al. (2005). Mutation in the 5′ alternatively spliced region of the XNP/ATR-X gene causes Chudley-Lowry syndrome. Eur. J. Hum. Genet. 13(2):176–183.
Abu-Safieh, L., Al-Anazi, S., et al. (2012). In search of triallelism in Bardet-Biedl syndrome. Eur. J. Hum. Genet. 20(4):420–427.
Adam, M.P., Hudgins, L., et al. (2011). Kabuki Syndrome. In: Pagon, R.A., et al., eds., GeneReviewsTM [Internet]. Seattle: University of Washington.
Allan, W., Herndon, C.N., et al. (1944). Some examples of the inheritance of mental deficiency: apparently sex-linked idiocy and microcephaly. Am. J. Ment. Defic. 48:325–334.
Armour, C.M., and Allanson, J.E. (2008). Further delineation of cardio-facio-cutaneous syndrome: clinical features of 38 individuals with proven mutations. J. Med. Genet. 45(4):249–254.
Badens, C., Lacoste, C., et al. (2006). Mutations in PHD-like domain of the ATRX gene correlate with severe psychomotor impairment and severe urogenital abnormalities in patients with ATRX syndrome. Clin. Genet. 70(1):57–62.
Baujat, G., and Cormier-Daire, V. (2007). Sotos syndrome. Orphanet. J. Rare Dis. 2:36.
Beales, P.L., Reid, H.A., et al. (2000). Renal cancer and malformations in relatives of patients with Bardet-Biedl syndrome. Nephrol. Dial. Transplant. 15(12):1977–1985.
Beales, P.L., Badano, J.L., et al. (2003). Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome. Am. J. Hum. Genet. 72(5):1187–1199.
Berry-Kravis, E.M., Hessl, D., et al. (2012). Effects of STX209 (Arbaclofen) on neurobehavioral function in children and adults with fragile X syndrome: a randomized, controlled, Phase 2 trial. Sci. Transl. Med. 4:152ra127
Betancur, C. (2011). Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 1380:42–77.
Clark, R.D., Graham, J.M., Jr., et al. (2009). FG syndrome, an X-linked multiple congenital anomaly syndrome: the clinical phenotype and an algorithm for diagnostic testing. Genet. Med. 11(11):769–775.
Cohen, M.M., Jr. (2003). Mental deficiency, alterations in performance, and CNS abnormalities in overgrowth syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 117C(1):49–56.
Curry, C.J., Stevenson, R.E., et al. (1997). Evaluation of mental retardation: recommendations of a Consensus Conference: American College of Medical Genetics. Am. J. Med. Genet. 72(4):468–477.
DeBarber, A.E., Eroglu, Y., et al. (2011). Smith-Lemli-Opitz syndrome. Expert Rev. Mol. Med. 13:e24.
Ding, N., Zhou, H., et al. (2008). Mediator links epigenetic silencing of neuronal gene expression with x-linked mental retardation. Mol. Cell 31 (3):347–359.
Douglas, J., Tatton-Brown, K., et al. (2005). Partial NSD1 deletions cause 5% of Sotos syndrome and are readily identifiable by multiplex ligation dependent probe amplification. J. Med. Genet. 42(9):e56.
Douzgou, S., and Petersen, M.B. (2011). Clinical variability of genetic isolates of Cohen syndrome. Clin. Genet. 79(6):501–506.
Dumitrescu, A.M., Liao, X.H., et al. (2004). A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am. J. Hum. Genet. 74(1):168–175.
Eichers, E.R., Lewis, R.A., et al. (2004). Triallelic inheritance: a bridge between Mendelian and multifactorial traits. Ann. Med. 36(4):262–272.
El Chehadeh, S., Aral, B., et al. (2010). Search for the best indicators for the presence of a VPS13B gene mutation and confirmation of diagnostic criteria in a series of 34 patients genotyped for suspected Cohen syndrome. J. Med. Genet. 47(8):549–553.
Forsythe, E., and Beales, P.L. (2013). Bardet-Biedl syndrome. Eur. J. Hum. Genet. 1:8–13.
Friesema, E.C., Grueters, A., et al. (2004). Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 364(9443):1435–1437.
Gerdes, J.M., Davis, E.E., et al. (2009). The vertebrate primary cilium in development, homeostasis, and disease. Cell 137(1):32–45.
Gibbons, R.J., Bachoo, S., et al. (1997). Mutations in transcriptional regulator ATRX establish the functional significance of a PHD-like domain. Nat. Genet. 17(2):146–148.
Gibbons, R.J., Pellagatti, A., et al. (2003). Identification of acquired somatic mutations in the gene encoding chromatin-remodeling factor ATRX in the alpha-thalassemia myelodysplasia syndrome (ATMDS). Nat. Genet. 34(4):446–449.
Gibbons, R.J., and Higgs, D.R. (2000). Molecular-clinical spectrum of the ATR-X syndrome. Am. J. Med. Genet. 97(3):204–212.
Gibbons, R.J., Picketts, D.J., et al. (1995). Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell 80(6):837–845.
Gibbons, R.J., Suthers, G.K., et al. (1992). X-linked alpha-thalassemia/mental retardation (ATR-X) syndrome: localization to Xq12-q21.31 by X inactivation and linkage analysis. Am. J. Hum. Genet. 51(5):1136–1149.
Gibson, W.T., Hood, R.L., et al. (2012). Mutations in EZH2 cause Weaver syndrome. Am. J. Hum. Genet. 90(1):110–118.
Graham, J.M., Jr., Visootsak, J., et al. (2008). Behavior of 10 patients with FG syndrome (Opitz-Kaveggia syndrome) and the p.R961W mutation in the MED12 gene. Am. J. Med. Genet. A 146A(23):3011–3017.
Guerrini, R., Shanahan, J.L., et al. (2000). A nonsense mutation of the ATRX gene causing mild mental retardation and epilepsy. Ann. Neurol. 47(1):117–121.
Hjortshoj, T.D., Gronskov, K., et al. (2010). Bardet-Biedl syndrome in Denmark—report of 13 novel sequence variations in six genes. Hum. Mutat. 31(4):429–436.
Hoffman, J.D., Ciprero, K.L., et al. (2005). Immune abnormalities are a frequent manifestation of Kabuki syndrome. Am. J. Med. Genet. A 135(3):278–281.
Howard, M.T., Malik, N., et al. (2004). Attenuation of an amino-terminal premature stop codon mutation in the ATRX gene by an alternative mode of translational initiation. J. Med. Genet. 41(12):951–956.
Huber, C., and Cormier-Daire, V. (2012). Ciliary disorder of the skeleton. Am. J. Med. Genet. C Semin. Med. Genet. 160(3):165–174.
Irons, M., Elias, E.R., et al. (1993). Defective cholesterol biosynthesis in Smith-Lemli-Opitz syndrome. Lancet 341(8857):1414.
Juberg, R.C., and Marsidi, I. (1980). A new form of X-linked mental retardation with growth retardation, deafness, and microgenitalism. Am. J. Hum. Genet. 32(5):714–722.
Katzaki, E., Pescucci, C., et al. (2007). Clinical and molecular characterization of Italian patients affected by Cohen syndrome. J. Hum. Genet. 52(12):1011–1017.
Kawame, H., Hannibal, M.C., et al. (1999). Phenotypic spectrum and management issues in Kabuki syndrome. J. Pediatr. 134(4):480–485.
Kim, L.S., Fishman, G.A., et al. (2007). Retinal dysfunction in carriers of bardet-biedl syndrome. Ophthalmic. Genet. 28(3):163–168.
Knuesel, M.T., Meyer, K.D., et al. (2009). The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. 23(4):439–451.
Knuesel, M.T., Meyer, K.D., et al. (2009). The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of mediator. Mol. Cell Biol. 29(3):650–661.
Kolehmainen, J., Black, G.C., et al. (2003). Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am. J. Hum. Genet. 72(6):1359–1369.
Kratz, L.E., and Kelley, R.I. (1999). Prenatal diagnosis of the RSH/Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. 82(5):376–381.
Kurotaki, N., Harada, N., et al. (2001). Molecular characterization of NSD1, a human homologue of the mouse Nsd1 gene. Gene 279(2):197–204.
Kurotaki, N., Imaizumi, K., et al. (2002). Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet. 30(4):365–366.
Lee, J.E., and Gleeson, J.G. (2011). Cilia in the nervous system: linking cilia function and neurodevelopmental disorders. Curr. Opin. Neurol. 24: 98–105.
Lubs, H.A., Stevenson, R.E., et al. (2012). Fragile X and X-linked intellectual disability: four decades of discovery. Am. J. Hum. Genet. 90(4):579–590.
Lyons, M.J., Graham, J.M., Jr., et al. (2009). Clinical experience in the evaluation of 30 patients with a prior diagnosis of FG syndrome. J. Med. Genet. 46(1):9–13.
Mazzucchelli, C., and Brambilla, R. (2000). Ras-related and MAPK signalling in neuronaplasticity and memory formation. Cell. Mol. Life Sci. 57: 604–611.
Mefford, H.C., Batshaw, M.L., et al. (2012). Genomics, intellectual disability, and autism. N. Engl. J. Med. 366(8):733–743.
Mervis, C.B., Becerra, A.M., et al. (2005). Intellectual abilities and adaptive behavior of children and adolescents with Kabuki syndrome: a preliminary study. Am. J. Med. Genet. A 132A(3):248–255.
Narumi, Y., Aoki, Y., et al. (2007). Molecular and clinical characterization of cardio-facio-cutaneous (CFC) syndrome: overlapping clinical manifestations with Costello syndrome. Am. J. Med. Genet. A 143A(8):799–807.
Neri, G., Zollino, M., et al. (1991). The Noonan-CFC controversy. Am. J. Med. Genet. 39(3):367–370.
Ng, S.B., Bigham, A.W., et al. (2010). Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 42(9):790–793.
Niihori, T., Aoki, Y., et al. (2006). Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat. Genet. 38(3):294–296.
Niikawa, N. (2004). Molecular basis of Sotos syndrome. Horm. Res. 62(Suppl 3):60–65.
Niikawa, N., Kuroki, Y., et al. (1988). Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patients. Am. J. Med. Genet. 31(3):565–589.
Niikawa, N., Matsuura, N., et al. (1981). Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J. Pediatr. 99(4):565–569.
Nowaczyk, M.J., Tan, M., et al. (2012). Smith-Lemli-Opitz syndrome: Objective assessment of facial phenotype. Am. J. Med. Genet. A 158A(5):1020–1028.
Opitz, J.M., and Kaveggia, E.G. (1974). Studies of malformation syndromes of man 33: the FG syndrome. An X-linked recessive syndrome of multiple congenital anomalies and mental retardation. Z. Kinderheilkd. 117(1):1–18.
Picketts, D.J., Higgs, D.R., et al. (1996). ATRX encodes a novel member of the SNF2 family of proteins: mutations point to a common mechanism underlying the ATR-X syndrome. Hum. Mol. Genet. 5(12):1899–1907.
Porter, F.D., and Herman, G.E. (2011). Malformation syndromes caused by disorders of cholesterol synthesis. J. Lipid Res. 52(1):6–34.
Quelin, C., Loget, P., et al. (2012). Phenotypic spectrum of fetal Smith-LemliOpitz syndrome. Eur. J. Med. Genet. 55(2):81–90.
Rauen, K.A., Schoyer, L., et al. (2010). Proceedings from the 2009 genetic syndromes of the Ras/MAPK pathway: From bedside to bench and back. Am. J. Med. Genet. A 152A(1):4–24.
Reynolds, J.F., Neri, G., et al. (1986). New multiple congenital anomalies/mental retardation syndrome with cardio-facio-cutaneous involvement—the CFC syndrome. Am. J. Med. Genet. 25(3):413–427.
Risheg, H., Graham, J.M., Jr., et al. (2007). A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat. Genet. 39(4):451–453.
Roberts, A., Allanson, J., et al. (2006). The cardiofaciocutaneous syndrome. J. Med. Genet. 43(11):833–842.
Rodriguez-Viciana, P., Tetsu, O., et al. (2006). Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 311(5765):1287–1290.
Ropers, H.H. (2010). Genetics of early onset cognitive impairment. Annu. Rev. Genomics Hum. Genet. 11:161–187.
Rump, P., Niessen, R.C., et al. (2011). A novel mutation in MED12 causes FG syndrome (Opitz-Kaveggia syndrome). Clin. Genet. 79(2):183–188.
Sarimski, K. (2003). Behavioural and emotional characteristics in children with Sotos syndrome and learning disabilities. Dev. Med. Child Neurol. 45(3):172–178.
Schaefer, G.B., Bodensteiner, J.B., et al. (1997). The neuroimaging findings in Sotos syndrome. Am. J. Med. Genet. 68(4):462–465.
Schwartz, C.E., May, M.M., et al. (2005). Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am. J. Hum. Genet. 77(1):41–53.
Schwartz, C.E., and Stevenson, R.E. (2007). The MCT8 thyroid hormone transporter and Allan-Herndon-Dudley syndrome. Best Pract. Res. Clin. Endocrinol. Metab. 21(2):307–321.
Schwartz, C.E., Tarpey, P.S., et al. (2007). The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J. Med. Genet. 44(7):472–477.
Schwartz, C.E., Ulmer, J., et al. (1990). Allan-Herndon syndrome. II. Linkage to DNA markers in Xq21. Am. J. Hum. Genet. 47(3):454–458.
Seifert, W., Kuhnisch, J., et al. (2011). Cohen syndrome-associated protein, COH1, is a novel, giant Golgi matrix protein required for Golgi integrity. J. Biol. Chem. 286(43):37665–37675.
Smith, D.W., Lemli, L., et al. (1964). A newly recognized syndrome of multiple congenital anomalies. J. Pediatr. 64:210–217.
Smith, R.D., Fineman, R.M., et al. (1980). Short stature, psychomotor retardation, and unusual facial appearance in two brothers. Am. J. Med. Genet. 7(1):5–9.
Stayton, C.L., Dabovic, B., et al. (1994). Cloning and characterization of a new human Xq13 gene, encoding a putative helicase. Hum. Mol. Genet. 3(11):1957–1964.
Tidyman, W.E., and Rauen, K.A. (2009). The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 19:230–236.
Tierney, E., Conley, S.K., et al. (2010). Analysis of short-term behavioral effects of dietary cholesterol supplementation in Smith-Lemli-Opitz syndrome. Am. J. Med. Genet. A 152A(1):91–95.
van Bokhoven, H. (2011). Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 45:81–104.
Villard, L., Fontes, M., et al. (2000). Identification of a mutation in the XNP/ATR-X gene in a family reported as Smith-Fineman-Myers syndrome. Am. J. Med. Genet. 91(1):83–85.
Villard, L., Gecz, J., et al. (1996). XNP mutation in a large family with Juberg-Marsidi syndrome. Nat. Genet. 12(4):359–360.
Villard, L., Lossi, A.M., et al. (1997). Determination of the genomic structure of the XNP/ATRX gene encoding a potential zinc finger helicase. Genomics 43(2):149–155.
Waters, A.M., and Beales, P.L. (2011). Ciliopathies: an expanding disease spectrum. Pediatr. Nephrol. 26(7):1039–1056.
Weatherall, D.J., Higgs, D.R., et al. (1981). Hemoglobin H disease and mental retardation: a new syndrome or a remarkable coincidence? N. Engl. J. Med. 305(11):607–612.
Wilkie, A.O., Pembrey, M.E., et al. (1991). The non-deletion type of alpha thalassaemia/mental retardation: a recognisable dysmorphic syndrome with X linked inheritance. J. Med. Genet. 28(10):724.
Winnepenninckx, B., Rooms, L., et al. (2003). Mental retardation: a review of the genetic causes. Br. J. Dev. Disabil. 49:29–44.