88 | THE NEUROBIOLOGY OF RESILIENCE
ADRIANA FEDER, MARGARET HAGLUND, GANG WU,
STEVEN M. SOUTHWICK, AND DENNIS S. CHARNEY
Resilience is the ability to adapt successfully to severe or chronic stress (Charney, 2004; Rutter, 2006). Resilient individuals are those who are able to demonstrate healthy psychological and physiological stress responses in the face of extreme stress or trauma exposure, thus maintaining “psychobiological allostasis” (Feder et al., 2009; Karatsoreos and McEwen, 2011). Historically, most research on the effects of severe stress focused on its deleterious effects on well-being. The study of resilience began in the 1970s with the study of children exposed to significant adversity during development (Masten, 2001), later extending to the study of trauma-exposed adults (Alim et al., 2008; Bonanno, 2004). More recent epidemiological studies are beginning to look at longitudinal trajectories of posttraumatic stress disorder (PTSD) symptoms and resilience over time, and have begun to distinguish a resistance trajectory, characterized by the absence of symptoms, from a resilience trajectory, characterized by some initial symptom development, followed by prompt recovery (Bonanno et al., 2011; Hobfoll et al., 2009).
In the last decade, there has been a great deal of interest in delineating the neurobiological and psychological profile of resilient individuals. Supplementing studies in humans, animal studies are greatly advancing our understanding of neural and molecular mechanisms underlying resilience to stress (Russo et al., 2012). Identifying the factors that set resilient individuals apart from those who are vulnerable to the effects of stress has clinical significance: understanding the neurobiology and psychology of resilience enables researchers and clinicians to develop new therapies for the prevention and treatment of stress-induced psychopathology in non-resilient individuals. Furthermore, motivated individuals can draw from our increasing understanding of the features of resilience to achieve greater personal resilience to stress (Southwick and Charney, 2012a).
This chapter outlines the current understanding of resilience, from genetic, epigenetic, developmental, psychological, and neurobiological perspectives, and presents promising new therapies for the promotion of resilience to stress.
PREVALENCE OF RESILIENCE
The majority of individuals are exposed to trauma during their lifetime: in the United States, lifetime risk of at least one significant traumatic event, such as sudden unexpected death of a loved one, injury in a motor vehicle accident, or experiencing assault, is estimated at 80–90% (Breslau et al., 1998; Bruce et al., 2001). The prevalence of resilience depends on the particular definition and measure(s) used, on the population studied, and importantly on the severity and chronicity of stress or trauma exposure. In the words of Rutter (2012), “resilience is an inference based on evidence that some individuals have a better outcome than others who have experienced a comparable level of adversity.”
Population studies in the United States have estimated the lifetime prevalence of PTSD at 8%, and the conditional probability of PTSD after exposure to trauma at 9% (Breslau et al., 1998; Kessler et al., 1995). Certain types of trauma, however, are significantly more likely to cause PTSD, particularly rape (65%) and combat exposure (38.8%) for men, and rape (45.9%) and physical abuse (48.5%) for women (Kessler et al., 1995). In general, traumatic events that are severe and unpredictable, intentionally caused by other human beings (such as assaultive trauma), and result in loss (of a loved one, property, or physical integrity) are most likely to lead to PTSD (Jordan et al., 1991; Yehuda, 2004). In a study of a high-risk primary care sample, the resilient group had a significantly lower lifetime prevalence of assaultive trauma (46.8% vs. 75.6%), as well as a significantly lower mean number of lifetime experienced trauma types (3.4% vs. 5.1%) than the group with current psychiatric disorders (Alim et al., 2008). Further, studies have shown that the degree of control that an individual has over a stressor or trauma is a key factor in modulating the impact of a stressor (Fleshner et al., 2011).
Recent longitudinal trajectories studies have employed latent growth mixture modeling, a statistical approach that identifies clusters of individuals following distinct courses of longitudinal symptom development without assuming a-priori trajectory types (Nagin and Odgers, 2010; Norris et al., 2009). In these studies, the prevalence of resilience has ranged widely depending on the population studied. For example, the resilience trajectory represented over 80% of the sample in a study of deployed US military service members (Bonanno et al., 2012), whereas it was 35.6% (resistance and resilience trajectories combined) in a sample under ongoing threat of terrorism and rocket attacks (Hobfoll et al., 2009). Of note, the second study used PTSD and depressive symptom measures to define resilience, whereas the first one only measured PTSD symptoms. Despite differences in prevalence across studies, research has shown that a significant percentage of individuals are resilient to the effects of extreme stress.
GENETICS OF RESILIENCE
Studies of gene-by-environment interactions have begun to identify how complex interactions between genetic contributions and an individual’s particular history of exposure to environmental stressors yield a unique profile of adaptability of an individual’s neurochemical stress responses and neural circuitry function upon exposure to new stressors. Further, as discussed in the following, epigenetic modifications are now understood to modulate stress reactivity by regulating gene expression at the molecular level.
It has long been known that stress-related disorders are at least partially heritable. For example, studies of combat-exposed men enrolled in the Vietnam Twin Registry have found that PTSD, panic disorder, and general anxiety disorder, frequently comorbid conditions, all have significant genetic contributions. It is estimated that up to 40% of the variance in occurrence of these disorders is accounted for by genetic factors, rather than by situation or trauma-related factors (Chantarujikapong et al., 2001; Scherrer et al., 2000; True et al., 1993). Genetic factors are not only important in determining an individual’s response to a stressful event, but also influence the likelihood of exposure to that event (Kremen et al., 2012).
A series of studies have identified a range of specific genetic polymorphisms affecting stress reactivity, and thus likely affecting differential vulnerability to stress exposure, of which some examples follow. Regulation of the hypothalamic-pituitary-adrenal (HPA) axis, a key hormonal system involved in adaptation to stress, is affected by genetic factors. Two separate studies have shown that the presence of certain alleles and haplotypes of the gene coding for the corticotropin-releasing hormone (CRH) type 1 receptor moderates the severity of depressive symptoms in adults with a history of childhood abuse (Bradley et al., 2008). Functional polymorphisms of the glucocorticoid receptor (GR) have also been identified: for example, the N363S variant of the GR gene increases cortisol responses to the Trier Social Stress Test, a public speaking and mental arithmetic task (Derijk and de Kloet, 2008). Genetic variation in FKBP5, a gene coding for a protein responsible for regulating GR sensitivity, has been found to affect the efficiency of recovery of HPA axis activation after exposure to the Trier Social Stress Test in healthy individuals (Ising et al., 2008), and to interact with severity of childhood abuse in predicting PTSD symptom severity in adults (Binder et al., 2008). More efficient recovery of HPA axis activation after exposure to environmental stressors is thought to be a key characteristic of resilient individuals. Of note, by combining genetic with gene expression and hormone studies, it is now possible to identify subtypes of PTSD differing, for example, in the nature of HPA axis abnormality (Mehta et al., 2011).
Genetic variations in the serotonergic system have also been found to affect susceptibility to stress-induced psychopathology. The short (S) allele of the serotonin (5-HT) transporter promoter gene (5HTTLPR), which compared with the long (L) allele results in decreased transporter availability and lower uptake of 5-HT from synaptic clefts, has been implicated in a number of studies in a maladaptive response to stress. Although individuals who are carriers for the S allele of 5HTTLPR show elevated risk for depression upon stress exposure in some but not all studies (Caspi et al., 2003; Kendler et al., 2005; Uher and McGuffin, 2010), this finding has been confirmed in a recent large metaanalysis (Karg et al., 2011). S allele carriers also show heightened activation of the amygdala (Hariri et al., 2005) and decreased coupling between the amygdala and the regulatory perigenual cingulate region while viewing fearful or aversive stimuli (Pezawas et al., 2005). Differential neural activation to threat stimuli might represent a biological marker of differential susceptibility to stress. In a recent fMRI study in healthy women, neural systems involved in fear responses and attention to threat showed higher activation during anticipation of a mild shock to the wrist in participants with the SS allele compared with L carriers (Drabant et al., 2012). Further, differential neural activation in medial prefrontal cortex (mPFC) and insula was associated with differential self-reports of anxiety and success at anxiety regulation. Interestingly, recent studies have found that youth homozygous for the short allele of 5HTTLPR were not only more vulnerable to negative parenting, but also more responsive to supportive parenting, showing higher levels of positive affect if parenting was supportive (Hankin et al., 2011). Thus some genetic variations might increase an individual’s responsivity to environmental influence, whether good or bad, potentially boosting or decreasing resilience depending on the quality of the environment during development (Homberg and Lesch, 2011). A recent prospective study in female college students found increased acute stress disorder symptoms after a college shooting in students with particular combinations of polymorphisms in the 5-HT transporter gene (Mercer et al., 2012). Variations in other genes affecting serotonin signaling are also being identified. For example, a common functional variation in the gene coding for the serotonin 1A autoreceptor was associated with differential amygdala reactivity to angry and fearful facial expressions in healthy adults, and indirectly with differential trait anxiety (Fakra et al., 2009).
Genetic polymorphisms affecting resilience to stress have been identified in the noradrenergic system as well as in the serotonergic system. Functional polymorphisms in the gene that produces catechol-O-methyltransferase (COMT), an enzyme that degrades dopamine (DA) and norepinephrine (NE), have been identified and found to affect response to stress. Individuals with the low functioning met allele for the enzyme, and hence higher circulating levels of DA and NE, tend to exhibit more anxious behaviors and lower resilience against negative moods (Heinz and Smolka, 2006). They also tend to avoid novelty-seeking behaviors, and while viewing aversive images, show heightened activity in the visuospatial attention system as well as increased reactivity and connectivity in corticolimbic circuits (Drabant et al., 2006), suggesting that they have a heightened awareness for potential threats (Smolka et al., 2005). Another group studying the COMT gene found that carriers of the low functioning met allele exhibited greater plasma endocrine and subjective stress responses in response to psychologically stressful tasks than did carriers of the high-functioning allele (Jabbi et al., 2007).
Polymorphisms in DA receptors and in the DA transporter gene have also been associated with vulnerability to depression and PTSD (Dunlop and Nemeroff, 2007). In a study of combat veterans, for instance, the A1+ allele of the D2 receptor was associated with more severe PTSD and higher levels of anxiety and depression (Lawford et al., 2006). Polymorphisms in other relevant systems have been identified. A polymorphism (Val66Met) in the gene for brain derived neurotrophic factor (BDNF), a widely expressed protein that stimulates neurogenesis and promotes learning and memory, has been linked with an increased likelihood of suffering stress-induced depression (Kim et al., 2007), and with increased amygdala reactivity in response to emotional stimuli in healthy females (Montag et al., 2008). A single nucleotide polymorphism (rs1617) located in the promoter region of the gene coding for neuropeptide Y (NPY), a peptide released during stress that appears to mitigate the negative effects of the stress response, alters NPY expression and was found to differentially affect stress responses and neural activation to emotional probes (Zhou et al., 2008).
A recent study identified associations between single nucleotide polymorphisms in the genes coding for the pituitary adenylate cyclase-activating polypeptide (PACAP) (involved in regulating the cellular stress response) and its PACAP-PAC1 receptor, and PTSD in females (Ressler et al., 2011). Additional genetic polymorphisms, gene-by-environment, and gene-by-gene-by-environment interactions involved in resilience to stress are currently being identified, the latter exemplified by a study of interactions between a history of childhood trauma and genetic polymorphisms in the CRH type 1 receptor and serotonin transporter genes on depressive symptoms in adults (Ressler et al., 2010). Thus our understanding of the genetics of resilience is likely to increase rapidly in coming years. As more of the genetic factors underlying resilience are uncovered, it may become possible for scientists to design gene or drug therapies specific for individuals with low-resilience genetic profiles.
THE DEVELOPMENT OF RESILIENCE: ROLE OF EARLY LIFE ENVIRONMENT
In addition to genetic makeup, another key contributor to resilience is developmental environment. The study of resilience from a developmental perspective dates back to the early 1970s (Masten and Obradovic, 2006). An important discovery to emerge from this research is that resilience is common, even in children who suffered severe adversity in early life. Studies of adolescents whose development was stunted in childhood due to traumatic experiences such as being orphaned have found that the majority of children rapidly demonstrate “developmental catch-up” when placed in safe and nurturing environments (Masten, 2001). It seems that, under the right circumstances, neural circuits involved in resilience are modifiable for many years after birth, possibly even into adulthood.
Nevertheless, severely traumatic experiences in early life can negatively affect the development of stress response systems, in some cases doing long-lasting damage. Studies of rodents and non-human primates indicate that animals abused by their mothers during the first few weeks after birth have delayed independence and a decreased ability to manage stress later in life, demonstrated by high levels of behavioral anxiety, a hyperactive HPA axis, and increased basal levels of the anxiogenic CRH in the cerebrospinal fluid (Claes, 2004; McCormack et al., 2006; Strome et al., 2002). Furthermore, non-human primates who suffered damage to their stress response systems as a result of abuse in infancy seem more likely to mistreat their own infants, perpetuating the effects of abuse across generations (Maestripieri et al., 2007).
Studies of human survivors of early life stress and abuse have also found long-lasting alterations in central nervous system circuits and structures involved in psychological well-being. Severe prenatal stress and early childhood abuse have been linked to increased HPA axis activity later in life, putting survivors at risk for the adverse effects of chronic hypercortisolemia (Heim et al., 2000; Janssen et al., 2007 Seckl and Meaney, 2006; Vythilingam et al., 2002). Developmental environment also influences the adult functioning of the locus coeruleus–norepinephrine (LC–NE) system, with severe stress in early life leading to its hyperfunctioning. For instance, one study found that police recruits with a history of childhood trauma had significantly higher levels of a salivary metabolite of NE in response to viewing aversive videos than did healthy controls (Otte et al., 2005). Other research has found that physical or sexual abuse in childhood can lead to smaller-than-average hippocampal volumes; decreased hippocampal volume is commonly seen in individuals with depression or chronic stress disorders (Janssen et al., 2007). Thus, it appears that early abuse can cause long-standing changes in brain structures and circuits associated with resilience.
Although early exposure to unpredictable and uncontrollable stress or trauma commonly leads to later psychopathology, exposure to moderately stressful events that can be mastered to an extent (e.g. family relocation, illness of a parent, or loss of a friendship) seems to enable children to effectively regulate their stress response systems. Children who have experience successfully coping with difficult situations reap benefits when faced with stressors later in life, experiencing less physiological and psychological distress (Boyce and Chesterman, 1990; Khoshaba and Maddi, 1999). It seems that the experience of mastering stress provides a form of immunity against later challenges. This phenomenon is known as stress inoculation, based on the analogy to vaccine-induced inoculation against disease (Rutter, 1993). Just as exposure to a low dose of a pathogen enables the body to mount a long-lasting immune response, exposure to moderate amounts of stress enables organisms to conquer and fight off future stressors.
Research in rodents and primates supports the stress inoculation hypothesis and provides insight into its neurobiological mechanisms. Young monkeys presented with a manageable stressor in the form of periodic short maternal separations from postnatal weeks 17 to 27 experience acute distress during the separation periods, manifested by behavioral agitation and temporarily increased cortisol levels. Later in life, however, the same monkeys demonstrate lower anxiety (e.g. increased exploratory behavior in novel environments) and lower basal stress hormone levels at nine months of age than monkeys who never underwent periods of maternal separation (Parker et al., 2004). Furthermore, these stress-inoculated monkeys demonstrate higher cognitive control assessed at 1.5 years, higher curiosity in a stress-free situation at 2.5 years, and larger ventromedial prefrontal cortex (PFC) volume assessed with neuroimaging at age 3.3 years compared with their non–stress-inoculated peers (Lyons et al., 2009; Parker et al., 2005). Poor control of prefrontal cognition has been associated with depression in humans (Harvey et al., 2005; Murphy et al., 2001). Of note, the size of the ventromedial PFC in humans predicts lower impulsivity, lower harm avoidance, and greater retention of learned fear extinction (Lyons et al., 2009).
Just as in humans, the degree of control that an animal has over a stressor plays a key role in determining whether the event will lead to subsequent vulnerability or resilience to stress. Animals administered unavoidable and unpredictable shocks tend to develop exaggerated fear responses, heightened anxiety states, and deficits in active coping when faced with subsequent stressors: this condition is known as learned helplessness (Overmier and Seligman, 1967). The phenomenon of learned helplessness is a well-known animal model for depression and is thought to lead to dysregulation of the serotonergic neurons in the dorsal raphé nuclei (Greenwood et al., 2003) as well as to reduce hippocampal cell proliferation. Because 5-HT has far-reaching effects in the limbic system, the dysregulations created by learned helplessness likely have serious negative effects on cognition and mood.
In contrast, animals that are administered shocks and given the ability to avoid them by modifying their behavior do not develop learned helplessness. Furthermore, animals that have at one time experienced behavioral control over predictable tail shocks are less likely than naïve animals to develop learned helplessness if they are subsequently exposed to unpredictable and inescapable shocks (Seligman and Maier, 1967). Similarly, human beings that have been “stress inoculated” to one type of stressor by successfully managing the stressor appear to acquire resilience to a broad range of other subsequent stressors (Masten and Obradovic, 2006). As noted by Rutter (2012), limited evidence from human studies suggests that steeling might occur via the acquisition of self-efficacy and mastery.
It is important to note that even among animals administered unpredictable and unavoidable shocks, not all develop learned helplessness. Similarly, in humans exposed to severe, unpredictable, and uncontrollable traumas, not all go on to develop PTSD or other anxiety or panic disorders. Clearly, genetic factors interact with environmental exposures to affect resilience. In some cases, a resilient genetic profile may be enough to overcome even the most adverse developmental circumstances.
EPIGENETICS
Prenatal and early postnatal environments regulate developmental processes at the molecular level via epigenetic mechanisms, affecting gene expression without involving changes in DNA sequence (Dudley et al., 2011). Initial studies in rodents showed that high maternal care early in development can lead to a “permanent increase” in GR expression in the hippocampus, resulting from lower GR promoter DNA methylation, and thereby lowering sensitivity to circulating glucocorticoids (Liu et al., 1997; Weaver et al., 2004). Further epigenetic changes can occur in response to experiences including drug use, social interactions, and exposure to stress during critical development periods (Dudley et al., 2011).
Stress reactivity in adult animals is partially a result of epigenetic changes stemming from differential early life experience, such as methylation levels of promoter regions in genes of central importance to stress responses (e.g., GR, brain-derived neurotrophic factor), and located in key regions of the brain (e.g., hippocampus, prefrontal cortex) (Roth et al., 2009; Weaver et al., 2004). Similarly, prenatal stress can also result in epigenetic changes, which in turn impact stress reactivity; on the other hand, early evidence suggests that some of these changes are potentially reversible by optimal postnatal experience (Dudley et al., 2011). Studies in humans have begun to identify the role of epigenetic changes, as exemplified by a report of differential methylation of the GR gene in newborn babies of mothers with prenatal depression (Oberlander et al., 2008). There has been a recent surge of animal and human studies investigating the role of epigenetic mechanisms in differential stress reactivity (Mifsud et al., 2011; Radley et al., 2011; Yehuda et al., 2011).
Having reviewed the role of early life environment and recent findings on epigenetic changes, in the following sections we outline what current research holds to be some of the most important factors, psychological and neurobiological, characterizing resilience to extreme stress in the adult.
PSYCHOBIOLOGICAL FEATURES
OF RESILIENCE
TRAITS AND BEHAVIORS
POSITIVE EMOTIONS
Positive emotions play an integral role in the capacity to tolerate stress. The broaden-and-build theory holds that positive emotions enable broader psychological and behavioral responses, ultimately building enduring resources (Fredrickson, 2001). Positive affect is associated with health-protecting factors including greater social connectedness and adaptive coping mechanisms (Steptoe et al., 2005). Positive emotions also appear to decrease autonomic arousal (Isen et al., 1987; Folkman and Moskowitz, 2000) and facilitate cardiovascular recovery after negative arousal in response to a stressor (Tugade and Fredrickson, 2004), leading to better physical health. Positive affect has been associated with reduced use of medical services; fewer stress-related illnesses; and decreased neuroendocrine, cardiovascular, and inflammatory reactivity (Carver et al., 1993; Danner et al., 2001; Steptoe et al., 2005; Scheier et al., 1989; Zeidner and Hammer, 1992).
Similarly, optimism has been repeatedly correlated with increased psychological well-being and health (Affleck and Tennen, 1996; Goldman et al., 1996) and with greater life satisfaction (Klohnen, 1996). An optimistic disposition is thought to be in large part inherited; however, motivated individuals can increase their optimism with practice. Optimists maintain positive emotions even in the face of adversity because they tend to view problems as temporary and limited in scope. Pessimists, on the other hand, tend to think of their problems as permanent and pervasive and consequently are more prone to depression (Table 88.1). Optimism has even been correlated with longevity: for example, a study of 180 nuns found that nuns whose diaries from youth reflected optimism lived significantly longer than nuns with more negative diaries (Danner et al., 2001). Optimists are thought to have robust brain reward circuits, which is discussed in further detail later in the chapter.
Dispositional gratitude, another positive emotion related to a generally appreciative outlook toward life, was shown in longitudinal studies to foster social support and protect individuals from stress and depression (Wood et al., 2008).
POSITIVE COGNITIVE REAPPRAISAL
Cognitive reappraisal, or the ability to find the silver lining in every cloud, is closely related to optimism and strongly associated with resilience (Gross, 2002; Southwick et al., 2005; Tugade and Fredrickson, 2004). The technique, also known as cognitive flexibility or cognitive reframing, refers to the purposeful, conscious transformation of emotional experience. Cognitive reframing is the reinterpretation of traumatic events to find their positive meaning, value, or the new opportunity that they provide. It stands in contrast to suppression, which signifies conscious attempts to forget traumatic events. Individuals who use cognitive reappraisal when dealing with trauma have less anger and physiologic arousal than those who use suppression techniques (Gross, 2002). It is thought that cognitive reappraisal works in part by attenuating biological stress responses (Davidson and McEwen, 2012; Disner et al., 2011).
The ability to find personal meaning in tragedy is extraordinarily helpful in successfully overcoming trauma. Viktor Frankl, psychiatrist and Holocaust survivor, attributes his survival of concentration camps largely to this process of “meaning making”; in fact, meaning making became the basis for the school of psychotherapy that he founded, known as logotherapy. On a long enforced march, weak from hunger and cold, he wrote, “I forced my thoughts to turn to another subject. Suddenly I saw myself standing on the platform of a well-lit, warm and pleasant lecture room. . . . I was giving a lecture on the psychology of the concentration camp!” (Frankl, 1959/2006: p. 73). The conscious construction of this narrative and of the meaning he would derive from his experiences provided Dr. Frankl with the psychological endurance to survive his days in concentration camps.
TABLE 88.1. Selected psychological resilience factors: attitudes and behaviors that can help maintain well-being during stress
|
1. Positive emotions and positive attitude Optimism is strongly related to resilience. Optimism is in part inherited but can be learned through therapy. Putative neurobiological mechanisms: strengthens reward circuits, decreases autonomic activity 2. Cognitive flexibility and reappraisal Finding meaning or value in adversity Traumatic experiences can be reevaluated through a more positive lens. Trauma can lead to growth: learn to reappraise or reframe adversity, finding its benefits; assimilate the event into personal history; accept its occurrence; and recover. Recognize that failure is an essential ingredient for growth. Putative neurobiological mechanisms: alters memory reconsolidation, strengthens cognitive control over emotions 3. Moral compass: embrace a set of core beliefs that few things can shatter Live by a set of guiding principles. For many, moral compass means religious or spiritual faith. Altruism strongly associated with resilience: selfless acts increase our own well being. Putative neurobiological mechanisms: spirituality/religiosity associated with strong serotonergic systems. Morality has neural basis, likely evolved because adaptive 4. Finding a resilient role model/mentor Role models and mentors can help teach resilience: imitation is powerful mode of learning. Putative neurobiological mechanisms: oxytocin mediates initial bonding/attachment. “Mirror”/Von Economo neurons involved in neuronal imprinting of human values 5. Facing fears: learning to move through fear Fear is normal and can be used as a guide for action. Facing and overcoming fears can increase self-esteem and sense of self-efficacy. Practice undertaking and completing challenging or anxiety-inducing tasks. Putative neurobiological mechanisms: promotes fear extinction, stress inoculation 6. Active coping: seeking solutions, managing emotions Resilient individuals use active rather than passive coping skills (dealing with problem and with emotions versus withdrawal, resignation, numbing). Can be learned: work on minimizing appraisal of threat, creating positive statements about oneself, focusing on aspects that can be changed Putative neurobiological mechanisms: prevents fear conditioning and learned helplessness, promotes fear extinction 7. Establishing and nurturing a supportive social network Establish and nurture a supportive social network. Very few can “go it alone”; resilient individuals derive strength from close relationships. Social support is safety net during stress. Putative neurobiological mechanisms: oxytocin mediates bonding/attachment, neural networks subserve automatic emotional responses and voluntary emotion regulation during social interactions 8. Physical exercise and attending to physical well-being Physical exercise has positive effects on physical and psychological hardiness. Effective at increasing mood and self-esteem Putative neurobiological mechanisms: promotes neurogenesis, improves cognition, attenuates HPA activity, aids in regulation of emotion, boosts immune system 9. Other resilience factors Training regularly (emotionally and physically), discipline Recognizing one’s own strengths and fostering them |
The importance of synthesizing traumatic events into one’s personal life narrative was described over a century ago by Pierre Janet, the French neurologist. Janet noted that his patients with posttraumatic pathology had failed to integrate their traumatic memories into a cohesive story. Janet stressed the necessity of cognitive reappraisal in preventing or overcoming posttraumatic stress; traumatic memories “needed to be modified and transformed, that is, placed in their proper context and reconstructed into neutral or meaningful narratives.” (van der Kolk and van der van der Kolk and van der Hart, 1989).
PURPOSE IN LIFE
A closely related concept to that of finding meaning is having a sense of purpose in life. Studies are beginning to document an association between higher self-reported purpose in life and resilience or recovery from psychiatric illness in traumatized populations (Alim et al., 2008). Purpose in life was significantly associated with psychological well-being in a study of veterans living with spinal cord injury (deRoon-Cassini et al., 2009). Another study reported greater perceptions of purpose and control in resilient OEF-OIF (Operation Enduring Freedom-Operation Iraqi Freedom) veterans compared with those with PTSD (Pietrzak and Southwick, 2011).
ACTIVE COPING STYLES
Active coping means deploying productive strategies for solving problems, managing stress, and regulating negative emotions that may arise in the aftermath of adverse events. Active coping behaviors include acknowledging and trying to solve problems, accepting that which cannot be changed, facing fears, using humor and physical exercise to alleviate stress, and seeking out social support and role models. Active coping has repeatedly been associated with hardiness and psychological resilience in various populations (Moos and Schaefer, 1993). In contrast, passive coping, including the blunting of emotions through substance use, denial, disengagement, or resignation, is associated with depression and lower levels of hardiness (Maddi, 1999). Several studies have shown an association between avoidance coping and psychopathology (Pietrzak et al., 2011; Thompson et al., 2011).
ACCEPTANCE
Acceptance is an adaptive coping strategy commonly found among people who are able to tolerate extreme and uncontrollable stress (Manne et al., 2003; Siebert, 1996). Acceptance involves recognizing the uncontrollable aspects of certain stressors, changing expectations about outcome based on reality, and focusing on controllable aspects of the stressor. Acceptance is not to be confused with resignation, which is giving up or coping passively. Acceptance has been linked with better physical and psychological health, lower levels of distress, and greater psychological adjustment after trauma exposure (Thompson et al., 2011; Wade et al., 2001). Recent evidence includes the finding that individuals who had an accepting coping style had fewer PTSD symptoms following the terrorist attacks of September 11, 2001 (Silver et al., 2002).
FACING FEARS
Facing fears is a key component of the active coping paradigm. Fear conditioning, which is discussed in greater detail later in the chapter, plays a major role in the development and maintenance of posttraumatic psychopathology. Individuals with PTSD avoid a wide variety of life’s opportunities (people, places, events, etc.) that may serve as reminders of the trauma; thus, conditioned fear is maintained rather than extinguished. In contrast, resilient individuals are more adept at managing fear, using it as a guide to critically appraise threat and to select appropriate action.
Active coping at the time of trauma or when exposed to traumatic reminders appears to attenuate fear conditioning, likely by redirecting activity in the lateral and central nuclei of the amygdala away from the brainstem and toward the motor circuits in the ventral striatum. This has the effect of reducing brainstem-mediated responses to fear, such as freezing behavior and autonomic and endocrine responses, and enabling productive action (LeDoux and Gorman, 2002). Resilient individuals have learned to face fears and to actively cope with them; by moving through fear, they avoid fear conditioning and are less likely to develop psychopathology and functional impairments in the aftermath of trauma.
HUMOR
Humor, frequently used by resilient individuals, has been identified as an adaptive coping style and mature defense mechanism (Vaillant, 1977). It appears that humor decreases the probability of developing stress-induced depression (Deaner and McConatha, 1993; Thorson and Powell, 1994). The use of humor has been studied in resilient Vietnam veterans (Haas and Hendin, 1984), surgical patients (Carver et al., 1993), cancer patients (Culver et al., 2002), and at-risk children (Werner and Smith, 1992; Wolin and Wolin, 1993) and has been shown to be protective against distress. Humor is thought to diminish the threatening nature and negative emotional impact of stressful situations (Folkman, 1997), fostering a more positive perspective on challenging circumstances. The use of humor also relieves tension and discomfort (Vaillant, 1992) and attracts social support. Humor is thought to activate a network of subcortical regions that are critically involved in the dopaminergic reward system (Mobbs et al., 2003).
PHYSICAL EXERCISE
Physical exercise has consistently been shown to have positive effects on physical hardiness, mood, and self-esteem. Individuals who exercise regularly report lower depression scores than those who do not exercise (Brosse et al., 2002; Camacho et al., 1991). Physical exercise has effects on a number of neurobiological factors that affect resilience. It attenuates the HPA axis response to stress, increases release of endorphins, and increases levels of plasma monoamines and the 5-HT precursor tryptophan. It is also thought to induce the expression of several genes related to neuroplasticity and neurogenesis, such as hippocampal BDNF (Cotman and Berchtold, 2002). Studies in animals have shown that exercise helps contain the stress response via central sympathetic, serotonergic, and dopaminergic reward pathways, resulting in protection from stress-induced immunosuppression, cytokine elevation, and affective dysregulation (Fleshner et al., 2011). A study of high-impact running in humans suggested that the activity has a beneficial impact on cognitive functioning: high-impact running was associated with improved vocabulary learning and retention as well as a lower likelihood of age-related cognitive diminution (Winter et al., 2006).
SOCIAL SUPPORT
The last active coping strategy we discuss is the seeking out of social support. Higher levels of social support have been associated with better mental and physical health outcomes following a wide variety of stressors (Resick, 2001). Conversely, lower levels of social support have been linked to PTSD and other psychiatric disorders (Tsai et al., 2012). In a study of OEF-OIF veterans, resilience was associated with self-reports of greater family support and understanding (Pietrzak and Southwick, 2011). Social support is thought to decrease appraisals of threat (Fontana et al., 1989), counteract feelings of loneliness (Bisschop et al., 2004), increase sense of self-efficacy, reduce functional impairment (Travis et al., 2004), and increase treatment compliance. Role models and mentors form a valuable part of a strong social support network.
Social isolation and lack of social support are associated with higher rates of mood and anxiety disorders, higher levels of stress, and increased morbidity and mortality from medical illness. Individuals who seek and nurture a supportive social network during times of stress tend to fare better during stress or adversity than individuals who are socially isolated.
MORAL COMPASS: A SET OF GUIDING
ETHICAL PRINCIPLES
Moral compass, or an internal framework of belief about right and wrong, is another feature common to resilient individuals (Southwick et al., 2005). This construct may, but does not have to, include adherence to a religious or spiritual system.
Religion and spirituality seem to have a protective effect on physical and psychological well-being. A metaanalysis of 126,000 individuals from 42 independent samples found religious practice or involvement to be associated with lower risk of mortality from all causes (McCullough et al., 2000). In addition, higher levels of religious belief have been correlated with lower incidence of depression and higher rates of remission from depression in a number of populations (Braam et al., 1997, 2002; Kasen et al., 2012; Koenig et al., 1998). Interestingly, one’s particular religious affiliation is not implicated in the overall relationship between religiousness and improved psychological and physical health.
There is some evidence suggesting that spiritual or self-transcendent experiences are associated with increased density of 5-HT1A receptors in the dorsal raphé nuclei, hippocampus, and neocortex (reviewed by Hasler et al., 2004). Chronic stress appears to lead to a down-regulation of 5HT1A receptors; spirituality and religiosity may enhance the functioning of the 5-HT system, fostering resilience and protecting against the development of posttraumatic mental illness.
Religious faith or spirituality is not an essential ingredient in a strong moral compass. In fact, morality appears to have a neural basis and is likely intrinsic to human nature. The idea that morality is inherent to human beings is ancient. Epictetus, a Greek philosopher living in Rome in the first century A.D. wrote: “Every one of us has come into this world with innate conceptions as to good and bad, noble and shameful . . . fitting and inappropriate” (Stockdale, 1991). Moral sense likely developed early in human evolution as our ancestors organized into societies: the successful functioning of society requires cooperation and trust between members. Many studies support the notion that principles of cooperation and reciprocity are ingrained within human nature. Games designed to test participants’ morality repeatedly find that human beings act according to the rule that fairness to others should supersede self-interest. For example, in games in which participants are given money to divide between themselves and strangers, most participants choose to divide the money equally among players. Furthermore, participants who play fairly in such games tend to show heightened activation of their dopaminergic reward systems on imaging studies (Fehr and Fischbacher, 2003). These findings provide support for the hypothesis that people know what is right and derive fulfillment from acting accordingly.
ALTRUISM
Altruism, or putting one’s moral compass into action, is a powerful contributor to resilience. For example, research on the behavior of citizens after bombing attacks during World War II showed that those who demonstrated altruism by caring for others suffered fewer trauma-related mood and anxiety symptoms than would be expected. Furthermore, individuals who had been symptomatic preattack experienced a meaningful decrease in psychological distress (Rachman, 1979). It appears that, similar to acting fairly, performing acts of altruism activates brain reward circuits. One study found that participants who gave money to charity, whether voluntarily or through taxation, showed heightened activation of the ventral striatum, an important part in reward pathways (Harbaugh et al., 2007).
Some individuals are able to find meaning in personal tragedy by embracing a survivor mission to help others. Rape survivors who go public with their experience to raise social awareness through events such as Take Back the Night, and the mothers who founded Mothers Against Drunk Driving after their children were injured or killed in drunk driving accidents, are examples of this phenomenon.
We now discuss some of the most important neurobiological factors associated with resilience. Individuals with the psychological features of resilience we have discussed likely have a neurobiological profile characterized by optimal levels of these factors.
NEUROBIOLOGICAL PROFILE OF RESILIENCE
A number of neurotransmitters, neuropeptides, and hormones have been implicated in acute and long-term adaptation to stress. A comprehensive review of the many functions and effects of these factors is beyond the scope of this chapter; we focus specifically on the factors’ role in mediating the stress response (Table 88.2, Fig. 88.1; see also Color Fig. 88.1 in separate insert).
CORTICOTROPIN-RELEASING HORMONE
The hypothalamus releases corticotropin-releasing hormone (CRH) in conditions of stress, activating the pituitary-adrenal axis and triggering the release of cortisol and dehydroepiandrosterone (DHEA). Corticotropin-releasing hormone also has important direct effects in the central nervous system; CRH containing neurons and CRH receptors are distributed widely throughout the brain and gut (Steckler and Holsboer, 1999). Activation of CRH neurons in the amygdala triggers fear-related behaviors, whereas activation of cortical CRH neurons reduces expectation of rewards. It seems that excessive stress in early life can result in abnormally elevated CRH activity in the adult brain (Strome et al., 2002).
Corticotropin-releasing hormone acts via two G-coupled receptors, CRH-1 and CRH-2. These receptors appear to have opposite effects in the response to stress (Bale et al., 2002; Grammatopoulos and Chrousos, 2002); the CRH-1 receptor primarily activates the behavioral, endocrine, and visceral responses to stress. Elevated hippocampal CRH-CRH-1 interaction may contribute to the structural and cognitive impairments associated with early-life stress (Ivy et al., 2010). CRH-1 receptor gene single nucleotide polymorphism rs110402 was found to moderate neural responses to emotional stimuli, thereby potentially increasing vulnerability for the development of psychiatric disorders (Hsu et al., 2012). The CRH-2 receptor generally serves to dampen these effects (Tache and Bonaz, 2007). However, activation of the CRH-2 receptor does produce some anxiogenic effects, for example, enhancing CRH-1 mediated suppression of feeding behavior (Bakshi et al., 2002). A more recent study shows that chronic activation of CRH-2 receptor by overexpressing its specific ligand (urocortin 3) promotes an anxiety-like state, yet with attenuated behavioral and HPA axis response to stress (Neufeld-Cohen et al., 2012). More research is necessary to determine the precise role of both receptors.
Abnormally high CRH levels in the cerebrospinal fluid have been linked to major depression and PTSD (Baker et al., 1999; Bremner et al., 1997; Nemeroff, 2002). Resilient individuals likely have the capacity to effectively regulate CRH levels and/or the relative activity of both receptor subtypes. Gender differences in corticotropin response to CRH challenge may explain differential gender susceptibility to psychopathology following early-life trauma (Desantis et al., 2011). Although initial studies showed promise for the use of CRH-1 receptor antagonists as potential new pharmacotherapies for anxiety and mood disorders as well as for stress-related gastrointestinal disorders such as irritable bowel syndrome (Tache and Bonaz, 2007; Zoumakis et al., 2006), subsequent clinical trials failed to show a beneficial effect of these compounds in patients with major depression (Binneman et al., 2008) or generalized anxiety disorder (Coric et al., 2010). A randomized controlled trial of a CRH-1 receptor antagonist in patients with PTSD is currently under way.
CORTISOL
Many forms of psychological stress increase the synthesis and release of cortisol, which leads to increased arousal, vigilance, inhibition of growth and reproduction, and containment of the immune response. In resilient individuals, the stress-induced increase in cortisol is effectively constrained via an elaborate negative feedback system involving GR and mineralocorticoid receptors (MR) (de Kloet et al., 2007). Excessive and sustained cortisol secretion can have serious adverse effects, including osteoporosis, immunosuppression, hypertension, metabolic syndrome, depression, and anxiety (Carroll et al., 2007; Whitworth et al., 2005).
Cortisol has important regulatory effects on the hippocampus, amygdala, and PFC. It plays a role in the formation, processing, and retrieval of memories, particularly fearful memories. Cortisol levels appear to be dysregulated in depression and PTSD. Patients with major depressive disorder tend to have higher than normal levels of cortisol and a blunted suppression of cortisol secretion in response to the dexamethasone suppression test (Yehuda, 2001). In contrast, basal circulating cortisol levels are generally lower in individuals with PTSD than in healthy individuals (Bierer et al., 2006; Gill et al., 2008; Griffin et al., 2005; Yang or Brand et al., 2006; Yehuda, 2006). Correspondingly, individuals with PTSD tend to show higher-than-average cortisol suppression during the dexamethasone suppression test, implying that their HPA-axis GRs are hypersensitive. Altered HPA-axis function is associated with impaired fear inhibition in subjects with PTSD (Jovanovic et al., 2010). Women with PTSD having lower basal salivary cortisol levels than men with PTSD may contribute to the gender difference in PTSD prevalence between men and women (Freidenberg et al., 2010).
That patients with PTSD might have lower-than average basal cortisol levels suggests that the disorder may develop in the setting of abnormally low cortisol; this hypothesis is supported by a number of studies showing that steroid administration inhibits traumatic memory formation (Bierer et al., 2006). Patients who are pretreated with stress doses of glucocorticoids before surgery and/or hospitalization in the intensive care unit are less likely to have traumatic memories of their hospital stay after discharge than patients treated with placebo (Brunner et al., 2006; Schelling et al., 2006; Weis et al., 2006). Low activity of 5-alpha reductase, a rate-limiting enzyme for cortisol, is associated with avoidance severity and predicts lack of response to psychological treatment for PTSD (Yehuda et al., 2009).
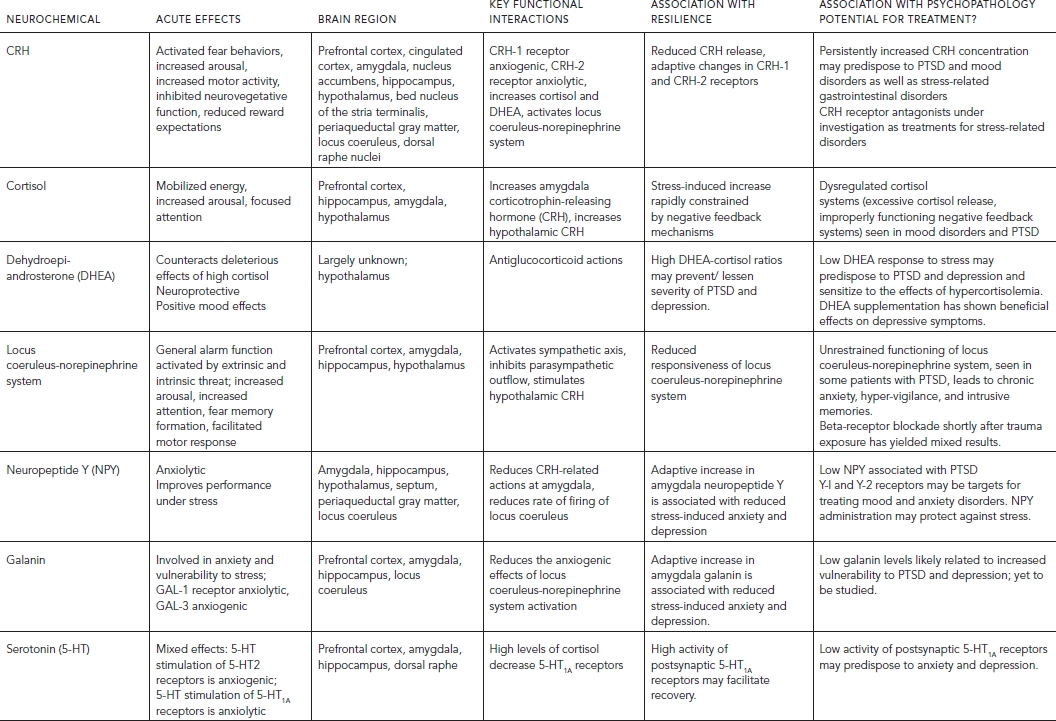

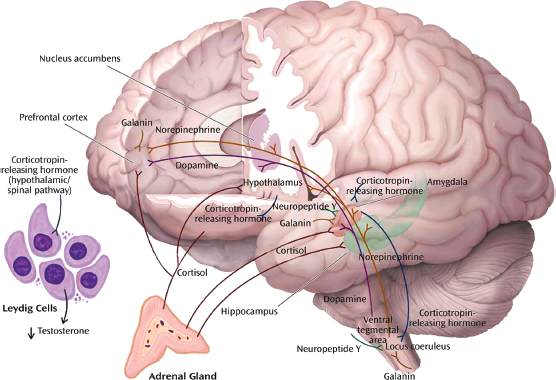
Figure 88.1 Neurochemical response patterns to acute stress. This figure illustrates some of the key brain structures involved in the neurochemical response patterns following acute psychological stress. The functional interactions among the different neurotransmitters, neuropeptides, and hormones are emphasized. The functional status of brain regions such as the amygdala (neuropeptide Y, galanin, corticotropin-releasing hormone [CRH], cortisol, and norepinephrine), hippocampus (cortisol and norepinephrine), locus coeruleus (neuropeptide Y, galanin, and CRH), and prefrontal cortex (dopamine, norepinephrine, galanin, and cortisol) depends upon the balance among multiple inhibitory and excitatory neurochemical inputs. Functional effects may vary depending on the brain region. For example, cortisol increases CRH concentrations in the amygdala and decreases concentrations in the paraventricular nucleus of the hypothalamus. As described in the text, these neurochemical response patterns may relate to resilience and vulnerability to the effects of extreme psychological stress. (Modified and reprinted with permission from Cambridge University Press, 2007.)
Although there are a great deal of data suggesting that patients with PTSD have lower basal cortisol than healthy controls and that PTSD may develop in the setting of low glucocorticoid levels, there is also conflicting evidence. Some studies have found no difference in cortisol levels between patients with PTSD and healthy controls (Metzger et al., 2008; Wheler et al., 2006); some have even found that patients with PTSD have higher-than-average basal cortisol levels, and that elevated cortisol at the time of trauma may predict subsequent PTSD development (Baker et al., 2005; Inslicht et al., 2006). A significant decrease in salivary cortisol levels has been found in PTSD treatment responders (Gerardi et al., 2010). In summary, though it is clear that cortisol is implicated in the encoding of traumatic memories and in the subsequent development of posttraumatic psychopathology, the exact nature of this effect is still under investigation. Undoubtedly though, dysregulation of cortisol secretion has adverse effects on resilience to stress.
DEHYDROEPIANDROSTERONE
Like cortisol, dehydroepiandrosterone (DHEA) is an adrenal steroid released under stress; in contrast to cortisol, DHEA protects against the negative effects of excessive stress. DHEA is secreted episodically and synchronously with cortisol (Rosenfeld et al., 1971) and has antiglucocorticoid activity in the brain (Browne et al., 1992). DHEA and its metabolites interfere with the normal uptake of GRs in the hippocampus (Bastianetto et al., 1999; Kimonides et al., 1998; Morfin and Starka, 2001), preventing corticosteroid-induced hippocampal neurotoxicity. DHEA derivatives also amplify long-term potentiation of hippocampal neurons, likely by modulating transmission at the N-methyld- aspartate (NMDA) receptor (Chen et al., 2006a). DHEA and fluoxetine were found to have synergistic effects in promoting hippocampal progenitor cell proliferation (Pinnock et al., 2009). The steroid’s neuroprotective and potentiating effects in the hippocampus help to facilitate learning and memory.
Studies have shown that DHEA is released under both acute and chronic stress, and that higher levels of DHEA are associated with PTSD and comorbid depression, whereas a higher DHEA-to-cortisol ratio may mitigate symptom severity (Gill et al., 2008; Maninger et al., 2010; Rasmusson et al., 2004). However, one study found no evidence for PTSD-related alterations in cortisol or DHEA secretion in response to stimulation by low doses of ACTH, indicating normal adrenocortical responsiveness in PTSD (Radant et al., 2009).
DHEA appears to enhance cognition and performance under stress. In a study of soldiers undergoing rigorous training as part of military survival school, DHEA-S (dehydroepiandrosterone sulfate) to cortisol ratios were highest in those soldiers who demonstrated the best performance during training. Thus, DHEA-S to cortisol ratios may index the degree to which an individual is buffered against the negative effects of stress (Morgan et al., 2004). In another study, DHEA and DHEA-S levels predicted superior performance in active duty soldiers undergoing an underwater navigation exam (Morgan et al., 2009). Recovery from severe stress in PTSD patients also appears to be facilitated by high levels of DHEA (Rasmusson et al., 2004; Yehuda, 2006; Yang or Brand et al., 2006).
Other studies have also found a negative correlation between plasma DHEA levels and depressive symptoms (Goodyer et al., 1998; Gallagher and Young, 2002; Haren et al., 2007; Young et al., 2002). Studies of DHEA supplementation have shown some beneficial effects on depressive and PTSD symptoms, but further research is needed (Rabkin et al., 2006; Taylor et al., 2012). Animal studies suggest that the antidepressant action of DHEA is mediated via GABA-ergic modulation of the mesolimbic system (Genud et al., 2009). In a recent study, low-dose DHEA supplementation in military men undergoing survival training was found to enhance anabolic balance but did not result in differences in subjective distress (Taylor et al., 2012). More research is needed to determine whether DHEA could enhance resilience if administered prior to trauma exposure.
THE LOCUS COERULEUS–NOREPINEPHRINE SYSTEM
Stress activates the locus coeruleus (LC), which results in increased norepinephrine (NE) release in projection sites of the LC, including the amygdala, PFC, and hippocampus. The LC is activated by a variety of stressors, intrinsic (e.g., hypoglycemia, hypotension) and extrinsic (environmental threats). Such activation serves as a general alarm function and is adaptive in a life-threatening situation. Activation of the LC contributes to the sympathetic nervous system and HPA axis stimulation and inhibits parasympathetic outflow and neurovegetative function, including eating and sleep. A high level of activation of the LC–NE system inhibits function of the PFC, thereby favoring instinctual responses over more complex cognition (Charney, 2004). A recent study in rats showed that during early adolescence, social stress in the form of a resident intruder led to elevated activity of LC neurons, promoting defensive behaviors in this particular age group (Bingham et al., 2011).
The activation of the HPA and LC–NE systems under acute stress facilitates the encoding and relay of aversively charged emotional memories, beginning at the amygdala. Animal studies have shown that injections of NE into the amygdala enhance memory consolidation; on the other hand, blocking NE activity during stress impedes the encoding of fearful memories. In rats, blocking the lateral nucleus of the amygdala to the effects of NE during reactivation of a fearful memory prevents the process of memory reconsolidation and appears to permanently impair that memory (Debiec and LeDoux, 2006).
If unchecked, persistent hyperresponsiveness of the LC-NE system contributes to chronic anxiety, fear, intrusive memories, and an increased risk of cardiovascular disease and hypertension. Findings from an imaging study in humans suggest that disinhibited endogenous NE signaling may contribute to the etiology of PTSD by eliciting exaggerated basolateral amygdala (BLA) responses to fear stimuli (Onur et al., 2009). Noradrenergic activation in the BLA is also involved in the facilitating effects of stress hormones (such as CRH) on the consolidation of emotional memory (Roozendaal et al., 2008). Interestingly, norepinephrine was shown to directly activate multipotent neural precursors in the hippocampus of adult mice via β3 adrenoreceptors, thus facilitating hippocampal neurogenesis; these findings may pave the way for the development of new types of antidepressants (Jhaveri et al., 2010).
NEUROPEPTIDE Y
Neuropeptide Y (NPY) is an abundant peptide that is widely distributed throughout the brain and acts via at least four G-protein coupled receptors (Y1, Y2, Y4, Y5) (Wu et al., 2011) (Baker and Herkenham, 1995; Karl et al., 2006; Makino et al., 2000 Pieribone et al., 1992; Risold and Swanson, 1997). The Y3 receptor has yet to be cloned and the y6 receptor is a truncated receptor in humans (Sah and Geracioti, 2012; Wu et al., 2011). Neuropeptide Y produces anxiolytic effects and appears to enhance cognitive functioning under stress. There are important functional interactions between NPY and CRH (Britton et al., 2000; Heilig et al., 1994; ): NPY counteracts the anxiogenic effects of CRH at various locations within the stress/anxiety circuit, including the amygdala, hippocampus, hypothalamus, and LC (Sajdyk et al., 2006). It may be that the balance between NPY and CRH neurotransmission is critical to maintaining a homeostatic emotional state during stress (Eva et al., 2006; Kask et al., 1997; Sajdyk et al., 2006).
High levels of NPY appear to protect against depression and anxiety, with the Y1 and Y2 receptors playing important roles in the peptide’s effects on mood (Heilig et al., 1993; Heilig, 1995, 2004). The Y1 receptor was found to be anxiolytic, whereas the Y2 receptor, a presynaptic autoreceptor, was found to be anxiogenic (reviewed in Wu et al., 2011). Neuropeptide Y activation of the Y1 receptor inhibits several metabolic and behavioral stress responses, including gastrointestinal distress, anxious behavior, and decreased sleep (Eva et al., 2006). Antidepressant drugs and electroconvulsive therapy increase NPY in rodent brains as they decrease depression; this effect is likely mediated by the Y1 receptor (Karl et al., 2006). A variety of antidepressant drugs increase NPY levels in humans as well (Husum and Mathe, 2002).
The potential role of NPY in fear conditioning is under investigation. In one study, central administration of NPY in rats inhibited fear-potentiated startle and enhanced its extinction; the latter effect was counteracted by the Y1 receptor antagonist BIBO3304 (Gutman et al., 2008). In another study, however, whereas fear conditioning was attenuated by exogenous NPY administration, BIBO3304 had no effect on conditioned fear; thus the role of endogenous NPY is more uncertain (Pickens et al., 2009).
Y2 knockout mice exhibit fewer anxious and depressed behaviors and reduced neuronal activation in response to the emotional stressors, and are more adventurous (Carvajal et al., 2006; Nguyen et al., 2009). Site-specific deletion of the Y2 receptor gene in the central and basolateral amygdala, but not in any other amygdaloid nucleus, results in anxiolytic and antidepressant-like effects, suggesting a possible mechanism for Y2 receptor-mediated regulation of anxiety and depression (Tasan et al., 2010). The Y1- and Y2-receptors may therefore be useful targets for treating mood and anxiety disorders in humans.
Elevated NPY levels are associated with better performance under stress (Sah and Geracioti, 2012). NPY treatment after exposure to stress significantly reduced prevalence rates of extreme behavioral responses and trauma-cue freezing responses in animals (Cohen et al., 2012). The Y1 receptor in the basolateral amygdala is implicated in the mediation effects of NPY on stress and may serve as a therapeutic target for the treatment of PTSD (Cui et al., 2008; Hendriksen et al., 2012). Preliminary work with special operations soldiers under extreme training stress indicates that high NPY levels are associated with better performance (Morgan et al., 2000). These findings suggest that the administration of NPY could be an effective intervention for the treatment of depression, anxiety disorders and PTSD, and for enhancing resilience to stress. However, it will first be necessary to design a drug delivery method that enables the peptide to penetrate into the CSF (Born et al., 2002); intranasal administration is currently being investigated.
GALANIN
Galanin, another abundant neuropeptide, has been shown to be involved in a number of physiological and behavioral functions, including anxiety, stress, and alcohol intake (Holmes et al., 2002; Moller et al., 1999). Understanding of galanin’s mechanisms of action and effects is still at an early stage; however, overall the peptide appears to mitigate the effects of stress.
Three receptors for galanin have been identified, and two of these (GAL-1 and GAL-3) have been found to affect stress and anxiety (Walton et al., 2006). The GAL-1 receptor, expressed in the amygdala, hypothalamus, and bed nucleus of the stria terminalis, acts as an auto-receptor (Gustafson et al., 1996; Sevcik et al., 1993; Xu et al., 2001) and appears to be involved in anxiolysis: knockout mice for the GAL-1 receptor show increased anxiety-like behaviors (Holmes et al., 2003) and fail to gain stress-resistant responses after galanin injection (Mitsukawa et al., 2009). The GAL-3 receptor, on the other hand, produces anxiogenic effects when activated (Swanson et al., 2005). In humans, certain polymorphisms of the gene for the GAL-3 receptor have been linked with anxiety and alcoholism (Belfer et al., 2006). Few studies have looked at the stress-related effects of the GAL-2 receptor. One study found that it is involved in depressive-like but not anxiety-like behavior (Le Maitre et al., 2011), whereas another study showed that it has both antidepressant-like and anxiolytic effects (Lu et al., 2008).
It has been suggested that galanin recruitment during periods of stress may diminish negative emotions caused by hyperactivity of the noradrenergic system (Karlsson and Holmes, 2006). Galanin appears to inhibit NE, serotonin (5-HT), and DA neurons from firing, reducing release of these neurotransmitters in forebrain target regions. Enhanced galanin expression in LC from running is associated with suppression of brain norepinephrine in runners and may contribute to the stress-protective effects of exercise (Sciolino and Holmes, 2012). Galanin also prevents toxicity-induced cell death (Counts and Mufson, 2010) and promotes neurogenesis (Abbosh et al., 2011), which may contribute to its antidepressant effect. Galanin was shown to be down-regulated in the CA1 of the hippocampus and the frontal cortex in a PTSD animal model with extreme behavioral response to stressors; immediate postexposure treatment with galnon, a galanin receptor agonist, significantly reduces prevalence rates of extreme responders and trauma-cue freezing responses (Kozlovsky et al., 2009). To our knowledge, galanin function has not been studied in patients exposed to traumatic stress or patients with PTSD or major depression.
SEROTONIN
Serotonin (5-HT) is well known as one of the neurotransmitters most relevant to mood. As a neuromodulator, 5-HT also regulates other neurotransmitter systems involved in mood and anxiety, and dysregulation of the 5-HT system may cause a cascade of changes in brain chemistry. The role of 5-HT as a neuromodulator is demonstrated in tryptophan depletion studies, whereby serotonin levels are lowered by ingestion of a tryptophan-free amino acid mixture. For example, tryptophan depletion led to failure to inhibit appropriate responses to punishing outcomes in healthy females (Robinson et al., 2012), and uncovered a potential vulnerability in never-depressed young adults at high familial risk for depression, who showed increased bias toward negative words while performing an affective go/no-go task in the depletion condition, compared to low-risk controls (Feder et al., 2011).
Acute stress results in increased 5-HT turnover in the PFC, nucleus accumbens, amygdala, and lateral hypothalamus (Kent et al., 2002). Serotonin release may have anxiogenic and anxiolytic effects, depending on the region of the forebrain involved and the receptor subtype activated. 5-HT1A receptors are anxiolytic and may be responsible for adaptive responses to aversive events, whereas anxiogenic effects appear to be mediated by 5-HT2A receptors (Charney and Drevets, 2002). Absence of 5HT1A receptor signaling during early stages of brain maturation predisposes an organism to affective dysfunction later in life (Vinkers et al., 2010). 5HT1A knock-out mice show an increase in anxiety-like behaviors (Heisler et al., 1998; Parks et al., 1998), including behavioral inhibition in ambiguous environments (settings that contain neutral and fear-conditioned cues). This type of behavior represents an inappropriate generalization of fearful behavior, a phenomenon that occurs in some human anxiety disorders, including panic disorder, specific phobias, and PTSD (Klemenhagen et al., 2006). Therefore, lower-than-normal activation of 5HT1A receptors may be involved in the pathophysiology of human anxiety disorders. On the other hand, antagonism of 5HT2A receptor has been shown to prevent the emergence of anxiety behavior and dysregulated stress response following early life stress (Benekareddy et al., 2011).
A scenario has been proposed in which early life stress increases CRH and cortisol levels, which, in turn, downregulate 5-HT1A receptors, resulting in a lower threshold for tolerating stressful life events. Consistent with findings in rodents and non-human primates, a negative association is found between plasma cortisol levels and 5HT1A receptor distribution in humans (Lanzenberger et al., 2010). Alternatively, 5-HT1A receptor density may be partly genetic. Data so far have been mixed. A positron emission tomography (PET) study scanning receptor status in individuals with PTSD found no differences in 5HT1A receptor distribution, binding potential, or tracer delivery (Bonne et al., 2005), whereas a more recent PET study in Rhesus monkeys showed that reduced 5HT1A receptor density during development might be a factor increasing vulnerability to stress-related neuropsychiatric disorders (Sciolino and Holmes, 2012).
DOPAMINE
Dopamine (DA) is released in some areas of the brain and inhibited in others during extreme stress. Stress activates DA release in the mPFC (Cabib et al., 2002) and inhibits DA release in the nucleus accumbens, one of the regions associated with human experience of pleasure and reward (Cabib and Puglisi-Allegra, 1996). Lesions of the amygdala in a conditioned stress model block stress-induced DA activation in the mPFC, implying amygdalar control over stress-induced DA release (Cabib et al., 2002; Goldstein et al., 1996). DA D1 receptors were shown to be responsible for stress-induced deficit of emotional learning and memory (Wang et al., 2012). The degree to which an individual’s DA system is activated by stress is in part genetically determined, and individuals whose genetic profile results in excessive mesocortical DA release after stressful events may have a tendency toward vulnerability to stress (Cabib et al., 2002). Variation in the dopamine transporter gene may contribute to one heritable path toward the development of PTSD (Drury et al., 2009). Polymorphisms in the dopamine D2 receptor gene might also contribute to susceptibility to PTSD (Voisey et al., 2009). As mentioned in the earlier discussion on NE, studies of individuals with low-functioning variants of the COMT gene, leading to less DA degradation, display higher anxiety and lower resistance to stress (Heinz and Smolka, 2006).
Although hyperactivity of the dopaminergic system may increase susceptibility to suffering negative effects of stress, it appears that underactivity of dopaminergic neurons in the PFC may be implicated in sustaining posttraumatic stress disorders. Lesions of dopaminergic neurons in the mPFC delay extinction of the conditioned fear response (Morrow et al., 1999), which can sustain PTSD symptoms. Dopamine transporter density is increased in PTSD patients, which may reflect higher dopamine turnover in PTSD (Hoexter et al., 2012).
Reduced levels of circulating DA have also been implicated in depression in a number of studies (Dunlop and Nemeroff, 2007). This link is supported by the fact that individuals with Parkinson’s disease (and decreased DA production) have high rates of depression. Postmortem studies of patients who were depressed have found reduced DA metabolites in the cerebrospinal fluid and in brain areas regulating mood and motivation. Polymorphisms in dopamine transporter and receptor genes in depressed patients appear to be associated with treatment response (Huuhka et al., 2008; Lavretsky et al., 2008).
BRAIN-DERIVED NEUROTROPHIC FACTOR
Brain-derived neurotrophic factor (BDNF) is an important neurotrophic factor that is active in brain regions including the hippocampus, cortex, and basal forebrain (Yamada and Nabeshima, 2003). BDNF helps support neuronal growth, differentiation, and survival (Huang and Reichardt, 2001). BDNF interacts with TrkB and p75 as its two main receptors (Castren and Rantamaki, 2010). The BDNF-TrkB pathway has been implicated in both PTSD in humans and in animal models of fear conditioning, extinction, and inhibitory learning (Mahan and Ressler, 2012). Inhibition of BDNF signaling in the amygdala can lead to impairment of the acquisition and consolidation of fear conditioning (Rattiner et al., 2004), and the consolidation of extinction (Chhatwal et al., 2006).
BDNF exerts different functions in different brain regions. Animal studies showed that, in the hippocampus, stress decreases BDNF expression, which is reversible by antidepressant treatment (Duman and Monteggia, 2006). Hippocampal BDNF expression has been shown to play a critical role in resilience to chronic stress (Taliaz et al., 2011). In the nucleus accumbens, however, stress increases BDNF expression, and this increase is associated with depression-like effects (Berton et al., 2006; Eisch et al., 2003).
There is also increasing evidence for an association between a single nucleotide polymorphism in the BDNF gene (Val66Met) and various psychiatric disorders including depression and PTSD (Mahan and Ressler, 2012). This polymorphism was found to alter BDNF stability and secretion, and might result in activation of the limbic system during memory formation and emotionally-relevant learning (Dennis et al., 2011; Donegan et al., 2003; Gonul et al., 2011; Mahan and Ressler, 2012). Studies in Val66Met knock-in mice revealed that mice with the Met/Met genotype exhibited increased anxiety-related behaviors (Chen et al., 2006b).
GLUTAMATE
Glutamate is the most widely distributed excitatory neurotransmitter in the brain. Glutamate functions via activation of its receptors, including ionotropic NMDA, Kainate, and AMPA receptors, as well as metabotropic receptors (Chung, 2012). Glutamate, together with its downstream product GABA, the chief inhibitory neurotransmitter in the nervous system, plays an important role in neuroplasticity, and in modulating cognitive and affective responses to stress (Harvey and Shahid, 2012).
Acute exposure to stress rapidly increases glutamate release in limbic and cortical brain regions, including the hippocampus, amygdala, and prefrontal cortex, areas associated with memory, learning, and affect. The effects of chronic stress on glutamate release are less well understood (Popoli et al., 2011). Numerous studies have found an important role of glutamate in the mediation of stress responsivity, formation of traumatic memories and the pathophysiology of PTSD (Cortese and Phan, 2005; Steckler and Risbrough, 2012). One study in mice found that the induction of DeltaFosB in the nucleus accumbens mediated resilience in response to chronic social defeat stress (Vialou et al., 2010). This effect is produced in part through induction of the GluR2 AMPA subunit of the glutamate receptor, decreasing nucleus accumbens neuron responsiveness to glutamate. In another study, deletion of AMPA receptor subunit GluR-A in mice was associated with increased depression-like symptoms such as learned helplessness, and decreased serotonin and norepinephrine levels (Chourbaji et al., 2008).
Glutamate system dysregulation has been implicated in the pathophysiology of mood disorders, and there is strong interest in drug development targeting the glutamatergic system (Murrough and Charney, 2010; Sanacora et al., 2008; Mathew et al., 2012; Mathews et al., 2012). The NMDA receptor antagonist ketamine has been shown to have antidepressant effects in patients with treatment-resistant depression (aan het Rot et al., 2010, Zarate et al., 2006). It is thought that NMDA receptor antagonist antidepressant effect may be related to a rapid increase in glutamate release, resulting in activation of AMPA receptors and ultimately resulting in changes in synaptic signaling and protein synthesis, and formation of new dendritic spine synapses in the PFC (Autry et al., 2011; Koike et al., 2011; Li et al., 2010). Moreover, antagonism of metabotropic glutamate receptors has also been shown to produce antidepressant-like effects (Belozertseva et al., 2007; Li et al., 2006).
ENDOCANNABINOIDS
Endocannabinoids including anandamide (AEA) and 2-arachidonoyl glycerol (2-AG) are substances naturally produced from within the body that activate cannabinoid receptors such as G protein-coupled CB1 and CB2 receptors. The endocannabinoid system is widely distributed throughout neural regions that regulate mood and emotion. The CB1 receptor and endocannabinoid ligands are found at high densities in regions including the prefrontal cortex, hypothalamus, amygdala, and hippocampus (Herkenham et al., 1991; Hill et al., 2007; Tsou et al., 1998; Moldrich and Wenger, 2000). Activation of CB1 receptors in these limbic areas can result in both excitatory and inhibitory neurotransmission, as well as the release of monoamines and neuropeptides (Azad et al., 2003; Di et al., 2003; Domenici et al., 2006). The CB2 receptor is located predominantly in peripheral immune cells and organs (Hillard, 2000; Cota et al., 2003). A growing body of evidence demonstrates that deficits in endocannabinoid signaling may result in depressive and anxiogenic behavioral responses, which could be ameliorated by the pharmacological augmentation of endocannabinoid signaling (Hill and Gorzalka, 2009a).
Preclinical studies showed that CB1 receptor knockout mice exhibited an increased susceptibility to the anhedonic effects of chronic stress (Martin et al., 2002), as well as increased passive stress coping behaviors in forced swim test and tail suspension test (Aso et al., 2008; Steiner et al., 2008), suggesting that the loss of CB1 receptor signaling promotes a depressive phenotype. CB1 receptor knockout mice also display increased anxiety-like behaviors and impaired extinction of aversive memories (Martin et al., 2002; reviewed in Hill and Gorzalka, 2009a).
Clinical studies in patients with medical conditions showed that the use of CB1 receptor antagonists rimonabant and taranabant can lead to adverse effects such as developing anxiety and depressive symptoms, suggesting that the disruption of endocannabinoid signaling may promote the emergence of mood and anxiety disorders (Christensen et al., 2007; Hill and Gorzalka, 2009a,b; Nissen et al., 2008). Reduction in circulating endocannabinoid ligands has also been seen in women with major depression (Hill et al., 2008). Little research has investigated the effects of endocannabinoids on symptoms of PTSD, but one study showed that the addition of the synthetic cannabinoid nabilone resulted in reduction of treatment-resistant nightmares in PTSD patients (Fraser, 2009). More research is warranted to elucidate the intricate endocannabinoid signaling in mood and emotion and to explore the therapeutic potential of pharmacological interventions acting on the endocannabinoid system in mood and anxiety disorders.
NEURAL CIRCUITRY OF REWARD: HOW REWARD PATHWAYS IMPACT RESILIENCE TO STRESS
A stable and well-functioning system of reward pathways and response to pleasant stimuli is a prerequisite for dealing successfully with stress and traumatic life experiences. The ability to respond appropriately to positive events and situations is vital to the preservation of reward expectation, optimism, and positive self-concept following stress or trauma. Resilient individuals likely have a robust reward system, which is strongly responsive to reward and/or resistant to change (Table 88.3, Fig. 88.2; see also Color Fig. 88.2 in separate insert).
The dopaminergic system is involved in mediating elements of the reward system, including motivation, incentive, and hedonic tone (Barrot et al., 2002; Wise, 2002). Dopaminergic neurons increase firing when rewards are unexpected or better than expected (Schultz, 2002) and decrease their rate of firing when rewards are absent or less than predicted. Imaging research suggests that the anticipation of reward involves mesolimbic DA pathways through the ventral striatum (Knutson and Cooper, 2005) and other subcortical limbic structures including the dorsal striatum, amygdala, and midbrain ventral tegmental area.
The roles of the orbitofrontal cortex (OFC) and mPFC in reward are somewhat more complex. The OFC, taking input from the nucleus accumbens, assists in discriminating the motivational value of rewarding stimuli (Martin-Soelch et al., 2001; Schultz, 2002). The mPFC appears to be involved in assessing the likelihood of receiving a given reward (as opposed to its magnitude) (Knutson and Cooper, 2005) and providing feedback along glutamatergic projections to the nucleus accumbens and the ventral tegmental area. The anterior insula is involved in producing aversion to risks in the pursuit of reward (Kuhnen and Knutson, 2005). In making decisions about behavior, it appears that a stimulus’s potential rewards are assessed in subcortical areas before being analyzed in the mPFC (Knutson and Cooper, 2005).

Figure 88.2 Neural circuits associated with reward, fear conditioning, and social behavior. The figure depicts a simplified summary of some of the brain structures and relevant neurochemistry mediating the neural mechanisms of reward (purple paths), fear conditioning and extinction (yellow paths), and social behaviors (blue paths). Only a subset of the many known interconnections among these various regions is shown, and relevant interneurons are not illustrated, yet it can be seen there is considerable overlap in the brain structures associated with these neural mechanisms. This suggests that there may be clinically relevant functional interactions among the circuits. For example, a properly functioning reward circuit may be necessary for the reinforcement of positive social behaviors. An overly responsive fear circuit or impaired extinction process may negatively influence functioning of the reward system. The assessment of these neural mechanisms must be considered in the context of their neurochemical regulation. Alterations in one neurotransmitter, neuropeptide, or hormone system will affect more than one circuit. Several receptors that may be targeted by new anti-anxiety and antidepressant drugs are illustrated. The functional status of these circuits has important influences on stress-related psychopathology and the discovery of novel therapeutics (see text). (Modified and reprinted with permission from Cambridge University Press, 2007)
Strength, persistence, and emotional value of reward memory are modulated by circuits formed by the amygdala, subiculum, bed nucleus of the stria terminalis, nucleus accumbens, and mPFC. Specifically, the cyclic adenosine monophosphate (cAMP) pathway and expression of cAMP response-element binding protein in the amygdala strengthen positive as well as negative associations (Marshall et al., 2001; Josselyn et al., 2001). Rodents have been shown to develop persistent anhedonia when increased cAMP response element binding is present in the nucleus accumbens (Barrot et al., 2002).
Stresses, chronic and acute, are correlated with decreased sensitivity to rewarding stimuli and increased sensitivity to aversive stimuli (Bogdan and Pizzagalli, 2006, Pliakas et al., 2001). During performance of a probabilistic reward task, high school student carriers of the S allele of the 5HTTLPR gene showed a larger stress-related reduction in responsiveness to reward as school finals approached, compared with L allele homozygotes (Nikolova et al., 2012). This effect was observed only in males. In a different study, reward learning under a stressful condition was differentially influenced in healthy females depending on their CRH receptor type 1 genotype, and was associated with differential activation to reward in the anterior cingulated cortex (ACC) and OFC (Bogdan et al., 2011). Although exposure to acute stress has the potential to alter reward circuits, decreasing positive response to reward, findings from these studies suggest that some people are more sensitive to the effects of stress, and other, potentially more resilient individuals show reduced stress sensitivity, maintaining highly functional reward systems under adverse conditions. In a recent fMRI study, adolescents and young adults with a parental history of depression showed lower OFC activation to rewarding stimuli and higher lateral OFC and insula activations to aversive stimuli, compared with controls. They also showed blunted ACC activation to both rewarding and aversive stimuli (Mc Cabe et al., 2012). These neural abnormalities suggest a potential mechanism for decreased resilience in individuals at high familial risk for depression.
We thus have a beginning understanding of the neurobiology underlying robust reward systems. There is also some evidence from animal models that provides insight into potential mechanisms. In rodent models, chronic exposure to stressful stimuli will lead to an enduring aversion to social contact. Researchers have found that knocking out BDNF in the mesolimbic DA system of the rodents seems to produce a reward system that has lost its plasticity, remaining resistant to change when the rats are subsequently presented with stressors. After undergoing this mesolimbic DA pathway-specific knockdown of BDNF, mice become inoculated to the effects of repeated social aggression (Berton et al., 2006). Studies in the social defeat paradigm in mice, whereby mice are exposed to a dominant intruder placed in their cage, have demonstrated that resilient mice, who show appropriate anxiety responses but no defeat, upregulate K+ channels in the VTA, thereby preventing an increase in neuronal excitability and associated BDNF release onto the nucleus accumbens observed by contrast in vulnerable mice (Krishnan et al., 2007).
These findings highlight the major role of the reward system’s plasticity in determining resilience and indicate possible mechanisms through which a resistant reward system plays a part in allowing resilient individuals to endure hardships without developing posttraumatic psychopathology.
THE NEURAL CIRCUITRY OF ANXIETY
AND FEAR: THE ROLE OF FEAR LEARNING IN RESILIENCE TO STRESS
FEAR CONDITIONING
Individuals with PTSD suffer distress when they encounter traumatic reminders. Over time, patients suffering from PTSD tend to “overgeneralize” fear cues: they begin to associate more and more features in their environment with their past traumas. Often, fear cues are objectively non-threatening and unrelated to trauma. The process through which previously neutral stimuli become fear inducing is known as fear conditioning. In the terminology of fear conditioning, previously neutral stimuli become “conditioned” stimuli by virtue of their association with aversive (unconditioned) stimuli (Blair et al., 2001).
Resilience to the effects of severe stress may be characterized in part by the capacity to avoid overgeneralizing fear-inducing triggers. Resilient individuals are also likely adept at extinguishing of fearful memories; in other words, those who are resistant to stress have an easier time letting go of frightening memories and associations.
Cue-specific conditioned stimuli are transmitted to the thalamus by external and visceral pathways. Afferents reach the lateral amygdala by means of two parallel circuits: a rapid subcortical path directly from the dorsal (sensory) thalamus and a slower regulatory cortical pathway encompassing the primary somatosensory cortices, the insula, and the anterior cingulate/PFC. Contextual conditioned stimuli are projected to the lateral amygdala from the hippocampus and the bed nucleus of the stria terminalis. The lateral nucleus of the amygdala is likely a site of fear memory storage as well as fear acquisition (Schafe et al., 2005). The existence of the long loop pathway (passing through cortical regions) indicates that sensory information relayed to the amygdala undergoes substantial higher-level processing, thereby enabling assignment of significance based on prior exposure to stimuli. This potential for higher-level processing of conditioned stimuli is important because it implies that there is potential for modifying the processing of fear-conditioned stimuli with cognitive therapies.
The lateral amygdala, once engaged, projects to the central and medial nuclei of the amygdala, which project indirectly to the acoustic startle pathway in the brain stem (Davis, 2006). The central nucleus of the amygdala appears to be the primary output nucleus involved in initial memory consolidation; infusions of anisomycin, a protein synthesis inhibitor, into the central nucleus during fear training disrupt consolidation of fear memory (Debiec and LeDoux, 2006). The central nucleus projects to the hypothalamus and brain stem, triggering the cascade of autonomic, endocrine, and behavioral responses associated with fear (Schafe et al., 2001).
The molecular processes underlying fear conditioning is an area of active investigation. Consolidation of fear conditioning is thought to occur via long-term potentiation of synapses in the lateral amygdala. Fear-conditioned rats show increases in an actin polymerization protein, profilin, in dendritic spines in the lateral amygdala; this finding could contribute to a greater understanding of the specific changes underlying synaptic plasticity (Schafe et al., 2005). However, our understanding of the cellular mechanisms underlying the process remains far from complete (Sigurdsson et al., 2007). The currently accepted model to explain how long-term potentiation in the lateral amygdala might lead to the consolidation of fear conditioning was proposed by LeDoux and colleagues (LeDoux, 2000). The model proposes that calcium entry through NMDA receptors and voltage-gated calcium channels initiates the molecular processes to consolidate synaptic changes into long-term memory; short-term memory requires only calcium entry through NMDA receptors (Blair et al., 2001).
Because NMDA receptors are critical in fear learning, which is mediated by glutamate acting in the central nucleus of the amygdala (Davis, 2006), we may be able to impair fear learning by blocking NMDA receptors in the amygdala during stress. This has been demonstrated in rodents (Rodrigues et al., 2001; Walker and Davis, 2000; ). Blockade of voltage-gated calcium channels may also prove useful in preventing PTSD in those who are exposed to stress. Of course, because these are mechanisms of initial fear consolidation, it would be necessary to initiate receptor blockade treatment prior to trauma exposure to prevent posttraumatic stress. This might be feasible and useful in populations that are certain to encounter severe stress, for example, soldiers to be deployed in combat, or disaster relief workers.
RECONSOLIDATION AND EXTINCTION
Existing memories, when retrieved or “reactivated” by remembering, become labile and prone to change. Reactivated memories may go down one of two pathways: they may either be strengthened by reconsolidation, or weakened by extinction (Eisenhardt and Menzel, 2007). The processes appear to be mutually exclusive, and may compete for cellular resources (Dudai and Eisenberg, 2004). The processes of memory reactivation, reconsolidation, and extinction involve NMDA and β-adrenergic receptors and require cAMP response element binding protein induction, indicating that nuclear protein synthesis is necessary (Kida et al., 2002). Reconsolidation and extinction are impaired by NMDA and β-adrenergic blockade, and enhanced by the NMDA partial agonist d-cycloserine (Charney and Drevets, 2002; Chhatwal et al., 2006; Debiec and LeDoux, 2006; Przybyslawski et al., 1999; Royer et al., 2000; Sotres-Bayon et al., 2007). Whether the process of reconsolidation or of extinction will be engaged appears to depend on the timing and type of memory retrieval; short memory reactivation sessions tend to lead to reconsolidation, whereas long and intensive extinction training is necessary to achieve extinction (J.L. Lee et al., 2006). Reconsolidation and extinction of a memory are affected by the age of the memory in question (Fulton, 2006). As would be expected, younger memories are more prone to being altered by either process, although intensifying the memory reactivation process helps enhance the lability of older memories (Suzuki et al., 2004).
Similarly, the process of extinction changes over time. Extinction induced immediately after fear acquisition (between 10 minutes and 1 hour later) is thought to occur by “erasing” the learned fear (Myers et al., 2006). However, extinction induced 24–72 hours after initial fear training appears to be a new form of learning altogether (Myers and Davis, 2007). The extinction that is achieved during this later time period may be thought of as the formation of a new, safe association with the original fear cue; this new association is placed, “like a blanket,” over the older fear association (Chhatwal et al., 2006). That the original fear association still exists underneath the blanket of safety is suggested by observations that extinguished fears can return spontaneously.
Memory consolidation, reconsolidation, and extinction involve different neural circuits. The central nucleus of the amygdala and hippocampus are involved in initial memory consolidation, the lateral amygdala in reconsolidation, and the basal lateral amygdala in extinction (Bahar et al., 2004; Debiec and LeDoux, 2006). The mPFC is a critical structure in extinction. Since the 1980s, studies have found that lesions of the mPFC result in an impairment of normal extinction (cited in Sotres-Bayon et al., 2006). Failure to achieve an adequate level of activation of the mPFC might lead to persistent fear responses (Herry and Garcia, 2002). Findings from an fMRI study in healthy volunteers suggest that the ability of fear responses to adapt flexibly as threat shifts from one stimulus to another is particularly mediated by the ventromedial PFC (Schiller et al., 2008). Individuals with the capacity to function well after high-stress experiences may have potent mPFC inhibition of amygdala responsiveness. In contrast, patients with PTSD have been shown to exhibit increased left amygdala activation during fear acquisition and decreased activity of the mPFC/anterior cingulate during extinction.
Reconsolidation and extinction of traumatic memories are implicated in vulnerability and resilience to extreme stress. Individuals who are able to attenuate learned fear through extinction are likely highly stress-resilient. Due to our growing understanding of the mechanisms of memory processing, there will likely be ever more possibilities for helping trauma survivors manage their traumatic memories. In particular, recent studies suggest that during reconsolidation, previously stored information becomes labile after retrieval and thus amenable to change as new information is incorporated (Schiller et al., 2010). Recent studies in healthy volunteers, whereby participants underwent extinction during the reconsolidation window, incorporating non-fearful information to the old fear memory, have successfully prevented the expression of fear responses (Schiller et al., 2010; 2012). This effect has been shown to last for at least one year (Schiller et al., 2010) and has been replicated by a different group (Oyarzun et al., 2012). A recent fMRI study in humans demonstrated that suppression of a fear memory by conducting extinction training during the reconsolidation window works by erasing the fear memory representation in the amygdala (Agren et al., 2012). These exciting findings might lead to new interventions for individuals with PTSD. Emerging evidence thus suggests that extinction training during reconsolidation might be a potentially effective treatment approach.
ADDITIONAL NEURAL CIRCUITRY RELEVANT IN RESILIENCE
Stress resilience has been related to a greater capacity for emotion regulation (Johnstone et al., 2007; Masten and Coatsworth, 1998), thought to be mediated by ventral and dorsal neural systems (Phillips et al., 2003a, 2003b). As discussed earlier, studies have linked genetic variation with differential amygdala reactivity to negative stimuli, as well as altered functional coupling between the PFC and the amygdala, a possible mechanism mediating differential vulnerability to stress. In addition, studies of offspring at high and low familial risk for depression have found differences in neural responses to emotional stimuli (Monk et al., 2008) and in attention to negative emotional stimuli during tryptophan depletion (Feder et al., 2011). In a recent study of acutely traumatized individuals, higher trait resilience measured by the Connor-Davidson Resilience Scale was positively correlated with activation in the right thalamus, and the inferior and middle frontal gyri (BA 47), additional areas involved in emotion regulation (Daniels et al., 2012).
The neural basis of a particular form of emotion regulation, cognitive reappraisal, has been examined in several imaging studies. Activation of the lateral PFC, thought to modulate amygdala activation and thus the intensity of emotional responses, has been linked to reappraisal success (Goldin et al., 2008; Ochsner et al., 2004; Wager et al., 2008). Further, greater use of reappraisal in everyday life has been found to correlate with lower amygdala and greater PFC activation to negative stimuli (Drabant et al., 2009).
Other imaging studies have begun to delineate additional neural circuits relevant for social behavior, for example the neural circuitry mediating the capacity for empathy, thought to be comprised of the mirror neuron system in interaction with limbic brain regions (Rizzolatti and Craighero, 2004; Schulte-Rüther et al., 2007). Future studies might uncover possible relationships between the capacity for empathy and resilience. As discussed, social contact and support are known to promote health and well-being (Charuvastra and Cloitre, 2008). Imaging studies are beginning to uncover the neural mechanisms mediating reduced responses to stress in the presence of social support (Coan et al., 2006).
IMPLICATIONS FOR PREVENTION AND TREATMENT OF STRESS-RELATED PSYCHOPATHOLOGY
Our growing understanding of genetic, developmental, psychological, and neurobiological resilience factors continues to lead to the development of therapeutic interventions for enhancing resilience to stress (Southwick and Charney, 2012b).
Cognitive-behavioral therapies emphasize cognitive reappraisal of negatively viewed circumstances into more positive perspectives, and have shown results across a broad range of populations (Beck and Dozois, 2011). Other therapies employing cognitive reappraisal include hardiness training (Maddi, 2008) and well-being therapy (Fava and Tomba, 2009), the latter also showing promising results in school interventions (Ruini and Fava, 2012). Positive psychology-based exercises, including reflecting daily on things that went well that day, were shown to increase positive affect and decrease depression levels in adults (Seligman et al., 2005). Similarly, gratitude interventions, whereby individuals are instructed to identify several things that they are grateful or thankful for in their life, have been associated with increased reports of well-being and positive affect in various populations (Wood et al., 2010).
Meaning-making interventions have been applied to help breast or colorectal cancer patients, resulting in increases in self-esteem, optimism, and self-efficacy, in comparison with a control group (Lee et al., 2006). Mindfulness training for individuals with residual depressive symptoms successfully increased their ability to experience positive emotions and was associated with a reduction in residual depressive symptoms, compared with a waitlist control condition (Geschwind et al., 2011). Research findings on psychological factors associated with resilience have been applied to design a resilience training program for the military (Reivich et al., 2011). Future studies should determine whether these interventions are successful in protecting individuals facing future stressors, thus boosting resilience in general and at-risk populations.
The military, along with first responder groups such as police or firefighters, has a strong tradition of stress inoculation training, in the form of training programs designed to maximize coping self-efficacy and mastery. This training is accomplished by means of exposure to manageable stressors including physical and mental challenges, providing opportunities for mastery by repeated practice and gradual exposure to increasingly challenging situations. A similar approach is applied in outdoor education programs for children and adolescents, such as Outward Bound.
As discussed in the preceding, recent research has made it possible to understand the process of fear extinction further, and current studies are focused on delineating windows of opportunity during memory retrieval when fear memories and responses are more receptive to extinction training, with the goal of developing novel therapeutic interventions (Karatsoreos and McEwen, 2011; Schiller et al., 2012). The principles of fear extinction have already been applied more broadly to the development of exposure therapy, a form of therapy with proven effectiveness in PTSD (Foa, 2011). Extensive investigation is also devoted to potential ways of enhancing stress-resistance and resilience with pharmacologic means, and the combination of pharmacologic and psychotherapy approaches.
Because antagonizing β receptors disrupts the process of consolidation, several trials have investigated the efficacy of β-blockers, in particular propranolol, in disrupting consolidation of traumatic memories shortly after trauma exposure. Although initial trials showed some promise (Pitman et al., 2002; Vaiva et al., 2003), later studies failed to prevent the emergence of PTSD (Hoge et al., 2012; Stein et al., 2007). One possible explanation is that propranolol was administered late in the consolidation window, or after consolidation was already complete (Cain et al., 2012). In a separate series of studies, the N-methyl D-aspartate (NMDA) receptor partial agonist D-cycloserine, which enhances memory reconsolidation and memory extinction, was shown to facilitate the effects of prolonged exposure therapy in humans with anxiety disorders (Hofmann et al., 2006; Ressler et al., 2004). Results in PTSD have been mixed and await further study (Cain et al., 2012).
Animal models are currently being used to investigate the use of other cognitive enhancers that might affect fear extinction, such as agents modulating GABAergic transmission (Kaplan and Moore, 2011). A major area of interest involves the study of the endogenous cannabinoid system, its role in modulating HPA axis activation during stress, and the potential for facilitating fear extinction by targeting endocannabinoid receptors (Hill and Tasker, 2012). Other receptors known to be involved in extinction, such as the BDNF receptor tyrosine kinase b, have been targeted in preclinical trials (Chhatwal et al., 2006).
Additional potential approaches involve targeting the HPA axis. Trials of hydrocortisone, a glucocorticoid receptor agonist, in intensive care unit patients with septic shock or undergoing cardiac surgery resulted in reduced PTSD symptoms and improved health-related quality of life in long-term survivors (Schelling et al., 2006). A pilot study of high-dose hydrocortisone administered immediately after trauma to patients recruited from the emergency department showed promising results (Zohar et al., 2011). Thus, it might be possible to prevent PTSD with treatments administered shortly after trauma exposure or even by pretreating individuals at high risk for trauma exposure. For individuals with chronic PTSD, current trials are investigating the effects of a CRH1 receptor antagonist and the glucocorticoid receptor antagonist mifepristone in separate trials.
Continued progress in understanding the genetic, developmental, biological, and psychological underpinnings of resilience and vulnerability to stress, as well as the interactions between these factors, will aid in moving the field toward the identification, prevention, and treatment of those at risk of developing stress-related psychopathology.
DISCLOSURES
Dr. Feder has no conflicts of interest or disclosures. Her research is funded by CDC/NIOSH and USAMRAA.
Dr. Haglund has no conflicts of interest or disclosures.
Dr. Wu has no conflicts of interest or disclosures.
Dr. Southwick has no conflicts of interest or disclosures. His research is funded by NIH/NIMH, CDC/NIOSH, USAMRAA and the Department of Veterans Affairs.
Dr. Charney and Mount Sinai School of Medicine have been named on a use patent application of Ketamine for the treatment of depression. If Ketamine were shown to be effective in the treatment of depression and received approval from the Food and Drug Administration for this indication, Dr. Charney and Mount Sinai School of Medicine could benefit financially.
Dr. Charney is funded by NIH, NIH/NIMH, NARSAD, USAMRAA.
Dr. Charney is on the following scientific advisory boards with no compensation:
| 2000 | Academic Advisory Board, Pfizer Postdoctoral Fellowship Program |
| 2001 | Scientific Advisory Board, Familial Dysautonomia Foundation |
| 2002 | National Education Alliance for Borderline Personality Disorder Scientific Advisory Board |
| 2002 | National Center for Posttraumatic Stress Disorder |
| 2002 | Senior Advisory Board, American Psychiatric Association/American Psychiatric Press Journal: Journal of Lifelong Learning in Psychiatry |
| 2005 | Institute of Medicine Committee on Stress of Military Deployment |
| 2006 | Institute of Medicine Subcommittee on PTSD |
| 2012 | Institute of Medicine Committee on DHS Workforce Resilience |
Dr. Charney is also on the following editorial boards with no compensation
| 1986 | Editorial Board, Journal of Affective Disorders |
| 1990 | International Editorial Board, Journal of Psychopharmacology |
| 1993 | Editorial Board, Journal of Serotonin Research |
| 1994 | Editorial Board, Depression and Anxiety |
| 1995 | Editorial Board, Seminars in Clinical Neuropsychiatry |
| 1996 | Editorial Board, CNS Spectrums |
| 1997 | Editorial Board, Human Psychopharmacology |
| 2012 | Editorial Board, CNS Spectrums |
REFERENCES
Aan het Rot, M., Collins, K.A., et al. (2010). Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol. Psychiatry 67:139–145.
Abbosh, C., Lawkowski, A., et al. (2011). GalR2/3 mediates proliferative and trophic effects of galanin on postnatal hippocampal precursors. J. Neurochem. 117:425–436.
Affleck, G., and Tennen, H. (1996). Constructing benefits from adversity: adaptational significance and dispositional underpinnings. J. Personality 64:899–922.
Agren, T., Engman, J., et al. (2012). Disruption of reconsolidation erases a fear memory trace in the human amygdala. Science 337:1550–1552.
Alim, T.N., Feder, A., et al. (2008). Trauma, resilience, and recovery in a high-risk African-American population. Am. J. Psychiatry 165:1566–1575.
Aso, E., Ozaita, A., et al. (2008). BDNF impairment in the hippocampus is related to enhanced despair behavior in CB1 knockout mice. J. Neurochem. 105:565–572.
Autry, A.E., Adachi, M., et al. (2011). NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475:91–95.
Azad, S.C., Eder, M., et al. (2003). Activation of the cannabinoid receptor type 1 decreases glutamatergic and GABAergic synaptic transmission in the lateral amygdala of the mouse. Learn Mem. 10:116–128.
Bahar, A., Dorfman, N., et al. (2004). Amygdalar circuits required for either consolidation or extinction of taste aversion memory are not required for reconsolidation. Eur. J. Neurosci. 19:1115–1118.
Baker, D.G., Ekhator, N.N., et al. (2005). Higher levels of basal serial CSF cortisol in combat veterans with posttraumatic stress disorder. Am. J. Psychiatry 162:992–994.
Baker, D.G., West, S.A., et al. (1999). Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. Am. J. Psychiatry 156:585–588.
Baker, R.A., and Herkenham, M. (1995). Arcuate nucleus neurons that project to the hypothalamic paraventricular nucleus: neuropeptidergic identity and consequences of adrenalectomy on mRNA levels in the rat. J. Comp. Neurol. 358:518–530.
Bakshi, V.P., Smith-Roe, S., et al. (2002). Reduction of stress-induced behavior by antagonism of corticotropin-releasing hormone 2 (CRH2) receptors in lateral septum or CRH1 receptors in amygdala. J. Neurosci. 22:2926–2935.
Bale, T.L., Picetti, R., et al. (2002). Mice deficient for both corticotropin-releasing factor receptor 1 (CRFR1) and CRFR2 have an impaired stress response and display sexually dichotomous anxiety-like behavior. J. Neurosci. 22:193–199.
Barrot, M., Olivier, J.D., et al. (2002). CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc. Natl. Acad. Sci. USA 99:11435–11440.
Bastianetto, S., Ramassamy, C., et al. (1999). Dehydroepiandrosterone (DHEA) protects hippocampal cells from oxidative stress-induced damage. Brain Res. Mol. Brain Res. 66:35–41.
Beck, A.T., and Dozois, D.J. (2011). Cognitive therapy: current status and future directions. Annu. Rev. Med. 62:397–409.
Belfer, I., Hipp, H., et al. (2006). Association of galanin haplotypes with alcoholism and anxiety in two ethnically distinct populations. Mol. Psychiatry 11:301–311.
Belozertseva, I.V., Kos, T., et al. (2007). Antidepressant-like effects of mGluR1 and mGluR5 antagonists in the rat forced swim and the mouse tail suspension tests. Eur. Neuropsychopharmacol. 17:172–179.
Benekareddy, M., Vadodaria, K.C., et al. (2011). Postnatal serotonin type 2 receptor blockade prevents the emergence of anxiety behavior, dysregulated stress-induced immediate early gene responses, and specific transcriptional changes that arise following early life stress. Biol. Psychiatry 70:1024–1032.
Berton, O., Mcclung, C.A., et al. (2006). Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science 311:864–868.
Bierer, L.M., Tischler, L., et al. (2006). Clinical correlates of 24-h cortisol and norepinephrine excretion among subjects seeking treatment following the World Trade Center attacks on 9/11. Ann. NY Acad. Sci. 1071:514–520.
Binder, E.B., Bradley, R.G., et al. (2008). Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA 299:1291–1305.
Bingham, B., Mcfadden, K., et al. (2011). Early adolescence as a critical window during which social stress distinctly alters behavior and brain norepinephrine activity. Neuropsychopharmacology 36:896–909.
Binneman, B., Feltner, D., et al. (2008). A 6-week randomized, placebo-controlled trial of CP-316,311 (a selective CRH1 antagonist) in the treatment of major depression. Am. J. Psychiatry 165:617–620.
Bisschop, M.I., Knegsman, D.M.W., et al. (2004). Chronic diseases and depression: the modifying role of psychosocial resources. Soc. Sci. Med. 59:721–733.
Blair, H.T., Schafe, G.E., et al. (2001). Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn. Mem. 8:229–242.
Bogdan, R., and Pizzagalli, D.A. (2006). Acute stress reduces reward responsiveness: implications for depression. Biol. Psychiatry 60:1147–1154.
Bogdan, R., Santesso, D.L., et al. (2011). Corticotropin-releasing hormone receptor type 1 (CRHR1) genetic variation and stress interact to influence reward learning. J. Neurosci. 31:13246–13254.
Bonanno, G.A. (2004). Loss, trauma, and human resilience: have we underestimated the human capacity to thrive after extremely aversive events? Am. Psychol. 59:20–28.
Bonanno, G.A., Mancini, A.D., et al. (2012). Trajectories of trauma symptoms and resilience in deployed U.S. military service members: prospective cohort study. Br. J. Psychiatry 200:317–323.
Bonanno, G.A., Westphal, M., et al. (2011). Resilience to loss and potential trauma. Annu. Rev. Clin. Psychol. 7:511–535.
Bonne, O., Bain, E., et al. (2005). No change in serotonin type 1A receptor binding in patients with posttraumatic stress disorder. Am. J. Psychiatry 162:383–385.
Born, J., Lange, T., et al. (2002). Sniffing neuropeptides: a transnasal approach to the human brain. Nat. Neurosci. 5:514–516.
Boyce, W.T., and Chesterman, E. (1990). Life events, social support, and cardiovascular reactivity in adolescence. J. Dev. Behav. Pediatr. 11:105–111.
Braam, A.W., Beekman, A.T.F., et al. (1997). Religiosity as a protective or prognostic factor of depression in later life; results from a community survey in The Netherlands. Acta Psychiatr. Scand. 96:199–205.
Braam, A.W., Van Den Eeden, P., et al. (2001). Religion as a cross-cultural determinant of depression in elderly Europeans: results from the EURODEP collaboration. Psychol. Med. 31:803–814.
Bradley, R.G., Binder, E.B., et al. (2008). Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Arch. Gen. Psychiatry 65:190–200.
Bremner, J.D., Licinio, J., et al. (1997). Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. Am. J. Psychiatry 154:624–629.
Breslau, N., Kessler, R.C., et al. (1998). Trauma and posttraumatic stress disorder in the community: the 1996 Detroit Area Survey of Trauma. Arch. Gen. Psychiatry 55:626–632.
Britton, K.T., Akwa, Y., et al. (2000). Neuropeptide Y blocks anxiogenic-like behavioral action of corticotropin-releasing factor in an operant conflict test and elevated plus maze. Peptides 21:37–44.
Brosse, A.L., Sheets, E.S., et al. (2002). Exercise and the treatment of clinical depression in adults: recent findings and future directions. Sports Med. 32:741–760.
Browne, E.S., Wright, B.E., et al. (1992). Dehydroepiandrosterone: antiglucocorticoid action in mice. Am. J. Med. Sci. 303:366–371.
Bruce, S.E., Weisberg, R.B., et al. (2001). Trauma and posttraumatic stress disorder in primary care patients. Prim. Care Companion J. Clin. Psychiatry 3:211–217.
Brunner, R., Schaefer, D., et al. (2006). Effect of high-dose cortisol on memory functions. Ann. NY Acad. Sci. 1071:434–437.
Cabib, S., and Puglisi-Allegra, S. (1996). Different effects of repeated stressful experiences on mesocortical and mesolimbic dopamine metabolism. Neurosci. 73:375–380.
Cabib, S., Ventura, R., et al. (2002). Opposite imbalances between mesocortical and mesoaccumbens dopamine responses to stress by the same genotype depending on living conditions. Behav. Brain Res. 129:179–185.
Cain, C.K., Maynard, G.D., et al. (2012). Targeting memory processes with drugs to prevent or cure PTSD. Expert Opin. Investig. Drugs 21:1323–1350.
Camacho, T.C., Roberts, R.E., et al. (1991). Physicalactivity and depression—Evidence from the Alameda County Study. Am. J. Epidem. 134:220–231.
Carroll, B.J., Cassidy, F., et al. (2007). Pathophysiology of hypercortisolism in depression. Acta Psychiatr. Scand. 115 (Suppl. 433):90–103.
Carvajal, C., Dumont, Y., et al. (2006). Emotional behavior in aged neuropeptide Y (NPY) Y2 knockout mice. J. Mol. Neurosci. 28:239–245.
Carver, C., Pozo, C., et al. (1993). How coping mediates the effect of optimism on distress: a study of women with early stage breast cancer. J. Pers. Soc. Psychology 65:375–390.
Caspi, A., Sugden, K., et al. (2003). Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301:386–389.
Castren, E., and Rantamaki, T. (2010). The role of BDNF and its receptors in depression and antidepressant drug action: reactivation of developmental plasticity. Dev. Neurobiol. 70:289–297.
Chantarujikapong, S.I., Scherrer, J.F., et al. (2001). A twin study of generalized anxiety disorder symptoms, panic disorder symptoms and post-traumatic stress disorder in men. Psychiatry Res. 103:133–145.
Charney, D.S., and Drevets, W.D. (2002). The neurobiological basis of anxiety disorders. In: Davis, K.L., Charney, D.S., Coyle, J.T., and Nemeroff, C., eds. Neuropsychopharmacology: The Fifth Generation of Progress. Philadelphia, PA: Lippincott Williams & Wilkins, pp. 901–930.
Charney, D.S. (2004). Psychobiological mechanisms of resilience and vulnerability: implications for successful adaptation to extreme stress. Am. J. Psychiatry 161:195–216.
Charuvastra, A., Cloitre, M. (2008). Social bonds and posttraumatic stress disorder. Annu. Rev. Psychol. 59:301–328.
Chen, L., Dai, X.N., et al. (2006a). Chronic administration of dehydroepiandrosterone sulfate (DHEAS) primes for facilitated induction of long-term potentiation via sigma 1 (sigma1) receptor: optical imaging study in rat hippocampal slices. Neuropharmacology 50:380–392.
Chen, Z.Y., Jing, D., et al. (2006b). Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 314:140–143.
Chhatwal, J.P., Stanek-Rattiner, L., et al. (2006). Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nat. Neurosci. 9:870–872.
Chourbaji, S., Vogt, M.A., et al. (2008). AMPA receptor subunit 1 (GluR-A) knockout mice model the glutamate hypothesis of depression. FASEB J. 22:3129–3134.
Christensen, R., Kristensen, P.K., et al. (2007). Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet 370:1706–1713.
Chung, C. (2012). New perspectives on glutamate receptor antagonists as antidepressants. Arch. Pharm. Res. 35:573–577.
Claes, S.J. (2004). Corticotropin-releasing hormone (CRH) in psychiatry: from stress to psychopathology. Ann. Med. 36:50–61.
Coan, J.A., Schaefer, H.S., et al. (2006). Lending a hand: social regulation of the neural response to threat. Psychol. Sci. 17:1032–1039.
Cohen, H., Liu, T., et al. (2012). The neuropeptide Y (NPY)-ergic system is associated with behavioral resilience to stress exposure in an animal model of post-traumatic stress disorder. Neuropsychopharmacology 37:350–363.
Coric, V., Feldman, H.H., et al. (2010). Multicenter, randomized, double-blind, active comparator and placebo-controlled trial of a corticotropin-releasing factor receptor-1 antagonist in generalized anxiety disorder. Depress. Anxiety 27:417–425.
Cortese, B.M., and Phan, K.L. (2005). The role of glutamate in anxiety and related disorders. CNS Spectr. 10:820–830.
Cota, D., Marsicano, G., et al. (2003). The endogenous cannabinoid system affects energy balance via central orexigenic drive and peripheral lipogenesis. J. Clin. Invest. 112:423–431.
Cotman, C.W., and Berchtold, N.C. (2002). Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 25:295–301.
Counts, S.E., and Mufson, E.J. (2010). Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. J. Neurochem. 113:649–660.
Cui, H., Sakamoto, H., et al. (2008). Effects of single-prolonged stress on neurons and their afferent inputs in the amygdala. Neuroscience 152:703–712.
Culver, J.L., Arena, P.L., et al. (2002). Coping and distress among women under treatment for early stage breast cancer: comparing African Americans, Hispanics and non-Hispanic whites. Psycho-Oncology 11:495–504.
Daniels, J.K., Hegadoren, K.M., et al. (2012). Neural correlates and predictive power of trait resilience in an acutely traumatized sample: a pilot investigation. J. Clin. Psychiatry 73:327–332.
Danner, D.D., Snowdon, D.A., et al. (2001). Positive emotions in early life and longevity: findings from the nun study. J. Pers. Soc. Psychol. 80:804–813.
Davidson, R.J., McEwen, B.S. (2012). Social influences on neuroplasticity: stress and interventions to promote well-being. Nat. Neurosci. 15:689–695.
Davis, M. (2006). Neural systems involved in fear and anxiety measured with fear-potentiated startle. Am. Psychol. 61:741–756.
de Kloet, E.R., Derijk, R.H., et al. (2007). Therapy insight: is there an imbalanced response of mineralocorticoid and glucocorticoid receptors in depression? Nat. Clin. Prac. 3:168–179.
Deaner, S.L., and McConatha, J.T. (1993). The relation of humor to depression and personality. Psychological Rep. 72:755–763.
Debiec, J., and LeDoux, J.E. (2006). Noradrenergic signaling in the amygdala contributes to the reconsolidation of fear memory: treatment implications for PTSD. Ann. NY Acad. Sci. 1071:521–524.
Dennis, N.A., Cabeza, R., et al. (2011). Brain-derived neurotrophic factor val66met polymorphism and hippocampal activation during episodic encoding and retrieval tasks. Hippocampus 21:980–989.
Derijk, R.H., and De Kloet, E.R. (2008). Corticosteroid receptor polymorphisms: determinants of vulnerability and resilience. Eur. J. Pharmacol. 583:303–311.
deRoon-Cassini, T.A., de St Aubin, E., et al. (2009). Psychological well-being after spinal cord injury: perception of loss and meaning making. Rehabil. Psychol. 54:306–314.
Desantis, S.M., Baker, N.L., et al. (2011). Gender differences in the effect of early life trauma on hypothalamic-pituitary-adrenal axis functioning. Depress. Anxiety 28:383–392.
Di, S., Malcher-Lopes, R., et al. (2003). Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J. Neurosci. 23:4850–4857.
Disner, S.G., Beevers, C.G., et al. (2011). Neural mechanisms of the cognitive model of depression. Nat. Rev. 12:467–477.
Domenici, M.R., Azad, S.C., et al. (2006). Cannabinoid receptor type 1 located on presynaptic terminals of principal neurons in the forebrain controls glutamatergic synaptic transmission. J. Neurosci. 26:5794–5799.
Drabant, E.M., Hariri, A.R., et al. (2006). Catechol O-methyltransferase val158met genotype and neural mechanisms related to affective arousal and regulation. Arch. Gen. Psychiatry 63:1396–1406.
Drabant, E.M., McRae, K., et al. (2009). Individual differences in typical reappraisal use predict amygdala and prefrontal responses. Biol. Psychiatry 65:367–373.
Drabant, E.M., Ramel, W., et al. (2012). Neural mechanisms underlying 5-HTTLPR-related sensitivity to acute stress. Am. J. Psychiatry 169:397–405.
Drury, S.S., Theall, K.P., et al. (2009). The role of the dopamine transporter (DAT) in the development of PTSD in preschool children. J. Trauma. Stress 22:534–539.
Dudai, Y., and Eisenberg, M. (2004). Rites of passage of the engram: reconsolidation and the lingering consolidation hypothesis. Neuron 44:93–100.
Dudley, K.J., Li, X., et al. (2011). Epigenetic mechanisms mediating vulnerability and resilience to psychiatric disorders. Neurosci. Biobehav. Rev. 35:1544–1551.
Duman, R.S., and Monteggia, L.M. (2006). A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59:1116–1127.
Dunlop, B.W., and Nemeroff, C.B. (2007). The role of dopamine in the pathophysiology of depression. Arch. Gen. Psychiatry 64:327–337.
Egan, M.F., Kojima, M., et al. (2003). The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112:257–269.
Eisch, A.J., Bolanos, C.A., et al. (2003). Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: a role in depression. Biol. Psychiatry 54:994–1005.
Eisenhardt, D., and Menzel, R. (2007). Extinction learning, reconsolidation and the internal reinforcement hypothesis. Neurobiol. Learn. Mem. 87:167–173.
Eva, C., Serra, M., et al. (2006). Physiology and gene regulation of the brain NPY Y1 receptor. Front. Neuroendocrinol. 27:308–339.
Fakra, E., Hyde, L.W., et al. (2009). Effects of HTR1A C(-1019)G on amygdala reactivity and trait anxiety. Arch. Gen. Psychiatry 66:33–40.
Fava, G.A., Tomba, E. (2009). Increasing psychological well-being and resilience by psychotherapeutic methods. J. Pers. 77:1903–1934.
Feder, A., Nestler, E.J., et al. (2009). Psychobiology and molecular genetics of resilience. Nat. Rev. Neurosci. 10:446–457.
Feder, A., Skipper, J., et al. (2011). Tryptophan depletion and emotional processing in healthy volunteers at high risk for depression. Biol. Psychiatry 69:804–807.
Fehr, E., and Fischbacher, U. (2003). The nature of human altruism. Nature 425:785–791.
Fleshner, M., Maier, S.F., et al. (2011). The neurobiology of the stress-resistant brain. Stress 14:498–502.
Foa, E.B. (2011). Prolonged exposure therapy: past, present, and future. Depress. Anxiety 28:1043–1047.
Folkman, S. (1997). Positive psychological states and coping with severe stress. Soc. Sci. Med. 45:1207–1221.
Folkman, S., and Moskowitz, J.T. (2000). Positive affect and the other side of coping. Am. Psychol. 55:647–654.
Fontana, A.F., Kerns, R.D., et al. (1989). Support, stress, and recovery from coronary heart disease: a longitudinal causal model. Health Psychol. 8:175–193.
Frankl, V.E. (2006). Man’s Search for Meaning. (I. Lasch, Trans). Boston: Beacon Press. (Original work published 1959).
Fraser, G.A. (2009). The use of a synthetic cannabinoid in the management of treatment-resistant nightmares in posttraumatic stress disorder (PTSD). CNS Neurosci. Ther. 15:84–88.
Fredrickson, B.L. (2001). The role of positive emotions in positive psychology. The broaden-and-build theory of positive emotions. Am. Psychol. 56:218–226.
Freidenberg, B.M., Gusmano, R., et al. (2010). Women with PTSD have lower basal salivary cortisol levels later in the day than do men with PTSD: a preliminary study. Physiol. Behav. 99:234–236.
Fulton, D. (2006). Reconsolidation: does the past linger on? J. Neuroscience 26:10935–10936; discussion 10936.
Gallagher, P., and Young, A. (2002). Cortisol/DHEA ratios in depression. Neuropsychopharmacol. 26:410.
Genud, R., Merenlender, A., et al. (2009). DHEA lessens depressive-like behavior via GABA-ergic modulation of the mesolimbic system. Neuropsychopharmacol. 34:577–584.
Gerardi, M., Rothbaum, B.O., et al. (2010). Cortisol response following exposure treatment for PTSD in rape victims. J. Aggress. Maltreat. Trauma 19:349–356.
Geschwind, N., Peeters, F., et al. (2011). Mindfulness training increases momentary positive emotions and reward experience in adults vulnerable to depression: a randomized controlled trial. J. Consult. Clin. Psychol. 79:618–628.
Gill, J., Vythilingam, M., et al. (2008). Low cortisol, high DHEA, and high levels of stimulated TNF-alpha, and IL-6 in women with PTSD. J. Trauma. Stress 21:530–539.
Goldin, P.R., McRae, K., et al. (2008). The neural bases of emotion regulation: reappraisal and suppression of negative emotion. Biol. Psychiatry 63:577–586.
Goldman, S.L., Kraemer, D.T., et al. (1996). Beliefs about mood moderate the relationship of stress to illness and symptom reporting. J. Psychosom. Res. 41:115–128.
Goldstein, L.E., Rasmusson, A.M., et al. (1996). Role of the amygdala in the coordination of behavioral, neuroendocrine, and prefrontal cortical monoamine responses to psychological stress in the rat. J. Neurosci. 16:4787–4798.
Gonul, A.S., Kitis, O., et al. (2011). Association of the brain-derived neurotrophic factor Val66Met polymorphism with hippocampus volumes in drug-free depressed patients. World J. Biol. Psychiatry 12:110–118.
Goodyer, I.M., Herbert, J., et al. (1998). Adrenal steroid secretion and major depression in 8- to 16-year-olds, III. Influence of cortisol/DHEA ratio at presentation on subsequent rates of disappointing life events and persistent major depression. Psychol. Med. 28:265–273.
Grammatopoulos, D.K., and Chrousos, G.P. (2002). Functional characteristics of CRH receptors and potential clinical applications of CRH-receptor antagonists. Trends Endocrin. Metab. 13:436–444.
Greenwood, B.N., Foley, T.E., et al. (2003). Freewheel running prevents learned helplessness/behavioral depression: Role of dorsal raphe serotonergic neurons. J. Neurosci. 23:2889–2898.
Griffin, M.G., Resick, P.A., et al. (2005). Enhanced cortisol suppression following dexamethasone administration in domestic violence survivors. Am. J. Psychiatry 162:1192–1199.
Gross, J.J. (2002). Emotion regulation: affective, cognitive, and social consequences. Psychophysiol. 39:281–291.
Gustafson, E.L., Smith, K.E., et al. (1996). Distribution of a rat galanin receptor mRNA in rat brain. Neuroreport. 7:953–957.
Gutman, A.R., Yang, Y., et al. (2008). The role of neuropeptide Y in the expression and extinction of fear-potentiated startle. J. Neurosci. 28:12682–12690.
Haas, A.P., and Hendin, H. (1984). Wounds of War: The Psychological Aftermath of Combat in Vietnam. New York: Basic Books.
Hall, J., Thomas, K.L., et al. (2001). Fear memory retrieval induces CREB phosphorylation and Fos expression within the amygdala. Eur. J. Neurosci. 13:1453–1458.
Hankin, B.L., Nederhof, E., et al. (2011). Differential susceptibility in youth: evidence that 5-HTTLPR X positive parenting is associated with positive affect “for better and worse.” Transl. Psychiatry 1(10):e44.
Harbaugh, W.T., Mayr, U., et al. (2007). Neural responses to taxation and voluntary giving reveal motives for charitable donations. Science 316:1622–1625.
Haren, M.T., Malmstrom, T.K., et al. (2007). Lower serum DHEAS levels are associated with a higher degree of physical disability and depressive symptoms in middle-aged to older African American women. Maturitas 57:347–360.
Hariri, A.R., Drabant, E.M., et al. (2005). A susceptibility gene for affective disorders and the response of the human amygdala. Arch. Gen. Psychiatry 62:146–152.
Harvey, B.H., and Shahid, M. (2012). Metabotropic and ionotropic glutamate receptors as neurobiological targets in anxiety and stress-related disorders: focus on pharmacology and preclinical translational models. Pharmacol. Biochem. Behav. 100:775–800.
Harvey, P.O., Fossati, P., et al. (2005). Cognitive control and brain resources in major depression: an fMRI study using the n-back task. NeuroImage 26:860–869.
Hasler, G., Drevets, W.C., et al. (2004). Discovering endophenotypes for major depression. Neuropsychopharmacol. 29:1765–1781.
Heilig, M. (1995). Antisense inhibition of neuropeptide Y (NPY)-Y1 receptor expression blocks the anxiolytic-like action of NPY in amygdala and paradoxically increases feeding. Regul. Pept. 59:201–205.
Heilig, M. (2004). The NPY system in stress, anxiety and depression. Neuropeptides 38:213–224.
Heilig, M., Koob, G.F., et al. (1994). Corticotropin-releasing factor and neuropeptide-y: role in emotional integration. Trends Neurosci. 17:80–85.
Heilig, M., McLeod, S., et al. (1993). Anxiolytic-like action of neuropeptide Y: mediation by Y1 receptors in amygdala, and dissociation from food intake effects. Neuropsychopharmacol. 8:357–363.
Heim, C., Newport, D.J., et al. (2000). Pituitary-adrenal and autonomic responses to stress in women after sexual and physical abuse in childhood. JAMA 284:592–597.
Heinz, A., and Smolka, M.N. (2006). The effects of catechol O- methyltransferase genotype on brain activation elicited by affective stimuli and cognitive tasks. Rev. Neurosci. 17:359–367.
Heisler, L.K., Chu, H.M., et al. (1998). Elevated anxiety and antidepressant-like responses in serotonin 5-HT1A receptor mutant mice. Proc. Natl. Acad. Sci. USA 95:15049–15054.
Hendriksen, H., Bink, D.I., et al. (2012). Re-exposure and environmental enrichment reveal NPY-Y1 as a possible target for post-traumatic stress disorder. Neuropharmacol. 63:733–742.
Herkenham, M., Lynn, A.B., et al. (1991). Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J. Neurosci. 11:563–583.
Herry, C., and Garcia, R. (2002). Prefrontal cortex long-term potentiation, but not long-term depression, is associated with the maintenance of extinction of learned fear in mice. J. Neurosci. 22:577–583.
Hill, M.N., Barr, A.M., et al. (2007). Electroconvulsive shock treatment differentially modulates cortical and subcortical endocannabinoid activity. J. Neurochem. 103:47–56.
Hill, M.N., and Gorzalka, B.B. (2009a). The endocannabinoid system and the treatment of mood and anxiety disorders. CNS Neurol. Disord. Drug Targets 8:451–458.
Hill, M.N., and Gorzalka, B.B. (2009b). Impairments in endocannabinoid signaling and depressive illness. JAMA 301:1165–1166.
Hill, M.N., Miller, G.E., et al. (2008). Serum endocannabinoid content is altered in females with depressive disorders: a preliminary report. Pharmacopsychiatry 41:48–53.
Hill, M.N., and Tasker, J.G. (2012). Endocannabinoid signaling, glucocorticoid-mediated negative feedback, and regulation of the hypothalamic-pituitary-adrenal axis. Neuroscience 204:5–16.
Hillard, C.J. (2000). Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat. 61:3–18.
Hobfoll, S.E., Palmieri, P.A., et al. (2009). Trajectories of resilience, resistance, and distress during ongoing terrorism: the case of Jews and Arabs in Israel. J. Consult. Clin. Psychol. 77:138–148.
Hoexter, M.Q., Fadel, G., et al. (2012). Higher striatal dopamine transporter density in PTSD: an in vivo SPECT study with [(99m)Tc]TRODAT-1. Psychopharmacology (Berl.) 224 (2):337–345.
Hofmann, S.G., Meuret, A.E., et al. (2006). Augmentation of exposure therapy with d-cycloserine for social anxiety disorder. Arch. Gen. Psychiatry 63:298–304.
Hoge, E.A., Worthington, J.J., et al. (2012). Effect of acute posttrauma propranolol on PTSD outcome and physiological responses during script-driven imagery. CNS Neurosci. Ther. 18:21–27.
Holmes, A., Yang, R.J., et al. (2002). Evaluation of an anxiety-related phenotype in galanin overexpressing transgenic mice. J. Mol. Neurosci. 18:151–165.
Holmes, F.E., Bacon, A., et al. (2003). Transgenic overexpression of galanin in the dorsal root ganglia modulates pain related behavior. Proc. Natl. Acad. Sci. USA 100:6180–6185.
Homberg, J.R., and Lesch, K.P. (2011). Looking on the bright side of serotonin transporter gene variation. Biol. Psychiatry 69:513–519.
Hsu, D.T., Mickey, B.J., et al. (2012). Variation in the corticotropin-releasing hormone receptor 1 (CRHR1) gene influences fMRI signal responses during emotional stimulus processing. J. Neurosci. 32:3253–3260.
Huang, E.J., and Reichardt, L.F. (2001). Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24:677–736.
Husum, H., and Mathe, A.A. (2002). Early life stress changes concentrations of neuropeptide Y and corticotropin-releasing hormone in adult rat brain. Lithium treatment modifies these changes. Neuropsychopharmacol. 27:756–764.
Huuhka, K., Anttila, S., et al. (2008). Dopamine 2 receptor C957T and catechol-o-methyltransferase Val158Met polymorphisms are associated with treatment response in electroconvulsive therapy. Neurosci. Lett. 448:79–83.
Inslicht, S.S., Marmar, C.R., et al. (2006). Increased cortisol in women with intimate partner violence-related posttraumatic stress disorder. Ann. NY Acad. Sci. 1071:428–429.
Isen, A.M., Daubman, K.A., and Nowicki, G.P. (1987). Positive affect facilitates creative problem-solving. J. Pers. Soc. Psychol. 52:1122–1131.
Ising, M., Depping, A.M., et al. (2008). Polymorphisms in the FKBP5 gene region modulate recovery from psychosocial stress in healthy controls. Eur. J. Neurosci. 28:389–398.
Ivy, A.S., Rex, C.S., et al. (2010). Hippocampal dysfunction and cognitive impairments provoked by chronic early-life stress involve excessive activation of CRH receptors. J. Neurosci. 30:13005–13015.
Jabbi, M., Kema, I.P., et al. (2007). Catechol O- methyltransferase polymorphism and susceptibility to major depressive disorder modulates psychological stress response. Psychiatr. Genet. 17:183–193.
Janssen, J., Hulshoff Pol, H.E., et al. (2007). Hippocampal volume and subcortical white matter lesions in late-life depression: comparison of early- and late-onset depression. J. Neurol. Neurosurg. Psychiatry 78(6):638–640.
Jhaveri, D.J., Mackay, E.W., et al. (2010). Norepinephrine directly activates adult hippocampal precursors via beta3-adrenergic receptors. J. Neurosci. 30:2795–2806.
Johnstone, T., van Reekum, C.M., et al. (2007). Failure to regulate: counterproductive recruitment of top-down prefrontal-subcortical circuitry in major depression. J. Neurosci. 27:8877–8884.
Jordan, B.K., Schlenger, W.E., et al. (1991). Lifetime and current prevalence of specific psychiatric disorders among Vietnam veterans and controls. Arch. Gen. Psychiatry 48:207–215.
Josselyn, S.A., Shi, C., et al. (2001). Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J. Neurosci. 21:2404–2412.
Jovanovic, T., Norrholm, S.D., et al. (2010). Fear potentiation is associated with hypothalamic-pituitary-adrenal axis function in PTSD. Psychoneuroendocrinology 35:846–857.
Kaplan, G.B., and Moore, K.A. (2011). The use of cognitive enhancers in animal models of fear extinction. Pharmacol. Biochem. Behav. 99:217–228.
Karatsoreos, I.N., and McEwen, B.S. (2011). Psychobiological allostasis: resistance, resilience and vulnerability. Trends Cogn. Sci. 15:576–584.
Karg, K., Burmeister, M., et al. (2011). The serotonin transporter promoter variant (5-HTTLPR): stress, and depression meta-analysis revisited: evidence of genetic moderation. Arch. Gen. Psychiatry 68:444–454.
Karl, T., Burne, T.H., et al. (2006). Effect of Y1 receptor deficiency on motor activity, exploration, and anxiety. Behav. Brain Res. 167:87–93.
Karlsson, R.M., and Holmes, A. (2006). Galanin as a modulator of anxiety and depression and a therapeutic target for affective disease. Amino Acids 31:231–239.
Kasen, S., Wickramaratne, P., et al. (2012). Religiosity and resilience in persons at high risk for major depression. Psychol. Med. 42:509–519.
Kask, A., Rago, L., et al. (1997). Alpha-helical CRF(9-41) prevents anxiogenic-like effect of NPY Y1 receptor antagonist BIBP3226 in rats. Neuroreport 8:3645–3647.
Kendler, K.S., Kuhn, J.W., et al. (2005). The interaction of stressful life events and a serotonin transporter polymorphism in the prediction of episodes of major depression: a replication. Arch. Gen. Psychiatry 62:529–535.
Kent, J.M., Mathew, S.J., et al. (2002). Molecular targets in the treatment of anxiety. Biol. Psychiatry 52:1008–1030.
Kessler, R.C., Sonnega, A., et al. (1995). Posttraumatic stress disorder in the National Comorbidity Survey. Arch. Gen. Psychiatry 52:1048–1060.
Khoshaba, D.M., and Maddi, S.R. (1999). Early experiences in hardiness development. Consul. Psychol. J. 51:106–116.
Kida, S., Josselyn, S.A., et al. (2002). CREB required for the stability of new and reactivated fear memories. Nat. Neurosci. 5:348–355.
Kim, J.M., Stewart, R., et al. (2007). Interactions between life stressors and susceptibility genes (5-HTTLPR and BDNF) on depression in Korean elders. Biol. Psychiatry 62:423–428.
Kimonides, V.G., Khatibi, N.H., et al. (1998). Dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEAS) protect hippocampal neurons against excitatory amino acid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 95:1852–1857.
Klemenhagen, K.C., Gordon, J.A., et al. (2006). In HT1A receptor. Neuropsychopharmacol. 31:101–111.
Klohnen, E.C. (1996). Conceptual analysis and measurement of the construct of ego-resiliency. J. Personal Soc. Psychology 70:1067–1079.
Knutson, B., and Cooper, J.C. (2005). Functional magnetic resonance imaging of reward prediction. Curr. Opin. Neurol. 18:411–417.
Koenig, H.G., George, L.K., et al. (1998). Religiosity and remission of depression in medically ill older patients. Am. J. Psychiatry 155:536–542.
Koike, H., Iijima, M., et al. (2011). Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav. Brain Res. 224:107–111.
Kozlovsky, N., Matar, M.A., et al. (2009). The role of the galaninergic system in modulating stress-related responses in an animal model of posttraumatic stress disorder. Biol. Psychiatry 65:383–391.
Kremen, W.S., Koenen, K.C., et al. (2012). Twin studies of posttraumatic stress disorder: differentiating vulnerability factors from sequelae. Neuropharmacol. 62:647–653.
Krishnan, V., Han, M.H., et al. (2007). Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131 (2):391–404.
Kuhnen, C.M., and Knutson, B. (2005). The neural basis of financial risk taking. Neuron 47:763–770.
Lanzenberger, R., Wadsak, W., et al. (2010). Cortisol plasma levels in social anxiety disorder patients correlate with serotonin-1A receptor binding in limbic brain regions. Int. J. Neuropsychopharmacol. 13:1129–1143.
Lavretsky, H., Siddarth, P., et al. (2008). The effects of the dopamine and serotonin transporter polymorphisms on clinical features and treatment response in geriatric depression: a pilot study. Int. J. Geriatr. Psychiatry 23:55–59.
Lawford, B.R., Young, R., et al. (2006). The D2 dopamine receptor (DRD2) gene is associated with co-morbid depression, anxiety and social dysfunction in untreated veterans with post-traumatic stress disorder. Eur. Psychiatry 21:180–185.
LeDoux, J.E. (2000). Emotion circuits in the brain. Annu. Rev. Neurosci. 23:155–184.
LeDoux, J.E., and Gorman, J.M. (2002). A call to action: overcoming anxiety through active coping. Am. J. Psychiatry 159:171–171.
Le Maitre, T.W., Xia, S., et al. (2011). Galanin receptor 2 overexpressing mice display an antidepressive-like phenotype: possible involvement of the subiculum. Neuroscience 190:270–288.
Lee, J.L., Milton, A.L., et al. (2006). Reconsolidation and extinction of conditioned fear: inhibition and potentiation. J. Neuroscience 26:10051–10056.
Lee, V., Robin Cohen, S., et al. (2006). Meaning-making intervention during breast or colorectal cancer treatment improves self-esteem, optimism, and self-efficacy. Soc. Sci. Med. 62:3133–3155.
Li, N., Lee, B., et al. (2010). mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists Science 329:959–964.
Li, X., Need, A.B., et al. (2006). Metabotropic glutamate 5 receptor antagonism is associated with antidepressant-like effects in mice. J. Pharmacol. Exp. Ther. 319:254–259.
Liu, D., Diorio, J., et al. (1997). Maternal care, hippocampal glucocorticoid receptors, and hypothalamic–pituitary–adrenal responses to stress. Science 277:1659–1662.
Lu, X., Ross, B., et al. (2008). Phenotypic analysis of GalR2 knockout mice in anxiety- and depression-related behavioral tests. Neuropeptides 42:387–397.
Lyons, D.M., Parker, K.J., et al. (2009). Developmental cascades linking stress inoculation, arousal regulation, and resilience. Front. Behav. Neurosci. 3:32.
Maddi, S.R. (1999). Hardiness and optimism as expressed in coping patterns. Consul. Psychology J. 51:95–105.
Maddi, S.R. (2008). The courage and strategies of hardiness as helpful in growing despite major, disruptive stresses. Am. Psychol. 63:563–564.
Maestripieri, D., Lindell, S.G., et al. (2007). Intergenerational transmission of maternal behavior in rhesus macaques and its underlying mechanisms. Dev. Psychobiol. 49:165–171.
Mahan, A.L., and Ressler, K.J. (2012). Fear conditioning, synaptic plasticity and the amygdala: implications for posttraumatic stress disorder. Trends Neurosci. 35:24–35.
Makino, S., Baker, R.A., et al. (2000). Differential regulation of neuropeptide Y mRNA expression in the arcuate nucleus and locus coeruleus by stress and antidepressants. J. Neuroendocrinol. 12:387–395.
Maninger, N., Capitanio, J.P., et al. (2010). Acute and chronic stress increase DHEAS concentrations in rhesus monkeys. Psychoneuroendocrinology 35:1055–1062.
Manne, S., Duhamel, K., et al. (2003). Coping and the course of mother’s depressive symptoms during and after pediatric bone marrow transplantation. J. Am. Acad. Child Adol. Psychiatry 42:1055–1068.
Martin, M., Ledent, C., et al. (2002). Involvement of CB1 cannabinoid receptors in emotional behaviour. Psychopharmacology (Berl.) 159:379–387.
Martin-Soelch, C., Leenders, K.L., et al. (2001). Reward mechanisms in the brain and their role in dependence: evidence from neurophysiological and neuroimaging studies. Brain Res. Brain Res. Rev. 36:139–149.
Masten, A.S. (2001). Ordinary magic. Resilience processes in development. Am. Psychol. 56:227–238.
Masten, A.S., Coatsworth, J.D. (1998). The development of competence in favorable and unfavorable environments. Lessons from research on successful children. Am. Psychol. 53:205–220.
Masten, A.S., and Obradovic, J. (2006). Competence and resilience in development. Ann. NY Acad. Sci. 1094:13–27.
Mathew, S.J., Shah, A., et al. (2012). Ketamine for treatment-resistant unipolar depression: current evidence. CNS Drugs 26:189–204.
Mathews, D.C., Henter, I.D., et al. (2012). Targeting the glutamatergic system to treat major depressive disorder: rationale and progress to date. Drugs 72:1313–1333.
McCabe, C., Woffindale, C., et al. (2012). Neural processing of reward and punishment in young people at increased familial risk of depression. Biol. Psychiatry 72:588–594.
McCormack, K., Sanchez, M.M., et al. (2006). Maternal care patterns and behavioral development of rhesus macaque abused infants in the first 6 months of life. Dev. Psychobiol. 48:537–550.
McCullough, M.E., Hoyt, W.T., et al. (2000). Religious involvement and mortality: a meta-analytic review. Health Psychology 19:211–222.
Mehta, D., Gonik, M., et al. (2011). Using polymorphisms in FKBP5 to define biologically distinct subtypes of posttraumatic stress disorder: evidence from endocrine and gene expression studies. Arch. Gen. Psychiatry 68:901–910.
Mercer, K.B., Orcutt, H.K., et al. (2012). Acute and posttraumatic stress symptoms in a prospective gene x environment study of a university campus shooting. Arch. Gen. Psychiatry 69:89–97.
Metzger, L.J., Carson, M.A., et al. (2008). Basal and suppressed salivary cortisol in female Vietnam nurse veterans with and without PTSD. Psychiatry Res. 161:330–335.
Mifsud, K.R., Gutierrez-Mecinas, M., et al. (2011). Epigenetic mechanisms in stress and adaptation. Brain Behav. Immun. 25:1305–1315.
Mitsukawa, K., Lu, X., et al. (2009). Bidirectional regulation of stress responses by galanin in mice: involvement of galanin receptor subtype 1. Neuroscience 160:837–846.
Mobbs, D., Greicius, M.D., et al. (2003). Humor modulates the mesolimbic reward centers. Neuron 40:1041–1048.
Moldrich, G., and Wenger, T. (2000). Localization of the CB1 cannabinoid receptor in the rat brain. An immunohistochemical study. Peptides 21:1735–1742.
Moller, C., Sommer, W., et al. (1999). Anxiogenic-like action of galanin after intra-amygdala administration in the rat. Neuropsychopharmacol. 21:507–512.
Monk, C.S., Klein, R.G., et al. (2008). Amygdala and nucleus accumbens activation to emotional facial expressions in children and adolescents at risk for major depression. Am. J. Psychiatry 165:90–98.
Montag, C., Reuter, M., et al. (2008). The BDNF Val66Met polymorphism affects amygdala activity in response to emotional stimuli: evidence from a genetic imaging study. NeuroImage 42:1554–1559.
Moos, R.H., and Schaefer, J.A. (1993). Coping resources and processes: current concepts and measures. In: Goldberger, L., and Breznits, S., eds. Handbook of Stress: Theoretical and Clinical Aspects. New York: Free Press, pp. 234–257.
Morfin, R., and Starka, L. (2001). Neurosteroid 7-hydroxylation products in the brain. Int. Rev. Neurobiol. 46:79–95.
Morgan, C.A., III, Rasmusson A., et al. (2009). Relationships among plasma dehydroepiandrosterone and dehydroepiandrosterone sulfate, cortisol, symptoms of dissociation and objective performance in humans exposed to underwater navigation stress. Biol. Psychiatry 66:334–340.
Morgan, C.A., III, Southwick, S., et al. (2004). Relationships among plasma dehydroepiandrosterone sulfate and cortisol levels, symptoms of dissociation, and objective performance in humans exposed to acute stress. Arch. Gen. Psychiatry 61:819–825.
Morgan, C.A., III, Wang, S., et al. (2000). Hormone profiles in humans experiencing military survival training. Biol. Psychiatry 47:891–901.
Morrow, B.A., Elsworth, J.D., et al. (1999). The role of mesoprefrontal dopamine neurons in the acquisition and expression of conditioned fear in the rat. Neurosci. 92:553–564.
Murphy, F.C., Rubinsztein, J.S., et al. (2001). Decision making cognition in mania and depression. Psycholog. Med. 31:679–693.
Murrough, J.W., Charney, D.S. (2010). Cracking the moody brain: lifting the mood with ketamine. Nat. Med. 16:1384–1385.
Myers, K.M., and Davis, M. (2007). Mechanisms of fear extinction. Mol. Psychiatry 12:120–150.
Myers, K.M., Ressler, K.J., et al. (2006). Different mechanisms of fear extinction dependent on length of time since fear acquisition. Learn Mem. 13:216–223.
Nagin, D.S., and Odgers, C.L. (2010). Group-based trajectory modeling in clinical research. Annu. Rev. Clin. Psychol. 6:109–138.
Nemeroff, C.B. (2002). Recent advances in the neurobiology of depression. Psychopharmacol. Bull. 36(Suppl 2):6–23.
Neufeld-Cohen, A., Kelly, P.A., et al. (2012). Chronic activation of corticotropin-releasing factor type 2 receptors reveals a key role for 5-HT1A receptor responsiveness in mediating behavioral and serotonergic responses to stressful challenge. Biol. Psychiatry 72 (6):437–447.
Nguyen, N.K., Sartori, S.B., et al. (2009). Effect of neuropeptide Y Y2 receptor deletion on emotional stress-induced neuronal activation in mice. Synapse 63:236–246.
Nikolova, Y., Bogdan, R., et al. (2012). Perception of a naturalistic stressor interacts with 5-HTTLPR/rs25531 genotype and gender to impact reward responsiveness. Neuropsychobiology 65(1):45–54.
Nissen, S.E., Nicholls, S.J., et al. (2008). Effect of rimonabant on progression of atherosclerosis in patients with abdominal obesity and coronary artery disease: the STRADIVARIUS randomized controlled trial. JAMA 299:1547–1560.
Norris, F.H., Tracy, M., et al. (2009). Looking for resilience: understanding the longitudinal trajectories of responses to stress. Soc. Sci. Med. 68:2190–2198.
Oberlander, T.F., Weinberg, J., et al. (2008). Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 3:97–106.
Ochsner, K.N., Ray, R.D., et al. (2004). For better or for worse: neural systems supporting the cognitive down- and up-regulation of negative emotion. NeuroImage 23:483–499.
Onur, O.A., Walter, H., et al. (2009). Noradrenergic enhancement of amygdala responses to fear. Soc. Cogn. Affect. Neurosci. 4:119–126.
Otte, C., Neylan, T.C., et al. (2005). Association between childhood trauma and catecholamine response to psychological stress in police academy recruits. Biol. Psychiatry 57:27–32.
Overmier, J.B., and Seligman, M.E. (1967). Effects of inescapable shock upon subsequent escape and avoidance responding. J. Comp. Physiol. Psychol. 63:28–33.
Oyarzun, J.P., Lopez-Barroso, D., et al. (2012). Updating fearful memories with extinction training during reconsolidation: a human study using auditory aversive stimuli. PLoS One 7:e38849.
Parker, K.J., Buckmaster, C.L., et al. (2005). Mild early life stress enhances prefrontal-dependent response inhibition in monkeys. Biol. Psychiatry 57:848–855.
Parker, K.J., Buckmaster, C.L., et al. (2004). Prospective investigation of stress inoculation in young monkeys. Arch. Gen. Psychiatry 61:933–941.
Parks, C.L., Robinson, P.S., et al. (1998). Increased anxiety of mice lacking the serotonin1A receptor. Proc. Natl. Acad. Sci. USA 95:10734–10739.
Pezawas, L., Meyer-Lindenberg, A., et al. (2005). 5-HTTLPR polymorphism impacts human cingulate-amygdala interactions: a genetic susceptibility mechanism for depression. Nat. Neurosci. 8:828–834.
Phillips, M.L., Drevets, W.C., et al. (2003a). Neurobiology of emotion perception I: the neural basis of normal emotion perception. Biol. Psychiatry 54:504–514.
Phillips, M.L., Drevets, W.C., et al. (2003b). Neurobiology of emotion perception II: implications for major psychiatric disorders. Biol. Psychiatry 54:515–528.
Pickens, C.L., Adams-Deutsch, T., et al. (2009). Effect of pharmacological manipulations of neuropeptide Y and corticotropin-releasing factor neurotransmission on incubation of conditioned fear. Neuroscience 164:1398–1406.
Pieribone, V.A., Brodin, L., et al. (1992). Differential expression of mRNAs for neuropeptide Y-related peptides in rat nervous tissues: possible evolutionary conservation. J. Neurosci. 12:3361–3371.
Pietrzak, R.H., Harpaz-Rotem, I., et al. (2011). Cognitive-behavioral coping strategies associated with combat-related PTSD in treatment-seeking OEF-OIF Veterans. Psychiatry Res. 189:251–258.
Pietrzak, R.H., and Southwick, S.M. (2011). Psychological resilience in OEF-OIF veterans: application of a novel classification approach and examination of demographic and psychosocial correlates. J. Affect. Disord. 133:560–568.
Pinnock, S.B., Lazic, S.E., et al. (2009). Synergistic effects of dehydroepiandrosterone and fluoxetine on proliferation of progenitor cells in the dentate gyrus of the adult male rat. Neuroscience 158:1644–1651.
Pitman, R.K., Sanders, K.M., et al. (2002). Pilot study of secondary prevention of posttraumatic stress disorder with propranolol. Biol. Psychiatry 51:189–192.
Pliakas, A.M., Carlson, R.R., et al. (2001). Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element-binding protein expression in nucleus accumbens. J. Neurosci. 21:7397–7403.
Popoli, M., Yan, Z., et al. (2011). The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 13:22–37.
Przybyslawski, J., Roullet, P., et al. (1999). Attenuation of emotional and nonemotional memories after their reactivation: role of beta adrenergic receptors. J. Neurosci. 19:6623–6628.
Rabkin, J.G., McElhiney, M.C., et al. (2006). Placebo controlled trial of dehydroepiandrosterone (DHEA) for treatment of nonmajor depression in patients with HIV/AIDS. Am. J. Psychiatry 163:59–66.
Rachman, S. (1979). Concept of required helpfulness. Behav. Res. Ther. 17:1–6.
Radant, A.D., Dobie, D.J., et al. (2009). Adrenocortical responsiveness to infusions of physiological doses of ACTH is not altered in posttraumatic stress disorder. Front. Behav. Neurosci. 3:40.
Radley, J.J., Kabbaj, M., et al. (2011). Stress risk factors and stress-related pathology: neuroplasticity, epigenetics and endophenotypes. Stress 14:481–497.
Rasmusson, A.M., Vasek, J., et al. (2004). An increased capacity for adrenal DHEA release is associated with decreased avoidance and negative mood symptoms in women with PTSD. Neuropsychopharmacol. 29:1546–1557.
Rattiner, L.M., Davis, M., et al. (2004). Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J. Neurosci. 24:4796–4806.
Reivich, K.J., Seligman, M.E., et al. (2011). Master resilience training in the U.S. Army. Am. Psychol. 66:25–34.
Resick, P.A. (2001). Clinical Psychology: A Modular Course. Philadelphia, PA: Taylor & Francis Group.
Ressler, K.J., Bradley, B., et al. (2010). Polymorphisms in CRHR1 and the serotonin transporter loci: gene x gene x environment interactions on depressive symptoms. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B:812–824.
Ressler, K.J., Mercer, K.B., et al. (2011). Post-traumatic stress disorder is associated with PACAP and PAC1 receptor. Nature 470:492–497.
Ressler, K.J., Rothbaum, B.O., et al. (2004). Cognitive enhancers as adjuncts to psychotherapy: use of d-cycloserine in phobic individuals to facilitate extinction of fear. Arch. Gen. Psychiatry 61:1136–1144.
Risold, P.Y., and Swanson, L.W. (1997). Chemoarchitecture of the rat lateral septal nucleus. Brain Res. Brain Res. Rev. 24:91–113.
Rizzolatti, G., Craighero, L. (2004). The mirror-neuron system. Annu. Rev. Neurosci. 27:169–192.
Robinson, O.J., Cools, R., et al. (2012). Tryptophan depletion disinhibits punishment but not reward prediction: implications for resilience. Psychopharmacology (Berl.) 219:599–605.
Rodrigues, S.M., Schafe, G.E., et al.. (2001). Intraamygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J. Neurosci. 21:6889–6896.
Roozendaal, B., Schelling, G., et al. (2008). Corticotropin-releasing factor in the basolateral amygdala enhances memory consolidation via an interaction with the beta-adrenoceptor-cAMP pathway: dependence on glucocorticoid receptor activation. J. Neurosci. 28:6642–6651.
Rosenfeld, R.S., Hellman, L., et al. (1971). Dehydroisoandrosterone is secreted episodically and synchronously with cortisol by normal man. J. Clin. Endocrinol. Metab. 33:87–92.
Roth, T.L., Lubin, F.D., et al. (2009). Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol. Psychiatry 65:760–769.
Royer, S., Martina, M., and et al. (2000). Bistable behavior of inhibitory neurons controlling impulse traffic through the amygdala: role of a slowly deinactivating K+ current. J. Neurosci. 20:9034–9039.
Ruini, C., Fava, G.A. (2012). Role of well-being therapy in achieving a balanced and individualized path to optimal functioning. Clin. Psychol. Psychother. 19:291–304.
Russo, S.J., Murrough, J.W., et al. (2012). Neurobiology of resilience. Nat. Neurosci. 15(11):1475–1484.
Rutter, M. (1993). Resilience: some conceptual considerations. J. Adolesc. Health 14:626–631.
Rutter, M. (2006). Implications of resilience concepts for scientific understanding. Ann. NY Acad. Sci. 1094:1–12.
Rutter, M. (2012). Resilience as a dynamic concept. Dev. Psychopathol. 24:335–344.
Sah, R., and Geracioti, T.D. (2012). Neuropeptide Y and posttraumatic stress disorder. Mol. Psychiatry:1–10.
Sajdyk, T.J., Fitz, S.D., et al. (2006). The role of neuropeptide Y in the amygdala on corticotropin-releasing factor receptor-mediated behavioral stress responses in the rat. Stress 9:21–28.
Sanacora, G., Zarate, C.A., et al. (2008). Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat. Rev. Drug Discov. 7:426–437.
Schafe, G.E., Doyere, V., et al. (2005). Tracking the fear engram: the lateral amygdala is an essential locus of fear memory storage. J. Neurosci. 25:10010–10014.
Schafe, G.E., Nader, K., et al. (2001). Memory consolidation of Pavlovian fear conditioning: a cellular and molecular perspective. Trends Neurosci. 24:540–546.
Scheier, M.F., Matthews, K.A., et al. (1989). Dispositional optimism and recovery from coronary artery bypass surgery: the beneficial effects on physical and psychological wellbeing. J. Pers. Soc. Psychol. 57:1024–1040.
Schelling, G., Roozendaal, B., et al. (2006). Efficacy of hydrocortisone in preventing posttraumatic stress disorder following critical illness and major surgery. Ann. NY Acad. Sci. 1071:46–53.
Scherrer, J.F., True, W.R., et al. (2000). Evidence for genetic influences common and specific to symptoms of generalized anxiety and panic. J. Affect. Disord. 57:25–35.
Schiller, D., Levy, I., et al. (2008). From fear to safety and back: reversal of fear in the human brain. J. Neurosci. 28:11517–11525.
Schiller, D., Monfils, M.H., et al. (2010). Preventing the return of fear in humans using reconsolidation update mechanisms. Nature 463:49–53.
Schiller, D., Raio, C.M., et al. (2012). Extinction training during the reconsolidation window prevents recovery of fear. J. Vis. Exp. 66:e3893.
Schulte-Rüther, M., Markowitsch, H.J., et al. (2007). Mirror neuron and theory of mind mechanisms involved in face-to-face interactions: a functional magnetic resonance imaging approach to empathy. J. Cogn. Neurosci. 19 (8):1354–1372.
Schultz, W. (2002). Getting formal with dopamine and reward. Neuron 36:241–263.
Sciolino, N.R., and Holmes, P.V. (2012). Exercise offers anxiolytic potential: a role for stress and brain noradrenergic-galaninergic mechanisms. Neurosci. Biobehav. Rev. 36 (9):1965–1984.
Seckl, J.R., and Meaney, M.J. (2006). Glucocorticoid “programming” and PTSD risk. Ann. NY Acad. Sci. 1071:351–378.
Seligman, M.E., and Maier, S.F. (1967). Failure to escape traumatic shock. J. Exp. Psychol. 74:1–9.
Seligman, M.E., Steen, T.A., et al. (2005). Positive psychology progress: empirical validation of interventions. Am. Psychol. 60:410–421.
Sevcik, J., Finta, E.P., et al. (1993). Galanin receptors inhibit the spontaneous firing of locus coeruleus neurons and interact with mu-opioid receptors. Eur. J. Pharmacol. 230:223–230.
Siebert, A. (1996). The Survivor Personality: Why Some People are Stronger, Smarter, and More Skillful at Handling Life’s Difficulties . . . and How You Can Be, Too. New York: Berkely.
Sigurdsson, T., Doyere, V., et al. (2007). Long-term potentiation in the amygdala: a cellular mechanism of fear learning and memory. Neuropharmacol. 52:215–227.
Silver, R.-C., Holman, E.A., et al. (2002). Nationwide longitudinal study of psychological responses to September 11. JAMA 288:1235–1244.
Smolka, M.N., Schumann, G., et al. (2005). Catechol-Omethyltransferase val158met genotype affects processing of emotional stimuli in the amygdala and prefrontal cortex. J. Neurosci. 25:836–842.
Sotres-Bayon, F., Bush, D.E., et al. (2007). Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacol. 32:1929–1940.
Sotres-Bayon, F., Cain, C.K., et al. (2006). Brain mechanisms of fear extinction: historical perspectives on the contribution of prefrontal cortex. Biol. Psychiatry 60:329–336.
Southwick, S.M., and Charney, D.S. (2012a). Resilience: The Science of Mastering Life’s Greatest Challenges. New York: Cambridge University Press.
Southwick, S.M. and Charney, D.S. (2012b). The science of resilience: implications for the prevention and treatment of depression. Science 338:78–82.
Southwick, S.M., Vythilingam, M., et al. (2005). The psychobiology of depression and resilience to stress: implications for prevention and treatment. Ann. Rev. Clin. Psychol. 1:255–291.
Steckler, T., and Holsboer, F. (1999). Corticotropin-releasing hormone receptor subtypes and emotion. Biol. Psychiatry 46:1480–1508.
Steckler, T., and Risbrough, V. (2012). Pharmacological treatment of PTSD—established and new approaches. Neuropharmacol. 62:617–627.
Stein, M.B., Kerridge, C., et al. (2007). Pharmacotherapy to prevent PTSD: results from a randomized controlled proof-of-concept trial in physically injured patients. J. Trauma. Stress 20:923–932.
Steiner, M.A., Wanisch, K., et al. (2008). Impaired cannabinoid receptor type 1 signaling interferes with stress-coping behavior in mice. Pharmacogenomics. J. 8:196–208.
Steptoe, A., Wardle, J., et al. (2005). Positive affect and health-related neuroendocrine, cardiovascular, and inflammatory processes. Proc. Natl. Acad. Sci. USA 102:6508–6512.
Stockdale, J.B. (1991). Courage Under Fire: Testing Epictetus’ Doctrines in a Laboratory of Human Behavior. Stanford, CA: Hoover Institution Press.
Strome, E.M., Wheler, G.H., et al. (2002). Intracerebroventricular corticotropin-releasing factor increases limbic glucose metabolism and has social context-dependent behavioral effects in nonhuman primates. Proc. Natl. Acad. Sci. USA 99:15749–15754.
Suzuki, A., Josselyn, S.A., et al. (2004). Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J. Neurosci. 24:4787–4795.
Swanson, C.J., Blackburn, T.P., et al. (2005). Anxiolytic- and antidepressant-like profiles of the galanin-3 receptor (Gal3) antagonists SNAP 37889 and SNAP 398299. Proc. Natl. Acad. Sci. USA 102:17489–17494.
Tache, Y., and Bonaz, B. (2007). Corticotropin-releasing factor receptors and stress-related alterations of gut motor function. J. Clin. Invest. 117:33–40.
Taliaz, D., Loya, A., et al. (2011). Resilience to chronic stress is mediated by hippocampal brain-derived neurotrophic factor. J. Neurosci. 31:4475–4483.
Tasan, R.O., Nguyen, N.K., et al. (2010). The central and basolateral amygdala are critical sites of neuropeptide Y/Y2 receptor-mediated regulation of anxiety and depression. J. Neurosci. 30:6282–6290.
Taylor, M.K., Padilla, G.A., et al. (2012). Effects of dehydroepiandrosterone supplementation during stressful military training: a randomized, controlled, double-blind field study. Stress 15:85–96.
Thompson, R.W., Arnkoff, D.B., et al. (2011). Conceptualizing mindfulness and acceptance as components of psychological resilience to trauma. Trauma Violence Abus. 12:220–235.
Thorson, J.A., and Powell, F.C. (1994). Depression and sense of humor. Psychological. Rep. 75:1473–1474.
Travis, L.A., Lyness, J.M., et al. (2004). Social support, depression, and functional disability in older adult primary care patients. Am. J. Geriatr. Psychiatry 12:265–271.
True, W.R., Rice, J., et al. (1993). A twin study of genetic and environmental contributions to liability for posttraumatic stress symptoms. Arch. Gen. Psychiatry 50:257–264.
Tsai, J., Harpaz-Rotem, I., et al. (2012). The role of coping, resilience, and social support in mediating the relation between PTSD and social functioning in veterans returning from Iraq and Afghanistan. Psychiatry 75:135–149.
Tsou, K., Brown, S., et al. (1998). Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83:393–411.
Tugade, M.M., and Fredrickson, B.L. (2004). Resilient individuals use positive emotions to bounce back from negative emotional experiences. J. Pers. Soc. Psychol. 86:320–333.
Uher, R., and Mcguffin, P. (2010). The moderation by the serotonin transporter gene of environmental adversity in the etiology of depression: 2009 update. Mol. Psychiatry 15:18–22.
Vaillant, G. (1977). Adaptation to Life. Boston, MA: Little Brown.
Vaillant, G. (1992). The historical origins and future potential of Sigmund Freud’s concept of the mechanisms of defence. Int. Rev. Psycho Analysis 19:35–50.
Vaiva, G., Ducrocq, F., et al. (2003). Immediate treatment with propranolol decreases posttraumatic stress disorder two months after trauma. Biol. Psychiatry 54:947–949.
van der Kolk, B.A., and van der Hart, O. (1989). Pierre Janet and the breakdown of adaptation in psychological trauma. Am. J. Psychiatry 146:1530–1540.
Vialou, V., Robison, A.J., et al. (2010). DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat. Neurosci. 13:745–752.
Vinkers, C.H., Oosting, R.S., et al. (2010). Early-life blockade of 5-HT(1A) receptors alters adult anxiety behavior and benzodiazepine sensitivity. Biol. Psychiatry 67:309–316.
Voisey, J., Swagell, C.D., et al. (2009). The DRD2 gene 957C>T polymorphism is associated with posttraumatic stress disorder in war veterans. Depress. Anxiety 26:28–33.
Vythilingam, M., Heim, C., et al. (2002). Childhood trauma associated with smaller hippocampal volume in women with major depression. Am. J. Psychiatry 159:2072–2080.
Wade, S.L., Borawski, E.A., et al. (2001). The relationship of caregiver coping to family outcomes during the initial year following pediatric traumatic injury. J. Consult. Clin. Psychol. 69:406–415.
Wager, T.D., Davidson, M.L., et al. (2008). Prefrontal-subcortical pathways mediating successful emotion regulation. Neuron 59:1037–1050.
Walker, D.L., and Davis, M. (2000). Involvement of NMDA receptors within the amygdala in short- versus long-term memory for fear conditioning as assessed with fear-potentiated startle. Behav. Neurosci. 114:1019–1033.
Walton, K.M., Chin, J.E., et al. (2006). Galanin function in the central nervous system. Curr. Opin. Drug Discov. Devel. 9:560–570.
Wang, Y., Wu, J., et al. (2012). Dopamine D1 receptors are responsible for stress-induced emotional memory deficit in mice. Stress 15:237–242.
Weaver, I.C., Cervoni, N., et al. (2004). Epigenetic programming by maternal behavior. Nat. Neurosci. 7:847–854.
Weis, F., Kilger, E., et al. (2006). Stress doses of hydrocortisone reduce chronic stress symptoms and improve health-related quality of life in high-risk patients after cardiac surgery: a randomized study. J. Thor. Cardiov. Surg. 131:277.
Werner, E., and Smith, R. (1992). Overcoming the Odds: High-risk Children from Birth to Adulthood. Ithaca, NY: Cornell University Press.
Wheler, G.H., Brandon, D., et al. (2006). Cortisol production rate in posttraumatic stress disorder. J. Clin. Endocrinol. Metab. 91:3486–3489.
Whitworth, J.A., Williamson, P.M., et al. (2005). Cardiovascular consequences of cortisol excess. Vasc. Health Risk Manag. 1:291–299.
Wiley, J.L., Beletskaya, I.D., et al. (2002). Resorcinol derivatives: a novel template for the development of cannabinoid CB(1)/CB(2) and CB(2)-selective agonists. J. Pharmacol. Exp. Ther. 301:679–689.
Winter, B., Breitenstein, C., et al. (2006). High impact running improves learning. Neurobiol. Learn Mem. 87(4):597–609.
Wise, R.A. (2002). Brain reward circuitry: insights from unsensed incentives. Neuron 36:229–240.
Wolin, S.J., and Wolin, S. (1993). The Resilient Self: How Survivors of Troubled Families Rise above Adversity. New York: Villard.
Wood, A.M., Froh, J.J., et al. (2010). Gratitude and well-being: a review and theoretical integration. Clin. Psychol. Rev. 30:890–905.
Wood, A.M., Maltby, J., et al. (2008). The role of gratitude in the development of social support, stress and depression: two longitudinal studies. J. Res. Pers. 42:854–871.
Wu, G., Feder, A., et al. (2011). Central functions of neuropeptide Y in mood and anxiety disorders. Expert. Opin. Ther. Targets 15:1317–1331.
Xu, Z.Q., Tong, Y.G., et al. (2001). Galanin enhances noradrenaline-induced outward current on locus coeruleus noradrenergic neurons. Neuroreport 12:1779–1782.
Yamada, K., and Nabeshima, T. (2003). Brain-derived neurotrophic factor/TrkB signaling in memory processes. J. Pharmacol. Sci. 91:267–270.
Yehuda, R. (2001). Biology of posttraumatic stress disorder. J. Clin. Psychiatry 62(Suppl 17):41–46.
Yehuda, R. (2004). Risk and resilience in posttraumatic stress disorder. J. Clin. Psychiatry 65(Suppl 1):29–36.
Yehuda, R., Bierer, L.M., et al. (2009). Cortisol metabolic predictors of response to psychotherapy for symptoms of PTSD in survivors of the World Trade Center attacks on September 11, 2001. Psychoneuroendocrinology 34:1304–1313.
Yehuda, R., Brand, S.R., et al. (2006). Clinical correlates of DHEA associated with post-traumatic stress disorder. Acta Psychiatr. Scand. 114:187–193.
Yehuda, R., Koenen, K.C., et al. (2011). The role of genes in defining a molecular biology of PTSD. Dis. Markers 30:67–76.
Yehuda, R., Yang, R.K., et al. (2006). Alterations in cortisol negative feedback inhibition as examined using the ACTH response to cortisol administration in PTSD. Psychoneuroendocrinol. 31:447–451.
Young, A.H., Gallagher, P., et al. (2002). Elevation of the cortisol-dehydroepiandrosterone ratio in drug-free depressed patients. Am. J. Psychiatry 159:1237–1239.
Zarate, C.A., Jr., Singh, J.B., et al. (2006). A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression Arch. Gen. Psychiatry 63:856–864.
Zeidner, M., and Hammer, A.L. (1992). Coping with missile attack—resources, strategies, and outcomes. J. Personality 60:709–746.
Zhou, Z., Zhu, G., et al. (2008). Genetic variation in human NPY expression affects stress response and emotion. Nature 452:997–1001.
Zohar, J., Yahalom, H., et al. (2011). High dose hydrocortisone immediately after trauma may alter the trajectory of PTSD: interplay between clinical and animal studies. Eur. Neuropsychopharmacol. 21:796–809.
Zoumakis, E., Grammatopoulos, D.K., et al. (2006). Corticotropin-releasing hormone receptor antagonists. Eur. J. Endocrinol. 155:S85–S91.