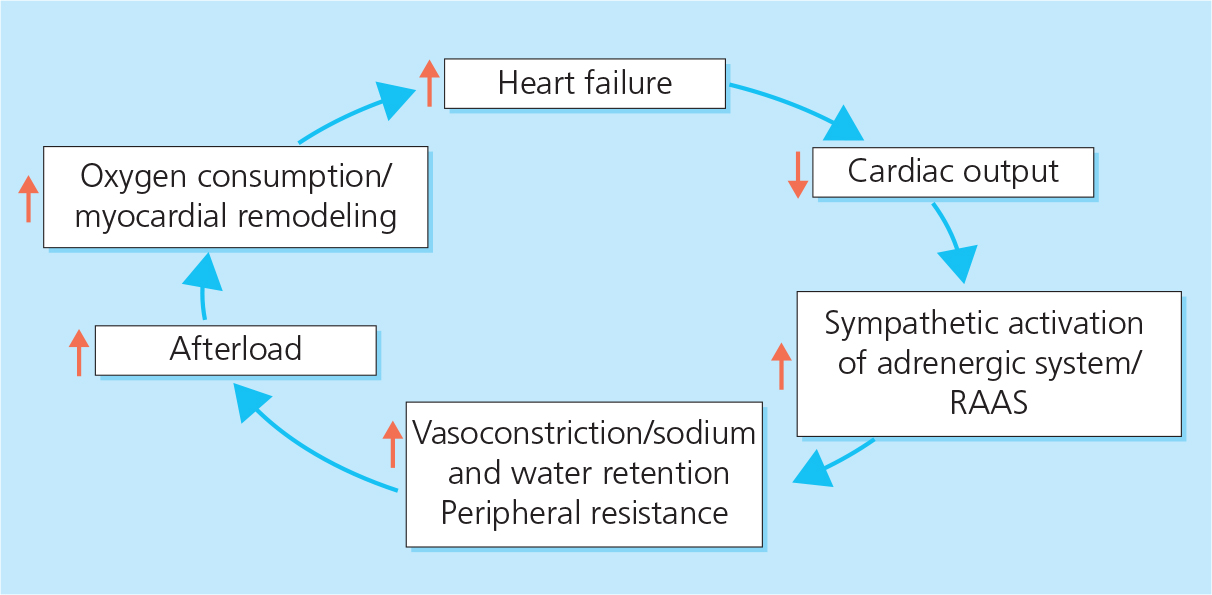
Figure 2.1 The vicious cycle of heart failure. RAAS, renin–angiotensin–aldosterone system.
| 2 | Pathophysiology and clinical stages |
Advances in the management of heart failure (HF) over the past 20 years have been informed by a better understanding of its pathophysiology. There are few situations in cardiology where treatment has been as closely linked to an appreciation of the underlying science.
Vicious cycle of heart failure. HF is a disease of inappropriate adaptation to injury. The body has a limited range of compensatory responses to circulatory impairment, mainly vasoconstriction and sodium and water retention (see below). In general, however, these adjustments to hypovolemia are poorly suited to pump failure, and increases in the preload and afterload of the failing heart lead to worsening HF (Figure 2.1).
Neurohormonal pathways activated in HF include the sympathetic nervous system (SNS), the renin–angiotensin–aldosterone system (RAAS) and the natriuretic peptide (NP) system. They play a significant role in the pathophysiology of HF, and pharmacological interventions have been developed accordingly (see Chapter 7).
Sympathetic nervous system. Sympathetic activation of the adrenergic system leads to vasoconstriction, which increases the resistance to blood flow and helps to maintain arterial pressure in the early stages of HF when cardiac output is reduced. However, vasoconstriction also increases the afterload on the heart, leading to a worsening of HF (see Figure 2.1).
Renin–angiotensin–aldosterone system. Enhanced sympathetic outflow also activates the RAAS (Figure 2.2). Renin release from the kidneys causes increased formation of angiotensin I from angiotensinogen and, via the action of angiotensin-converting enzyme (ACE), angiotensin II. Angiotensin II causes systemic vasoconstriction and acts on the adrenal cortex to produce aldosterone, leading to sodium and water retention. In addition, aldosterone (which may be released even in the setting of ACE inhibition) contributes to myocardial and vascular fibrosis. Sympathetic stimulation also releases antidiuretic hormone, which leads to retention of free water and contributes to dilutional hyponatremia.
Figure 2.2 Neurohormonal pathways in heart failure. ACE, angiotensin-converting enzyme; ADH, antidiuretic hormone; AI/AII, angiotensin I/II.
Natriuretic peptide system. The natriuretic peptide family consists of A (atrial) and B (brain) type natriuretic peptides (ANP and BNP), which are produced by cardiomyocytes in response to atrial and ventricular stretch, and C type natriuretic peptide, which is secreted by endothelial and renal cells. NPs, mainly BNP, lead to increased sodium excretion and vasodilation, especially in the early phases of HF. BNP also has anti-remodeling properties. The biological action of BNP is mediated through membrane-bound natriuretic peptide receptors (NPRs) and the peptide is degraded by neutral endopeptidase (including neprilysin). It is postulated that HF is a state of relative BNP deficiency caused by both lack of biologically active peptide and resistance at a receptor level. In end-stage HF, the peptides may not be released because of myocyte loss.
Other pathways also reflect an inappropriate response to injury. Cytokine release is increased in HF, leading to a variety of consequences including apoptosis. The role of these as contributors to the progression of HF, rather than a correlate, is debated. Certainly, the failure of tumor necrosis factor (TNF) inhibitors to improve outcome argues against a causative role.
Remodeling of the myocardium. Global and local responses to maladaptive stimuli lead to myocardial remodeling, namely increased myocardial volume and mass and a net loss of myocytes. The heart has the ability to change the force of contraction, and therefore stroke volume, in response to changes in venous return (the Frank–Starling mechanism). A reduction in stroke volume due to myocardial injury can be overcome by left ventricular (LV) enlargement. This is not a response that can keep occurring indefinitely – eventually a loss of LV function will occur due to reduced interaction between contractile elements, caused by their separation.
LV hypertrophy (LVH) maintains wall stress as the LV enlarges. However, it is also eventually maladaptive as the hypertrophied myocardium exceeds the growth of its blood supply.
Autonomic reflexes. The increase of sympathetic tone associated with HF leads to disturbance of the autonomic reflexes. Persistent elevation of the heart rate is maladaptive in the ventricle: disturbances of LV relaxation are common, so the shortening of diastole that occurs with tachycardia (the duration of systole remains stable) is not well tolerated.
Insulin resistance is an important metabolic sequel to HF. It contributes to the disturbance of myocyte energy metabolism, leading to the description of the failing heart as ‘an engine out of fuel’. Causes of insulin resistance include the underlying etiologies of HF (central obesity, diabetes mellitus) and loss of skeletal muscle (see below).
Peripheral vasoconstriction, as described above, symptomatically may contribute to cold sensitivity.
Loss of skeletal muscle is an important manifestation of HF, reflecting inactivity, consequences of circulating substances such as tumor growth factor (TGF)-β and reduced cardiac output. In its most advanced manifestation, loss of skeletal muscle may lead to cachexia. The consequences of this process include contributions to insulin resistance as well as loss of the skeletal muscle circulatory bed. The loss of this vasculature represents an additional decrement in the amount of vasculature that can undergo vasodilation (and therefore unload the LV).
Cardiorenal interactions. Reduced renal perfusion in HF (due to reduced stroke volume and vasoconstriction) is an important contributor to sodium and fluid overload. The exact links between cardiac and renal function have yet to be resolved. More marked disturbances of renal function, leading to coexisting renal failure, may also occur and pose problems for volume control.
The clinical syndrome of HF represents the final manifestation of advanced disease. Although progress has been made in the management of this entity, the greatest hope of avoiding the adverse outcome of HF is to intervene at an earlier subclinical stage, when there is more likelihood of reversing the process. The American Cardiology Association (ACC)/American Heart Association (AHA) guidelines divide progression of the disease into four preclinical and clinical stages (Table 2.1).
|
TABLE 2.1 The clinical stages and functional classes of heart failure |
|||
ACC/AHA stage |
Clinical status |
NYHA class |
Functional status |
A |
Preclinical: risk factors for HF but no structural heart disease or symptoms |
I |
No limitation in any activities; no symptoms from ordinary activities |
B |
Preclinical: structural evidence of heart disease, but no symptoms |
I |
No limitation in any activities; no symptoms from ordinary activities |
C |
Clinical: structural evidence of heart disease, and symptoms or signs of HF |
I, II, III |
No, slight (e.g. mild shortness of breath) or marked limitation of any activity due to symptoms; comfortable only at rest |
D |
Clinical: structural evidence of advanced heart disease, and marked symptoms and signs of HF |
II, III, IV |
Slight or marked limitation of any activity; symptoms at rest |
|
Adapted from the American College of Cardiology (ACC)/American Heart Association (AHA) 2001 guidelines and The Criteria Committee of the New York Heart Association (NYHA), Nomenclature and Criteria for Diagnosis of Diseases of the Heart and Great Vessels, 9th edn. Boston: Little, Brown & Co, 1994:253–6. |
|||
It is important to distinguish these from functional classes, as described in the New York Heart Association (NYHA) classification system, which is based on severity of symptoms and exercise capacity and can be used to assess response to treatment (see Table 2.1).
ACC/AHA stages A and B are preclinical; these patients fall into NYHA functional class I. ACC/AHA stage C reflects patients with symptoms or signs of HF, so these patients may be classified functionally in any of the NYHA classes I to III. The functional status of patients in ACC/AHA class D (with marked symptoms and signs of HF) is usually limited (NYHA class II to IV).
The association between functional class, cardiac function (LVEF) and prognosis is discussed in more detail in Chapter 9.
Key points – pathophysiology and clinical stages
• Heart failure (HF) is a disease of response to injury, which is initially appropriate and becomes inappropriate.
• Inappropriate non-cardiac responses include activation of a variety of hormonal reflexes (vasoconstriction, and sodium and water retention). Reversal of these inappropriate responses is critical to the management of HF.
• Remodeling is a cardiac response that is initially appropriate and becomes inappropriate as the heart enlarges excessively and hypertrophies.
• Symptoms develop in HF as a result of a complex web of interactions, involving not only the heart, hormonal changes and skeletal muscle, but also the vasculature (especially the endothelium), lungs and other organs.
• In addition to the classification of HF by clinical manifestation (acute vs chronic, systolic vs diastolic), etiology and precipitants (addressed in other chapters), HF can be classified by clinical stage (the course of the disease) and functional class (functional status based on symptom severity and exercise capacity).
References
Hunt SA, Baker DW, Chin MH et al. ACC/AHA guidelines for the evaluation and management of chronic heart failure in the adult: executive summary report of the American College of Cardiology/American Heart Association task force on practice guidelines. Circulation 2001;104:2996–3007.
National Heart Foundation of Australia. New York Heart Association (NYHA) grading of symptoms in congestive heart failure. In: Guidelines for the Prevention, Detection and Management of Chronic Heart Failure in Australia, 2011. www.heartfoundation.org.au/images/uploads/publications/Chronic_ Heart_Failure_Guidelines_2011.pdf, last accessed 12 May 2017.
Piepoli MF, Guazzi M, Boriani G et al. Exercise intolerance in chronic heart failure: mechanisms and therapies. Part I. Eur J Cardiovasc Prev Rehabil 2010;17:637–42.
Sica DA. Sodium and water retention in heart failure and diuretic therapy: basic mechanisms. Cleve Clin J Med 2006;73(Suppl 2):S2–7.
Triposkiadis F, Karayannis G, Giamouzis G et al. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol 2009;54:1747–62.
Volpe M, Carnovali M, Mastromarino V. The natriuretic peptides system in the pathophysiology of heart failure: from molecular basis to treatment. Clin Sci (Lond) 2016;130:57–77.
