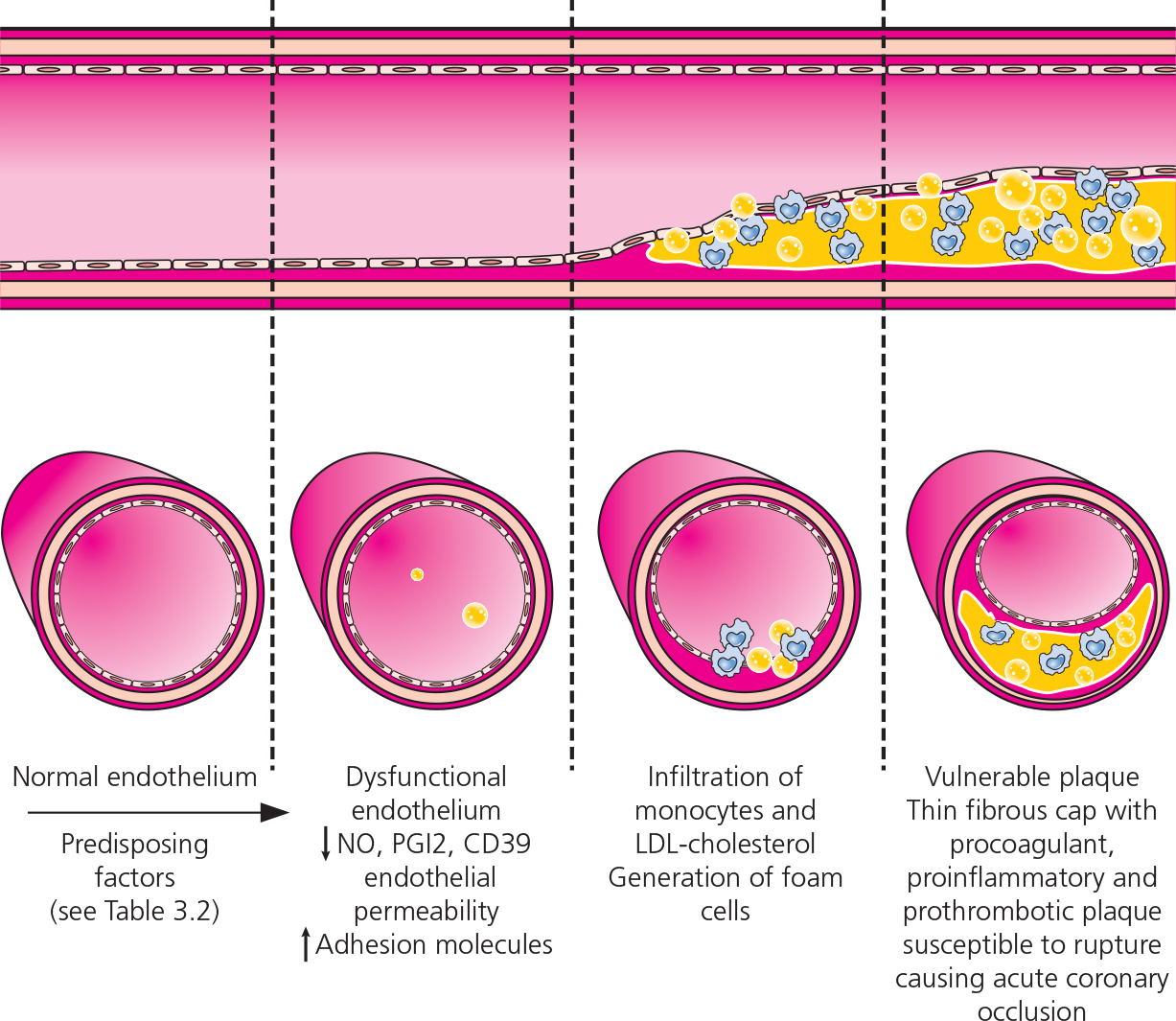
Figure 3.1 Coronary artery disease: development of an atherosclerotic lesion (plaque) in a coronary artery wall. CD39, cluster of differentiation 39; LDL, low-density lipoprotein; NO, nitric oxide; PGI2, prostaglandin I2 (prostacyclin).
| 3 | Causes |
In the majority of patients with chronic heart failure (HF), clinical deterioration is caused by one factor or a combination of factors (Table 3.1). In de novo cases (acute HF), acute cardiac injury (either due to myocardial infarction [MI] or myocarditis) is the prevalent cause, although a sudden rise in afterload (uncontrolled blood pressure) or preload (intravenous fluid loading), extreme tachycardia or hypotension (as in acute anemia or sepsis) can result in cardiac compromise. Knowledge of the etiology is crucial in determining the optimal therapeutic strategy.
|
TABLE 3.1 Causes of heart failure |
|
Common • Ischemic heart disease • Arterial hypertension • Valvular heart disease • Primary cardiomyopathy • Diabetes mellitus |
Rare • Thyroid disease • Severe anemia • Cardiotoxicity • Peripartum cardiomyopathy • Stress-provoked cardiomyopathy |
Less common • Infection and inflammation (myocarditis) • Persistent arrhythmia (tachycardiomyopathy) • Congenital heart disease • Severe lung disease (cor pulmonale) • Substance abuse (alcohol) |
|
Ischemic heart disease is the commonest cause of left ventricular (LV) dysfunction and HF. It is characterized by a constant or intermittent decrease in coronary perfusion, caused by significant narrowing of the lumen of coronary arteries. This leads to a mismatch between the rate of oxygen delivery and the rate of its utilization by the cardiac muscle.
Coronary ischemia is usually due to coronary artery disease (CAD), in which an atherosclerotic lesion (plaque) develops within the wall of the coronary artery; when unstable, the plaque ruptures causing acute coronary occlusion and subsequent MI (Figure 3.1). Predisposing factors for the development of CAD are shown in Table 3.2.
Figure 3.1 Coronary artery disease: development of an atherosclerotic lesion (plaque) in a coronary artery wall. CD39, cluster of differentiation 39; LDL, low-density lipoprotein; NO, nitric oxide; PGI2, prostaglandin I2 (prostacyclin).
|
TABLE 3.2 Predisposing factors for developing coronary artery disease |
|
|
• Older age • Male sex • Family history • Smoking |
• Hyperlipidemia • Arterial hypertension • Diabetes mellitus • Obesity |
Ischemic and non-ischemic cardiomyopathy. Ischemic cardiomyopathy is the term widely used to encompass coronary ischemia and MI with resulting myocardial scarring. In some patients, however, the degree of LV dysfunction is out of proportion to the magnitude of CAD. Such patients require further testing (invasive or non-invasive), as those with non-ischemic cardiomyopathy (i.e. not related to CAD) often present with typical angina, while those with CAD may have silent (asymptomatic) ischemia.
Over the past 25 years, mortality rates from acute coronary syndromes (MI and unstable angina) have declined, mainly as a result of improved and timely therapy to open the occluded artery (e.g. fibrinolysis, angioplasty, stenting). However, at the same time the rate of new presentation with HF has increased, indicating that many survivors of MI are left with or subsequently develop significant LV systolic dysfunction; some reports have indicated that more than 60% of individuals over 65 will develop HF within 2 years of MI.
The treatment of patients with non-ischemic cardiomyopathy (caused by infection, drugs, alcohol, arrhythmia etc.) and coexistent obstructive CAD is challenging, because the natural history of these patients is not predictable. The prognosis in many cases depends on the extent of CAD (number of vessels involved) and the presence of inducible coronary ischemia and myocardial scarring.
Hypertension frequently develops between 35 and 60 years of age. In the developed world, arterial hypertension affects around 20% of the adult population. Although most have no identifiable cause (i.e. idiopathic hypertension), a family history of hypertension is common, so the proposed etiology includes interaction between polygenic mutations and environmental triggers. Secondary causes, which contribute to the development of hypertension in about 5% of cases, include primary adrenal disease (hyperaldosteronism, hypercortisolemia, pheochromocytoma), hyperthyroidism, renal causes (including renal artery stenosis and parenchymal disease), sleep-disordered breathing, drugs, alcohol and others.
Hypertension and heart failure. Elevated arterial blood pressure (BP) is a common cause of LV hypertrophy (LVH) and may lead to LV diastolic and systolic dysfunction and overt HF. The development of LVH is associated with adverse outcomes and worse prognosis. The risk of developing HF increases with the degree of BP elevation; for example, a person with a BP of 160/100 mmHg or greater is twice as likely to develop HF as a person with a BP below 140/90 mmHg. Moderate elevations contribute to risk in the long term.
Pathological changes found in patients with LVH include an increase in the size of cardiac myocytes, progressive fibrosis, vascular changes of small arterioles with medial hypertrophy, and perivascular fibrosis.
The magnitude of LVH in response to a trigger such as hypertension depends not only on a direct response to the shear stress of raised pressure, but also the effect of changes in the levels of neurohormones, growth factors and cytokines. The reason why some hypertensive individuals progress directly to symptomatic LV systolic dysfunction without evidence of a significant cardiac event (MI) or diastolic HF is unknown.
Optimal BP control is an essential element of therapy in hypertensive heart disease. Evidence is emerging that tighter BP control (< 130/80 mmHg) is associated with reduction of LVH and cardiovascular events. Nonetheless, the role of tighter BP control remains controversial in the population as a whole and in certain subgroups, in particular diabetics and the elderly. Secondary causes of hypertension (including sleep-disordered breathing and renal artery stenosis) need to be ruled out in individuals with resistant hypertension and recurrent presentation with acute HF and elevated BP.
Aortic valve stenosis. The most common cause of aortic valve stenosis is age-related valve degeneration, a result of ongoing inflammation with lipid deposition and progressive calcification. Congenital bicuspid aortic valve stenosis may be found in middle-aged populations. Rheumatic fever remains an important cause of aortic valve stenosis in areas of lower socioeconomic status.
Progressive aortic stenosis leads to exertional symptoms, such as dyspnea, chest pain and syncope, and the development of congestive HF. The emergence of symptoms heralds a worse prognosis (50% survival for 2–3 years) and dictates the need for aortic valve replacement. Historically, poor surgical candidates were managed medically or palliated with balloon valvuloplasty. However, the emergence of transcatheter aortic valve implantation (TAVI) with acceptable initial results provides an alternative for patients for whom corrective surgery is not appropriate.
Aortic valve stenosis and left ventricular dysfunction. Patients with severe aortic valve stenosis and LV systolic dysfunction (ejection fraction < 40%) are a challenging group to manage. They often present with a relatively low pressure gradient on echocardiography but with severe symptoms of HF. Importantly, the cause of HF (i.e. primary valvular disease versus primary myopathic process) has to be determined first, as the therapeutic options are different (surgery versus medical treatment). Patients without appropriate contractile reserve (on stress echocardiography) generally have a poor prognosis with either medical or surgical management, but may still benefit from aortic valve replacement.
Aortic valve regurgitation can be acute or chronic. Acute regurgitation, which presents with acute dyspnea and HF, is a cardiac emergency. Causes include perforation of aortic cusps in the course of bacterial endocarditis, proximal extension of a dissecting aortic aneurysm, trauma or dehiscence of an aortic prosthesis. Chronic regurgitation results from congenital, infective (syphilis), rheumatic or degenerative causes. Symptoms include progressive dyspnea, angina and symptoms of HF. Symptomatic patients have a much higher mortality rate than asymptomatic patients, and the severity of preoperative symptoms is a strong determinant of survival after valve replacement.
Mitral valve regurgitation is one of the commonest valvular diseases leading to HF. It poses a major diagnostic and therapeutic dilemma, as the distinction between organic (primary) and functional (secondary) causes, and thus the need for surgery, is not always obvious (Table 3.3).
|
TABLE 3.3 Causes of mitral valve regurgitation |
|
Primary (organic) • Rheumatic heart disease • Valve prolapse • Valvular degenerative disease • Chordal rupture due to endocarditis, trauma or myocardial infarction |
Secondary (functional) • Apical displacement of the papillary muscles (due to global LV remodeling) • Dilation of mitral annulus (in dilated cardiomyopathy) • Inferior LV wall remodeling (after infarction with displacement of the posterior papillary muscle) |
LV, left ventricular. |
|
Acute regurgitation – rupture of the papillary muscle or chordae tendineae due to endocarditis, cardiac ischemia or trauma – is a cardiac emergency. It manifests with severe dyspnea at rest, chest pain and symptoms of pulmonary edema. Chronic regurgitation leads to progressive breathlessness, initially on effort, with the development of symptoms of HF. Atrial arrhythmias are common. Symptomatic patients with severe primary mitral regurgitation are treated with surgery (valve repair or replacement).
Secondary mitral valve regurgitation (in patients with cardiomyopathy and CAD) is primarily a disease of the LV myocardium. Therapies that promote the reversal of LV remodeling, such as myocardial revascularization, beta-blocker therapy and cardiac resynchronization, should be instituted first. New percutaneous techniques for poor surgical candidates have emerged but require long-term data. Surgery should be considered in selected cases.
Mitral valve stenosis. The prevalence of mitral valve stenosis has declined in developed countries as a result of early recognition and treatment of rheumatic fever (its main cause). However, the resurgence of the disease has been observed in areas of lower socioeconomic status. Mitral valve stenosis causes progressive dyspnea and palpitations. Untreated, it leads to severe pulmonary hypertension and may result in right-sided HF characterized by peripheral edema and abdominal distension. Percutaneous (valvuloplasty) or surgical (valve replacement) treatment is required.
Tricuspid valve regurgitation often accompanies advanced left-sided heart or pulmonary disease, due to annular dilation and non-closure of the valve leaflets. Rare causes of structural tricuspid valve regurgitation include endocarditis, rheumatic heart disease and trauma. Isolated severe tricuspid valve regurgitation may lead to right ventricular dysfunction and symptoms of right-sided HF. Symptoms of left-sided heart or pulmonary disease are prominent in most secondary cases.
Pulmonary valve stenosis is usually congenital, either isolated or part of a more complex congenital heart disease (e.g. tetralogy of Fallot). Symptoms include dyspnea and progressive right-sided HF.
Pulmonary valve regurgitation often occurs after surgical repair of a narrowed right ventricular outflow tract (with or without native valve excision) in patients with congenital heart disease (e.g. tetralogy of Fallot). Isolated significant pulmonary valve regurgitation is rare and caused by either endocarditis or hepatic carcinoid. Symptoms include right-sided HF and arrhythmia.
Patient prosthesis mismatch. All prosthetic valves are, to some degree, stenotic. Underestimating the size of the prosthesis at the time of aortic valve replacement may lead to persistence of LVH and retard negative LV remodeling. Patient prosthesis mismatch during aortic valve replacement contributes to increased cardiac mortality rates, especially in patients with depressed LV function.
Cardiomyopathies are cardiac diseases in which the myocardium is a primary target of the pathological processes, characterized by abnormal chamber size and wall thickness or functional contractile dysfunctions. In ‘primary cardiomyopathies’ the cause is myocardial specific, whereas ‘secondary cardiomyopathies’ have a pre-existing cardiac (valvular disease, coronary ischemia, congenital heart disease or arrhythmia) or systemic (arterial hypertension, infective, hormonal or metabolic) cause.
Several classifications of cardiomyopathies have been proposed. In 2006, the American Heart Association introduced a classification based on the etiology (genetic, acquired or mixed). In 2008, the European Society of Cardiology published its revised definition based on phenotype (dilated, hypertrophic, restrictive, arrhythmogenic right ventricular dysplasia and unclassified; Table 3.4). In 2013, the World Heart Federation proposed the MOGE(S) nosology system, which integrates both phenotype description and genetic information:
• M – morphology
• O – organs involved
• G – genetic inheritance
• E – etiology
• S – stage plus NYHA functional status.
|
TABLE 3.4 Classification of primary cardiomyopathies |
||
Genetic |
Mixed |
Acquired |
• Hypertrophic • Arrhythmogenic RV cardiomyopathy/dysplasia • Isolated LV non-compaction • Glycogen storage diseases – Danon disease – Pompe disease • Conduction defects • Mitochondrial myopathies • Ion channel disorders – Long QT syndrome – Brugada syndrome – Short QT syndrome – CPVT – Asian SUNDS |
• Dilated • Restrictive (non-hypertrophied and non-dilated) |
• Inflammatory (myocarditis) • Stress-provoked (tako-tsubo) • Peripartum • Tachycardia-induced • Infants of insulin-dependent diabetic mothers |
CPVT, catecholaminergic polymorphic ventricular tachycardia; LV, left ventricular; RV, right ventricular; SUNDS, sudden unexpected nocturnal death syndrome. Adapted from Maron BJ et al. 2006. American Heart Association Scientific Statement. |
||
The MOGE(S) system is supported by an online app at http://moges.biomeris.com/moges.html.
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant genetic disorder of the heart muscle, in which a multitude of mutated genes encode compromised proteins of the cardiac sarcomere. The reported prevalence in the general population is about 0.2% (1 in 500), although true prevalence may be higher because it is often asymptomatic initially.
At least 11 abnormal sarcomeric genes are implicated, which may partly explain the wide clinical presentation of the disease. Double or triple gene mutations in affected individuals have been identified and are associated with a malignant phenotype. Genetic modifiers could influence the effects of the primary sarcomere mutation leading to either earlier or delayed onset of clinical disease. The penetrance of a sarcomere mutation increases with age; diagnosis tends to be earlier in males. A sedentary lifestyle, and comorbidities such as hypertension, obesity, diabetes and obstructive sleep apnea may have cumulative effects on disease penetrance and severity in older individuals. Patients may present with arrhythmia (including sudden cardiac death), syncope or HF.
The disorder is characterized by an abnormal thickening of the left ventricle (LV septum thickness > 1.5 cm; Figure 3.2), usually in the absence of other conditions that increase ventricular pressure loading (e.g. systemic hypertension, aortic valve disease, the athlete’s heart).
Figure 3.2 Hypertrophic cardiomyopathy: echocardiogram showing abnormal thickening of the septum (arrow). LV, left ventricle.
The four morphological variants described on echocardiography are diffuse, subaortic, midventricular and apical type. Although common (∼25% of cases at rest), LV outflow obstruction with resulting mitral valve dysfunction (systolic anterior motion of anterior mitral valve leaflet resulting in varying degrees of mitral regurgitation) is not a consistent feature and the term hypertrophic cardiomyopathy is now generally accepted as the name for this condition. Cardiac MRI is the best means of assessing the myocardial abnormality (fibrosis).
In pregnancy, there is no convincing evidence that HCM increases risk, but all patients should be offered obstetric and cardiologic care at specialized centers experienced in treating such conditions. Normal delivery is possible, but patients with severe outflow obstruction require special care and possibly Cesarean section.
All patients with HCM require cardiologic referral, and syncope requires urgent investigation. Family members should be screened.
HCM phenocopy. Several other genetic disorders include cardiac hypertrophy as a component. Although cardiac hypertrophy in these conditions may appear similar to HCM on echocardiography the differences are apparent on tissue histology as well as in the pathogenesis of the disease.
Examples of such disorders include:
• hereditary transthyretin-related amyloidosis (ATTR)
• Fabry disease
• glycogen storage disease (PRKAG2, LAMP2, Pompe)
• respiratory chain enzyme defects (mitochondrial cardiomyopathies).
Arrhythmogenic right ventricular cardiomyopathy (ARVC)/dysplasia is an uncommon genetic condition characterized by fibrofatty infiltration of the right ventricle. Both autosomal dominant and recessive inheritance have been described and a number of genes that encode desmosomal proteins have been implicated. It is an important cause of sudden cardiac death, particularly in the young, and is usually due to a ventricular arrhythmia, or patients may manifest signs of right-sided HF and eventually biventricular failure.
The diagnosis of ARVC requires the presence of two out of four major criteria:
• RV dysfunction, dilation or aneurysm formation
• conduction abnormalities (broad QRS in V1–V3, epsilon waves)
• tissue diagnosis
• family history.
Isolated left ventricular non-compaction is an idiopathic form of cardiomyopathy due to intrauterine arrest of myocardial compaction. It was originally described in infants but more recently reported in adults. Both sporadic and familial forms are recognized. There is a spectrum of clinical presentation, and recent clinical reports have suggested that the disorder is associated with the important complications of thromboembolism, congestive HF, ventricular arrhythmias and sudden cardiac death.
Glycogen storage diseases are rare genetic causes of cardiomyopathy. They include Danon disease (also known as lysosomal glycogen-storage disease with normal acid maltase) and Pompe disease (an autosomal recessive disorder characterized by the deficiency of acid alpha-glucosidase, a lysosomal hydrolase).
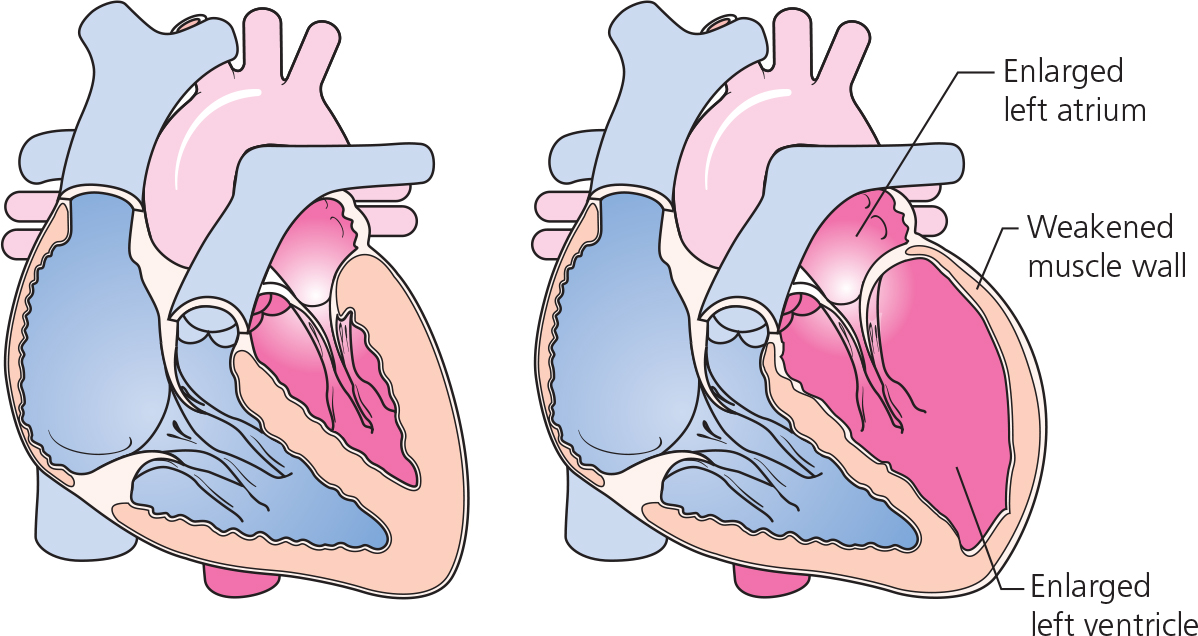
Dilated cardiomyopathy is characterized by an increase in LV end-diastolic diameter (> 2.7 cm/m2) (Figure 3.3) and reduced LV systolic function (ejection fraction < 45%). Most cases of dilated cardiomyopathy are idiopathic, but 35–50% of cases have a family history. In addition, about 10% of asymptomatic relatives have evidence of unrecognized LV dysfunction.
Mutations have been identified in 33 genes (31 autosomal, two X-chromosome linked). Autosomal dominant inheritance is most common with incomplete penetrance and variable expression. These are low frequency mutations that are found only in about one-third of cases. Some cases ‘cross over’ with ARVC, HCM and long QT syndrome (LQTS), or have multiple mutations (oligogenic inheritance).
Familial dilated cardiomyopathy (FDC) can be further divided into two subtypes – isolated FDC and FDC with cardiac conduction defects (FDC-CD). Titin (TTN) truncating mutations are most frequent (∼25%) in isolated FDC. FDC-CD typically presents in the second decade of life, with mild cardiac conduction defects that often progress to complete heart block. Mutations in Lamin A/C, desmin and SCN5A genes have been identified.
The disease comprises a preclinical/early phase with three stages (no cardiac expression, isolated ventricular dilation or arrhythmic cardiomyopathy) and a clinical phase with two phenotypes (hypokinetic non-dilated cardiomyopathy and dilated cardiomyopathy).
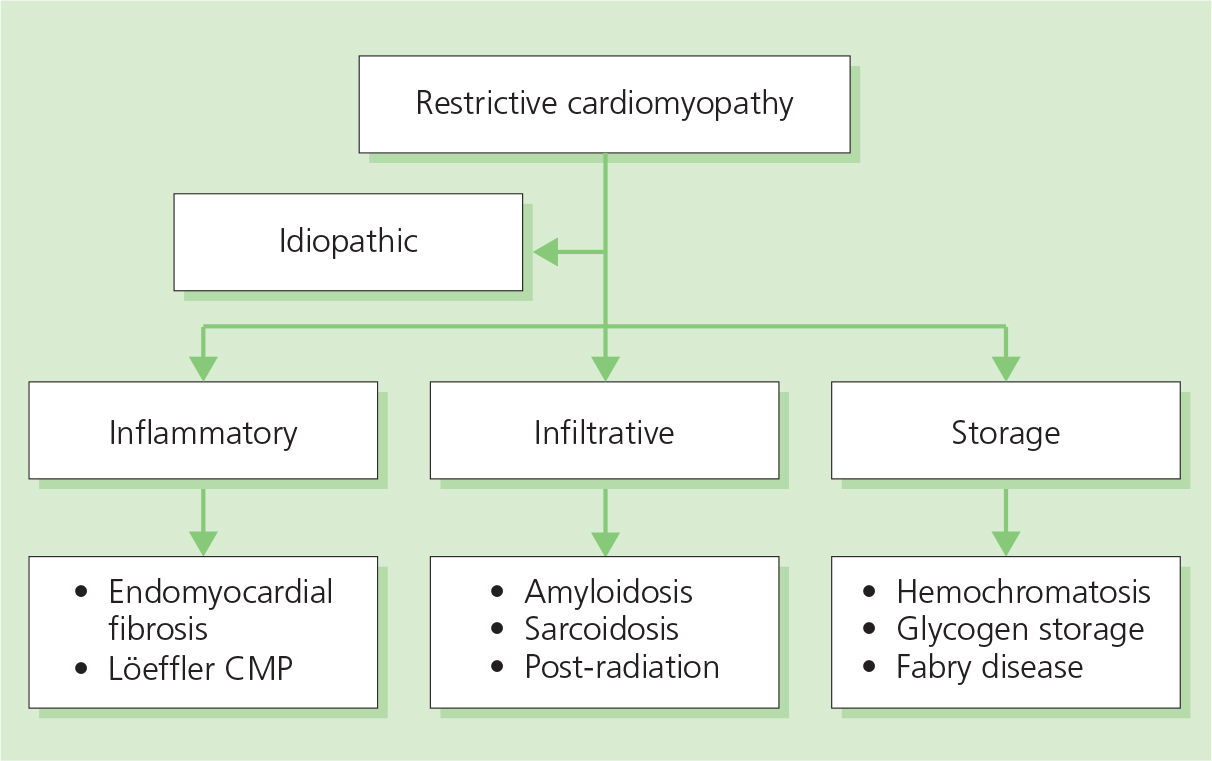
Restrictive cardiomyopathies are heterogeneous myocardial diseases with a common clinical picture of progressive symptoms of HF, including dyspnea, fatigue and exercise intolerance and varying degrees of peripheral edema, due to abnormal (restrictive) diastolic filling which results in a marked increase in ventricular end-diastolic pressures. Systolic function is usually preserved at least until the very advanced stage of the disease. A normal LV thickness differentiates this group from hypertrophic cardiomyopathies (see Figure 3.2); however, advanced stages of some of the diseases (e.g. cardiac amyloid) may present with marked LVH.
Restrictive cardiomyopathies are caused by inflammatory, infiltrative or storage diseases (Figure 3.4), or are idiopathic. Some forms are triggered by mutations in the genes responsible for synthesis of the sarcomeric proteins, similar to those already identified in HCM (see page 30).
The prognosis is poor, with death or heart transplantation usually within the first few years after diagnosis. The treatment to control HF is particularly difficult because of diuretic-resistant edema, medication intolerance and drug toxicity leading to hypotension and arrhythmias. Underlying conduction disease also limits treatment choices and requires pacing in some cases.
Cardiac amyloidosis is caused by the extracellular deposition of misfolded proteins leading to organ dysfunction. Secondary amyloidosis due to a chronic inflammatory condition is seldom associated with cardiac involvement. Cardiac amyloidosis is characterized by a primary myocardial infiltration and the vast majority of clinically significant cases are caused by either amyloid light-chain (AL)- or transthyretin-related (ATTR) disease.
AL amyloidosis typically presents with progressive deposition of abnormal protein (monoclonal immunoglobulin light chain) in multiple tissues and organs including the heart, kidneys, nervous system, skin and liver. Less than 5% have disease limited to the heart, while 20% of cases are caused by hematologic disorders (lymphoma, myeloma or macroglobulinemia).
ATTR amyloidosis is caused by deposition of wild-type (formerly senile systemic amyloidosis) or mutant transthyretin protein (inherited) amyloid fibrils. ATTR wild-type disease is a slow progressive cardiomyopathy found mainly in patients over 80 years of age. The disease occurs more often in males and may present with isolated cardiac infiltration and carpal tunnel syndrome. Hereditary (inherited) ATTR disease with prominent cardiac involvement is caused by a mutation in the transthyretin gene, and is frequently found in African-Americans.
Mayo Clinic staging allows assessment of the severity of cardiac involvement, prognosis and response to treatment in AL disease. This system is based on the levels of cardiac biomarkers (N-terminal B-type natriuretic peptide [NT-proBNP] > 332 ng/L; B-type natriuretic peptide [BNP] > 100 ng/L and cardiac troponin [TnI] > 0.035 μg/L). Patients are stratified into three groups:
• high risk (stage III) – all biomarkers are elevated
• intermediate risk (stage II) – at least one biomarker is elevated
• low risk (stage I) – normal biomarker levels.
Recently, the level of serum free light chains (sFLC) has also been added to the score.
Management of cardiac amyloidosis includes supportive therapy for HF and arrhythmia, and treatment of underlying amyloid disease. Specific management of AL amyloidosis includes treatment of the hematologic disorder with a melphalan-based regimen. New bortezomib-based chemotherapy has been shown to improve response rates in patients with significant cardiac involvement (see Fast Facts: Multiple Myeloma and Plasma Cell Dyscrasias).
In isolated cases, cardiac transplantation may be considered, although this is rarely an option because of the involvement of other organs and deposition of amyloid in the donor heart. Combined heart and stem cell transplantation in young adults with preserved kidney, liver and autonomic function has been successfully performed.
Fabry disease is an X-linked recessive lysosomal storage disorder caused by the absence of alpha-galactosidase A activity and the resultant accumulation of globotriaosylceramide and related glycosphingolipids. Severe forms of the disease (the classic phenotype) have no or just detectable residual enzyme activity, while variant forms have low enzyme levels. Typically, males are affected, but female carriers may have varying forms of cardiac and cerebrovascular disease due to random X-chromosome inactivation.
The disease is progressive in nature and the classic form has its onset in childhood. The symptoms are initially subtle or non-specific. By adulthood, specific organ involvement is fully established. Affected individuals present with renal failure requiring dialysis, cerebrovascular disease with transient ischemic attacks, strokes and complications of vascular disease, cutaneous angiokeratomas and characteristic corneal opacities. Cardiac involvement includes LVH, valvular disease with predominant mitral insufficiency, ascending aortic aneurysm formation, CAD and conduction disease. Patients may present with symptoms of HF, arrhythmia or MI. Specific enzyme-replacement therapy (ERT) should be started early, ideally before end-organ damage. Treatment otherwise remains supportive.
Cardiac sarcoid. Sarcoidosis is a systemic disease with chronic inflammation and non-caseating granulomas in affected organs. Women are at slightly higher risk, with rates peaking at 20 to 29 years of age. Extracardiac manifestations involve the skin (erythema nodosum), eyes (uveitis), lungs (interstitial infiltrates and adenopathy), kidneys (nephrocalcinosis, progressive renal failure) and parotid gland (swelling) and cause hypercalcemia. Cardiac involvement has been reported in about 5% of patients with systemic sarcoidosis, in about 20% of autopsies (subclinical cardiac sarcoid) and in up to 80% of Japanese patients.
Conduction disease is the commonest cardiac presentation; complete heart block is not uncommon. Ventricular arrhythmia is seen in up to a quarter of patients, atrial tachyarrhythmia less frequently. Cardiomyopathy and HF may develop in undiagnosed cases. It is initially characterized by normal LV volumes with wall motion abnormalities, focal myocardial thinning and aneurysm formation, usually at the base of the septum. If untreated, sarcoidosis leads to progressive LV systolic dysfunction. The initial presentation could also mimic MI.
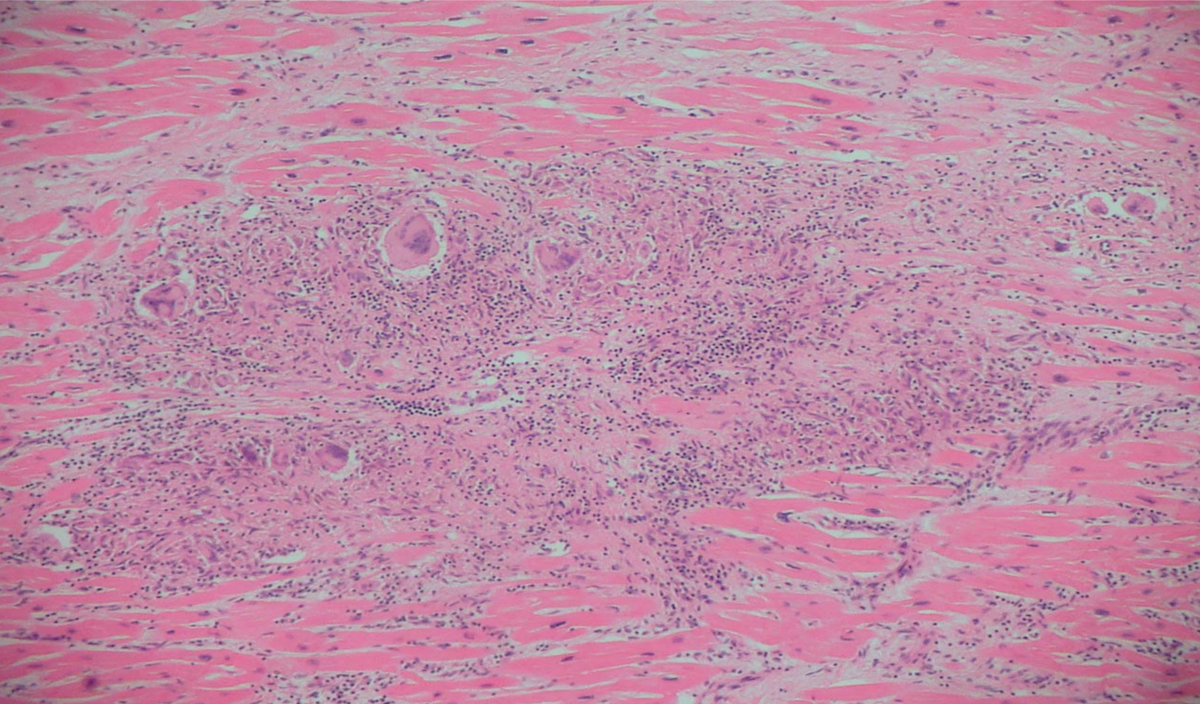
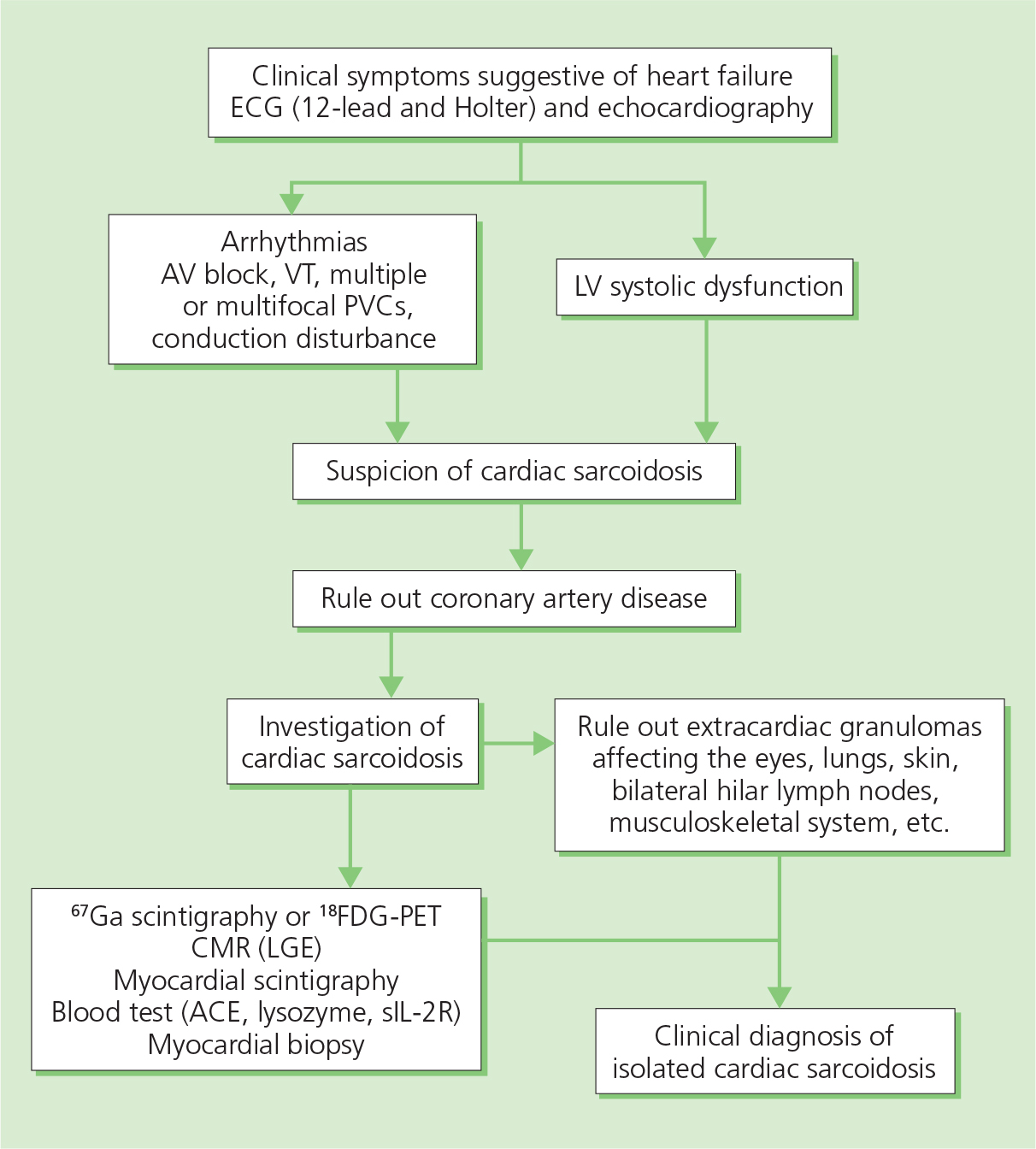
Diagnosis is based either on the presence of non-caseating granuloma in a myocardial biopsy (Figure 3.5) or histological confirmation of extracardiac sarcoidosis (e.g in the lungs or lymph nodes) with evidence of cardiac involvement (cardiomyopathy, heart block, ventricular arrhythmias, or positive cardiac FDG-PET, MRI or gallium scan) (Figure 3.6). The serum level of angiotensin-converting enzyme (ACE) is not specific and results may be affected by inhibitors.
Figure 3.6 Diagnostic work-up in patients with sarcoidosis and suspected cardiac involvement. ACE, angiotensin-converting enzyme; CMR (LGE), cardiac magnetic resonance with late gadolinium enhancement; FDG-PET, fluorodeoxyglucose positron emission tomography; 67Ga, gallium radiotracer; LV, left ventricular; PVC, premature ventricular contraction; sIL-2R, soluble interleukin-2 receptor; VT, ventricular tachycardia.
The introduction of new imaging modalities has helped to assess the activity of the disease (FDG-PET), the extent of scarring (MRI), the response to therapy and prognosis of the disease (FDG-PET, MRI).
Treatment of cardiac sarcoidosis includes disease-specific therapy with oral steroids (and immunosuppressive drugs) and supportive therapy, with HF medications, pacing and an implanted defibrillator in cases of advanced conduction disease, life-threatening arrhythmia and cardiomyopathy with reduced systolic function.
Hemochromatosis is an autosomal recessive disorder in which the HFE gene mutation C282Y (chromosome 6) reduces the affinity of transferrin for its receptor, resulting in the accumulation of iron in multiple organs including the liver, heart, pancreas, pituitary, joints and skin. C282Y homozygotes account for most clinical diagnoses of hereditary hemochromatosis among people of northern European descent.
Iron deposition may lead to restricted or dilated cardiomyopathies. Conduction disease including sinus node disease may coexist. In up to 15% of cases cardiac involvement may precede the involvement of other organs. The therapy of the condition includes iron removal (phlebotomy) as soon as the diagnosis is made. Established cardiac disease is rarely responsive to therapy.
Endomyocardial fibrosis is an idiopathic disorder characterized by the development of restrictive cardiomyopathy. It is sometimes considered part of a spectrum of a single disease process that includes Löeffler endocarditis (non-tropical eosinophilic endomyocardial fibrosis or fibroblastic parietal endocarditis with eosinophilia). It can cause progressive HF, thromboembolism, myocardial ischemia, arrhythmias and, rarely, pericarditis. Prognosis is poor. Therapy includes management of the underlying cause, the treatment of HF, arrhythmia and thromboembolism, and heart transplantation in selected cases.
Inflammatory cardiomyopathy is characterized by a generalized inflammation of the heart muscle in response to a specific trigger. Viral infection is the commonest cause of the immune and autoimmune mechanisms that contribute to the myocardial injury.
Myocarditis is considered the most common pathological form of dilated cardiomyopathy. Typical manifestation of this disease includes HF, chest pain or arrhythmias. The non-invasive imaging used in the diagnosis includes echocardiography, cardiac MRI and FDG-PET. The endomyocardial biopsy, although diagnostic, is usually performed only in acute cases with severe cardiac dysfunction or recurrent ventricular arrhythmia where suspicion of an immunosuppression responsive process (giant cell [Figure 3.7] or eosinophilic necrotizing myocarditis) is high.
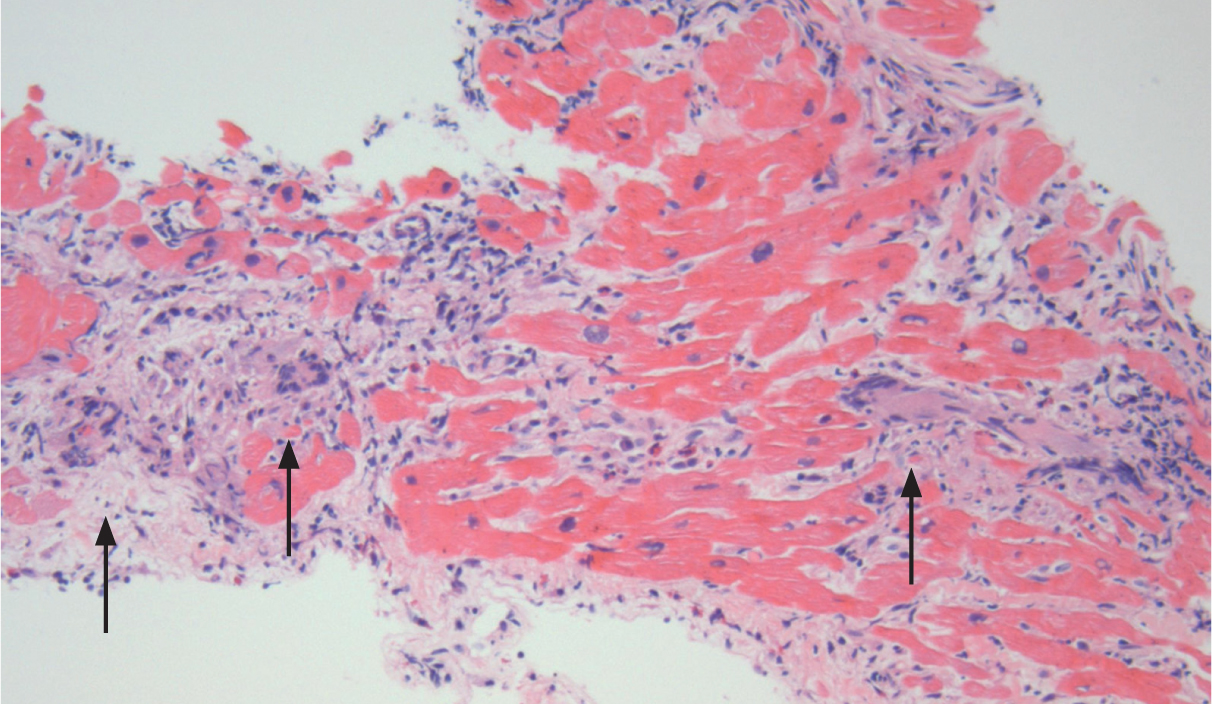
Figure 3.7 Giant cell myocarditis, i.e. myocytolysis with formation of giant cells (arrows). Biopsy in such cases is diagnostic.
In 50–60% of treated cases, LV function will improve (half will recover), hence the final assessment of the response to therapy (including consideration of a prophylactic automatic implantable cardioverter-defibrillator) can be safely delayed for 3–6 months.
Treatment of inflammatory cardiomyopathy includes standard HF medications and immunosuppressive therapy in selected cases. If severe dysfunction of the left ventricle ensues, devices or even cardiac replacement therapy may be considered.
Chronic viral cardiomyopathy has been identified as a separate entity but the role of antiviral therapy in this condition has not yet been proved.
Clozapine is a rare, potentially reversible, cause of myocarditis in patients with schizophrenia. It affects about 1% of cases, mainly within the first 2 months of therapy. Detection of LV dysfunction on routine echocardiography is important, and the use of cardiac troponins and inflammatory markers such as C-reactive protein help in early diagnosis.
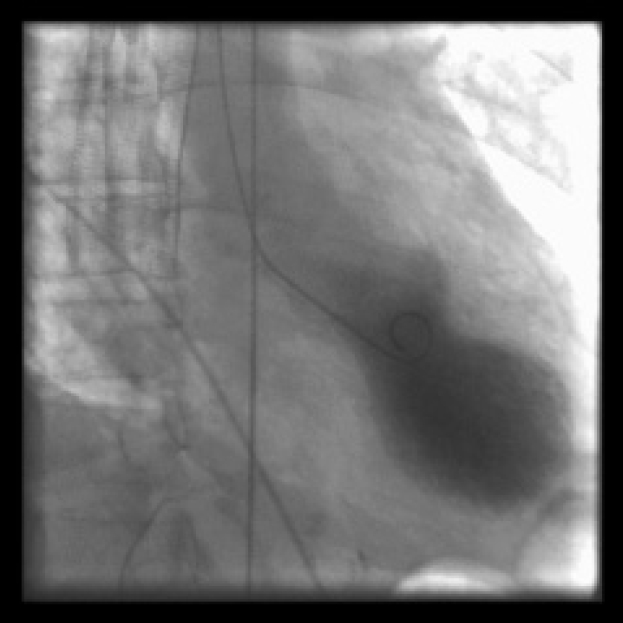
Stress-provoked (tako-tsubo) cardiomyopathy is an increasingly recognized cause of reversible LV cardiomyopathy, which occurs in around 2% of acute coronary syndromes. It occurs more often in women, usually after menopause, typically in circumstances of acute and severe emotional stress, hence it has been referred to as the ‘broken heart syndrome’. Tako-tsubo cardiomyopathy is characterized by typical ischemic chest pain, ST segment elevation and an increase in cardiac enzymes, mimicking a full-thickness MI. Often, the coronary arteries show little or no disease. The left ventricle shows a characteristic abnormality on echocardiography and sometimes on angiography, that of apical hypo- or akinesis (otherwise it looks like a true infarct) and significant dilation (ballooning) (Figure 3.8). Given the way it presents, treatment is usually the same as that for an infarct: beta-blockers, ACE inhibitors, statins and an antiplatelet agent. Although it can present dramatically with cardiogenic shock or HF, with supportive treatment in-hospital mortality is usually low. The overall long-term prognosis is excellent.
Figure 3.8 Left ventriculography during systole in a patient with tako-tsubo cardiomyopathy, showing ballooning of the apex, usually associated with apical hypo- or akinesis.
Peripartum cardiomyopathy is rare in developed countries and in white populations, but is ten times more common in African and African-American women (e.g. affecting 1 in 300 live births in Haiti). The diagnostic criteria of peripartum cardiomyopathy include:
• HF presenting in the last month of pregnancy and 5 months after delivery
• lack of other causes of cardiomyopathy
• no pre-existing heart disease before pregnancy.
Risk factors are shown in Table 3.5.
|
TABLE 3.5 Risk factors for peripartum cardiomyopathy |
|
|
• Multiparity • Advanced age (> 30 years) • Twin pregnancies |
• Pre-eclampsia • Gestational hypertension • African origin |
Peripartum cardiomyopathy has an uncertain etiology associated with pregnancy. It is potentially fatal if not recognized and treated early. Recent work has focused on the beneficial effects of prolactin inhibition with bromocriptine. In general, however, the prognosis is good and relapses are uncommon.
Tachycardia-induced cardiomyopathy. A long-standing, persistently raised ventricular rate due to a supraventricular rhythm is a well-recognized cause of non-ischemic cardiomyopathy and HF. Its recognition is important, as treatment often reverses HF and LV damage.
Patients may present with exertional dyspnea, orthopnea and peripheral edema, but initially it is often difficult to determine whether the arrhythmia caused the HF or vice versa. The diagnosis can usually be inferred if LV function improves after successful treatment of the arrhythmia.
The most common tachycardias are atrial fibrillation and flutter; less commonly, atrial tachycardia and re-entrant atrial arrhythmias. Rarely, persistent ventricular tachycardia may present as HF. Paroxysmal tachycardias are rarely implicated as a cause of non-ischemic cardiomyopathy.
The common link is a persistently elevated ventricular rate, often more than 100 beats per minute, sometimes over a long period of time, even years. However, the heart rate itself is not a good predictor of HF. Although there is experimental evidence showing that the faster the heart rate the more likely LV impairment becomes, it is usually the slower but incessant arrhythmias that are associated with non-ischemic cardiomyopathy in humans.
Tachycardia-induced cardiomyopathy can occur at any age but it is rare in childhood. Atrial fibrillation and flutter are common in older age groups. The aim of treatment is to control the ventricular rate (either through rhythm or rate control), which should alleviate symptoms of HF, improve LV function and prevent future relapses (see Fast Facts: Cardiac Arrhythmias).
Athlete’s heart. Long-term physical training, particularly in competitive athletes, can lead to a physiological increase in LV mass. To differentiate between athlete’s heart and HCM (see pages 31–3), training should be stopped for 4–6 weeks to demonstrate regression of the LV thickening. If HCM is diagnosed, and particularly if there is an outflow gradient, the patient should be advised to stop taking part in extreme or competitive sports. Otherwise, a return to sporting activities can be allowed.
HF has been called the ‘forgotten complication’ of diabetes mellitus. Indeed, this association appears to be bidirectional, as diabetes is associated with HF, and HF with impaired glucose tolerance and diabetes. Moreover, the development of HF is associated with worse outcome in people with diabetes than in non-diabetic subjects, particularly in those with ischemic heart disease.
This observation has led to the hypothesis that the diabetic heart is in some way compromised to the extent that it is less able to sustain injury from an exogenous insult such as MI. Indeed, sophisticated imaging techniques have demonstrated a reduction of diastolic and systolic function in apparently healthy subjects with type 2 diabetes mellitus. This is a frequent observation, with various manifestations reported in 20% to more than 50% of apparently healthy diabetic subjects. These findings are also present in patients who have similar metabolic diseases, including impaired glucose tolerance and obesity; a unifying feature may be insulin resistance. This subclinical dysfunction is multifactorial, with contributions from disturbed metabolism, fibrosis, microvascular disease (both structural and functional) and diabetic autonomic neuropathy. It is further magnified by the presence of hypertension, and is most common in individuals with poor glycemic control.
As these findings have been detected using highly sensitive new techniques, there has been some concern that they represent an echo-cardiographic curiosity rather than a disease. However, follow-up studies have shown an association with the subsequent development of HF.
Given the increasing frequency of both obesity and type 2 diabetes mellitus, the subclinical cardiomyopathy associated with these conditions may prove to be extremely important. At present, no clear treatment has been described, although there is evidence that dysfunction is reduced with improved glycemic control, weight reduction and exercise training (see Fast Facts: Diabetes Mellitus). The extent to which fibrosis may contribute to uncomplicated diabetes mellitus remains controversial.
Cardiotoxicity is one of the most serious side effects of cancer therapy (chemotherapy, and mediastinal and neck radiation). It may manifest as cardiomyopathy, pericarditis, congestive HF, valvular heart disease or premature CAD.
Causes. Anthracycline chemotherapy (e.g. doxorubicin) is by far the most common cause of type 1 myocardial damage (Table 3.6). Acute toxicity is unlikely with current regimens. Chronic toxicity (development of cardiomyopathy within 1 year of therapy) and late onset (development of cardiomyopathy from years to decades after therapy) are all dose dependent. Other agents that have been associated with cardiotoxicity include cyclophosphamide, ifosfamide, cisplatin, carmustine, busulfan, mechlorethamine and mitomycin.
|
TABLE 3.6 Chemotherapy-related cardiac dysfunction |
||
Type 1 (myocardial damage) |
Type 2 (myocardial dysfunction) |
|
Cardiotoxic agent |
Doxorubicin |
Trastuzumab |
Clinical course, response to CRCD therapy |
May stabilize, but underlying damage is permanent and irreversible; recurrence in months or years may be related to sequential cardiac stress |
High likelihood of recovery (to or near baseline cardiac status) in 2–4 months (reversible) |
Dose effects |
Cumulative, dose related |
Not dose related |
Mechanism |
Free radical formation, oxidative stress/damage |
Blocked ErbB2 signaling |
Ultrastructure |
Vacuoles: myofibrillar disarray and dropout; necrosis (changes resolve over time) |
No apparent ultrastructural abnormalities |
Non-invasive cardiac testing |
Decreased EF by ultrasound or nuclear determination; global decrease in wall motion |
Decreased EF by ultrasound or nuclear determination; global decrease in wall motion |
Effect of rechallenge |
High probability of recurrent and progressive dysfunction, may result in intractable heart failure and death |
Increasing evidence for the relative safety of rechallenge; additional data needed |
Effect of late sequential stress |
High likelihood of sequential stress-related cardiac dysfunction |
Low likelihood of sequential stress-related cardiac dysfunction |
CRCD, chemotherapy-related cardiac dysfunction; EF, ejection fraction. |
||
Treatment with tyrosine kinase inhibitors (trastuzumab) could lead to the development of type 2 myocardial damage. The targeted proteins are regulatory enzymes responsible for cell division, growth and apoptosis, so it is not surprising that cardiac toxicities have been encountered. Cardiac damage is not dose dependent and discontinuation of the drug reverses it. The current use of multiple chemotherapeutic regimens in the same patient often leads to a combination of type 1 and 2 injury, with a somewhat uncertain prognosis.
Risk factors for cardiac toxicity are shown in Table 3.7.
|
TABLE 3.7 Risk factors for cardiac toxicity |
|
• Cumulative dose (> 5% risk if anthracycline dose > 400 mg/m2) • Female sex • Age < 18 years, > 65 years • Renal disease • History of radiotherapy or use of other cytotoxic agents • Cardiac disease with high wall stress • Hypertension • Genetic factors |
Cardiac surveillance of cancer therapy should include an annual history and physical examination. A baseline echocardiogram should be performed, including three- and two-dimensional evaluation of LVEF and global longitudinal strain (GLS) assessment of myocardial deformation. Subsequent frequency should be based on age at treatment, cumulative dose and concomitant chest irradiation (for an anthracycline-based regimen). Cardiac biomarkers (troponins and natriuretic peptides) and cardiac MRI can also be helpful (Table 3.8).
|
TABLE 3.8 Detection of cardiac toxicity (using imaging and biomarker levels) |
|
• Echocardiography – 3D LVEF, 2D LVEF and GLS assessment of myocardial deformation – decrease in LVEF > 10% (below EF of 50%) – decrease in strain (GLS) > 15% from baseline • Cardiac MRI – drop in LVEF and detection of cardiac fibrosis • Biomarkers – TnI, hsTNI – elevations identify patients who may benefit from ACE inhibitor treatment – BNP and NT-proBNP can be used in surveillance of high-risk patients |
ACE, angiotensin-converting enzyme; BNP, B-type natriuretic peptide; GLS, global longitudinal strain; hsTNI, high-sensitive troponin I; LVEF, left ventricular ejection fraction; MRI, magnetic resonance imaging; NT-proBNP, N-terminal pro-B-type natriuretic peptide; TNI, troponin I. |
Management. If LVEF decreases by more than 10% to a value below the lower limit of normal (an LVEF of 50%), angiotensin-converting enzyme (ACE) inhibitors (or angiotensin-receptor blockers [ARBs]) in combination with beta-blockers are recommended to prevent further LV dysfunction or the development of symptomatic HF, unless contraindicated. ACE inhibitors (or ARBs) and beta-blockers are also recommended in patients with symptomatic HF or asymptomatic cardiac dysfunction unless contraindicated.
There are a number of cardiac and non-cardiac factors not directly related to the biology of HF that may worsen the symptoms of HF and lead to acute hospitalization (Table 3.9). Identification, prevention and early treatment of these form the target for disease management programs.
|
TABLE 3.9 Common causes of worsened heart failure |
|
|
Cause and effect |
Action |
Dietary indiscretion |
|
|
• Excessive sodium and water intake are well-recognized precipitating factors for readmission |
• Restrict salt intake to < 2 g/day • Limit fluid intake to 1.5–2.0 L/day |
Excess fluid |
|
|
• Excessive administration of intravenous fluids may worsen HF in certain clinical conditions (e.g. sepsis, after surgery) |
• Careful monitoring of volume status |
Medication non-adherence |
|
|
• Loss of therapeutic effect |
• Address the reasons for non-adherence (e.g. patient fear of side effects, financial constraints or service access issues) |
|
• Ongoing cardiac remodeling |
• Step up HF therapy and optimize current therapy • Introduce new agents • Consider ancillary therapy (pacemakers) or mechanical cardiac support • Check for the presence of associated non-cardiac precipitants (e.g. infection, metabolic derangement, toxins, drugs) |
Atrial arrhythmias |
|
|
• Loss of atrial contraction contributes to decreased LV filling and cardiac output • Uncontrolled atrial rhythm with fast ventricular response further limits diastolic time and worsens hemodynamics • Sudden deterioration may be accompanied by symptoms of coronary ischemia and low BP |
• Treat the arrhythmia (see Fast Facts: Cardiac Arrhythmias) |
Myocardial ischemia/infarction |
|
|
• Acute coronary ischemia is a common precipitant of HF deterioration |
• Immediate medical therapy to stabilize the ruptured plaque (heparins, antiplatelet agents, statins) • Careful evaluation for percutaneous or surgical revascularization • Coronary angiography and intervention for patients with HF complicating acute coronary syndromes, and for patients with angina and non-ischemic cardiomyopathy |
|
• An important precipitant of HF • Particularly frequent in elderly patients with LVH and diastolic dysfunction |
• Antihypertensive therapy • Search for secondary causes, e.g. excessive salt or liquorice intake, drugs (NSAIDs) or renal artery stenosis |
Acute or progressive renal dysfunction |
|
|
• Indicates progression of cardiac dysfunction and low output • Leads to sodium and water retention and progressive volume overload • Coexisting factors may worsen HF (rise in BP, poor metabolism of drugs leading to cardiotoxicity or progressive anemia) |
• See Chapter 4, Cardiorenal syndrome |
Cardiac pacing |
|
|
• Patients with indications for permanent pacing (advanced AV block) may develop progressive HF symptoms • Pacemaker-dependent patients (with prolonged RV pacing) fare worst |
• Implantation of LV lead |
Pulmonary disease |
|
|
• Patients with COPD are more susceptible to pulmonary infection and worsening HF |
• See Chapter 4, Pulmonary disease |
Anemia |
|
|
• Impairs oxygen delivery to tissues and if abrupt (in cases of bleeding) may lead to organ hypoperfusion and sympathetic activation |
• See Chapter 4, Anemia |
Thyroid disease |
|
|
• Both hypo- and hyperthyroidism exacerbate HF |
• Evaluate thyroid function in all acute presentations |
|
• The following drugs can precipitate worsening HF: antiarrhythmics (flecainide, sotalol), non-dihydropiridine calcium-channel blockers, glitazones, NSAIDs, COX-2 inhibitors, TCAs, theophylline, beta-agonist bronchodilators, OTC drugs with pseudoephedrine, corticosteroids, chemotherapeutics (anthracycline, taxanes etc.) |
• Evaluate and monitor the patient’s cardiac and non-cardiac drug regimen |
AV, atrioventricular; BP, blood pressure; COPD, chronic obstructive pulmonary disease; COX-2 cyclooxygenase-2; HF, heart failure; LV, left ventricle; LVH, left ventricular hypertrophy; NSAID, non-steroidal anti-inflammatory drug; OTC, over the counter; RV, right ventricular; TCA, tricyclic antidepressant. |
|
Key points – causes
• Ischemic heart disease is the commonest cause of left ventricular dysfunction and heart failure (HF).
• Arterial hypertension is an important cause of new presentation but also a powerful precipitant of acute worsening of existing HF.
• Aortic valve disease is an important cause of HF. Surgery is the best therapy for suitable candidates; assessment of severity in the context of existing cardiac dysfunction can be challenging, but helps to define individuals who would benefit most from surgery.
• Surgery is always required for organic mitral valve regurgitation, but if the cause is functional then surgery may not help and may even be deleterious.
• All patients with a new diagnosis of cardiomyopathy require cardiologic referral.
• Cardiotoxicity is one of the most serious side effects of cancer therapy; cardiac function must be assessed at baseline and during therapy.
• Timely detection and treatment of cardiac and non-cardiac factors that may worsen HF, and in some cases preventative measures, constitute an inherent part of modern HF management.
Arbustini E, Narula N, Tavazzi L et al. The MOGE(S) classification of cardiomyopathy for clinicians. J Am Coll Cardiol 2014;64:304–18. Also see http://moges.biomeris.com/moges.html.
Chen MH, Kerkelä R, Force T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation 2008;118:84–95.
Drazner MH. The progression of hypertensive heart disease. Circulation 2011;123:327–34.
Felker GM, Shaw LK, O’Connor CM. A standardized definition of ischemic cardiomyopathy for use in clinical research. J Am Coll Cardiol 2002;39:210–18.
Gianni M, Dentali F, Grandi AM et al. Apical ballooning syndrome or takotsubo cardiomyopathy: a systematic review. Eur Heart J 2006;27:1523–9.
Gurbel PA, Tantry US, Huber K. Fast Facts: Acute Coronary Syndromes. Oxford: Health Press Limited, 2014.
Hahn VS, Lenihan DJ, Ky B. Cancer therapy-induced cardiotoxicity: basic mechanisms and potential cardioprotective therapies. J Am Heart Assoc 2014;3:e000665.
Heart Failure Society of America, Lindenfeld J, Albert NM et al. HFSA 2010 Comprehensive Heart Failure Practice Guideline. J Card Fail 2010;16:e1–194.
Houmsse M, Tyler J, Kalbfleisch S. Supraventricular tachycardia causing heart failure. Curr Opinion Cardiol 2011;26:261–9.
Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet 2010;375:752–62.
Kaye G, Lemery R. Fast Facts: Cardiac Arrhythmias, 3rd edn. Oxford: Health Press Limited, 2017.
Layland JJ, Liew D, Prior DL. Clozapine-induced cardiotoxicity: a clinical update. Med J Aust 2009;190:190–2.
Marcus FI, McKenna WJ, Sherrill D et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation 2010;121:1533–41.
Maron BJ, McKenna WJ, Danielson GK et al. American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. Eur Heart J 2003;24:1965–91.
Maron BJ, Ommen SR, Semsarian C et al. Hypertrophic cardiomyopathy, present and future, with translation into contemporary cardiovascular medicine. J Am Coll Cardiol 2014;64:83–99.
Maron BJ, Towbin JA, Thiene G et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006;113:1807–16.
Mogensen J, Arbustini E. Restrictive cardiomyopathy. Curr Opinion Cardiol 2009;24:214–20.
Picano E, Pibarot P, Lancellotti P et al. The emerging role of exercise testing and stress echocardiography in valvular heart disease. J Am Coll Cardiol 2009;54:2251–60.
Pinto YM, Elliott PM, Arbustini E et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardio-myopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J 2016;37:1850–8.
Ramaraj R, Sorrell VL. Peripartum cardiomyopathy: causes, diagnosis, and treatment. Cleve Clin J Med 2009;76:289–96.
Ramasamy K, Lonial S. Fast Facts: Multiple Myeloma and Plasma Cell Dyscrasias, 2nd edn. Oxford: Health Press Limited, 2017.
Selvanayagam JB, Hawkins PN, Paul B et al. Evaluation and management of the cardiac amyloid. J Am Coll Card 2007;50:2101–10.
Vahanian A, Baumgartner H, Bax J et al. Guidelines on the management of valvular heart disease: the Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology. Eur Heart J 2007;28:230–68.
Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Engl J Med 2011;364:1643–56.
Zaidi A, Sharma S. The athlete’s heart. Br J Hosp Med (Lond) 2011;72:275–81.