2.1 Crystal Structures and Symmetry
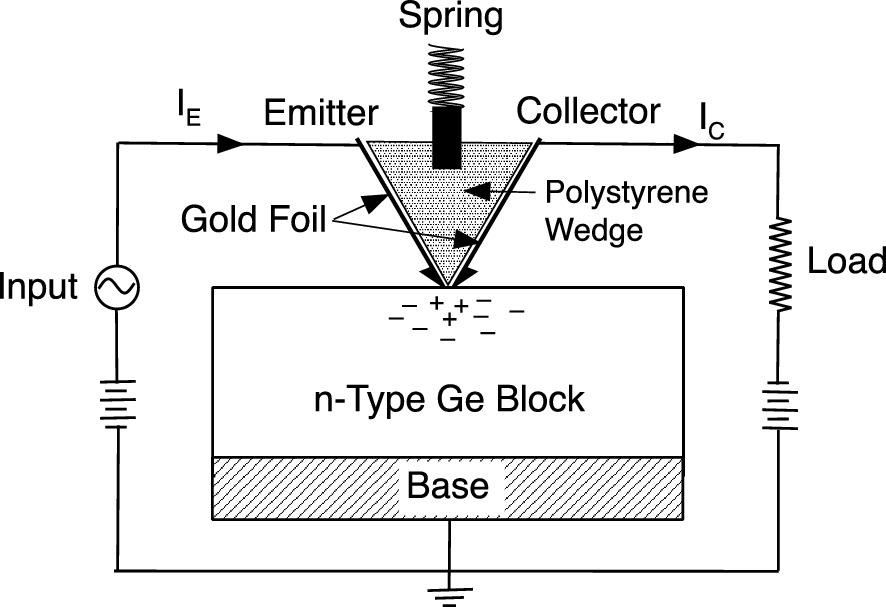
Schematic of the point-contact bipolar transistor invented by Bardeen and Brattain of Bell Telephone Laboratories in 1947. The polystyrene wedge covered with gold foil on two sides was held against the Ge with a spring to form emitter and collector contacts. The n-type Ge slab forms the base of the transistor. Minority carrier (hole) injection was demonstrated for the first time
2.1.1 Crystal Structures
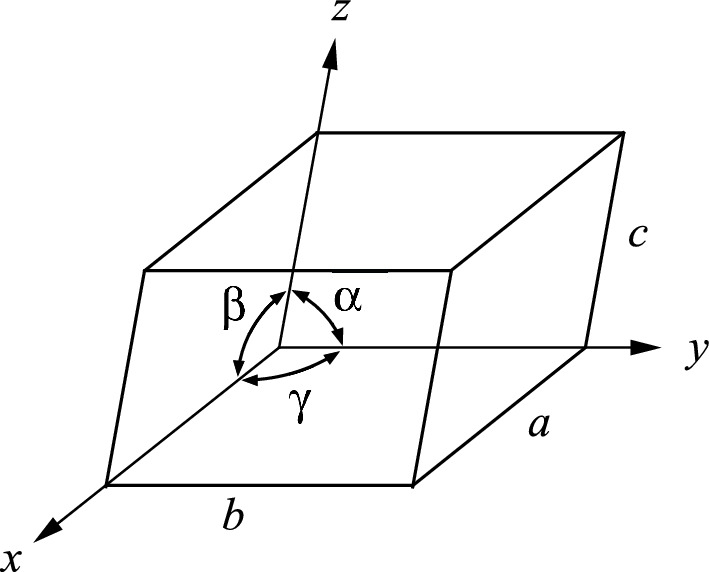
Definition of axes, dimensions and angles for a general unit cell
Seven crystal classes and related Bravais lattices
System | Unit cell | Bravais lattice |
|---|---|---|
Cubic | a = b = c, α = β = γ = 90° | P (primitive), I (body-centered), F (face-centered) |
Hexagonal | a = b ≠ c, α = β = 90°, γ = 120° | P (rhombohedral) |
Tetragonal | a = b ≠ c, α = β = γ = 90° | P, I |
Orthorhombic | a ≠ b ≠ c, α = β = γ = 90° | P, I, F, C (base-centered) |
Monoclinic | a ≠ b ≠ c, α = β = 90° ≠ γ | P, C |
Trigonal | a = b = c, 120° > α = β = γ ≠ 90° | P (rhombohedral) |
Triclinic | a ≠ b ≠ c, α ≠ β ≠ γ | P |

Four basic 3D Bravais lattices most relevant to semiconductor crystals. The face-centered cubic and hexagonal crystal structures are particularly important in semiconductor physics
2.1.2 Crystallographic Notation—Miller Indices
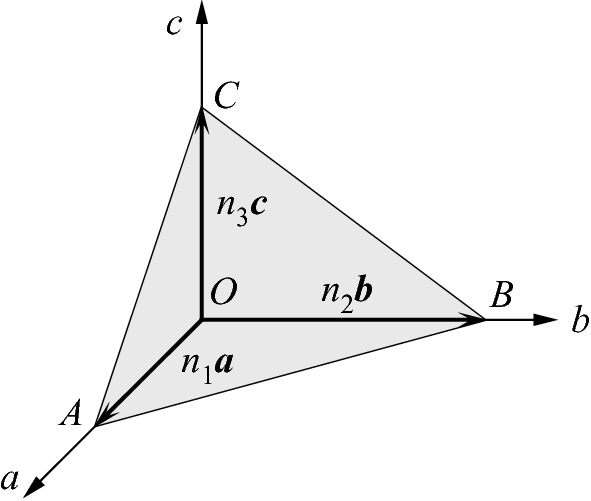
Description of the construction of Miller indices for the plane passing through the lattice points A, B, and C. a, b, and c are unit vectors and n1, n2, and n3 are integers

 , 1, and
, 1, and  ; the smallest three integers having the same ratio are (362). If a plane cuts an axis (say, the a-axis) on the negative side of the origin, the corresponding index is negative, indicated by placing a bar (minus sign) above the index as (
; the smallest three integers having the same ratio are (362). If a plane cuts an axis (say, the a-axis) on the negative side of the origin, the corresponding index is negative, indicated by placing a bar (minus sign) above the index as ( ). For an intercept at infinity, the corresponding index is zero. Figure 2.5 illustrates the Miller indices of some important plans in a cubic crystal. The indices (h00) may denote a plane parallel to both b- and c-axes, e.g., the (100) plane, and the indices (hk0) represent a plane parallel to the c-axis. The normal to the plane with indices (hkl) is the direction [hkl]. In a cubic crystal there are six cubic faces: (100), (010), (001), (
). For an intercept at infinity, the corresponding index is zero. Figure 2.5 illustrates the Miller indices of some important plans in a cubic crystal. The indices (h00) may denote a plane parallel to both b- and c-axes, e.g., the (100) plane, and the indices (hk0) represent a plane parallel to the c-axis. The normal to the plane with indices (hkl) is the direction [hkl]. In a cubic crystal there are six cubic faces: (100), (010), (001), ( ), (
), ( ), and (
), and ( ). This set of equivalent planes of a cubic crystal may be denoted as a group by {100}. The family of plane directions in the cubic crystal may be expressed as 〈100〉.
). This set of equivalent planes of a cubic crystal may be denoted as a group by {100}. The family of plane directions in the cubic crystal may be expressed as 〈100〉.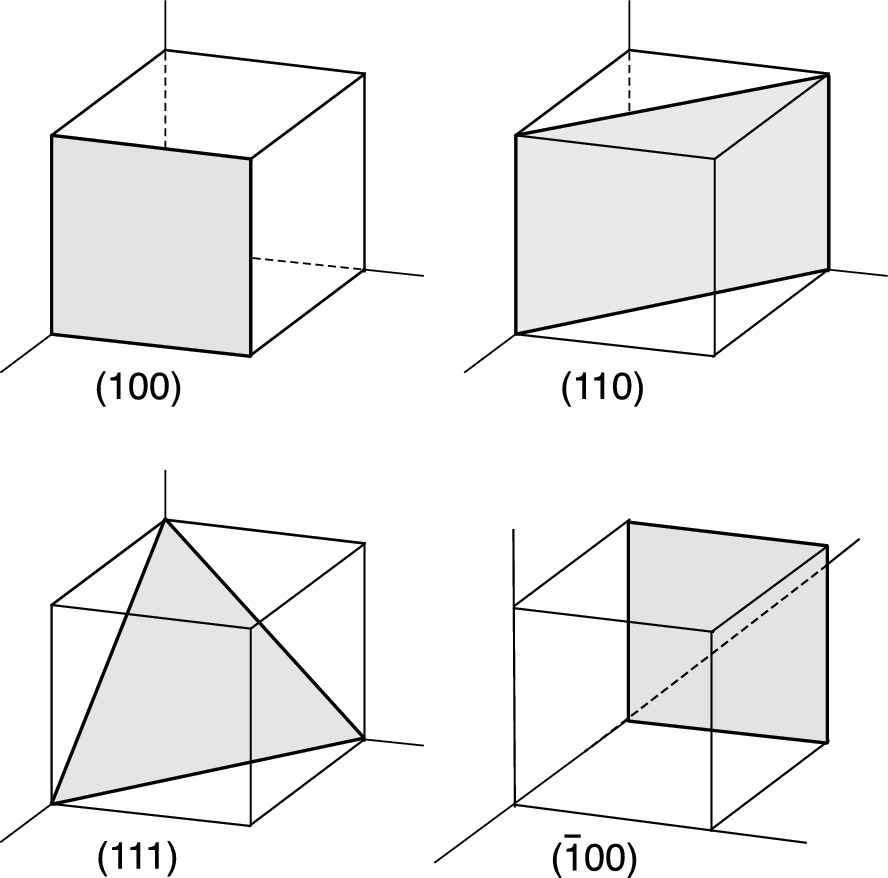
Miller indices of some important planes in a cubic crystal
 according to
according to
Four unit vectors used to formulate Miller indices for a hexagonal crystal

2.1.3 Lattice Symmetries


Symmetry operations in a cubic crystal. a Reflection at a plane, b inversion through a point, c rotation about an axis, and d rotation-reflection about an axis
![$$ \left[ {\begin{array}{*{20}c} 1 & 0 & 0 \\ 0 & 1 & 0 \\ 0 & 0 & { - 1} \\ \end{array} } \right]\left[ {\begin{array}{*{20}c} x \\ y \\ z \\ \end{array} } \right] = \left[ {\begin{array}{*{20}c} x \\ y \\ { - z} \\ \end{array} } \right] $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_Equ3.png)
The presence of a mirror plane in a crystal is represented by the symbol m. The inversion symmetry operation (Fig. 2.7b) may be described by the coordinate transformation x′ = –x, y′ = –y, and z′ = –z. It may also be expressed by the transformation of the position vector r′ = –r at the inversion point. The symbol of the inversion symmetry is  . The other symmetry operation is the rotation about an axis. The axis of rotation is named an n-fold axis if the angle ϕ of rotation to carry a unit cell into itself is equal to ϕ = 2π/n. In this example, Fig. 2.7c, a 90° rotation about the axis will bring point P to point Q or a fourfold symmetry. All possible rotation operations are one-, two-, three-, four-, and sixfold symmetry, which correspond to rotations by 2π, π, 2π/3, π/2, and π/3 radians. These are the only five allowed rotation symmetry operations since they are consistent with a crystal’s requirements for translational symmetry.
. The other symmetry operation is the rotation about an axis. The axis of rotation is named an n-fold axis if the angle ϕ of rotation to carry a unit cell into itself is equal to ϕ = 2π/n. In this example, Fig. 2.7c, a 90° rotation about the axis will bring point P to point Q or a fourfold symmetry. All possible rotation operations are one-, two-, three-, four-, and sixfold symmetry, which correspond to rotations by 2π, π, 2π/3, π/2, and π/3 radians. These are the only five allowed rotation symmetry operations since they are consistent with a crystal’s requirements for translational symmetry.
The symmetry operations may be combined with one another in various ways. For example, there is a rotation-reflection compound operation to carry the lattice into itself. As shown in Fig. 2.7d, a 90° (fourfold) rotation about an axis parallel to the z-axis through the center of the cube combining with a mirror reflection at the x–y plane will bring points P and Q to S and R, respectively. This combined operation is represented by the symbol  for the n-fold rotation symmetry. Conversely, every crystal may be described by a particular combination of symmetry elements that strongly influence its material properties. More specifically, this combination determines the nature of independent components of the tensors describing macroscopic material properties.
for the n-fold rotation symmetry. Conversely, every crystal may be described by a particular combination of symmetry elements that strongly influence its material properties. More specifically, this combination determines the nature of independent components of the tensors describing macroscopic material properties.
 of the material using the symmetry concept and tensor representation. By definition,
of the material using the symmetry concept and tensor representation. By definition,  or
or![$$ \left[ {\begin{array}{*{20}c} {D_{x} } \\ {D_{y} } \\ {D_{z} } \\ \end{array} } \right] = \left[ {\begin{array}{*{20}c} {\epsilon_{11} } & {\epsilon_{12} } & {\epsilon_{13} } \\ {\epsilon_{21} } & {\epsilon_{22} } & {\epsilon_{23} } \\ {\epsilon_{31} } & {\epsilon_{32} } & {\epsilon_{33} } \\ \end{array} } \right]\left[ {\begin{array}{*{20}c} {E_{x} } \\ {E_{y} } \\ {E_{z} } \\ \end{array} } \right] $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_Equ4.png)

Rotation symmetry operations along a [111] and b ![$$ [\bar{1}11] $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_IEq12.png) axes of a cubic crystal
axes of a cubic crystal
![$$ \left[ {\begin{array}{*{20}c} {D_{y} } \\ {D_{z} } \\ {D_{x} } \\ \end{array} } \right] = \left[ {\begin{array}{*{20}c} {\epsilon_{11} } & {\epsilon_{12} } & {\epsilon_{13} } \\ {\epsilon_{21} } & {\epsilon_{22} } & {\epsilon_{23} } \\ {\epsilon_{31} } & {\epsilon_{32} } & {\epsilon_{33} } \\ \end{array} } \right]\left[ {\begin{array}{*{20}c} {E_{y} } \\ {E_{z} } \\ {E_{x} } \\ \end{array} } \right] $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_Equ5.png)


 and
and  . The threefold rotation symmetry exists along the other rotation axes. A rotation of 120° about
. The threefold rotation symmetry exists along the other rotation axes. A rotation of 120° about ![$$ [\bar{1}11] $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_IEq15.png) will bring x → −y, y → z, z → −x (Fig. 2.8b), or
will bring x → −y, y → z, z → −x (Fig. 2.8b), or![$$ \left[ {\begin{array}{*{20}c} { - D_{y} } \\ {D_{z} } \\ { - D_{x} } \\ \end{array} } \right] = \left[ {\begin{array}{*{20}c} {\epsilon_{11} } & {\epsilon_{12} } & {\epsilon_{13} } \\ {\epsilon_{21} } & {\epsilon_{22} } & {\epsilon_{23} } \\ {\epsilon_{31} } & {\epsilon_{32} } & {\epsilon_{33} } \\ \end{array} } \right]\left[ {\begin{array}{*{20}c} { - E_{y} } \\ {E_{z} } \\ { - E_{x} } \\ \end{array} } \right] $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_Equ8.png)
 and
and  . In order to fulfill both these results obtained from the two different rotation axes, we conclude that
. In order to fulfill both these results obtained from the two different rotation axes, we conclude that  for i ≠ j and
for i ≠ j and  . Thus,
. Thus,![$$ \epsilon = \left[ {\begin{array}{*{20}c} {\epsilon_{11} } & 0 & 0 \\ 0 & {\epsilon_{11} } & 0 \\ 0 & 0 & {\epsilon_{11} } \\ \end{array} } \right] $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_Equ9.png)
Therefore, the simple cubic crystal has an isotropic dielectric constant  .
.

2.2 Ionic Bond and Inter-atomic Forces
 ). The other way of constructing the NaCl structure is to position alternatively with Na+ or Cl− ions at the lattice sites of a simple cubic crystal. Each ion is surrounded by six nearest-neighbor ions of the opposite type in 〈100〉 directions. The CsCl structure is based on the simple cubic crystal structure with a basis containing one Cs+ ion at (0,0,0) and one Cl− ion at (
). The other way of constructing the NaCl structure is to position alternatively with Na+ or Cl− ions at the lattice sites of a simple cubic crystal. Each ion is surrounded by six nearest-neighbor ions of the opposite type in 〈100〉 directions. The CsCl structure is based on the simple cubic crystal structure with a basis containing one Cs+ ion at (0,0,0) and one Cl− ion at ( ). The structure looks like BCC but, strictly speaking, is not since different ions occupy the two lattice sites at the body center and corners. There are eight nearest-neighbor ions of the opposite type in 〈111〉 directions.
). The structure looks like BCC but, strictly speaking, is not since different ions occupy the two lattice sites at the body center and corners. There are eight nearest-neighbor ions of the opposite type in 〈111〉 directions.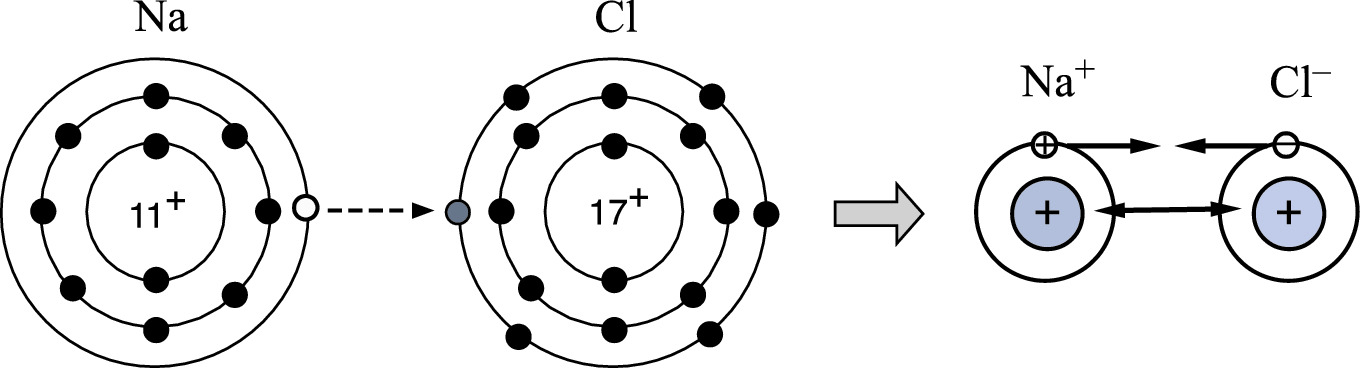
Electron configuration in NaCl. The outer ring represents the shell of n = 3. The dashed arrow indicates the electron transfer from Na to Cl resulting in closed shell configurations in Na and Cl ions. The Coulomb attraction between two ions and repulsive force between cores are also shown
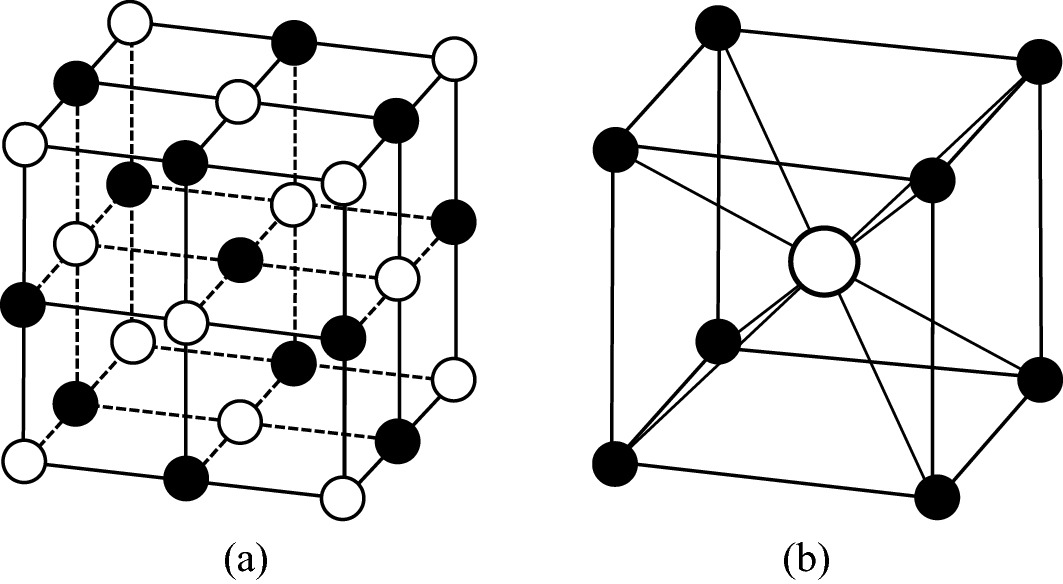
Two typical ionic crystal structures: a NaCl and b CsCl. The ionic radii of Na+, Cs+, and Cl− are 0.875 Å, 1.455 Å, and 1.475 Å, respectively
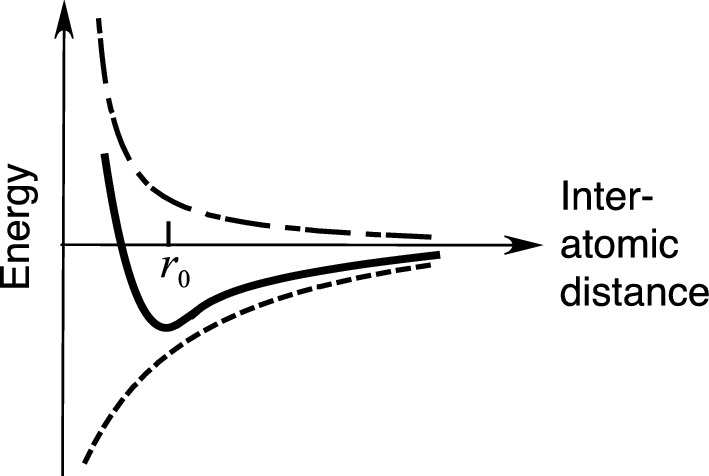
Total potential energy as a function of the separation of two ions. The dot-dashed and dashed curves correspond to contributions of repulsive and Coulomb attraction forces, respectively. r0 is the equilibrium separation of ions







For stability of the crystal, n must be greater than unity. Of course it should be possible to determine it from an exact quantum mechanical treatment of the problem. However, from a fit to experimental quantities, n has been determined to be in the range 6–10. Therefore, the Coulomb attraction force becomes the major contributor to the cohesive energy.

Hard spheres arranged in a close-packed layer structure

Two layers of close-packed spheres
2.3 Covalent Bond and sp3 Hybrid Orbit
2.3.1 General Properties of Covalent Bond Formation
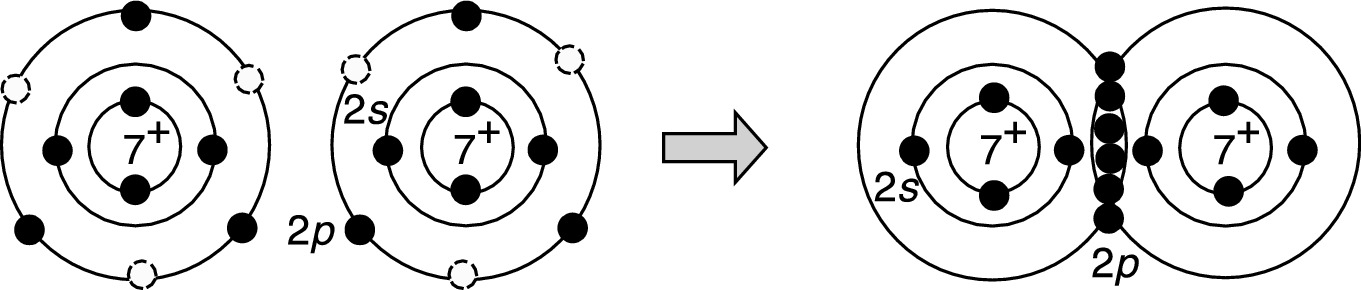
Schematic of the formation of covalent bonding in a nitrogen molecule. The circles in each nitrogen atom represent the unfilled 2p valence electron states

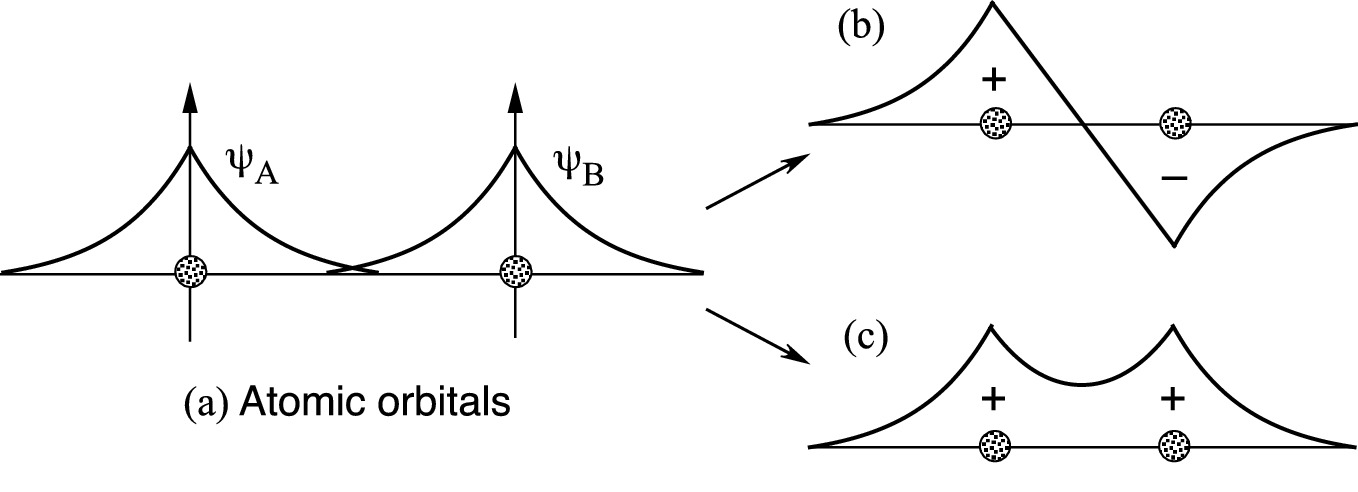
Formation of bonding and anti-bonding combinations of atomic orbitals in diatomic hydrogen molecules with a single bonding electron. a The wave functions of two nearby hydrogen atoms. b The anti-symmetric wave function and c symmetric wave function formed in a H2+ ion
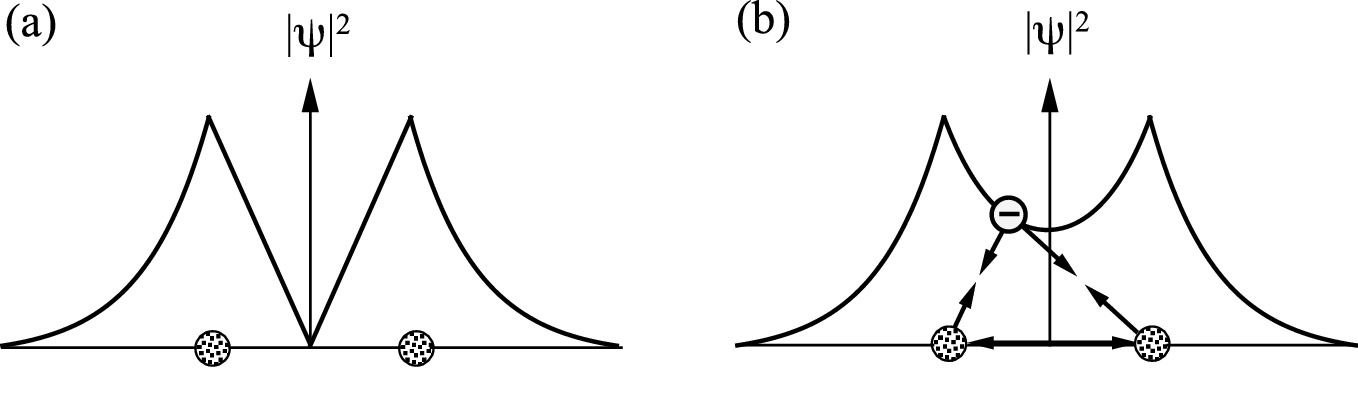
Probability density of the electron distribution in a H2+ ion with a anti-symmetric and b symmetric wave functions

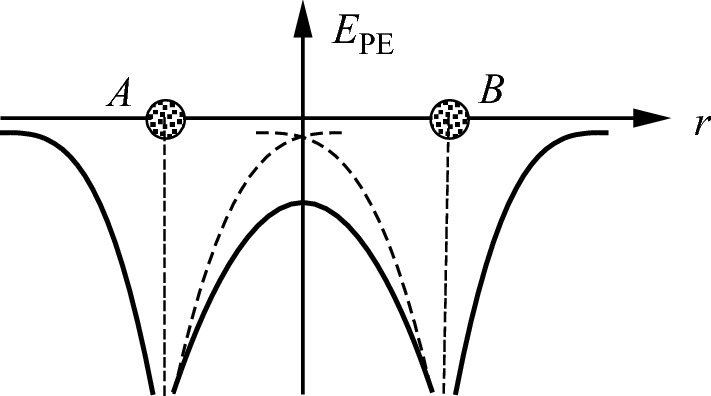
Potential energy of the H2+ ion (solid curves). The dashed curve is the potential energy of a single hydrogen nucleus
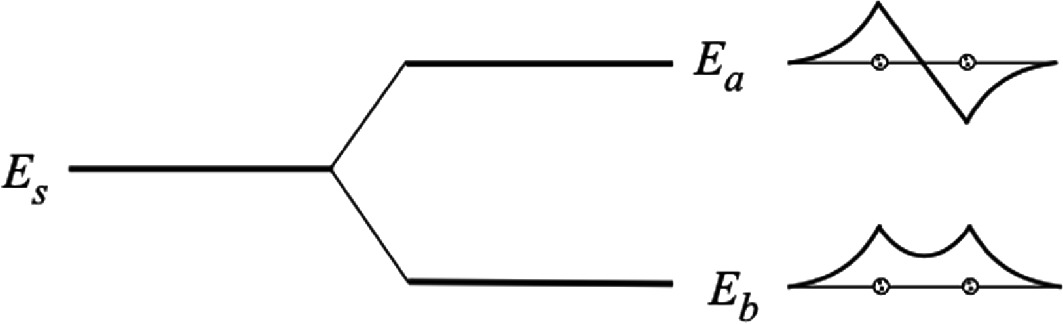
Energy level diagram shows the relative position of bonding (Eb) and anti-bonding (Ea) energies with respect to isolated ψ(1s) state energy (Es). The related wave functions are also shown
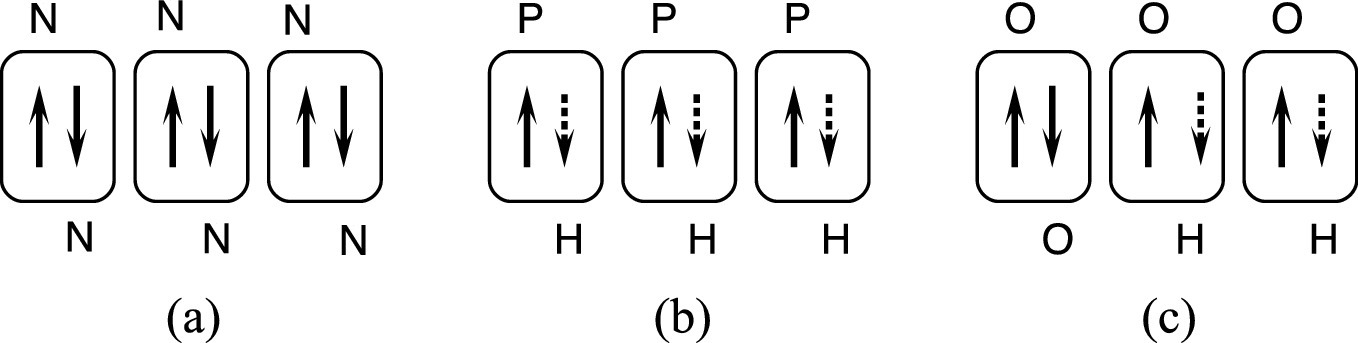
Electron-pair bond formed in between a pair of electrons in p orbital with anti-parallel spin in a N2, b PH3, and c H2O. The dotted arrows indicate hydrogen electrons which make pair-bonds with p electrons of phosphorous and oxygen
We further examine this idea in water (H2O) and phosphine (PH3) (Fig. 2.19). The oxygen has an electron configuration 1s22s22p4, and phosphorous has an electron configuration 1s22s22p63s23p3. Since the s electrons in the outer shell already have their spins paired, only the p electrons need to be considered. For a phosphorous atom, there are three electrons that have unpaired spins, whereas for an oxygen atom, two of the four electrons already have paired spins leaving two electrons having unpaired spins. Therefore, a phosphorous atom is capable of making three electron-pair bonds with three hydrogen atoms in PH3, and an oxygen atom is capable of forming two electron-pair bonds in H2O.
2.3.2 Directional Property of Covalent Bonds
 where R, Θ, and Φ are the radial, azimuthal, and angular wave functions, respectively. The algebraic expressions for some of the wave functions are given below.
where R, Θ, and Φ are the radial, azimuthal, and angular wave functions, respectively. The algebraic expressions for some of the wave functions are given below.


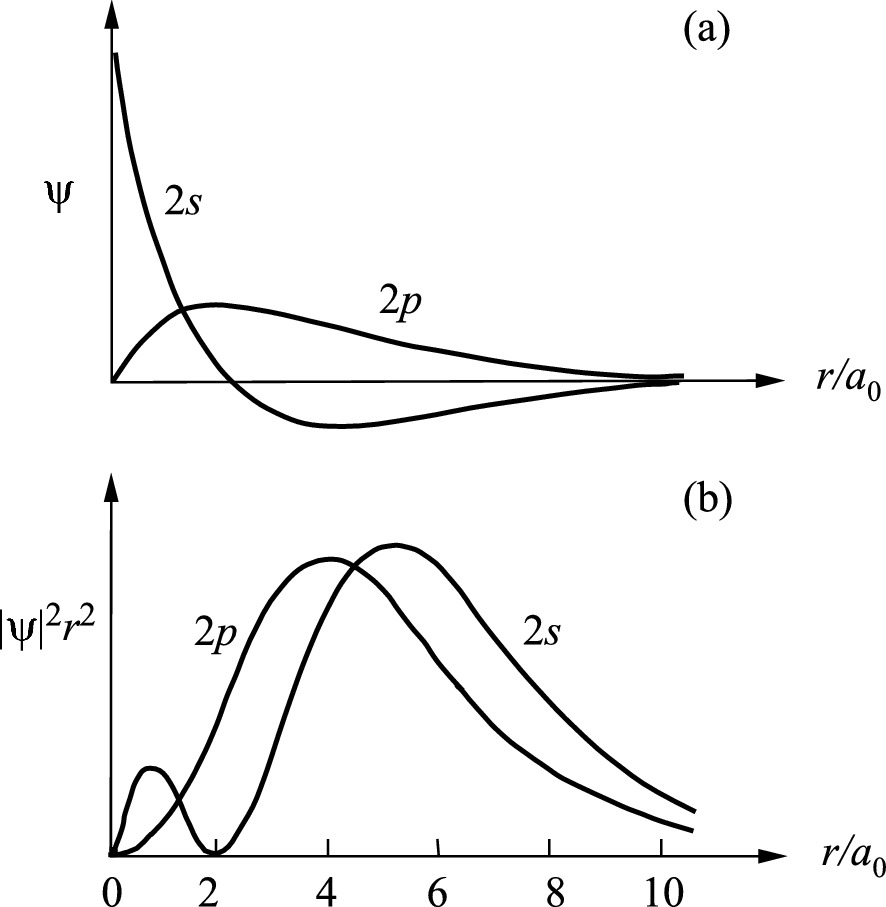
a Radial wave functions Rnl(r) and b radial probability density |ψ|2r2 of the 2s (ψ200) and 2p (ψ210) states of the hydrogen atom as a function of radial distance r in the unit of Bohr radius a0

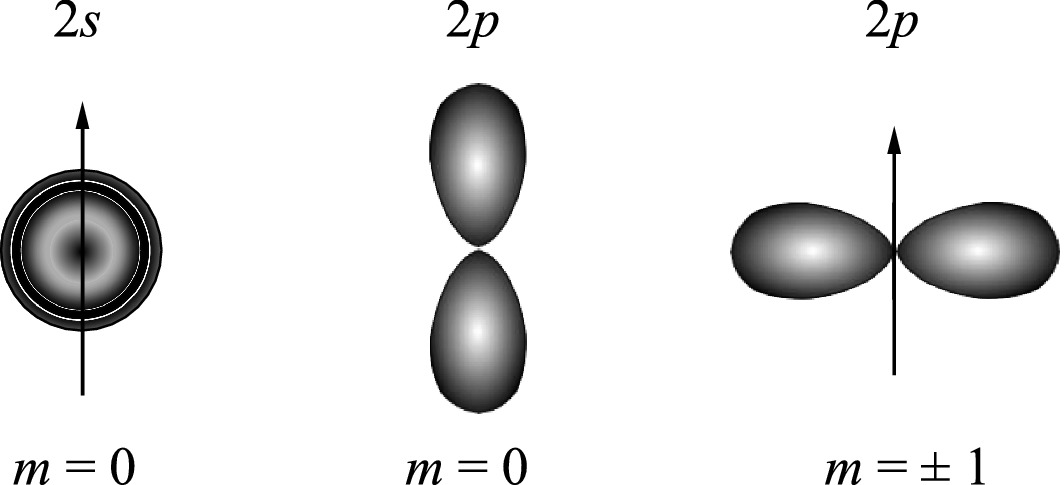
Electron density distributions of 2s and 2p states for a hydrogen atom. The polar (z) axis is oriented vertically

Figure 2.20 shows the electron wave functions and the corresponding radial probability densities (|ψ|2r2) of 2s and 2p states in a hydrogen atom. Note that the radial probability density distributions of s and p states overlap heavily. More 2s electrons than 2p electrons are occupying states at larger r. As we will see in the next section, these results have important consequences in determining crystal structures of semiconductors. Consider the case of mixing s and p orbital wave functions. The s orbits (l = 0) of an atom always have a spherical symmetry, i.e., ψs = g(r). For p orbits (l = 1), the wave functions are angularly dependent as described in (2.18c). Consequently, the spatial distribution of the wave functions leads to a strong orientation dependency in covalent bonding.



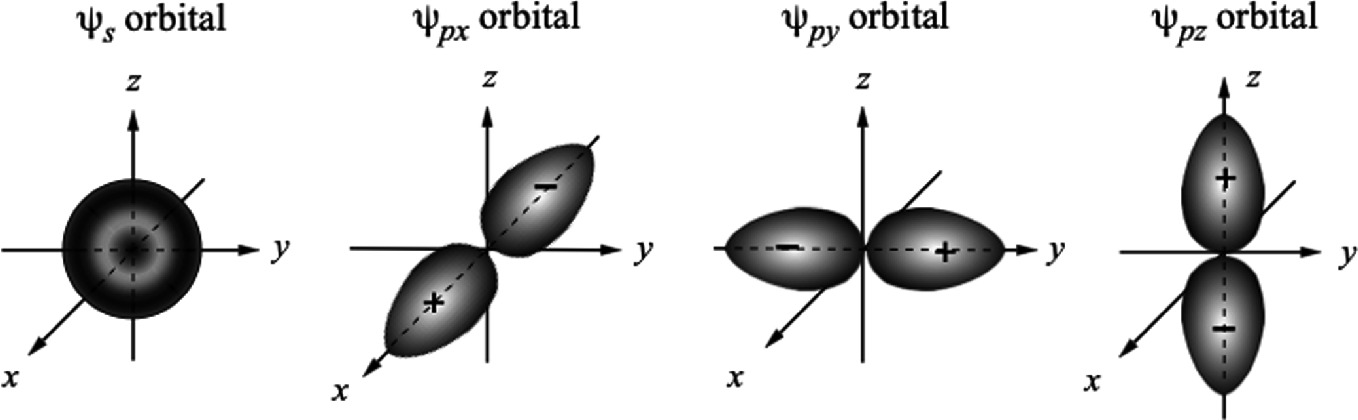
Spherical s orbital and three p orbitals each directed along a different Cartesian axis
2.3.3 sp3 Hybrid Orbit
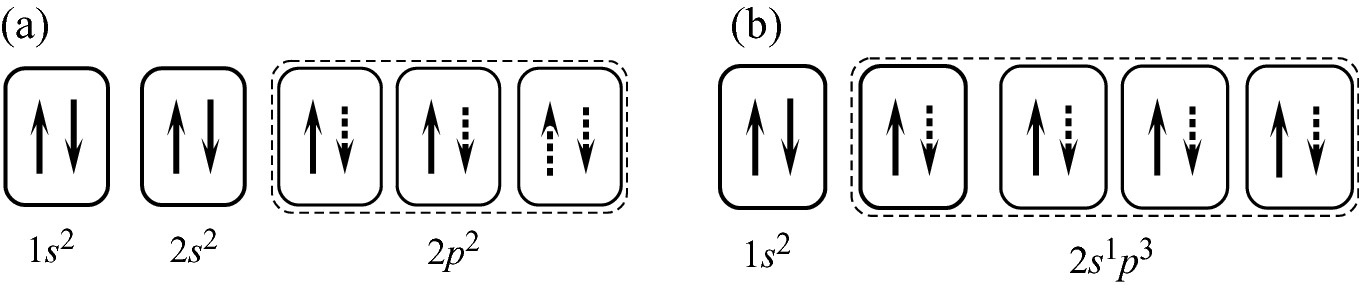
Electron-pair bond formed in a carbon atom, and b CH4. The dotted boundary indicates the bonding orbitals. The dotted arrows are sites available for electron-pair bonds
![$$ \left\{ {\begin{array}{*{20}c} {\psi_{111} = \frac{1}{2}\left[ {\psi_{s} + \psi_{x} + \psi_{y} + \psi_{z} } \right]} \\ {\psi_{{1\bar{1}\bar{1}}} = \frac{1}{2}\left[ {\psi_{s} + \psi_{x} - \psi_{y} - \psi_{z} } \right]} \\ {\psi_{{\bar{1}1\bar{1}}} = \frac{1}{2}\left[ {\psi_{s} - \psi_{x} + \psi_{y} - \psi_{z} } \right]} \\ {\psi_{{\bar{1}\bar{1}1}} = \frac{1}{2}\left[ {\psi_{s} - \psi_{x} - \psi_{y} + \psi_{z} } \right]} \\ \end{array} } \right. $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_Equ27.png)

a A set of four sp3 orbitals forms a tetrahedron with each sp3 orbital containing a large lobe and a small lobe. The large lobes point to the corners of the tetrahedron and can be used to share electrons in covalent bonds. b The sp3 hybrid orbitals of carbon in a CH4 molecule, each having an electron distribution directed toward one of the four alternating corners of a cube
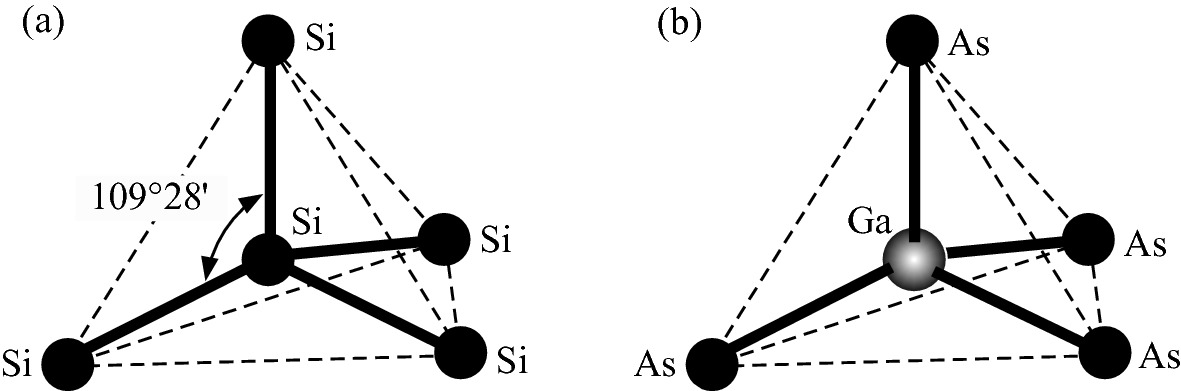
Tetrahedron bond structure of a Si and b GaAs
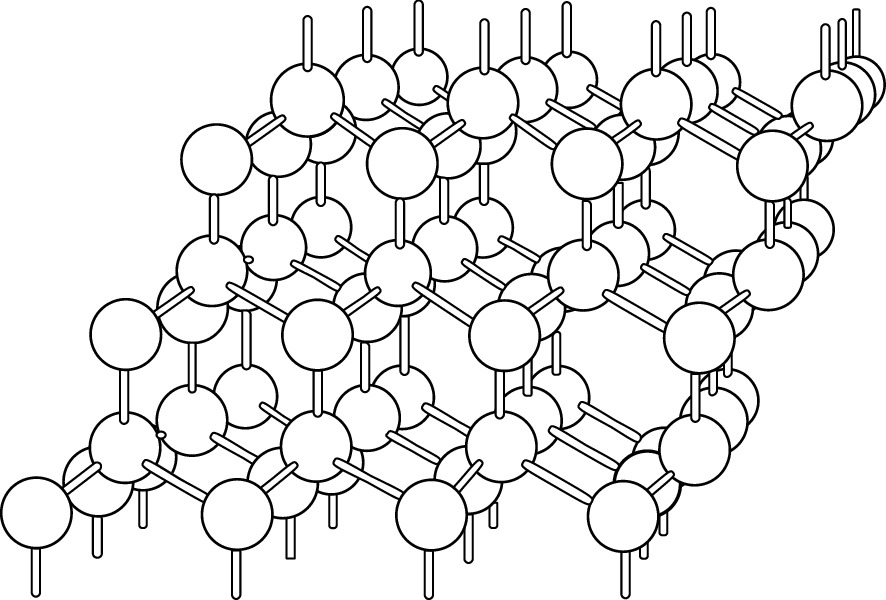
An infinite tetrahedron arrangement of a covalent bonded diamond cubic structure.
Reprinted with permission from [1], copyright Oxford University Press
2.3.4 Mixed Ionic-Covalent Bonds




Crystal structure and ionicity for III–V compound semiconductors. (W = wurtzite structure; ZB = zinc-blende structure) [2]
III–V | Structure | Ionicity | III–V | Structure | Ionicity | III–V | Structure | Ionicity |
|---|---|---|---|---|---|---|---|---|
AlN | W | 0.449 | GaN | W | 0.500 | InN | W | 0.578 |
AlP | ZB | 0.307 | GaP | ZB | 0.374 | InP | ZB | 0.421 |
AlAs | ZB | 0.274 | GaAs | ZB | 0.310 | InAs | ZB | 0.357 |
AlSb | ZB | 0.426 | GaSb | ZB | 0.261 | InSb | ZB | 0.321 |
2.4 Major Semiconductor Crystal Structures
2.4.1 Diamond Structure
 . The unit cell cube has four atoms in its interior, an atom in the center of each of its faces (each of which is to be thought of as ‘shared’ with an adjoining cube in a large crystal), and eight atoms at the cube vertices (each of which is shared with seven other elementary cubes). Thus, the number of atoms in a crystal is eight times the number of elementary cubes; or, equivalently, there are eight atoms in a cube of size a3.
. The unit cell cube has four atoms in its interior, an atom in the center of each of its faces (each of which is to be thought of as ‘shared’ with an adjoining cube in a large crystal), and eight atoms at the cube vertices (each of which is shared with seven other elementary cubes). Thus, the number of atoms in a crystal is eight times the number of elementary cubes; or, equivalently, there are eight atoms in a cube of size a3.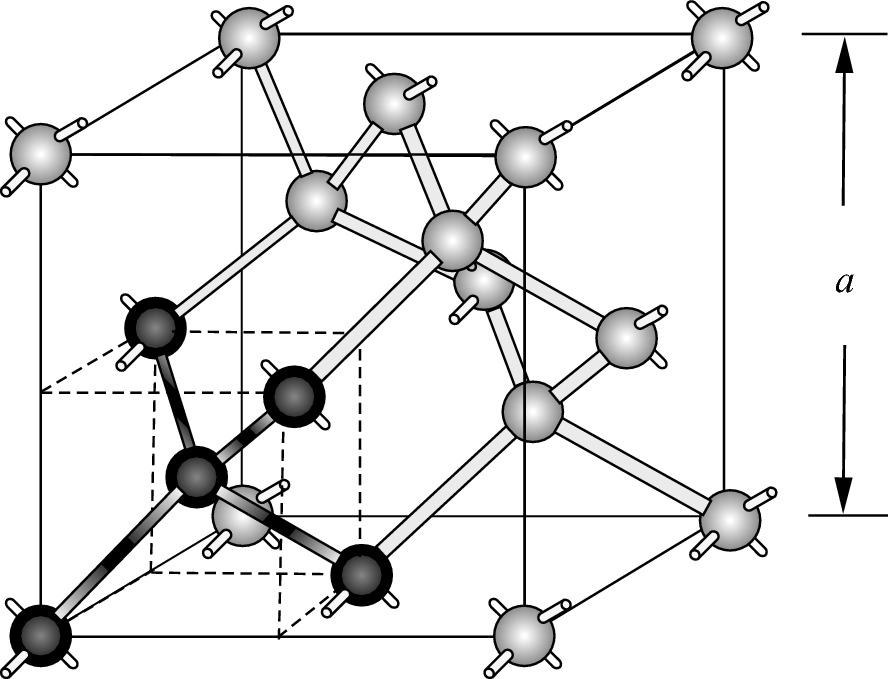
Diamond structure. Each atom is symmetrically surrounded in an imaginary cube (enclosed by dotted lines) by four nearest neighbors
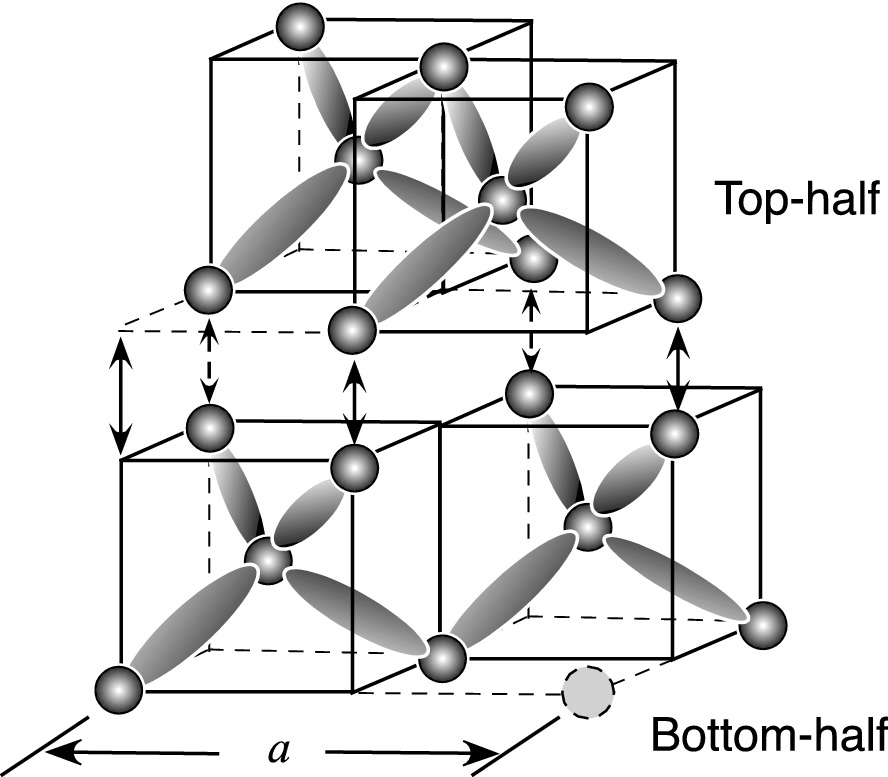
Construction of a diamond lattice structure using tetrahedral structures as building blocks
2.4.2 Zinc-Blende Structure

Zinc-blende crystal structure. Two types of atoms are arranged alternately with each atom surrounded by four nearest neighbors of different types of atoms
The diamond structure of elemental semiconductors has the highest symmetry among all semiconductors. However, it lacks many of the symmetrical features enjoyed by primitive cubic. Compared to the diamond crystal structure, the symmetry of zinc-blende structure is further reduced. The physical structures of (111) and ( ) surfaces are the same for diamond structure but different for zinc-blende crystal. For example, in GaAs the (111) consists entirely of Ga atoms, while (
) surfaces are the same for diamond structure but different for zinc-blende crystal. For example, in GaAs the (111) consists entirely of Ga atoms, while ( ) is a sheet of As atoms. Because of the different surface constituents, these two types of surface show very different chemical properties. Further, due to the nature of partially ionic bonding in zinc-blende structure, electric dipoles exist between the neighboring Ga and As atoms along 〈111〉. Although, at equilibrium, the net internal electric field is zero as the individual dipole moments get canceled out, compressions along 〈111〉 will cause atoms to move closer and induce a net internal electric field. This piezoelectric effect in compound semiconductors enhances carrier scattering which reduces the carrier mobility. More detailed discussions on the physical properties of III–V compounds with zinc-blende structure will be given in Chap. 4.
) is a sheet of As atoms. Because of the different surface constituents, these two types of surface show very different chemical properties. Further, due to the nature of partially ionic bonding in zinc-blende structure, electric dipoles exist between the neighboring Ga and As atoms along 〈111〉. Although, at equilibrium, the net internal electric field is zero as the individual dipole moments get canceled out, compressions along 〈111〉 will cause atoms to move closer and induce a net internal electric field. This piezoelectric effect in compound semiconductors enhances carrier scattering which reduces the carrier mobility. More detailed discussions on the physical properties of III–V compounds with zinc-blende structure will be given in Chap. 4.
2.4.3 Wurtzite Structure
 .
.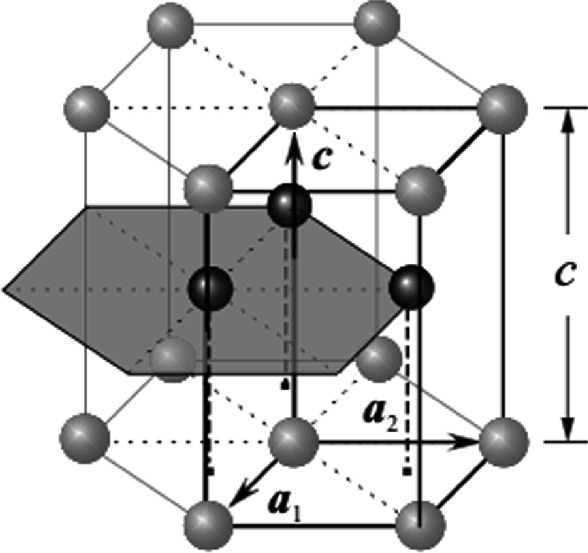
Unit cell of the hexagonal close-packed (HCP) structure. The three atoms from the second hexagonal inside the unit cell are shown as dark spheres. The primitive cell is outlined and defined by unit vectors a1, a2, and c
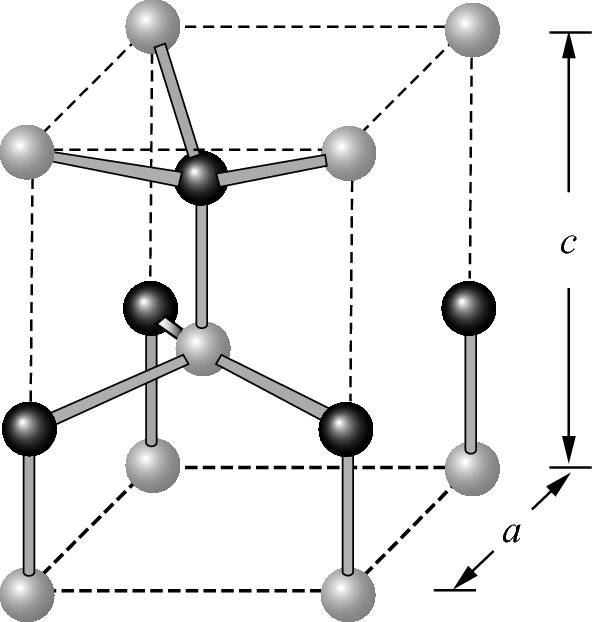
Primitive unit cell of the wurtzite structure
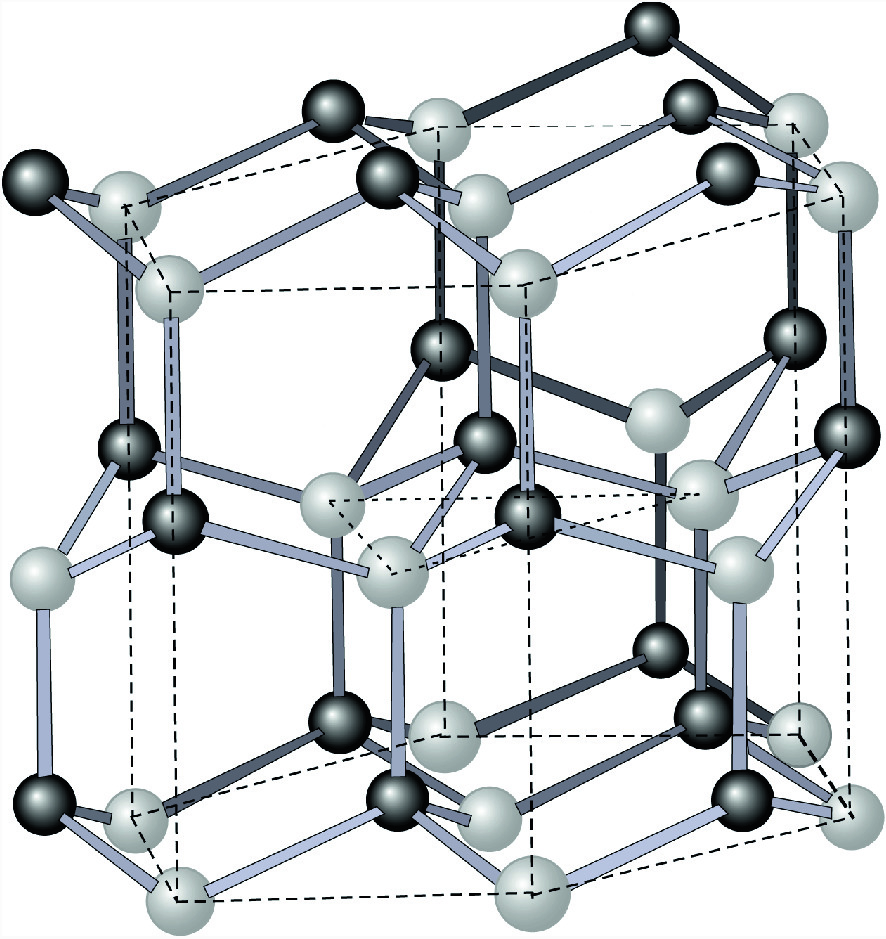
Unit cell of the wurtzite structure. The hexagonal boundary is outlined with dashed lines
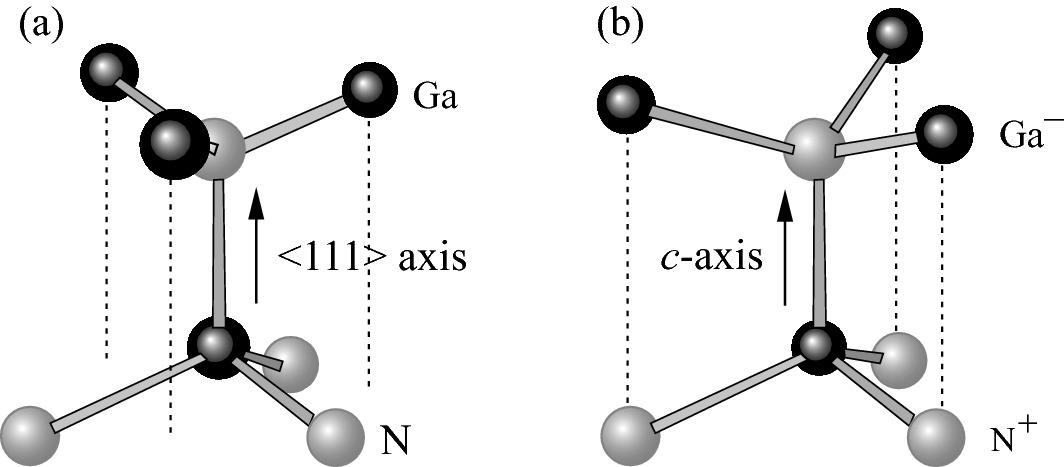
Second-nearest-neighbor bond arrangements along a 〈111〉 axis in zinc-blende and b c-axis in wurtzite GaN crystals
 m-plane and
m-plane and  a-plane, the net spontaneous polarization field is zero. These are non-polar planes with c-axis parallel to the layer surface as shown in Fig. 2.34. Any crystal plane inclined between a non-polar plane and c-plane is a semi-polar plane including
a-plane, the net spontaneous polarization field is zero. These are non-polar planes with c-axis parallel to the layer surface as shown in Fig. 2.34. Any crystal plane inclined between a non-polar plane and c-plane is a semi-polar plane including  and
and 
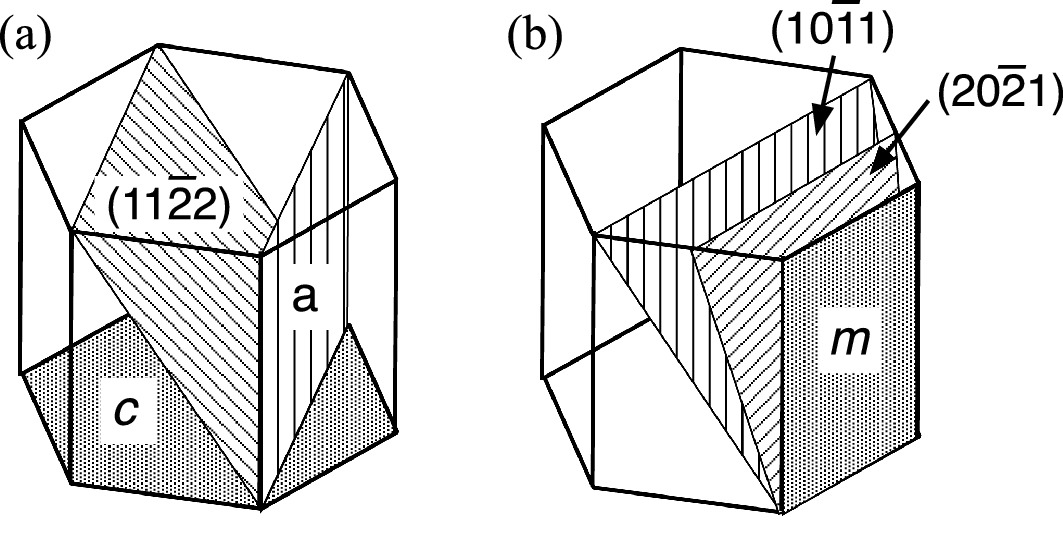
Non-polar and semi-polar crystal planes in wurtzite III-N crystals. a  a plane and
a plane and  semi-polar plane and b
semi-polar plane and b  m-plane and
m-plane and  and
and  semi-polar planes
semi-polar planes
2.5 Reciprocal Lattice, Diffraction Condition, and Brillouin Zone
2.5.1 Crystal Diffraction

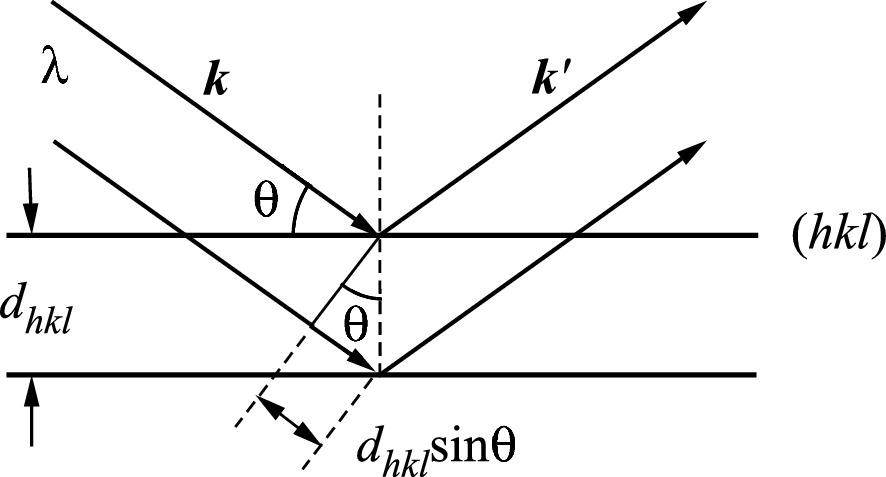
Reflection of a parallel beam of X-rays from two adjacent (hkl) planes of atoms in a crystal. The path difference between two X-rays has to be an integer multiplication of the path difference of 2dhkl sin θ for constructive reinforcement
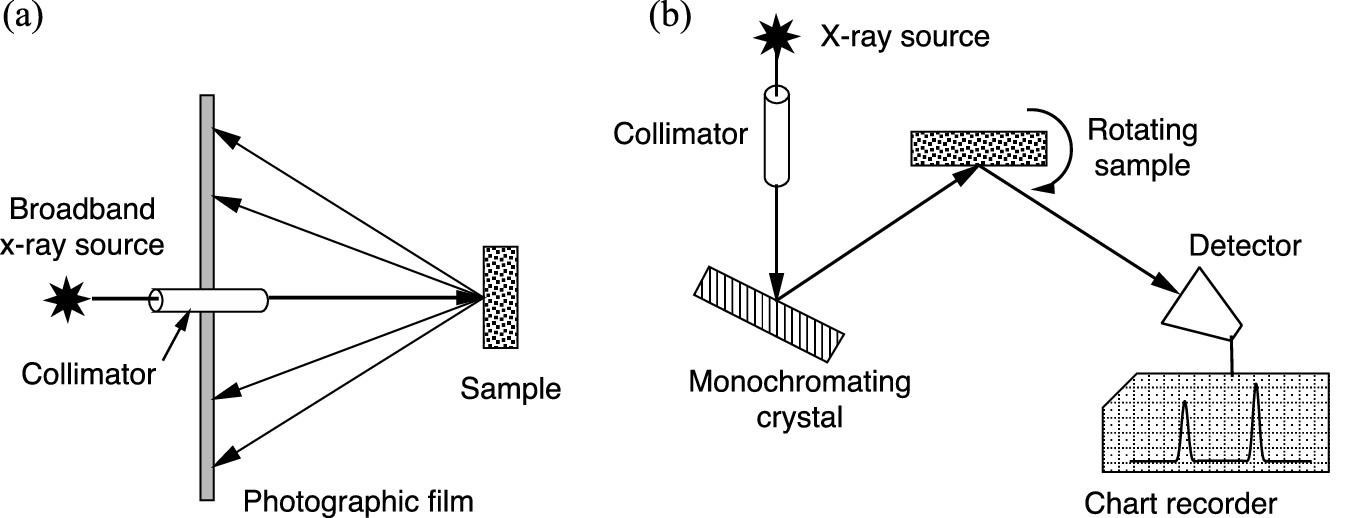
a Laue diffraction method arrangement using broadband X-ray source. b A rotating crystal X-ray diffraction arrangement using monochromatic X-rays selected by Bragg reflection from a separate single crystal
2.5.2 Reciprocal Lattice

It is obvious that this vector has the dimension of inverse length and can be used to define the so-called reciprocal lattice. Thus, every crystal structure has both a real-space lattice and a reciprocal lattice associated with it.





 is the translation vector. We can use the same equation with a minor modification as shown below:
is the translation vector. We can use the same equation with a minor modification as shown below:










![$$ \begin{aligned} \rho \left( {\varvec{r} + \varvec{T}} \right) = \mathop \sum \limits_{G} c_{G} \exp \left( {i\varvec{G} \cdot \varvec{r}} \right)exp\left( {i\varvec{G} \cdot \varvec{T}} \right) = \rho \left( \varvec{r} \right) \quad \quad \quad \quad \quad \quad \qquad \qquad \qquad \quad & \\ \exp \left( {i\varvec{G} \cdot \varvec{T}} \right) = \exp \left[ {i\left( {hh^{\prime} + kk^{\prime} + ll^{\prime}} \right)} \right] = \exp \left( {iu} \right) = { \sin }\left( {2u\pi } \right) \qquad \qquad \qquad \quad & \\ + { \cos }\left( {2u\pi } \right) = 1 \qquad \qquad \qquad \quad \\ \end{aligned} $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_Equg.png)

The periodicity is maintained in the 3D reciprocal lattice.
2.5.3 Properties of the Reciprocal Lattice Vector
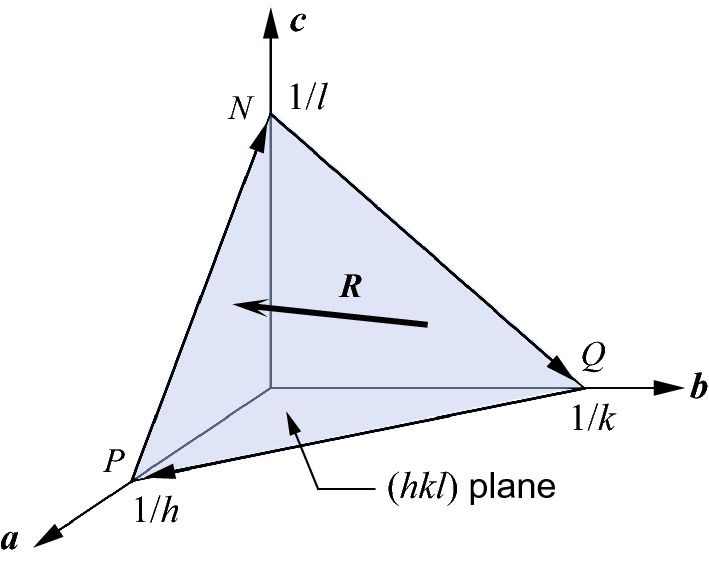
Geometry of the direct lattice (hkl) plane and related vectors



![$$ \begin{aligned} \varvec{R} \cdot \varvec{G}_{hkl} & = \left[ {\left( {\frac{A}{h}} \right)\varvec{a} + \left( {\frac{B}{k}} \right)\varvec{b} + \left( {\frac{C}{l}} \right)\varvec{c}} \right] \cdot \left( {h\varvec{a}^{*} + k\varvec{b}^{*} + l\varvec{c}^{*} } \right) \\ & \quad = \left( {\frac{A}{h}} \right)h + \left( {\frac{B}{k}} \right)k + \left( {\frac{C}{l}} \right)l = A + B + C = 0 \\ \end{aligned} $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_Equ49.png)
Therefore, R ⊥ Ghkl. The reciprocal lattice vector defined by Ghkl = ha* + kb* + lc* lies perpendicular to the direct lattice (hkl) plane.
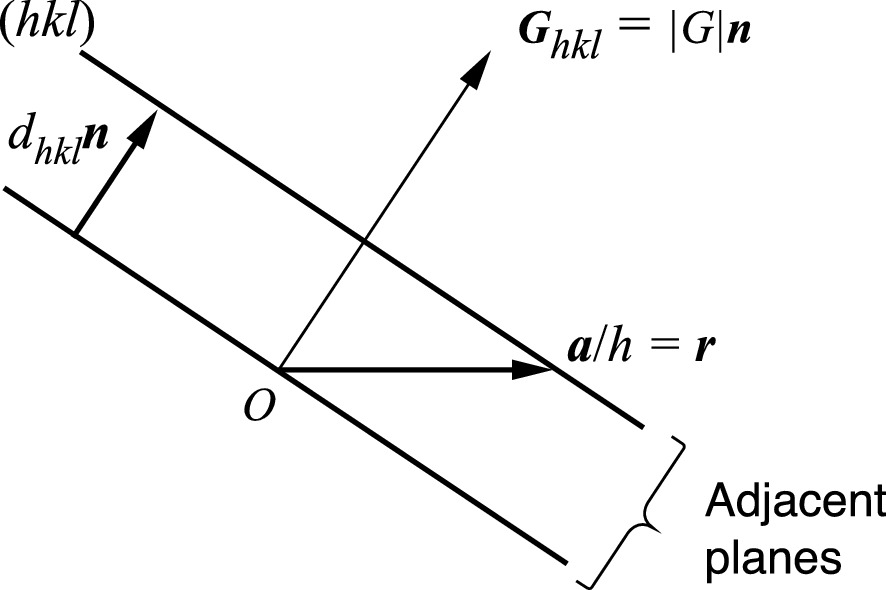
Construction for the determination of the spacing between successive (hkl) planes. n is the surface normal unit vector


2.5.4 Diffraction Condition

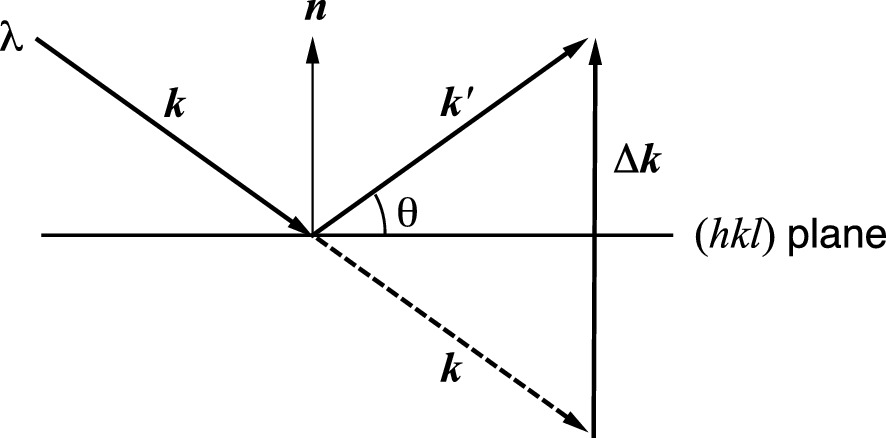
The change in wave vector when undergoing an elastic scattering at the (hkl) plane

The change Δk is in the direction of the surface normal and perpendicular to the (hkl) planes.



Thus the scattering vector Δk is equal to a reciprocal lattice vector Ghkl. This is the 3D Bragg diffraction condition.
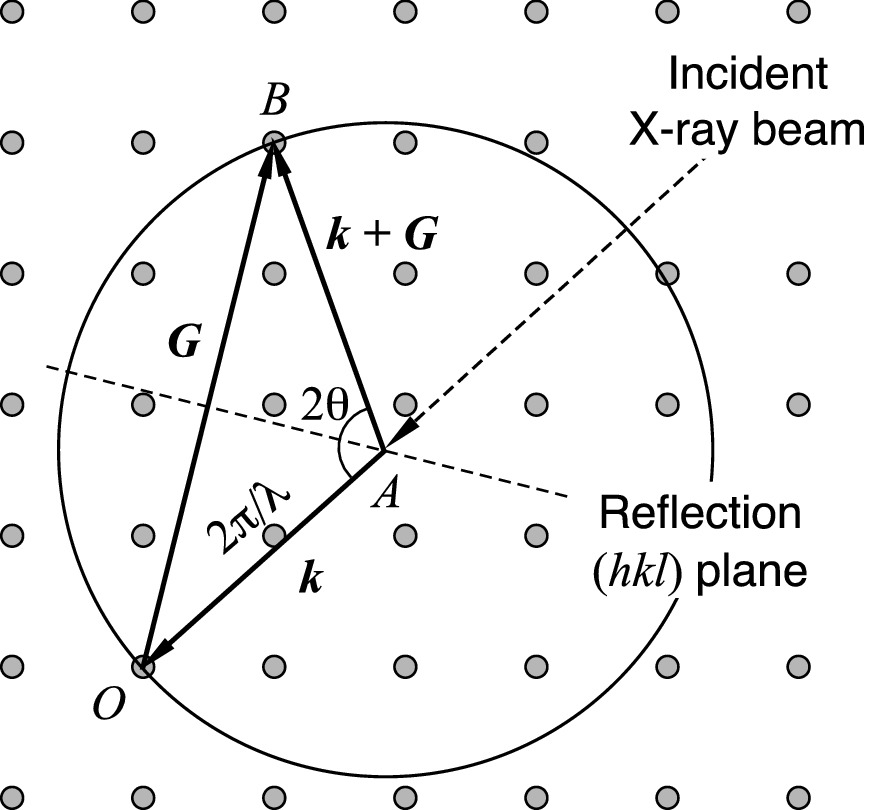
Ewald construction for the Laue diffraction condition from a crystal in the reciprocal lattice. The incident X-ray beam has a wavelength λ

Schematic diagram of the RHEED arrangement showing the incident electron beam at an angle θ ≤ 5° to the sample surface. The elongated spots indicate the intersection of the Ewald sphere with diffraction rods. (Reprint with permission from [3], copyright AIP Publishing.)
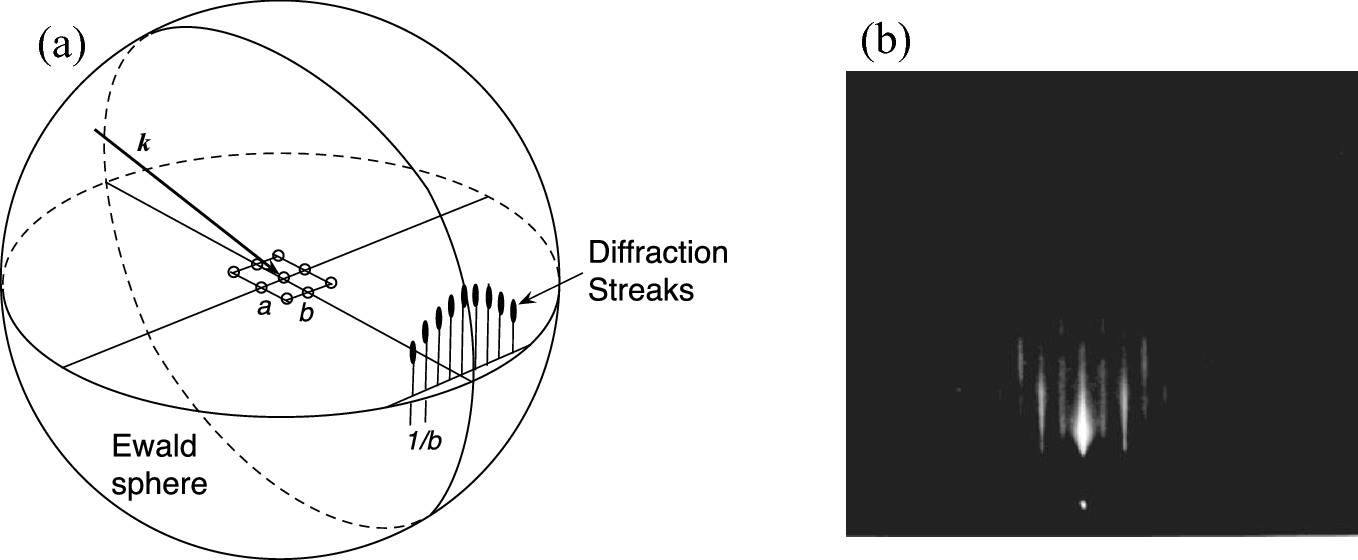
a Ewald sphere construction for a reconstructed semiconductor surface in reciprocal lattice space. The real-space lattice atoms are shown as circles. (Reprint with permission from [4], copyright Elsevier) b Diffraction pattern from the GaAs (001) − (2 × 4) surface along the ![$$ \left[ {\bar{1}\bar{1}0} \right] $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_IEq38.png) azimuth
azimuth

Thus, at 5 keV, λ = 0.0174 nm. The radius of the Ewald sphere is |k| = 2π/λ = 362 nm−1. If the surface lattice network has a lattice constant a of 5 Å, then the distance between adjacent rods in reciprocal space will be 2π/a = 12.57 nm−1. As a result, the intersection of the nearly flat Ewald sphere and rods occurs along their length, resulting in a streaked diffraction pattern as illustrated in Fig. 2.42a. However, due to the finite surface roughness, the short diffraction streaks transform into elongated lines. Figure 2.42b is a typical RHEED pattern for a smooth (001) surface of GaAs.
2.5.5 The Brillouin Zone




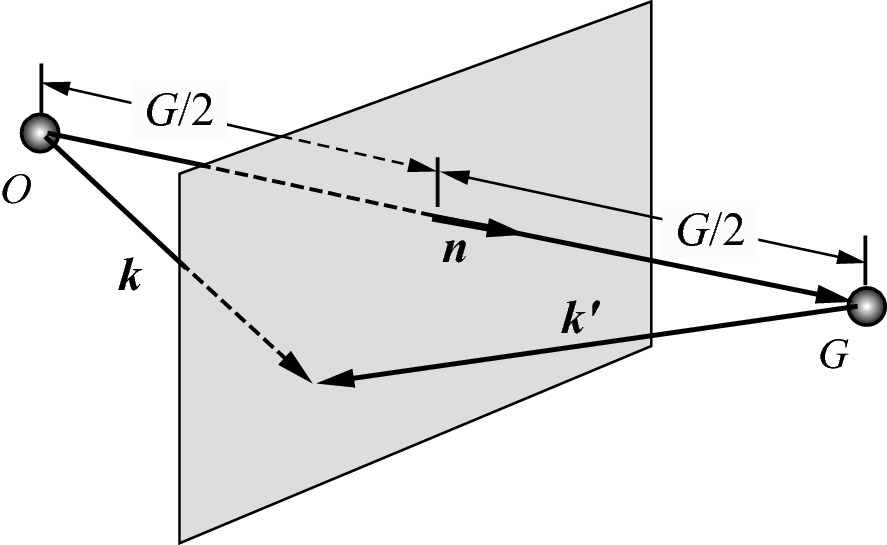
Geometrical representation of the Bragg diffraction condition. Diffraction only occurs for an incident wave vector whose tip lies on the Bragg plane if its origin is tied to a reciprocal lattice point
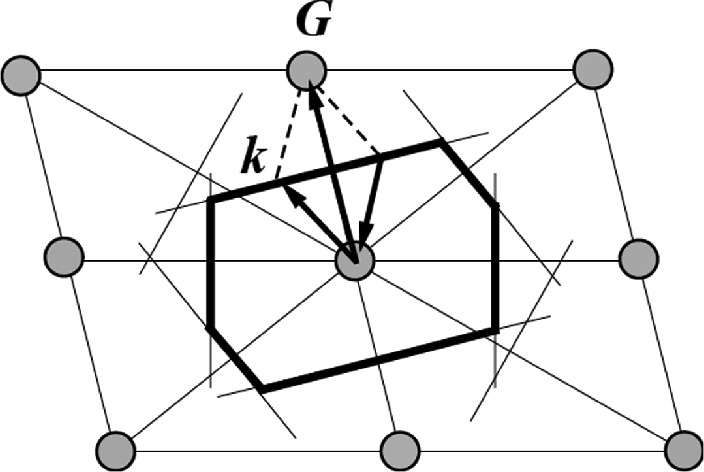
First Brillouin zone formed in the reciprocal space for a 2D oblique lattice. It is shown bisecting the midpoints of six shortest reciprocal lattice vectors from the center of this lattice structure
- (a)
Example for a 2D square lattice

Construction of the first (dashed line boundary) and second (solid line boundary) Brillouin zones (BZ) in a 2D reciprocal lattice space. The square direct lattice has a lattice spacing a



To construct the smallest rectangle, we calculate kx and ky with the smallest possible u and v. At u = 0 and v = ± 1, we have ky = ± π/a. Similarly, at v = 0 and u = ± 1, we have kx = ± π/a. The four constant k’s form a square. This is the first Brillouin zone as shown in Fig. 2.45. Alternatively, bisecting the midpoints of four shortest reciprocal lattice vectors from the origin can also form the first Brillouin zone. For the second Brillouin zone, kx ± ky = ± 2π/a for u = ± 1 and v = ± 1. The higher-order Brillouin zones can be constructed through the same procedures. For example, using u = 0, v = ± 2 and v = 0, u = ± 2, the third zone boundaries are defined.



Thus, 2a sin θ = 2a · (λ/2a) = λ, fulfilling the Bragg law.
- (b)
3D Brillouin zone of a FCC crystal
As we will discuss in Chap. 3, the Brillouin zone concept is essential for the development of the energy band structure of semiconductors. The construction of Brillouin zones in 3D semiconductor crystals follows a procedure similar to that for the 2D case. Since most semiconductor crystals have either zinc-blende or wurtzite lattice structure, we shall investigate the Brillouin zones of the basic FCC and simple hexagonal structures.

Primitive unit cell of an FCC crystal structure is formed by three translation vectors a′, b′, and c′. These translation vectors point from the origin toward the surface atoms diagonally

 .
.
![$$ \left[ {\bar{1}11} \right] $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_IEq40.png) ,
, ![$$ \left[ {1\bar{1}1} \right] $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_IEq41.png) , and
, and ![$$ \left[ {11\bar{1}} \right] $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_IEq42.png) ] directions of a cubic lattice, respectively. We can plot (2.65) as shown in Fig. 2.47 and realize that these are the primitive translation vectors of a BCC lattice! Thus, the reciprocal of an FCC lattice is a BCC lattice. The volume of the primitive cell in the reciprocal lattice is a* · b* × c* = 4(2π/a)3.
] directions of a cubic lattice, respectively. We can plot (2.65) as shown in Fig. 2.47 and realize that these are the primitive translation vectors of a BCC lattice! Thus, the reciprocal of an FCC lattice is a BCC lattice. The volume of the primitive cell in the reciprocal lattice is a* · b* × c* = 4(2π/a)3.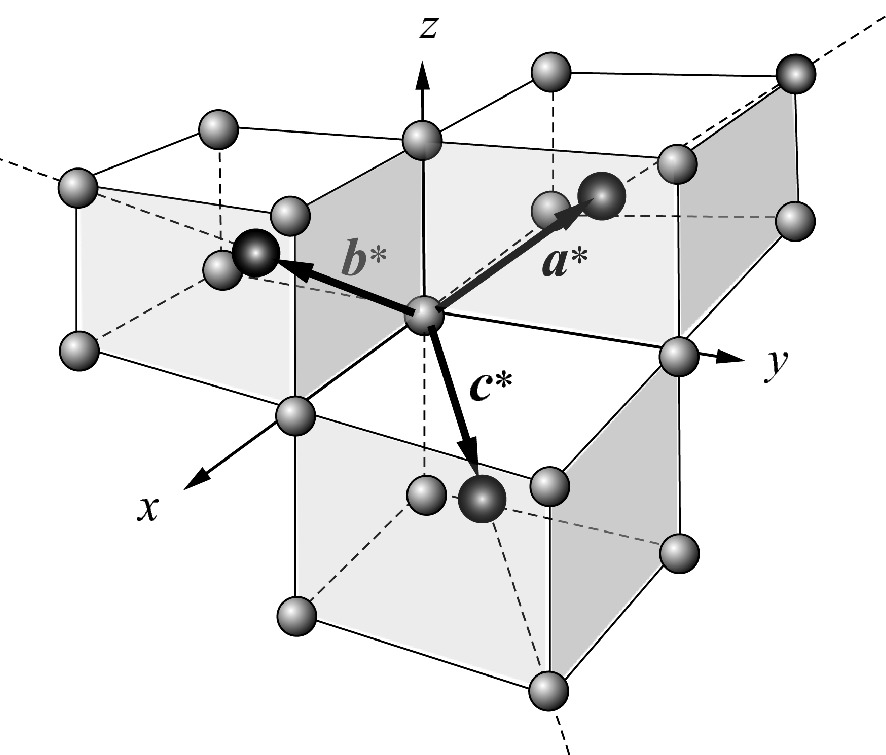
Reciprocal translation vectors a*, b*, and c* of the FCC crystal structure. The lattice points at the body centers are drawn as larger dark spheres
![$$ \varvec{G}_{hkl} = h\varvec{a}^{*} + k\varvec{b}^{*} + l\varvec{c}^{*} = \left( {\frac{2\pi }{a}} \right)\left[ {\left( { - h + k + l} \right)\varvec{a} + \left( {h - k + l} \right)\varvec{b} + \left( {h + k - l} \right)\varvec{c}} \right] $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_Equ70.png)
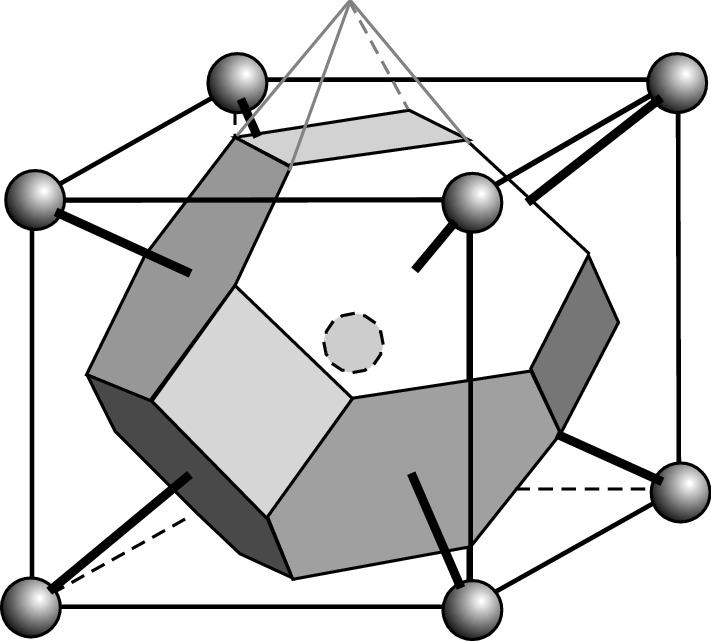
First Brillouin zone of the face-centered cubic lattice in reciprocal space. Six pyramid-shaped volumes (one shown in gray lines) are excluded since they are intersected by second nearest neighbors

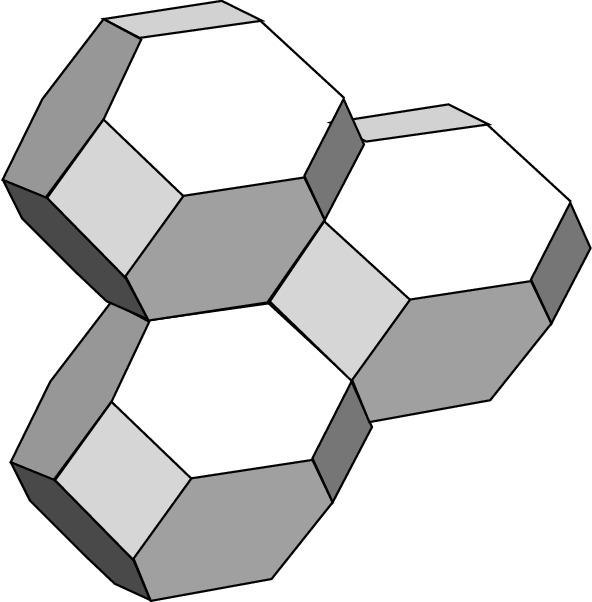
Way in which the first Brillouin zone cells of an FCC lattice can fill the whole space
- (c)
3D Brillouin zone of a simple hexagonal crystal
 and c* = 2π/c. Figure 2.50 shows seven points of the reciprocal lattice of a simple hexagonal lattice. If we take the central point O as origin, the other six lie at the corners of a regular hexagon of side a* =
and c* = 2π/c. Figure 2.50 shows seven points of the reciprocal lattice of a simple hexagonal lattice. If we take the central point O as origin, the other six lie at the corners of a regular hexagon of side a* =  and are the six nearest neighbors. The first Brillouin zone is formed by connecting bisectors of the lines joining O to the other six nearest neighbors. It is a hexagon whose sides have length
and are the six nearest neighbors. The first Brillouin zone is formed by connecting bisectors of the lines joining O to the other six nearest neighbors. It is a hexagon whose sides have length  = 4π/3a and hence covers a unit cell area of
= 4π/3a and hence covers a unit cell area of  =
=  . It should be noted that the hexagon forming the first Brillouin zone has the same orientation as the hexagons of the direct lattice.
. It should be noted that the hexagon forming the first Brillouin zone has the same orientation as the hexagons of the direct lattice.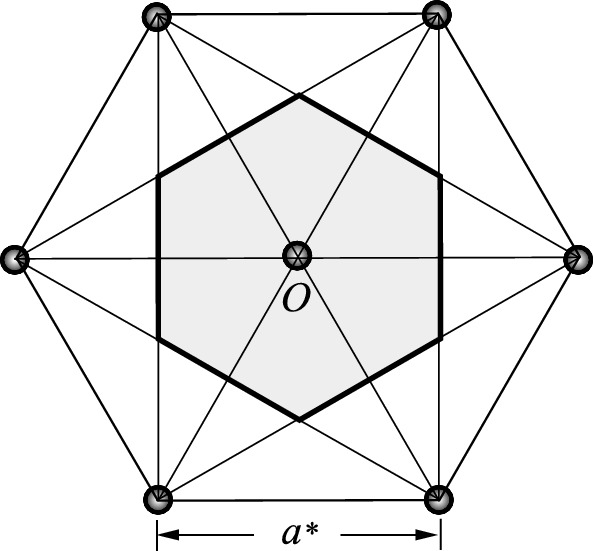
Construction of the first Brillouin zone for the plane simple hexagonal lattice. The reciprocal lattice points are shown at six corners and the origin
 . The first Brillouin zone is shown in Fig. 2.51, together with some points of the reciprocal lattice.
. The first Brillouin zone is shown in Fig. 2.51, together with some points of the reciprocal lattice.
First Brillouin zone for the simple hexagonal lattice. Some lattice points of the reciprocal lattice are also shown
- 1.
Both AsH3 and SiCl4 are common gases for semiconductor growth and processing. Discuss their molecular bonding properties.
- 2.The ionic radii of Na, Cs, and Cl are 0.875 Å, 1.455 Å, and 1.475 Å, respectively.
- (a)
For NaCl, the FCC structure is the preferred crystal structure. Verify that this structure prevents anion–anion or cation–cation contact.
- (b)
Verify that the simple cubic structure has a higher atomic packing density than the FCC structure in CsCl crystals.
- (a)
- 3.Calculate the atomic packing density for the following crystal structures:
- (a)
Simple FCC
- (b)
Simple hexagonal close-packed (HCP)
- (c)
Diamond
- (d)
Wurtzite.
- (a)
- 4.
Assume the cube containing a tetrahedron bond structure has a length a on each side. Calculate the bond length of the tetrahedron bond in terms of length a. Also, determine the angle between two tetrahedron bonds.
- 5.
The primitive unit cell of a hexagonal close-packed (HCP) structure can be defined by unit vectors a and c. Prove that, using the incompressible spheres model, the ratio between c and a is
 .
. - 6.
The primitive unit cell of a wurtzite structure can be defined by unit vectors a and c (Fig. 2.31). Derive the relations between a, c, and the bond length.
- 7.
Prove that the reciprocal lattice of a simple hexagonal lattice is another simple hexagonal lattice. Determine the new lattice constants along the a-axis and c-axis.
- 8.
- (a)
Derive the reciprocal lattice of the BCC structure. What is the new crystal structure?
- (b)
What is the angle between G110 and G101?
- (a)
- 9.
Make a plot of the first two Brillouin zones of a primitive rectangular two-dimensional lattice with axes a, b = 2a.
- 10.
Derive the dielectric constant tensor for a simple tetragonal crystal.
- 11.
The hexagonal crystal is called an optically uniaxial crystal and has two principal dielectric constants:
 along the c-axis and
along the c-axis and  in the basal plane. Show that the dielectric constant tensor of hexagonal crystals can be expressed as
in the basal plane. Show that the dielectric constant tensor of hexagonal crystals can be expressed as
![$$ \epsilon = \left[ {\begin{array}{*{20}c} {\epsilon_{11} } & 0 & 0 \\ 0 & {\epsilon_{11} } & 0 \\ 0 & 0 & {\epsilon_{33} } \\ \end{array} } \right]. $$](../images/325043_1_En_2_Chapter/325043_1_En_2_Chapter_TeX_Equi.png)
