Nephrology
ANATOMY
Kidneys
 Paired bean-shaped retroperitoneal organs, each with an adrenal gland on the upper pole (kidney’s “beanie hat”; Fig. 15-1A).
Paired bean-shaped retroperitoneal organs, each with an adrenal gland on the upper pole (kidney’s “beanie hat”; Fig. 15-1A).
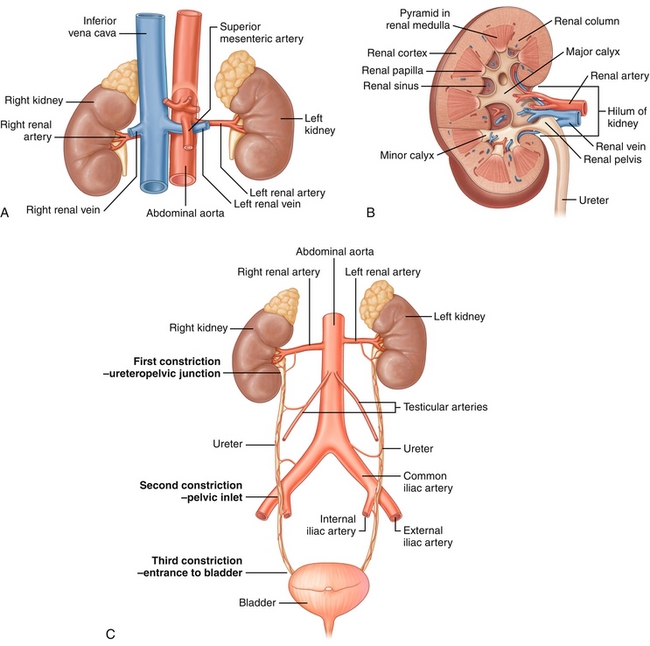
Figure 15-1 A, Kidneys and their relationship to the inferior vena cava and aorta. B, Coronal view of a kidney, detailing the interior anatomy. C, The urinary tract, including the kidneys, ureters, and bladder. (From Drake RL, Vogl AW, Mitchell AWM. Gray’s Anatomy for Students. 2nd ed. Philadelphia: Elsevier; 2009.)
The outermost part of the kidney is the cortex, and the inner part is the medulla (Fig. 15-1B).
Blood Supply
Perfused by renal arteries, which branch off the aorta. Both renal arteries further branch into smaller vessels to perfuse the glomeruli (site of blood filtration in each nephron); each glomerulus has an afferent and efferent arteriole, which modulate blood flow and glomerular filtration pressure.
After the glomeruli and efferent arteriole, the blood flows through peritubular capillaries, sites in which secretion and reabsorption of various substances occur. These capillaries run down the nephron into the medulla. Because the medulla is the last to be perfused, it has the least oxygen tension and is susceptible to hypoxia and hypoxic injury. These peritubular capillaries also serve an endocrine role, mediating release of erythropoietin when local hypoxia occurs to stimulate red blood cell production.
The kidneys are drained by renal veins that drain into the inferior vena cava (IVC). Because the IVC is on the right side, the left renal vein is longer and is therefore preferred for transplantation (more plumbing to work with).
Of note, the left gonadal vein drains into the left renal vein (as opposed to the right gonadal vein draining into the IVC). Because the renal vein is of smaller caliber compared with the IVC (higher resistance; resistance is proportional to 1/r4), there is greater potential for venous congestion in the left gonadal vein—hence, greater risk for testicular varicoceles on the left compared with the right side.
Urinary Collecting System
The renal pelvis has projections into the kidney to facilitate urine collection; the smallest projections are termed minor calices, with groups of them joining to become major calices.
The area where the minor calyx interfaces with the renal medulla is called the renal papilla. This becomes important in renal papillary necrosis (see later).
The major calices then unite and become the renal pelvis, which drains into the ureter.
The ureters drain urine from the renal pelvis into the bladder for storage (Fig. 15-1C).
Ureters have three sites of relative constriction in which obstruction of urine flow can easily occur (e.g., limiting kidney stone passage): (1) ureteropelvic junction (UPJ), where the renal pelvis meets the ureter; (2) pelvic inlet, where the ureter crosses over and is mildly compressed by the iliac arteries; and (3) ureterovesical junction (UVJ), where the ureters insert into the bladder (stones can get stuck where the ureter starts, where it enters the pelvis or where the ureter ends).
The ureters pass under the uterine artery (female) and vas deferens (male).
Mnemonic: Water (ureters) under the bridge (uterine artery, vas deferens). This is important to know during surgery to ensure the ureters are not damaged. It should be intuitive that the ureters are under these structures; the kidneys are retroperitoneal.
PHYSIOLOGY
Overview and Terminology
The kidneys have important functions:
1. Endocrine: (1) Synthesizes erythropoietin to stimulate red blood cell production; (2) converts 25-OH vitamin D into its active form, 1,25-(OH)2 vitamin D by the 1-α- hydroxylase enzyme in the proximal tubule; and (3) secretes the enzyme renin into circulation to start the renin-angiotensin-aldosterone (RAA) axis to increase blood pressure.
2. Homeostatic: (1) Eliminates waste products and water as urine; and (2) controls blood pH, electrolyte concentrations, and volume status.
There are some terms that must be covered before an in-depth discussion of renal physiology can occur; familiarize yourself with these before moving forward (Fig. 15-2):
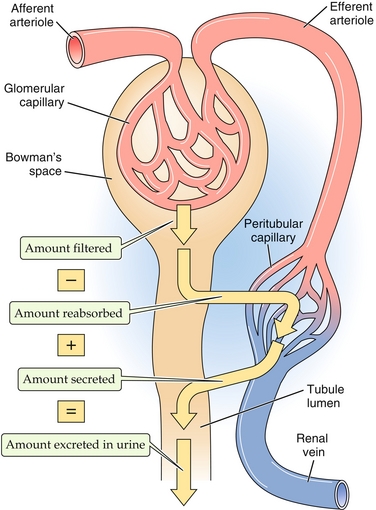
Figure 15-2 Terminology of the nephron. (1) Filtration, when a substance enters the nephron via the glomerulus through Bowman’s space. (2) Secretion, when a substance is pumped into the nephron at a site other than the glomerulus. (3) Reabsorption, when a substance is moved from the nephron back into the peritubular capillaries (and therefore back into the general circulation). (From Boron WF, Boulpaep EL. Medical Physiology. 2nd ed. Philadelphia: Elsevier; 2008.)
 Filtration: When a substance in question goes into Bowman’s space via the glomerulus (the kidney’s filter)
Filtration: When a substance in question goes into Bowman’s space via the glomerulus (the kidney’s filter)
Secretion: When a substance is delivered into the nephron from the peritubular capillaries (after the glomerulus)
Reabsorption: When something is returned into peritubular capillaries from the nephron
Clearance (of a substance X into the urine): The total plasma volume that is completely cleared of substance X per unit time (i.e., mL/min). A high amount of X in the urine (high urine concentration of X [U]X and/or a high urine flow rate, V; large numerator) in association with a low plasma concentration of X [P]X, (not much left to excrete; small denominator) indicates high clearance.
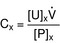
where CX = clearance of substance X, [P]X = plasma concentration of X, [U]X = urine concentration of X, and 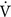 = urine flow rate.
= urine flow rate.
Glomerular Filtration Rate (GFR)
The amount of fluid being filtered through the glomerulus per unit time, normally 100 to 120 mL/min, but significantly reduced in renal failure. Inulin (not to be confused with insulin) is used to calculate GFR because it is a substance that is only filtered through the glomerulus and not significantly secreted or reabsorbed. Because infusing patients with inulin is unwieldy in the real world, creatinine (breakdown product of creatine phosphate, used as a phosphate source by muscle to quickly regenerate adenosine triphosphate [ATP] when needed), which is always present in the blood, is used as an endogenous surrogate for GFR. There are formulas that can approximate GFR based on creatinine levels (higher creatinine levels in the blood = lower GFR, because the creatinine is not being removed and is building up).
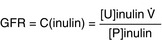
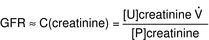
Clearance and GFR

When substance X is creatinine, the latter two processes are relatively small and presumed negligible; hence, creatinine clearance is traditionally used as an estimate of so-called pure glomerular filtration.
CX > GFR: Indicates that more of the substance got into the urine than by filtration alone (therefore, there was net tubular secretion of the substance as well).
CX < GFR: Indicates that less of the substance got into the urine than expected from filtration (therefore, was net tubular reabsorption of the substance as well).
CX = GFR: Exactly the same amount of substance got into the urine as was expected from filtration. This does not mean that no secretion or reabsorption occurred, but rather if it did, they happened to the same extent (no net secretion or reabsorption).
Effective renal plasma flow (ERPF): Estimates total amount of plasma flowing through the kidney, based on the clearance ofpara-aminohippuric acid (PAH). This substance is used because it has an incredibly high clearance; it is filtered (at the glomerulus) and secreted (by the proximal tubule of the kidney). Essentially 100% of the PAH is cleared from the bloodstream into the urine. Renal plasma flow is important because the glomeruli filter plasma; red blood cells are normally too big to be filtered (unless the kidney is diseased; nephritic syndromes have passage of blood through the glomeruli).
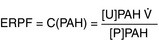
Renal blood flow (RBF): Related to renal plasma flow (RPF), because plasma is a component of blood. This also takes into account the red blood cells (RBCs) that are flowing through; knowing the plasma flow makes it an easy conversion, based on the hematocrit (HCT; the percentage of the blood that is RBCs).
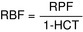
Logically, this makes sense, because if there were no RBCs in the blood (HCT = 0), it would all be plasma and the RBF would be the same as the RPF. With a normal hematocrit (e.g., 45%, [0.45]) the RBF would be higher than the RPF because it is taking into account red blood cells and plasma.
Filtration fraction (FF): Demonstrates what percentage of the plasma going through the glomerulus is actually put into the nephron. Normally, this is approximately 20%. Therefore, it is the simple fraction of how much is put into the nephron through the glomerulus (GFR) divided by all the plasma that went through the glomerulus (RPF).
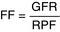
The Glomerulus and Filtration
The glomerulus is the interface between the blood and the nephron. The goal of the glomerulus is to prevent cells and protein in the blood from entering the nephron, creating a plasma ultrafiltrate, which is only comprised of electrolytes and low-molecular-weight solutes. The glomerulus has a specialized filtration barrier that helps ensure minimal loss of larger solutes with filtration (Fig. 15-3). From the blood to Bowman’s space, these barriers, in order, are as follows:
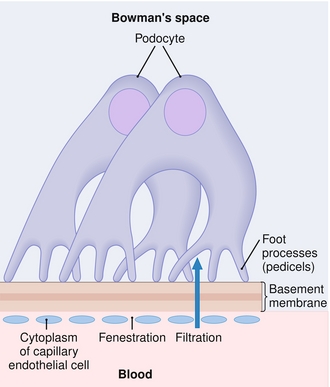
Figure 15-3 Layers through which a filtered molecule must pass, from the glomerular capillary bed (bottom) to Bowman’s space inside the nephron (top). (From Page CP, Hoffman B, Curtis M, Walker M. Integrated Pharmacology. 3rd ed. Philadelphia: Elsevier; 2006.)
1. Fenestrated capillary endothelium: Fenestrations (holes) in the capillary endothelium act as a size barrier, preventing cells from entering Bowman’s space.
2. Basement membrane: This basement membrane has three layers from the lamina rara interna (interna, internal, nearest to the capillary), lamina densa, and lamina rara externa (externa, external). This area is heavily negatively charged because of the presence of heparan sulfate (not to be confused with heparin, an anticoagulant). Because almost all blood proteins are negatively charged, this acts as a charge barrier to prevent proteins from filtration.
3. Podocytes: On the epithelium of Bowman’s space, the podocytes are attached to the basement membrane by means of foot processes, which have small slits between them that are also coated in heparan sulfate to ensure further that no proteins are filtered.
If the charge barrier is lost, nephrotic syndrome (where large proteins such as albumin are filtered and lost in the urine can occur.
Mnemonic: Nephrotic-oncotic rhymes; protein makes up oncotic pressure and severe protein loss in the urine is observed in nephrotic syndromes.
If inflammation allows entire cells to pass through, red blood cells can be filtered and lost in the urine, leading to nephritic syndrome. These syndromes will be covered later, in the pathology section.
The nephrons can only filter what the glomerulus is able to deliver. The blood that flows through the renal arteries eventually makes it to the glomeruli, first encountering the afferent arteriole, then the glomerular capillaries, and exiting out the efferent arteriole (the afferent arteriole comes before the efferent arteriole, A before E, just like the alphabet) into the peritubular capillaries, which feed the nephron oxygen and are conduits for reabsorption and secretion (Fig. 15-4). The fluid that passes through the glomerular capillaries goes into Bowman’s space and subsequently the nephron. Just like all capillary beds, the driving forces for fluid moving out of the capillary are the Starling forces of osmotic and hydrostatic pressure from the blood in the capillary bed and the interstitium surrounding the capillary bed.
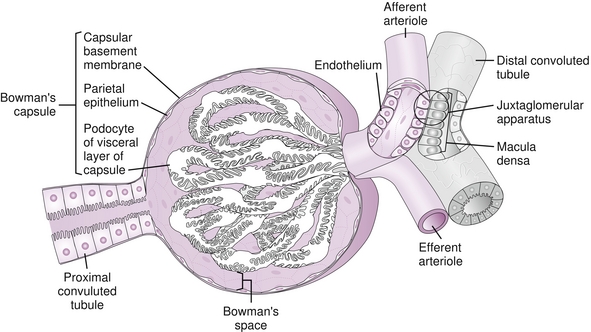
Figure 15-4 Blood flows into the glomerular capillary bed through the afferent arteriole, into the glomerular capillary bed, and out the efferent arteriole (A before E, just like the alphabet) into the peritubular capillaries. (From Bargmann W. Histologie und Mikronscopische Anatomie des Menshen. Stuttgart, Germany: Georg Thieme; 1977:86.)
As the fluid moves from afferent arteriole to efferent arteriole, fluid and not protein is pushed into Bowman’s space, which would normally reduce hydrostatic pressure as fluid left the capillaries. However, glomerular capillary hydrostatic pressure is constant along the entire capillary bed (unique to glomerular capillaries) because of vasoconstriction of the efferent arteriole acting as a dam that backs up the pressure into the glomerulus. Because fluid is leaving (and not protein), the glomerular capillary oncotic pressure will increase along the capillary bed until forces reach equilibrium. Therefore, alterations in any of the Starling forces will change the glomerular filtration rate (Table 15-1):
Table 15-1
Starling Force Alterations: Effects on Renal Plasma Flow (RPF), Glomerular Filtration Rate (GFR), and Filtration Fraction (FF)

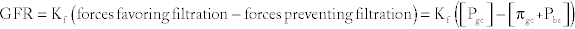
where Kf = filtration coefficient, Pgc = hydrostatic pressure in the glomerular capillaries (pushing fluid into Bowman’s space), πgc = oncotic pressure in glomerular capillaries (osmotic gradient pulling fluid into glomerular capillaries), Pbs = hydrostatic pressure in Bowman’s space, and πbs = oncotic pressure in Bowman’s space (omitted because proteins are not filtered; normally, this is zero).
Pgc increases with efferent arteriolar vasoconstriction and afferent arteriolar vasodilation and decreases with efferent arteriolar vasodilation and afferent arteriolar vasoconstriction.
πgc decreases with protein wasting (malnutrition, nephrotic syndrome).
Pbs increases with urinary obstruction (back pressure from obstruction goes to Bowman’s space).
πbs increases with nephrotic syndrome, because protein now leaks into Bowman’s space.
Changes in Starling forces may also alter the filtration fraction; higher glomerular capillary hydrostatic pressures (e.g., with efferent arteriole constriction) will “push” more plasma into Bowman’s space and a higher filtration fraction will result. Lower glomerular capillary oncotic pressures (e.g., hypoalbuminemia) will cause the capillaries to have less “pull” to keep fluid in and will result in a higher filtration fraction because a larger proportion of plasma is now going into Bowman’s space. The opposite conditions (efferent vasodilation, high capillary oncotic pressure) will cause a lower filtration fraction. Changes in the afferent arteriole do not modify filtration fraction because it is before the glomerular capillary bed (Fig. 15-5A); dilation of the afferent arteriole, for example, increases renal plasma flow and GFR to the same degree, so the fraction of plasma is unchanged (numerator and denominator increase the same).
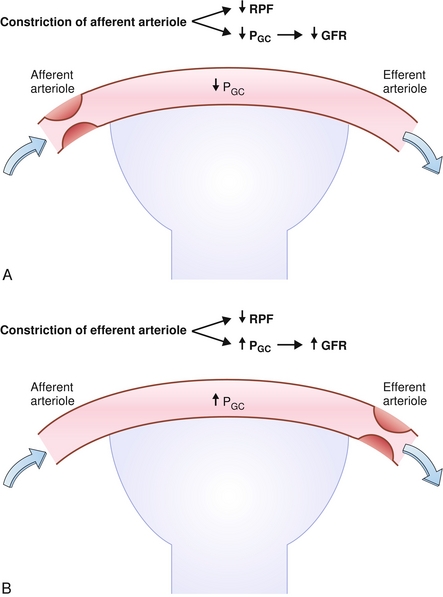
Figure 15-5 Effects of afferent arteriole constriction (A) and B, efferent arteriole constriction on renal plasma flow (RPF), glomerular hydrostatic pressure (PGC) and glomerular filtration rate (GFR). (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
Angiotensin II (AT II): Preferentially constricts the efferent arteriole (Fig. 15-5B). Part of the RAA axis, AT II is a vasoconstrictor that the body uses to maintain blood pressure when the patient becomes hypotensive. By preferentially constricting the efferent arteriole, it increases glomerular pressure (acts as a dam, preventing blood exiting the glomerulus) and therefore attempts to maintain GFR, in spite of hypotension. With high levels of AT II (e.g., in severe hypotension), the afferent arteriole will constrict as well, which lowers the GFR because the blood is needed elsewhere (e.g., the brain).
Prostaglandins: Vasodilate the afferent arteriole, increasing renal plasma flow. Nonsteroidal anti-inflammatory drugs (NSAIDs) prevent prostaglandin synthesis and in turn cause decreased renal plasma flow. In hypotensive or hypovolemic patients who already have low flow to the kidney, taking NSAIDs can further cut off blood supply to the kidney, leading to damage.
Remember that when a substance is filtered, it does not necessarily end up in the urine; it can be reabsorbed into the bloodstream during passage through the nephron. Similarly, when a substance is not filtered, it can still end up in the urine; it can be secreted into the nephron from the bloodstream. Next, the nephron and its segments and pumps will be discussed.
The Nephron and Its Segments
See Figure 15-6.
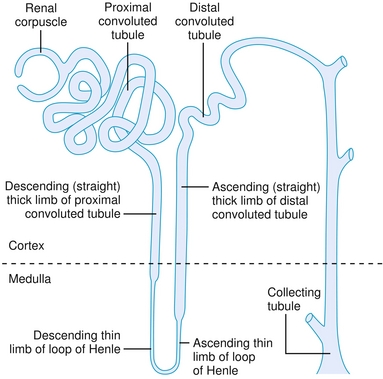
Figure 15-6 Overview of the nephron. (From Telser AG, Young JK, Kate M, Baldwin KM. Elsevier’s Integrated Histology. Philadelphia: Elsevier; 2007.)
Proximal Tubule
The only site of the nephron that has isosmotic reabsorption; there are small gaps between cells that allow water to move back into the peritubular capillaries along with solutes, so the osmolality does not change from the urinary to blood space.
Reabsorbs all glucose and amino acids if present at normal levels. However, high levels overwhelm the ability of these transporters, leading to glucose being present in the urine (glucosuria) at blood glucose levels over 200 mg/dL (with all transporters fully saturated at 350 mg/dL), or protein in the urine (proteinuria), with large amounts of filtered protein.
Na+ reabsorbed via the Na+/H+ exchanger, with H+ being secreted into the lumen in exchange. This H+ will eventually be recycled (put back into the proximal tubule cell and secreted again into the lumen (Fig. 15-7) in the following fashion: (1) the Na+/H+ exchanger secretes H+ into the lumen to reabsorb Na+, (2) this H+ combines with bicarbonate to make H2CO3; (3) H2CO3 becomes H2O and CO2, catalyzed by carbonic anhydrase (carbonic anhydrase inhibitors such as acetazolamide block this step, shutting down this recycling); (4) CO2, a freely diffusible gas, diffuses back into the cell; and (5) again with carbonic anhydrase, the CO2 and H2O become H2CO3, breaking down into H+ and 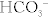 . The
. The 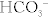 is pumped into the peritubular capillaries and reabsorbed and the H+ is put back into the tubular lumen to restart the process. This Na+/H+ exchanger is upregulated by AT II (see earlier).
is pumped into the peritubular capillaries and reabsorbed and the H+ is put back into the tubular lumen to restart the process. This Na+/H+ exchanger is upregulated by AT II (see earlier).
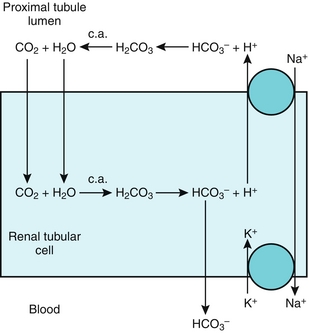
Figure 15-7 The proximal tubule. Shown is the H+ recycling system mediated by the Na+/H+ exchanger and carbonic anhydrase enzyme to help reabsorb bicarbonate. This process is inhibited by carbonic anhydrase (c.a.) inhibitors such as acetazolamide. (From Goljan EF, Sloka KI: Rapid Review Laboratory Testing in Clinical Medicine. St. Louis: Mosby Elsevier; 2008:32.)
The Na+/H+ exchanger functions well because the H+ secreted into the lumen is buffered (mostly by phosphate) and therefore the H+ gradient continues to be favorable by ensuring that the concentration of free H+ remains low. However, there is an additional method whereby the kidneys can get rid of additional H+ and generate additional 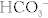 when too much acid is present (e.g., during respiratory acidosis). The proximal tubule cells also can metabolize glutamine by removing the ammonia (NH3) off it, leaving α-ketoglutarate behind, which can enter into the Krebs cycle and be metabolized to CO2 and water. The CO2 and H2O can be changed into
when too much acid is present (e.g., during respiratory acidosis). The proximal tubule cells also can metabolize glutamine by removing the ammonia (NH3) off it, leaving α-ketoglutarate behind, which can enter into the Krebs cycle and be metabolized to CO2 and water. The CO2 and H2O can be changed into 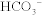 and H+ by carbonic anhydrase activity. This H+ can be secreted into the lumen with NH3, creating
and H+ by carbonic anhydrase activity. This H+ can be secreted into the lumen with NH3, creating 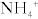 ; the bicarbonate left over is reabsorbed into the bloodstream, and in effect, a new
; the bicarbonate left over is reabsorbed into the bloodstream, and in effect, a new  is generated and the ammonium will be excreted (Fig. 15-8).
is generated and the ammonium will be excreted (Fig. 15-8).
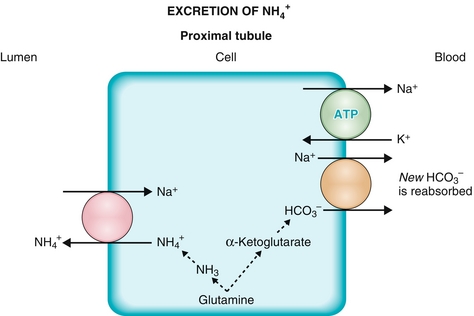
Figure 15-8 The proximal tubule. Shown is the method for generating new bicarbonate to be put into the bloodstream via the breakdown of glutamine and secretion of ammonium into the urine. (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
Phosphate is reabsorbed in this nephron segment. Parathyroid hormone (PTH) inhibits this process, hence PTH promotes phosphaturia (see Chapter 9).
Absorbs and secretes organic cations and anions, such as uric acid and lactic acid.
The proximal tubule has the 1-α-hydroxylase enzyme, which converts vitamin D into its active form, calcitriol (1,25-dihydroxycholecalciferol) by adding a hydroxyl group to the position 1 of 25-hydroxyvitamin D; this activity is upregulated by parathyroid hormone (PTH). Calcium homeostasis is covered in detail in Chapter 9.
Thin Loop of Henle
The descending thin limb is highly permeable to water, whereas the ascending limb is permeable to solutes.
There is an osmotic gradient (corticopapillary gradient) where the medulla has a much higher osmolarity of the interstitial fluid than the cortical area; the thin loop of Henle takes advantage of this because water and solutes are permeable in this part of the nephron (the part of the nephron furthest toward the medulla). It uses a countercurrent multiplication system to generate and maintain this gradient.
The net effect of the thin loop of Henle is that more solutes are lost (moved out of the tubular lumen) than water (because the ascending limb is permeable to solutes), leading to hyposmotic fluid generation.
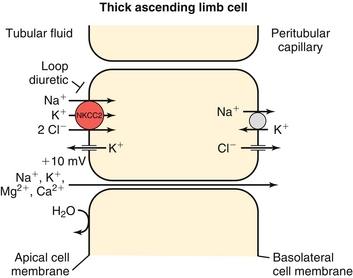
Thick Ascending Loop of Henle
Has tight junctions which prevent reabsorption of water; only solutes are modified here.
The thick ascending loop of Henle uses the NKCC2 (Na+-K+-Cl−-Cl−) cotransporter (inhibited by loop diuretics; Fig. 15-9). This ATP-driven pump pumps two cations (Na+ and K+) and two anions (Cl− and Cl−) into the thick ascending limb cell, but the potassium can leak back into the lumen via a potassium channel (see Fig. 15-9). Therefore, although the pump itself does not directly generate an electric gradient, it does do so indirectly by allowing a cation back into the tubular lumen. The sodium and chloride are reabsorbed into the peritubular capillaries. Because the cation leaked back into the lumen, there is a more net positive voltage in the tubular lumen, aiding in the reabsorption of other cations, especially calcium and magnesium, via a paracellular (between cells) route. Shutting this pump off with a loop diuretic then causes decreased magnesium and calcium reabsorption as well.
Distal Convoluted Tubule
Home to a NaCl cotransporter (inhibited by thiazide diuretics), which normally reabsorbs sodium and chloride in equal amounts (Fig. 15-10).
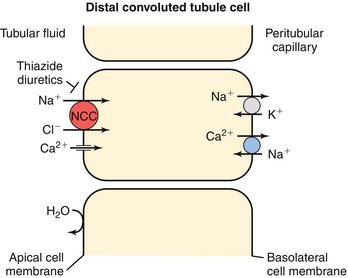
Figure 15-10 The distal convoluted tubule, showing the NaCl cotransporter blocked by thiazide diuretics such as hydrochlorothiazide. (From Wecker L, Crespo L, Dunaway G, et al. Brody’s Human Pharmacology. 5th ed. Philadelphia: Elsevier; 2009.)
There is also a calcium channel on these cells, which takes up calcium from the urine. Because the voltage gradient for reabsorption is only so large, whenever sodium is reabsorbed into this cell, less calcium can be reabsorbed (both cations “fighting” for the same electrochemical gradient). Therefore, blockage of the NaCl cotransporter with a thiazide diuretic will allow increased calcium reabsorption (opposite of loop diuretics).
Collecting Duct
Has alpha-intercalated cells (Fig. 15-11A) responsible for secretion of acid (remember: alpha, acid) into the lumen and principal cells, responsible for the reabsorption of sodium at the expense of secreting potassium; the activity of both cells is upregulated by aldosterone.
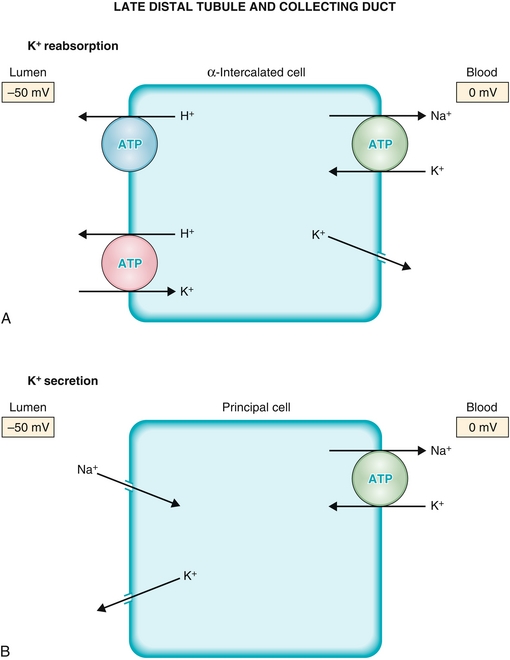
Figure 15-11 The late distal tubule and collecting duct. A, Alpha-intercalated cells, which secrete acid into the urine. B, Principal cells which reabsorb sodium at the expense of excreting potassium. Both these cells increase their activity in response to aldosterone by synthesizing new proteins, such as increasing the number of epithelial sodium channels (ENaCs) in the principal cells. (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
The principal cells (Fig. 15-11B) are the final place in the nephron at which sodium and potassium balance can be adjusted before excretion. Any increased sodium delivery to the principal cells causes sodium to move from the lumen of the nephron into the principal cell through the epithelial Na+ channel (ENaC) because of an increased electrochemical gradient (increased difference in sodium concentration from tubular lumen to distal convoluted tubule cell). This Na+ is subsequently pumped into the blood by the Na+/K+ ATPase, but recall that this pumps sodium out and potassium in. Therefore, the more sodium is pumped out, the more potassium is moved into the principal cell. This potassium then flows through a potassium channel into the tubular lumen, with eventual excretion in the urine. Aldosterone upregulates this (see discussion of RAA axis) in an effort to increase intravascular volume by taking up more sodium, but in the process it causes K+ secretion and also stimulates H+ secretion in the alpha-intercalated cells.
The collecting duct also has an important effect on water balance because it can cause increased water reabsorption in the presence of antidiuretic hormone (diuretics increase urine output, antidiuretic hormone decreases it by reabsorbing water). Increased water reabsorption leads to concentrated urine. The function of ADH and its mechanism of action are covered later (see “Antidiuretic Hormone and Free Water Clearance”).
Body Fluid Compartments and Maintenance
The human body, by weight, is mostly water. This water is present inside the cells (intracellular) and outside the cells (extracellular). The 60-40-20 rule is that 60% of an average person’s body weight is water, with two-thirds (40% of body weight) of that water in the intracellular fluid and one-third (20% of body weight) is in the extracellular fluid. The extracellular compartment is divided into plasma (or intravascular; i.e., in the bloodstream) and interstitial fluid, the area between the cells and capillary beds. Of the extracellular fluid, 25% is plasma volume and 75% is interstitial volume. See Figure 15-12.
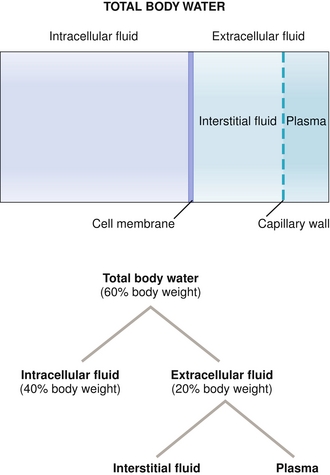
Figure 15-12 Distribution of fluid in the body. Note the 60-40-20 rule—60% of a person’s body weight is water (more if lean, less if fat), 40% of the body weight (two-thirds of the water) is intracellular fluid, and 20% of the body weight (one-third of the water) is extracellular fluid. Of the extracellular fluid, 75% is interstitial fluid and 25% is plasma. (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
Total body water (60% total body weight): Represents all fluid compartments (intracellular plus extracellular compartments); can be measured by using deuterated water, D2O (water with deuterium, hydrogen with an extra neutron, taking place of the hydrogen atoms), because water will come to equilibrium with the rest of the body.
Intracellular fluid (ICF; two-thirds of total body water): Impossible to measure directly because you cannot measure the amount of water inside each cell. Therefore, measurement of this is indirect by taking the total body water and subtracting the extracellular water, leaving you with only intracellular water. The major cation here is potassium and the major anion is phosphate.
Extracellular fluid (ECF; one-third of total body water): Represents plasma volume and interstitial volume together (because both of these compartments are outside the cells). ECF can be measured by using sugars that cannot be metabolized by the body, such as mannitol or inulin. These sugars cannot get into the cells but can go into plasma and the interstitial compartment. The major cation in the extracellular compartment is sodium because cells have Na+/K+ ATPases that continually pump sodium out of the cells in exchange for pumping potassium into the cells.
Plasma volume (25% of the ECF): Contains albumin, the major anion of plasma, which also provides oncotic pressure to keep fluid inside the vessels. Therefore, radioactive albumin can measure the plasma volume (or Evans blue, a blue dye that attaches strongly to albumin).
Interstitial fluid (75% of the ECF): Just like plasma, except no plasma proteins or cells, because the capillaries do not normally permit their passage. Cannot be directly measured, but if the ECF and plasma volume are known, then the ECF minus the plasma volume must be the interstitial volume.
The normal osmolarity of these compartments is 290 mOsm/L. The osmolarity of the intracellular and extracellular compartments should be identical because any changes will lead to osmotic water shifts to balance the osmolarities. For example, if someone urinates extremely hypotonic fluid (almost urinating water, as in diabetes insipidus), the ECF will increase in osmolarity because water but not solute is being lost, in effect concentrating the blood. The ICF, now relatively lower in osmolarity, will have water move from the ICF (lower osmolarity) to the ECF (higher osmolarity) to balance the osmolarity; this is termed osmosis. On the other hand, if a patient had diarrhea, which is isosmotic to the body’s fluids (because the colon does not have pumps to generate a gradient and therefore reaches equilibrium with the cells), fluid would be lost but no changes in osmolarity would occur and therefore no fluid shifts.
It is important to remember that all changes begin in the ECF because the cells themselves cannot generate water or solute (therefore, changes do not begin in the ICF), but urinary or gastrointestinal (GI) losses can take fluid and electrolytes out of the interstitial and plasma volumes, just as drinking or eating sodium will put fluid or sodium into your interstitial or plasma volumes. Because with loss of fluid (volume contraction) or gain of fluid (volume expansion) there can also be changes in solute, the concentration of the fluid lost or gained is important. It is important to understand that osmolarity and volume do not necessarily move together; it is possible to lose volume and have higher or lower osmolarity, just as, it is possible to gain volume and have higher or lower osmolarity. It just depends on the concentration of the fluid lost or gained.
Refer to Figure 15-13 when analyzing the following:
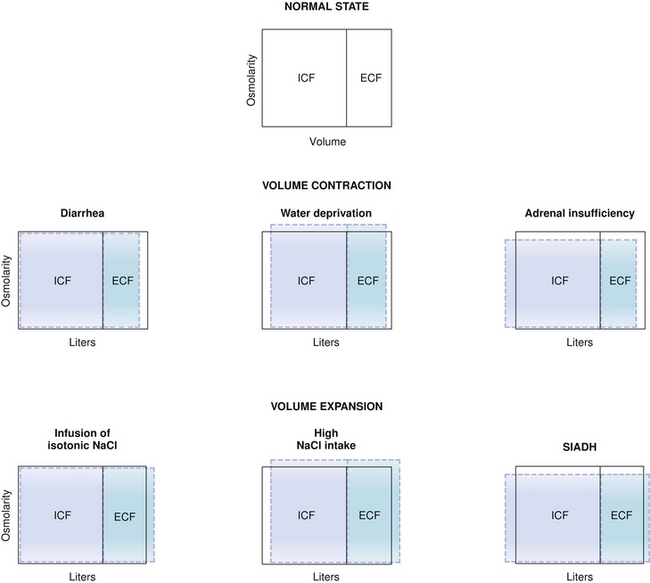
Figure 15-13 Changes to ICF and ECF volumes after various disturbances (middle row, volume contraction; bottom row, volume expansion). ECF, extracellular fluid; ICF, intracellular fluid. (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
Loss of isosmotic fluid (e.g., diarrhea) will take fluid just from the ECF (with no osmotic gradient generated because the ECF osmolarity will be unchanged).
Loss of more water than solute (e.g., diabetes insipidus or insensible fluid loss, as in sweating or breathing) will lead to the ECF becoming hyperosmotic, with an osmotic gradient now favoring water moving from the ICF (osmolarity = 290 mEq/L) to the ECF (osmolarity > 290 mEq/L) until equilibrium is reached.
Loss of more solute than water (e.g., adrenal insufficiency, leading to decreased ability to reabsorb sodium in the kidney because of lack of aldosterone) will cause the ECF to become hyposmotic, favoring water movement from ECF (osmolarity > 290 mEq/L) to ICF (osmolarity = 290 mEq/L) until equilibrium is reached.
Gain of isosmotic fluid (e.g., 0.9% normal saline intravenous [IV] infusion) will lead to ECF expansion with no osmotic gradient because the ECF will still be 290 mEq/L.
Gain of more solute than water will cause the ECF to be hyperosmotic (e.g., 3% normal saline IV infusion), leading to water moving from ICF to ECF until equilibrium is established.
Gain of less solute than water (e.g., syndrome of inappropriate antidiuretic hormone secretion [SIADH], increased ADH secretion, leading to abnormally high amounts of water reclamation from the kidney) will cause the ECF to become hyposmotic, leading to water moving into the ICF until equilibrium is established.
Renin-Angiotensin-Aldosterone Axis
The RAA axis plays a critical role in the following: (1) maintenance of blood pressure; (2) altering urinary sodium excretion; and (3) altering renal blood flow parameters. The RAA axis is initially triggered when the juxtaglomerular apparatus (JG cells) in the afferent arteriole of the kidney sense a decrease in perfusion (hypovolemia, hypotension). See Figure 15-14 when referring to the following explanation of the axis pathway.
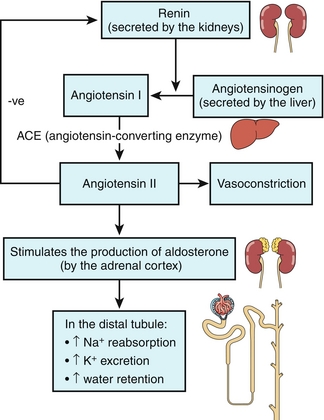
Figure 15-14 The renin-angiotensin-aldosterone axis. (From Jones TC. Crash Course: Renal and Urinary Systems. 3rd ed. Philadelphia: Elsevier; 2007.)
1. The JG cells, in response to decreased blood flow (decreased stretch of the afferent arteriole), secrete the proteolytic enzyme renin into the bloodstream. Another important stimulus for renin release includes sympathetic outflow via stimulation of the β1-adrenergic receptors on the JG cells and subsequent renin release.
2. Renin cleaves angiotensinogen in the bloodstream (produced by the liver) into angiotensin I.
3. Angiotensin I becomes angiotensin II by angiotensin-converting enzyme (ACE) found mostly in the pulmonary endothelium.
4. Angiotensin II is a potent vasoconstrictor, increasing blood pressure by increasing systemic vascular resistance. It preferentially constricts the efferent arteriole in the kidney to maintain GFR in low blood pressure states by increasing glomerular capillary hydrostatic pressure. In addition, it increases the activity of the Na+/H+ exchanger in the proximal tubule of the nephron to increase sodium and water reabsorption, leading to increased blood pressure. Finally, it is a potent dipsogen, meaning that it stimulates thirst to try to get the patient to consume more fluid to replenish intravascular volume.
5. Angiotensin II stimulates release of the hormone aldosterone from the zona glomerulosa of the adrenal gland cortex.
6. Aldosterone acts on the distal tubules to increase sodium reabsorption (at the expense of potassium excretion), resulting in expanded blood volume and increased blood pressure. Because aldosterone is a steroid hormone, it works by increasing transcription of new sodium channels (luminal side) and the Na+/K+ ATPase pumps (basolateral side) to assist in reclaiming sodium. This means that there is a lag on the order of hours before aldosterone can have an effect because it takes time to make the new proteins.
Antidiuretic Hormone and Free Water Clearance
Antidiuretic hormone (the “no pee” hormone; just as the diuretic medication class causes urination, anti-diuretic hormone stops it) is produced by the posterior pituitary gland. ADH increases water reclamation, leading to low urine volume and concentrated urine (decreased free water clearance). ADH increases water permeability at the late distal tubule and collecting duct; these cells have V2 receptors that are coupled to the Gs signaling pathway, leading to protein kinase A–mediated phosphorylation of proteins, with various effects:
Primary effect: Fusion of preformed aquaporin 2 (AQP2) channels into the cell membrane, leading to increased water reclamation, but leaving the solutes and therefore leading to concentrated urine.
Additional effects: Increased water reclamation would destroy the osmotic gradient in the loop of Henle because it would get overly diluted with all the water moving into the interstitial space. Therefore, ADH also increases the NKCC2 cotransporter in the thick ascending limb to ensure that more solutes are available to maintain the gradient and increase urea permeability to maintain the gradient further.
The two primary stimuli for ADH release are increased plasma osmolarity and low intravascular volume (the volume that effectively circulates in the blood vessels), such as what might happen when someone stops drinking water (while insensible hypotonic fluid loss from sweating and respiration are ongoing). The syndrome of inappropriate ADH secretion (SIADH) when ADH is secreted abnormally, as well as diabetes insipidus (DI), in which the ability to make ADH is impaired (central DI) or the receptor on the kidney does not function correctly (nephrogenic DI), were discussed earlier (“Pathology”).
ADH hormone is the primary determinant of free water clearance (CH2O). Free water clearance is simply a way to express if the urine is hypotonic (dilute urine) with respect to the plasma, or hypertonic (concentrated urine). Plasma osmolarity is about 290 mOsm/L and the urine concentration can range from 50 mOsm/L (very hypotonic, very positive free water clearance, because the urine is almost all water) to 1200 mOsm/L (very hypertonic, very negative free water clearance).
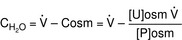
CH2O is positive: Means that free water is being removed in the urine and therefore the urine must be more dilute than plasma. Imagine the most extreme case, in which the urine is almost all water (50 mOsm/L). The kidney must have essentially no ADH activity and therefore no water in the distal nephron and collecting duct is reclaimed. Therefore, because the urine is essentially all water, free water is being removed from the body.
CH2O is negative: Means that free water is not being removed; the urine must be more concentrated than plasma. The term negative free water clearance may be confusing; just remember this is essentially free water “kept” by the body as opposed to free water “cleared” by the body.
CH2O is zero: Means that the urine has the exact same osmolarity as plasma, isosmotic. This can occur in renal failure when the kidney loses the ability to concentrate or dilute urine because of extensive damage, or with loop diuretic usage because the thick ascending limb with the NaKCC channels is the site of free water generation (remember that the proximal tubule has isosmotic reabsorption). Because loop diuretics inhibit this channel, it will lead to zero free water generation.
Other Endocrine Functions of the Kidneys
The kidney also functions as an endocrine organ in addition to the RAA axis described earlier. It also plays a role in (1) red blood cell synthesis via erythropoietin production and (2) activation of vitamin D via 1-α-hydroxylase enzyme in the proximal tubule of the kidney.
Erythropoietin stimulates the hematopoietic stem cells in the bone marrow to increase red blood cell synthesis. The stimulus for this release is hypoxia in the peritubular capillary endothelium (a small amount of erythropoietin can also be created in the liver, a remnant from when it was mostly generated there in utero). Of interest, hepatocellular carcinoma (liver cancer) is associated with erythrocytosis, or excess red blood cell production, for this reason.
Vitamin D metabolism and effects are discussed in depth in Chapter 9. The kidney is responsible for the final step of turning vitamin D into its active form, calcitriol (1,25-dihydroxycholecalciferol), by adding a hydroxyl group to position 1 of 25-hydroxyvitamin D by 1-α-hydroxylase enzyme in the proximal tubule of the kidney. Active vitamin D increases intestinal absorption of both calcium and phosphate. The activity of 1-α-hydroxylase is primarily dependent on PTH.
pH Homeostasis and Acid-Base Disturbances
The body is normally kept at a pH of 7.40 ± 0.05 (7.35 to 7.45). Whenever the pH is lower than this, it is termed acidemia; if the pH is higher than this, it is termed alkalemia. Acids are continually being created in the body, fixed acids (e.g., sulfuric and phosphoric acid, acids that cannot be breathed out and must be excreted by the kidney) and CO2 (carbon dioxide), which is a volatile acid because it can be breathed out. The reason that our pH does not vary wildly over the course of the day is due to buffers, which resist changes in pH (remember your chemistry courses), with bicarbonate being the most important. It is especially important because of carbonic anhydrase, which catalyzes the (bolded) reaction between H+ and  to H2O and CO2 (
to H2O and CO2 (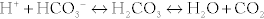 ). Because CO2 is highly diffusible, it can move from the blood to the alveoli of the lungs with ease; this CO2, a volatile acid, can then be removed from the body simply by exhaling. When we talk about the various acid-base disturbances, this reaction is why hyperventilation is a compensation for metabolic acidosis; the body is attempting to expel CO2 to get rid of acid as a compensatory mechanism. The kidneys play a role in fixed acid balance, because they reabsorb bicarbonate and excrete H+, and can even generate ammonia to help buffer H+. This is why individuals with renal failure develop a metabolic acidosis; they have lost this important regulatory mechanism. Therefore, it should be apparent that there are two main organs responsible for acid-base balance, the lungs (managing the volatile acid CO2) and the kidneys (managing the fixed acids).
). Because CO2 is highly diffusible, it can move from the blood to the alveoli of the lungs with ease; this CO2, a volatile acid, can then be removed from the body simply by exhaling. When we talk about the various acid-base disturbances, this reaction is why hyperventilation is a compensation for metabolic acidosis; the body is attempting to expel CO2 to get rid of acid as a compensatory mechanism. The kidneys play a role in fixed acid balance, because they reabsorb bicarbonate and excrete H+, and can even generate ammonia to help buffer H+. This is why individuals with renal failure develop a metabolic acidosis; they have lost this important regulatory mechanism. Therefore, it should be apparent that there are two main organs responsible for acid-base balance, the lungs (managing the volatile acid CO2) and the kidneys (managing the fixed acids).
There are four basic processes that account for all acid-base disturbances (although more than one may be present at any time)—metabolic acidosis or alkalosis, and respiratory acidosis or alkalosis. See Figure 15-15 and follow the explanations.
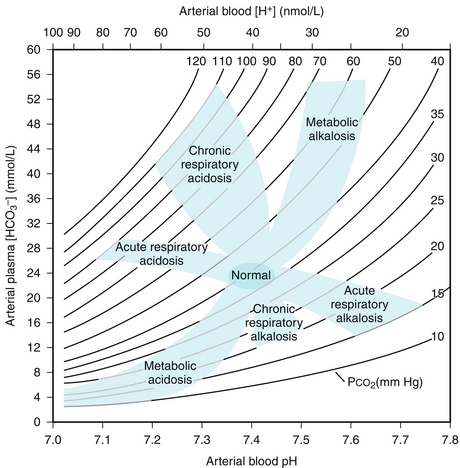
Figure 15-15 Acid-base nomogram showing the normal pH range and various acid-base disturbances that can occur. (From Taussig LM, Landau LI. Pediatric Respiratory Medicine. 2nd ed. Philadelphia: Elsevier; 2008.)
Metabolic Acidosis: Caused by a decrease in bicarbonate because of increased fixed acids (e.g., lactate) or loss of bicarbonate (e.g., through the stool in diarrhea). Metabolic acidosis is further divided into an anion gap metabolic acidosis (when the calculated anion gap is ≥ 12) or a non–gap metabolic acidosis (when the calculated anion gap is normal, or < 12). The anion gap is the difference between unmeasured anions and cations. It is derived from the fact that the total number of cations is equal to the total number of anions in the plasma (for electroneutrality, total cations = total anions). That is,
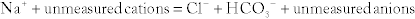
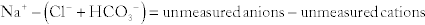
The quantity [unmeasured anions = unmeasured cations] is defined as the anion gap. In high anion gap metabolic acidosis, the acid HA enters circulation as an acid H+ and an anion A− (e.g., lactic acid = H+ + lactate−). The acid H+ consumes an equivalent number of basic 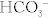 and leaves behind an anion A− in the plasma that is normally not measured. This excess unmeasured A− causes the anion gap to increase, or high anion gap metabolic acidosis. A non–gap metabolic acidosis can be caused by diarrhea (colonic secretion of bicarbonate in exchange for chloride; hence, no change in anion gap because for every bicarbonate lost, there will be a chloride gained, and the measured anions [chloride + bicarbonate] will therefore not change), or by renal tubular acidosis (equivalent loss of urinary bicarbonate and sodium in proximal renal tubular acidosis; increased urinary sodium loss with reduced acid H+ secretion in distal renal tubular acidosis. In both types of renal tubular acidosis, there is no change in anion gap, defined by equation above).
and leaves behind an anion A− in the plasma that is normally not measured. This excess unmeasured A− causes the anion gap to increase, or high anion gap metabolic acidosis. A non–gap metabolic acidosis can be caused by diarrhea (colonic secretion of bicarbonate in exchange for chloride; hence, no change in anion gap because for every bicarbonate lost, there will be a chloride gained, and the measured anions [chloride + bicarbonate] will therefore not change), or by renal tubular acidosis (equivalent loss of urinary bicarbonate and sodium in proximal renal tubular acidosis; increased urinary sodium loss with reduced acid H+ secretion in distal renal tubular acidosis. In both types of renal tubular acidosis, there is no change in anion gap, defined by equation above).
Metabolic alkalosis: Caused by loss of acids or retention/gain of bicarbonate. Kidney loss of acids is predominantly regulated by aldosterone. Any condition with increased aldosterone production can induce metabolic alkalosis because of increased acid H+ secretion at the collecting tubule. Hyperaldosteronism can be primary (a tumor or hyperplasia of the adrenal glands causing increased aldosterone release) or secondary (secondary to renal artery stenosis, making the JG cells secrete more renin and increase activation of the RAA axis). Kidney retention of bicarbonate predominantly occurs when there is no chloride delivery distally to the collecting tubules. Bicarbonate secretion at this nephron segment requires an exchange for a urine chloride. In volume-depleted states, most of the filtered sodium chloride is reabsorbed in the proximal tubules, which severely limits chloride delivery to the collecting tubules to exchange for a bicarbonate secretion. Finally, there’s one cause of metabolic alkalosis that is caused by the gain of bicarbonate, excessive ingestion of the antacid calcium carbonate. The body will respond by attempting to retain CO2 (volatile acid) and so the respiratory rate will be slow.
Respiratory acidosis: Caused by hypoventilation because CO2 (volatile acid) is not being adequately expelled by the lungs. There can be many reasons for this, from the blood-alveolar interface (fibrosis, edema) to the muscles of inspiration (diaphragmatic paralysis from phrenic nerve damage), to obstruction of the airway, to loss of inspiratory drive from a central nervous system (CNS) insult (especially in the medulla, where the respiratory center is located) or medications (especially opiates). The kidneys will respond by attempting to excrete more acid and generate more bicarbonate.
Respiratory alkalosis: Caused by (you guessed it) hyperventilation, blowing off too much of the volatile acid CO2, leading to alkalosis. Any stimulus to breathe above normal will cause this, from a panic attack to hypoxemia (the normal drive to breathe is usually CO2, but can be O2 if severe hypoxemia exists). The kidneys will attempt to compensate by retaining fixed acids and getting rid of bicarbonate by decreasing reabsorption.
The body will attempt to compensate to adjust the pH back to normal, because all enzymes and physiologic processes were intended to function at a normal pH (Table 15-2). Changes in this pH can cause abnormal organ function. However, compensation takes time; the lungs are more easily able to respond to a metabolic acidosis or alkalosis because changes in respiratory rate occur quickly. The kidneys, on the other hand, have to adjust the reabsorption and secretion of acids and bases, which takes longer. It is also important to remember that compensation is never complete. This is helpful because even if a metabolic acidosis is well-compensated, the pH will never return to 7.40; it will be slightly less, allowing you to determine that the initial disturbance was an acidosis.
Table 15-2
Acid-Base Disturbances: Their Effects pH, Pco2, and 
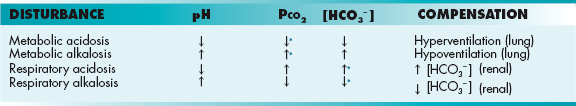
*Denotes a compensatory response to attempt to keep pH as close to normal as possible, but compensation is never complete.
Understanding whether an acidosis or alkalosis is properly compensated is difficult to remember because each has specific criteria. In general, if the pH is close to normal, it is compensating appropriately, whereas if the pH is significantly abnormal, it is uncompensated. It is important to remember that the normal Pco2 is 40 mm Hg and the normal bicarbonate concentration is 24 mEq/L. There are also two formulas to remember as well:
Winter’s formula for metabolic acidosis:
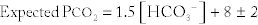
Because the expected change for metabolic acidosis is hyperventilation to get rid of CO2, if the bicarbonate level goes from 24 (normal) to 16 mEq/L (low), then the Pco2 should be = 1.5 (16) + 8 ± 2 = 32 ± 2 (30 − 34) mm Hg, which is less than the normal value of 40 mm Hg, indicating that CO2 has been blown off to compensate for the metabolic acidosis.
Metabolic alkalosis: Pco2 should increase 0.7 mm Hg for every 1-mEq/L increase in 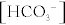 . This is because the increased bicarbonate will cause alkalosis, leading the lungs to compensate by retaining CO2; if it has retained 0.7 mm Hg for every 1-mEq/L increase in
. This is because the increased bicarbonate will cause alkalosis, leading the lungs to compensate by retaining CO2; if it has retained 0.7 mm Hg for every 1-mEq/L increase in  , there has been adequate compensation.
, there has been adequate compensation.
This is a lot of information. Therefore, a systematic approach can help narrow down the possibilities; refer to Table 15-3 for causes of various acid-base disturbances once you have learned to identify them.
Because compensation is never complete, even if the acid-base disturbance is fully compensated, the initial problem will always dictate the direction of the pH. If it is less than 7.40, the initial process was an acidosis (metabolic or respiratory). If it is greater than 7.40, the initial process was an alkalosis (metabolic or respiratory).
If an acidosis: The pCO2 will determine if the problem is respiratory or metabolic. If the Pco2 (an acid) is high, the acidosis is caused by the increased pCO2 and therefore it is a respiratory acidosis. If the Pco2 is normal or low, it indicates that the problem cannot be with CO2 and therefore it is a metabolic acidosis (and the bicarbonate will be low); in the latter case, in which the Pco2 is low, it indicates that the lungs are compensating.
If an alkalosis: Again, the pCO2 will determine if the problem is respiratory or metabolic. If the Pco2 is low, it indicates that the acid CO2 is being removed from the body excessively to cause the alkalosis, and the issue is a respiratory alkalosis. On the other hand, if the Pco2 is normal or high, it indicates that the CO2 is not the primary problem (and in the case where the Pco2 is high, the lungs are compensating), and the problem must be a metabolic alkalosis (and the bicarbonate level will be high).
3. Bonus step—if it is a metabolic acidosis, calculate the anion gap to assess if the anion gap is elevated:
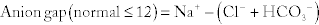
The cation potassium is not included in the calculation.
Another important item to remember is that cells have K+/H+ exchangers, meaning that they can swap K+ for H+ and vice versa. This is important in acidosis because the excess H+ in the bloodstream gets taken into the cells to help buffer, but in turn the cells release K+ into the bloodstream, leading to hyperkalemia. The opposite effect can occur with alkalosis.
PATHOLOGY
Hyponatremia and Hypernatremia
A normal sodium concentration is from 135 to 145 mEq/L. A sodium concentration under 135 mEq/L is hyponatremia and over 145 mEq/L is hypernatremia. It is important to consider the overall volume status of the patient as well as whether or not this is an acute or chronic process. Sodium concentration is based not only on gain or loss of sodium but also on gain or loss of free water; disturbances in either of these can lead to sodium concentration abnormalities, but changes in total body water are more common. Important regulatory hormones implicated include antidiuretic hormone and aldosterone.
Hyponatremia
Hyponatremia can be caused by the following: (1) net sodium loss in excess of net free water loss; (2) net free water gain in excess of net sodium gain; or (3) free water shift. Severe, symptomatic hyponatremia ([Na+] < 120 mEq/L) is almost always caused by SIADH. The third cause of hyponatremia is traditionally referred to as pseudohyponatremia; it is typically observed in a hyperosmotic hyperglycemic state in which intracellular free water shifts extracellularly to maintain osmotic balance. The extracellular free water shift induces a dilutional state for sodium—hence, hyponatremia. The total body sodium, however, is not reduced—hence, the term pseudohyponatremia. Hyponatremia associated with hyperglycemia can be corrected by the control of hyperglycemia alone. The hyponatremia associated with hyperglycemia may be corrected as follows. For each 100 mg/dL of glucose over normal (e.g., normal is ≈ 100 mg/dL), add 2.4 mEq/L of sodium as a correction. Hyponatremia can be further classified into hypovolemic, euvolemic, or hypervolemic.
Hypovolemic hyponatremia: Caused by hypotonic to hypertonic fluid loss plus concomitant pure free water or relatively hypotonic fluid replacement. Stated another way, hypovolemic hyponatremia occurs when the patient has lost volume and sodium, but has lost more sodium. Examples include hypotonic fluid loss (diarrhea, sweating respiration), in which the urine sodium would be low (kidney trying to actively reabsorb sodium and water), as well as hypertonic fluid loss (diuretics, aldosterone insufficiency), in which the urine sodium would be high (kidney cannot reabsorb the sodium or water).
Euvolemic hyponatremia: Usually caused by excess free water reabsorption with the syndrome of inappropriate ADH (SIADH). Causes of SIADH are many, including malignancy, pulmonary or CNS lesions, antipsychotic, antidepressant, or antiepileptic drugs, pain medications, acute nausea and vomiting, and pain. A classic example is a smoker with small cell carcinoma of the lung (also known as oat cell carcinoma, a nickname given to it because of the histologic appearance of the cells), which can secrete ADH (among other hormones; see Chapter 9). Hyponatremia caused by SIADH is considered euvolemic, even though the body is reabsorbing large amounts of water because the accumulation of volume can stimulate the intravascular pressure-sensing receptors (baroreceptors) to induce a natriuretic effect to enhance sodium and water excretion. Other causes of euvolemic hyponatremia include excessive ingestion of free water and poor oral intake. The former overwhelms the maximal ability of the kidneys to excrete water and the latter is caused by the need of the kidneys to pull out solutes with free water excretion. Limited solute intake (poor oral intake as in alcoholics [“beer potomania”] or “tea and toast diet”) limits the ability of the kidneys to excrete free water. The difference between SIADH and the others is that with SIADH, the urine will be concentrated (reabsorbed all the water), but the urine will be dilute in the other conditions. Other causes of euvolemic hyponatremia are hypothyroidism, hypocortisolism, and nephrogenic syndrome of inappropriate diuresis.
Hypervolemic hyponatremia: In hypervolemic hyponatremia, the hypervolemia is caused by volume overload from heart, liver, or kidney failure or hypoalbuminemia (nephrotic syndrome), leading to significant interstitial fluid overload.
Acute hyponatremia results in decreased osmoles in the intravascular space, leading to water rushing into the cells (osmotic gradient). This precipitates cellular swelling and cerebral edema, leading to altered mental status, headache, vomiting, and seizures. The treatment of hyponatremia can involve the following: (1) restrict water and allow the kidneys to fix the problem by urinating out the excess water; (2) give salt-containing fluids IV or sodium tablets to correct the sodium; or (3) give medications (e.g., ADH receptor antagonists, vaptans, demeclocycline [ADH antagonist]) to increase free water excretion. Care must be taken not to correct hyponatremia too quickly. This is because with chronic hyponatremia (with decreased intravascular osmoles), the body has made intracellular adjustments to the fewer osmoles; increasing the osmoles in the bloodstream rapidly by introducing a large sodium load from IV fluids will pull water out of the cells because of the osmotic gradient. This pull of water out of the cells is particularly destructive to myelin, potentially causing a syndrome called osmotic demyelinating syndrome (ODS), previously called central pontine myelinolysis (CPM), which is a condition that may cause permanent neurologic damage or death.
Hypernatremia
Hypernatremia can occur from the following: (1) gain of sodium; (2) loss of free water (more common); or less commonly (3) intracellular free water shift. It may not be intuitive that loss of water could cause hypernatremia. Wouldn’t people just drink water? That’s true and is why hypernatremia is often seen in those with altered mental status (e.g., nursing home patients) or intubated patients, people who cannot get access to free water. Another way to lose water is to have diabetes insipidus, which causes polyuria (increased urine production) of very dilute urine. Symptoms of hypernatremia include altered mental status and coma.
In central diabetes insipidus (DI), the problem is centrally located in the posterior pituitary gland. If it fails to secrete ADH, hypernatremia will result by stopping water reabsorption in the distal nephron and subsequently results in a large loss of free water. This is sometimes seen after head trauma. Responds to desmopressin (DDAVP; synthetic ADH) administration.
Nephrogenic diabetes insipidus occurs when there is a problem with the receptors at the kidney level: there is ADH but the kidney can’t use it. This can be caused by chronic lithium use, hypokalemia, or hypercalcemia, or mutations of the ADH receptors. Does not respond to desmopressin administration.
Differentiation of the two types of DI can be done by administration of desmopressin, a synthetic ADH analogue. In central DI, the body will respond to desmopressin because the problem is a lack of ADH and not a problem with the receptor pathway; the urine osmolarity should increase (become more concentrated as fluid is reabsorbed from the collecting duct) by 50%. If it does not increase by 50%, it indicates that the problem may be with the kidney’s ability to use ADH, and therefore suggests nephrogenic DI. Interestingly, the treatment of nephrogenic DI is a thiazide diuretic; this is counterintuitive, but as salt and water is lost (instead of just water) with diuretic use, the increased RAA axis activation will cause increased sodium (and water) reabsorption in earlier parts of the nephron (e.g., upregulation of the Na+/H+ exchanger in the proximal tubule by angiotensin II), leading to a net decrease in water loss.
If primary polydipsia is thought to be in the differential diagnosis of dilute polyuria (although this causes hyponatremia, it would also lead to dilute urine), then fluid restriction can lead to a diagnosis. If the patient does not take in fluids with primary polydipsia, the urine will concentrate normally (not the case with diabetes insipidus).
Hypokalemia and Hyperkalemia
Changes in potassium levels alter the resting membrane potential, leading to abnormal cellular activity. Potassium homeostasis is controlled by the kidneys, with aldosterone being the key regulatory hormone, leading to excretion of potassium in the urine. As mentioned in the acid-base section, cells also have a H+/K+ exchanger, leading to changes in potassium levels with changes in pH (acidosis causing cells to take in H+ in exchange for putting K+ into the bloodstream, alkalosis causing cells to give H+ to the bloodstream in exchange for taking in K+).
Hyperkalemia
Hyperkalemia is defined as potassium level higher than 5.0 mEq/L.
Causes
Hyperkalemia can be caused by many factors, with the main causes involving the following: (1) decreased renal excretion; (2) cell lysis; and (3) transcellular movement. Causes of decreased renal excretion include renal failure (inability to excrete potassium), hypoaldosteronism (because aldosterone causes potassium loss in urine), and potassium-sparing diuretic use (prevents elimination of potassium). Cell lysis such as rhabdomyolysis (skeletal muscle breakdown) or high cell turnover, such as in some leukemias and lymphomas, can cause hyperkalemia because it is spilling the intracellular potassium into the bloodstream. Lysis of cells during blood draws (hemolysis) can lead to elevated potassium of the blood sample as well, so it is important to keep in mind. Transcellular movement, as noted, can occur with acidosis as the excess H+ in the bloodstream moves into the cells in exchange for K+ because insulin and sympathetic drive both activate the Na+/K+ ATPases in cells (promoting K+ uptake in cells); loss of either of these can cause hyperkalemia.
Findings
Electrocardiographic findings include peaked T waves (from vigorous accelerated repolarization), PR interval prolongation, QRS widening, and eventually a sinusoidal tracing. Ventricular arrhythmias can also occur from abnormal excitability of the heart. Muscle weakness can occur as well because of higher resting membrane potential, leading to sodium channels not being able to reset fully (repolarization not complete).
Treatment
Treatment is threefold: (1) reduce myocardial irritability to prevent arrhythmia and death; (2) move potassium intracellularly to temporarily reduce potassium; and (3) promote potassium loss through the urine and stool. Reduction of myocardial irritability is via calcium administration, which helps stabilize the cell membranes. Potassium can be moved intracellularly by increasing Na+/K+ ATPase activity via insulin (and glucose, to prevent hypoglycemia) and sympathetic stimulation (usually albuterol) or by causing an alkalosis and promoting H+/K+ exchange across the cell via bicarbonate administration. Finally, potassium must eventually be removed from the body, usually via potassium-wasting diuretics (e.g., furosemide), potassium-binding resins that bind potassium in the intestines (sodium polystyrene sulfonate [Kayexalate]), or dialysis.
Hypokalemia
Hypokalemia is defined as potassium level less than 3.5 mEq/L.
Causes
The causes of hypokalemia, in general, are the opposite of the causes of hyperkalemia, and involve the following: (1) increased renal excretion; (2) transcellular movement; and (3) gastrointestinal loss. Increased renal excretion is seen with hyperaldosteronism from any cause, hypercortisolism because high levels of cortisol can act on the aldosterone receptor, and potassium-wasting diuretic use. Hypokalemia can also be seen in states of increased diuresis, such as in diabetes from glucosuria leading to polyuria. Hypokalemia can be seen with alkalosis because the cells give up some H+ to help replenish the lost serum H+ and exchange it by taking in potassium. Finally, the gastrointestinal fluids are generally potassium-rich (stomach acid and stool), so vomiting and diarrhea can lead to potassium loss, further exacerbated by volume loss, leading to RAA axis stimulation and increased potassium loss via aldosterone action.
Findings
Electrocardiographic findings include the presence of a U wave (a small hump after the T wave), and altered membrane potentials can also lead to arrhythmias with hypokalemia. There is muscle weakness caused by a more negative membrane resting potential (hypokalemia or hyperkalemia causes muscle weakness).
Urinalysis and Urine Microscopy
Understanding basic urinalysis (UA) and urine microscopy will help you understand the pathology of the kidney. A typical urinalysis has many findings, which give clues to the function or dysfunction of the kidney:
Color
Gross appearance of the urine; can be yellow (concentrated urine), clear (dilute urine), tea-colored, or red (blood or myoglobin in the urine).
Specific Gravity
The specific gravity of plasma is 1.010; any higher and the urine is more concentrated than plasma, any less and the urine is more dilute than plasma. In renal failure, the kidney loses the ability to concentrate or dilute urine and the specific gravity can be stuck at 1.010.
pH
The pH can have a wide range, depending on the acid-base status of the patient. In a urinary tract infection or with urolithiasis (kidney stones), a high pH can signify the presence of ammonia from urease-splitting bacteria, such as Proteus.
Protein
Normally, the urine should be relatively free of protein. Proteinuria can occur when the glomerulus does not adequately prevent large proteins such as albumin from being filtered (nephrotic syndrome) or the proximal tubules fail to absorb normally filtered low-molecular-weight proteins. Routine urinalysis is not sensitive enough to detect small amounts of albumin. In addition, it does not detect Bence-Jones proteins, seen in multiple myeloma. Detection for the latter may be done with prior addition of sulfosalicylic acid to the urine. More sensitive testing for a low degree of albuminuria (microalbuminuria) may be done separately to detect early glomerular injury in diabetic patients.
Glucose
Normally, glucose is filtered and then reabsorbed in the proximal tubule. With hyperglycemia, the glucose can overwhelm the transporters and lead to glucose in the urine, called glucosuria.
Ketones
Present when the body is using fatty acids for energy; can be a clue to diabetic ketoacidosis (DKA) or starvation, with subsequent fat catabolism for nutrition. In DKA, there are two main ketones produced—acetoacetate and β-hydroxybutyrate, but UA cannot detect the latter.
Bilirubin
Can be seen with increased levels of conjugated (water-soluble) bilirubin, such as in liver or gallbladder disease.
Urobilinogen
Formed by bacterial modification of bilirubin in the gut. If present, it means that bilirubin is making it to the intestines; therefore, it is less likely an obstructive jaundice because if there is no bile getting to the gut, there can be no urobilinogen produced to be excreted in the urine.
Blood
The blood dipstick will turn positive with blood or myoglobin in the urine. Blood in the urine can be from broken down red blood cells being excreted in the urine from hemolysis, red blood cell casts from nephritic syndrome, or red blood cells from a lower urinary tract bleed. Myoglobin can be from rhabdomyolysis (skeletal muscle breakdown, where myoglobin is stored). Differentiation of these entities can be done by microscopy; with myoglobinuria, there will be no red blood cells on microscopy.
Nitrites
Many gram-negative bacteria reduce nitrates to nitrites, which can be seen on UA. The most common urinary tract infection (UTI) pathogen, Escherichia coli, is a nitrate reducer. However, this reduction reaction takes time to occur and may be negative, even in the setting of a UTI, if the patient is urinating frequently or has an infection by an organism that is not a nitrate reducer. Therefore, it is specific (positive result almost guarantees UTI) but not sensitive (negative result does not rule out UTI).
Leukocyte Esterase
Released by white blood cells. The presence of leukocyte esterase indicates the presence of white blood cells and suggests a UTI or other inflammatory condition of the urinary tract (e.g., renal tuberculosis, Chlamydia urethritis), stones, or stents that irritate the ureters.
Microscopy allows the analysis of any cells, casts, or crystals in the urine. This is important because it can give insight into some of the urinalysis abnormalities described.
Cells
Red blood cells may indicate glomerular bleeding, lower urinary tract bleeding, or contamination from menstruation in women. White blood cells (WBCs) indicate the presence of an infection or inflammation. Bacteria can indicate a urinary tract infection, but can also be a contaminant if the specimen was not collected in a clean fashion.
Casts
Red blood cell casts indicate glomerular bleeding, or acute glomerulonephritis. White blood cell casts indicate an ongoing intrinsic kidney infection (pyelonephritis) or allergic reaction (interstitial nephritis). Granular casts indicate varying degrees of degenerated cellular casts (e.g., degenerated RBC or WBC casts) and are thus nonspecific. Muddy brown casts indicate the presence of dead necrotic renal tubular cells seen with acute tubular necrosis. Waxy casts are acellular and indicate advanced kidney disease. Finally, fatty casts are seen in nephrotic syndrome (see explanation of nephrotic syndrome).
Kidney Stones (Urolithiasis)
Kidney stones come in many types and sizes (Table 15-4). Clinically, symptomatic stones cause hematuria and sudden-onset flank pain, in which patients continue to move because they are trying to find a less painful position (but cannot). The pain is often so severe as to cause nausea and vomiting. Stones can cause obstruction and hydronephrosis from the increased back pressure, potentially leading to kidney damage. Inadequate fluid intake is a predisposing factor for all stones because the concentration of the compounds making up the stones is higher, promoting precipitation and stone formation. Most stones (except uric acid) can be seen by radiography and are termed radiopaque, although cystine stones are only faintly radiopaque; ultrasound can show obstruction (hydronephrosis) but not the stone itself.
Table 15-4
Types of Stones and Their Characteristics

*Mnemonic: magnesium ammonium phosphate crystals can cause the entire renal pelvis to fill with stones and destroy the kidney, killing it and putting a coffin lid on it!
Calcium Stones
Most common (80%), comprised of calcium oxalate (adults), or calcium phosphate (children). Predisposing factors include anything that causes hypercalcemia or increases oxalate concentration. One classic scenario is the presence of stones after ethylene glycol (antifreeze) ingestion because the last metabolite is oxalate (oxalic acid). Other predisposing factors include vegan diets (some vegetables such as spinach are high in oxalates) and inflammatory bowel disease (poor fat absorption causes calcium to bind to fat instead of oxalate in the gut, leading to oxalate absorption). Treatment for recurrent stones is hydrochlorothiazide because it increases calcium reabsorption in the distal nephron, thereby decreasing its concentration in the urine.
Magnesium Ammonium Phosphate (Struvite)
Ammonium can be produced by organisms that split urea with their urease enzyme, typically Proteus spp. The ammonium is a base and therefore the urine will become alkaline (high urine pH), which is a key finding. Unfortunately, these stones can get so large that they fill the entire renal pelvis, termed staghorn calculus because it looks like antlers when the stones fill the entire renal pelvis. Treatment involves antibiotics to kill the urease-producing organism and potentially surgery for very large stones.
Uric Acid Stones
The only truly radiolucent (not seen on x-ray) stone, often seen in those with high uric acid levels (hyperuricemia), such as patients with gout or those with high cell turnover rates (because uric acid is a purine nucleotide breakdown byproduct) in conditions such as leukemia. Treatment is allopurinol, which inhibits xanthine oxidase and prevents uric acid production in the purine breakdown pathway. Initial treatment can also include high fluid intake and alkalinization of the urine to make the uric acid into the ionized (soluble) form. These stones are radiolucent; remember, Uric acid stones are radiolUcent and therefore Unseen on X-ray.
Glomerular Diseases
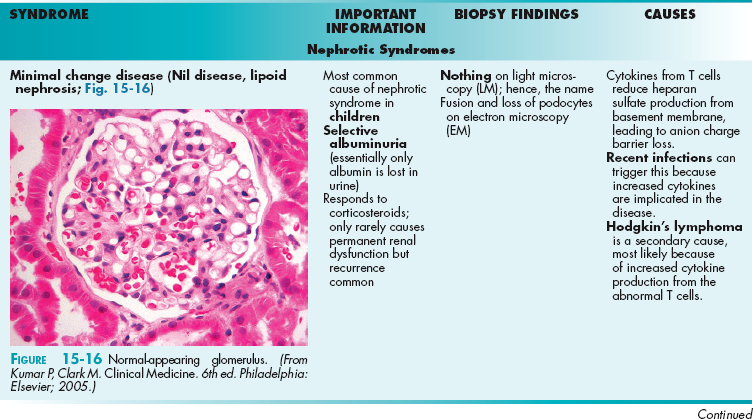
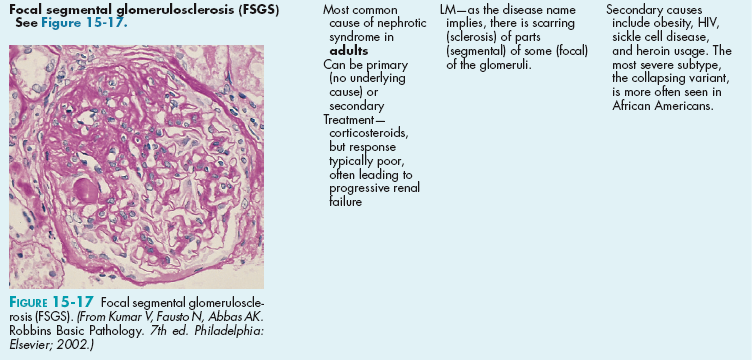
The glomerulus is implicated in a wide variety of pathology. Generally, these diseases will allow protein (nephrotic syndromes) or red blood cells and some protein (nephritic syndromes) to pass through the glomerulus, leading to hematuria and/or proteinuria. There are many nephrotic syndromes and many nephritic syndromes. The terminology describing the syndromes (e.g., focal segmental glomerulosclerosis, a nephrotic syndrome) can be confusing but is explained here; all these are histologic determinations after a percutaneous kidney biopsy has been obtained:
Focal versus diffuse: Defines the number of glomeruli on the biopsy affected. If less than half the glomeruli on the biopsy are affected, it is focal. If more than half are affected, it is diffuse.
Segmental versus global: Defines how much of each individual glomerulus is affected; for each affected glomerulus, if only part is affected, then it is segmental. If the entire glomerulus is affected, it is global. (Therefore, focal segmental glomerulosclerosis means that less than half of the glomeruli are affected [focal] and, of those affected, only part of each glomerulus is affected [segmental].)
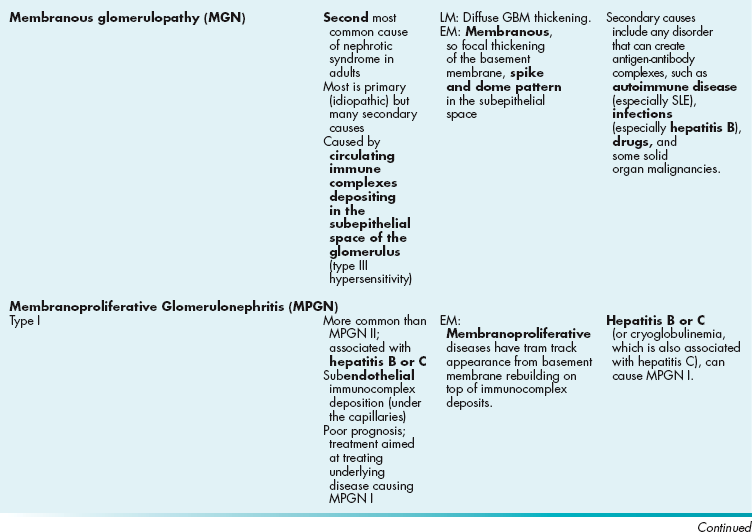
Membranous versus proliferative versus membranoproliferative: In membranous, the glomerular basement membrane becomes thickened in parts; those thickenings appear as spikes and domes on microscopy because of the bulging membrane. Proliferative indicates that the cells are proliferating and numerous nuclei will be seen on microscopy from the added cell count. Membranoproliferative just indicates that there is membranous thickening and proliferation. This leads to a so-called tram track appearance because the basement membrane is rebuilt on top of the damaged deposits.
The best way to remember nephrotic syndrome is the mnemonic nephrotic-oncotic (rhymes) because the oncotic pressure is based on protein, which is what is lost in nephrotic syndromes, leading to decreased oncotic pressure and subsequent edema.
Nephrotic syndromes are usually caused by cytokines from immune cells that cause loss of the negative charge barrier (loss of heparan sulfate leading to loss of negative repelling forces between podocytes, with subsequent fusion of the podocytes) or by an intrinsic abnormality in various podocyte proteins. This leads to the passage of negatively charged proteins in the urine, especially albumin, leading to hypoalbuminemia, low intravascular oncotic pressure, and edema formation. The edema is further exacerbated by the decreased effective arterial volume (from the hypoalbuminemia), leading to RAA axis activation, sodium retention, and worsening edema. There is also evidence that there is an intrinsic increase in sodium reabsorption independent of the RAA axis in nephrotic syndrome.
The liver reacts to hypoalbuminemia and loss of other large proteins by increasing its synthetic function, but unfortunately it also increases the synthesis of apolipoproteins, leading to hypercholesterolemia. Loss of cholesterol in the urine leads to fatty casts with a so-called Maltese cross appearance under polarized light.
Loss of other proteins is significant as well. The loss of ATIII, an anticoagulant molecule (among other anticoagulants), leads to hypercoagulability and increased risk for renal vein thrombosis, lower extremity deep venous thrombosis, and pulmonary embolism.
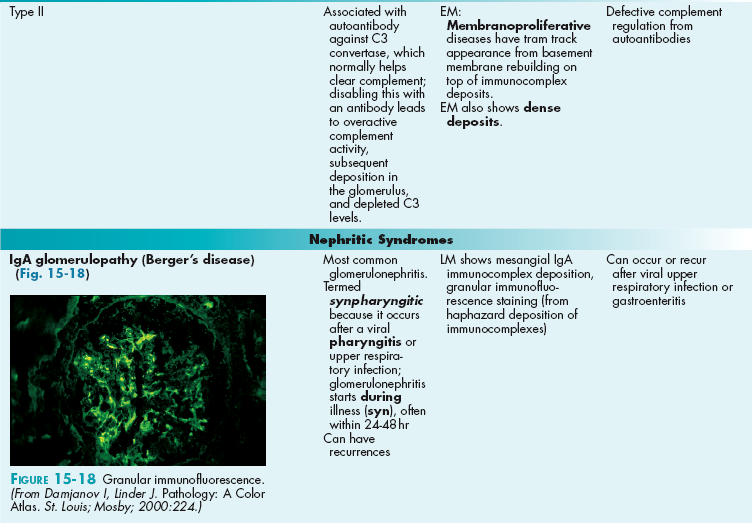
In nephritic syndromes, red blood cells can pass through the glomerulus (and also usually protein, because if large red blood cells can pass through, so can smaller proteins).
It is important to remember that when red blood cells pass through the glomerulus, they become red blood cell casts (protein-coated) and can be seen as such in the urine on microscopy. Imagine that the red blood cell gets banged up when it passes through the nephron and needs a cast. This can differentiate blood that has passed through the kidney (glomerulonephritis) from bleeding at a site after the kidney, such as a lower urinary tract bleed (bleeding from the ureter, bladder, urethra).
The larger permeability of the glomerulus in this case is attributed to inflammation and damage by neutrophils, usually a response to complement deposition in the glomerulus (type III hypersensitivity) or antibodies directed at the basement membrane of the glomerulus itself (type II hypersensitivity). See Chapter 6 for an explanation of the various hypersensitivities.
The severe glomerular inflammation decreases glomerular performance and leads to decreased glomerular filtration rate with oliguria (decreased urinary output). Remember that this is a problem with the glomerulus; the tubules in the nephron work just fine, but because the glomerulus is not functioning properly, the performance of the whole kidney suffers.
As a result of the decreased GFR, the functioning tubules attempt to increase sodium reabsorption to attempt to correct the decreased GFR, potentially leading to hypertension and edema.
Nephrotic and Nephritic Syndromes (Table 15-5)


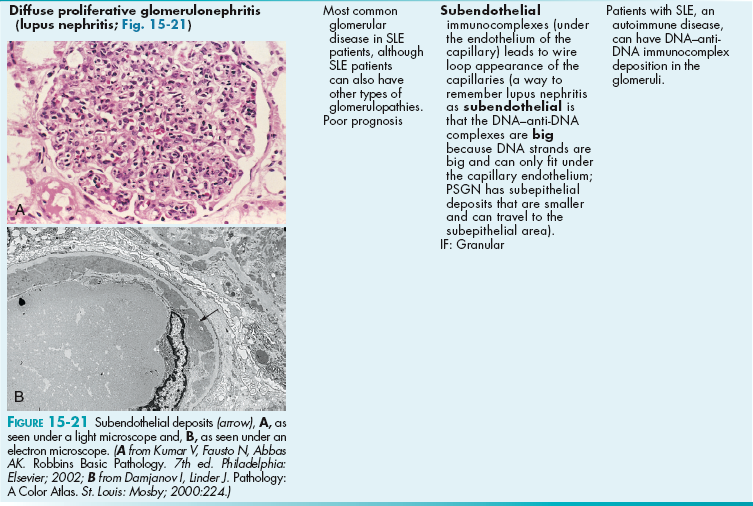
Other Systemic Diseases Causing Glomerular Damage
Diabetes is a very common cause of renal damage (diabetes causes the triad of retinopathy, neuropathy, and retinopathy as part of its microvascular disease) and is the most common cause of chronic renal failure in the United States. Disease severity is based on glycemic control; good glycemic control will prevent or retard the diabetic glomerulopathy from occurring. The pathogenesis is nonenzymatic glycosylation (NEG) of the basement membrane, which is a fancy term for just sugar attaching itself to the proteins of the basement membrane (without the aid of enzymes; hence, nonenzymatic), preventing the negative charge barrier from functioning properly, leading to proteinuria.
NEG of basement membrane: Thickening, loss of charge barrier, proteinuria.
NEG of arterioles: Efferent arterioles affected first, before afferent arterioles, leading to hyaline arteriolosclerosis (narrowing and hardening). This causes increased hydrostatic pressure on the glomerulus, with subsequent glomerular hyperfiltration (initial increased GFR) and damage. This is why ACE inhibitors are nephroprotective in diabetics.
Early kidney damage can be detected by checking for microalbuminuria, a sensitive test for small amounts of albumin leaking in the urine.
On biopsy, Kimmelstiel-Wilson nodules can be present (Fig. 15-22). These are focal nodules of pink hyaline material that forms in the glomerulus from NEG.
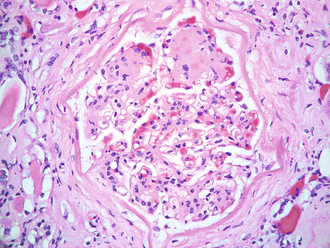
Amyloidosis describes any disease in which abnormal proteins deposit in tissues, leading to organ damage. These amyloid plaques are improperly folded proteins in a beta-pleated sheet configuration.
AL amyloidosis (Amyloid Light chains): Seen in multiple myeloma, in which Bence-Jones proteins are present (immunoglobulin light chains, hence AL).
AA amyloidosis (Amyloid-Associated): Caused by any disorder that has long-standing cell breakdown with chronic inflammation, including autoimmune diseases such as rheumatoid arthritis and ankylosing spondylitis, but also chronic infections such as tuberculosis.
There are other miscellaneous diseases that require brief mention:
Alport syndrome (mostly X-linked recessive): Hereditary mutation of type IV collagen, preventing proper basement membrane (BM) formation in many areas, leading to splitting of the BM in the glomerulus and nephritic syndrome. Also implicated in eye problems (lenticonus), deafness, and nerve disorders for the same reason.
Thin basement membrane disease (autosomal dominant): Characterized by thin BMs, leading to proteinuria and microscopic hematuria. Also known as benign familial hematuria because the disease itself does not lead to serious renal problems.
Tubular and Interstitial Disorders
This section will discuss acute tubular necrosis (ATN), drug-induced interstitial nephritis, renal papillary necrosis, and diffuse cortical necrosis.
Acute tubular necrosis (ATN) occurs when tubules of nephrons die from lack of blood flow (ischemic ATN), the most common type, or from drugs that cause kidney damage to the point of necrosis (nephrotoxic ATN). In both cases, the nephrons that are shed will be lost in the urine, leading to microscopic findings of muddy brown casts.
Ischemic ATN: The kidney requires a constant supply of blood; any condition that significantly diminishes blood flow to the kidney can result in ischemic ATN. Usually, hypovolemia will be the precipitating factor, such as in hypovolemic shock from hemorrhage. Treatment is aimed at correcting the underlying cause of the low renal perfusion.
Nephrotoxic ATN: There are many nephrotoxic agents. The most common two are aminoglycoside antibiotics (e.g., gentamycin), and iodinated radiocontrast agents, such as those used in contrast-enhanced CT scans. Treatment is aimed at adjusting or discontinuing the responsible medication(s) and providing supportive care.
Drug-induced interstitial nephritis is triggered by a drug (as the name implies) and causes inflammation in the interstitium of the kidney, often starting 1 to 2 weeks after beginning a drug. The mnemonic FARE can be used as a memory aid to remember the symptoms: Fever, Arthralgias, Rash and Eosinophilia. Common offenders include penicillins and sulfa drugs. Treatment is simply stopping the inducing drug.
Renal papillary necrosis is characterized by necrosis of the renal papilla, which is the innermost tip of the medulla and therefore the last section of the kidney to receive oxygen and nutrients (recall that the peritubular capillaries run downward after the glomerulus). Renal papillary necrosis occurs most often after chronic acetaminophen, aspirin, or NSAID drug use, called analgesic nephropathy. In those with sickle cell disease, sickle formation in the ischemic medullary papilla leads to occlusion and infarction. Infections such as acute pyelonephritis and systemic diseases such as diabetes mellitus can also cause this.
Diffuse cortical necrosis (Fig. 15-23) is a rare occurrence that causes necrosis of the outer parts of the kidneys (the cortex). This occurs almost exclusively during obstetric emergencies such as abruptio placentae but can also be seen in sepsis. It is thought that disseminated intravascular coagulation (DIC) is implicated in this disease, but the mechanism of cortical death is unknown. In Figure 15-23, coagulation necrosis has occurred on the cortical area of the kidney only.
Figure 15-23 Diffuse cortical necrosis, displaying coagulation necrosis in the outer part (cortex) of the kidney. (From Kumar V, Abbas AK, Fausto N, Aster J. Robbins & Cotran Pathologic Basis of Disease. 8th ed. New York: Elsevier; 2009.)
Renal Tubular Acidosis (RTA)
Renal tubular acidosis (RTA) refers to metabolic acidosis caused by the inability of the kidneys to secrete acid or reabsorb bicarbonate adequately to regulate body pH. The diagnosis of RTA implies that the degree of tubular dysfunction in acid-base regulation is out of proportion to the degree of renal failure.
Type 1 RTA
(distal RTA): The collecting tubules (specifically the α intercalated cells) in the distal nephron are unable to secrete H+ and reclaim K+. This leads to a normal anion gap hyperkalemic metabolic acidosis and also a risk of developing calcium oxalate kidney stones because of the alkalotic urine (remember that the kidneys are unable to acidify the urine, so the urine is alkalotic and the body is acidotic from the retained acids).
Type 2 RTA
In type 2 RTA (proximal RTA), the proximal tubules are unable to reabsorb 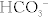 (bicarbonate) from the urine, leading to bicarbonate wasting in the urine and therefore metabolic acidosis from loss of base. This can be a stand-alone disease, but more often occurs in Fanconi syndrome, in which the entire proximal tubule is dysfunctional. Recalling the normal functions of the proximal tubule makes the symptoms logical—rickets from hypovitaminosis D and inability to reabsorb phosphate, acidosis from bicarbonate wasting, and decreased sodium reabsorption, leading to RAA axis activation and hypokalemia from increased distal nephron sodium reuptake.
(bicarbonate) from the urine, leading to bicarbonate wasting in the urine and therefore metabolic acidosis from loss of base. This can be a stand-alone disease, but more often occurs in Fanconi syndrome, in which the entire proximal tubule is dysfunctional. Recalling the normal functions of the proximal tubule makes the symptoms logical—rickets from hypovitaminosis D and inability to reabsorb phosphate, acidosis from bicarbonate wasting, and decreased sodium reabsorption, leading to RAA axis activation and hypokalemia from increased distal nephron sodium reuptake.
Whereas the previous types of RTA were characterized by tubular dysfunction, type 4 RTA (aldosterone failure) is not a problem with the tubules at all. It is a consequence of hypoaldosteronism (low aldosterone) or aldosterone resistance, such as in those taking spironolactone. Recall that aldosterone allows the distal nephron to reabsorb sodium at the expense of secreting potassium and hydrogen ions. Lack of aldosterone therefore would cause sodium not to be reabsorbed (hypotension), and also results in the inability to excrete potassium (hyperkalemia) and hydrogen ions (acidosis). Interestingly, the acidosis may be exacerbated in patients with concurrent hyperkalemia caused by reduced ammonia secretion. Cells have a H+/K+ exchanger; the hyperkalemia causes excess K+ to move into the cell, with H+ exiting; this intracellular alkalosis inhibits ammoniagenesis. The reduced urinary level of ammonia associated with hypoaldosterone-induced hyperkalemia leads to reduced H+ buffering in the urine—hence, acidic urine and low urine pH (typically < 5.5).
You may wonder about type 3 RTA. It’s simply a combination of types 1 and 2 RTA.
Azotemia and Renal Failure
Azotemia: Must establish if it is prerenal, renal, or postrenal (Table 15-6)

The term azotemia implies that there are high serum levels of nitrogen-containing waste products, especially urea (measured as blood urea nitrogen [BUN]). Azotemia in general is caused by a lack of filtration by the kidney, but that can be caused by blood not reaching the kidney (prerenal azotemia); the kidney itself is damaged and unable to filter properly (renal azotemia), or urine produced by the kidney is unable to leave the body and backs up into the ureters and kidney, preventing proper function (postrenal azotemia; Fig. 15-24). Differentiating among these three disorders is important because it gives a clue about the underlying cause of the decreased renal function and how best to manage it.
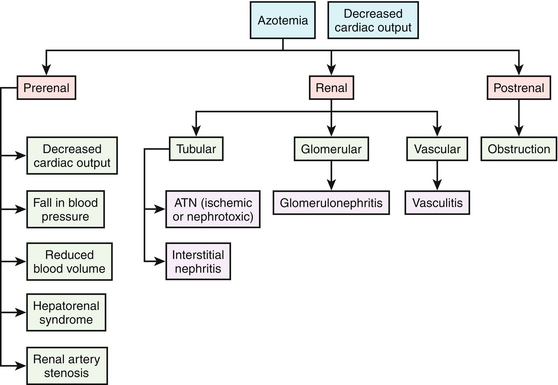
The determination of prerenal azotemia (problem is before the kidney) versus renal azotemia (problem is the kidney) versus postrenal azotemia (problem is after the kidney) is often helped by the BUN-to-creatinine (Cr) ratio, urine sodium level, and fractional excretion of sodium, FeNa. Remember before reading the explanations that urea can be reabsorbed by the kidney, but creatinine cannot; this will become important in understanding BUN/Cr ratios.
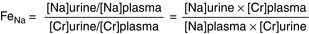
FeNa is simply a measure of the percentage of sodium filtered by the nephron that is excreted in the urine. In azotemia, if the FeNa is low (< 1%), the nephron is actively trying to reclaim all the sodium possible; if it is higher, it is possible that the nephrons are dysfunctional and unable to reclaim sodium properly.
Prerenal Azotemia
In prerenal azotemia, there is not enough blood going to the normally functioning kidney to allow for an adequate GFR, leading to buildup of waste products. The reduced renal perfusion may be caused by hypovolemia, poor cardiac output (e.g., cardiomyopathy, congestive heart failure, severe aortic valvular dysfunction, pericardial effusions), renal artery stenosis, or medications that cause afferent arteriolar vasoconstriction or efferent arteriolar vasodilation. Other causes can include hepatorenal syndrome, a serious disorder caused by cirrhosis, and subsequent hemodynamic changes that can lead to poor renal perfusion. The serum creatinine level will increase because of the decreased overall GFR, but all the creatinine that is filtered is excreted because creatinine is not reabsorbed. However, urea can go back into the bloodstream because it is freely diffusible and can be reabsorbed. The low flow through the nephron caused by low perfusion allows more time for urea to move back into the bloodstream, leading to significantly increased BUN with a smaller increase in creatinine and then leading to a BUN/Cr ratio > 15:1. Next, because of the low-volume state, the RAA axis is fully active, leading to low urine sodium from increased sodium reclamation to promote volume expansion. The urine will be concentrated because ADH is also present to reabsorb the water.
Renal Azotemia
In renal azotemia, there is intrinsic renal parenchymal dysfunction, which can be tubular (ATN [most common], interstitial nephritis), glomerular (glomerulonephritis), or vascular (vasculitis). Although the BUN and Cr levels are both elevated, the BUN/Cr ratio is similar to what is seen in a normal patient (< 15:1) because both BUN and Cr are elevated to the same degree. This is because the functioning nephrons are not in a low-flow state, like in prerenal azotemia. Because the damaged kidney does not reabsorb sodium as efficiently, the FeNa and urine sodium levels will be elevated.
Postrenal Azotemia
In postrenal azotemia, there is enough blood flow to the kidney, and the kidney is (at least initially) functioning normally, but there is a urinary tract obstruction preventing formed urine from leaving the body. This can be anywhere in the lower urinary tract (ureteral obstruction if bilateral), bladder outlet obstruction, urethral obstruction) and is usually caused by benign prostatic hyperplasia (BPH) in older men. Treatment is aimed at the underlying cause, draining the urine (e.g., Foley catheterization in those with BPH). The BUN/Cr ratio is more than 15:1, similar to prerenal azotemia, but for a different reason. The urinary blockage increases back pressure in the kidney, decreasing GFR, but there is a low-flow state because the urine cannot exit out of the kidney, leading to urea reabsorption and BUN increase. Prolonged blockage, however, leads to intrinsic renal damage and renal failure, with laboratory values similar to those of renal azotemia.
Urinary Tract Infections
Almost all UTIs are ascending infections; that is, the normally present bacteria in the perineal area (mostly E. coli from fecal material) start around the urethra and climb up the urethra into the bladder (causing cystitis), eventually trying to climb further up into the ureters and kidneys (pyelonephritis; Fig. 15-25). Because the urethra is significantly shorter in women, women are at higher risk for UTIs. Both cystitis and pyelonephritis are characterized by painful urination (dysuria), frequency, and urgency, as well as bacteriuria (bacteria in the urine). Because the kidneys are highly vascular, when bacteria reach them in pyelonephritis, the bacteria have the opportunity to enter the bloodstream, leading to fever, chills, nausea, and vomiting and the possibility for sepsis (termed urosepsis because the sepsis is of urinary origin). In addition, with pyelonephritis, the inflamed kidneys will be tender, leading to costovertebral (CVA) angle tenderness and flank pain. With cystitis, on the other hand, there is generally no fever because the bacteria are confined to the bladder.
Figure 15-25 The urinary tract, showing what occurs when each part becomes infected. (Modified from Kumar V, Abbas AK, Fausto N, Aster J. Robbins & Cotran Pathologic Basis of Disease. 8th ed. New York: Elsevier; 2009.)
Because the urethra is part of the urinary tract, inflammation can occur here too, termed urethritis. However, this is usually caused by a sexually transmitted infection, characterized by dysuria and urethral discharge. Usually, this is caused by chlamydia or gonorrhea. Gonorrhea will display gram-negative diplococci on staining of the discharge, whereas chlamydia, because it is intracellular, will not stain (stain will not reach the bacteria).
Finally, the prostate can become inflamed, termed prostatitis. The main clinical clue here is an exquisitely tender prostate on digital rectal exam.
Although E. coli is the most common pathogen in UTIs (> 80%), there are other bacteria that can be implicated:
There are many risk factors for UTIs. The most common is female gender, caused by a short urethra. Secondly, older men are at risk caused by BPH. Urination flushes out the bacteria climbing up the urethra; with BPH, the decreased ability to void urine leads to increased risk for UTIs. Also, catheterization is a risk factor because there is an instrument placed into the bladder that can serve as a nidus for infection.
Malignancies and Benign Tumors of the Urinary Tract
There are a few important malignancies of the urinary tract: (1) renal cell carcinoma; (2) transitional cell carcinoma; and (3) nephroblastoma.
Renal Cell Carcinoma (RCC)
This is the most common malignancy affecting the kidney, with smoking being the most common risk factor because the kidney filters the carcinogenic substances. Another rare but important risk factor is Von Hippel Lindau disease. Patients with this disease can develop bilateral RCC. Histologically, the most common appearance of RCC is that of clear cell carcinoma caused by the histologic appearance of the vacuolated cells (Fig. 15-26B). The tumor spreads hematogenously (unlike most carcinomas, which spread via lymphatics) by traveling through the renal vein to the IVC. It often metastasizes to lung and bone as well. In the gross section of the RCC shown (Fig. 15-26A), tumor in the renal vein can be seen causing thrombosis. Hemorrhagic areas of the tumor lead to hematuria and flank pain; the size often allows for a palpable mass to be felt. Because the kidney is involved in the endocrine system, paraneoplastic syndromes can occur from ectopic hormone production, especially erythropoietin, causing secondary polycythemia.
Figure 15-26 Renal cell carcinoma. A, Gross image of RCC. B, Histologic appearance, showing the vacuolated appearance of clear cell carcinoma. (A from Kumar V, Abbas AK, Fausto N, Aster J. Robbins & Cotran Pathologic Basis of Disease. 8th ed. New York: Elsevier; 2009; B from Stevens A, Lowe J, Scott I. Core Pathology. 3rd ed. Philadelphia: Elsevier; 2008.)
Transitional Cell Carcinoma
The area from the renal calices to the renal pelvis, ureters, and bladder is lined with transitional epithelium (to allow stretch), and therefore malignancies in this area are termed transitional cell carcinoma. The unique feature of this type of malignancy is the field effect, in which multiple unique tumors can occur together. This is because the carcinogens responsible are in the urine and bathe the entire urinary tract in carcinogens, leading to the development of multiple foci of malignancy in the urinary tract. Those carcinogens include smoking (just like RCC), but also aniline dyes used in the manufacture of some plastics, and also medications such as cyclophosphamide (a chemotherapeutic agent and immunosuppressant).
Nephroblastoma (Wilms’ Tumor)
A rare malignancy occurring in children (although rare, it is the most common primary renal tumor in children), occurring between the ages of 2 to 4 years (Fig. 15-27). Although mostly sporadic, it can be associated with the WAGR syndrome, an acronym standing for Wilms’ tumor, Aniridia (absent iris), Genitourinary abnormalities, and Retardation. It also is associated with Beckwith-Wiedemann syndrome, which is characterized by hemihypertrophy of the extremities (e.g., one but not both arms with hypertrophy) and large organs. This tumor is derived from mesonephric mesoderm and has numerous parts histologically: (1) primitive blastema cells (undifferentiated cells); (2) primitive glomeruli and tubules; and (3) rhabdomyoblasts (creating muscle and connective tissue strands). Grossly, the tumor appears as a large, tan-colored mass. Like RCC, there is often a palpable abdominal mass. Unique to nephroblastoma is that some of the cells can secrete renin, leading to hypertension (see Fig. 15-27).
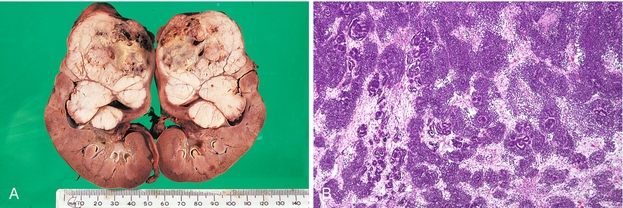
Figure 15-27 Nephroblastoma (Wilms’ tumor). A, Gross image of bilateral nephroblastoma. B, Histologic appearance, showing primitive glofmeruli as a prominent feature and connective tissue strands. (A from Stevens A, Lowe J, Scott I. Core Pathology. 3rd ed. Philadelphia: Elsevier; 2008; B courtesy Nephron, Wikimedia Commons/CC-BY-SA-3.0/GFDL; http://en.wikipedia.org/wiki/File:Wilms_tumour_-_low_mag.jpg.)
Angiomyolipomas
Angiomyolipomas are benign tumors of the kidney and, as the name implies, are composed of blood vessels (angio), smooth muscle (myo), and fat (lipo). A commonly tested association is with tuberous sclerosis, a rare genetic disease that causes benign tumor formation in multiple organs, developmental delay, and characteristic skin findings—adenoma sebaceum, a facial rash of reddish bumps that are angiofibromas, and areas of hypomelanosis called ash leaf spots, because the lack of melanin causes an ashen appearance.
Congenital and Inherited Renal Disorders
There are a few congenital and inherited renal disorders that are important to know, especially polycystic kidney disease (PKD) in the autosomal recessive (fatal) form and autosomal dominant form and horseshoe kidney.
Polycystic Kidney Disease
Autosomal Dominant PKD
This disease is characterized by a generalized abnormality of collagen, leading to numerous problems, especially multiple bilateral renal cysts that develop during adolescence and adulthood, with the eventual development of renal failure later in life (Fig. 15-28A). These cysts can rupture, leading to a localized peritonitis and flank pain if on the outside of the kidney, or hematuria, if rupturing into the renal pelvis. Because it is a generalized collagen defect, other organs are affected—berry aneurysms in the circle of Willis from arterial wall weakening, resulting in subarachnoid hemorrhage if ruptured, weakening of the colonic wall, leading to diverticulosis, and mitral valve prolapse from valvular collagen abnormalities.
Autosomal Recessive PKD
Causes bilateral cystic disease that occurs during development in utero. Unfortunately, this severe disease is usually fatal. Normally, the kidneys of the fetus generate urine, which the fetus expels, becoming amniotic fluid. This fluid protects the fetus and also helps the lung develop when the fluid and accompanying growth factors fill the lungs. Without functioning kidneys, oligohydramnios (decreased amniotic fluid) occurs, leading to Potter’s sequence—loss of the protective cushion with subsequent trauma leading to facial abnormalities, pulmonary hypoplasia because of decreased growth factors, and renal insufficiency. Also associated with congenital hepatic fibrosis.
Horseshoe Kidney
May be associated with Turner’s syndrome (45,X), characterized by fusion of the kidneys at the lower pole, forming a U shape. As the kidneys ascend to their normal place in the retroperitoneum, the horseshoe kidney will get stuck on the inferior mesenteric artery (Fig. 15-28B).
Chronic Kidney Disease
Chronic kidney disease (CKD) is becoming more and more common, with most causes linked to diabetes, hypertension, and glomerular diseases. CKD implies any chronic impairment in kidney function, but chronic renal failure (CRF) implies severe dysfunction. A normal GFR is 100 to 120 mL/min, but patients with CRF will have a GFR less than 10 mL/min, with some producing no urine at all. The hallmark of irreversible renal damage on imaging is small, shrunken kidneys. Remembering the vital functions of the kidneys already described, the changes in CRF can be understood:
Fluid retention: Without the kidney’s ability to regulate sodium and water balance, fluid overload and edema develop.
Electrolyte disturbances: Especially hyperkalemia, because the kidney normally is responsible for the excretion of potassium. Also, because the kidney normally excretes fixed acids to maintain pH, a metabolic acidosis can occur. Phosphoric acid is one of those fixed acids, so hyperphosphatemia develops.
Uremia: A consequence of nitrogenous waste products building up, leading to altered metal status and uremic pericarditis and platelet dysfunction, because the byproducts prevent normal platelet aggregation. With severe uremia, it is possible for urea to be secreted in sweat in such high amounts that urea crystals develop on the skin, termed uremic frost for the white color.
Anemia: The kidneys use the hypoxic stimulus of the peritubular capillaries to release erythropoietin, which stimulates red blood cell production. Loss of this hormone leads to anemia.
Renal Osteodystrophy
Encompasses a triad of (1) osteomalacia, (2) osteitis fibrosa cystica, and (3) osteoporosis. Recall that the kidneys are responsible for the hydroxylation of vitamin D into its active form, 1,25-calcitriol, by 1-α-hydroxylase in the proximal tubule. In renal failure, vitamin D cannot be activated because of decreased GFR (vitamin D doesn’t make it to the proximal tubule), and therefore osteomalacia occurs from hypovitaminosis D. This leads to decreased mineralization of bone. Because vitamin D also acts to raise the calcium level by promoting GI absorption, hypocalcemia causes sustained PTH release to attempt to correct it. This is termed secondary hyperparathyroidism (see Chapter 9). The resultant hyperparathyroidism induces a high turnover bone disease (associated with increased bone reabsorption, lytic lesions, and hemorrhage into bones), known as osteitis fibrosa cystica. Overly aggressive suppression of secondary hyperparathyroidism may lead to the opposite of high turnover bone disease, termed adynamic bone disease. Finally, the impaired mineralization and increased demineralization lead to osteoporosis and predisposes patients to fractures. Osteoporosis is further exacerbated by metabolic acidosis because H+ ions are not effectively excreted by the kidneys. More often, renal patients suffer from mixed bone lesions.
Treatment
Treatment is multimodal. Dialysis targets many of the problems with volume and electrolyte disturbances, but pharmacologic therapy is important as well. Phosphate-binding agents (e.g., sevelamer) prevent phosphate absorption from the gut, erythropoietin can treat low erythropoietin levels and anemia, activated vitamin D can correct low levels of active 1,25-(OH)2 vitamin D, and low-sodium diets can help slow volume overload.
PHARMACOLOGY
The main diuretic classes, from most proximally acting to most distally acting, are carbonic anhydrase inhibitors, osmotic agents, loop diuretics, thiazide diuretics, and potassium-sparing diuretics (Fig. 15-29). Each class will be individually covered in detail. All diuretics increase the rate of urination, but each works in a different manner. Any diuretic that provides increased Na+ to the collecting tubule’s principal cells will cause potassium wasting because some of that sodium will be absorbed at the expense of K+ caused by the Na+/K+ ATPase activity there.
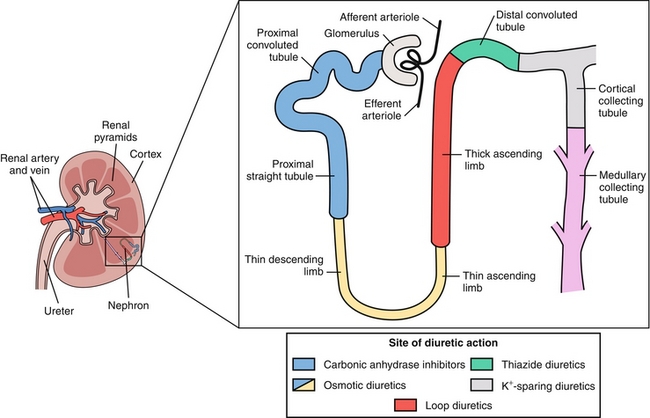
Figure 15-29 Overview of the pharmacologic agents that act on the kidney and their primary location of action. (From Wecker L, Crespo L, Dunaway G, et al. Brody’s Human Pharmacology. 5th ed. Philadelphia: Elsevier; 2009.)
Carbonic Anhydrase Inhibitors (Acetazolamide)
Carbonic anhydrase catalyzes this reaction:
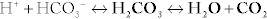
{kind=link}
This reaction is why there is a continuous gradient for H+ secretion and bicarbonate reabsorption into the proximal nephron by the Na+/H+ exchanger. The gradient is normally maintained by the action of carbonic anhydrase because once the H+ is secreted into the lumen, it binds with 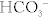 in the lumen; with the aid of carbonic anhydrase, it can become CO2 and H2O. The CO2 can then diffuse back into the proximal tubular cells, dissociating again to H+ and
in the lumen; with the aid of carbonic anhydrase, it can become CO2 and H2O. The CO2 can then diffuse back into the proximal tubular cells, dissociating again to H+ and 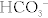 , with the H+ recycled into the lumen and the
, with the H+ recycled into the lumen and the 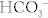 (bicarbonate) reabsorbed into the bloodstream.
(bicarbonate) reabsorbed into the bloodstream.
Blocking carbonic anhydrase, therefore, leads to stopping this recycling and bicarbonate wasting in the urine (Fig. 15-30), causing a metabolic acidosis. Blockage of this pathway, because it ultimately impedes the Na+/H+ exchanger, also prevents Na+ reabsorption.
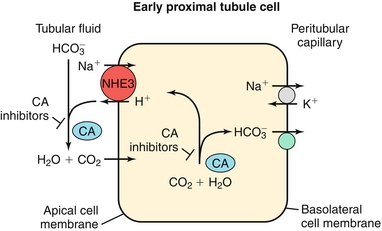
Figure 15-30 Proximal tubule of the nephron and the action of carbonic anhydrase inhibitors. (From Wecker L, Crespo L, Dunaway G, et al. Brody’s Human Pharmacology. 5th ed. Philadelphia: Elsevier; 2009.)
Uses: For glaucoma (decreases aqueous humor secretion, because the aqueous humor is bicarbonate-rich); for altitude sickness (high altitudes), because the resultant metabolic acidosis causes compensatory hyperventilation by the lungs (respiratory alkalosis) and improved oxygenation; and for alkalization of the urine, as with toxic ingestions of acids (to keep the acid in the conjugate base; charged form to promote excretion).
Side effects: Causes a hyperchloremic (non–anion gap) metabolic acidosis. It is a sulfa drug (contraindicated in those with sulfa allergies).
Osmotic Agents
Mannitol is a nonabsorbable sugar that when given intravenously, will be filtered through the glomeruli and act as an osmotic agent to keep water in the lumen of the nephron. This leads to increased urination (polyuria). This is similar to the mechanism of polyuria in diabetes, except endogenous glucose acts as the osmotic agent. While in the bloodstream, the mannitol acts as an osmotic agent there too, causing fluid to move into the extracellular fluid from the intracellular tissue (increased osmolarity of the ECF), this is why it is beneficial in the treatment of cerebral edema by drawing out the excess water.
Uses: Decreasing cerebral edema, promoting diuresis and therefore renal clearance of certain drug overdoses
Side effects: Giving mannitol leads to a large expansion of extracellular fluid because it draws water out of the cells via osmosis, potentially leading to or exacerbating existing congestive heart failure and pulmonary edema in those with poor cardiac and/or renal function. With the increased diuresis, eventual dehydration is also a concern.
Loop Diuretics
Loop diuretics (e.g., furosemide, bumetanide, ethacrynic acid) are potent, high-volume diuretics that block the NKCC2 cotransporter in the thick ascending limb of the loop of Henle, with far-reaching implications (see Fig. 15-9). First, it causes increased delivery of sodium, potassium, and chloride to the distal nephron. The distal nephron can absorb sodium at the expense of potassium (enhanced by aldosterone), leading to further potassium loss in the urine. Therefore, loop diuretics are potassium-wasting diuretics. Normally some of the potassium that is pumped into the thick ascending limb cells leaks back into the lumen of the nephron via a K+ channel, leading to a more positive charge in the lumen. This positive charge enhances the paracellular reabsorption of calcium and magnesium (also positively charged). Therefore, blocking the NKCC2 pump prevents this, leading to a decreased ability to reabsorb calcium and magnesium and potentially leading to hypocalcemia and hypomagnesemia.
Uses: Any edematous state in which unloading volume is advantageous (e.g., congestive heart failure [CHF], hypoalbuminemia from any cause [hepatic failure or loss in urine, as in nephrotic syndrome]), hypercalcemia since loop diuretics have calciuretic effects.
Side effects: A common mnemonic is OH DANG—ototoxicity, hypo(-kalemia, -calcemia, -magnesemia), dehydration, allergy (sulfa drug, except for ethacrynic acid), nephritis (drug-induced interstitial), gout. The reason for ototoxicity is that the ear uses the NKCC2 transporter to maintain proper endolymph and perilymph electrolyte concentrations; blockage can lead to damage.
Thiazide Diuretics
Thiazide diuretics (e.g., hydrochlorothiazide [HCTZ], chlorthalidone, metolazone) work in the distal convoluted tubule of the nephron on the NaCl cotransporter (see Fig. 15-10). Blockage of this transporter leads to increased sodium loss, but also to increased potassium loss because of increased sodium delivery to the collecting tubule (where again, sodium is reclaimed at the expense of potassium caused by Na+/K+ ATPase activity). Therefore, it is a potassium-wasting diuretic. As shown in Figure 15-10, sodium and calcium are both cations and are therefore competing for uptake in the distal convoluted tubule cell (both can’t be reabsorbed because cell voltage would become too positive; there is not enough of a gradient). Blockage of sodium uptake here by thiazide diuretics leads to increased calcium reabsorption.
Uses: Hypertension, hypercalciuria (thiazides will help reclaim calcium from the urine, the opposite effect as a loop diuretic), nephrogenic diabetes insipidus. It is ironic that a thiazide diuretic (which produces more urine) would be used to treat nephrogenic diabetes insipidus because lack of ADH responsiveness already leads to increased urination of pure water. With thiazide diuretics, there is now water and sodium loss, leading the body to activate the RAA axis and increase proximal tubule sodium and water reabsorption (via an AT II stimulatory effect on Na+/H+ exchange), which limits water delivery distally to the collecting tubules for potential loss in the urine.
Side effects: Hypokalemia, hypercalcemia, hyperglycemia, hyperlipidemia, hyperuricemia.
Potassium-Sparing Diuretics
Potassium-sparing diuretics (e.g., spironolactone, eplerenone, amiloride, triamterene; Fig. 15-31) all work on the principal cells because these cells have the final input on potassium excretion. Whenever there is excess sodium delivered here, it will move through the epithelial (luminal side) sodium channel (ENaC) into the cell and be pumped out into the peritubular capillaries via the Na+/K+ ATPase in exchange for potassium. That potassium will then leak into the lumen of the nephron and be excreted. Aldosterone increases activity of the Na+/K+ ATPase pump and ENaC channel synthesis to facilitate volume repletion by reabsorbing more sodium, at the expense of more potassium. This cell’s activity can be impeded by two main mechanisms, (1) blocking the ENaC (amiloride, triamterene) or (2) blocking aldosterone’s upregulation of these cells (spironolactone, eplerenone).
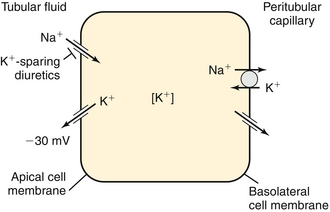
Figure 15-31 Collecting duct and the site of action of potassium-sparing diuretics. (From Wecker L, Crespo L, Dunaway G, et al. Brody’s Human Pharmacology. 5th ed. Philadelphia: Elsevier; 2009.)
Uses: Spironolactone and eplerenone are used in hyperaldosteronism to prevent the effects of the abnormally elevated aldosterone on the kidney, as well as finding use as a general diuretic. Amiloride and triamterene are also used as diuretics if hypokalemia is a concern.
Side effects: Too much of a good thing—preventing potassium excretion by shutting down the principal cell can lead to hyperkalemia. Also, spironolactone is nonspecific and can also block the testosterone receptor, leading to gynecomastia in men; eplerenone is aldosterone-specific and does not have this side effect.