Endocrinology
OVERVIEW OF THE ENDOCRINE SYSTEM
The endocrine system uses hormones to transfer information between different tissues. It is a finely regulated machine that uses feedback loops and sensors to ensure constant homeostasis within the body; the endocrine system plays some form of regulatory role in almost all physiologic processes. It has effects on development, growth, and metabolism and works with almost every organ system, including the nervous and immune system. In contrast to neurotransmitters, which work in the synapse between the neuron endplate and the receptors they act on, hormones are secreted into the circulation and can work on tissues far away from the source of origin.
Hormones
Hormones are secreted by the endocrine glands into the bloodstream. With the exception of the endocrine functions of the reproductive organs, kidneys, and adrenal medulla (see Chapters 16, 15, and 7, respectively), by the end of this chapter the mechanism of action of each of the hormones in Figure 9-1 will be understood, as well as how they are regulated, what pathology can occur, and what medications act on these systems.

Figure 9-1 Overview of the endocrine system. Items present in this figure but covered in other chapters include reproductive endocrinology (Chapter 16), renal endocrinology (Chapter 15), and adrenal medulla endocrinology (Chapter 7). (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
Regulation
Hormones must be precisely regulated to ensure that homeostasis is maintained. The most common method of hormone regulation is through negative feedback loops. Essentially, these loops all function in a similar manner: some downstream effect or product of the released hormone inhibits further hormone release. That way, when the downstream product or change is made (the goal of the hormone), it will decrease hormone production because the intended goal of the hormone was already met. An example of this is parathyroid hormone (PTH): the main stimulus for parathyroid hormone is low ionized calcium levels—when parathyroid hormone has its effects on the body that ultimately increase calcium concentration, that calcium inhibits further PTH secretion because the job of the hormone has been accomplished and further PTH activity would cause hypercalcemia.
A rare method of regulation is the positive feedback loop, whereby the end product causes more production of hormone, causing more production of end product, creating a self-promoting loop. This occurs during the menstrual cycle for a brief period to promote ovulation and also occurs during parturition (labor) when oxytocin helps to create uterine contractions; both are covered in Chapter 16.
Types of Hormones
Hormones fall into one of two main types: peptides/protein hormones and steroid hormones. There are also hormones derived from a single amino acid tyrosine (e.g., the catecholamines, such as epinephrine), covered in Chapters 7 and 8.
Terminology of Pathophysiology
Endocrinopathies are generally termed primary, secondary, or tertiary (when applicable). The example used for this to demonstrate the terminology is hypothyroidism. Hypothyroidism will be described in detail later, but briefly the hypothalamus generates thyrotropin-releasing hormone (TRH) to stimulate the pituitary to secrete thyroid-stimulating hormone (TSH) to stimulate the thyroid to generate thyroid hormone. When the organ itself fails and causes hypothyroidism, it is termed primary; when the stimulus for the organ fails (the pituitary), it is termed secondary; when the stimulus for the stimulus for the organ fails (the hypothalamus), it is termed tertiary (Fig. 9-2).

Figure 9-2 Hypothalamic-pituitary-thyroid axis. Primary hypothyroidism occurs when the endocrine gland itself is the problem; secondary when the stimulus for the gland (the pituitary) is the problem; tertiary when the next stimulus upstream is dysfunctional. Note that there is an exception with tertiary hyperparathyroidism, in which the hyperparathyroidism is due to long-standing hyperplasia of the glands from renal failure, and the endocrine gland has lost the ability to respond to calcium levels appropriately. (From Brenner GM, Stevens GW. Pharmacology. 4th ed. Philadelphia: Elsevier; 2012.)
 Primary dysfunction, such as in primary hypothyroidism, means the endocrine organ itself (i.e., the thyroid) is dysfunctional, such as an autoimmune destruction of the thyroid gland.
Primary dysfunction, such as in primary hypothyroidism, means the endocrine organ itself (i.e., the thyroid) is dysfunctional, such as an autoimmune destruction of the thyroid gland.
Secondary dysfunction, such as in secondary hypothyroidism, means the direct stimulus for that organ is abnormal (i.e., the pituitary gland failing to produce TSH to stimulate the thyroid; even if the thyroid is functioning normally, it will fail to produce thyroid hormone in the absence of a stimulus); an example would be a tumor that has destroyed the pituitary’s function.
Tertiary dysfunction, such as tertiary hypothyroidism, means that the problem is one step further downstream: in the case of tertiary hypothyroidism, it is the hypothalamus failing to secrete TRH, which would normally stimulate the pituitary to secrete TSH to stimulate the thyroid.
Multiple Endocrine Neoplasia Syndromes
The multiple endocrine neoplasia (MEN) syndromes are syndromes featuring tumors of endocrine organs and are inherited in an autosomal dominant fashion. There are three types: MEN I, IIa, and IIb; each is a distinctive syndrome, but they have some overlap in the locations in which the tumors occur. These can be confirmed with genetic testing (Table 9-1).

MEN I (MEN I gene): Characterized by the three Ps of pituitary, pancreas, and parathyroid. The pituitary lesion is most commonly a prolactin-secreting adenoma. The pancreatic lesion is a neuroendocrine tumor (as this is an endocrine neoplasia disorder), such as a gastrinoma (Zollinger-Ellison syndrome). Hyperparathyroidism due to hyperplasia of all four parathyroid glands is the most common feature of MEN I.
MEN IIa and IIb (RET protooncogene activation mutation): Have two of the three features in common—both can develop pheochromocytomas (catecholamine-secreting tumors of the medulla of the adrenal gland), and both can develop medullary carcinoma of the thyroid. However, the main difference is that those with MEN IIa can develop primary hyperparathyroidism like those with MEN I, whereas those with MEN IIb develop ganglioneuromas of various mucosal sites, such as the mouth and gastrointestinal (GI) tract, and often have a marfanoid habitus.
Remember: There is MEN I, MEN IIa and MEN IIb. MEN I shares 1 condition (parathyroid hyperplasia) with MEN IIa. MEN IIa shares 2 conditions (pheochromocytoma, medullary carcinoma of the thyroid) with MEN IIb.
Hypothalamus and Pituitary Gland
Hypothalamus and Pituitary Function Overview
The hypothalamus is below the thalamus (hence hypothalamus) and is responsible for regulation of numerous aspects of the body (covered in Chapter 13); this chapter will concern itself with the important role the hypothalamus has on stimulating or inhibiting the pituitary gland, and therefore regulating pituitary gland activity. The pituitary gland has anterior and posterior lobes, which each make a different set of hormones (Table 9-2; Fig. 9-3).
Table 9-2
Hormones of the Pituitary Gland
| Hypothalamus | Pituitary Anterior: “Flat Peg” Mnemonic Posterior: ADH and Oxytocin |
End Organ |
| Gonadotropin-releasing hormone (GnRH) | Follicle-stimulating hormone (FSH) | Gonads |
| Gonadotropin-releasing hormone (GnRH) | Luteinizing hormone (LH) | Gonads |
| Corticotropin-releasing hormone (CRH) | Adrenocorticotropic hormone (ACTH) | Adrenal cortex •Stimulates cortisol (zona fasciculata) and androgen (zona reticularis) secretion |
| Thyrotropin-releasing hormone (TRH) | Thyroid-stimulating hormone (TSH) | Thyroid |
| (dopamine inhibits) | Prolactin | Breasts |
| Endorphins | ||
| Growth hormone-releasing hormone (GHRH) (somatostatin inhibits) |
Growth hormone (GH) | Entire body •Important for growth |
| Antidiuretic hormone (posterior pituitary) | Kidneys | |
| Oxytocin (posterior pituitary) | Uterus, breasts |

Figure 9-3 The hypothalamus and pituitary. The anterior pituitary is an endocrine gland; the veins from the hypothalamus directly drain into the anterior pituitary to ensure that the signals from the hypothalamus get there. The posterior pituitary contains the axons of neurons that have their cell bodies in the hypothalamus (supraoptic nuclei and paraventricular nuclei). (From Carroll RG. Elsevier’s Integrated Physiology. Philadelphia: Elsevier; 2006.)
The anterior pituitary (previously called the adenohypophysis because adeno refers to “gland”) is made up of endocrine glandular cells, which secrete their products into the bloodstream; their hormones can be remembered by the mnemonic FLAT PeG (FSH, LH, ACTH, TSH, prolactin, (endorphins), GH). Each of these hormones will be covered individually because their function and regulation differ greatly. Because the function of the anterior pituitary is so closely linked with the hypothalamus, the venous blood from the hypothalamus directly drains into the anterior pituitary (called the hypothalamic-hypophysial portal system, similar to the goal of the portal vein draining the intestines into the liver). This close connection ensures that any hormones that the hypothalamus secretes to instruct the anterior pituitary on what to do immediately go in high concentration directly there.
The posterior pituitary (previously called the neurohypophysis) is actually neural tissue, with the cell bodies in the hypothalamus and the axons running down into the posterior pituitary. When stimulated, the neurons release their products into the bloodstream, just like a neuron would release its neurotransmitters into a synapse. The posterior pituitary releases antidiuretic hormone (ADH) and oxytocin, each primarily synthesized by its own set of neuron cell bodies. ADH is mainly synthesized by the supraoptic nuclei, and oxytocin is mainly synthesized by the paraventricular nuclei; but both sets of neurons make both hormones to some degree. A helpful way to remember that the paraventricular cells make oxytocin is that oxytocin causes contraction of the uterus, just like ventricles of the heart contract (although the paraventricular nuclei are actually named for their location near the third ventricle of the brain). The anatomic structure of the posterior pituitary becomes important in head trauma, where the axons in the pituitary stalk (infundibulum) can become disrupted, causing central diabetes insipidus from the loss of ADH secretion.
Pituitary Hormones Covered Elsewhere
Follicle-stimulating hormone (FSH), luteinizing hormone (LH), and oxytocin: see Chapter 16.
Adrenocorticotropic hormone (ACTH): see later section, “Adrenal Glands (Cortex and Medulla)”
Antidiuretic hormone (ADH): see Chapter 15.
Growth Hormone
Growth hormone, as the name implies, is the main hormone regulating growth; the rapid growth in puberty is a result of increased levels of this hormone (Fig. 9-4). This hormone is secreted in a pulsatile fashion, with starvation and hypoglycemia acting as two potent stimuli for secretion. Stimulation is from the hypothalamus through growth hormone–releasing hormone (GHRH) and is inhibited by somatostatin (which is also secreted by the hypothalamus) and end-products such as somatomedins, which are released when growth hormone (GH) has its effect on the end organ on which it is working. The effects of GH are widespread, and after reviewing these effects, the reasons that hypoglycemia and starvation are stimuli for GHRH secretion will become apparent (because initially, starvation and hypoglycemia seem counterintuitive for GH secretion, but GH has a secondary role in maintaining blood sugar levels during periods of starvation and hypoglycemia):
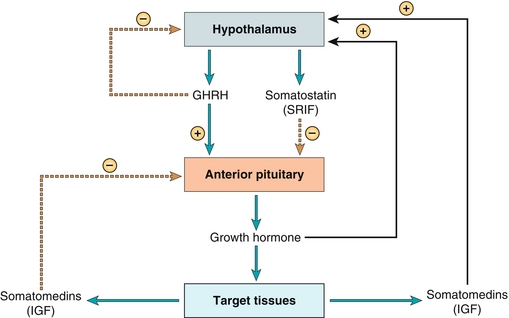
Figure 9-4 Growth hormone. The stimulus is growth hormone-releasing hormone (GHRH, from the hypothalamus), which is released with starvation and hypoglycemia; the main negative feedback is via the end products (somatomedins), such as IGF-1. (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
Insulin antagonism: Growth hormone causes insulin resistance with a resultant increase in blood sugar and subsequently increased insulin release by the pancreas. This relative insulin resistance also causes increased lipolysis. Therefore, GH simply acts during starvation and hypoglycemia to make blood sugar available to keep the body running (e.g., ensuring that enough blood glucose is available to allow the brain to function because the brain uptakes glucose in an insulin-independent fashion).
Increased linear growth: This is not mediated by GH directly, but rather because GH stimulates production of insulin-like growth factor-1 (IGF-1, previously known as somatomedin C) in the liver. IGF-1 acts on osteoblasts and chondrocytes to allow for long bone growth, increasing height in those who do not have their growth plates fused yet.
Increased synthetic function and organ growth: Causes increased protein synthesis and growth in nearly all of the body’s organs, especially muscle. Coupled with the lipolysis from the insulin resistance, this has the net effect of decreasing fat and increasing lean muscle mass, as well as causing organomegaly.
Just like all hormones, two general pathologic processes can occur: too much hormone or too little hormone.
Growth hormone excess: This can cause one of two conditions—gigantism or acromegaly. Almost invariably this is due to a GH-secreting pituitary adenoma. Making the distinction between gigantism and acromegaly involves simply finding out the patient’s age to determine whether their growth plates have likely fused or not. Younger patients who have growth plates that will allow for increased long bone growth (via IGF-1, made in the liver by GH stimulation) will have an extraordinarily tall stature as well as other stigmata of GH excess. Those with fused growth plates will not have increased linear growth, but will have all the other problems that come with GH excess, essentially relating to enlarged organs (cardiomyopathy, the most common cause of death), diabetes (from insulin resistance), and classic coarsening of facial features and enlarged hands, head, and feet (probe about hat size changes, ring size changes, and shoe size changes, or ask for an old photo). Diagnosis of this can be made by increased IGF-1 levels as well as glucose suppression testing—because hypoglycemia is a stimulus for GH release, giving a glucose load should decrease GH in a normally functioning pituitary gland; failure to do so suggests a GH-secreting tumor in the pituitary. Treatment is surgical removal of the tumor. Nonresectable tumors can be treated with somatostatin analogues such as octreotide, which will provide negative feedback to attempts to decrease GH levels.
Growth hormone deficiency: Can be caused by many processes. Consider the stimuli and each step of growth hormone activity—problems at any point can cause GH deficiency (e.g., hypothalamus failing to secrete GHRH, pituitary failing to secrete GH, liver failing to produce IGF-1, receptor dysfunction). The symptoms are intuitive: poor growth and delayed puberty. Treatment is simply giving GH!
Prolactin
Prolactin, as the name implies, is pro-lactation. It allows for maternal breast milk production and has a trophic effect on the breast. This hormone normally is only high during pregnancy and lactation, but can be increased pathologically with prolactin-secreting tumors, with medications that block dopamine, or with hypothyroidism. The effects of prolactin are twofold:
Breast development and support of milk production: Allows for breast-feeding by promoting mammary duct proliferation and synthesis of milk proteins.
Negative feedback on gonadotropin-releasing hormone (GnRH): this is often referred to as “nature’s birth control”; in breast-feeding mothers, the persistently high prolactin levels downregulate GnRH to attempt to inhibit ovulation to space out children appropriately.
Although prolactin deficiency can occur from pituitary insufficiency, there are many causes of hyperprolactinemia, as alluded to previously. Hyperprolactinemia can lead to galactorrhea (milk secretion from the nipples) as well as infertility or loss of libido due to GnRH downregulation.
Hypothyroidism: As can be seen in Figure 9-5A, thyrotropin-releasing hormone (TRH) from the hypothalamus results in increased prolactin secretion. Those with hypothyroidism have increased TRH to attempt to stimulate the thyroid to increase thyroid hormone output. Treatment is repleting thyroid hormone.
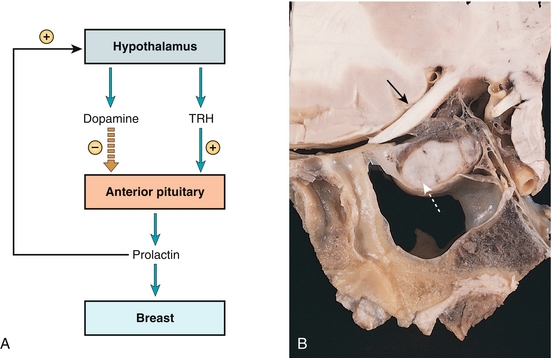
Figure 9-5 A, Prolactin. The stimulus is thyrotropin-releasing hormone, and the inhibition is via dopamine. Prolactin promotes breast development and milk production and also downregulates gonadotropin-releasing hormone (GnRH) secretion from the hypothalamus. B, Prolactinoma, a benign, prolactin-secreting tumor of lactotroph cells in the anterior pituitary. Close proximity to the optic chiasm leads to compressive symptoms such as bitemporal hemianopsia. Increased prolactin secretion can lead to galactorrhea or severely downregulated GnRH secretion, which leads to infertility and/or loss of libido. (A, from Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009. B, from Burger PC, Scheithauer BW, Vogel KS. Surgical Pathology of the Nervous System. 4th ed. London: Churchill Livingstone; 2002:444.)
Medications that block dopamine: Dopamine receptor antagonists (classically the antipsychotic class of medications, which are D2-blocking agents) block the dopamine receptors throughout the body, including the anterior pituitary. This shuts off the negative feedback on prolactin secretion, leading to hyperprolactinemia. Treatment is stopping the offending medication, if possible.
Tumors of the lactotroph cells of the anterior pituitary (tumors of the cells that make prolactin): Called prolactinomas (white dashed arrow in Fig. 9-5B), these can cause hyperprolactinemia. Prolactinoma is the most common pituitary tumor. Because the pituitary is so close to the optic chiasm (black arrow in Fig. 9-5B), the tumor can press on it, causing bitemporal hemianopsia (see Chapter 13 for visual field defect explanations), which is classic for this tumor if large. Treatment is either surgical removal of the tumor (if large) or giving a dopamine agonist such as bromocriptine or cabergoline to increase negative feedback on the cells to decrease size and output. Serum prolactin levels in prolactinoma are higher than in the other causes, and a serum prolactin level of more than 200 ng/mL is essentially diagnostic of a prolactinoma.
Causes of Anterior Pituitary Dysfunction
The anterior pituitary supplies numerous hormones to the body, and damage to the pituitary (or to the hypothalamus, which sends regulatory signals to the pituitary) can lead to hypopituitarism. The main causes of damage to the anterior pituitary gland are mass effect (tumors, empty sella syndrome) and infarction (Sheehan syndrome, sickle cell anemia).
Tumors affecting the pituitary: Masses can arise from the pituitary, such as nonfunctioning pituitary adenomas, which eventually overtake the entirety of the gland, or tumors that exert a mass effect on the pituitary, such as craniopharyngiomas. A craniopharyngioma is a remnant of Rathke pouch, which normally is a depression in the roof of the developing mouth that gives rise to the anterior pituitary. Because of mass effect, a craniopharyngioma can press on and destroy the pituitary gland.
Empty sella syndrome: Mainly affects obese women with hypertension. This is thought to represent intracranial hypertension. An anatomic defect allows the subarachnoid space to extend into the sella turcica, partially filling it with cerebrospinal fluid (CSF), which then flattens the pituitary gland.
Sheehan syndrome (postpartum necrosis of the pituitary): During pregnancy, the mother’s anterior pituitary increases in size significantly owing to increased hormone requirements. However, the blood supply is not increased to the same degree, leaving the anterior pituitary gland relatively ischemic during this period. If the mother becomes hypotensive, such as from hemorrhage during childbirth, the ischemia can turn into infarction and cause pituitary infarction.
Sickle cell anemia: Sickle cell patients often have vasoocclusive crises in which sickled cells cause ischemic injury to the target organ. This can occur in the pituitary and cause dysfunction.
Thyroid Gland
Anatomy, Embryology, and Histology
The thyroid gland is located in the anterior neck, below and lateral to the thyroid cartilage (“Adam’s apple,” which is a misnomer because, although more prominent in men, it is also present in women). It is a bilobed gland, connected by the isthmus (Fig. 9-6A). This gland was initially an outgrowth near the base of the tongue, migrating down the neck into its current position, and leaving the foramen cecum of the back of the tongue as a remnant. This embryologic movement can malfunction, leading to either a thyroglossal duct cyst or ectopic thyroid tissue.

Figure 9-6 Thyroid anatomy and blood supply. A, The thyroid’s anatomical position in the neck. B, Blood supply of the thyroid with the superior thyroid artery originating from the external carotid artery and the inferior thyroid artery, which is a branch of the thyrocervical trunk (itself the first branch from the subclavian artery). (A, from Douglas G, Nicol F, Robertson C. Macleod's Clinical Examination. 12th ed. Edinburgh: Elsevier; 2009. B, from Drake RL, Vogl AW, Mitchell AWM. Gray’s Anatomy for Students. 2nd ed. Philadelphia: Elsevier; 2009.)
Thyroglossal duct cyst: Normally, the thyroglossal duct (the passageway for the thyroid migrating down to its position in the neck) atrophies and closes. Failure of this closure can lead to a cyst, presenting as a midline mass. Because the thyroglossal duct is attached (at the foramen cecum) to the tongue, this cyst will move with tongue movement. The cyst can be asymptomatic (most common) or become infected; rarely, this can cause cancer (thyroglossal duct carcinoma). Because the mass is midline, this can be differentiated from a branchial cleft cyst, which is lateral (see Chapter 4).
Ectopic thyroid tissue: Can present in many forms, but simply represents thyroid tissue that did not migrate in a normal fashion. A lingual thyroid can occur when the thyroid fails to descend, leading to a mass at the base of the tongue and potentially causing dysphagia (difficulty swallowing).
The thyroid is perfused by two major arteries: the superior thyroid artery (a branch of the external carotid artery) and the inferior thyroid artery (a branch of the thyrocervical trunk, which comes off of the beginning of the subclavian artery; Fig. 9-6B). It is drained by the superior, middle, and inferior thyroid veins.
The histologic image in Figure 9-7 is of thyroid tissue. You should be able to recognize thyroid tissue histologically and understand why it appears the way it does.

Figure 9-7 Histologic appearance of the thyroid. A, Microscopic view of thyroid tissue, demonstrating thyroid follicles filled with colloid. B, Illustration of the histologic change in the thyroid follicular epithelium from cuboidal to columnar with increased stimulation from thyroid-stimulating hormone (TSH). (A, from Kierszenbaum AL, Tres LL. Histology and Cell Biology. 3rd ed. St. Louis: Elsevier; 2011. B, from Kester M, Karpa KD, Vrana KE. Elsevier’s Integrated Review Pharmacology. 2nd ed. St. Louis: Elsevier; 2011.)
Follicular epithelial cells: The rim of cells surrounding the colloid in the center. These are metabolically active cells that pull iodine from the bloodstream (via Na+/I− symporters) and begin the synthesis of new thyroid hormone. When the thyroid is stimulated by TSH, these cells take thyroid hormone from the colloid and prepare it for release into the bloodstream. When very active, these cells appear columnar instead of cuboidal.
Colloid: The pink “centers” of the glands contain colloid, which contains formed thyroid hormone and partially formed thyroid hormone attached to a protein called thyroglobulin. They will remain stored until the thyroid is stimulated to release hormone.
C cells: Also known as parafollicular cells (because they are between the colloid follicles), these are neural crest in origin, secreting calcitonin to decrease blood calcium level. This also becomes important in medullary thyroid cancer (covered later in the section “Pathology”), a cancer of the parafollicular C cells.
Thyroid Hormone Physiology
The thyroid gland is responsible for regulation of metabolism and also plays an important role in growth and development—these overarching functions play a role in almost every process in the body; dysfunction therefore leads to widespread symptoms. The thyroid gland generates thyroid hormone, both T4 (with four iodine molecules attached, inactive) and T3 (with three iodine molecules attached, active) into the bloodstream, with T3 having direct effects on the cells and T4 being converted to T3 in the peripheral tissue by 5′-deiodinase enzyme to become active. The stimulus for thyroid hormone production is TSH, produced by the pituitary. The pituitary releases TSH when the hypothalamus secretes TRH. Thyroid hormone has negative feedback on both the hypothalamus (TRH) and the pituitary (TSH), ensuring precise regulation of hormones. Interestingly, TRH is also a stimulatory signal for prolactin release; this becomes important in conditions such as primary hypothyroidism, in which the hypothalamus generates increased TRH to promote TSH secretion from the pituitary to stimulate the thyroid: this will lead to increased prolactin and even the potential for galactorrhea and infertility. In addition, the C cells of the thyroid secrete calcitonin, which prevents bone breakdown and stimulates osteoblasts, and therefore leads to decreased blood calcium levels (calcitonin tones down the calcium level).
Synthesis of thyroid hormone is relatively complex but can be broken down into a few steps (Fig. 9-8):

Figure 9-8 Thyroid hormone synthesis and physiology. A, Thyrotropin-secreting hormone (TRH) is secreted by the hypothalamus, triggering the anterior pituitary to release thyroid-stimulating hormone (TSH), which stimulates the thyroid to make and release thyroid hormone (T3 and T4). B, Thyroid hormone is synthesized in a series of steps (see text for details). (A, from Damjanov I. Pathophysiology. Philadelphia: Elsevier; 2008. B, from Kester M, Karpa KD, Vrana KE. Elsevier’s Integrated Review Pharmacology. 2nd ed. St. Louis: Elsevier; 2011.)
1. Iodide (I−) is taken into the follicular cells by Na+/I− cotransport.
2. This iodide is oxidized into iodine (I2) at the same time as it is moved into the colloid.
3. The I2 then is attached onto the amino acid tyrosine (termed organification) into either MIT (monoiodotyrosine, one iodine molecule) or DIT (diiodotyrosine, two iodine molecules); these are stored on thyroglobulin in the colloid.
4. The MIT and DIT can combine into either T3 (MIT + DIT, because there are three iodine molecules total), or T4 (DIT + DIT, because there are four iodine molecules total). More T4 is generated than T3.
5. When stimulated (TRH from hypothalamus → TSH from pituitary → TSH stimulation of thyroid), the thyroglobulin with its attached molecules will be endocytosed into the follicular cell, and T3 and T4 will be released into the bloodstream. This T3 and T4 will then act in a negative feedback loop on the pituitary and hypothalamus to decrease thyroid stimulation to ensure the level of thyroid hormone does not become too high.
All of the synthetic steps are catalyzed by thyroid peroxidase. Therefore, blockage of this enzyme with medications such as propylthiouracil or methimazole will stop thyroid hormone synthesis, which can be effective in the treatment of hyperthyroidism (too much thyroid hormone production).
Large amounts of iodine administration can have very different effects depending on the initial iodine status of the individual. This can be seen with iatrogenic iodide administration (iodinated contrast material, intravenous or oral iodide administration) or with medications that contain large amounts of iodide (e.g., amiodarone, an antiarrhythmic drug).
Wolff-Chaikoff effect: In a normal individual, giving large amounts of iodine at once will lead to transient hypothyroidism, which is counterintuitive. This is because the thyroid has an autoregulatory function; when it sees large amounts of iodide, it shuts down to prevent massive thyroid hormone generation and release. Initially it overreacts, leading to transient hypothyroidism before readjusting to normal. This can be a potential therapeutic agent in severe hyperthyroidism to quickly shut down the thyroid.
Jod-Basedow phenomenon: A very different outcome occurs in those with chronic iodine deficiency. These individuals have hypothyroidism (iodine deficiency is the most common cause of hypothyroidism in developing countries) owing to lack of iodine available to make thyroid hormone. Therefore, their Na+/I− cotransporters are heavily upregulated to take up any iodine in the blood, and TSH levels are high to stimulate the thyroid gland to make hormone. When these individuals are given large amounts of iodine, they quickly create large amounts of thyroid hormone from the highly stimulated thyroid, leading to transient hyperthyroidism before readjusting.
Once the thyroid hormone (T3 and T4) is in the bloodstream, it circulates mostly bound to thyroxine-binding globulin (TBG, not to be confused with thyroglobulin, which is only present in the thyroid). The rest is bound to albumin or free in the bloodstream (only a tiny fraction). This free (unbound) T3 and T4 is the thyroid hormone that can have action on the body’s cells. It is important to remember that T3 essentially has all of the thyroid hormone activity, and therefore T4 must be converted by the tissues to T3 (by removing one iodine molecule via 5′-deiodinase enzyme) to be active (Fig. 9-9). The effects of thyroid hormone are widespread, including:
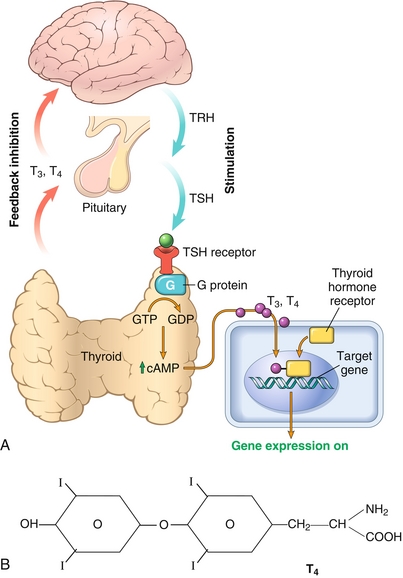
Figure 9-9 A, Action of TSH on the thyroid and action of thyroid hormone on the target cell. TSH binds to a G-protein-coupled receptor to exert its action on the thyroid. Thyroid hormone, however, binds to intracellular nuclear receptors to alter gene expression, allowing for the synthesis of new proteins. B, Structure of T4. Notice that there are four iodine molecules; when one is removed by 5′-deiodinase, it will become T3. (A, from Kumar V, Abbas AK, Aster J. Robbins Basic Pathology, 9th ed. St. Louis: Elsevier; 2012. B, from Kester M, Karpa KD, Vrana KE. Elsevier’s Integrated Review Pharmacology. 2nd ed. St. Louis: Elsevier; 2011.)
Increasing metabolic rate: Increases Na+/K+-ATPase activity, increasing ATP use and therefore increasing metabolic rate.
Increasing catecholamine sensitivity: Increases synthesis of β1 receptors on cardiac tissue, leading to increased sensitivity of the body to catecholamines, increasing cardiac output (CO) by increasing the heart rate (HR) and stroke volume (SV) (recall that CO = HR × SV). This is to support the increased oxygen requirement by the increased metabolic rate to allow for increased ATP generation.
Proper growth and development: Allows for proper differentiation and maturation of cells; this is why hypothyroidism in the neonate can be so devastating, with symptoms just as expected from before—low metabolic rate leading to low body temperature, poor musculature, excessive sleeping, improper development. Thyroid hormone is also important to brain maturation, leading to irreversible mental retardation in the neonatal period. The other classic clue to neonatal hypothyroidism is a protruding umbilicus (umbilical hernia due to improper development of anterior abdominal wall). Note that historically, neonatal hypothyroidism was termed cretinism, but the former term is now preferred.
Laboratory Testing
The first study in evaluating thyroid function is checking a thyroid stimulating hormone (TSH) level (Table 9-3); recall that TSH is produced by the pituitary and has negative feedback from thyroid hormone released from the thyroid. Therefore, a normal result ensures that the patient has normal thyroid function. Abnormal results require further workup:

High TSH: Indicates that the pituitary is attempting to stimulate the thyroid to make more thyroid hormone, indicating primary hypothyroidism (such as from autoimmune destruction of the thyroid gland in Hashimoto thyroiditis). As an aside, TSH-secreting tumors essentially do not exist because there is an α subunit and a β subunit generated by different cells. Therefore, uncontrolled secretion of one subunit would not be able to generate a functional hormone.
Low TSH: Indicates that the pituitary gland is not attempting to stimulate the thyroid to make thyroid hormone, indicating one of two overarching possibilities: (1) the body has too much thyroid hormone (such as in patients taking excess thyroid hormone medication, or in autoimmune stimulation of the thyroid gland as in Graves disease), or (2) the problem is upstream of the thyroid—either pituitary dysfunction with failure to secrete TSH, or hypothalamic dysfunction in which the pituitary and thyroid may be perfectly fine, but the hypothalamus is not giving the signal through TRH to allow the pituitary to generate TSH to stimulate the thyroid gland.
Because an abnormal TSH usually does not give a single diagnosis, further workup is required, which usually involves getting free T3 and T4 levels. Lab tests that quantify total T3 or T4 are generally less reliable than free levels. Lab tests that quantify free T3 and free T4 are typically most useful. TBG levels can vary, making total T3 and T4 levels unreliable; the free T3 and T4 will allow determination of the amount of hormone available to the cells.
Decreased TBG: Seen with increased testosterone (think that increased testosterone means your body wants to make more muscle, breaking down TBG to make more available protein). Because TBG is made in the liver, hepatic failure also causes decreased TBG.
Increased TBG: Seen with increased estrogen (in pregnancy or with oral contraceptive use), which inhibits hepatic breakdown of TBG.
If the cause of hyperthyroidism is not clear, more advanced testing such as radioactive iodine (123I) uptake can occur, which can show whether the thyroid is “hungry” for iodine (actively uptaking iodine to make thyroid hormone, high uptake). (Note that 123I is used for scanning and diagnostic purposes because it is not toxic to the thyroid, but 131I causes radioactivity that destroys the thyroid and can be used for therapy of hyperthyroidism—don’t mix these up!) This test is helpful when the TSH is low but the free T3/T4 is high because iodine uptake scans will show whether the entire thyroid is taking up iodine (diffuse overproduction), if a specific spot in the thyroid is taking up too much iodine (such as small nodules in the thyroid that are autonomously making thyroid hormone independent of stimulation, termed “hot” nodules, which are rarely cancerous), or if none of the iodine is taken up (therefore the thyroid hormone is coming from pills the patient is taking, and not the thyroid).
Pathology
Thyroid pathology is incredibly common; other than malignancies, it can be broken down into two groups: (1) conditions that cause increased hormone production or secretion, and (2) conditions that cause decreased hormone production or secretion.
Conditions that cause increased hormone production or secretion lead to symptoms consistent with thyroid hormone over activity. Any hypermetabolic state characterized by elevated levels of free T3 and T4 is known as thyrotoxicosis. Because thyroid hormone is involved in metabolic regulation, thyrotoxicosis will lead to increased basal metabolic rate, which will lead to weight loss and heat intolerance with diaphoresis as well as increased catecholamine sensitivity, leading to tachycardia; the increased sympathetic drive can even lead to atrial fibrillation. Increased stimulation of the gut causes hyperdefecation, which is not diarrhea, but rather is numerous episodes of defecation from hypermotility.
Lastly, although it is possible to get lid lag (when the patient looks down, the upper eyelid does not move downward quickly) with all forms of hyperthyroidism (because the superior tarsal muscle is smooth muscle innervated by the sympathetic nervous system, which is sensitized with thyrotoxicosis), only Graves disease actually causes exophthalmos.
Graves disease: The most common cause of hyperthyroidism, in which B cells produce immunoglobulin G (IgG) autoantibodies that stimulate the thyroid’s TSH receptors (making this a type II hypersensitivity reaction). There is lymphocytic infiltration of the orbital tissue, and autoantibodies also stimulate fibroblasts to secrete hyaluronic acid (a glycosaminoglycan). These two factors increase the orbital volume, leading to exophthalmos (eyes bulging out), which can eventually be so severe as to cause blindness. The increased glycosaminoglycan deposition in the skin also leads to pretibial myxedema. It is important to note that these IgG antibodies (like all IgG antibodies) can cross the blood–placenta barrier in pregnant women, leading to potential transient hyperthyroidism in the neonate until they clear the IgG.
Toxic multinodular goiter (Plummer syndrome): The term goiter refers to enlargement of the thyroid, and multinodular goiter means that there are many nodules in the thyroid causing this goiter. These diffuse nodules are present throughout the thyroid and are hyperfunctioning, working independently of TSH, sometimes owing to mutations in the TSH receptor that allow for constitutive activity. Symptoms of hyperthyroidism are present, but there is no exophthalmos and no pretibial myxedema because those are immune mediated and not hormone mediated, and this is not an autoimmune phenomenon.
Exogenous thyroid intake: Patients taking thyroid hormone in an effort to lose weight will have signs and symptoms of hyperthyroidism.
Struma ovarii: A rare syndrome—essentially an ovarian teratoma (which can generate many tissue types) that can lead to hyperthyroidism if there is presence of large amounts of thyroid tissue.
Early thyroiditis syndromes: Any time the thyroid is inflamed, it can initially leak a surge of thyroid hormone from the colloid. Afterward, hypothyroidism can occur because the stored hormone has been lost.
Conditions that cause decreased hormone production or secretion lead to symptoms that are almost exactly the opposite of hyperthyroidism. The lower metabolic rate leads to weight gain, cold intolerance, fatigue, and dry skin and brittle hair. Decreased catecholamine sensitivity can lead to bradycardia. Decreased gut motility leads to constipation. Interestingly, decreased synthesis of low-density lipoprotein (LDL) receptors leads to increased circulating levels of cholesterol, causing hypercholesterolemia. As explained earlier in the section “Thyroid Physiology,” primary hypothyroidism (thyroid gland dysfunction from inflammation or destruction) will lead to increased levels of TRH from the hypothalamus to promote TSH release from the pituitary to stimulate the thyroid gland to make more hormone; this increased TRH can lead to galactorrhea as a symptom because TRH is also a stimulus for prolactin release.
Hashimoto thyroiditis: The most common cause of hypothyroidism in developed countries. This is classically seen in middle-aged women but can be seen in both genders at almost any age. In Hashimoto thyroiditis, the body attacks the thyroid directly via CD8 + cytotoxic T cells, leading to cell destruction and release of contents into the bloodstream. These released proteins, such as thyroglobulin and thyroid peroxidase, are usually not present in the bloodstream and are seen as foreign; the B cells then make antithyroglobulin and antithyroid peroxidase antibodies as a response. These antibodies can cause further destruction of the gland. Therefore, the damage to the gland is both cell mediated (type IV hypersensitivity) and antibody mediated (type II hypersensitivity). With the initial inflammation of the gland, there can be initial hyperthyroidism because formed hormone leaks from the cell; this is known as hashitoxicosis.
Iodine deficiency: The most common cause of hypothyroidism and goiter worldwide—chronically high TSH levels cause sustained stimulation and growth of the gland (goiter) to attempt to extract any iodine present in the blood.
Subacute granulomatous (painful) thyroiditis (DeQuervain thyroiditis): Often triggered by a viral infection, especially upper respiratory tract infection. There is either cross-reactivity with a viral antigen or inflammation of the thyroid leading to exposure of a thyroid antigen with subsequent painful inflammation of the thyroid. This inflammation can lead to initial hyperthyroidism as preformed hormone is released, but afterward the thyroid can take weeks to return to normal, and hypothyroidism can occur during this time.
Subacute lymphocytic (painless) thyroiditis: A variant of this is the most commonly tested, which is postpartum thyroiditis. This is thought to potentially represent a subset of Hashimoto thyroiditis but is characterized by postpartum hyperthyroidism secondary to follicle rupture with subsequent hypothyroidism. Many of these patients will recover normal thyroid function over weeks to months, but antigen exposure from the follicle rupture may contribute to development of Hashimoto thyroiditis (up to 50% will eventually develop Hashimoto thyroiditis in the future).
Riedel thyroiditis: Very rare disease characterized by painless fibrosis in the thyroid gland and nearby structures in the neck. This can cause tracheal obstruction from the hard, rocklike fibrous deposition.
Thyroid cancer has four separate types: papillary, follicular, medullary, and anaplastic.
Papillary carcinoma (> 85%): The most common thyroid cancer (papillary is popular!), with the major risk factors being exposure to ionizing radiation and family history. Luckily, this usually has an excellent prognosis. Histologically, this is characterized by two items: Orphan Annie eye nuclei (Fig. 9-10B), so called because there appears to be nothing inside the nuclei, and (right image) psammoma bodies, which are concentrically calcified structures; psammoma is derived from the Greek word psammos meaning “sand.” Psammoma bodies (Fig. 9-10C) are found in many cancers (mnemonic PSaMMoma: Papillary thyroid cancer, Serous cancers of the ovary, Meningioma, Mesothelioma), but of the thyroid cancers, psammoma bodies are only found in papillary thyroid carcinoma (remember: papillary is popular and has psammoma bodies) (see Fig. 9-10).

Figure 9-10 A, Gross image of papillary carcinoma. B, Histology of papillary carcinoma, showing classic “Orphan Annie eye” nuclei. C, Psammoma bodies, found in papillary thyroid cancer, but also in serous cancers of the ovary, meningioma, and mesothelioma. (A and B, from Kumar V, Abbas AK, Fausto N, Aster J. Robbins & Cotran Pathologic Basis of Disease. 8th ed. New York: Elsevier; 2009. C, from The AFIP Atlas of Tumor Pathology, courtesy The Armed Forces Institute of Pathology. Accessed January 10, 2013, from http://fr.wikipedia.org/wiki/Fichier:Psammoma.jpg.)
Follicular carcinoma (10%): The second most common thyroid cancer, this occurs in older (40–60 years of age) women, and the prognosis is directly related to whether metastases are present: when no metastatic disease is present, the prognosis is very good; when metastatic lesions are found, the prognosis is much worse. One unique aspect of this carcinoma is that metastasis occurs by hematogenous spread, which is not characteristic of carcinomas (usually carcinomas spread by the lymphatics, and sarcomas spread by hematogenous routes). Histologically, follicular carcinoma reveals colloid follicles (hence the name), unlike the other carcinomas. This needs to be differentiated from a follicular adenoma, which is the most common benign tumor and is surrounded by a complete capsule; it is an adenoma (benign) and not a carcinoma (malignant) because the complete encapsulation prevents metastatic spread. However, the only way to definitively tell the two apart is after resection and pathologic evaluation, so all of these are typically removed (Fig. 9-11).

Figure 9-11 A, Gross image of follicular carcinoma. B, Histology of follicular carcinoma, showing some disorganized colloid follicles. (From Kumar V, Abbas AK, Fausto N, Aster J. Robbins & Cotran Pathologic Basis of Disease. 8th ed. New York: Elsevier; 2009.)
Medullary carcinoma (5%): This is a neoplasm of the parafollicular C cells, which create calcitonin. This is the type of thyroid cancer associated with MEN IIa and IIb. Because they secrete calcitonin, calcitonin can be used as a tumor marker for recurrence of disease and can be used in immunohistochemical staining of the biopsy to diagnose disease. The excess secretion of calcitonin builds up and is deposited as amyloid in the stroma of the thyroid, which can be seen on histology (Fig. 9-12B). In patients with this rare cancer, checking family history and signs of other MEN IIa and IIb–associated pathology is important (see Fig. 9-12).

Figure 9-12 A, Gross image of medullary carcinoma. B, Histology of medullary carcinoma, showing amyloid stroma from calcitonin deposition. (From Kumar V, Abbas AK, Fausto N, Aster J. Robbins & Cotran Pathologic Basis of Disease. 8th ed. New York: Elsevier; 2009. Courtesy Joseph Corson, MD, Brigham and Women’s Hospital, Boston, MA.)
Anaplastic carcinoma: Undifferentiated, aggressive, and almost uniformly fatal. Some patients will have a past history of a prior thyroid cancer.
Pharmacology
The pharmacology of the thyroid gland essentially is aimed at either blocking thyroid hormone synthesis and release or providing supplementary thyroid hormone, depending on what needs correcting.
Propylthiouracil (PTU) and methimazole: Inhibit organification of iodide and therefore decrease thyroid hormone synthesis; useful in hyperthyroidism to prevent thyrotoxicosis. Can cause agranulocytosis and a skin rash. When taken during pregnancy, methimazole is associated with aplasia cutis congenita (congenital absence of skin, which manifests as a scarlike appearance in a neonate). For this reason, it should be avoided in pregnancy (methimazole is for men).
Levothyroxine (synthetic T4) and triiodothyronine (also known as liothyronine, synthetic T3): Replacement thyroid hormones for hypothyroid conditions. T4 can be converted in the periphery to T3 and is favored because the half-life of T4 is considerably longer than that of T3 (owing to greater albumin binding affinity), and therefore blood levels are more stable over time. Toxicity gives symptoms of hyperthyroidism (tachycardia, heat intolerance, tremors).
Iodine: Iodine can be given in thyrotoxicosis to shut down the thyroid through the Wolff-Chaikoff effect. It is important to know that any iodine load can cause this, including unintended administration (such as using iodinated contrast for computed tomography scanning, or the iodine-rich antiarrhythmic drug amiodarone). In addition, 123I is used for diagnostic scanning of the thyroid to visualize uptake. 131I is used in ablation of the thyroid because the thyroid tissue concentrates the iodine and is subsequently destroyed by the 131I radiation.
Adrenal Glands (Cortex and Medulla)
Anatomy, Embryology, and Histology
The adrenal glands sit on top of each kidney (superior to the kidney, therefore also called the suprarenal glands by some); therefore, like the kidney, the adrenal glands are retroperitoneal structures. Each adrenal gland has a cortex and medulla, which have distinctly different functions (Fig. 9-13).

Figure 9-13 Anatomy of the adrenal glands, demonstrating their location on top of the kidney. (From Drake RL, Vogl AW, Mitchell AWM. Gray’s Anatomy for Students. 2nd ed. Philadelphia: Elsevier; 2009.)
Adrenal cortex: The outer layer of the adrenal gland, consisting of three layers (zona glomerulosa, zona fasciculata, and zona reticularis), each of which secretes different steroid hormones because of differences in enzyme activity. Mnemonic: from outside to in, the layers can be remembered by GFR (just like the filtration rate of the kidney!) – zona glomerulosa, zona fasciculata, zona reticularis.
Adrenal medulla: The inner layer of the adrenal gland, the adrenal medulla is neuroectodermal in origin. The medulla is responsible for generation of epinephrine and norepinephrine to activate the sympathetic nervous system.
It can also be seen in Figure 9-13 that because the left kidney is farther away from the inferior vena cava, the left adrenal vein drains into the left renal vein, whereas the right adrenal vein drains into the inferior vena cava. This is the same way that the gonadal (testicular or ovarian) veins drain.
Adrenal Gland Physiology
The adrenal cortex, as mentioned earlier, has three layers, each with different steroid hormone synthetic abilities. The zona glomerulosa secretes mineralocorticoids such as aldosterone (salt retention); the zona fasciculata secretes glucocorticoids such as cortisol (blood sugar elevation); and the zona reticularis secretes androgens (sex steroids). This leads to the mnemonic: the deeper you go, the sweeter it gets (salt → sugar → sex!) (Fig. 9-14).
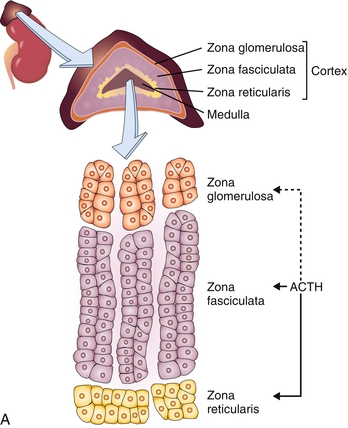

Figure 9-14 A, Illustration of the zones of the adrenal cortex, including the zona glomerulosa (which makes mineralocorticoids such as aldosterone), zona fasciculata (which makes glucocorticoids such as cortisol), and zona reticularis (which makes androgens such as DHEA sulfate). B, Image showing the general mechanism of action of all the products of the adrenal cortex and medulla, as well as the stimulus for their release. (A, from Damjanov I. Pathophysiology. Philadelphia: Elsevier; 2008. B, from Colledge NR, Walker BR, Ralston SH. Davidson’s Principles and Practice of Medicine. 21st ed. Philadelphia: Elsevier; 2010.) ACE, angiotensin-converting enzyme; JGA, juxtaglomerular apparatu; MR, mineralocorticoid (aldosterone) receptor; MSH, melanocyte-stimulating hormone.
Zona glomerulosa: Responsible for mineralocorticoid (aldosterone) secretion—it is the only part of the three layers of the adrenal cortex that does not secrete its hormone because of adrenocorticotrophic hormone (ACTH).* Instead, as covered in depth in Chapter 15, the trigger for aldosterone release is through the renin-angiotensin-aldosterone axis, with angiotensin II causing aldosterone release. The actions of aldosterone are mainly on the principal cells of the distal nephron, increasing sodium reclamation from the nephron to increase intravascular volume, at the expense of increasing potassium secretion. See Chapter 15 for details. It also promotes H+ secretion through the α-intercalated cells into the lumen of the nephron to promote acid excretion.
Zona fasciculata: Responsible for glucocorticoid (cortisol) secretion: The stimulus for its release is ACTH from the pituitary, and it is often a hormone secreted in response to stress. There is also a normal daily cortisol spike just before waking in the morning (termed a diurnal pattern). Cortisol has numerous effects on the body, but the overarching activities of cortisol are a catabolic and diabetogenic effect, an anti-inflammatory effect, and causing catecholamine sensitivity.
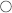 Catabolism: Provides increased glucose synthesis through gluconeogenesis by breakdown of fat and muscle (catabolism), which is important to maintain blood sugar levels and provide for survival during fasting (ensure a glucose source is available even if no glucose is taken in). In addition, it causes insulin resistance (diabetogenic effect) to ensure that glucose is not taken up unnecessarily by cells in times of starvation.
Catabolism: Provides increased glucose synthesis through gluconeogenesis by breakdown of fat and muscle (catabolism), which is important to maintain blood sugar levels and provide for survival during fasting (ensure a glucose source is available even if no glucose is taken in). In addition, it causes insulin resistance (diabetogenic effect) to ensure that glucose is not taken up unnecessarily by cells in times of starvation.
Inhibition of bone formation: Decreases osteoblastic function and collagen synthesis, leading to cessation of bone formation; this is adaptive in periods of starvation but is the reason that when prescription steroids are used chronically (or another form of hypercortisolism), osteoporosis can develop or worsen.
Anti-inflammatory: Increases synthesis of lipocortin, which inhibits the normal function of phospholipase A2 of cleaving arachidonic acid off of lipid membranes to become inflammatory mediators such as prostaglandins and leukotrienes. It also modulates the immune system more directly by preventing T lymphocyte proliferation.
Catecholamine sensitivity: Is said to be permissive to catecholamines, meaning that cortisol is required at some level to permit the catecholamines epinephrine and norepinephrine to work effectively by upregulating α1 receptors, promoting vasoconstriction. This becomes extremely important in the pathology of the adrenal glands because this key effect can lead to blood pressure disturbances when too much or too little cortisol is present.
Zona reticularis: responsible for androgen secretion, most notably dehydroepiandrosterone (DHEA) and androstenedione. Although this is not important in males (who have testes that are androgen factories), this is the major androgenic source in females.
Pathology
Decreased Adrenal Cortex Function
The adrenal cortex can malfunction in a variety of ways, mostly from various causes of primary adrenal insufficiency (adrenal gland hypofunction), but also from pituitary dysfunction (decreased ACTH) or from hypothalamic dysfunction (decreased corticotropin-releasing hormone, or CRH). Because primary adrenal insufficiency is the most common, the various causes will be covered in detail—it can be further divided into acute and chronic.
Acute adrenal insufficiency: Secondary to either (1) stopping prescribed corticosteroids without a tapering period, or (2) bilateral adrenal hemorrhage (Waterhouse-Friderichsen syndrome). Prolonged (> 1 week) use of corticosteroids leads to hypothalamic-pituitary-adrenal axis hypofunction because the exogenous steroids have replaced the need for the adrenal gland to make its own steroids. This can lead to acute adrenal insufficiency and potential hypotension, which is why tapering of steroids is always necessary for longer duration use. Bilateral adrenal hemorrhage can occur during sepsis, especially with Neisseria meningitides meningococcemia; the endotoxin that it secretes can lead to disseminated intravascular coagulation (DIC) and adrenal hemorrhage, called Waterhouse-Friderichsen syndrome. This condition is almost uniformly fatal.
Chronic adrenal insufficiency (Addison disease): This is most commonly autoimmune but can be due to miliary tuberculosis spreading to the adrenal glands in developing countries with a high prevalence of tuberculosis (TB). Loss of adrenal gland function leads to decreased aldosterone, cortisol, and androgen production. This leads to (1) hyponatremia with a hyperkalemic metabolic acidosis due to loss of aldosterone, (2) hypoglycemia due to loss of cortisol, and (3) hypotension due to loss of both aldosterone and cortisol. Because cortisol normally provides negative feedback on the pituitary, loss of cortisol leads to increased ACTH secretion. ACTH has a breakdown product of α-melanocyte-stimulating hormone (α-MSH); this increased melanocyte stimulation leads to hyperpigmentation (Fig. 9-15). The last clue to Addison disease is eosinophilia due to loss of cortisol’s immunomodulatory role, which usually also causes apoptosis of eosinophils. Treatment is replacement of both mineralocorticoids and glucocorticoids.
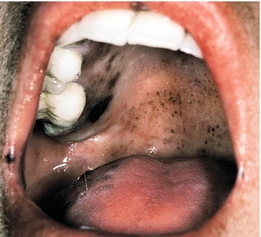
Figure 9-15 Hyperpigmentation of the buccal mucosa in a patient with Addison disease. This is due to increased ACTH from the pituitary, which subsequently breaks down to α-melanocyte-stimulating hormone (as well as other molecules), stimulating melanocyte production of melanin with resultant hyperpigmentation. (From Douglas G, Nicol F, Robertson C. Macleod's Clinical Examination.12th ed. Edinburgh: Elsevier; 2009.)
Congenital Adrenal Hyperplasia
Like all steroid hormones, the adrenal cortex uses cholesterol as a base substrate to generate aldosterone (the major mineralocorticoid), cortisol (the major glucocorticoid), and the adrenal androgens (DHEA and androstenedione). The various modifications require numerous enzymatic steps, but only the key enzymes shown in Figure 9-16 are important to commit to memory because these are the enzymes that can be deficient in various types of congenital adrenal hyperplasia (CAH). With these syndromes, any substrates before the blocked enzyme will build up and take alternative pathways, and of course, substrates after the blocked enzyme will not be used (similar to a roadblock; all the cars take another detour pathway because they need to go somewhere). These disorders are called CAH because the decreased cortisol causes increased ACTH secretion from the pituitary, leading to a tropic effect on the adrenal gland, which causes hyperplasia.
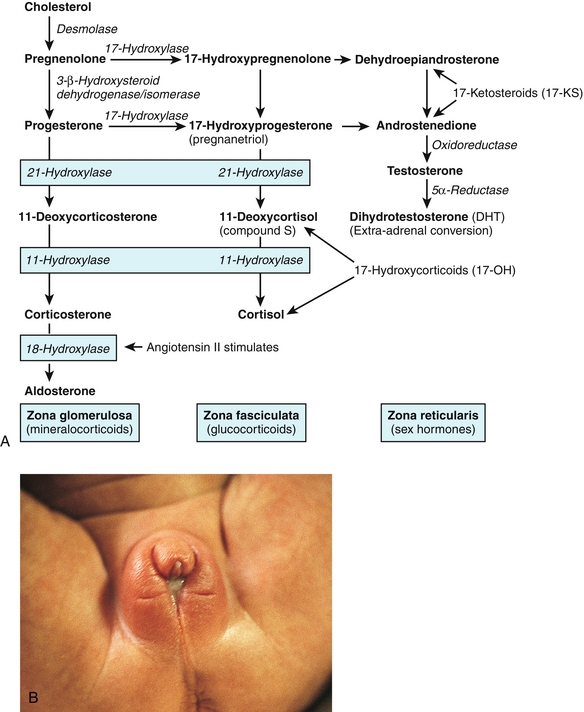
Figure 9-16 A, Adrenal cortex hormone synthetic pathways, including key enzymes that can be deficient in various forms of congenital adrenal hyperplasia. B, Ambiguous genitalia in a female with 21-hydroxylase deficiency. (From Goljan EF. Rapid Review Pathology Revised Reprint. 3rd ed. Philadelphia: Elsevier; 2011. Courtesy Patrick C. Walsh, MD, Johns Hopkins School of Medicine.)
21-Hydroxylase deficiency: The most common cause of CAH. In Fig. 9-16, it can be seen that both aldosterone and cortisol require this enzyme; loss of this enzyme therefore leads to hypoaldosteronemia and hypocortisolism, causing similar symptoms of Addison disease in the neonate (hyperkalemic metabolic acidosis, hypotension). However, with the “roadblock” at the 21-hydroxylase step, the products will instead follow the androgen pathway, and increased androgen levels will occur. Therefore, females will have ambiguous genitalia, and males will have precocious puberty from the increased male sex steroids.
11β-Hydroxylase deficiency: Also required in the synthesis of both aldosterone and cortisol, but the main difference here is that 11-deoxycortisol (proximal to the block) will build up. This compound has mineralocorticoid (aldosterone-like) activity. Therefore, affected individuals will have the same issue with increased androgens but now will also have stigmata of increased mineralocorticoid activity: hypokalemic metabolic alkalosis and hypertension.
17α-Hydroxylase deficiency: 17α-Hydroxylase is required to make anything other than aldosterone. Therefore, with this deficiency, only aldosterone can be made. This leads to predictable results: hypokalemic metabolic alkalosis, hypertension (from hyperaldosteronism), and decreased androgens and cortisol.
Increased Aldosterone Secretion
Increased aldosterone secretion can occur from increased production of any part of the renin-angiotensin-aldosterone axis and generally includes juxtaglomerular (JG) cell tumors (secreting renin), renal artery stenosis (causing kidney JG cells to sense less perfusion and increase renin), or aldosterone-secreting tumors.
Primary hyperaldosteronism (Fig. 9-17) (Conn syndrome): Caused by an aldosterone-secreting adenoma in the zona glomerulosa of the adrenal gland cortex, leading to findings of increased aldosterone activity: a hypokalemic metabolic alkalosis and potentially hypertension. The potassium derangements often lead to weakness. Alkalosis can lead to tetany (calcium normally is bound to albumin; with alkalosis, less H+ is bound to albumin and can therefore accept more Ca2 +, leading to a decreased ionized calcium level and tetany). The main distinguishing factor of primary versus secondary hyperaldosteronism is that in primary hyperaldosteronism, the aldosterone is being secreted independent of renin, and therefore renin levels will be low.

Figure 9-17 Diagram depicting the action of aldosterone on the kidney, with hyperaldosteronism causing increased potassium and acid (H+) excretion, sodium retention, and resulting hypertension. (From Kumar V, Abbas AK, Fausto N, Aster J. Robbins & Cotran Pathologic Basis of Disease. 8th ed. New York: Elsevier; 2009.)
Secondary hyperaldosteronism: Can be caused by renal artery stenosis (kidney thinking it is hypoperfused) or a JG cell tumor, or even simply by having actual hypoperfusion (such as in congestive heart failure). Symptoms can be similar, but renin levels will be high in all these conditions.
Treatment involves addressing the underlying cause, but as a temporizing measure, spironolactone, an aldosterone-receptor antagonist and K+-sparing diuretic, can be given to block the effects of the excess aldosterone on the kidney.
Increased Cortisol Secretion
Increased cortisol secretion can occur from increased amounts of anything along the hypothalamic-pituitary-adrenal axis (CRH from hypothalamus, ACTH from pituitary or ectopic site, adrenal gland tumor producing cortisol). The findings in hypercortisolism (Fig. 9-18) (Cushing syndrome) can be striking: cortisol causes fat and protein catabolism, leading to wasted extremities, but the increased insulin causes upregulation of lipoprotein lipase, promoting central fat deposition (so-called truncal obesity). The decreased collagen synthesis leads to hemorrhagic purple-red striae in the abdomen when the skin stretches because it cannot accommodate the increased truncal fat. Other findings include moon facies (fat deposition in the facial area), a buffalo hump (dorsocervical [upper back] fat deposition), and even electrolyte derangements seen in hyperaldosteronism. With very high levels, cortisol can attach to the mineralocorticoid receptor and have aldosterone-like activity. It is important to first elicit the history from the patient: iatrogenic hypercortisolism can occur in patients taking chronic glucocorticoid therapy (such as patients with autoimmune diseases or transplants).

Figure 9-18 A, Purple-red striae, centripetal obesity, and moon facies in a patient with hypercortisolism. B, The different forms of hypercortisolism. (1) An ACTH-secreting tumor from the pituitary causing overproduction of cortisol (Cushing disease). (2) Primary cortisol-secreting tumor of the adrenal gland causing overproduction of cortisol. (3) Tumor outside of the pituitary causing ACTH secretion and overproduction of cortisol. (4) Iatrogenic hypercortisolism from long-term prescription corticosteroid use. (A, from Lloyd RV, Douglas BR, Young WF. Atlas of Nontumor Pathology: Endocrine Diseases. Washington, DC: American Registry of Pathology; 2002. B, adapted from Kumar V, Abbas AK, Aster J. Robbins Basic Pathology, 9th ed. St. Louis: Elsevier; 2012.)
Adrenal Cushing syndrome: Excess cortisol is being produced because of an adrenal gland tumor, causing high cortisol levels. In this case, the ACTH level will be low because the tumor will be providing large amounts of cortisol and therefore large amounts of negative feedback on the pituitary.
Pituitary Cushing syndrome (Cushing disease): Only an ACTH-secreting tumor is termed Cushing disease, yet any condition that causes hypercortisolism is referred to as Cushing syndrome. An ACTH-secreting tumor in the pituitary is the most common cause of hypercortisolism after an iatrogenic cause is ruled out. Because the tumor secretes ACTH, it will lead to high levels of both ACTH and cortisol.
Ectopic (paraneoplastic) Cushing syndrome: Most often seen in small cell carcinoma of the lung (which can also secrete ADH), this will lead to high levels of both ACTH and cortisol.
Both pituitary and ectopic Cushing syndrome cause increased ACTH and cortisol, so there must be a way to distinguish the two. The answer is a dexamethasone suppression test. In this test, a low dose of dexamethasone (a cortisol analogue) is given, and a high dose of dexamethasone is given to see if negative feedback is still active to some extent. In the low-dose case, neither pituitary nor ectopic Cushing syndrome will show a decrease in cortisol production. However, the high dose test can differentiate the two—the pituitary still has some negative feedback ability (because it is used to having negative feedback) and will show cortisol suppression with high-dose dexamethasone. Because the ectopic ACTH-secreting tumor does not have a negative feedback loop, it shows no suppression with either low or high dose.
Increased Catecholamine Secretion
Increased catecholamine secretion can occur from tumors of the adrenal medulla or the sympathetic nervous chain. This can be caused by a pheochromocytoma, paraganglioma, or neuroblastoma, depending on the age of the patient.
Pheochromocytoma (Fig. 9-19): Occurs in adults and is a neoplasm of the chromaffin cells, which secrete catecholamines such as epinephrine and norepinephrine. Although these are mostly benign (meaning they cannot metastasize), they cause the five Ps of hyperadrenergic states: pressure (hypertension), pain (headache), perspiration (sweating is sympathetic cholinergic), palpitations (tachycardia from β1-receptor activation on the heart), and pallor (from α1 vasoconstriction). These symptoms are not constant, but rather occur sporadically. There is also the rule of 10% (which is not quite accurate, but easy to remember):

Figure 9-19 Gross image of a pheochromocytoma, a tumor of the chromaffin cells of the adrenal medulla. (From Kumar V, Abbas AK, Fausto N, Aster J. Robbins & Cotran Pathologic Basis of Disease. 8th ed. New York: Elsevier; 2009. Courtesy Jerrold R. Turner, MD, PhD, Department of Pathology, University of Chicago Hospitals, Chicago, IL.)
10% are extraadrenal in the sympathetic chain (paraganglioma)
10% (actually 30%) are familial (neurofibromatosis, MEN IIa/IIb, von Hippel–Lindau)
… and of course, you’re 10 times more likely to see this on an exam than in real life.
Diagnosis is by measuring 24-hour urine excretion of catecholamines and total metanephrines (metabolites of catecholamines). Treatment is surgery, but medical interventions to block the excess sympathetic outflow are also required before removal. The medications phenoxybenzamine (an irreversible α1 antagonist; important that it is irreversible because the high levels of catecholamines can outcompete a reversible agent) and a beta blocker are commonly given.
Neuroblastoma (Fig. 9-20) occurs in children and is the fourth most common childhood cancer (after acute lymphocytic leukemia, lymphoma, and medulloblastoma, making it the most common solid extracranial tumor). This is associated with N-MYC amplification and can commonly metastasize to liver, skin, and bones. Young children (< 1 year of age) have a good prognosis, with the tumor often spontaneously disappearing; older children have a very poor prognosis. Histologically, this is a “small cell” tumor (see Fig. 9-20) with Homer-Wright rosettes; a rosette is a rose-shaped decoration, meaning that all the cells come together in grouped clusters like a bunch of roses. Neuroblastoma may be first suspected by finding an abdominal mass on physical examination. Diagnosis can be confirmed by urinary homovanillic acid (HVA) levels (a breakdown product of dopamine). Treatment is surgery.

Figure 9-20 A, Gross image of neuroblastoma. B, Histology of neuroblastoma including Homer-Wright rosettes where cells come together like a cluster of roses. (From Kumar V, Abbas AK, Fausto N, Aster J. Robbins & Cotran Pathologic Basis of Disease. 8th ed. New York: Elsevier; 2009. Courtesy Arthur Weinberg, MD, University of Texas Southwestern Medical School, Dallas, TX.)
Endocrine Pancreas
Anatomy, Embryology, and Histology
The pancreas is a dual-function organ, having both endocrine (hormone release into the blood) and exocrine (excreting contents such as enzymes into the pancreatic duct and then the duodenum) functions. The exocrine function of the pancreas is covered in Chapter 10, but the endocrine function will be covered here.
The pancreas lies deep (posterior) to the stomach and is a retroperitoneal organ except for part of the tail of the pancreas (Fig. 9-21). The ductal system is important to the exocrine function of the pancreas, but because endocrine hormones are put into the blood and not into ducts, they do not play a role in the endocrine function of the pancreas.

Figure 9-21 Position of the pancreas as it lies in the abdomen (deep to the stomach). (From Drake RL, Vogl AW, Mitchell AWM. Gray’s Anatomy for Students. 2nd ed. Philadelphia: Elsevier; 2009.)
Embryologically, the pancreas is made up of dorsal and ventral buds that came from the foregut (Fig. 9-22). These can fuse abnormally and lead to an annular pancreas (see Fig. 9-22) that encircles the duodenum. This can lead to duodenal stenosis if the annular pancreas is causing compression.
Figure 9-22 A, Embryology of the pancreas, showing both dorsal and ventral buds coming together; in this image, they come together incorrectly, causing an annular pancreas. B, Annular pancreas causing compression of the duodenum. (A, from Schoenwolf GC, Bleyl SB, Brauer PR, Francis-West PH. Larsen’s Human Embryology. 4th ed. Philadelphia: Elsevier; 2008. B, from Carlson B. Human Embryology and Developmental Biology. 4th ed. Philadelphia: Elsevier; 2008.)
Although the endocrine pancreas accounts for only 2% of the mass of the pancreas, it is necessary to sustain life. The endocrine pancreas is contained within islets of Langerhans (Fig. 9-23), which can be seen in the histologic image as a lighter-colored and more scantly cellular area. These islets of Langerhans have alpha (α), beta (β), and delta (δ) cells which are the main cells that secrete glucagon, insulin, and somatostatin/gastrin, respectively. The alpha cells, which secrete glucagon, are on the outside (alpha – away from center). The beta cells, which secrete insulin, are towards the center.

Figure 9-23 A, Histology of the pancreas, with zoom view of an islet of Langerhans, demonstrating the different cell types and their distribution throughout the islet. B, Animated representation of the islet of Langerhans, demonstrating the peripheral α cells (glucagon), the central β cells (insulin) and the randomly dispersed δ cells (somatostatin, gastrin). (A, from Kierszenbaum AL, Tres LL. Histology and Cell Biology. 3rd ed. St. Louis: Elsevier; 2011. B, from Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
Endocrine Pancreas Physiology
Insulin (From the B Cells of the Islets of Langerhans)
Insulin plays a key role in the regulation of blood glucose levels. Although there are many stimuli for its release, the most important is elevated glucose in the blood (hyperglycemia), such as after a meal. Insulin is released from the beta cell of the islet of Langerhans in response to elevated blood glucose by the following steps: (1) glucose goes into the beta cell via GLUT-2 glucose transporters; (2) glucose is metabolized into adenosine triphosphate (ATP); (3) this ATP closes the potassium channel that previously was hyperpolarizing the cell, leading to depolarization of the cell; (4) this depolarization triggers a calcium channel to open, facilitating calcium influx into the cell; and (5) the calcium influx into the cell triggers release of preformed insulin into the bloodstream (Fig. 9-24A). In this preformed insulin-containing granule is also a cleavage byproduct of proinsulin called C-peptide (connecting peptide)—this will become important in pathology because C-peptide is present with endogenous insulin, but is not present in commercially available injectable insulin; C-peptide levels can determine whether the insulin is coming from the body or from a needle.

Figure 9-24 A, Illustration of an insulin-secreting β cell in the islets of Langerhans, showing the mechanism of insulin secretion. Glucose moves into the cell via GLUT-2. It is then metabolized into ATP, which closes the K+ channel, stopping the tonic hyperpolarization of the cell. This relative depolarization triggers calcium influx into the cell via a voltage-gated Ca2 + channel, and insulin is then released. B, Action of insulin on target cells, causing cell growth, anabolic effects, and GLUT-4 glucose channel fusion into the membrane to enhance glucose uptake into insulin-dependent cells. (From Kumar V, Fausto N, Abbas AK. Robbins & Cotran Pathologic Basis of Disease. 7th ed. Philadelphia: WB Saunders; 2004.)
The mechanism of action of the beta cell also becomes important with sulfonylurea medications, which attach to the sulfonylurea receptor on the potassium channel. Triggering the sulfonylurea receptor closes the potassium channel (much like ATP did), leading to depolarization and calcium influx, and triggering insulin release from the beta cells. Diazoxide, on the other hand, keeps this channel open and prevents insulin release, and can be used in the treatment of insulinoma (described later).
The insulin, now released into the bloodstream, acts on its insulin receptor, starting a cascade mediated by MAP kinase and PI-3 K. All these physiologic effects are what would be expected when a lot of sugar is available for the body to use. Insulin is the “hormone of plenty” and has an anabolic effect overall. The tissues most dependent on insulin for glucose uptake are skeletal muscle and adipose tissue. Some parts of the body, such as the brain and red blood cells, use a different glucose channel (GLUT-1) that is not insulin-dependent because these always need glucose regardless of availability (red blood cells do not have nuclei or mitochondria and can only use glucose as a fuel).
Fusion of GLUT-4 glucose channels into the cell membrane: Promotes glucose uptake into cells, reducing serum blood sugar level.
Promotes glycogen creation, prevents gluconeogenesis: This is due to the abundance of sugar with higher insulin levels—promotes storage of sugar as glycogen and prevents gluconeogenesis because there is no need to make glucose if there is adequate glucose available.
Increases lipoprotein lipase activity (which brings fat into cells) and inhibits hormone-sensitive lipase activity (which brings fat into the bloodstream). This decreases lipolysis by promoting fat deposition in the adipocytes, which makes sense because there is glucose available, which is a preferable fuel. Glucagon has the opposite effect on these two hormones.
Promotes K+ uptake into cells by stimulating the Na+/K+-ATPase.
Glucagon (From the α Cells of the Islets of Langerhans)
Glucagon (Fig. 9-25) has many effects opposing to insulin (and is the “hormone of starvation” rather than the hormone of plenty). Therefore, the major stimulus for glucagon secretion is hypoglycemia, and the response of the cells of the body is to act to correct it.
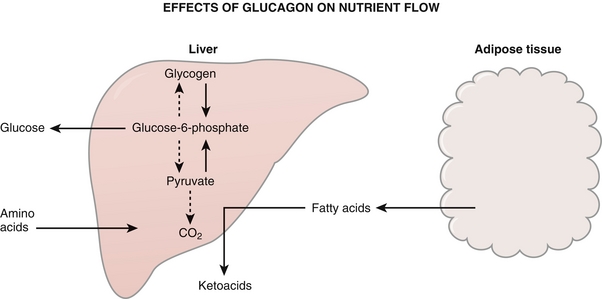
Figure 9-25 Effects of glucagon. Glucagon is the “hormone of starvation” and acts to promote release of fatty acids from adipocytes to fuel the starving body. It also increases gluconeogenesis and causes breakdown of glycogen to keep blood sugar levels up. (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
Increasing glycogenolysis and gluconeogenesis: Glucagon promotes breakdown of glycogen as well as gluconeogenesis. This is through decreased production of fructose 2,6-bisphosphate, which, in turn, decreases phosphofructokinase activity. The end result of this is slowing metabolism of glucose, favoring gluconeogenesis.
Increasing availability of fatty acids: Glucagon decreases lipoprotein lipase activity (less importing of fat into adipocytes) and increases hormone-sensitive lipase activity (increases exportation of fat from adipocytes into the bloodstream). These will then be turned into ketoacids after metabolism and account for the ketonuria and ketonemia in patients who are starving (or have absolute insulin deficiency such as in diabetic ketoacidosis).
Diabetes
Of all the functions of the pancreas, the most commonly seen pathology involves disorders of insulin production (type 1 diabetes) and disorders of insulin sensitivity (type 2 diabetes) (Fig. 9-26). Both of these will lead to the cardinal symptoms of polyuria (osmotic diuresis) and polydipsia (thirst secondary to fluid loss from osmotic diuresis), but type 2 diabetes may often be asymptomatic and simply discovered on laboratory screening.

Figure 9-26 A, Systemic complications of diabetes. B, Glove-and-stocking distribution of peripheral neuropathy in diabetes. C, Diabetic retinopathy. D, Foot ulcer as a result of peripheral neuropathy and peripheral vascular disease. (A, from Kumar V, Abbas AK, Aster J. Robbins Basic Pathology. 9th ed. St. Louis: Elsevier; 2012. B, from Douglas G, Nicol F, Robertson C. Macleod's Clinical Examination.12th ed. Edinburgh: Elsevier; 2009. C, from Swartz MH. Textbook of Physical Diagnosis. 4th ed. Philadelphia: Elsevier; 2002. D, from Becker JM, Stucchi AF. Essentials of Surgery. Philadelphia: Elsevier; 2006.)
Type 1 diabetes mellitus (10%): Characterized by autoimmune destruction of the beta cells of the islets of Langerhans, leading to absolute insulin deficiency. This usually affects younger people, and their body habitus is typically thin owing to lack of insulin to deposit fat into adipose tissue. This is associated with HLA-DR3 and HLA-DR4 in whites, HLA-DR7 in African Americans, and HLA-DR9 in Japanese. The absolute insulin deficiency can lead to diabetic ketoacidosis (DKA), a potentially fatal disease if untreated.
Type 2 diabetes mellitus (90%): The most common type of diabetes, by far, characterized by insulin resistance secondary to decreased insulin receptor synthesis from increased adiposity. Defects in the receptor and signaling pathways also develop, further limiting the effect of insulin in these patients. Initially, patients may be hyperinsulinemic as a compensation for their decreased insulin sensitivity, but eventually the beta cells will fail from the increased demand, and they will have a relative insulin deficiency. The relative insulin deficiency does not lead to ketoacidosis (enough insulin present to prevent widespread lipolysis) but can lead to a hyperosmolar nonketotic state (HNS; previously called hyperosmolar hyperglycemic nonketotic coma, HHNKC). HNS is more descriptive because it does not necessarily cause coma. Type 2 diabetes mellitus has a much stronger link to family history than type 1 diabetes mellitus.
Diagnosis of Diabetes
The following are the current criteria used for diagnosis of diabetes. The diagnosis is not met with a single value on a single day; one of the four criteria must be met on two separate days.
Random plasma glucose ≥ 200 mg/dL with symptoms of diabetes (polyuria, polydipsia)
Fasting plasma glucose ≥ 126 mg/dL
Two-hour plasma glucose level after 75 g of oral glucose administered ≥ 200 mg/dL
HbA1c of 6.5% or higher (HbA1c is a measure of the level of glycosylation of hemoglobin and is a marker of long-term glucose control over the past few months because red blood cells usually live 120 days)
If there is a suspicion of type 1 diabetes and the diagnosis is not clear, there are autoantibodies that can be tested as well. These include islet cell autoantibodies, anti-insulin antibodies, and antibodies to glutamic acid decarboxylase.
Chronic Effects of Hyperglycemia
There are many problems with long-term hyperglycemia, whether from type 1 or type 2 diabetes. The classic triad of microvascular disease is retinopathy, neuropathy, and nephropathy, but there is also macrovascular disease (accelerated atherosclerosis), which leads to increased incidence of strokes and myocardial infarctions. These complications occur through three pathologic processes: nonenzymatic glycosylation, osmotic damage, and diabetic microangiopathy.
Nonenzymatic glycosylation (Fig. 9-27B): This is when hyperglycemia allows sugar to attach to proteins (without the aid of an enzyme, hence nonenzymatic). This causes dysfunction of the protein; in blood vessels, this leads to increased vessel permeability and predisposes it to atherosclerosis. Not only does this play a role in all three microvascular diseases (retinopathy, neuropathy, and nephropathy) as well as macrovascular disease, but it also gives a way to track glycemic control over the long term, by HbA1c, which is a measure of the glycosylation of hemoglobin. In diabetic patients, the goal is usually an HbA1c of less than 7%.

Figure 9-27 A, Osmotic damage via the aldose reductase pathway. B, Diabetic nephropathy with Kimmelsteil-Wilson nodules of type IV collagen, as well as thickening of the arteriole (hyaline arteriolosclerosis) (arrow) from nonenzymatic glycosylation. (A, from Clark M. Crash Course: Metabolism and Nutrition. Philadelphia: Elsevier; 2005. B, from Kumar P, Clark M. Clinical Medicine. 6th ed. Edinburgh: Elsevier; 2005.)
Osmotic damage: Glucose can be converted into sorbitol by aldose reductase. Although some organs have the ability to metabolize sorbitol, the eye and the nerves do not. This sorbitol gets trapped inside the cell and can cause osmotic damage when water rushes into the cell to balance the osmotic forces. This leads to Schwann cell damage in neurons and promotes neuropathy through demyelination. In the eye, this can lead to cataracts (in the lens) and retinopathy (in the retina).
Diabetic microangoiopathy: With increased blood sugar, the endothelial cells that line the blood vessels take in additional glucose. Some of this glucose is made into glycoproteins, which are placed on the surface of the endothelial cells and cause thickening and weakening of the basement membrane (type IV collagen). This weakening can lead to increased permeability, causing proteinuria in the kidney (diabetic glomerulosclerosis with “Kimmelstiel-Wilson nodules,” which are actually type IV collagen) and potential for edema and hemorrhage in the retina (diabetic retinopathy). Because there are blood vessels that nourish the nerves (vasa nervorum), damage here can also lead to diabetic neuropathy.
Acute Emergencies in Diabetes
As mentioned previously, type 1 diabetes predisposes to the potential development of DKA, whereas type 2 diabetes predisposes to the potential development of HNS. These are both acute emergencies that need to be recognized and treated.
Diabetic ketoacidosis: Often seen in patients who are noncompliant with insulin dosage when treated for type 1 or due to a secondary illness causing increased stress on the body. This may even be the first manifestation of previously undiagnosed type 1 diabetes mellitus. The absolute insulin deficiency leads to hyperglycemia from decreased cell uptake of glucose coupled with high levels of gluconeogenesis from high glucagon levels; the hyperglycemia causes subsequent dehydration from osmotic diuresis. Despite the abundance of serum glucose, without insulin, there is a lack of intracellular glucose. The body shifts toward lipolysis (using fat for energy), with ketoacids such as acetoacetate and β-hydroxybutyrate being produced as a byproduct of β-oxidation of fatty acids; this is because there is no insulin to inhibit hormone-sensitive lipase or stimulate lipoprotein lipase, which would decrease fat metabolism. Laboratory findings include hyperkalemic acidosis with increased anion gap (due to ketones and potentially lactic acidosis if in shock), pseudohyponatremia (glucose osmoles draw in water and “dilute” the sodium, making it appear as though there is hyponatremia), and ketonuria and ketonemia. Clinical findings include Kussmaul respirations (deep rapid breathing from profound acidosis—respiratory compensation), dehydration with tachycardia (from osmotic diuresis), and a “fruity” odor on the breath from acetone being secreted in the saliva. Treatment is fluids and insulin, as well as potassium supplementation. In addition to promoting glucose uptake, insulin upregulates the Na+/K+-ATPase, promoting cellular uptake of potassium. Hypokalemia with insulin administration must be carefully monitored in this condition because the patients usually have a global potassium deficit from osmotic diuresis, even if they appear hyperkalemic from the acidosis moving K+ out of the cells (by H+/K+ exchangers).
Hyperosmolar nonketotic state (HNS): Associated with type 2 diabetes mellitus, with severely high blood sugar (usually > 600 mg/dL) and subsequent hyperosmolarity. This leads to significant dehydration from osmotic diuresis, as well as osmolar shifts, which cause changes in mental status. There is no ketosis because of the presence of insulin, which inhibits lipolysis. This syndrome goes by numerous names, and hyperosmolar hyperglycemic state (HHS) and hyperosmolar nonketotic coma (HONKC) may also be seen.
Islet Cell Tumors
Insulinoma: A benign tumor of the β cells of the islets of Langerhans, with a strong correlation to MEN I syndrome (pituitary, pancreas, parathyroid). This causes chronically elevated insulin levels, leading to hypoglycemia. As mentioned before, because this is endogenous insulin, there will be high levels of insulin as well as high levels of C-peptide. High levels of C-peptide will not be seen with injectable insulin, and therefore the two conditions can be differentiated based on this fact. Treatment is surgery or streptozocin, which is a medication that is toxic to beta cells. Diazoxide can also help prevent release of insulin and prevent hypoglycemia. Octreotide can also be considered because it will decrease insulin release as well.
Glucagonoma: A malignant tumor of the α cells of the islets of Langerhans, with hyperglycemia from the increased gluconeogenesis and glycogenolysis caused by the glucagon. In addition, there is a characteristic rash called necrolytic migratory erythema (NME). Treatment is surgery and octreotide (a somatostatin analogue), which decreases glucagon production by the malignant tumor.
Somatostatinoma: A malignant tumor of the δ cells of the islets of Langerhans. Somatostatin inhibits almost every GI-related hormone, including gastrin, cholecystokinin (CCK), and secretin. Inhibition of CCK causes the gallbladder to fail to contract, leading to cholelithiasis. Inhibition of secretin can cause steatorrhea because secretin causes bicarbonate secretion from the pancreas, allowing lipases to work—lack of this can cause the digestive enzymes to be ineffective, leading to malabsorption and steatorrhea.
VIPoma: A malignant tumor causing release of vasoactive intestinal peptide— this peptide in high amounts causes what is called pancreatic cholera. A cholera-like syndrome characterized by diffuse diarrhea with dehydration, hypokalemic acidosis (GI loss of potassium and bicarbonate), and widespread vasodilation (hence vasoactive).
Zollinger-Ellison syndrome (gastrinoma): A malignant tumor that secretes gastrin, leading to increased acid production with widespread gastroduodenal ulcers. The increased acid denatures digestive enzymes, leading to malabsorption and diarrhea. Any time multiple or refractory duodenal ulcers are present, you should have a high suspicion for this syndrome. The diagnosis is also suggested by elevated gastrin levels (but proton-pump inhibitor medication will also cause elevated gastrin as a reflex from the decreased stomach acidity).
Pharmacology of Diabetes Mellitus
Insulin
For those with type 1 diabetes, insulin injections are required to sustain life. For those with type 2 diabetes, insulin may be required because there is progressive beta cell failure over time and subsequent insulin deficiency. Insulin is also the most common medication used to treat pregnant women with gestational diabetes because insulin does not cross the blood–placenta barrier. Insulin is injected subcutaneously or through an insulin pump. Because insulin promotes fatty acid uptake into adipocytes, local lipodystrophy can occur. The most common side effect of insulin injections is hypoglycemia, especially if a patient forgets to take a meal after dose administration.
Sulfonylurea Drugs—Glyburide, Glipizide, Glimepiride
Sulfonylurea drugs act to increase the body’s pancreatic production of insulin. Recall that normally glucose enters the beta cell and is metabolized to ATP. ATP then closes a potassium channel that was previously hyperpolarizing the cell (see Fig. 9-24A). This depolarization leads to calcium influx and insulin release. The same potassium channel has a sulfonylurea receptor that sulfonylurea can bind to and close the channel independent of ATP, leading to insulin release. Of course, this will only work if there are functional beta cells in the pancreas available to secrete insulin (will not work in type 1 diabetes). The most common side effect of sulfonylurea drugs is hypoglycemia.
Biguanides—Metformin
Metformin has numerous actions to reduce hyperglycemia by decreasing hepatic gluconeogenesis and acts as an insulin-sensitizing agent by promoting glucose uptake in the body’s cells. Because this medication prevents hyperglycemia rather than causing hypoglycemia, it is said to be euglycemic in that it cannot be a cause of hypoglycemia. Also, because there is no direct action of metformin on the pancreas, this can be used in patients without beta cell function to help lower injected insulin requirements. The most serious side effect is lactic acidosis—lactate uptake in the liver is decreased with metformin because lactate is changed into glucose through gluconeogenesis (a process that metformin inhibits). Although this is inconsequential in healthy individuals because the excess lactate can be cleared by the kidneys, patients with renal failure (decreased lactate clearance) or respiratory disease causing relative hypoxia (leading to increased lactate through anaerobic metabolism) can develop lactic acidosis.
Thiazolidinediones—Pioglitazone (-Glitazone Drugs)
These medications are insulin sensitizers and bind to a nuclear receptor called the peroxisome proliferator-activated receptor gamma receptor (PPAR-γ). Activation of PPAR-γ leads to increased synthesis of GLUT-4 glucose channels and other genes involved in insulin sensitivity. Side effects include water retention, leading to edema, weight gain, and potentially heart failure from fluid overload. Older preparations of thiazolidinediones caused hepatotoxicity—this has not been found in newer preparations (but may be found on an exam regardless).
Incretin Mimetics (-Tides: Exenatide, Liraglutide) and DPP-4 Inhibitors (-Gliptins: Sitagliptin, Linagliptin, Saxagliptin)
The incretin pathway (Fig. 9-28) is activated when nutrients reach the intestine and act to stimulate insulin release before the glucose actually makes it to the bloodstream to better regulate sugar. These receptors are found in the intestines, so intravenous nutrition will not activate this pathway. DPP-4 inhibitors such as sitagliptin prevent breakdown of incretins, whereas incretin analogues such as exenatide mimic the incretin GLP-1 and activate the pathway directly. As an interesting aside, exenatide is derived from Gila monster saliva and can potentially cause nausea and pancreatitis.

Figure 9-28 The incretin pathway and the drugs that act on it. (Drawn in Inkscape by Ilmari Karonen based on w:Image:Incretins and DPP 4 inhibitors.jpg from http://casesblog.blogspot.com/2006/11/dpp-4-inhibitors-for-treatment-of.html (uploaded by author). Accessed January 10, 2013, from http://en.wikipedia.org/wiki/File:Incretins_and_DPP_4_inhibitors.svg.)
α-Glucosidase Inhibitors—Acarbose, Miglitol
Acarbose inhibits the intestinal brush border α-glucosidase, an enzyme necessary for digestion of carbohydrates, leading to decreased blood sugar by decreased absorption. However, this leftover osmotic agent causes osmotic diarrhea and flatulence (from bacterial metabolism of carbohydrates, which forms H2 and CO2 gas). Because this enzyme breaks down branches of carbohydrates, monosaccharides will still be absorbed normally. Anyone who is lactose intolerant and cannot break down and absorb lactose knows that the osmotic diarrhea and flatulence from bacterial breakdown of the disaccharides is unpleasant; because of these side effects, this medication is rarely used.
Parathyroid Glands
Anatomy, Embryology, and Histology
The four parathyroid glands are the key mediators of calcium and phosphate homeostasis and therefore play an important role in bone growth and development. As the name implies, they are near the thyroid (usually on the posterior aspect, or inside the thyroid parenchyma; Fig. 9-29). The two superior parathyroid glands arise from the fourth pharyngeal pouch; the two inferior parathyroid glands arise from the third pharyngeal pouch (counterintuitive because they swap places!). The embryologic development of these two pouches can fail to develop properly in DiGeorge syndrome.
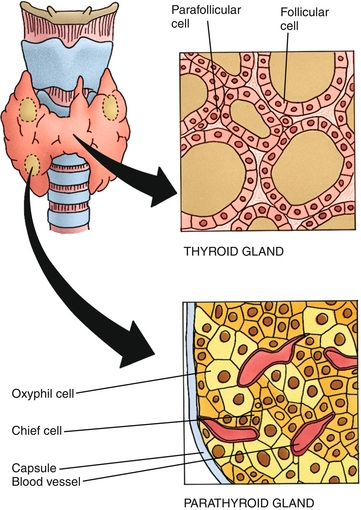
Figure 9-29 Anatomy of the parathyroid glands and illustrations of their histology. (From Gartner LP, Hiatt JL. Color Textbook of Histology. 3rd ed. Philadelphia: Saunders; 2007:313.)
DiGeorge syndrome: Failure of the third and fourth pharyngeal pouches to develop (see Chapter 4). The mnemonic to remember the associated defects is CATCH-22—Cardiac abnormalities (especially tetralogy of Fallot), Abnormal facies, Thymic aplasia, Cleft palate, and Hypoparathyroidism with hypocalcemia due to failure of parathyroid development; this disease is a deletion on chromosome 22.
The histology of the gland shows two distinct cell morphologies: chief cells and oxyphil cells. The chief cells are small, round, eosinophilic cells that create parathyroid hormone, which is stored until stimulated for release (the Chief cells have a role in Calcium). The role of oxyphil cells is unknown.
Overview of Calcium Homeostasis
Calcium (and phosphorus) homeostasis (Table 9-4; Fig. 9-30) are maintained by hormones (parathyroid hormone, calcitonin, vitamin D) and the organs that react to these hormones (bones, kidneys, and intestines). These hormones mainly regulate the way that each of these organs handles calcium; for example, bone can be deposited or reabsorbed, the intestines can absorb more or less dietary calcium, and the kidneys can reabsorb more or less calcium from the tubules. The major player in calcium homeostasis is parathyroid hormone, which is secreted in response to low serum ionized calcium (the word ionized describes the calcium that is free as ions, rather than bound to albumin or other proteins). Follow Figure 9-30 with the explanation here:


Figure 9-30 Calcium homeostasis. (From Colledge NR, Walker BR, Ralston SH. Davidson’s Principles and Practice of Medicine. 21st ed. Philadelphia: Elsevier; 2010.)
1. The parathyroid gland senses decreased ionized calcium and secretes parathyroid hormone (PTH). Parathyroid hormone has direct actions on the bones and kidneys.
2. PTH activity has three effects on the kidneys: (1) PTH increases expression of 1α-hydroxylase in the proximal tubule of the nephron, which will convert vitamin D into its active form; (2) also in the proximal tubule, PTH inhibits phosphate reabsorption (inhibits Na+-phosphate cotransporter) causing phosphaturia; this is because if both calcium and phosphate were increased in the blood, they would precipitate as calcium phosphate; (3) PTH increases calcium reabsorption in the distal convoluted tubule to increase serum calcium levels. As an aside, PTH works through the Gs-signaling pathway, which generates increased cyclic adenosine monophosphate (cAMP) as a signaling molecule—the actions on the renal tubular cells cause increased cAMP production, leading to increased urinary cAMP.
3. PTH activity directly stimulates the osteoblasts. This may be counterintuitive when thinking about osteoblasts, which form bone by depositing calcium. The reason for this apparent paradox is that the osteoblasts in turn signal osteoclasts through RANKL to reabsorb bone and therefore increase serum calcium. This initial stimulation of osteoblasts, however, is why small bursts of PTH can be used clinically to just stimulate osteoblasts and treat osteoporosis.
4. Vitamin D, when in its active form [1,25(OH)-vitamin D, “calcitriol”], has its main effect on the intestines, increasing synthesis of a calcium-binding protein (calbindin D-28 K) to promote increased absorption of dietary calcium. Calcitriol also increases dietary absorption of phosphate. Vitamin D also has effects on the kidneys (increasing both calcium and phosphate reabsorption) and on bone to stimulate resorption of old bone and mineralization of new bone (bone remodeling). The net effect of vitamin D is increasing calcium and phosphate levels through the kidneys, gut, and bone (and subsequently promoting bone remodeling).
Vitamin D can be obtained through dietary sources or formed in the skin through sunlight (major source). This inactive vitamin D then goes to the liver and, through hepatic 25-hydroxylase, becomes 25(OH)-vitamin D, which is still inactive. It is not until the renal activity of 1α-hydroxylase turns it into 1,25(OH)-vitamin D, or calcitriol, that it is active. Therefore, hepatic dysfunction (lack of sufficient 25-hydroxylase activity) or renal dysfunction (lack of sufficient 1α-hydroxylase activity) can lead to a functional vitamin D deficiency.
Calcitonin is also involved in calcium homeostasis, but plays at most a minor role. Calcitonin tones down calcium (lowers serum calcium) by inhibiting bone resorption (inhibiting osteoclast activity). Calcitonin is secreted by the parafollicular C cells.
Laboratory Testing and Pitfalls
Understanding the laboratory testing necessary to diagnose disorders of calcium and phosphate homeostasis is important. First, it is important to ensure that the calcium level is truly accurate: changes in albumin and changes in acid-base status can alter total and ionized calcium levels, respectively. Total calcium measures the amount of calcium in the blood regardless of whether it is attached to albumin or not—a free calcium only takes into account the free (ionized) calcium that is active.
Hypoalbuminemia: Because calcium is attached to albumin, lower albumin levels can lead to a decreased calcium level (1 g/dL of albumin binds 0.8 mg/dL of calcium at physiologic pH). Therefore, in the asymptomatic patient, hypoalbuminemia is the most common cause of low calcium on a lab test. However, this will not change the ionized calcium level. The ionized (free, unbound) calcium has not changed, so the patient will not have symptoms related to abnormal calcium levels.
Acid-base disturbances: Albumin is a large, negatively charged protein. These negatively charged sites can bind H+ or Ca2 +. In acidemia, in which there is excess H+, the H+ will take up most of those sites, displacing calcium and leading to increased ionized calcium levels because the calcium will have nowhere to bind. Conversely, in alkalemia, in which there are fewer H+, there are more open sites on albumin for Ca2 + to bind, and therefore proportionally more calcium will bind to albumin, leading to decreased ionized calcium. Because in both these cases the calcium is just being moved between the bound and unbound state and is not being gained or lost from the body, the total calcium does not change in acid-base disturbances. However, the ionized calcium has changed, so the patient may have symptoms related to abnormal ionized calcium levels (Fig. 9-31).

{kind=link}
{kind=link}
The etiology of the calcium disturbance can be further elucidated by the use of additional lab testing, depending on the pathology. In the next section, each pathologic cause of altered calcium or phosphate levels will be addressed as well as the diagnosis and treatment. It is also important to note that for these disturbances, as well as for endocrine disturbances in general, “normal ranges” for hormones must be taken in their appropriate context. For instance, a normal calcium level is 8.5 to 10 mg/dL and a normal PTH level is 10 to 60 pg/mL. However, if a patient’s calcium is 11 mg/dL (high) and PTH level is 50 pg/mL (“normal” range), it is abnormal because in the context of hypercalcemia, the normal-range PTH level is much too high; it should be below 10 pg/mL because there should be negative feedback on the parathyroid glands if they are functioning normally. It is important to keep this in mind to prevent being misled.
A Primary hypoparathyroidism: Calcium is low and the parathyroid gland is not attempting to fix it (low PTH).
B Hypoalbuminemia: Decreased albumin leads to decreased total calcium but ionized (free) calcium is normal, and therefore PTH is normal range.
C Secondary hyperparathyroidism: High PTH is unable to correct calcium; this can be seen in renal failure because vitamin D cannot be made to help correct Ca2 +. Note that this could also represent pseudohypoparathyroidism, described later.
D Respiratory alkalosis: Total calcium is normal, but alkalosis causes increased binding of calcium to albumin, leading to decreased ionized calcium and subsequent increased PTH level to increase ionized calcium.
E Primary hyperparathyroidism: High calcium, yet PTH is still high.
F Hypercalcemia of malignancy: High calcium, PTH low because PTHrP is being created instead (or direct osteolytic bone metastases).
Refer to Figure 9-32 when revisiting each pathologic disease to understand why the lab values are as described previously.

Figure 9-32 Schematic showing serum calcium and serum PTH; normal is shown by the box in the center. A represents primary hypoparathyroidism, B represents hypoalbuminemia, C represents secondary hyperparathyroidism, D represents respiratory alkalosis, E represents primary hyperparathyroidism, and F represents hypercalcemia of malignancy. (From Goljan EF. Rapid Review Pathology Revised Reprint. 3rd ed. Philadelphia: Elsevier; 2011.)
Pathology Causing Increased Calcium Levels
When hypercalcemia occurs, the typical symptoms are remembered by “stones, bone, groans, and psychiatric overtones.”
Stones: Calcium-containing renal stones are the most common manifestation of hypercalcemia because the increased calcium in the urine promotes stone formation. Gallstones can also occur.
Bones: depending on the cause of the hypercalcemia, different bone manifestations can occur. In hyperparathyroidism, the elevated PTH levels cause hypercalcemia, and the effects of sustained high PTH levels on the osteoblasts (which signal the osteoclasts) lead to a syndrome called osteitis fibrosa cystica, in which there are cystic and hemorrhagic bone lesions from high osteoclastic activity and subperiosteal resorption of bone, especially in the fingers. High PTH levels also cause osteoporosis for similar reasons. Lastly, there can be a “salt-and-pepper” appearance of the skull (Fig. 9-33B) from diffuse osteoclastic activity in the skull.

Figure 9-33 A, Effects of hyperparathyroidism with hypercalcemia. B, Salt-and-pepper skull from hyperparathyroidism. C, Comparative radiographs of fingers (left finger, hyperparathyroidism; right finger, normal). (A, from Kumar V, Cotran RS, Robbins SL. Robbins Basic Pathology. 7th ed. Philadelphia: Elsevier; 2002. B, from Weissman BN. Imaging of Arthritis and Metabolic Bone Disease. Philadelphia: Elsevier; 2009. C, from Underwood JCE, Cross SS. General and Systematic Pathology. 5th ed. Edinburgh: Elsevier; 2009.)
Groans: The groans are from GI problems such as abdominal pain, nausea, and vomiting. This is because hypercalcemia is a cause of pancreatitis (from enzyme activation), and calcium also causes increased gastrin production and subsequent acid secretion in the stomach, potentially leading to peptic ulcer disease.
Psychiatric overtones: Common, but can have varied findings, ranging from depression and anxiety to psychosis and coma.
The differential diagnosis of increased calcium levels is long and can be remembered by the mnemonic CHIMPANZEES. However, it is worth noting that primary hyperparathyroidism and malignancies account for almost all causes (> 90%) of hypercalcemia and should come to mind first when hypercalcemia is found.
Calcium ingestion (milk-alkali syndrome)
Hyperparathyroidism (most common cause in absence of malignancy)
Multiple myeloma (from bone breakdown)
Addison disease (due to volume contraction causing increased reabsorption of fluid and electrolytes, including calcium, from the proximal tubule of the nephron)
Neoplasms (either those secreting parathyroid hormone–related peptide [PTHrP] or from bone metastases)
Excessive vitamin D intake or vitamin A intake
Excessive thyroid hormone (hyperthyroidism causes increased bone turnover)
Sarcoidosis (or other granulomatous disease, due to increased macrophage activity; macrophages also have 1α-hydroxylase activity and therefore, in granulomatous diseases with sustained macrophage activity, hypercalcemia secondary to increased 1,25-vitamin D levels can occur)
Because the two main causes of hypercalcemia are hyperparathyroidism and malignancy, they will be covered in additional detail.
Hyperparathyroidism (Fig. 9-34) can be primary (gland hyperfunction), secondary (due to hypovitaminosis D or renal failure), or tertiary (prolonged secondary hyperparathyroidism causes the glands to become autonomous). Primary hyperparathyroidism is usually due to a single hyperfunctioning adenoma (80%) but can also be due to diffuse hyperplasia of all four glands (20%). Recall that primary hyperparathyroidism is associated with MEN I and IIa.

Figure 9-34 Hyperparathyroidism and the effects downstream of increased PTH and increased calcium. (From Damjanov I. Pathophysiology. Philadelphia: Elsevier; 2008.)
Laboratory diagnosis of primary hyperparathyroidism: Hypercalcemia in the setting of normal to high PTH is diagnostic of hyperparathyroidism because it shows that the parathyroid glands are secreting PTH to increase calcium when it is inappropriate to do so.
Imaging diagnosis of primary hyperparathyroidism: A technetium-99 m-sestamibi radionuclide scan can indicate whether or not there is a single hyperfunctioning adenoma, which takes up the radiotracer more avidly than the other normal parathyroid glands. In either case (hyperplasia or adenoma) the treatment is surgical.
Secondary hyperparathyroidism: Due to hypovitaminosis D, either because of dietary deficiency or secondary to renal failure. Because active 1,25(OH)-vitamin D is essential to the ability of the body to increase calcium levels, in deficiency of vitamin D or in renal failure, in which the 25(OH)-vitamin D does not get to the proximal tubule to be 1-hydroxylated, the PTH will have sustained release because the calcium never increases to give negative feedback to the glands. Therefore, the laboratory studies will show high PTH with low calcium and high phosphorous.
Tertiary hyperparathyroidism: With prolonged secondary hyperparathyroidism, the parathyroid glands become autonomous, and even if the underlying cause is fixed (usually with renal transplantation), the PTH will remain high. The sustained high PTH has caused these hyperfunctioning parathyroid glands to lose their negative feedback.
Hypercalcemia of malignancy: Can occur in two settings: (1) a paraneoplastic syndrome in which PTHrP is secreted by the cancerous cells, and (2) direct bone destruction leading to increased calcium levels. The best way to distinguish between these two entities is a PTHrP level. With hypercalcemia of malignancy caused by PTHrP secretion, the PTH will be low, the PTHrP will be high, and the calcium will be high. The classic cause of this is small cell carcinoma of the lung. In addition, diffuse lytic metastases to bone can cause destruction, leading to high calcium but low PTH and PTHrP levels.
Treatment of any form of hypercalcemia is correction of the underlying cause. However, acute treatment is aimed at (1) diluting the calcium, (2) increasing urinary excretion of calcium, and (3) protecting bone.
Dilution of the serum calcium occurs from large amounts of intravenous hydration.
Intravenous furosemide (Lasix) causes calcium to be excreted in the urine (see Chapter 15 for mechanism)
Bisphosphonates deposit in the bone and prevent osteoclastic release of calcium by preventing breakdown of bone.
Pathology Causing Decreased Calcium Levels
Again, when dealing with a low serum calcium level, it is most important to first correct the calcium for hypoalbuminemia, if present. This is easily the most common cause of hypocalcemia in an asymptomatic patient. However, a patient who truly has hypocalcemia will often display some of the following symptoms:
Tetany: The decreased calcium level causes hyperexcitability of the muscles; this can be assessed clinically by Trousseau sign of latent tetany and Chvostek sign. Trousseau sign involves inducing carpal (or pedal) spasm by inflating a blood pressure cuff on the arm (or leg) above the patient’s systolic pressure. This action allows the limb to develop a mild acidosis, and the subsequent hyperexcitable neurons will fire and trigger muscle contraction. Chvostek sign is tapping on the inferior portion of the zygoma, in the area of the facial nerve, which will subsequently cause facial twitching.
Arrhythmias can occur because hypocalcemia leads to QT prolongation of the electrocardiogram, increasing the susceptibility of the patient to torsades de pointes, which occurs when a premature beat attempts to fire during the repolarization of the heart (termed an R-on-T phenomenon because the QRS complex represents ventricular depolarization, which is abnormally occurring during the repolarization of the heart).
The main cause of hypocalcemia is hypoparathyroidism. Other causes include DiGeorge syndrome (covered earlier in the section “Anatomy, Embryology, and Histology”) and pseudohypoparathyroidism. Pseudohypoparathyroidism has many subtypes, the most commonly tested being Albright hereditary osteodystrophy (pseudohypoparathyroidism type Ia). This is an autosomal dominant genetic condition in which the Gs subunit of the PTH receptor is broken, leading to ineffective action of PTH on the kidney.
Hypoparathyroidism: Can be caused iatrogenically by previous neck surgery, but the most common nonsurgical cause is autoimmune. This is characterized by low PTH levels in the face of low calcium. Treatment is supplementation with calcium and active vitamin D.
Pseudohypoparathyroidism: There is a dysfunctional PTH receptor on the kidney, but the parathyroid gland itself functions normally. Therefore, there will be low calcium (as though the patient had hypoparathyroidism, hence the name pseudohypoparathyroidism), but the PTH will be high because the gland is attempting to correct the calcium level. One classic sign of pseudohypoparathyroidism is the knuckle-knuckle-dimple-dimple sign due to shortening of the fourth and fifth metacarpals (Fig. 9-35). This is in contrast to Turner syndrome, in which there is a knuckle-knuckle-dimple-knuckle sign.

Figure 9-35 Pseudohypoparathyroidism showing classic “knuckle-knuckle-dimple-dimple” sign. (From Swash M, Glynn M, Hutchison R. Hutchison’s Clinical Methods. 22nd ed. Edinburgh: Elsevier; 2007.)
Hypovitaminosis D: Lack of vitamin D leads to two different conditions, depending on whether the patient is a child or an adult. In children, it leads to rickets (Fig. 9-36), which causes insufficient bone mineralization for the growing bones, leading to skeletal deformities, especially genu varum (bowing of the femurs). In adults, lack of vitamin D leads to ineffective mineralization of new bone, so bone remodeling does not occur properly. This causes osteoporosis and weakening of the bones.

Figure 9-36 A, Bow legs (genu varum) in a patient with rickets. B, Osteomalacia in an adult, showing pathologic fractures. (A, from Lissauer T, Clayden G. Illustrated Textbook of Paediatrics. 3rd ed. Edinburgh: Elsevier; 2007. B, from Dandy DJ, Edwards DJ. Essential Orthopaedics and Trauma. 5th ed. Philadelphia: Elsevier; 2009.)
*ACTH does stimulate cholesterol desmolase, which is the starting point for aldosterone production. However, the main trigger for aldosterone release is angiotensin II.