Embryology
DESCRIPTION OF ANATOMY
This chapter will make use of various anatomic terms, shown below. Familiarize yourself with them before beginning.
 Cranial: Toward the head (cranium), similar to the term superior (Fig. 4-1A).
Cranial: Toward the head (cranium), similar to the term superior (Fig. 4-1A).
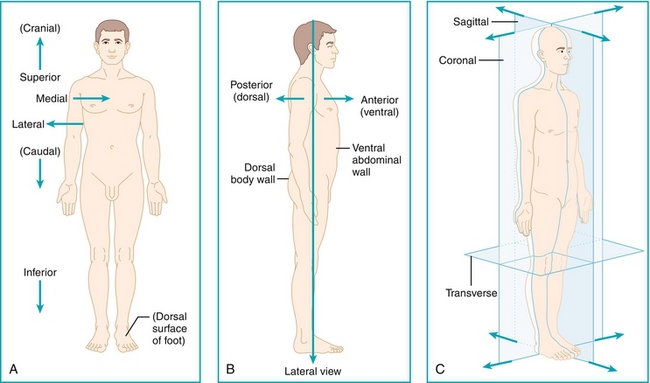
Figure 4-1 Anatomic position and terminology. (From Bogart BI, Ort V. Elsevier’s Integrated Anatomy and Embryology. Philadelphia: Elsevier; 2007.)
Caudal: Toward the tail, similar to the term inferior (Fig. 4-1A).
Medial: Toward the midline (Fig. 4-1A).
Lateral: Away from the midline (Fig. 4-1A).
Dorsal: Toward the back of the body (Fig. 4-1B).
Ventral: Toward the front of the body (Fig. 4-1B).
Sagittal, coronal, transverse: Bodily planes that transect the body, each perpendicular to the others (Fig. 4-1C).
EARLY EMBRYOLOGY: THE FIRST MONTH
The First Week: From Fertilization to Implantation
Day 0: Fertilization occurs when a sperm enters the egg, usually in the ampulla of the fallopian tube. The acrosome reaction occurs when a sperm meets the egg and releases enzymes that penetrate the zona pellucida (the outer “shell” of the egg).
Day 1:The single-cell zygote undergoes rapid mitotic cell divisions. Recall that the egg initially was arrested in metaphase II after ovulation, but completes this division only after fertilization (see Chapter 16). There is no time for cell growth during this rapid dividing phase, so it is termed cleavage because the cells are cleaved into more numerous (but smaller) cells. Each cleavage doubles the cell number (as each individual cell participates).
Day 3: After there are 16 or 32 cells, it is now termed a morula (Latin for “mulberry” because there are now many cells resembling a berry).
Days 4 to 5: Na+/K+-ATPase pumps deliver sodium into the interior of the morula, creating an osmotic gradient and subsequently forming a fluid-filled cavity inside the morula. The embryoblast cells (inside, in a cluster) and the trophoblast cells (outside) are now distinct. Altogether, this is called the blastocyst. As can be seen in Figure 4-2, the embryonic pole is the portion of the blastocyst in which the embryoblast cells are located. The embryoblast will go on to make the embryo (as expected), subsequently separating into the epiblast (dorsal) and hypoblast (ventral). The trophoblast will eventually make the placenta, including the cytotrophoblast and syncytiotrophoblast.
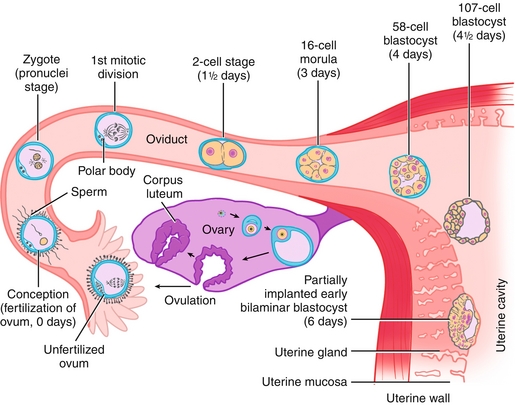
Figure 4-2 Schematic showing the fertilization of an egg by a sperm (usually in the ampulla of the fallopian tube), creating a zygote. Over the next week, this zygote will divide while moving down the fallopian tube toward the uterus, finally implanting into the endometrium. Ovulation occurs 14 days before the onset of menses. Once ovulation occurs, the egg has a viable life of approximately 1 day. Therefore, for pregnancy to occur, a viable sperm must fertilize the egg during this short window of time to form a zygote and begin embryogenesis. (From Schoenwolf GC, Bleyl SB, Brauer PR, Francis-West PH. Larsen’s Human Embryology. 4th ed. Philadelphia: Elsevier; 2008.)
Day 6: The blastocyst implants into the endometrium. If this implantation occurs in an abnormal location (e.g., the fallopian tube), it is termed an ectopic pregnancy, which is a medical emergency.
Twinning
Understanding twinning can be confusing because twinning is described in terms of the number of zygotes (monozygotic or dizygotic), the number of chorions (monochorionic or dichorionic), and the number of amnions (monoamniotic or diamniotic) (Fig. 4-3). The zygote part is easy: recall that a zygote is made when an egg and sperm combine. Therefore, when twins arise from one egg and one sperm, they are considered monozygotic. If two eggs were each fertilized by an individual sperm, the twins would be considered dizygotic. The chorion refers to a membrane that is formed by structures including both trophoblastic layers (cyto- and syncytiotrophoblast) and will eventually form the placenta; therefore, monochorionic twins will share a placenta. The amnion refers to the amniotic sac; those that are monoamniotic will not be separated from one another (share the same “pool” of amniotic fluid) and could potentially be conjoined.
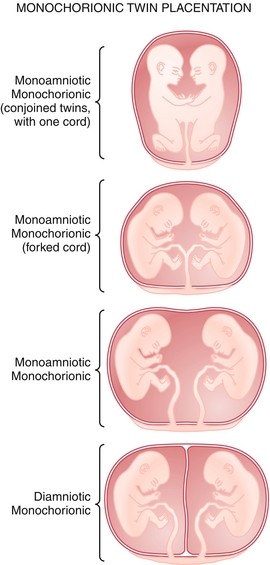
Figure 4-3 Diagram showing different shared structures in twinning. (Redrawn from Benirschke K, Driscoll SG. Pathology of the Human Placenta. New York: Springer-Verlag; 1974.)
Because dizygotic twins are formed from two different eggs and two different sperm, they are not genetically identical, and are called fraternal twins. All dizygotic twins therefore have their own placenta (dichorionic) and amniotic sac (diamniotic), and they each develop in the normal fashion independent of one another—they are simply two pregnancies simultaneously. The case with a monozygotic twin can be more complicated.
Monozygotic twins occur when a single zygote splits and forms two embryos. They are termed identical twins because each forms from the same zygote and therefore they have the same genetic makeup. However, depending on how early the split occurs, they may or may not share a chorion or amnion. The later the embryo splits, the more structures will be shared—imagine that the zygote had immediately split, that it had not started to form any structures yet and therefore each could do so on its own (dichorionic, diamniotic). If the split occurred later, structures would have already begun to form and therefore must be shared. The important landmarks to remember are that the chorion will form at day 3 and the amnion will form at day 8. Splits that occur after these landmarks will share those structures. Remember that all of the following examples are only applicable to monozygotic twins.
Dichorionic, diamniotic: The split must have occurred before day 3 to have two separate chorions. Because the amnion forms after the chorion, if the twin is dichorionic, it must also be diamniotic. Therefore, these monozygotic twins will not share a placenta (dichorionic), nor will they share the same amniotic fluid sac (diamniotic).
Monochorionic, diamniotic: The split must have occurred between days 3 and 8 because the chorion has formed (and is therefore shared), but the amnion has not. These fetuses therefore share a single placenta (monochorionic), but each has its own amniotic fluid sac (diamniotic), and they are spatially separated from one another.
Monochorionic, monoamniotic: The split must have occurred after day 8 because both structures are shared. Therefore, these fetuses will share a placenta (monochorionic) and amniotic fluid sac (monoamniotic) and will potentially be conjoined if the split was late enough. There is also a phenomenon called twin-twin transfusion syndrome that only occurs in monochorionic, monoamniotic twins in which, because of placental anastomoses, one twin gets proportionally more blood than the other. This leads to a large twin (that got most of the blood) and a small twin (that got less of the blood). It is usually a fatal condition for both twins.
The Second Week: The Rule of Twos
The second week has the rule of twos (Fig. 4-4).
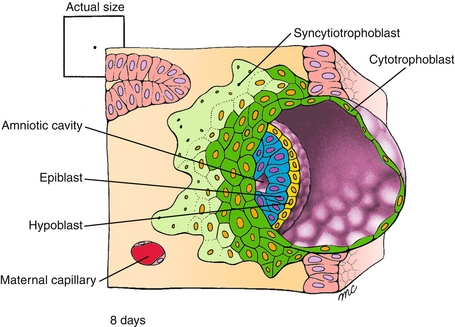
Figure 4-4 The status of the embryo by the beginning of the second week. Note that the trophoblast (which makes placental tissue) has differentiated into two layers: the cytotrophoblast and syncytiotrophoblast. The embryoblast (which makes embryonic tissue) has differentiated into two layers: the hypoblast and epiblast. There are also two cavities that can be seen: the amniotic cavity (formed from the epiblast cells) and the yolk sac (formed from the hypoblast cells). The two sets of two layers and the two cavities are the basis for the rule of twos in week 2. (From Schoenwolf GC, Bleyl SB, Brauer PR, Francis-West PH. Larsen’s Human Embryology. 4th ed. Philadelphia: Elsevier; 2008.)
The trophoblast has now differentiated into two layers: the cytotrophoblast (cellular) and the syncytiotrophoblast (a syncytium, in which the cells have fused together). The cytotrophoblast divides through mitotic division and will generate the chorionic villi; this has embryogenic importance because it allows maximal surface area of contact with maternal blood in the placenta. It also has clinical importance because chorionic villus sampling is a method to diagnose chromosomal or genetic disorders in the fetus. The syncytiotrophoblast does not divide through mitotic division; this generates human chorionic gonadotropin (hCG) which is used clinically. The beta subunit, β-hCG, is the substance that pregnancy tests use. It is also important in the diagnosis of ectopic pregnancy (see Chapter 16).
The embryoblast has now differentiated into two layers: the epiblast (dorsal structures) and hypoblast (ventral structures); together, the epiblast and hypoblast are known as the bilaminar disk.
There are also two cavities now: the amniotic cavity (formed from the epiblast cells) and the yolk sac (formed from the hypoblast cells).
Lastly, at about day 10, the syncytiotrophoblast will begin to secrete hCG. As mentioned previously, β-hCG is used clinically to detect pregnancy and assess for an ectopic pregnancy as well. Because ovulation occurs 14 days before menses, by the time a patient is supposed to have menses, the pregnancy test should be accurate. Dipstick pregnancy tests are qualitative, meaning positive or negative (no numerical values). Blood tests can be quantitative, meaning they give an actual value. In early pregnancy, the β-hCG should double every 48 hours; this is used as a benchmark to determine whether an ectopic pregnancy is potentially present. This is because the syncytiotrophoblast, if not implanted into the endometrium (and implanted elsewhere, such as in a fallopian tube), would not have enough blood supply to increase the β-hCG twofold in 48 hours. This suggests an ectopic pregnancy or other nonviable pregnancy. Please refer to Figure 4-5 for a summary of embryogenesis thus far.
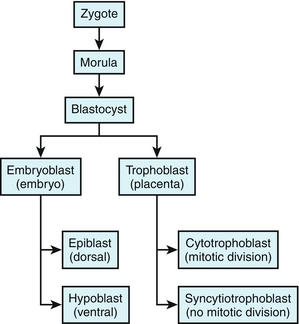
Figure 4-5 Summary of embryogenesis thus far by the end of week 2. After fertilization, a zygote is formed, which subsequently undergoes multiple cleavages to become a morula, then later, a blastocyst. The blastocyst differentiates into the embryoblast and trophoblast. These further differentiate by week 2.
The Third Week: Gastrulation and the Ectoderm, Endoderm, and Mesoderm
The third week has the rule of threes (three germ layers after gastrulation).
Gastrulation is the important step that produces the three germ layers: the ectoderm, endoderm, and mesoderm. Understanding the ectoderm, endoderm, and mesoderm and the organs they produce is helpful in understanding organogenesis and even development. This concept also becomes important in adult life because malignancies of tissues derived from the mesoderm (muscle, bone) are termed sarcomas (myosarcomas and osteosarcomas, respectively), whereas malignancies of tissues derived from ectoderm or endoderm are termed carcinomas.
The process of gastrulation begins with the formation of a primitive streak, which is essentially just a midline invagination that forms (Fig. 4-6A). Subsequently, the epiblast cells move through the primitive streak: the bottom-most layer becomes endoderm, the middle layer becomes mesoderm, and the top layer of epiblast cells that did not migrate becomes ectoderm (Fig. 4-6B). These three germ layers will eventually give rise to various parts of the developing embryo.
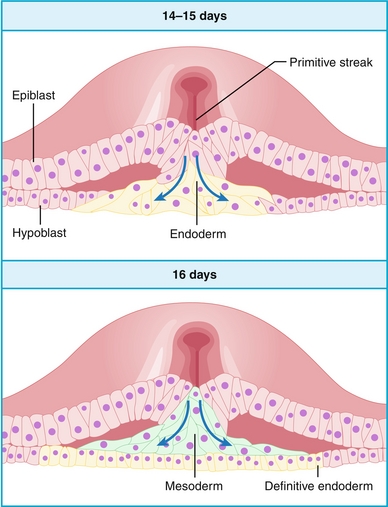
Figure 4-6 The process of gastrulation, beginning with formation of the primitive streak (A), and epiblast cells migrating inward (B). (From Bogart BI, Ort V. Elsevier’s Integrated Anatomy and Embryology. Philadelphia: Elsevier; 2007.)
Ectoderm
Consists of surface ectoderm, neuroectoderm, and neural crest cells.
Surface ectoderm: Makes the surface layer of many organs, including the epidermis of the skin as well as sense organ structures such as the olfactory epithelium and epithelium of the mouth. It also makes glandular structures, such as the adenohypophysis (which itself is an outpouching of the roof of the mouth, known as Rathke pouch), and other glands including the mammary, sweat, and salivary glands.
Neuroectoderm: Makes neural structures: essentially all central nervous system structures (brain and spinal cord), but also the retina because the retina is neural tissue.
Neural crest cells: The neural crest cells create essentially all peripheral nervous system structures. This also includes melanocytes, parafollicular (C) cells of the thyroid, and chromaffin cells of the adrenal medulla, which become important in melanoma, medullary thyroid carcinoma, and pheochromocytoma, respectively. Neural crest cells also play an important role in the formation of conotruncal endocardial cushions, which are essential for the proper development of the heart (genetic disorders that involve problems in neural crest cell migration are often associated with cardiac abnormalities). Lastly, many structures in the face (including teeth and bony structures) are derived from neural crest cells. Therefore, conditions such as DiGeorge syndrome (see later) involve both craniofacial abnormalities and cardiac defects.
Endoderm
Mainly responsible for developing into structures of the gut (such as the liver, pancreas, and other organs of, or relating to, the gastrointestinal system) and some endocrine glands (thyroid follicles, parathyroid glands).
Mesoderm
Further separates into sclerotome, myotome, and dermatome, which develop into bone, muscle, and skin structures, respectively. Although the skin is mostly of mesodermal origin, the epidermis is derived from surface ectoderm.
The rule of threes for week 3 also encompasses the formation of the three body axes: the cranial-caudal axis (the primitive streak is organized top to bottom, head to rump), the medial-lateral axis (because the primitive streak is midline), and the dorsal-ventral axis (front to back).
The end of the third week also marks the beginning of the embryonic period, during which organs begin to form. Before the end of the third week, teratogens typically have an all-or-nothing phenomenon, causing natural abortion of the embryo or no harm at all—there is nothing in between (such as birth defects). However, starting in the next week, with organogenesis, teratogens can cause errors in formation and lead to significant birth defects.
The Fourth Week: Neural Tube Closure and Beginning of Organogenesis
Week 4 also marks the rule of fours as organogenesis begins to take place:
Four chambers of the heart have developed and begin to beat (heart begins to beat at 22 or 23 days, when it has grown to such a size that it cannot get adequate nutrition by diffusion alone).
Although the third week marked the beginning of neural plate development, which will give rise to the spinal cord, the fourth week marks the closure of the neural tube. This closure can be normal or abnormal: abnormal cranial closure leads to anencephaly, whereas abnormal caudal closure leads to spina bifida in one of its three main forms.
Anencephaly occurs when the cranial (head) end of the neural tube does not close and leads to an absence of the forebrain and therefore is not compatible with life. Most patients with anencephaly die in utero or within hours to days of birth. Because there is no forebrain, there is no possibility to generate conscious thought, but because the brainstem is developed normally, primitive reflexes may be present.
Spina bifida occurs when the caudal (bottom) end of the neural tube does not close, usually near the L5–S1 area. This incomplete closure can lead to malformation of the vertebrae overlying the spinal cord and to a passageway for the spinal cord to protrude out of the back. The severity of the defect is related to the size of the opening. This can result in a spectrum of disease, ranging from completely asymptomatic to permanent paralysis.
Spina bifida occulta (Fig. 4-7A): This defect usually does not have symptoms or signs because the incomplete closure is so minor that the spinal cord cannot protrude out of the defect (occulta is Latin for “hidden”). However, some common findings include a small tuft of hair or hyperpigmented skin over the affected area. Because there is a small vertebral fusion defect, this can be seen on lumbar spine radiographs.
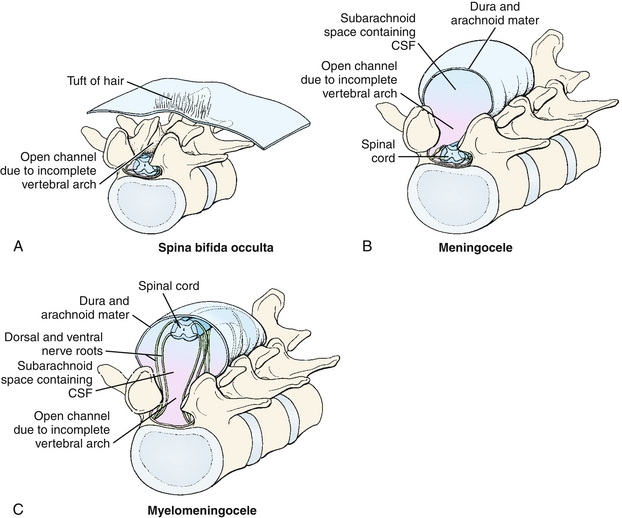
Figure 4-7 Three severity types of spina bifida. A, Spina bifida occulta, in which the defect is not large enough to allow either the meninges or the spinal cord to herniate; a tuft of hair or hyperpigmented skin overlying the defect is often found. B, Spina bifida with meningocele, in which the defect is large enough to accommodate only the meninges and not the spinal cord. C, Spina bifida with myelomeningocele, in which the defect is large enough to accommodate both the meninges and spinal cord, resulting in spinal cord damage and paralysis and loss of sensation distal to the defect. Note that both spina bifida with meningocele and myelomeningocele are termed spina bifida cystica as a category. (From Schoenwolf GC, Bleyl SB, Brauer PR, Francis-West PH. Larsen’s Human Embryology. 4th ed. Philadelphia: Elsevier; 2008.)
Spina bifida with meningocele (Fig. 4-7B): A meningocele occurs when the meninges (the three layers [dura mater, arachnoid mater, and pia mater] that surround the spinal cord) protrude through a defect in the vertebrae, but the spinal cord does not protrude. Therefore, the chance of neurologic dysfunction is still relatively low. A meningeal cyst (sac filled with cerebrospinal fluid [CSF]) may be visible at the site of the defect.
Spina bifida with myelomeningocele (Fig. 4-7C): In this severe form, the defect is large enough that the spinal cord and meninges both protrude through the vertebral defect and are damaged. This leads to neurologic problems below the level of the cord damage, typically resulting in paralysis and loss of sensation of the legs.
Folate deficiency increases the risk for the previously mentioned neural tube closure defects. Because the neural tube closes by the fourth week (before many women even know they are pregnant), folate supplementation in women of reproductive age, especially during the prenatal period, is important in preventing both anencephaly and spina bifida. Anencephaly, myelomeningocele, and meningocele (but not spina bifida occulta) can be suspected in utero if high levels of alpha-fetoprotein (fetal albumin) are detected in maternal serum. This laboratory finding makes sense because alpha-fetoprotein will diffuse into the amnion. Polyhydramnios (excess amniotic fluid) may also be present as CSF leaks into the amnion through the defect.
EMBRYOLOGY: WEEK 5 AND BEYOND
The first month of embryogenesis is important to understand in detail. However, the rest of embryogenesis can be talked about in generalities.
Weeks 4 to 8: Organogenesis and Teratogenicity
Week 4 was described in detail because it marked the closure of the neural tube and the beginning of organogenesis (hence the rule of fours, with four cardiac chambers and four limb buds). Because this is a critical stage when the organs of the body begin to be constructed, teratogen exposure in this period is especially deleterious and can result in significant fetal malformations. Recall that the first 3 weeks marked the all-or-nothing period when an exposure to a teratogen would either cause abortion or nothing.
The most common teratogens are alcohol, smoking, and medications. Chapter 5 covers maternal infections that can lead to birth defects. High-yield teratogens are covered here.
Substances of Abuse
Alcohol: Fetal alcohol syndrome represents the most common birth defect caused by a teratogen. Along with mental retardation, these patients have the classic smooth philtrum (Fig. 4-8A), a thin upper lip, a saddle-shaped nose, and maxillary hypoplasia.
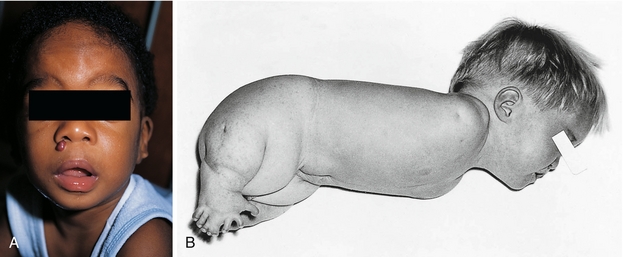
Figure 4-8 A, Fetal alcohol syndrome, displaying the characteristic absent philtrum between the nose and the patient’s characteristically thin upper lip. There is also a saddle-shaped nose and maxillary hypoplasia. B, Thalidomide causing amelia (agenesis of the limb) in the upper extremities and phocomelia (shortening) in the lower extremities. (A, from Lissauer T, Clayden G. Illustrated Textbook of Paediatrics. 4th ed. Edinburgh: Elsevier; 2011. B, from Turnpenny P, Ellard S. Emery’s Elements of Medical Genetics. 14th ed. Philadelphia: Elsevier; 2011. Courtesy Emeritus Professor R. W. Smithells, University of Leeds, UK.)
Cocaine: By preventing reuptake of catecholamines, cocaine is a potent sympathomimetic. This causes increased vasoconstriction (from α1-agonist activity of catecholamines) and therefore decreased blood flow to the placenta, leading to hypoxia in the fetus. This can lead to placental abruption (abruptio placentae), which is an obstetric emergency. It can also lead to generalized problems such as mental retardation and a number of birth defects, presumably also due to hypoxia in the fetus.
Opioids (heroin): Opioids are not teratogenic but are included here to emphasize that fact. However, opioid use in the mother can lead to neonatal opioid withdrawal.
Smoking: Smoking has not been shown to cause specific birth defects, but just as smoking causes damage to the endothelial cells in the rest of the body (predisposing the mother to heart attack and stroke), the placental vasculature is similarly damaged. This leads to a poorer blood supply to the fetus and can cause intrauterine growth restriction and preterm labor.
Common Medications
Angiotensin-converting enzyme (ACE) inhibitors: Recall that ACE changes angiotensin I to angiotensin II, which has effects on the renal system. Use of ACE inhibitors during pregnancy has been linked to renal agenesis or other renal problems. Because amniotic fluid is essentially fetal urine, fetal renal dysfunction leads to oligohydramnios. This warning also applies to angiotensin II receptor–blocking drugs.
Antiepileptic medications: Most antiepileptic medications are teratogenic to some degree. Phenytoin has a specific syndrome called fetal hydantoin syndrome, which consists of facial defects and mental retardation. Many antiepileptic medications have folate antagonist activity (such as valproic acid); therefore, as expected, many are associated with neural tube defects.
Aminoglycoside antibiotics: Aminoglycoside antibiotics have ototoxicity and nephrotoxicity as side effects in adults; these side effects are amplified in the fetus if the mother takes these while pregnant. This can lead to sensorineural (cranial nerve [CN] VIII) deafness and renal damage.
Lithium: Used in treatment of bipolar disorder, this is linked to a specific defect termed Ebstein anomaly, in which there is downward displacement of the tricuspid valve through “atrialization” of part of the right ventricle (right atrium becomes too large, right ventricle becomes too small).
Retinoic acid (vitamin A) and other retinoids: Often prescribed as isotretinoin as a treatment for cystic acne, all retinoids in supraphysiologic amounts can cause severe birth defects. The developing embryo uses Hox genes to control anterior-posterior development and uses a gradient of retinoic acid concentration to trigger development in the proper order. Large amounts of retinoids disrupt this and lead to significant growth defects, spontaneous abortion, and cleft palate.
Tetracycline antibiotics: Most tetracycline antibiotics chelate calcium and therefore can deposit in developing bone. In the fetus and even in children, this can lead to discoloration of teeth from deposition and chelation of calcium.
Warfarin: Warfarin has anticoagulant activity by blocking the vitamin K epoxide reductase enzyme, preventing the synthesis of active vitamin K and therefore blocking clotting factor synthesis. Therefore, lack of fetal clotting factors can lead to fetal hemorrhage and abortion, as well as bone deformities.
Historical Medications
These medications are no longer prescribed, but are commonly tested.
Diethylstilbestrol (DES): Prescribed up until 1971 to supposedly prevent miscarriages (it did not), this drug was subsequently found to cause clear cell carcinoma of the vagina in the female offspring and increased risk for cryptorchidism and sex hormone abnormalities in male offspring. The mothers who took DES are at increased risk for breast cancer as well.
Thalidomide*: This medication was supposed to decrease morning sickness in pregnant women. However, it caused birth defects in the offspring, specifically phocomelia, which is a shortening of the limbs. Commonly, these patients are referred to as “flipper babies” because the shortened limbs have the appearance of flippers (Fig. 4-8B).
Weeks 9 to Birth: The “Fetal Period” of Organ Maturation
During this stage, all organs have at least begun to form. However, there is still much development and maturation that will occur. Because the focus is now on growth, teratogens have less effect compared with during the period of organogenesis. However, teratogens can still have a significant impact on development.
FETAL CIRCULATION AND ERYTHROPOIESIS
Structure of the Placenta and Umbilical Cord
The placenta acts as the interface between the mother and fetus (Fig. 4-9). Through the placenta, oxygen and other nutrients are transferred, but so are potentially harmful things such as drugs, toxins, pathogens, and immunoglobulin G (IgG) antibodies (IgM pentamers are too large to traverse the placenta). The placenta is termed a fetomaternal organ because part of the placenta develops from the fetus (through trophoblastic cells) and part develops from the endometrium of the mother.
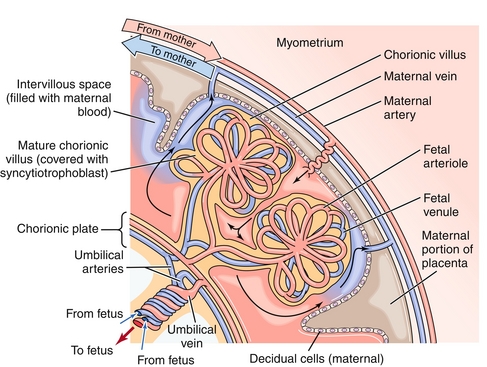
Figure 4-9 The placenta. The maternal blood leaves through spiral arteries into the intervillous space; the maternal blood is no longer in an artery at this time. This blood bathes the fetal vessels and allows the fetal hemoglobin to “steal” oxygen from the maternal hemoglobin. (From Boron WF, Boulpaep EL. Medical Physiology. 2nd ed. Philadelphia: Elsevier; 2008.)
The umbilical cord consists of two umbilical arteries and one umbilical vein. The umbilical arteries will become the medial umbilical ligaments when they close after birth. Do not confuse this with the median umbilical ligament that forms when the urachus closes (discussed later; logically the umbilical arteries, because there are two, could not be median because two things cannot be in the exact middle!).
Fetal Erythropoiesis
Now that the path of red blood cells and their oxygenation has been covered, the actual sites of synthesis and types of hemoglobin will be briefly discussed.
Fetal hemoglobin (HbF, α2γ2) binds oxygen with greater affinity than maternal hemoglobin (that is, the oxygen dissociation curve is shifted to the left). Normally, in adult hemoglobin, 2,3-diphosphoglycerate (2,3-DPG) is synthesized by red blood cells during breakdown of glucose. 2,3-DPG decreases the affinity of oxygen for hemoglobin. However, fetal hemoglobin cannot bind 2,3-DPG, and this results in fetal hemoglobin having a higher affinity for oxygen than adult hemoglobin.
The bone marrow does not synthesize red blood cells in the fetus until the 28th week. Before then, other sites provide that role because the bone marrow has not developed and matured enough to be the primary source of erythropoiesis. The way to remember where the fetus synthesized red blood cells developmentally is that the young liver synthesizes blood (YLSB) in the fetus:
Overview of Fetal Circulation
The fetal circulation shares many similarities to the adult circulation but also has important differences. The main differences are in terms of oxygen concentration and the presence of physiologic shunts.
The oxygen in the fetus was obtained from the oxygen in the mother; fetal hemoglobin (HbF) has a higher affinity for oxygen than adult hemoglobin (HbA), which was discussed previously in the section, “Fetal Erythropoiesis.” Therefore, at the interface of the maternal and fetal blood in the placenta, oxygen exchange occurs from mother to fetus. This newly oxygenated fetal blood returns through the umbilical vein toward the fetal heart. Therefore, oxygen tension is highest in the umbilical vein because it was just oxygenated at the placenta (Fig. 4-10).
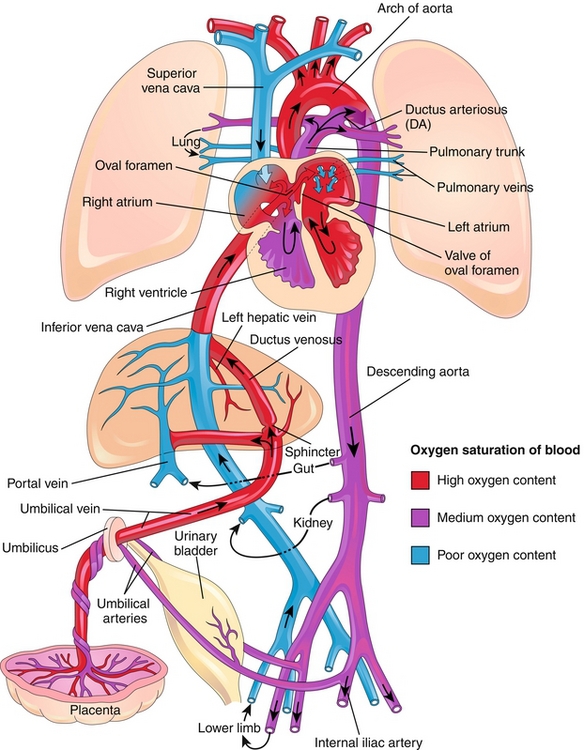
Figure 4-10 The fetal circulation. Note that the umbilical vein has high oxygen content as it is taking blood that was oxygenated at the placenta back to the heart. The two umbilical arteries have low oxygen content. There exist three major shunts to help fetal circulation bypass the liver and lungs. (1) The ductus venosus to bypass the liver; (2) the foramen ovale, permitting passage of blood from the right atrium directly to the left atrium to bypass the lungs; and (3) the ductus arteriosus, permitting the passage of blood from the pulmonary artery to the aorta to bypass the lungs. (From Moore KL, Persaud TVN. Before We Are Born. 8th ed. Philadelphia: Elsevier; 2011.)
The first physiologic shunt is encountered in the liver. The ductus venosus shunts about half of the oxygenated blood away from the liver because the fetal liver does not need 100% of the blood to be adequately oxygenated. This blood moves into the inferior vena cava, subsequently moving into the right atrium. The fetal lungs do not oxygenate blood because they are not breathing air, but rather are breathing amniotic fluid. Therefore, the fetal lungs do not have a large oxygen requirement, and most of the blood will bypass the pulmonary circulation. It does so through the second and third shunts: the foramen ovale and the ductus arteriosus. The foramen ovale is a passageway between the right and left atria and allows blood to bypass the lungs and move directly to the left heart. The ductus arteriosus is a passageway between the pulmonary artery and aorta, allowing blood that did go into the right ventricle and pulmonary artery to again bypass the lungs by moving directly into the aorta.
The ductus arteriosus is distal to the aortic arch, so blood that moves through here will go into the descending aorta and to various end organs, or back to the placenta for reoxygenation. On the other hand, the blood that went into the left ventricle can go into the aortic arch, oxygenating the upper limbs and brain. Interestingly, more deoxygenated blood returning from the brain through the superior vena cava will move into the right ventricle, into the pulmonary artery, ductus arteriosus, and descending aorta to be reoxygenated. The oxygenated blood preferentially moves through the foramen ovale instead, helping to oxygenate the brain. In this way, deoxygenated blood is efficiently moved into either of the two umbilical arteries to be reoxygenated at the placenta.
Fetal Circulation Summary
Fetal blood is oxygenated at the placenta and moves toward the right heart through the umbilical vein.
Fifty percent of this oxygenated blood bypasses the liver through the ductus venosus.
Once at the right heart, there are two possible shunts bypassing the lung: the foramen ovale and the ductus arteriosus.
Oxygenated blood preferentially moves through the foramen ovale to allow the left heart to pump oxygenated blood into the aortic arch and therefore the brain.
Oxygen-poor blood returning from the superior vena cava preferentially moves into the right ventricle, pulmonary artery, and ductus arteriosus pathway to be placed into the descending aorta and to then move into either of two umbilical arteries to reoxygenate the blood at the placenta.
Changes to Fetal Circulation at Birth
At birth, changes occur that drastically change the fetal circulation and transition it to the adult circulation. Recall that in utero the fetus did not need to breathe for oxygenation because oxygen was provided through the placenta. (Note that the fetus in utero does “breathe” to cycle amniotic fluid containing growth factors through the lungs to promote development.) Once born, the baby now takes its first breath, causing multiple changes:
Reactive hypoxic vasoconstriction relieved: Recall that low oxygen tension causes vasoconstriction in the pulmonary vasculature. Once the baby takes its first breath, the increased oxygen tension in the lungs causes significant vasodilation and subsequent dropping in pulmonary vascular resistance and therefore pressure.
Decreased pulmonary pressure: Once born, the drop in pulmonary pressure causes a decrease in right heart pressures; now the left heart pressure is higher than the right, which closes the foramen ovale. Failure of this closure causes a patent foramen ovale, which is a type of atrial septal defect (ASD) and can cause a persistent left-to-right shunt (see Chapter 8 for details).
Beginning of closure of the ductus arteriosus: The ductus arteriosus is no longer needed because there is no need to shunt blood away from the pulmonary vasculature. The ductus arteriosus will close functionally soon after birth and will subsequently close anatomically through fibrosis, becoming the ligamentum arteriosum. The closure occurs through a decrease in prostaglandins, which normally help keep the ductus open. In some congenital cardiac disease states, the presence of a patent ductus arteriosus is important, and prostaglandin E1 (alprostadil) is given to keep the ductus open. If the ductus arteriosus is improperly open, a nonsteroidal anti-inflammatory drug such as indomethacin can be used to prevent prostaglandin production and close the ductus arteriosus.
Closure of the ductus venosus: Because the neonate no longer needs to shunt blood away from the liver, the ductus venosus will close and subsequently become the ligamentum venosum.
CARDIAC EMBRYOLOGY
Formation of the Cardiac Loop
At about 22 to 23 days, the fetal heart will begin to beat. It does this because it has now grown to a size at which diffusion alone does not meet the requirements for oxygen and nutrition delivery. Although the heart is beating, the chambers are not yet in their proper orientation (Fig. 4-11); the “twisting” of the cardiac loop allows for the chambers to be moved into their rightful place. The proper movement of this is dependent on many genes and proper migration of neural crest cells. This is why there are many syndromes that include both cardiac abnormalities and craniofacial abnormalities: proper migration of neural crest cells is required for both the heart and the jaw and face to form correctly. Specifically for the heart, the neural crest cells are important in the formation of “twisting” aorticopulmonary septa that divide the truncus arteriosus (common outflow tract of the RV and LV) into the ascending aorta and pulmonary artery. Failure of proper migration of neural crest cells to the truncus arteriosus region is implicated in transposition of the great vessels and tetralogy of Fallot (see Chapter 8).
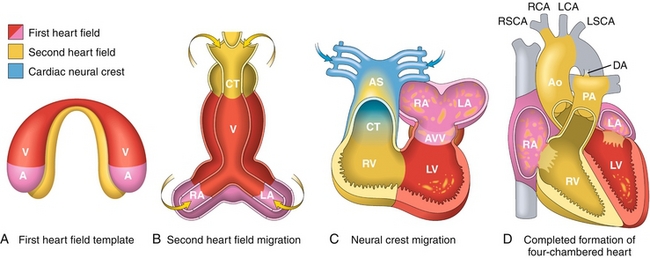
Figure 4-11 Stages of fetal cardiac development; the image shown here ranges from (A) day 15, to (B) day 21, to (C) day 28, and to (D) day 50. A, atria; Ao, aorta; AS, aortic sac; AVV, atrioventricular valves; CT, conotruncus; DA, ductus arteriosus; LA, left atrium; LCA, left carotid artery; LSCA, left subclavian artery; LV, left ventricle; PA, pulmonary artery; RA, right atrium; RCA, right carotid artery; RSCA, right subclavian artery; RV, right ventricle; V, ventricle. (From Kumar V, Abbas AK, Fausto N, Mitchell R. Robbins Basic Pathology. 8th ed. Philadelphia: Elsevier; 2007. Modified by permission from Srivastava D. Making or breaking the heart: From lineage determination to morphogenesis. Cell 2006;126:1037.)
Septation of the Heart: Interatrial Septum Formation
Understanding how the interatrial septum forms makes understanding atrial septal defects much easier. In the fetus, some passageway between the right and left atrium is always present to allow oxygenated blood to bypass the lungs by directly moving from the right to left atrium. Follow along with the explanation for Figure 4-12:
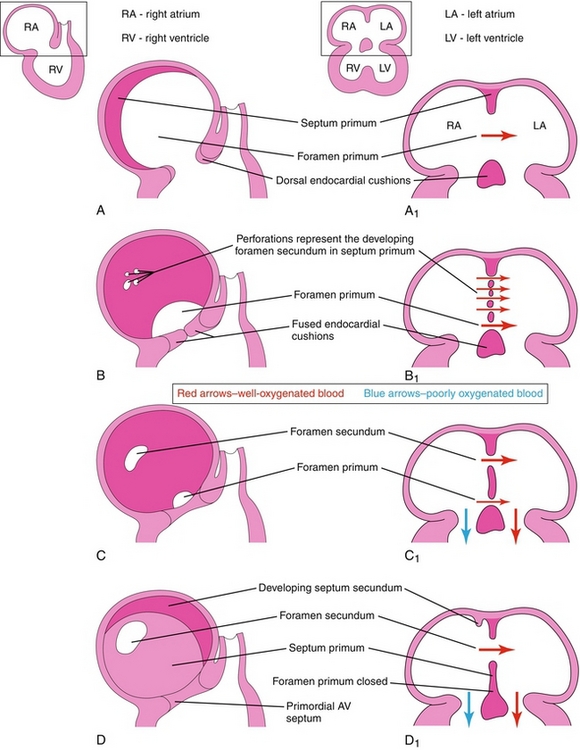
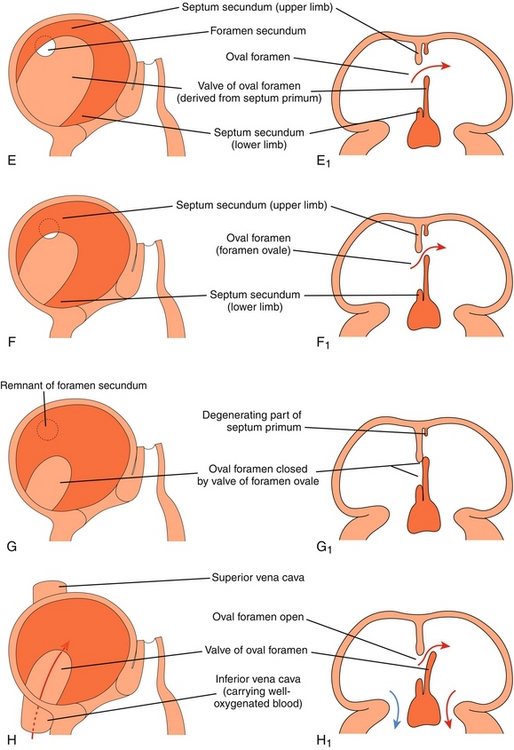
Figure 4-12 Formation of the atrial septum (see text for explanation). (From Moore KL, Persaud TVN. Before We Are Born. 7th ed. Philadelphia: Elsevier; 2007.)
A The foramen primum is the first (primum) hole (foramen) between the two atria. The septum primum is the first septum to form, forming across the foramen primum to close it.
B The foramen primum is now almost closed off by the septum primum, but the septum primum develops perforations in it. These perforations are now a new (second) set of holes.
C The perforations in the second set of holes coalesce, becoming the foramen secundum.
D The foramen primum is now closed. The second septum, the septum secundum, can now be seen developing.
E The septum secundum leaves a space between the atria, termed the oval foramen (foramen ovale).
F The lower limb of the septum primum forms the valve of the oval foramen (which will later close the oval foramen when the baby is born because the left atrial pressure increase will shut the one-way valve).
G Demonstration of the one-way valve: if left atrial pressure (as in a neonate) is higher than right atrial pressure, the valve should shut the oval foramen.
H Failure of the oval foramen to close leads to a patent foramen ovale, a type of atrial septal defect.
Septation of the Heart: Interventricular Septum Formation
The fetal ventricle starts as a single chamber. The muscular interventricular septum begins by forming at the apex of the heart and grows upward. Because it has not yet fully formed, there is an interventricular foramen at this time. By the end of week 7, the foramen closes when the membranous part of the interventricular septum forms. This forms by the joining of nearby tissues, including the endocardial cushion and the bulbar ridges.
VASCULAR EMBRYOLOGY
Blood vessels develop through two separate processes: vasculogenesis and angiogenesis. Vasculogenesis involves angioblasts grouping to form the major vessels (e.g., dorsal aorta). Angiogenesis involves new vessels growing from existing ones and is the major source of vascular development.
The aortic arches (Table 4-1) are six paired embryologic arteries that supply their corresponding branchial arch and eventually form major adult vascular structures (Fig. 4-13). They emanate from the distal part of the truncus arteriosus.
Table 4-1
| Fetal Arterial Structure | Adult Structure | Comment |
| 1st aortic arch | Maxillary artery | |
| 2nd aortic arch | Hyoid artery Stapedial artery |
|
| 3rd aortic arch | Common carotid artery Internal carotid artery |
“Carotid arch” |
| 4th right aortic arch | Brachiocephalic artery | |
| 4th left aortic arch | Arch of the aorta | |
| 5th aortic arch | N/A | 5th arch regresses early |
| 6th right aortic arch | Right pulmonary artery | |
| 6th left aortic arch | Left pulmonary artery Ligamentum arteriosum |
|
| Truncus arteriosus | Ascending aorta Pulmonary trunk |
Aorticopulmonary septum develops to divide the truncus |
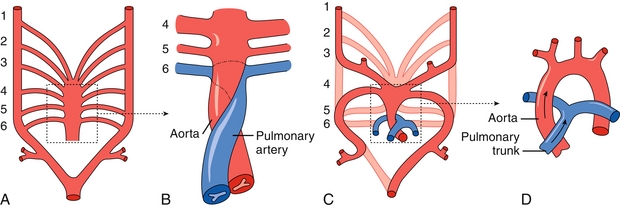
Figure 4-13 A, Six paired aortic arches emanating from the truncus arteriosus. B, The aorticopulmonary septum divides the truncus arteriosus into the ascending aorta and pulmonary trunk. The sixth aortic arch contributes to the right and left pulmonary arteries. C, The third aortic arch forms the common carotid and internal carotid arteries. The left fourth arch forms the arch of the aorta. The right fourth arch forms the brachiocephalic artery. The fifth arch regresses early in development. D, Mature aortic arch. (From Moore NA, Roy WA. Rapid Review Gross and Developmental Anatomy. 3rd ed. Philadelphia: Elsevier; 2010.)
NEUROEMBRYOLOGY
The cephalic portion of the neural tube eventually dilates into three structures: the forebrain (prosencephalon), midbrain (mesencephalon), and hindbrain (rhombencephalon). The forebrain is further divided into the telencephalon (future cerebral hemispheres) and the diencephalon (future thalamus/hypothalamus). Logically, the fetal midbrain (mesencephalon) will develop into the adult midbrain. The hindbrain is also divided into two parts: the metencephalon (future pons and cerebellum) and the myelencephalon (future medulla) (Fig. 4-14).
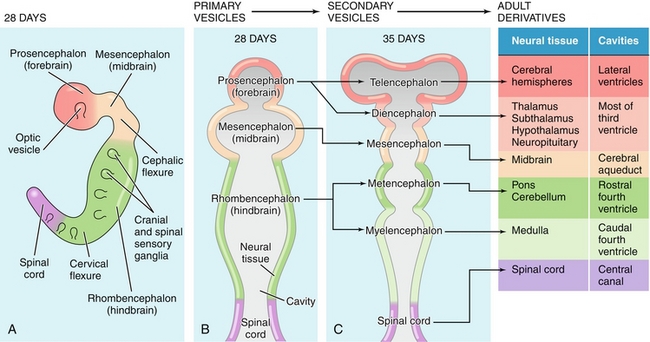
Figure 4-14 Development of the cephalic neural tube into the adult brain. (From Boron WF, Boulpaep EL. Medical Physiology. 2nd ed. Philadelphia: Elsevier; 2008.)
Somites
Primitive masses of mesoderm that flank the neural tube. This mesoderm is termed paraxial mesoderm because of its position lateral to the neural tube. Somites eventually form the vertebrae as well as some of the cartilage and musculature of the back.
Pituitary Gland
The posterior pituitary is a neural structure formed from the downward growth of the diencephalon. The anterior pituitary, on the other hand, develops from an outpouching of the oral cavity called Rathke pouch. Persistence of Rathke pouch may lead to craniopharyngiomas. These benign suprasellar tumors may compress the pituitary, causing endocrine abnormalities or compress the optic chiasm causing visual disturbance (e.g., bitemporal hemianopsia).
Notochord
Longitudinal structure lying ventral to the neural tube that helps differentiate the ventral from dorsal axis of the spinal cord and body. It polarizes the spinal cord with the help of the protein sonic hedgehog homologue, which induces the formation of motor neurons along the ventral aspect of the spinal cord. The notochord persists in the adult as the nucleus pulposus of the intervertebral discs.
Holoprosencephaly
Failure of development of midline structures due to incomplete cleavage of the prosencephalon into the telencephalon. In its most severe form, it is incompatible with life and results in cyclopia, absent nose, and fused cerebral hemispheres. In milder forms, midline structures may be affected, but two cerebral hemispheres develop. For example, a mildly affected individual may present with a single incisor. Holoprosencephaly may be associated with sonic hedgehog gene mutations.
Dandy-Walker Syndrome
A spectrum of genetic conditions presenting as loss of the cerebellar vermis and eventually dilation of the fourth ventricle. Symptoms may be surprisingly absent at birth, although absence of the cerebellar vermis often causes ataxia. As dilation of the fourth ventricle progresses, a protuberance of the occiput can be seen on physical exam. Increased intracranial pressure may also result. The use of a ventricular shunt can drain the excess CSF, treat the hydrocephalus, and normalize the intracranial pressure.
Arnold-Chiari Malformation
Congenital herniation of the cerebellar tonsils through the foramen magnum (Fig. 4-15). This herniation may occlude the passage of CSF leading to hydrocephalus. It is almost always present in cases of spina bifida. Furthermore, this malformation is highly associated with syringomyelia because of altered CSF flow dynamics.
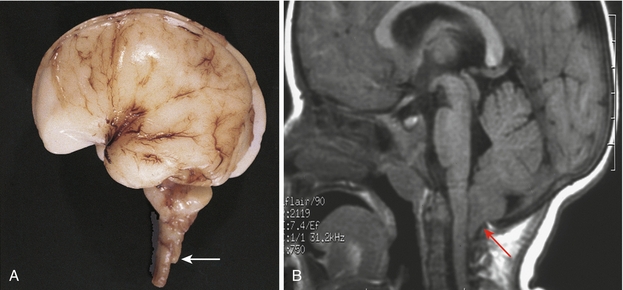
Figure 4-15 The Arnold-Chiari malformation. The cerebellar tonsils (arrow) are inferior to the foramen magnum. (From Moore KL, Persaud TVN. Before We Are Born. 7th ed. Philadelphia: Elsevier; 2007. A, Courtesy Dr. Marc R. Del Bigio, Department of Pathology [Neuropathology], University of Manitoba, Winnipeg, Manitoba, Canada. B, Courtesy Dr. R. Shane Tubbs and Dr. W. Jerry Oakes, Children’s Hospital Birmingham, Birmingham, AL.)
GASTROINTESTINAL EMBRYOLOGY
Table 4-2 reviews the embryologic origins of the gastrointestinal structures as discussed in Chapter 10.
Allantois
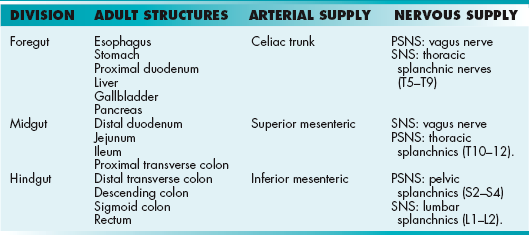
An outpouching of the hindgut that is nonfunctional in the human embryo and is obliterated early. It connects to the apex of the developing bladder at the proximal end and travels through the umbilical cord (Fig. 4-16). The proximal portion of the allantois is termed the urachus, which spans from the umbilicus to the bladder. Once obliterated, the urachus is termed the median umbilical ligament. It continues to have no physiologic use but may be used as a surgical landmark to indicate the midline of the abdomen.
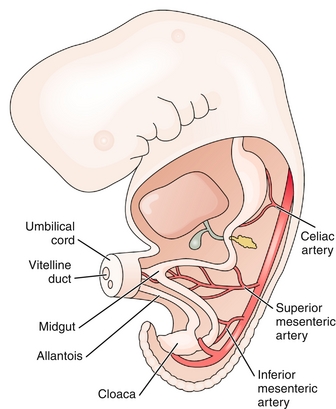
Figure 4-16 An embryo at week 5. The urachus is the proximal portion of the allantois that connects to the developing bladder (cloaca). It will obliterate during the fetal period to form the median umbilical ligament. The vitelline duct connects the yolk sac to the midgut and provides nourishment to the embryo. (From Becker J, Stucchi AF. Essentials of Surgery. Philadelphia: Elsevier; 2006.)
Vitelline Duct (Omphalomesenteric Duct)
Tube that connects the embryologic yolk sac to the midgut and provides nourishment to the embryo. It is obliterated in the seventh week but may partially persist in the form of Meckel diverticulum (see Chapter 10).
Cloaca
The terminal portion of the hindgut, and the early embryologic cavity into which the gastrointestinal and genitourinary systems empty. The cloaca will later divide into the rectum and the urogenital sinus.
Congenital Diaphragmatic Hernia
Incomplete formation of pleuroperitoneal membrane of the diaphragm (usually in the left posterolateral portion) allows abdominal contents to herniate into the thorax. This herniation causes pressure on the lung buds and leads to pulmonary hypoplasia and pulmonary hypertension. Neonates will experience respiratory distress. This condition is associated with about a 50% mortality rate.
Omphalocele
Failure of the gastrointestinal viscera to enter the abdominal cavity after physiologic herniation during the early fetal period. The result is a midline, peritoneal-covered sac protruding through the umbilicus and containing abdominal organs (Fig. 4-17A). Although the condition is treatable, it is often associated with more severe problems, including heart defects, neural tube defects, and chromosomal abnormalities. Omphalocele can be distinguished by the common (and benign) umbilical hernia, which is covered by skin (not just peritoneum).
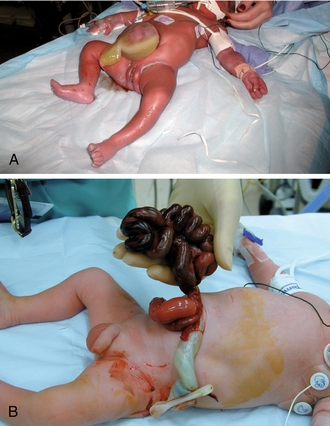
Figure 4-17 A, Omphalocele. Note the midline protrusion of abdominal viscera surrounded by peritoneum. B, Gastroschisis. Note the lateral protrusion of viscera without surrounding peritoneum. (From Schoenwolf GC, Bleyl SB, Brauer PR, Francis-West PH. Larsen’s Human Embryology. 4th ed. Philadelphia: Elsevier; 2008.)
Gastroschisis
Incomplete fusion of the body wall leading to protrusion of gastrointestinal viscera. The protrusion is lateral to the umbilicus and is not covered by peritoneum (Fig. 4-17B). This condition is not associated with chromosomal abnormalities or other serious malformations.
RENAL EMBRYOLOGY
Renal embryology has three distinct phases that chronologically occur in a cranial to caudal sequence (Fig. 4-18).
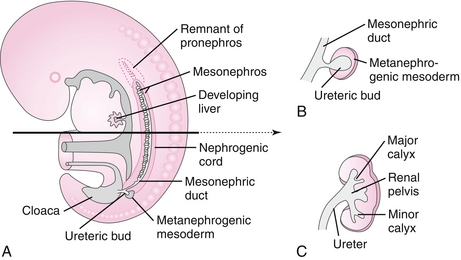
Figure 4-18 The developing kidney. A, Renal development has three stages that progress in a cranial to caudal fashion. The pronephros regresses early. B, The ureteric bud of the mesonephros will form the collecting ducts, renal pelvis, and ureters of the adult kidney. C, The metanephros will form the majority of the adult kidney: Bowman capsules, proximal tubules, loops of Henle, and distal tubules. (From Moore NA, Roy WA. Rapid Review Gross and Developmental Anatomy. 3rd ed. Philadelphia: Elsevier; 2010.)
Mesonephros
This system begins to develop nephron-like structures that also eventually regress. A structure persists, however, called the ureteric bud, which develops from the mesonephric ducts. This bud penetrates the metanephros to form the renal pelvis, collecting duct system, and ureters. Additionally, in males, the mesonephric ducts (wolffian ducts) persist and will later form the male reproductive tract.
Metanephros
This system will form the nephrons and parenchyma of the definitive kidney. Metanephric tissue, under the influence of the collecting duct system, begins to form into recognizable nephrons including Bowman capsules, proximal tubules, loops of Henle, and distal tubules. By the 12th week of gestation, distal tubules (metanephros) have connected with collecting ducts (ureteric bud), and glomeruli have formed. At this point, urine production can begin. Importantly, the placenta, not the fetal kidney, is responsible for clearing the body of waste. Fetal urine is excreted into the amniotic sac, where it is swallowed and recycled. This explains why renal agenesis leads to oligohydramnios because fetal urine is a major component of amniotic fluid.
Potter Sequence (Oligohydramnios Sequence)
A decrease in the volume of amniotic fluid (oligohydramnios) causes facial deformations (due to mechanical stress) and pulmonary hypoplasia (due to decreased nutrients and alveolar hydrostatic pressure). Oligohydramnios has many causes but is usually due to renal, ureteral, or urethral disease (classically bilateral renal agenesis). Knowing Potter sequence is high yield because it shows the importance of the fetal urologic system in producing amniotic fluid and the role of amniotic fluid as a mechanical cushion and promoter of growth.
REPRODUCTIVE EMBRYOLOGY
The reproductive system begins developing as paired gonadal ridges where primordial germ cells migrate and begin to form the primitive sex cords known as the indifferent gonad. The genetic determination of sexual differentiation begins at fertilization when the ovum (X chromosome) is joined with a sperm containing either another X chromosome (XX) or a Y chromosome (XY). The phenotypic determination of sexual differentiation, however, begins with the gonads. The gonads will determine maturation of the duct system (wolffian or müllerian), external genitalia, and eventually secondary sexual characteristics.
The female phenotype is considered the default, and phenotypic “maleness” requires the presence of the testis-determining factor, the SRY gene (sex-determining region on Y).
Male Embryology
The SRY gene encodes a transcription factor that causes the indifferent gonad to turn into the testes. The testes begin to produce testosterone, which influences the mesonephric duct (wolffian duct) to form the epididymis, vas deferens, and seminal vesicles. Testosterone is further converted into dihydrotestosterone (DHT) by the enzyme 5α-reductase. This powerful androgen virilizes the genital tubercle and the rest of the male genitalia (Table 4-3). Meanwhile, antimüllerian hormone is produced, which causes the regression of the müllerian ducts.

Female Embryology
The absence of an SRY gene allows the indifferent gonads to turn into the ovaries by default. The ovaries begin to produce estrogen, which influences the müllerian ducts to form the fallopian tubes, uterus, cervix, and upper vagina. Estrogen also causes the genital tubercle to form the lower vagina and the labioscrotal swelling to form the vulva.
Urogenital Sinus
By the end of the embryonic period (week 8), the ventral part of the cloaca has been partitioned off into the urogenital sinus, which will eventually form the bladder and proximal urethra. In men, it will also form the prostate and bulbourethral glands, whereas in women, it will form the Bartholin glands and glands of Skene.
Hypospadias
Incomplete fusion of the urethral folds leading to a urethral meatus on the inferior portion of the penis. Surgery can be curative.
Epispadias
Rare malformation in which defective migration of the genital tubercle results in a urethral meatus on the dorsum of the penis. It is highly associated with exstrophy of the bladder, in which epispadias is also a component. Only rarely can epispadias occur in isolation.
Exstrophy of the Bladder
Incomplete migration of the primitive streak mesoderm (abdominal wall) around the cloacal membrane leading to bladder mucosa extending outside of the body. It is always associated with epispadias.
Micropenis
Insufficient androgen stimulation from any part of the hypothalamic-pituitary-gonadal axis results in incomplete growth of the penis.
Cryptorchidism
Three percent of males will be born with an undescended testicle. Most of these will descend within the first months of life, and no treatment is indicated. Rarely, physiologic descent does not occur, and surgery is indicated to prevent complications such as reduced fertility or testicular cancer. Of note, any cause of an intraabdominal testicle (e.g., androgen insensitivity syndrome) puts a patient at increased risk for testicular cancer.
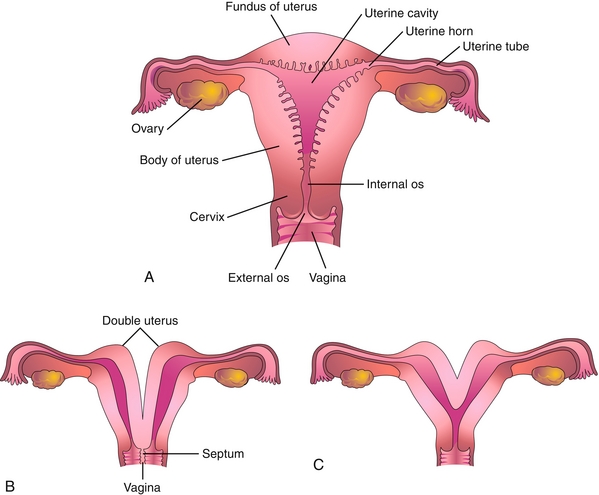
Uterine Anomalies
If the paired müllerian ducts fail to fuse, the result is a double uterus, double cervix, and double vagina (uterus didelphys). Partial fusion results in the more common bicornuate uterus, in which two uterine cavities share a single cervix and vagina (Fig. 4-19).
HEAD AND NECK EMBRYOLOGY
The most distinctive feature of head and neck embryology is the development of the branchial (pharyngeal) arches, which will develop into the musculoskeletal components of the region (Table 4-4). There are six paired arches composed of a “sandwich” with ectoderm on the outside, endoderm on the inside, and neural crest cells in the middle (Fig. 4-20). Each branchial arch is supplied by its numerically corresponding aortic arch and a cranial nerve (which unfortunately does not correspond numerically). The branchial arches are separated from each other by branchial clefts. These clefts do not contribute to the adult structure, except for the first, which will form the external acoustic meatus (Table 4-5). On the endodermal side, between the pharyngeal arches, lie the pharyngeal pouches (Table 4-6). These pouches form many important structures of the head and neck.
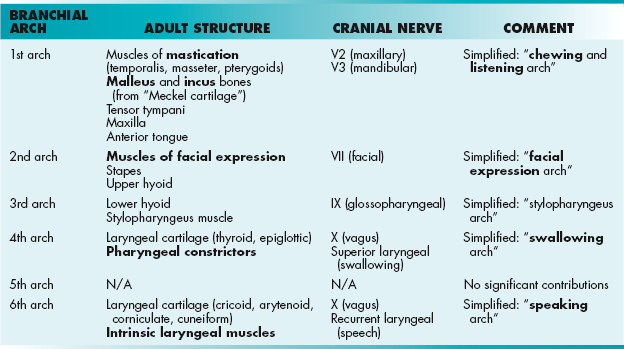
Table 4-5
| Branchial Cleft | Adult Structure | Comment |
| 1st cleft | External auditory meatus | |
| 2nd–4th cleft | Obliterated | Failure to obliterate these clefts leads to a branchial cleft cyst |
Table 4-6
| Branchial Pouch | Adult Structure |
| 1st pouch | Tympanic membrane Middle ear cavity Eustachian tube |
| 2nd pouch | Palatine tonsil Tonsillar fossa |
| 3rd pouch | Inferior parathyroid gland Thymus |
| 4th pouch | Superior parathyroid gland |
| 5th pouch | C cells of thyroid |
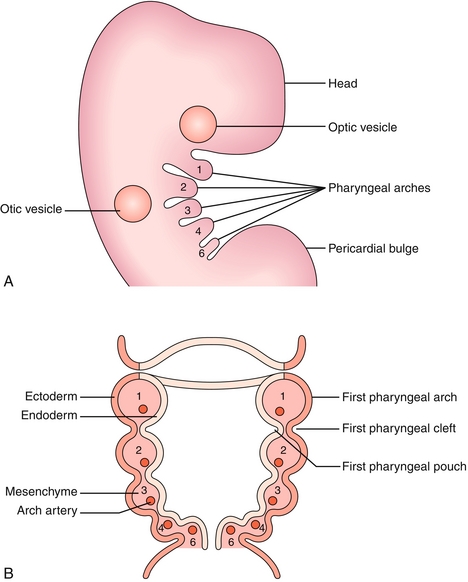
Figure 4-20 Branchial apparatus including arches, clefts, and pouches. A, Lateral view. B, Cross-section. (From Turnpenny P, Ellard S. Emery’s Elements of Medical Genetics. 14th ed. Philadelphia: Elsevier; 2011. Redrawn from Graham A, Smith A. Patterning the pharyngeal arches. Bioessays 2001;23:54-61, with permission of Wiley-Liss Inc., a subsidiary of John Wiley & Sons, Inc.)
Treacher Collins Syndrome
Lack of neural crest cell migration into the first branchial arch causes syndromic facial malformations, including micrognathia and conductive hearing loss. This can be remembered because Meckel cartilage develops from the first branchial arch. It forms the malleus and incus and also guides development of the mandible.
Pierre Robin Syndrome
Lack of neural crest cell migration into the first branchial arch causes syndromic facial malformations, including micrognathia and cleft palate. This can be remembered because the hard palate is made partially of the maxillary bone, a first arch derivative.
DiGeorge Syndrome
Failure of differentiation of the third and fourth pharyngeal pouches leading to absent parathyroid glands (causing hypocalcemia) and thymic aplasia (causing T cell immunodeficiency). This syndrome is accompanied by facial abnormalities (similar to first arch syndromes) and cardiac anomalies (especially tetralogy of Fallot). Mnemonic: CATCH-22—Cardiac anomalies, Abnormal facies, Thymic aplasia, Cleft palate, Hypoparathyroidism/Hypocalcemia due to a deletion on chromosome 22.
Branchial Cleft Cyst
Failure of obliteration of one of the branchial clefts leads to a cystic structure of the lateral neck along the anterior border of the sternocleidomastoid (most commonly the second branchial cleft is implicated).
Tongue
The tongue receives contributions from branchial arches 1 to 4, which explains its complex innervation pattern (Fig. 4-21).
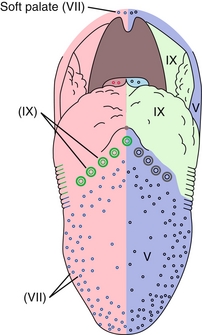
Figure 4-21 Cranial nerve innervation of the tongue for taste (left) and general sensation (right). (From Nolte J. The Human Brain. 6th ed. Philadelphia: Elsevier; 2008.)
General Sensation: The anterior two thirds of the tongue (the body) derives from the first pharyngeal arch; therefore, sensory innervation is through CN V3 (mandibular nerve). The posterior one third of the tongue (the base) derives from the third (and partially the fourth) pharyngeal arch; therefore, sensory innervation is through CN IX (glossopharyngeal).
Taste: Taste to the anterior two thirds of the tongue is through CN VII (chorda tympani branch), whereas taste to the posterior one third, like general sensation, is through CN IX.
Motor: The intrinsic and extrinsic musculature of the tongue (genioglossus, hyoglossus, styloglossus, and palatoglossus) is derived from occipital somites. Motor innervation is through CN XII (hypoglossal nerve) except for the palatoglossus muscle (innervated by CN X).
Thyroid
Thyroid tissue begins to proliferate on the pharyngeal floor and migrates down the midline of the anterior neck (Fig. 4-22). It remains connected to the base of the tongue by the thyroglossal duct, which will eventually be obliterated, leaving only the foramen cecum, a small indentation, at the base of the tongue.
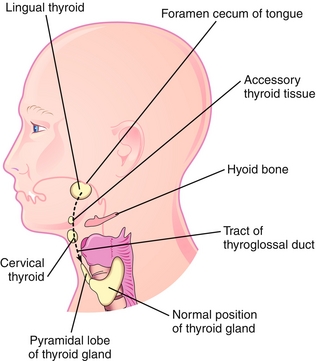
Figure 4-22 The thyroid gland migrates from the foramen cecum (on the tongue) to its normal position inferior to the thyroid cartilage. Note the tract of the thyroglossal duct and the potential locations of ectopic thyroid tissue and thyroglossal duct cysts. (From Moore KL, Persaud TVN. Before We Are Born. 8th ed. Philadelphia: Elsevier; 2012.)
Thyroglossal duct cyst: Failure of obliteration of the thyroglossal duct causes a midline cystic dilation anywhere along the migratory pathway of the thyroid. This cyst will move with swallowing.
Ectopic thyroid: Failure of descent of the thyroid tissue along the neck leads to ectopic tissue. Most commonly, the tissue is found on the base of the tongue, behind the foramen cecum; however, it can be anywhere along the migratory path. It is subject to the same disease states as the normal thyroid, including hyperthyroidism and thyroid cancer.
GROWTH AND DEVELOPMENT
Neonatal Medicine
Apgar Score
A grading system, named for Dr. Virginia Apgar, to assess the general health of a newborn at 1 minute and 5 minutes of life (Table 4-7). For each of the five categories (each of which start with the letters of her name), 0 to 2 points may be given. Ten is a perfect score, but it is rarely given because most newborns will lose 1 point for acrocyanosis.

Low Birth Weight
Infants are typically born at term (38 to 40 weeks) with a birth weight of approximately 2500 to 3800 g (5 lb, 8 oz to 8 lb, 6 oz). Infants who weigh less than 2500 g at birth are defined as low birth weight, and they are at risk for numerous complications. Because of the delicate vasculature in the subependymal germinal matrix of the cerebral ventricles, they are prone to intraventricular hemorrhage, which can lead to neurologic devastation in some cases. They may have abnormal blood vessels in the retina, which can lead to scarring and blindness (called retinopathy of prematurity). With their suboptimally developed immune systems, they are susceptible to infections, which can be severe. Small infants are also prone to respiratory distress syndrome because of insufficient surfactant production. They are at risk for necrotizing enterocolitis, a disease in which the bowel dies and may bleed and perforate. They are also more likely than babies of normal weight to have a persistently patent ductus arteriosus, which can lead to heart failure, as described in Chapter 8.
Developmental Milestones
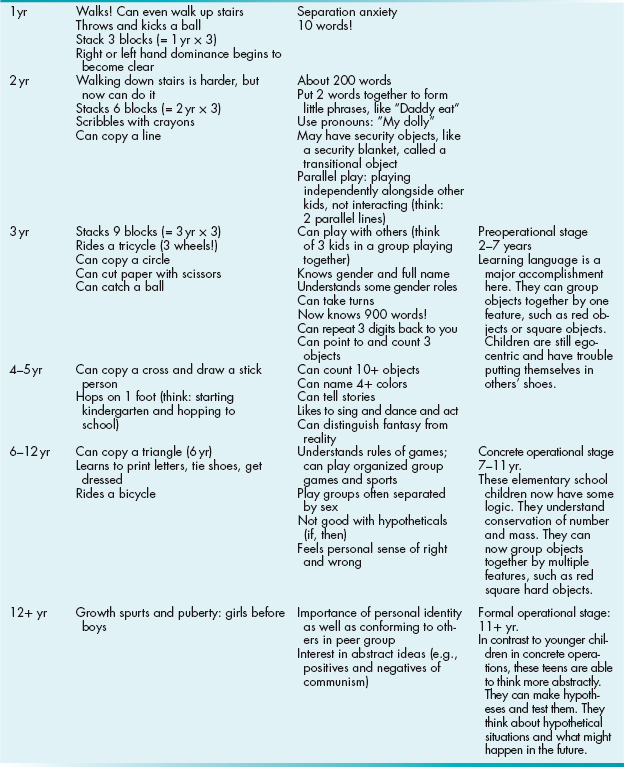
Unfortunately, these can be difficult to commit to memory. The best strategy is usually to think of your own personal experience with normal infants and children and their abilities at certain ages. A list of testable milestones along with a few mnemonics are listed in Table 4-8. The right-most column describes Jean Piaget’s stages of cognitive development. He made these observations of how children’s cognitive function develops by watching his own children.

*Thalidomide does still find use today in the treatment of multiple myeloma.