Gastroenterology
ANATOMY
Abdominal Wall
A surgeon cutting through the lateral abdominal wall would cut through anatomic structures in the following order: skin → superficial fascia (Camper’s and Scarpa’s fascia) → external oblique → internal oblique → transversus abdominis → transversalis fascia → extraperitoneal fascia → parietal peritoneum (Fig. 10-1).

Figure 10-1 The abdominal wall. (From Drake RL, Vogl AW, Mitchell AWM. Gray’s Anatomy for Students. 2nd ed. Philadelphia: Elsevier; 2009.)
Mnemonic: Beauty is only skin deep at fat Camp.
A midline incision would cut through the skin → Camper’s fascia → Scarpa’s fascia → linea alba (aponeuroses of rectus sheaths) → transversalis fascia → extraperitoneal fascia → parietal peritoneum.
Gastrointestinal Tract
The upper gastrointestinal (GI) tract begins at the mouth and extends to the ligament of Treitz (actually not a ligament at all, but the suspensory muscle of the duodenum, which participates in GI motility). The lower GI tract includes everything distal to this point.
Of note, an upper GI bleed is a bleed proximal to the ligament of Treitz. It will usually present with hematemesis or melena (although it may present as hematochezia if very brisk). A lower GI bleed is distal to the ligament of Treitz and will usually present as hematochezia. Overall, two thirds of all GI bleeds are from an upper GI source.
Embryologic Divisions
The GI tract can also be divided embryologically into the foregut, midgut, and hindgut, which correspond best with the adult vascular supply. The foregut consists of the first half of the duodenum as well as the esophagus, stomach, liver, gallbladder, and pancreas. The vascular supply of the foregut comes from the celiac trunk (which gives rise to the left gastric, common hepatic, and splenic arteries; Figs. 10-2 and 10-3). Parasympathetic innervation comes from the vagus nerve, and sympathetic innervation comes from the thoracic splanchnic nerves (T5-T11).
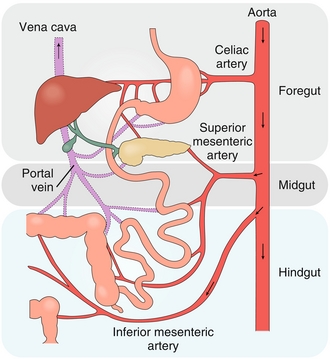
Figure 10-2 Embryologic divisions of the GI tract. (From Brown T. Rapid Review Physiology. 2nd ed. Philadelphia: Elsevier; 2011.)
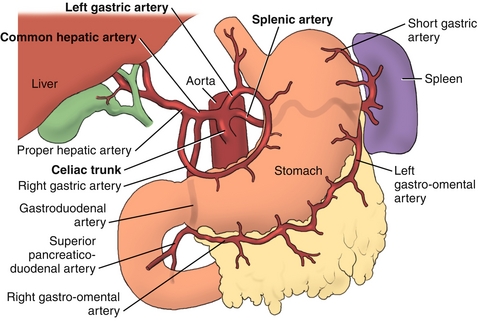
Figure 10-3 Branches of the celiac trunk. (From Morton DA, Albertine KH, Peterson KD. Gray’s Dissection Guide for Human Anatomy. 2nd ed. Philadelphia: Elsevier; 2006.)
The midgut includes the distal duodenum, jejunum, ileum, and proximal two thirds of the transverse colon. The midgut is supplied by the superior mesenteric artery and, like the foregut, is innervated by the vagus nerve and thoracic splanchnics (T11-12).
The hindgut consists of the distal transverse colon, descending colon, sigmoid colon, and rectum. It is supplied by the inferior mesenteric artery and has parasympathetic innervation by the pelvic splanchnics (S2-S4) and sympathetic innervation from the lumbar splanchnics (L1-L2).
The entire GI tract has venous drainage that passes through the liver via the portal circulation. Physiologically, this allows the liver to participate in digestion. Clinically, it is relevant for first-pass metabolism of medications and explains why GI cancers tend to metastasize to the liver. It is so vascular that it is the most common organ to have metastases from other malignancies. Don’t be fooled—a malignancy found in the liver is more likely to be of metastatic than primarily hepatic origin, especially if there are multiple growths.
Retroperitoneum
The peritoneum is a membranous tissue that lines the abdominal viscera (visceral peritoneum) and abdominal wall (parietal peritoneum). Several abdominal structures, however, are situated behind the peritoneum (at least in part). These can be remembered with the mnemonic SAD-PUCKER—Suprarenal glands (adrenals), Aorta–inferior vena cava (IVC), Duodenum (second and third parts), Pancreas, Ureters, Colon (ascending and descending), Kidney, Esophagus, and Rectum.
Portal Triad
The triad of hepatic artery, portal vein, and bile duct. Portal triads form the six points of a hexagon that make up the liver lobule (Fig. 10-4).

Hepatic Vasculature
Nutrient-rich blood from the GI tract enters the liver via the portal vein, whereas oxygen-rich blood enters the liver via the common hepatic artery (see Fig. 10-4). The blood passes through sinusoids lined by single-layered sheets of hepatocytes. The sinusoids are fenestrated, allowing easy access of the products of digestion into hepatocytes. The sinusoids drain into central veins, which empty into the hepatic veins and finally to the IVC. Note that blood drains away from the portal triad toward the central vein, whereas bile flows in the opposite direction toward the portal triad and bile ducts.
Biliary Tree and Bile Formation
Bile is created by the hepatocytic breakdown of cholesterol (the primary method of eliminating cholesterol from the body). Bile is secreted from the apical surface of hepatocytes and travels via bile canaliculi into bile ductules. Bile ductules lead to the left and right hepatic ducts, which combine to form the common hepatic duct. The cystic duct exits the gallbladder and joins the common hepatic duct to form the common bile duct (CBD). The CBD and pancreatic duct empty into the second part of the duodenum via the ampulla of Vater, which leads to the major duodenal papilla. The sphincter of Oddi controls the flow of bile and pancreatic enzymes into the duodenum (Fig. 10-5).
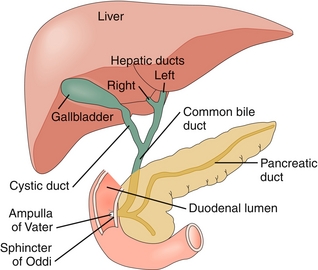
Figure 10-5 Anatomy of the biliary tree. (From Brown T. Rapid Review Physiology. 2nd ed. Philadelphia: Elsevier; 2011.)
When reviewing the biliary pathology section below, consider the clinical importance of each step in the flow of bile. For example, primary biliary cirrhosis (PBC) is caused by the destruction of canaliculi, and primary sclerosing cholangitis (PSC) is caused by the destruction of the intra- and extrahepatic bile ducts. Cholecystitis usually results from obstruction of the gallbladder neck or cystic duct. Cholangitis is usually secondary to obstruction of the common bile duct, and obstruction of the pancreatic duct is one cause of pancreatitis.
 Of clinical importance, the liver uses cholesterol to produce bile acids. Statins are a class of medications that interfere with hepatic synthesis of cholesterol by inhibiting HMG-CoA reductase (3-hydroxy-3-methyl-glutaryl-CoA reductase). The conversion of HMG-CoA to mevalonic acid is the first committed step in the hepatic synthesis of cholesterol (Fig. 10-6). Inhibition of this step causes hepatocytes to absorb cholesterol from the circulation by increasing hepatocyte low-density-lipoprotein (LDL) receptors. Decreased synthesis of cholesterol and increased hepatic uptake of serum cholesterol leads to a reduction of serum LDL.
Of clinical importance, the liver uses cholesterol to produce bile acids. Statins are a class of medications that interfere with hepatic synthesis of cholesterol by inhibiting HMG-CoA reductase (3-hydroxy-3-methyl-glutaryl-CoA reductase). The conversion of HMG-CoA to mevalonic acid is the first committed step in the hepatic synthesis of cholesterol (Fig. 10-6). Inhibition of this step causes hepatocytes to absorb cholesterol from the circulation by increasing hepatocyte low-density-lipoprotein (LDL) receptors. Decreased synthesis of cholesterol and increased hepatic uptake of serum cholesterol leads to a reduction of serum LDL.

Digestive Tract Anatomy
The gut wall is composed of mucosa, submucosa, muscularis propria, and serosa (Fig. 10-7).
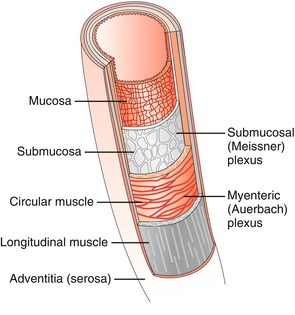
Figure 10-7 Layers of the gut wall. (From Brown T. Rapid Review Physiology. 2nd ed. Philadelphia: Elsevier; 2011.)
Mucosa
The innermost layer of the GI tract. It consists of the epithelial layer, lamina propria, and muscularis mucosa. Invaginations increase the surface area of the epithelial layer, which is involved primarily in absorption and secretion. The lamina propria contains supportive structures, and the muscularis mucosa shapes the underlying epithelium.
Submucosa
Connective tissue that supports the mucosa. Notably, it contains the submucosal plexus (Meissner’s plexus), which innervates the muscularis mucosa and aids in GI motility.
PHYSIOLOGY
Gastric Histology
See Figure 10-8.

Figure 10-8 Histologic and anatomic regions of the stomach. (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
Parietal Cells
Located in the body and fundus of the stomach. Secrete H+ ions via H+/K+-ATPase. They also secrete intrinsic factor, which binds to vitamin B12 and facilitates vitamin B12 absorption in the ileum. The H+/K+-ATPase on parietal cells is the target of proton pump inhibitors (PPIs).
Chief Cells
Secrete pepsinogen (proenzyme for protein digestion active only below pH of 6). Chief cells also secrete gastric lipase, which plays a small role in lipid digestion.
Histology of the Intestine
Villi
Finger-like projections into the intestinal lumen, which increase the surface area for digestion.
Intestinal Crypts
Lying between villi, crypts contain stem cells that replace sloughed enterocytes and secrete digestive enzymes.
Enterocytes
Intestinal columnar epithelial cells. Their apical surface contains a brush border of microvilli, which greatly increases the intestinal surface area and participates in the digestion and absorption of nutrients.
Goblet Cells
These cells lie on intestinal villi and produce a protective and lubricating mucus. The more distal in the small intestine, the more goblet cells are present.
Paneth Cells
Located in intestinal crypts below stem cells, these cells exocytose enzymes that destroy GI microorganisms and participate in the innate immunity of the small intestine.
Duodenal (Brunner) Glands
Submucosal glands of the upper duodenum that secrete bicarbonate and pepsinogen to neutralize gastric acid and facilitate digestion.
Peyer’s Patches
Lymph nodules in the lamina propria–submucosa of the ileum. This histologic feature helps distinguish ileum from jejunum. They participate in the adaptive immune function of the gut. It is logical that these lymphoid follicles are in the ileum because it is closest to the bacteria-rich colon. Clinically, hypertrophy of Peyer’s patches has been associated with intussusception (see later).
Gastrointestinal Hormones
Gastrin
Prepares for the gastric phase of digestion (Table 10-1). Secreted by antral G cells, gastrin increases gastric motility and parietal cell activity (secretion of intrinsic factor and H+ ions). Serum levels will be extremely high in gastrinoma (Zollinger-Ellison syndrome) or if the patient is on a PPI, which raises gastric pH and initiates a feedback loop.
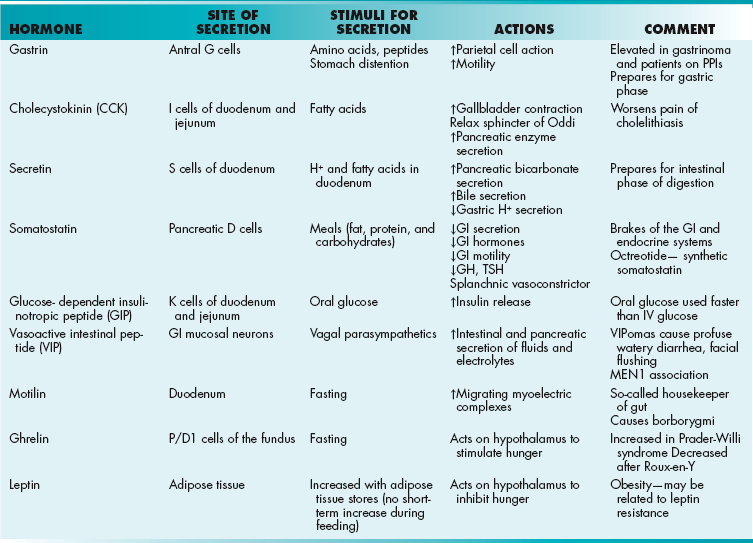
Cholecystokinin (CCK)
Greek for “gallbladder move”. CCK stimulates gallbladder contraction, relaxation of the sphincter of Oddi, and pancreatic enzyme secretion. Release is stimulated by fatty acids in the GI tract. CCK is secreted by I cells of the duodenum and jejunum. Clinically this hormone is responsible for pain after fatty meals if cholelithiasis is present because it causes the gallbladder to contract against the blocked cystic duct.
Secretin
Prepares for the intestinal phase of digestion by stimulating secretions from the pancreas and gallbladder while inhibiting gastric secretions. Like CCK, it is secreted in the duodenum. Unlike CCK, secretin tends to stimulate pancreatic bicarbonate secretion, whereas CCK stimulates pancreatic enzyme secretion.
Somatostatin
Acts as the “brakes” of the GI and endocrine systems. Somatostatin decreases the secretion of gastric acid, GI motility, and gallbladder motility. Hormonally, it inhibits insulin, glucagon, thyroid-stimulating hormone, and growth hormone release. It also is a splanchnic vasoconstrictor. D cells in the duodenum and pancreas control its secretion.
Glucose-Dependent Insulinotropic Peptide (GIP)
As the name suggests, this hormone, GIP, increases pancreatic beta cell release of insulin in response to an oral glucose load. Clinically, this means that the body uses an oral glucose load more rapidly than an intravenous glucose load. K cells of the small intestine secrete GIP.
Vasoactive Intestinal Peptide (VIP)
Vasoactive intestinal peptide (VIP) is released from neurons within the GI mucosa. It increases the intestinal and pancreatic secretion of fluids and electrolytes. It also causes relaxation of smooth muscle. Clinically, a VIPoma (often of the pancreas) causes profuse watery diarrhea and facial flushing secondary to fluid secretion and vasodilation, respectively. VIPomas are associated with multiple endocrine neoplasia type 1(MEN1 syndrome; see Chapter 9).
Motilin
During periods of fasting, the upper duodenum secretes motilin to initiate migrating myoelectric complexes. The resulting muscular contractions push indigestible substances (e.g., fiber) through the digestive tract. Motilin functions as a sort of housekeeper of the gut by making room for subsequent meals. Clinically, this hormone is responsible for the rumbling sounds (borborygmi) created by the GI tract when fasting.
Ghrelin
During periods of fasting, P/D1 cells of the fundus secrete ghrelin. Ghrelin is thought to act predominantly on the hypothalamus, and it contributes to the sensation of hunger. Not surprisingly, it is found at elevated levels prior to meals and is decreased after feeding. Clinically, high levels of ghrelin contribute to the insatiable appetite of patients with Prader-Willi syndrome (partial deletion of chromosome 15). Conversely, bariatric surgeries that remove the fundus or exclude it from digestion (e.g., Roux-en-Y surgery) lead to lower levels of circulating ghrelin and contribute to weight loss.
Mnemonic: When hungry, your stomach growls because of ghrelin.
Saliva
The parotid, submandibular, and sublingual glands secrete saliva. Saliva provides mechanical lubrication to facilitate swallowing of food boluses. Furthermore, it contains α-amylase and lingual lipase, which initiate digestion of carbohydrates and lipids, respectively. Saliva also contains 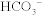 , which helps neutralize refluxed gastric contents and inhibits bacterial growth within the mouth. Clinically, patients with Sjögren syndrome produce less saliva and as a result are prone to dental caries. They may also suffer complications of gastroesophageal reflux disease because of decreased salivary buffering activity. Saliva production is stimulated by both parasympathetic and sympathetic activity, although the parasympathetic system is dominant.
, which helps neutralize refluxed gastric contents and inhibits bacterial growth within the mouth. Clinically, patients with Sjögren syndrome produce less saliva and as a result are prone to dental caries. They may also suffer complications of gastroesophageal reflux disease because of decreased salivary buffering activity. Saliva production is stimulated by both parasympathetic and sympathetic activity, although the parasympathetic system is dominant.
DIGESTION AND ABSORPTION
Digestion is the process whereby ingested food is broken down into constituent parts capable of being absorbed. The mechanical activity of chewing and movement of food boluses through the GI tract contribute to the process of digestion, but the major activity takes place at the molecular level.
Carbohydrates
Consist of polysaccharides, disaccharides (including lactose and sucrose), and monosaccharides (glucose, galactose, and fructose). Only monosaccharides are capable of being absorbed by the small intestine. Carbohydrate digestion begins with α-amylase in the saliva, which is quickly inactivated in the acidic gastric environment. Once in the duodenum, however, pancreatic amylase continues the process and catalyzes the glycosidic bonds in carbohydrates to form disaccharides. Intestinal brush border enzymes (lactase, sucrase, and maltase) finish the conversion of disaccharides into monosaccharides. Glucose and galactose are absorbed in the small intestine via secondary active transport (Na+ cotransport), whereas fructose is absorbed down its concentration gradient using a transport protein (facilitated diffusion).
Of clinical importance, lactose intolerance is a disaccharidase deficiency caused by a relative lack of brush border lactase. Undigested lactose remains in the intestinal lumen, resulting in osmotic diarrhea along with symptoms of cramping and bloating because of fermentation by colonic bacteria. An analogous osmotic diarrhea may occur in patients ingesting large amounts of sorbitol (a sweetener often found in sugar-free gum or candy). This monosaccharide is very slowly absorbed and, in excess, can cause an osmotic diarrhea by drawing water into the lumen.
Dietary Lipids
Consist of cholesterol, triglycerides, and phospholipids. Digestion begins with lingual lipase and gastric lipase hydrolyzing triglycerides into free fatty acids and glycerol. This phase is only responsible, however, for a small fraction of lipid digestion. Once in the duodenum, bile salts emulsify the lipids into micelles, hydrophobic products of lipid digestion surrounded by the amphipathic bile salts. This structure greatly increases their surface area for digestion by pancreatic lipase. When micelles approach the brush border, their lipid contents are absorbed into enterocytes and repackaged as chylomicrons. Chylomicrons then distribute lipids to the rest of the body. Bile salts, on the other hand, remain in the intestinal lumen until they are resorbed in the ileum and recycled via the enterohepatic circulation.
Protein
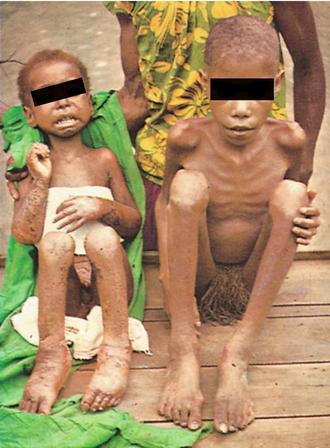
Protein digestion begins in the stomach with chief cell secretion of pepsinogen, which is converted to pepsin in the acidic environment. Pepsin initiates protein digestion by hydrolyzing internal peptide bonds. In the duodenum, however, pancreatic 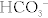 inactivates pepsin, and pancreatic trypsinogen takes over. This enzyme is converted to its active form (trypsin) by the action of brush border enterokinases. Trypsin not only hydrolyzes peptide bonds, but also catalyzes other pancreatic proteases to become active. The end result is protein breakdown into tripeptides and amino acids, which are absorbed via Na+ or H+ cotransport. Clinically, protein malnutrition contributes to poor wound healing. Kwashiorkor refers to severe protein-energy malnutrition despite sufficient caloric intake. The lack of protein causes decreased osmotic pressure in blood vessels, resulting in generalized edema (called anasarca). Kwashiorkor can be distinguished from marasmus (starvation resulting from inadequate intake of all nutrients). Marasmus is characterized by more severe muscle wasting and emaciation without associated edema (Fig. 10-9).
inactivates pepsin, and pancreatic trypsinogen takes over. This enzyme is converted to its active form (trypsin) by the action of brush border enterokinases. Trypsin not only hydrolyzes peptide bonds, but also catalyzes other pancreatic proteases to become active. The end result is protein breakdown into tripeptides and amino acids, which are absorbed via Na+ or H+ cotransport. Clinically, protein malnutrition contributes to poor wound healing. Kwashiorkor refers to severe protein-energy malnutrition despite sufficient caloric intake. The lack of protein causes decreased osmotic pressure in blood vessels, resulting in generalized edema (called anasarca). Kwashiorkor can be distinguished from marasmus (starvation resulting from inadequate intake of all nutrients). Marasmus is characterized by more severe muscle wasting and emaciation without associated edema (Fig. 10-9).
PATHOLOGY
Esophageal Pathology
Gastroesophageal Reflux Disease (GERD)
GERD is inappropriate relaxation of the lower esophageal sphincter (LES), causing burning retrosternal pain (especially after meals or when laying flat). It also may present as hoarseness, sour taste in the mouth (water brash), worsening asthma, or chronic cough. Diagnosis is made clinically, but attention must be paid for red flag symptoms such as dysphagia (difficulty swallowing), odynophagia (painful swallowing), and weight loss. If these symptoms are present, additional testing (endoscopy, barium swallow) should be performed to rule out complications such as stricture, Barrett’s esophagus, adenocarcinoma, or esophagitis. Treatment of GERD involves diet modification (avoidance of caffeine, alcohol, and spicy foods), H2 blockers, or PPIs. Nissen fundoplication, a surgical procedure in which the gastric fundus is wrapped around the lower esophagus to increase pressure at the lower esophageal sphincter, can be performed in refractory cases.
 Esophageal stricture: Chronic gastric acid exposure results in scarring of the esophagus, eventually narrowing the lumen. Dysphagia is the predominant symptom because food has difficulty passing the stricture.
Esophageal stricture: Chronic gastric acid exposure results in scarring of the esophagus, eventually narrowing the lumen. Dysphagia is the predominant symptom because food has difficulty passing the stricture.
Barrett’s esophagus: Intestinal (columnar) metaplasia in place of the normal stratified squamous epithelium. This histologic finding predisposes patients to adenocarcinoma. Of note, gastric metaplasia may also occur but is not associated with adenocarcinoma.
Hiatal Hernias
A sliding hiatal hernia occurs when the gastroesophageal junction (GEJ) herniates through the esophageal hiatus (Fig. 10-10B). The diaphragm is no longer able to contribute to the GEJ pressure gradient, thereby predisposing the patient to GERD; most hiatal hernias, however, are asymptomatic. A paraesophageal hiatal hernia occurs when the gastric fundus herniates through the esophageal hiatus (Fig. 10-10C). Because the GEJ is not affected, this anatomic arrangement does not lead to GERD but does put patients at risk for incarceration and strangulation of the fundus. Fortunately, paraesophageal hernias are significantly less common than the more benign sliding hernias.
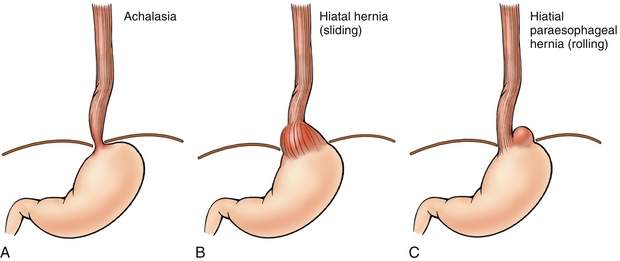
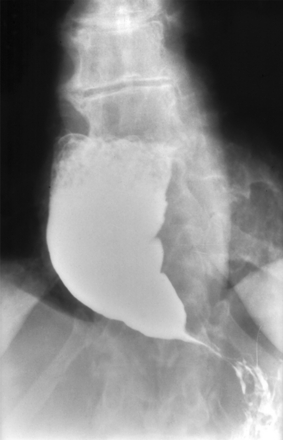
Achalasia
A dysfunction of the esophageal myenteric plexus causing increased LES tone and impaired peristalsis, which can be revealed on barium swallow (Figs. 10-10A and 10-11). The failure to relax leads to food accumulation proximal to the lesion and subsequent dilation of the esophagus. This combination of pathology results in the bird’s beak appearance of achalasia on a barium swallow study. Although usually idiopathic, Chagas disease, caused by Trypanosoma cruzi, causes damage to the myenteric plexus resulting in achalasia. T. cruzi also causes megacolon via destruction of the colonic myenteric plexus. Endoscopic pneumatic dilation of the LES is a common treatment modality for achalasia. Botulinum toxin can also be injected into the LES to induce relaxation.
Mallory-Weiss Tear
Partial-thickness esophageal laceration caused by forceful retching (e.g., after alcohol consumption or from bulimia). Presents as painful, blood-streaked emesis. No treatment is necessary in most cases.
Boerhaave Syndrome
Esophageal perforation often caused by retching or instrumentation (endoscopy). Presents as chest pain and painful swallowing. Physical exam may reveal crepitus from subcutaneous air, and chest x-ray may reveal a widened mediastinum or air in the mediastinum. A pleural effusion may also be present because of irritation from the luminal contents. Diagnosis is made with water-soluble contrast esophagraphy. Barium contrast, which will damage and inflame thoracic structures outside the GI tract, should be avoided. Computed tomography (CT) may also be confirmatory.
Esophagitis
Nonspecific inflammation of the esophagus, most often caused by GERD. So-called pill esophagitis is caused by certain medications becoming lodged in the esophagus (e.g., nonsteroidal anti-inflammatory drugs (NSAIDs), bisphosphonates, iron, and KCl). In AIDS patients, consider infectious causes such as cytomegalovirus, Candida, or herpes simplex virus.
Zenker’s Diverticulum
Esophageal diverticulum causing regurgitation, dysphagia, and halitosis (from trapped rotting food; Fig. 10-12).

Plummer-Vinson Syndrome
Iron deficiency anemia leading to formation of esophageal webs (causing dysphagia and glossitis—smooth, shiny, red tongue; Fig. 10-13). It is unclear exactly why iron deficiency produces this condition in some patients, but most symptoms respond to iron supplementation. Esophageal webs can also be treated endoscopically.

Gastric and Duodenal Pathology
Gastritis
Gastric inflammation and associated mucosal injury found on biopsy. However, overall a term that is not well-defined and has several classification systems. A distinction is often made between acute and chronic gastritis. Histopathologically, acute gastritis is neutrophil- predominant whereas chronic gastritis demonstrates a mixture of mononuclear cells. Causes of gastritis include Helicobacter pylori infection, alcohol, NSAIDs, and autoimmune conditions.
Of note, pernicious anemia is characterized by anti-intrinsic factor antibodies and antiparietal cell antibodies. The result is a chronic atrophic gastritis and loss of intrinsic factor, leading to vitamin B12 (cobalamin) deficiency and megaloblastic anemia.
Peptic Ulcer Disease
Erosions of gastric mucosa associated with gnawing epigastric abdominal pain. Ulcers are most commonly caused by H. pylori infection or heavy NSAID use. Patients with active H. pylori infection will have positive stool antigen and urease breath test. The urease breath test involves having a patient consume radiolabeled urea. In the presence of urease from H. pylori, the urea will be broken down into CO2 and ammonia. The radiolabeled CO2 can then be detected on exhalation. Physiologically, H. pylori uses urease to neutralize gastric acid with ammonia and create a more hospitable environment. H. pylori serum studies indicate only that infection was present at one time, but are often used in diagnosis in the appropriate clinical setting (i.e., classic symptoms). Endoscopy, however, remains the gold standard.
NSAIDs cause ulcers by inhibiting the production of protective prostaglandins. In general, NSAIDs inhibit cyclooxygenase-1 (COX-1) and COX-2. Because COX-1 is responsible for the production of protective prostaglandins in the stomach, selective COX-2 inhibitors such as celecoxib are thought to have fewer GI side effects and be less likely to contribute to gastritis or ulcer formation. Unfortunately, selective inhibition of COX-2 leads to a generalized prothrombotic state, which increases the risk of stroke and myocardial infarction.
Complications of peptic ulcers include perforation and bleeding. Malignancies also tend to ulcerate, so one should always biopsy ulcers during endoscopy to rule out cancer. Zollinger-Ellison syndrome (gastrinoma) should be considered in patients with recurrent or refractory ulcers and in patients with MEN1 syndrome.
Perforation: Perforation of an abdominal viscus (e.g., perforated peptic ulcers) classically presents as acute onset abdominal pain and peritonitis. An x-ray may reveal free air under the diaphragm (pneumoperitoneum; Fig. 10-14). Prompt surgical consultation is essential.

Figure 10-14 Perforated viscus causing air under the diaphragm on chest x-ray. (From Lim EKS. Medicine and Surgery. Philadelphia: Elsevier; 2007.)
Bleeding ulcer: If the ulcer overlies a vein or artery (e.g., gastroduodenal or gastric artery) brisk bleeding may occur. Patients with bleeding ulcers most frequently present with melena, although brisk or large-volume bleeding can result in hematemesis and even hematochezia.
Ménétrier’s Disease
Thought to be secondary to increased epidermal growth factor receptor, which causes extreme hypertrophy of gastric rugae. Symptoms include pain, weight loss, and even protein-losing gastroenteropathy, caused by increased permeability of the gastric mucosa. Biopsy is diagnostic and reveals mucous cell hyperplasia and gland atrophy.
Gastric Cancer
Often presents as weight loss, abdominal pain, and early satiety. Diagnosis is made by endoscopy with biopsy. Gastric cancer is usually adenocarcinoma. Risk factors include smoked foods, chronic gastritis, and H. pylori infection. Biopsy may reveal characteristic signet ring cells, in which mucin has pushed the nucleus to the periphery, giving the cells the appearance of signet rings (Fig. 10-15). A palpable supraclavicular lymph node (Virchow’s node) should always be concerning for malignancy and gastric cancer should be high on the differential. Because gastric cancer tends to ulcerate, all gastric ulcers should be biopsied to exclude malignancy. Linitis plastica (leather bottle stomach) is a form of gastric cancer in which the malignancy diffusely infiltrates the gastric wall and causes severe thickening and rigidity of the stomach. The lack of distensibility makes early satiety and vomiting an important feature. Prognosis for this condition is extremely poor. Gastric MALT lymphoma is highly associated with H. pylori and responds very well to eradication, perhaps making it the only cancer that can be treated with antibiotics.

Small Bowel and Colon
Small Bowel Obstruction
Presents as colicky abdominal pain, vomiting, and obstipation (failure to pass stool or gas; Fig. 10-16). The pain is colicky because pain is felt specifically when peristalsis causes contraction against the obstruction. An abdominal x-ray usually reveals dilated loops of bowel, often with multiple air fluid levels (Fig. 10-17). All patients should be made NPO (nothing by mouth), and given IV fluids and medications to control pain and nausea. A nasogastric tube will decompress the bowel and may even resolve the obstruction. Surgical intervention may be necessary, however.
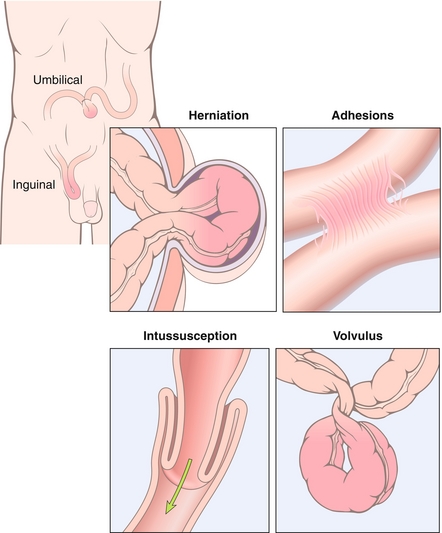
Figure 10-16 Causes of small bowel obstruction. (From Kumar V, Abbas AK, Fausto N, Mitchell R. Robbins Basic Pathology. 8th ed. Philadelphia: Elsevier; 2007.)
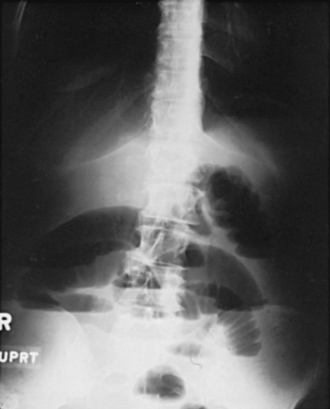
Figure 10-17 X-ray of dilated loops of bowel caused by an SBO. (From Lawlor MW. Rapid Review USMLE Step 2. Philadelphia: Elsevier; 2006.)
Adhesions: The most likely cause of obstruction in patients with previous abdominal surgery. Treated with surgical lysis of adhesions in severe cases. Incomplete bowel obstruction may respond to bowel rest (keeping the patient NPO) and nasogastric (NG) tube decompression.
Hernia incarceration: Most common cause of obstruction in adults without history of abdominal surgery (see later).
Intussusception: One piece of small bowel (intussusceptum) telescopes into an adjacent section (intussuscipiens). Most common cause of intestinal obstruction in young children (two thirds of cases occur in children younger than 1 year). It occurs commonly after viral illnesses, because of lymphatic hypertrophy at the lead point. In adults, the lead point is more likely to be a polyp or tumor. Patients present with severe, colicky abdominal pain. Small children often present with alternating periods of crying and lethargy, and parents may describe their child drawing his or her legs up during painful episodes. Hematochezia is an inconsistent symptom, although many patients will have hemoccult-positive stool, and currant jelly stool (actually dead mucosa that has sloughed off of ischemic bowel) is an extremely late finding. Physical exam may reveal a palpable, sausage-shaped abdominal mass. Ultrasonography, which is currently the first-line diagnostic test in most centers, shows a bulls-eye lesion demonstrating one region of bowel surrounding another. Diagnosis and treatment can be accomplished via an air- or contrast-based enema.
Volvulus: Twisting of a loop of bowel around its mesentery, which leads to physical obstruction of the bowel lumen and ischemia of the bowel from interruption of the vascular supply. Symptoms include abdominal pain, distention, obstipation and vomiting. Sigmoid volvulus most frequently occurs in debilitated older patients. Cecal volvulus most frequently occurs in middle-aged adults because of increased mobility of the right colon. Midgut volvulus occurs in the first months of life and is caused by congenital malrotation of the gut.
Celiac Disease
A hypersensitivity to wheat and rice. More specifically, sensitivity to the glycoprotein gliadin found in gluten. Antigliadin antibodies target gliadin and create a local inflammatory reaction that damages the GI mucosa. Patients present with abdominal pain, vomiting, and diarrhea associated with ingestion of gluten. Laboratory tests often reveal antitissue transglutaminase antibody and may also be consistent with iron deficiency anemia (microcytic) or folate deficiency (macrocytic) because mucosal damage can lead to malabsorption of these micronutrients. Intestinal biopsy reveals villous atrophy and crypt hyperplasia (Fig. 10-18). Treatment involves a strict gluten-free diet, which can often lead to resolution of symptoms. Of note, dermatitis herpetiformis is a commonly tested dermatologic manifestation of celiac disease—the presence of herpetic lesions, especially on the elbows and other extensor surfaces (Fig. 10-19).

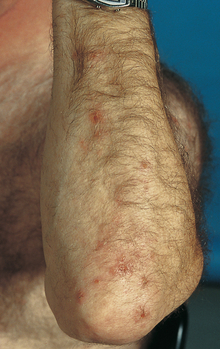
Tropical Sprue
Clinically and histologically indistinguishable from celiac disease, tropical sprue is thought to be caused by an infectious source, although the organism is not known. It is distinguished from celiac because it is found predominantly in the tropics and does not respond to a gluten-free diet. Antibiotics are the treatment of choice.
Whipple’s Disease
Symptoms are similar to tropical sprue and celiac disease; however, biopsy reveals lipid vacuolation and periodic acid–Schiff (PAS)–positive macrophages. The bacilli Tropheryma whipplei is the causative organism. Treatment is with long-term antibiotics.
Appendicitis
The underlying cause is appendiceal obstruction. Obstruction is often from a fecalith in adults. In children, however, lymphoid hyperplasia, especially after a viral illness, is commonly the inciting event. Acute appendicitis presents as an acute onset of ill-defined periumbilical pain that migrates to the right lower quadrant and becomes sharply defined. This migration occurs because the inflammation is sensed initially by the poorly defined visceral receptors of the appendix, which synapse in the spinal cord at the same level as the periumbilical region of the anterior abdominal wall. Once the inflammation spreads to the parietal peritoneum, however, the more distinguishing parietal receptors accurately localize the pain to the right lower quadrant. Signs and symptoms of appendicitis include fever, vomiting, and positive psoas or obturator signs (pain with hip flexion or internal rotation, respectively). Blood work is of limited use in aiding diagnosis because of its poor sensitivity and specificity for the disease, but may reveal leukocytosis (a normal WBC count does not exclude the diagnosis). CT scanning is usually highly accurate in making the diagnosis, but ultrasound is a reasonable initial diagnostic modality when limiting radiation exposure is a high priority (e.g., in children). Ultrasound can also help distinguish appendicitis from pelvic pathology, such as an ectopic pregnancy or ovarian torsion. MRI may also be used, but it is less common due to the greater speed and availability of CT scans. Treatment is with appendectomy and antibiotics. Complications include perforation, which may lead to peritonitis and/or abscess formation.
Inflammatory Bowel Disease
Ulcerative Colitis (UC)
UC is an autoimmune inflammatory condition of the colon. Patients present with relapsing episodes of bloody diarrhea, mucus-covered stools, and cramping. Colonoscopy reveals inflammation that begins in the rectum and spreads continuously as far as the cecum. Pseudopolyps, which are areas of normal mucosa that appear polypoid in comparison to surrounding ulcerated or friable mucosa, may be seen on colonoscopy. Barium enema reveals a lead pipe colon, so-called because of the absence of haustral markings (Fig. 10-20). Histology demonstrates inflammation and ulceration of the mucosa and submucosa. Unlike in Crohn’s disease, ulcerations do not involve the full thickness of the bowel and there is no granuloma formation. Extraintestinal manifestations of the disease include primary sclerosing cholangitis, polyarthritis, uveitis, pyoderma gangrenosum, and erythema nodosum. Patients with UC are at increased risk for colon cancer and should be screened with colonoscopy starting 8 years after diagnosis. Treatment involves 5-ASA compounds (e.g., sulfasalazine), steroids, and biologics, including infliximab. Colectomy with ileoanal anastomosis is usually curative for colitis, but does not treat extraintestinal manifestations.

Crohn’s Disease
An autoimmune inflammatory condition of the entire GI tract but most commonly affecting the ileocecal region. Patients may present with abdominal pain, nausea, vomiting, and diarrhea (often with passage of blood and mucus). Colonoscopy reveals skip lesions—ulcers that may be present anywhere from mouth to rectum. Histology reveals transmural (full-thickness) inflammation and may include noncaseating granulomas. Complications include abscesses, strictures, fistulas, and fissures. Furthermore, Crohn’s disease itself or surgical treatment with bowel resection may cause a deficiency in vitamin B12 and bile salts because these are absorbed in the ileum. A relative lack of bile salts can also lead to impaired absorption of fat-soluble vitamins (A, D, E, and K). Also, the presence of bile salts in the colon is cathartic and may cause diarrhea. Therefore, cholestyramine (a bile acid–binding resin) can be used to bind bile salts and prevent their cathartic effect. Extraintestinal manifestations are similar to UC; however, primary sclerosing cholangitis is comparatively rare. The risk of colon cancer is elevated in Crohn’s disease, although less so than in UC. Treatment is similar to UC, but surgery is not curative and recurrent bowel resection may lead to short bowel syndrome—malabsorption and steatorrhea caused by decreased intestinal length. Both UC and Crohn’s disease are associated with the HLA-B27 major histocompatibility complex (Table 10-2).
Table 10-2
Comparison of Crohn’s Disease and Ulcerative Colitis
| Feature | Crohn’s Disease | Ulcerative Colitis |
| Most frequent distribution | Ileocecal region | Rectum to cecum |
| Entire GI tract involvement? | Yes | No |
| Spread | Skip lesions | Continuous |
| Biopsy findings | Transmural inflammation Granulomas |
Partial-thickness inflammation |
| Cancer risk | + | +++ |
| PSC risk | + | +++ |
| Colectomy curative? | No (symptoms often recur) | Yes (of GI symptoms only) |
| HLA-B27 association | Yes | Yes |
| Other | Abscesses, strictures, fistulas, fissures are complications | Lead pipe colon seen on barium enema; pseudopolyps seen on endoscopy |
Colonic Polyps
A polyp’s risk of malignancy is related to size, degree of dysplasia, and architecture. Polyps can be divided into two types based on histology:
Tubular adenoma: Accounts for approximately 80% of polyps (Fig. 10-21A). Defined by the presence of tubular glands on histology (branching adenomatous epithelium). More likely to be pedunculated (on a stalk) on gross appearance. Less likely to become cancerous.
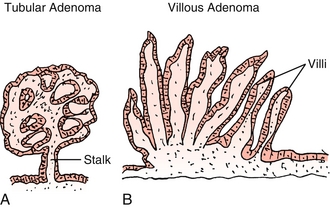
Figure 10-21 A, Tubular adenoma. B, Villous adenoma. (From Damjanov I. Pathology Secrets, 3rd ed. Philadelphia: Elsevier; 2008.)
Villous adenoma: Defined by presence of villous projections on histology–glands that project from the surface directly to the center of the polyp (Fig. 10-21B). More likely to be sessile (no stalk) on gross appearance. More likely to become cancerous.
Colon Cancer
Patients may be asymptomatic, with cancer found on screening colonoscopy. Patients may also present with hematochezia, obstruction, or small-caliber stools. In general, left-sided cancers tend to obstruct because the colonic lumen is narrower and stool is more solid. Right-sided cancers, on the other hand, tend to bleed. Bleeding may be occult, however, so GI malignancy should be considered in patients with iron deficiency anemia or positive fecal occult blood tests. Colon cancer is the third leading cause of cancer death in North America. Most are adenocarcinomas. Current guidelines recommend screening for colon cancer every 10 years starting at age 50. In patients with a family history of colon cancer, screening should begin 10 years before the age at which the youngest affected first-degree relative was diagnosed.
Lynch Syndrome
Also called hereditary nonpolyposis colorectal cancer. Autosomal dominant mutation of DNA repair genes, leading to increased risk of colon, endometrial, and ovarian cancers. In Lynch syndrome, colon cancer may occur without being preceded by a polyp.
Familial Adenomatous Polyposis (FAP)
FAP is caused by the autosomal dominant mutation of adenomatous polyposis coli (APC) gene. The APC gene leads to hundreds of polyps in the colon and throughout the GI tract, with the lifetime risk of malignancy approaching 100% without treatment. Treat with prophylactic colectomy.
Diarrhea
Osmotic
Occurs when water is drawn into the bowel because of an increase in luminal osmoles. Osmotic laxatives such as magnesium hydroxide (Milk of Magnesia) act in this manner. In lactose intolerance (disaccharidase deficiency), lactose is the offending osmole. Sorbitol is poorly absorbed and may cause osmotic diarrhea in patients consuming a lot of low-calorie sweetener (e.g., sugar-free gum). Osmotic diarrhea should resolve with withdrawal of the offending agent.
Secretory
An increase in intestinal secretion of ions and water, causing extremely watery stools. Most notably, Vibrio cholerae produces the cholera toxin, which irreversibly activates the enterocyte’s Gs subunit. Gs then stimulates cyclic adenosine monophosphate (cAMP)–mediated secretion of ions and water, causing so-called rice water diarrhea.
Exudative
Diarrhea with the presence of blood or pus in the stool is considered exudative. It may be secondary to IBD or infectious in origin. Shigella and Salmonella spp., enterohemorrhagic Escherichia coli, and Entamoeba histolytica are the common culprit pathogens.
Deranged motility: Anything that reduces gut transit time may lead to an increase in the frequency of loose stools. Surgical resection, carcinoid syndrome, hyperthyroidism, and IBS all cause diarrhea partially through increasing transit time. GI irritants (e.g., blood) also have decreased transit time and can increase the frequency of stools.
Diverticular Disease
An outpouching of mucosa and submucosa through the muscularis propria that is covered by the serosa (technically pseudodiverticula, because they do not contain all layers of the wall; Fig. 10-22). These outpouchings are extremely common and generally asymptomatic. They tend to occur in the sigmoid colon, in which luminal pressure is the highest, and specifically around the vasa recta, where the penetrating blood vessel creates a local area of weakness. Anything that increases colonic pressure is a risk factor, including low-fiber diets, obesity, and straining with defecation.
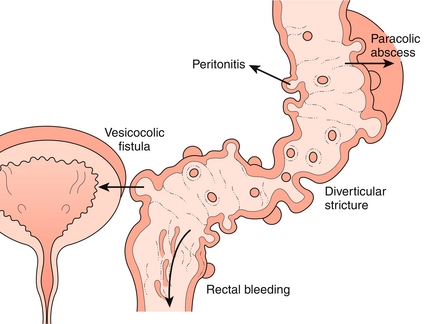
Diverticulitis
Occurs when there is a perforation of a diverticulum leading to inflammation and focal necrosis. Clinically, patients tend to present with more than 24 hours of left lower quadrant abdominal pain and tenderness and sometimes low-grade fever, nausea, and vomiting. Because diverticulitis tends to recur, many patients will report similar previous episodes. The history and physical exam alone may be enough to diagnose a recurrence of diverticulitis. Colonoscopy is contraindicated during active inflammation, but CT is diagnostic and will reveal complications. Although the omentum tends to wall off the inflammatory process, complications include peritonitis secondary to abscess formation, fistulas, and strictures. Uncomplicated diverticulitis can be treated with antibiotics directed against colonic flora, predominantly gram-negative rods and anaerobes. The combination of ciprofloxacin and metronidazole is commonly used.
Bleeding Diverticulosis
Diverticula tend to occur around the vasa recta, where the penetrating blood vessels create local weakness. The associated vasa recta are then only protected by the overlying mucosa and are at risk for bleeding, even from trivial intraluminal trauma. Bleeding can be profound and presents as hematochezia. Diagnosis and treatment can be achieved via colonoscopy.
Hemorrhoids
These are dilated hemorrhoidal veins in the submucosal layer of the rectum. Hemorrhoids are present in approximately 25% of adults. Constipation contributes to their formation. Improvement may be seen with a high-fiber diet and stool softeners, which can reduce straining during defecation. Do not confuse hemorrhoids with skin tags. A perianal skin tag may represent a sentinel tag from an anal fissure, a painful tear in the rectal mucosa below the dentate line. Unlike a hemorrhoid, an anal fissure will be exquisitely tender on defecation or digital rectal exam.
Internal Hemorrhoids
Located above the dentate (pectinate) line, internal hemorrhoids cause bleeding from the superior hemorrhoidal veins. Because innervation is visceral, internal hemorrhoids are painless. They may, however, prolapse and become palpable. Treatment is with band ligation.
External Hemorrhoids
Located below the pectinate line, external hemorrhoids arise from dilation of the inferior hemorrhoidal veins and are covered with squamous epithelium. They are innervated by somatic receptors. The somatic innervation means they may be pruritic or painful, especially if thrombosed.
External hemorrhoids are painful. Internal hemorrhoids are painless.
Laboratory Tests
Aspartate Aminotransferase and Alanine Aminotransferase
Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are liver enzymes important in amino acid metabolism, but are used clinically as markers of hepatocyte damage because they can be detected in the serum. The normal reference range for AST and ALT is 5-40 U/L. Levels tend to be extremely high >1000 U/L in acute viral hepatitis and more modestly elevated >100 U/L in chronic forms of hepatitis. The ratio of ALT to AST can also be useful in diagnosing the cause of liver inflammation. Whenever liver inflammation is present the rise in ALT tends to be greater than the rise in AST. In alcoholic hepatitis, however, AST is twice as elevated as ALT (think AST—Smirnoff vodka). Importantly, the degree of enzymatic elevation does not necessarily correlate with the severity of liver failure. A patient with end-stage liver disease, for example, may only have a slight elevation of AST and ALT levels because most of the liver has already been destroyed. A patient with hepatitis A, on the other hand, may have AST and ALT values >1000 U/L but maintain a fully functional liver after recovery.
Alkaline Phosphatase (AP)
AP is a ubiquitous enzyme that, as the name suggests, removes phosphates, most efficiently in alkaline environments. This enzyme is particularly concentrated in the biliary tree, bone, and placenta. The AP level is often elevated in cholestatic syndromes and in bile duct obstruction (e.g. cholangitis).
Gamma-Glutamyltransferase
Another enzyme of amino acid metabolism that is elevated in diseases of the biliary tree. The gamma-glutamyltransferase (GGT) level is elevated along with AP but is more specific to biliary disease (e.g., will not be increased in bone disease). It is also raised with alcohol ingestion.
Prothrombin Time and International Normalized Ratio
Because the liver makes most clotting factors, an elevation of the prothrombin time (PT) and international normalized ratio (INR) suggests impaired hepatic synthetic function such as in cirrhosis.
Bilirubin
In the spleen, heme released from senescent red blood cells is broken down to unconjugated bilirubin. The insoluble, inexcretable, unconjugated form travels to the liver bound to albumin, where it is conjugated with glucuronic acid to form conjugated (direct) bilirubin. This form is then excreted from the body via the bile. In the gut, bacteria convert conjugated bilirubin to urobilinogen, part of which is excreted in the feces as stercobilin; the rest is resorbed and secreted in the urine as urobilin. Conditions that limit the metabolic function of the liver (e.g., cirrhosis) cause elevations in the unconjugated bilirubin level. Furthermore, an increase in the production of bilirubin (e.g. intravascular or extravascular hemolysis) will also elevate unconjugated bilirubin. On the other hand, a patient with a cholestatic process such as choledocholithiasis will be able to conjugate bilirubin but will be unable to excrete it, resulting in a rise in conjugated bilirubin levels.
Interestingly, hemoglobin is a colorful molecule and, along with its breakdown products, is responsible for the red color of blood, yellow color of urine (urobilin), brown color of feces (stercobilin), and yellow discoloration of jaundice (bilirubin). A patient with a cholestatic process (e.g., choledocholithiasis, cholangiocarcinoma) will be unable to excrete bilirubin into the feces and will have clay-colored stools. A patient who cannot conjugate bilirubin will also subsequently lack urobilin so, although the patient may be jaundiced from unconjugated bilirubin, the urine will not appear more yellow because unconjugated bilirubin will not pass through the glomerular basement membrane because it is bound to albumin.
Hepatic Pathology
Nonalcoholic Steatosis
Elevation of the AST and ALT levels, with a hypoechoic liver on imaging and no apparent cause except for obesity and/or metabolic syndrome. Treatment involves diet, exercise, and management of metabolic comorbidities, such as diabetes.
Nonalcoholic Steatohepatitis (NASH)
NASH is nonalcoholic steatosis plus biopsy-proven histologic evidence of inflammation.
Alcoholic Hepatitis
Chronic alcohol consumption leading to fatty infiltration, similar to nonalcoholic steatosis. Eventually, transaminase levels will rise secondary to hepatocyte damage. The AST-to-ALT ratio may be more than 2:1. Mallory bodies (eosinophilic keratin filament inclusion bodies) may be present on biopsy. These are more commonly found in alcoholic hepatitis than in NASH.
Hemochromatosis
Autosomal recessive disorder of the HFE gene (acronym for high Fe2 +) leading to dysregulated overabsorption of iron and excessive hemosiderin accumulation, leading to free radical damage. Patients present with cirrhosis, bronzed skin, diabetes (pancreatic involvement), and cardiomyopathy. Diagnosis is suspected with high serum iron and ferritin levels and confirmed with genetic testing. Liver biopsy with Prussian blue staining reveals hemosiderin deposits. Treatment is aimed at normalizing iron stores via repeated phlebotomy. Secondary hemochromatosis is caused by excessive iron intake, usually after repeated red blood cell transfusions, such as for β-thalassemia.
Wilson’s Disease
Autosomal recessive disorder of Wilson’s disease protein, an ATPase responsible for transporting copper into bile and important in the formation of ceruloplasmin (the transport protein for copper). The inability to excrete copper leads to an increase in the free serum copper level although the total serum copper may be high, low, or normal, given the relative lack of ceruloplasmin to transport copper. Consequently, copper accumulates in the liver (cirrhosis), brain (cognitive deterioration and parkinsonism), and cornea (Kayser-Fleischer rings). Diagnosis is based on a decrease in levels of serum ceruloplasmin, an elevation in urinary copper levels, and copper deposits seen on liver biopsy. Treatment involves chelation with d-penicillamine.
α1-Antitrypsin Deficiency
Autosomal recessive defect of α1-antitrypsin (A1AT), a protein that inactivates proteases and protects tissues from their enzymatic destruction. A1AT deficiency leads to early emphysema (see Chapter 17) and accumulation of A1AT in the hepatocyte endoplasmic reticulum, leading to cirrhosis. In a nonsmoker with early-onset emphysema, A1AT deficiency should be considered as the cause of the cirrhosis. Histology reveals PAS-positive granules.
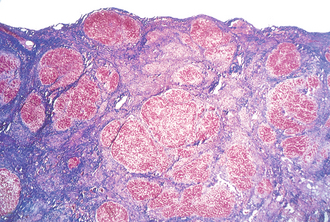
Cirrhosis
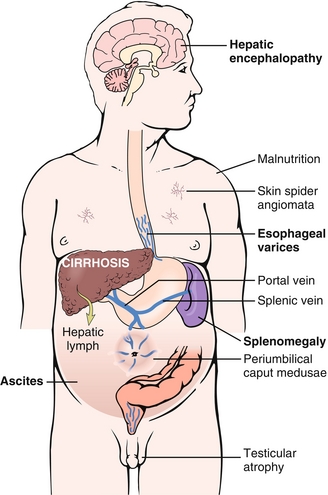
The final common pathway of liver disease and a pathologic diagnosis based on the presence of regenerative nodules surrounded by fibrosis (Fig. 10-23). Sequelae of cirrhosis are secondary to impaired metabolic function of the liver and portal hypertension because the fibrosed liver cannot handle as much blood flow through the portal circulation and shunts it to the caval circulation. Impaired hepatic breakdown of estrogen may also lead to gynecomastia, testicular atrophy, and spider angioma (via weakening of vascular walls). Impaired synthesis of clotting factors causes a coagulopathy reflected by an elevation of PT and INR values. Finally, hepatic encephalopathy may occur, leading to altered mental status and asterixis (flapping tremor of negative myoclonus). Although the mechanism of hepatic encephalopathy is not entirely clear, it is associated with an elevated serum ammonia level (Fig. 10-24).
Portal Hypertension
Defined as a pressure gradient greater than 10 mm Hg between the portal vein (entering the liver) and hepatic veins (leaving the liver). The hypertension is usually intrahepatic (sinusoidal) secondary to cirrhosis (as described above). Rarely, however, it may be posthepatic secondary to hepatic vein thrombosis (Budd-Chiari syndrome) or prehepatic secondary to portal vein thrombosis. The sequelae of portal hypertension are outlined here.
Esophageal varices: Present in 30% to 60% of cirrhotics. Dilated periesophageal veins often begin as asymptomatic, but may spontaneously rupture, leading to massive upper GI bleeding. A GI bleed in a cirrhotic patient should be considered variceal until proven otherwise, although other causes are possible. Prophylaxis may be accomplished with nonselective beta blockers (e.g., propranolol) or endoscopic banding of varices. Management of acute bleeds involves resuscitation and IV octreotide, a synthetic somatostatin and splanchnic vasoconstrictor that mitigates bleeding. Broad-spectrum antibiotics (e.g., ceftriaxone) are used prophylactically to prevent bacteremia caused by esophageal flora. Endoscopic banding should be attempted within 24 hours.
Caput medusae: Dilation of periumbilical veins and abdominal wall veins causing the appearance of the head (caput) of Medusa on the abdominal wall.
Splenomegaly: Enlarged spleen secondary to collateral flow via the splenic vein.
Ascites: Portal hypertension causes increased portal hydrostatic pressure. This, combined with decreased osmotic pressure from impaired albumin production, leads to fluid collection in the abdomen. This process can lead to generalized edema (anasarca).
Hepatorenal syndrome: Systemic vasodilation leading to poor renal perfusion. Hepatorenal syndrome is a late sequela.
Budd-Chiari syndrome (hepatic vein thrombosis) is often idiopathic but is associated with hypercoagulable states, especially polycythemia vera. It presents with the classic triad of abdominal pain, ascites, and hepatomegaly. To remember how to distinguish Budd-Chiari from Arnold-Chiari (a neurologic malformation), think Budd for blockage of the hepatic vein.
Hepatic Neoplasms
Hemangioma
Most common hepatic tumor caused by a benign proliferation of blood vessels. Often asymptomatic and incidentally discovered, or may cause abdominal pain. Rarely, hemangiomas will rupture, causing massive blood loss. There is an uncertain link between hemangiomas and hormone use, and some physicians recommend avoiding hormonal contraception if hemangiomas are present. Lesions should not be biopsied because this might precipitate bleeding.
Hepatocellular Adenoma
Benign hepatic neoplasm highly associated with the use of hormonal contraception. These tumors tend to be avascular and will not bleed if biopsied. Resection of these lesions is the norm to avoid rare complications such as rupture or transformation to hepatocellular carcinoma.
Hepatocellular Carcinoma (Hepatoma, HCC)
Most common primary hepatic malignancy and most frequently secondary to cirrhosis (e.g., viral, alcoholic). Of note, HCC may develop in patients with hepatitis B before cirrhosis occurs, although this is not the case in hepatitis C. Overall, however, there is a stronger association between hepatitis C and HCC. Symptoms include jaundice, ascites, and hepatomegaly, but patients with cirrhosis should be monitored, because early HCC may be asymptomatic. The tumor marker for HCC is alpha-fetoprotein (AFP), but diagnosis should be confirmed with imaging and often biopsy.
Of note, AFP acts as the fetal form of albumin. Its level is elevated in primary HCC and metastatic disease to the liver and in yolk sac tumors. In neural tube defects, the AFP level is also elevated in the mother’s blood because the fetal skin is not fully intact, and AFP leaks into the mother’s circulation. In Down syndrome, the levels are decreased, presumably because the size of the fetus and yolk sac are decreased.
Disorders of Bilirubin Metabolism
Gilbert Syndrome
Common and benign cause of jaundice caused by reduced activity of glucuronyltransferase, the enzyme responsible for the conjugation of bilirubin. Without conjugation, the bilirubin remains insoluble and incapable of being excreted in bile. Those affected may have mild jaundice, especially in times of stress or dehydration, or an increase in the indirect bilirubin level found incidentally on laboratory studies. There are no sequelae, and treatment is unnecessary.
Crigler-Najjar Syndrome
Rare autosomal recessive condition leading to near-total absence of functional glucuronyltransferase and inability to conjugate (and therefore excrete) bilirubin. Type 1 is most severe and leads to kernicterus and early death if aggressive phototherapy and/or plasmapheresis are not initiated. Type 2 is a significant but incomplete absence of glucuronyltransferase, and survival to adulthood is the norm. Laboratory tests reveal elevated indirect bilirubin levels.
Dubin-Johnson Syndrome
Rare autosomal recessive condition characterized by inability to excrete conjugated bilirubin into the bile. Characteristically the liver will be black in these patients because of pigment deposition in lysosomes. Most patients are asymptomatic or have only mild jaundice. They rarely require treatment. Rotor syndrome is even milder and the patient’s liver does not turn black.
GALLBLADDER AND PANCREAS
Gallstones
Risk factors for cholesterol stones include the four Fs—fat, female, forty, and fertile. These factors combine to create a high-cholesterol, low–gallbladder motility environment. Progesterone specifically inhibits gallbladder motility in an analogous manner to its inhibition of uterine smooth muscle contraction during pregnancy. Pigment stones are composed of bilirubin precipitate and are found secondary to high red blood cell (RBC) turnover. For example, a patient with sickle cell anemia and right upper quadrant (RUQ) pain is likely to have symptomatic pigment stone cholelithiasis. In general, stones are best diagnosed with ultrasound because they have a different density from that of the surrounding structures (Fig. 10-25). Although pigment stones are generally radiopaque, conventional x-rays are less helpful because the much more common cholesterol stones are generally radiolucent. Asymptomatic gallstones found on ultrasound require no treatment. Elective surgery is generally recommended for symptomatic cholelithiasis, whereas cholecystitis, inflammation of the gallbladder and surrounding tissues from an impacted stone, requires antibiotics and urgent surgical attention. Stone impaction within the common bile duct (choledocholithiasis) can lead to cholangitis (ascending infection of the biliary tree). These patients often require endoscopic stone removal via retrograde cholangiopancreatography as well as IV antibiotics.
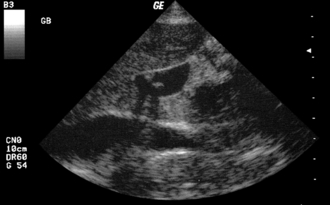
Cholelithiasis
The presence of stones in the gallbladder. When symptomatic, it causes colicky RUQ pain after fatty meals when the gallbladder contracts against a stone lodged in the cystic duct under the stimulation of cholecystokinin. Murphy’s sign will not be present.
Cholecystitis
An inflammatory process in the gallbladder often secondary to stasis from a stone in the cystic duct or (less likely) biliary sludging. Secondary infection often occurs, especially with Escherichia coli, Enterococcus, or Klebsiella. Clinically presents as constant or worsening RUQ pain, fever, and positive Murphy’s sign. Labs may reveal leukocytosis elevated liver enzymes, and elevated AP.
Cholangitis
Bacterial infection of the biliary tree. Clinically severe presentation of fever, RUQ pain, and jaundice (Charcot’s triad). May progress to include hypotension and altered mental status (Reynolds’ pentad). Expect elevation in AST, ALT, conjugated bilirubin, and alkaline phosphatase levels. Elevated amylase and lipase levels suggest associated pancreatitis. In this context, abdominal ultrasound revealing gallstones is highly suggestive of cholangitis. Endoscopic retrograde cholangiopancreatography (ERCP) would be diagnostic and therapeutic.
Acute Pancreatitis
Symptoms of epigastric abdominal pain radiating to the back, with elevated amylase and lipase levels (lipase is more specific). Usually caused by gallstones present in the ampulla causing reflux of pancreatic enzymes, leading to autodigestion and inflammation of the pancreas. Alcohol use is also a common cause. Other causes include hypertriglyceridemia, hypercalcemia, trauma, and medications, especially antiretrovirals and diuretics.
Chronic Pancreatitis
Patients may present with exocrine insufficiency (malabsorption) or endocrine insufficiency (diabetes); pain may or may not be a feature. Malabsorption often leads to a deficiency of fat-soluble vitamins (A, D, E, and K). Amylase and lipase levels are often normal. Chronic pancreatitis usually is the result of alcoholism, because ethanol can alter zymogen activation. It may also be a complication of cystic fibrosis caused by thickened pancreatic secretions. Of note, chronic pancreatitis is not a complication of gallstones. Abdominal radiography may reveal calcification of the pancreas (Fig. 10-26).
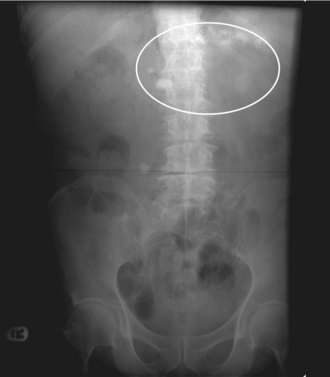
Figure 10-26 X-ray showing calcifications of chronic pancreatitis. (From Lim EKS. Medicine and Surgery. Philadelphia: Elsevier; 2007.)
In an alcoholic suffering from intractable steatorrhea (oily, fat containing stool), consider chronic pancreatitis. A lack of pancreatic enzymes, including lipase, and subsequent impaired digestion contributes to this malabsorptive syndrome.
Pancreatic Adenocarcinoma
Patients often present with painless jaundice, weight loss, early satiety, and/or abdominal pain radiating to the back. Physical exam may reveal a palpable gallbladder or abdominal mass. Labs show elevated bilirubin and alkaline phosphatase levels if the biliary tree is obstructed by the mass. These findings can be summarized as an obstructive process leading to failure of excretion of bile and bilirubin. Diagnosis requires imaging (often CT) and may be confirmed with biopsy (often performed endoscopically). Pancreatic cancer has a poor prognosis because it often presents late (early stages are asymptomatic) and it responds poorly to chemotherapy. A pancreaticoduodenectomy (Whipple procedure) is potentially curable if the entire mass is resected; however, metastases, especially to the liver, are often already present at the time of diagnosis. Masses in the head of the pancreas have a better prognosis because they are more likely to obstruct early and therefore can be detected early.
Hernias
A hernia is the protrusion of a peritoneal sac through a musculoaponeurotic barrier, usually a fascial defect. Inguinal hernias are common examples and often present as palpable swellings within defined regions. They may or may not be painful and may or may not be able to be reduced (pushed back into place); however, almost all hernias should be repaired because of the risk of strangulation (compromised blood supply leads to ischemia and infarction).
Inguinal Triangle (Hesselbach’s Triangle)
The anatomic region bounded by the inguinal ligament, inferior epigastric artery, and edge of the rectus abdominis muscle. Inguinal hernias within this region are considered direct.
Direct Inguinal Hernia
A protrusion of bowel or omentum medial to the inferior epigastric artery (Fig. 10-27A). The parietal peritoneum forms the hernia sac for direct hernias. Direct hernias proceed directly through the abdominal musculature and do not pass through the inguinal canal. For this reason, they rarely are found in the scrotum. They are most common in men because of weakening of the abdominal wall.
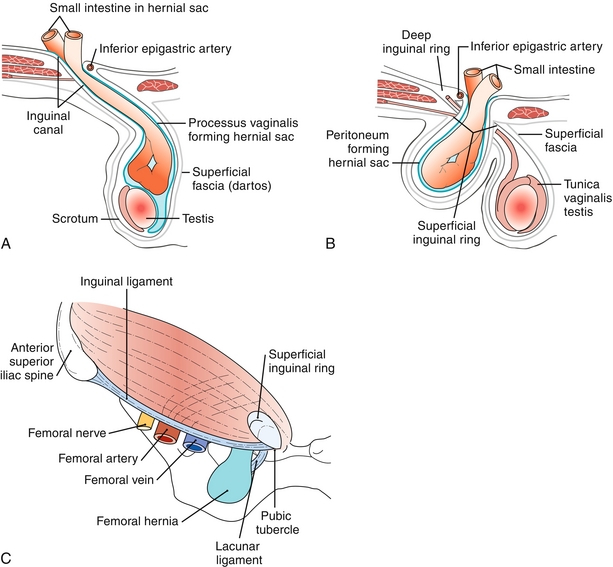
Figure 10-27 A, Indirect inguinal hernia passing lateral to the inferior epigastric artery and into the scrotum. B, Direct inguinal hernia passing medial to the inferior epigastric artery and not entering the scrotum. C, Femoral hernia passing inferior to the inguinal ligament. (A, B from Moore NA, Roy WA. Rapid Review Gross and Developmental Anatomy. 3rd ed. Philadelphia: Elsevier; 2010; C from Kontoyannis A, Sweetland H. Crash Course: Surgery E-Book. 3rd ed. Philadelphia: Elsevier; 2008.)
Indirect Inguinal Hernia
A protrusion of bowel or omentum through the internal inguinal ring, the inguinal canal, and the external inguinal ring (Fig. 10-27B). The hernia sac is formed by the processus vaginalis within the scrotum. Indirect inguinal hernias are more common in infants and children because of the congenital persistence of the processus vaginalis. They occur lateral to the inferior epigastric artery. Note that indirect hernias pass through the internal ring, INto the scrotum, and are common in infancy.
Femoral Hernia
A protrusion of bowel or omentum inferior to the inguinal ligament through the femoral canal (Fig. 10-27C). Femoral hernias are more common in females secondary to the wider structure of the bony pelvis.
PEDIATRIC GASTROENTEROLOGY AND HEPATOLOGY
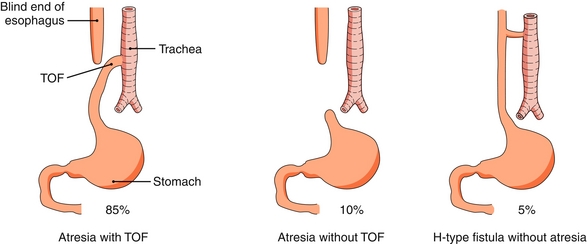
Tracheoesophageal Fistula
Congenital abnormal connection between the esophagus and trachea. Most frequently atresia of the upper esophagus and fistula between the lower esophagus and trachea (Fig. 10-28). Often presents at birth with copious salivation, choking, and cyanosis with feeding. There is also an inability to pass a nasogastric tube. It is one of the VACTERL anomalies (see Chapter 4).
Pyloric Stenosis
Hypertrophy of muscles in the pylorus leads to narrowing and lengthening of the canal and inability to pass gastric contents to the small intestine. Presents around the fourth week of life as projectile, postprandial, nonbilious vomiting associated with a voracious appetite. A right upper quadrant abdominal mass (palpable olive) may also be felt on physical exam. Diagnosis is accomplished with ultrasound or barium radiography. Surgery is the definitive treatment.
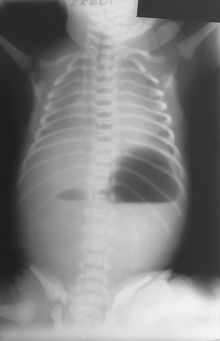
Duodenal Atresia
Congenital intestinal obstruction caused by failure to canalize the duodenum. Presents as bilious vomiting in the first days of life, earlier than pyloric stenosis. Abdominal radiography reveals the double-bubble sign, two air-filled spaces representing the stomach and duodenum (Fig. 10-29). Duodenal atresia is present in approximately 8% of neonates with Down syndrome.
Meconium Ileus
Presents as failure to pass meconium and almost pathognomonic of cystic fibrosis. Abnormal GI secretions cause thickened meconium that can obstruct the lumen of the small bowel. Ileus is actually a misnomer because this is a mechanical obstruction, not a motility disorder.
Congenital Aganglionic Megacolon (Hirschsprung Disease)
Presents as failure to pass meconium. This condition is caused by a failure of embryologic neural crest cell migration, leading to the absence of the submucosal and myenteric plexus in a segment of distal colon. A digital rectal exam may temporarily dilate the affected section, leading to a large forceful bowel movement (squirt sign). A barium enema will display a narrowing at the affected site and dilation of proximal segments to compensate for the inability to move stool forward. Biopsy is diagnostic, and surgery is usually curative.
Imperforate Anus
Congenital anal atresia often presents as failure to pass meconium and absence of an anus on physical exam. Dilation of the colon will also be present. It is associated with Down syndrome and is one of the VACTERL anomalies.
Necrotizing Enterocolitis
A condition of premature infants in which colonic bacteria take advantage of a neonate’s relatively weak immune system to invade the bowel wall. The bacterial invasion leads to pneumatosis intestinalis (air in the bowel wall) and bowel necrosis.
Neonatal Hyperbilirubinemia
Physiologic jaundice: Unconjugated hyperbilirubinemia caused by the neonatal liver’s inability to compensate fully for the rapid turnover of red blood cells during the transition from fetal hemoglobin to adult hemoglobin. Jaundice usually occurs after 24 to 48 hours and improves or resolves after 1 week. Kernicterus is a potential complication and occurs when bilirubin is deposited in the brain, leading to mental disability. It is a concern only if bilirubin levels are extremely high (> 20 mg/dL). If bilirubin levels are concerning, phototherapy can be performed; this converts bilirubin to a water-soluble form that can be excreted, without the need for conjugation.
Nonphysiologic jaundice: The most severe form of neonatal jaundice is hemolytic disease of the newborn. It occurs when a sensitized Rh-negative mother gives birth to an Rh-positive fetus. These neonates will have an unconjugated hyperbilirubinemia immediately after birth and will have a positive direct Coombs’ test. Traumatic injuries during birth may also result in hematoma formation and red blood cell breakdown, leading to jaundice. Neonatal jaundice may also indicate enzymatic deficiencies such as those in Gilbert, Crigler-Najjar, Rotor, or Dubin-Johnson syndrome.
Reye’s Syndrome
Thought to be secondary to aspirin use in children with viral illnesses, this condition leads to mitochondrial dysfunction. Although many systems are affected, liver failure and encephalopathy are the most distinguishing features. Therefore, aspirin is generally contraindicated in children.
Meckel’s Diverticulum
A true diverticulum (includes all layers of the GI gut wall) caused by a remnant of the vitelline duct (embryologic connection to yolk sac). Commonly remembered by the rule of 2. Found in 2% of the population, usually occurs within 2 feet of the ileocecal valve, 2 inches in length, containing two types of ectopic tissue (gastric and pancreatic), and with 2% of those affected becoming symptomatic, usually before 2 years of age. It often presents as a sudden onset of painless melena or hematochezia discovered in the diaper of an otherwise healthy and happy baby. Less commonly, the presence of gastric mucosa may cause intestinal ulcerations that mimic acute appendicitis. A technetium scan is used for diagnosis because it detects the ectopic gastric mucosa. Treatment is surgical removal.
PHARMACOLOGY
Antiemetics
Ondansetron
Serotonin 5-HT3 antagonist. Initially used for nausea in patients on chemotherapy but now is a ubiquitous antiemetic. This drug is generally well tolerated with few complications; however, QTc prolongation is a potential side effect.
Metoclopramide
Antiemetic property is from its effects as a dopamine antagonist, but its muscarinic activity increases upper GI motility. This is why it is also used as a prokinetic agent in conditions such as diabetic gastroparesis. Metoclopramide has an overlapping mechanism of action with antipsychotics, so extrapyramidal side effects (e.g., dystonia, parkinsonism, irreversible tardive dyskinesia) may occur. Metoclopramide should be avoided in patients with suspected small bowel obstruction (SBO) because the prokinetic activity may actually worsen the symptoms or precipitate perforation.
Antacids
Proton Pump Inhibitors
Directly inhibit H+-K+-ATPase on gastric parietal cells, thereby lowering gastric H+ secretion and raising pH (Fig. 10-30). Medications end in the suffix “-prazole” (e.g., omeprazole, pantoprazole). Note that a higher gastric pH will cause increased gastrin release via a feedback loop.
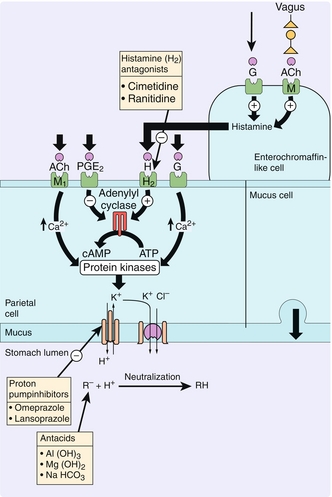
H2 Blockers
Antagonize histamine-2 (H2) receptors on parietal cells, causing decreased cAMP activation of H+,K+-ATPase (see Fig. 10-30). Medications end in the suffix “-tidine” (e.g., cimetidine, ranitidine, famotidine). Of note, cimetidine inhibits multiple cytochrome P-450 enzymes, leading to elevated levels of interacting medications. Other noninteracting H2 blockers are generally preferred.
Cytoprotective Agents
Bismuth Sucralfate
Bind the ulcer base, protecting it against the acidic environment and digestive enzymes.
Misoprostol
Prostaglandin E1 analogue, which promotes gastric secretion of mucus, bicarbonate, and gastrin. Used to prevent NSAID-induced ulcers. Because misoprostol is a prostaglandin E1 analogue, it is also used as a cervical ripening agent for induction of labor. It can also be used to maintain patent ductus arteriosus in congenital heart anomalies by the same mechanism of action.
Inflammatory Bowel Disease Management
Sulfasalazine
A precursor to the NSAID 5-aminosalicylic acid (5-ASA). This compound is used in the treatment of inflammatory bowel disease (IBD) because it is not converted to its active form until reaching the ileum. Its adverse effects on the gastric mucosa are thus avoided. Furthermore, it is poorly absorbed in the small bowel so it can reach sufficiently high concentrations to be locally anti-inflammatory, without causing systemic side effects.
Others
Glucocorticoids may be needed to manage IBD. If still unresponsive, immunomodulators such as 6-mercaptopurine, azathioprine, and methotrexate should be considered (see Chapter 11). Anti–tumor necrosis factor (TNF) biologic therapy such as adalimumab and infliximab is generally reserved for refractory cases (see Chapter 12).
Other Agents
Statins
HMG-CoA reductase inhibitors, which interfere with hepatic cholesterol synthesis by inhibiting the transformation of HMG-CoA into mevalonic acid, the rate-limiting step in cholesterol synthesis. To have sufficient cholesterol to produce bile acids, hepatocytes must upregulate cholesterol receptors. Increased LDL receptors lead to a decrease in serum LDL levels. Side effects are rare but include hepatotoxicity and myositis, so ALT, AST, and creatine kinase levels are monitored during treatment.
Cholestyramine
Bile acid binding resin that renders bile salts unabsorbable. Initially this drug was used to lower cholesterol by increasing the loss of bile salts, thereby increasing the hepatic conversion of cholesterol to bile salts. Currently cholestyramine is generally used for those with disease of the terminal ileum (e.g., Crohn’s disease). Bile salts that are not absorbed in the terminal ileum act as an irritant in the colon. Cholestyramine binds these irritants and prevents their cathartic effect. Side effects include GI upset and cholesterol gallstones. Because cholestyramine interferes with lipid absorption, it may interfere with the absorption of fat-soluble vitamins and medications.
Loperamide
Decreases GI motility via opioid receptor–induced cholinergic inhibition of the enteric nervous system. Like all opioids, loperamide is an agonist at the opioid receptor. It does not cross the blood-brain barrier, however, so it does not have analgesic properties.
Octreotide
Synthetic somatostatin used in the treatment of esophageal varices because of its effects as a splanchnic vasoconstrictor. Additionally, because of its inhibitory effects on the endocrine system, it is used in the treatment of VIPoma, gastrinoma, glucagonoma, carcinoid syndrome, and acromegaly.
Orlistat
Pancreatic lipase inhibitor that prevents the digestion of triglycerides into free fatty acids. It is used in the treatment of obesity. Because triglycerides are no longer digested or absorbed, they are excreted in the feces. This may cause the predictable side effect of steatorrhea when large quantities of fat are consumed.