Cardiology
HEART AND BLOOD VESSELS
Overview
The heart is responsible for pumping oxygen- and nutrient-rich blood to all the organs, as well as removing waste from them. If at any point in a person’s life the heart stops doing this for more than a few minutes, irreversible organ damage can result. The heart, blood, and blood vessels make up the cardiovascular system.
The heart has four chambers that blood flows through: right atrium, right ventricle, left atrium, and left ventricle. It is important to understand the path that the blood takes through these chambers of the heart to understand how problems with each of these chambers will lead to symptoms. Each time blood leaves a ventricle, blood is leaving the heart through an artery. Therefore, arteries, such as the aorta and pulmonary artery, take blood away from the heart (A-A). Veins, such as the superior and inferior vena cava, return blood back to the heart (T-V). Each time blood flows from an atrium to a ventricle or from a ventricle to an artery, blood passes through a valve, which if working correctly, ensures that blood flows only in the correct direction. Study the following steps of blood flow with the image of the circulatory system provided in Figure 8-1A.

Figure 8-1 A, Cardiovascular system schematic with blue representing deoxygenated blood, red representing oxygenated blood, and red-blue gradients representing capillary beds, which take up the oxygen. B, General cardiac anatomy. (A, from Drake RL, Vogl AW, Mitchell AWM. Gray’s Anatomy for Students. 2nd ed. Philadelphia: Elsevier; 2009. B, from Bogart BI, Ort VH. Elsevier's Integrated Anatomy and Embryology. Philadelphia: Elsevier; 2007.)
1. After the body’s tissues and organs have taken up the oxygen and delivered their metabolic waste to the bloodstream, deoxygenated blood returns toward the heart through the superior and inferior vena cava (veins) to the right atrium.
2. From the right atrium, the blood travels through the tricuspid valve into the right ventricle.
3. From the right ventricle, the blood travels through the pulmonic valve to the pulmonary artery, which travels away from the heart to the lungs to drop off all of the CO2 waste the tissues have made from metabolism and replenish the oxygen supply in the blood.
4. After gas exchange in the lung occurs, the oxygenated blood returns to the left atrium through the pulmonary veins.
5. From the left atrium, the blood travels to the left ventricle through the mitral valve (also known as the bicuspid valve). Always “tri” something before you “bi” it; the tricuspid valve is before the mitral (bicuspid) valve!
6. From the left ventricle, the blood travels to the aorta (artery) through the aortic valve to be distributed to the tissues, and the cycle repeats.
Anatomy of the Heart
The heart has three layers, from inside to out: the endocardium, myocardium, and epicardium.
 The endocardium is a single layer of endothelial cells that envelops the inside of the heart, including the valves, similar to the endothelial cells that line blood vessels. This is important in terms of talking about ischemia of the heart, which can either be transmural (across all layers of the heart including the endocardium) or subendocardial (just affecting the endocardium), as well as in infective endocarditis, which is an infection that takes hold at the endothelium covering the heart valves.
The endocardium is a single layer of endothelial cells that envelops the inside of the heart, including the valves, similar to the endothelial cells that line blood vessels. This is important in terms of talking about ischemia of the heart, which can either be transmural (across all layers of the heart including the endocardium) or subendocardial (just affecting the endocardium), as well as in infective endocarditis, which is an infection that takes hold at the endothelium covering the heart valves.
The myocardium (myo = muscle) is the thick, striated muscular layer of the heart that is responsible for the pumping activity of the heart. These muscles are perfused through coronary arteries, and in a myocardial infarction (heart attack) the death of parts of cardiac muscle can cause the heart to fail.
The epicardium is actually the visceral pericardium, which will be discussed next.
The heart is surrounded by a pericardium, a double-walled sac that has a serosal visceral (inner) layer and a fibrous parietal (outer) layer; between the visceral and parietal pericardium there exists a small amount of pericardial fluid, which allows the heart to beat with minimal friction, similar to how a car uses oil to decrease friction between moving parts. This becomes important clinically with pericarditis, which is inflammation of the pericardium, usually due to infection, uremia, or autoimmune disease. It is also important in pericardial effusion and tamponade, when too much fluid builds up between the two layers (effusion) and can even act to “strangle” the heart if enough fluid and pressure builds up such that the heart’s ability to fill with blood is impeded (tamponade).
The heart relies on an electrical conduction system to ensure rapid, coordinated contraction of the heart (Fig. 8-2A).
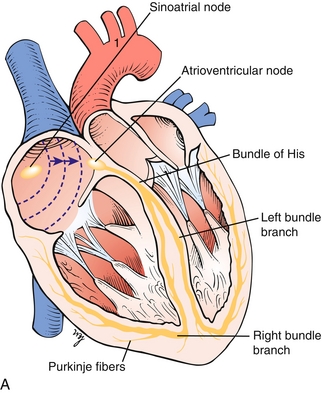

Figure 8-2 A, The cardiac conducting system, in which the signal is normally generated in the sinoatrial (SA) node and travels through the atria to the atrioventricular (AV) node; it then transmits to the bundle of His and subsequently to the left and right bundle branches. B, Image depicting the normal sequence of depolarization of the heart: from the SA node to the AV node, to the bundle of His, with initial septal depolarization, and then moving through the right ventricle via the right bundle branch and left ventricle via the left bundle branch. (A, from Moore NA, Roy WA. Rapid Review Gross and Developmental Anatomy. 3rd ed. Philadelphia: Elsevier; 2010. B, from Boron WF, Boulpaep EL. Medical Physiology. 2nd ed. Philadelphia: Elsevier; 2008.)
Sinoatrial (SA) node: The “pacemaker” of the heart, which sets the normal heart rate by generating electrical signals.
Atrioventricular (AV) node: Named because it lies between the atria and ventricles; in the normal heart this area is the only place where the electrical signal can be transmitted to the ventricles, through the bundle of His, which further branches into the left and right bundle branches. Malfunction of the AV node and bundle of His leads to various types of AV block (heart block), which are described later.
Left bundle branch: Responsible for coordinated contraction of the left ventricle. If these fibers are damaged and lose conducting ability, a left bundle branch block results. (Although too detailed for the purposes of Step 1, the left bundle further separates into the anterior, posterior, and septal branches, and blockage of these can lead to anterior fascicular block [anterior branch] or posterior fascicular block [posterior branch].)
Right bundle branch: Responsible for coordinated contraction of the right ventricle. Can be damaged and lead to a right bundle branch block, although a right bundle branch block can be normal in some individuals.
The coronary arteries are responsible for perfusion of the heart, and atherosclerosis and blockage of these vessels can lead to angina or myocardial infarction, respectively. The left and right coronary arteries come off the aorta at the aortic valve cusps (Fig. 8-3).
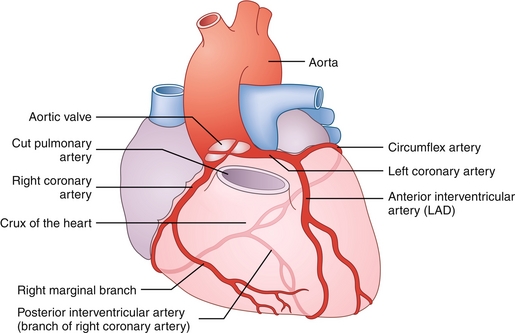
Figure 8-3 Coronary arterial vasculature, anterior view. The right and left coronary arteries can be seen coming off of the aortic valve cusps. The right coronary artery supplies the sinoatrial node and most often the posterior aspect of the heart; the left coronary artery divides into the left anterior descending (LAD) and the circumflex arteries. (From Bogart BI, Ort VH. Elsevier's Integrated Anatomy and Embryology. Philadelphia: Elsevier; 2007.)
Left coronary artery (LCA): Divides into the left anterior descending (LAD) artery, which supplies the anterior left ventricle, and the circumflex artery, which supplies the lateral and posterior left ventricle. The LAD is the most commonly occluded vessel in patients with myocardial infarction.
Right coronary artery (RCA): Supplies the right ventricle. In most people (85%), the posterior descending artery (PDA) comes off of the right coronary artery, which perfuses the inferior and posterior ventricles (in the other 15% of people, the PDA usually derives from the left circumflex). The right coronary artery also supplies the SA node and AV node, which can lead to arrhythmias and heart block if damaged.
Anatomy of the Circulatory System
The circulatory system can be divided into two components: the pulmonary circulation and the systemic circulation.
Pulmonary circulation: Made up of the right heart, pulmonary arteries, capillaries feeding the lungs, and pulmonary veins. Responsible for taking deoxygenated blood from the right heart to the lungs for oxygenation and then moving the newly oxygenated blood to the left heart so that it may be pumped into the systemic circulation.
Systemic circulation: Made up of the left heart, systemic arteries, capillaries, and veins. The systemic circulation is everything outside of the pulmonary circulation; the goal of this circulation is to take oxygenated blood to the body to deliver oxygen and nutrients and return that newly deoxygenated blood to the right heart.
The circulatory system uses arteries, arterioles, capillaries, venules, and veins—each serves a different purpose. Blood flows through these structures in the order written above (Fig. 8-4).
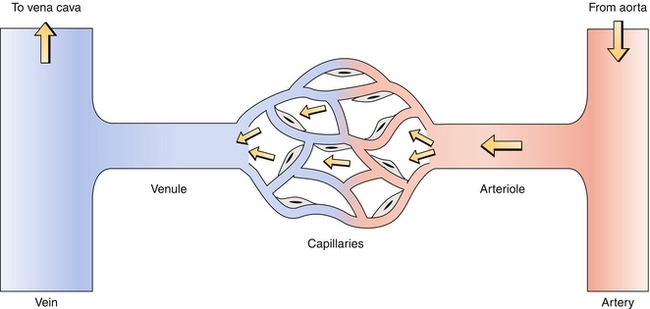
Figure 8-4 In the systemic circulation, oxygenated blood from the arterial system enters a capillary bed, oxygen is extracted by the tissues, and deoxygenated blood enters the venous system and returns to the heart. (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
1. Arteries are thick walled because they receive blood from ventricles and therefore must withstand large changes in pressure. Examples include the pulmonary artery and aorta. The arteries have three layers, starting at the inside layer called the intima (intima—inside), which has a single layer of endothelial cells; the media (media—middle), a thick elastic and muscular layer; and the adventitia, the outer layer which houses the vasa vasorum (“vessel of vessels,” blood vessels that nourish the artery itself), nerves, and lymphatics.
2. Arterioles are the site of highest resistance to blood flow and are regulated by α1 receptors for constriction and β2 receptors for dilation of their smooth muscle, helping the body regulate distribution of blood to various organs.
3. Capillaries have a single layer of endothelial cells, allowing for diffusion and exchange of gases, fluid, and nutrients between the blood and tissues. The amount of fluid exchange that occurs is dictated by Starling forces, which describe the net driving force for fluid to come into or out of capillaries. The Starling forces are determined by the hydrostatic pressure and the oncotic pressure of both the capillaries and the interstitium that surrounds them. The oncotic pressure is the osmotic pressure derived from proteins that cannot traverse the capillary–interstitium interface; the oncotic pressure of the capillaries (πc) is mainly determined by albumin, the main plasma protein. Low albumin (hypoalbuminemia), therefore, causes decreased capillary oncotic pressure and can promote fluid moving from capillary to interstitium as the osmotic drive keeping fluid in the capillary is lost. Hydrostatic pressure is simply the pressure exerted by the fluid.
 Pnet = (forces promoting fluid leaving capillary) – (forces promoting fluid returning to capillary)
Pnet = (forces promoting fluid leaving capillary) – (forces promoting fluid returning to capillary)
Pnet = [(Pc + πi) – (Pi + πc)]
Pc: hydrostatic pressure in capillaries—attempts to push fluid out of the capillary
πi: interstitial oncotic pressure—attempts to pull fluid out of the capillary
Pi: hydrostatic pressure in interstitium—attempts to push fluid back into the capillary
πc: capillary oncotic pressure—attempts to pull fluid out of the interstitium
4. Venules and veins are also thin walled because they are meant to be capacitance vessels, meaning that they can hold a large amount of blood. The veins, like the arteries, have three layers (intima, media, adventitia) but have a thin media because they are not muscular. Sympathetic tone can cause these veins to constrict, promoting venous return back to the heart to assist in increasing cardiac output. Veins have one-way valves that help ensure that blood can return to the heart even in the face of gravity.
PHYSIOLOGY
Cardiovascular Terminology and Formulas
Systole: The phase during which the heart contracts, pumping the blood in the ventricles into the arteries through the pulmonic valve (right ventricle) and aortic valve (left ventricle).
Diastole: The phase during which the heart relaxes and allows the ventricles to fill with blood from the atria through the tricuspid valve (right ventricle) or mitral valve (left ventricle). During exercise, the time spent in diastole is decreased to allow for a greater heart rate.
Mean arterial pressure (MAP): 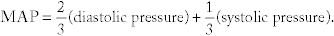 This is the average pressure in the artery. It is important to note that because the heart spends more time in diastole (2⁄3) than systole (1⁄3), the mean arterial pressure is not a simple average of the two pressures.
This is the average pressure in the artery. It is important to note that because the heart spends more time in diastole (2⁄3) than systole (1⁄3), the mean arterial pressure is not a simple average of the two pressures.
Stroke volume (SV): 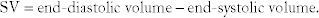 The absolute amount of blood ejected from the heart; how much blood the heart started with after filling (end-diastolic volume, EDV) subtracted by how much it ended up with after contraction (end-systolic volume, ESV).
The absolute amount of blood ejected from the heart; how much blood the heart started with after filling (end-diastolic volume, EDV) subtracted by how much it ended up with after contraction (end-systolic volume, ESV).
Ejection fraction (EF): 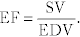 This describes what percentage of blood was ejected from the heart. A normal EF is ≥ 55%; in systolic heart failure, this number is significantly reduced.
This describes what percentage of blood was ejected from the heart. A normal EF is ≥ 55%; in systolic heart failure, this number is significantly reduced.
Cardiac output (CO): 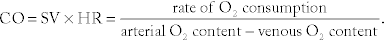 The CO is the volume of blood being pumped per minute; this can increase considerably during exercise in the healthy heart and is decreased in heart failure. Initially in exercise, the stroke volume increases to augment cardiac output; as exercise continues, the heart rate increases to meet the increased demand. Increasing either the stroke volume or the heart rate can increase cardiac output. The second equation is more tricky. Imagine with a low cardiac output, the blood is moving more slowly through the tissues, giving them more time to extract oxygen, leading to a large arterial-venous oxygen content difference (large denominator). Conversely, with a high cardiac output, so much oxygenated blood is being circulated through the tissues that the tissues do not need to extract all the oxygen from the blood each time it passes through.
The CO is the volume of blood being pumped per minute; this can increase considerably during exercise in the healthy heart and is decreased in heart failure. Initially in exercise, the stroke volume increases to augment cardiac output; as exercise continues, the heart rate increases to meet the increased demand. Increasing either the stroke volume or the heart rate can increase cardiac output. The second equation is more tricky. Imagine with a low cardiac output, the blood is moving more slowly through the tissues, giving them more time to extract oxygen, leading to a large arterial-venous oxygen content difference (large denominator). Conversely, with a high cardiac output, so much oxygenated blood is being circulated through the tissues that the tissues do not need to extract all the oxygen from the blood each time it passes through.
Total peripheral resistance (TPR): 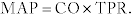 The total peripheral resistance is the resistance in the entire systemic circulation (not the pulmonary circulation). This is largely determined by the arterioles, which constrict owing to α1-receptor activation to increase TPR, and which dilate owing to β2-receptor activation to decrease TPR.
The total peripheral resistance is the resistance in the entire systemic circulation (not the pulmonary circulation). This is largely determined by the arterioles, which constrict owing to α1-receptor activation to increase TPR, and which dilate owing to β2-receptor activation to decrease TPR.
Poiseuille’s equation: 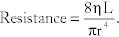 Where η is viscosity, L is length, and r is radius. This law describes how much resistance there is in a circuit; radius is to the 4th power and is therefore the largest determinant of resistance. Decreasing the radius of an arteriole by 2, for example, raises the resistance by 24, or 16 times. Increased viscosity can be seen if there are more red blood cells (polycythemia) or more protein (such as in multiple myeloma).
Where η is viscosity, L is length, and r is radius. This law describes how much resistance there is in a circuit; radius is to the 4th power and is therefore the largest determinant of resistance. Decreasing the radius of an arteriole by 2, for example, raises the resistance by 24, or 16 times. Increased viscosity can be seen if there are more red blood cells (polycythemia) or more protein (such as in multiple myeloma).
Preload: ventricular end-diastolic volume, based on how much blood has come back to the heart by venous return. Sympathetic tone increases venous return and preload; venodilators such as nitroglycerin decrease preload.
Afterload: mean arterial pressure, essentially how much pressure the heart has to work against to eject blood from the ventricles. Increased afterload, such as from hypertension, aortic stenosis, or vasoconstriction, causes the heart to work harder and consume more oxygen to eject blood.
ELECTROPHYSIOLOGY
The electrical conducting system described in the anatomy section is the basis for an organization of the electrical signal leading to a coordinated and effective contraction. This electrical activity is due to action potentials that normally originate in the SA node because it generates these potentials with the greatest frequency; the AV node and the bundle of His are the next quickest and will generate action potentials if the SA node is malfunctioning.
Pacemaker cells such as the SA node and AV node do not require outside stimuli to generate an action potential—they are self-depolarizing because of activity of the funny sodium channels (“funny” in that the channels are open before an action potential occurs, different from most sodium channels, which only open during an action potential). When the heart rate is slow, fewer of these channels will be open, and the sodium current If will be lower, such as after parasympathetic stimulation by the vagus nerve (through inhibitory M2 receptors); more channels will be open when the heart rate is fast (Fig. 8-5A).
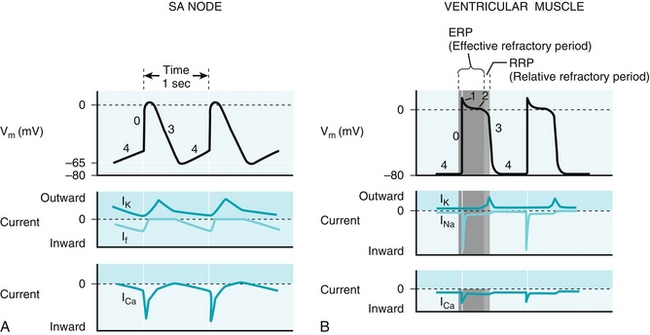
Figure 8-5 A, Electrophysiology of the sinoatrial (SA) node, where “funny” sodium channels are open before an action potential occurs; the sodium leak causes an action potential when the threshold potential is reached. This then triggers calcium (not sodium) influx to depolarize the cell. B, Electrophysiology of ventricular myocytes; these are, in the normal state, not self-depolarizing and must have a depolarizing stimulus to cause an action potential. Like most cells (but unlike the pacemaker cells), the main cation entering to initially depolarize the cell is sodium via sodium channels. The depolarization is sustained by calcium from L-type calcium channels, and potassium efflux repolarizes the cell. IK, ICa, INa, If, currents through potassium, calcium, sodium and “funny” sodium channels, respectively; Vm, membrane voltage. (From Boron WF, Boulpaep EL. Medical Physiology. 2nd ed. Philadelphia: Elsevier; 2008.)
Phase 0: Once the funny sodium channels have leaked enough sodium to reach the threshold potential, voltage-gated calcium channels open to cause further depolarization. The upstroke of phase 0 in the AV node is slower than in myocytes to allow the ventricles time to fill fully before the AV node transmits the signal to them.
Phase 3 (note that phases 1 and 2 are not present in pacemaker cells): The calcium channels close, and potassium channels open to repolarize the cell by causing potassium efflux.
Phase 4: The funny sodium channels open, slowly depolarizing the cell until the threshold potential is reached, when the process will restart.
The cardiac myocytes are responsible for the contraction of the heart, and normally depolarize when they have received the signal from the conducting system of the heart or nearby myocytes. The depolarization of myocytes is more complex, consisting of phases 0, 1, 2, 3, and 4 (Fig. 8-5B).
Phase 0: Once the threshold potential has been reached, sodium channels open and cause a quick and transient influx of sodium, rapidly depolarizing the cell. Note that in cardiac myocytes, the ion causing the upstroke of depolarization is sodium, unlike in pacemaker cells, which use calcium.
Phase 1: The sodium channels are now closed, and some potassium efflux occurs, leading to a small drop in voltage.
Phase 2: L-type calcium channels (L for long-lasting) open, leading to a plateau where potassium efflux is balanced with calcium influx. This “trigger calcium” triggers release of stored calcium from within the sarcoplasmic reticulum in the cardiac myocyte and will lead to contraction.
Phase 3: Calcium channels have inactivated, and potassium efflux dominates, leading to repolarization of the cell.
Phase 4: Resting state. Unlike the pacemaker cells such as the SA and AV node, these cells do not spontaneously depolarize under normal conditions and wait for another depolarizing signal before the process restarts.
Cardiac Myocytes
The details of muscle contraction in general are covered in Chapter 12, but important points regarding cardiac myocyte contraction will be covered here.
Troponin has three types: troponin T, troponin I, and troponin C. Troponin I inhibits contraction, and troponin C binds to calcium (I—inhibits, C—calcium). When calcium binds to troponin C, it stops troponin I’s inhibition and allows for actin and myosin to interact and for contraction to occur. Therefore, more calcium means more actin and myosin interaction and increased strength of contraction (increased contractility, inotropy).
Calcium influx during phase 2 of myocyte depolarization causes stored calcium to be released from the sarcoplasmic reticulum (SR), in much greater amounts. This is referred to as calcium-induced calcium release. After this calcium release, myocytes pump calcium either (a) out of the cell, or (b) into the SR. Anything that increases calcium influx from the outside or increases calcium storage in the SR will increase contractility by providing more intracellular calcium to the myocytes. Digitalis prevents calcium efflux from the cell, causing increased retrieval into the SR and increased inotropy. Sympathetic activation of β1 receptors causes increased sarcoplasmic reticulum Ca2 + ATPase activity (SERCA, the pump that pumps calcium back into the SR), leading to increased calcium stored in the sarcoplasmic reticulum and therefore increased contractility. Both of these will be covered in-depth later in the section “Pharmacology,” but it is important to keep the concept in mind that more calcium availability leads to increased contractility.
The actin (thin filaments) and myosin (thick filaments) overlap more favorably when stretched; therefore, more blood returning to the heart (increased preload) will fill the heart more, causing the myocytes to stretch and in turn creating a stronger contraction. However, do not confuse this with contractility, which is ability to contract at a given preload and is dependent on intracellular calcium levels. More preload will lead to a stronger contraction, but not increased contractility. This concept will be further explained in the next section.
Frank-Starling Curve
The Frank-Starling curve (Fig. 8-6) shows that the force of contraction of the myocytes is dependent on the initial length of cardiac muscle fiber, the degree of stretch, which is determined by the preload.
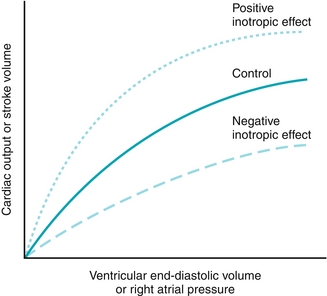
Figure 8-6 Frank-Starling curve. The solid green curve represents the “control” curve, which describes the strength of contraction of the myocytes for a given preload (ventricular end-diastolic volume). When there is a change in contractility (inotropy), which is defined as strength of contraction for a given preload, a new curve occurs. (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
More venous return = more preload → larger ventricular end-diastolic volume → more muscle fiber stretch → stronger contraction.
Ensures that if more blood returns to the heart, then the heart can pump that extra blood by increasing stroke volume.
Contractility is the force of contraction at a given preload, and therefore increasing preload does nothing to contractility. Increased contractility would be a whole new curve (such as the positive inotropic effect curve shown in Figure 8-6), not moving along the existing curve (with increased preload without change in contractility, it would move to the right on the same curve). Contractility increases with positive inotropes (β1 stimulation, digitalis) and decreases with negative inotropes (β blockade, heart failure, acid-base disturbances).
Cardiac Cycle
The cardiac cycle is described by one round of diastole and systole. During this time, the ventricle will fill (diastole) from the preload offered to it by the atrium by flowing through one of the atrioventricular (AV) valves: the tricuspid valve for the right heart, the mitral (bicuspid) valve for the left heart (the left atrial pressure is shown by the black line at the bottom of Fig. 8-7A). The atrium will contract near the end of filling to help push more blood into the ventricle, called atrial kick. After filling, the AV valve will close, and the ventricle will contract and eject the blood out of the pulmonic (right heart) or aortic (left heart) valve into the pulmonary artery or aorta, respectively. A closer look at the cardiac cycle and the heart sounds will follow.
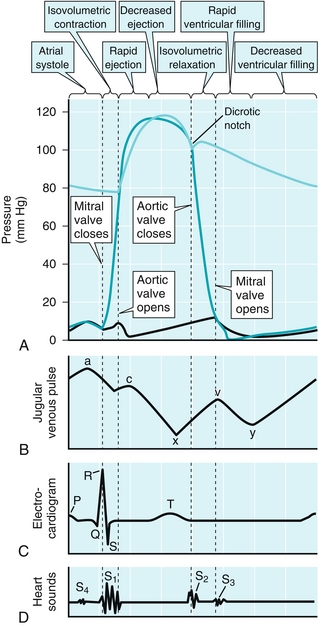
Figure 8-7 Cardiac cycle. A, Pressure: showing left-heart tracing only, where the dark green represents ventricular pressures, light green represents aortic pressure, and black line represents left atrial pressure (which is estimated by the pulmonary capillary wedge pressure). B, Jugular venous pulse: shows pressures in the jugular vein, which feeds into the superior vena cava and into the right atrium. The “a” wave rise in pressure is due to atrial kick; the “c” wave rise is due to the contraction of the ventricle; the following “x” descent represents blood refilling the relaxes and empty atria; the “v” wave represents venous filling (mnemonic: “villing” from the veins); the “y” descent represents ventricular filling once the tricuspid valve opens. C, Electrocardiogram: shows ECG findings of the cardiac cycle. “P” wave: atrial depolarization. “QRS” complex: ventricular depolarization. “T” wave: ventricular repolarization. D, Heart sounds: normal sounds include S1: closure of tricuspid/mitral valves, and S2: closure of aortic/pulmonic valves. (From Boron WF, Boulpaep EL. Medical Physiology. 2nd ed. Philadelphia: Elsevier; 2008.)
The aortic pressure is shown in the figure by the light green line on top of Figure 8-7A. When the left ventricular pressure (shown by the darker green line on Fig. 8-7A) is higher than the aortic pressure, the aortic valve is pushed open by the pressure gradient; this valve remains open until the left ventricle has ejected the stroke volume for that beat. A similar waveform (but with lower pressures because the pulmonary circulation has lower pressure than the systemic circulation) would be present for the right heart.
The venous pressure is depicted by the solid black line on Figure 8-7B and will be described for the right heart; this is usually shown as “jugular venous pressure,” which is an estimation of right atrial pressure. A similar waveform occurs for the left side of the heart as well. The normal venous waveform has a, c, and v waves as well as x and y descents. The a wave represents the atrial kick (atrial contraction) for the right atrium, which helps fill the right ventricle, but also transmits some pressure back to the jugular veins. The c wave is the next upward deflection in pressure, corresponding to contraction of the right ventricle, causing the tricuspid to bow inward toward the right atrium, transiently increasing pressure. The right atrium, having finished contracting, is now relaxing and filling, causing the x descent during relaxation (as blood moves from the venous system into the empty atrium). The v wave occurs during venous filling (v for “villing”). At first, it can be seen that atrial pressure decreases with relaxation (x descent) but subsequently rises as the atrium becomes full. Now the filled right atrium can move the blood into the right ventricle. During ventricular diastole, the tricuspid valve opens, and the blood in the atrium will empty into the right ventricle, leading to the y descent.
Atrial fibrillation leads to loss of the a wave because there is no coordinated atrial contraction. Tricuspid stenosis will lead to a prominent a wave because the atrium will be contracting against a valve that cannot open sufficiently, and the atrial kick will cause an increase in venous pressure because the blood cannot go into the ventricles.
AV regurgitation (mitral or tricuspid) will show a c-v wave where there is a large upward deflection in pressure between the c and v waves. This is because during ventricular contraction, some of the blood will move back into the atrium, causing a large rise in pressure.
A stenotic aortic valve would manifest by larger ventricular pressures than aortic pressures because the aortic valve, which cannot open fully, cannot receive all the blood that the ventricle is attempting to pump into it owing to the increased resistance through the valve opening. (In Fig. 8-7A, this would manifest as the dark green line, which represents ventricular pressures, being significantly higher than the light green line, which represents aortic pressures.)
Figure 8-8 demonstrates a pressure-volume loop, which occurs during each cardiac cycle. The loop shown represents the left ventricle only; follow each step described in Figure 8-8 (dashed lines show how the pressure-volume loop would change with changes in physiology):
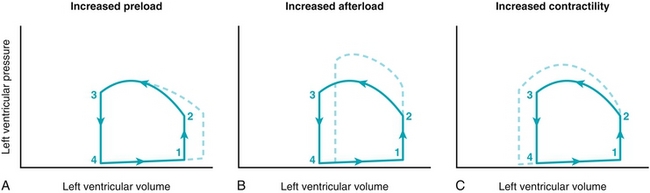
Figure 8-8 A pressure-volume loop that demonstrates how left ventricular volume (x-axis) and left ventricular (y-axis) pressure are related in conditions of increased preload (A), increased afterload (B), such as in aortic stenosis or hypertension, and increased contractility (C), such as in exercise. (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
Isovolumetric contraction (1 to 2): At the end of diastole (point 1), the ventricle is filled (end-diastolic volume), and the mitral valve closes (the aortic valve is already closed). The ventricle begins to contract against the closed mitral and aortic valves, increasing pressure but not changing volume (isovolumetric) because both valves are closed. When the ventricular pressure exceeds aortic pressure (point 2), the aortic valve will open.
Ventricular ejection (2 to 3): The aortic valve opens (point 2), and blood leaves the ventricle into the aorta. Once the blood is ejected, the aortic valve will close (point 3). The blood remaining is the end-systolic volume. Recall from earlier that the stroke volume is simply the end-diastolic volume (filled ventricle) minus the end-systolic volume (emptied ventricle); therefore, this curve also shows the stroke volume. With increased afterload the ventricle has to generate higher pressures to open the aortic valve (it is working harder, pushing against a higher pressure), causing a decreased stroke volume and increased myocardial oxygen demand (Fig. 8-8B). With increased contractility, the heart will generate a larger stroke volume; thus, even with the same end-diastolic (filling) volume, there will be a smaller end-systolic volume.
Isovolumetric relaxation (3 to 4): The aortic valve is closed (point 3), and the mitral valve is still closed, and the ventricle now relaxes. Because there are no valves open, the volume inside the ventricle cannot change (isovolumetric).
Ventricular filling (4 to 1): Once the pressure in the ventricle is lower than that of the left atrium (point 4), the mitral valve will open and fill the ventricle. The amount the ventricle fills (end-diastolic volume) is dependent on the preload (amount of blood in the left atrium waiting to fill the ventricle), the compliance of the ventricle (whether or not it is “stiff,” e.g., from hypertrophy), and the presence of the atrial kick. Figure 8-8A depicts a higher preload, leading to a higher end-diastolic volume.
Heart Sounds and Murmurs
Heart sounds are generated by the closing of valves. Understanding when valves normally open and close during the cardiac cycle will make it easy to understand how abnormal valve or heart function will cause abnormal heart sounds. Refer to the figure when various valves are mentioned to understand where they are heard best. A mnemonic commonly used to remember where to auscultate for each valve, is APT M (apartment M) for aortic, pulmonic, tricuspid, mitral (Fig. 8-9A).
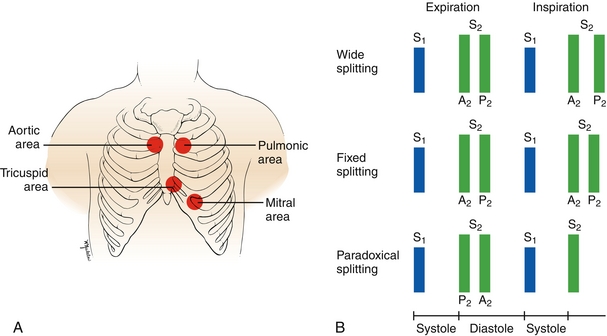
Figure 8-9 A, Areas on the chest to auscultate to best hear sounds generated by each valve. B, S1 sound (generated by mitral and tricuspid valve closure) and S2 sound (generated by aortic and pulmonic valve closure) in pathology. The S2 sound can be normally split during inspiration only, but pathologic splitting such as wide, fixed, or paradoxical splitting can occur. (A, from Swartz M. Textbook of Physical Diagnosis: History and Examination. 5th ed. Philadelphia: Saunders; 2006. B, from Seidel HM, Ball JW, Dains JE, et al. Mosby’s Physical Examination Handbook. 6th ed. Philadelphia: Elsevier; 2006.)
S1
This is the sound made normally by the closure of the tricuspid and mitral valves. This sound marks the end of diastole because filling of the ventricles is complete. These two events occur nearly simultaneously, so there is never any splitting of this sound.
S2
This is the sound made normally by closure of the pulmonic and aortic valves, referred to as P2 and A2, respectively. This sound marks the end of systole because ejection of blood from the ventricle has now been completed. During inspiration, intrathoracic pressure becomes more negative, “sucking” more blood back into the right atrium from the vena cava, increasing venous return. This increases right ventricular preload, leading to a larger right ventricular stroke volume, prolonging the duration in which the pulmonic valve is open (needs more time to eject all that extra blood). This moves P2 after A2 and causes the S2 sound to be split, termed physiologic splitting (a completely normal entity). On expiration, this does not occur, and P2 and A2 occur at the same time, causing the S2 sound to not be split. There are some examples of when splitting can be pathologic (Fig. 8-9B):
Fixed splitting: Described when splitting occurs on both inspiration and expiration. This occurs with an atrial septal defect (ASD) because there is a hole between the two atria. Because there is always a left-to-right shunt present (because left atrial pressures are higher than right atrial pressures), the right ventricle always has increased preload, and the pulmonic valve will always close after the aortic valve.
Wide splitting: Described when the P2–A2 split is longer than usual, seen with anything that causes a delay in right ventricular emptying. With pulmonic stenosis, the valve is narrowed, and therefore it takes the blood longer to fully eject, leading to a delay in valve closure. In right bundle branch block, the fast His-Purkinje system to the right ventricle is blocked, and right ventricular contraction is delayed, causing the pulmonic valve to close after the aortic valve because the left ventricle will contract and finish ejecting blood first.
Paradoxical splitting: Described when the aortic valve paradoxically closes after the pulmonic valve, seen with anything that causes a delay in left ventricular emptying. This causes the split to paradoxically occur during expiration rather than inspiration. This is because the pulmonic valve is closing earlier than the aortic valve owing to pathology, but during inspiration the increased volume moving through the pulmonic valve allows the valves to open for the same amount of time. As you may have guessed, aortic stenosis and left bundle branch block can cause this, for the same reason described with wide splitting, just now affecting the left ventricle.
Normally, there are no other sounds other than S1 and S2 during the cardiac cycle. However, there can be extra sounds, called S3 and S4 sounds (also called “gallops”). See Figure 8-7D for the location of these heart sounds in relation to the S1 and S2 sounds.
S3
Although this can be a normal finding in children, it is always abnormal in adults and usually signifies volume overload. When the tricuspid valve and mitral valve open during diastole, the extra volume rushes into the ventricle, tensing the chordae tendineae (tendons that tether the valve to the heart) of the affected valve (depending on which side of the heart is overloaded with volume), causing the extra sound. This sound is heard during the rapid ventricular filling phase in early diastole and is therefore positioned after the S2 sound.
S4
At the very end of diastole, the atria contract, called the “atrial kick,” to try to squeeze in the last bit of blood before the mitral and tricuspid valves close (S1 sound). If the ventricle is stiff and noncompliant (such as from hypertrophy from hypertension), there will be no room for the extra blood, and the S4 sound will be generated. Because the atrial kick occurs just before the end of diastole and closure of the AV valves, this sound is heard just before the S1 sound.
Blood flow through the heart is normally laminar and silent. When there is turbulent blood flow through the heart, a murmur is heard (turbulent flow through a narrowed artery is instead called a bruit). Understanding the cardiac cycle and when valves should be opened or closed makes understanding murmurs easy. Stenosis is a problem with opening a valve because it has been narrowed, whereas regurgitation (or insufficiency) is a problem with keeping a valve closed.
During systole, the aortic and pulmonic valves should open to allow for ejection of blood from the ventricles, and therefore the murmur of aortic or pulmonic stenosis will be heard during systole. Because the tricuspid and mitral valves should be closed, the murmur of tricuspid or mitral regurgitation will also be heard during systole (Fig. 8-10).
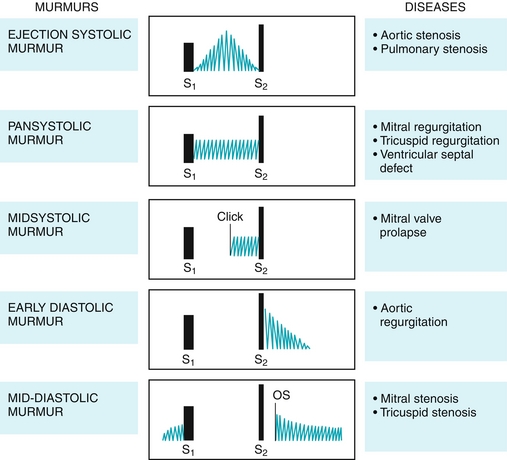
Figure 8-10 Heart murmurs, their location during the cardiac cycle, and their characteristics. OS: opening snap, seen in stenosis. (From Damjanov I. Pathophysiology. Philadelphia: Elsevier; 2008.)
Aortic or pulmonic stenosis (Fig. 8-11): The murmur is described as “crescendo–decrescendo” or “diamond shaped” because as the contraction of the heart progresses, the pressure builds up and drops again, leading to a murmur that increases and then decreases in intensity. Individuals with aortic stenosis will often complain of chest pain because of increased myocardial oxygen demand from the increased afterload. They may also experience exertional syncope because the demands for increased cardiac output during exercise cannot be met with the high resistance through the aortic valve and will experience syncope owing to cerebral hypoperfusion. Classically, on physical exam these patients will have pulsus parvus et tardus, which is Latin for a weak (parvus) and late (tardus) pulse. Predisposing factors to aortic stenosis include congenital bicuspid aortic valve (two cusps instead of three) and rheumatic heart disease causing damage to the aortic valve.

Figure 8-11 A, Aortic stenosis. B, Pulmonic stenosis. C, Mitral/tricuspid regurgitation. D, Mitral valve prolapse. (A and B, from Lissauer T, Clayden G. Illustrated Textbook of Paediatrics. 4th ed. Edinburgh: Elsevier; 2011.)
Mitral or tricuspid regurgitation: The murmur is described as “holosystolic” or “pansystolic” because it occurs at the same intensity for the duration of systole. This is because the left and right atria have low pressure and can accept blood even at low pressures if the valve is incompetent.
Mitral valve prolapse: The most frequent valvular lesion, where an abnormally thickened valve “prolapses” into the left atrium during systole. The sudden tensing of the chordae tendineae that stops the movement of the valve causes a “click” to be heard, and subsequently, a murmur is present. This is most often a benign lesion.
Ventricular septal defect: If there is a passage between the left and right ventricles, during systole the left ventricle (which has higher pressures) will eject blood into the right ventricle, leading to a left-to-right shunt. This leads to a holosystolic murmur, similar to that of mitral regurgitation. It is classically described as “harsh” sounding, whereas regurgitant murmurs are “high pitched.”
During diastole, the tricuspid and mitral valves should be open to allow for ventricular filling through the atria, and therefore murmurs of tricuspid or mitral stenosis will be heard during diastole. Conversely, the aortic and pulmonic valves should be closed to prevent backward flow of blood from the arteries into the ventricles, and therefore the murmurs of aortic or pulmonic regurgitation will be heard during diastole. In contrast to systolic murmurs, which can be benign, diastolic murmurs are always pathologic (see Fig. 8-10).
Mitral or tricuspid stenosis: The murmur is described as beginning with an opening “snap” when the stenotic valve finally opens, followed by a turbulent rumbling murmur. Most commonly this is caused by rheumatic fever, which most often affects the mitral valve.
Aortic or pulmonic regurgitation: The murmur is described as early decrescendo because after the ventricles eject the blood, the valves should shut—failure to do so causes blood to crash back into the ventricle during diastole, leading to the murmur. Because in aortic regurgitation, the blood crashes back into the left ventricle instead of staying in the systemic circulation, there is a wide pulse pressure from decreased diastolic blood pressure. This drop in diastolic blood pressure leads to many clinical signs, including a large-volume pulse that collapses in diastole (termed a Corrigan water-hammer pulse); de Musset sign, in which the head bobs with the heartbeat; and Quincke sign, in which the capillary bed in the nail can be seen to be pulsating from the wide pulse pressure.
Lastly, the murmur of a patent ductus arteriosus (PDA) is a continuous murmur (present in systole and diastole) and is described as “machine like” (Fig. 8-12). See Chapter 4 for a detailed description, but briefly, the ductus arteriosus is normally open when the fetus is in utero to act as a bypass tract from the pulmonary artery to the aorta because the lungs did not need significant blood flow (the fetus can’t breathe in utero). After birth, the increased oxygen tension in the lungs (because the baby now needs to breathe) causes constriction of the ductus arteriosus and eventually fibrosis, becoming the ligamentum arteriosum. Although a postnatal PDA is not normal, it may be beneficial to the baby if there are other congenital heart disease lesions present—vasodilating prostaglandins keep this open, and therefore medications that block prostaglandin synthesis (such as indomethacin) will promote closure, whereas giving prostaglandin E analogues such as misoprostol will help keep it open.
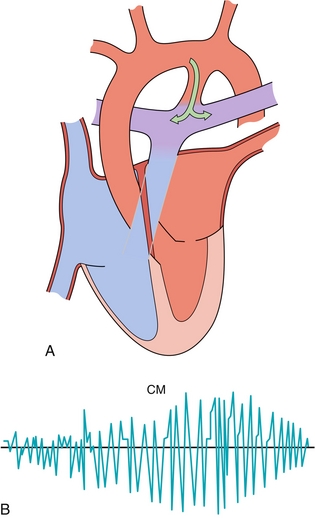
Figure 8-12 A, Patent ductus arteriosus, connecting the aorta to the pulmonary artery, resulting in a left-to-right shunt (oxygenated blood being put back into the pulmonary circulation). B, Continuous machine-like murmur (CM) heard in patients with a patent ductus arteriosus. (A, from Stevens A, Lowe J, Scott I. Core Pathology. 3rd ed. Philadelphia: Elsevier; 2008.)
Murmur Summary
Although it is good to know about all the murmurs, left-sided lesions are much more likely to be tested on Step 1 (or relevant clinically). Furthermore, right-sided murmurs will usually have a question stem that hints at intravenous drug use (tricuspid regurgitation). Mitral stenosis will often have a question stem that hints at rheumatic fever. Aortic regurgitation will usually be your diastolic murmur without other clues. The main goal will then be distinguishing the two left-sided systolic murmurs: aortic stenosis (crescendo–decrescendo, radiates to carotids) and mitral regurgitation (holosystolic, radiates to axilla).
Pressure in the Cardiovascular System
Pressures in the heart can be measured with a pulmonary artery (Swan-Ganz) catheter (Fig. 8-13A) and can provide information about the clinical status of a patient.

Figure 8-13 A, A pulmonary artery (Swan-Ganz) catheter to assess pressures in the cardiovascular system. The catheter is introduced into the patient and can measure right atrial pressure, move into the right ventricle to measure right ventricular pressure, move into the pulmonary artery to measure pulmonary arterial pressure, and wedge into the distal pulmonary arteries to get a pulmonary capillary wedge pressure (PCWP), which approximates the left ventricular pressure. B, Schematic showing pressure outputs as the pulmonary artery catheter moves through the right atrium, right ventricle, and pulmonary artery, and finally with the balloon inflated in a distal pulmonary artery. (From Colledge NR, Walker BR, Ralston SH. Davidson’s Principles and Practice of Medicine. 21st ed. Philadelphia: Elsevier; 2010.)
Right atrial pressure is equal to central venous pressure because the central veins (the superior and inferior vena cava) feed the right atrium. This is a low-pressure area because the veins are low-pressure vessels. Right atrial pressure will be increased when there is too much volume in the veins or difficulty feeding that volume to the heart (such as in right ventricular failure, when the ventricle cannot pump the blood given to it).
Right (RV) ventricular pressure is normally lower than left ventricular pressure because the pulmonary circulation is normally a lower pressure circuit than the systemic circulation. RV pressure can increase if it is pushing against a higher pressure (afterload), such as in pulmonic stenosis or pulmonary hypertension. This lower pressure in the right heart compared with the left is the reason that if there is a shunt, such as an atrial or ventricular septal defect, the shunt initially moves from left heart to right heart (high pressure to low pressure).
The pulmonary artery pressure can be increased in pulmonary hypertension (from lung disease or pulmonary vessel disease such as pulmonary embolism) or from left heart failure (because the fluid will back up into the lungs if the left ventricle cannot pump it out).
The pulmonary capillary wedge pressure (PCWP) is our way of measuring left atrial pressure because the catheter cannot fit through the small capillary beds of the lungs. This can be increased if the left atrial pressure is high, such as in left-sided heart failure, but also in problems with the mitral valve: stenosis because the left atrium cannot empty, regurgitation because blood will be forced back into the atrium from the ventricle.
In cardiac tamponade (covered in depth later), the heart is being essentially strangled by fluid that is trapped in the pericardial sac. This leads to equalization of pressure across all four chambers—the high pressure exerted on the heart is from all sides because the pericardium surrounds the heart. This is rapidly fatal if not corrected.
ELECTROCARDIOGRAPHY
An electrocardiogram (Fig. 8-14; ECG, or often referred to as EKG from the German spelling) is a useful tool to assess the electrical activity of the heart. When the cardiac myocytes go from polarized (resting state) to depolarized from an action potential, an electrical current is generated that can be measured. The USMLE Step 1 will only test ECG basics, but it is worth your while to invest time in reading an ECG book to gain a deeper understanding—after you’re done crushing Step 1.
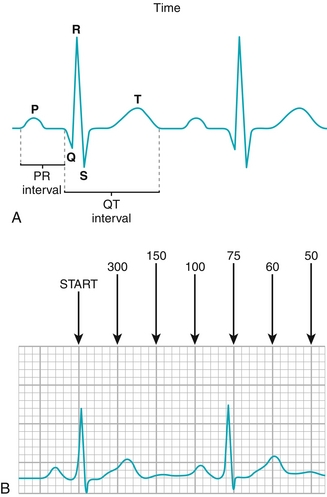
Figure 8-14 A, ECG tracing from a single heartbeat with the output labeled. P wave: atrial depolarization; PR interval: time between atrial depolarization and ventricular depolarization; QRS complex: ventricular depolarization; ST segment: time after ventricular depolarization but before repolarization; T wave: ventricular repolarization. Note atrial repolarization is never found on the ECG tracing because it occurs during the QRS complex (ventricular depolarization). B, The 300-150-100-75 method of counting heart rate. Count the number of large boxes (denoted by the thicker lines on the tracing, comprising five smaller boxes) that occur from the R wave (peak of the QRS complex). If the amount of large boxes separating the two R waves is 1, the heart rate is 300; 2, the heart rate is 150, and so on. Each large box represents 0.2 second (200 msec) and each small box represents 0.04 second (40 msec). (A, from Pazdernik T, Kerecsen L. Rapid Review Pharmacology. 3rd ed. Philadelphia: Elsevier; 2010.)
P wave: Represents atrial depolarization. After the P wave, there is a PR segment—this is the conduction delay at the AV node, which allows the ventricles time to fill fully before contraction (normally 0.12 to 0.20 second). Therefore, diseases of the AV node often manifest as PR-segment prolongation or abnormalities.
QRS complex: Represents ventricular depolarization. The terminology of the QRS complex can be difficult, but basically: if the first deflection is downard, it is a Q wave; if the first deflection is upward, a Q wave is not present. The first upward reflection is always an R wave. Any downward deflection after the R wave is known as an S wave. The atria repolarize during the QRS complex but do not make a tracing because the ventricles have many more myocytes and dominate the ECG. Normally the QRS duration is less than 0.12 second, but if the fast His-Purkinje conduction system is malfunctioning, then the QRS complex becomes abnormally widened, such as in bundle branch blocks.
T wave: After the ventricles depolarize during the QRS complex, there is a brief delay (ST segment) followed by repolarization, which is the T wave. There is sometimes a U wave that follows the T wave, which can be normal, but is not a common finding. It may also be seen in conditions such as hypokalemia or bradycardia.
An ECG printout has two main components: the 12-lead ECG, which looks at the heart from different vectors or angles, and the rhythm strip, which is useful for noting abnormalities in rhythm (arrhythmias) or rate. Basic rhythm strip abnormalities will be presented and explained. It is important to be able to assess the heart rate—each “little box” on the ECG strip is 0.04 second (40 msec), and each “big box” is made up of five “little boxes” and is therefore 0.20 second (200 msec). To assess the heart rate, count the number of “big boxes” between QRS complexes—and then remember 300-150-100-75-60-50. If there is one big box between beats, the rate is 300; two is 150, and so forth (see Fig. 8-13B).
Normal Sinus Rhythm
This is a normal ECG (Fig. 8-15). To be classified as sinus, a P wave (atrial contraction) must precede every QRS complex, denoting that the atria are conducting the impulse that was generated by the SA node through the AV node. In addition, it is “normal” because there is neither bradycardia nor tachycardia present: the heart rate is between 60 and 100. In this ECG, using the big box counting method, the heart rate is 75.

Sinus Tachycardia
This ECG is sinus because there are P waves before each QRS complex, but using the big box counting method, we see that the heart rate is 150 (Fig. 8-16). Therefore, it is sinus tachycardia.

Atrial Flutter
In atrial flutter (Fig. 8-17), there is an abnormal reentry of electrical activity around the tricuspid valve annulus, causing a self-sustaining loop of activity. This usually occurs at an atrial rate of 300, which is too fast for the AV node to conduct to the ventricles. Most commonly, the impulses have a 2:1 conduction block, meaning the ventricular rate is about 150 if the atrial rate is 300 because only half of the impulses are conducted.

Figure 8-17 Atrial flutter. (From Goldberger AL, Goldberger ZD, Shvilkin A. Clinical Electrocardiography: A Simplified Approach. 8th ed. Philadelphia: Saunders; 2012.)
Patients with atrial flutter most commonly have some form of structural heart disease. The flutter waves in the image are denoted with an F and often have a “saw tooth” appearance, and each wave should be nearly identical.
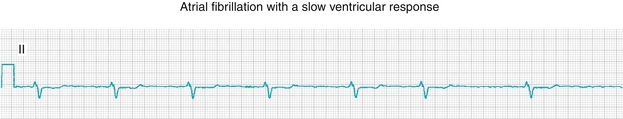
Atrial Fibrillation
In atrial fibrillation (Fig. 8-18), there is random activation of the atria that is unpredictable and chaotic, leading to no discernible P waves because there is no coordinated depolarization; the impulses that are transmitted through the AV node to the ventricles occur without a pattern, leading to an irregularly irregular rhythm. This is in contrast to a regularly irregular rhythm in which the irregularity is predictable and occurs at regular intervals (e.g., an irregular beat every fifth beat, which can occur with atrioventricular blocks). This is the most common chronic arrhythmia and can be dangerous because without a coordinated atrial contraction, stasis of blood can occur with resulting clot formation, potentially leading to strokes if the thrombi occur in the left atrium, with embolization of clots to the brain. Therefore, these patients require anticoagulation with either aspirin or warfarin, depending on their risk factors.
Ventricular Tachycardia
In ventricular tachycardia (Fig. 8-19), there is an abnormality in the patient’s ventricles, often an old scar from a myocardial infarction, that causes abnormal conduction and a reentry circuit in the ventricle leading to rhythmic depolarization of the ventricles. In the ECG tracing in Figure 8-19, the patient’s rhythm changes from sinus rhythm to ventricular tachycardia. Treatment is to “reset” the heart by defibrillation, which depolarizes the entire heart at once to reset any abnormal circuits.

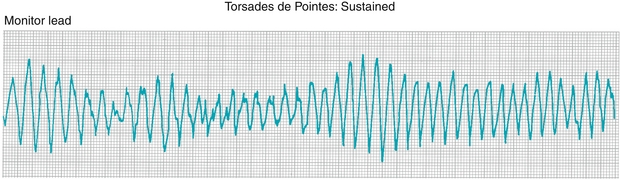
Torsades De Pointes
French for “twisting of the points,” torsades de pointes (Fig. 8-20) is a specific type of polymorphic ventricular tachycardia (in contrast to the previous example of ventricular tachycardia, where each beat looked the same and was therefore monomorphic). This most often occurs when a ventricular depolarization is triggered (a QRS complex) while the heart is attempting to repolarize (during the T wave). Therefore, the inciting event is often termed an “R-on-T phenomenon.” Anything that prolongs the QT interval makes this more likely to occur—such as congenital long QT syndromes, hypocalcemia, hypomagnesemia, hypokalemia, or medications that prolong the QT interval. In fact, treatment of torsades de pointes includes rapid intravenous infusion of magnesium.
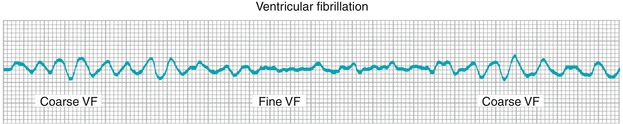
Ventricular Fibrillation
This disordered and rapid depolarization of the ventricles (Fig. 8-21) causes cardiac output to drop to essentially zero—making this rhythm life threatening and rapidly fatal if untreated. Cardiopulmonary resuscitation (CPR) and defibrillation in a rapid manner are life saving.
Wolff-Parkinson-White (WPW) Syndrome
Also known as ventricular preexcitation syndrome, WPW syndrome (Fig. 8-22) is caused by an accessory pathway (known as the bundle of Kent) between the atria and ventricles. Normally, the AV node is the only passageway for electrical conduction—these patients have an extra pathway. This extra pathway causes the ventricles to begin depolarizing before they normally would, leading to a premature upsloping in the QRS complex known as a delta wave. Because now there are two routes to transmit impulses through the ventricle, it is possible to have a reentry circuit in which an electrical impulse can travel in a circuitous path in one of two directions: (1) orthodromic, meaning that the signal travels antegrade down the AV node (as normal), but then retrograde through the bundle of Kent; (2) antidromic, meaning the signal travels antegrade through the bundle of Kent and then retrograde through the AV node (Table 8-1).
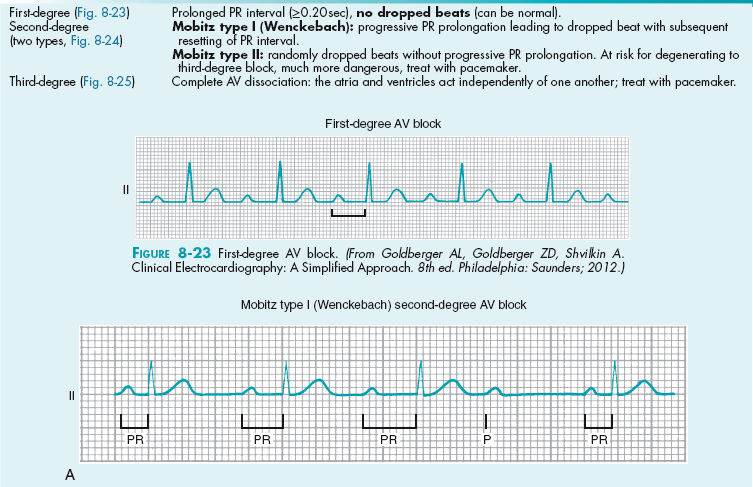


Figure 8-22 Wolff-Parkinson-White (WPW) syndrome. A, ECG demonstrating “delta waves” where there is an upslope before the QRS complex due to premature ventricular depolarization by early transmission of the action potentials via the accessory pathway (bundle of Kent). B, Demonstration of the accessory pathway; recall that normally there is no other method for conducting impulses from the atria to the ventricles other than via the AV node. (A, from Goldberger AL, Goldberger ZD, Shvilkin A. Clinical Electrocardiography: A Simplified Approach. 8th ed. Philadelphia: Saunders; 2012. B, from Colledge NR, Walker BR, Ralston SH. Davidson’s Principles and Practice of Medicine. 21st ed. Philadelphia: Elsevier; 2010.)
Regulation of Blood Pressure
Regulation of blood pressure occurs with two main mechanisms: (1) the baroreceptor reflex and (2) the renin-angiotensin-aldosterone axis.
The baroreceptor reflex uses pressure sensors (baro = pressure) in both the aortic arch and carotid body to monitor blood pressure and modulate parasympathetic and sympathetic tone accordingly. These baroreceptors are constantly sending signals to the brainstem, but the rate of these signals will change with the pressure exerted on their receptors. With an increase in blood pressure, the rate of these parasympathetic signals will increase; with a decrease in blood pressure, the rate will decrease in an effort to normalize the blood pressure disturbance. The carotid sinus baroreceptors send their information to the brainstem using the glossopharyngeal nerve (cranial nerve [CN] IX), whereas the aortic arch baroreceptors use the vagus nerve (CN X) as their afferent nerve. See Figure 8-26 to see how a change in blood pressure leads to a reaction from the baroreceptors.

Figure 8-26 Baroreceptor pathway. The carotid sinus sends its signals via the glossopharyngeal nerve (CN IX), whereas the aortic arch sends its signals via the vagus nerve (CN X) to the brainstem. In both cases, this signal is sent via the nucleus tractus solitarius, which distributes the signal to the body to effect changes in blood pressure. (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
Increase in blood pressure: Causes more stretch on the pressure sensors because the arteries have more pressure to exert on the arterial wall. This causes the baroreceptors to enact changes that will decrease blood pressure. The increased stretch leads to increased firing of the baroreceptors which (1) increase parasympathetic tone through the vagus nerve (CN X) on the SA node to decrease heart rate, and also (2) decrease sympathetic tone.
Decrease in blood pressure: Causes less stretch on the pressure sensors because the arteries have less pressure to exert on the arterial wall. This causes the baroreceptors to enact changes that will increase blood pressure. The decreased stretch causes decreased firing of the baroreceptors; this in turn causes (1) decreased vagal tone to the SA node, resulting in an increased heart rate; as well as (2) increased sympathetic tone to increase heart rate, contractility, and vasoconstriction. This helps mediate the body’s response to acute decreases in blood pressure, such as what occurs during hemorrhage.
Carotid sinus massage puts pressure on the baroreceptors in the carotid body, leading to the body “thinking” there is high blood pressure, causing increased parasympathetic tone (as well as decreased sympathetic tone) and therefore a decrease in heart rate. This was previously proposed as a treatment for supraventricular tachycardia (SVT), but is falling out of favor, owing to risk for inducing embolic stroke by massaging cholesterol-laden carotid arteries.
The renin-angiotensin-aldosterone axis is explained in detail in Chapter 9, but briefly, the juxtaglomerular (JG) cells of the kidney also sense blood pressure. Any decrease in blood flow to the kidney will cause the JG cells to secrete the enzyme renin into the bloodstream. The bloodstream always has angiotensinogen in it, and renin cleaves this angiotensinogen into angiotensin I. Angiotensin I gets cleaved into angiotensin II by angiotensin-converting enzyme (ACE), which an ACE inhibitor blocks. Angiotensin II is a potent vasoconstrictor, increasing SVR and therefore blood pressure, and also mediates aldosterone release from the zona glomerulosa of the adrenal gland. Aldosterone increases sodium reuptake from the kidney, leading to an expansion in blood volume and thus an increase in blood pressure.
PATHOLOGY
Congenital Heart Disease
There are many congenital heart diseases, classified broadly into two categories: (1) cyanotic heart disease and (2) acyanotic heart disease. Cyanotic heart diseases are characterized by right-to-left shunts, where deoxygenated blood is put into the systemic circulation with subsequent early cyanosis; acyanotic lesions have left-to-right shunts, causing oxygenated blood to be circulated back into the lungs and therefore not causing early cyanosis. Definitive treatment for cyanotic lesions is almost always surgical.
It is necessary to remember the five cyanotic heart diseases (Fig. 8-27); a common mnemonic is the five Ts of Truncus arteriosus, Transposition of the great vessels, Tricuspid atresia, Tetralogy of Fallot, and Total anomalous pulmonary venous return (TAPVR). Another way to remember this using the “five-finger method” will be described:

Figure 8-27 Cyanotic congenital heart disease. A, Truncus arteriosus. B, Transposition of the great vessels. C, Tricuspid atresia with hypoplasia of the right ventricle. D, Tetralogy of Fallot. E, Total anomalous pulmonary venous return. (From Marcdante KJ, Kliegman RM, Behrman RE, Jenson HB. Nelson Essentials of Pediatrics. 6th ed. Philadelphia: Elsevier; 2010.)
Two Fingers Up, Fingers Crossed—Transposition of the Great Vessels
Fingers crossed because they are transposed.
The aorta and pulmonary artery are transposed (switched!): the aorta comes off the right ventricle, whereas the pulmonary artery comes off the left ventricle. This causes two completely separate circulations, whereby the right ventricle is pumping deoxygenated blood to the body through the aorta, which returns to the right ventricle; the left ventricle is pumping oxygenated blood to the lungs through the pulmonary artery, which returns to the left ventricle.
For the infant to survive, a shunt that connects the two systems must be present to bridge the two circulations, such as a patent ductus arteriosus or patent foramen ovale (an opening between the right and left atria).
Prostaglandin E (misoprostol) can be given to ensure that the ductus arteriosus remains open.
A commonly tested association of infantile transposition is maternal diabetes!
Three Fingers Up—Tricuspid Atresia
Absence of the tricuspid valve, leading to no connection between the right atrium and right ventricle. Because the right ventricle is not receiving blood to pump, it is hypoplastic (very small, underdeveloped).
An atrial septal defect (ASD) and a ventricular septal defect (VSD) must both be present to maintain blood flow—from the right atrium, the blood must flow through the ASD to the left atrium to the left ventricle and through the VSD to the right ventricle to allow access to the lungs!
Four Fingers Up—Tetralogy of Fallot
Four fingers for tetralogy (tetra prefix means four, for four abnormalities).
Developmental defect is anterosuperior displacement of the infundibular septum leading to four abnormalities that can be remembered with the PROVe mnemonic (PROVe that they have tetralogy of Fallot):
Pulmonary stenosis: Degree of stenosis correlates with prognosis.
Right ventricular hypertrophy: A consequence of the high resistance against blood flow out of the right ventricle from the pulmonary stenosis. Leads to “boot-shaped heart” (cour-en-sabot) on radiograph.
The pulmonary stenosis causes high resistance out of the right ventricle, causing right ventricular hypertrophy and deoxygenated blood to instead move into the left ventricle through the VSD but also directly from the right ventricle to the aorta because the overriding aorta often has connection to both ventricles.
Patients suffer cyanotic spells called “Tet spells”—children learn to improve their cyanosis by squatting, which increases systemic vascular resistance by putting pressure on the femoral arteries, leading to comparatively less resistance in the pulmonary circulation and therefore more blood going into the lungs through the stenotic pulmonary valve.
This is the most common and most commonly tested cyanotic congenital heart disease.
Five Fingers Up—Total Anomalous Pulmonary Venous Return
Five fingers for the five words of TAPVR.
Normally, the pulmonary veins drain into the left atrium; in TAPVR, the pulmonary veins drain to the right atrium. It is called total because all four pulmonary veins anomalously return to the right atrium.
A shunt between the two atria such as an ASD or patent foramen ovale must be present, or oxygenated blood will never reach the left heart.
The acyanotic heart defects include VSD (most common), ASD, and PDA in descending order of frequency; these cause left-to-right shunts, whereby oxygenated blood is put back into the right heart (Fig. 8-28). Although these do not typically cause problems in the neonate, eventually it can lead to Eisenmenger syndrome.

Figure 8-28 A, Ventricular septal defect (VSD). B, Atrial septal defect (ASD). C, Coarctation of the aorta. (A and B, from Marcdante KJ, Kliegman RM, Behrman RE, Jenson HB. Nelson Essentials of Pediatrics. 6th ed. Philadelphia: Elsevier; 2010. C, from Lissauer T, Clayden G. Illustrated Textbook of Paediatrics. 4th ed. Edinburgh: Elsevier; 2011.)
Eisenmenger syndrome: With a left-to-right shunt, the right heart receives additional blood, which is pumped through the pulmonary circulation. This additional flow causes hypertrophy of both the right heart and the pulmonary vasculature. The hypertrophy of the pulmonary vasculature causes increased pulmonary resistance (recall resistance is proportional to 1/r4). Eventually, the pulmonary resistance increases enough that the left-to-right shunt now becomes a right-to-left shunt, leading to deoxygenated blood being shunted into the systemic circulation, with resulting cyanosis.
Lastly, coarctation of the aorta is a narrowing of the aorta most strongly associated with Turner syndrome (45,XO) and bicuspid aortic valves (Fig. 8-28C).
Narrowing causes increased afterload for the left ventricle, resulting in left ventricular hypertrophy. Because the narrowing is after the aortic arch vessels, there is no impediment to perfusing the “upper half” of the body (head and upper limbs).
Long-standing coarctation can lead to dilation of intercostal vessels that act as collateral circulation, meaning that the blood flows in the reverse direction from intercostal vessels to aorta to allow blood to reach the legs; this causes a characteristic “rib notching” appearance on radiograph due to erosion of the ribs.
Classic findings are weak femoral pulses and differential cyanosis. In neonates, the coarctation is “preductal,” meaning the narrowing occurs before the ductus arteriosus; therefore, the lower half of the body can be cyanotic due to deoxygenated blood from the pulmonary artery being shunted through the ductus arteriosus into the aorta. Because the narrowing is after the aortic arch, the upper half of the body is perfused normally with oxygenated blood from the left ventricle: pink top, blue bottom = differential cyanosis.
Atherosclerosis
1. Fatty streaks (Figs. 8-29 and 8-30) are the beginning of atherosclerosis and start with endothelial dysfunction. The endothelium of the arteries normally makes vasodilating substances such as nitric oxide and also prevents fatty deposition. Chemical disruption such as toxins in tobacco smoke or high levels of blood sugar in diabetes cause the endothelium to have lower defenses against lipid deposition. Physical disruption such as high blood pressure and turbulent flow such as at arterial branch points also promote lipid deposition. Low-density lipoprotein (LDL) cholesterol can now slip under the endothelium; high cholesterol exacerbates this simply because there is more LDL cholesterol available in the blood to deposit. Macrophages then eat this LDL cholesterol, becoming foam cells that are too full to leave and are now stuck.
Figure 8-29 Early atherosclerosis, demonstrating damaged, activated endothelium taking in monocytes (which become macrophages), which then phagocytose lipids, becoming foam cells. Atherosclerosis, often referred to as “hardened arteries” by lay people, causes roughly half of all deaths in developed countries by causing cardiovascular disease and stroke. Risk factors include smoking, hypertension, diabetes, and dyslipidemia. The most common site for atherosclerosis is the abdominal aorta, followed by the coronary arteries, popliteal arteries, and carotid arteries. The stages of plaque development include (1) fatty streak formation, and (2) atherosclerotic plaque progression and rupture. (From Colledge NR, Walker BR, Ralston SH. Davidson’s Principles and Practice of Medicine. 21st ed. Philadelphia: Elsevier; 2010.)

Figure 8-30 Advanced atherosclerosis leading to significant stenosis (which in the coronary arteries can lead to stable angina), or complications such as plaque rupture with thrombus formation (which in the coronary arteries can lead to myocardial infarction if the thrombus occludes blood supply, or unstable angina if the thrombus is severe but nonocclusive). (From Colledge NR, Walker BR, Ralston SH. Davidson’s Principles and Practice of Medicine. 21st ed. Philadelphia: Elsevier; 2010.)
2. Plaque progression occurs when the smooth muscle of the media migrates toward the intima, promoted by platelet-derived growth factor (PDGF) secreted by the resident foam cells. These smooth muscle cells produce extracellular matrix and lead to a fibrous plaque, which narrows the affected artery. Those plaques with large amounts of lipid, prolonged inflammation, or a thin fibrous layer are at risk for rupture. When plaques rupture, they can create thrombi, which can have devastating consequences, such as an atherosclerotic coronary artery rupturing and causing a clot to form that completely occludes the artery, leading to myocardial infarction.
The main complications of this atherosclerotic process include aneurysm formation (especially in the abdominal aorta, where atherosclerosis is the main risk factor), ischemia (leading to angina in the coronary arteries, peripheral vascular disease, and claudication in the peripheral vasculature), and clot formation (leading to severe consequences such as myocardial infarction or stroke).
Hypertension
Known to the public simply as high blood pressure, hypertension is a major risk factor for numerous causes of morbidity and mortality, including coronary artery disease, strokes, heart failure, and kidney failure (Table 8-2). Because hypertension is often asymptomatic, it is termed the “silent killer.” About 90% to 95% of hypertension is essential (primary) hypertension, and the remaining 5% to 10% is secondary hypertension.
Table 8-2
| Classification | Systolic (mm Hg) | Diastolic (mm Hg) |
| Normal | < 120 | < 80 |
| Prehypertensive | 120–139 | 80–89 |
| Stage I hypertension | 140–159 | 90–99 |
| Stage II hypertension | ≥ 160 | ≥ 100 |
Essential hypertension comes from a mix of genetic factors, obesity, and dietary factors. Secondary hypertension has discrete causes that are often curable if recognized, so the causes of secondary hypertension are important to keep in mind (note that most of these will be covered in detail in Chapter 9):
Renovascular hypertension: In Chapter 9, the renin-angiotensin-aldosterone (RAA) system is explained in detail. Briefly, whenever the kidney’s JG cells “see” less blood, they will think the blood pressure is low and act to increase it. If the renal arteries are stenotic, which in older adults occurs because of atherosclerosis and in younger women because of fibromuscular dysplasia, the JG cells will “see” less blood and secrete renin to activate the RAA system and increase the blood pressure. The increased angiotensin II causes vasoconstriction, and the increased aldosterone causes sodium retention. Diagnosis is by plasma renin activity; in the face of high blood pressure, renin should be low, but it will be high in renovascular hypertension. Aldosterone levels will be high in both renovascular hypertension and primary aldosteronism, but renin levels will be low in the latter.
Primary aldosteronism (Conn syndrome): Caused by an aldosterone-secreting (mineralocorticoid) tumor in the zona glomerulosa of the adrenal gland. Primary aldosteronism causes increased blood pressure by increased sodium retention by the kidney (see Chapter 9 for a description of other effects). Levels of aldosterone are high, but renin levels are low because the aldosterone is being secreted directly by the tumor.
Pheochromocytoma: A catecholamine-secreting tumor in the medulla of the adrenal gland, this causes hypertension and sporadic episodes of palpitations, diaphoresis, and headache owing to sudden rushes of epinephrine and norepinephrine.
Cushing syndrome: Increased glucocorticoid levels as a result of (1) an exogenous cause, such as prescription steroids; (2) an adrenocorticotropic hormone (ACTH)-secreting tumor in the pituitary gland (Cushing disease); (3) an ectopic ACTH-secreting tumor (e.g., small cell carcinoma of the lung); or (4) a corticosteroid-secreting tumor of the zona fasciculata of the adrenal gland. ACTH stimulates corticosteroid secretion by the adrenal gland. The reason for increased blood pressure in Cushing syndrome is twofold: glucocorticoids such as cortisol in high levels can attach to aldosterone receptors (thus functioning as mineralocorticoids) as well as cause sensitization to epinephrine (which vasoconstricts the peripheral vasculature). Patients often have typical stigmata of Cushing syndrome such as moon facies (a round face), central obesity, hemorrhagic purple-red striae, and hirsutism.
Regardless of etiology, hypertension causes increased afterload on the left ventricle, potentially causing heart failure over time. In addition, direct arterial damage from the high pressures leads to accelerated atherosclerosis, which can promote myocardial infarction, stroke, and organ failure such as renal failure.
Ischemic Heart Disease
Ischemic heart disease occurs when there is an imbalance of myocardial oxygen supply and demand, leading to inadequate oxygenation of the myocardium. This can lead to angina, myocardial infarction, or chronic ischemic heart disease.
Angina is the squeezing or pressure-like sensation in the chest that patients have during myocardial ischemia. There are three types of angina that must be understood.
Stable angina: Occurs during exertion, is relieved by rest, but is not worsening and is therefore stable. Typically secondary to stable nonocclusive atherosclerotic plaques in the coronary arteries (to be symptomatic, severity of stenosis typically at least 70%).
Unstable angina: Angina that is worsening (such as with less exertion) or is occurring at rest. Unstable angina is often due to nonocclusive coronary arterial thrombi, which could potentially become completely occlusive; therefore, this is a medical emergency because it may progress to a myocardial infarction.
Prinzmetal (variant) angina: This angina usually occurs at rest and is not due to atherosclerosis but rather vascular spasm.
Chronic ischemic heart disease: with chronic oxygen deprivation, progressive congestive heart failure (CHF) can be due to replacement of ischemic cardiac myocytes with noncontractile fibrous tissue over time.
Myocardial Infarction
Myocardial infarction (MI) is typically from thrombus formation from a disrupted atherosclerotic plaque in a coronary artery, resulting in ischemia and death of myocardial tissue. Whereas in stable and unstable angina no heart tissue dies from the ischemia, in MI there is coagulation necrosis. These MIs can either be transmural or subendocardial (Fig. 8-31).

Figure 8-31 Difference between transmural infarction and subendocardial infarction. The endocardium is the most oxygen-deprived area of the heart and is at risk for infarction with hypoperfusion. (From Damjanov I. Pathophysiology. Philadelphia: Elsevier; 2008.)
Transmural MI: Necrosis involving the entire thickness of myocardium due to occlusion of a coronary artery leading to ST-segment elevation on ECG. It is transmural because the coronary artery branches are responsible for perfusing all three layers of the heart; blockage causes cessation of blood flow to all these layers.
Subendocardial MI: Necrosis involving only the innermost layer of the heart. The subendocardial area is the farthest from the coronary arteries and therefore is the last to be perfused with the fewest collaterals supplying it; in addition, the contracting myocardium exerts high pressure in this area, reducing blood flow further. This is usually due to hypoperfusion, from either generalized hypotension or a coronary artery that is not completely occluded but rather is narrowed enough to cause death of the most starved myocardial tissue. There is ST-segment depression on ECG.
Diagnosis of MI is by history, ECG, and lab tests. The ECG is very helpful and can show ST-segment changes as well as pathologic Q waves (Q waves > 1⁄3 of the height of the QRS complex; can be a permanent change after an MI). When myocytes die, their cellular contents are released into the bloodstream and aid in the diagnosis of an MI. Troponin I rises within 4 hours and stays elevated for 1 week, whereas CK-MB (creatine kinase) rises essentially as fast as troponin (range of 3 to 8 hours) but normalizes after about 3 days. CK-MB is also less specific than troponin elevation because it may be released from cells other than just cardiac myocytes, such as skeletal myocytes.
There are many complications of MI, from the second it happens to weeks afterward (Fig. 8-32).
Figure 8-32 Selected complications of myocardial infarction. A, Ventricular aneurysm without rupture, leading to stasis of blood in the aneurysm and mural thrombus formation. B, Papillary muscle rupture, septal rupture, and free wall rupture. (From Damjanov I. Pathophysiology. Philadelphia: Elsevier; 2008.)
Arrhythmia: Fatal arrhythmias are a common cause of death before hospitalization (in the hospital, arrhythmias are usually immediately recognized and treated because of cardiac monitoring and access to defibrillation). Damaged cell membranes and sections of dead myocardium all promote abnormal conduction and arrhythmias. Ventricular fibrillation is the most common fatal arrhythmia in acute MI (see Fig. 8-21).
Myocardial dysfunction: With the death of myocardium and possible ongoing ischemia, the ability of the heart to function as a pump is compromised. With large MIs, cardiogenic shock can occur immediately, causing severely decreased cardiac output and hypotension, but delayed heart failure can also occur.
Papillary muscle rupture: With some MIs involving the left ventricle, the papillary muscle can rupture, leading to acute mitral regurgitation because the chordae tendineae no longer function to prevent regurgitation.
Ventricular free wall rupture: Usually occurs when macrophages begin to remove necrotic areas, causing a weakening, typically between days 3 and 7 after an MI. This leads to rapid bleeding into the pericardium, resulting in pericardial tamponade and death.
Ventricular aneurysm: A late complication occurring between 1 and 2 months after the MI; weakening of the ventricular wall leads to fibrosis and dilation without rupture (unlike ventricular free wall rupture, which is a relatively early complication with rupture). This is a late complication because fibrosis takes time, just like scar formation. The fibrotic aneurysmal area cannot contract, leading to congestive heart failure and potential clot formation in the ventricle from stasis of blood (mural thrombus, like the mural on a wall, covered in clot).
Fibrinous pericarditis: Early after an MI (within the first week), the inflammation caused by the necrosis of myocardium can cause increased vessel permeability and exudate formation. This is directly caused by inflammation, unlike Dressler syndrome.
Dressler syndrome (autoimmune fibrinous pericarditis): When myocardium dies, it releases cellular contents that the immune system has not seen before (previously sequestered in cells); the body can see these as foreign and develop an autoimmune response to them. This leads to autoimmune pericarditis that usually occurs 1 to 2 months after the MI.
Congestive Heart Failure
Heart failure is inability of the heart to supply sufficient blood flow to meet the demands of the body, causing specific symptoms and signs depending on which side of the heart is affected. Heart failure can occur from either (or both) of two types of dysfunction: systolic dysfunction and diastolic dysfunction.
Systolic dysfunction: A problem with ejection of blood during systole, characterized by a low ejection fraction (normal, ≥ 55%). There is no problem in filling; the heart is full, but cannot eject the blood that it needs to. This can be because the heart either cannot contract well (poor contractility, secondary to ischemia or infarction, or cardiomyopathy) or is contracting against too great of an afterload (aortic stenosis, severe hypertension).
Diastolic dysfunction: A problem with filling in diastole, typically with a preserved (normal or near-normal) ejection fraction. This is typically due to a stiff ventricle, either from hypertrophy, fibrosis, or restrictive diseases (restrictive cardiomyopathy, pericardial tamponade). In this case, the stiff ventricle cannot relax enough to fill adequately, but the ventricle can eject the little blood it receives without difficulty.
Heart failure can also be divided into left heart failure, right heart failure, and biventricular failure. Left heart failure is often referred to as a disease of symptoms because the symptoms of dyspnea, paroxysmal nocturnal dyspnea (PND), and orthopnea are prominent. Conversely, right heart failure is often referred to as a disease of signs because peripheral edema, jugular venous distention, and hepatomegaly are prominent.
Left heart failure occurs when the left ventricle cannot eject sufficient blood into the aorta, causing the blood to back up into the lungs, producing pulmonary edema. Systolic dysfunction is a common cause of left heart failure because of ischemia and myocardial infarction, although diastolic dysfunction can occur as well. Most of the manifestations of left heart failure are symptoms rather than signs. The higher pressures in the pulmonary system lead to increased afterload for the right ventricle, which can subsequently lead to right heart failure (Fig. 8-33).

Figure 8-33 Left heart failure, demonstrating blood “backing up” into the lungs, causing pulmonary edema, pulmonary hypertension, and cor pulmonale (right heart failure from pulmonary hypertension). (From Damjanov I. Pathophysiology. Philadelphia: Elsevier; 2008.)
Pulmonary edema: Transudation of fluid into alveoli due to increased hydrostatic pressure in the pulmonary capillaries because the left heart can no longer pump that fluid out. This fluid also causes the physical exam finding of bibasilar inspiratory crackles because inspiration forces air into fluid-filled alveoli that are edematous. Often, there may be small hemorrhages, which is why patients with pulmonary edema often cough up a pink frothy liquid. Macrophages eat the blood hemorrhaged into the alveoli but retain the hemosiderin; microscopically, these hemosiderin-laden macrophages are called heart-failure cells.
Orthopnea: Difficulty breathing while lying supine, owing to increased venous return because gravity is no longer impeding blood from returning to the heart. This exacerbates the volume overload because the left ventricle cannot pump the added fluid, leading to worsening pulmonary edema. Often, patients will sleep with multiple pillows, propping themselves upright in an attempt to reduce venous return to the heart.
Paroxysmal nocturnal dyspnea (PND): This also causes difficulty breathing while lying supine, but takes hours to occur, usually after the patient has fallen asleep. Unlike in orthopnea, when blood already in the vasculature returns to the heart, in PND the interstitial edema outside of the vasculature reaccumulates into the vasculature and returns to the heart.
S3/S4 sound: The S3 sound, as previously discussed, occurs during the rapid-filling phase of diastole and is a marker for fluid overload. The S4 sound occurs when the atrial kick attempts to force more blood into a stiff or overfilled ventricle. Both sounds can be present in left heart failure.
Right heart failure (Fig. 8-34) occurs when the right ventricle cannot eject sufficient blood into the pulmonary artery, causing blood to back up into the rest of the body (not the lungs). It is worth noting that the most common cause of right heart failure is left heart failure. Left heart failure causes fluid overload in the lungs, increasing the afterload for the right ventricle, eventually contributing to the failure of the right ventricle. When the right ventricle fails due to pulmonary hypertension in the absence of left ventricular dysfunction, it is termed cor pulmonale; examples include increased pulmonary resistance (afterload) from destruction of pulmonary vasculature from lung diseases such as chronic obstructive pulmonary disease, obstructive sleep apnea, or blood clots blocking branches of the pulmonary artery (pulmonary embolism). Rather than symptoms, most of the manifestations of right heart failure are signs.
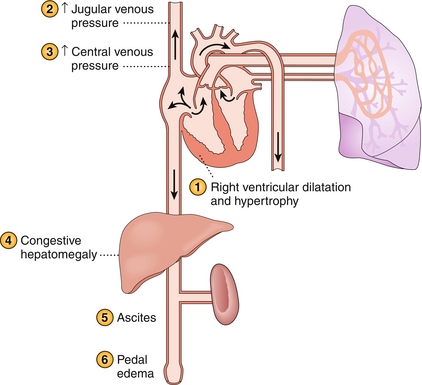
Figure 8-34 Right heart failure, demonstrating blood “backing up” into the body, including the jugular veins (jugular venous distension on physical exam), the liver (congestive hepatomegaly), and the capillary beds of the systemic circulation (dependent edema). (From Damjanov I. Pathophysiology. Philadelphia: Elsevier; 2008.)
Jugular venous distension (JVD): Backup of blood from the right ventricle moves into the right atrium and the veins feeding it. The increased venous hydrostatic pressure can be measured at the internal jugular vein.
Edema: The increased hydrostatic pressure also forces fluid out of the capillary beds into the interstitial tissue in the periphery (similar to how increased pulmonary hydrostatic pressure in left heart failure causes pulmonary edema). Usually, the edema is dependent, meaning it occurs in the areas where gravity further increases hydrostatic pressure (ankles for those sitting or upright, sacrum for those supine).
Hepatomegaly: Because the hepatic veins drain into the vena cava, blood will back up into the liver as well, causing liver congestion. In severe cases, this can cause ascites or even cirrhosis if the pressure in the liver impedes portal vein drainage into the liver. Because the now-congested liver holds much blood, the physical exam maneuver “hepatojugular reflux” can be used to accentuate the JVD in those with heart failure. This hepatomegaly can be painful.
Cardiomyopathies
There are three cardiomyopathies (Fig. 8-35): dilated, hypertrophic, and restrictive (in order of decreasing prevalence). Any of these can cause symptoms of heart failure, described previously.

Figure 8-35 Cardiomyopathies. A, Normal heart. B, Dilated cardiomyopathy. C, Hypertrophic cardiomyopathy. D, Restrictive cardiomyopathy. ASH, asymmetrical septal hypertrophy; SAM, systolic anterior motion (of the mitral valve). (From Colledge NR, Walker BR, Ralston SH. Davidson’s Principles and Practice of Medicine. 21st ed. Philadelphia: Elsevier; 2010.)
Dilated cardiomyopathy is the most common, accounting for 90% of cases. See Figure 8-35B.
Etiology: Most commonly idiopathic, but also due to alcohol and drugs (both illicit drugs such as cocaine and medications such as the chemotherapeutic agent doxorubicin), pregnancy, and after myocarditis (most commonly viral myocarditis from coxsackievirus, but also from Chagas disease from the trypanosome Trypanosoma cruzi).
Findings: Globally dilated heart (eccentric hypertrophy) with poor contractility leading to systolic failure and low ejection fraction. Dilation of the valve ring seats causes valves to no longer line up correctly, leading to mitral and tricuspid regurgitation. Poor ejection leads to fluid overload leading to S3 and S4 heart sounds. Typical symptoms of heart failure are present.
Treatment: Medical therapy is similar to congestive heart failure (see earlier section “Congestive Heart Failure”), but cardiac transplantation may be necessary if refractory to treatment.
Hypertrophic cardiomyopathy (previously called idiopathic hypertrophic subaortic stenosis) is a common cause of syncope and sudden death in young athletes and in those with Friedreich ataxia (Fig. 8-35C).
Etiology: Most commonly this is familial, with autosomal dominant inheritance.
Findings: There is asymmetrical hypertrophy near the ventricular septum (concentric hypertrophy), leading to possible obstruction of flow out of the left ventricle. The narrowed outflow tract leads to a Venturi phenomenon, in which the high velocity of blood being ejected through the narrowed opening causes the anterior leaflet of the mitral valve to be pulled toward the septum during systole (termed systolic anterior motion [SAM] of the mitral valve), closing the outflow tract. The narrowing also causes a systolic crescendo–decrescendo murmur similar to that of aortic stenosis.
The intensity of the murmur of hypertrophic cardiomyopathy is inversely related to the preload. Increased preload causes the ventricle to fill more, increasing the diameter of the outflow tract and therefore decreasing the intensity of the murmur. A poorly filled ventricle will have a much more narrow outflow tract and a louder murmur. The Valsalva maneuver (forcible exhalation against a closed glottis) causes a transient rise in intrathoracic pressure, which prevents venous return, causing decreased preload and a more harsh murmur. Squatting compresses lower extremity veins, increasing venous return and thus preload, decreasing the murmur intensity; standing will cause venous pooling from gravity and decreased preload. This concept is important because the murmur sounds similar to that of aortic stenosis; but in aortic stenosis, the increased preload leads to a louder murmur (more blood being ejected through the stenotic valve), whereas in hypertrophic cardiomyopathy, the increased preload leads to a softer murmur (larger ventricular volumes lead to a larger outflow tract and less obstruction).
Treatment: Ensuring that the ventricular chamber volumes are always relatively high to lessen the obstruction. Therefore, beta blockers or calcium channel blockers are used to ensure that the heart rate does not increase (which would decrease filling time and lead to smaller chamber volumes) and that the contractility does not increase (which would lead to smaller ventricular volumes from increased stroke volumes and worsening obstruction).
Restrictive cardiomyopathy is characterized by rigid, noncompliant ventricles, which may be able to contract normally (normal systolic function and therefore normal ejection fraction) but cannot fill correctly owing to their rigidity (diastolic dysfunction) (Fig. 8-35D).
Etiology: most commonly amyloidosis, a systemic disease in which abnormal proteins are deposited in the body’s organs and can be detected on biopsy with a Congo red stain, which demonstrates apple-green birefringence. In addition, sarcoidosis, hemochromatosis, and postirradiation fibrosis are common causes of this uncommon cardiomyopathy.
Findings: Symptoms of heart failure are present. Fibrosis of the conduction system can lead to arrhythmias.
Treatment: Unfortunately, there is no treatment for restrictive cardiomyopathy except to treat the condition responsible for it. Treatment of volume overload with diuretics can be beneficial for symptomatic relief.
Rheumatic Fever
Rheumatic fever (RF) occurs in about 3% of cases of untreated group A streptococcus (GAS) pharyngitis (“strep throat”); with the advent of antibiotic treatment, RF has become rare in developed countries. (Note: although poststreptococcal glomerulonephritis can occur after either a skin or pharynx infection with GAS and cannot be prevented with antibiotics, only a pharyngeal infection with GAS can cause RF, and this can be prevented with antibiotics.) This is primarily a disease of childhood and adolescence because children more frequently have strep throat. These bacteria have a protein called M protein; when the immune system makes antibodies to the M protein, they can cross-react with proteins on the heart, leading to symptoms (a type II hypersensitivity reaction). The criteria for diagnosis are called the Jones criteria, and the mnemonic JONES can help you remember the main parts of this:
Joints: This is classically described as a migratory arthritis that affects many joints but leaves no permanent damage. Most patients with RF will have this symptom.
Obvious (the heart!): Rheumatic fever affects the heart, so it can damage the heart. This can range from endocarditis leading to valve damage and risk for future infective endocarditis, to myocarditis (inflammation of the cardiac muscle itself, which can be fatal), to pericarditis. RF can thus easily become rheumatic heart disease (with permanent damage to the heart)!
Nodules: Subcutaneous nodules that are painless and firm can appear, typically over bones or tendons.
Erythema marginatum: An erythematous macular rash that spreads out, leaving a clearing in the middle.
Sydenham chorea (St. Vitus dance): Chorea means abnormal movements, and Sydenham chorea affects all limbs and can occur months after initial infection.
Infective Endocarditis
Endocarditis is inflammation of the endocardium, usually involving the heart valves. Although there are noninfectious causes of endocarditis such as Libman-Sacks endocarditis (LSE) from immune complex deposition in autoimmune diseases such as systemic lupus erythematosus (SLE—remember SLE causes LSE!), infectious endocarditis is the most common endocarditis and the most highly tested on the USMLE Step 1.
Infectious endocarditis can be either acute or subacute, which is mostly dependent on the virulence of the organism causing the infection. Although damaged valves are easier for bacteria to latch onto because of irregularities in the surface, normal valves can be affected by virulent bacteria. Any time there is a patient with fever and a heart murmur (valve damaged from the infection), consider endocarditis.
Acute: Usually caused by Staphylococcus aureus, which can infect normal valves and aggressively destroy them. This is characterized by large vegetations, rapid onset, and patients who are very sick. Most commonly this is due to IV drug use because unsterile injection technique provides a passageway for S. aureus to move directly into the veins. Because the veins bring blood back to the right atrium, the tricuspid valve is the first valve the S. aureus encounters and is the most commonly affected valve in acute infectious endocarditis in IV drug users.
Subacute: Usually caused by less virulent bacteria such as viridans group streptococci, which are found as normal oral flora in humans and routinely find their way in the bloodstream during toothbrushing and dentist visits. Because they are low virulence, they require damaged valves to attach and cause progressive and less dramatic symptoms compared with acute endocarditis. The most common valve affected in subacute infectious endocarditis is the mitral valve because it is often damaged by rheumatic fever, followed by the aortic valve.
Other bacteria deserve mention, although they are much less common. Streptococcus bovis endocarditis can be found in patients with colorectal cancer. Staphylococcus epidermidis is implicated in endocarditis involving prosthetic valves because of their ability to generate a biofilm on the prosthesis. There is a group of bacteria that do not grow on standard bacterial cultures (because they need a special environment to survive that is not provided by the culture medium) and therefore cause endocarditis with “negative” cultures, the HACEK group of bacteria (Haemophilus, Aggregatibacter, Cardiobacterium, Eikenella, Kingella).
There are many symptoms and signs of endocarditis, although all may not be present in each patient. A common mnemonic is endocarditis FROM JANE (because Janeway lesions can be present in endocarditis!):
Fever: Most common symptom, due to immune response to infection of valve.
Roth spots (Fig. 8-36A): Due to immune complex deposition in the retina (Roth = retina), causing inflammation and white exudate formation with hemorrhages, visible on funduscopic examination.
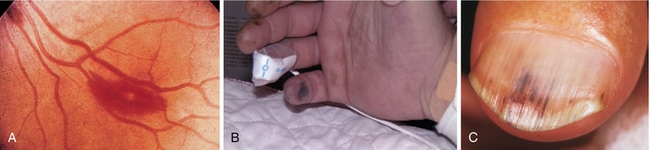
Figure 8-36 Clinical findings in endocarditis. A, Roth spots. B, Osler nodes. C, Splinter hemorrhages. (A, from Douglas G, Nicol F, Robertson C. MacLeod’s Clinical Examination. 11th ed. Philadelphia: Elsevier; 2005. B, from Goldman L, Ausiello D. Cecil’s Textbook of Medicine. 23rd ed. Philadelphia: Elsevier; 2008. C, Swartz, M: Textbook of Physical Diagnosis: History and Examination. 5th ed. Philadelphia: Saunders; 2006:147.)
Osler nodes (Fig. 8-36B): Painful, violaceous nodules on the extremities caused by immune complex deposition (Osler nodes are painful, Ouch!).
Murmur: New heart murmur due to infection and vegetation of the valve.
Janeway lesion: Nontender, small hemorrhagic lesions often found on the extremities, can be differentiated from Osler nodes because Janeway lesions are nontender.
Anemia: Anemia of chronic disease secondary to chronic inflammation.
Nail-bed (splinter) hemorrhages (Fig. 8-36C): Linear reddish brown lesions that look like splinters found under the nail bed.
Emboli: Small pieces of the vegetation on the valve can break off, leading to pulmonary emboli (if the right heart is affected) or strokes and peripheral organ infarction (if the left heart is affected).
Diagnosis is by history and physical exam, echocardiography to visualize the vegetation or valvular abnormality, and blood cultures to isolate the organism. Treatment is antibiotics, usually for at least a month!
Pericardial Disease
Pericarditis is the most common disease of the pericardium, characterized by inflammation that can be caused by a wide variety of agents. It can be either infectious (more common) or noninfectious.
Infectious: Coxsackie B virus (most common), tuberculosis, other bacteria.
Noninfectious: Uremia (secondary to renal failure), autoimmune (SLE, Dressler syndrome), nearby inflammation (post-MI, myocarditis), metastatic cancer.
The diagnosis of pericarditis can be made both by history and physical exam but also by an ECG.
Findings: Classically, patients will have chest pain relieved by sitting forward; on cardiac auscultation, they may have a pericardial friction rub because the inflamed pericardium no longer provides a smooth surface for the heart to beat against.
ECG: Because of diffuse inflammation, the ECG shows diffuse ST-segment elevation and PR-segment depression. The ST-segment elevation can be differentiated from that of a transmural MI because it encompasses the entire heart, whereas a MI affects only sections of the heart supplied by the occluded artery.
Although pericarditis is often self-limited and benign, possible complications include pericardial effusion (with possible cardiac tamponade) and constrictive pericarditis.
Pericardial effusion (Fig. 8-37): The inflammation can lead to fluid accumulation in the pericardial sac. Because the pericardium is fibrous and does not stretch easily, acute accumulation of fluid in the pericardium can be dangerous and lead to cardiac tamponade. Chronic fluid accumulation in the pericardium allows for stretching of the pericardium; therefore, larger volumes can build up without causing tamponade. A hemorrhagic effusion is most likely due to tuberculous or malignant pericarditis. With a large effusion, electrical alternans (Fig. 8-37B) can occur; the heart is essentially swinging in a large effusion and causes the recordings to alternate depending on whether the heart is moving toward or away from the chest wall.
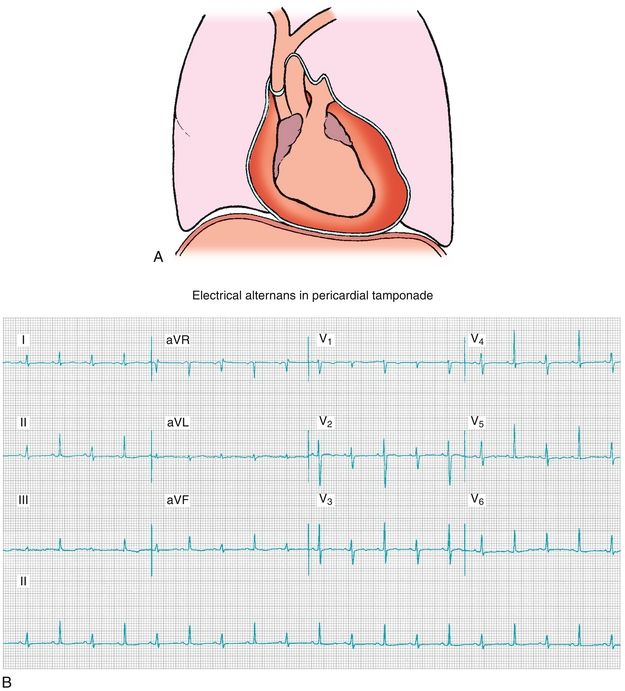
Figure 8-37 Pericardial effusion. A, Image showing significant fluid in the pericardial space. B, Electrical alternans, an ECG finding in which a larger effusion causes the heart to “swing” back and forth in the fluid. (A, from Dandy DJ, Edwards DJ. Essential Orthopaedics and Trauma. 5th ed. Philadelphia: Elsevier; 2009. B, from Goldberger AL, Goldberger ZD, Shvilkin A. Clinical Electrocardiography: A Simplified Approach. 8th ed. Philadelphia: Saunders; 2012.)
Constrictive pericarditis: The inflammation can lead to scarring, where the two pericardial layers adhere to each other and fill with fibrous tissue, preventing filling of the heart.
Cardiac tamponade is a life-threatening emergency. The heart becomes compressed by fluid in the pericardium, preventing filling of the ventricles and subsequently decreased cardiac output. This can be secondary to an effusion or bleeding due to trauma (such as knife through the heart), ventricular wall rupture, or extension of an aortic dissection into the heart (Fig. 8-37A).
Beck’s triad (constellation of three findings in tamponade): hypotension from decreased cardiac output, jugular venous distention from blood backing up into the veins because the heart cannot fill, and muffled heart sounds because the heart is being strangled with fluid.
Pulsus paradoxus can also be present in tamponade and is an exaggeration of a normal physiologic response. During inspiration, the negative intrathoracic pressure increases venous return to the right heart, filling the right ventricle with more volume. This larger filling of the right ventricle causes the septum between the right and left ventricles to bow toward the left ventricle, reducing left ventricular filling and causing a small decrease in blood pressure. In cardiac tamponade, because there is so little room in the heart, this effect is exaggerated, and the blood pressure drops by more than 10 mm Hg during inspiration, called pulsus paradoxus.
Treatment of pericarditis involves treating the underlying cause when possible (such as dialysis in uremic pericarditis) as well as using anti-inflammatory medications to prevent the inflamed pericardium from developing an effusion or scarring. With tamponade, immediate pericardiocentesis (needle drainage of the fluid) is indicated.
Vasculitides
Vasculitis refers to inflammation of the blood vessels, which can be classified as small vessel vasculitis (affecting small vessels such as capillaries), medium vessel vasculitis (affecting smaller muscular arteries), and large vessel vasculitis (affecting the large elastic arteries such as the aorta) (Table 8-3).


Cardiac and Vascular Tumors
Cardiac tumors are uncommon. There are essentially two tumors to remember: (1) myxomas, which are found in adults; and (2) rhabdomyomas, which are found in children.
Myxomas are almost always found in the left atrium and have the potential to act as a “ball valve” where during diastole, when blood flows from left atrium to left ventricle, the tumor moves with it and blocks flow. This flow obstruction can lead to a transient drop in cardiac output and syncopal episodes due to cerebral hypoperfusion. There is a potential for intravascular tumor embolism (small pieces entering the bloodstream), potentially leading to strokes or other vascular occlusive processes.
Rhabdomyomas are associated tuberous sclerosis and are a tumor of the striated muscle itself. It is benign and may regress.
Vascular tumors include the following:
Kaposi sarcoma is a tumor caused by human herpesvirus 8 (HHV-8); the malignant cells arise from the lymphatic endothelium. Although there are types that do not require an immunocompromised state, the most commonly tested association is with AIDS. The lesions are purple-red macules that subsequently enlarge to papules and nodules and can occur on the skin, intraorally, and in the gastrointestinal tract. AIDS patients who have Kaposi sarcoma often see improvement in these lesions after antiretroviral treatment.
Capillary (“strawberry”) hemangiomas classically are facial lesions found in infants; they grow rapidly and then slowly fade. Almost all will regress by 9 years of age. No treatment is necessary unless they cause problems such as blockage of nostrils or visual impairment.
Angiomyolipomas are, as the name implies, tumors that include blood vessels (angio), muscle (myo), and fat (lipo). These are associated with tuberous sclerosis, just as rhabdomyomas (mentioned earlier) are associated with it.
Angiosarcomas are malignant (sarcoma) neoplasms of endothelial cells and are rare; angiosarcoma of the liver is associated with exposure to vinyl chloride, which is used to make polyvinyl chloride (PVC), the most commonly produced plastic. It is also associated with arsenic and thorium dioxide.
PHARMACOLOGY
Vasopressors
Vasopressors (Fig. 8-38) are drugs that work primarily through increasing systemic vascular resistance by clamping down blood vessels; they are essentially “antihypotensive” agents, most of them sympathomimetic agents with α1 activity. Many of these drugs also have direct positive inotropic effects through β1-receptor agonist activity to increase cardiac output to further correct hypotension. Common vasopressors include epinephrine, norepinephrine, isoproterenol, and dopamine. The following section will rely on an understanding of sympathetic adrenergic receptors; please review Chapter 7 if necessary. For the purposes of this section, receptors can be simplified as having α1-receptor activity, causing increased systemic vascular resistance and therefore increasing blood pressure; β1-receptor activity, causing increased cardiac output through increased inotropy (contractility) and chronotropy (rate); and β2-receptor activity, causing vasodilation, decreased systemic vascular resistance, and therefore decreased blood pressure.

Norepinephrine has predominant α1-vasoconstricting effects but also has β1 activity. Therefore, when a patient’s hypotension is due to vasodilation (such as in septic shock), norepinephrine is the drug of choice because it will increase systemic vascular resistance and increase cardiac output, both contributing to increasing the blood pressure. Because the baroreceptors will see a large increase in blood pressure, vagal tone will be increased, potentially leading to reflex bradycardia, especially if the patient is taking a beta blocker, which will block the chronotropic effect of norepinephrine’s β1 stimulation.
Epinephrine is an excellent β1 and β2 agonist (with significant α activity at higher doses). Therefore, it has effects predominantly on increasing cardiac output through β1 activity but can cause vasodilation due to its β2 activity. The vasodilatory effects can actually cause a decrease in blood pressure if the cardiac output increase does not compensate for the vasodilation. The reason for the increased heart rate is twofold: (1) the decreased blood pressure triggers baroreceptor-mediated increases in heart rate, and (2) the direct β1 receptor chronotropic effect. Clinically, epinephrine is used in anaphylaxis and asthma for β2 bronchodilation, but it is also used in cardiac arrest because of the cardiac stimulant effect.
Isoproterenol is a pure β agonist (β1 and β2) with essentially zero α activity. Therefore, it will increase cardiac output but cause vasodilation, potentially dropping blood pressure like epinephrine. However, unlike epinephrine, isoproterenol has no α effect, and therefore the drop in blood pressure with isoproterenol is more profound.
Dopamine is a precursor to norepinephrine and has different effects at different dosages. At low doses, dopamine interacts with dopamine receptors, which cause vasodilation in the kidneys and gut; at medium doses, it has β1 activity, increasing cardiac output; at high doses, it has significant α1 activity and acts as a potent vasoconstrictor.
Phenylephrine is a pure α agonist and therefore causes significant vasoconstriction and increases blood pressure; the baroreceptors will sense this increase in blood pressure and increase vagal tone to the heart, often leading to reflex bradycardia.
Vasodilators
Vasodilators act to decrease total peripheral resistance (when causing smooth muscle relaxation in arterioles) and/or decrease preload (when causing venous relaxation). Widely used vasodilator drugs include angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs), nitrates, hydralazine, and calcium channel blockers.
Angiotensin-Converting Enzyme (ACE) Inhibitors (-Pril Drugs, Lisinopril, Enalapril, Captopril)
Mechanism of action: ACE inhibitors work by blocking angiotensin-converting enzyme, preventing the conversion of angiotensin I to angiotensin II and halting the RAA axis (Fig. 8-39). Angiotensin II (AT II) is a potent vasoconstrictor and also causes aldosterone release from the adrenal gland, leading to increased blood pressure and increased sodium reclamation by the kidney. By blocking AT II formation, the vasoconstriction effect is removed and blood pressure falls. In addition, ACE typically also removes bradykinin, a vasodilator, from the bloodstream; increased bradykinin levels cause vasodilation and a further decrease in blood pressure.

Figure 8-39 The renin-angiotensin-aldosterone system and the drugs that block various parts of this pathway. (From Brenner GM, Stevens GW. Pharmacology. 4th ed. Philadelphia: Elsevier; 2012.)
Clinical uses: ACE inhibitors are typically used in hypertension due to the vasodilation effect; in heart failure because AT II causes maladaptive remodeling in the failed heart, with ACE inhibition therefore slowing the progression of heart failure; and in diabetes because AT II preferentially vasoconstricts the efferent arteriole (the arteriole leaving the glomerulus), and blocking that vasoconstriction decreases glomerular pressure, slowing the progression of kidney damage in diabetes.
Side effects: Increased bradykinin levels cause cough in about 20% of patients. Rarely, life-threatening angioedema can occur.
Contraindications: Bilateral renal artery stenosis is a contraindication because the kidneys are receiving such small amounts of blood that removing the vasoconstricting effect of AT II on the glomerular arterioles will lead to a large drop in glomerular filtration rate (GFR) and precipitate renal failure. Pregnancy is a contraindication because the fetus requires a functioning RAA axis for renal development, and ACE inhibitor use is associated with fetal renal agenesis. Renal insufficiency is a contraindication because ACE inhibitors can mildly decrease GFR; those with renal failure cannot afford that drop and can develop hyperkalemia from decreased renal potassium excretion.
Angiotensin II Receptor Blockers (ARBs) (-Sartan Drugs, Losartan, Valsartan)
Identical to ACE inhibitors except that instead of blocking ACE, they leave the enzyme open and allow AT II to be formed. Instead, ARBs block the AT II receptor directly. This allows bradykinin to be cleared from the bloodstream and therefore does not cause cough as a side effect and is a recommended alternative for patients who develop an ACE inhibitor–associated cough.
Hydralazine
Mechanism of action: Hydralazine increases cyclic guanosine monophosphate (cGMP) levels, which reduces calcium release from the sarcoplasmic reticulum of smooth muscle, ultimately resulting in smooth muscle relaxation. Therefore, hydrAlazine acts as an Arteriolar dilator, reducing blood pressure and afterload by decreasing systemic vascular resistance.
Clinical uses: Used for severe hypertension (usually in hospitals, not as home therapy) and often used in hypertension during pregnancy because it is not teratogenic (the beta blocker labetalol and the α2 agonist methyldopa are other pregnancy-safe antihypertensives).
Side effects: Because it is a direct arteriolar dilator, it will lower blood pressure, triggering the baroreceptors to increase sympathetic flow and decrease vagal tone of the heart, leading to reflex tachycardia. Cerebral vasodilation is implicated in migraines, so headache is also a potential side effect.
Contraindications: Angina or coronary artery disease is a contraindication because the reflex tachycardia can increase myocardial oxygen consumption and lead to worsening disease.
Nitrates (Nitroglycerin, Isosorbide Dinitrate, Isosorbide Mononitrate)
Mechanism of action: Nitrates release nitric oxide (NO), which activates guanylate cyclase in vascular smooth muscle cells and causes elevated cyclic guanosine monophosphate (cGMP) levels (similar to hydralazine, described previously), causing smooth muscle relaxation (vasodilation). However, nitrates have greater effect on veins and therefore cause decreased preload by reducing venous return rather than reducing afterload. This reduced preload causes decreased myocardial oxygen consumption, improving ischemia. At higher doses, nitrates act on arterioles as well and reduce afterload. NO is normally produced by the endothelial cells of the blood vessel, but the action of NO is on the smooth muscle cells.
Clinical uses: Angina is the most common indication.
Side-effects: As with hydralazine, if the dose is sufficient to cause arteriolar dilation, reflex tachycardia and headache can occur. In addition, tolerance to nitrates occurs quickly, and there must be a daily break from the medication or efficacy will diminish.
Nitroprusside
Mechanism of action: Nitroprusside has an iron core with cyanide (CN−) and nitric oxide (NO) groups attached. The NO can detach and cause increased cGMP levels similar to hydralazine and nitrates, in this case causing both arteriolar and venous dilation and therefore decreasing preload and afterload.
Clinical uses: Treatment of malignant hypertension because it acts quickly (the NO group is readily available) and is easily titrated.
Side effects: Cyanide toxicity can occur with prolonged use because the CN− groups can detach as well. As with any drug mentioned that decreases afterload, reflex tachycardia can occur as the baroreceptors sense decreased blood pressure and enact compensatory changes. (See “Toxicology” section of Chapter 7 for discussion of management of cyanide toxicity.)
Contraindications: Relative contraindications include renal insufficiency because the metabolites of cyanide (such as thiocyanate) are renally excreted and can build up as well as hepatic insufficiency because the liver can metabolize free cyanide into thiocyanate.
Calcium Channel Blockers (Verapamil, Nifedipine, Diltiazem)
Mechanism of action: Block L-type calcium channels in cardiac and smooth muscle. Recall that the L-type calcium channels mediate phase 2 of the myocyte action potential, which normally allows calcium influx and calcium-induced calcium release to initiate myocyte contraction. Inhibition of these channels results in decreased contractility (inotropy). Vascular smooth muscle also uses L-type calcium channels, and their blockage leads to muscular relaxation and therefore vasodilation. Each calcium channel blocker has different affinity for either cardiac or vascular smooth muscle:
Cardiac L-type channels: Verapamil > diltiazem > nifedipine (Verapamil likes the Ventricles!)
Vascular L-type channels: nifedipine > diltiazem > verapamil (the exact opposite as the above)
Clinical uses: Because nifedipine has vascular preference, it is used to treat Raynaud disease to prevent the vasoconstriction of the distal digits. It is used in hypertension because it is a vasodilator. Verapamil works on the ventricle and is therefore used to treat arrhythmias and angina—it would be a poor choice to use nifedipine for angina because it acts on the blood vessels, causing vasodilation and reflex tachycardia, making angina worse!
Side effects: Cardiac side effects include AV block from decreased L-type calcium channel activity preventing signal transmission, and cardiac depression. Vascular side effects include peripheral edema, headache, and dizziness.
Sympatholytics
β-receptor Antagonists (Beta Blockers) (-Lol Drugs, β1 Selective: Metoprolol, Atenolol, Carvedilol, Esmolol; β Nonselective: Propranolol, Nadolol)
Mechanism of action: β1-receptor stimulation increases contractility (inotropy), heart rate (chronotropy), and AV conduction, whereas β2-receptor stimulation dilates the bronchial tree and causes peripheral vasodilation. β1 blockade then causes decreased inotropy, heart rate, and AV conduction and is therefore very useful in a variety of conditions. β2 blockade is less clinically useful because preventing bronchial dilation is not helpful, although preventing cerebral vasodilation makes β2 blockade attractive for migraine headache prophylaxis. Therefore, most beta blockers in use are β1 selective.
Clinical uses: Used to treat hypertension; used to treat heart failure because sympathetic activity on the heart also causes maladaptive remodeling of the failed heart, similar to angiotensin II; used to treat coronary artery disease because beta blockade will prevent sympathetic stimulation of the heart, which would increase myocardial oxygen demand.
Side effects: Because beta blockade causes decreased AV conduction, it can precipitate heart block; nonselective beta blockers can cause bronchospasm due to prevention of β2-mediated bronchodilation and can also cause erectile dysfunction due to prevention of β2-mediated vasodilation. β1 blockade may also mask the adrenergic symptoms of hypoglycemia. Overdose can be treated with glucagon, which directly stimulates myocardial cyclic adenosine monophosphate (cAMP) production; β1-receptor activation works through the same pathway.
Contraindications: Asthma is a contraindication to the use of nonselective beta blockers.
α2 Agonists (Clonidine, Methyldopa)
Mechanism of action: The α2 receptor acts as negative feedback for the sympathetic nervous system. Using an α2-receptor agonist causes a net decrease in sympathetic outflow.
Clinical uses: Previously used to treat hypertension (although methyldopa is sometimes still used in the pregnant hypertensive patient); now clonidine is mostly used in treatment of opioid withdrawal because sympathetic surges are responsible for some of the side effects.
Side effects: Without sympathetic stimulation, as expected, side effects include bradycardia, dry mouth (xerostomia), and sedation. Because it downregulates the sympathetic nervous system, abrupt cessation of α2 blockers can cause a sympathetic surge and resulting hypertension.
Diuretics
Diuretics (Fig. 8-40) are explained in detail in Chapter 15 but are covered here briefly.
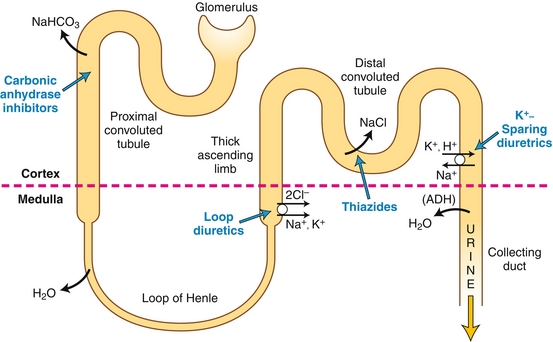
Thiazide Diuretics (-Thiazide, Hydrochlorothiazide, Chlorthalidone)
Mechanism of action: Inhibits the sodium chloride cotransporter in the distal convoluted tubule of the nephron, leading to natriuresis (sodium excretion in the urine) and diminished intravascular volume, and thus to decreased blood pressure. Compared with loop diuretics, the thiazide diuretics are longer lasting but cause less diuresis. Thiazide diuretics also increase calcium resorption due to opening of a voltage-gated calcium channel, leading to decreased calcium excretion in the urine and higher serum levels of calcium.
Clinical uses: Because the thiazide diuretics are longer lasting but less potent, they are useful for the treatment of hypertension because rapid diuresis is not necessary. For those with increased calcium in the urine leading to calcium oxalate kidney stones, hydrochlorothiazide can prevent stone formation by increasing calcium resorption.
Side effects: Hypokalemia and metabolic alkalosis due to increased sodium delivery to the distal nephron as well as increased aldosterone secondary to the body’s attempt at restoring intravascular volume (mechanism explained in Chapter 15), hyperuricemia, and hypertriglyceridemia. As mentioned previously, it increases calcium reclamation in the distal nephron and therefore can lead to hypercalcemia.
Contraindications: Thiazides are sulfa drugs and are contraindicated in sulfa allergy (although this is contentious).
Loop Diuretics (Furosemide [Lasix], Bumetanide, Ethacrynic Acid)
Mechanism of action: Inhibits the NaKCC (Na+, K+, Cl−, Cl−) cotransporter in the thick ascending loop of Henle, leading to significant natriuresis. Loop diuretics can cause rapid, high-volume diuresis; in fact, Lasix is so named because it lasts only 6 hours (lasts-six, Lasix)! (Technically, furosemide only lasts 4 hours, but they didn’t want to change the brand name.)
Clinical uses: Loop diuretics are used for high-volume diuresis, such as in pulmonary edema or decreasing fluid overload in heart failure. Because of its short duration of activity, it is not useful for the treatment of hypertension. Because of its calcium-wasting effect, it can be used to treat hypercalcemia.
Side effects: As with thiazide diuretics, loop diuretics are potassium-wasting diuretics and therefore have the potential to cause hypokalemia and a metabolic alkalosis. Because the resorption of calcium and magnesium requires functional NaKCC cotransporter activity, hypomagnesemia and hypocalcemia can occur. Endolymph uses an NaKCC cotransporter as well; blockage can prevent normal electrolyte balance in the ear, causing ototoxicity.
Contraindications: Loop diuretics (except for ethacrynic acid) are sulfa drugs and are contraindicated in sulfa allergy (although this is contentious).
Potassium-Sparing Diuretics (Amiloride, Spironolactone, Triamterene)
Mechanism of action: Unlike the loop and thiazide diuretics, potassium-sparing diuretics work in mechanisms that prevent potassium loss in the urine. Amiloride blocks the sodium channel in the principal cell in the collecting duct, causing natriuresis. Normally, this sodium channel reclaims sodium at the expense of potassium, and increased sodium delivery here by other diuretics leads to potassium loss. Spironolactone causes blockage of the aldosterone receptor, preventing upregulation of the activity of the principal cell and leading to decreased sodium reclamation.
Clinical uses: Amiloride is used in the treatment of heart failure when hypokalemia is a concern, such as concomitant loop diuretic use. Spironolactone is also used in severe heart failure as well as in aldosterone-secreting tumors before surgical correction.
Side effects: Hyperkalemia is possible with potassium-sparing diuretics owing to decreased ability to excrete potassium. Spironolactone also blocks the testosterone receptor, causing the side effects of androgen antagonism such as gynecomastia (eplerenone is an aldosterone receptor antagonist that does not have any antiandrogen effects).
Antiarrhythmics
Various arrhythmias can occur in the heart, most of them causing a fast heart rate (tachyarrhythmia); arrhythmias treatable with antiarrhythmic medications include atrial fibrillation and flutter, ventricular tachycardia, and paroxysmal supraventricular tachycardia (PSVT) (Table 8-4). These medications are grouped into four classes (I to IV) based on their mechanism of action; class I antiarrhythmics are further classified into Ia, Ib, and Ic.
Table 8-4
Summary Table of Antiarrhythmic Medications
| Class | Mechanism of action | Medications, side-effects, contraindications |
| Ia | Blocks fast Na+ channels (phase 0) Slows conduction velocity Prolongs action potential duration |
All: risk for torsades de pointes from prolonging refractory period Procainamide: lupus-like syndrome (rash/arthralgia/fever) Quinidine: “cinchonism”—headaches, tinnitus, altered mental status |
| Ib | Blocks fast Na+ channels (phase 0) Slows conduction velocity Binds to ischemic/diseased areas Shortens action potential duration |
All: local anesthetic toxicity (CNS symptoms: confusion, seizures) |
| Ic | Blocks fast Na+ channels (phase 0) Slows conduction velocity Does not change action potential duration |
All: contraindicated in diseased hearts owing to proarrhythmic effect |
| II | β-Adrenergic receptor antagonist Decreases sympathetic tone in heart Decreases heart rate Decreases AV nodal conduction |
All: beta blocker side effects (heart block from too much AV node depression, impotence, bradycardia, bronchospasm, sedation) |
| III | Blocks K+ channels (phase 3) Prolongs action potential Prolongs refractory period *Amiodarone has class I, II, III, IV effects |
Amiodarone: check the PFTs (pulmonary fibrosis), TFTs (iodine in drug causes thyroid abnormalities), and LFTs (hepatotoxicity). Photosensitivity and blue/gray skin and corneal deposits. |
| IV | Calcium channel blocker Decreases heart rate Decreases AV nodal conduction |
All: edema, heart block, constipation, flushing |
Class I (Fig. 8-41): Are all fast sodium channel blockers that affect phase 0 (depolarization) of the myocyte action potential.

Figure 8-41 Effects of class I antiarrhythmics on the action potential. (From Pazdernik T, Kerecsen L. Rapid Review Pharmacology. 3rd ed. Philadelphia: Elsevier; 2010.)
Class Ia: Disopyramide, Quinidine, Procainamide—Double Quarter Pounder
Class Ib: Lidocaine, Mexiletine, Tocainide—Lettuce Mayo Tomato
Class Ic: Encainide, Moracizine, Flecainide, Propafenone—Eat More Fries Please
 There are two Ms in these mnemonics: Moracizine and More share the first two letters to differentiate.
There are two Ms in these mnemonics: Moracizine and More share the first two letters to differentiate. Class II: Are all beta blockers.
Class III: Are all potassium channel blockers that affect phase 3 (repolarization) of the myocyte action potential.
Class I antiarrhythmics are local anesthetics—blocking the Na+ channels in nerves slows action potentials (APs) in pain fibers, blocking pain; blocking Na+ channels in cardiac myocytes slows cardiac conduction, blocking some arrhythmias. Therefore, all class I medications will slow the initial phase 0 depolarization and slow conduction velocity in the conducting system but are further divided based on whether or not they Prolong (class Ia), Shorten (Ib), or Do not change (Ic) the overall action potential duration (and I’ll be PiSseD if you forget which is which)!
Class Ia: prolongs AP duration and therefore also prolongs the refractory period (QT interval on ECG), prolonging the time when cells cannot depolarize again. Therefore, this can stop arrhythmias that are due to cells depolarizing too frequently such as in atrial flutter, atrial fibrillation, PSVT, and ventricular tachycardia. However, because torsades de pointes can begin when a depolarization occurs during the repolarization period, there is an increased risk for torsades de pointes.
Class Ib: shortens AP duration but preferentially binds to ischemic or abnormal cells and helps stabilize ventricular arrhythmias (ventricular tachycardia) in situations in which the heart is diseased or ischemic (e.g., post-MI). Because the refractory period is not prolonged, torsades de pointes is not a concern.
Class Ic: does not change AP duration but binds to Na+ channels most avidly out of all class I medications, especially binding Na+ channels in the AV node, and is therefore useful for treatment of supraventricular tachycardia. However, class Ic medications are contraindicated in those with diseased hearts because it has been shown to have a proarrhythmic effect in these patients.
Class II antiarrhythmics are all beta blockers (β-adrenergic antagonists), which help stop sympathetic activity on the heart. Recall that the amount of funny sodium channels open (phase 4 of pacemaker cells) depends on sympathetic tone: beta blockade decreases heart rate and decreases AV nodal conduction velocity (increasing PR interval on ECG). They are useful for treating PSVT, atrial fibrillation, and atrial flutter by decreasing the ability to transmit the signal to the ventricles through the AV node.
Class III antiarrhythmics (Fig. 8-42) are potassium channel blockers; K+ channels are responsible for repolarization of the cells (phase 3), with prolongation causing prolonged action potential duration and prolonged refractory period (QT interval). Again, prolonged QT interval can predispose to torsades de pointes. One of the medications in this class, amiodarone, actually has class I, II, III, and IV activity and is therefore helpful for many different tachyarrhythmias—but with many side effects.
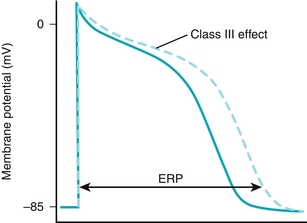
Figure 8-42 Effects of class III antiarrhythmics on the action potential. (From Pazdernik T, Kerecsen L. Rapid Review Pharmacology. 3rd ed. Philadelphia: Elsevier; 2010.)
Class IV antiarrhythmics (Fig. 8-43) are L-type calcium channel blockers that, similar to beta blockers, decrease conduction velocity and heart rate. Also similar to beta blockers, they are used in the treatment of PSVT. The blockade of calcium decreases rapidity of SA node depolarization, which depends on calcium channels to fully depolarize.
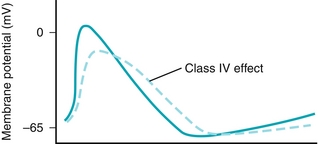
Adenosine
Adenosine is a medication that essentially stuns the AV node causing a complete (third-degree) heart block for a very short duration (half-life of adenosine is seconds). In any arrhythmia that uses the AV node as a focus for reentry, such as AV nodal reentrant tachycardia (AVNRT), the most common supraventricular tachycardia, stunning the AV node causes the reentry circuit to cease and stops the arrhythmia. The underlying conduction abnormality will still be present, however, and therefore the arrhythmia can always return.
Mechanism of action: adenosine is an endogenous purine nucleoside that acts on adenosine receptors that are coupled to inhibitory G-protein-coupled receptors (Gi), inhibiting cAMP formation and subsequently hyperpolarizing the cell by promoting K+ efflux from the cell. This hyperpolarization prevents action potentials for a brief period of time and effectively stuns the AV node. The hyperpolarization of arterioles of the heart causes transient coronary artery vasodilation as well. Because caffeine binds to the adenosine receptor as well, those who consume caffeine may require higher doses.
Clinical uses: Termination of some types of supraventricular tachycardia.
Digoxin (cardiac glycosides)
Digoxin is a medication that causes increased inotropy by increasing the amount of calcium available to the myocytes for contraction.
Mechanism of action (Fig. 8-44): During myocyte contraction, there are high levels of Na+ (from depolarization) and Ca2 + (for contraction). The Na+ is normally removed from the cell by the Na+/K+-ATPase; some of this can reenter the cell through the Na+/Ca2 + exchanger and help drive Ca2 + out of the cell. Digoxin blocks the Na+/K+-ATPase by binding to the K+ site, indirectly shutting down the Na+/Ca2 + exchanger by destroying the sodium gradient that it needs to function. The Ca2 + is put back into the sarcoplasmic reticulum instead of being removed from the cell, and subsequently more calcium will be available for contraction (increased inotropy).

Figure 8-44 Mechanism of action of digoxin. (1) Digoxin binds to the K+ site on the Na+/K+-ATPase, blocking sodium efflux from the cell; (2) increased intracellular sodium concentration occurs; (3) this decreases the activity of the Ca2 +/Na+ exchanger because the sodium gradient is no longer favorable; (4) increased intracellular calcium occurs, which is reclaimed into the sarcoplasmic reticulum; (5) increased calcium release from the sarcoplasmic reticulum leads to increased actin–myosin interaction and increased contractility. (From Costanzo LS. Physiology. 4th ed. New York: Elsevier; 2009.)
Clinical uses: Increases inotropy in congestive heart failure (decreases symptoms, but does not improve mortality). Also used for atrial fibrillation to slow the ventricular rate. The reason that it is effective in atrial fibrillation is because the increased inotropy from the medication results in less sympathetic tone because the contractility is already high. Less sympathetic tone leads to slower AV conduction and therefore slows the ventricular rate in atrial fibrillation.
Side effects: Digoxin toxicity is common, owing to its low therapeutic index. Systemic symptoms include cholinergic symptoms such as diarrhea, nausea, and vomiting; yellow vision is also possible. Cardiac symptoms include arrhythmias and hyperkalemia. Digitalis toxicity is treated with anti-Dig antibody fragments (Fab) and correcting potassium (if it is abnormal).
Contraindications: Because digoxin binds to the K+ site on the Na+/K+-ATPase, hypokalemia can result in digitalis toxicity because the K+ is no longer there to compete for the binding site. Digitalis is renally excreted, and therefore renal insufficiency is a relative contraindication.